

umwelt-online: 85/337/EWG Umweltverträglichkeitsprüfungs-Richtline (2/2)
| |
zurück | |
4.3. Bewertung der Sicherheit für den Verbraucher
4.3.1. Vorschlag für die tolerierbare tägliche Aufnahme (ADI) des Zusatzstoffs
Wenn dies angezeigt ist, sollte eine ADI vorgeschlagen werden.
Die ADI (ausgedrückt als mg des Zusatzstoffs oder der von ihm stammenden Stoffe pro Person und Tag) wird hergeleitet, indem man den NOEL (No-Observed-Effect-Level) durch einen geeigneten Sicherheitsfaktor teilt und mit einem mittleren menschlichen Körpergewicht (KG) von 60 kg multipliziert. Dieser als mg/kg KG pro Tag ausgedrückte NOEL kann anhand toxikologischer oder pharmakologischer Ergebnisse gewählt werden. In einigen Fällen ist möglicherweise eine ADI relevanter, die auf den mikrobiologischen Eigenschaften des Zusatzstoffes beruht. Die Entscheidung hängt davon ab, welche Eigenschaft unter dem Gesichtspunkt der gesundheitlichen Gefährdung des Verbrauchers eher relevant ist.
Bei der Wahl des Sicherheitsfaktors für die Festlegung der ADI eines bestimmten Zusatzstoffs sind folgende Punkte zu beachten:
Es ist üblich, bei der ADI-Berechnung einen Sicherheitsfaktor von mindestens 100 anzuwenden (einen Faktor von 10 zur Berücksichtigung potenzieller Interspezies-Schwankungen und einen weiteren Faktor von 10 zur Berücksichtigung möglicher individueller Unterschiede bei der Reaktion des Menschen). Liegen Humandaten zum Wirkstoff vor, so kann unter Umständen auch ein niedrigerer Sicherheitsfaktor akzeptiert werden.
4.3.2. Vorschlag für Höchstmengen für Rückstände (MRL) des Zusatzstoffs
Bei der MRL-Berechnung geht man davon aus, dass die Aufnahme von essbaren Geweben, Milch- und Eiprodukten die einzige Quelle einer potenziellen Exposition des Menschen ist. Ist dies nicht der Fall, so sind die übrigen Quellen zu berücksichtigen.
Einige dieser Stoffe werden bereits als Futtermittelzusatzstoffe und zu anderen Zwecken verwendet. In diesen Fällen ist zu erwarten, dass die gleichen MRL berechnet werden. Es kann auch der Fall eintreten, dass aufgrund streng wissenschaftlicher Überlegungen für die diversen Verwendungszwecke unterschiedliche MRL berechnet werden, und zwar dann, wenn Aufnahmeweg, Menge, Anwendungshäufigkeit und - dauer sich so von denjenigen bei der Verwendung als Futtermittelzusatzstoff unterscheiden, dass es Hinweise darauf gibt, dass die Kinetik und/oder der Metabolismus unterschiedliche Rückstandsprofile zur Folge hat. Es wird davon ausgegangen, dass in solchen Fällen die niedrigste MRL Anwendung findet.
Zur Festlegung einer MRL muss der chemische Charakter des von dem Zusatzstoff stammenden Stoffes, der zur Bestimmung der Rückstandsmengen im Gewebe verwendet werden soll, definiert werden. Dieser wird als Markerrückstand bezeichnet. Bei diesem Rückstandsbestandteil braucht es sich nicht zwangsläufig um den toxikologisch relevanten Rückstand zu handeln, er muss vielmehr als ein Indikator gewählt werden, der geeignet ist, den signifikanten Gesamtrückstand zu repräsentieren. Das Verhältnis zwischen Markerrückstand und Gesamtrückständen in Zusammenhang mit der ADI (d. h. Verhältnis Markerrückstand / gesamte radioaktive Rückstände, Markerrückstand / sämtliche biologisch aktiven Rückstände) ist für alle in den Depletionsstudien betrachteten Zeitpunkte zu bestimmen. Insbesondere sollte dieses Verhältnis für den Zeitpunkt bekannt sein, der zur MRL-Bestimmung gewählt wird. Ferner muss für diesen Markerrückstand ein geeignetes Analyseverfahren zur Verfügung stehen, damit die Einhaltung der MRL überprüft werden kann.
Bei der Festlegung von MRL (ausgedrückt als g/kg Markerrückstand pro kg essbares Feuchtgewebe oder -produkt) unter Zugrundelegung einer ADI sind folgende Werte für den täglichen Lebensmittelverzehr des Menschen anzuwenden:
| Säuger | Vögel | Fische | |
| Muskel | 300 g | 300 g | 300 g * |
| Leber | 100 g | 100 g | |
| Niere | 50 g | 10 g | |
| Fett | 50 g ** | 90 g *** | |
| + Milch | 1.500 g | ||
| + Ei | 100 g | ||
| *) Muskel und Haut im natürlichen Verhältnis. **) Beim Schwein: 50 g Fett und Haut im natürlichen Verhältnis. ***) Fett und Haut im natürlichen Verhältnis. |
|||
Die einzelnen MRL für verschiedene Gewebe sollen die Depletionskinetik der Rückstände in den betreffenden Geweben bei der für die Verwendung vorgesehenen Tierart widerspiegeln. Erforderlich ist ein Analyseverfahren, dessen Quantifizierungsgrenze unter der MRL liegt (siehe Kapitel II Ziffer 2.5.3).
Besteht die Möglichkeit, dass ein Stoff Rückstände in Geweben und Erzeugnissen bildet, so ist die MRL dergestalt vorzuschlagen, dass die Gesamtmenge des täglich aufgenommenen toxikologisch (oder mikrobiologisch) signifikanten Rückstands13 unter der ADI liegen sollte (siehe obige Tabelle).
Die MRL sollte erst nach Betrachtung und Einbeziehung sämtlicher sonstiger potenzieller Quellen einer Exposition des Verbrauchers gegenüber Rückstandsbestandteilen festgesetzt werden.
Bei bestimmten Zusatzstoffen könnten in Milch, Eiern oder Fleisch Rückstände auftreten, die unterhalb der MRL liegen, aber mit der für bestimmte Verfahren der Lebensmittelherstellung erforderlichen Lebensmittelqualität unvereinbar sind, so z.B. bei der für die Käseherstellung verwendeten Milch. Es kann angezeigt sein, für solche Zusatzstoffe zusätzlich zur MRL einen "maximalen mit der Lebensmittelherstellung vereinbaren Rückstand" in Erwägung zu ziehen.
Unter bestimmten Umständen ist eine MRL nicht erforderlich, beispielsweise
4.3.3. Vorschlag für die Wartezeit
Die Wartezeit wird nach Maßgabe der MRL festgesetzt. Hierbei handelt es sich um den Zeitraum nach dem Absetzen der vorgeschlagenen Formulierung des Zusatzstoffs, welcher erforderlich ist, damit die Rückstandsmengen bis unter die MRL sinken können (95 % Konfidenzintervall).
Zur Bestimmung einer Wartezeit kann ein bestimmtes essbares Gewebe als Surrogat für andere verwendet werden; es wird häufig als Zielgewebe bezeichnet.
4.4. Bewertung der Sicherheit der Arbeitnehmer
Eine Exposition von Arbeitnehmern kann hauptsächlich inhalativ oder topisch bei der Herstellung, Handhabung oder Anwendung des Zusatzstoffs erfolgen; so sind landwirtschaftliche Arbeitskräfte beispielsweise bei der Handhabung oder Mischung des Zusatzstoffs exponiert. Es sollten zusätzliche Informationen über die Art und Weise der Handhabung geliefert werden. Eine Bewertung des für die Arbeitnehmer bestehenden Risikos ist beizufügen.
Erfahrungen im Herstellungsbetrieb sind häufig eine wichtige Informationsquelle für die Beurteilung des für die Arbeitnehmer bestehenden Risikos aufgrund der inhalativen oder topischen Exposition gegenüber dem Zusatzstoff. Besondere Beachtung verdienen Zusatzstoffe bzw. mit Zusatzstoffen behandelte Futtermittel und/oder tierische Ausscheidungen, die in getrockneter Pulverform vorliegen bzw. eine solche Form annehmen können, sowie Zusatzstoffe mit möglichem allergenem Potenzial.
4.4.1. Toxikologische Risikobewertung im Hinblick auf die Sicherheit der Arbeitnehmer
4.4.1.1. Wirkungen auf das Atmungssystem
Es ist nachzuweisen, dass flugfähige Staubmengen keine Gesundheitsgefährdung für die Arbeitnehmer darstellen. Erforderlichenfalls umfasst dieser Nachweis Inhalationsstudien an Labortieren, veröffentlichte epidemiologische Daten und/oder die im eigenen Betrieb des Antragstellers gewonnenen Daten und/oder Tests auf Reizung und Sensibilisierung des Atmungssystems.
4.4.1.2. Wirkungen auf Augen und Haut
Sofern verfügbar, sind direkte Nachweise darüber vorzulegen, dass in bekannten Situationen beim Menschen keine Reizung und/oder Sensibilisierung hervorgerufen wird. In Ergänzung hierzu sind Ergebnisse validierter Tierversuche auf Haut- und Augenreizung sowie auf Sensibilisierungspotenzial einzureichen.
4.4.1.3. Systemische Toxizität
Die zur Erfüllung der Sicherheitsanforderungen gewonnenen Toxizitätsdaten (auch zur Toxizität bei wiederholter Verabreichung, Mutagenität, Kanzerogenität und zu Reproduktionsuntersuchungen) sind heranzuziehen, um andere Aspekte der Arbeitnehmersicherheit zu bewerten. Dabei ist daran zu erinnern, dass die Kontamination der Haut und/oder das Einatmen des Zusatzstoffs die häufigsten Expositionswege sind.
4.4.2. Expositionsbewertung
Es sind Informationen darüber vorzulegen, auf welche Weise die Anwendung des Zusatzstoffs zur Exposition führen kann - durch Einatmen, über die Haut oder durch orale Aufnahme. Diese Angaben schließen eine quantitative Bewertung ein, sofern eine solche vorliegt, beispielsweise zu der typischen Konzentration in der Luft, zur Hautkontamination oder zur oralen Aufnahme. Liegen keine quantitativen Angaben vor, so sind ausreichende Informationen zu liefern, damit eine angemessene Expositionsbewertung erfolgen kann.
4.4.3. Maßnahmen zur Expositionsbegrenzung
Anhand der Informationen, die die Toxizitäts- und Expositionsbewertung geliefert hat, ist eine Schlussfolgerung bezüglich der gesundheitlichen Risiken (systemische Toxizität, Reizung oder Sensibilisierung) zu ziehen, die bei Anwendung von Maßnahmen zur Expositionsbegrenzung, die unter den gegebenen Umständen vernünftig sind, für die Anwender bestehen. Ist das Risiko nicht akzeptabel, so sind Schutzmaßnahmen zur Eindämmung oder Beseitigung der Exposition zu treffen. Eine Neuformulierung des Produkts oder eine Änderung der Verfahren zur Herstellung, Anwendung und/oder Entsorgung des Zusatzstoffs sind die bevorzugten Lösungen. Der Einsatz persönlicher Schutzausrüstungen sollte nur als letzter Ausweg in Betracht gezogen werden, und zwar zum Schutz vor etwaigen Restrisiken, die nach Einführung von Schutzmaßnahmen verbleiben.
4.5. Bewertung der Umweltrisiken
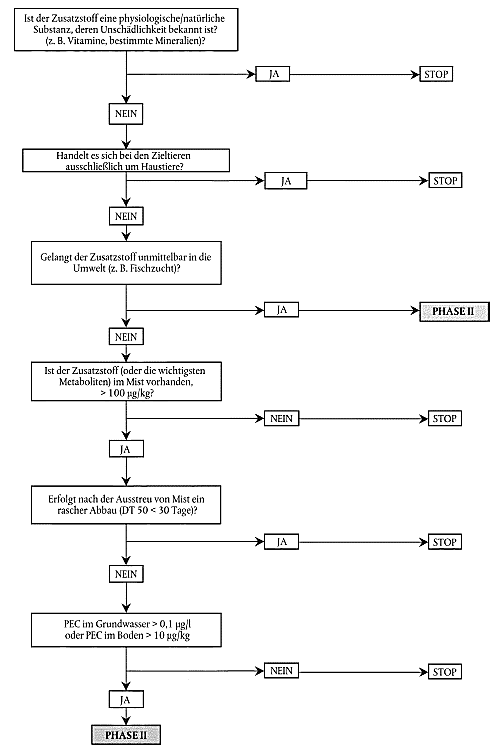
Es ist wichtig, die Auswirkungen von Futtermittelzusatzstoffen auf die Umwelt zu betrachten, da diese typischerweise über einen langen Zeitraum hinweg verabreicht werden (sogar lebenslang), da große Gruppen von Tieren betroffen sein können und da zahlreiche Zusatzstoffe schlecht resorbiert und somit nahezu unverändert ausgeschieden werden. Dennoch besteht in bestimmten Fällen nur ein geringerer Bedarf an einer Bewertung der Umweltverträglichkeit. Es ist nicht angezeigt, im Rahmen dieser allgemeinen Leitlinien strenge Regeln aufzustellen. Bei der Bestimmung der Auswirkungen von Futtermittelzusatzstoffen auf die Umwelt sollte schrittweise vorgegangen werden (siehe Entscheidungsbaum), wobei in der ersten Phase diejenigen Zusatzstoffe, die keiner weiteren Prüfung bedürfen, eindeutig identifizierbar sind. Für die übrigen Zusatzstoffe ist eine zweite Phase von Studien erforderlich (Phase II A) zwecks Gewinnung zusätzlicher Informationen, auf deren Grundlage sich weitere Studien (Phase II B) als notwendig erweisen können. Gegebenenfalls sind die Studien gemäß der Richtlinie 67/548/EWG durchzuführen.
4.5.1. Phase-I-Bewertung
Zweck der Phase-I-Bewertung ist es, zu beurteilen, ob eine signifikante Auswirkung des Zusatzstoffs oder seiner Metaboliten wahrscheinlich ist oder nicht, wobei man sich im Wesentlichen auf Daten stützt, die bereits für andere Zwecke erhoben wurden.
Auf Phase II kann in zwei Fällen verzichtet werden:
Die im schlimmsten Fall zu erwartende Umweltkonzentration im Boden entsteht wahrscheinlich dadurch, dass der während der maximalen Ausscheidung der Hauptrückstandsbestandteile (des Zusatzstoffs oder seiner wichtigsten Metaboliten) anfallende Mist auf dem Acker ausgestreut wird. Die PEC ist für jeden Hauptbestandteil des Rückstands im Dung und für jedes relevante Medium zu bewerten. Für den Boden kann dann auf weitere Untersuchungen verzichtet werden, wenn die PEC für die summierten wichtigsten Rückstandsbestandteile im Mist nicht größer ist als 100 µg/kg oder wenn die wichtigsten Rückstandsbestandteile im Mist problemlos zu natürlichen Stoffen oder zu Konzentrationen unter 100 µg/kg abgebaut werden (Abbauzeit DT 50 < 30 Tage) (sofern die entsprechenden Daten vorliegen) oder wenn die PEC im Boden (in 5 cm Tiefe) weniger als 10 µg/kg beträgt.
Die im schlimmsten Fall zu erwartende Umweltkonzentration in Wasser kann dadurch entstehen, dass verschüttete Futtermittel oder Exkremente, die den Zusatzstoff und seine Metaboliten enthalten, in Gewässer gelangen oder dass in den Exkrementen oder im Boden enthaltene Stoffe in das Grundwasser sickern. Wird die PEC für die Kontamination von Gewässern zuverlässig auf unter 0,1 µg pro Liter geschätzt, so ist eine Phase-II-A-Bewertung der Umweltauswirkungen des Zusatzstoffs auf Wasser nicht erforderlich.
Kann der Antragsteller nicht nachweisen, dass der vorgeschlagene Zusatzstoff in eine dieser Ausnahmekategorien fällt, oder wird der Zusatzstoff unmittelbar in die Umwelt freigesetzt (z.B. Aquakultur), so ist in der Regel eine Phase-II-Bewertung erforderlich.
Umweltrisiko durch Futtermittel-Zusatzstoffe
Entscheidungsbaum für Phase I
4.5.2. Phase-II-Bewertung
Phase-II-Bewertung in zwei Abschnitten - Phase II a und Phase II B
Das Bioakkumulationspotenzial des Zusatzstoffs und/oder seiner wichtigsten Metaboliten sowie der Einfluss dieses Potenzials auf den vorausgesagten Sicherheitsabstand sind zu bewerten. Es wird von einer potenziell nicht signifikanten Bioakkumulation ausgegangen, wenn z.B. Kow (Verteilungskoeffizient) < 3 ist. Kann ein entsprechender Sicherheitsabstand nicht festgelegt werden, so sind geeignete Phase-II-BStudien erforderlich.
4.5.2.1. Phase II A
Zweck der Phase-II-A-Bewertung ist die Ermittlung des Umweltrisikos durch
Zur Bestimmung des Risikos wird folgendes sequenzielle Verfahren empfohlen:
Für die Zwecke der Risikobewertung in Phase II a ist für jedes Umweltmedium der höchste auf diese Weise errechnete PEC-Wert zu verwenden.
Ist im Steady state mit einer hohen Persistenz im Boden (DT90 > 1 Jahr) bei Konzentrationen über 10 g/kg Boden zu rechnen, so kann sich eine Phase-II-B-Bewertung als notwendig erweisen.
4.5.2.2. Phase II B (eingehendere toxikologische Studien)
Bleiben bei einem Zusatzstoff nach der Phase-II-A-Bewertung Zweifel hinsichtlich seiner Umweltauswirkungen bestehen, so sind eingehendere Untersuchungen der Effekte auf die biologischen Spezies in dem Umweltmedium erforderlich, in dem die Phase-II-A-Studien mögliche Probleme erkennen ließen. In diesem Fall müssen weitere Tests durch-geführt werden, um die chronischen und spezifischeren Wirkungen auf entsprechende Tier-, Pflanzen und Mikrobenarten zu bestimmen. Möglicherweise wurde bei der Phase-II-A-Bewertung der PEC-Wert zu hoch angesetzt. Um dies zu beweisen, können Messungen der Umweltkonzentrationen und der Persistenz des Zusatzstoffs und/oder seiner wichtigsten Metaboliten unter Feldbedingungen erforderlich sein.
Geeignete zusätzliche Ökotoxizitätstests sind in einer Reihe von Veröffentlichungen beschrieben, unter anderem in den OECD-Leitlinien. Es kann sich als notwendig erweisen, drei Kategorien von Umweltspezies zu betrachten: Tiere, Pflanzen und Mikroorganismen. Die Tests sind sorgfältig auszuwählen, um sicherzustellen, dass sie sich für die Bedingungen eignen, unter denen der Zusatzstoff und/oder seine Metaboliten in die Umwelt freigesetzt bzw. dort verbreitet werden.
Die Bewertung der Auswirkungen auf den Boden kann Folgendes umfassen:
Die Bewertung der Auswirkungen auf Gewässer kann Folgendes umfassen:
5. Kapitel V: Muster einer Monographie
5.1. Identität des Zusatzstoffs
5.1.1. Vorgeschlagene Handelsbezeichnung(en).
5.1.2. Art des Zusatzstoffs gemäß seiner hauptsächlichen Wirkung. Etwaige andere Verwendungszwecke des Wirkstoffs sind anzugeben.
5.1.3. Qualitative und quantitative Zusammensetzung (Wirkstoff, sonstige Bestandteile, Verunreinigungen, Chargenstreuung). Handelt es sich bei dem Wirkstoff um ein Gemisch aus eindeutig definierbaren Wirkstoffkomponenten, so sind die Hauptkomponenten gesondert zu beschreiben und ihr Anteil im Gemisch ist anzugeben.
5.1.4. Physikalische Beschaffenheit, Verteilung der Teilchengöße, Teilchenform, Dichte, Schüttdichte; bei Flüssigkeiten: Viskosität, Oberflächenspannung.
5.1.5. Herstellungsverfahren einschließlich etwaiger spezifischer Behandlungsverfahren.
5.2. Spezifizierung des Wirkstoffs
5.2.1. Generische Bezeichnung, chemische Bezeichnung nach der IUPACNomenklatur, sonstige internationale generische Bezeichnungen und Abkürzungen. CAS-Nummer (CAS: Chemical Abstracts Service).
5.2.2. Strukturformel, Summenformel und Molekulargewicht. Ist der Wirkstoff ein Fermentationserzeugnis: qualitative und quantitative Zusammensetzung der wichtigsten Bestandteile, mikrobiologischer Ursprung (Name und Anschrift der Kultursammlung, wo der Stamm hinterlegt ist).
5.2.3. Reinheit
Qualitative und quantitative Zusammensetzung der Wirkstoffe und der zusammen mit ihnen auftretenden Verunreinigungen und toxischen Substanzen, Bestätigung des Fehlens von Produktionsorganismen.
5.2.4. Relevante Eigenschaften
Physikalische Eigenschaften chemisch definierter Stoffe: Dissoziationskonstante, pKa, elektrostatische Eigenschaften, Schmelzpunkt, Siedepunkt, Dichte, Dampfdruck, Löslichkeit in Wasser und organischen Lösungsmitteln, Kw und Koc, Massen- und Absorptionsspektren, NMR-Daten, eventuelle Isomeren und sonstige relevante physikalische Eigenschaften.
5.3. Physikalisch-chemische, technologische und biologische Eigenschaften des Zusatzstoffs
5.3.1. Stabilität des Zusatzstoffs bei Exposition gegenüber Umweltfaktoren wie Licht, Temperatur, pH-Wert, Feuchtigkeit und Sauerstoff. Vorschlag für die Haltbarkeitsdauer.
5.3.2. Stabilität während der Herstellung von Vormischungen und Futtermitteln, insbesondere Stabilität bei den zu erwartenden Prozessbedingungen (Hitze, Feuchtigkeit, Druck/Schub, Zeit). Etwaige Abbau- oder Zersetzungsprodukte.
5.3.3. Stabilität während der Lagerung von Vormischungen und verarbeiteten Futtermitteln unter definierten Bedingungen. Vorschlag für die Haltbarkeitsdauer.
5.3.4. Sonstige relevante physikalisch-chemische, technologische oder biologische Eigenschaften, wie Dispergierbarkeit unter günstigen Bedingungen, um in Vormischungen und Futtermitteln homogene Gemische zu erhalten, Staubbildung verhindernde und antistatische Eigenschaften, Dispergierbarkeit in Flüssigkeiten.
5.4. Überwachungsmethoden
5.4.1. Beschreibung der Methoden zur Bestimmung der Kriterien gemäß den Ziffern 2.1.3, 2.1.4, 2.2.3, 2.2.4, 2.3.1, 2.3.2, 2.3.3 und 2.3.4.
5.4.2. Beschreibung der qualitativen und quantitativen Analyseverfahren zur Bestimmung der Markerrückstände des Wirkstoffs in Zielgeweben und tierischen Erzeugnissen.
5.4.3. Falls die genannten Methoden veröffentlicht worden sind, reicht ein Quellennachweis unter Umständen aus, und die entsprechenden Veröffentlichungen sind in Kopie vorzulegen.
5.4.4. Angaben zu den optimalen Lagerungsbedingungen für die Referenzstandards.
5.5. Biologische Eigenschaften des Zusatzstoffs
5.5.1. Bei Kokzidiostatika und anderen Arzneimitteln: Einzelheiten zu den prophylaktischen Wirkungen (z.B. Morbidität, Mortalität, Anzahl der Oozyten, Bewertung der Schädigungen).
5.5.2. Für Zusatzstoffe, die für zootechnische Zwecke bestimmt sind und nicht unter 5.5.1 fallen: Einzelheiten zu den Wirkungen auf Futteraufnahme, Körpergewicht, Futterverwertung, Erzeugnisqualität und -ergiebigkeit sowie auf sonstige Parameter von Nutzen für das Tier, die Umwelt, den Erzeuger oder den Verbraucher.
5.5.3. Für technologische Zusatzstoffe: relevante technologische Wirkungen.
5.5.4. Eventuelle schädliche Wirkungen, Gegenanzeigen oder Warnhinweise (Zieltier, Verbraucher, Umwelt), einschließlich biologischer Wechselwirkungen, unter Angabe einer Begründung. ADI- oder MRL-Werte, die für andere Verwendungszwecke des Zusatzstoffs festgelegt wurden, sind anzugeben.
5.6. Genaue quantitative und qualitative Angabe etwaiger Rückstände in Zielgeweben, die bei bestimmungsgemäßer Anwendung des Zusatzstoffs in tierischen Erzeugnissen gefunden wurden
5.7. Gegebenenfalls sind die ADI, die festgelegten MRL-Werte und die Wartezeit anzugeben.
5.8. Sonstige relevante Eigenschaften zur Identifizierung des Zusatzstoffs
5.9. Anwendungsbedingungen
5.10. Datum
6. Kapitel VI: Muster einer technischen Spezifikation
1. Identität des Zusatzstoffs1.1. Art des Zusatzstoffs
1.2. Physikalische Beschaffenheit
1.3. Qualitative und quantitative Zusammensetzung
1.4. Verfahren für die Analyse des Zusatzstoffs und der Rückstände
1.5. Gemeinschaftliche Registrierungsnummer (EG-Nummer)
1.6. Verpackung
2. Spezifizierung des Wirkstoffs
2.1. Generische Bezeichnung, chemische Bezeichnung, CAS-Nummer
- Generische Bezeichnung
- Chemische Bezeichnung (IUPAC)
- CAS-Nummer
2.2. Summenformel
3. Physikalisch-chemische, technologische und biologische Eigenschaften des Zusatzstoffs
3.1. Stabilität des Zusatzstoffs
3.2. Stabilität während der Herstellung von Vormischungen und Futtermitteln
3.3. Stabilität während der Lagerung von Vormischungen und Futtermitteln
3.4. Sonstige Eigenschaften
4. Anwendungsbedingungen
4.1. Tierart oder -kategorie, gegebenenfalls Höchstalter
4.2. Mindest- und Höchstgehalt in Futtermitteln
4.3. Gegenanzeigen, Wechselwirkungen
4.4. Warnhinweise
5. Für das Inverkehrbringen verantwortliche Person
5.1. Name
5.2. Anschrift
5.3. Registrierungsnummer
6. Hersteller
6.1. Name
6.2. Anschrift
6.3. Zulassungsnummer oder Registrierungsnummer des Unternehmens oder des Zwischenhändlers
7. Datum
7. Kapitel VII: Verlängerung der Zulassung von Zusatzstoffen, deren Zulassung an eine für das Inverkehrbringen verantwortliche Person gebunden ist
1. Allgemeines
Es ist ein aktualisiertes Dossier und eine ebensolche Monographie nach den neuesten Leitlinien zu erstellen; ferner ist ein Verzeichnis sämtlicher wie auch immer gearteter Änderungen seit der Genehmigung des Inverkehrbringens oder seit der letzten Verlängerung vorzulegen.
Es muss bestätigt werden, dass die Monographie und das Sicherheitsdatenblatt dergestalt angepasst wurden, dass sie sämtliche neuen Angaben enthalten, die für den Zusatzstoff relevant sind oder infolge der Änderung dieser Leitlinien nunmehr verlangt werden.
Ferner sind Informationen über den Zulassungsstatus weltweit und das Verkaufsvolumen vorzulegen.
2. Identität des Wirkstoffs und des Zusatzstoffs
Es ist nachzuweisen, dass der Zusatzstoff bzw. seine Zusammensetzung, Reinheit oder Wirkungsweise gegenüber dem bereits zugelassenen Zusatzstoff nicht verändert wurde. Etwaige Änderungen am Herstellungsprozess sind anzugeben.
3. Wirksamkeit
Es ist nachzuweisen, dass der Zusatzstoff die beanspruchte Wirkung unter den zum Zeitpunkt der Verlängerungsbeantragung üblichen Bedingungen der tierischen Erzeugung behält. Zu diesem Zweck ist über die allgemeinen Erfahrungen mit der Anwendung des Zusatzstoffs und der Leistungsüberwachung zu berichten.
4. Mikrobiologie
Besonderes Augenmerk ist auf die mögliche Entwicklung einer Resistenz gegen antimikrobielle Substanzen während der Langzeitanwendung unter Praxisbedingungen zu richten. Daher müssen die Tests unter Feldbedingungen in Betrieben durchgeführt werden, in denen der Zusatzstoff so lange wie möglich routinemäßig angewandt wurde. Als Testorganismen ist eine Auswahl der üblichen Darmbakterien zu verwenden, darunter relevante endogene und exogene grampositive sowie gramnegative Organismen.
Ergeben die Tests gegenüber den ursprünglichen Zahlen eine Veränderung im Resistenzmuster, so sind die resistenten Bakterien auf Kreuzresistenz gegenüber den zur Behandlung von Infektionskrankheiten bei Mensch und Tier eingesetzten relevanten Antibiotika zu prüfen. Am wichtigsten sind Antibiotika, die zur gleichen Gruppe gehören wie der Zusatzstoff; aber auch andere Antibiotikagruppen sind in den Versuch einzubeziehen.
Ein Bericht über die Ergebnisse der entsprechenden Überwachungsprogramme ist beizufügen.
5. Sicherheit
Es ist nachzuweisen, dass der Zusatzstoff nach dem gegenwärtigen Kenntnisstand unter den genehmigten Bedingungen für die Zieltierart, die Verbraucher, die Anwender und die Umwelt sicher bleibt. Es ist eine Aktualisierung der Sicherheitsangaben für den Zeitraum seit der Genehmigung des Inverkehrbringens oder seit der letzten Verlängerung vorzulegen, die Angaben zu folgenden Punkten enthält:
so ist dies klar zu begründen.
8. Kapitel VIII: Neuer Antragsteller, der Bezug nimmt auf die Erstzulassung eines Zusatzstoffs, dessen Zulassung an eine für das Inverkehrbringen verantwortliche Person gebunden ist.
Da auf die Bewertung der für die ursprüngliche Zulassung vorgelegten Daten zurückgegriffen werden kann, braucht ein Dossier für einen Antrag gemäß Artikel 9c Absatz 3 nur die im Folgenden aufgeführten Anforderungen zu erfüllen.
Ein Zusatzstoff kann dann als identisch für diesen Zweck gelten, wenn die qualitative und quantitative Zusammensetzung und die Reinheit des Wirkstoffs und der sonstigen Bestandteile im Wesentlichen die gleichen sind, die Zubereitung die gleiche ist und die Anwendungsbedingungen identisch sind.
Bei derartigen Produkten ist es in der Regel nicht erforderlich, die pharmakologischen, toxikologischen und Wirksamkeitsstudien zu wiederholen; statt dessen kann ein verkürzter Antrag eingereicht werden. Dieser muss Sachverständigenberichte umfassen.
Teil II
Mikroorganismen und Enzyme 16
_____________________
1) ABl. Nr. L 270 vom 14.12.1970 S. 1.
2) ABl. Nr. L 310 vom 05.11.1986 S. 19.
3) ABl. Nr. L 106 vom 17.04.2001 S. 1.
4) ABl. Nr. L 15 vom 17.01.1987 S. 29.
5) ABl. Nr. L 196 vom 16.08.1967 S. 1
6) ABl. Nr. L 136 vom 08.06.2000 S. 90.
7) ABl. Nr. L 76 vom 22.03.1991 S. 35.
8) ABl. Nr. L 187 vom 16.07.1988 S. 14.
9) Method Validation - a Laboratory Guide, EURACHEM Secretariat, Laboratory of the Government Chemist, Teddington, UK, 1996.
10) Beim Markerrückstand handelt es sich um einen Rückstand, dessen Konzentration in einem bekannten Verhältnis zu der Geschwindigkeit steht, mit der die Konzentration des Gesamtrückstands im Zielgewebe zur MRL depletiert.
11) Das Zielgewebe ist das essbare Gewebe, welches für die Überwachung des Gesamtrückstands im Zieltier ausgewählt wurde.
12) Für die Bestimmung der Wartezeit werden folgende Mindestanzahlen gesunder Tiere empfohlen, von denen bei jeder Schlachtung oder jedem Zeitpunkt Proben zu nehmen sind:
13) Vorschlag für die Berechnung: [500 g Fleisch (bestehend aus 300 g Muskel, 100 g Leber, 50 g Niere, 50 g Fett) oder 500 g Geflügel (bestehend aus 300 g Muskel, 100 g Leber, 10 g Niere, 90 g Fett) oder 300 g Fisch] + 1 500 g Milch + 100 g Ei.
14) Gemäß der Richtlinie 89/107/EWG des Rates vom 21. Dezember 1988 zur Angleichung der Rechtsvorschriften der Mitgliedstaaten über Zusatzstoffe, die in Lebensmitteln verwendet werden dürfen (ABl. Nr. L 40 vom 11.02.1989 S. 27).
15) OECD Guidelines for Testing of Chemicals.
16) Siehe Richtlinie 94/40/EG der Kommission (ABl. Nr. L 208 vom 11.08.1994 S. 15), geändert durch die Richtlinie 95/11/EG.
| |
ENDE | |
(Stand: 11.03.2019)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion
...
X
⍂
↑
↓