

umwelt-online: Verordnung (EG) Nr. 152/2009 zur Festlegung der Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln (3)
|
zurück | |
K. Bestimmung des Lactosegehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Lactosegehalts von Futtermitteln, die mehr als 0,5 % Lactose enthalten.
2. Prinzip
Die Zucker werden in Wasser gelöst. Die Lösung wird mit Hefe - Saccharomyces cerevisiae - vergoren, wobei die Lactose nicht angegriffen wird. Nach Klärung und Filtration wird der Gehalt an Lactose im Filtrat nach der Luff-Schoorl-Methode bestimmt.
3. Reagenzien
3.1 Saccharomycescerevisiae-Suspension: 25 g frische Hefe werden in 100 ml Wasser suspendiert. Im Kühlschrank ist die Suspension höchstens 1 Woche haltbar.
3.2 Carrez-Lösung I: 21,9 g Zinkacetat, Zn (CH3COO)2⋅ 2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.3 Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4 Fe(CN)6⋅ 3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.4 Reagenz nach Luff-Schoorl:
Die Zitronensäurelösung ( 3.4.2) wird unter vorsichtigem Rühren in die Natriumcarbonatlösung ( 3.4.3) gegossen. Nach Zugabe der Kupfersulfatlösung ( 3.4.1) wird auf 1 l mit Wasser aufgefüllt. Über Nacht absetzen lassen und dann filtrieren. Die Konzentration des so erhaltenen Reagenz (Cu 0,05 mol/l; Na2CO3 1 mol/l) muss geprüft werden. Der pH-Wert muss bei etwa 9,4 liegen.
3.4.1 Kupfersulfatlösung: 25 g Kupfersulfat, CuSO4 ⋅5H2O, eisenfrei, werden in 100 ml Wasser gelöst,
3.4.2 Zitronensäurelösung: 50 g Zitronensäure, C6H8O7 ⋅ H2O, werden in 50 ml Wasser gelöst,
3.4.3 Natriumcarbonatlösung: 143,8 g Natriumcarbonat, wasserfrei, werden in ca. 300 ml warmem Wasser gelöst. Die Lösung wird abkühlen gelassen.
3.5 Bimssteinkörner mit Salzsäure ausgekocht, mit Wasser gewaschen und getrocknet.
3.6 Kaliumiodidlösung (Massenkonzentration = 30 %).
3.7 Schwefelsäure 3 mol/l.
3.8 Natriumthiosulfatlösung 0,1 mol/l.
3.9 Stärkelösung: Eine Aufschlämmung von 5 g löslicher Stärke in 30 ml Wasser wird zu 1 l siedendem Wasser hinzugefügt und 3 min lang im Sieden gehalten; dann wird abgekühlt und gegebenenfalls 10 mg Quecksilberiodid als Konservierungsmittel hinzugefügt.
4. Geräte
Wasserbad mit Thermostat, auf 38 bis 40 °C eingestellt.
5. Verfahren
Von der Probe wird 1 g auf 1 mg genau eingewogen und mit 25 bis 30 ml Wasser in einen 100-ml-Messkolben gebracht. Der Messkolben wird 30 min lang in ein Bad mit siedendem Wasser gestellt und auf eine Temperatur von etwa 35 °C abgekühlt. Anschließend werden 5 ml Hefesuspension ( 3.1) zugefügt. Dann wird der Kolben geschüttelt, 2 h lang in einem Wasserbad bei 38 bis 40 °C stehen gelassen und auf etwa 20 °C abgekühlt.
Danach werden 2,5 ml Carrez-Lösung I ( 3.2) hinzugefügt, und es wird 30 s lang gerührt. Dann werden 2,5 ml Carrez-Lösung II (3.3) hinzugefügt, und es wird nochmals 30 s lang gerührt. Nun wird mit Wasser auf 100 ml aufgefüllt, gemischt und filtriert. Vom Filtrat wird eine Menge von höchstens 25 ml, die möglichst 40 bis 80 mg Lactose enthält, in einen 300-ml-Erlenmeyerkolben abpipettiert. Gegebenenfalls wird mit Wasser auf 25 ml aufgefüllt.
Auf dieselbe Weise wird ein Blindversuch mit 5 ml Hefesuspension ( 3.1) ausgeführt. Dann wird der Gehalt an Lactose nach der Luff-Schoorl-Methode wie folgt bestimmt: Nach Zugabe von genau 25 ml des Reagenz nach Luff-Schoorl ( 3.4) und von 2 Körnchen Bimsstein ( 3.5) wird unter Schütteln mit der Hand über freier Flamme von mittlerer Höhe erhitzt, so dass die Flüssigkeit in etwa 2 min zum Sieden kommt. Anschließend wird der Erlenmeyerkolben sofort auf ein Drahtnetz mit einer Asbestscheibe, in die ein Loch von etwa 6 cm Durchmesser gestanzt wurde, gestellt; vorher wird unter dem Drahtnetz eine Flamme entzündet und so eingestellt, dass der Kolben nur am Boden erhitzt wird. Dann wird der Kolben mit einem Rückflusskühler verbunden. Von diesem Augenblick an wird der Kolben genau 10 min sieden gelassen und anschließend sofort in kaltem Wasser abgekühlt. Nach etwa 5 min wird wie folgt titriert:
Zu der Flüssigkeit werden 10 ml Kaliumiodidlösung ( 3.6) und unmittelbar danach, aber vorsichtig (wegen der Gefahr des übermäßigen Schäumens), 25 ml Schwefelsäure ( 3.7) hinzugegeben. Dann wird mit Natriumthiosulfatlösung (3.8) zunächst bis zum Auftreten einer mattgelben Farbe titriert und nach Zugabe von Stärkelösung ( 3.9) als Indikator die Titration zu Ende geführt.
Die gleiche Titration wird in einer Mischung aus genau 25 ml Reagenz nach Luff-Schoorl ( 3.4) und 25 ml Wasser nach Zugabe von 10 ml Kaliumiodidlösung ( 3.6) und 25 ml Schwefelsäure ( 3.7) durchgeführt, jedoch ohne vorheriges Erhitzen.
6. Berechnung der Ergebnisse
Aus der unten angeführten Tabelle wird die Menge Lactose in mg abgelesen, die der Differenz der Ergebnisse beider Titrationen, ausgedrückt in ml Natriumthiosulfatlösung (0,1 mol/l), entspricht.
Das Ergebnis entspricht dem Gehalt an wasserfreier Lactose und wird als Prozentsatz der Probe ausgedrückt.
7. Bemerkung
Wenn mehr als 40 % vergärbare Zucker vorhanden sind, werden mehr als 5 ml Hefesuspension ( 3.1) verwendet.
Tabelle für 25 ml Reagenz nach Luff-Schoorl
ml Na2 S2 O3 0,1 mol/l, 2 min erhitzen, 10 min sieden
| Na2 S2 O3 0,1 mol/l |
Glucose, Fructose, Invertzucker C6 H12 O6 |
Lactose C12 H22 O11 |
Maltose C12 H22 O11 |
Na2 S2 O3 0,1 mol/l |
|||
| ml | mg | Differenz | mg | Differenz | Mg | Differenz | ml |
| 1 | 2,4 | 2,4 | 3,6 | 3,7 | 3,9 | 3,9 | 1 |
| 2 | 4,8 | 2,4 | 7,3 | 3,7 | 7,8 | 3,9 | 2 |
| 3 | 7,2 | 2,5 | 11,0 | 3,7 | 11,7 | 3,9 | 3 |
| 4 | 9,7 | 2,5 | 14,7 | 3,7 | 15,6 | 4,0 | 4 |
| 5 | 12,2 | 2,5 | 18,4 | 3,7 | 19,6 | 3,9 | 5 |
| 6 | 14,7 | 2,5 | 22,1 | 3,7 | 23,5 | 4,0 | 6 |
| 7 | 17,2 | 2,6 | 25,8 | 3,7 | 27,5 | 4,0 | 7 |
| 8 | 19,8 | 2,6 | 29,5 | 3,7 | 31,5 | 4,0 | 8 |
| 9 | 22,4 | 2,6 | 33,2 | 3,8 | 35,5 | 4,0 | 9 |
| 10 | 25,0 | 2,6 | 37,0 | 3,8 | 39,5 | 4,0 | 10 |
| 11 | 27,6 | 2,7 | 40,8 | 3,8 | 43,5 | 4,0 | 11 |
| 12 | 30,3 | 2,7 | 44,6 | 3,8 | 47,5 | 4,1 | 12 |
| 13 | 33,0 | 2,7 | 48,4 | 3,8 | 51,6 | 4,1 | 13 |
| 14 | 35,7 | 2,8 | 52,2 | 3,8 | 55,7 | 4,1 | 14 |
| 15 | 38,5 | 2,8 | 56,0 | 3,9 | 59,8 | 4,1 | 15 |
| 16 | 41,3 | 2,9 | 59,9 | 3,9 | 63,9 | 4,1 | 16 |
| 17 | 44,2 | 2,9 | 63,8 | 3,9 | 68,0 | 4,2 | 17 |
| 18 | 47,1 | 2,9 | 67,7 | 4,0 | 72,2 | 4,3 | 18 |
| 19 | 50,0 | 3,0 | 71,7 | 4,0 | 76,5 | 4,4 | 19 |
| 20 | 53,0 | 3,0 | 75,7 | 4,1 | 80,9 | 4,5 | 20 |
| 21 | 56,0 | 3,1 | 79,8 | 4,1 | 85,4 | 4,6 | 21 |
| 22 | 59,1 | 3,1 | 83,9 | 4,1 | 90,0 | 4,6 | 22 |
| 23 | 62,2 | 88,0 | 94,6 | 23 | |||
L. Bestimmung des Stärkegehalts
Polarimetrisches Verfahren
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Stärke und ihrer Abbauprodukte mit hoher Molmasse in Futtermitteln zum Zweck der Prüfung ihrer Übereinstimmung mit dem angegebenen Energiegehalt (Anhang VII) und mit der Richtlinie 96/25/EG des Rates1.
2. Prinzip
Die Methode basiert auf einer doppelten Bestimmung. Bei der ersten Bestimmung wird die Probe mit verdünnter Salzsäure behandelt. Nach Klärung und Filtration wird die optische Drehung der Lösung polarimetrisch gemessen.
Bei der zweiten Bestimmung wird die Probe mit 40 %igem Ethanol extrahiert. Nach Behandlung des Filtrats mit Salzsäure wird geklärt, filtriert und die optische Drehung unter den gleichen Bedingungen wie bei der ersten Bestimmung gemessen.
Der Unterschied zwischen den beiden Messungen, multipliziert mit einem bekannten Faktor, ergibt den Stärkegehalt der Probe.
3. Reagenzien
3.1 Salzsäure, Lösung (Massenanteil = 25 %) Dichte: 1,126 g/ml.
3.2 Salzsäure, Lösung (Massenkonzentration = 1,13 %).
Die Konzentration muss durch Titration mit Natriumhydroxidlösung (0,1 mol/l) in Gegenwart von Methylrot (Massenkonzentration = 0,1 %) in Ethanol (Volumenkonzentration = 94 %) geprüft werden. Für die Neutralisierung von 10 ml werden 30,94 ml NaOH (0,1 mol/l) benötigt.
3.3 Carrez-Lösung I: 21,9 g Zinkacetat, Zn(CH3COO)2⋅ 2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.4 Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat (II), K4Fe(CN)6⋅ 3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.5 Ethanol, Lösung (Volumenkonzentration = 40 %), Dichte: 0,948 g/ml bei 20 °C.
4. Geräte
4.1 250-ml-Erlenmeyerkolben mit Schliffstopfen und Rückflusskühler.
4.2 Polarimeter oder Saccharimeter.
5. Verfahren
5.1 Vorbereitung der Probe
Die Probe muss so fein gemahlen werden, dass sie vollständig durch ein Rundlochsieb mit einem Lochdurchmesser von 0,5 mm passiert werden kann.
5.2 Bestimmung der gesamten optischen Drehung (P oder S) (siehe Bemerkung 7.1)
Von der Probe werden 2,5 g auf 1 mg genau in einen 100-ml-Messkolben eingewogen und 25 ml Salzsäure ( 3.2) hinzugefügt. Der Kolben wird geschüttelt, bis sich der Stoff gleichmäßig verteilt hat; dann werden weitere 25 ml Salzsäure ( 3.2) hinzugegeben. Der Kolben wird in ein Bad mit kochendem Wasser gestellt und während der ersten 3 min kräftig und regelmäßig geschüttelt, um die Bildung von Klumpen zu verhindern. Die in dem Wasserbad enthaltene Wassermenge muss ausreichen, um das Wasser auch dann noch über dem Siedepunkt zu halten, wenn der Kolben eingetaucht wird. Während des Schüttelns darf der Kolben nicht aus dem Wasser herausgenommen werden. Nach genau 15 min wird der Kolben aus dem Wasserbad entfernt, 30 ml kaltes Wasser hinzugefügt und unverzüglich auf 20 °C abgekühlt.
Danach werden 5 ml Carrez-Lösung I ( 3.3) hinzugefügt, und es wird ca. 30 s lang geschüttelt. Anschließend werden 5 ml Carrez-Lösung II ( 3.4) hinzugefügt, und es wird nochmals 30 s geschüttelt. Dann wird mit Wasser zur Marke aufgefüllt, gemischt und filtriert. Ist das Filtrat nicht vollständig klar (was selten vorkommt), muss die Bestimmung mit größeren Mengen Carrez-Lösungen I und II, z.B. 10 ml, wiederholt werden.
Anschließend wird die optische Drehung der Lösung in einem 200-mm-Rohr mit einem Polarimeter oder einem Saccharimeter gemessen.
5.3 Bestimmung der optischen Drehung (P' oder S') der in 40 %igem Ethanol löslichen Substanzen
Von der Probe werden 5 g auf 1 mg genau in einen 100-ml-Messkolben eingewogen und etwa 80 ml Ethanol ( 3.5) hinzugefügt (siehe Bemerkung 7.2). Anschließend wird der Kolben 1 h bei Raumtemperatur stehen gelassen und währenddessen 6-mal kräftig geschüttelt, damit sich die Testprobe gründlich mit dem Ethanol vermischt. Dann wird mit Ethanol ( 3.5) zur Marke aufgefüllt, geschüttelt und filtriert.
Von dem Filtrat werden 50 ml (= 2,5 g der Probe) in einen 250-ml-Erlenmeyerkolben abpipettiert und 2,1 ml Salzsäure (3.1) hinzugefügt; der Kolben wird kräftig geschüttelt, an einen Rückflusskühler angeschlossen und in ein Bad mit siedendem Wasser gestellt. Nach genau 15 min wird der Erlenmeyerkolben aus dem Wasserbad herausgenommen und der Inhalt in einen 100-ml-Messkolben mit einer kleinen Menge von kaltem Wasser überspült; anschließend wird auf eine Temperatur von 20 °C abgekühlt.
Danach wird mit den Carrez-Lösungen I ( 3.3) und II ( 3.4) geklärt, mit Wasser zur Marke aufgefüllt, geschüttelt und filtriert und die optische Drehung gemessen, wie unter 5.2 zweiter und dritter Absatz beschrieben.
6. Berechnung der Ergebnisse
Der Stärkegehalt (%) wird wie folgt berechnet:
6.1 Polarimetrische Messung
| P | = Optische Drehung insgesamt in Winkelgrad | |
| P' | = Optische Drehung in Winkelgrad der in 40 %igem Ethanol löslichen Substanzen | |
| [a] | 20 D |
= Spezifische optische Drehung von reiner Stärke. Für diesen FaktorD werden die nachstehenden, allgemein anerkannten Werte angewendet:
+ 185,9°: Reisstärke, |
6.2 Saccharimetrische Messung
| S | = Optische Drehung insgesamt in Saccharimeter-Grad | |
| S' | = Optische Drehung in Saccharimeter-Grad der in 40 %igem Ethanol löslichen Substanzen | |
| N | = Gewicht (g) von Saccharose in 100 ml Wasser, das bei Messung mit einem 200-mm-Rohr eine optische Drehung von 100 Saccharimeter-Grad bewirkt:
16,29 g für die französischen Saccharimeter, |
|
| [α] | 20° D |
= Spezifische optische Drehung von reiner Stärke (siehe 6.1). |
6.3. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 0,4 % (absolut) bei Stärkegehalten von weniger als 40 % und 1 % (relativ) bei Stärkegehalten von 40 % und mehr nicht überschreiten.
7. Bemerkungen
7.1 Enthält die Probe mehr als 6 % Carbonate, berechnet als Calciumcarbonat, so müssen diese vor der Bestimmung der gesamten optischen Drehung durch Behandlung mit der genau notwendigen Menge verdünnter Schwefelsäure zerstört werden.
7.2 Bei Erzeugnissen mit hohem Lactosegehalt, z.B. bei Molkenpulver oder Magermilchpulver, wird nach Zusatz von 80 ml Ethanol ( 3.5) wie folgt verfahren: Der Messkolben wird an einen Rückflusskühler angeschlossen und 30 min lang in ein Wasserbad von 50 °C gestellt. Nach dem Abkühlen wird das Verfahren, wie unter 5.3 beschrieben, fortgeführt.
7.3 Folgende Futtermittel-Ausgangserzeugnisse führen, sofern sie in größeren Mengen in Futtermitteln vorhanden sind, erwiesenermaßen zu Interferenzen bei der Bestimmung des Stärkegehalts durch das polarimetrische Verfahren und könnten so falsche Ergebnisse zur Folge haben:
_________
1) ABl. L 125 vom 23.05.1996 S. 35.
M. Bestimmung des Rohaschegehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Rohaschegehalts von Futtermitteln.
2. Prinzip
Die Probe wird bei 550 °C verascht; der Rückstand wird gewogen.
3. Reagenzien
Ammoniumnitrat, Lösung (Massenkonzentration = 20 %).
4. Geräte
4.1 Heizplatte.
4.2 Elektrischer Muffelofen mit Thermostat.
4.3 Veraschungsschale aus Quarzglas, Porzellan oder Platin, entweder rechteckig (ca. 60 × 40 × 25 mm) oder rund (Durchmesser: 60 bis 75 mm, Höhe: 20 bis 40 mm).
5. Verfahren
Etwa 5 g (2,5 g bei Stoffen, die zur Blasenbildung neigen) der Probe werden auf 1 mg genau in eine vorher bei 550 °C geglühte, abgekühlte und tarierte Veraschungsschale eingewogen. Die Schale wird auf der Heizplatte allmählich bis zum Verkohlen des Stoffes erhitzt. Die Veraschung erfolgt gemäß 5.1 oder 5.2.
5.1 Die Schale wird in den auf 550 °C eingestellten Muffelofen gebracht und darin bei dieser Temperatur so lange gehalten, bis sich eine weiße, hellgraue oder rötliche Asche bildet, die offensichtlich frei von Kohlepartikeln ist. Dann wird die Schale in einen Exsikkator gestellt und unmittelbar nach dem Abkühlen gewogen.
5.2 Die Schale wird 3 h lang in den auf 550 °C eingestellten Muffelofen gebracht. Dann wird die Schale in einen Exsikkator gestellt und unmittelbar nach dem Abkühlen gewogen. Anhand einer zweiten Veraschung von 30 min ist zu prüfen, ob das Gewicht der Asche konstant geblieben ist (der Gewichtsverlust zwischen 2 aufeinanderfolgenden Wägungen darf 1 mg nicht überschreiten).
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird berechnet, indem das Leergewicht abgezogen wird. Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkungen
7.1 Bei schwer zerstörbaren Stoffen werden der abgekühlten Rohasche nach einer ersten Veraschung von mindestens 3 h einige Tropfen 20 %iger Ammoniumnitratlösung oder Wasser zugefügt (jedoch vorsichtig, um ein Zerstäuben oder Verkleben der Asche zu vermeiden). Nach Trocknung im Trockenschrank wird die Veraschung weitergeführt. Der Vorgang wird gegebenenfalls bis zur vollständigen Veraschung wiederholt.
7.2 Bei Stoffen, die der unter 7.1 beschriebenen Behandlung widerstehen, ist wie folgt vorzugehen: Nach einer Veraschung von 3 h wird die Asche mit warmem Wasser aufgenommen und durch einen kleinen aschefreien Filter abfiltriert. In der ursprünglichen Schale werden Filter und dessen Inhalt verascht. Das Filtrat wird in die abgekühlte Schale gebracht, bis zur Trockne eingedampft, verascht und gewogen.
7.3 Bei Ölen und Fetten wird eine Probe von 25 g in eine Schale entsprechender Abmessung genau eingewogen. Es wird dann durch Anzünden des Stoffes mit einem Streifen aus aschefreiem Filterpapier verkohlt. Nach der Verkohlung wird mit möglichst geringer Menge Wasser angefeuchtet. Anschließend wird getrocknet und die Veraschung, wie unter 5 beschrieben, zu Ende geführt.
N. Bestimmung des Gehalts an in Salzsäure unlöslicher Asche
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an in Salzsäure unlöslichen, mineralischen Bestandteilen von Futtermitteln. Abhängig von der Art der Probe sind 2 Verfahren vorgesehen.
1.1 Verfahren A: anzuwenden bei organischen Futtermittel-Ausgangserzeugnissen und bei den meisten Mischfuttermitteln.
1.2 Verfahren B: anzuwenden bei Mineralstoffen und Mineralstoffmischungen sowie bei allen Mischfuttermitteln, deren Gehalt an in Salzsäure unlöslicher Asche bei der Bestimmung nach Verfahren a mehr als 1 % beträgt.
2. Prinzip
2.1 Verfahren A: Die Probe wird verascht. Die Asche wird in Salzsäure zum Sieden gebracht und der unlösliche Rückstand abfiltriert und gewogen.
2.2 Verfahren B: Die Probe wird mit Salzsäure behandelt. Die Lösung wird abfiltriert, der Rückstand wird verascht, und die so erhaltene Asche nach Verfahren a weiterbehandelt.
3. Reagenzien
3.1 Salzsäure 3 mol/l.
3.2 Trichloressigsäure, Lösung (Massenkonzentration = 20 %).
3.3 Trichloressigsäure, Lösung (Massenkonzentration = 1 %).
4. Geräte
4.1 Heizplatte.
4.2 Elektrischer Muffelofen mit Thermostat.
4.3 Veraschungsschalen aus Quarzglas, Porzellan oder Platin, entweder rechteckig (ca. 60 × 40 × 25 mm) oder rund (Durchmesser: 60 bis 75 mm, Höhe: 20 bis 40 mm).
5. Verfahren
5.1 Verfahren A
Die Einwaage wird unter den Bedingungen, die für die Bestimmung des Rohaschegehalts beschrieben sind, verascht. Es kann auch die bei dieser Analyse erhaltene Asche verwendet werden.
Die Asche wird zusammen mit 75 ml Salzsäure ( 3.1) in ein Becherglas von 250 bis 400 ml Inhalt überführt. Es wird vorsichtig zum Sieden erhitzt und 15 min weiter in schwachem Sieden gehalten. Die warme Lösung wird durch einen aschefreien Papierfilter filtriert und der Rückstand mit warmem Wasser bis zum Ausbleiben der sauren Reaktion ausgewaschen. Der Filter mit dem Rückstand wird nach dem Trocknen bei einer Temperatur von mindestens 550 °C und höchstens 700 °C in einer tarierten Veraschungsschale verascht und anschließend im Exsikkator gekühlt und gewogen.
5.2 Verfahren B
Von der Analysenprobe werden 5 g auf 1 mg genau in ein Becherglas von 250 bis 400 ml Fassungsvermögen eingewogen. Dann werden nacheinander 25 ml Wasser und 25 ml Salzsäure ( 3.1) zugegeben, gemischt, und es wird bis zum Nachlassen des Aufbrausens gewartet. Zuletzt werden weitere 50 ml Salzsäure ( 3.1) zugegeben. Nach dem Nachlassen der eventuell erneut auftretenden Gasentwicklung wird das Becherglas 30 min lang oder, wenn notwendig, länger in ein siedendes Wasserbad gestellt, bis die vollständige Hydrolyse eventuell vorhandener Stärke eingetreten ist. Es wird warm durch einen aschefreien Filter filtriert. Der Filter wird mit etwa 50 ml warmem Wasser ausgewaschen (siehe Bemerkung 7). Anschließend wird der Filter mit Rückstand in eine Veraschungsschale gestellt, getrocknet und bei einer Temperatur von mindestens 550 °C und höchstens 700 °C verascht. Die Asche wird mit 75 ml Salzsäure ( 3.1) in ein Becherglas von 250 bis 400 ml Fassungsvermögen überführt und anschließend, wie unter 5.1 zweiter Absatz beschrieben, weiterbehandelt.
6. Berechnung der Ergebnisse
Das Gewicht des Rückstands wird berechnet, indem das Leergewicht abgezogen wird. Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
7. Bemerkung
Treten bei der Filtration Schwierigkeiten auf, so wird die Bestimmung wiederholt. Hierbei werden anstelle von 50 ml Salzsäure ( 3.1) 50 ml 20 %iger Trichloressigsäurelösung ( 3.2) zugegeben; der Filter wird mit einer warmen 1 %igen Trichloressigsäurelösung ( 3.3) ausgewaschen.
O. Bestimmung des Gehalts an Carbonaten
1. Zweck und Anwendungsbereich
Die Methode erlaubt es, in den meisten Futtermitteln den Gehalt an Carbonaten, herkömmlicherweise als Calciumcarbonat bezeichnet, zu bestimmen.
In bestimmten Fällen (z.B. bei Eisencarbonat) ist jedoch eine besondere Methode anzuwenden.
2. Prinzip
Die Carbonate werden mit Salzsäure zersetzt; die frei gewordene Kohlensäure wird in einem graduierten Rohr aufgefangen. Das erhaltene Gasvolumen wird mit demjenigen verglichen, welches unter den gleichen Bedingungen von einer bekannten Menge Calciumcarbonat freigesetzt wird.
3. Reagenzien
3.1 Salzsäure, Dichte 1,10 g/ml.
3.2 Calciumcarbonat.
3.3 Schwefelsäure, etwa 0,05 mol/l, mit Methylrot angefärbt.
4. Geräte
Apparatur nach Scheibler-Dietrich (siehe Skizze) oder gleichwertiges Gerät.
5. Verfahren
Je nach dem Carbonatgehalt der Probe wird eine Einwaage wie folgt eingewogen:
Die Einwaage wird in die Spezialflasche (4) der Apparatur gebracht, die mit einem Röhrchen aus unzerbrechlichem Material versehen ist, das 10 ml Salzsäure ( 3.1) enthält. Anschließend wird die Flasche an die Apparatur angeschlossen. Dann wird der Drei-Weg-Hahn (5) so gedreht, dass das Rohr (1) mit der Außenluft in Verbindung steht. Mit dem beweglichen Rohr (2), das mit angefärbter Schwefelsäure ( 3.3) gefüllt und mit dem graduierten Rohr (1) verbunden ist, wird das Niveau der Flüssigkeit auf den Nullpunkt der Skala eingestellt. Der Hahn (5) wird so gedreht, dass das Rohr (1) mit dem Rohr (3) verbunden ist. Anschließend wird der Nullpunkt kontrolliert.
Die Salzsäure ( 3.1) wird durch Kippen der Flasche (4) langsam über die Einwaage laufen gelassen. Durch Absenken des beweglichen Rohrs (2) wird der Druck ausgeglichen. Die Flasche (4) wird so lange hin und her bewegt, bis die Kohlensäureentwicklung ganz aufgehört hat.
Der Druckausgleich erfolgt, indem die Flüssigkeit in den Rohren (1) und (2) wieder auf gleiches Niveau gebracht wird. Nach einigen Minuten - sobald das Volumen konstant ist - kann abgelesen werden.
Es ist ein Vergleichsversuch mit 0,5 g Calciumcarbonat ( 3.2) unter denselben Bedingungen durchzuführen.
6. Berechnung der Ergebnisse
Der Gehalt an Carbonaten, ausgedrückt als Calciumcarbonat, wird nach folgender Formel berechnet:
| X = | V x 100 |
| V1 x 2m |
wobei!
X = Massenanteil (%) an Carbonaten in der Probe, ausgedrückt als Calziumkarbonat,
V = ml CO2, von der Einwaage freigesetzt,
V1 = ml CO2, aus 0,5 g CaCO3 freigesetzt,
m = Probeneinwaage in g.
7. Bemerkungen
7.1 Beträgt die Einwaage mehr als 2 g, werden vor Beginn des Versuchs 15 ml destilliertes Wasser in die Flasche (4) zugegeben und gemischt. Beim Vergleichsversuch ist dieselbe Wassermenge zu verwenden.
7.2 Bei Verwendung eines Apparats, dessen Volumen von dem der Scheibler-Dietrich-Apparatur abweicht, sind die Einwaage, die Vergleichsmenge und die Berechnung der Ergebnisse entsprechend anzupassen.
Apparatur nach Scheibler-Dietrich zur Bestimmung von CO2
P. Bestimmung des Gesamtphosphorgehalts
Fotometrische Methode
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Gehalts an Gesamtphosphor in Futtermitteln. Sie ist besonders für die Untersuchung von Futtermitteln mit einem geringen Gehalt an Phosphor geeignet. In bestimmten Fällen (phosphorreiche Erzeugnisse) kann eine gravimetrische Methode angewandt werden.
2. Prinzip
Die Probe wird entweder auf trockenem Wege (bei organischen Futtermitteln) oder auf nassem Wege (bei Mineralstoffen und flüssigen Futtermitteln) aufgeschlossen und in Säure gelöst. Die Lösung wird mit dem Vanadat-Molybdat-Reagenz behandelt. Die Extinktion der gelb gefärbten Lösung wird bei 430 nm im Spektralfotometer gemessen.
3. Reagenzien
3.1 Calciumcarbonat.
3.2 Salzsäure, ρ20 = 1,10 g/ml (ca. 6 mol/l).
3.3 Salpetersäure, ρ20 = 1,045 g/ml.
3.4 Salpetersäure, ρ20 = 1,38 bis 1,42 g/ml.
3.5 Schwefelsäure, ρ20 = 1,84 g/ml.
3.6 Vanadat-Molybdat-Reagenz: 200 ml Ammoniumheptamolybdatlösung ( 3.6.1) werden mit 200 ml Ammoniummonovanadatlösung ( 3.6.2) und 134 ml Salpetersäure (3.4) in einem l-l-Messkolben gemischt und mit Wasser zur Marke aufgefüllt.
3.6.1 Ammoniumheptamolybdatlösung: 100 g Ammoniumheptamolybdat, (NH4) 6Mo7 O244H2O, werden in heißem Wasser gelöst. 10 ml Ammoniakwasser (D: 0,91 g/ml) werden hinzugefügt, und das Volumen wird mit Wasser auf 1 l aufgefüllt.
3.6.2 Ammoniummonovanadatlösung: 2,35 g Ammoniummonovanadat, NH4 VO3, werden in 400 ml heißem Wasser gelöst. 20 ml verdünnte Salpetersäure (7 ml HNO3 ( 3.4) + 13 ml H2O) werden unter ständigem Rühren langsam hinzugefügt, und das Volumen wird mit Wasser auf 1 l aufgefüllt.
3.7 Phosphor-Standardlösung 1 mg je ml: 4,387 g Kaliumdihydrogenphosphat, KH2PO4, werden in Wasser gelöst. Das Volumen wird mit Wasser auf 1 l aufgefüllt.
4. Geräte
4.1 Veraschungsschalen aus Quarzglas, Platin oder Porzellan.
4.2 Elektrischer Muffelofen mit Thermostat auf 550 °C eingestellt.
4.3 Kjeldahl-Kolben, 250 ml.
4.4 Messkolben und Präzisions-Pipetten.
4.5 Spektralfotometer.
4.6 Reagenzgläser, ca. 16 mm Durchmesser, mit Stopfen graduiert bis zu 14,5 mm Durchmesser; Fassungsvermögen: 25 bis 30 ml.
5. Verfahren
5.1 Vorbereitung der Lösung
Je nach der Art der Probe wird die Lösung, wie unter 5.1.1 oder 5.1.2 angegeben, vorbereitet.
5.1.1 Allgemeines Verfahren
Von der Probe werden 1 g oder mehr auf 1 mg genau in einen Kjeldahl-Kolben eingewogen und mit 20 ml Schwefelsäure ( 3.5) versetzt; dann wird geschüttelt, um das Material vollständig mit der Säure zu benetzen und um zu verhindern, dass es an den Wandungen des Kolbens anhaftet; anschließend wird der Kolben erhitzt und 10 min im Sieden gehalten. Nach geringem Abkühlen werden 2 ml Salpetersäure ( 3.4) hinzugefügt; es wird schwach erhitzt und wieder etwas abgekühlt. Dann wird noch ein wenig Salpetersäure ( 3.4) hinzugegeben und erneut zum Sieden erhitzt. Das Verfahren wird so oft wiederholt, bis die Lösung farblos geworden ist. Hierauf wird abgekühlt, etwas Wasser zugesetzt und die Flüssigkeit in einen 500-ml-Messkolben überführt, wobei der Kjeldahl-Kolben mit heißem Wasser ausgespült wird. Nach Abkühlen wird mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.2 Verfahren für Proben, die organische Stoffe enthalten, aber frei sind von Calcium- bzw. Magnesiumdihydrogenphosphaten
Von der Probe werden 2,5 g auf 1 mg genau in eine Veraschungsschale eingewogen und mit 1 g Calciumcarbonat ( 3.1) vollständig vermischt. Im Muffelofen wird bei 550 °C verascht, bis die Asche weiß oder grau ist (kleine Mengen Kohle stören nicht). Die Asche wird in ein 250-ml-Becherglas gebracht, 20 ml Wasser und so viel Salzsäure ( 3.2) zugefügt, bis das Aufbrausen ausbleibt. Zuletzt werden weitere 10 ml Salzsäure ( 3.2) zugegeben. Dann wird das Becherglas auf ein Sandbad gestellt und bis zur Trockne eingedampft, um die Kieselsäure unlöslich zu machen. Der Rückstand wird in 10 ml Salpetersäure ( 3.3) aufgenommen und 5 min lang auf dem Sandbad oder der Heizplatte zum Sieden erhitzt, wobei der Rückstand nicht ganz trocken werden darf. Die Flüssigkeit wird in einen 500-ml-Messkolben übergespült, wobei das Becherglas mehrmals mit heißem Wasser ausgewaschen wird. Nach Abkühlen wird mit Wasser zur Marke aufgefüllt, homogenisiert und filtriert.
5.2 Entwicklung der Färbung und Messung der Extinktion
Ein aliquoter Teil des nach 5.1.1 oder 5.1.2 erhaltenen Filtrats wird verdünnt, um eine Konzentration an Phosphor von höchstens 40 μg/ml zu erhalten. Von dieser Lösung werden 10 ml in ein Reagenzglas ( 4.6) pipettiert und 10 ml Vanadat-Molybdat-Reagenz ( 3.6) hinzugefügt. Das Reagenzglas wird geschüttelt und dann mindestens 10 min bei einer Temperatur von 20 °C stehen gelassen. Anschließend wird die Extinktion im Spektralfotometer bei 430 nm gegen eine Lösung von 10 ml Wasser und 10 ml Vanadat-Molybdat-Reagenz ( 3.6) gemessen.
5.3 Kalibrationskurve
Aus der Standardlösung ( 3.7) werden Lösungen hergestellt, die 5, 10, 20, 30 bzw. 40 μg Phosphor je ml enthalten. Zu jeweils 10 ml dieser Lösungen werden 10 ml Vanadat-Molybdat-Reagenz ( 3.6) hinzugefügt. Das Reagenzglas wird geschüttelt und dann mindestens 10 min bei einer Temperatur von 20 °C stehen gelassen. Dann wird die Extinktion entsprechend den unter 5.2 genannten Bedingungen gemessen. Die Kalibrationskurve wird aufgestellt, indem die Extinktionswerte auf der Ordinate und die entsprechende Phosphormenge auf der Abszisse aufgetragen werden. Bei Phosphorkonzentrationen von 0 bis 40 μg/ml ist die Kurve linear.
6. Berechnung der Ergebnisse
Der Phosphorgehalt der Analysenprobe wird mithilfe der Kalibrationskurve bestimmt. Das Ergebnis ist als Prozentsatz der Probe auszudrücken.
Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf die folgenden Werte nicht überschreiten:
Q. Bestimmung des Chlorgehalts aus Chloriden
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Chlorsgehalts aus wasserlöslichen Chloriden, herkömmlicherweise als Natriumchlorid bezeichnet. Sie ist bei allen Futtermitteln anwendbar.
2. Prinzip
Die Chloride werden in Wasser gelöst. Handelt es sich um Erzeugnisse, die organische Stoffe enthalten, wird eine Klärung vorgenommen. Die Lösung wird mittels Salpetersäure schwach angesäuert, und die Chloride werden mit einer Silbernitratlösung als Silberchlorid gefällt. Der Silbernitratüberschuss wird mit einer Ammoniumthiocyanatlösung nach der Volhard-Methode zurücktitriert.
3. Reagenzien
3.1 Ammoniumthiocyanatlösung 0,1 mol/l.
3.2 Silbernitratlösung 0,1 mol/l.
3.3 Gesättigte Ammonium-Eisen(III)-Sulfatlösung (NH4)Fe(SO4)2.
3.4 Salpetersäure, Dichte: 1,38 g/ml.
3.5 Diethylether.
3.6 Aceton.
3.7 Carrez-Lösung I: 21,9 g Zinkacetat, Zn (CH3COO)2⋅ 2H2O, und 3 g Eisessig werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.8 Carrez-Lösung II: 10,6 g Kaliumhexacyanoferrat(II), K4Fe(CN)6⋅ 3H2O, werden in Wasser gelöst und mit Wasser auf 100 ml aufgefüllt.
3.9 Aktivkohle, chloridfrei und keine Chloride adsorbierend.
4. Geräte
Mechanisches Schüttelgerät, ca. 35 bis 40 min-1.
5. Verfahren
5.1 Vorbereitung der Lösung
Je nach der Art der Probe wird, wie unter 5.1.1, 5.1.2 oder 5.1.3 beschrieben, eine Lösung hergestellt.
Gleichzeitig ist ein Blindversuch ohne die zu analysierende Probe durchzuführen.
5.1.1 Proben, die frei von organischen Stoffen sind
Eine Menge von höchstens 10 g der Analysenprobe mit einem Gehalt von höchstens 3 g Chlor in Form von Chloriden wird auf 1 mg genau abgewogen und in einen 500-ml-Messkolben mit 400 ml Wasser von etwa 20 °C gebracht. Dann wird 30 min lang in dem Schüttelgerät rotiert, zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.2 Proben, die organische Stoffe enthalten (mit Ausnahme der unter 5.1.3 angeführten Erzeugnisse)
Etwa 5 g der Analysenprobe werden auf 1 mg genau eingewogen und mit 1 g Aktivkohle in einen 500-ml-Messkolben gebracht. Hierzu werden 400 ml Wasser von etwa 20 °C und 5 ml Carrez-Lösung I ( 3.7) gegeben, 30 s gerührt und anschließend 5 ml Carrez-Lösung II ( 3.8) zugesetzt. Dann wird 30 min lang in dem Schüttelgerät rotiert, zur Marke aufgefüllt, homogenisiert und filtriert.
5.1.3 Backfutter, Leinkuchen und Leinmehl, Erzeugnisse mit einem hohen Leinmehlgehalt oder sonstige Erzeugnisse mit einem hohen Gehalt an Pflanzen schleim oder kolloidalen Stoffen(z.B. verkleisterte Stärke)
Die Lösung wird, wie unter 5.1.2 beschrieben, zubereitet, jedoch nicht filtriert. Es wird dekantiert (falls erforderlich, zentrifugiert); dann werden 100 ml der überstehenden Lösung in einen 200-ml-Messkolben pipettiert. Es wird mit Aceton ( 3.6) gemischt und mit demselben Lösungsmittel zur Marke aufgefüllt, homogenisiert und filtriert.
5.2 Titration
Von dem Filtrat, das nach 5.1.1, 5.1.2 oder 5.1.3 erhalten wurde, werden (je nach dem zu erwartenden Chlorgehalt) 25 bis 100 ml in einen Erlenmeyerkolben pipettiert. Die aliquote Menge darf höchstens 150 mg Chlor (Cl) enthalten. Falls erforderlich, wird mit Wasser auf mindestens 50 ml verdünnt. Dann werden 5 ml Salpetersäure ( 3.4), 20 ml der gesättigten Ammonium-Eisen(III)-Sulfatlösung ( 3.3) und 2 Tropfen Ammoniumthiocyanatlösung ( 3.1) aus einer bis zum Nullpunkt gefüllten Bürette hinzugesetzt. Anschließend wird aus einer Bürette Silbernitratlösung ( 3.2) zugegeben, bis ein Überschuss von 5 ml vorhanden ist. Dann werden 5 ml Diethylether ( 3.5) zugegeben, und es wird kräftig geschüttelt, um den Niederschlag zum Ausflocken zu bringen. Der Überschuss an Silbernitrat wird mit Ammoniumthiocyanatlösung ( 3.1) titriert, bis eine rotbraune Farbe auftritt, die 1 min beständig ist.
6. Berechnung der Ergebnisse
Die Menge Chlor (X), ausgedrückt als % Natriumchlorid, wird nach folgender Formel berechnet: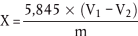
wobei:
V1 = zugegebene Silbernitratlösung 0,1 mol/l, in ml,
V2 = Ammoniumthiocyanatlösung 0,1 mol/l, die für die Titration verbraucht werden, in ml,
m = Probeneinwaage in g.
Falls der Blindversuch einen Verbrauch an Silbernitratlösung 0,1 mol/l anzeigt, wird dieser Wert von dem Volumen (V1- V2) abgezogen.
7. Bemerkungen
7.1 Die Titration kann auch mit der potenziometrischen Methode erfolgen.
7.2 Bei sehr fettreichen Erzeugnissen ist eine vorherige Entfettung mit Diethylether oder Petrolether durchzuführen.
7.3 Bei Fischmehl kann die Titration nach der Mohr-Methode durchgeführt werden.
_________
1) Für die Trocknung von Getreide, Mehl, Grütze und Grieß muss der Trockenschrank eine Wärmekapazität haben, mit der er nach Beladung mit der Höchstzahl gleichzeitig zu trocknender Proben die voreingestellte Temperatur von 131 °C in weniger als 45 min wieder erreicht. Die Lüftung muss so beschaffen sein, dass die Ergebnisse nach 2-stündiger Trocknung einer sein gesamtes Fassungsvermögen auslastenden Anzahl an Weichweizenproben von den Ergebnissen nach 4-stündiger Trocknung um weniger als 0,15 % abweichen.
| Analysemethoden zur Untersuchung von Futtermitteln auf ihren Gehalt an zugelassenen Zusatzstoffen | Anhang IV |
A. Bestimmung des Vitamin-A-Gehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Vitamin-A-(Retinol-)Gehalts in Futtermitteln und Vormischungen. Unter Vitamin a wird der nach dieser Methode ermittelte Gehalt an alltrans-Vitamin-A-Alkohol und seinen cis-Isomeren verstanden. Der Vitamin-A-Gehalt wird in Internationalen Einheiten (IE) je kg angegeben. Eine IE entspricht der Aktivität von 0,300 µg alltrans-Vitamin-A-Alkohol oder 0,344 µg alltrans-Vitamin-A-Acetat oder 0,550 µg alltrans-Vitamin-A-Palmitat.
Die Bestimmungsgrenze beträgt 2.000 IE Vitamin A/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxidlösung hydrolysiert, und das Vitamin a wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft; der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-A-Gehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt. Die chromatografischen Bedingungen werden so gewählt, dass keine Ruftrennung zwischen alltrans-Vitamin-AAlkohol und seinen cis-Isomeren erfolgt.
3. Reagenzien
3.1 Ethanol, σ = 96 %.
3.2 Petrolether, Siedeintervall 40 bis 60 °C.
3.3 Methanol.
3.4 Kaliumhydroxid-Lösung, c = 50 g/100 ml.
3.5 Natriumascorbat-Lösung, c = 10 g/100 ml (siehe Bemerkung 7.7).
3.6 Natriumsulfid, Na2S ⋅ x H2O (x = 7 bis 9)
3.6.1 Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (für x = 9) (siehe Bemerkung 7.8).
3.7 Phenolphthalein-Lösung, c = 2 g/100 ml in Ethanol ( 3.1).
3.8 2-Propanol.
3.9 Mobile Phase für die HPLC: Mischung von Methanol ( 3.3) und Wasser, z.B. 980 + 20 (V+V). Das Mischungsverhältnis ist der jeweils verwendeten Säule anzupassen.
3.10 Stickstoff, sauerstofffrei.
3.11 All-trans-Vitamin-A-Acetat, reinst, mit zertifizierter Aktivität, z.B. 2,80 × 106 IE/g
3.11.1 Stammlösung von all-trans-Vitamin-A-Acetat: 50 mg Vitamin-A-Acetat ( 3.11) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und zur Marke mit demselben Lösungsmittel auffüllen. Der Sollgehalt dieser Lösung beträgt 1 400 IE Vitamin a je ml. Der genaue Gehalt ist gemäß 5.6.3.1 zu bestimmen.
3.12 All-trans-Vitamin-A-Palmitat, reinst, mit zertifizierter Aktivität, z.B. 1,80 × 106 IE/g
3.12.1 Stammlösung von all-trans-Vitamin-A-Palmitat: 80 mg Vitamin-A-Palmitat ( 3.12) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und mit demselben Lösungsmittel zur Marke auffüllen. Der Sollgehalt dieser Lösung beträgt 1 400 IE Vitamin a je ml. Der genaue Gehalt ist gemäß 5.6.3.2 zu bestimmen.
3.13 2,6-Ditertbutyl-4-methylphenol (BHT) (siehe Bemerkung 7.5).
4. Geräte
4.1 Vakuum-Rotationsverdampfer.
4.2 Braunglasgeräte
4.2.1 Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse.
4.2.2 Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml.
4.2.3 Scheidetrichter, konische Form, 1.000 ml, mit Schliffstopfen.
4.2.4 Spitzkolben, 250 ml, mit Schliffhülsen.
4.3 Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung.
4.4 Phasentrennungsfaltenfilter, Durchmesser 185 mm (z.B. Schleicher & Schuell 597 HY 1/2).
4.5 HPLC-Einrichtung mit Injektionssystem
4.5.1 HPLC-Trennsäule, 250 × 4 mm, C18, 5 oder 10 µm Korngröße, oder vergleichbare Säule (Leistungskriterium: nur ein Peak für alle Retinol-Isomeren unter diesen HPLC-Bedingungen).
4.5.2 UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung.
4.6 Spektralfotometer mit Quarz-Küvetten von 10 mm Schichtdicke.
4.7 Wasserbad mit Magnetrührer.
4.8 Extraktionsapparat (Abbildung 1) bestehend aus:
4.8.1 1-l-Standzylinder mit Schliff und Schliffstopfen,
4.8.2 Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr sollte ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann.
5. Verfahren
Anmerkung:
Vitamin a ist empfindlich gegenüber Licht (UV-Strahlung) und Oxidation. Es muss deshalb unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder von mit Aluminiumfolie umhüllten Glasgeräten) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion muss das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen rechtzeitig lüften).
5.1 Vorbereitung der Probe
Die Probe wird unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-A-Verluste auftreten können.
5.2 Verseifung
Je nach Vitamin-A-Gehalt 2 bis 25 g der Probe auf 1 mg genau in einen 500-ml-Steh- oder Erlenmeyerkolben ( 4.2.1) einwiegen. Nacheinander unter Schwenken 130 ml Ethanol ( 3.1), etwa 100 mg BHT (3.13), 2 ml Natriumascorbat-Lösung ( 3.5) und 2 ml Natriumsulfid-Lösung ( 3.6) zusetzen. Den Kolben mit einem Rückflusskühler ( 4.3) verbinden und in ein Wasserbad mit Magnetrührer ( 4.7) geben. Bis zum Sieden erhitzen und 5 min am Rückflusskühler kochen. Anschließend 25 ml Kaliumhydroxidlösung ( 3.4) durch den Rückflusskühler ( 4.3) zugeben und unter ständigem Rühren und schwachem Stickstoffstrom 25 min weiterkochen. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3 Extraktion
Die Verseifungslösung wird mit insgesamt 250 ml Wasser quantitativ in einen 1.000-ml-Scheidetrichter ( 4.2.3) oder in den Extraktionsapparat (4.8) überspült. Der Verseifungskolben wird mit 25 ml Ethanol ( 3.1) und anschließend mit 100 ml Petrolether ( 3.2) nachgewaschen, und die Spülflüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen muss etwa 2:1 betragen. 2 min lang kräftig schütteln und dann 2 min lang absetzen lassen.
5.3.1 Extraktion mithilfe eines Scheidetrichters ( 4.2.3)
Nach Phasentrennung (siehe Bemerkung 7.3) wird die Petroletherphase in einen anderen Scheidetrichter ( 4.2.3) überführt. Diese Extraktion 2-mal mit je 100 ml Petrolether ( 3.2) und 2-mal mit je 50 ml Petrolether ( 3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter zunächst unter leichtem Schwenken (Vermeidung von Emulsionsbildung) 2-mal mit jeweils 100 ml Wasser gespült und dann durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung ( 3.7) farblos bleibt (4-maliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch einen trockenen Phasentrennungsfaltenfilter ( 4.4) in einen 500-ml-Messkolben ( 4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether ( 3.2) nachwaschen, mit Petrolether ( 3.2) zur Marke auffüllen und gut schütteln.
5.3.2 Extraktion mithilfe eines Extraktionsapparats ( 4.8)
Nach Phasentrennung (siehe Bemerkung 7.3) wird der Schliffstopfen des Standzylinders ( 4.8.1) durch den Schliffeinsatz ( 4.8.2) ersetzt und das verschiebbare Rohr so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1.000-ml-Scheidetrichter ( 4.2.3) überführt. 100 ml Petrolether ( 3.2) werden in den Standzylinder gegeben, der mit einem Stopfen verschlossen und gründlich geschüttelt wird. Nach der Phasentrennung wird die obere Phase wie zuvor in den Scheidetrichter überführt. Diese Extraktion wird noch 1-mal mit 100 ml Petrolether (3.2) und 2-mal mit je 50 ml Petrolether ( 3.2) wiederholt, und die Petroletherphasen werden in den Scheidetrichter überführt.
Die vereinigten Petroletherextrakte, wie unter 5.3.1 erläutert, waschen und wie dort beschrieben weiterverfahren.
5.4 Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (gemäß 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben ( 4.2.4) pipettiert. Das Lösungsmittel wird unter vermindertem Druck und bei einer Badtemperatur von höchstens 40 °C am Rotationsverdampfer ( 4.1) fast bis zur Trockne eingedampft. Danach wird unter Stickstoff ( 3.10) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom ( 3.10) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10 bis 100 ml) Methanol ( 3.3) aufgenommen (die Konzentration von Vitamin a muss etwa 5 bis 30 IE/ml betragen).
5.5 Bestimmung durch HPLC
Die Abtrennung von Vitamin a erfolgt mithilfe einer Umkehrphasen-C18-Säule ( 4.5.1), und die Konzentration wird mithilfe eines UV-Detektors (325 nm) oder eines Fluoreszenzdetektors (Anregung: 325 nm, Emission: 475 nm) ( 4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z.B. 20l) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase ( 3.9) eluiert. Die mittlere Peakhöhe (-fläche) von mehreren Einspritzungen derselben Probenlösung wird ermittelt. In gleicher Weise werden auch die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen ( 5.6.2) bestimmt.
HPLC - Bedingungen
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| HPLC-Trennsäule (4.5.1): | 250 × 4 mm, C18, 5 oder 10m Korngröße, oder vergleichbare Säule |
| Mobile Phase ( 3.9): | Mischung von Methanol ( 3.3) und Wasser, z.B. 980 + 20 (V+V). |
| Durchflussrate: | 1 bis 2 ml/min |
| Detektor ( 4.5.2): | UV-Detektor (325 nm) oder Fluoreszenzdetektor (Anregung: 325 nm, Emission: 475 nm) |
5.6 Kalibrierung
5.6.1 Herstellender Gebrauchsstandardlösungen
Von der Vitamin-A-Acetat-Stammlösung ( 3.11.1) oder der Vitamin-A-Palmitat-Stammlösung ( 3.12.1) 20 ml in einen 500-ml-Steh- oder Erlenmeyerkolben ( 4.2.1) pipettieren und, wie unter 5.2 beschrieben, aber ohne Zugabe von BHT, hydrolysieren. Anschließend mit Petrolether ( 3.2) gemäß 5.3 extrahieren und mit Petrolether ( 3.2) auf 500 ml auffüllen. 100 ml dieses Extrakts werden am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft. Das verbleibende Lösungsmittel wird im Stickstoffstrom ( 3.10) abgeblasen, und der Rückstand wird in 10,0 ml Methanol ( 3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 560 IE Vitamin a je ml. Der genaue Gehalt ist nach 5.6.3.3 zu bestimmen. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
Von dieser Gebrauchsstandardlösung werden 2,0 ml in einen 20-ml-Messkolben pipettiert. Es wird mit Methanol ( 3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser verdünnten Gebrauchsstandardlösung beträgt 56 IE Vitamin a je ml.
5.6.2 Herstellender Kalibrierlösungen und Erstellung der Kalibrationskurve
Von der verdünnten Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 5,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert. Es wird mit Methanol ( 3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,8 IE, 5,6 IE, 14,0 IE bzw. 28,0 IE Vitamin a je ml.
Von jeder Kalibrierlösung werden mehrmals 20 μl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) und unter Berücksichtigung der Ergebnisse der UV-Kontrolle ( 5.6.3.3) wird eine Kalibrationskurve erstellt.
5.6.3 UV- Kontrolleder Standardlösungen
5.6.3.1 Vitamin - a - Acetat - Stammlösung
Von der Vitamin-A-Acetat-Stammlösung ( 3.11.1) werden 2,0 ml in einen 50-ml-Messkolben ( 4.2.2) pipettiert, und es wird mit 2-Propanol ( 3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin a je ml. Von dieser verdünnten Vitamin-A-Acetat-Lösung werden 3,0 ml in einen 25-ml-Messkolben pipettiert; es wird mit 2-Propanol ( 3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin a je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol ( 3.8) im Spektralfotometer ( 4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
| (E | 1 % 1 cm |
E für Vitamin-A-Acetat = 1.530 bei 326 nm in 2-Propanol) |
5.6.3.2 Vitamin - a - Palmitat - Stammlösung
Von der Vitamin-A-Palmitat-Stammlösung ( 3.12.1) werden 2,0 ml in einen 50-ml-Messkolben ( 4.2.2) pipettiert, und es wird mit 2-Propanol ( 3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin a je ml. Von dieser verdünnten Vitamin-A-Palmitat-Lösung werden 3,0 ml in einen 25-ml-Messkolben pipettiert; es wird zur Marke mit 2-Propanol ( 3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin a je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol ( 3.8) im Spektralfotometer ( 4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
| (E | 1 % 1 cm |
E für Vitamin-A-Palmitat = 957 bei 326 nm in 2-Propanol) |
5.6.3.3 Vitamin - a - Gebrauchsstandardlösung
Von der gemäß 5.6.1 hergestellten, unverdünnten Vitamin-A-Gebrauchsstandardlösung werden 3,0 ml in einen 50-ml-Messkolben ( 4.2.2) pipettiert, und es wird mit 2-Propanol ( 3.8) zur Marke aufgefüllt. 5,0 ml dieser Lösung werden in einen 25-ml-Messkolben pipettiert; es wird mit 2-Propanol ( 3.8) zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin a je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol ( 3.8) im Spektralfotometer ( 4.6) zwischen 300 und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E325 × 18,3
| (E | 1 % 1 cm |
E für Vitamin-A-Alkohol = 1 821 bei 325 nm in 2-Propanol) |
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-A-Peaks der Probenlösung wird anhand der Kalibrationskurve ( 5.6.2) die Konzentration der Probenlösung in IE/ml bestimmt.
Der Vitamin-A-Gehalt w (in IE/kg) der Probe wird nach folgender Formel berechnet: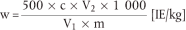
wobei:
c = Vitamin-A-Konzentration der Probenlösung ( 5.4) in IE/ml,
V1 = Volumen der Probenlösung ( 5.4) in ml,
V2 = Volumen des unter 5.4 entnommenen aliquoten Teils in ml,
m = Probeneinwaage in g.
7. Bemerkungen
7.1 Bei Proben mit niedriger Vitamin-A-Konzentration kann es zweckmäßig sein, die Petroletherextrakte von 2 Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen.
7.2 Die Einwaage für die Analyse darf höchstens 2 g Fett enthalten.
7.3 Bei schlechter Phasentrennung können etwa 10 ml Ethanol ( 3.1) zur Zerstörung der Emulsion zugegeben werden.
7.4 Bei Lebertran oder anderen reinen Fetten ist die Verseifungsdauer auf 45 bis 60 min zu erhöhen.
7.5 Statt BHT kann Hydrochinon verwendet werden.
7.6 Bei Verwendung einer Normalphasensäule ist die Trennung der Retinolisomeren möglich. Allerdings müssen in diesem Fall für die Berechnungen die Peakhöhen (-flächen) aller cis- und trans-Isomeren addiert werden.
7.7 Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden.
7.8 Statt Natriumsulfid-Lösung können etwa 50 mg EDTa verwendet werden.
7.9 Bei der Bestimmung des Gehalts an Vitamin a in Milchaustausch-Futtermitteln sind folgende Aspekte zu berücksichtigen:
Um festzustellen, ob die angewandte Analysemethode zuverlässige Ergebnisse für diese spezifische Matrix (Milchaustausch-Futtermittel) liefert, ist ein Wiederfindungstest an einer weiteren Probeneinwaage durchzuführen. Ist die Wiederfindungsrate kleiner als 80 %, muss das Analyseergebnis um die Wiederfindung korrigiert werden.
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 15 % des höheren Werts nicht überschreiten.
9. Ergebnisse eines Ringversuchs1
| Vormischungen | Fertigfuttervor- mischungen |
Meralfutter | Einweiß- konzentrat |
Ferkelauf- zuchtfutter |
|
| L | 13 | 12 | 13 | 12 | 13 |
| n | 48 | 45 | 47 | 46 | 49 |
| Mittelwert [IE/kg] |
17,02 × 106 | 1,21 × 106 | 537 100 | 151 800 | 18 070 |
| sr [IE/kg] | 0,51 × 106 | 0,039 × 106 | 22 080 | 12 280 | 682 |
| r [IE/kg] | 1,43 × 106 | 0,109 × 106 | 61 824 | 34 384 | 1 910 |
| VKr[%] | 3,0 | 3,5 | 4,1 | 8,1 | 3,8 |
| SR[IE/kg] | 1,36 × 106 | 0,069 × 106 | 46 300 | 23 060 | 3 614 |
| R [IE/kg] | 3,81 × 106 | 0,193 × 106 | 129 640 | 64 568 | 10 119 |
| VKR[%] | 8,0 | 6,2 | 8,6 | 15 | 20 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
r = Wiederholbarkeit
R = Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
Abbildung 1: Extraktionsapparat ( 4.8)
B. Bestimmung des Vitamin-E-Gehalts
1. Zweck und Anwendungsbereich
Die Methode erlaubt die Bestimmung des Vitamin-E-Gehalts von Futtermitteln und Vormischungen. Der Vitamin-E-Gehalt wird in mg DL-α-Tocopherol-Acetat je kg angegeben. 1 mg DL-α-Tocopherol-Acetat entspricht 0,91 mg DL-α-Tocopherol (Vitamin E).
Die Bestimmungsgrenze beträgt 2 mg Vitamin E/kg. Diese Grenze wird nur mit dem Fluoreszenzdetektor erreicht. Bei einem UV-Detektor beträgt die Bestimmungsgrenze 10 mg/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxidlösung hydrolysiert, und das Vitamin E wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft; der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-E-Gehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeitschromatografie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt.
3. Reagenzien
3.1 Ethanol, σ = 96 %.
3.2 Petrolether, Siedeintervall 40 bis 60 °C.
3.3 Methanol.
3.4 Kaliumhydroxid-Lösung, c = 50 g/100 ml.
3.5 Natriumascorbat-Lösung, c = 10 g/100 ml (siehe Bemerkung 7.7).
3.6 Natriumsulfid, Na2S × x H2O (x = 7 bis 9)
3.6.1 Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (für x = 9) (siehe Bemerkung 7.8).
3.7 Phenolphthalein-Lösung, c = 2 g/100 ml in Ethanol ( 3.1).
3.8 Mobile Phase für die HPLC: Mischung von Methanol ( 3.3) und Wasser, z.B. 980 + 20 (V+V). Das Mischungsverhältnis ist der jeweils verwendeten Säule anzupassen.
3.9 Stickstoff, sauerstofffrei.
3.10 DL-α-Tocopherol-Acetat, reinst, mit zertifizierter Aktivität
3.10.1 DL-α-Tocopherol-Acetat-Stammlösung: 100 mg DL-α-Tocopherol-Acetat ( 3.10) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In Ethanol ( 3.1) lösen und mit demselben Lösungsmittel zur Marke auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol-Acetat. (UV-Kontrolle siehe 5.6.1.3; Stabilisierung siehe Bemerkung 7.4).
3.11 DL-α-Tocopherol, reinst, mit zertifizierter Aktivität
3.11.1 DL-α-Tocopherol-Stammlösung: Von DL-α-Tocopherol ( 3.11) werden 100 mg auf 0,1 mg genau in einen 100-ml Messkolben eingewogen. In Ethanol ( 3.1) lösen und mit demselben Lösungsmittel zur Marke auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol. (UV-Kontrolle siehe 5.6.2.3; Stabilisierung siehe Bemerkung 7.4).
3.12 2,6-Di-tert-butyl-4-methylphenol (BHT) (siehe Bemerkung 7.5).
4. Geräte
4.1 Rotationsfilmverdampfer.
4.2 Braunglasgeräte
4.2.1 Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse.
4.2.2 Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml.
4.2.3 Scheidetrichter, konische Form, 1.000 ml, mit Schliffstopfen.
4.2.4 Spitzkolben, 250 ml, mit Schliffhülsen.
4.3 Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung.
4.4 Phasentrennungsfaltenfilter, Durchmesser 185 mm (z.B. Schleicher & Schuell 597 HY 1/2).
4.5 HPLC-Einrichtung mit Injektionssystem.
4.5.1 HPLC-Trennsäule, 250 × 4 mm, C18, 5 oder 10 μm Korngröße, oder vergleichbare Säule,
4.5.2 UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung.
4.6 Spektralfotometer mit Quarz-Küvetten von 10 mm Schichtdicke.
4.7 Wasserbad mit Magnetrührer.
4.8 Extraktionsapparat (Abbildung 1) bestehend aus:
4.8.1 1-l-Standzylinder mit Schliff und Schliffstopfen,
4.8.2 Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr muss ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann.
5. Verfahren
Anmerkung:
Vitamin E ist empfindlich gegenüber Licht (UV-Strahlung) und Oxidation. Daher muss unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder von mit Aluminiumfolie umhüllten Glasgeräten) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion muss das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen immer wieder lüften).
5.1 Vorbereitung der Probe
Die Probe wird unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-E-Verluste auftreten können.
5.2 Verseifung
Je nach Vitamin-E-Gehalt 2 bis 25 g der Probe auf 0,01 g genau in einen 500-ml-Steh- oder Erlenmeyerkolben ( 4.2.1) einwiegen. Nacheinander unter Schwenken 130 ml Ethanol ( 3.1), etwa 100 mg BHT ( 3.12), 2 ml Natriumascorbat-Lösung ( 3.5) und 2 ml Natriumsulfid-Lösung ( 3.6) zusetzen. Den Kolben mit dem Rückflusskühler ( 4.3) verbinden und in ein Wasserbad mit Magnetrührer ( 4.7) geben. Bis zum Sieden erhitzen und 5 min am Rückflusskühler kochen. Anschließend 25 ml Kaliumhydroxidlösung ( 3.4) durch den Rückflusskühler ( 4.3) zugeben, und unter ständigem Rühren und schwachem Stickstoffstrom 25 min weiterkochen. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3 Extraktion
Die Verseifungslösung wird mit insgesamt 250 ml Wasser quantitativ in einen 1 000-ml-Scheidetrichter ( 4.2.3) oder in den Extraktionsapparat ( 4.8) überspült. Der Verseifungskolben wird mit 25 ml Ethanol ( 3.1) und anschließend mit 100 ml Petrolether ( 3.2) nachgewaschen, und die Spülflüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen muss etwa 2:1 betragen. 2 min kräftig schütteln und dann 2 min absetzen lassen.
5.3.1 Extraktion mithilfe eines Scheidetrichters ( 4.2.3)
Nach Phasentrennung (siehe Bemerkung 7.3) wird die Petroletherphase in einen anderen Scheidetrichter ( 4.2.3) überführt. Diese Extraktion 2-mal mit je 100 ml Petrolether ( 3.2) und 2-mal mit je 50 ml Petrolether ( 3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter unter leichtem Schwenken (Vermeidung von Emulsionsbildung) 2-mal mit jeweils 100 ml Wasser gespült und dann durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung ( 3.7) farblos bleibt (4-maliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch einen trockenen Phasentrennungsfaltenfilter ( 4.4) in einen 500-ml-Messkolben ( 4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether ( 3.2) nachwaschen, mit Petrolether ( 3.2) zur Marke auffüllen und gut schütteln.
5.3.2 Extraktion mithilfe eines Extraktionsapparats (4.8)
Nach Phasentrennung (siehe Bemerkung 7.3) wird der Stopfen des Standzylinders ( 4.8.1) durch den Schliffeinsatz (4.8.2) ersetzt und das verschiebbare Rohr so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1.000-ml-Scheidetrichter ( 4.2.3) überführt. 100 ml Petrolether ( 3.2) werden in den Standzylinder gegeben, der mit einem Stopfen verschlossen und gründlich geschüttelt wird. Nach der Phasentrennung wird die obere Phase wie zuvor in den Scheidetrichter überführt. Diese Extraktion wird noch 1-mal mit 100 ml Petrolether ( 3.2) und 2-mal mit je 50 ml Petrolether ( 3.2) wiederholt, und die Petroletherphasen werden in den Scheidetrichter überführt.
Die vereinigten Petroletherextrakte, wie unter 5.3.1 erläutert, waschen und wie dort beschrieben weiterverfahren.
5.4. Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (gemäß 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben ( 4.2.4) pipettiert. Das Lösungsmittel wird unter vermindertem Druck und bei einer Badtemperatur von höchstens 40 °C am Rotationsverdampfer ( 4.1) fast bis zur Trockne eingedampft. Danach wird unter Stickstoff ( 3.9) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom ( 3.9) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10 bis 100 ml) Methanol ( 3.3) aufgenommen (die Konzentration von DL-α-Tocopherol muss etwa 5 bis 30 μg/ml betragen).
5.5 Bestimmung durch HPLC
Die Abtrennung von Vitamin E erfolgt mithilfe einer Umkehrphasen-C18-Säule ( 4.5.1), und die Konzentration wird mithilfe eines Fluoreszenzdetektors (Anregung: 295 nm, Emission: 330 nm) oder eines UV-Detektors (292 nm) ( 4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z.B. 20 µl) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase ( 3.8) eluiert. Die mittlere Peakhöhe (-fläche) von mehreren Einspritzungen derselben Probenlösung wird ermittelt. In gleicher Weise werden auch die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen ( 5.6.2) bestimmt.
HPLC - Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| HPLC-Trennsäule ( 4.5.1): | 250 × 4 mm, C18, 5 oder 10m Korngröße, oder vergleichbare Säule |
| Mobile Phase ( 3.8): | Mischung von Methanol ( 3.3) und Wasser z.B. 980 + 20 (V+V) |
| Durchflussrate: | 1 bis 2 ml/min |
| Detektor ( 4.5.2) | Fluoreszenzdetektor (Anregung: 295 nm; Emission 330 nm) oder UV-Detektor (292 nm) |
5.6 Kalibrierung (DL-α-Tocopherol-Acetat oder DL-α-Tocopherol)
5.6.1 DL-α-Tocopherol - Acetat - Standard
5.6.1.1 Herstellender Gebrauchsstandardlösung
Von der DL-α-Tocopherol-Acetat-Stammlösung ( 3.10.1) werden 25 ml in einen 500-ml-Steh- oder Erlenmeyerkolben ( 4.2.1) pipettiert und, wie unter 5.2 beschrieben, hydrolysiert. Anschließend mit Petrolether ( 3.2) gemäß 5.3 extrahieren und mit Petrolether auf 500 ml auffüllen. Von diesem Extrakt werden 25 ml am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft. Das verbleibende Lösungsmittel wird im Stickstoffstrom ( 3.9) abgeblasen, und der Rückstand wird in 25,0 ml Methanol ( 3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 45,5 µg DL-α-Tocopherol je ml; das entspricht 50 µg DL-α-Tocopherol-Acetat je ml. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
5.6.1.2 Herstellender Kalibrierlösungen und Erstellung der Kalibrationskurve
Von der Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 4,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert; es wird mit Methanol ( 3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,5 µg/ml, 5,0 µg/ml, 10,0 µg/ml bzw. 25,0 µg/ml DL-α -Tocopherol-Acetat, d. h. 2,28 µg/ml, 4,55 µg/ml, 9,10 µg/ml bzw. 22,8 µg/ml DL-α -Tocopherol.
Von jeder Kalibrierlösung werden mehrmals 20 µl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) wird eine Kalibrationskurve erstellt.
5.6.1.3 UV- Kontrolle der DL -α-Tocopherol - Acetat - Stammlösung (3.10.1)
Von der DL-α-Tocopherol-Acetat-Stammlösung ( 3.10.1) werden 5,0 ml mit Ethanol auf 25,0 ml aufgefüllt. Das UV-Spektrum dieser Lösung wird gegen Ethanol ( 3.1) im Spektralfotometer ( 4.6) zwischen 250 und 320 nm gemessen.
Das Extinktionsmaximum muss bei 284 nm liegen:
| E | 1 % 1 cm |
= 43,6 bei 284 nm in Ethanol |
Bei der vorliegenden Verdünnung muss eine Extinktion von 0,84 bis 0,88 erzielt werden.
5.6.2 DL -α -Tocopherol - Standard
5.6.2.1 Herstellender Gebrauchsstandardlösung
Von der DL-α-Tocopherol-Stammlösung ( 3.11.1) werden 2 ml in einen 50-ml-Messkolben pipettiert und in Methanol ( 3.3) gelöst; es wird mit Methanol zur Marke aufgefüllt. Der Sollgehalt dieser Lösung beträgt 40 µg DL-α-Tocopherol je ml; das entspricht 44,0 µg DL-α-Tocopherol-Acetat je ml. Die Gebrauchsstandardlösung muss vor Gebrauch frisch hergestellt werden.
5.6.2.2 Herstellen der Kalibrierlösungen und Erstellung der Kalibrationskurve
Von der Gebrauchsstandardlösung werden 1,0 ml, 2,0 ml, 4,0 ml bzw. 10,0 ml in jeweils einen 20-ml-Messkolben pipettiert; es wird mit Methanol (3.3) zur Marke aufgefüllt und gemischt. Der Sollgehalt dieser Lösungen beträgt 2,0 µg/ml, 4,0 µg/ml, 8,0 µg/ml bzw. 20,0 µg/ml DL-α -Tocopherol, d. h. 2,20 µg/ml, 4,40 µg/ml, 8,79g/ml bzw. 22,0 µg/ml DL-α -Tocopherol-Acetat.
Von jeder Kalibrierlösung werden mehrmals 20 µl eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Anhand der so ermittelten mittleren Peakhöhen (-flächen) wird eine Kalibrationskurve erstellt.
5.6.2.3 U V-Kontrolle der DL-α-Tocopherol - Stammlösung ( 3.11.1)
Von der DL-α-Tocopherol-Stammlösung ( 3.11.1) werden 2,0 ml mit Ethanol auf 25,0 ml aufgefüllt; das UV-Spektrum dieser Lösung wird gegen Ethanol ( 3.1) im Spektralfotometer ( 4.6) zwischen 250 und 320 nm gemessen. Das Extinktionsmaximum muss bei 292 nm liegen:
| E | 1 % 1 cm |
= 75,8 bei 292 nm in Ethanol |
Bei der vorliegenden Verdünnung muss eine Extinktion von 0,6 erzielt werden.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-E-Peaks der Probenlösung wird anhand der Kalibrationskurve ( 5.6.1.2 oder 5.6.2.2) die Konzentration der Probenlösung in µg/ml (berechnet als α-Tocopherol-Acetat) bestimmt.
Der Vitamin-E-Gehalt w (in mg/kg) der Probe wird nach folgender Formel berechnet: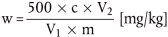
wobei:
c = Vitamin-E-Konzentration (als-Tocopherol-Acetat) der Probenlösung ( 5.4) in µg/ml,
V1 = Volumen der Probenlösung ( 5.4) in ml,
V2 = Volumen des unter 5.4 entnommenen aliquoten Teils in ml,
m = Probeneinwaage in g.
7. Bemerkungen
7.1 Bei Proben mit niedriger Vitamin-E-Konzentration kann es zweckmäßig sein, die Petroletherextrakte von 2 Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen.
7.2 Die Einwaage für die Analyse darf höchstens 2 g Fett enthalten.
7.3 Bei schlechter Phasentrennung können etwa 10 ml Ethanol ( 3.1) zur Zerstörung der Emulsion zugegeben werden.
7.4 Nach der spektralfotometrischen Vermessung der DL-α-Tocopherol-Acetat- oder der DL-α-Tocopherol-Lösung nach 5.6.1.3 bzw. 5.6.2.3 empfiehlt es sich, der Lösung ( 3.10.1 bzw. 3.10.2) etwa 10 mg BHT ( 3.12) zuzusetzen und die Lösung im Kühlschrank aufzubewahren (Haltbarkeit höchstens 4 Wochen).
7.5 Statt BHT kann Hydrochinon verwendet werden.
7.6 Bei Verwendung einer Normalphasensäule ist die Trennung von α-,β -,γ- und δ-Tocopherol möglich.
7.7 Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden.
7.8 Statt Natriumsulfid-Lösung können etwa 50 mg EDTa verwendet werden.
7.9 Vitamin-E-Acetat hydrolysiert unter alkalischen Bedingungen sehr schnell und ist deshalb sehr oxidationsanfällig, insbesondere in Gegenwart von Spurenelementen wie Eisen oder Kupfer. Bei der Bestimmung des Vitamin-EGehalts in Vormischungen, bei denen ein Gehalt von 5.000 mg/kg überschritten wird, könnte es zu einem Abbau von Vitamin E kommen. Deshalb wird zur Bestätigung eine HPLC-Methode mit einem enzymatischen Aufschluss der Vitamin-E-Formel ohne einen alkalischen Verseifungsschritt empfohlen.
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen an ein und derselben Probe darf 15 % des höheren Werts nicht überschreiten.
9. Ergebnisse eines Ringversuchs1
| Vormischungen | Fertigfuttervor- mischungen |
Mineralfutter | Eiweiß- konzentrat |
Ferkelaufzucht- futter |
|
| L | 12 | 12 | 12 | 12 | 12 |
| n | 48 | 48 | 48 | 48 | 48 |
| Mittelwert [mg/kg] | 17 380 | 1 187 | 926 | 315 | 61,3 |
| sr[mg/kg] | 384 | 45,3 | 25,2 | 13,0 | 2,3 |
| r [mg/kg] | 1 075 | 126,8 | 70,6 | 36,4 | 6,4 |
| VKr[ %] | 2,2 | 3,8 | 2,7 | 4,1 | 3,8 |
| SR [mg/kg] | 830 | 65,0 | 55,5 | 18,9 | 7,8 |
| R [mg/kg] | 2 324 | 182,0 | 155,4 | 52,9 | 21,8 |
| VKR[ %] | 4,8 | 5,5 | 6,0 | 6,0 | 12,7 |
L = Anzahl der Laboratorien
n = Anzahl der Einzelwerte
sr = Standardabweichung der Wiederholbarkeit
sR = Standardabweichung der Vergleichbarkeit
r = Wiederholbarkeit
R = Vergleichbarkeit
VKr = Variationskoeffizient der Wiederholbarkeit
VKR = Variationskoeffizient der Vergleichbarkeit
Abbildung 1: Extraktionsapparat ( 4.8)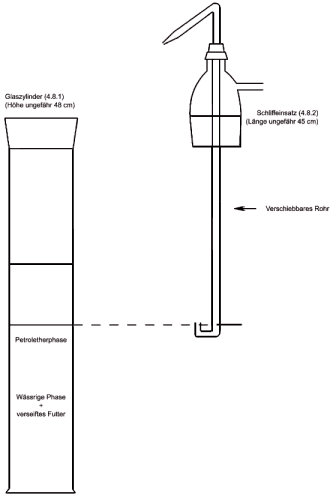
__________
1) Durchgeführt von der Fachgruppe Futtermittel des Verbands deutscher landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA).
|
weiter . | |
(Stand: 10.06.2022)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion
...
X
⍂
↑
↓