 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, Arbeitsschutz; Arbeits- und Sozialrecht

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, Arbeitsschutz; Arbeits- und Sozialrecht | |
Wissenschaftliche Begründung für eine neu in die Anlage zur Berufskrankheiten-Verordnung aufzunehmende Berufskrankheit
"Erkrankungen des Blutes, des blutbildenden und des lymphatischen Systems durch Benzol"
Vom 1. September 2007
(GMBl. Nr. 49-51 vom 12.11.2007 S. 974)
Siehe Fn. *
Zur Übersicht in Anlage 1 der BKV siehe =>
Zum Merkblatt 1303 siehe =>
Angesichts der Heterogenität der bisher unter der Nummer 1303 der Anlage zur Berufskrankheiten-Verordnung "Erkrankungen durch Benzol, seine Homologe oder Styrol" zusammengefassten Krankheitsbilder und der sie verursachenden Chemikalien ist es erforderlich, die durch Benzol verursachten Erkrankungen des Blutes, des blutbildenden und des lymphatischen Systems aus dieser Nummer auszugliedern und als eigene Berufskrankheit zu umreißen.
Die Berufskrankheit umfasst sowohl toxische Schädigungen (aplastische Anämie, Leukopenien, Thrombozytopenien und ihre Kombinationen) als auch maligne Erkrankungen (Leukämien, Non-Hodgkin-Lymphome, myelodysplastische Syndrome und myeloproliferative Erkrankungen).
Der Morbus Hodgkin ist nicht Gegenstand dieser wissenschaftlichen Begründung.
In der Begutachtungspraxis und bei den Rechtsanwendern herrschte und herrscht Konsens, dass "infolge seines pathophysiologischen Schädigungsmusters mit einer Alteration des hämatopoetischen Stammzellpooles Benzol alle malignen hämolymphatischen Systemerkrankungen, deren Zeltreihen sich von der omnipotenten Stammzelle ableiten, verursachen kann: Expositionsbedingungen mit langjähriger, chronischer beruflicher Benzolbelastung müssen vorliegen" (Schönberger et al. 1998, 2003). In 2001 erschienen in der deutschsprachigen arbeitsmedizinischen Literatur Artikel (Hoffmann et al. 2001, Tannapfel et al. 2001), die demgegenüber in Frage stellten, ob Benzol in reifen Lymphozyten eine ausreichende kanzerogene Wirkung entfalten kann, um periphere Non-Hodgkin-Lymphome zu verursachen; diesen folgte eine Entgegnung durch Woitowitz et al. (2003). Der Ärztliche Sachverständigenbeirat, Sektion Berufskrankheiten, hat diese Wissenschaftliche Begründung zum Anlass einer Überprüfung und Klarstellung genommen. Die Non-Hodgkin-Lymphome wurden daher im vorliegenden Text mit besonderer Ausführlichkeit behandelt.
Die Empfehlung des Ärztlichen Sachverständigenbeirats wird wie folgt begründet:
1. Aktueller Erkenntnisstand
1.1 Chemischphysikalische Charakteristik der ursächlich schädigenden Einwirkung
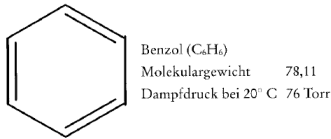
Strukturformel von Benzol
Benzol ist der einfachste aromatische Kohlenwasserstoff. Es ist in praktisch allen fossilen Brennstoffen enthalten und fällt u. a. bei der Destillation von Kohle und Erdöl sowie bei der Pyrolyse von organischem Material an.
1.2 Vorkommen und Gefahrenquellen
Benzol wurde in großem Umfang als Löse- und Reinigungsmittel in u. a. Druckereien, Waffenfabriken, metallverarbeitenden und anderen Betrieben sowie als Verdünner von flüssigen Klebern - insbesondere für die Herstellung von Schuhen - verwendet. Wegen seiner krebserregenden Wirkungen ist der Gebrauch inzwischen stark eingeschränkt. Allerdings gehen noch viele organische Synthesen von Benzol aus. Als erdölbedingter Begleitstoff ist Benzol praktisch immer in Autokraftstoffen als Antiklopfmittel enthalten. Wegen seiner geringeren Flüchtigkeit reichert es sich in "Reinigungsbenzin" an (Henschler 1992). Mit Benzolexposition ist daher u. a. in der erdölverarbeitenden Industrie, bei der Herstellung und dem Vertrieb von Kraftstoffen sowie beim Betrieb, der Wartung und der Reparatur von Verbrennungsmotoren zu rechnen.
Benzol als Handelsprodukt ist fast immer ein Gemisch. Häufig enthalten die im technischen Bereich verwendeten Homologe des Benzols zudem reines Benzol. Obwohl heute am Arbeitsplatz nur noch Zubereitungen verwendet werden dürfen, die weniger als 0,1 % Massengehalt Benzol enthalten und auch der Benzolgehalt in Kraftstoffen auf unter 1 % beschränkt ist, muss man im Hinblick auf Berufskrankheiten-Fragestellungen bedenken, dass in früheren Jahrzehnten der Benzolgehalt von technischen Benzingemischen auch ohne entsprechende Kennzeichnung bis zu 30 % oder höher betragen konnte.
Im Rahmen von retrospektiven Expositionsabschätzungen ist daher der Benzolgehalt der verwendeten Benzin- und Lösemittelgemische von großer Bedeutung.
Einen Überblick zu den wichtigsten benzolhaltigen Produkten, Beschichtungsstoffen, Oberflächenbehandlungsmitteln und zur Verwendung von Benzol in zahlreichen Industrie- und Gewerbebereichen bieten die "Anwendungshinweise zur retrospektiven Beurteilung der Benzolexposition" in der jeweils aktuellen Fassung des BGIA-Ringbuchs unter der Nr. 9105 (BGIA 2006), wobei in der derzeitigen Fassung für die retrospektive Schätzung bei besonders intensivem Hautkontakt mit Benzol kein ausreichender Hinweis gegeben werden kann.
1.3 Kenntnisse zur Wirkung am Menschen
Seit dem Ende des 19. Jahrhunderts ist bekannt, dass sich Benzol bei wiederholter, langdauernder, aber unter Umständen auch bei einmaliger massiver Einwirkung als Blutgift erweist. In Deutschland wurden daher bereits im Jahre 1925 Erkrankungen durch Benzol in die Liste der Berufskrankheiten aufgenommen. Die zugrunde liegende toxische Wirkung des Benzols auf das blutbildende Knochenmark äußert sich in der Regel in einer Reduktion der Zahl der Blutkörperchen im peripheren Blut. Diese Verminderung kann die weißen Blutzellen (Leukozyten), die roten Blutzellen (Erythrozyten) und die Blutplättchen (Thrombozyten) einzeln oder in Kombination betreffen (Lan et al. 2004). Während die knochenmarksdepressive Wirkung des Benzols in den meisten Fällen reversibel ist und sich das Blutbild nach Abbruch der Exposition (u. U. erst nach Jahren) normalisiert, können vereinzelt schwere irreversible Erkrankungen auftreten, die als aplastische Anämie bezeichnet werden, und in den Geltungsbereich dieser neuen Berufskrankheit fallen.
Seit mehreren Jahrzehnten ist erwiesen, dass es sich bei Benzol auch um einen humankanzerogenen Arbeitsstoff handelt. Benzol wurde 1971 von der Senatskommission zur Prüfung gesundheitsschädlicher Arbeitsstoffe der Deutschen Forschungsgemeinschaft als K1 Stoff und 1982 von der International Agency for Research an Cancer (IARC) der Weltgesundheitsorganisation (WHO) als gesichert krebserzeugend für den Menschen eingestuft.
Die krebserzeugende Wirkung wird von Stoffwechselprodukten verursacht, die im Organismus beim Benzolabbau gebildet werden. Als systemisches Kanzerogen verursacht Benzol im Tierversuch Tumoren unterschiedlicher Lokalisation (NTP 1986, Maltorsi et al. 1989, Huff et al. 1989). Entsprechend gibt es auch beim Menschen Hinweise auf benzolbeding,te Risikoerhöhungen für Krebserkrankungen der Niere, des Magens, des Dickdarms und der Lunge (Mehluran 2004). Da die Datenlage bezüglich solcher
Tumorlokalisationen jedoch bislang unzureichend und einheitlich ist, sind diese ausdrücklich nicht Gegenstand der vorliegenden Empfehlung.
1.3.1 Pathomechanismen
1.3.1.1 Aufnahme und Stoffwechsel
Die Aufnahme von Benzol am Arbeitsplatz erfolgt überwiegend inhalativ. Benzol wird gut resorbiert und verteilt sich schnell im gesamten Organismus. Da es sich bei Benzol um einen hautresorptiven Gefahrstoff handelt (Kennzeichnung mit "H" in der aktuellen Liste der MAK- und BAT-Werte der DPG) ist darüber hinaus die Aufnahme durch die Haut zu berücksichtigen. Eine zusammenfassende Auswertung der zur dermalen Exposition vorliegenden Literatur ergab, dass für offenen Hautkontakt eine dermale Penetrationsrate von bis zu 1 mg/cm2/h, für semiokklusive oder okklusive Verhältnisse (durchtränkte Arbeitskleidung oder Benzol auf der Innenseite von Schutzhandschuhen) allerdings die doppelte Penetrationsrate mit 2 mg/cm2/h angesetzt werden kann (Korinth et al. 2005). Dies stellt eine Worstcase-Abschätzung dar. Es ist zu berücksichtigen, dass die Studienpopulationen in den vorliegenden epidemiologischen Studien über Benzol alle auch dermal exponiert waren, obwohl in der Regel nur die inhalative Expositionskonzentration angegeben ist. Daher sollte die dermale Exposition vornehmlich dann besondere Berücksichtigung finden, wenn der Hautkontakt das übliche Maß überschritten hat.
Abb. 1: Benzol-Metabolismus in der Leber nach Lovern et al. (2001).
Die Bildung kanzerogener Metabolite erfolgt ausgehend von den hier gebildeten oxidativen Metaboliten Hydrochinon, Brenzkatechin und 1,2,4-Trihydroxybenzol, katalysiert durch Peroxidasen. Die sehr kurzlebigen Intermediate sind nicht benannt.
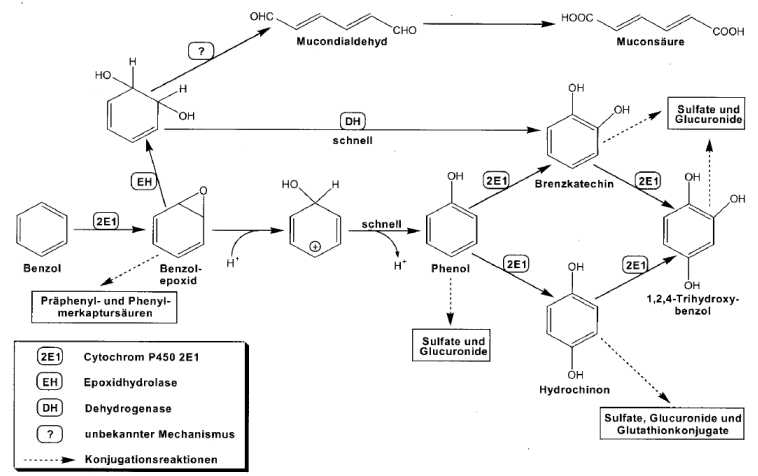
Die Oxidation von Benzol zu reaktiven Zwischenprodukten ist eine Voraussetzung für dessen Toxizität. Primäres Organ dieses Stoffwechsels ist die Leber. Eine Übersicht über den hepatischen Benzolstoffwechsel zeigt Abb. 1.
Als erstes Zwischenprodukt wird das Benzolepoxid gebildet, woraus spontan Phenol entsteht. In einem zweiten Stoffwechselweg entsteht aus Benzolepoxid das Brenzkatechin (Catechol). Es kann dabei intermediär eine Ringöffnung erfolgen; als Produkt entsteht die trans,trans-Muconsäure (Inoue et al. 1989). Ein reaktives Zwischenprodukt ist hierbei das trans,trans-Muconaldehyd, welches bislang lediglich in vitro, aber nicht in vivo nachgewiesen worden ist (Latriano et al. 1986). Die ringoffenen Metabolite machen allerdings nur einen geringen, jedoch in Abhängigkeit von der Expositionshöhe unterschiedlichen, Anteil der Stoffwechselprodukte von Benzol aus (Rothman et al. 1998).
Phenol hydroxyliert weiter zu Hydrochinon, Brenzkatechin (Catechol), 1,4-Benzochinon und Trihydroxybenzol (Lovern et al. 2001). Benzochinon und Hydrochinon können über epigenetische Mechanismen klastogen wirken: 1,4-Benzochinon hemmt die Bildung von Tubulin (Irons et al. 1981) und Hydrochinon die Topoisomerase II (Eastmond et al. 2005). Phenolische Metaboliten des Benzols führen zur Bildung reaktiver Sauerstoffspezies (ROS) und verursachen oxidative DNA-Schäden. Die Oxidation und nachfolgende Hydroxylierung wird durch Cytochrom P450-abhängige Monooxygenasen katalysiert, insbesondere Cytochrom P450 2E1 (CYP2E1). Die Bedeutung dieses Isoenzyms für die gesundheitsschädlichen Wirkungen von Benzol wird dadurch unterstrichen, dass eine fehlende CYP2E1-Aktivität in transgenen Mäusen zu einer Reduktion der Benzoltoxizität führte (Valentine et al. 1996). Ethanol verstärkt (induziert) zudem den Benzolmetabolismus, insbesondere durch Steigerung der CYP2E1-Aktivität (Nakajima et al. 1985, Sato und Nakajima 1985, Johansson und Ingelman-Sundberg 1988). Somit ist der CYP2E1 vermittelte Metabolismus ein wesentlicher Schritt für die Benzoltoxizität und Kanzerogenität.
Bei steigender Benzolbelastung wird ein zunehmend geringerer Anteil in Form von Metaboliten mit dem Harn ausgeschieden, während der als unverändertes Benzol abgeatmete Teil zunimmt. Die lineare Extrapolation von hohen auf niedrige Benzolkonzentrationen führt daher zu einer Unterschätzung der krebserzeugenden Wirkung niedriger Expositionen (Lovern et al. 2001).
Die oxidativen Stoffwechselprodukte unterliegen in der Phase II des Fremdstoffwechsels verschiedenen entgiftenden Konjugationsschritten, die von Sulfatasen, Glukuronidasen oder Glutathion-S-transferasen (GST) katalysiert werden. Hierdurch entstehen Phenylsulfat, verschiedene Glukuronkonjugate und Phenylmerkaptursäuren.
Die Frage, warum Benzol, jedoch nicht Phenol, hämatotoxisch ist, dürfte eine Folge der lokalen Verteilung der Stoffwechselaktivität im Lebergewebe sein. Exogen zugeführtes Phenol wird bereits im Gastrointestinaltrakt (bei oraler Aufnahme) oder im periportalen Bereich konjugiert und damit entgiftet. Im Körper aus Benzol gebildetes Phenol wird jedoch in der Leber erst in einem nachgeschalteten Areal, nämlich zentrilobulär, gebildet, so dass es der Konjugation weitgehend entgeht (Medinsky et al. 1996).
Die Gewebeverteilung der Stoffwechselaktivität ist heterogener als bislang angenommen. In der Leber wird nur 2 % des Benzols, allerdings bei größerer metabolischer Kapazität, zu Hydrochinon umgewandelt, im Lungengewebe dagegen 39 % (Chaney und Carlson 1995). Offenbar muss daher dem Stoffwechsel in der Lunge größere Beachtung geschenkt werden. Systematische Untersuchungen zur Organverteilung möglicher alkylierender Wirkungen von Benzol, die zur Klärung dieser Fragen beitragen können, sind selten. Die Analyse von Benzol-Addukten in B6C3F1 Mäusen nach Injektion von 25-880 mg/kg KG mit 32 P-Postlabelling führte zur Identifizierung von N2-(4-Hydroxyphenyl)-2"-deoxyguanosin-3"-phosphat. Das Addukt war in Knochenmarkszellen und in peripheren mononuklearen Blutzellen nachweisbar (Bodell et al. 1996).
Auffälligstes Zielorgan für die toxischen und krebserzeugenden Wirkungen von Benzol ist das blutbildende Knochenmark. Bei dem blutbildenden Knochenmark handelt es sich toxikokinetisch um ein "tiefes Kompartiment", welches für Schadstoffe schwer zugänglich ist. Zur Illustration sei darauf hingewiesen, dass in der Chemikalien- und Arzneistoffprüfung für tierexperimentelle Studien, die Zielzellen im Knochenmark nutzen (z.B. Mikrokerntest aus dem Mäusefemur), der Nachweis verlangt wird, dass die Testsubstanz in das Knochenmark gelangen kann. Im Umkehrschluss ist daher davon auszugehen, dass Metabolite, die im Knochenmark toxische Wirkungen entfalten, jedes hydrophile Kompartiment des Körpers erreichen. Das aus Benzol gebildete Benzolepoxid war bei (allerdings) 400 mg/kg hoher inhalativer Exposition von Ratten stabil genug, um das Knochenmark zu erreichen (Lindstrom et al. 1997). Nach Benzolexposition von Mäusen und Ratten wurden Hämoglobinaddukte und Albuminaddukte des Benzolepoxids im Blut, Knochenmark und Harn der Tiere nachgewiesen (Krewet et al. 1993, McDonald et al. 1994). Creek et al. (1997) wiesen mit 14 C-markiertem Benzol in Mäusen nach, dass eine Bindung an die DNA und an Proteine bereits bei so niedrigen Benzolkonzentrationen in der Atemluft erfolgt, wie sie der umweltbedingten Belastung des Menschen entspricht. Bei niedrigen Benzolkonzentrationen war die DNA-Bindung linear zur applizierten Konzentration, die Proteinbindung annähernd linear. Albuminaddukte des Epoxids konnten im Harn benzolexponierter Menschen nachgewiesen werden (Bechtold et al. 1992). Eine Identifizierung dieser Bindungsprodukte war allerdings nicht möglich.
Die in der Leber gebildeten Benzolmetaboliten verteilen sich somit im gesamten Körper. Nachdem sie unter anderem das Knochenmark erreicht haben, werden Hydrochinon, Brenzkatechin (Catechol) und 1,2,4-Trihydroxybenzol durch Peroxidasen zu reaktiven Intermediaten umgewandelt, die an die DNA und an andere Makromoleküle binden können. 1,4-Benzochinon wird als die wichtigste Ausgangsverbindung für die Hämatotoxizität und Leukämogenität angesehen. Aus Benzochinon oder Hydrochinon entstehen durch Peroxidasekatalysierte Elektronenübertragung freie Radikale, die über vielfältige Mechanismen DNA schädigen können. Wegen der Kurzlebigkeit dieser Intermediate und dem resultierenden Fehlen nennenswerter Mengen an DNA-Addukten gelang es bislang nicht, einen dieser Metabolite zu identifizieren. Abb. 2 gibt eine Übersicht über den hepatischen und den extrahepatischen (insbesondere knochenmarksständigen) Metabolismus von Benzol wieder.
Abb. 2: Benzol-Metabolismus nach Rothman et al. (1997).
CYP2E1 = Cytochrom P450 2E1,
NQO1 = Chinonoxidoreduktase 1,
MPO = Myeloperoxidase
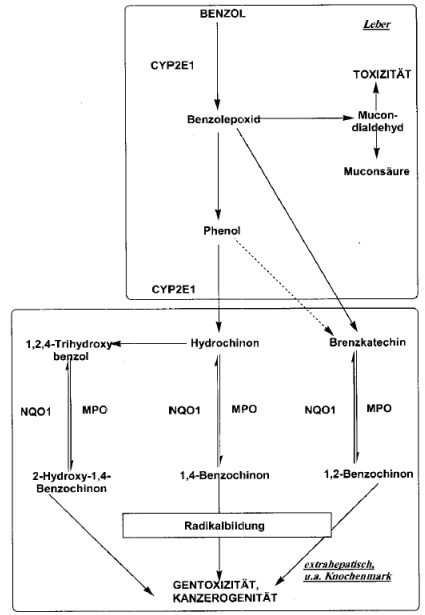
Wegen der großen exprimierten Mengen in verschiedenen weißen Blutzellen wird der Myeloperoxidase (MPO) eine wesentliche Rolle bei dieser Aktivierung zugeschrieben. Die Myeloperoxidase ist ein neutrophiles Protein und wird zu den Hämproteinen gerechnet. Sie ist in neutrophilen Granulozyten und in Monozyten lokalisiert, von wo sie u. a. bei der Phagozytose Freigesetzt wird. Physiologisch katalysiert MPO im Rahmen körpereigener Abwehrreaktionen die Reaktion von Wasserstoffperoxid und Chlorid zu dem hoch toxischen Hypochlorid, das u. a. bakterizid und nematozid wirkt. In Anwesenheit von Wasserstoffperoxid kann MPO jedoch auch phenolische Verbindungen zu radikalischen Intermediaten umsetzen.
Myeloperoxidase liegt sowohl in CD34+ Vorläuferzellen, wie auch in peripheren Neutrophilen und Monozyten inaktiv und streng kompartimentiert in Granula vor und wird beispielsweise während entzündlicher Reaktionen aktiviert. Durch Myeloperoxidase katalysierte Reaktionen benötigen H2O2als Co-Substrat (Winterbourn und Kettle 2004). H2O2 und Myeloperoxidase werden parallel an Orten der Immunabwehr durch Zellen des natürlichen (angeborenen) Immunsystems induziert; insbesondere auch in peripheren Zellen, u. a. in Monozyten und Neutrophilen. Auch im Knochenmark bedarf es erst der Induktion der Myeloperoxidase. Ob und wie Myeloperoxidase durch Benzol oder seine Metabolite aktiviert wird, ist bislang nicht bekannt. Weiterhin kann nicht ausgeschlossen werden, dass auch andere Peroxidasen Benzolmetabolite aktivieren können.
Myeloperoxidase katalysiert eine Vielzahl von Oxidationsreaktionen, die für die antimikrobielle Abwehr wesentlich sind. Vornehmlich werden halogenierte (chlorierte) Verbindungen oxidiert, wobei Hypochlorsäure als Hauptprodukt der Myeloperoxidase entsteht. Allerdings ist dies auch mit einer entzündungsfördernden Wirkung verbunden; daher wird die Myeloperoxidase mit der Pathogenese einer Reihe von entzündlichen Erkrankungen, u. a. dem Reperfusionsschaden nach Ischämie, dem Atemnotsyndrom RDS (respiratory distress syndrome), der Glomerulonephritis, der Arthritis, dem peptischen Ulcus und dem Magenkarzinom in Verbindung gebracht. Verschiedene seltene genetische Varianten der MPO ohne oder mit verminderter Aktivität werden beschrieben. Auswirkungen auf die Empfindlichkeit gegenüber Benzol sind nicht bekannt (Winterbourn et al. 2000).
Das Enzym NAD(P)H-Chinonoxidoreduktase 1 (NQO1) reduziert 1,4-Benzochinon zurück zum Hydrochinon. Entsprechendes gilt auch für die Benzochinon-Analoga von Brenzkatechin und Trihydroxybenzol. Dies stellt einen wesentlichen Entgiftungsmechanismus für die gentoxischen Chinone dar. Normalerweise ist im Knochenmark des Menschen keine Aktivität der NQO1 nachweisbar. Benzolmetabolite induzieren jedoch die NQO1-Aktivität drastisch (Moran et al. 1999).
Die Toxizität im Vorläuferzell-(Stammzell-)Kompartiment des Blutsystems ist für die Entwicklung der mit der Benzolexposition assoziierten hämatolymphatischen Störungen von wesentlicher, wenn auch nicht ausschließlicher, Bedeutung (Bonnet und Dick 1997). Die Empfindlichkeit der Stammzellen für die durch Benzolmetabolite induzierte Toxizität wird einerseits auf die hohe Expression von Myeloperoxidase (Strobl et al. 1993, Schattenberg et al. 1994) zurückgeführt. Andererseits wird diese Wirkung durch eine schwache Entgiftung bei mangelnder Expression von NQO1 (Ross et al. 1996, Trush et al. 1996, Ganousis et al. 1992) verstärkt. Möglicherweise spielt beim Menschen auch Glutathion für die Entgiftung eine Rolle (Li et al. 1994).
Zur Veranschaulichung der entscheidenden Stoffwechselvorgänge und der Rolle der Enzyme CYP2E1, Myeloperoxidase (MPO) und Chinonoxidoreduktase (NQO1) ist in Abb. 3 eine stark vereinfachte Zusammenfassung der Stoffwechselwege wiedergegeben.
Weitere Peroxidasen, die zur Oxidation der phenolischen Metabolite beitragen, sind die Prostaglandin-H-Synthase (Gaido und Wierda 1987) und die eosinophile Peroxidase. Die Prostaglandinsynthase, die als Teilaktivitäten die Cyclooxygenase und die Hydroperoxidase einschließt, kommt in praktisch allen Zellen vor, insbesondere auch in Makrophagen und Lymphozyten. Phenol und Hydrochinon sind Cofaktoren und Aktivatoren der Prostaglandin-H-Synthase. Phenole werden von dem Enzym als Reduktionsmittel bei der Oxidation von Arachidonsäure benutzt. Dabei entstehen Semichinon und Benzochinon (Eling et al. 1990, Marnett 1990). Neben der Bildung dieser gentoxischen Produkte ist von Bedeutung, dass Produkte von Prostaglandinsynthasekatalysierten Reaktionen, insbesondere Prostaglandin E2, eine in epithelialen Tumorsystemen nachgewiesene promovierende Wirkung entfalten (Levy 1997, Williams et al. 1999).
Ein Beleg für die Bedeutung der Prostaglandin-H-Synthase für die toxische und kanzerogene Wirkung von Benzol ist die wiederholte Beobachtung, dass Inhibitoren der Prostaglandin-H-Synthase, insbesondere Indometazin, in vivo die toxische Wirkung von Benzol auf das blutbildende System hemmen (Gaido und Wierda 1987, Kalf et al. 1989).
Sowohl die Myeloperoxidase als auch die Prostaglandin-H-Synthase sind im Organismus des Menschen und anderer Spezies Schlüsselenzyme im alltäglichen Entzündungsgeschehen. Entzündungsreaktionen lassen sich experimentell durch Phorbolacetat (PMA) oder bakterielles Lipopolysaccharid (LPS) stimulieren. Die Interaktion zwischen Entzündungsreaktionen und der gentoxischen Wirkung von Benzol konnte so in Knochenmarkszellen wie auch in peripheren Lymphozyten in vitro und in vivo nachgewiesen werden. Durch Gabe von PMA bei den Invitro-Versuchen wurde ein "oxidative burst" in den Knochenmarkszellen und Lymphozyten induziert. Gentoxische Effekte wurden durch Bestimmung von 8-Oxodesoxyguanosin und dem Nachweis von DNA-Strangbrüchen mittels Comet-Assay quantifiziert. Die kombinierte Verabreichung von PMA bzw. LPS und Benzol verursachte signifikant höhere gentoxische Effekte in vitro und in vivo als in der Summe der durch PMA/ LPS oder Benzol allein induzierten Wirkung (Tuo et al. 1999).
Abb. 3 : Stark vereinfachtes Stoffwechselschema von Benzol mit einer Fokussierung auf die für die tumorerzeugende Wirkung wesentliche Bildung von Benzochinonverbindungen. Aus den Benzochinonen entstehen im Knochenmark oder im lymphatischen Gewebe gentoxische Radikale. Näheres siehe Abschnitt 1.3.1.2: Molekularbiologischonkologische Aspekte

Genetische Polymorphismen der beteiligten Enzyme spielen eine wesentliche Rolle für das individuelle Risiko der Benzoltoxizität. Das Enzym NAD(P)H-Chinonoxidoreduktase 1 (NQO1) reduziert das Chinon (siehe oben). Ein Polymorphismus (C gegen T an der Position 609) führt bei homozygoten Merkmalsträgern zu einem vollständigen Verlust der Enzymaktivität (Moran et M. 1999). Eine erhöhte Hämatotoxizität bei benzolexponierten Arbeitern mit diesem Polymorphismus wurde beschrieben: Eine Fallkontrollstudie an 11.117 benzolexponierten Beschäftigten in China zeigte ein erhöhtes Risiko für Hämatotoxizität für Personen mit dem T/T-Genotyp (homozygot, beide Allele mutiert) für die NQO1. Der Polymorphismus des CYP2E1 alleine zeigte keinen Effekt; Personen mit der Kombination einer hohen CYP2E1-Expression und homozygoter Defizienz von NQO1 hatten jedoch das 7,6-fache Risiko eines Malignoms des blutbildenden Systems (Rothman et al. 1997).
In einer Fallkontroll-Studie wurden erwachsene Patienten mit akuter Leukämie mit gematchten Kontrollen verglichen. Eine niedrige NAD(P)H-Chinonoxidoreduktase 1-Aktivität war signifikant assoziiert mit einem erhöhten Risiko für akute Leukämie, und zwar sowohl für die myeloische als auch für die lymphatische Form. Die Autoren konnten diesen Befund an einem zweiten Kollektiv verifizieren (Smith et al. 2001). Ein modulierender Einfluss auf den Benzolmetabolismus mit postulierten Folgen für benzolinduzierte Gesundheitsrisiken wurde auch für die Polymorphismen von CYP2D6, GSTT1 und NQO1 festgestellt (Rossi et al. 1999).
1.3.1.2 Molekularbiologischonkologische Aspekte
Krebs hat eine komplexe Entstehung, die häufig multikausal erfolgt. Eine genetische Veränderung im Erbgut (DNA), die Initiation, ist der erste Schritt der Krebsentstehung. Die initiierte Zelle unterliegt einer klonalen Selektion, d. h. ihre Vermehrung wird in einer Abfolge von Schritten gegenüber der Vermehrung anderer (gesunder) Zellen bevorteilt. Jede Stufe der Kanzerogenese beinhaltet erneute genetische Veränderungen (z.B. die Einschaltung von "Krebsgenen").
Nach Hanahan und Weinberg (2000) muss ein Gewebe mindestens sechs essentielle Eigenschaften aufweisen, um als Krebs angesehen zu werden. Es sind dies die Unabhängigkeit von externen Wachstumssignalen, die Unempfindlichkeit gegenüber wachstumshemmenden Signalen, die weitgehende Ausschaltung der Apoptose, eine uneingeschränkte Fähigkeit der Genomreplikation, die eigenständige Induktion der Blutgefäßbildung sowie die Fähigkeit, in gesunde Gewebe einzudringen und Metastasen zu bilden. Die vielfältigen miteinander interagierenden Signalwege einer gesunden Zelle ergeben eine große Zahl von Angriffspunkten für ein Kanzerogen. Diesen stehen umfangreiche Schutzfunktionen und Systemredundanzen gegenüber, so dass in der Regel mehrere "hits" notwendig sind, um lediglich eine bestimmte kanzerogene Eigenschaft herbeizuführen.
Gleichzeitig oder sukzessive einwirkende krebserzeugende Noxen können am gleichen oder an verschiedenen dieser Angriffspunkte wirken. So kann ein krebserzeugender Stoff initiierend (einen genetischen Schaden verursachend) oder promovierend sein (eine bestehende Läsion zu einem Tumor fortentwickeln). Benzol - wie die Mehrzahl der krebserzeugenden Stoffe - ist in seinen Stoffwechselprodukten ein "komplettes" Kanzerogen, das sowohl initiierend als auch promovierend auf verschiedenen Stufen der Krebsentstehung wirksam ist. Wegen der irreversiblen, chromosomenschädigenden (klastogenen) Effekte, die Benzol verursacht, wirken in zeitlicher Abfolge aufgenommene Dosen kumulativ und mindestens additiv.
Invitro-Untersuchungen zu chromosomenschädigenden Wirkungen von Benzol mit Mikroorganismen waren negativ (DFG 1992) bis auf einen vereinzelten Befund mit dem Ames Test und gasdichter Vorinkubation (Glatt et al. 1989). Hinsichtlich dieser Untersuchung bleibt unklar, welche mutagenen Benzol-Metabolite evaporieren können.
Die Validität der Invitro-Untersuchungen zu Benzol und seiner Metaboliten leidet durchweg unter den sehr hohen eingesetzten Konzentrationen, die im Bereich zytotoxischer Konzentrationen lagen. 1,4-Benzochinon und Hydrochinon induzierten Mikrokerne in humanen Lymphozyten in vitro (Yager et al. 1990). Hydrochinon und Trihydroxybenzol (THB) führten in HL-60 Zellen neben THB-DNA-Addukten zu oxidativem Stress (Kolachana et al. 1993, Hedli et al. 1996). Phenol, Hydrochinon und Benzochinon bildeten DNA-Addukte in vitro (Reddy et al. 1990, Levay et al. 1993, Bodell et al. 1993, Pathak et al. 1995, Levay et al. 1996). Bis zu 5 mM Benzol induzierten weder mit noch ohne metabolischem Aktivierungssystem Mikrokerne in menschlichen Lymphozyten (Zarani et al. 1999).
"Falsch negative" Ergebnisse mit Benzol in vitro können auf komplexe Interaktionen in verschiedenen Kompartimenten hinweisen (Leber und Knochenmark), die in vitro nicht nachgestellt werden können. Wegen der hohen Komplexizität des Benzolmetabolismus wurde gefolgert, dass Invitro-Untersuchungen hier generell von limitierter Aussagefähigkeit seien (Eastmond 2000).
Tatsächlich bewirkt Benzol in zahlreichen tierexperimentellen Untersuchungen die Induktion von Chromosomenaberrationen, Mikrokernen und Schwesterchromatidaustausch (SCE) (zusammengefasst in DFG 1992, Snyder et al. 1993). Dagegen ist die Bildung von DNA-Addukten nicht eindeutig nachweisbar (Snyder et al. 1993). Die inhalative Exposition von Mäusen gegen 40 bis 1000 ppb Benzol über sechs Wochen führte zu einer dosisabhängigen Vermehrung von Mutationen im hprt-Locus in Lymphozyten (Ward et al. 1992). Vermehrte Mutationen im lacl-Transgen wurden nach zwölfwöchiger Exposition von Mäusen gegen 300 ppm Benzol in Lunge und Milz, aber nicht in der Leber festgestellt (Mullin et al. 1995). Bei oraler Benzolexposition wurde in Mäusen eine dosisabhängige Bildung von Mikrokernen im Knochenmark beobachtet. Die Fluoreszenzin-situ-Hybridisierung (FISH) zeigte vornehmlich Chromosomenbrüche im Euchromatin, dagegen nur in geringem Ausmaß im Heterochromatin. Ein Test auf Induktion von Aneuploidie zeigte nur einen marginalen Effekt (Eastmond et al. 2001). Neuere Untersuchungen belegen, dass die Benzolmetaboliten trans,trans-Muconaldehyd und Hydrochinon die interzelluläre Kommunikation von Zellen (gap junctions) wirksam inhibieren (Rivedal und Witz 2005).
Promovierende Wirkungen von Benzol manifestieren sich durch Einflüsse auf die Regulation des Zellzyklus. Ein zentraler Faktor in der Zellzyklusregulation von hämatopoetischen Vorläuferzellen ist GM-CSF (granulocyte/macrophage colonystimulating factor). In der Peripherie aktiviert GM-CSF TH1-Zellen. Veränderungen in der Empfindlichkeit bestimmter Zellpopulationen gegen GM-CSF finden sich häufig bei myeloproliferativen Störungen (Review: Irons 2000). Mit Hydrochinon behandelte CD34+ Voräuferzellen wurden empfindlich gegenüber GM-CSF (Irons et al. 1992, Gross et al. 1997). 0,5 - 4 µM Hydrochinon induzierten - ähnlich wie LTD4 - die Granulozyten-Differenzierung zu 32D-Myelözyten (Hazel und Kalf 1996). LTD vermittelte die intrazelluläre Signaltransduktion von GMCSF.
Die Tumorentstehung ist somit ein mehrstufiger Prozess, der von einer ersten Genmutation ausgeht und dann über weitere Schritte zu erhöhter Wachstumsautonomie führt (Aktivierung von Protoonkogenen oder Inaktivierung von Tumorsuppressor-Genen, Apoptoseresistenz, Verlust der Zellzykluskontrolle, klonale Expansion). Eine Zelle, die einen initialen Schaden im Knochenmark erlitten hat, kann unter Umständen weitere Teilungs- bzw. Reifungsschritte durchlaufen und erst später zusätzliche Schädigungen erleiden, die letztlich das Tumorgeschehen unumkehrbar machen. Selbst wenn ein Tumor sich von Zellen eines bestimmten Reifungsgrades ableitet, lässt sich nicht mit Sicherheit nachweisen, in welchen Reifungsstadien die jeweiligen Schritte der Kanzerisierung erfolgt sind. Zudem zirkulieren einige periphere Lymphozyten wieder in das Knochenmark (Homing-Phänomen).
Prädisponierend für die klonale Expansion maligne transformierter Zellen ist ihr "Microenvironmcnt"; mithin der umgebende Zellverband und die Signale, die von ihm ausgehen. Dies können u. a. wachstumshemmende aber auch proliferationsfördernde Signale sein. Pharmakokinetisch gesehen ist das Knochenmark kein geeignetes Ziel kanzerogener Benzolmetabolite, da es als "tiefes Kompartiment" von diesen Verbindungen nur verzögert und in geringer Konzentration erreicht wird. Da die hämatopoetischen Stammzellen jedoch eine besonders hohe Teilungsaktivität aufweisen, ist das blutbildende Knochenmark dennoch als Zielorgan von der krebserzeugenden Wirkung der Benzolmetabolite betroffen. Im Allgemeinen sind Zellen mit hoher Teilungsaktivität besonders vulnerabel, da sich hier gentoxische Schäden besonders leicht manifestieren und von vornherein eine hohe Wachstumsautonomie besteht.
Das "Microenvironment" für lymphatische, insbesondere TH1- und TH2-Zellen wird entscheidend nicht nur von Entzündungsprozessen, sondern insbesondere auch von den immunologischen humoralen Typ I- und zellulären Typ IV-Immunreaktionen nach Coombs und Gell determiniert. Nach Kontakt mit antigenpräsentierenden Zellen werden bei der Typ IV-Reaktion TH1-Zellklone zu massiver Expansion angeregt. Im Falle einer Typ I-Reaktion auf Antigene werden ebenso die prä-B-Zellen zur Proliferation angeregt. Beim zweiten Kontakt mit dem Antigen werden ausgereifte B- oder T-Zellen aktiviert. Die determinierenden Faktoren des "Microenvironment" der prä-B, prä-T, B- und T-Zellen sind demnach maßgeblich "antigenpräsentierende Zellen" bzw. Antigene.
Charakteristika von Zellen, die zur malignen Entartung neigen, sind Proliferationsneigung und die intrinsische Instabilität. In der frühen Entwicklungsphase von T- und B-Zellen werden "Antigen-Rezeptor-Variable" Domänen durch somatische Rekombination (VDJ Rekombinationen) generiert. Die somatische Rekombination ist örtlich und zeitlich hoch reguliert. Insgesamt ist die Bedeutung dieser Mechanismen in der Pathogenese maligner Erkrankungen des hämatopoetischen Systems noch nicht bis in alle Einzelheiten geklärt. Es ist jedoch davon auszugehen, dass gentoxische und epigenetische Phänomene sowie Wechselwirkungen mit Entzündungs- und Immunreaktionen bei der kanzerogenen Wirkung des Benzols auf das lymphatische System, d. h. bei der Entstehung von NHL, eine entscheidende Rolle spielen (EPA 1998).
Tierexperimentell lassen sich Zusammenhänge zwischen entzündlichen Reaktionen und Benzol-Toxizität zeigen. Oxidative Benzolschäden können mit PMA (Phorbol-12-Acetat-13-Myristat) oder LPS (bakterielle Lipopolysaccharide) induziert werden. Sie sind besonders ausgeprägt im Knochenmark von Mäusen, können aber auch in peripheren Blutzellen nachgewiesen werden (Tuo et al. 1999).
1.3.1.3 Zytogenetische Untersuchungen an Lymphozyten benzolexponierter Menschen
In zahlreichen Studien wurden vermehrte chromosomale Veränderungen in den Lymphozyten von benzolexponierten Beschäftigten erfasst. In peripheren Lymphozyten von 20 englischen Fabrikarbeitern, die 1 bis 20 Jahre gegen Benzol exponiert waren, wurden instabile strukturelle Chromosomenanomalien, Ringchromosomen und dizentrische Chromosomen nachgewiesen, die signifikant häufiger waren als in Vergleichspopulationen (Tough et al. 1970). Bei 25 Personen, die vor Jahren eine schwere Knochenmarkschädigung durch Benzol erlitten hatten, wurden in den Lymphozyten Chromosomenanomalien (Brüche sowie stabile und instabile Aberrationen) in einer signifikant höheren Häufigkeit nachgewiesen als bei gleichaltrigen Kontrollpersonen. Diese Veränderungen persistierten über Jahrzehnte; bei den einzelnen Personen waren im Verlauf sowohl Zunahmen, gleichbleibende Häufigkeiten oder Abnahmen der Anomalien zu verzeichnen. Die Autoren bemerkten, dass bei Patienten nach Röntgen- oder y-Bestrahlung ähnliche Lymphozytenschäden zu beobachten seien (Forni 1971).
In den peripheren Lymphozyten von 52 Arbeitern, die über Zeiträume zwischen einem Monat und 26 Jahren niedrigen Benzolkonzentrationen (Schichtmittelwert 2,1 ppm) ausgesetzt waren, wurden eine Verdopplung von Chromosomenbrüchen, eine Verdrcifachung von Translokationen und weitere anomale Chromosomenveränderungen im Vergleich zum Kontrollkollektiv registriert (Picciano 1979).
Bei 20 gegenüber Benzol und Toluol exponierten Arbeiterinnen einer Schuhfabrik zeigten die Lymphozyten vermehrte DNA-Strangbrüche und Schwesterchromatidaustausch (SCE) (Popp et al. 1992). In einer weiteren Studie wurden 49 Arbeiter in drei Gruppen eingeteilt, die über 0 - 2, 2 - 10 und mehr als zehn Jahre, gegen 3 bis 69 mg/m3 Benzol exponiert waren. In den exponierten Gruppen waren Chromosomenaberrationen und Schwesterchromatidaustausch in peripheren Lymphozyten signifikant häufiger als bei Kontrollen. Mit einer Absenkung der Benzolkonzentration am Arbeitsplatz sank die Häufigkeit der Chromosomenaberrationen auf die Hälfte bzw. ein Drittel des ursprünglichen Wertes, lag aber noch immer über dem Kontrollwert (Tompa et al. 1994).
Rothman et al. (1995, 1996) wiesen bei 24 benzolexponierten chinesischen Arbeitern in der Gummiindustrie Mutationen in Erythrozyten-Vorläuferzellen in signifikant erhöhter Häufigkeit nach als bei den 23 Kontrollpersonen. Die Analyse der Mutationsmuster zeigte eine Induktion von Genverlust-Genverdopplungsmutationen, aber keine Gen-Inaktivierungsmutationen. Die Autoren verweisen auf die wichtige Rolle derartiger mitotischer Rekombination bei der Entwicklung von Leukämien und Lymphomen.
In einer Studie an benzolexponierten chinesischen Arbeitern wurde eine Häufung von Deletionen oder Verlusten von Chromosom 5 und 7 beschrieben (Zhang et al. 1998a). Vergleichbares wird für periphere Lymphozyten berichtet, die in Kultur gegen Hydrochinon und 1,2,4-Trihydroxybenzol exponiert wurden (Zhang et al. 1998b). Hydrochinon induzierte selektiv Verluste von Chromosom 7 und Deletionen in 5q31 in humanen CD34+ CD19- Knochenmarkszellen (Stillman et al. 2000). Numerische und strukturelle Aberrationen von Chromosom 8 und 21 waren ebenfalls mit Benzol-Exposition assoziiert, wobei eine Zunahme der klastogenen Effekte bei höherer Benzolkonzentration aufgezeigt wurde (Smith et al. 1998).
Verschiedene Ansätze wurden unternommen, benzolinduzierte Chromosomenschäden zu charakterisieren, um im Vergleich zu spontanen und therapieinduzierten Leukämien typische Muster zu identifizieren. Die Benzolmetabolite Hydrochinon und Trihydroxybenzol verursachten eine Aneuploidie an den Chromosomen 1, 5, 6, 7, 8, 9, 11, 12 und 21 (Zhang et al. 2005). Besonders interessant ist in dieser Hinsicht der Vergleich mit therapieinduzierten hämatopoetischen Erkrankungen, da für Benzol ein ähnlicher Wirkmechanismus postuliert wird wie für Topoisomerasehemmer. Etwa 12 % der Patienten, die mit Topoisomerase II-Hemmstoffen behandelt werden, erkranken an therapieinduzierten myeloischen und lymphatischen Leukämien (Merlat et al. 1999, Pegram et al. 2000, Rowley und Olney 2002). Topoisomerase II ist ein zentrales Enzym bei der DNA-Replikation und ist u. a. beteiligt an der Chromosomenkondensation, der Bewegung der Replikationsgabel und der Segregation replizierter Chromosomen. Topoisomerase II-Hemmstoffe wie Etoposid verhindern die Dissoziation der Topoisomerase II von der DNS. Solche Stoffe führen zu einem "DNA-Topoisomerase-Komplex" und so nachfolgend zu Doppelstrangbrüchen und/oder Translokationen. Dies wird als wesentlicher Wirkmechanismus der Induktion therapieinduzierter Leukämien durch Etoposid-Toxine angesehen.
Die Hemmung von Topoisomerasen wurde auch für phenolische Benzolmetabolite gezeigt und im Zusammenhang mit klastogenen Benzoleffekten diskutiert (Chen und Eastmond 1995, Hutt und Kalf 1996, Baker et al. 2001, Lindsey et al. 2004). Allerdings ist der Nachweis der Hemmung von Topoisomerase II durch Benzolmetabolite stark abhängig von den Inkubationsbedingungen. Nach Aktivierung mit Myeloperoxidase und Wasserstoffperoxid - ohne die sonst übliche Zugabe von DTT als Oxidationsschutz - war eine Hemmung der Topoisomerase II schon ab 50 nM Hydrochinon nachweisbar (Eastmond et al. 2005). Dies unterstreicht eine Beteiligung dieses Mechanismus an der kanzerogenen Wirkung von Benzol. Allerdings ist die für Topoisomerasehermcr typische Translokation 12g23/21g22 in benzolexponierten Kollektiven nicht auffällig häufig.
Chromosomale Translokationen spielen bei der Induktion sowohl spontaner als auch therapieinduzierter Leukämien eine wesentliche Rolle. Translokationen sind mit herkömmlichen Färbemethoden oft schwer zu identifizieren und können zur Bildung chimärer Proteine führen. Sind solche chimären Proteine in zellulären Signalwegen involviert, insbesondere in solchen, die den Zellzyklus regulieren, kann dies zu klonaler Expansion geschädigter, responsiver Zellen führen. Die für die chronisch myeloische Leukämie (CML) charakteristische Verkürzung von q22 (Philadelphia Chromosom, Nowell und Hungerford 1960 ergänzt) beruht darauf, dass ein kleinerer Teil von Chromosom 9 auf Chromosom 22 übertragen und vice versa ein größeres Fragment von Chromosom 22 auf Chromosom [t(9;22)] (Klein 1981). Dies führt zu dem BCR-ABL Fusions-Gen und Expression einer chimären Tyrosin-Protein-Kinase mit einem hyperaktiven N-terminalen Ende. Tyrosinkinasen sind Bestandteile intrazellulärer Signalwege, die die Wirkung von Wachstumsfaktoren vermitteln. Diese chimäre Tyrosin-Protein-Kinase kann so die klonale Expansion von Tumorzellen bewirken.
Eine Metaanalyse möglicherweise durch Benzol verursachter Leukämien zeigte, dass t(8;21) regelmäßig bei AML-M2 (akute myeloische Leukämie) beobachtet wurde und t(9;22) bei CML (chronische myeloische Leukämie). Zudem traten aneuploide Effekte relativ häufig auf; insbesondere Verlust von Chromosom 7 und überzählige Chromosomen 8 und 9. Bei den meisten benzolinduzierten Chromosomenaberrationen handelt es sich allerdings offenbar um Chromosomenbrüche und "Gaps" (Zhang et al. 2002). Dies wird auch in einer Studie an zwei Kollektiven bestätigt, in der Chromosomenschäden in benzolexponierten Arbeitern mit FISH untersucht wurden (Eastmond et al. 2001).
Insgesamt lässt sich nach Benzol-Exposition kein spezifisches chromosormales Schädigungsmuster nachweisen.
1.3.1.4 Übertragbarkeit tierexperimenteller Befunde auf den Menschen
Es ist im Tierversuch ausreichend, wenn der Fundamentalprozess der Kanzerisierung nachgewiesen wird. Eine 1:1-Deckungsgleichheit zwischen Tier und Mensch gibt es nicht. Es muss keine Organgleichheit der Wirkung bestehen. Sie ist aus toxikokinetischen Gründen auch nicht wahrscheinlich, da sich die Organismen hinsichtlich ihrer Größe, der unterschiedlichen Durchblutung einzelner Organe etc. unterscheiden. Der identische kanzerogene Mechanismus kann in verschiedenen Spezies also unterschiedliche Zellarten und Gewebe betreffen.
Dennoch lassen sich Gemeinsamkeiten in der Verursachung von Tumoren im Tierexperiment und beim Menschen finden. Einige Organe, die in der Ratte und Maus bei Benzolexposition besonders häufig von der Tumorverursachung betroffen sind, insbesondere die Zymbaldrüse, haben eine hohe Peroxidase-Aktivität (Maltoni et al. 1982, Maltoni und Scarnato 1979). Auch Versuche mit Mäusen, die heterozygot defizient für p53 sind, weisen auf Parallelen in der Benzol-Toxizität zwischen Mensch und Tier hin: Der Verlust der Heterozygotie (Loss of heterozygosity: LOH) wird als zentrales Ereignis in der Krebsentstehung bei Mensch und Tier angesehen; insbesondere wenn Tumorsuppressor-Gene betroffen sind. In Mäusen, die heterozygot defizient für p53 sind, können Tumoren besonders leicht ausgelöst werden. Das p53-Gen supprimiert Tumoren gleichermaßen in Mäusen und Menschen. In 13 von 16 Tumoren, die durch Benzol verursacht wurden, war der Verlust des verbliebenen p53 Allels nachweisbar. Insgesamt war der Verlust des p53 Wildtypallels in Thymus-Lymphomen und Sarkomen häufig und in Blasenkarzinomen (3 von 25) weniger häufig. Wegen der Parallelen in der Rolle von p53 bei Menschen und Mäusen werden p53 heterozygot defiziente Mäuse als ein geeignetes Tiermodell zur Verursachung von mesenchymalen und benzolinduzierten Tumoren angesehen (French et al. 2001, Boley et al. 2001).
Zwischen dem Stoffwechsel von Benzol im Menschen und im Nagetier bestehen keine grundlegenden Unterschiede.
Das quantitative Verhältnis toxischer Produkte (Hydrochinon und Mucondialdehyd) zu weniger toxischen Produkten (Phenylglukuroniden) ist bei F344/N-Ratten, B6C3F1-Mäusen und benzolexponierten Menschen vergleichbar (Yu und Weisel 1996). Es werden bei den verschiedenen Spezies nicht nur dieselben Metabolite gebildet, sondern sogar in vergleichbaren Proportionen. Auch die molekularen Wirkungen von Benzol an der Erbinformation unterscheiden sich nicht zwischen den Nagetieren und dem Menschen. Hierbei ist zu bedenken, dass Zellkulturexperimente mit Benzolmetaboliten auch mit menschlichen Zellen durchgeführt wurden.
In japanischen Studien (Inoue et al. 1986, 1988) wurde die Ausscheidung von Benzolmetaboliten im Harn bei Personen gemessen, die beruflich über acht Stunden gegen 100 ppm Benzol exponiert waren. Das Ausscheidungsmuster war demjenigen in zahlreichen Studien an Nagern vergleichbar. Daher wird das Nagermodell (Ratten, Mäuse) als geeignet erachtet, um den Metabolismus von Benzol im Menschen zu modellieren.
Die Vergleichbarkeit von Nagetier und Mensch in Bezug auf sowohl die Gentoxizität als auch den Stoffwechsel (Biotransformation) ergibt sich aus der Zusammenfassung der Übersichtsarbeit von Whysner et al. (2004):
"Rodent and human data were compared, and benzene genotoxicity results in both were similar for the available tests. Also, the biotransformation of benzene was qualitatively similar in rodents, humans and nonhuman primates, further indicating that rodent and human genotoxicity data were compatible."
Übereinstimmend mit den Befunden beim Menschen führt eine Benzol-Exposition im Tierexperiment mit Mäusen zu einer deutlichen Verminderung hämatopoetischer und lymphoider Zellen. Auch in transgenen Tg.AC Mäusen supprimierte Benzol (0,02 % im Trinkwasser über 28 Tage) bereits hämatopoetische Vorläuferzellen. Parallele Untersuchungen zur Genexpression mit "cDNA Microarrays" zeigten zudem eine signifikant erhöhte Genexpression von bax, c fos, E124, hsfl, IkBa und p57. Hierbei war cfos 22-fach (cfos codiert für einen Transkriptionsfaktor, der als Reaktion auf Verletzungen gebildet wird) und ei24 18-fach überexprimiert (ei24 codiert für einen p53 responsiven Faktor der mit DNS-Schädigungen assoziiert ist) (Nwosu ct al. 2004). Tg.AC Mäuse weisen nach Benzol-Exposition Parallelen zur Benzol-Toxizität beim Menschen auf und sind daher beim gegenwärtigen Erkenntnisstand ein geeignetes Tiermodell.
1.3.1.5 Zusammenfassung der experimentelltoxikologischen und hämatologischonkologischen Erkenntnisse
Eine arbeitsbedingte Benzolexposition ist in der Lage, sowohl nichtmaligne als auch maligne Erkrankungen des Blut- und Lymphsystems zu verursachen. Die primäre Wirkung der Benzolexposition ist eine Unterdrückung der Blutbildung infolge toxischer Knochenmarksdepression. Diese äußert sich klinisch in einer Verringerung der Zahl der Zellen im peripheren Blutbild, wobei das rote Blutbild, das weiße Blutbild und die Blutplättchen sowohl einzeln (Anämie, Leukopenie, Thrombopenie), als auch gemeinsam (Panzytopenie) betroffen sein können. Die aplastische Anämie, das myelodysplastische Syndrom (MDS) und die myeloproliferativen Erkrankungen stellen Übergangsformen zur malignen Erkrankung dar, wobei die beiden letzteren heute bereits als bösartige Erkrankung aufgefasst werden.
Benzol selbst zeigt in toxikologischen Untersuchungen keine eindeutig erbgutschädigende (mutagene) oder kanzerogene (krebserzeugende) Wirkung. Vielmehr sind Stoffwechselprodukte für die durch Benzolexposition verursachten Krebserkrankungen und toxischen Wirkungen verantwortlich. Phenolische Benzol-Metabolite entstehen in der Leber, aber auch in anderen Organen und verteilen sich im gesamten Körper. Benzol verursacht entsprechend im Tierversuch systemisch sowohl solide Tumoren als auch Tumoren des Blutsystems. Da nicht primär Lebertumoren entstehen, werden nachfolgende Reaktionen für die Bildung der letztlich kanzerogenen Intermediate verantwortlich gemacht. Dies sind Reaktionen, die von Peroxidasen, insbesondere Myeloperoxidase (MPO) und Prostaglandin-H-Synthasen, vermittelt werden. Sie führen zu labilen radikalischen Verbindungen, die über verschiedene Mechanismen klastogen und kanzerogen wirken, wie z.B. über die Hemmung der Topoisomerase II, über radikalische Reaktionen oder über die Induktion von oxidativem Stress. Die Organotropie der Krebserkrankungen nach Benzolexposition des Menschen wird u. a. dadurch erklärt, dass Benzol in Geweben mit hoher Peroxidaseaktivität - insbesondere durch MPO - zum ultimal wirksamen Kanzerogen aktiviert wird.
Zelluläre Vorgänge sind durch das "Microenvironment" der Zellen determiniert; beispielsweise durch teilungshemmende oder -fördernde Signale. Da oxidativen Enzymen wie der MPO eine zentrale Rolle bei der Benzol-Aktivierung zugeschrieben wird, ist dieses "Microenvironment" charakterisiert durch die Bedingungen, die Voraussetzung sind für die Aktivität des Enzyms. MPO-katalysierte Reaktionen benötigten H2O2als Co-Substrat. MPO ist beim gesunden Menschen inaktiv. Das Enzym ist in peripheren Neutrophilen und Monozyten enthalten und wird parallel zu seinem Co-Substrat H2O2an Orten der Immunabwehr induziert und freigesetzt. Dies gilt ebenso für das Knochenmark wie für periphere Gewebe. Weiterhin ist oft ein Proliferationsreiz notwendig, um die maligne transformierten Zellen zur Teilung anzuregen. Dies gilt nicht für die Stammzellen, die stärker wachstumsautonom sind. Ein Proliferationsreiz für aktivierbare stammzellennahe periphere Blutzellen ist gegeben durch Kontakt mit spezifischen Antigenen und "Antigen präsentierenden Zellen" (APC) während der allergischen Typ I- oder Typ IV-Reaktion. Dies führt bei prä-B-, prä-T-, B- und T-Lymphozyten zur Reifung und massiven klonalen Expansion. Teilungsfähige Zellen befinden sich daher auch im peripheren Blut und Lymphgewebe. Wesentliche Unterschiede im Benzolmetabolismus zwischen Mensch und Tier können nicht nachgewiesen werden. Obwohl das Knochenmark toxikokinetisch ein im Vergleich zum peripheren Blut für chemische Verbindungen schwerer erreichbares "tiefes Kompartiment" darstellt und lymphatische Tumoren (insbesondere Non-Hodgkin-Lymphome) generell häufiger sind als myeloische, wird dies im Knochenmark durch eine im Vergleich zum peripheren Gewebe wesentlich höhere Zahl teilungsaktiver Zellen, insbesondere wachstumsautonomer Stammzellen, kompensiert.
Die akute myeloische Leukämie unterscheidet sich zwar aus pathologischer Sicht klar von der chronischen lymphatischen Leukämie. Dies rechtfertigt jedoch keine pathogenetischen Schlussfolgerungen. Beim Menschen wird die Generierung und Entgiftung gentoxischer Metabolite des Benzol durch Enzympolymorphismen individuell moduliert. Hämatotoxische und immunsuppressive Wirkungen werden bereits bei Benzolexposition unter 1 ppm beschrieben (Lan et al. 2004, 2005). Dies ist deswegen bemerkenswert, weil bei der Pathogenese der NHL immunsuppressive Effekte bekannt sind und offenkundig eine zentrale Rolle spielen. Aktuelle epidemiologische Auswertungen bestätigen die Bedeutung sowohl der gleichen Mechanismen wie bei Leukämien (Chromosomenschäden durch DNA-Doppelstrangbrüche) als auch der Immuntoxizität für die Entstehung von Non-Hodgkin-Lymphomen (Smith et al. 2007).
Die Vulnerabilität nicht nur der Stammzellen des Knochenmarks sondern insbesondere auch der reiferen immunkompetenten lymphatischen Zellformen geht aus folgendem Zitat aus einer aktuellen Publikation der IARC (Smith et al. 2004) hervor:
"Lymphocytes are unique in being the only cell TYPEs in which a specific mechanism has evolved that can rearrange and mutate genomic DNA. Both T and B lymphocytes undergo genetic recombination and mutation as part of their normal life cycle.... DNA instability is generated during the development of lymphocytes and different components are active at different stages of lymphoid development; and the lymphoid system has developed specifically to deal with environmentally encountered infectious agents and is directly shaped by the infections it has encountered.... Thus, doublestrand breaks generated by derangement of physiological processes or by environmentally encountered DNA-damaging agents may be central features in the causation of lymphomaspecific translocations."
Hieraus erklärt sich möglicherweise auch eine besondere Empfindlichkeit des juvenilen Organismus, in welchem der Aufbau des Immunsystems besonders aktiv erfolgt, für die leukämieinduzierende Wirkung von Benzol (Steffen et al. 2004, Crosignani et al. 2004).
Fazit
Benzol ist ein komplettes systemisches Kanzerogen mit tumorinitiierenden, -promovierenden und epigenetischen Wirkungen. Benzol besitzt die Fähigkeit, das breite Spektrum prämaligner und maligner Erkrankungen des Blut- und Lymphsystems (Aplastische Anämie, Myelodysplastische Syndrome, Leukämien, Non-Hodgkin-Lymphome, Mycloproliferative Erkrankungen) zu verursachen. Für die krebserzeugende Wirkung sind verschiedene Stoffwechselprodukte (Metabolite) des Benzols verantwortlich. Nach heutiger Kenntnis und internationalem wissenschaftlichen Konsens werden die Hemmung der Topoisomerase durch Benzochinon-Radikale und die gentoxische Wirkung reaktiver Sauerstoffspezies (Radikale) als die beiden wichtigsten Mechanismen angesehen. Hinzu kommen insbesondere immuntoxische Wirkungen.
Oxidative Stoffwechselenzyme, die die Generierung gentoxischer Produkte aus den phenolischen Metaboliten des Benzol katalysieren, liegen im Organismus ubiquitär vor. Dies gilt insbesondere für die Myeloperoxidase (MPO) und Prostaglandin-H-Synthasen. Entsprechend treten im Tierexperiment neben lymphatischen und myeloischen auch solide Tumoren auf. Physiologisch werden die oxidativen Enzyme während entzündlicher Vorgänge aktiviert (Enzyminduktion).
Im Tierexperiment treten Tumoren insbesondere in peroxidasereichen Geweben auf. Die beim Menschen beobachtete Organotropie des Benzols für das blutbildende Knochenmark wird mit der dort vorliegenden hohen Peroxidase-Aktivität, insbesondere der Myeloperoxidase (MPO), begründet. Ein weiterer Grund ist die hohe Teilungsaktivität der hämatopoetischen Stammzellen. Im lymphatischen Gewebe besteht während peripherer entzündlicher Reaktionen, insbesondere im Verlauf von Typ I- und Typ IV-Immunreaktionen nach Coombs und Gell, infolge biochemischer Signale ("Microenvironment") ebenfalls eine hohe Teilungsaktivität (Reifung und klonale Expansion von T-Zellen und B-Zellen nach Kontakt mit Antigenen).
Das Ausmaß der individuellen Gefährdung durch eine arbeitsbedingte Benzolexposition wird nachweislich durch Enzympolymorphismen (vor allem NQO1, aber auch MPO, CYP2E1) und durch weitere Faktoren wie z.B. erhöhte Aufnahme durch körperliche Aktivität, junges Expositionsalter, beeinflusst. Im Erkrankungsfall sind, konkurrierende arbeitsbedingte und nicht arbeitsbedingte Ursachen einer malignen Erkrankung des myeloischen oder lymphatischen Systems in Betracht zu ziehen, insbesondere ionisierende Strahlung und Viruserkrankungen.
1.3.2 Krankheitsbilder und Diagnosen
1.3.2.1 Unter dieser Berufskrankheit erfasste Krankheitsbilder
Gegenstand dieser wissenschaftlichen Begründung sind
Während die Leukopenie, Thrombozytopenie und Panzytopenie nach Ende der Benzolexposition meist reversibel sind, handelt es sich bei der aplastischen Anämie um eine irreversible Erkrankung.
Die Krebsvorstufen (Präkanzerosen) wurden früher zum Teil als Präleukämie bezeichnet. Heute zählen die myelodysplastischen Syndrome und myeloproliferativen Erkrankungen aufgrund molekularbiologischer Erkenntnisse zu den malignen Erkrankungen, da es sich um monoklonale Erkrankungen handelt.
In der Vergangenheit existierten mehrere konkurrierende Einteilungen (Klassifikationen) der hämatologischen Erkrankungen. Unterschiede der in Deutschland, in anderen europäischen Ländern und in den USA gebräuchlichen Systemen erschwerten die Vergleichbarkeit hämatologischer und epidemiologischer Untersuchungen. Im Jahre 1997 wurden unter der Federführung der Weltgesundheitsorganisation die unterschiedlichen Einteilungen der malignen hämatologischen Krankheitsbilder zu einem gemeinsamen internationalen System, der WHO-Klassifikation, zusammengeführt (Stein und Hiddemann 1999, Vardiman et al. 2002).
Die in der vorliegenden wissenschaftlichen Begründung verwendeten Krankheitsbezeichnungen basieren auf der WHO-Klassifikation, die künftig auch in Berufskrankheiten-Verfahren ausschließlich verwendet werden sollte. Schwierigkeiten ergeben sich bei der Bewertung epidemiologischer Studien aus der Vergangenheit; hierauf wird im Abschnitt 2. Validität und Reliabilität der vorliegenden epidemiologischen Erkenntnisse ausführlich eingegangen.
Auch nach der auf molekularbiologischen Erkenntnissen fußenden WHO-Klassifikation gibt es Überschneidungen der klassischen, im klinischen Alltag gebräuchlichen Bezeichnungen "Leukämie" und "Lymphom" mit neueren Einteilungskategorien. So handelt es sich bei der chronisch myeloischen Leukämie (CML) sowohl um eine Leukämie als auch um eine myeloproliferative Erkrankung; die chronisch lymphatische Leukämie (CLL) ist sowohl eine Leukämie als auch ein Non-Hodgkin-Lymphom.
Abb. 4 verdeutlicht diese Überlappungen:
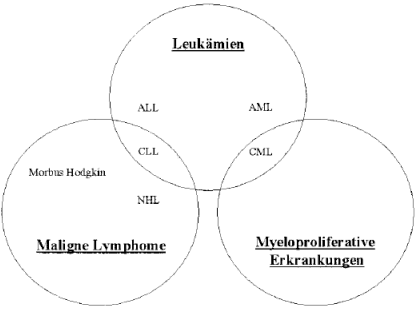
Überlappungen der klinischen Diagnosen Leukämie, maligne Lymphome und myeloproliferative Erkrankungen
ALL = akute lymphatische Leukämie;
CLL = chronische lymphatische Leukämie;
AML = akute myeloische Leukämie;
CML = chronische myeloische Leukämie;
NHL = Non-Hodgkin-Lymphom
1.3.2.2 Hämatologische Aspekte
Die Blutzellen des Menschen werden in drei Gruppen eingeteilt, nämlich in rote Blutzellen (Erythrozyten), weiße Blutzellen (Leukozyten) und Blutplättchen (Thrombozyten). Die Bildung aller drei Zellformen (Hämatopoese) erfolgt während der embryonalen Entwicklung vorwiegend in der Milz und Leber, dann während des ganzen Lebens fast ausschließlich im Knochenmark. Eine Ausnahme stellt die Entwicklung des lymphatischen Systems dar, die in ihren späteren Phasen außerhalb des Knochenmarks (extramedullär) erfolgt, zunächst im Thymus und dann auch im lymphatischen Gewebe einschließlich der Lymphknoten.
Vorläuferzellen der drei Zellformen sind undeterminierte (pluripotente) und determinierte hämatopoetische Stammzellen. Bei der Teilung von Stammzellen werden aus einem Teil der Tochterzellen wieder Stammzellen, wodurch deren Konstanz gewährleistet bleibt. Ob die Stammzellen ruhen oder sich über den Zellteilungszyklus vermehren, hängt vom
Bedarf an bestimmten Funktionszellen in der Peripherie ab und wird durch stimulierende und hemmende humorale Faktoren reguliert. Das Knochenmark ist eine komplexe Matrix mit Stammzellen, Progenitorzellen der Blutzellen und Stromazellen. Die Stromazellen liefern die Wachstumsfaktoren, die für die Proliferation und Differenzierung der Stamm- und Progenitorzellen wesentlich sind.
Wichtig für das Verständnis verschiedener Systemerkrankungen, insbesondere maligner Erkrankungen des Blutsystems, ist die Tatsache, dass ein Austausch von Stammzellen zwischen verschiedenen Organen und auch eine Besiedlung des Knochenmarks mit Stammzellen aus extramedullären Blutbildungsstätten, insbesondere aus Keimzentren des lymphatischen Systems, erfolgt. Hierdurch bedingte fließende Übergänge zwischen verschiedenen Zellformen und Lokalisationen sind kennzeichnend für pathologische Störungen wie die myeloproliferativen Erkrankungen. Sie können aber auch therapeutisch genutzt werden, etwa bei der Knochenmarkstransplantation.
Die weißen Blutzellen befähigen den Organismus, physikalische und chemische Fremdeinwirkungen - insbesondere Infektionen - abzuwehren und durch die Antikörperbildung gegen körper- oder artfremde Antigene immunologisch zu reagieren (sog. zelluläre und humorale Immunität). Maligne Erkrankungen des Blutsystems betreffen meist die weißen Zellen, so dass den Leukämien und malignen Lymphomen auch im Zusammenhang mit der krebserzeugenden Wirkung von Benzol besondere Bedeutung zukommt. Dies schließt nicht aus, dass die seltenen malignen Erkrankungen der beiden anderen Systeme ggf. auch für die Benzolproblematik relevant sind.
Die Unterscheidung zwischen Leukämien und Lymphomen ist klinischer Natur und erlaubt keine Rückschlüsse auf die Pathogenese. Der Begriff "Leukämie" wurde zuerst 1847 von VIRCHOW geprägt, der mit diesem Begriff die extreme Vermehrung unreifer weißer Blutzellen bezeichnete. Allerdings muss die Leukämie nicht immer von einer Vermehrung weißer Zellen im Blut gekennzeichnet sein. Bei der Erkrankung treten mitunter eine Leukozytose, eine Leukopenie oder sogar ein agranulozytäres Blutbild auf.
Während der Begriff Leukämie von einem klinischen Erscheinungsbild des peripheren Blutbildes ausgeht, kennzeichnet das Lymphom eine abnorme Schwellung des lymphatischen Gewebes, insbesondere von Lymphknoten oder Lymphknotenpaketen.
Obwohl sich in den letzten Jahrzehnten die Differenzierung der malignen Erkrankungen des weißen Blutbildes durch die Bestimmung histologischer, biochemischer und neuerdings auch molekularbiologischer Merkmale erheblich weiterentwickelt hat, wurde national und international die Grundeinteilung in die historischen klinischmorphologischen Gruppen Leukämie und Lymphom beibehalten. Bei der Anwendung der neueren Klassifikationen sollte aber wie oben angeführt bedacht werden, dass es Überlappungen zwischen der klinischen, der histologischen und der molekularbiologischen Einteilung gibt. So handelt es sich etwa bei der chronischen lymphatischen Leukämie (CLL) um ein malignes Non-Hodgkin-Lymphom (Chiorazzi et al. 2005, WHO-Klassifikation). Einzelne Malignome können sich auch im Krankheitsverlauf stadienabhängig von einem durch Lymphome geprägten zu einem leukämischen Erscheinungsbild wandeln.
Abb. 5: Stammbaum der Zeltformen des hämatolymphatischen Systems
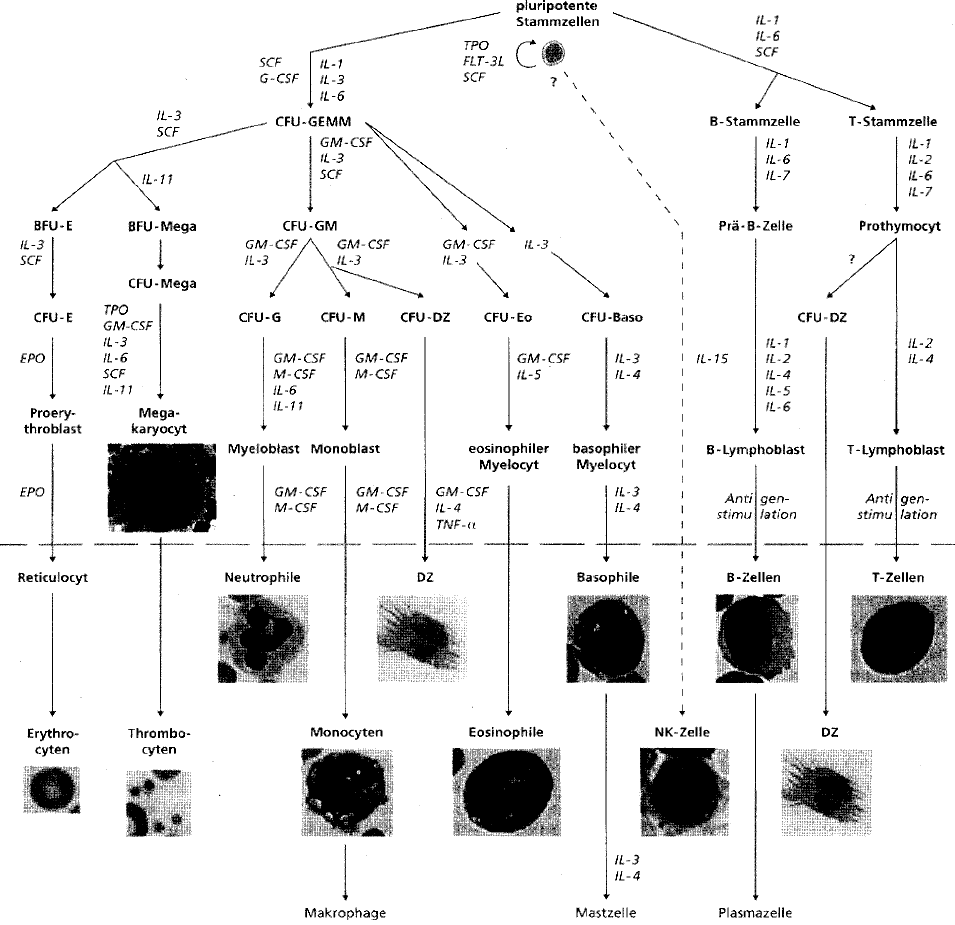
Abbildung aus Schmidt, Unsicker Lehrbuch Vorklinik, 2003 © Deutscher Ärzte-Verlag
Bei den Leukämien unterscheidet man zwischen der akuten (AML) und der chronischen myeloischen Leukämie (CML) sowie der akuten (ALL) und der chronischen lymphatischen Leukämie (CLL). Die weitere Unterteilung erfolgt heute in der WHO-Klassifikation auf der Grundlage einer Vielzahl unterschiedlicher genetischer Veränderungen, die teilweise sehr eng mit einem bestimmten Phänotyp der Leukämie assoziiert sind. Solche genetischen Veränderungen führen zu einem gestörten Proliferations- und Differenzierungsverhalten oder zu einem gestörten Ablauf des programmierten Zelltodes (Apoptose). Die zugunsten des Zellwachstums verschobene Imbalance zwischen Proliferation und Apoptose resultiert in der klonalen Expansion der leukämischen Zelle mit Verdrängung der normalen hämatopoetischen Zellpopulation (Feuring-Buske et al. 2002).
Die akute myeloische Leukämie (AML) hat in Deutschland eine Häufigkeit von zwei bis drei Erkrankungen pro 100.000
Einwohner und Jahr. Der Altersmedian liegt bei 60 Jahren. Im höheren Alter ist ein starker Anstieg der Inzidenz bis auf über 15 Erkrankungen/100.000 Einwohner/Jahr zu beobachten. Exogene Faktoren spielen bei der Entstehung der AML offenbar eine große Rolle; etwa 10-20 % aller Fälle sind therapieinduziert durch eine Behandlung mit Chemotherapeutika.
Die AML entsteht durch die maligne Transformation früher hämatopoetischer Progenitorzellen. Für die Mehrzahl dieser Erkrankungen ist die multipotente hämatopoetische Stammzelle als Ausgangszelle des malignen Klons anzusehen. Es gibt jedoch auch Ausnahmen, die von reiferen Progenitorzellen ausgehen, etwa die Promyelozytenleukämie.
Die chronisch myeloische Leukämie (CML) entsteht durch die klonale Proliferation einer maligne transformierten multipotenten hämatopoetischen Stammzelle. Charakteristisch ist insbesondere die Philadelphia-Chromosomentranslokation t(9;22)(g34;g11). Die Inzidenz der CML liegt in Deutschland bei einer Erkrankung pro 100.000 Einwohner und Jahr und hat einen Anteil von 15-20 % an allen Erwachsenenleukämien. Das mediane Erkrankungsalter liegt bei 53 Jahren. Auch bei dieser Leukämieform kommen exogene Risikofaktoren in Betracht; die erhöhte Inzidenz der CML nach den Atombombenabwürfen von Hiroshima und Nagasaki belegt die Bedeutung ionisierender Strahlung. Die chronisch myeloische Leukämie wird nach der aktuellen WHO-Definition (Vardiman et al. 2002) zu den myeloproliferativen Erkrankungen gerechnet.
Die Inzidenz der akuten lymphatischen Leukämie (ALL) liegt bei 1,1 pro 100.000 Einwohner und Jahr. Betroffen sind vor allem Kinder unter fünf Jahren (5,3/100.000). Danach fä11t die Inzidenz kontinuierlich ab, um nach dem 35. Lebensjahr wieder anzusteigen auf einen zweiten Häufigkeitsgipfel im Alter von über 80 Jahren (2,3/100.000).
Als Risikofaktoren für die ALL gelten im Kindesalter verschiedene Erbkrankheiten mit chromosomalen Auffälligkeiten, etwa die Trisomie 21 (Down-Syndrom). Daneben werden, insbesondere beim Erwachsenen, exogene Faktoren wie ionisierende Strahlung, Benzol, Zytostatika und Viren (insbesondere HTLV1) als Risikofaktoren betrachtet.
Die ALL kann durch maligne Transformation verschiedener Differenzierungsstufen der Lymphopoese entstehen. Bei ca. 75 % aller Patienten mit ALL finden sich klonale chromosomale Veränderungen. Bei einigen Formen der ALL finden sich Mutationen des p53-Tumorsuppressorgens.
Die chronisch lymphatische Leukämie (CLL) ist die häufigste Leukämieform der Erwachsenen in der westlichen Welt mit einem Anteil von 25-30 %. Die Inzidenz der CLL steigt mit zunehmendem Lebensalter und liegt insgesamt bei 3/100.000 Einwohnern pro Jahr. Der Häufigkeitsgipfel liegt zwischen dem 60. und 65. Lebensjahr. Nach der aktuellen WHO-Definition (Stein und Hiddemann 1999) wird die chronische lymphatische Leukämie zu den Non-Hodgkin-Lymphomen (NHL) gerechnet.
Bei den myeloproliferativen Erkrankungen handelt es sich um monoklonale Erkrankungen der myeloischen Stammzellen mit autonomer Proliferation einer oder mehrerer hämatopoetischer Zellreihen (Leuko-, Erythro-, Thrombozytose). Die myeloproliferativen Erkrankungen umfassen nach der WHO-Klassifikation sieben Krankheitsentitäten (Vardiman et al. 2002), insbesondere:
Die myelodysplastischen Syndrome (MDS) sind erworbene klonale Stammzellerkrankungen mit qualitativen und quantitativen Veränderungen aller Zeltreihen der Hämatopoese, peripherer Zytopenie, zellreichem dysplastischem Knochenmark und oft erhöhtem Blastenanteil (Vardiman et al. 2002).
Aktuelle Daten zur Häufigkeit von Leukämien, Non-Hodgkin-Lymphomen und anderen malignen Erkrankungen in Deutschland sind von der Arbeitsgemeinschaft bevölkerungsbezogener Krebsregister in Deutschland in Zusammenarbeit mit dem Robert-Koch-Institut veröffentlicht worden (GEKID 2006).
Insbesondere die aplastische Anämie und das myelodysplastische Syndrom (MDS) sind als Frühstadien einer malignen Erkrankung anzusehen. Da es sich bei MDS um klonale Stammzellenerkrankungen handelt, stellt es per se eine maligne Erkrankung des hämatopoetischen Systems dar (Janssen et al. 1989, Haase et al. 1997). Verlaufsuntersuchungen haben gezeigt, dass innerhalb von fünf Jahren ca. 15 % aller aplastischen Anämien in ein MDS oder eine AML übergehen, wobei das Auftreten klonaler Chromosomenanomalien einerseits als Malignitätskriterium zu werten ist und andererseits mit einer Erkrankungsprogression in Richtung MDS und AML assoziiert ist (De Planque et al. 1988, Maciejewski et al. 2002). Myelodysplasien gehen in ca. 30 % in eine akute myeloische Leukämie über. Das Risiko für einen Übergang in eine AML und die Geschwindigkeit der leukämischen Transformation ist in hohem Maße vom Karyotyp abhängig (Greenberg et al. 1997).
1.3.2.3 Toxikologische Aspekte der Benzolwirkung
Das primäre Organ der Blutbildung ist das (rote) Knochenmark. Im Embryo und im Säuglingsalter findet die Blutbildung auch in der Leber und in der Milz statt. Beim Erwachsenen kann die Blutbildung in diesen Organen bei erhöhtem Bedarf und bei verschiedenen Erkrankungen reaktiviert werden. Wie bereits beschrieben wandert ein Teil der Blutzellen im Laufe ihrer Reifung in den Thymus und in das lymphatische Gewebe aus. Zellteilungsvorgänge lymphatischer Zellen finden nach dem Auswandern auch extramedullär z.B. in Lymphknoten statt. Hier werden Vorläufer-Lymphozyten antigenabhängig stimuliert und differenzieren zu antikörperbildenden Lymphozyten aus. Diese Zellen sind somit während der Teilungsphase für Noxen, die mit der Erbsubstanz (DNA) gentoxisch interagieren, vulnerabel. Somit darf bezüglich des Mechanismus der Krebsentstehung das Knochenmark nicht isoliert betrachtet werden.
Bei wiederholter, langdauernder, u. U. auch bei einmaliger, sehr massiver Einwirkung erweist sich Benzol als Blutgift - eine Wirkung, die bereits im 19. Jahrhundert bekannt war. Charakteristisch ist die Hemmung der Bildung der Blutzellen, die sich klinisch als Anämie, Leukopenie oder Thrombopenie zeigt. Diese klinischen Erscheinungsbilder der toxischen Wirkung von Benzol können allein oder in Kombination auftreten. Häufig geht der Depression der einzelnen Systeme eine mehr oder weniger vorübergehende Überproduktion voraus. Daneben kommt es zu einer malignen Entartung von Blutzellen. Eine solche maligne Erkrankung des Blutsystems kann sich klinisch sowohl aus einem der vorangehend genannten toxischen Krankheitsbilder als auch ohne toxisches Vorstadium entwickeln.
Obwohl Fälle von benzolverursachter Leukämie ohne vorangehender toxischer Reduzierung der Zeltzahlen des weißen Blutbildes berichtet wurden (Yin et al. 1989), ist die Hämatoxizität ein wesentlicher Risikofaktor für die Leukämieentstehung. Durch vermehrten Zelltod in den Vorläuferzell-Populationen und abnormer regulatorischer Signaltransduktion können geschädigte Zeltklone einen Proliferationsvorteil erlangen. Die klonale Hämatopoese wird als Vorstufe des myelodysplastischen Syndroms (MDS) und der akuten myeloischen Leukämie (AML) angesehen. Zelltod und Zytopenie fördern die Expansion der verbliebenen Stammzellen, wodurch die Wahrscheinlichkeit erhöht wird, dass eine Zelle mit einer genetischen Anomalie in den Generationszyklus gelangt (EPA 1998). Derartige Effekte wurden bereits bei 10-9-molaren Konzentrationen des Benzolmetaboliten Hydrochinon festgestellt (Irons et al. 1992). Darüber hinaus kommt es zu einer Vermehrung myeloperoxidase-(MPO)-positiver Zellen im Knochenmark und infolgedessen zu einer Verstärkung der Produktion gentoxischer Benzolmetabolite (Irons und Stillman 1993).
Benzol wirkt toxisch auf das blutbildende Knochenmark und kann eine Knochenmarksdepression und eine aplastische Anämie verursachen (Goldstein 1988, Aksoy 1988, Kipen et al. 1988). Der toxische Effekt ist sogar bei Benzolkonzentrationen unterhalb 1 ppm signifikant (Lan et al. 2004). Beim Menschen reagieren die Lymphozyten am empfindlichsten auf diese toxische Wirkung (Aksoy et al. 1971, Moszczynski und Lisiewicz 1982). Benzolexponierte Beschäftigte in China wiesen eine hoch signifikant geringere Lymphozytenkonzentration im Blut auf als nichtbenzolexponierte Kontrollen. Die Verminderung der Zellzahl korrelierte mit der Konzentration von Benzolmetaboliten im Urin (Rothman et al. 1996). Die toxische Wirkung auf die lymphozytären Zellen beeinträchtigt die Produktion von Zytokinen, insbesondere IL-3; hierdurch sollen Knochenmarksdepression und Leukämieinduktion verstärkt werden (EPA 1998). Die toxische Wirkung des Benzols auf die Blutbildung betrifft jedoch nicht nur die Lymphozyten, sondern auch die erythrozytären, granulozytären und thrombozytären Zellformen.
Ein signifikanter, dosisabhängiger Trend für diese Schädigungen weißer Blutzellen zeigt sich bei Personen mit einer inhalativen Benzolexposition unter 1 ppm. Vermindert waren die Gesamtzahl der weißen Blutzellen, ferner Granulozyten, Lymphozyten, CD4+ T-Zellen, B-Zellen, NK-Zellen und Thrombozyten (Lan et al. 2004). Ausgenommen waren bemerkenswerterweise CD8+ T-Zellen und Monozyten. Dies deutet auf einen selektiven Effekt in Bezug auf die T-Zellen: T-Helfer Zellen (Th1 und Th2 Zellen) sind CD4+, während cytotoxische T-Zellen CD8+ sind. Monozyten waren lediglich in der Gruppe mit der höchsten Exposition vermindert. Da die CD4+ Zellen proportional zur Benzolexposition abnehmen und die CD8+ Zellen unverändert bleiben, vermindert sich das Verhältnis von CD4+/CD8+. Diese Beobachtung ist wesentlich, weil das gleiche Phänomen im Zusammenhang mit der Exposition gegen Kohlenwasserstoffverbindungen in der "Phenol-Abteilung" einer petrochemischen Anlage beschrieben wurde (Zeman et al. 1990).
Somit ist belegt, dass bei relativ niedriger Benzolexposition immuntoxische Effekte auf periphere Lymphozyten nachweisbar sind.
Eine weitere unabhängige Bestätigung für immuntoxische Benzolwirkungen hinsichtlich der Verminderung der Thrombozyten stellt die Verminderung des Thrombozyten-Faktors (PF)4 und des "Connective Tissue Activating Peptide" (CTAP)-III im Serum von benzolexponierten Personen aus einer Schuhfabrik dar. Diese nachgewiesene Hämatotoxizität wurde als immunsuppressiver Effekt bewertet (Vermeulen et al. 2005).
Da die Entwicklung eines Tumors mehrstufig erfolgt, ist der genaue Ort einer "richtungsgebenden" Einwirkung einer Noxe wie Benzol (bzw. eines Benzolmetaboliten) nicht exakt zu bestimmen. Eine entscheidende Mutation kann z.B. im Knochenmark erfolgen, jedoch zunächst ohne Konsequenzen bleiben, da ein Wachstumsfaktor betroffen ist, der erst in einem späteren Reifungszustand, z.B. nach Auswanderung in das lymphatische Gewebe, relevant ist.
Als Beispiel sei das zur Diagnosegruppe der Non-Hodgkin-Lymphomen (NHL) zählende multiple Myelom (MM) aufgeführt. Beim MM wird davon ausgegangen, dass die maligne Transformation einen post-Keimzentrums-B-Lymphozyten der sekundären Immunantwort, also einen sehr weit differenzierten Lymphozyten, betrifft (Sahota et al. 1996). Diese MM-Vorläuferzellen differenzieren zu Plasmazellen, die dann im Knochenmark mittels "Homing-Rezeptoren" eine sessile maligne Population bilden, wobei das Knochenmarkstroma eine wichtige supportive Rolle inne hat und die Apoptose von Plasmazellen verhindert (Bakkus et al. 1994, Uchiyarna et al. 1993). Die aktuellen hämatologischen Daten sprechen also dafür, dass die maligne Transformation beim MM extramedullär stattfindet, wobei zu bedenken ist, dass eine wesentliche Komponente der Pathogenese, die klonale Expansion, innerhalb des Knochenmarks abläuft. Es ist davon auszugehen, dass die klonale Expansion weiterer genetischer Anomalien bedarf, die dann intramedullär entstehen (Dalton et al. 2001). Obwohl das multiple Myelom nach der aktuellen WHO-Klassifikation zu den peripheren Non-Hodgkin-Lymphomen zählt, erfolgt ein wesentlicher Teil seiner Pathogenese im blutbildenden Knochenmark.
Somit ist die Unterstellung nicht gerechtfertigt, dass es sich bei peripheren Non-Hodgkin-Lymphomen um Erkrankungen handle, deren Entstehung ausschließlich außerhalb des Knochenmarks angesiedelt sei (Hoffmann et al. 2001, Tannapfel et al. 2001). Vielmehr besteht ein intensiver Austausch zwischen dem peripheren lymphatischen Gewebe und dem blutbildenden Knochenmark; gerade beim multiplen Myelom wird der Einfluss des "Microenvironment" des Knochenmarks auf die Pathogenese peripherer Non-Hodgkin-Lymphome deutlich.
1.3.3 Epidemiologische Befunde
Die toxische Wirkung von Benzol auf das Knochenmark (Knochenmarksdepression) mit resultierender Verminderung der Zellzahlen ist seit dem Ende des 19. Jahrhunderts bekannt und bildete die Grundlage für die Festlegung als Berufskrankheit in 1925. Da die toxischen Wirkungen von Benzol auf das hämatolymphatische System sowohl in der internationalen und nationalen wissenschaftlichen Literatur als auch in der bisherigen Berufskrankheiten-Rechtsanwendung unter der Nummer 1303 unstrittig sind, wird hinsichtlich der Toxikologie, Kasuistik und Epidemiologie dieser nichtkanzerogenen Benzolwirkung auf die ausführliche Darstellung in den toxikologischarbeitsmedizinischen Begründungen für MAK-Werte der Deutschen Forschungsgemeinschaft verwiesen (DFG 1971, 1992).
In einer Untersuchung des National Cancer Institute (NCI), National Institutes of Health (NIH) sowie mehrerer Universitäten der USA mit vergleichbaren chinesischen Einrichtungen wurden 250 benzolexponierte Beschäftigte mit 140 Kontrollpersonen verglichen (Lan et al. 2004). Dabei wurden ausgeprägte akute toxische Wirkungen von Benzol bereits bei Expositionskonzentrationen deutlich unterhalb 1 ppm festgestellt. Die durchschnittliche Zahl der weißen Blutkörperchen (Leukozyten) betrug bei den Kontrollen 6.480/µl Blut, bei <1 ppm Benzol (durchschnittlich 0,57ppm) 5.540 (p<0,0001), bei 1-10 ppm 5.660, bei > 10 ppm 4.770. Die Trendanalyse über alle Personen ergab p<0,0001. Besonders ausgeprägt war die Reduktion der
Granulozyten, signifikant aber auch der Abfall der Lymphozyten, der Thrombozyten und des Hämoglobin. Auffallend war eine selektive Verminderung der CD4+-T-Lymphozyten bei nahezu CD8+-unveränderten T-Lymphozyten und infolgedessen veränderter CD4+/CD8+-Relation. Dieser immuntoxische Effekt wurde von den Autoren im Rahmen weiterer differenzierter Analysen untermauert (Lan et al. 2005).
Benzol ist, wie bereits dargestellt, sowohl von der International Agency for Research an Cancer (IARC) (1987) und der Europäischen Union als auch von der Deutschen Forschungsgemeinschaft (DFG) (Henschler 1971 - MAK-Begründung) als Humankanzerogen eingestuft. Die epidemiologische Evidenz beruht in erster Linie auf Studien zum Auftreten von Leukämien, insbesondere der akuten myeloischen Leukämie (AML). Hinsichtlich der unstrittigen leukämieverursachenden Wirkung wird ebenfalls auf die ausführlichen Darstellungen der DFG, der EU und der IARC verwiesen.
Angesichts der in der Einleitung zu dieser wissenschaftlichen Begründung zitierten Artikel in der deutschen arbeitsmedizinischen Literatur wird in der nachstehenden Erörterung anhand der Literatursichtung untersucht, ob genügend epidemiologische Evidenz besteht, dass Benzol auch Non-Hodgkin-Lymphome (NHL) verursachen kann.
Betrachtet man die Ergebnisse und Schlussfolgerungen der zahlreichen, bisher durchgeführten Kohorten- und Fall-Kontroll-Studien, so lassen sich bis auf wenige Ausnahmen keine Risikoerhöhungen für die jeweils unterschiedlich nach ICD oder WHO (siehe Abschnitt 2.) als NHL zusammengefassten Hauptgruppen der lymphoproliferativen Malignome durch die vermutete Benzolexposition nachweisen. So kommen Möhner und Heuchert (2000) in ihrer Meta-Analyse, in der sie 29 Kohorten- und 20 Fall-Kontroll-Studien aus dem Zeitraum 1966 bis 1999 kritisch analysieren, zu dem Schluss, dass lediglich in einigen wenigen Studien Assoziationen zwischen einer Benzolexposition und einem erhöhten Erkrankungsrisiko bezüglich der B-Zell-Lymphome beobachtet werden. Jedoch sei es aus diesen Studien schwierig, den Effekt von Benzol von den anderen Lösungsmitteln zu trennen (vgl. auch Miligi et al. 2006). Ihrer Meinung nach spricht die Meta-Analyse nicht für einen Zusammenhang zwischen einer Benzolexposition und einem erhöhten Risiko, an einem Non-Hodgkin-Lymphom zu erkranken. Andererseits schließen die Autoren einen Zusammenhang auch nicht völlig aus, sie halten ihn aber aus epidemiologischer Sicht eher für unwahrscheinlich.
Wong und Raabe (2000) haben in ihrer Meta-Analyse 26 teilweise nicht veröffentlichte Kohortenstudien von Beschäftigten in der Erdölindustrie aus USA, England, Kanada, Australien, Italien und Finnland im Hinblick auf einen Zusammenhang zwischen Benzolexposition und NHL untersucht. Der Beobachtungszeitraum erstreckte sich von 1937 bis 1996. Die Analyse umfasste 308.000 Beschäftigte. Insgesamt wurden 506 NHL Todesfälle registriert und 561,88 erwartet. Sie berechneten unter Einschluss aller Studien ein SMR von 0,9 mit einem 95 %-Konfidenzintervall von 0,82 bis 0,98, das somit auf eine statistisch gesicherte Untersterblichkeit an NHL hinweist, was verwunderlich ist. In den sieben Kohorten mit Beschäftigten der Benzinauslieferung bzw. in der Rohölverarbeitung (insgesamt 82.000 Personen) waren alle SMRs unter Eins (die Wahrscheinlichkeit dafür ist 0,5' = 0,008, wenn man davon ausgeht, dass, wenn Benzol
keinen Einfluss auf die Todesursache NHL hat, Werte größer und kleiner Eins gleichwahrscheinlich sind). Es ist somit zu vermuten, dass eine Untererfassung für NHL vorliegt. In den 19 Kohorten der Raffineriearbeiter wurde in zwölf ein SMR größer Eins gefunden; jedoch waren alle SMRs statistisch nicht signifikant (Die Wahrscheinlichkeit für diese Anzahl beträgt 0,18.). Eine Quantifizierung der Benzolexposition konnte für die Arbeiter von den Autoren anhand der vorliegenden Information nicht vorgenommen werden. Mutmaßlich war die Mehrheit der Beschäftigten nur einer geringen Benzolkonzentration ausgesetzt, so dass NHL nicht induziert wurden (siehe auch Punkt 3 der Seite 996). Der Schlussfolgerung der Autoren "Petroleum workers were not at an increased risk of NHL as a result of their exposure to benzene or other benzenecontaining petroleum products in their work environment" kann aus den genannten Gründen nicht zugestimmt werden.
Schnatter, Rosamilia und Wojcik (2005) schließen aus ihrem Literaturstudium bis Oktober 2004, in das sie nach vorgegebenen Kriterien zehn Kohortenstudien und 13 populationsbasierte Fall-Kontroll-Studien, darunter drei eingebettete Fall-Kontroll-Studien, aufgenommen hatten, dass zwar in allen Studien und Industriebereichen übereinstimmend eine Beziehung zwischen Benzolexposition und AML nachweisbar war, wobei in Bereichen mit hoher Exposition auch höhere Risiken auftraten. Eine derart eindeutige Beziehung ließ sich jedoch nicht für CLL, ALL und CML nachweisen. Nur in den eingebetteten Fall-Kontroll-Studien fanden sie eine Beziehung zwischen Benzolexposition und CLL angedeutet. Jedoch schränkt ihrer Meinung nach die geringe Fallzahl eine definitive Aussage über den Zusammenhang zwischen der Benzolexposition und dem Risiko für ALL und CLL ein.
Auch Lamm, Engel und Byrd (2005), die eine bis April 2005 reichende Literaturrecherche anhand spezieller Kriterien in der National Library of Medicine durchführten, fanden in den 21 Studien zu NHL und Benzolexposition elf mit einem Risikoschätzer < 1, zwei mit einem Risikoschätzer = 1 und nur acht mit einem erhöhten Risiko. In zwei Studien (Fabbro-Peray et al. 2001 und Yin et al. 1996) ergaben sich signifikante Risikoerhöhungen, jedoch sind ihrer Ansicht nach diese Studien mit multiplen Expositionen und daher nicht verwertbar. Nach Ausschluss von vier Studien mit vermuteter Koexposition errechneten die Autoren in der Meta-Analyse ein OR von 0,96 (95 % CI: 0,86 - 1,06) - ohne Ausschluss OR = 1,04 (0,94 - 1,14).
Smith, Jones und Smith (2007) kommen anhand der systematischen Durchsicht von 43 Fall-Kontroll-Studien von Personen mit mutmaßlicher beruflicher Benzolexposition und 26 Kohortenstudien aus Raffinerien zu dem Ergebnis, dass die Mehrzahl der Studienergebnisse für einen Zusammenhang zwischen Benzolexposition und NHL spricht, auch wenn die Risikoerhöhungen häufig nicht im statistischen Sinne signifikant sind.
Wesentliche Faktoren, die die Aussage epidemiologischer Studien limitieren, sind Verdünnungseffekte durch unzureichend erfasste Exposition. Als Marker dafür, dass ein solcher Verdünnungseffekt in einer Studie nicht auftritt, wird ein nachgewiesenes erhöhtes Risiko für AML bzw. ANLL (akute nichtlymphatische Leukämie) angesehen. Untersuchungen, die nicht in der Lage waren, diese Risiken nachzuweisen, verfügen mit weit überwiegender Wahrscheinlichkeit nicht über genügend Power, um die selteneren bösartigen Neubildungen des blutbildenden Systems nachzuweisen.
Erkennt man als Ausdruck einer wesentlichen Benzolexposition einen Anstieg des relativen Risikos (SMR, SIR bzw. RR) für AML bzw. ANLL von >2,0 an, unabhängig davon, ob dieser Wert statistisch signifikant ist (Signifikanz ist u. a. eine Frage des Stichprobenumfanges), dann trifft dieser Sachverhalt für vier der insgesamt 31 in der Anlage A aufgeführten und in die Bewertung einbezogenen Kohortenstudien zu. Bei zwei weiteren Studien betrifft die Risikoverdoppelung die "Leukämien" insgesamt, so dass diese Studien nicht dem vorgegebenen Kriterium entsprechen. Diese sechs Kohortenstudien sind in Tabelle 1 mit den Risikoschätzern auch nach Untergruppen unterteilt - aufgeführt. Die Risikoverdoppelung für ANLL wurde als Selektionskriterium gewählt, da ein Wert dieser Größenordnung auf einen Effekt hindeutet, der mit großer Wahrscheinlichkeit nicht auf Artefakten beruht.
Das Multiple Myelom (MM) als eine eigene Hauptgruppe der NHL mit drei Untergruppen wird in allen vier Studien separat ausgewiesen. Die geschätzten relativen Risiken reichen von RR = 0,4 (China Kohorte) bis RR = 3,22 (Health Watch Kohorte) bzw. 2,12 (Pliofilm Kohorte). Die Risikoschätzer dieser vier Studien sind im statistischen Sinn als homogen zu bewerten (X2 = 0,82; p = 0,84). Als gewichteter Schätzer ergibt sich ein Wert von RR = 2,29 (95 %-CI: 1,21 - 4,34) - siehe Tabelle 2. Diese Risikoschätzung stimmt gut mit derjenigen aus der Meta-Analyse von Infante (2006) überein, der nach etwas anderen Kriterien sieben Studien mit gegenüber Benzol exponierten Personen in die Analyse einbezogen hat und ein gewichtetes relatives Risiko von 2,13 (95 % Cl: 1,31 - 3,46) erhält.
Die Non-Hodgkin-Lymphome (NHL), eingegrenzt beschrieben als ICD-9 200 (Lymphosarkom und Reticulosarkom) und 202 (other neoplasms of lymphoid tissues), werden in drei Studien (Pliofilm-, China- und Health Watch Kohorte) separat ausgewiesen mit relativen Risiken von 1,0 (Pliofilm Kohorte) und 3,0 (China Kohorte). Als gewichteter Schätzer ergibt sich ein Wert von RR = 1,63 (95 %-CI: 0,98 - 2,71). Die Risikoschätzer sind als homogen zu bewerten: X2= 0,64; p = 0,42.
Betrachtet man alle "Lymphome" ohne Unterteilung nach ihren Untergruppen, so liegen die relativen Risiken zwischen 1,13 (Pliofilm Kohorte) und RR = 3,0 (China Kohorte). Die Risikoschätzer sind im statistischen Sinne als homogen zu bewerten (Homogenitätstest X2= 2,55; p = 0,466). Als gewichtetes Mittel der Risikoschätzer aus den Einzelstudien ergibt sich ein Wert von RR = 1,55 (95 %-CI: 1,08 bis 2,23).
 | weiter . | |