 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI/GUV-I / DGUV-I

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk; BGI/GUV-I / DGUV-I | |
BGI/GUV-I 505-46 / DGUV Information 213-546 - Verfahren zur getrennten Bestimmung der Konzentrationen von lungengängigen anorganischen Fasern in Arbeitsbereichen - Rasterelektronenmikroskopisches Verfahren
Von den Unfallversicherungsträgern anerkannte Analysenverfahren zur Feststellung der Konzentrationen krebserzeugender Arbeitsstoffe in der Luft in Arbeitsbereichen
Deutsche Gesetzliche Unfallversicherung (DGUV) Information
(Ausgabe 04/2004; 02/2014)
Archiv:
BGI 505-46 (04/2004)![]()
Verfahren zur getrennten Bestimmung der Konzentrationen von lungengängigen anorganischen Fasern in Arbeitsbereichen - Rasterelektronenmikroskopisches Verfahren
Von den Unfallversicherungsträgern anerkannte Analysenverfahren zur Feststellung der Konzentrationen krebserzeugender Arbeitsstoffe in der Luft in Arbeitsbereichen
Verfahren 01
Probenahme mit Pumpe und Abscheidung der Partikel auf einem goldbeschichteten Kernporenfilter, Präparation des Filters und Auswertung mit Rasterelektronenmikroskop (REM) und energiedispersiver Röntgenmikroanalyse (EDXA)
Fasern - 01 - REM/EDXA (erstellt:
Januar 1991, zurückgezogen:
April 2004)
Verfahren 02
Probenahme mit Pumpe und Abscheidung der Partikel auf einem goldbeschichteten Kernporenfilter, Präparation des Filters und Auswertung mit Rasterelektronenmikroskop (REM) und energiedispersiver Röntgenmikroanalyse (EDXA)
Fasern - 02 - REM/EDXA (erstellt: April 2004, zurückgezogen:
März 2013)
Verfahren 03
Probenahme mit Pumpe und Abscheidung der Partikel auf einem goldbeschichteten Kernporenfilter, Präparation des Filters und Auswertung mit Rasterelektronenmikroskop (REM) und energiedispersiver Röntgenmikroanalyse (EDXA)
Fasern - 03 - REM/EDXA (erstellt:
März 2013, ersetzt Verfahren BGI 505-46-02 vom April 2004)
Teil dieses Verfahrens sind die im "Allgemeinen Teil" (BGI/GUV-I 505-0) beschriebenen Anforderungen und Grundsätze.
Verfahren 03
Probenahme mit Pumpe und Abscheidung der Partikel auf einem goldbeschichteten Kernporenfilter, Präparation des Filters und Auswertung mit Rasterelektronenmikroskop (REM) und energiedispersiver Röntgenmikroanalyse (EDXA)
Erprobtes und von den Unfallversicherungsträgern anerkanntes, diskontinuierliches Verfahren zur Bestimmung anorganischer Fasern der Länge L > 5 µm, der Breite 0,2 µm < D < 3 µm und des Länge-zu-Breite-Verhältnisses L/D > 3 (Kriterien nach WHO [2]) in der Luft in Arbeitsbereichen.
Es sind personenbezogene oder ortsfeste Probenahmen für Messungen zur Beurteilung von Arbeitsbereichen möglich.
Die Probenahme erfolgt durch Abscheidung von Partikeln aus der mit einer Pumpe angesaugten Luft auf einem goldbeschichteten Kernporenfilter.
Die Auswertung wird mit dem Rasterelektronenmikroskop (REM) unter Verwendung der energiedispersiven Röntgenmikroanalyse (EDXA) durchgeführt.
Dieses Verfahren benutzt das in der VDI-Richtlinie 3492 [1] beschriebene Bestimmungsprinzip. Es ergänzt das phasenkontrastmikroskopische Verfahren BGI/GUV-I 505-31 [3] in solchen Fällen, in denen
Dieses Verfahren ermöglicht den Nachweis und die Identifizierung von Asbestfasern, Calciumsulfatfasern und anderen anorganischen Fasern mit einer Breite von D > 0,2 µm. Faserförmige Partikel mit D < 0,2 µm werden separat ausgewiesen, aber bei der Berechnung des Messergebnisses nicht berücksichtigt, da entsprechend den Erläuterungen (13) in TRGS 900, Abschnitt 3, die hier beschriebene Methode als Alternative zur lichtmikroskopischen Methode BGI/GUV-I 505-31 [3] eingesetzt werden kann, wenn zusätzlich zur Quantifizierung die Identifizierung der faserförmigen Partikel erforderlich ist. Mit der Methode BGI/GUV-I 505-31 sind Fasern dünner als etwa 0,2 µm nicht erkennbar. Dazu kommt, dass eine Identifizierung so dünner Fasern mit EDXA im Allgemeinen nicht gewährleistet ist.
Das infrarotspektroskopische Verfahren in Kombination mit der Phasenkontrastmikroskopie (BGI 505-30 [4]) erlaubt eine direkte quantitative Massenanteilbestimmung von Asbest im Feinstaub bzw. nach entsprechender Aufarbeitung in Materialproben und kann bei Bedarf wertvolle Zusatzinformationen in Ergänzung der Verfahren BGI/GUV-I 505-31 und BGI/GUV-I 505-46 liefern.
Messungen im Sinne der Asbest-Richtlinien der Länder [5] werden nach [1] vorgenommen. Sie sind nicht Gegenstand dieses Verfahrens.
Kurzfassung
Mit diesem Verfahren wird die über die Probenahmedauer gemittelte Konzentration von anorganischen Fasern der Länge L > 5 µm, der Breite 0,2 µm < D < 3 µm und des Länge-zu-Breite-Verhältnisses L/D > 3 [2] in der Luft im Arbeitsbereich personenbezogen oder ortsfest bestimmt.
| Messprinzip: | Mit Hilfe einer Pumpe wird ein definiertes Luftvolumen durch ein goldbeschichtetes Kernporenfilter gesaugt. Die abgeschiedenen Fasern werden mittels Rasterelektronenmikroskopie (REM) und energiedispersiver Röntgenmikroanalyse (EDXA) gezählt und analysiert. |
| Nachweisgrenze: | Die Nachweisgrenze ist wesentlich vom Probeluftvolumen abhängig. Für ein Probeluftvolumen von 40 l/cm2 Filterfläche liegt die statistische Nachweisgrenze unter Standardauswertebedingungen bei 15.000 Fasern/m3. Ergebnisse werden jedoch bereits oberhalb der analytischen Empfindlichkeit von 5.000 Fasern/m3 ausgewiesen (siehe Abschnitt 5.3). |
| Selektivität: | Die Unterscheidung nach Chrysotilfasern, Amphibolasbestfasern, Calciumsulfatfasern, gegebenenfalls Produktfasern (siehe Abschnitt 2.2.2), und sonstigen anorganischen Fasern ist möglich. |
| Vorteile: | Die Analyse ist faserspezifisch (Morphologie) und faserartspezifisch (Material). |
| Nachteile: | Dieses Verfahren ist mit einem hohen apparativen und zeitlichen Aufwand verbunden. |
| Apparativer Aufwand: | Einrichtung zur Goldbeschichtung (z.B. Sputtereinrichtung) 1), Probenahmeapparatur, Kaltveraschungseinrichtung, Rasterelektronenmikroskop, energiedispersives Röntgenmikroanalysensystem. |
1 Geräte, Betriebsmittel und Zubehör
Zur Durchführung einer Messung nach dem hier beschriebenen Verfahren sind die in Abschnitt 1.1 bis 1.5 genannten Geräte, Betriebsmittel und Zubehörteile erforderlich. Es ist sicherzustellen, dass diese vor ihrer Verwendung sauber und faserfrei sind. Die Überprüfung erfolgt anlassbezogen mit Hilfe von Laborblindproben (z.B. bei neuer Filtercharge oder geänderten Betriebsmitteln). Dazu wird der komplette analytische Verfahrensgang auf ein unbeaufschlagtes Messfilter angewandt.
1.1 Geräte für die Probenahme
- Probenahmekopf
Dieser besteht im Wesentlichen aus einem zylindrischen Ansaugtubus, einem Filterhalter mit Messfilter und einem Absaugstutzen.
Ansaugtubus und Filterhalter sollen aus korrosionsbeständigem Material gefertigt sein. Ein dichter Sitz des eingelegten Messfilters muss gewährleistet sein. Die Länge des Ansaugtubus vor dem Filter muss das 1,5- bis 3,0-fache des effektiven Filterdurchmessers deff (Durchmesser der durchströmten kreisförmigen Filterfläche) betragen [2].
Abb. 1 Prinzipskizze für einen geeigneten Probenahmekopf
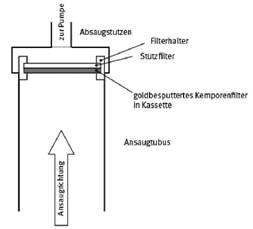
Geeignet sind die für Fasermessungen in Arbeitsbereichen zur phasenkontrastmikroskopischen Auswertung üblichen Probenahmeköpfe [3]. Abbildung 1 zeigt die Prinzipskizze eines geeigneten Probenahmekopfes mit einer Kassette, die als Filterhalter für das auf einem Stützfilter liegende Messfilter dient und nach der Probenahme mit Abdeckungen verschlossen und als Transportbehälter (Filterkapsel) verwendet wird (siehe beispielhaft auch Probenahmesystem PGP-FAP in Abbildung 2).
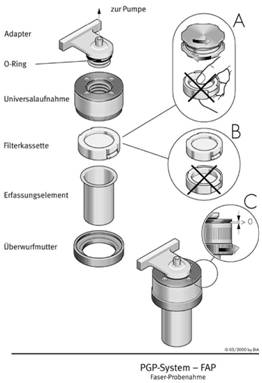
Abb. 2 Probenahmekopf mit Filterkassette - System BIA [5]
(A: Filteroberfläche nicht berühren,
B: Kassette mit Filteroberfläche nach unten einlegen,
C: Überwurfring nicht zu fest anschrauben.)
- Probenahmepumpe
Als Probenahmepumpe wird eine Pumpe verwendet, die durch das Filter je cm2 in der Regel wenigstens 0,24 l/min saugen kann. An staubarmen Arbeitsplätzen können auch höhere Volumenströme verwendet werden.
Der Luftstrom muss pulsationsfrei sein, so dass eine sichere Messung des Volumenstroms möglich ist. Sofern eine batteriebetriebene Pumpe verwendet wird, muss die Kapazität der Batterie für einen kontinuierlichen Einsatz während der gesamten gewählten Probenahmedauer ausreichen.
- Strömungsmesser
Ein geeignetes kalibriertes Messgerät, das die Messung des Luftvolumenstroms mit einer Messgenauigkeit von besser als 5 % ermöglicht.
1.2 Geräte für die Filterpräparation
1.3 Geräte für die Auswertung
Rasterelektronenmikroskop, z.B. Fa. Carl Zeiss Microscopy, 73447 Oberkochen, mit EDX-Analyseeinrichtung, z.B. Fa. Thermo Scientific, 63303 Dreieich, zur Faserzählung und Faseridentifizierung. Das Gerät muss den Mindestanforderungen hinsichtlich Erkennbarkeit und Identifizierung der Fasern genügen (siehe Abschnitt 4.3).
1.4 Betriebsmittel
Messfilter:
Kernporenfilter aus Polycarbonat. Filterdurchmesser je nach Probenahmesystem (üblich sind 25 mm und 37 mm), Nennporenweite 0,4 µm oder 0,8 µm, mit Gold beschichtet (Vorderseite ca. 40 nm, Rückseite ca. 20 nm), z.B. Fa. APC, 65760 Eschborn.
Stützfilter:
Membranfilter aus Celluloseester mit einem mittleren Porendurchmesser > 3 µm (kein faserhaltiges Material, wie z.B. Glasfaserfilter oder Karton).
Sauerstoff:
technisch rein, zum Betrieb der Kaltveraschungsanlage.
Goldtarget:
zum Beschichten der Kernporenfilter.
Argon:
technisch rein, zum Betrieb der Goldbeschichtungsanlage 1).
Kohlenstofflack, Leit-C, Kohle-Tabs oder dergleichen:
zum elektrisch leitfähigen Aufkleben des zu untersuchenden Filters auf den Probenträger des REM, z.B. Fa. Plano, 35578 Wetzlar.
Flüssigstickstoff:
zur Kühlung des EDX-Detektors und des Feldeffekt-Transistors (FET) des EDX-Analysensystems.
1.5 Zubehör
Filterbehälter:
Die in der Filterhalterung eingespannten Filter werden staubdicht in einem Behälter (Filterkapsel) transportiert.
Pinzette:
mit abgerundeten Spitzen zur Handhabung der Filter.
Skalpell:
zum Zerteilen der beaufschlagten Filter.
Stereomikroskop:
zur visuellen Prüfung der Filterbelegung oder Feststellung von Beschädigungen, Vergrößerung ca. 20-fach.
Testpräparat:
mit Chrysotil- und Krokydolithfasern belegtes goldbeschichtetes Kernporenfilter, das zur Überprüfung der Sichtbarkeit und des EDX-Spektrums am REM auch Fasern einer Breite von < 0,2 µm enthält (Präparation wie in Abschnitt 3.1).
Kalibrierstandard:
zur Kalibrierung des Abbildungsmaßstabes des REM.
Testobjekt (Elementstandard), insbesondere Cu:
zur Energiekalibrierung des EDX-Analysensystems.
Referenzmaterialien je nach Aufgabenstellung (z.B. für die Bestimmung von Produktfasern; Asbestreferenzmaterialien, erhältlich z.B. beim Institute of Occupational Medicine (IOM), Edinburgh, Schottland).
2 Probenahme
2.1 Vorbereiten der Messfilter
Die Kernporenfilter müssen vor der Probenahme mit Hilfe einer Bedampfungsanlage oder einer Sputtereinrichtung mit Gold beschichtet werden. Geeignete goldbeschichtete Kernporenfilter sind auch kommerziell erhältlich. Diese Goldbeschichtung ist eine Voraussetzung für eine aufladungsfreie Abbildung im REM und hat gegenüber einer nachträglichen Beschichtung folgende Vorteile:
Zur Minimierung von Kontrastschwankungen ist eine gleichmäßige Goldschichtdicke erforderlich. Eine ausreichende Schichtdicke kann angenommen werden, wenn die Filteroberfläche im Laufe der Beschichtung die dunkle Farbe verliert und den typischen metallischen Goldglanz annimmt sowie bei der Betrachtung im Durchlicht mit einem grünen Schimmer erscheint.
Die Dicke der Goldschicht soll auf der zu beaufschlagenden Seite des Messfilters (stärker reflektierende, glänzende Seite) ca. 40 nm betragen. Eine Beschichtung der Filterrückseite mit einer ca. 20 nm dicken Goldschicht trägt zur Stabilisierung des Messfilters bei und kann eine Verbesserung der Kontrastverhältnisse bewirken. Sofern für die Beschichtungsanlage keine integrierte Schichtdickenmessvorrichtung zur Verfügung steht, kann die Schichtdicke auf einfache Weise im REM mittels der EDX-Einrichtung kontrolliert werden. Dazu werden Filter bekannten Durchmessers mit Gold beschichtet und die Massen des aufgebrachten Goldes durch Differenzwägungen bestimmt. Vergleicht man die Höhen der Goldpeaks für diese Testfilter bei konstantem Strahlstrom mit den Massen, ergibt sich ein linearer Zusammenhang, aus dem sich die Schichtdicke bestimmen lässt [1].
Kernporenfilter einer neuen Charge werden vor der weiteren Verwendung hinsichtlich ihrer Verunreinigung durch anorganische Fasern am REM überprüft. Dazu werden auf zwei goldbeschichteten Filtern der zu prüfenden Charge mit REM/EDXA unter den in Abschnitt 4 genannten Bedingungen je 0,5 mm2 ausgewertet. Dabei darf insgesamt höchstens eine anorganische Faser mit L > 5 µm gefunden werden.
Das für die Probenahme vorbereitete Messfilter wird so in die Filterhalterung eingelegt, dass es plan auf dem Stützfilter aufliegt, beim Einlegen nicht beschädigt wird und ein dichter Sitz gewährleistet ist. Die Berührung der Filteroberfläche mit bloßen Fingern ist zu vermeiden. Die Filterkassetten werden bereits im Labor mit Messfiltern und faserfreien Stützfiltern bestückt und verschlossen. Falls unterhalb des Stützfilters ein Stützgitter eingesetzt werden soll, kann das Erzielen der Leckfreiheit des Probenahmekopfes problematisch sein. Das Messfilter darf nicht direkt auf dem Stützgitter aufliegen.
2.2 Durchführen der Probenahme
2.2.1 Luftproben
Bei der Manipulation der Filter ist eine Berührung mit bloßen Fingern zu vermeiden. Der Probenahmekopf mit eingelegtem Messfilter (z.B. 25 mm Durchmesser) wird unmittelbar vor Beginn der Probenahme geöffnet. Bei Verwendung von Filterkassetten (z.B. 37 mm Durchmesser) werden diese unmittelbar vor der Probenahme geöffnet und in den Probenahmekopf eingelegt - auch hierbei ist eine Berührung der Filteroberfläche zu vermeiden. Die Probenahme erfolgt mit nach unten weisendem Ansaugtubus. Abbildung 2 zeigt ein Beispiel für einen Probenahmekopf mit Filterkassette.
Vor Beginn der Probenahme wird der Volumenstrom so eingestellt, dass je cm2 effektiver Filterfläche 0,24 bis 0,3 l/min (entsprechend 4 cm/s bis 5 cm/s Filteranströmgeschwindigkeit) gefördert werden. Beispielsweise wird bei Verwendung eines Filterhalters mit 30 mm effektivem Durchmesser ein Luftvolumenstrom von 1,7 l/min bis 2,1 l/min benötigt. Der spezifische Volumenstrom soll 0,24 l/(cm2 * min) nicht unterschreiten. Im Einzelfall (z.B. bei hohen Staubkonzentrationen) kann die Anströmgeschwindigkeit auf bis zu 2 cm/s abgesenkt werden. Nur wenn keine Grobstaubpartikel in der Luft im Arbeitsbereich erwartet werden, kann empfohlen werden, den spezifischen Volumenstrom auf bis zu etwa 1,2 l/(cm2 * min) (entsprechend bis zu etwa 20 cm/s Anströmgeschwindigkeit) zu erhöhen. Die Messung des Probeluftvolumenstroms erfolgt für das komplette Probenahmesystem (mit Messfilter und Stützfilter bestückter Probenahmekopf, Schlauchverbindung und Pumpe) mit Hilfe eines geeigneten und kalibrierten Volumenstrommessgerätes.
Die Durchflussrate darf am Ende der Probenahme um nicht mehr als 10 % von der anfänglichen Durchflussrate abweichen. Für die Berechnung des Probeluftvolumens wird der Mittelwert aus Anfangs- und Endvolumenstrom herangezogen. Die Dauer der Probenahme richtet sich bei gegebenem Volumenstrom nach der Staubkonzentration. Keinesfalls darf die Belegung zu dicht werden. Die goldglänzende Filteroberfläche muss mit bloßem Auge noch erkennbar sein. Sobald die Oberfläche anfängt matt zu werden, sollte die Probenahme beendet werden. Unmittelbar nach Beendigung der Probenahme wird die Probenahmepumpe abgeschaltet, die Probenahmedauer notiert und die Filterkassette mit dem beaufschlagten Messfilter entnommen und staubdicht verschlossen. Ort, Zeit und Dauer der Probenahme sind so zu wählen, dass die Exposition repräsentativ erfasst wird [7].
In der Regel gibt eine Probenahmedauer von 2 bis 3 h bei einer Filteranströmgeschwindigkeit von 5 cm/s eine auswertbare Filterbelegung. Bei nur geringen Staubkonzentrationen ohne Grobstaubpartikel ist eine Probenahmedauer auch von 8 Stunden und länger oder eine höhere Filteranströmgeschwindigkeit bis zu 20 cm/s möglich. Im Zweifelsfall können mehrere Filter mit gestaffelter Probenahmedauer (z.B. 1 h, 2 h, 4 h usw.) belegt werden, so dass darunter mindestens eine auswertbare Probe erwartet werden kann. Im Ausnahmefall kann es unvermeidlich sein, eine Probenahmedauer von weniger als 1 h zu wählen, z.B. bei hohen Staubkonzentrationen oder vielen Grobstaubpartikeln. Bei Kurzzeitexpositionen hat sich bewährt, denselben Probenträger während mehrerer Kurzzeitphasen einzusetzen. Auch wenn nur eine einzelne Kurzzeitexposition auftritt, sollte diese durch eine Probenahme von mindestens 1 h erfasst werden.
Tabelle 1 zeigt den Zusammenhang zwischen der erreichbaren Nachweisgrenze und dem spezifischen Probeluftvolumen bei der Auswertung von 0,5 mm2 Filterfläche. Durch Vergrößerung der Auswertefläche kann die Nachweisgrenze herabgesetzt werden (z.B. auf 300 Fasern/m3 bei 1 mm2 Auswertefläche und 1.000 l/cm2). Abbildung 3 zeigt den Zusammenhang zwischen der Nachweisgrenze und dem Probeluftvolumen.
Abb. 3 Nachweisgrenze (NWG) und analytische Empfindlichkeit (AE) in Fasern/m3 in Abhängigkeit vom Probeluftvolumen in m3 für 37 mm- und 25 mm-Filter bei Auswertung von 0,5 mm2 bei einer angenommenen effektiven Filterfläche von 707 mm2 bzw. 380 mm2, vergleiche Abschnitt 4.7 und 5.2
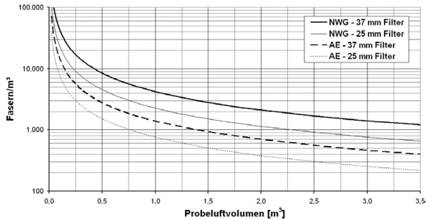
Tabelle 1: Abhängigkeit der Nachweisgrenze vom spezifischen Probeluftvolumen bei Auswertung von 0,5 mm 2 (bezogen auf 37 mm-Filter mit effektiver Filteroberfläche von 707 mm2)
| Nachweis- grenze [Fasern/m3] | Absolutes Probeluft- volumen [l] | Spezifisches Probeluftvolu- men [l/cm2] | Probenahmedauer [h] bei Anströmgeschwindigkeit | ||
| 5 cm/s | 10 cm/s | 20 cm/s | |||
| 15.000 | 283 | 40 | 2,2 | 1,1 | 0,6 *) |
| 10.000 | 424 | 60 | 3,3 | 1,7 | 0,8 *) |
| 7.500 | 566 | 80 | 4,4 | 2,2 | 1,1 |
| 6.000 | 707 | 100 | 5,6 | 2,8 | 1,4 |
| 4.000 | 1.060 | 150 | 8,3 | 4,2 | 2,1 |
| 1.000 | 4.241 | 600 | 33 | 17 | 8,3 |
| 600 | 7.069 | 1.000 | 56 | 28 | 14 |
| *) Die Probenahmedauer sollte 1 h nicht unterschreiten. | |||||
2.2.2 Produktfasern
Unter Produktfasern sind Fasern zu verstehen, die Materialien zuzuordnen sind, die bei dem jeweiligen Arbeitsprozess zum Einsatz kommen oder die in der Umgebung des Probenahmeortes vorhanden sind. Um feststellen zu können, wie hoch insbesondere die Konzentration von Produktfasern in einem Arbeitsbereich ist, müssen zusätzlich zu den Filterproben auch Proben der infrage kommenden Materialien entnommen und als Referenzproben zusammen mit den Filterproben dem Auswertelabor zur Verfügung gestellt werden. Die Proben sollen jeweils wenigstens ein Volumen von 1 cm3 besitzen und repräsentativ für das Material auch hinsichtlich vorhandener Kontaminationen oder der Fasern im Verbund (Kompositwerkstoffe) sein. Nach Möglichkeit sind dem Auswertelabor Kopien der Sicherheitsdatenblätter, zumindest aber vorhandene Produktinformationen (Hersteller, Produktname, Kennzeichnung usw.), zur Verfügung zu stellen.
Die analytischen Möglichkeiten des hier beschriebenen Verfahrens zur Abgrenzung von Produktfasern beschränken sich auf anorganische Fasern. Eine Unterscheidung verschiedener Arten organischer Fasern voneinander ist in der Regel nicht möglich.
2.3 Abstimmungserfordernisse zwischen Probenahme und Auswertung
Probenahme und Auswertung der Messfilter werden in der Regel entweder durch verschiedene Abteilungen und/oder verschiedene Personen innerhalb der jeweiligen Organisation oder verschiedener Firmen durchgeführt.
Ein Informationsfluss zwischen der probenehmenden und auswertenden Stelle, der über die bloße Mitteilung von Luftvolumen und Probenbezeichnung hinausgeht, ist daher im Zuge der Qualitätssicherung unerlässlich. Weitergehende Informationen umfassen neben den nachfolgend aufgeführten Punkten auch die Art der Messung. Werden bei der Auswertung Abweichungen, die möglicherweise während der Probenahme aufgetreten sind, erkannt, sind sie unverzüglich der probenehmenden Stelle oder Person mitzuteilen. Dazu gehören z.B.:
Bei der Probenahme erkennbar auftretende Veränderungen sind dem Labor mitzuteilen, z.B.:
Im Zuge der Auswertung kann dann geprüft werden, ob am Filter Besonderheiten (Filtersitz, Partikel, Faserform und Faserart, Inhomogenitäten) auftreten.
3 Probenvorbereitung
3.1 Präparation der beaufschlagten Filter
Das beaufschlagte Messfilter wird durch Betrachtung zumindest des Filterrandes und der Filtermitte mittels Stereomikroskop und/oder REM bei geringer Vergrößerung daraufhin geprüft, ob die Partikelbelegung des Filters gleichmäßig ist. Sofern dabei eine ungleichmäßige Belegung festgestellt wird, ist das Filter für eine quantitative Auswertung nicht geeignet. Es wird empfohlen, mit dem Stereomikroskop auch den Rand, an dem das Filter in der Filterhalterung angepresst war, auf mögliche Undichtigkeiten während der Probenahme zu untersuchen. Zusätzlich muss das Stützfilter auf mögliche Verfärbungen hin kontrolliert werden, die ebenfalls auf Undichtigkeiten hindeuten. Außerdem muss die Filteroberfläche des Präparates unter streifender Beleuchtung (z.B. mit einer Halogenlampe) makroskopisch auf Wischspuren, Fingerabdrücke oder Beschädigungen hin untersucht werden.
Sofern keine organischen Fasern zu bestimmen sind, kann durch eine Kaltveraschung das organische Material auf dem Filter weitgehend entfernt werden. Dies erleichtert die Auswertung der Probe im REM wesentlich.
Hierzu wird das beladene Filter mit der Staubseite nach oben mit einem doppelseitig klebenden Kohle-Tab elektrisch leitend auf einem REM-Probenträger montiert und in das Kaltveraschungsgerät eingebracht. Aufgrund der elektrischen Leitfähigkeit von Filter und Probenträger ist der Angriff des O2-Plasmas auf das auf dem Filter befindliche organische Material sehr wirksam. Bei Geräten mit kapazitiver Kopplung des Plasmas muss die Leistungsaufnahme so geregelt werden, dass keine Überschläge auftreten, die zu Löchern in der Goldschicht des Filters führen. Nach ca. 30 bis 60 min ist die Veraschung in der Regel abgeschlossen.
Die so vorbereitete Filterprobe ist nun ohne weitere Präparation (insbesondere ohne Nachbeschichten der Probe) für die Untersuchung im REM bereit.
Werden zu Beginn der Auswertung mit dem REM noch zu viele störende organische Partikel gefunden, sollte die Kaltveraschung wiederholt werden.
Es ist empfehlenswert, das ganze Filter für die REM-Auswertung zu montieren. Wenn es erforderlich ist, das Filter zu teilen, sollte dies mit einem scharfen gekrümmten Skalpell mit abrollendem (wiegendem) Schnitt geschehen. Dabei ist darauf zu achten, die Staubbelegung auf der Filteroberfläche nicht zu beeinträchtigen. Wird ein solches Teilstück verwendet, muss es sowohl den Rand- als auch den Mittelbereich des Filters beinhalten (z.B. Sektor). Bei Verwendung von Kohle-Tabs zur Montage auf dem Probenträger kann es unter Umständen nach einiger Zeit zu Veränderungen in der Oberflächenbeschaffenheit der Filter kommen, so dass es sinnvoll ist, ein Teilstück des Filters als Rückstellprobe aufzubewahren.
3.2 Vorbereitung der Referenzproben
Von den Referenzproben (Materialproben) werden Streupräparate hergestellt. Stückige Materialien werden zerkleinert (z.B. gebrochen, geschnitten, gesägt, geraspelt, geschabt), aus Fasermatten oder ähnlichen Produkten wird mit der Pinzette etwas Material entnommen. Diese so zerkleinerten Materialien werden so wie auch die Partikel von Pulvern oder Pudern auf Kohle-Tabs aufgebracht, mit einem Spatel leicht festgedrückt und der lose aufliegende Rest vorsichtig entfernt (unter dem Abzug abklopfen oder abblasen). Die Kohle-Tabs werden auf den REM-Probenträgern befestigt.
4 Auswertung mit dem Rasterelektronenmikroskop
4.1 Allgemeine Verfahrenshinweise
Vergrößerung
Unter Vergrößerung wird das Verhältnis der Länge eines Objektes auf dem Bild, das ausgewertet wird, zur tatsächlichen Länge dieses Objektes verstanden. Es handelt sich demzufolge um die Ist-Vergrößerung und nicht um die am Gerät angezeigte Nenn-Vergrößerung. Die Faserzählung ist bei einer Ist-Vergrößerung von 2.000 : 1 bis 2.500 : 1 durchzuführen. Hiervon kann abgewichen werden, wenn gezeigt werden kann, dass gleichwertige Ergebnisse erzielt werden.
In regelmäßigen Zeitabständen und nach Wartungs- und Reparaturarbeiten am REM muss mit Hilfe eines geeigneten Testpräparates (z.B. Kreuzgitter) die Vergrößerung überprüft werden.
Beschleunigungsspannung
Die Anregungsspannung für die Aufnahme der EDX-Spektren soll mindestens 15 kV betragen. Sowohl der Mg-Kα-Peak von Chrysotil als auch der Fe-Kα-Peak von Krokydolithfasern müssen sicher detektiert werden können. Für die stabile Abbildung organischer Fasern kann es notwendig sein, mit einer Beschleunigungsspannung von 10 kV oder weniger zu arbeiten. Morphologische Details werden durch eine deutlich niedrigere Beschleunigungsspannung (z.B. 3 kV) wesentlich besser erkennbar (siehe Abbildung 4).
Abb. 4: Anorganische Faser auf Kernporenfilter (0,4 µm Poren) bei 15 kV und bei 3 kV


Kippwinkel
Die Probe darf für die Auszählung nicht gekippt werden (Kippwinkel 0°).
Sichtbarkeit dünner Fasern
Alle REM-Parameter (insbesondere Vergrößerung, Beschleunigungsspannung, Strahldurchmesser, Arbeitsabstand, Rasterzeit) müssen so gewählt werden, dass auch sehr dünne und kontrastschwache Fasern noch sichtbar sind. Dazu wird bei der für die Zählung gewählten Vergrößerung eine gerade noch sichtbare Faser auf einem Testpräparat mit Chrysotilfasern nach Abschnitt 1.5 ausgesucht. Die Breite D dieser Faser wird anschließend bei einer Vergrößerung von mindestens 10.000 : 1 bestimmt. Ist sie < 0,2 µm, so ist die Mindestanforderung an die Sichtbarkeit von faserförmigen Partikeln erfüllt. Ist diese Mindestanforderung auch nach mehreren Versuchen nicht erfüllt, müssen die REM-Parameter entsprechend angepasst und die Sichtbarkeitskontrolle wiederholt werden. Diese Kontrolle muss einmal pro Arbeitswoche sowie nach Wartungs- und Reparaturarbeiten am REM vorgenommen werden.
EDX-Analyse
Für die Aufnahme der EDX-Spektren muss ein Detektor verwendet werden, der für die Analyse leichter Elemente geeignet ist (Linienspektrum mit Elementen der Ordnungszahl Z > 11). Die Einstellungen der Betriebsparameter müssen so gewählt werden, dass eine Chrysotilfaser von < 0,2 µm Durchmesser innerhalb einer maximalen Messzeit von 100 s ein hinreichend ausgeprägtes Röntgen-Emissionsspektrum liefert (vergleiche Abschnitt 4.3). Das bedeutet, dass für die Mg- und für die Si-Linie bei Chrysotil ein Verhältnis der Intensitäten S > U erreicht werden muss ( S = Signalhöhe über Untergrund, U = Untergrundsignal bei dieser Energielage). Gleichzeitig ist auf die notwendige Randbedingung S > 3 * √U für die entsprechende Energielage zu achten [1]. In der Regel muss für die EDX-Analyse die Vergrößerung am REM > 5.000 : 1 eingestellt werden.
Für die EDX-Analyse ist ein möglichst großer Raumwinkel des Detektorsystems erwünscht. Die Identifizierbarkeit von Fasern einer Breite um 0,2 µm ist nur bei Raumwinkeln > 10-3 sr gesichert.
In regelmäßigen Zeitabständen und nach Wartungs- und Reparaturarbeiten an der EDX-Anlage muss eine Überprüfung und gegebenenfalls Kalibrierung der Energielage und der Intensitätsverhältnisse der Peaks durchgeführt werden.
Auswertung
Die nach dem Test zur Sichtbarkeit dünner Fasern und zur EDX-Analyse (siehe oben) festgelegten Parameter sind für die Auswertung der Filterproben beizubehalten. Für die EDX-Analyse und für die Ermittlung der Faserabmessungen darf bei höherer Vergrößerung als der für die Faserzählung verwendeten gearbeitet werden.
Die Elementpeaks werden in die Kategorien A, B und C eingeteilt, die wie folgt definiert sind [1]:
| - Kategorie A: | 3U < S |
| - Kategorie B: | U < S < 3U |
| - Kategorie C: | S < U und signifikanter Nachweis des Elements (gerätespezifischer Nachweis in der Regel über Auswertesoftware 2); generell gilt für signifikanten Nachweis: S > 3 * √U mit der Signalhöhe S über dem Untergrund und der Höhe des Untergrunds U). |
4.2 Regeln für die Faserzählung
Grundlage für die Faserzählung sind die Kriterien nach WHO [2].
Abb. 5 Ermittlung der Länge und Breite einer Faser
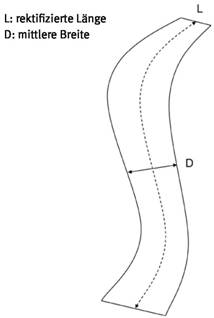
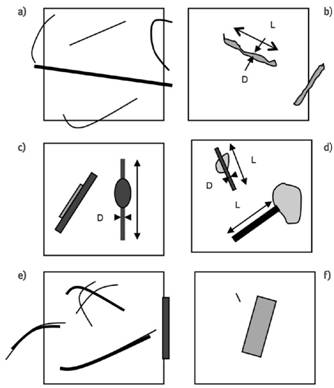
Abbildung 6 zeigt schematisch Beispiele für die Anwendung der Faserzählregeln.
Abb. 6 Beispiele zur Anwendung der Faserzählregeln
(die Länge der Bildkante entspricht 38 µm):
a) 2 1/2 Fasern: 5 Enden im Zählfeld.
b)1 1/2 Fasern.
c) 3 Fasern:
Die beiden aneinanderliegenden Fasern sind deutlich unterscheidbar; die Ausbauchung wird für die Breitenbetrachtung ignoriert.
d) 2 Fasern:
Der sichtbare Teil der Faser wird berücksichtigt.
e) 4 1/2 Fasern:
Agglomerat aus 3 Fasern, die Aufspleißungen werden ignoriert.
Die Enden der an der rechten Kante liegenden Faser werden als außerhalb befindlich angesehen.
f) 0 Fasern:
Die faserförmigen Partikeln sind zu kurz oder zu dick.
4.3 Faseridentifizierung
Nach diesem Verfahren werden Chrysotilfasern, Amphibolasbestfasern, Calciumsulfatfasern, gegebenenfalls anorganische Produktfasern und sonstige anorganische Fasern unterschieden. In die Gruppe "sonstige anorganische Fasern" werden alle Fasern eingestuft, die nicht als Asbest, Calciumsulfatfasern oder gegebenenfalls Produktfasern identifiziert werden können, jedoch ein Linienspektrum mit Elementen der Ordnungszahl Z > 11 (Ausnahme: Kohlenstofffasern) ergeben. Zu organischen Fasern siehe Abschnitt 4.5.
Eine Faseridentifizierung, bei der zwischen Chrysotil, Amphibolasbesten, Calciumsulfat, gegebenenfalls anorganischen Produktfasern und sonstigen anorganischen Fasern unterschieden wird, lässt sich anhand einer halbquantitativen Beurteilung der Elementspektren durchführen. Beispiele für Spektren anorganischer Fasern zeigen die Abbildungen im Anhang 1 (die Goldpeaks werden durch die Goldbeschichtung des Kernporenfilters verursacht). Da die Peakhöhen von dem verwendeten Detektor und den Geräteparametern abhängen, müssen unter den gegebenen Bedingungen eigene Referenzspektren von den Faserarten, z.B. unter Zuhilfenahme von Standardproben, erstellt werden.
Bei der praktischen Durchführung der Analyse ist darauf zu achten, dass der Elektronenstrahl stabil (ohne Drift) auf die Faser gerichtet ist und dabei eventuell anhaftende oder benachbarte Partikel möglichst weit von seinem Auftreffpunkt entfernt liegen.
Zur Identifizierung der Fasern sind die in den Tabellen 2 und 3 genannten Kriterien anzuwenden. Zur Benennung und Beschreibung verschiedener Faserarten wird die in Anhang 3 aufgeführte Fasersystematik empfohlen.
Bei Messungen im Bereich der Verwendung mineralischer Rohstoffe stehen ergänzende Kriterien zur Abgrenzung der Asbestminerale von anderen Mineralen ähnlicher Zusammensetzung zur Verfügung [10].
Tabelle 2: Identifizierungskriterien für Faserstäube
| Faserart | Identifizierungskriterien | Bemerkungen |
| Chrysotil |
| Chrysotil aus Asbestzement: zusätzlicher Ca-Peak durch Bindemittelreste möglich.
Chrysotil aus Magnesiumestrich: zusätzlicher Cl-Peak möglich. |
| Amphibolasbeste | Siehe Tabelle 3 und analytische Hinweise in [8] | Da es sich bei den Amphibolasbesten um eine Mineralgruppe handelt, unterscheiden sich die Spektren hinsichtlich detektierter Elemente und deren Peakhöhen. |
| Calciumsulfat |
| |
| Talk | Mg- und Si-Peak deutlich | Bei Talk ist das Verhältnis von Si- zu Mg-Signal größer als bei Chrysotil. |
| Aluminiumsilikatfasern | Al- und Si-Peak deutlich | Al und Si in etwa gleichen Anteilen, keine weiteren Elemente. |
| Produktfasern | Vergleiche Abschnitt 4.4 | Bei gebrauchten Produktfasern können zusätzlich zu den typischen Peaks andere Elementpeaks, z.B. von Fe, auftreten. |
| Sonstige anorganische Fasern | Alle Fasern, die ein anderes Elementspektrum für mindestens ein Element mit Z > 11 ergeben (Ausnahme: Kohlenstofffasern) | Typische Hauptelemente können sein:
Na, Mg, Al, Si, K, Ca, Fe, weiterhin können auftreten: Ti, Mn, Ba, Zr, B. |
Tabelle 3: Unterscheidungsmerkmale für Amphibolasbeste
| Amphibolasbest | Identifizierungskriterien | Bemerkungen |
| Aktinolith |
| Übergänge zu Tremolit vorhanden. |
| Amosit |
| |
| Anthophyllit |
| Eine Abgrenzung zu Fe-haltigem Talk ist mit dieser Methode in der Regel nicht möglich. |
| Krokydolith |
| |
| Tremolit |
| Übergänge zu Aktinolith vorhanden. |
(Anmerkung: Ist eine exakte Unterscheidung notwendig, sind die ergänzenden Identifikationskriterien [10] anzuwenden und muss die EDXA für die quantitative Elementbestimmung mit geeigneten Standards kalibriert sein.)
Fasern, welche nicht einer der in den Tabellen 2 und 3 genannten Faserarten zuzuordnen sind, werden für die Berechnung des Analysenergebnisses nicht berücksichtigt.
Die Identifizierung nach den hier genannten Kriterien ist nicht in jedem Fall eindeutig [8]. Im Zweifelsfall sind Kenntnisse über die im Arbeitsbereich verwendeten Materialien zu berücksichtigen. In allen Fällen sind die M- und L-Linien der Goldbeschichtung des Filters zusätzlich im Spektrum vorhanden und führen zu Überlagerungen unter anderem mit der K-Linie des Schwefels.
Die Identifizierungskriterien sind zu modifizieren, wenn
Fehlen in dem Elementspektrum Peaks für Elemente mit Z > 11 (mit Ausnahme der Goldpeaks), kann dies als Hinweis auf unter Umständen nach der Veraschung verbliebenes organisches Material gewertet werden. Auch beim Vorliegen sehr dünner anorganischer Fasern (D < 0,2 µm) ist damit zu rechnen, dass keine auswertbaren oder unvollständigen Elementsignale erscheinen.
4.4 Anorganische Produktfasern
Für die Einstufung von Fasern als anorganische Produktfasern müssen die Spektren der Fasern auf dem Messfilter weitgehend (nach den im Folgenden genannten Kriterien) mit denen im Produkt übereinstimmen. Deshalb werden für das in Frage kommende Produkt Referenzproben nach Abschnitt 3.2 hergestellt und darin an unterschiedlichen Stellen Elementspektren dünner Fasern als Referenzspektren aufgenommen. Die Aufnahme dieser Referenzspektren muss - unter Umständen mit Ausnahme der verwendeten Vergrößerung - unter den gleichen Gerätebedingungen erfolgen, wie die Auswertung des Messfilters.
Zur Charakterisierung dieser Referenzspektren werden für jede Materialprobe Elementspektren für jeweils zehn dünne Fasern aufgenommen [1]. Dabei wird für das Produkt
Beim Vergleich der für Fasern auf dem Messfilter gefundenen Spektren mit den Referenzspektren müssen
um die gefundene Faser als Produktfaser zu interpretieren. Bei Produktfasern aus künstlichen Mineralfasern muss zusätzlich die Morphologie (parallele Kanten) mit derjenigen der Fasern im Produkt vereinbar sein.
Zur Benennung und Beschreibung verschiedener Faserarten wird die in Anhang 3 aufgeführte Fasersystematik empfohlen. Eine Übersicht über den Chemismus anorganischer Fasern befindet sich in Anhang D der VDI-Richtlinie 3492 [1].
Bei der Identifizierung von Kohlenstoff- /Carbonfasern als Produktfasern sind zwei Aspekte zu beachten. Zum Einen handelt es sich bei den auf den Probenfiltern gefundenen Fasern dieser Art um splitterförmige Fasern (siehe Abschnitt 4.5). Zum Anderen können diese Fasern anhand des Elementspektrums nur eingeschränkt von anderen organischen Fasern oder Partikeln aus der Matrix von Kohlenstoff-/Carbonfaser-Verbundwerkstoffen unterschieden werden.
4.5 Splitterförmige Fasern
Neben den in technischen Produkten primär vorhandenen alveolengängigen Fasern können in unterschiedlichen Materialien auch splitterförmige Fasern auftreten, die erst durch mechanische Zerkleinerung aus größeren Partikeln/Stücken entstehen. Das Entstehen von Fasern der kritischen Abmessungen geschieht dabei zufällig. Neben faserförmigen Partikeln entstehen zumeist überwiegend anders geformte Partikel.
Der Anteil der alveolengängigen Fasern am zersplitterten Material liegt typischerweise bei deutlich weniger als 0,1 Masse-%. Beispiele hierfür sind körniges Siliziumcarbid und nicht alveolengängige Carbonfasern (typischer Faserdurchmesser vor Zerkleinerung: 7 bis 9 µm) oder Textilglasfasern (Durchmesser 10 bis 25 µm). Alveolengängige Fasern, die erst durch mechanische Bearbeitung aus nicht faserförmigen Materialien oder aus Fragmenten und Bruchstücken dicker Fasern entstanden sind, werden als splitterförmige Fasern bezeichnet.
Zur Identifizierung splitterförmiger Fasern wird in Analogie zu dem in Abschnitt 4.4 genannten Verfahren zur Bestimmung der Produktfaserkonzentration vorgegangen. Die Referenzspektren werden dann an den gröberen Partikeln bzw. dicken Fasern aufgenommen, aus denen die splitterförmigen Fasern generiert werden können.
4.6 Organische Fasern
Sollen bei der Untersuchung auch organische Fasern berücksichtigt werden, darf die Probe nicht verascht werden. Die optimale Beschleunigungsspannung muss anhand der Probe ermittelt werden (mitunter weniger als 10 kV), um eine stabile Abbildung zu erzielen. Dabei ist sicherzustellen, dass Fasern der Dicke 0,2 µm oder, wenn so dünne Fasern in der Probe nicht vorkommen, die dünnsten vorkommenden Fasern sicher erkannt werden können. Es sollen fensterlose Detektoren oder Detektoren mit ultradünnen Fenstern verwendet werden, mit denen leichte Elemente ab Z = 6 (Kohlenstoff) nachgewiesen werden können. Fasern werden dann als organische Fasern eingestuft, wenn die Hauptpeaks Elementen mit Z < 11 zuzuordnen sind (Ausnahme: Kohlenstofffasern). In speziellen Fällen können auch Cl oder S zu beobachten sein.
Eine Unterscheidung zwischen verschiedenen Arten organischer Fasern ist mit dieser Methode nicht möglich. Unter Umständen können morphologische Charakteristiken brauchbare Hinweise liefern [11].
4.7 Berechnen der Faseranzahlkonzentration und Darstellung des Messergebnisses
Das Verfahren liefert als Analysenergebnis die Faseranzahlkonzentrationen Ci (Fasern der Gruppe i je m3 Luft) an anorganischen Fasern in der Luft in Arbeitsbereichen mit den Kriterien 0,2 µm < D < 3 µm, L > 5 µm und L/D > 3.
C1: Chrysotilfasern
C2: Amphibolasbestfasern
C3: Calciumsulfatfasern
C4: Produktfasern (falls gemessen)
C5: sonstige anorganische Fasern
Die Faseranzahlkonzentration für die Fasergruppe i (i = 1 bis 5) wird wie folgt berechnet:
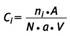
Hierbei bedeuten:
| Ci = | Faseranzahlkonzentration für die Fasergruppe i in Fasern/m3 |
| ni = | nach den Zählregeln ermittelte gewichtete Faserzahl für die Fasergruppe i ("Zählergebnis") |
| A = | wirksame Filterfläche in mm2 |
| N = | Anzahl der ausgewerteten Zählfelder |
| a = | Fläche eines Zählfeldes in mm2 |
| V = | Probeluftvolumen in m3 mit V = Q * t (Q: Probeluftvolumenstrom in m3/h, t: Probenahmedauer in h) |
Fasern mit D < 0,2 µm werden, falls gefunden, ohne Zuordnung zu einer Faserart gesondert aufgeführt.
Die gesonderte Erfassung der Fasern mit D < 0,2 μm ist notwendig, da das internationale Referenzverfahren zur Bestimmung der Faserkonzentration auf einem lichtmikroskopischen Verfahren basiert, mit dem nur Fasern mit D > 0,2 μm erkannt werden. Aus diesem Grund gehen auch nur Fasern mit D > 0,2 μm in das Messergebnis ein.
Die Darstellung des Messergebnisses für die Bestimmung von anorganischen Fasern richtet sich nach dem Messauftrag. Umfasst dieser nicht die Bestimmung der Asbestfaserkonzentration, wird kein Analysenergebnis für Chrysotil und Amphibolasbest ausgewiesen. Da inzwischen überwiegend Messaufträge in Arbeitsbereichen mit künstlichen Mineralfasern vorliegen, würden ansonsten die für Asbest ausgewiesenen Nachweisgrenzen zu Irritationen führen.
Werden jedoch Asbestfasern in der Probe identifiziert, müssen diese mit ausgewiesen werden.
Produktfasern sind hingegen nur dann auszuweisen, wenn der Messauftrag dies vorsieht und eine Probe vom Referenzmaterial vorliegt.
Beim Zählergebnis "0 Fasern" ist für die Faserzahlkonzentration in der Luftprobe als Messergebnis "< analytische Empfindlichkeit" auszuweisen (siehe Abschnitt 5.3). Ist das Zählergebnis "1/2 Faser", so wird es als "1 Faser" ausgewiesen.
Tabelle 4 stellt beispielhaft die Messergebnisse für Zählergebnisse zwischen 0 und 3 Fasern dar.
Wird die Summe von zwei oder mehr Faserklassen ausgewiesen, wird hierfür die Summe der berechneten Messergebnisse je Faserart addiert.
Tabelle 4: Messergebnis für Zählergebnisse zwischen 0 und 3 Fasern bei Probenahme unter Standardbedingungen (NWG 15.000 Fasern/m 3)
| Zählergebnis | Messergebnis [Fasern/m3] | 95 %-Vertrauensbereich [Fasern/m3] |
| 0 | < 5.000 | 0 - 15.000 |
| 1/2 | 5.000 | 130 - 27.860 |
| 1 | 5.000 | 130 - 27.860 |
| 2 | 10.000 | 1.200 - 36.130 |
| 3 | 15.000 | 3.090 - 43.840 |
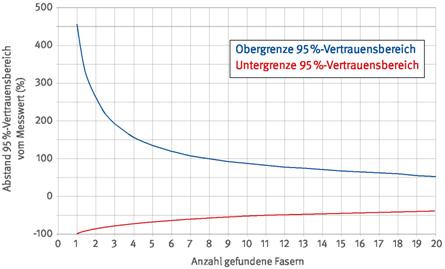
Im Analysenbericht sind grundsätzlich das Messergebnis, die Nachweisgrenze (NWG) und der 95 %-Vertrauensbereich auszuweisen.
Der relative Abstand der Ober- und Untergrenze des 95 %-Vertrauensbereichs vom Messwert hängt allein von der Zahl der gefundenen Fasern ab (siehe auch Anhang 2). Dieser Zusammenhang ist in Abbildung 7 dargestellt.
Abb. 7 95 %-Vertrauensbereich in Abhängigkeit von der Zahl der gefundenen Fasern (siehe auch Anhang 2)
4.8 Analysenbericht
Die Grundlage für die Erstellung des Analysenberichtes ist das in Anlehnung an [1] und [2] während der Auswertung geführte Auswerteprotokoll (Urprotokoll), bei dem zu allen gefundenen Fasern die Elemente aufzuführen sind.
Neben den bereits in DIN EN 17025 aufgeführten Angaben muss der Analysenbericht mindestens folgende Informationen enthalten:
Zusätzlich können Abbildungen und Spektren der untersuchten Fasern zweckdienlich sein.
4.9 Bildung von Mittelwerten
In bestimmten Situationen kann es erforderlich sein, aus zwei oder mehreren Messwerten einen Mittelwert zu bilden (̄C ). Hierzu benötigt man die Faserzahl ni (gefundene Fasern) und das ausgewertete Probeluftvolumen Vi jeder einzelnen Probe. Dafür werden mehrere Einzelwerte zusammengefasst, indem die Faserzahlen ni und die ausgewerteten Probeluftvolumina Vi aufsummiert werden.
Das ausgewertete Probeluftvolumen Vi einer Probe berechnet sich anhand der ausgewerteten Fläche einer Probe (siehe auch Abschnitt 4.7).
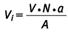
Es gilt dann
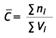
und für die Nachweisgrenze
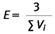
Zur Berechnung des 95 %-Vertrauensbereiches werden die Werte ∑ni und ∑Vi verwendet.
5 Beurteilung des Verfahrens
5.1 Messunsicherheit
Abweichungen der Messgröße (Faseranzahlkonzentration) vom wahren Wert können außer bei der Probenahme entstehen bei:
Die optimale Faserbelegungsdichte liegt im Bereich von etwa 100 bis 1.000 Fasern/mm2. Auch hohe Anteile nicht faserförmiger Partikel beeinträchtigen die Auswertung, da sie diese stören und Fasern überdecken können. Dadurch ist sowohl die Erkennbarkeit als auch die Elementanalyse behindert.
Der 95 %-Vertrauensbereich für die Faseranzahlkonzentration aufgrund der Zählstatistik kann mittels der Poisson-Verteilung abgeschätzt werden (siehe Tabelle im Anhang 2). Zur Berechnung des Vertrauensbereiches werden in der in Abschnitt 4.7 angegebenen Formel die zu der gefundenen Faserzahl ni gehörenden Vertrauensgrenzen λU und λO eingesetzt.
5.2 Nachweisgrenze
Die Nachweisgrenze des Zählverfahrens ist unterschritten, wenn bei der REM-Auswertung keine Faser gefunden wurde. Unter Verwendung der Poisson-Statistik ergibt sich aus der oberen Vertrauensgrenze des 95 %-Vertrauensbereiches zu x = 0 mit λO ∼ 3,0 die Nachweisgrenze aus der Formel in Abschnitt 4.7 (als Zählergebnis wird n = 3 eingesetzt). Sie ist somit probenabhängig und abhängig vom Auswerteaufwand und beträgt beispielsweise für ein spezifisches Probenvolumen von 40 l/cm2 bei einer Auswertefläche von 0,5 mm2 ca. 15.000 Fasern/m3. Für ein spezifisches Probenvolumen von 1 m3/cm2, das nur unter optimalen Bedingungen bei sehr geringen Staubkonzentrationen auswertbare Proben liefert, wäre die Nachweisgrenze ca. 300 Fasern/m3 für 1 mm2 und ca. 600 Fasern/m3 für 0,5 mm2 Auswertefläche. Unter den oben genannten Probenahmebedingungen (Anströmgeschwindigkeit etwa 5 cm/s) würde dies eine Probenahmedauer von etwa 56 h bedeuten.
5.3 Analytische Empfindlichkeit (nach VDI 3492 [1])
Unter der analytischen Empfindlichkeit versteht man im Sinne dieses Verfahrens die berechnete Faseranzahlkonzentration in der Luftprobe, die einer einzelnen auf der Filterprobe gezählten Faser entspricht (siehe auch VDI 3492 [1] und ISO 14966 [13]). Der Wert der analytischen Empfindlichkeit ergibt sich, indem in die Formel aus Abschnitt 4.7 für das Zählergebnis der Wert 1 eingesetzt wird.
5.4 Selektivität
Das Verfahren ist selektiv nach den in den Abschnitten 4.2 und 4.3 genannten Kriterien für Chrysotilfasern, Amphibolasbestfasern, Calciumsulfatfasern, gegebenenfalls Produktfasern und sonstige anorganische Fasern.
Fehlinterpretationen, insbesondere bei der Identifizierung von Asbestfasern, sind möglich, wenn
6 Literatur
| [1] | VDI 3492 Messen von Innenraumluftverunreinigungen - Messen von Immissionen - Messen anorganischer faserförmiger Partikel - Rasterelektronenmikroskopisches Verfahren Beuth Verlag, Berlin 2013 |
| [2] | Determination of airborne fibre number concentrations A recommended method, by phasecontrast optical microscopy (membrane filter method) World Health Organisation (WHO), Genf 1997 |
| [3] | BGI/GUV-I 505-31 Verfahren zur Bestimmung von lungengängigen Fasern - Lichtmikroskopisches Verfahren DGUV, Berlin 2013 |
| [4] | BGI 505-30 Verfahren zur Bestimmung der Massenanteile von Chrysotilasbest und Amphibolasbesten Carl Heymanns Verlag, Köln 1991 |
| [5] | Richtlinie für die Bewertung und Sanierung schwach gebundener Asbestprodukte in Gebäuden (Asbest-Richtlinie) Mitteilungen DIBt, 3/1996, S. 88 |
| [6] | Messung von Gefahrstoffen BIA-Arbeitsmappe, Kennzahl 3030 Erich Schmidt Verlag, Bielefeld 1997 |
| [7] | TRGS 402 Ermittlung und Beurteilung der Konzentrationen gefährlicher Stoffe in der Luft in Arbeitsbereichen BArbBl 11/1997, S. 27 |
| [8] | Messung von Gefahrstoffen. BIA-Arbeitsmappe, Kennzahl 7487 Erich Schmidt Verlag, Bielefeld 1997 |
| [9] | Sachs, L. Angewandte Statistik. Anwendung statistischer Methoden Springer-Verlag, Berlin 1984 |
| [10] | Mattenklott, M. Identifizierung von Asbestfasern in Stäuben, Pulvern und Pudern mineralischer Rohstoffe Gefahrstoffe - Reinhaltung der Luft 58, 1998, S. 15 - 22 |
| [11] | Latzke, P.M., Hesse, R. Textile Fasern. Rasterelektronenmikroskopie der Chemie- und Naturfasern Deutscher Fachverlag, Frankfurt 1988 |
| [12] | TRGS 521 Faserstäube BArbBl 5/2002, S. 96 |
| [13] | ISO14966 Atmosphärische Luft - Bestimmung der Faserzahlkonzentration anorganischer faserförmiger Partikel - Rasterelektronenmikroskopisches Verfahren, Beuth Verlag, Berlin 2002, geändert durch ISO 14966 Technical Corrigendum 1, 2007 |
| [14] | DIN EN 1094-1 Feuerfeste Erzeugnisse für Wärmedämmzwecke - Teil 1: Terminologie, Klassifizierung und Prüfverfahren für Erzeugnisse aus Hochtemperaturwolle zur Wärmedämmung Beuth Verlag, Berlin 2008 |
| Beispiele für Spektren anorganischer Fasern | Anhang 1 |
Alle Spektren:
Abb. A1.1 Chrysotilspektrum (hier Beispiel eines Fehaltigen Chrysotils, Verhältnis S/U ca. 1 für Fe)
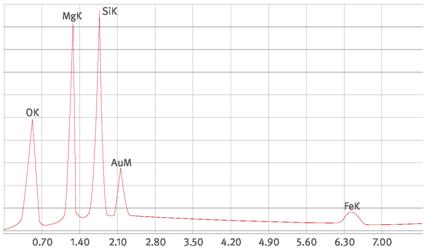
Abb. A1.2 Krokydolithspektrum
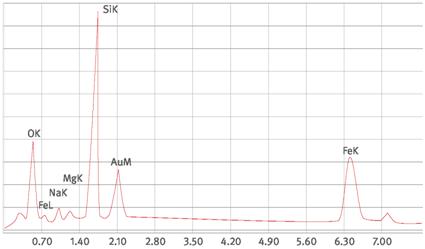
Abb. A1.3 Aktinolithspektrum
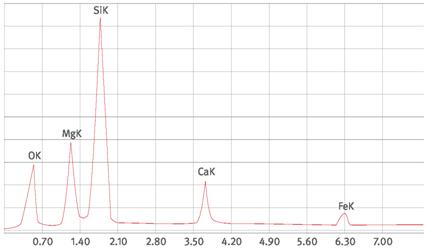
Abb. A1.4 Tremolitspektrum
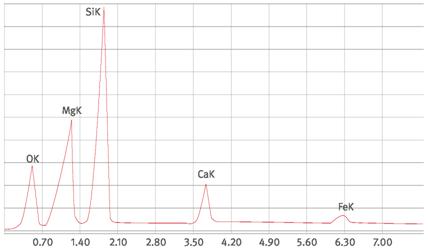
Abb. A1.5 Talkspektrum
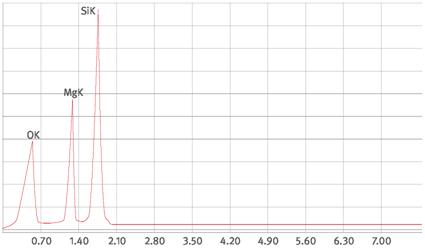
Abb. A1.6 Spektrum einer Aluminiumsilikatfaser
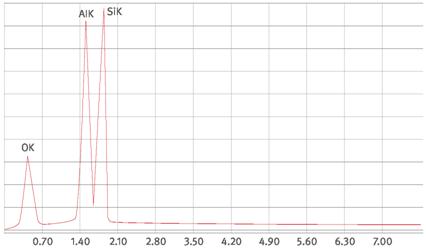
Abb. A1.7 Spektrum einer Steinwollfaser
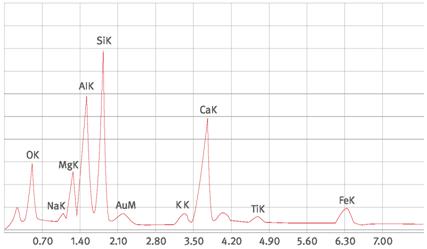
Abb. A1.8 Spektrum einer Hochtemperaturglasfaser
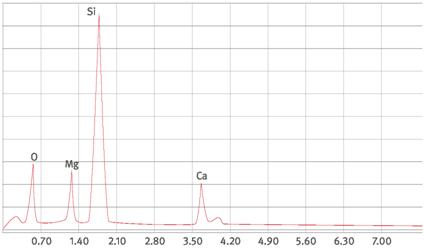
| Vertrauensbereichsgrenzen für das Zählergebnis | Anhang 2 |
Tabelle: Untere und obere Grenzen λU und λO des 95 %-Vertrauensintervalls eines Zählergebnisses x bei Anwendung der Poisson-Statistik
| x | λU | λO | x | λU | λO | |
| 1 | 0,025 | 5,572 | 11,5 | 5,844 | 20,323 | |
| 1,5 | 0,108 | 6,416 | 12 | 6,201 | 20,962 | |
| 2 | 0,242 | 7,225 | 12,5 | 6,560 | 21,597 | |
| 2,5 | 0,416 | 8,006 | 13 | 6,922 | 22,230 | |
| 3 | 0,619 | 8,767 | 13,5 | 7,287 | 22,861 | |
| 3,5 | 0,845 | 9,511 | 14 | 7,654 | 23,490 | |
| 4 | 1,090 | 10,242 | 14,5 | 8,024 | 24,116 | |
| 4,5 | 1,350 | 10,960 | 15 | 8,395 | 24,740 | |
| 5 | 1,623 | 11,668 | 15,5 | 8,769 | 25,363 | |
| 5,5 | 1,908 | 12,368 | 16 | 9,145 | 25,983 | |
| 6 | 2,202 | 13,059 | 16,5 | 9,523 | 26,602 | |
| 6,5 | 2,504 | 13,744 | 17 | 9,903 | 27,219 | |
| 7 | 2,814 | 14,423 | 17,5 | 10,285 | 27,834 | |
| 7,5 | 3,131 | 15,095 | 18 | 10,668 | 28,448 | |
| 8 | 3,454 | 15,763 | 18,5 | 11,053 | 29,060 | |
| 8,5 | 3,782 | 16,426 | 19 | 11,439 | 29,671 | |
| 9 | 4,115 | 17,085 | 19,5 | 11,827 | 30,280 | |
| 9,5 | 4,453 | 17,739 | 20 | 12,217 | 30,888 | |
| 10 | 4,795 | 18,390 | 20,5 | 12,607 | 31,495 | |
| 10,5 | 5,141 | 19,038 | 21 | 12,999 | 32,101 | |
| 11 | 5,491 | 19,682 | 21,5 | 13,393 | 32,705 |
| x | λU | λO | x | λU | λO | |
| 22 | 13,787 | 33,308 | 33,5 | 23,130 | 46,928 | |
| 22,5 | 14,183 | 33,910 | 34 | 23,546 | 47,512 | |
| 23 | 14,580 | 34,511 | 34,5 | 23,962 | 48,094 | |
| 23,5 | 14,978 | 35,111 | 35 | 24,379 | 48,676 | |
| 24 | 15,377 | 35,710 | 35,5 | 24,796 | 49,258 | |
| 24,5 | 15,777 | 36,308 | 36 | 25,214 | 49,839 | |
| 25 | 16,179 | 36,905 | 36,5 | 25,632 | 50,420 | |
| 25,5 | 16,581 | 37,501 | 37 | 26,051 | 51,000 | |
| 26 | 16,984 | 38,096 | 37,5 | 26,471 | 51,579 | |
| 26,5 | 17,388 | 38,690 | 38 | 26,891 | 52,158 | |
| 27 | 17,793 | 39,284 | 38,5 | 27,312 | 52,736 | |
| 27,5 | 18,199 | 39,876 | 39 | 27,733 | 53,314 | |
| 28 | 18,606 | 40,468 | 39,5 | 28,154 | 53,892 | |
| 28,5 | 19,013 | 41,059 | 40 | 28,577 | 54,469 | |
| 29 | 19,422 | 41,649 | 40,5 | 28,999 | 55,045 | |
| 29,5 | 19,831 | 42,238 | 41 | 29,422 | 55,621 | |
| 30 | 20,241 | 42,827 | 41,5 | 29,846 | 56,197 | |
| 30,5 | 20,652 | 43,415 | 42 | 30,270 | 56,772 | |
| 31 | 21,063 | 44,002 | 42,5 | 30,694 | 57,346 | |
| 31,5 | 21,475 | 44,589 | 43 | 31,119 | 57,921 | |
| 32 | 21,888 | 45,174 | 43,5 | 31,545 | 58,495 | |
| 32,5 | 22,301 | 45,760 | 44 | 31,970 | 59,068 | |
| 33 | 22,716 | 46,344 | 44,5 | 32,397 | 59,641 |
| x | λU | λO | x | λU | λO | |
| 45 | 32,823 | 60,214 | 56,5 | 42,74 | 73,29 | |
| 45,5 | 33,250 | 60,786 | 57 | 43,17 | 73,85 | |
| 46 | 33,678 | 61,358 | 57,5 | 43,61 | 74,41 | |
| 46,5 | 34,106 | 61,929 | 58 | 44,04 | 74,98 | |
| 47 | 34,534 | 62,500 | 58,5 | 44,48 | 75,54 | |
| 47,5 | 34,962 | 63,071 | 59 | 44,91 | 76,11 | |
| 48 | 35,391 | 63,641 | 59,5 | 45,35 | 76,67 | |
| 48,5 | 35,821 | 64,211 | 60 | 45,79 | 77,23 | |
| 49 | 36,250 | 64,781 | 60,5 | 46,22 | 77,79 | |
| 49,5 | 36,681 | 65,350 | 61 | 46,66 | 78,36 | |
| 50 | 37,111 | 65,919 | 61,5 | 47,10 | 78,92 | |
| 50,5 | 37,54 | 66,49 | 62 | 47,54 | 79,48 | |
| 51 | 37,97 | 67,06 | 62,5 | 47,97 | 80,04 | |
| 51,5 | 38,40 | 67,62 | 63 | 48,41 | 80,60 | |
| 52 | 38,84 | 68,19 | 63,5 | 48,85 | 81,17 | |
| 52,5 | 39,27 | 68,76 | 64 | 49,29 | 81,73 | |
| 53 | 39,70 | 69,33 | 64,5 | 49,73 | 82,29 | |
| 53,5 | 40,13 | 69,89 | 65 | 50,17 | 82,85 | |
| 54 | 40,57 | 70,46 | 65,5 | 50,60 | 83,41 | |
| 54,5 | 41,00 | 71,02 | 66 | 51,04 | 83,97 | |
| 55 | 41,43 | 71,59 | 66,5 | 51,48 | 84,53 | |
| 55,5 | 41,87 | 72,16 | 67 | 51,92 | 85,09 | |
| 56 | 42,30 | 72,72 | 67,5 | 52,36 | 85,65 |
| x | λU | λO | x | λU | λO | |
| 68 | 52,80 | 86,21 | 79,5 | 62,99 | 99,01 | |
| 68,5 | 53,25 | 86,77 | 80 | 63,44 | 99,57 | |
| 69 | 53,69 | 87,32 | 80,5 | 63,88 | 100,12 | |
| 69,5 | 54,13 | 87,88 | 81 | 64,33 | 100,68 | |
| 70 | 54,57 | 88,44 | 81,5 | 64,77 | 101,23 | |
| 70,5 | 55,01 | 89,00 | 82 | 65,22 | 101,78 | |
| 71 | 55,45 | 89,56 | 82,5 | 65,66 | 102,34 | |
| 71,5 | 55,89 | 90,11 | 83 | 66,11 | 102,89 | |
| 72 | 56,34 | 90,67 | 83,5 | 66,56 | 103,44 | |
| 72,5 | 56,78 | 91,23 | 84 | 67,00 | 104,00 | |
| 73 | 57,22 | 91,79 | 84,5 | 67,45 | 104,55 | |
| 73,5 | 57,66 | 92,34 | 85 | 67,89 | 105,10 | |
| 74 | 58,11 | 92,90 | 85,5 | 68,34 | 105,66 | |
| 74,5 | 58,55 | 93,46 | 86 | 68,79 | 106,21 | |
| 75 | 58,99 | 94,01 | 86,5 | 69,24 | 106,76 | |
| 75,5 | 59,44 | 94,57 | 87 | 69,68 | 107,31 | |
| 76 | 59,88 | 95,13 | 87,5 | 70,13 | 107,87 | |
| 76,5 | 60,32 | 95,68 | 88 | 70,58 | 108,42 | |
| 77 | 60,77 | 96,24 | 88,5 | 71,03 | 108,97 | |
| 77,5 | 61,21 | 96,79 | 89 | 71,47 | 109,52 | |
| 78 | 61,66 | 97,35 | 89,5 | 71,92 | 110,07 | |
| 78,5 | 62,10 | 97,90 | 90 | 72,37 | 110,63 | |
| 79 | 62,55 | 98,46 | 90,5 | 72,82 | 111,18 |
| x | λU | λO |
| 91 | 73,27 | 111,73 |
| 91,5 | 73,72 | 112,28 |
| 92 | 74,16 | 112,83 |
| 92,5 | 74,61 | 113,38 |
| 93 | 75,06 | 113,93 |
| 93,5 | 75,51 | 114,48 |
| 94 | 75,96 | 115,03 |
| 94,5 | 76,41 | 115,58 |
| 95 | 76,86 | 116,13 |
| 95,5 | 77,31 | 116,68 |
| 96 | 77,76 | 117,23 |
| 96,5 | 78,21 | 117,78 |
| 97 | 78,66 | 118,33 |
| 97,5 | 79,11 | 118,88 |
| 98 | 79,56 | 119,43 |
| 98,5 | 80,01 | 119,98 |
| 99 | 80,46 | 120,53 |
| 99,5 | 80,91 | 121,08 |
| 100 | 81,36 | 121,63 |
| Systematik anorganischer und organischer Fasern (Abbildungen geändert und aktualisiert nach Anlage 1 der TRGS 521 vom Mai 2002 [12]) | Anhang 3 |
Abb. A3.1 Gesamtübersicht der Fasern
Abb. A3.2 Systematik der natürlichen anorganischen Fasern
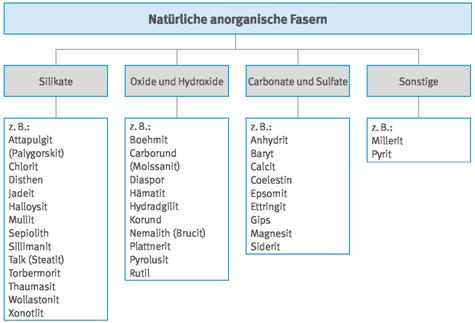
Abb. A3.3 Systematik der natürlichen organischen Fasern
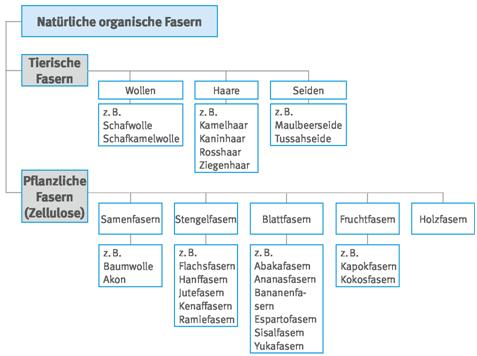
Abb. A3.4 Systematik der künstlich hergestellten anorganischen kristallinen Fasern
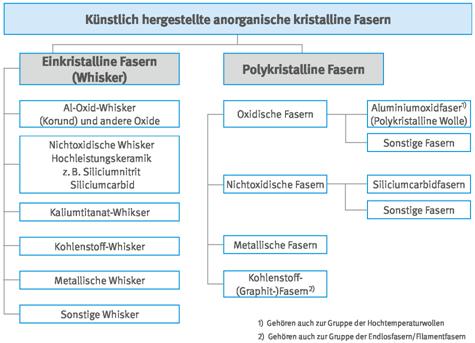
Abb. A3.5 Systematik der künstlich hergestellten anorganischen amorphen (glasigen) Fasern
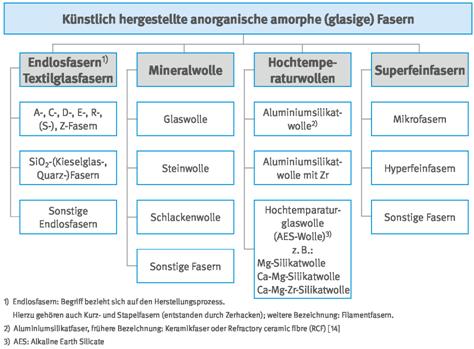
Abb. A3.6 Systematik der künstlich hergestellten organischen Fasern
____________
| 1) | Nicht erforderlich, wenn für die Messung kommerziell erhältliche goldbeschichtete Kernporenfilter verwendet werden. |
| 2) | Bei Zuordnung eines Elements zu einem Signal im Spektrum ist die mögliche Interferenz mit Linien anderer Elemente zu beachten. |
 | ENDE | |