

umwelt-online: TensV - Tensidverordnung (2)
| |
zurück | |
3.3 Gewinnung der Tenside für den Auswahltest
3.3.1 Prinzip
Aus der homogenen Probe wird ein Äthanolextrakt gewonnen, der die Tenside, die Seife und andere alkohollösliche Bestandteile der Wasch- oder Reinigungsmittel-Probe enthält.
Der Extrakt wird im Isopropanol-Wasser-Gemisch gelöst und diese Lösung durch eine auf 323 (K) (50 °C) geheizte Austauscherkombination aus stark saurem Kationenaustauscher und schwach basischem makroporösen Anionenaustauscher gegeben.
Nach Abdampfen des Durchlaufs erhält man die nichtionischen Tenside.
Durch fraktionierte Elution trennt man die aus der Seife stammenden Fettsäuren und die nichttensidischen Anionen ab und gewinnt dann mit einer wäßrig-isopropanolischen Lösung von Ammoniumhydrogencarbonat die anionischen Tenside als Ammoniumsalze. Sie werden als solche dem Abbautest unterworfen.
3.3.2 Vorbereitung der Extraktion
Um die Kapazität der Ionenaustauscher nicht zu überschreiten, ist die Einwaage so zu bemessen, daß in der zur Extraktion angewendeten Probe nicht mehr als 5 g anionische synthetische Tenside und 3 q Seife vorliegen. Sollte die dabei isolierte Menge an nicht-ionischen Tensiden für den Abbautest nicht ausreichen, so ist die Aufarbeitung ein zweites Mal durchzuführen.
Flüssige Produkte sind zur Trockne einzudampfen.
Abbildung 5
Beheizte Austauschersäule

3.3.3 Gewinnung der alkohollöslichen Bestandteile
Zur trockenen Einwaage gibt man 500 ml Äthanol nach Nummer 3.2.2 und kocht eine Stunde am Rückfluß. Zur Vermeidung des Stoßens empfiehlt es sich, einen Rührer einzusetzen. Die noch heiße alkoholische Lösung wird durch eine auf 323 (K) (50 °C) aufgeheizte Nutsche nach Nummer 3.2.11 mit einem grobporigen Filter gegeben. Anschließend spült man Kolben und Nutsche mit etwa 100 ml heißem Äthanol nach. Filtrat und Waschalkohol werden vereinigt. Die Lösung wird auf dem siedenden Wasserbad zur Trockne eingedampft.
3.3.4 Vorbereitung der Ionenaustauscher
Kationenaustauscher
60 ml KAT nach Nummer 3.2.7 werden in ein Becherglas gegeben und mit 200 ml Salzsäure nach Nummer 3.2.9 übergossen. Man läßt unter gelegentlichem Umrühren mindestens 2 Stunden stehen. Man dekantiert die Säure und spült den KAT mit entsalztem Wasser nach Nummer 3.2.1 in die Säule, in die man zuvor einen Glaswollebausch als Stützschicht eingeführt hat. Die Säule wird bei einer Durchlaufgeschwindigkeit von 5 bis 10 ml/min mit entsalztem Wasser chloridfrei gewaschen. Das Wasser verdrängt man mit 200 ml Isopropanol-Wassergemisch nach Nummer 3.2.4, wonach die KAT-Säule betriebsbereit ist.
Anionenaustauscher
Man übergießt im Becherglas 60 ml AAT nach Nummer 3.2.8 mit 200 ml entsalztem Wasser nach Nummer 3.2.1 und läßt den Austauscher mindestens 2 Stunden quellen. Man spült den AAT mit entsalztem Wasser in die Säule. Auch hier dient ein Glaswollebausch als Stützschicht für den Austauscher. Die Säule wird mit 1 N Ammoniumhydrogencarbonatlösung nach Nummer 3.2.5 chloridfrei gewaschen. Dazu werden etwa 1000 ml Lösung benötigt. Anschließend wird mit 200 ml entsalztem Wasser nachgewaschen. Schließlich verdrängt man das Wasser mit 200 ml Isopropanol-Wassergemisch nach Nummer 3.2.4 mit einer Durchflußgeschwindigkeit von 5 bis 10 ml/min.
3.3.5 Durchführung des Ionenaustausches
Man verbindet beide Austauschersäulen derart miteinander, daß sich die KAT-Säule vor der AAT-Säule befindet. Mittels eines Thermostaten werden die Austauschersäulen auf 323 (K) (50 °C) aufgeheizt.
Man löst den nach Nummer 3.3.3 gewonnenen Trockenrückstand in 1000 ml Isopropanol-Wassergemisch nach Nummer 3.2.4 und erwärmt auf etwa 333 (K) (60 °C). Die heiße Lösung gibt man durch die Säulenkombination mit einer Durchlaufgeschwindigkeit von 5 ml/min. Dann wäscht man mit 200 ml des heißen Isopropanol-Wassergemisches nach Nummer 3.2.4 nach.
Zur Gewinnung der nichtionischen synthetischen Tenside werden Durchlauf und Waschalkohol vereint und auf dem Wasserbad nach Nummer 3.2.14 oder im Vakuumrotationsverdampfer eingedampft. Der Rückstand enthält die BiAS. Letzteren bringt man mit entsalztem Wasser nach Nummer 3.2.1 auf ein definiertes Volumen und bestimmt in einem Aliquot den BiAS-Gehalt nach Nummer 4.3. Diese Lösung wird als Stammlösung für die Abbauprüfung der nichtionischen synthetischen Tenside verwendet. Sie muß bei Temperaturen unter 278 (K) (5 °C) aufbewahrt werden.
Zur Gewinnung der anionischen synthetischen Tenside nimmt man die KAT-Säule ab. Aus der AAT-Säule löst man zuerst die aus der Seife stammenden Fettsäuren mit 600 ml 323 (K) (50 °C) warmer Äthanol-CO2-Lösung nach Nummer 3.2.3 heraus. Das Eluat wird verworfen. Das in der AAT-Säule befindliche Äthanol wird mit 200 ml entsalztem Wasser nach Nummer 3.2.1 verdrängt. Sodann werden die nichttensidischen Anionen mit 500 ml Ammoniumhydrogencarbonat-Lösung in Isopropanol-Wassergemisch nach Nummer 3.2.5 herausgelöst; das Eluat wird ebenfalls verworfen. Erst dann können die MBAS mit 600 ml Ammoniumhydrogencarbonat-Lösung in Isopropanol-Wassergemisch nach Nummer 3.2.6 aus der AAT-Säule eluiert werden. Man dampft dieses Eluat auf dem siedenden Wasserbad nach Nummer 3.2.14 oder im Vakuumrotationsverdampfer ein. Als Rückstand verbleibt das Ammoniumsalz der MBAS. Der Rückstand wird mit entsalztem Wasser nach Nummer 3.2.1 auf ein definiertes Volumen gebracht. In einem Aliquot wird der MBAS-Gehalt nach Nummer 4.2 bestimmt. Die Lösung dient als Stammlösung für die Abbauprüfung der anionischen synthetischen Tenside. Sie muß bei Temperaturen unter 278 (K) (5 °C) aufbewahrt werden.
3.3.6 Behandlung- der verwendeten Austauscher
Der Kationenaustauscher wird nach Gebrauch verworfen.
Der Anionenaustauscher ist nach Durchgabe von 200 ml Isopropanol-Wassergemisch nach Nummer 3.2.4 wieder einsatzbereit.
3.4 Gewinnung der Tenside für den Bestätigungstest
3.4.1 Prinzip
Damit die Aufarbeitung der Wasch- oder Reinigungsmittel-Probe handlich bleibt, wird die einzusetzende Menge auf 1000 g begrenzt. Es kann daher nötig werden, die Aufarbeitung zweimal durchzuführen, um die für die biologische Abbauprüfung benötigten Mengen zu erhalten. Erfahrungsgemäß ist diese chargenweise Gewinnung arbeitstechnisch vorteilhafter als das Arbeiten im größeren Maßstab.
Die vorgeschriebenen Austauschermengen sind auf eine nutzbare Kapazität von 600 bis 700 mmol Tenside und Seife ausgelegt.
Zeigt die analytische Vorprüfung die Abwesenheit bestimmter Tensidgruppen, so kann der entsprechende Verfahrensschritt entfallen. Ein Alkoholextrakt ist in jedem Falle erforderlich.
3.4.2 Gewinnung der alkohollöslichen Bestandteile
Nach Eintragen von 1000 g des zu untersuchenden Wasch- oder Reinigungsmittels in 5 l Äthanol nach Nummer 3.2.2 wird das Gemisch 1 Stunde unter Rühren und Rückfluß zum Sieden erhitzt. Dabei ist es zweckmäßig, mit Teilmengen in mehreren Ansätzen zu arbeiten. Die noch heiße alkoholische Lösung wird über eine auf 323 (K) (50 °C) aufgeheizte Nutsche nach Nummer 3.2.11 mit einem grobporigen Filter gegeben und scharf abgesaugt. Anschließend spült man Kolben und Nutsche mit etwa 200 ml heißem Äthanol nach. Filtrat und Spülalkohol werden in einer Saugflasche nach Nummer 3.2.12 aufgefangen.
Bei pastösen und flüssigen Produkten wägt man soviel ein, daß nicht mehr als 55 g anionisches Tensid und 35 g Seife vorliegen. Diese Einwaage wird zur Trockne gebracht. Der Rückstand wird in 2 l Äthanol nach Nummer 3.2.2 aufgenommen und wie vorstehend beschrieben verfahren.
Das äthanolische Filtrat dampft man zweckmäßig mit einem Vakuumrotationsverdampfer zur Trockne ein und nimmt den Rückstand in 5 l Isopropanol-Wassergemisch nach Nummer 3.2.4 auf.
3.4.3 Vorbereitung der Ionenaustauscher Kationenaustauscher
600 ml KAT nach Nummer 3.2.7 werden in ein 3 l-Becherglas gegeben und darin mit 2 l Salzsäure nach Nummer 3.2.9 übergossen. Man läßt mindestens 2 Stunden unter gelegentlichem Umrühren stehen, dekantiert die Säure, spült den KAT mit entsalztem Wasser in die Säule nach Nummer 3.2.13, in die man zuvor einen Glaswollebausch gelegt hat. Die Säule wird mit entsalztem Wasser nach Nummer 3.2.1 bei einer Durchlaufgeschwindigkeit von 10 bis 30 ml/min chloridfrei gewaschen. Das Wasser verdrängt man schließlich mit 2 l Isopropanol-Wassergemisch nach Nummer 3.2.4 . Damit ist die KAT-Säule betriebsbereit.
Anionenaustauscher
Man übergießt im Becherglas 600 ml AAT nach Nummer 3.2.8 mit 2 l entsalztem Wasser nach Nummer 3.2.1 und läßt den Austauscher mindestens 2 Stunden lang quellen. Man spült den AAT mit entsalztem Wasser in die Säule. Auch hier dient ein Glaswollebausch als Stützschicht für den Austauscher. Die Säule wird mit 1 N Ammoniumhydrogencarbonat-Lösung nach Nummer 3.2.5 chloridfrei gewaschen. Dazu werden etwa 2 bis 3 l Lösung benötigt. Anschließend wird mit 2 1 entsalztem Wasser nachgewaschen. Schließlich verdrängt man das Wasser mit 2 l Isopropanol-Wassergemisch nach Nummer 3.2.4 , Durchflußgeschwindigkeit 10 bis 30 ml/min.
3.4.4 Durchführung des Ionenaustausches
Man verbindet beide Austauschersäulen derart miteinander, daß sich die KAT-Säule vor der AAT-Säule befindet. Mittels eines Thermostaten nach Nummer 3.2.16 werden die Austauschersäulen auf 323 (K) (50 °C) aufgeheizt. Man erwärmt die nach Nummer 3.4.2 erhaltenen 5 l Lösung auf 333 (K) (60 °C) und gibt sie über die Austauscherkombination mit einer Durchflußgeschwindigkeit von 20 ml/min. Danach wäscht man die Säulen mit 1000 ml heißem Isopropanol-Wassergemisch nach Nummer 3.2.4 nach.
Zur Gewinnung der nichtionischen synthetischen Tenside werden Durchlauf und Waschalkohol vereint und zur Trockne eingedampft, wozu ein Vakuumrotationsverdampfer empfohlen wird. Der Rückstand enthält die BiAS. Man bringt ihn mit entsalztem Wasser nach Nummer 3.2.1 auf ein definiertes Volumen und bestimmt in einem Aliquot den BiAS-Gehalt nach Nummer 4.3. Die Lösung dient als Stammlösung für die Abbauprüfung der nichtionischen synthetischen Tenside. Sie muß bei Temperaturen unter 278 (K) (5 °C) aufbewahrt werden.
Zur Gewinnung der anionischen synthetischen Tenside (MBAS) nimmt man die KAT-Säule ab und eluiert aus der AAT-Säule zunächst mit 5 l 323 (K) (50 °C) warmer Äthanol-CO2-Lösung nach Nummer 3.2.3 die aus der Seife stammenden Fettsäuren. Das Eluat wird verworfen.
Anschließend eluiert man mit 5 l Ammoniumhydrogencarbonat-Lösung nach Nummer 3.2.6 die MBAS aus der AAT-Säule. Man dampft das Eluat auf dem siedenden Wasserbad nach Nummer 3.2.14 oder im Vakuumrotationsverdampfer zur Trockne ein. Im Rückstand verbleibt das Ammoniumsalz der MBAS, eventuell auch nichttensidische Anionen, die im Bestätigungstest nicht stören. Man bringt den Rückstand mit entsalztem Wasser nach Nummer 3.2.1 auf ein definiertes Volumen und bestimmt in einem Aliquot den MBAS-Gehalt nach Nummer 4.2. Die Lösung dient als Stammlösung für die Abbauprüfung der anionischen synthetischen Tenside; sie muß bei Temperaturen unter 278 (K) (5 °C) aufbewahrt werden.
3.4.5 Behandlung der verwendeten Austauscher
Der Kationenaustauscher wird nach Gebrauch verworfen.
Der Anionenaustauscher ist nach Durchgabe von 2 l Isopropanol-Wassergemisch nach Nummer 3.2.4 wieder einsatzbereit.
4 Analytische Bestimmung kleiner Konzentrationen anionischer und nichtionischer Tenside für die biologische Abbauprüfung
4.1 Chemikalien und Geräte
4.1.1 Pufferlösung pH 10:
Man löst 24 g Natriumhydrogencarbonat (NaHCO3) p. a. und 27 g Natriumcarbonat wasserfrei (Na2CO3) p. a. in entsalztem Wasser und füllt zu 1000 ml auf.
4.1.2 Neutrale Methylenblaulösung:
0,35 g Methylenblau nach dem Deutschen Arzneibuch (DAB 7)* werden mit entsalztem Wasser zu 1000 ml gelöst. Die Lösung muß mindestens 24 Stunden vor Aufstellung der Eichkurve zubereitet worden sein. Die Extinktion der Chloroformphase der Blindprobe, gemessen gegen Chloroform, darf den Wert von 0,015 pro 1 cm Schichtdicke nicht übersteigen.
4.1.3 Saure Methylenblaulösung:
0,35 g Methylenblau nach DAB 7 werden in 500 ml entsalztem Wasser gelöst und mit 6,5 ml H2SO4 der Dichte 1,84 versetzt. Die Lösung wird mit entsalztem Wasser auf 1000 ml aufgefüllt. Die frisch angesetzte Lösung muß mindestens 24 Stunden vor der Aufstellung der Eichkurve zubereitet worden sein. Die Extinktion der Chloroformphase der Blindprobe, gemessen gegen Chloroform, darf den Wert von 0,015 pro 1 cm Schichtdicke nicht übersteigen.
4.1.4 Chloroform, CHCl3 frisch destilliert
4.1.5 Dodecylbenzolsulfonsäuremethylester
4.1.6 Äthanolische Kaliumhydroxidlösung, KOH 0,1 N
4.1.7 Äthanol rein COH5OH
4.1.8 Schwefelsäure H2SO4 1 N
4.1.9 Phenolphthalein-Lösung:
Man löst 1 g Phenolphthalein in 50 ml Äthanol und gibt unter ständigem Rühren 50 ml entsalztes Wasser hinzu. Ein etwaiger Niederschlag wird abfiltriert.
4.1.10 Scheidetrichter, Inhalt 250 ml
4.1.11 Meßkolben, Inhalt 50 ml
4.1.12 Meßkolben, Inhalt 500 ml
4.1.13 Meßkolben, Inhalt 1000 ml
4.1.14 Wägepipette
4.1.15 Rundkolben mit Schliff und aufgesetztem Rückflußkühler, Inhalt 250 ml
Siedeperlen
4.1.16 pH-Meter
4.1.17 Photometer mit Meßmöglichkeit bei 650 nm und 1 bis 5 cm-Küvetten
4.1.18 Watte, Reinheitsanforderungen nach DAB 7
4.1.19 Entsalztes Wasser
4.1.20 Essigsäureäthylester CH3CO × O × C2H5 rein, frisch destilliert
4.1.21 Natriumhydrogencarbonat NaHCO3 p. a.
4.1.22 Salzsäure HCl 1 %
4.1.23 Methanol CH3OH
frisch destilliert, in Glasflaschen aufzubewahren
4.1.24 Bromkresolpurpur-Lösung:
0,1 g Bromkresolpurpur werden in 100 ml Methanol nach Nummer 4.1.23 gelöst
4.1.25 Fällungsreagenz:
Das Fällungsreagenz ist eine Mischung von 2 Volumteilen der Lösung a nach Nummer 4.1.26 und 1 Volumteil der Lösung B nach Nummer 4.1.27. Die Mischung ist in einer braunen Flasche aufzubewahren und etwa eine Woche haltbar.
4.1.26 Lösung A:
1,7 g Wismut (III)-nitrat, reinst, DAB 7 (Bi(OH)2 NO3) werden in 20 ml Eisessig nach Nummer 4.1.28 gelöst und mit entsalztem Wasser auf 100 ml aufgefüllt.
65 g Kaliumjodid (KJ) p. a. werden in etwa 100 ml entsalztem Wasser gelöst. Beide Lösungen werden in einen 1000 ml-Meßkolben gegeben, 200 ml Eisessig hinzugefügt und mit entsalztem Wasser zur Marke aufgefüllt. Diese Lösung ist etwa eine Woche haltbar.
4.1.27 Lösung B:
290 g Bariumchlorid BaCl2 × 2 H2O gelöst in 1000 ml entsalztem Wasser.
4.1.28 Eisessig, 99 bis 100 % CH3COOH (Eisessig geringerer Konzentration ist ungeeignet)
4.1.29 Ammoniumtartrat-Lösung:
12,4 g Weinsäure p. a. und 12,4 ml Ammoniaklösung etwa 25 % NH3 (d = 0,91), reinst, werden vereinigt und mit entsalztem Wasser auf 1000 ml aufgefüllt.
4.1.30 Ammoniaklösung NH3 1 %
4.1.31 Standardacetatpuffer-Lösung:
40 g Natriumhydroxid NaOH p. a. werden in einen 1000 ml-Meßkolben gegeben und mit etwa 500 ml entsalztem Wasser gelöst. Dann werden 120 ml Eisessig zugefügt. Nach gründlichem Mischen und Abkühlen wird mit entsalztem Wasser zur Marke aufgefüllt.
4.1.32 Pyrrolidindithiocarbamat-Lösung 0,0005 N (abgekürzt Carbat-Lösung):
Man löst 103,0 mg Pyrrolidindithiocarbonsäure-Natriumsalz in etwa 500 ml entsalztem
Wasser, gibt dann 10 ml n-Amylalkohol p. a. und 0,5 g Natriumhydrogencarbonat hinzu und füllt mit entsalztem Wasser auf 1000 ml auf.
4.1.33 Kupfersulfat-Stammlösung:
1,249 g Kupfer (II)-sulfat (CuSO4 × 5 H2O) p. a. und 50 ml 1 N-Schwefelsäure werden in etwa 200 ml entsalztem Wasser gelöst und zu 1000 ml aufgefüllt. (Keine verwitterten Kristalle verwenden)
4.1.34 Kupfersulfat-Eichlösung:
50,0 ml der Stammlösung nach Nummer 4.1.33 und 10 ml 1 N-Schwefelsäure werden mit entsalztem Wasser zu 1000 ml aufgefüllt.
4.1.35 Natriumchlorid NaCl p. a.
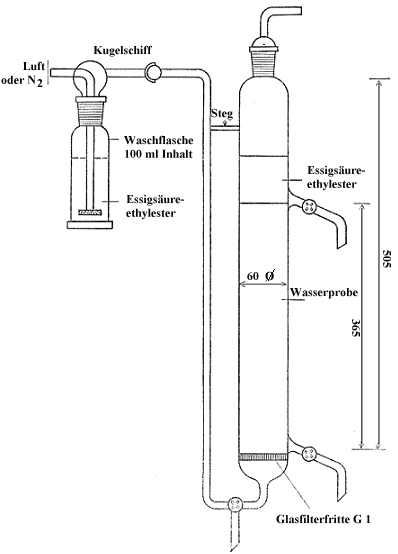
4.1.36 Tensid-Ausblasegerät ( Abb. 6):
Der Durchmesser von Fritte und Zylinder soll etwa gleich groß sein.
Abbildung 6
Tensid-Ausblasegerät
4.1.37 Kationenaustauscher (H-Form) 50 bis 100 mesh.
4.1.38 Magnetrührwerk mit Magnetstab 25 bis 30 mm.
4.1.39 Goochtiegel, Durchmesser des perforierten Bodens 25 mm, type G 4.
4.1.40 Rundfilter aus Glasfaserpapier, Ø = 27 mm, Faserdurchmesser 0,5 bis 1,5 µm.
4.1.41 Saugflaschen mit Vorstoß und Gummimanschette für Filtertiegel, Inhalt 250 und 500 ml.
4.1.42 Polyäthylenspritzflasche, Inhalt 500 ml, für Eisessig.
4.1.43 Austauschersäule für 10 ml Austauscher.
4.1.44 Registrierendes Potentiometer mit Platin/Kalomel- oder Platin/Silberchlorid-Meßkette, Meßbereich 250 mV mit automatischer Bürette, 20 bis 25 ml Inhalt oder alternativ eine manuelle Einrichtung zur Potentiometrie.
4.1.45 methanolische Salzsäure HCl 10 %.
4.2 Bestimmung anionischer Tenside als MBAS
4.2.1 Prinzip, Störungen
Die Methode beruht darauf, daß der kationische Farbstoff Methylenblau mit anionischen Substanzen blaue Salze bildet, die mit Cloroform extrahierbar sind. Zur Ausschaltung von Störungen wird zunächst aus alkalischer Lösung extrahiert und der Extrakt dann mit saurer Methylenblaulösung geschüttelt. Die abgetrennte organische Phase wird im Absorptionsmaximum bei 650 nm gemessen. Zur Auswertung der Messung bedarf es einer Eichkurve. Die dazu benötigte Eichlösung wird aus der Standardsubstanz Dodecylbenzolsulfonsäuremethylester (Tetrapropylentyp, MG 340) nach Verseifen zum Kaliumsalz hergestellt. Die MBAS wird als Natriumdodecylbenzolsulfonat angegeben.
Die unter Nummer 4.2.3 angegebene Formel berücksichtigt dies.
Die Analysenproben dürfen nicht durch eine Schaumschicht hindurch entnommen werden.
Die für die Analysen verwendeten Geräte sind nach gründlicher Reinigung mit Wasser, mit alkoholischer Salzsäure nach Nummer 4.1.45 und anschließend wieder mit Wasser ausgiebig zu spülen.
4.2.2 Ausführung
Das zu untersuchende Wasser aus dem Zu- und Ablauf der Belebungsanlage ist unmittelbar nach der Probeentnahme zu filtrieren. Die ersten 100 ml des Filtrats sind zu verwerfen.
In einen Scheidetrichter nach Nummer 4.1.10 ist ein abgemessenes Volumen der filtrierten, soweit erforderlich, neutralisierten Probe zu geben, deren Gehalt an MBAS zwischen 20 und 150 mg liegen soll. Bei geringerem Gehalt an MBAS können bis zu 100 ml der Probe verwendet werden. Werden weniger als 100 ml verwendet, so ist mit entsalztem Wasser nach Nummer 4.1.19 auf 100 ml aufzufüllen. Der Probe sind 10 ml Pufferlösung nach Nummer 4.1.1, 5 ml neutrale Methylenblaulösung nach Nummer 4.1.2 und 15 ml Chloroform nach Nummer 4.1.4 zuzusetzen. Die Mischung ist gleichmäßig und nicht zu heftig 1 Minute zu schütteln. Nach Phasentrennung ist die Chloroformschicht in einen zweiten Scheidetrichter nach Nummer 4.1.10 abzulassen, der 110 ml entsalztes Wasser und 5 ml saure Methylenblaulösung nach Nummer 4.1.3 enthält. Die Mischung ist gleichmäßig und nicht zu heftig 1 Minute zu schütteln. Die Chloroformschicht ist durch ein mit Chloroform nach Nummer 4.1.4 angefeuchtetes Wattefilter nach Nummer 4.1.18 in einen Meßkolben nach Nummer 4.1.11 zu filtern.
Die Extraktion der alkalischen und der sauren Lösung ist je dreimal auszuführen, wobei für die zweite und dritte Extraktion je 10 ml Chloroform nach Nummer 4.1.4 anzuwenden sind. Die durch die gleiche Watte filtrierten und vereinigten Chloroformextrakte sind im Meßkolben nach Nummer 4.1.11 mit Chloroform nach Nummer 4.1.4 bis zur Marke aufzufüllen. Die hierfür notwendige Menge Chloroform muß zum Nachwaschen der Watte benutzt werden. Die Farbintensität der Chloroformlösung ist mit einem Photometer nach Nummer 4.1.17 bei 650 nm und mit 1- bis 5-cm-Küvetten zu messen.
4.2.3 Aufstellung der Eichkurve
Man wägt aus einer Wägepipette 400 bis 450 mg Dodecylbenzolsulfonsäuremethylester nach Nummer 4.1.5 auf 0,1 mg genau in einen Rundkolben nach Nummer 4.1.15 ein und fügt 50 ml äthanolische Kaliumhydroxidlösung nach Nummer 4.1.6 und einige Siedeperlen hinzu. Nach Aufsetzen des Rückflußkühlers wird 1 Stunde gekocht. Nach dem Erkalten spült man Kühler und Schliff mit etwa 30 ml Äthanol nach Nummer 4.1.7 ab und gibt diese Spülflüssigkeit zum Kolbeninhalt hinzu. Anschließend titriert man die Lösung mit Schwefelsäure nach Nummer 4.1.8 gegen Phenolphthalein nach Nummer 4.1.9 bis zur Entfärbung, überführt diese Lösung in einen Meßkolben nach Nummer 4.1.13, füllt mit entsalztem Wasser zur Marke auf und mischt durch.
Von dieser Tensid-Stammlösung wird eine weitere Verdünnung hergestellt. Man entnimmt 25 ml, überführt sie in einen Meßkolben nach Nummer 4.1.12, füllt mit entsalztem Wasser zur Marke auf und mischt durch.
|
Diese Eichlösung enthält pro ml |
E × 1,023 ______________ 20000 |
mg MBAS, wobei E die Einwaage in mg bedeutet |
Für die Eichkurve werden je 1, 2, 4, 6, 8 ml dieser Eichlösung entnommen und im Scheidetrichter nach Nummer 4.1.10 jeweils auf 100 ml mit entsalztem Wasser aufgefüllt. Dann verfährt man weiter wie unter Nummer 4.2.2 angegeben.
4.3 Bestimmung nichtionischer Tenside als BiAS
4.3.1 Prinzip, Störungen
Die Tenside werden durch Ausblasen in eine Essigsäureäthylester-Phase überführt und isoliert. In der angewendeten Probenmenge sollte der Gehalt an nichtionischen Tensiden im Bereich zwischen 250 bis 800 feg liegen.
Nach Phasentrennung und Abdampfen des Lösungsmittels werden die nichtionischen Tenside vom Typ der Alkylenoxid-Addukte in wäßriger Lösung mit modifiziertem Dragendorffschen Reagenz nach Nummer 4.1.25 gefällt.
Anionische Tenside gelangen beim Ausblasen mit in die Essigsäureäthylester-Phase. Sie stören jedoch bis zu einer Konzentration von 5 mg/l die Bestimmung nicht. Kationische Tenside werden miterfaßt. Sie müssen gegebenenfalls durch Kationenaustausch abgetrennt werden.
Der Niederschlag wird abfiltriert, mit Eisessig nach Nummer 4.1.28 gewaschen und in Ammoniumtartrat-Lösung nach Nummer 4.1.29 gelöst. Das in Lösung befindliche Wismut wird bei pH 4 bis 5 mit Pyrrolidindithiocarbamat-Lösung nach Nummer 4.1.32 mit der Meßkette nach Nummer 4.1.44 potentiometrisch titriert.
Das Titrationsergebnis wird mit dem empirischen Eichfaktor 54 zur Umrechnung auf die Bezugssubstanz Nonylphenol mit 10 Molen Äthylenoxid multipliziert.
4.3.2 Ausführung
Anreicherung und Isolierung der Tenside
Die Wasserprobe wird durch ein grobporiges Papierfilter filtriert. In das Ausblasgerät nach Nummer 4.1.36 ( Abb.6) wird eine abgemessene Probemenge gegeben, die 250 bis 800 arg nichtionische Tenside enthalten soll.
Zur Verbesserung des Isoliereffektes werden 100 g Natriumchlorid nach Nummer 4.1.35 und 5 g Natriumhydrogencarbonat nach Nummer 4.1.21 benötigt.
Beträgt das angewandte Probenvolumen über 500 ml, so werden diese Salze in fester Form in das Ausblasgerät gegeben und unter Durchleiten von Stickstoff oder Luft gelöst. Kommt ein geringeres Probenvolumen zur Anwendung, werden die Salze in etwa 400 ml Wasser gelöst und dann zugegeben.
In jedem Fall wird mit Wasser bis zum oberen Ablaßhahn aufgefüllt.
Man überschichtet mit 100 ml Essigsäureäthylester nach Nummer 4.1.20.
Die Waschflasche in der Gasstromzuleitung (Stickstoff oder Luft) wird zu etwa :a mit Essigsäureäthylester gefüllt.
Man leitet einen Gasstrom von 50 bis 601 je Stunde durch die Apparatur; der Einbau eines Strömungsmessers ist zu empfehlen. Die Gasmenge muß so bemessen sein, daß die Phasen erkennbar getrennt bleiben und an der Phasengrenzfläche keine Turbulenz entsteht. Damit wird eine Vermischung der Phasen und ein Inlösunggehen des Essigsäureäthylesters vermieden. Nach 5 Minuten wird der Gasstrom abgestellt.
Ist die organische Phase durch Lösen in Wasser um mehr als 20 % vermindert worden, muß der Ansatz verworfen werden.
Die organische Phase wird vollständig in den Scheidetrichter nach Nummer 4.1.10 abgelassen. Die im Scheidetrichter etwa abgesetzte wäßrige Phase - es sollen nur wenige ml sein - wird in das Ausblasegerät zurückgegeben. Die Essigsäureäthlylester-Phase wird durch ein trockenes grobporiges Filter in ein 250 ml Becherglas filtriert.
Man gibt erneut 100 ml Essigsäureäthylester in das Ausblasegerät und leitet weitere 5 Minuten Stickstoff oder Luft durch. Die organische Phase wird in den bereits bei der ersten Abtrennung benutzten Scheidetrichter abgelassen. Die wäßrige Phase wird verworfen und die organische Phase über das gleiche Filter zu der ersten Essigsäureäthylester-Charge gegeben. Scheidetrichter und Filter werden mit 20 ml Essigsäureäthylester nachgespült.
Der Essigsäureäthylester-Extrakt wird auf dem Wasserbad unter dem Abzug zur Trockne eingedampft. Es hat sich bewährt, während des Eindampfvorganges einen leichten Luftstrom auf die Oberfläche der Lösung zu richten, um die Verdampfung zu beschleunigen.
Fällen und Filtrieren:
Der Trockenrückstand wird in 5 ml Methanol nach Nummer 4.1.23 aufgenommen. Man fügt 40 ml entsalztes Wasser nach Nummer 4.1.19 und 0,5 ml Salzsäure nach Nummer 4.1.22 hinzu und rührt die Lösung mit dem Magnetrührer nach Nummer 4.1.38.
In diese Lösung gibt man aus einem Meßzylinder 30 ml Fällungsreagenz nach Nummer 4.1.25. Der Niederschlag bildet sich bei fortgesetztem Rühren. Nach 10 Minuten bricht man das Rühren ab und läßt mindestens 5 Minuten stehen.
Danach filtriert man durch einen Goochtiegel nach Nummer 4.1.39, dessen Boden mit einem Glasfaser-Filterpapier nach Nummer 4.1.40 belegt ist. Das Filter wird zuvor mit etwa 2 ml Eisessig angefeuchtet und angesaugt. Becherglas, Magnetstab und Tiegel werden gründlich mit Eisessig nachgewaschen, wozu etwa 40 bis 50 ml notwendig sind. Das Waschen mit Eisessig wird durch Anwendung einer Polyäthylenspritzflasche nach Nummer 4.1.42 sehr erleichtert. Es ist nicht erforderlich, den am Becherglas fest anhaftenden Niederschlag quantitativ auf das Filter zu bringen, da die Lösung des Niederschlages vor der Titration wieder in das Fällungs-Becherglas gegeben und der verbleibende Niederschlag dann gelöst wird.
Lösen des Niederschlags:
Der Niederschlag wird im Filtertiegel nach Nummer 4.1.39 gelöst. Damit ein Verspritzen der Lösung in der Saugflasche nach Nummer 4.1.41 verhindert wird, setzt man den Filtertiegel in einem gläsernen Vorstoß auf die 250 ml-Saugflasche. Um Fehler durch Verschleppen von Fällungsreagenz auszuschließen, darf die Gummimanschette aus dem Filtrationsschritt hier nicht verwendet werden. Die Gummimanschetten für das Filtrieren und für den Lösungsschritt sind getrennt aufzubewahren.
Der Niederschlag wird durch Zugabe von heißer Ammoniumtartratlösung nach Nummer 4.1.29 in 3 Portionen von je 10 ml gelöst. Der Inhalt der Saugflasche wird in das Fällungsbecherglas gegeben; weitere 20 ml Ammoniumtartratlösung läßt man die Wandungen des Fällungsbecherglases hinablaufen, um Reste des Niederschlages zu lösen.
Filtertiegel, Vorstoß und Saugflasche werden gründlich mit 100 bis 150 ml Wasser gewaschen und dieses Wasser in das Fällungsbecherglas gegeben.
Titration:
Man rührt die Lösung mit dem Magnetrührwerk nach Nummer 4.1.38, setzt einige Tropfen Bromkresolpurpur-Lösung nach Nummer 4.1.24 zu und stellt mit Ammoniaklösung nach Nummer 4.1.30 auf Farbumschlag nach violett ein, da die Lösung durch Essigsäurereste, die vom Nachwaschen herrühren, schwach sauer sein kann.
Man gibt 10 ml Standardacetatpuffer-Lösung nach Nummer 4.1.31 hinzu, führt die Elektroden nach Nummer 4.1.44 ein und titriert mit eingetauchter Bürettenspitze potentiometrisch mit der Carbat-Lösung nach Nummer 4.1.32 bis über den Potentialsprung hinaus.
Titrationsgeschwindigkeit 2 ml/min, Papiervorschub etwa 2 cm/ml.
Als Endpunkt gilt der Schnittpunkt der Tangenten, die man an die beiden Äste der Potentialkurve legt. Eine gelegentlich zu beobachtende Verflachung des Potentialsprungs läßt sich durch leichtes Abschmirgeln der Platin-Elektrode beheben.
4.3.3 Blindversuch
Parallel zu den eigentlichen Bestimmungen läuft ein Blindversuch mit, bei dem 5 ml Methanol nach Nummer 4.1.23 und 40 ml entsalztem Wasser eingesetzt und nach Nummer 4.3.2 ab "Fällen und Filtrieren" weiter verarbeitet werden. Der Verbrauch im Blindversuch sollte unter 1 ml Meßlösung liegen, andernfalls sind die Reagenzien neu anzusetzen und die Bestimmungen zu wiederholen (Die Ursache von überhöhten Blindwerten kann die Anwesenheit von Schwermetallen in den Reagenzien sein.). Das Ergebnis des Blindversuchs ist bei dei Berechnung zu berücksichtigen.
Kontrolle des Faktors der Carbat-Lösung:
Zur Faktorkontrolle der Carbat-Lösung werden vor den Bestimmungen oder bei Serienanalysen einmal täglich 10 ml der Kupfersulfat-Eichlösung nach Nummer 4.1.34 nach Zugabe von 100 ml entsalztem Wasser und 10 ml Standardacetatpuffer-Lösung nach Nummer 4.1.31 titriert. Der Faktor f der Carbat-Lösung wird nach folgender Formel errechnet:
f = |
10
a |
a: Verbrauch an Carbat-Lösung in ml
Mit diesem Faktor sind die Titrationsergebnisse zu multiplizieren.
4.3.4 Abtrennung störender kationischer Tenside
Kationische Tenside werden bei der Fällung miterfaßt und täuschen einen Gehalt an nicht-ionischen Tensiden vor. Sie müssen deshalb, sofern sie vorhanden sein sollten, wie nach-stehend beschrieben, abgetrennt werden.
Ist der Bestimmung eine Aufarbeitung nach Nummer 3 vorangegangen, entfällt die Abtrennung störender kationischer Tenside nach Nummer 4.3.4.
Der Abdampfrückstand des Essigsäureäthylester-Extraktes nach Nummer 4.3.2 wird in etwa 20 ml Methanol nach Nummer 4.1.23 aufgenommen. Diese Lösung gibt man über eine Austauschersäule nach Nummer 4.1.43, gefüllt mit 10 ml Kationenaustauscher nach Nummer 4.1.37.
Die Durchflußgeschwindigkeit wird auf schnelle Tropfenfolge des Ablaufes eingestellt. Nachgewaschen wird mit etwa 50 bis 60 ml Methanol.
Bei Vorliegen höher äthoxylierter Tenside (ÄO-Kette > 25 ÄO/Mol) wende man anstelle des Methanols die Mischung Methanol : Methylenchlorid 80 : 20 (v/v) an.
Die methanolische Lösung wird auf dem Wasserbad zur Trockne eingedampft. Der Trockenrückstand wird nach Nummer 4.3.2 ab "Fällen und Filtrieren" weiterverarbeitet.
Der Kationenaustauscher muß vor jeder Verwendung regeneriert werden. Das Regenerieren erfolgt mit 10 Obiger methanolischer Salzsäure nach Nummer 4.1.45. Es wird solange mit Methanol nachgewaschen, bis der Ablauf gegen Methylrot nicht mehr sauer reagiert. Der regenerierte Kationenaustauscher wird unter Methanol aufbewahrt.
4.3.5 Berechnung der Ergebnisse
Da jedes nichtionische Tensid einen von der Länge seiner Äthylenoxidkette abhängigen Eichfaktor hat, bezieht man auf eine Standardsubstanz. Hierfür ist das Nonylphenol mit im Mittel 10 Äthylenoxid-Einheiten (NP 10) festgelegt. Für diese Substanz wurde der empirische Umrechnungsfaktor 54 ermittelt. Man erhält damit die Menge an nichtionischem Tensid in µ g, ausgedrückt als NP 10, die in der angewandten Wassermenge enthalten war. Es gilt also
(b - c) × f × 54 = µg nichtionische Tenside oder
(b - c) × f × 0,054 = mg nichtionische Tenside
Es sind:
b = Verbrauch an Carbat-Lösung der Probe in ml
c = Verbrauch an Carbat-Lösung des Blindversuchs in ml
f = Faktor der Carbat-Lösung
4.3.6 Angabe der Ergebnisse
Die Ergebnisse sind in mg/l wie folgt anzugeben:
weniger als 1 mg/l mit zwei Dezimalen
mehr als 1 mg/l mit einer Dezimale
5 Bestimmung des Gehaltes an Seife
Der Gehalt an Seife wird nach der in den "Deutschen Einheitsmethoden zur Untersuchung von Fetten, Fettprodukten und verwandten Stoffen (DGF-Einheitsmethoden) Abteilung H (Tenside), III 7 a " 1) angegebenen Analysenmethode bestimmt.
6 Bestimmung des gelösten Sauerstoffs
Der gelöste Sauerstoff wird nach der in den "Deutschen Einheitsverfahren zur Wasser-, Abwasser- und Schlamm-Untersuchung (DEV) Gruppe G2/1" 2) angegebenen Analysenmethode bestimmt.
7 Bestimmung der Oxidierbarkeit
Die Oxidierbarkeit wird nach einer der in den "Deutschen Einheitsverfahren zur Wasser-, Abwasser- und Schlamm-Untersuchung (DEV) Gruppe H 4"2) angegebenen Analysenmethode bestimmt.
8 Bestimmung der organischen Substanz des Belebtschlamms
Die organische Substanz des Belebtschlamms wird nach der in den "Deutschen Einheitsverfahren zur Wasser-, Abwasser- und Schlamm-Untersuchung (DEV) Gruppe S 3" 3) angegebenen Analysenmethode bestimmt.
____________________
*) Verordnung über das Deutsche Arzneibuch (DAB 7) vom 7. August 1968 (BGBl. I S. 913), zuletzt geändert durch die Dritte Verordnung zur Änderung des Deutschen Arzneibuchs 7. Ausgabe (DAB 7) vom 22. Juli 1975 (BGBl. I S. 1962)
1) Ausgabe 1965, verlegt bei der Wissenschaftlichen Verlagsgesellschaft mbH, Stuttgart
2) 5. Lieferung Ausgabe 1968, verlegt bet Verlag Chemie GmbH, Weinheim/Bergstr.
3) 7. Lieferung Ausgabe 1975, verlegt bei Verlag Chemie GmbH, Weinheim/Bergstr.
| |
ENDE | |
(Stand: 27.06.2018)
Alle vollständigen Texte in der aktuellen Fassung im Jahresabonnement
Nutzungsgebühr: 90.- € netto (Grundlizenz)
(derzeit ca. 7200 Titel s.Übersicht - keine Unterteilung in Fachbereiche)
Die Zugangskennung wird kurzfristig übermittelt
? Fragen ?
Abonnentenzugang/Volltextversion