Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2009, Chemikalien - EU Bund
| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2009, Chemikalien - EU Bund |
Verordnung (EG) Nr. 761/2009 der Kommission vom 23. Juli 2009 zur Änderung der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zwecks Anpassung an den technischen Fortschritt
(ABl. Nr. L 220 vom 24.08.2009 S. 1)
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Agentur für chemische Stoffe, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission 1 insbesondere auf Artikel 13 Absatz 3,
in Erwägung nachstehender Gründe:
(1) In der Verordnung (EG) Nr. 440/2008 der Kommission 2 sind die im Sinne der Verordnung (EG) Nr. 1907/2006 anzuwendenden Prüfmethoden zur Bestimmung der physikalisch-chemischen Eigenschaften, der Toxizität und der Ökotoxizität von Stoffen festgelegt.
(2) Es ist angezeigt, die Verordnung (EG) Nr. 440/2008 zu aktualisieren, um bestimmte Prüfmethoden zu ändern und verschiedene von der OECD angenommene neue Testmethoden hinzuzufügen. Interessenträger wurden zu diesem Vorschlag gehört. Die betreffenden Änderungen dienen der Anpassung der Prüfmethoden an den wissenschaftlichen und technischen Fortschritt.
(3) Die Bestimmungen über den Dampfdruck sollten mit Blick auf die Einbeziehung der neuen Effusionsmethode überprüft werden.
(4) Es ist angezeigt, zur Messung des längengewichteten mittleren geometrischen Durchmessers von Fasern eine neue Methode hinzuzufügen.
(5) Es empfiehlt sich, die Verordnung (EG) Nr. 440/2008 zu aktualisieren und vorrangig eine neue Invitro-Methode für Hautreizungstests einzubeziehen, um die Zahl der Versuchstiere in Einklang mit der Richtlinie 86/609/EWG des Rates vom 24. November 1986 zur Annäherung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Tiere 3 zu verringern. Obgleich der Entwurf einer Invitro-Methode für Hautreizungstests auf Ebene der OECD noch debattiert wird, ist es in diesem Ausnahmefall angezeigt, Methode B 46 in die Verordnung einzubeziehen. Methode B 46 sollte so schnell wie möglich, d. h. sobald innerhalb der OECD Einigung erzielt wurde oder weitere Informationen, die eine Überprüfung rechtfertigen, verfügbar werden, aktualisiert werden.
(6) Die Bestimmungen über den Algeninhibitionstest müssen mit Blick auf die Einbeziehung zusätzlicher Arten und die Erfüllung der Anforderungen an die Gefahrenbewertung und die Klassifizierung von Chemikalien überprüft werden.
(7) Es ist angezeigt, als neue Methode zur Messung der aeroben Mineralisierung in Oberflächengewässern einen Simulationstest zur Prüfung der biologischen Abbaubarkeit sowie als neue Methode zur Bewertung der Toxizität für die Gattung Lemna einen Wachstumsinhibitionstest hinzuzufügen.
(8) Die Verordnung (EG) Nr. 440/2008 ist daher entsprechend zu ändern.
(9) Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des mit Artikel 133 der Verordnung (EG) Nr. 1907/2006 eingesetzten Ausschusses
- hat folgende Verordnung erlassen:
Der Anhang der Verordnung (EG) Nr. 440/2008 wird wie folgt geändert:
1. Teil A wird wie folgt geändert:
a) Kapitel A.4 erhält die Fassung von Kapitel A.4 in Anhang I der vorliegenden Verordnung;
b) es wird ein neues Kapitel A.22 (Anhang II der vorliegenden Verordnung) hinzugefügt.
2. Teil B wird wie folgt geändert:
Es wird ein neues Kapitel B.46 (Anhang III der vorliegenden Verordnung) hinzugefügt.
3. Teil C wird wie folgt geändert:
a) Kapitel C.3 erhält die Fassung von Kapitel C.3 in Anhang IV der vorliegenden Verordnung;
b) es werden die neuen Kapitel C.25 und C.26 (Anhänge V und VI der vorliegenden Verordnung) hinzugefügt.
Diese Verordnung tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
Brüssel, den 23. Juli 2009
2) ABl. L 142 vom 31.05.2008 S. 1.
3) ABl. L 358 vom 18.12.1986 S. 1.
| Anhang I |
A.4 Dampfdruck
1. Methode
Diese Methode entspricht der Prüfrichtlinie OECD TG 104 (2004).
1.1 Einleitung
Diese geänderte Fassung von Methode A.4 (1) beinhaltet als zusätzliche Methode die "Effusionsmethode: isotherme Thermogravimetrie"; diese Methode wurde entwickelt für Substanzen mit sehr niedrigen Drücken (bis zu einem Mindestdruck von 10-10 Pa). Angesichts der Verfahrenserfordernisse, insbesondere bei der Ermittlung des Dampfdrucks für Substanzen mit niedrigem Dampfdruck, werden auch andere Verfahren zur Anwendung dieser Methode im Hinblick auf sonstige Einsatzbereiche neu bewertet.
Bei thermodynamischem Gleichgewicht hängt der Dampfdruck einer reinen Substanz ausschließlich von der Temperatur ab. Die zugrunde liegenden Prinzipien werden an anderer Stelle erläutert (2)(3).
Kein einzelnes Messverfahren ist für sämtliche Dampfdrucke von unter 10-10 Pa bis zu 105 Pa geeignet. Entsprechend umfasst diese Beschreibung acht Methoden zur Messung des Dampfdrucks, die in verschiedenen Dampfdruckbereichen eingesetzt werden können. Die vorgesehenen Einsatzmöglichkeiten und Messbereiche der einzelnen Methoden sind in Tabelle 1 zusammengestellt. Die Methoden können nur im Zusammenhang mit Verbindungen eingesetzt werden, bei denen unter den Testbedingungen kein Abbau erfolgt. In Fällen, in denen die Versuchsmethoden aus technischen Gründen nicht eingesetzt werden können, kann der Dampfdruck auch geschätzt werden; eine empfohlene Schätzmethode wird in der Anlage beschrieben.
1.2 Begriffsbestimmungen und Einheiten
Als Dampfdruck einer Substanz wird der Sättigungsdruck über einer festen oder flüssigen Substanz bezeichnet.
In den Messungen sollte die Einheit Pascal (Pa) als SI-Einheit für Druckwerte verwendet werden. Im Folgenden sind weitere, früher verwendete Einheiten jeweils mit den entsprechenden Umrechnungsfaktoren zusammengestellt:
| 1 Torr | = 1 mm Hg | = 1,333 x 102 Pa |
| 1 Atmosphäre | = 1,013 x 105 Pa | |
| 1 bar | = 105 Pa |
Die SI-Einheit der Temperatur ist Kelvin (K). Angaben in Grad Celsius werden mit folgender Formel in Kelvin umgerechnet:
T= t + 273,15
wobei T = Kelvin oder thermodynamische Temperatur und t = Temperatur in Grad Celsius
| Messmethode | Substanzen | Geschätzte Wiederholbarkeit | Geschätzte Reproduzierbarkeit | Empfohlener Bereich | |
| Fest | Flüssig | ||||
| Dynamische Methode | niedriger Schmelz- punkt | ja | bis zu 25 %
1 bis 5% | bis zu 25 %
1 bis 5% | 103 Pa bis 2 x 103 Pa 2 x 103 Pa bis 105 Pa |
| Statische Methode | ja | ja | 5 bis 10 % | 5 bis 10 % | 10 Pa bis 105 Pa 10-2 Pa bis 105 Pa 1 |
| Isoteniskopmethode | ja | ja | 5 bis 10 % | 5 bis 10 % | 102 Pa bis 105 Pa |
| Effusionsmethode: Dampfdruckgleichgewicht | ja | ja | 5 bis 20 % | bis zu 50 % | 10-3 bis 1 Pa |
| Effusionsmethode: Knudsen-Zelle | ja | ja | 10 bis 30 % | - | 10-10 bis 1 Pa |
| Effusionsmethode: isotherme Thermogravimetrie | ja | ja | 5 bis 30 % | bis zu 50 % | 10-10 bis 1 Pa |
| Gassättigungsmethode | ja | ja | 10 bis 30 % | bis zu 50 % | 10-10 bis 103 Pa |
| Rotationsmethode | ja | ja | 10 bis 20 % | - | 10-4 bis 0,5 Pa |
| 1) In Verbindung mit einem Kapazitätsmanometer. | |||||
1.3 Prinzip der Prüfmethode
Im Allgemeinen wird der Dampfdruck bei verschiedenen Temperaturen gemessen. In einem begrenzten Temperaturbereich ist der Logarithmus des Dampfdrucks einer reinen Substanz eine lineare Umkehrfunktion der thermodynamischen Temperatur gemäß der vereinfachten Clapeyron-Clausius-Gleichung:
| log p = | ΔHv | + constant |
|
| ||
| 2,3RT |
wobei
| p | = Dampfdruck in Pascal |
| ΔHv | = Verdampfungswärme in J mol-1 |
| R | = universale Gaskonstante, 8,314 J mol-1 K-1 |
| T | = Temperatur in K |
1.4 Referenzsubstanzen
Referenzsubstanzen brauchen nicht unbedingt verwendet zu werden. Sie dienen in erster Linie zur gelegentlichen Überprüfung der Leistungsfähigkeit einer Methode und sollen Vergleiche der mit unterschiedlichen Methoden erzielten Ergebnisse ermöglichen.
1.5 Beschreibung der Methode
1.5.1 Dynamische Methode (Cottrell-Verfahren)
1.5.1.1 Prinzip
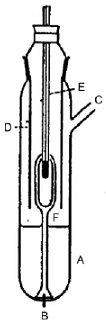
Der Dampfdruck wird durch die Messung der Siedetemperatur einer Substanz bei verschiedenen vorgegebenen Drücken zwischen etwa 103 und 105 Pa gemessen. Diese Methode wird auch für die Bestimmung der Siedetemperatur empfohlen. Für diesen Zweck kann die Methode bei Temperaturen bis zu 600 K eingesetzt werden. Wegen des hydrostatischen Drucks der Flüssigkeitssäule liegen die Siedetemperaturen von Flüssigkeiten bei einer Tiefe von 3 bis 4 cm etwa 0,1 °C höher als an der Oberfläche. Beim Cottrell-Verfahren (4) wird das Thermometer über der Flüssigkeit in den Dampf gebracht und die siedende Flüssigkeit kontinuierlich über die Thermometerkugel gepumpt. Eine dünne Flüssigkeitsschicht, die sich bei Atmosphärendruck im Gleichgewicht mit dem Dampf befindet, bedeckt die Kugel. Das Thermometer gibt dann den echten Siedepunkt an; Fehler durch Überhitzung oder hydrostatischen Druck werden ausgeschlossen. Die ursprünglich von Cottrell verwendete Pumpe ist in Abbildung 1 dargestellt. Rohr A enthält die siedende Flüssigkeit. Ein Platindraht B, der in den Boden eingesiegelt wurde, begünstigt ein gleichmäßiges Siedeverhalten. Das seitliche Rohr C führt zu einem Kondensator, und der Spritzschutz D verhindert, dass das kalte Kondensat in das Thermometer E gelangt. Wenn die Flüssigkeit in A siedet, werden die entstehenden Blasen und die Flüssigkeit mit dem Trichter abgetrennt und über die beiden Arme der Pumpe F über die Thermometerkugel gegossen.
| Abbildung 1
| Abbildung 2
|
Cottrell-Pumpe (4)
A: Thermoelement
B: Vakuum-Puffervolumen
C: Druckmesser
D: Unterdruck
E: Messpunkt
F: Heizelement ca. 150 W
1.5.1.2 Apparatur
In Abbildung 2 ist eine sehr genaue Apparatur dargestellt, die auf dem Cottrell-Prinzip beruht. Die Apparatur besteht aus einem Rohr mit einem Siedebereich im unteren Teil, einem Kühler im mittleren Bereich und einem Auslass und einem Flansch im oberen Bereich. Die Cottrell-Pumpe befindet sich im Siedebereich, der mit einer elektrischen Patrone beheizt wird. Die Temperatur wird mit einem ummantelten Thermoelement oder einem Widerstandsthermomenter gemessen, das über den oben befindlichen Flansch in das Gerät geführt wird. Der Auslass ist mit einem Druckregelsystem verbunden. Letzteres besteht aus einer Vakuumpumpe, einem Puffervolumen, einem Druckwächter zur Druckregulierung unter Stickstoffeinleitung und einem Druckmesser.
1.5.1.3 Verfahren
Die Substanz wird in den Siedebereich gebracht. Bei nicht pulverigen Feststoffen können Probleme auftreten, die sich gelegentlich aber durch Beheizung des Kühlmantels beheben lassen. Die Apparatur ist am Flansch versiegelt, und die Prüfsubstanz wird entgast. Schäumende Substanzen können mit dieser Methode nicht gemessen werden.
In der Apparatur wird der niedrigste gewünschte Druck eingestellt und die Heizung eingeschaltet. Dann wird der Temperatursensor mit einem Aufzeichnungsgerät verbunden.
Das Gleichgewicht ist dann erreicht, wenn eine gleichbleibende Siedetemperatur bei konstantem Druck aufgezeichnet wird. Insbesondere ist darauf zu achten, dass während des Siedevorgangs nicht gegen die Apparatur gestoßen wird. Außerdem muss auf dem Kühler eine vollständige Kondensation erfolgen. Bei der Bestimmung des Dampfdrucks von niedrigschmelzenden Feststoffen ist darauf zu achten, dass der Kondensator nicht verblockt.
Nach der Aufzeichnung dieses Gleichgewichtspunkts wird ein höherer Druck eingestellt. Der Prozess wird auf diese Weise fortgesetzt, bis ein Druck von 105 Pa erreicht ist (insgesamt etwa 5 bis 10 Messpunkte). Zur Kontrolle sind die Gleichgewichtspunkte bei abnehmenden Drücken zu reproduzieren.
1.5.2 Statische Methode
1.5.2.1 Prinzip
Bei der statischen Methode (5) wird der Dampfdruck bei thermodynamischem Gleichgewicht und einer gegebenen Temperatur bestimmt. Diese Methode eignet sich für Substanzen und für aus mehreren Bestandteilen bestehende Flüssigkeiten und Feststoffe im Druckbereich von 10-1 bis 105 Pa sowie bei entsprechend sorgfältiger Vorgehensweise auch im Bereich von 1 bis 10 Pa.
1.5.2.2 Apparatur
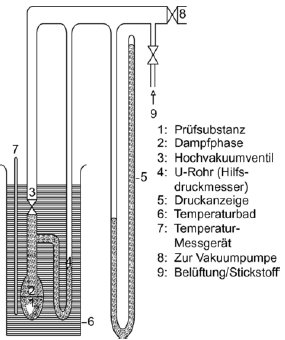
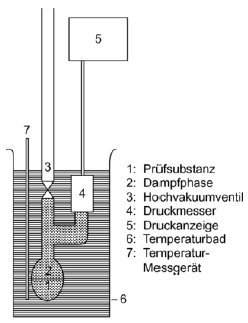
Die Apparatur besteht aus einem Bad mit konstanter Temperatur (Genauigkeit ± 0,2 K), einem mit einer Unterdruckleitung verbundenen Probenbehälter, einem Druckmesser und einem System zur Druckregelung. Die Probenkammer (Abbildung 3a) ist über ein Ventil und einen Differenzdruckmesser (ein U-Rohr mit einer geeigneten Manometerflüssigkeit) verbunden, das als Nullpunktanzeige dient. Im Differenzdruckmesser können Quecksilber, Silikone und Phthalate verwendet werden; maßgeblich sind der jeweilige Druckbereich und das chemische Verhalten der Prüfsubstanz. Aus Gründen des Umweltschutzes sollte nach Möglichkeit jedoch auf Quecksilber verzichtet werden. Die Prüfsubstanz darf sich nicht merklich in der im U-Rohr enthaltenen Flüssigkeit auflösen oder mit dieser reagieren. Statt eines U-Rohrs kann auch ein Druckmesser verwendet werden (Abbildung 3b). Für den Druckmesser kann Quecksilber im Bereich des Atmosphärendrucks bis zu einem Mindestdruck von 102 Pa eingesetzt werden; Silikon-Flüssigkeiten und Phthalate sind für Drücke unter 102 Pa bis zu 10 Pa geeignet. Unter 102 Pa können sonstife Druckmesser eingesetzt werden; Kapazitätsmanometer mit Heizmembran können sogar bei Drücken unter 10-1 Pa verwendet werden. Die Temperatur wird an der Außenwand des Probenbehälters oder im Behälter selbst gemessen.
1.5.2.3 Verfahren
In der in Abbildung 3a beschriebenen Apparatur wird das U-Rohr mit der gewählten Flüssigkeit gefüllt; vor Durchführung der Messungen ist die Flüssigkeit bei höherer Temperatur zu entgasen. Die Prüfsubstanz wird in die Apparatur gebracht und bei niedrigerer Temperatur entgast. Bei aus mehreren Bestandteilen bestehenden Proben sollte die Temperatur so niedrig sein, dass die Zusammensetzung des jeweiligen Materials erhalten bleibt. Durch Rühren kann das erforderliche Gleichgewicht schneller herbeigeführt werden. Die Probe kann dann mit flüssigem Stickstoff oder mit Trockeneis abgekühlt werden. Dabei ist allerdings sicherzustellen, dass die Luft oder die Pumpflüssigkeit nicht kondensieren. Bei geöffnetem Ventil über der Probe wird über mehrere Minuten ein Sog ausgeübt, um die eingeschlossene Luft zu entfernen. Wenn erforderlich, wird die Entgasung mehrmals wiederholt.
| Abbildung 3a
| Abbildung 3b
|
Wenn die Probe bei geschlossenem Ventil beheizt wird, erhöht sich der Dampfdruck. Dadurch ändert sich das Gleichgewicht der Flüssigkeit im U-Rohr. Um die Änderung auszugleichen, wird Stickstoff oder Luft in die Apparatur geleitet, bis die Differenzdruckanzeige wieder auf null steht. Der dazu erforderliche Druck kann an einem Druckmesser oder an einem genaueren Messgerät abgelesen werden. Dieser Druck entspricht dem Dampfdruck der Substanz bei der Messtemperatur. Bei der in Abbildung 3b dargestellten Apparatur wird der Dampfdruck direkt abgelesen.
Der Dampfdruck wird in geeigneten geringen Temperaturintervallen (insgesamt etwa 5 bis 10 Messpunkte) bis zur gewünschten Höchsttemperatur gemessen.
Zur Kontrolle sind Messungen bei niedrigen Temperaturen durchzuführen. Wenn die in den mehrfachen Messungen ermittelten Werte nicht auf der Kurve liegen, die sich bei den höheren Temperaturen ergibt, kann dies auf eine der folgenden Ursachen zurückzuführen sein:
1.5.3 Isoteniskopmethode
1.5.3.1 Prinzip
Das Isoteniskop (6) beruht auf dem Prinzip der statischen Methode. Bei dieser Methode wird eine Probe in einen Kolben gebracht, in dem eine gleichbleibende Temperatur besteht, und der mit einem Druckmesser und einer Vakuumpumpe verbunden ist. Verunreinigungen mit höherer Flüchtigkeit als die Prüfsubstanz werden durch Entgasen bei reduziertem Druck entfernt. Der Dampfdruck der Probe bei bestimmten Temperaturen wird durch einen mit einem inerten Gas ausgeübten bekannten Druck ausgeglichen. Das Isoteniskop wurde entwickelt, um den Dampfdruck bestimmter flüssiger Kohlenwasserstoffe zu messen, kann aber auch zur Untersuchung von Feststoffen eingesetzt werden. Für Systeme mit mehreren Bestandteilen ist diese Methode im Allgemeinen nicht geeignet. Die Ergebnisse weisen nur leichte Fehler bei Proben mit nicht flüchtigen Verunreinigungen auf. Die Methode wird für den Bereich 102 bis 105 Pa empfohlen.
1.5.3.2 Apparatur
In Abbildung 4 wird ein Messsystem dargestellt. Eine vollständige Beschreibung ist ASTM D 2879-86 (6) zu entnehmen.
1.5.3.3 Verfahren
Bei Flüssigkeiten dient die Substanz selbst als Flüssigkeit im Differenzdruckmesser. In das Isoteniskop wird so viel Flüssigkeit eingebracht, dass der Kolben und der kurze Fuß des Druckmessers gefüllt sind. Das Isoteniskop wird mit einem Vakuumsystem verbunden und mit Hilfe des Vakuumsystems vollständig entleert. Anschließend wird das Isoteniskop mit Stickstoff gefüllt. Die Absaugung und die Reinigung des Systems wird zweimal wiederholt, um den verbliebenen Sauerstoff zu entfernen. Das befüllte Isoteniskop wird in horizontal ausgerichtet, damit sich die Probe in Kolben und Druckmesser als dünne Schicht ausbreitet. Der Systemdruck wird auf 133 Pa reduziert, und die Probe wird allmählich erwärmt, bis eben der Siedepunkt erreicht ist (Abtrennung gelöster Gase). Danach wird das Isoteniskop so ausgerichtet, dass die Probe wieder in den Kolben zurückfließt und den kurzen Fuß des Druckmessers ausfüllt. Dabei wird ein Druck von 133 Pa aufrechterhalten. Die vorgezogene Spitze des Probenkolbens wird mit kleiner Flamme erwärmt, bis sich der freigesetzte Probendampf hinreichend ausgedehnt hat, um einen Teil der Probe aus dem oberen Bereich des Kolbens und des Druckmesserarms in den Druckmesser hinein zu verdrängen und somit einen stickstofffreien Raum mit Dampf gefüllt hat. Darauf wird das Isoteniskop in ein Bad mit konstanter Temperatur gebracht, und Druck und Stickstoffzufuhr werden so eingestellt, dass sie mit dem Druck und dem Stickstoffanteil der Probe übereinstimmen. Wenn das erwünschte Gleichgewicht erreicht ist, entspricht der Stickstoffdruck dem Dampfdruck der Prüfsubstanz.
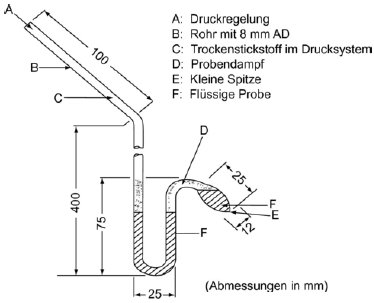
Bei Feststoffen sowie je nach Druck- und Temperaturbereich werden Manometerflüssigkeiten wie z.B. Silikon-Flüssigkeiten oder Phthalate verwendet. Die entgaste Manometerflüssigkeit wird in eine dafür vorgesehene Rufwölbung am langen Arm des Isoteniskops gebracht. Anschließend wird der zu prüfende Feststoff in den Probenkolben gegeben und bei einer höheren Temperatur entgast. Darauf wird das Isoteniskop so geneigt, dass die Manometerflüssigkeit in das U-Rohr fliegen kann.
1.5.4 Effusionsmethode: Dampfdruckgleichgewicht (7)
1.5.4.1 Prinzip
Eine Probe der Prüfsubstanz wird in einem kleinen Ofen erhitzt und in eine Gasglocke gebracht, in der ein Unterdruck hergestellt wurde. Der Ofen wird mit einem Deckel verschlossen, in dem sich kleine Löcher mit einem bestimmten Durchmesser befinden. Der aus der Substanz erzeugte Dampf entweicht durch eines dieser Löcher und wird unmittelbar auf die Schale einer hoch empfindlichen Waage geleitet, die sich ebenfalls im Vakuum der Glasglocke befindet. Bei manchen Ausführungen ist die Waagschale von einem Kühlgefäß umgeben, über das die Wärme thermisch abgeleitet und unter Abstrahlung der Wärme so gekühlt wird, dass sich der entweichende Dampf darauf niederschlägt. Die Energie des Dampfstrahls wirkt als Kraft auf die Waage. Der Dampfdruck kann auf zweierlei Weise ermittelt werden: entweder direkt aus der auf die Waagschale wirkenden Kraft oder mit Hilfe der Hertz-Knudsen-Gleichung aufgrund der Verdampfungsgeschwindigkeit (2):
p = G ((2πRT x 103)/M)0,5
wobei
G = Verdampfungsgeschwindigkeit (kg s-1 m-2)
M = Molmasse (g mol-1)
T = Temperatur (K)
R = Universale Gaskonstante (J mol-1 K-1)
P = Dampfdruck (Pa)
Der empfohlene Bereich liegt zwischen 10-3 und 1 Pa.
1.5.4.2 Apparatur
Abbildung 5 veranschaulicht das Grundprinzip der Apparatur:
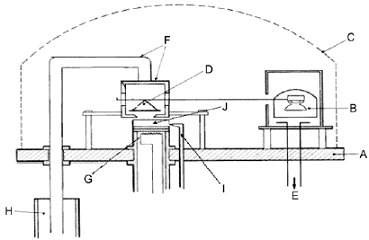 | A: Bodenplatte B: Bewegliches Kühlinstrument C: Gasglocke D: Waage mit Waagschale E: Vakuummessgerät F: Kühlgehäuse und Kühlschiene G: Verdampfungsofen H: Dewargefäß mit flüssigem Stickstoff I: Messung der Probentemperatur J: Prüfsubstanz |
1.5.5 Effusionsmethode: Knudsen-Zelle
1.5.5.1 Prinzip
Die Methode beruht auf der Schätzung der Masse der Prüfsubstanz, die im Ultravakuum pro Zeiteinheit aus einer Knudsen-Zelle (8) als Dampf durch eine Mikrobohrung strömt. Die Masse des ausströmenden Dampfs kann entweder aufgrund des Masseverlustes der Zelle oder durch Kondensation des Dampfs bei niedriger Temperatur und anschließende chromatographische Messung der Menge der verflüchtigten Substanz bestimmt werden. Der Dampfdruck wird mit Hilfe der Hertz-Knudsen-Gleichung (siehe Abschnitt 1.5.4.1) unter Berücksichtigung von Korrekturfaktoren berechnet, die von den Parametern der jeweiligen Apparatur abhängen (9). Empfohlen wird diese Methode für den Bereich 10-10 bis 1 Pa (10)(11)(12)(13)(14).
1.5.5.2 Apparatur
Abbildung 6 veranschaulicht das Grundprinzip der Apparatur:
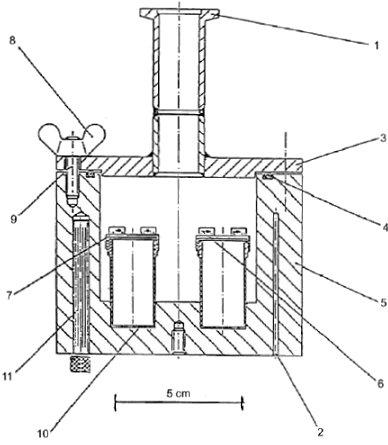 | 1: Verbindung zum Vakuum 2: Bohrungen des Platinwiderstand-Thermometers 3: Deckel Vakuumtank 4: O-Ring 5: Aluminum-Vakuumtank 6: Vorrichtung zum Einsetzen und Abnehmen der Effusionszellen 7: Deckel mit Schraubgewinde 8: Flügelmuttern oder Temperaturmessung und Kontrolle 9: Schrauben 10: Edelstahl-Effusionszellen 11: Heizpatrone |
1.5.6 Effusionsmethode: isotherme Thermogravimetrie
1.5.6.1 Prinzip
Die Methode beruht auf der Bestimmung der Geschwindigkeit einer beschleunigten Verdampfung der Prüfsubstanz bei höheren Temperaturen und bei Umgebungsdruck durch Thermogravimetrie (10)(15)(16)(17)(18)(19)(20). Die Verdampfungsgeschwindigkeiten vT ergeben sich daraus, dass die jeweils ausgewählte Verbindung einer Atmosphäre mit einem langsam strömenden inerten Gas ausgesetzt wird; bei gegebenen isothermen Temperaturen T (ausgedrückt in Kelvin) wird dann über bestimmte Zeitspannen der Gewichtsverlust überwacht. Die Dampfdrücke pT werden aus den Werten für vT aufgrund der linearen Beziehung zwischen dem Logarithmus des Dampfdrucks und dem Logarithmus der Verdampfungsgeschwindigkeit bestimmt. Wenn erforderlich, kann durch eine Regressionsanalyse von log pT bezogen auf 1/T eine Extrapoherung auf Temperaturen von 20 und 25 °C erfolgen. Diese Methode kommt für Substanzen mit Mindestdampfdrücken bis zu 10-10 Pa (10-12 mbar) in Betracht; um Fehlinterpretationen der gemessenen Gewichtsverluste zu vermeiden, sollte die Reinheit annähernd 100 % betragen.
1.5.6.2 Apparatur
In Abbildung 7 wird das allgemeine Prinzip der Apparatur dargestellt:
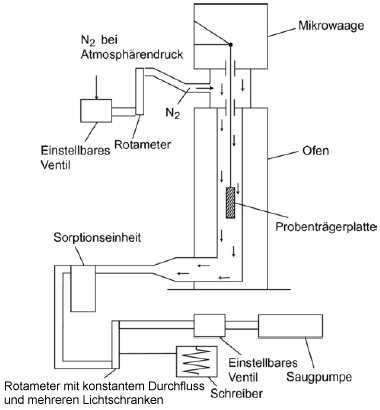
Die in einer Kammer mit Temperaturüberwachung an einer Mikrowaage aufgehängte Probenträgerplatte wird von trockenem Stickstoffgas umströmt, in dem die verdampften Moleküle der Prüfsubstanz geführt werden. Nach dem Verlassen der Kammer wird der Gasstrom durch eine Sorptionseinheit gereinigt.
1.5.6.3 Verfahren
Die Prüfsubstanz wird als homogene Schicht auf eine aufgeraute Glasplatte aufgebracht. Bei Feststoffen wird die Platte gleichförmig mit einer Lösung befeuchtet, in der die Prüfsubstanz in einem geeigneten Lösungsmittel gelöst wurde; anschließend wird die Lösung in einer inerten Atmosphäre getrocknet. Für die Messung wird die beschichtete Platte in den Thermogravimetrie-Analysator gehängt, dort wird der Gewichtsverlust kontinuierlich als zeitabhängige Funktion bestimmt.
Aus dem Gewichtsverlust Am der Probenplatte wird die Verdampfungsgeschwindigkeit vT bei einer bestimmten Temperatur mit der folgenden Formel berechnet:
vT = Δm/F×t (gcm-2 h-1)
wobei F = Oberfläche der beschichteten Prüfsubstanzen (im Allgemeinen die Oberfläche der Probenplatte) und t = Dauer des Gewichtsverlusts Δm
Der Dampfdruck pT wird ausgehend von der Funktion der Verdampfungsgeschwindigkeit vT wie folgt berechnet:
Log pT = C + D log vT
wobei C und D spezifische Konstanten für die jeweilige Apparatur sind und vom Durchmesser der Messkammer sowie vom Gasstrom abhängen. Diese Konstanten sind einmal zu bestimmen, indem eine Reihe von Verbindungen mit bekanntem Dampfdruck gemessen und dann durch Regressionsanalyse log pT gegenüber log vT ermittelt wird (11)(21)(22).
Die Beziehung zwischen dem Dampfdruck pT und der Temperatur T in Kelvin wird wie folgt ausgedrückt:
Log pT = A + B 1/T
Dabei sind A und B Konstanten, die sich aus der Regressionsanalyse von log pT gegenüber 1/T ergeben. Mit dieser Formel kann der Dampfdruck für jede beliebige Temperatur extrapoliert werden.
1.5.7 Gassättigungsmethode (23)
1.5.7.1 Prinzip
Das inerte Gas wird bei Raumtemperatur und mit einer bekannten Durchflussgeschwindigkeit so langsam durch bm über eine Probe der Prüfsubstanz geleitet, dass eine Sättigung eintreten kann. Die Herbeiführung der Sättigung in der Gasphase ist von entscheidender Bedeutung. Die mitgeführte Substanz wird eingeschlossen (im Allgemeinen mit einem Sorptionsmittel); anschließend wird die Menge der abgetrennten Substanz bestimmt. Alternativ zum Dampfeinschluss mit anschließender Analyse kommen in den Gasstrom integrierte Analyseverfahren wie z.B. die Gaschromatographie zur quantitativen Bestimmung des mitgeführten Materials in Betracht. Der Dampfdruck wird ausgehend von der Annahme berechnet, dass sich der Gasstrom entsprechend dem idealen Gasgesetz verhält und dass der Gesamtdruck eines Gasgemischs mit der Summe der Drücke der einzelnen enthaltenen Gase übereinstimmt. Der Teildruck der Prüfsubstanz, d. h. der Dampfdruck, wird aus dem bekannten Volumen des gesamten Gasstroms und aus dem Gewicht des mitgeführten Materials berechnet.
Das Gassättigungsverfahren ist für feste und flüssige Substanzen geeignet. Es kann bei Mindestdampfdrücken bis zu 10-10 Pa genutzt werden (10)(11)(12)(13)(14). Bei Dampfdrücken unter 103 Pa ist diese Methode die zuverlässigste Methode. Über 103 Pa werden die Dampfdrücke im Allgemeinen überschätzt; dies ist wahrscheinlich auf die Bildung von Aerosolen zurückzuführen. Da die Dampfdruckmessungen bei Raumtemperatur erfolgen, brauchen keine Extrapolierungen hoher Temperaturen vorgenommen zu werden; entsprechend entfallen die häufig mit der Extrapolierung hoher Temperaturen verbundenen schwerwiegenden Fehler.
1.5.7.2 Apparatur
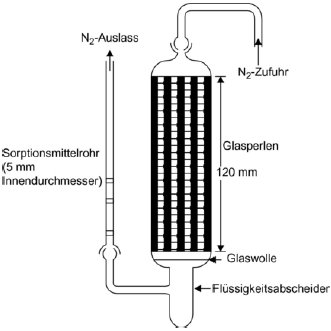
Für das Verfahren wird ein Behälter mit konstanter Temperatur benötigt. In Abbildung 8 ist ein Behälter mit Haltern für jeweils drei feste und flüssige Proben skizziert, in dem jeweils drei wahlweise feste oder flüssige Proben analysiert werden können. Die Temperatur wird mit einer Genauigkeit von mindestens ± 0,5 °C überwacht.
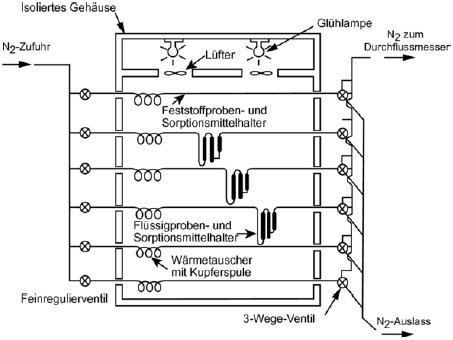
Im Allgemeinen wird Stickstoff als inertes Trägergas verwendet; gelegentlich können aber auch sonstige Gase erforderlich sein (24). Das Trägergas muss trocken sein. Der Gasstrom wird in sechs Teilströme getrennt, die jeweils durch Kegelventile (mit Bohrungen von ca. 0,79 mm) geregelt und über ein Kupferrohr mit einem Innendurchmesser von 3,8 mm in den Behälter geleitet werden. Nach dem Temperaturausgleich strömt das Gas durch die Probe und verlässt den Behälter durch die Adsorptionsfalle.
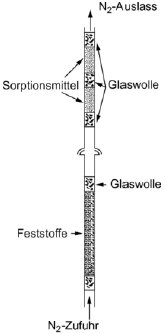
Feste Proben werden in ein mit Glaswollestopfen verschlossenes Glasrohr mit einem Innendurchmesser von 5 mm gegeben (siehe Abbildung 9). In Abbildung 10 sind ein Halter für feste Proben und ein Sorptionssystem dargestellt. Die am besten reproduzierbare Methode zur Messung des Dampfdrucks von Flüssigkeiten besteht darin, die zu prüfende Flüssigkeit auf Glasperlen oder auf ein inertes Sorptionsmittel wie z.B. Kieselerde aufzubringen und den Halter mit diesen Perlen zu packen. Alternativ kann das Trägergas auch über eine grobe Fritte und eine Blase durch die Säule mit der flüssigen Prüfsubstanz geleitet werden.
| Abbildung 9
| Abbildung 10
|
Das Sorptionssystem enthält einen vorderen und einen hinteren Sorptionsabschnitt. Bei sehr niedrigen Dampfdrücken werden nur geringe Anteile vom Sorptionsmittel abgetrennt; dabei kann die Adsorption auf der Glaswolle und im Glasrohr zwischen der Probe und dem Sorptionsmittel ein ernsthaftes Problem sein.
Mit festem CO2 gekühlte Abscheider stellen eine weitere wirksame Möglichkeit zur Aufnahme des verdampften Materials dar. Diese Abscheider verursachen keinerlei Gegendruck auf die Sättigungssäule; außerdem lässt sich das abgetrennte Material leicht quantitativ bestimmen.
1.5.7.3 Verfahren
Die Durchflussgeschwindigkeit des ausströmenden Trägergases wird bei Raumtemperatur gemessen. Während der Messung wird die Durchflussgeschwindigkeit häufig geprüft, um sicherzustellen, dass das Gesamtvolumen des Trägergases zuverlässig ermittelt wird. Vorzugsweise erfolgt eine kontinuierliche Überwachung mit einem Mengendurchflussmesser. Die Sättigung der Gasphase kann eine beträchtliche Kontaktzeit und entsprechend verhältnismäßig geringe Gasdurchflüsse erfordern (25).
Im Anschluss an die Messungen werden der vordere und der hintere Sorptionsabschnitt getrennt analysiert. Die Verbindungen in den einzelnen Abschnitten werden durch Zugabe eines Lösungsmittels desorbiert. Die entstehenden Lösungen werden einer quantitativen Analyse unterzogen, um die jeweils aus den Abschnitten desorbierten Gewichte zu bestimmen. Die Wahl der Analysemethode (sowie die Wahl des Sorptionsmittels und des desorbierenden Lösungsmittels) hängt von der Beschaffenheit des zu prüfenden Materials ab. Die Desorptionsleistung wird bestimmt, indem eine bekannte Probenmenge auf das Sorptionsmittel gespritzt, die Substanz desorbiert und anschließend die zurückgewonnene Menge analysiert wird. Die Desorptionsleistung muss mit der gleichen oder zumindest annähernd gleichen Probenkonzentration wie in der eigentlichen Prüfung bestimmt werden.
Um sicherzustellen, dass das Trägergas mit der Prüfsubstanz gesättigt ist, werden drei verschiedene Durchflussgeschwindigkeiten eingestellt. Wenn sich der berechnete Dampfdruck auch bei unterschiedlichen Durchflüssen nicht ändert, wird angenommen, dass das Gas gesättigt ist.
Der Dampfdruck wird mit folgender Gleichung berechnet:
p = W / V x RT / M
wobei
p = Dampfdruck (Pa)
W = Gewicht der verdampften Prüfsubstanz (g)
V = Volumen des gesättigten Gases (m3)
R = universale Gaskonstante 8,314 0 mol-1 K-1)
T = Temperatur (K)
M = Molmasse der Prüfsubstanz (g mol-1)
Die gemessenen Volumina sind unter Berücksichtigung von Druck- und Temperaturunterschieden zwischen Durchflussmesser und Sättigungssäule zu korrigieren.
1.5.8 Rotationsmethode
1.5.8.1 Prinzip
Bei dieser Methode wird ein Rotationsviskosimeter eingesetzt, bei dem das Messelement aus einer kleinen Stahlkugel besteht, die in einem Magnetfeld aufgehängt durch Drehfelder in Drehungen versetzt wird (26)(27)(28). Aufnehmerspulen ermöglichen eine Messung der Drehzahl. Wenn die Kugel eine bestimmte Drehzahl erreicht hat (in der Regel etwa 400 Umdrehungen pro Sekunde), wird die Erregung unterbrochen, und infolge der Gasreibung erfolgt eine Verzögerung. Der Rückgang der Drehzahl wird zeitabhängig gemessen. Aus der druckabhängigen Verlangsamung der Stahlkugel wird der Dampfdruck abgeleitet. Diese Methode wird für den Bereich von 10-4 bis 0, 5 Pa empfohlen.
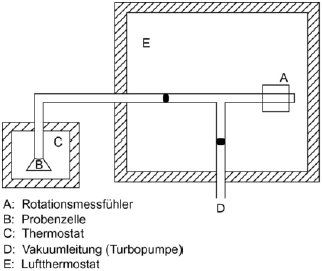
1.5.8.2 Apparatur
In Abbildung 11 ist die Apparatur schematisch dargestellt. Der Messfühler wird in ein Gehäuse mit konstanter Temperatur gebracht, in dem die Temperatur mit einer Genauigkeit von 0,1 °C geregelt wird. Der Probenbehälter wird in ein getrenntes Gehäuse gesetzt, dessen Temperatur ebenfalls mit einer Genauigkeit von 0,1 °C geregelt wird. Alle übrigen Teile der Apparatur werden auf einer höheren Temperatur gehalten, um eine Kondensatbildung auszuschließen. Die gesamte Apparatur ist mit einem Hochvakuumsystem verbunden.
2. Daten und Abschlussbericht
2.1 Daten
Der Dampfdruck sollte mit den genannten Methoden jeweils bei mindestens zwei Temperaturen gemessen werden. Im Bereich 0 bis 50 °C werden Messungen vorzugsweise bei drei Temperaturen durchgeführt, um zu prüfen, ob die Dampfdruckkurve linear verläuft. Bei der Effusionsmethode (Knudsen-Zelle und isotherme Thermogravimetrie) und bei der Gassättigungsmethode wird statt des üblichen Bereichs von 0 bis 50 °C ein Temperaturbereich von 120 bis 150 °C empfohlen.
2.2 Prüfbericht
Der Prüfbericht muss folgende Informationen enthalten:
Wenn ein Übergang (Änderung des Aggregatzustandes oder Zersetzung) beobachtet wird, sollten folgende Informationen vermerkt werden:
Alle für die Auswertung der Ergebnisse maggeblichen Informationen und Bemerkungen sind zu protokollieren; dies gilt insbesondere für Verunreinigungen und für die physikalische Beschaffenheit der Prüfsubstanz.
3. LITERATUR
1. Amtsblatt der Europäischen Gemeinschaften L 383 A, S. 26-47 (1992).
2. Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Nein dre, B., an d Vodar, B., Eds., Butterworths, London.
3. Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3' ed., Vol I, Part I. Chapter IX, Interscience Publ., New York.
4. Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York.
5. NF T 20-048 AFNOR (September 1985). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within a range from 10-1 to 105 Pa - Static method.
6. ASTM D 2879-86, Standard test method for vapour pressure - temperature relationship and initial decomposition temperature of liquids by isoteniscope.
7. NF T 20-047 AFNOR (September 1985). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within range from 10-3 to 1 Pa - Vapour pressure balance method.
B. Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593.
9. Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, S. 1173.
10. Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, S. 521-532.
11. Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000).
12. Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), S. 22-28.
13. Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), S. 269-278.
14. Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twentynine halogenated dibenzo-pdioxins, Thermochimia Acta 112 Issue 1 (1987), S. 117-122.
15. Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973), S. 137-147.
16. Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974), S. 393-400.
17. Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982), S. 161-168.
18. Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based an Evaporation Rate; Pesticide Science 45 (1995), S. 27-31.
19. Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science 53 (1998), S. 300-310.
20. Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998), S. 1512-20.
21. Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range - 25 °C to 150 °C.
22. Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002).
23. 40 CFR, 796. (1993). S. 148-153, Office of the Federal Register, Washington DC.
24. Rordorf B.F. (1985). Thermochimica Acta 85, S. 435.
25. Westcott et al. (1981). Environ. Sci. Technol. 15, S. 1375.
26. Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), S. 2440.
27. Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), S. 642.
28. Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), S. 1715.
| Schätzmethode | Anlage |
Einleitung
Geschätzte Dampfdrücke können verwendet werden,
Schätzmethode
Der Dampfdruck von Flüssigkeiten und Feststoffen kann aufgrund der modifizierten Watson-Korrelation geschätzt werden (a). In diesem Fall braucht als Prüfwert nur der normale Siedepunkt bestimmt zu werden. Diese Methode kommt im Druckbereich 105 Pa bis 10-5 Pa in Betracht.
Detaillierte Informationen zu dieser Methode sind dem "Handbook of Chemical Property Estimation Methods" zu entnehmen (b). Außerdem wird auf die OECD Environmental Monograph No. 67 verwiesen (c).
Berechnungsverfahren
Der Dampfdruck wird wie folgt berechnet:
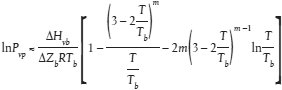
Tb wobei
| T | = maßgebliche Temperatur |
| Tb | = normaler Siedepunkt |
| Pvp | = Dampfdruck bei Temperatur |
| ΔHvb | = Verdampfungswärme |
| ΔZb | = Kompressibilitätsfaktor (geschätzt 0,97) |
| m | = empirischer Faktor, abhängig von der physikalischen Beschaffenheit bei der maßgeblichen Temperatur |
Außerdem ist folgende Berechnung vorzunehmen:
![]()
wobei KF ein empirischer Faktor ist, der die Polarität der Substanz berücksichtigt; im Anhang sind die Faktoren KF für verschiedene Verbindungen zusammengestellt.
Verhältnismäßig häufig sind Daten verfügbar, bei denen ein Siedepunkt bei reduziertem Druck angegeben wird. In diesen Fällen wird der Dampfdruck wie folgt berechnet:
![]()
wobei Tl = Siedepunkt bei reduziertem Druck Pl
Bericht
Bei der Schätzmethode sollte die vorgenommene Berechnung im Bericht umfassend dokumentiert werden.
Literatur
a) Watson, K.M. (1943). Ind. Eng. Chem, 35, 398.
b) Lyman, W.J., Reehl, W.F., Rosenblatt, D. H. (1982). Handbook of Chemical Property Estimation Methods, McGraw Hill.
c) OECD Environmental Monograph No. 67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993).
| Anhang II |
A.22 Längengewichteter mittlerer geometrischer Durchmesser von Fasern
1. Methode
1.1 Einleitung
Mit dieser Testmethode wird ein Verfahren zur Messung des längengewichteten mittleren geometrischen Durchmessers (GWGMD - Length Weighted Geometric Mean Diameter) von künstlichen Mineralfasern (MMMF - Man Made Mineral Fibres) beschrieben. Da der LWGMD der Population mit einer Wahrscheinlichkeit von 95 % zwischen den 95 %-Vertrauensintervallen (LWGMD ± 2 Standardfehler) der Probe liegt, entspricht der im Bericht angegebene Wert (der Testwert) der unteren 95 %-Vertrauensgrenze der Probe (d. h. LWGMD - 2 Standardfehler). Diese Methode basiert auf einer aktualisierten Fassung Quni 1994) des Entwurfs einer HSEIndustrieverfahrensweisung, die am 26. September 1993 in Chester zwischen ECHA und HSE vereinbart und für und aus einem zweiten laborinternen Versuch entwickelt wurde (1, 2). Diese Messmethode kann zur Kennzeichnung des Faserdurchmessers von Schüttgutstoffen oder Produkten verwendet werden, die MMMF enthalten, z.B. feuerfeste Keramikfasern (PCF - Refractory Ceramic Fibres), künstliche Glasfasern (MMVF - Man-Made Vitreous Fibres), kristalline und polykristalline Fasern.
Die Längengewichtung dient zur Kompensation der Auswirkungen auf die Durchmesserverteilung, zu denen es durch den Bruch langer Fasern bei der Probenahme oder beim Umgang mit dem Material kommt. Die Größenverteilung der Durchmesser der MMMF wird durch geometrische statistische Verfahren (geometrisches Mittel) gemessen, da diese Durchmesser normalerweise Größenverteilungen aufweisen, die näherungsweise dem Lognormal entsprechen.
Die Messung der Länge und des Durchmessers ist ein mühsamer, zeitaufwändiger Prozess, werden jedoch nur jene Fasern gemessen, die eine unendlich dünne Linie in einem REM-Sichtfeld berühren, so ist die Wahrscheinlichkeit, eine bestimmte Faser auszuwählen, proportional zu deren Länge. Da damit die Länge in den Berechnungen der Längengewichtung berücksichtigt wird, muss lediglich der Durchmesser gewichtet werden; der LWGMD - 2SF kann dann auf die beschriebene Weise berechnet werden.
1.2 Begriffsbestimmungen
Partikel: Ein Objekt mit einem Länge-Breite-Verhältnis von weniger als 3:1.
Faser: Ein Objekt mit einem Länge-Breite-Verhältnis (Seitenverhältnis) von mindestens 3:1.
1.3 Umfang und Einschränkungen
Durch diese Methode sollen die Durchmesserverteilungen untersucht werden, deren mittlerer Durchmesser zwischen 0,5 µm und 6 µm liegt. Größere Durchmesser können mithilfe geringerer REM-Vergrößerungsfaktoren gemessen werden, allerdings stößt diese Methode bei feineren Faserverteilungen zunehmend an seine Grenzen; bei mittleren Durchmessern unter 0,5 µm wird die Messung mit Transmissions-Elektronenmikroskopen (TEM) empfohlen.
1.4 Prinzip der Testmethode
Aus der Fasermatte oder aus losen Fasern wird eine bestimmte Anzahl repräsentativer Kernproben entnommen. Die Länge der losen Fasern wird durch Brechen verringert und es wird eine repräsentative Teilprobe in Wasser dispergiert. Aliquote Teile werden extrahiert und durch ein Polycarbonatfilter mit einer Porengröße von 0,2 µm gefiltert und zur Untersuchung unter einem Rasterelektronenmikroskop (SEM) vorbereitet. Die Faserdurchmesser werden mit einem Rastervergrößerungsfaktor von x 10.000 oder mehr (1) nach einem Line-Intercept-Verfahren gemessen, das eine unverfälschte Schätzung des mittleren Durchmessers ergibt. Das untere 95 %-Vertrauensintervall (auf der Basis eines einseitigen Tests) wird so berechnet, dass ein Schätzwert für den niedrigsten Wert des mittleren geometrischen Faserdurchmessers des Materials entsteht.
(1) Dieser Vergrößerungsfaktor bezieht sich auf 3 µm-Fasern; bei 6µm-Fasern ist ein Vergrößerungsfaktor von x 5.000 möglicherweise geeigneter.
1.5 Beschreibung der Testmethode
1.5.1 Sicherheits-Vorsichtsmaßnahmen
Die Belastung des menschlichen Organismus durch Schwebfasern ist zu minimieren; daher ist bei der Arbeit mit den trockenen Fasern ein Abzugsschrank oder eine Glove-Box zu verwenden. In periodischen Abständen ist die Belastung des menschlichen Organismus zu überwachen, um die Wirkung der Schutzverfahren zu überprüfen. Bei der Handhabung von MMMF sind Einweghandschuhe zu tragen, um Hautreizungen zu verringern und eine Querkontaminierung zu vermeiden.
1.5.2 Geräte/Apparatur
1.5.3 Testverfahren
1.5.3.1 Probenahme
Bei Fasermatten und Platten wird ein 25 mm-Kernbohrer oder Korkbohrer zur Entnahme von Proben aus dem Querschnitt verwendet. Diese Proben sind gleichmäßig über die Breite a der Schmalseite der Matte anzuordnen oder, wenn lange Abschnitte der Matte zur Verfügung stehen, aus beliebigen Stellen zu entnehmen. Die gleichen Hilfsmittel können auch für die Entnahme von Zufallsproben aus losen Fasern verwendet werden. Es sind sechs Stichproben möglichst so zu entnehmen, das räumliche Schwankungen im losen Material damit wiedergegeben werden.
Die sechs Kernproben sind in einem Stempel mit 50 mm Durchmesser unter 10 MPa zu zerkleinern. Das Material wird mit einem Spachtel durchmischt und bei 10 MPa erneut gepresst. Anschließend wird das Material aus dem Stempel entnommen und in einer verschlossenen Glasflasche gelagert.
1.5.3.2 Vorbereitung der Probe
Falls erforderlich, können organische Bindemittel entzogen werden, indem die Faser ca. eine Stunde lang bei 450 °C in einen Ofen eingelegt wird.
Die Probe nach dem Coneand-Quarter-Verfahren in vier gleiche Teile unterteilen (dieser Schritt sollte in einem Staubschrank erfolgen).
Mit einem Spatel eine geringe Menge (< 0,5 g) der Probe in 100 ml frisch destilliertes Wasser geben, das durch einen 0,2 µm-Membranfilter gefiltert wurde (andere Beschaffungsquellen von hochreinem Wasser sind ebenfalls zulässig, wenn sie nachgewiesenermagen von ausreichender Qualität sind). Die Probe mithilfe einer mit 100 W betriebenen Ultraschallsonde, die so eingestellt ist, dass eine Kavitation eintritt, gründlich dispergieren (Steht keine Sonde zur Verfügung, ist wie folgt vorzugehen: 30 Sekunden lang mehrmals schütteln und umkehren; fünfminütige Ultraschallbehandlung in einem Tisch-Ultraschallbad; dann wieder 30 Sekunden lang mehrmals schütteln und umkehren).
Sofort nach der Dispersion der Faser mehrere aliquote Teile (z.B. drei aliquote Teile mit 3, 6 und 10 ml) mit einer breiten Pipette (Aufnahmevermögen 2-5 ml) entnehmen.
Jeder aliquote Teil wird durch einen 0,2 µm-Polycarbonatfilter mit MEC-Zusatzfilter mit 5 µm-Poren unter Verwendung eines 25-mm-Glasfiltertrichters mit zylindrischem Vorratsbehälter vakuumgefiltert. Rund 5 ml des gefilterten destillierten Wassers wird in den Trichter gegeben und den aliquoten Teil langsam in das Wasser gegeben, wobei die Pipettenspitze unterhalb des Meniskus gehalten wird. Pipette und Vorratsbehälter müssen nach dem Pipettieren gründlich gespült werden, da dünne Fasern dazu neigen, sich eher an der Oberfläche anzusammeln.
Filter vorsichtig entnehmen und vom Zusatzfilter trennen, bevor er zum Trocknen in einen Behälter eingelegt wird.
Einen Viertel- oder halben Filterquerschnitt der Filterrückstände mit ruckartigen Bewegungen mit einem Skalpell Typ 24 ausschneiden. Den ausgeschnittenen Querschnitt mit Kohlenstoffklebeband oder Kohlenstoffkleber vorsichtig auf dem Träger (Stub') des REM befestigen. An mindestens drei Stellen ist Kolloidalsilber zur Verbesserung des elektrischen Kontakts der Filterränder und des "Stub" aufzubringen. Wenn der Klebstoff bzw. das Kolloidalsilber getrocknet ist, durch Sputter-Beschichtung ca. 50 nm Gold oder Gold/Palladium auf die Oberfläche des Filterrückstands aufbringen.
1.5.3.3 Kalibrierung und Betrieb des REM
1.5.3.3.1 Kalibrierung
Die Kalibrierung des REM ist mindestens einmal wöchentlich (idealerweise täglich) anhand eines freigegebenen Kalibriergitters zu kontrollieren. Die Kalibrierung ist anhand eines freigegebenen Normals zu kontrollieren; stimmt der Messwert (REM) nicht auf ± 2 % mit dem zertifizierten Wert überein, muss die Kalibrierung des REM nachjustiert und erneut kontrolliert werden.
Das REM muss bei Verwendung einer Probenmatrix mindestens die Auflösung eines sichtbaren Mindestdurchmessers von 0,2 µm bei einem Vergrößerungsfaktor x 2000 ermöglichen.
1.5.3.3.2 Betrieb
Das REM ist mit einem Vergrößerungsfaktor von 10.000 1 unter Bedingungen zu betrieben, die eine gute Auflösung und eine akzeptable Bildqualität bei geringer Abtastgeschwindigkeit von beispielsweise 5 Sekunden je Aufnahme ergeben. Da die Betriebsvoraussetzungen unterschiedlicher REM sich voneinander unterscheiden können, sind bei Materialien mit relativ geringer Atommasse Beschleunigungsspannungen von 5-10 keV zu verwenden, um eine möglichst gute Sichtbarkeit und Auflösung zu erreichen; dabei ist eine geringe Punktgröße und ein kurzer Arbeitsabstand einzustellen. Wird ein Linear-Trverse-Verfahren angewandt, ist eine Neigung von 0° zu verwenden, um Refokussierung auf ein Minimum zu beschränken; weist das REM eine euzentrische Stufe auf, ist der euzentrische Arbeitsabstand zu verwenden. Ein geringerer Vergrößerungsfaktor kann verwendet werden, wenn das Material keine kleinen Fasern (mit geringem Durchmesser) enthält und große Faserdurchmesser (> 5 µm) vorliegen.
___________________
1) Siehe vorherige Fußnote zu 3 µm-Fasern.
1.5.3.4 Größenbestimmung
1.5.3.4.1 Bewertung der Probe bei geringer Vergrößerung
Zunächst ist die Probe mit einem geringen Vergrößerungsfaktor auf Anzeichen von Klumpenbildung größerer Fasern zu untersuchen und die Faserndichte zu ermitteln. Bei übermäßiger Klumpenbildung wird empfohlen, eine neue Probe herzustellen.
Aus Gründen der statistischen Genauigkeit muss eine bestimmte Mindestzahl Fasern gemessen werden, eine hohe Faserndichte ist dabei erstrebenswert, da die Untersuchung leerer Felder zeitaufwändig ist und keinen Beitrag zum Analyseergebnis liefert. Ist der Filter jedoch überladen, ist die Messung aller messbaren Fasern erschwert und es besteht die Gefahr, dass kleinere Fasern durch größere Fasern überdeckt und dadurch übersehen werden.
Eine Verfälschung in Richtung einer zu hoch geschätzten LWGMD kann dann eintreten, wenn Faserdichten von mehr als 150 Fasern je Millimeter Linear-Traverse vorliegen. Durch geringe Faserkonzentrationen nimmt andererseits die Dauer der Analyse zu, weshalb es oft kostengünstiger ist, eine Probe herzustellen, deren Faserdichte näher am Optimum liegt, statt ständig die Faserzahlen in Filtern geringer Konzentrationen zu zählen. Die optimale Faserdichte soll durchschnittlich ca. 1 oder 2 zählbare Fasern je Sichtfeld bei einer 5000-fachen Vergrößerung ergeben. Allerdings ist die optimale Dichte von der Größe (Durchmesser) der Fasern abhängig, also muss der Bediener mit entsprechendem Sachverstand entscheiden, ob die Faserdichte dem Optimum nahekommt oder nicht.
1.5.3.4.2 Längengewichtung der Faserdurchmesser
Es werden nur diejenigen Fasern gezählt, die eine (unendlich) dünne Linie auf dem Raster des REM berühren (oder kreuzen). Hierzu wird eine waagerechte (oder vertikale) Linie durch die Rastermitte gezogen.
Alternativ dazu wird ein einzelner Punkt in der Mitte des Rasters angeordnet und ein kontinuierlicher Abtastvorgang in einer Richtung über den Filter hinweg gestartet. Der Durchmesser jeder Faser mit einem Seitenverhältnis von mehr als 3:1, die diesen Punkt berührt oder kreuzt, wird gemessen und aufgezeichnet.
1.5.3.4.3 Bestimmung der Fasergröße
Es wird empfohlen, mindestens 300 Fasern zu messen. Jede Faser wird nur ein einziges Mal am Schnittpunkt mit der auf dem Bild gezeichneten Linie oder Punkt (oder nahe dem Schnittpunkt, wenn die Faserkanten verdeckt sind) gemessen. Werden Fasern mit uneinheitlichen Querschnitten festgestellt, ist eine Messung am durchschnittlichen Faserdurchmesser zugrundezulegen. Bei der Festlegung des Randes und der Messung des geringsten Abstands zwischen den Faserrändern ist vorsichtig vorzugehen. Die Größenbestimmung kann online oder offline an gespeicherten Bildern oder Fotoaufnahmen erfolgen. Die Verwendung von halbautomatischen Bildmesssystemen, bei denen die Daten direkt in eine Tabellenkalkulation geladen werden, ist zu empfehlen, da diese Verfahren Zeit sparen, Abschriftfehler vermeiden und eine automatische Berechnung ermöglichen.
Die Enden langer Fasern sind bei geringer Vergrößerung zu prüfen, damit sichergestellt ist, dass sie sich nicht in den Sichtbereich des Messfeldes rollen und nur einmal gemessen werden.
2. Daten
2.1 Behandlung der Ergebnisse
Die Faserdurchmesser weisen normalerweise keine Normalverteilung auf. Mit Hilfe einer Log-Transformation kann jedoch eine Verteilung ermittelt werden, die näherungsweise der Normalverteilung entspricht.
Das arithmetische Mittel (mittlerer 1nD) und die Standardabweichung (SDlnD) der 1nD-Werte (log to base e) der n Faserdurchmesser (D) wird berechnet.
mittl.lnD = ΣlnD / n (1)
SDlnD = {[Σ(lnD - mittl. lnD)]2/ (n-1)}0,5 (2)
Die Standardabweichung wird durch die Quadratwurzel der Anzahl Messungen (n) dividiert und daraus der Standardfehler (SE,J ermittelt.
SElnd = SD / n0,5 (3)
Das Zweifache des Standardfehlers wird vom Mittelwert abgezogen und der Exponentialwert dieses Wertes (Mittelwert minus dem Zweifachen des Standardfehlers) berechnet; dies ergibt den geometrischen Mittelwert minus zwei geometrischen Standardfehlern.
LWGMD - 2SE = e(mitt1.1ndD-2SElnd) (4)
3. Berichtserstellung
Testbericht
Der Testbericht muss mindestens folgende Angaben enthalten:
4. LITERATUR
1. B. Tylee SOP MF 240. Health and Safety Executive. February 1999.
2. G. Burdett und G. Revell. Development of a standard method to measure the lengthweigthed geometric mean fibre diameter. Results of the Second interlaboratory exchange. IR/LIMF/94107. Project 1142.75 HPD. Health and Safety Executive. Research and Laboratory Services Division, 1994.
| Anhang III |
B.46. Invitro-Hautreizung: Test an rekonstruierten Modellen menschlicher Epidermis
1. Methode
1.1 Einleitung
Der Begriff Hautreizung bezeichnet das Auslösen einer reversiblen Hautschädigung nach Applikation einer Prüfsubstanz für die Dauer von bis zu 4 Stunden (Definition des Globalen Harmonisierten Systems (GHS) zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN))(1). Die vorliegende Prüfmethode entspricht einem in vitro-Verfahren, das es je nach Informationsanforderungen gestattet, als vollwertiger Ersatztest (Standalone-Test) im Rahmen einer Teststrategie und nach einem evidenzbasierten Bewertungsansatz (Weightof-Evidence approach) das Hautreizungspotenzial von Substanzen zu bestimmen (2).
Hautreizungen wurden bisher meist anhand von Versuchstieren bewertet (siehe Prüfmethode B.4) (3). Um dem Tierschutzaspekt Rechnung zu tragen, sieht Methode B.4 vor, dass Hautverätzungenreizungen auch im Wege einer sequenziellen Teststrategie bestimmt werden können, bei der validierte in vitro- und ex vivo-Methoden angewandt werden, die den Tieren Schmerzen und Leiden ersparen. Drei validierte in vitro-Prüfinethoden bzw. Prüfrichtlinien - B.40, B.40bis und TG 435 (4, 5, 6) - sind für den Hautverätzungen betreffenden Teil der sequenziellen Teststrategie der Methode B.4 sinnvoll.
Die vorliegende Prüfmethode beruht auf Modellen rekonstruierter menschlicher Epidermis, die in ihrer allgemeinen Ausgestaltung (Verwendung von menschlichen Keratinozyten als zelluläres Ausgangsmaterial, repräsentativem Gewebe und Zytoarchitektur) die biochemischen und physiologischen Eigenschaften der menschlichen Oberhaut, d. h. der Epidermis, weitgehend nachvollziehen. Das im Rahmen dieser Prüfmethode beschriebene Verfahren gestattet die Identifizierung der Gefahren von Reizstoffen gemäß Kategorie 2 des UN-GHS-Systems. Die Prüfmethode umfasst auch eine Reihe von Leistungsnormen für die Bewertung ähnlicher und modifizierter Methoden für Tests auf Basis rekonstruierter menschlicher Epidermis (7).
Für zwei in vitro-Prüfmethoden (8, 9, 10, 11, 12, 13, 14, 15, 16, 17), die als EpiSkinTM und EpiDermTM im Handel erhältlich sind und Modelle rekonstruierter menschlicher Epidermis verwenden, wurden Prävalidierungs-, Optimierungs- und Validierungsstudien abgeschlossen. Die angeführten Referenzen beruhten auf 1138-Kennzeichnung. Bestimmte Aspekte der Umrechnung zum Zwecke des GHS werden unter Referenz 25 angesprochen. Methoden, die unter Leistungsgesichtspunkten mit der Methode EpiSkinTM(validierte Referenzmethode 1) vergleichbar sind, werden als vollwertige Ersatztests für den in-vivo-Test am Kaninchen zwecks Einstufung von Substanzen mit Hautreizungspotenzial in Kategorie 2 des GHS-Systems empfohlen. Methoden, die unter Leistungsgesichtspunkten mit der Methode EpiDermTM(validierte Referenzmethode 2) vergleichbar sind, werden lediglich für Reihenuntersuchungen (Screening) oder im Rahmen einer sequenziellen Teststrategie mit evidenzbasiertem Bewertungsansatz zur Einstufung von Substanzen mit Hautreizungspotenzial in Kategorie 2 des GHS-Systems empfohlen. Bevor ein vorgeschlagener Hautreizungstest an Modellen von in vitro rekonstruierter menschlicher Epidermis für regulatorische Zwecke verwendet werden kann, sollten nach den in der vorliegenden Prüfmethode vorgegebenen Leistungsnormen (siehe Anhang) Verlässlichkeit, Relevanz (Genauigkeit) und Grenzen des Tests für den vorgeschlagenen Verwendungszweck bestimmt werden, um sicherzustellen, dass er mit der validierten Referenzmethode 1 vergleichbar ist.
Zwei andere Methoden für Tests an in vitro rekonstruierter menschlicher Epidermis - der modifizierte EpiDermTM-Test (modifizierte Referenzmethode 2) und der SkinEthic RHETM-Test (Me-Too-Methode 1) - wurden entsprechend den Anforderungen für die vorliegende Prüfinethode validiert und erbringen ähnliche Ergebnisse wie die validierte Referenzmethode 1 (18).
1.2 Definitionen
Für die vorliegende Prüfmethode gelten die folgenden Definitionen:
Genauigkeit: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Mag der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode.
Chargenkontroffsubstanz: Die beim Gewebe eine mittlere Zellviabilität hervorrufende Referenzsubstanz.
Zellviabilität: Parameter zur Messung der Gesamtaktivität einer Zellpopulation, z.B. Fähigkeit zellulärer mitochondrialer Dehydrogenasen, den Vitalfarbstoff MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolyl-Blau) zu reduzieren, der je nach gemessenem Endpunkt und angewandtem Testkonzept der Gesamtzahl und/oder der Vitalität lebender Zellen entspricht.
ET50: Die Expositionszeit, die erforderlich ist, um die Zellviabilität bei Anwendung der Markersubstanz in vorgegebener fester Konzentration um 50 % zu reduzieren; siehe auch IC50.
Falschnegativ-Rate: Der Anteil aller positiven Substanzen, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Die Falschnegativ-Rate ist ein Leistungsindikator der Prüfmethode.
Falschpositiv-Rate: Der Anteil aller negativen (nicht wirkenden) Substanzen, die fälschlicherweise als positiv identifiziert werden. Die Falschpositiv-Rate ist ein Leistungsindikator der Prüfmethode.
Unendliche Dosis: Die Menge der auf die Haut aufgetragenen Prüfsubstanz, die über die zur vollständigen und gleichmäßigen Bedeckung der Hautoberfläche erforderliche Menge hinausgeht.
GHS (Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien): Ein System zur Klassifizierung von Substanzen und Gemischen nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenbögen, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1); in der EU umgesetzt durch die Verordnung (EG) Nr. 1272/2008.
IC50: Die Konzentration, bei der eine Markersubstanz die Viabilität der Gewebe nach einer vorgegebenen Expositionszeit um 50 % (IC50) reduziert; siehe auch ET50.
Leistungsnormen: Auf einer validierten Referenzmethode beruhende Normen, auf deren Grundlage die Vergleichbarkeit einer vorgeschlagenen, mechanistisch und funktionell ähnlichen Prüfinethode bewertet werden kann. Sie umfassen i) wesentliche Elemente der Prüfmethode; ii) ein Mindestverzeichnis von Referenzsubstanzen, ausgewählt aus den Substanzen, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden; und iii) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Genauigkeits- und Zuverlässigkeitswerte, die die vorgeschlagene Prüfinethode bei der Bewertung anhand des Mindestverzeichnisses von Referenzsubstanzen demonstrieren sollte.
Zuverlässigkeit: Mag der Verlässlichkeit der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien in einem bestimmten Zeitintervall bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet.
Empfindlichkeit: Der Anteil aller positiven/wirkenden Stoffe, die durch den Test korrekt eingestuft werden. Die Empfindlichkeit ist ein Mag der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz.
Spezifität: Der Anteil aller negativen/wirkungslosen Stoffe, die durch den Test korrekt eingestuft werden. Die Spezifität ist ein Mag der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz.
Hautreizung: Das Auslösen einer reversiblen Hautschädigung nach Applikation einer Prüfsubstanz für die Dauer von bis zu 4 Stunden. Eine Hautreizung ist eine lokal auftretende, nicht immunogene Reaktion, die kurz nach der Stimulation eintritt (24). Ihr Hauptmerkmal ist ihr umkehrbarer Prozess, der mit Entzündungsreaktionen und den bei Entzündungen gängigsten klinischen Reizsymptomen (Erythema, Ödeme, Juckreiz und Schmerzen) einhergeht.
1.3 Anwendungsbereich und Grenzen
Die unter diese Prüfmethode fallenden Tests an rekonstruierter menschlicher Epidermis sind dadurch begrenzt, dass Stoffe nur als Hautreizstoffe im Sinne von Kategorie 2 des UN-GHS-Systems eingestuft werden können. Da keine Einstufung in die wahlfreie Kategorie 3 des UN-GHS-Systems möglich ist, bleiben alle restlichen Stoffe unklassifiziert (d. h. ohne Kategorieneinstufung). Je nach Regelungsbedarf und bei etwaiger künftiger Einbeziehung neuer Endpunkte, bei Verbesserungen oder bei Entwicklung neuer "Me-Too"-Tests muss die vorliegende Prüfmethode möglicherweise überprüft werden.
Die vorliegende Prüfmethode gestattet die Gefahrenidentifizierung bei reizenden Monosubstanzen (19), liefert jedoch keine zweckdienlichen Informationen zur Hautverätzung. Gase und Aerosole können nicht getestet werden, und Gemische waren bisher noch nicht Gegenstand einer Validierungsstudie.
1.4 Testprinzip
Die Prüfsubstanz wird topisch aufgetragen auf ein dreidimensionales Modell rekonstituierter menschlicher Epidermis, bestehend aus normalen menschlichen epidermalen Keratinozyten, die zu einem mehrschichtigen, stark differenzierten Modell menschlicher Epidermis kultiviert wurden. Letztere besteht aus angeordneten Basal-, Stachel- und Körnerzellschichten und einer mehrlagigen Hornschicht (stratum corneum), die zwischen den Zellen lamellare Fettschichten aufweist, welche nach in-vivoähnlichen Mustern angeordnet sind.
Das Prinzip der Testung an Modellen rekonstruierter menschlicher Epidermis beruht auf der Prämisse, dass Reizsubstanzen die Hornschicht diffusionsbedingt durchdringen können und die Zellen in den darunterliegenden Schichten schädigen. Die Zellviabilität wird durch Dehydrogenase-Konversion des Vitalfarbstoffs MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolyl-Blau; EINECS-Nr. 206-069-5, CAS-Nr. 298-93-1)], zu einem blauen Formazan-Salz gemessen, das nach seiner Extaktion aus Geweben quantifiziert wird (20). Reizstoffe werden anhand ihrer Fähigkeit erkannt, die Zellviabilität unter vorgegebene Schwellenwerte zu senken (d. h. ≤50 % bei Reizstoffen der UN-GHS-Kategorie 2). Stoffe, die Zellviabilitäten über dem vorgegebenen Schwellenwert generieren, werden nicht klassifiziert (d. h. bei > 50 % keine Kategorieneinstufung).
Modelle rekonstruierter menschlicher Epidermis eignen sich zur Untersuchung von Feststoffen, Flüssigkeiten, halbfesten Stoffe und Wachsen. Flüssigkeiten können wässrig oder nichtwässrig, Feststoffe wasserlöslich oder nicht wasserlöslich sein. Feststoffe sollten nach Möglichkeit als Feinpulver getestet werden. Da 58 sorgfältig ausgesuchte Substanzen, die ein breites Spektrum chemischer Klassen repräsentieren, in die Validierung der Tests an Modellen rekonstruierter menschlicher Epidermis einbezogen wurden, wird davon ausgegangen, dass die Methoden allgemeingültig, d. h. für alle chemischen Klassen geeignet sind (16). Die Validierung betrifft 13 Reizsubstanzen der Kategorie 2 des GHS-Systems. Es wird darauf hingewiesen, dass nicht verätzende Säuren, Laugen, Salze und andere anorganische Stoffe nicht validiert wurden und dass einige bekannte Klassen organischer Reizsubstanzen wie Hydroperoxide, Phenole und Tenside nicht oder nur begrenzt inbegriffen waren.
1.5 Leistungsnachweis
Bevor eine validierte Methode, die den Anforderungen der vorliegenden Prüfinethode genügt, routinemäßig angewandt werden kann, sollten Laboratorien anhand von zehn der in Tabelle 1 aufgeführten Substanzen die technische Leistungsfähigkeit der Methode demonstrieren. Im Rahmen der vorliegenden Prüfinethode gilt die wahlfreie Kategorie 3 des UN-GHS-Systems nicht als Kategorie. Für im Rahmen der Prüfinethode entwickelte neuartige analoge Prüfinethoden ("Me-Too"-Tests), die den validierten Referenzmethoden strukturell und funktionell vergleichbar sind, oder für Änderungen validierter Methoden sollten zum Nachweis der Vergleichbarkeit der Zuverlässigkeit und Genauigkeit der neuen Prüfinethode, bevor diese für Regulierungszwecke eingesetzt wird, die im Anhang zur vorliegenden Prüfinethode beschriebenen Leistungsnormen zugrunde gelegt werden.
Tabelle 1 : Leistungssubstanzen als Untergruppe der im Anhang aufgelisteten Referenzsubstanzen
| Substanz | CAS-Nummer | Invivo-Punktzahl | Physikalischer Zustand | GHS-Kategorie |
| Naphthalen-Essigsäure | 86-87-3 | 0 | fest | keine Kat. |
| Isopropanol | 67-63-0 | 0,3 | flüssig | keine Kat. |
| Methylstearat | 112-61-8 | 1 | fest | keine Kat. |
| Heptyl-Butyrat | 5870-93-9 | 1,7 | flüssig | wahlfrei Kat. 3 |
| Hexyl-Salicylat | 6259-76-3 | 2 | flüssig | wahlfrei Kat. 3 |
| Cyclamenaldehyd | 103-95-7 | 2,3 | flüssig | Kat.2 |
| 1-Bromohexan | 111-25-1 | 2,7 | flüssig | Kat.2 |
| Butylmethacrylat | 97-88-1 | 3 | flüssig | Kat.2 |
| 1-Methyl-3-Phenyl-l-Piperazin | 5271-27-2 | 3,3 | fest | Kat.2 |
| Heptanal | 111-71-7 | 4 | flüssig | Kat.2 |
1.6 Beschreibung der Methode
Es folgt eine Beschreibung der Elemente und Verfahrensschritte eines Hautreizungstests an Modellen rekonstruierter menschlicher Epidermis. Modelle rekonstruierter menschlicher Epidermis können kultiviert, präpariert oder im Handel erworben werden (z.B. EpiSkinTM, EpiDermTM und SkinEthic RHETM). Standard-Testprotokolle für EpiSkinTM, EpiDermTM und SkinEthic RHETM sind über http://ecvam.jrc.ec.europa.eu erhältlich (21, 22, 23). Die Testungen sollten wie folgt durchgeführt werden:
1.6.1 Elemente des Modells rekonstruierter menschlicher Epidermis
1.6.1.1 Allgemeine Modellbedingungen
Die Epithelschicht sollte aus normalen menschlichen Keratinozyten gebildet werden. Unter der funktionsfähigen Hornschicht (stratum corneum) sollten mehrere Lagen lebensfähiger Epithelzellen (Basalzellschicht, Stachelzellschicht, Körnerzellschicht) vorhanden sein. Die Hornschicht sollte mehrlagig sein und das zur Erzeugung einer funktionsfähigen Barriere, die robust genug ist, um das schnelle Eindringen zytotoxischer Markersubstanzen wie Natriumdodecylsulfat (SDS) oder Triton X-100 zu verhindern, essenzielle Lipidprofil aufweisen. Die Barrierefunktion kann entweder durch Bestimmung der Konzentration, in der eine Markersubstanz die Viabilität der Gewebe nach einer vorgegebenen Expositionsdauer um 50 % verringert (IC50), oder durch Bestimmung der Expositionszeit bewertet werden, die erforderlich ist, um die Zellviabilität bei Anwendung der Markersubstanz in einer vorgegebenen festen Konzentration um 50 % zu reduzieren (ET50). Das Modell muss rückhaltefähig genug sein, um zu verhindern, dass Material rund um die Hornschicht in lebensfähiges Gewebe eindringt, was zu einer mangelhaften Modellierung der Hautexposition führen würde. Das Hautmodell sollte nicht mit Bakterien, Viren, Mycoplasma oder Pilzen kontaminiert sein.
1.6.1.2 Bedingungen für das Funktionsmodell
1.6.1.2.1 Viabilität
Die Viabilität wird vorzugsweise anhand des MTT-Tests bestimmt (20). Die optische Dichte (OD) des aus dem mit der Negativkontrolle (NK) behandelten Gewebe extrahierten (solubilisierten) Farbstoffs sollte mindestens dem Zwanzigfachen der OD des Extraktionslösemittels allein entsprechen. Es ist protokollarisch festzuhalten, dass das mit der NK behandelte Gewebe während der Expositionszeit nachweislich in der Kultur stabil ist (d. h. es sollte vergleichbare Viabilitätsmesswerte aufweisen).
1.6.1.2.2 Barrierefunktion
Die Hornschicht und ihre Fettzusammensetzung sollten in der Lage sein, das schnelle Eindringen zytotoxischer Markersubstanzen wie SDS oder Triton X-100, wie durch IC50 oder ET50 bestimmt, zu verhindern.
1.6.1.2.3 Morphologie
Die histologische Untersuchung der rekonstruierten Haut/Epidermis sollte durch entsprechend qualifiziertes Personal erfolgen, das nachweisen muss, dass das Modell eine der menschlichen Haut/Epidermis ähnliche Struktur (einschließlich mehrlagiger Hornschicht) aufweist.
1.6.1.2.4 Reproduzierbarkeit
Die Methode sollte unter gleichen Modellbedingungen, vorzugsweise anhand einer geeigneten Chargenkontroll-(Referenz-)substanz (siehe Anhang), im Zeitverlauf nachweislich reproduzierbar sein.
1.6.1.2.5 Qualitätskontrollen (QK) des Modells
Jede Charge des verwendeten Epidermis-Modells sollte bestimmten Freigabekriterien genügen, von denen die Viabilitätskriterien (Absatz 1.6.1.2.1) und die Kriterien für die Barrierefunktion (Absatz 1.6.1.2.2) die wichtigsten sind. Der Hersteller des Hautmodells (oder - bei Verwendung eines hauseigenen Modells - der Prüfer) sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für IC50 oder ET50 festlegen. Das Labor sollte die Barriereeigenschaften der Gewebe nach deren Bezug überprüfen. Nur mit geeigneten Geweben erzielte Ergebnisse kommen für eine zuverlässige Vorhersage von Reizwirkungen in Frage. Beispiele für Akzeptanzspannen für die validierten Referenzmethoden:
Tabelle 2 : Beispiele für Chargenfreigabekriterien im Rahmen der Qualitätskontrolle
| Untere Akzeptanzgrenze | Mittelwert | Obere Akzeptanzgrenze | |
| Validierte Referenzmethode1 (18-stündige Behandlung mit SDS) | 1 IC50 = 1,0 mg/ml | IC50 = 2,32 mg/ml | IC50 = 3,0 mg/ml |
| Validierte Referenzmethode 2 (1 % Triton X-100) | ET50 = 4,8 Std. | ET50 = 6,7 Std. | ET50 = 8,7 Std. |
1.6.1.3 Applikation der Prüf- und Kontrollsubstanzen
Bei jeder Behandlung und für die Kontrollen sollte eine ausreichende Anzahl Gewebereplikate (mindestens drei je Test) verwendet werden. Bei flüssigen und festen Substanzen sollte eine ausreichende Menge Prüfsubstanz gleichmäßig auf die gesamte Hautoberfläche aufgetragen werden; unendliche Dosen (siehe 1.2 - Definitionen) sind zu vermeiden, d. h. es sollten mindestens 25 µL/cm2 bzw. 25 mg/cm2 verwendet werden. Bei festen Stoffen sollte die Epidermis-Oberfläche vor der Applikation mit deionisiertem oder destilliertem Wasser angefeuchtet werden, um guten Hautkontakt zu gewährleisten. Feststoffe sollten nach Möglichkeit als Feinpulver getestet werden. Am Ende der Expositionszeit sollte die Prüfsubstanz mit wässriger Pufferlösung oder 0,9 % NaCl von der Haut abgewaschen werden. Je nachdem, welches Modell rekonstruierter menschlicher Epidermis verwendet wurde, kann die Expositionszeit 15 bis 60 Minuten betragen und die Inkubationstemperatur zwischen 20 und 37 °C liegen. Für Einzelheiten siehe Standardvorgehensweise (Standard Operating Procedures) für die drei Methoden (21, 22, 23).
Bei jeder Studie sollten parallel Negativkontrollen (NK) und Positivkontrollen (PK) verwendet werden, um nachweisen zu können, dass die Viabilität (NK), die Barrierefunktion und die resultierende Empfindlichkeit (PK) der Gewebe innerhalb einer vorgegebenen historischen Akzeptanzspanne liegen. Als PK-Substanz wird 5 % wässrige SDS empfohlen. Als NK-Substanzen empfehlen sich Wasser oder phosphatgepufferte Kochsalzlösung (PBS).
1.6.1.4 Zellviabilitätsmessungen
Wichtigstes Element des Testprotokolls ist es, dass Viabilitätsmessungen nicht unmittelbar nach dem Kontakt mit den Prüfsubstanzen, sondern nach einer ausreichend langen Inkubationszeit der abgespülten Gewebe (nach der Behandlung) in frischem Medium erfolgen. Während dieser Zeit kann sich das Gewebe von milden Reizwirkungen erholen bzw. deutliche zytotoxische Effekte können sich herausbilden. Während der Phase der Testoptimierung (9, 10, 11, 12, 13) hat sich eine Inkubationszeit von 42 Stunden nach der Behandlung als optimal erwiesen und wurde daher bei der Validierung der Referenztestmethoden zugrunde gelegt.
Der MTT-Konversionstest ist eine validierte quantitative Methode, die zur Messung der zellulären Aktivität angewandt werden sollte. Sie ist zur Anwendung in einem dreidimensionalen Gewebemodell geeignet. Die Hautprobe wird für drei Stunden in eine MTT-Lösung angemessener Konzentration (z.B. 0,3-1 mg/mL) gegeben. Das präzipitierte blaue Formazan-Produkt wird anschließend mit Hilfe eines Lösemittels (z.B. Isopropanol, Säure-Isopropanol) aus dem Gewebe extrahiert, und die Formazan-Konzentration wird durch Bestimmung der OD bei 570 nm mittels eines Bandpasses von maximal ± 30 nm gemessen.
Optische Eigenschaften der Prüfsubstanz oder ihre chemische Wirkung auf den MTT können mit dem Test interferieren und (weil die Prüfsubstanz die Farbbildung sowohl verhindern oder umkehren als auch hervorrufen kann) zu einer falschen Viabilitätsschätzung führen. Dies kann der Fall sein, wenn eine bestimmte Prüfsubstanz nicht komplett von der Haut abgewaschen wurde oder wenn sie in die Epidermis eindringt. Wirkt sich die Prüfsubstanz unmittelbar auf den MTT aus, ist sie natürlich gefärbt oder verfärbt sie sich während der Gewebebehandlung, so sollten zum Nachweis und zur Behebung einer etwaigen Interferenz der Prüfsubstanz mit der Viabilitätsmesstechnik zusätzliche Kontrollen verwendet werden. Für eine genaue Beschreibung des Tests der direkten MTT-Reduktion siehe das Testprotokoll für die validierten Referenzmethoden (21, 22, 23). Eine infolge dieser Interferenzen eintretende unspezifische Färbung (non speafu colour, NSC) sollte 30 % der NK (bei Behebung) nicht überschreiten. Beträgt die NSC > 30 %, so gilt die Prüfsubstanz als nicht mit dem Test vereinbar.
1.6.1.5 Testakzeptanzkriterien
Bei jedem Test an gültigen Chargen (siehe Absatz 1.6.1.2.5) sollten mit der Negativkontrolle behandelte Gewebe OD-Werte aufweisen, die für die Gewebequalität nach abgeschlossenen Beförderungs- und Annahmevorgängen und sämtlichen Schritten des Reizungsprotokolls aussagekräftig sind. Die OD-Werte der Kontrollen sollten nicht unter historisch etablierten niedrigeren Grenzwerten liegen. Gleichermaßen sollten mit der Positivkontrolle, d. h. 5 % wässrige SDS, behandelte Gewebe die verbleibende Empfindlichkeit der Gewebe und ihre Fähigkeit reflektieren, unter den Bedingungen jedes einzelnen Tests (z.B. Viabilität ≤40 %bei dervalidierten Referenzmethode 1 und s 20 % bei der validierten Referenzmethode 2) auf eine Reizsubstanz zu reagieren. Es sollten entsprechende und geeignete Maße der Variabilität zwischen Gewebereplikaten festgelegt werden (Beispiel: soweit Standardabweichungen verwendet werden, sollten sie ≤18 % betragen).
2. Daten
2.1 Daten
Für jede Behandlung sollten Daten aus einzelnen Replikattestproben (z.B. OD-Werte und Daten über die berechnete prozentuale Zellviabilität für jede Prüfsubstanz, einschließlich Einstufung), gegebenenfalls auch Daten aus Wiederholungsversuchen, tabellarisch mitgeteilt werden. Darüber hinaus sollten für jeden Versuch der Mittelwert ± Standardabweichung und für jede geprüfte Substanz festgestellte Interaktionen mit dem MTT-Reagens und Testfarbstoffen übermittelt werden.
2.2 Auswertung der Ergebnisse
Die für die einzelnen Testproben erzielten OD-Werte können zur Berechnung der prozentualen Viabilität im Vergleich zur Negativkontrolle, die auf 100 % festgesetzt ist, herangezogen werden. Der Schwellenwert der prozentualen Viabilität, der zwischen Reizstoff und nicht klassifizierten Prüfsubstanzen unterscheidet, und das (die) statistische(n) Verfahren zur Auswertung der Ergebnisse und zur Identifizierung von Reizstoffen sollten genau definiert und protokolliert werden und nachweislich geeignet sein. Die Schwellenwerte für die Vorhersage der Reizung im Rahmen der validierten Referenzmethoden sind nachstehend angegeben:
Die Prüfsubstanz gilt als hautreizend im Sinne von Kategorie 2 des UN-GHS-Systems,
3. Berichterstattung
3.1 Testbericht
Der Testbericht sollte folgende Informationen enthalten:
Prüf- und Kontrollsubstanzen:
Gründe für die Verwendung des betreffenden Hautmodells und Protokolls.
Testbedingungen:
Ergebnisse:
Schlussfolgerungen.
4. REFERENZEN
| Bewertung der Leistungsmerkmale der für Hautreizungstests vorgeschlagenen Modelle in vitrorekonstruierter menschlicher Epidermis | Anlage |
Einleitung
Zur Bewertung der Zuverlässigkeit und Genauigkeit der für die Anwendung im Rahmen der vorliegenden Prüfmethode vorgeschlagenen Verfahren sollten Substanzen verwendet werden, die das gesamte Reizwertspektrum nach Draize repräsentieren. Bei der Bewertung anhand der 20 empfohlenen Referenzsubstanzen (Tabelle 2) sollten die vorgeschlagenen Verfahren Zuverlässigkeits- und Genauigkeitswerte aufzeigen, die denen der validierten Referenzmethode 1 (Tabelle 3) entsprechen (1). Die angestrebten Zuverlässigkeits- und Genauigkeitsnormen sind in den nachstehenden Abschnitten II und III vorgegeben. Nicht eingestufte Substanzen und (in Kategorie 2 des UN-GHS-Systems) eingestufte Substanzen relevanter chemischer Klassen werden einbezogen, um die Zuverlässigkeit und Leistung (Empfindlichkeit, Spezifität, Falschnegativ-Falschpositiv-Raten und Genauigkeit) der vorgeschlagenen Prüfinethode mit denen der validierten Referenzmethode 1 vergleichen zu können. Die Zuverlässigkeit der Prüfmethode sowie ihre Fähigkeit, Reizstoffe der Kategorie 2 des UN-GHS-Systems korrekt zu identifizieren, sollten bestimmt werden, bevor die Methode zur Testung neuer Substanzen eingesetzt wird.
Leistungsnormen
Die Leistungsnormen umfassen die drei folgenden Elemente: i) wesentliche Elemente der Prüfinethode, ii) Referenzsubstanzen und iii) vorgegebene Genauigkeits- und Zuverlässigkeitswerte (2). Diese Normen beruhen auf den im Anschluss an die ECVAM-Hautreizungsvalidierungsstudie aufgestellten Leistungsnormen (3).
I) Wesentliche Elemente der Prüfmethode Allgemeine Modellbedingungen
Die Epithelschicht sollte aus normalen menschlichen Keratinozyten gebildet werden. Unter der funktionsfähigen Hornschicht (stratum comeum) sollten mehrere Lagen lebensfähiger Epithelzellen (Basalzellschicht, Stachelzellschicht, Körnerzellenschicht) vorhanden sein. Die Hornschicht sollte mehrlagig sein und das zur Erzeugung einer funktionsfähigen Barriere, die robust genug ist, um das schnelle Eindringen zytotoxischer Markersubstanzen wie Natriumdodecylsulfat (SDS) oder Triton X-100 zu verhindern, essenzielle Lipidprofil aufweisen. Die Barrierefunktion kann entweder durch Bestimmung der Konzentration, in der eine Markersubstanz die Viabilität der Gewebe nach einer vorgegebenen Expositionsdauer um 50 % verringert (ICso), oder durch Bestimmung der Expositionszeit bewertet werden, die erforderlich ist, um die Zellviabilität bei Anwendung der Markersubstanz in einer vorgegebenen festen Konzentration um 50 % zu reduzieren (ET"). Das Modell muss rückhaltefähig genug sein, um zu verhindern, dass Material rund um die Hornschicht in lebensfähiges Gewebe eindringt, was zu einer mangelhaften Modellierung der Hautexposition führen würde. Das Hautmodell sollte nicht mit Bakterien, Viren, Mycoplasma oder Pilzen kontaminiert sein.
Bedingungen für das Funktionsmodell Viabilität
Die Viabilität wird vorzugsweise anhand des MTT-Tests bestimmt (4). Die optische Dichte (OD) des aus dem mit der Negativkontrolle (NK) behandelten Gewebe extrahierten (solubilisierten) Farbstoffs sollte mindestens dem Zwanzigfachen der OD des Extraktionslösemittels allein entsprechen. Es ist protokollarisch festzuhalten, dass das mit der NK behandelte Gewebe während der Expositionszeit nachweislich in der Kultur stabil ist (d. h. es sollte vergleichbare Viabilitätsmesswerte aufweisen).
Barrierefunktion
Die Hornschicht und ihre Fettzusammensetzung sollten in der Lage sein, das schnelle Eindringen zytotoxischer Markersubstanzen wie SDS oder Triton X-100, wie durch ICs, oder ET., bestimmt, zu verhindern.
Morphologie
Die histologische Untersuchung der rekonstruierten Haut/Epidermis sollte durch entsprechend qualifiziertes Personal erfolgen, das nachweisen muss, dass das Modell eine der menschlichen Haut/Epidermis ähnliche Struktur (einschließlich mehrlagiger Hornschicht) aufweist.
Reproduzierbarkeit
Die Methode sollte unter gleichen Modellbedingungen, vorzugsweise anhand einer geeigneten Chargenkontroll-(Referenz-)substanz (siehe Definitionen in Abschnitt 1.2), im Zeitverlauf nachweislich reproduzierbar sein.
Qualitätskontrollen (QK) des Modells
Jede Charge des verwendeten Epidermis-Modells sollte bestimmten Freigabekriterien genügen, von denen die Viabilitätskriterien und die Kriterien für die Barrierefunktion die wichtigsten sind. Der Hersteller des Hautmodells (oder - bei Verwendung eines hauseigenen Modells - der Prüfer) sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für IC50 oder ET50 festlegen. Das Labor sollte die Barriereeigenschaften der Gewebe nach deren Bezug überprüfen. Nur mit geeigneten Geweben erzielte Ergebnisse kommen für eine zuverlässige Vorhersage von Reizwirkungen in Frage. Beispiele für Akzeptanzspannen für die validierten Referenzmethoden:
Tabelle 1 : Beispiele für Chargenfreigabekriterien im Rahmen der Qualitätskontrolle
| Untere Akzeptanzgrenze | Mittelwert | Obere Akzeptanzgrenze | |
| Validierte Referenzmethode 1 (18-stündige Behandlung mit SDS) | IC50 = 1,0 mg/ml | IC50 = 2,32 mg/ml | IC50 = 3,0 mg/ml |
| Validierte Referenzmethode 2 (1 % Triton X-100) | ET50 = 4,8 Std. | ET50 = 6,7 Std. | ET50 = 8,7 Std. |
II) Referenzsubstanzen
Um festzustellen, ob Genauigkeit und Zuverlässigkeit vorgeschlagener neuartiger Prüfinethoden für Testungen an in vitrc rekonstruierten Modellen menschlicher Epidermis, die den validierten Referenzmethoden strukturell und funktionel nachweislich ähneln oder die sich nur unwesentlich von einer validierten Referenzmethode unterscheiden, unter Leistungsgesichtspunkten mit der validierten Referenzmethode 1 vergleichbar sind, werden Referenzsubstanzen verwendet (1). Die in Tabelle 2 aufgelisteten 20 Referenzsubstanzen umfassen Substanzen unterschiedlicher chemisches Klassen sowie Substanzen der Kategorie 2 des UN-GHS-Systems. Die Liste umfasst 10 Substanzen der Kategorie 2 des UN-GHS-Systems, 3 Substanzen der Kategorie 3 des UN-GHS-Systems sowie 7 nicht eingestufte Substanzen. Im Rahmen dieser Prüfmethode gilt die wahlfreie Kategorie 3 nicht als Kategorie. Diese Referenzsubstanzen entsprechen des Mindestanzahl Substanzen, die zur Beurteilung der Genauigkeit und Zuverlässigkeit einer vorgeschlagenen Methode fib Hautreizungstests an Modellen rekonstruierter menschlicher Epidermis verwendet werden sollten. In Fällen, in denen eine in der Liste geführte Substanz nicht verfügbar ist, könnten andere Substanzen, für die angemessene in-vivo-Referenzdaten vorliegen, verwendet werden. Falls erwünscht, kann die Mindestliste der Referenzsubstanzen um zusätzliche Substanzen anderer chemischer Klassen, für die angemessene in-vivo-Referenzdaten vorliegen, ergänzt werden, um die Genauigkeit der vorgeschlagenen Prüfmethode noch genauer beurteilen zu können.
Tabelle 2 : Referenzsubstanzen zur Bestimmung der Genauigkeits- und Zuverlässigkeitswerte für Modelle
rekonstruierter menschlicher Epidermis
| Substanz 0 | CAS-Nr. | EINECS-Nr. | Physikalischer Zustand | Invivo- Punktezahl | GHS-in-vitro- Kat. | GHS-in-vivo-Kat. |
| 1-Bromo-4-Chlorobutan | 6940-78-9 | 230-089-3 | L | 0 | Kat. 2 | keine Kat. |
| Diethyl-Phthalat | 84-66-2 | 201-550-6 | L | 0 | keine Kat. | keine Kat. |
| Naphthalin-Essigsäure | 86-87-3 | 201-705-8 | S | 0 | Keine Kat. | keine Kat. |
| Allyl-Phenoxyacetat | 7493-74-5 | 231-335-2 | L | 0,3 | keine Kat. | keine Kat. |
| Isopropanol | 67-63-0 | 200-661-7 | L | 0,3 | keine Kat. | keine Kat. |
| 4-Methyl-Thio-Benzaldehyd | 3446-89-7 | 222-365-7 | L | 1 | Kat. 2 | keine Kat. |
| Methylstearat | 112-61-8 | 203-990-4 | S | 1 | keine Kat. | keine Kat. |
| Hepty-Butyrat | 5870-93-9 | 227-526-5 | L | 1,7 | keine Kat. | wahlfrei Kat. 3 |
| Hexyl-Salicylat | 6259-76-3 | 228-408-6 | L | 2 | keine Kat. | wahlfrei Kat. 3 |
| Tri-Isobutylphosphat | 126-71-6 | 204-798-3 | L | 2 | Kat.2 | wahlfrei Kat. 3 |
| 1-Decanol | 112-30-1 | 203-956-9 | L | 2,3 | Kat.2 | Kat.2 |
| Cyclamenaldehyd | 103-95-7 | 203-161-7 | L | 2,3 | Kat.2 | Kat.2 |
| 1-Bromhexan | 111-25-1 | 203-850-2 | L | 2,7 | Kat.2 | Kat.2 |
| 2-Chloromethyl-3,5-Dimethyl- 4-Methoxypyridin- Hydrochlorid | 86604-75-3 | 434-680-9 | S | 2,7 | Kat.2 | Kat.2 |
| a-Terpineol | 98-55-5 | 202-680-6 | L | 2,7 | Kat.2 | Kat.2 |
| Di-n-Propyldisulfid | 629-19-6 | 211-079-8 | L | 3 | keine Kat. | Kat. 2 |
| Butylmethacrylat | 97-88-1 | 202-615-1 | L | 3 | Kat.2 | Kat.2 |
| Benzenethiol, 5-(1,1-Dimethylethyl)-2- Methyl | 7340-90-1 | 438-520-9 | L | 3,3 | Kat.2 | Kat.2 |
| 1-Methyl-3-Phenyl-1-Piperazin | 5271-27-2 | 431-180-2 | S | 3,3 | Kat.2 | Kat.2 |
| Heptanal | 111-71-7 | 203-898-4 | L | 4 | Kat.2 | Kat.2 |
| *) Die 20 Referenzsubstanzen umfassen eine repräsentative Auswahl aus den 58 Substanzen, die ursprünglich zur Validierung der Referenzmethode 1 (EpiSkinTM) verwendet wurden. Eine vollständige Liste der Präfsubstanzen und der Auswahlkriterien liegt vor (5). | ||||||
Die in Tabelle 2 aufgelisteten Substanzen entsprechen einer repräsentativen Auswahl aus 58 Substanzen, die in der internationalen ECVAM-Hautreizungsvalidierungsstudie (1) verwendet werden. Die Wahl erfolgte nach folgenden Kriterien:
III) Vorgegebene Genauigkeits- und Zuverlässigkeitswerte
Die Leistung (Empfindlichkeit, Spezifität, Falschnegativ-Rate, Falschpositiv-Rate und Genauigkeit) der vorgeschlagenen Prüfinethode sollten denen der validierten Referenzmethode 1 (Tabelle 3) vergleichbar sein, d. h. die Empfindlichkeit sollte gleich oder höher als (≥) 80 %, die Spezifität gleich oder höher als (≥) 70 % und die Genauigkeit gleich oder höher als (≥) 75 % sein. Die Leistung sollte anhand sämtlicher Klassifizierungen, die in den verschiedenen Teilnehmerlaboratorien für die 20 Substanzen erzielt wurden, berechnet werden. Die Klassifizierung der verschiedenen Substanzen in den einzelnen Laboratorien sollte auf Basis des durchschnittlichen Viabilitätswertes bei den verschiedenen Tests (mindestens drei gültige Tests) erfolgen.
Tabelle 3 : Leistung der validierten Referenzmethode 1 *
| Prüfmethode | Anzahl Substanzen | Empfind- lichkeit | Spezifität | Falsch- negativ-Rate | Falsch- positiv-Rate | Genauigkeit |
| Validierte Referenzmethode 1 1 | 58 | 87,2% 2 | 71,1 % 3 | 12,8% | 29,9% | 74,7% |
| validierte Referenzmethode 1 1 | 20 | 90% | 73,3% | 10% | 26,7% | 81,7% |
| 1) EpiSkinTM.
2) Auf Basis von 13 Reizsubstanzen der Kategorie 2 des GHS-Systems. 3) Auf Basis von 45 Reizsubstanzen der Kategorie 3 des GHS-Systems oder nicht GHS-eingestufte Substanzen. *) Tabelle 3 zeigt die Leistung der validierten Referenzmethode 1 in Bezug auf ihre Fähigkeit, für die 58 bzw. 20 Referenzsubstanzen (Tabelle 2) Reizsubstanzen (der Kategorie 2 des UN-GHS-Systems) und nicht klassifizierte Substanzen (keine Kategorie, einschließlich der wahlfreien Kategorie 3) korrekt zu identifizieren. | ||||||
Die Zuverlässigkeit der vorgeschlagenen Prüfinethode sollte mit der Zuverlässigkeit der validierten Referenzmethoden vergleichbar sein.
Intra-Labor-Reproduzierbarkeit
Eine Bewertung der Intra-Labor-Variabilität sollte eine Übereinstimmung der von einem einzigen Labor bei verschiedenen unabhängigen Tests der 20 Referenzsubstanzen erzielten Klassifizierungen (Kategorie 2/keine Kategorie) von ≥90 % ergeben.
Inter-Labor-Reproduzierbarkeit
Eine Bewertung der Inter-Labor-Reproduzierbarkeit ist nicht wesentlich, wenn die vorgeschlagene Prüfinethode nur in einem Labor angewandt werden soll. Beim Methodentransfer zwischen Laboratorien sollte die Übereinstimmung der bei verschiedenen unabhängigen Tests der 20 Referenzsubstanzen, die vorzugsweise zwischen mindestens drei Laboratorien durchgeführt werden, erzielten Klassifizierungen (Kategorie 2/keine Kategorie) ≥80 % betragen.
Literatur
1) Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovi6, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandärovä, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. and Zuang, V. (2007). The ECVAM International Validation Study an In Vitro Tests for Acute Skin Irritation: Report an the Validity of the EPISKIN and EpiDerm Assays and an the Skin Integrity Function Test. ATLA 35, S. 559-601.
2) OECD (2005) Guidance Document No. 34 an the validation and international acceptance of new or updated test methods for hazard assessment. OECD, Paris.
3) ECVAM (2007) Performance Standards for applying human skin models to in vitro skin irritation. Abrufbar unter Download Study Documents: http://ecvam.jrc.ec.europa.eu. Letzter Zugriff am 27.10.2008.
4) Mosman, T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods 65, S. 55-63.
5) Eskes, C., Cole, T., Hoffinann, S., Worth, A., Cockshott, A., Gerner, I. & Zuang. V (2007) ECVAM International Validation Study an In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals. ATLA 35, S. 603-619.
| Anhang IV |
C.3 Süsswasseralgen und Cyanobakterien: Wachstumsinhibitionstest
1. Methode
Diese Methode entspricht der Prüfrichtlinie OECD TG 201 (2006) (1).
1.1 Einleitung
Die Prüfmethoden werden regelmäßig unter Berücksichtigung des wissenschaftlichen Fortschritts überarbeitet und aktualisiert. Prüfmethode C.3 musste unter Einbeziehung weiterer Arten und unter Berücksichtigung der Anforderungen an die Risikobewertung und Klassifizierung chemischer Stoffe überarbeitet werden. Die Überarbeitung wurde auf der Grundlage umfassender praktischer Erfahrungen sowie des wissenschaftlichen Fortschritts im Zusammenhang mit Untersuchungen zur Algentoxizität und der weit reichenden Anwendung entsprechender Rechtsvorschriften seit Annahme der ursprünglichen Fassung vorgenommen.
1.2 Begriffsbestimmungen
Im Zusammenhang mit dieser Prüfmethode werden die folgenden Begriffsbestimmungen zugrunde gelegt und folgende Abkürzungen verwendet:
Biomasse: Trockengewicht lebenden Materials einer Population bezogen auf ein vorgegebenes Volumen (z.B. mg Algen/Liter Testlösung); gewöhnlich wird "Biomasse" definiert als Masse; in Verbindung mit dieser Prüfung wird der Begriff allerdings zur Bezeichnung einer Masse pro Volumen verwendet. Typischerweise werden Surrogate für die betreffende Biomasse (z.B. Zellgehalt oder Fluoreszenz) gemessen; entsprechend bezieht sich der Begriff "Biomasse" auch auf diese Surrogatparameter.
Variationskoeffizient: ein dimensionsloses Mag für die Veränderlichkeit eines Parameters; definiert als Verhältnis der Standardabweichung vom Mittelwert; der Variationskoeffizient kann auch als Prozentwert ausgedrückt werden. Der mittlere Variationskoeffizient der durchschnittlichen spezifischen Wachstumsrate sollte bei Wiederholungs-Kontrollkulturen wie folgt berechnet werden:
ECx: Konzentration der Prüfsubstanz aufgelöst im Prüfmedium, bei der das Wachstum des Testorganismus binnen einer bestimmten Expositionsdauer (die ausdrücklich anzugeben ist, wenn die Expositionsdauer nicht mit der vollständigen oder gewöhnlichen Dauer der Prüfung übereinstimmt) um x % (z.B. 50 %) abnimmt; um eindeutig anzugeben, ob ein EC-Wert aus der Wachstumsrate (growth rate) oder aus dem Zellertrag (yield) abgeleitet wurde, werden die Kurzbezeichnungen "Er C" und "Ey C" verwendet.
Nährmedium: gesamtes synthetisches Kulturmedium, in dem die zu prüfenden Algen wachsen, wenn sie der Prüfsubstanz ausgesetzt werden; die Prüfsubstanz wird im Allgemeinen im Prüfinedium aufgelöst.
Wachstumsrate (durchschnittliche spezifische Wachstumsrate): logarithmische Zunahme der Biomasse während der Expositionsdauer.
Niedrigste Konzentration mit beobachteter Wirkung (LOEC): niedrigste geprüfte Konzentration, bei der beobachtet wurde, dass die Substanz binnen einer bestimmten Expositionsdauer gegenüber der Kontrollprobe eine statistisch signifikante Wachstumsreduzierung bewirkt (bei p < 0,05); allerdings müssen sämtliche Testkonzentrationen über der LOEC schädliche Folgen haben, die mindestens den bei der LOEC beobachteten schädlichen Folgen gleichwertig sind. Wenn diese beiden Bedingungen nicht erfüllt werden können, ist umfassend darzulegen, warum die LOEC (und entsprechend die NOEC) gewählt wurde.
Höchste geprüfte Konzentration ohne beobachtete schädliche Wirkung (NOEC): Testkonzentration unmittelbar unterhalb der LOEC.
Reaktionsvariable: Variable für die geschätzte Toxizität, abgeleitet aus beliebigen gemessenen Parametern zur Beschreibung der Biomasse durch verschiedene Berechnungsmethoden; bei dieser Methode sind Wachstumsrate und Zellertrag Reaktionsvariablen, die aus der direkten Messung der Biomasse oder einer Messung eines der genannten Surrogate abgeleitet werden.
Spezifische Wachstumsrate: Reaktionsvariable, die sich aus dem Quotienten der Differenz der natürlichen Logarithmen eines beobachteten Parameters (bei dieser Prüfinethode die Biomasse) und dem betreffenden Zeitraum ergibt.
Zellertrag: Wert einer Messvariable am Ende der Expositionsdauer abzüglich des Wertes der Messvariablen zu Beginn der Expositionsdauer als Maß für die Zunahme der Biomasse während der Prüfung.
1.3 Anwendbarkeit der Prüfmethode
Diese Prüfmethode ist am einfachsten bei wasserlöslichen Substanzen anzuwenden, die unter den Prüfbedingungen voraussichtlich im Wasser gelöst bleiben. Zum Prüfen von flüchtigen, stark adsorbierenden, farbigen und schlecht in Wasser löslichen Substanzen sowie von Substanzen, die sich auf die Verfügbarkeit von Nährstoffen oder Mineralien im Prüfmedium auswirken können, sind am beschriebenen Verfahren unter Umständen gewisse Änderungen vorzunehmen (z.B. die Herstellung eines geschlossenen Systems oder eine besondere Vorbereitung der Prüfgefäße). Hinweise zu verschiedenen Änderungen sind den Quellen (2), (3) und (4) zu entnehmen.
1.4 Prinzip der Prüfmethode
Mit dieser Prüfung sollen die Auswirkungen einer Substanz auf das Wachstum von Süßwassermikroalgen und/oder Cyanobakterien bestimmt werden. Exponentiell wachsende Testorganismen werden über einen Zeitraum von im Allgemeinen 72 Stunden in Batch-Kulturen der Prüfsubstanz ausgesetzt. Trotz der verhältnismäßig kurzen Testdauer können die Auswirkungen über mehrere Generationen beurteilt werden.
Die Systemreaktion besteht in der Verringerung des Wachstums einer Reihe von Algenkulturen (Versuchseinheiten), die einer Prüfsubstanz in unterschiedlichen Konzentrationen ausgesetzt wurden. Diese Reaktion wird dann in Abhängigkeit von der Expositionskonzentration gegenüber dem durchschnittlichen Wachstum in zur Wiederholung verwendeten Kontrollkulturen bewertet, die der betreffenden Substanz nicht ausgesetzt waren. Um die Systemreaktion auf toxische Auswirkungen (optimale Empfindlichkeit) umfassend beschreiben zu können, wird ein unbegrenztes exponentielles Wachstum der Kulturen bei hinreichender Ernährung und kontinuierlicher Beleuchtung über einen ausreichenden Zeitraum ermöglicht, damit anschließend die Verringerung der spezifischen Wachstumsrate gemessen werden kann.
Wachstum und Wachstumshemmung werden durch zeitabhängige Messung der Biomasse der Algen bestimmt. Die Biomasse der Algen wird als Trockengewicht pro Volumen ausgedrückt (z.B. in mg Algen/Liter Testlösung). Das Trockengewicht ist jedoch schwer zu messen; daher werden Surrogatparameter verwendet. Häufigster Surrogatparameter ist der Zellgehalt. Weitere Surrogatparameter sind das Zellvolumen, die Fluoreszenz, die optische Dichte usw. Ein Faktor für die Umrechnung zwischen dem gemessenen Surrogatparameter und der Biomasse sollte bekannt sein.
Der Endpunkt des Tests ist die Wachstumshemmung, ausgedrückt als logarithmische Zunahme der Biomasse (durchschnittliche spezifische Wachstumsrate) während der Expositionsdauer. Aus den in einer Reihe von Testlösungen erfassten durchschnittlichen spezifischen Wachstumsraten wird die Konzentration bestimmt, bei der sich eine spezifizierte Hemmung der Wachstumsrate von x % (z.B. 50 %) ergibt; diese Konzentration wird als ErC. bezeichnet (z.B. Er C50).
Bei der Anwendung dieser Methode im Rahmen des Gemeinschaftsrechts sollte die Ergebnisberechnung aus den im nachfolgenden Abschnitt 2.2 dargelegten Gründen auf einer durchschnittlichen spezifischen Wachstumsrate basieren. Bei dieser Prüfmethode wird als weitere Reaktionsvariable der Zellertrag verwendet; diese Variable kann erforderlich sein, damit in manchen Ländern bestimmte maßgebliche Rechtsvorschriften erfüllt werden. Der Zellenrag wird definiert als Biomasse am Ende der Expositionsdauer abzüglich der Biomasse zu Beginn der Expositionsdauer. Aus dem in einer Reihe von Testlösungen erfassten Zellertrag wird die Konzentration berechnet, bei der sich eine spezifizierte Hemmung des Zellertrags (z.B. um 50 %) ergibt; diese Konzentration wird als Ey Cx angegeben (z.B. Ey C50).
Außerdem können die niedrigste Konzentration mit beobachteter Wirkung (LOEC) und die höchste geprüfte Konzentration ohne beobachtete schädliche Wirkung (NOEC) statistisch bestimmt werden.
1.5 Informationen zur Prüfsubstanz
Informationen zur Prüfsubstanz, die bei der Bestimmung der Prüfbedingungen hilfreich sein könnten, sind z.B. die Strukturformel, die Reinheit, die Lichtbeständigkeit, die Beständigkeit unter den Prüfbedingungen, das Lichtabsorptionsverhalten, pKa und die Ergebnisse von Transformationsstudien (u. a. die Ergebnisse von Studien zur biologischen Abbaubarkeit in Wasser).
Die Wasserlöslichkeit, der oktanol/Wasser-Verteilungskoeffizient (P") und der Dampfdruck der Prüfsubstanz sollten bekannt sein, eine validierte Methode zur Quantifizierung der Substanz in den Testlösungen mit bekannten Wiederfmdungsraten und mit bekannter Nachweisgrenze sollte verfügbar sein.
1.6 Referenzsubstanz
Um das Prüfverfahren zu testen, können Referenzsubstanzen wie z.B. das im internationalen Ringtest (4) verwendete 3,5-Dichlorphenol geprüft werden. Für Grünalgen kann auch Kaliumdichromat als Referenzsubstanz verwendet werden. Nach Möglichkeit sollten Referenzsubstanzen mindestens zweimal jährlich getestet werden.
1.7 Validität des Tests
Damit ein Test als gültig gewertet werden kann, sollten die folgenden Kriterien erfüllt sein:
1.8 Beschreibung der Methode
1.8.1 Apparatur
Testgefäße und sonstige Apparaturen, die mit den Testlösungen in Berührung kommen, sollten vollständig aus Glas oder einem sonstigen chemisch inerten Material bestehen. Die Komponenten sollten gründlich gespült werden, um sicherzustellen, dass keine organischen oder anorganischen Verunreinigungen das Algenwachstum oder die Zusammensetzung der Testlösungen beeinträchtigen können.
Als Prüfgefäße kommen im Allgemeinen Glaskolben mit Abmessungen in Betracht, die während des Tests ein hinreichendes Kulturvolumen und einen hinreichenden CO2-Transfer aus der Umgebungsluft gewährleisten (siehe Abschnitt 1.8.9 Absatz 2). Das Flüssigkeitsvolumen muss für analytische Bestimmungen hinreichend sein (siehe Abschnitt 1.8.11 Absatz 5).
Außerdem werden unter Umständen die folgenden Geräte benötigt:
1.8.2 Testorganismen
Für die Tests können mehrere Arten frei treibender Mikroalgen und Cyanobakterien verwendet werden. Die in Anlage 1 genannten Stämme haben sich für das in dieser Prüfmethode beschriebene Testverfahren als geeignet erwiesen.
Wenn sonstige Arten verwendet werden, sollte der betreffende Stamm und/oder Ursprung angegeben werden. Außerdem ist sicherzustellen, dass das exponentielle Wachstum der ausgewählten Testalgen während der gesamten Testdauer unter den jeweiligen Bedingungen aufrechterhalten werden kann.
1.8.3 Nälirmedium
Als Nährmedien werden wahlweise das OECD-Medium und das AAP-Medium empfohlen. Die Zusammensetzungen dieser Medien sind Anlage 2 zu entnehmen. Beide Medien haben unterschiedliche pH-Ausgangswerte und unterschiedliche Pufferkapazitäten (zur Regulierung des pH-Anstiegs). Daher können die Testergebnisse je nach verwendetem Medium unterschiedlich ausfallen; dies gilt insbesondere für die Prüfung ionisierender Substanzen.
Für bestimmte Zwecke muss unter Umständen das Nährmedium modifiziert werden (z.B. für die Prüfung von Metallen und Chelatbildnern oder für Prüfungen bei unterschiedlichen pH-Werten). Die Verwendung modifizierter Nährmedien ist im Einzelnen zu erläutern und zu begründen (3)(4).
1.8.4 Biomasse-Ausgangskonzentration
Die Ausgangsbiomasse der Prüfkulturen muss bei allen Prüfkulturen identisch und so gering sein, dass während der Inkubationsdauer ein exponentielles Wachstum erzielt werden kann, ohne eine Erschöpfung des Nährmediums befürchten zu müssen. Die Ausgangsbiomasse sollte höchstens 0,5 mg/l Trockengewicht betragen. Folgende Ausgangs-Zellkonzentrationen werden empfohlen:
| Pseudokirchneriella subcapitata: | 5 x 103-104 | Zellen/ml |
| Desmodesmus subspicatus | 2-5 x 103 | Zellen/ml |
| Navicula pelliculosa | 104 | Zellen/ml |
| Anabaena flosaquae | 104 | Zellen/ml |
| Synechococcus leopoliensis | 5 x 104-105 | Zellen/ml |
1.8.5 Konzentrationen der Prüfsubstanz
Der Konzentrationsbereich, in dem wahrscheinlich Auswirkungen auftreten, kann aufgrund von Tests zur Bestimmung des Konzentrationsbereichs ermittelt werden. Für den definitiven Test sollten mindestens fünf Konzentrationen in einer geometrischen Reihe mit einem Faktor von höchstens 3,2 ausgewählt werden. Bei Prüfsubstanzen mit einer flacheren Konzentrations-Reaktionskurve kann ein höherer Faktor gerechtfertigt sein. Die Konzentrationsreihen sollten vorzugsweise einen Bereich abdecken, in dem das Algenwachstum um 5-75 % gehemmt wird.
1.8.6 Wiederholungen und Kontrollen
Das Prüfprotokoll sollte für jede Testkonzentration drei Wiederholungen vorsehen. Wenn die NOEC nicht bestimmt werden muss, kann das Prüfprotokoll dahingehend geändert werden, dass die Anzahl der Konzentrationen erhöht und die Anzahl der Wiederholungen verringert wird. Es müssen mindestens drei Kontrollwiederholungen durchgeführt werden, im Idealfall die doppelte Anzahl der Wiederholungen für jede Testkonzentration.
Für analytische Bestimmungen der Prüfsubstanzkonzentrationen kann eine eigene Reihe von Testlösungen hergestellt werden (siehe Abschnitt 1.8.11 Absätze 4 und 6).
Wenn zur Auflösung der Prüfsubstanz ein Lösungsmittel verwendet wird, sind in das Prüfprotokoll weitere Kontrollen mit dem Lösungsmittel in der Konzentration einzubeziehen, die auch in den Prüfkulturen verwendet wird.
1.8.7 Herstellung der Impfkultur
Um eine Anpassung der Testalgen an die Testbedingungen zu ermöglichen und um sicherzustellen, dass sich die Algen in der Phase des exponentiellen Wachstums befinden, wenn sie zur Impfung der Testlösungen verwendet werden, wird 2-4 Tage vor Testbeginn im Prüfmedium eine Impfkultur hergestellt. Die Algenbiomasse sollte so angepasst werden, dass ein exponentielles Wachstum der Impfkultur bis zum Testbeginn erfolgen kann. Die Impfkultur ist unter den gleichen Bedingungen wie die Prüfkulturen zu inkubieren. Die Zunahme der Biomasse der Impfkultur ist zu messen, um sicherzustellen, dass das Wachstum unter den gegebenen Kulturbedingungen für den jeweiligen Teststamm im normalen Bereich liegt. In Anlage 3 wird ein Beispiel für ein Verfahren zur Herstellung einer Algenkultur beschrieben. Um gleichzeitige Zellteilungen während des Tests zu vermeiden, kann unter Umständen ein zweiter Schritt zur Vermehrung der Impfkultur erforderlich sein.
1.8.8 Herstellung der Testlösungen
Alle Testlösungen müssen das Nährmedium und die Ausgangsbiomasse der Testalgen in denselben Konzentrationen enthalten. Die Testlösungen in den ausgewählten Konzentrationen werden gewöhnlich durch Mischen einer Stammlösung der Prüfsubstanz mit dem Nährmedium und der Impfkultur hergestellt. Stammlösungen werden im Allgemeinen durch Auflösung der betreffenden Substanz im Prüfinedium hergestellt.
Wenn Substanzen mit geringer Wasserlöslichkeit zum Prüfinedium hinzugegeben werden sollen, können Lösungsmittel (z.B. Aceton, t-Butyl-Alkohol und Dimethylformamid) als Träger verwendet werden (2)(3). Die Lösungsmittelkonzentration sollte höchstens 100 µl/l betragen, und für alle Kulturen der Testreihen (einschließlich der Kontrollkulturen) sollte die gleiche Konzentration verwendet werden.
1.8.9 Inkubation
Die Testgefäße sind mit luftdurchlässigen Stopfen zu versehen. Anschließend werden die Gefäße geschüttelt und in die Kulturapparatur gebracht. Während des Tests müssen die Algen suspendiert bleiben, und der CO2-Transfer muss erleichtert werden. Dazu sollten die Gefäße ständig geschüttelt oder umgerührt werden. Die Temperatur der Kulturen sollte bei einer Toleranz von ± 2 °C ständig auf 21 bis 24 °C geregelt werden. Bei anderen als den in Anlage 1 genannten Arten (z.B. bei tropischen Arten) sind unter Umständen höhere Temperaturen angemessen, wenn die Validitätskriterien erfüllt sind. Es wird empfohlen, die Kolben zufällig verteilt und täglich neu in den Inkubator einzusetzen.
Der pH-Wert des Kontrollmediums darf während des Tests höchstens um 1, 5 Einheiten ansteigen. Bei Metallen und Verbindungen, die bei pH-Werten etwa im Bereich der im Test verwendeten pH-Werte teilweise ionisieren, muss die pH-Verschiebung möglicherweise begrenzt werden, um reproduzierbare und gut definierte Ergebnisse zu erhalten. Eine Verschiebung des pH-Werts um < 0,5 ist technisch möglich und kann hervorgerufen werden, um einen angemessenen CO2-Transfer aus der Umgebungsluft in die Testlösung zu erzielen (z.B. durch stärkeres Schütteln). Eine weitere Möglichkeit besteht in der Verringerung des CO2-Bedarfs durch Verringerung der Ausgangsbiomasse oder in einer Verkürzung der Testdauer.
Die Oberfläche, auf der die Kulturen inkubiert werden, sollte kontinuierlich und gleichförmig fluoreszierend beleuchtet werden (z.B. mit den Lichtfarben kaltweiß ("coolwhite') oder Tageslicht (dayligli ). Algen- und Cyanobakterienstämme stellen unterschiedliche Anforderungen an die Lichtverhältnisse. Die Lichtintensität sollte entsprechend den verwendeten Testorganismen gewählt werden. Bei den empfohlenen Grünalgenarten ist die Lichtintensität auf der Höhe der Testlösungen im Bereich 60-120 µE × m-2 . s-1 zu wählen (bei Messung im photosynthetisch wirksamen Spektralbereich von 400-700 nm mit einem geeigneten Rezeptor). Einige Arten, insbesondere Anabaena flosaquae, wachsen bei geringeren Lichtintensitäten und können durch größere Lichtintensitäten beschädigt werden. Bei diesen Arten sollte die durchschnittliche Lichtintensität im Bereich 40-60 µE × m-2 . s-1 liegen. (Bei in Lux kalibrierten Lichtmessgeräten entspricht der Bereich 4.440-8.880 lx für die Lichtfarbe "coolwhite" etwa der empfohlenen Lichtintensität von 60-120 µE × m-2 . s-1) Die Lichtintensität darf um höchstens ± 15 % von der durchschnittlichen Lichtintensität über dem Inkubationsbereich abweichen.
1.8.10 Testdauer
Im Allgemeinen dauert der Test 72 Stunden. Allerdings sind auch kürzere oder längere Testzeiten möglich, sofern alle in Abschnitt 1.7 genannten Validitätskriterien eingehalten werden.
1.8.11 Messungen und analytische Bestimmungen
Die Algenbiomasse in den einzelnen Kolben wird während der Testdauer mindestens einmal täglich überprüft. Wenn die Messungen an kleinen Volumina vorgenommen werden, die aus der Testlösung pipettiert wurden, sollten die betreffenden Volumina nicht ersetzt werden.
Die Biomasse wird durch manuelle Zählung der Zellen unter dem Mikroskop oder mit einem elektronischen Teilchenzähler (Einstellung auf Zellgehalt und/oder Biovolumen) ermittelt. Alternative Verfahren wie z.B. die Messung mit einem Durchflusszytometer, in vitro und/oder in vivo durchgeführte Chlrorophyll-Fluoreszenzmessungen (6)(7), oder Messungen der optischen Dichte kommen in Betracht, wenn in dem für den jeweiligen Test maßgeblichen Biomassebereich eine befriedigende Korrelation mit der Biomasse nachgewiesen werden kann.
Der pH-Wert der Lösungen wird am Anfang und am Ende des Tests gemessen.
Wenn ein Analyseverfahren zur Bestimmung der Prüfsubstanz im maßgeblichen Konzentrationsbereich verfügbar ist, sollten die Testlösungen analysiert werden, um die Ausgangskonzentrationen und die Aufrechterhaltung der Expositionskonzentrationen während der Tests zu prüfen bzw. sicherzustellen.
Die Analyse der Prüfsubstanzkonzentration am Anfang und am Ende des Tests bei einer niedrigen und einer hohen Testkonzentration sowie bei einer Konzentration im zu erwartenden EC50-Bereich kann hinreichend sein, wenn anzunehmen ist, dass die Expositionskonzentrationen während des Tests um weniger als 20 % von den Nennwerten abweichen. Wenn die Konzentrationen eher nicht im Bereich von 80-120 % der Nennkonzentrationen bleiben werden, wird die Analyse sämtlicher Testkonzentrationen am Anfang und am Ende der Tests empfohlen. Bei flüchtigen, instabilen oder stark adsorbierenden Prüfsubstanzen werden während der Expositionsdauer weitere Probenahmen zur Durchführung von Analysen in Abständen von jeweils 24 Stunden empfohlen, um den Verlust der Prüfsubstanz besser bestimmen zu können. Bei diesen Substanzen werden zusätzliche Wiederholungen benötigt. In jedem Fall müssen die Prüfsubstanzkonzentrationen für alle Testkonzentrationen bei den Wiederholungen jeweils nur in einem Gefäß (bzw. in dem Gesamtinhalt aller Gefäße einer Wiederholung) bestimmt werden.
Ausdrücklich für die Analyse von Expositionskonzentrationen während der Testdauer hergestellte Prüfmedien sollten auf die gleiche Weise behandelt werden, wie die für die Tests verwendeten Medien, d. h. die Medien sollten mit Algen geimpft und unter identischen Bedingungen inkubiert werden. Wenn eine Analyse der gelösten Prüfsubstanz erforderlich ist, müssen die Algen unter Umständen vom Medium getrennt werden. Die Trennung sollte vorzugsweise durch Zentrifugierung mit niedriger Beschleunigungskraft erfolgen, die eben noch hinreichend für die Ausfällung der Algen ist.
Wenn nachgewiesen wird, dass die Konzentration der Prüfsubstanz während der gesamten Testdauer in befriedigender Weise im Bereich von ± 20 % der Nennkonzentration oder der gemessenen Ausgangskonzentration aufrechterhalten werden konnte, können die Ergebnisse auch ausgehend von den Nennwerten bm von den gemessenen Ausgangswerten analysiert werden. Wenn die Abweichung von der Nennkonzentration oder von der gemessenen Ausgangskonzentration mehr als ± 20 % beträgt, sollte bei der Analyse der Ergebnisse von der geometrischen mittleren Konzentration während der Expositionsdauer oder von Modellen ausgegangen werden, welche den Rückgang der Prüfsubstanzkonzentration beschreiben (3)(8).
Der Test zur Hemmung des Algenwachstum ist ein dynamischeres Prüfsystem als die meisten sonstigen aquatischen Toxizitätstests mit kürzerer Testdauer. Entsprechend sind die tatsächlichen Expositionskonzentrationen unter Umständen schwer zu bestimmen; dies gilt besonders für adsorbierende Substanzen, die bei niedrigen Konzentrationen getestet werden. In diesen Fällen bedeutet das Verschwinden der Substanz aus der Lösung infolge der Adsorption in die zunehmende Algenbiomasse nicht, dass das Testsystem die Substanz "verliert". Bei der Analyse des Testergebnisses sollte geprüft werden, ob ein Rückgang der Prüfsubstanzkonzentration im Laufe des Tests mit einer Abnahme der Wachstumshemmung einhergeht. Wenn dies der Fall ist, kann der Einsatz eines geeigneten Modells zur Beschreibung des Rückgangs der Prüfsubstanzkonzentration (8) in Betracht gezogen werden. Ansonsten ist es unter Umständen nicht angemessen, bei der Analyse der Ergebnisse von den Ausgangskonzentrationen (Nennkonzentrationen oder gemessenen Konzentrationen) auszugehen.
1.8.12 Sonstige Beobachtungen
Durch Beobachtung unter dem Mikroskop sollte sichergestellt werden, dass die Impfkultur normal und ges- und wirkt; außerdem können unter dem Mikroskop am Ende des Tests gegebenenfalls Auffälligkeiten an den Algen festgestellt werden (die z.B. auf die Einwirkung der Prüfsubstanz zurückzuführen sein könnten).
1.8.13 Limit-Test
Unter gewissen Umständen, z.B. wenn ein vorläufiger Test darauf hindeutet, dass die Prüfsubstanz bei Konzentrationen bis zu 100 mg ×l-1 bzw bis zur Löslichkeitsgrenze im Prüfinedium (maßgeblich ist die jeweils niedrigere Konzentration) keine toxische Wirkung hat, kann ein Limit-Test durchgeführt werden, in dem die Reaktionen einer Kontrollgruppe und einer Behandlungsgruppe (100 mg ×l-1 bzw. eine mit der Löslichkeitsgrenze identische Konzentration) verglichen werden. Es wird nachdrücklich empfohlen, diese Tests durch Analysen der Expositionskonzentration zu verifizieren. Alle oben beschriebenen Testbedingungen und Validitätskriterien gelten für Limit-Tests; allerdings sollten mindestens sechs Wiederholungen mit behandelten Proben durchgeführt werden. Die Reaktionsvariablen der Kontrollgruppe und der Behandlungsgruppe können mit einem statistischen Test zum Vergleich der Mittelwerte analysiert werden (z.B. mit einem Student-Test). Wenn die Varianzen der beiden Gruppen nicht übereinstimmen, sollte ein für nicht identische Varianzen angepasster t-Test (Student-Test) durchgeführt werden.
1.8.14 Änderung für stark kolorierte Substanzen
Die Bestrahlung (Lichtintensität) sollte im obersten Ende des bei dieser Testmethode vorgeschriebenen Bereichs liegen: 120 µE m-2 s-1 oder höher.
Der Strahlengang sollte durch eine Verringerung des Volumens der Testlösungen (im Bereich von 5-25 ml) verkürzt werden.
Es sollte (z.B. durch mäßiges Schütteln) für ausreichende Bewegung gesorgt werden, um zu gewährleisten, dass die Algen häufig einer hohen Bestrahlung an der Oberfläche der Kultur exponiert sind.
2. Daten
2.1 Grafische Darstellung der Wachstumskurven
Die in den Prüfgefäßen enthaltene Biomasse kann in Einheiten des für die Messung verwendeten Surrogatparameters ausgedrückt werden (z.B. als Zellgehalt oder Fluoreszenz).
Die geschätzte Biomassekonzentration der Prüfkulturen und der Kontrollkulturen ist zusammen mit den mindestens zu jeder vollen Stunde zu erfassenden Konzentrationen des Prüfmaterials und den Messzeitpunkten in Tabellen zusammenzustellen und für die Erstellung von Wachstumskurven zu verwenden. In diesem ersten Stadium können sowohl die logarithmischen Skalen als auch die linearen Skalen hilfreich sein; während der Testdauer sind jedoch die logarithmischen Skalen vorgeschrieben, die im Allgemeinen eine bessere Darstellung von Abweichungen der Wachstumsstrukturen ermöglichen. Bei exponentiellem Wachstum ergibt sich bezogen auf eine logarithmische Skala eine Gerade; die Steigung der Gerade gibt Aufschluss über die jeweilige Wachstumsrate.
Anhand der Darstellungen ist sicherzustellen, dass die Kontrollkulturen während der gesamten Testdauer tatsächlich mit der erwarteten Geschwindigkeit exponentiell wachsen. Alle Datenpunkte sowie die Darstellungen sind kritisch zu überprüfen, und Ausgangsdaten und Verfahren sind auf mögliche Fehler zu kontrollieren. Insbesondere sind alle Datenpunkte zu prüfen, die aufgrund eines systematischen Fehlers abzuweichen scheinen. Wenn Verfahrensfehler zweifelsfrei festgestellt und/oder als sehr wahrscheinlich betrachtet werden können, wird der betreffende Datenpunkt als Ausreißer gekennzeichnet und nicht in die anschließende statistische Analyse einbezogen. (Eine Algenkonzentration von null in einem der beiden bzw. der drei für die Wiederholungen verwendeten Gefäße kann darauf hindeuten, dass das Gefäß nicht ordnungsgemäß geimpft oder nicht angemessen gereinigt wurde.) Die Gründe für den Ausschluss eines als Ausreißer eingestuften Datenpunkts sind im Prüfbericht genau anzugeben. Als Begründungen werden ausschließlich (selten) Verfahrensfehler, nicht aber einfach ungenaue Messungen anerkannt. Statistische Verfahren zur Bestimmung von Ausreißern sind bei dieser Art von Problemen von begrenztem Nutzen und können die Beurteilung durch Fachleute nicht ersetzen. Ausreißer (die als solche gekennzeichnet wurden) sollten vorzugsweise in den Datenpunkten verbleiben, die später in grafischen oder tabellarischen Darstellungen verwendet werden.
2.2 Reaktionsvariablen
Mit der Prüfung sollen die Auswirkungen der Prüfsubstanz auf das Algenwachstum bestimmt werden. Diese Prüfmethode beschreibt zwei Reaktionsvariablen, da in den Mitgliedsländern unterschiedliche Präferenzen und rechtliche Anforderungen bestehen. Damit die Testergebnisse in allen Mitgliedsländern anerkannt werden können, sollten die Auswirkungen mit Hilfe der beiden im Folgenden beschriebenen Reaktionsvariablen a) und b) beurteilt werden.
Bei der Anwendung dieser Methode im Rahmen des Gemeinschaftsrechts sollte die Ergebnisberechnung aus den nachfolgend dargelegten Gründen auf einer durchschnittlichen spezifischen Wachstumsrate basieren. Es wird darauf hingewiesen, dass die mit diesen beiden Reaktionsvariablen berechneten Toxizitätswerte nicht vergleichbar sind; der entsprechende Unterschied muss bei der Verwendung der Testergebnisse berücksichtigt werden. Die mit der durchschnittlichen spezifischen Wachstumsrate (Er Cx) berechneten Werte für ECx werden im Allgemeinen höher sein als die anhand des Zellertrags (Ey Cx) ermittelten Werte, wenn die für diese Testmethode vorgesehenen Bedingungen eingehalten werden; dies ist auf die unterschiedliche mathematische Grundlage der beiden Berechnungsverfahren zurückzuführen. Die auftretenden Unterschiede sollten jedoch nicht als Anzeichen für eine unterschiedliche Empfindlichkeit der beiden Reaktionsvariablen betrachtet werden; beide Werte sind einfach mathematisch verschieden. Das Konzept der durchschnittlichen spezifischen Wachstumsrate beruht auf dem im Allgemeinen exponentiellen Verlauf des Algenwachstums bei nicht begrenzten Kulturen, bei denen die Toxizität aufgrund der Auswirkungen auf die Wachstumsrate geschätzt wird, ohne jedoch von der absoluten Höhe der jeweiligen Wachstumsrate der Kontrollprobe, von der Steigung der Konzentration-Reaktionskurve oder von der Testdauer abhängig zu sein. Auf der Reaktionsvariable Zellertrag beruhende Ergebnisse hingegen hängen von allen übrigen genannten Variablen ab. Ey Cx ist von der spezifischen Wachstumsrate der in den einzelnen Tests verwendeten Algenarten sowie von der maximalen spezifischen Wachstumsrate abhängig, die je nach Art sowie sogar zwischen den einzelnen Algenstämmen unterschiedlich sein kann. Diese Reaktionsvariable sollte nicht verwendet werden, um die Empfindlichkeit von Algenarten oder auch nur verschiedener Algenstämme gegenüber Giftstoffen zu vergleichen. Die Verwendung der durchschnittlichen spezifischen Wachstumsrate zur Schätzung der Toxizität wird in der Wissenschaft bevorzugt; bei dieser Prüfmethode werden jedoch auch Toxizitätsschätzungen aufgrund des Zellertrags berücksichtigt, um den derzeitigen rechtlichen Anforderungen einiger Länder Rechnung zu tragen.
2.2.1 Durchschnittliche Wachstumsrate
Die durchschnittliche spezifische Wachstumsrate in einem bestimmten Zeitraum wird mit Hilfe der folgenden Formel als logarithmische Zunahme der Biomasse für die Gefäße mit den verschiedenen Kontrollproben und mit den behandelten Proben berechnet:
![]()
wobei
µi-j: durchschnittliche spezifische Wachstumsrate zwischen Zeitpunkt i und Zeitpunkt j;
Xi: Biomasse zum Zeitpunkt i;
Xj: Biomasse zum Zeitpunkt j.
Für jede Behandlungsgruppe und für jede Kontrollgruppe sind die mittlere Wachstumsrate sowie die Varianzschätzungen zu berechnen.
Die durchschnittliche spezifische Wachstumsrate wird über die gesamte Testdauer (im Allgemeinen Tage 0-3) berechnet; dabei ist eher als ein gemessener Ausgangswert die geimpfte Nennbiomasse als Ausgangswert anzunehmen, da mit diesem Wert gewöhnlich eine höhere Genauigkeit erzielt wird. Wenn die zur Messung der Biomasse verwendete Ausrüstung eine hinreichend genaue Bestimmung der Biomasse mit dem geringen Inokulum zulässt (z.B. ein Durchflusszytometer), kann die gemessene Konzentration der Ausgangsbiomasse angenommen werden. Außerdem ist während der gesamten Testdauer (Tage 0-1, 1-2 und 2-3) die abschnittsbezogene Wachstumsrate als tägliche spezifische Wachstumsrate zu ermitteln und zu prüfen, ob das Wachstum der Kontrollgruppe konstant bleibt (siehe Gültigkeitskriterien, Abschnitt 1.7). Eine spezifische Wachstumsrate, die an einem bestimmten Tag signifikant niedriger ist als die durchschnittliche spezifische Gesamtwachstumsrate, kann Anzeichen für eine "Lag"-Phase sein. Bei den Kontrollkulturen kann eine "Lag"-Phase minimiert und praktisch ausgeschlossen werden, wenn die Vorkultur in geeigneter Weise vermehrt wird; bei den behandelten Kulturen hingegen kann eine "Lag"-Phase Anzeichen für eine Erholung nach anfänglicher toxischer Belastung oder Anzeichen für eine geringere Belastung infolge eines Verlustes der Prüfsubstanz (u. a. durch Sorption in die Algenbiomasse) nach der anfänglichen Behandlung sein. Entsprechend kann die abschnittsbezogene Wachstumsrate bewertet werden, um die Auswirkungen der Prüfsubstanz während der Expositionsdauer zu beurteilen. Erhebliche Unterschiede zwischen der abschnittsbezogenen Wachstumsrate und der durchschnittlichen Wachstumsrate deuten auf Abweichungen vom konstanten exponentiellen Wachstum hin und erfordern eine genaue Überprüfung der Wachstumskurven.
Die prozentuale Hemmung der Wachstumsrate der einzelnen Wiederholungen mit behandelten Proben ist mit folgender Formel zu berechnen:
![]()
wobei
%Iy: Hemmung der durchschnittlichen spezifischen Wachstumsrate in Prozent;
µC: mittlere durchschnittliche spezifische Wachstumsrate (µ) der Kontrollgruppe;
µT: durchschnittliche spezifische Wachstumsrate der Wiederholung mit einer behandelten Probe.
Wenn die Testlösungen mit Lösungsmitteln hergestellt werden, sollten eher die Kontrolllösungen mit dem Lösungsmittel als die Kontrolllösungen ohne das Lösungsmittel zur Berechnung der prozentualen Hemmung verwendet werden.
2.2.2 Zellertrag
Der Zellertrag wird für alle Gefäße mit Kontrolllösungen und behandelten Lösungen als Biomasse bei Ende des Tests abzüglich der Biomasse am Anfang des Tests berechnet. Für die Testkonzentrationen und für die Kontrolllösungen ist jeweils ein mittlerer Zellertrag zu berechnen; die Varianzen sind jeweils zu schätzen. Die prozentuale Hemmung des Zellertrags (%Iy) kann für die Wiederholungen mit behandelten Proben wie folgt berechnet werden:
![]()
wobei
%Iy: prozentuale Hemmung des Zellertrags;
YC: mittlerer Zellertrag der Kontrollgruppe;
Y,: Zellertrag der Wiederholung mit einer behandelten Probe.
2.3 Grafische Darstellung der Konzentrations-Reaktionskurve
Die prozentuale Hemmung ist bezogen auf den Logarithmus der Prüfsubstanzkonzentration grafisch darzustellen und sorgfältig zu überprüfen; dabei sind alle Datenpunkte zu verwerfen, die in der ersten Phase als Ausreißer identifiziert wurden. Die Datenpunkte sind nach visueller oder rechnergestützter Interpolation zu einer gleichmäßigen Kurve zu verbinden, um einen ersten Eindruck von der Beziehung zwischen den verschiedenen Konzentrationen und Reaktionen zu erhalten. Anschließend ist mit einer differenzierteren Methode (vorzugsweise mit einer rechnergestützten statistischen Methode) fortzufahren. Abhängig von der beabsichtigten Verwendung der Daten, der Qualität (Genauigkeit) und dem Umfang der Daten sowie von der Verfügbarkeit von Werkzeugen zur Analyse der Daten kann (gelegentlich durchaus berechtigt) entschieden werden, die Datenanalyse in diesem Stadium zu beenden und einfach die wesentlichen Werte für EC" und EC" (und/oder EC") aus der nach visueller Interpolation erstellten Kurve abzulesen (siehe auch folgender Abschnitt zu stimulierenden Auswirkungen). Die folgenden Gründe können dafür sprechen, von der Verwendung einer statistischen Methode abzusehen:
2.4 Statistische Verfahren
Ziel ist die Ermittlung einer quantitativen Konzentrations-Reaktionsbeziehung durch Regressionsanalyse. Im Anschluss an eine linearisierte Transformation der Reaktionsdaten (z.B. in Einheiten nach dem Probit-, Logit- oder Weibull-Modell) (9) kann eine gewichtete lineare Regression vorgenommen werden; nichtlineare Regressionsverfahren, mit denen die unvermeidlichen Unregelmäßigkeiten der Daten und Abweichungen von gleichförmigen Verteilungen besser verarbeitet werden können, werden jedoch bevorzugt. Gegen null bzw. gegen die vollständige Hemmung können diese Unregelmäßigkeiten durch die Transformation vergrößert werden und die Analyse beeinträchtigen (9). Es wird darauf hingewiesen, dass Standard-Analysmethoden mit Probit-, Logit- oder Weibull-Transformationen für quantitative Daten (z.B. Mortalität oder Überlebensraten) vorgesehen sind und zur Verwendung in Verbindung mit Wachstums- oder Biomassedaten entsprechend modifiziert werden müssen. Spezifische Verfahren zur Bestimmung von ECx-Werten aus kontinuierlichen Daten sind den Quellen (10)(11) and (12) zu entnehmen. In Anlage 4 wird der Einsatz nichtlinearer Regressionsanalysen näher erläutert.
Für jede zu analysierende Reaktionsvariable sind aufgrund der Konzentration-Reaktionsbeziehung Punktschätzungen für die EC. -Werte zu ermitteln. Nach Möglichkeit sollten für jede Schätzung die 95%-Konfidenzintervalle bestimmt werden. Die Qualität der Übereinstimmung der Reaktionsdaten mit dem Regressionsmodell sollte grafisch oder statistisch bewertet werden. Die Regressionsanalyse sollte mit den Reaktionen der einzelnen Wiederholungen (und nicht mit den Mittelwerten der Behandlungsgruppe) durchgeführt werden. Wenn eine nichtlineare Kurvenanpassung jedoch schwierig oder wegen zu großer Streuung der Daten nicht möglich ist, kann die Regression auch bezogen auf die Mittelwerte der Gruppe vorgenommen werden, um so auf einfache Weise die Auswirkungen erwarteter Ausreißer zu reduzieren. Wenn von dieser Möglichkeit Gebrauch gemacht wird, sollte dies im Prüfbericht als Abweichung vom normalen Verfahren vermerkt und darauf hingewiesen werden, dass mit Kurvenanpassungen der einzelnen Wiederholungen kein befriedigendes Ergebnis erzielt wurde.
Schätzwerte für EC" und für die Konfidenzintervalle können auch durch lineare Interpolation mit einem Bootstrapping-Algorithmus (13) erzielt werden, wenn die verfügbaren Regressionsmodelleimethoden für die betreffenden Daten nicht geeignet sind.
Um die LOEC und entsprechend die NOEC zu schätzen sowie um die Auswirkungen der Prüfsubstanzen auf die Wachstumsrate zu ermitteln, müssen die Mittelwerte der behandelten Proben mit Varianzanalyseverfahren (ANOVA) verglichen werden. Der Mittelwert der einzelnen Konzentrationen ist dann mit einer geeigneten Methode zur Durchführung von Mehrfachvergleichen bzw. zur Durchführung von Trendtests mit dem Mittelwert der Kontrollgruppe zu vergleichen. Dunnett- und Williams-Tests können hilfreich sein (14)(15)(16)(17)(18). Die ANOVA-Annahme der Varianzhomogenität muss einer Überprüfung unterzogen werden. Die entsprechende Bewertung kann anhand einer grafischen Darstellung oder aufgrund eines formalen Tests vorgenommen werden (18). Geeignet sind Levene- und Bartlett-Tests. Wenn die Annahme der Varianzhomogenität nicht erfüllt ist, kann gelegentlich eine Korrektur durch logarithmische Datentransformation erfolgen. Bei außerordentlicher Varianzheterogenität, die durch Transformation nicht korrigiert werden kann, sollten Analysen durch Methoden wie z.B. Jonckheere-Trendtests (Stepdown) erwogen werden. Weitere Hinweise zur Bestimmung von NOEC-Werten sind Quelle (12) zu entnehmen.
Aufgrund neuer Forschungsergebnisse wird empfohlen, das Konzept der NOEC aufzugeben und durch Punktschätzungen von EC. -Werten zu ersetzen, die durch Regression ermittelt wurden. Für diesen Algentest wurde noch kein geeigneter Wert für x definiert. Ein Bereich von 10 bis 20 % scheint geeignet (abhängig von der auswählten Reaktionsvariable); vorzugsweise sollten sowohl EC" als auch EC" protokolliert werden.
2.5 Wachstumsstimulation
Gelegentlich wird bei niedrigen Konzentrationen eine Wachstumsstimulation (negative Hemmung) beobachtet. Diese Wachstumsstimulation kann entweder auf eine Hormesis ("to)iische Stimulation") oder darauf zurückzuführen sein, dass im verwendeten Minimalmedium mit dem zu prüfenden Material zusätzliche stimulierende Wachstumsfaktoren zum Tragen kommen. Die Zugabe anorganischer Nährstoffe sollte keinerlei unmittelbare Auswirkungen haben, weil das Prüfmedium während der gesamten Testdauer ohnehin überschüssige Nährstoffe enthalten sollte. Eine Stimulation mit niedriger Dosierung kann bei Berechnungen von EC" gewöhnlich übergangen werden, wenn die Stimulation nicht ungewöhnlich stark ist. Bei ungewöhnlich starker Stimulation oder wenn ein ECx Wert für einen niedrigen x-Wert berechnet werden soll, sind möglicherweise jedoch besondere Verfahrensweisen erforderlich. Die Löschung von Stimulationsreaktionen aus der Datenanalyse sollte nach Möglichkeit vermieden werden, und wenn die verfügbare Software zur Kurvenanpassung nicht in der Lage ist, geringere Stimulationen zu verarbeiten, kann eine lineare Interpolation mit einem Bootstrapping-Algorithmus vorgenommen werden. Bei ungewöhnlich starker Stimulation kann der Einsatz eines Hormesis-Modells erwogen werden (19).
2.6 Nicht toxische Wachstumshemmung
Licht absorbierende Testmaterialien können zu einer Verringerung der Wachstumsrate führen, weil die Abdunklung den Anteil des verfügbaren Lichts verringert. Diese physikalischen Auswirkungen sollten durch geeignete Modifikation der Testbedingungen von toxischen Auswirkungen unterschieden und getrennt protokolliert werden. Entsprechende Hinweise sind den Quellen (2) und (3) zu entnehmen.
3. Abschlussbericht
3.1 Prüfbericht
Der Prüfbericht muss folgende Informationen enthalten: Prüfsubstanz:
Im Test verwendete Art:
Prüfbedingungen:
Ergebnisse:
4. LITERATUR
1. OECD TG 201 (2006) Freshwater Alga and Cyanobacteria, Growth Inhibition Test.
2. ISO 1998: Water quality - Guidance for algal growth inhibition tests with poorly soluble materials, volatile compounds, metals and waster water. ISO/DIS 14442.
3. OECD 2000: Guidance Document an Aquatic Toxicity Testing of Difficult Substances and mixtures. Environmental Health and Safety Publications. Series an Testing and Assessment, no. 23.
4. ISO 1998: Water quality - Sampling - Part 16: General Guidance for Biotesting. ISO 5667-16.
5. ISO 1993: Water quality - Algal growth inhibition test. ISO 8692.
6. Mayer, P., Cuhel, R. and Nyholm, N. (1997). A simple in vitro fluorescence method for biomass measurements in algal growth inhibition tests. Water Research 31, S. 2525-2531.
7. Slovacey, R.E. and Hanna, P.J. In vivo fluorescence determinations of phytoplancton chlorophyll, Limnology & Oceanography 22,5 (1977), S. 919-925.
8. Simpson, S.L., Roland, M.G.E., Stauber, J.L. and Batley, G.E. (2003) Effect of declining toxicant concentrations an algal bioassay endpoints. Environ. Toxicol. Chem 22, S. 2073-2079.
9. Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, S. 713-718.
10. Nyholm, N. Serensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, S. 157-167.
11. Bruce, R.D., and Versteeg, D.J. (1992). A statistical procedure for modelling continuous toxicity data. Env. Toxicol. Chem. 11, S. 1485-1494.
12. OECD. (2004). Guidance Document an Statistical Analysis of Ecotoxicity Data.
13. Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment center Technical Report 05-88. USEPA, Duluth, MN.
14. Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50, S. 1096-1121.
15. Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics 20: 482-491.
16. Wilhams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, S. 103-117.
17. Wilhams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28, S. 510-531.
18. Draper, NK and Smith, H. (1981). Apphed Regression Analysis, second edition. Wiley, New York.
19. Brain P. and Cousens R. (1989). An equation to describe doseresponses where there is stimulation of growth at low doses. Weed Research 29, S. 93-96.
| Stämme, die sich für den Test als geeignet erwiesen haben | Anlage 1 |
Grünalgen
Kieselalgen
Cyanobakterien
Herkunft der Stämme
Die empfohlenen Stämme sind als artenreine Algenkulturen aus folgenden Sammlungen verfügbar (in alphabetischer Reihenfolge):
ATCC: American Type Culture Collection
10801 University Boulevard
Manassas, Virginia 20110-2209
USA
CCAP, Culture Collection of Algae and Protozoa
Institute of Freshwater Ecology,
Windermere Laboratory
Far Sawrey, Amblerside
Cumbria
LA22 OLP
UK
SAG: Sammlung Algenkulturen
Pflanzenphysiologisches Institut
Universität Göttingen
Nikolausberger Weg 18
3400 Göttingen
DEUTSCHLAND
UTEX Culture Collection of Algae
Section of Molecular, Cellular and Developmental Biology
School of Biological Sciences
the University of Texas at Austin
Austin, Texas 78712
USA
Aussehen und Merkmale der empfohlenen Arten
| P. subcapitata | D. subspicatus | N. pelliculosa | A. flosaquae | S. leopoliensis | |
| Aussehen | Gekrümmte und gedrehte einzelne Zellen | Oval, meist einzelne Zellen | Stäbchen | Ketten ovaler Zellen | Stäbchen |
| Größe (L x B) µm | 8-14 x 2-3 | 7-15 x 3-12 | 7,1 x 3,7 | 4,5 x 3 | 6 x 1 |
| Zellvolumen (µm3/Zelle) | 40-60 1 | 60-80 1 | 40-50 1 | 30-40 1 | 2,5 2 |
| Zelltrockengewicht (mg/Zelle) | 2-3 x 10-5 | 3-4 x 10-5 | 3-4 x 10-5 | 1-2 x 10-5 | 2-3 x 10-9 |
| Wachstumsrate 3(Tag-1) | 1,5-1,7 | 1,2-1,5 | 1,4 | 1,1-1,4 | 2,0-2,4 |
| (1) Gemessen mit einem elektronischen Teilchenzähler. (2) Aus der Größe berechnet. (3) Am häufigsten beobachtete Wachstumsrate im OECD-Medium bei einer Lichtintensität von ca. 70 µE × m-2 × s-1 und einer Temperatur von 21 °C. | |||||
Spezifische Empfehlungen zur Kultivierung und zur Handhabung der für den Test empfohlenen Arten
Pseudokirchneriella subcapitata und Desmodesmus subspicatus
Diese Grünalgen sind in unterschiedlichen Kulturmedien im Allgemeinen leicht zu kultivieren. Informationen zu geeigneten Medien sind von den Stellen zu beziehen, welche die Sammlungen unterhalten. Die Zellen kommen im Allgemeinen einzeln vor, mit einem elektronischen Teilchenzähler oder unter einem Mikroskop kann die Zelldichte problemlos bestimmt werden.
Anabaena flosaquae
Für Stammkulturen können verschiedene Nährmedien verwendet werden. Insbesondere muss vermieden werden, dass die Batch-Kultur bei der Erneuerung das Stadium des exponentiellen Wachstums überschreitet; eine Rückgewinnung wäre ansonsten schwierig.
Anabaena flosaquae bildet Ansammlungen verschachtelter Zellketten. Die Größe dieser Ansammlungen kann je nach Kulturbedingungen unterschiedlich sein. Unter Umständen müssen diese Ansammlungen getrennt werden, wenn die Biomasse unter dem Mikroskop bzw. mit einem elektronischen Teilchenzähler bestimmt werden soll.
Zum Trennen der Ketten mit dem Ziel, Schwankungen der Zählergebnisse zu verringern, können Teilproben einer Ultraschallbehandlung unterzogen werden. Eine unnötig lange Ultraschallbehandlung zum Trennen der Ketten in kürzere Stücke kann die Zellen zerstören. Intensität und Dauer der Ultraschallbehandlung müssen bei allen Behandlungen identisch sein.
Mit dem Hämozytometer sind hinreichend Zellen zu zählen (mindestens 400), um auftretende Schwankungen korrigieren zu können und die Zuverlässigkeit der mikroskopischen Dichtebestimmungen zu erhöhen.
Zur Bestimmung des Gesamtvolumens der Anabaena-Zellen nach dem Trennen der Zellketten durch vorsichtige Ultraschallbehandlung kann ein elektronischer Teilchenzähler verwendet werden. Die Ultraschallenergie ist so anzupassen, dass die Zellen nicht beschädigt werden.
Mit einem Wirbelmischer oder durch ein ähnliches geeignetes Verfahren ist sicherzustellen, dass die zur Impfung der Prüfgefäße verwendete Algensuspension gut durchgemischt und homogen beschaffen ist.
Die Prüfgefäße sind auf einen Schütteltisch (mit kreisförmiger oder gerader Schüttelbewegung) zu bringen, der mit etwa 150 Umdrehungen pro Minute bewegt wird. Alternativ kann bei Anabaena die Verklumpungstendenz auch durch intermittierendes Schütteln verringert werden. Wenn eine Verklumpung auftritt, ist darauf zu achten, dass repräsentative Proben für Messungen der Biomasse vorliegen. Unter Umständen müssen die Gefäße vor der Probenahme heftig geschüttelt werden, um die Algenklumpen aufzulösen.
Synechococcus leopoliensis
Für Stammkulturen können verschiedene Nährmedien verwendet werden. Informationen zu geeigneten Medien sind von den Stellen zu beziehen, welche die Sammlungen unterhalten.
Synechococcus leopoliensis wächst in einzelnen stäbchenförmigen Zellen. Die Zellen sind sehr klein; dies erschwert Messungen der Biomasse durch Zählungen unter dem Mikroskop. Elektronische Teilchenzähler, die für die Zählung von Teilchen mit einer Größe von bis zu etwa 1 µm ausgelegt sind, können hilfreich sein. Invitro-Fluoreszenzmessungen kommen ebenfalls in Betracht.
Navicula pelliculosa
Für Stammkulturen können verschiedene Nährmedien verwendet werden. Informationen zu geeigneten Medien sind von den Stellen zu beziehen, welche die Sammlungen unterhalten. Das Medium muss Silikat enthalten.
Navicula pelliculosa kann unter bestimmten Wachstumsbedingungen Ansammlungen bilden. Wegen der Bildung von Lipiden neigen die Algenzellen gelegentlich zur Akkumulierung im Oberflächenfilm. In diesem Fall sind besondere Verfahrensweisen erforderlich, wenn repräsentative Teilproben zur Bestimmung der Biomasse genommen werden sollen. Unter Umständen ist ein heftiges Schütteln z.B. mit einem Wirbelmischer erforderlich.
| Nährmedien | Anlage 2 |
Für die Tests kann eines der beiden folgenden Nährmedien verwendet werden: OECD-Medium: Originalmedium gemäß OECD TG 201 sowie gemäß ISO 8692 US. EPA-Medium AAP, auch gemäß ASTM.
Bei der Herstellung dieser Medien sollten chemische Stoffe in Reagenzien- oder Analysequalität und entionisiertes Wasser verwendet werden.
Zusammensetzung des AAP-Mediums (US. EPA) und des Mediums gemäß OECD TG 201:
| Bestandteil | EPA | OECD | ||
| mg/l | mm | mg/l | mm | |
| NaHCO3 | 15,0 | 0,179 | 50,0 | 0,595 |
| NaNO3 | 25,5 | 0,300 | ||
| NH4 Cl | 15,0 | 0,280 | ||
| MgCl2×6(H2O) | 12,16 | 0,0598 | 12,0 | 0,0590 |
| CaCl22(H2O) | 4,41 | 0,0300 | 18,0 | 0,122 |
| MgSO47(H2O) | 14,6 | 0,0592 | 15,0 | 0,0609 |
| K2HPO4 | 1,044 | 0,00599 | ||
| KH2 PO4 | 1,60 | 0,00919 | ||
| FeCl3 ×6(H2O) | 0,160 | 0,000591 | 0,0640 | 0,000237 |
| Na2 EDTA× 2(H2O) | 0,300 | 0,000806 | 0,100 | 0,000269 * |
| H3BO3 | 0,186 | 0,00300 | 0,185 | 0,00299 |
| MnCl2×4(H2O) | 0,415 | 0,00201 | 0,415 | 0,00210 |
| ZnC12 | 0,00327 | 0,000024 | 0,00300 | 0,0000220 |
| CoCl2×6(H2O) | 0,00143 | 0,000006 | 0,00150 | 0,00000630 |
| Na2MnO4 .2(H2O) | 0,00726 | 0,000030 | 0,00700 | 0,0000289 |
| CuCl2 ×2(H2O) | 0,000012 | 0,00000007 | 0,00001 | 0,00000006 |
| pH | 7,5 | 8,1 | ||
| *) Das Molverhältnis von EDTA zu Eisen ist geringfügig größer als eins. Daher kann das Eisen nicht ausfällen, und die Chelatbildung von Schwermetallionen wird minimiert. | ||||
Im Test mit der Kieselalge Navicula pelliculosa sind beide Medien mit Na2SiO3×9H2O aufzufüllen, bis eine Konzentration von 1,4 mg Sill erreicht ist.
Der pH-Wert des Mediums wird ermittelt, wenn sich das Carbonatsystem des Mediums und der Partialdruck des CO2 in der Umgebungsluft im Gleichgewicht befinden. Die ungefähre Beziehung zwischen dem pH-Wert bei 25 °C und der molaren Bicarbonat-Konzentration lässt sich mit folgender Formel berechnen:
pHeq = 11,30 + log [HCO3]
Bei 15 mg NaHCO3 pHeq = 7,5 (U.S. EPA-Medium) und 50 mg NaHCO3/l, pHeq = 8,1 (OECD-Medium).
Stoffliche Zusammensetzung des Prüfmediums
| Element | EPA mg/l | OECD mg/l |
| C | 2,144 | 7,148 |
| N | 4,202 | 3,927 |
| P | 0,186 | 0,285 |
| K | 0,469 | 0,459 |
| Na | 11,044 | 13,704 |
| Ca | 1,202 | 4,905 |
| Mg | 2,909 | 2,913 |
| Fe | 0,033 | 0,017 |
| Mn | 0,115 | 0,115 |
Herstellung des OECD-Mediums
| Nährstoff | Konzentration in der Stammlösung |
| Stammlösung 1: Makronährstoffe | |
| NH4Cl | 1,5 g×l-1 |
| MgCl2 ×6H2O | 1,2 g×l-1 |
| CaCl2×2H2O | 1,8 g×l-1 |
| MgSO4×7H2O | 1,5 g×l-1 |
| KH2PO4 | 0,16 g×l-1 |
| Stammlösung 2: Eisen | |
| FeCl36 H2O | 64 mg g×l-1 |
| Na2 EDTA×2H2O | 100 mg×l-1 |
| Stammlösung 3: Spurenelemente | |
| H3BO3 | 185 mg×l-1 |
| MnCl2×4H2O | 415 mg×l-1 |
| ZnCl2 | 3 mg×l-1 |
| CoCl26×H2O | 1,5 mg×l-1 |
| CuCl22×H2O | 0,01 mg×l-1 |
| Na2MoO4×2H2O | 7 mg×l-1 |
| Stammlösung 4: Bicarbonat | |
| NaHCO3 Na2SiO3 9H2O | 50 g×l-1 |
Die Stammlösungen sind durch Membranfiltration (mittlerer Porendurchmesser 0,2 µm) oder durch Autoklavieren (120 °C, 15 min) zu sterilisieren. Anschließend werden die Lösungen bei einer Temperatur von 4 °C dunkel gelagert.
Die Stammlösungen 2 und 4 dürfen nicht autoklaviert werden, sondern sind durch Membranfiltration zu sterilisieren. Zum Herstellen des Nährmediums wird eine geeignete Menge der Stammlösungen 1-4 wie folgt zu Wasser hinzugegeben: Zu 500 ml sterilisiertem Wasser werden folgende Mengen hinzugegeben:
- 10 ml Stammlösung 1
- 1 ml Stammlösung 2
- 1 ml Stammlösung 3
- 1 ml Stammlösung 4
Anschließend wird sterilisiertes Wasser bis zu einem Volumen von 1.000 ml hinzugegeben.
Danach muss hinreichend Zeit zur Herstellung eines Gleichgewichts zwischen dem Medium und dem CO2-Gehalt der Umgebungsluft belassen werden; wenn erforderlich, ist das Nährmedium einige Stunden mit steriler gefilterter Luft zu sprudeln.
Herstellung des AAP-Mediums
A1.1. Von den Stammlösungen aus A1.2.1-A1.2.7 ist jeweils 1 ml zu etwa 900 ml entionisiertem oder destilliertem Wasser hinzuzugeben; anschließend wird die Flüssigkeit bis zu einem Volumen von 11 aufgefüllt.
A1.2. Stammlösungen mit Makronährstoffen werden hergestellt, indem die folgenden Stoffmengen jeweils zu 500 ml entionisiertem oder destilliertem Wasser hinzugegeben werden. Die Reagenzien A1.2.1, A1.2.2, A1.2.3 und A1.2.4 können zu einer einzigen Stammlösung kombiniert werden.
A1.2.1. NaNO3- 12,750 g.
A1.2.2. MgCl26H2O - 6,082 g.
A1.2.3. CaCl22H2O - 2,205 g.
A1.2.4. Micronutrient Stock Solution - (siehe A1.3).
A1.2.5. MgSO47H2O - 7,350 g.
A1.2.6. K2 HPO4- 0,522 g.
A1.2.7. NaHCO3- 7,500 g.
A1.2.8. Na2 SiO3.9H2O - Siehe Hinweis A1.1.
Hinweis A1.1. Ausschließlich für im Test eingesetzte Kieselalgen-Arten zu verwenden; kann unmittelbar (202,4 mg) hinzugegeben oder mittelbar durch Auffüllen mit einer Stammlösung bis zum Erreichen einer Endkonzentration von 20 mg/l Si im Medium hinzugegeben werden.
A1.3. Die Mikronährstoff-Lösung wird hergestellt, indem folgende Mengen in 500 ml entionisiertem oder destilliertem Wasser aufgelöst werden:
A1.3.1. H3BO3- 92,760 mg.
A1.3.2. MnCl24H2O - 207,690 mg.
A1.3.3. ZnCl2- 1,635 mg.
A1.3.4. FeCl3.6H2O - 79,880 mg.
A1.3.5. CoCl2-6H2O - 0,714 mg.
A1.3.6. Na2 MoO42H2O - 3,630 mg.
A1.3.7. CuCl22H2O - 0,006 mg.
A1.3.8. Na2 EDTA 2H2O - 150,000 mg.
[Dinatrium(ethylendinitril)tetraacetat]
A1.3.9. Na2 SeO4. 5H2O - 0,005 mg Siehe Hinweis A1.2.
Hinweis A1.2: Darf ausschließlich im Medium für Stammkulturen mit Kieselalgen-Arten verwendet werden.
A1.4. Der pH-Wert ist auf 7,5 ± 0,1 einzustellen (mit 0,1 N oder 1,0 N NaOH oder HCl).
A1.5. Wenn ein Teilchenzähler verwendet werden soll, wird das Medium durch einen 0,22-µm-Membranfilter in ein steriles Gefäß gefiltert; ansonsten erfolgt die Filtration in ein steriles Gefäß durch einen 0,45-µm-Filter.
A1.6. Das Medium ist bis zur Verwendung bei einer Temperatur von etwa 4 °C dunkel zu lagern.
| Beispiel eines Verfahrens zur Kultivierung der Algen | Anlage 3 |
Allgemeine Bemerkungen
Durch die Kultivierung nach dem folgenden Verfahren sollen Algenkulturen für Toxizitätsprüfungen hergestellt werden.
Um sicherzustellen, dass die Algenkulturen nicht mit Bakterien verunreinigt sind, sind geeignete Methoden zu verwenden. Wenngleich u. U. axenische Kulturen erwünscht sein mögen, sind doch Algenkulturen mit einer einzigen Alge herzustellen und zu verwenden.
Sämtliche Schritte sind unter sterilen Bedingungen auszuführen, um Verunreinigungen mit Bakterien und anderen Algen zu vermeiden.
Ausrüstung und Materialien
Siehe Prüfmethode: Apparatur.
Verfahren zur Herstellung von Algenkulturen Herstellung der Nährlösungen (Medien)
Alle Nährsalze des Mediums werden als konzentrierte Stammlösungen hergestellt und dunkel und kühl gelagert. Diese Lösungen werden durch Filtration oder durch Autoklavieren hergestellt.
Das Medium wird durch Zugabe der jeweils erforderlichen Menge der Stammlösung zu sterilem destilliertem Wasser hergestellt; dabei ist darauf zu achten, dass keine Verunreinigungen entstehen können. Bei festen Medien ist 0,8 % Agar hinzuzugeben.
Stammkultur
Die Stammkulturen bestehen aus kleinen Algenkulturen, die regelmäßig in frische Medien übertragen werden und als Ausgangs-Prüfmaterial fungieren. Wenn die Kulturen nicht regelmäßig verwendet werden, sind die Kulturen auf Agarröhrchen (Slopes) abzustreichen. Diese werden anschließend mindestens einmal alle zwei Monate in frische Medien gebracht.
Die Stammkulturen werden in konischen Kolben mit dem jeweils geeigneten Medium kultiviert (Volumen etwa 100 ml). Wenn die Algen bei 20 °C unter ständiger Beleuchtung inkubiert werden, ist eine wöchentliche Überimpfung erforderlich.
Dabei ist von der "alten" Kultur mit sterilen Pipetten so viel in einen Kolben mit frischem Medium zu geben, dass bei schnell wachsenden Arten die Ausgangskonzentration etwa 100 mal geringer ist als die Konzentration der alten Kultur.
Die Wachstumsrate einer Art kann anhand der Wachstumskurve bestimmt werden. Wenn diese bekannt ist, kann die Dichte geschätzt werden, bei der die Kultur in das neue Medium gegeben werden sollte. Die Dichteschätzung muss erfolgen, bevor die betreffende Kultur in die Absterbephase gelangt.
Vorkultur
Mit der Vorkultur sollen Algen zur Impfung der Prüfkulturen hergestellt werden. Die Vorkultur wird unter den Prüfbedingungen inkubiert und noch während der Phase des exponentiellen Wachstums verwendet (in der Regel nach einer Inkubationsdauer von 2 bis 4 Tagen). Algenkulturen mit deformierten oder anomalen Zellen sind zu verwerfen.
| Datenanalyse durch nichtlineare Regression | Anlage 4 |
Allgemeine Bemerkungen
In den Algentests und in sonstigen Tests zur Ermittlung des mikrobiologischen Wachstums - des Wachstums einer Biomasse - stellt die Reaktion naturgemäß eine kontinuierliche oder metrische Variable dar. eine Prozessrate, wenn von einer Wachstumsrate ausgegangen wird, und ein zeitbezogenes Integral, wenn die Biomasse zugrunde gelegt wird. Beide Parameter werden auf die entsprechende mittlere Reaktion von zur Wiederholung verwendeten nicht behandelten Kontrollproben bezogen, die am stärksten auf die jeweils gegebenen Bedingungen reagieren; bei Algentests sind Licht und Temperatur die Hauptfaktoren. Das System kann verteilt oder homogen sein, und die Biomasse kann als kontinuierlicher Parameter betrachtet werden; einzelne Zellen brauchen nicht berücksichtigt zu werden. Die Varianzverteilung des Reaktionstyps dieser Systeme hängt ausschließlich von den Versuchsbedingungen ab (die in der Regel durch logarithmischnormale oder normale Fehlerverteilungen gekennzeichnet sind). In dieser Hinsicht besteht ein Unterschied gegenüber typischen Reaktionen in Bioassays mit quantitativen Daten, bei denen die Toleranz (typischerweise mit binomischer Verteilung) der einzelnen Organismen häufig als maßgebliche Varianzkomponente betrachtet wird. Die Kontrollreaktionen liegen hier bei Null oder im Bereich des Hintergrundwerts.
Im einfachsten Fall nimmt die normalisierte oder relative Reaktion r gleichmäßig von 1 (Hemmung Null) bis 0 (vollständige Hemmung) ab. Sämtliche Reaktionen sind mit einem Fehler verbunden, und offensichtlich negative Hemmungen können rechnerisch ausschließlich das Ergebnis zufälliger Fehler sein.
Regressionsanalyse Modelle
Durch eine Regressionsanalyse soll die Konzentrations-Reaktionskurve als mathematische Regressionsfunktion Y = f (C) bzw. häufiger als F (Z) beschrieben werden; dabei ist Z = log C. Umgekehrt ermöglicht C = f-1(Y) die Berechnung von ECx-Werten einschließlich EC50, EC10 und EC20 sowie der jeweiligen 95- %-Konfidenzintervalle. Verschiedene einfache mathematische Funktionen haben sich als geeignet zur Beschreibung von Konzentrations-Reaktionsbeziehungen in Tests zur Ermittlung der Hemmung des Algenwachstums erwiesen. Zu diesen Formeln zählen die logistische Gleichung, die nicht symmetrische Weibull-Verteilung und die logarithmische Normalverteilung als Sigma-Kurven, bei denen sich jeweils ein asymptotischer Verlauf gegen eins bei C -> 0 bzw. gegen Null bei C -> unendlich ergibt.
Der Einsatz kontinuierlicher Schwellenwert-Funktionsmodelle (z.B. des Kooijman-Modells der "Hemmung des Populationswachstums" (Kooijman u. a. 1996) wurde kürzlich als Alternative zu asymptotischen Modellen vorgeschlagen. Dieses Modell geht davon aus, dass bei Konzentrationen unter einem bestimmten Schwellenwert ECO+ keine Auswirkungen mehr gegeben sind; dieser Schwellenwert wird mit Hilfe einer einfachen kontinuierlichen Funktion mit undifferenziertem Ausgangspunkt durch Extrapolation der Reaktions-Konzentrationsbeziehung so geschätzt, dass die Konzentrationsachse geschnitten wird.
Die Analyse kann in einer einfachen Minimierung der Restquadratsummen (bei Annahme einer konstanten Varianz) oder der gewichteten Quadrate (bei Ausgleich einer Varianzheterogenität) bestehen.
Verfahren
Das Verfahren kann wie folgt beschrieben werden: Eine geeignete Funktionsgleichung Y = f (C) wird gewählt und durch nichtlineare Regression an die Daten angepasst. Vorzugsweise sind die Messungen der einzelnen Kolben und nicht die Mittelwerte der Wiederholungen zu verwenden, um möglichst viele Informationen aus den Daten zu gewinnen. Bei einer hohen Varianz haben praktische Erfahrungen hingegen gezeigt, dass die Mittelwerte der Wiederholungen eine sicherere mathematische Schätzung ermöglichen, die weniger von systematischen Fehlern bei den Daten als von den einzelnen ermittelten Datenpunkten abhängt.
Die angepasste Kurve und die gemessenen Daten werden grafisch dargestellt; anschließend ist zu prüfen, ob die Kurve in angemessener Weise angepasst wurde. Eine Analyse der Restwerte könnte für diesen Zweck besonders hilfreich sein. Wenn die gewählte Funktionsbeziehung zur Anpassung der Konzentrations-Reaktionskurve die Kurve nicht vollständig beschreibt oder einen wesentlichen Teil der Kurve wie z.B. die Reaktion bei niedrigen Konzentrationen nicht gut beschreibt, ist eine andere Option zur Kurvenanpassung zu wählen (z.B. eine nicht symmetrische Kurve wie etwa die Weibull-Funktion anstelle einer symmetrischen Kurve). Negative Hemmungen können z.B. in Verbindung mit der logarithmischen Normalverteilung problematisch sein und ebenfalls den Einsatz einer alternativen Regressionsfunktion erfordern. Es wird nicht empfohlen, diesen negativen Werten einen Wert von Null oder einen kleinen positiven Wert zuzuweisen, weil ansonsten die Fehlerverteilung beeinträchtigt werden könnte. Angemessen sind unter Umständen getrennte Kurvenanpassungen für bestimmte Bereiche der Kurve (z.B. für den Bereich mit der niedrigen Hemmung, wenn EClow x-Werte geschätzt werden sollen). Aus der angepassten Formel sind (mit der Umkehrfunktion C = f-1(Y)) charakteristische Schätzwerte für ECx zu ermitteln und mindestens für ECso sowie für einen oder zwei Werte von EClow x zu protokollieren. Praktische Erfahrungen haben gezeigt, dass die Genauigkeit der Algentests im Allgemeinen eine angemessen exakte Schätzung der Konzentration bei einer Hemmung von etwa 10 % ermöglicht, wenn hinreichende Datenpunkte verfügbar sind (sofern nicht eine Unregelmäßigkeit in Form einer Stimulation auch bei niedrigen Konzentrationen gegeben ist). Die Genauigkeit einer EC20-Schätzung ist häufig beträchtlich größer als die Genauigkeit geschätzter EC10-Werte, weil die EC20-Werte gewöhnlich im annähernd linearen Bereich der zentralen Konzentration-Reaktionskurve vorgenommen werden. Gelegentlich ist die Beurteilung von EC10-Werten wegen der Wachstumsstimulation problematisch. Werte für EClo sind also im Allgemeinen mit hinreichender Genauigkeit zu erhalten; in jedem Fall empfiehlt sich jedoch, auch die EC20-Werte zu erfassen.
Gewichtungsfaktoren
Die Versuchsvarianz ist nicht grundsätzlich konstant und beinhaltet typischerweise eine proportionale Komponente; daher wird vorzugsweise regelmäßig auch eine gewichtete Regression vorgenommen. Die Gewichtungsfaktoren für diese Analysen werden im Allgemeinen als umgekehrt proportional zur Varianz angenommen:
Wi = 1/Var(ri)
Viele Regressionsprogramme beinhalten die Option zur Durchführung gewichteter Regressionsanalysen mit Gewichtungsfaktoren, die aus einer Tabelle ausgewählt werden können. Die Normalisierung der Gewichtungsfaktoren kann auf bequeme Weise erfolgen, indem die Gewichtungsfaktoren so mit n/Σwi(n = Anzahl der Datenpunkte) multipliziert werden, dass sich die Summe 1 ergibt.
Normalisierung der Reaktionen
Die Normalisierung durch die mittlere Kontrollreaktion bringt einige grundsätzliche Probleme mit sich und bedingt eine eher komplizierte Varianzstruktur. Mit der Division der Reaktionen durch die mittlere Kontrollreaktion zur Ermittlung der prozentualen Hemmung wird ein weiterer Fehler eingeführt, der auf den in den Mittelwerten der Kontrollen enthaltenen Fehler zurückzuführen ist. Wenn dieser Fehler nicht vernachlässigbar gering ist, sind Gewichtungsfaktoren in Verbindung mit der Regression und mit den Konfidenzintervallen bezogen auf die Kovarianz der Kontrollen zu korrigieren (17). Eine hohe Genauigkeit der geschätzten Mittelwerte der Kontrollreaktionen ist wichtig, um die Gesamtvarianz der relativen Reaktion zu minimieren. Diese Varianz lässt sich wie folgt beschreiben.
(Dabei steht das tiefgestellte i für die Konzentration i und die tiefgestellte 0 für die Kontrollen.)
Yi = relative Reaktion = ri/ro = 1 - I = f (Ci)
bei gegebener Varianz:
Var (Yi) = Var (ri/ro) ≡(∂ Yi/ ∂ri)2 - Var(ri) + (∂Yi/ ∂ro)2-Var (ro)
entsprechend gilt
(∂Yi / ∂ ri) = 1/ro und (∂ Yi/ ∂ro) = ri/ ro2
bei normal verteilten Daten und mi und mo Wiederholungen:
Var(ri) = σ2 /mi
für die Gesamtvarianz der relativen Reaktion Yi gilt somit:
Var(Yi) = σ2/(ro2 mi) + ri2 σ2 / ro4 mo
Der Fehler im Mittelwert der Kontrollen ist umgekehrt proportional zur Quadratwurzel der Anzahl der durchschnittlichen Kontrollwiederholungen; gelegentlich ist die Einbeziehung historischer Daten gerechtfertigt, um den Fehler auf diese Weise erheblich zu reduzieren. Ein alternatives Verfahren besteht darin, die Daten nicht zu normalisieren und die absoluten Reaktionen einschließlich der Daten der Kontrollreaktionen nicht anzupassen, sondern den Kontroll-Reaktionswert als zusätzlichen Parameter einzuführen, der durch nichtlineare Regression anzupassen ist. In Verbindung mit einer normalen Regressionsgleichung mit zwei Parametern erfordert diese Methode die Anpassung von drei Parametern; daher sind mehr Datenpunkte erforderlich als bei der nichtlinearen Regression von Daten, die mit einer vordefinierten Kontrollreaktion normalisiert werden.
Umkehrung der Konfidenzintervalle
Die Berechnung nichtlinearer Konfidenzintervalle durch Schätzung mit der Umkehrfunktion gestaltet sich eher komplex und ist in den üblichen statistischen Computer-Programmen als reguläre Option nicht enthalten. Ungefähre Konfidenzintervalle können mit Standardprogrammen zur Berechnung nichtlinearer Regressionen durch Neu-Parametrisierung ermittelt werden (Bruce und Versteeg, 1992); dabei wird die mathematische Formel mit den angestrebten Punktschätzungen (z.B. EC" und EC50) als zu schätzenden Parametern neu entwickelt. Vorausgesetzt wird I = f (a, ß, Konzentration. Die Definitionsbeziehungen f (a, ß, EC10) = 0,1 und f (a, ß, EC50) = 0,5 werden verwendet, um f (a, ß, Konzentration) durch eine äquivalente Funktion g (EC10, EC50, Konzentration) zu ersetzen.
Für eine direktere Berechnung (Andersen u. a. 1998) kann die ursprüngliche Formel verwendet und eine Taylor-Expansion um die Mittelwerte für r; und r0 angenommen werden.
In letzter Zeit werden zunehmend auch Methoden mit Bootstrapping-Algorithmen verwendet. Diese Methoden beruhen auf Schätzungen einer empirischen Varianzverteilung ausgehend von den gemessenen Daten und von häufigen Stichproben unter Einsatz eines Zufalls-Nummerngenerators.
Literatur
Kooijman, S.A.L.M.; Hanstveit, A.O.; Nyholm, N. (1996): Noeffect concentrations in algal growth inhibition tests. Water Research 30, S. 1625-1632.
Bruce, R.D. and Versteeg, D.J.(1992) A Statistical Procedure for Modelling Continuous Ecotoxicity Data. Env. Toxicol. Chem. 11, S. 1485-1494.
Andersen, J.S., Holst, H., Spliid, H., Andersen, H., Baun, A. & Nyholm, N. (1998): Continuous ecotoxicological data evaluated relative to a control response. Journal of Agricultural, Biological and Environmental Statistics 3, S. 405-420.
-------------------------------------------------------------------------
| Anhang V |
C.25. Aerobe Mineralisation in Oberflächenwasser - Simulationstest zur biologischen Abbaubarkeit
1. Methode
Diese Methode entspricht der Prüfrichtlinie OECD TG 309 (2004) (1).
1.1 Einleitung
Mit diesem Test sollen der zeitliche Verlauf des biologischen Abbaus einer niedrig konzentrierten Prüfsubstanz in aerobem natürlichem Wasser gemessen und die Beobachtungen als kinetische Geschwindigkeit quantifiziert werden. Dieser Simulationstest besteht aus einem im Labor durchgeführten Schüttelkolben-Batch-Test, in dem die Rate des aeroben biologischen Abbaus organischer Substanzen in Proben natürlichen Oberflächenwassers (Süßwasser, Brackwasser oder Meerwasser) ermittelt wird. Der Test beruht auf ISO/DIS 14592-1 (2) und beinhaltet außerdem Elemente der Prüfmethoden C.23 und C.24 (3)(4). Bei langer Testdauer wird anstelle der Batch-Untersuchung ein semikontinuierlicher Test durchgeführt, um den Abbau der Testorganismen zu verhindern. Das Hauptziel des Simulationstests besteht in der Mineralisation der Prüfsubstanz in Oberflächenwasser, aufgrund der Mineralisation wird dann die Abbaukinetik beschrieben. Ein fakultatives untergeordnetes Ziel des Tests besteht in der Ermittlung von Informationen zum primären Abbau und zur Bildung wesentlicher Transformationsprodukte. Die Bestimmung der Transformationsprodukte sowie nach Möglichkeit die Quantifizierung der jeweiligen Konzentrationen sind besonders wichtig bei Substanzen, die sehr langsam mineralisiert werden (z.B. wenn die Halbwertszeiten für den Abbau sämtlicher 14C-Rückstände mehr als 60 Tage betragen). Zur Bestimmung und Quantifizierung wichtiger Transformationsprodukte sollten aus analytischen Gründen die Prüfsubstanzen im Allgemeinen in höheren Konzentrationen (z.B. > 100 µg/l) verwendet werden.
Niedrige Konzentrationen sind in diesem Test Konzentrationen (z.B. von weniger als 1 µg/l auf 100 µg/l), die so gering sind, dass sichergestellt ist, dass die Kinetik des biologischen Abbaus im Test die in der natürlichen Umgebung zu erwartende Kinetik widerspiegelt. Gegenüber der Gesamtmasse an biologisch abbaubaren Kohlenstoffsubstraten, die in im Test verwendetem natürlichem Wasser vorkommen, fungiert die niedrig konzentrierte Prüfsubstanz als untergeordnetes Substrat. Dies bedeutet, dass die zu erwartende Kinetik des biologischen Abbaus als Kinetik erster Ordnung einzustufen ist (Kinetik "ohne Wachstum') und dass die Prüfsubstanz durch "Cometabolismus" abgebaut werden kann. Eine Kinetik erster Ordnung impliziert, dass die Abbaurate (mg/l/Tag) proportional zur im Laufe der Zeit abnehmenden Substratkonzentration ist. Bei echter Kinetik erster Ordnung ist die Konstante der spezifischen Abbaurate (k) unabhängig von der Dauer und von der Konzentration. Die Konstante k ändert sich also während eines Versuchs nicht nennenswert, und wesentliche Änderungen treten auch nach Konzentrationssteigerungen von einem Versuch zum nächsten nicht auf. Per Definition entspricht die Konstante der spezifischen Abbaurate der relativen Konzentrationsänderung pro Zeiteinheit: k = (1/C) ×(dC/dt). Unter den vorgegebenen Bedingungen ist im Allgemeinen eine Kinetik erster Ordnung zu erwarten; unter gewissen Bedingungen kann jedoch auch eine sonstige Kinetik gegeben sein. Abweichungen von der Kinetik erster Ordnung können z.B. festzustellen sein, wenn die Geschwindigkeit der biologischen Transformation stärker durch ein Massentransfer-Phänomen wie z.B. die Diffusionsrate als durch die Geschwindigkeit der biologischen Reaktion begrenzt wird. Die Daten können jedoch nahezu immer durch eine Pseudo-Kinetik erster Ordnung beschrieben werden, wobei eine konzentrationsabhängige Konstante für die Abbaurate angenommen wird.
Informationen zur biologischen Abbaubarkeit der Prüfsubstanz bei höheren Konzentrationen (z.B. aufgrund von Standard-Screening-Tests) sowie Informationen zur abiotischen Abbaubarkeit, zu Transformationsprodukten und zu maggeblichen physikalisch-chemischen Merkmalen sollten vor dem Test verfügbar sein, um den Versuch planen und die Ergebnisse auswerten zu können. Die Verwendung von mit 14C-markierten Prüfsubstanzen und die Bestimmung der Phasenverteilung von 14C am Ende des Tests ermöglichen die Bestimmung der biologischen Endabbaubarkeit. Bei nicht markierten Prüfsubstanzen kann der biologische Endabbau nur geschätzt werden, wenn eine höhere Konzentration getestet wird und alle wesentlichen Transformationsprodukte bekannt sind.
1.2 Begriffsbestimmungen
Primärer biologischer Abbau Strukturänderung (Transformation) einer chemischen Substanz durch Mikroorganismen infolge des Verlusts der chemischen Identität.
Funktionaler biologischer Abbau Strukturänderung (Transformation) einer chemischen Substanz durch Mikroorganismen infolge des Verlusts eines spezifischen Merkmals.
Aerober biologischer Endabbau: Zersetzung einer chemischen Substanz durch Mikroorganismen unter Sauerstoffeinwirkung in Kohlendioxid und Wasser sowie in Mineralsalze sonstiger vorhandener Elemente (Mineralisation) und Erzeugung neuer Biomasse und organischer mikrobiologischer Biosyntheseprodukte.
Mineralisation Zersetzung einer chemischen Substanz oder eines organischen Materials durch Mikroorganismen unter Sauerstoffeinwirkung in Kohlendioxid und Wasser sowie in Mineralsalze sonstiger vorhandener Elemente.
"Lag"-Phase: Zeitraum vom Beginn eines Tests bis zur Anpassung der abbauenden Mikroorganismen und bis der biologische Abbau einer chemischen Substanz oder eines organischen Materials einen nachweisbaren Umfang angenommen hat (z.B. 10 % des maximalen theoretischen biologischen Abbaus bzw. je nach Genauigkeit des Messverfahrens auch weniger).
Maximaler Umfang des biologischen Abbaus: In Prozent erfasster Umfang des biologischen Abbaus einer chemischen Substanz oder eines organischen Materials in einem Test, über den hinaus während des Tests kein weiterer biologischer Abbau mehr erfolgt.
Primärsubstrat: Zusammenstellung natürlichen Kohlenstoffs und natürlicher Energiequellen, in der eine mikrobiologische Biomasse wachsen und kultiviert werden kann.
Sekundärsubstrat: Niedrig konzentrierte Substratkomponente, durch deren Abbau den maßgeblichen Mikroorganismen im Vergleich zur Kohlenstoff- und Energieversorgung infolge des Abbaus der wesentlichen Substratkomponenten (Primärsubstrate) nur unerhebliche Mengen an Kohlenstoff und Energie zugeführt werden.
Konstante der Abbaurate: Konstante der Abbaukinetik erster Ordnung k (d-'); diese Konstante gibt Aufschluss über die Geschwindigkeit von Abbauprozessen; bei Batch-Versuchen wird k aufgrund des anfänglichen Abschnitts der Abbaukurve geschätzt, die am Ende der "Lag"-Phase ermittelt wird.
Halbwertszeit, t1/2 (d): Parameter zur Beschreibung der Geschwindigkeit einer Reaktion erster Ordnung; Zeitabstand, der einem Konzentrationsrückgang um den Faktor 2 entspricht. Der Zusammenhang zwischen der Halbwertszeit und der Konstante der Abbaurate lässt sich mit der Formel t1/2 =1n2/k beschreiben.
Abbauzeit, DT50 (d): Parameter zur Quantifizierung des Ergebnisses von Tests zur Ermittlung der biologischen Abbaubarkeit; Zeitintervall einschließlich der "Lag"-Phase, in dem ein biologischer Abbau von 50 % erreicht wird.
Nachweisgrenze (LOD = Limit of Detection) und Quantifizierungsgrenze (LOQ = Limit of Quantification): Als Nachweisgrenze (LOD) wird die Konzentration einer Substanz bezeichnet, unter der die Substanz nicht mehr von analytischen Artefakten unterschieden werden kann. Als Quantifizierungsgrenze (LOQ) gilt die Konzentration einer Substanz, unter der die Konzentrationen nicht mehr mit annehmbarer Genauigkeit bestimmt werden kann.
Gelöster organischer Kohlenstoff (DOC = Dissolved organic Carbon): Der Anteil des in einer Wasserprobe enthaltenen organischen Kohlenstoffs, der nicht durch die spezifizierte Phasentrennung entfernt werden kann (etwa durch 15-minütiges Zentrifugieren mit 40.000 ms-2 oder durch Membranfiltration mit einer Porengröße von 0,2 µm-0,45 µm).
TOA (Total organic 14C Activity): 14C-Gesamtaktivität von organischem Kohlenstoff
DOA (Dissolved organic 14C Activity): 14C-Aktivität von gelöstem organischem Kohlenstoff
POA (Particulate organic 14C Activity): 14C-Aktivität von als Feststoff vorliegendem organischem Kohlenstoff
1.3 Anwndbarkeit der Prüfmethode
Dieser Simulationstest ist bei nicht flüchtigen oder leicht flüchtigen organischen Substanzen durchzuführen, die bei niedrigen Konzentrationen getestet werden. Bei Verwendung von zur Atmosphäre offenen Kolben (z.B. nur mit einem Wattepropf verschlossen) können Substanzen mit Henry-Konstanten von unter etwa 1 Pa × m3/mol (ca. 10-5 atm × m3/mol) in der Praxis als nicht flüchtig betrachtet werden. Mit geschlossenen Kolben mit einem gewissen Luftraum können leicht flüchtige Substanzen (mit Henry-Konstanten < 100 Pa × m3/mol bzw. < 10-3 atm × m3/mol) verlustfrei getestet werden. Bei mit 14C markierten Substanzen können Verluste auftreten, wenn beim Abtrennen des CO2 nicht die erforderlichen Vorsichtsmaßnahmen getroffen werden. In diesen Fällen muss unter Umständen CO2 in einem internen Absorber mit Alkali gebunden oder ein externes CO2-Absorbersystem verwendet werden (direkte 14CO2-Bestimmung, wie in Anhang 3 beschrieben). Damit die Kinetik des biologischen Abbaus bestimmt werden kann, muss die Konzentration der Prüfsubstanz unter der Wasserlöslichkeit der Prüfsubstanz liegen. Es wird jedoch darauf hingewiesen, dass die in der FachLiteratur genannten Werte für die Wasserlöslichkeit erheblich höher sein können als die Löslichkeit der Prüfsubstanz in natürlichen Gewässern. Fakultativ kann die Löslichkeit von Prüfsubstanzen mit besonders schlechter Wasserlöslichkeit auch unter Verwendung von Material aus den zu prüfenden natürlichen Gewässern bestimmt werden.
Die Methode kann zur Simulierung des biologischen Abbaus in von groben Partikeln freiem Oberflächenwasser ("pelagischer Test") oder in trübem Oberflächenwasser z.B. an einem Wasser-/Sediment-Übergang ("Test mit suspendierten Sedimenten") eingesetzt werden.
1.4 Prinzip der Prüfmethode
Der Test wird als Batch-Test durchgeführt, indem die Prüfsubstanz entweder ausschließlich mit Oberflächenwasser ("pelagischer Test') oder in mit suspendierten Feststoffen/Sedimenten mit 0,01 bis 1 gjl Trockengewicht aufbereitetem Oberflächenwasser ("Test mit suspendierten Sedimenten') inkubiert wird, um ein Wasservolumen mit suspendierten Feststoffen oder neu suspendierten Sedimenten zu simulieren. Die Konzentration der suspendierten Feststoffe/Sedimente im unteren Bereich dieses Intervalls ist typisch für die meisten Oberflächengewässer. Die Testkolben werden im Dunkeln bei Umgebungstemperatur unter Schütteln bei aeroben Bedingungen inkubiert. Die Prüfsubstanz sollte in mindestens zwei Konzentrationen verwendet werden, um die Abbaukinetik zu bestimmen. Die Konzentrationen sollten sich um den Faktor 5 bis 10 voneinander unterscheiden und den in der Umwelt zu erwartenden Konzentrationsbereich abdecken. Die maximale Konzentration der Prüfsubstanz sollte höchstens 100 µg/l betragen; maximale Testkonzentrationen unter 10 µg/l sind jedoch zu bevorzugen, um sicherzustellen, dass beim biologischen Abbau eine Kinetik erster Ordnung gegeben ist. Die niedrigste Konzentration sollte höchstens 10 µg/l betragen; niedrigste Konzentrationen von 1-2 µg/l oder von weniger als 1 µg/l sind zu bevorzufen. Im Allgemeinen kann eine angemessene Analyse derart niedriger Konzentrationen mit handelsüblichen mit' C markierten Substanzen durchgeführt werden. Aus analysetechnischen Gründen kann die Konzentration der Prüfsubstanz häufig nicht mit der erforderlichen Genauigkeit gemessen werden, wenn die Prüfsubstanz in einer Konzentration von ≤100 µg/l vorliegt (siehe Abschnitt 1.7.2 Absatz 2). Höhere Konzentrationen der Prüfsubstanz (> 100 µg/l und gelegentlich > 1 mg/l) können zur Bestimmung und zur Quantifizierung wichtigerer Transformationsprodukte sowie dann verwendet werden, wenn ein spezifisches Analyseverfahren mit niedriger Nachweisgrenze nicht verfügbar ist. Wenn hohe Konzentrationen einer Prüfsubstanz getestet werden sollen, können die Ergebnisse unter Umständen nicht zur Schätzung der Konstante des Abbaus erster Ordnung und zur Schätzung der Halbwertszeit verwendet werden, weil der Abbau wahrscheinlich nicht der Kinetik erster Ordnung folgt.
Nach dem Abbau werden in geeigneten Zeitabständen entweder die 14C-Restwerte oder die Restkonzentration der Prüfsubstanz gemessen, wenn eine spezifische chemische Analyse erfolgt. Die 14C-Markierung der stabilsten Molekülbereiche gewährleistet, dass die Gesamtmineralisation bestimmt wird; erfolgt die Markierung mit 14C bei weniger stabilen Molekülbereichen oder werden spezifische Analyseverfahren eingesetzt, kann nur der primäre biologische Abbau ermittelt werden. Der stabilste Bereich beinhaltet jedoch nicht zwangsläufig den funktionsrelevanten Teil des Moleküls (der einem spezifischen Merkmal wie z.B. der Toxizität oder der Bioakkumulation zugeordnet werden kann). In diesem Fall ist unter Umständen die Verwendung einer im funktionsrelevanten Teil mit 14C markierten Prüfsubstanz angemessen, wenn die Aufhebung eines spezifischen Merkmals überwacht werden soll.
1.5 Informationen zur Prüfsubstanz
Für diesen Test können sowohl radioaktiv markierte als auch nicht markierte Prüfsubstanzen verwendet werden. Die Markierung mit 14C wird empfohlen; markiert werden sollten im Allgemeinen die stabilsten Bereiche der jeweiligen Moleküle (siehe auch Abschnitt 1.4). Bei Substanzen mit mehreren aromatischen Ringen sollte vorzugsweise mindestens ein Kohlenstoffmolekül pro Ring mit 14C markiert werden. Außerdem sollte vorzugsweise mindestens ein Kohlenstoffmolekül auf beiden Seiten leicht abbaubarer Brücken mit 14C markiert werden. Die chemische und/oder radiochemische Reinheit der Prüfsubstanz sollte > 95 % sein. Bei radioaktiv markierten Substanzen ist eine spezifische Aktivität von mindestens ca. 50 µCi/mg (1,85 MBq) zu bevorzugen, um die 14C-Messungen in Tests mit niedrigen Ausgangskonzentrationen zu erleichtern. Zu den Prüfsubstanzen sollten folgende Informationen verfügbar sein:
Wenn Substanzen mit geringer Wasserlöslichkeit in Meerwasser getestet werden, kann die Kenntnis der Aussalzungskonstante (oder "Setschenow-Konstante') Ks von Vorteil sein; diese Konstante wird mit folgender Formel definiert: log (S/S') = Ks Cm; dabei stehen S und S' für die Löslichkeit der Substanz in Süßwasser bzw. in Meerwasser und Cm für die molare Salzkonzentration.
Wenn der Test als "Test mit suspendierten Sedimenten" durchgeführt wird, sollten auch die folgenden Informationen vorliegen:
Außerdem können die folgenden Informationen hilfreich sein:
1.6 Referenzsubstanz
Als Referenzsubstanz sollte eine unter aeroben Bedingungen im Allgemeinen leicht abzubauende Substanz (z.B. Anilin oder Natriumbenzoat) verwendet werden. Gewöhnlich werden Anilin und Natriumbenzoat in weniger als zwei Wochen.abgebaut. Mit den Referenzsubstanzen soll sichergestellt werden, dass die mikrobiologische Aktivität des Testwassers in einem bestimmten Rahmen liegt, d. h. dass das Wasser eine aktive mikrobiologische Population enthält.
1.7 Qualitätskriterien
1.7.1 Wiederfindungsraten
Unmittelbar nach der Zugabe der Prüfsubstanz sollte durch Messungen der 14C-Aktivität bzw. - bei nicht markierten Substanzen - durch chemische Analysen in jeweils mindestens zwei Proben jeweils die Ausgangs-Testkonzentration geprüft werden. Die entsprechenden Messungen geben Aufschluss über die Anwendbarkeit und über die Wiederholbarkeit der Analysemethode sowie über die Homogenität der Verteilung der Prüfsubstanz. Im Allgemeinen wird in den anschließenden Datenanalysen eher die gemessene 14C-Ausgangsaktivität oder die Prüfsubstanzkonzentration gemessen als die Nennkonzentration, da bei den Messungen der 14C-Ausgangsaktivität oder der Prüfsubstanzkonzentration die auftretenden Sorptions- und Dosierungsfehler kompensiert werden. Bei mit' 4C markierten Prüfsubstanzen wird der Umfang der Wiederfindung am Ende des Versuchs als Massenbilanz angegeben (siehe Abschnitt 1.8.9.4 letzter Absatz). Im Idealfall sollte die Massenbilanz bei radioaktiver Markierung im Bereich 90 % bis 110 % liegen; die erforderliche Analysegenauigkeit sollte bei nicht markierten Prüfsubstanzen durch eine anfängliche Wiederfindung von 70 % bis 110 % gegeben sein. Diese Bereiche sollten jedoch als Idealbereiche betrachtet und nicht als Kriterien für die Annehmbarkeit eines Tests angenommen werden. Die Analysegenauigkeit kann für die Prüfsubstanz auch bei einer niedrigeren Konzentration als der Ausgangskonzentration sowie für wichtigere Transformationsprodukte bestimmt werden.
1.7.2 Wiederholbarkeit und Empfindlichkeit der Analysemethode
Die Wiederholbarkeit der zur Quantifizierung der Prüfsubstanz sowie ggf. der Transformationsprodukte eingesetzten Analysemethode (einschließlich der Methoden zur Überprüfung der Wirksamkeit der Erstextraktion) sollte durch fünf Wiederholungsanalysen der einzelnen Extrakte des Oberflächenwassers geprüft werden.
Die Nachweisgrenze (LOD) der zur Analyse der Prüfsubstanz und der Transformationsprodukte verwendeten Methode sollte nach Möglichkeit bei mindestens 1 % der in das Testsystem eingebrachten Ausgangsmenge liegen. Die Quantifizierungsgrenze (LOQ) sollte kleiner oder gleich 10 % der verwendeten Konzentration sein. Die chemischen Analysen vieler organischer Substanzen und der jeweiligen Transformationsprodukte setzen häufig voraus, dass die Prüfsubstanz in verhältnismäßig hohen Konzentrationen (d. h. > 100 µg/l) eingebracht wird.
1.8 Beschreibung der Prüfmethode
1.8.1 Apparatur
Der Test kann in konischen oder zylindrischen Kolben mit geeignetem Fassungsvermögen (z.B. 0,5 oder 1,01) durchgeführt werden, die mit Silikon- oder Gummistopfen verschlossen sind; alternativ können auch Serumflaschen mit CO2-dichten Deckeln (z.B. mit Butylkautschuk-Septum) verwendet werden. Eine weitere Möglichkeit besteht in der Durchführung der Tests mit mehreren Kolben und in der Entnahme des gesamten Inhalts von jeweils mindestens zwei Kolben je Probenintervall (siehe Abschnitt 1.8.9.1 letzter Absatz). Für nicht flüchtige Prüfsubstanzen, die nicht radioaktiv markiert wurden, sind gasdichte Stopfen oder Deckel nicht erforderlich. Lose Wattepfropfen, die eine Verunreinigung aus der Luft verhindern, können verwendet werden (siehe Abschnitt 1.8.9.1 Absatz 2). Leicht flüchtige Substanzen sollten in einem Biometersystem unter leichten Rühren der Wasseroberfläche getestet werden. Um sicherzustellen, dass keine Verunreinigung durch Bakterien erfolgt, können die Gefäße auch durch Erwärmen oder durch Autoklavieren vor der Verwendung sterilisiert werden. Außerdem wird die folgende Standard-Laborausrüstung verwendet:
1.8.2 Stammlösungen der Prüfsubstanz
Zur Herstellung von Stammlösungen der Prüfsubstanzen und der Referenzsubstanzen wird entionisiertes Wasser verwendet (siehe Abschnitt 1.8.7 Absatz 1). Das entionisierte Wasser sollte frei von Substanzen sein, die auf Mikroorganismen toxisch wirken könnten; außerdem sollte der Anteil an gelöstem organischem Kohlenstoff (DOC) höchstens 1 mg/l betragen (6).
1.8.3 Entnahme und Transport von Oberflächenwasser
Die Stellen, an denen das Oberflächenwasser entnommen wird, sollten grundsätzlich abhängig vom Zweck des jeweiligen Tests ausgewählt werden. Bei der Auswahl von Entnahmestellen sind mögliche Einträge aus Landwirtschaft, Industrie und Haushalten zu berücksichtigen. Wenn bekannt ist, dass eine aquatische Umgebung in den letzten vier Jahren mit der Prüfsubstanz oder mit analog aufgebauten Substanzen verunreinigt wurde, sollte dort nur dann Wasser für die Tests entnommen werden, wenn ausdrücklich die Abbauraten an bereits belasteten Stellen untersucht werden sollen. An der Entnahmestelle sollten der pH-Wert und die Temperatur des Wassers gemessen werden. Die Tiefe, in der das Wasser entnommen wurde, sowie das Aussehen der Wasserproben (z.B. Farbe und Trübung) sollten ebenfalls protokolliert werden (siehe Abschnitt 3). Die Sauerstoffkonzentration und/oder das Redoxpotenzial in Wasser und in der Sedimentoberfläche sollten gemessen werden, um die aerobe Qualität der Proben nachzuweisen, wenn diese nicht aufgrund des Aussehens oder bereits vorliegender Erfahrungen mit der Entnahmestelle offensichtlich bm bekannt sind. Das Oberflächenwasser sollte in einem gründlich gespülten Behälter transportiert werden. Während des Transports sollte die Probentemperatur die Testtemperatur nicht erheblich überschreiten. Bei einer Transportdauer von mehr als 2-3 Stunden wird eine Kühlung auf 4 °C empfohlen. Die Wasserprobe darf nicht gefroren sein.
1.8.4 Lagerung und Vorbereitung des Oberflächenwassers
Der Test sollte vorzugsweise binnen eines Tages nach der Probenahme begonnen werden. Die ggf. erforderliche Lagerung des Wassers sollte auf ein Minimum beschränkt werden; Lagerzeiten von mehr als vier Wochen sind unter keinen Umständen annehmbar. Die Wasserprobe sollte bis zur Verwendung bei einer Temperatur von 4 °C aufbewahrt werden. Vor der Verwendung sollten grobe Teilchen entfernt werden (z.B. durch Filtration durch einen Nylonfilter mit einer Maschenweite von 100 µm oder mit einem groben Papierfilter oder durch Ausfällung).
1.8.5 Vorbereitung von mit Sedimenten aufbereitetem Wasser (fakultativ)
Für den Test mit dem suspendierten Sediment wird ein Oberflächensediment zu den Kolben mit dem wie in Abschnitt 1.8.4 beschrieben zur Abtrennung grober Teilchen gefilterten natürlichen Wasser hinzugegeben, um eine Suspension herzustellen; die Konzentration der suspendierten Feststoffe sollte zwischen 0,01 und 1 g/l betragen. Das Oberflächensediment sollte aus der Stelle stammen, aus der auch die Wasserprobe genommen wurde. Abhängig von der jeweiligen aquatischen Umgebung kann das Oberflächensediment entweder durch einen hohen Anteil an organischem Kohlenstoff (2,5-7,5 %) und durch eine feine Textur oder durch einen geringen Anteil an organischem Kohlenstoff (0,5-2,5 %) und eine grobe Textur gekennzeichnet sein (3). Das Oberflächensediment kann wie folgt hergestellt werden: Mit einem durchsichtigen Kunststoffröhrchen werden mehrere Sedimentkerne gezogen; unmittelbar nach der Probenahme werden anschließend die oberen aeroben Schichten abgeschnitten (von der Oberfläche bis zu einer Tiefe von maximal 5 mm) und zusammengegeben. Die so hergestellte Sedimentprobe sollte in einem Behälter mit großem Luftraum transportiert werden, um die aerobe Umgebung des Sediments aufrechtzuerhalten. (Bei einer Transportdauer von mehr als 2-3 Stunden ist das Sediment auf 4 °C zu kühlen.) Die Sedimentprobe sollte dann im für den Test zu verwendenden Wasser im Verhältnis 1:10 suspendiert und bis zur Verwendung belüftet bei einer Temperatur von 4 °C gelagert werden. Die ggf. erforderliche Lagerung des Sediments sollte auf ein Minimum beschränkt werden; Lagerzeiten von mehr als vier Wochen sind unter keinen Umständen annehmbar.
1.8.6 Semikontinuierliches Verfahren (fakultativ)
Eine längere Inkubation (über mehrere Monate) kann erforderlich sein, wenn ein erheblicher Abbau der Prüfsubstanz erst nach einer langen Verzögerung messbar ist. Wenn die Notwendigkeit einer längeren Inkubation aus früheren Tests einer Substanz bekannt ist, kann der Test mit einem semikontinuierlichen Verfahren begonnen werden, bei dem regelmäßig ein Teil des im Test verwendeten Wassers bzw. der Suspension erneuert wird (siehe Anhang 2). Alternativ kann auch der normale Batch-Test zu einem semikontinuierlichen Test abgewandelt werden, wenn binnen einer Testdauer von etwa 60 Tagen im Batch-Test (siehe Abschnitt 1.8.8.3 Absatz 2) kein Abbau der Priifsubstanz festgestellt wurde.
1.8.7 Zugabe der Prüfsubstanz (bzw. der Referenzsubstanz)
Bei Substanzen mit hoher Wasserlöslichkeit (> 1 mg/l) und geringer Flüchtigkeit (Henry-Konstanten < 1 Pa × m3/mol oder < 10-5 atm × m3/mol) kann eine Stammlösung in entionisiertem Wasser hergestellt werden (siehe Abschnitt 1.8.2); die jeweils erforderliche Menge der Stammlösung wird zu den Prüfgefäßen hinzugegeben, bis die gewünschte Konzentration erreicht ist. Das Volumen einer hinzugefügten Stammlösung sollte auf das geringstmögliche Mag begrenzt werden (möglichst < 10 % des endgültigen Flüssigkeitsvolumens). Ein weiteres Verfahren besteht in der Auflösung der Prüfsubstanz in einem größeren Volumen des im Test verwendeten Wassers; diese Möglichkeit kommt als Alternative zur Verwendung organischer Lösungsmittel in Betracht.
Wenn zwingend erforderlich, sollten Stammlösungen nicht flüchtiger Substanzen mit geringer Wasserlöslichkeit unter Einsatz eines flüchtigen organischen Lösungsmittels hergestellt werden; die Menge des zum Testsystem hinzugegebenen Lösungsmittels sollte jedoch maximal 1 % (v/v) betragen und die mikrobiologische Aktivität nicht beeinträchtigen. Außerdem sollte das Lösungsmittel keine Auswirkungen auf die Stabilität der Prüfsubstanz im Wasser haben. Das Lösungsmittel sollte bis zu einem äußerst geringen Volumen so weit abgetrennt werden, dass die DOC-Konzentration des im Test verwendeten Wassers bzw. der verwendeten Suspension nicht erheblich erhöht wird. Dies sollte durch eine substanzspezifische Analyse bzw. nach Möglichkeit durch eine DOC-Analyse sichergestellt werden (6). Dabei ist sorgfältig darauf zu achten, dass die Menge des übertragenen Lösungsmittels auf das absolut erforderliche Minimum begrenzt wird; außerdem muss gewährleistet sein, dass sich die vorhandene Prüfsubstanz im Endvolumen des im Test verwendeten Wassers auflösen kann. Für die Einbringung der Prüfsubstanz in die Prüfgefäße können auch andere Verfahren verwendet werden (siehe Quellen (7) und (8)). Wenn zur Einbringung der Prüfsubstanz ein organisches Lösungsmittel verwendet wird, sollten Lösungsmittelkontrollen mit dem im Test verwendeten Wasser (ohne weitere Zusätze) sowie das im Test verwendete Wasser unter Zugabe einer Referenzsubstanz in ähnlicher Weise wie die aktiven Prüfgefäße behandelt werden, zu denen die Prüfsubstanz in einem Trägerlösungsmittel hinzugegeben wurde. Mit den Lösungsmittelkontroollen sollen anhand des Abbaus der Referenzsubstanz mögliche Beeinträchtigungen der mikrobiologischen Population durch das Lösungsmittel untersucht werden.
1.8.8 Prüfbedingungen
1.8.8.1 Testtemperatur
Die Inkubation sollte (vorzugsweise) im Dunkeln oder bei diffuser Beleuchtung und kontrollierter Temperatur (± 2 °C) erfolgen; dies kann die in der betreffenden Außenumgebung gegebene Temperatur oder eine Standardtemperatur von 20-25 °C sein. Die in der Außenumgebung gegebene Temperatur kann entweder die tatsächliche Probentemperatur zum Zeitpunkt der Probenahme oder eine durchschnittliche Temperatur in der Außenumgebung der Entnahmestelle sein.
1.8.8.2 Vermischung
Durch Vermischung unter kontinuierlichem Schütteln oder Rühren muss sichergestellt werden, dass die Teilchen und Mikroorganismen suspendiert bleiben. Außerdem begünstigt die ständige Mischung den Sauerstoffeintrag aus dem Luftraum über der Flüssigkeit und somit die Aufrechterhaltung angemessener aerober Bedingungen. Dazu können die Kolben auf einen Schütteltisch (mit einer Frequenz von etwa 100 Umdrehungen pro Minute) gebracht oder mit einem Magnetrührer gerührt werden: Die Proben sind in einem kontinuierlichen Prozess zu schütteln. Das Schütteln oder Rühren muss möglichst vorsichtig erfolgen; trotzdem muss eine homogene Suspension aufrechterhalten werden.
1.8.8.3 Testdauer
Die Testdauer sollte im Allgemeinen höchstens 60 Tage betragen, wenn nicht das semikontinuierliche Verfahren unter regelmäßiger Erneuerung der Testsuspension eingesetzt wird (siehe Abschnitt 1.8.6 und Anhang 2). Die Testdauer des Batch-Tests kann jedoch auf maximal 90 Tage ausgedehnt werden, wenn binnen der ersten 60 Tage der Abbau der Prüfsubstanz begonnen hat. Der Abbau wird in geeigneten Zeitabständen aufgrund der 14C-Restaktivität oder der gebildeten 14CO2-Menge (siehe Abschnitt 1.8.9.4) und/oder durch chemische Analyse (Abschnitt 1.8.9.5) überwacht. Die Inkubationsdauer muss so lange sein, dass der Abbauprozess bewertet werden kann. Der Abbau sollte vorzugsweise in einem Umfang von über 50 % erfolgt sein; bei langsam abbauenden Substanzen muss der Umfang des Abbaus hinreichend sein (im Allgemeinen mehr als 20 %), um die Schätzung einer Konstante der kinetischen Abbaurate zu ermöglichen.
Der pH-Wert und die Sauerstoffkonzentration des Testsystems sind regelmäßig zu messen, wenn nicht aus ähnlichen Tests mit Wasser- und Sedimentproben, die aus derselben Stelle entnommen wurden, Erfahrungen vorliegen und diese Messungen daher nicht mehr erforderlich sind. Unter gewissen Bedingungen kann der Metabolismus von Primärsubstraten in stark erhöhten Konzentrationen im Wasser bzw. im Sediment dazu führen, dass so viel CO2 entwickelt und so viel Sauerstoff abgebaut wird, dass sich die Versuchsbedingungen während der Testdauer erheblich ändern.
1.8.9 Verfahren
1.8.9.1 Vorbereitung der Kolben für den pelagischen Test
Ein geeignetes Volumen des im Test zu verwendenden Wassers wird in die Testkolben gebracht; die Kolben sind bis zu etwa einem Drittel zu füllen. Eine Füllmenge von etwa 100 ml darf nicht unterschritten werden. Auch wenn mehrere Kolben verwendet werden (um ggf. auch den gesamten Inhalt eines Kolbens prüfen zu können), beträgt das Volumen des zu verwendenden Wassers etwa 100 ml, da sich zu geringe Probenvolumina auf die Dauer der "Lag"-Phase auswirken könnten. Die Prüfsubstanz wird aus einer Stammlösung hinzugegeben, wie in den Abschnitten 1.8.2 und 1.8.7 beschrieben. Die Prüfsubstanz sollte in mindestens zwei Konzentrationen verwendet werden, die sich mindestens um den Faktor 5 bis 10 unterscheiden; anhand dieser Konzentrationen sollte die Abbaukinetik bestimmt und die Konstante der kinetischen Abbaurate ermittelt werden. Die beiden ausgewählten Konzentrationen sollten unter 100 µg/l liegen und sich vorzugsweise im Bereich < 1-10 µg/l bewegen.
Die Kolben sind mit für Luft und für CO2 undurchlässigen Stopfen oder Deckeln zu verschließen. Bei nicht flüchtigen und nicht mit 14C markierten Prüfchemikalien können lose Wattepropfen zum Schutz vor Verunreinigungen aus der Luft verwendet werden (siehe Abschnitt 1.8.1), wenn alle wichtigeren Abbauprodukte bekanntermaßen nicht flüchtig sind und wenn die CO2 -Konzentration indirekt bestimmt wird (siehe Anhang 3).
Die Kolben werden bei der jeweils ausgewählten Temperatur inkubiert (siehe Abschnitt 1.8.8.1). Proben zur chemischen Analyse oder für 14C-Messungen sind am Anfang des Tests zu entnehmen (d. h. noch vor Beginn des biologischen Abbaus; siehe Abschnitt 1.7.1); anschließend folgen während der Testdauer weitere Probenahmen in geeigneten Zeitabständen. Die Probenahme kann entweder durch Entnahme von Teilproben (z.B. 5-ml-Aliquoten) aus den einzelnen Wiederholungen oder durch Entnahme jeweils des gesamten Inhalts eines Kolbens bei der Probenahme erfolgen. Die Mineralisation der Prüfsubstanz kann wahlweise direkt oder indirekt bestimmt werden (siehe Anhang 3). In der Regel werden während der Abbauphase (d. h. nach Ende der "Lag"-Phase) mindestens fünf Probenahmestellen benötigt, um die Konstante der Abbaurate zuverlässig bestimmen zu können, wenn nicht bei rasch abbaubaren Substanzen begründet werden kann, dass drei Probenahmestellen hinreichend sind. Bei Substanzen, die nicht schnell abgebaut werden, können leicht weitere Messungen während der Abbauphase vorgenommen werden; daher sollten mehr Datenpunkte zur Bestimmung von k verwendet werden. Ein bestimmter zeitlicher Rahmen für die Probenahme kann nicht vorgegeben werden, da sich der biologische Abbau von Fall zu Fall unterschiedlich vollzieht; bei langsamem Abbau wird allerdings empfohlen, wöchentlich eine Probe zu entnehmen. Wenn die Prüfsubstanz rasch abbaubar ist, sollten Proben während der ersten drei Tage einmal täglich und anschließend jeden zweiten oder dritten Tag entnommen werden. Unter gewissen Umständen (z.B. bei sehr rasch hydrolysierenden Substanzen) müssen Proben unter Umständen stündlich genommen werden. Eine Vorstudie vor Durchführung des eigentlichen Tests wird empfohlen, um die geeigneten Intervalle für die Probenahme zu bestimmen. Wenn Proben für weitere spezifische Analysen verfügbar sein müssen, sollten mehr Proben entnommen und dann am Ende des Versuchs in rückläufiger Reihenfolge (d. h. die letzten Proben zuerst) die zu analysierenden Proben ausgewählt werden. (Hinweise zur Stabilität der Proben während der Lagerung sind Abschnitt 1.8.9.5 Absatz 2 zu entnehmen.).
1.8.9.2 Anzahl der Kolben und Proben
Testkolben werden in folgendem Umfang benötigt:
Bei der Gestaltung des Prüfprotokolls sollte die relative Bedeutung häufigerer Wiederholungen bei einer höheren Anzahl an Probenahmen berücksichtigt werden. Die genaue Anzahl der erforderlichen Kolben hängt von der jeweiligen Methode zur Messung des Abbaus ab (siehe Abschnitt 1.8.9.1 Absatz 3 und Abschnitt 1.8.9.4 sowie Anhang 3).
Aus allen Testkolben sollten bei der Probenahme jeweils zwei Teilproben (z.B. 5-ml-Aliquoten) genommen werden. Wenn mehrere Kolben eingesetzt werden, um den gesamten Inhalt eines Kolbens als Probe verwenden zu können, sollten mindestens zwei Kolben pro Probenahme entnommen werden (siehe Abschnitt 1.8.9.1 Absatz 1).
1.8.9.3 Vorbereitung der Kolben für Tests mit suspendierten Sedimenten (fakultativ)
Die erforderliche Menge des im Test zu verwendenden Wassers und das ggf. erforderliche Sediment werden zum Prüfgefäß hinzugegeben (siehe Abschnitt 1.8.5). Die Vorbereitung der Kolben für Tests mit suspendierten Sedimenten unterscheidet sich nicht von der Vorbereitung für den pelagischen Test (siehe Abschnitte 1.8.9.1 und 1.8.9.2). Vorzugsweise sind Serumflaschen oder ähnlich geformte Kolben zu verwenden. Die geschlossenen Kolben werden horizontal auf eine Schüttelvorrichtung gesetzt. Offene Kolben zur Untersuchung von nicht mit 14C markierten und nicht flüchtigen Substanzen müssen selbstverständlich aufrecht eingesetzt werden. In diesem Fall werden ein Magnetrührer sowie der Einsatz von glasbeschichteten Magnetrührstäben empfohlen. Wenn erforderlich, sind die Flaschen zu belüften, um die erforderlichen aeroben Bedingungen herzustellen.
1.8.9.4 Radiochemische Bestimmungen
Die entstandene 14CO2-Menge wird indirekt und direkt gemessen (siehe Anhang 3). Indirekt wird der 14CO2-Anteil aufgrund der Differenz zwischen der anfänglichen 14C-Aktivität des im Test verwendeten Wassers bzw. der verwendeten Suspension und der gesamten Restaktivität zum Zeitpunkt der Probenahme bestimmt, nachdem die Probe auf einen pH-Wert von 2-3 angesäuert und das enthaltene CO2 abgetrennt wurde. Auf diese Weise wird anorganischer Kohlenstoff entfernt, und die Restaktivität ergibt sich aus dem gemessenen organischen Material. Die indirekte 14CO2 Bestimmung kommt nicht in Betracht, wenn während der Transformation der Prüfsubstanz wichtigere flüchtige Transformationsprodukte entstehen (siehe Anhang 3). Nach Möglichkeit sollte die 14CO2 Bildung direkt gemessen werden; die Messung sollte bei der Probenahme für mindestens einen Testkolben erfolgen. So können die Massenbilanz und der biologische Abbau überprüft werden; diese Vorgehensweise beschränkt sich allerdings auf Tests mit verschlossenen Kolben.
Wenn das entstandene 14CO2 während des Tests direkt emessen wird, sollten bei Beginn des Tests mehrere Kolben für diesen Zweck vorgesehen werden. Die direkte 14CO2-Bestimmung wird empfohlen, wenn während der Transformation der Prüfsubstanz wichtigere flüchtige Transformationsprodukte entstehen. Bei jeder Messung werden die zusätzlichen Testkolben auf einen pH-Wert von 2-3 angesäuert, und das 14CO2 wird in einem internen oder externen Absorber gebunden (siehe Anhang 3).
Fakultativ können auch die Konzentrationen von mit 14C markierten Prüfsubstanzen sowie von wichtigeren Transformationsprodukten durch Radiochromatographie (z.B. Dünnschichtchromatographie, RAD-TLC) oder HPLC unter radiochemischem Nachweis bestimmt werden.
Die Phasenverteilung der verbliebenen Radioaktivität (siehe Anhang 1) und die verbliebene Prüfsubstanz sowie die noch vorhandenen Transformationsprodukte können ebenfalls bestimmt werden.
Am Ende des Tests sollte die Massenbilanz durch direkte 14CO2-Messung mit eigenen Testkolben bestimmt werden, aus denen während des Tests die benötigten Proben entnommen werden.
1.8.9.5 Spezifische chemische Analyse
Wenn eine empfindliche spezifische Analysemethode verfügbar ist, kann der primäre biologische Abbau statt durch radioaktive Markierung auch aufgrund einer Messung der gesamten Restkonzentration der Prüfsubstanz ermittelt werden. Wird eine radioaktiv markierte Prüfsubstanz eingesetzt (um die Gesamtmineralisation zu messen), können spezifische chemische Analysen parallel durchgeführt werden, um weitere hilfreiche Informationen zu erhalten und das Verfahren bewerten zu können. Ferner können spezifische chemische Analysen zur Messung der beim Abbau der Prüfsubstanz gebildeten Transformationsprodukte eingesetzt werden; dies wird für Substanzen empfohlen, bei denen die Mineralisation mit Halbwertszeiten von über 60 Tagen erfolgt ist. Die Konzentration der Prüfsubstanz und die Transformationsprodukte sollten jeweils bei der Probenahme gemessen und protokolliert werden (als Konzentration und als Prozentanteil). Im Allgemeinen sollten die Transformationsprodukte bestimmt werden, die bei ≥ 10 % der verwendeten Konzentration jeweils bei der Probenahme nachgewiesen werden; ansonsten ist in angemessener Weise zu begründen, warum die Transformationsprodukte nicht bestimmt wurden. Transformationsprodukte, deren Konzentration während der Studie kontinuierlich ansteigt, sollten ebenfalls bestimmt werden, selbst wenn die jeweiligen Konzentrationen den genannten Höchstwert nicht überschreiten, da aus diesen Transformationsprodukten auf eine Persistenz geschlossen werden könnte. Analysen der Transformationsprodukte in sterilen Kontrolllösungen sollten in Erwägung gezogen werden, wenn eine rasche abiotische Transformation der Prüfsubstanz (z.B. durch Hydrolyse) für möglich gehalten wird. Die Notwendigkeit einer Quantifizierung und Bestimmung von Transformationsprodukten sollte im Einzelfall geprüft werden; die entsprechenden Begründungen sind zu protokollieren. Extraktionen unter Verwendung organischer Lösungsmittel sollten gemäß den Anweisungen zum betreffenden Analyseverfahren durchgeführt werden.
Alle Proben sollten bei 2 bis 4 °C luftdicht gelagert werden, wenn die Analyse binnen 24 Stunden (vorzugsweise) durchgeführt wird. Für eine längere Lagerung sollten die Proben auf Temperaturen unter -18 °C gefroren oder chemisch konserviert werden. Eine Ansäuerung zur Konservierung der Proben wird nicht empfohlen, da angesäuerte Proben unter Umständen instabil sind. Wenn die Proben nicht binnen 24 Stunden analysiert und länger gelagert werden, sollte eine Untersuchung zur Stabilität unter Lagerungsbedingungen durchgeführt werden, um die Stabilität der maßgeblichen Chemikalien bei einer Temperatur von -18 °C oder bei sonstiger Konservierung der Chemikalien nachzuweisen. Erfolgt die Analyse unter Extraktion mit einem Lösungsmittel oder durch Festphasenextraktion (SPE), sollte die Extraktion unmittelbar nach der Probenahme oder nach maximal 24-stündiger Lagerung der gekühlten Probe vorgenommen werden:
Je nach Empfindlichkeit der Analysemethode können größere Probenvolumina erforderlich sein als in Abschnitt 1.8.1 genannt. Der Test kann auf bequeme Weise mit Testvolumina von einem Liter in Kolben mit einem Volumen von 2-3 Litern durchgeführt werden, aus denen dann Proben mit einem Volumen von etwa 100 ml entnommen werden.
2. Daten und Abschlussbericht
2.1 Behandlung der Ergebnisse
2.1.1 Grafische Darstellung der Daten
Die Zeitpunkte der Probenahme sind jeweils auf volle Stunden zu runden (sofern die betreffenden Substanzen nicht binnen Minuten oder weniger Stunden abgebaut werden); eine Rundung auf Tage ist nicht zulässig. Die geschätzte Restaktivität der Prüfsubstanzen (mit`C markierte Substanzen) bzw. die Restkonzentration (nicht markierte Substanzen) ist bezogen auf den zeitlichen Verlauf sowohl linear als auch semilogarithmisch darzustellen (siehe Abbildungen 1a und 1b). Wenn ein Abbau erfolgt ist, sind die Ergebnisse der Kolben FT mit den Ergebnissen der Kolben Fs zu vergleichen. Wenn die Mittelwerte der Ergebnisse der Kolben mit der Prüfsubstanz (FT) und der sterilen Kolben (Fs) um weniger als 10 % voneinander abweichen, kann davon ausgegangen werden, dass der festgestellte Abbau vorwiegend abiotisch erfolgt. Wird in den Kolben Fs ein geringerer Abbau beobachtet, können die Zahlen verwendet werden, um die bei den Kolben FT ermittelten Werte zu korrigieren (durch Subtraktion) und entsprechend den Umfang des biologischen Abbaus zu schätzen. Wenn fakultative Analysen der wichtigeren Transformationsprodukte vorgenommen werden, sollten ergänzend zur grafischen Darstellung des Abbaus der Prüfsubstanz auch die Entstehung und der Abbau dieser Transformationsprodukte grafisch dargestellt werden.
Die Dauer der "Lag"-Phase tL wird aufgrund der Abbaukurve (semilogarithmische Darstellung) durch Extrapolierung des linearen Abschnitts bis zum Abbau null oder alternativ durch Bestimmung der Zeit bis zu einem Abbau von etwa 10 % geschätzt (siehe Abbildungen la und lb). Aus der semilogarithmischen Darstellung sind die Konstante der Rate erster Ordnung (k) sowie die betreffende Standardabweichung durch lineare Regression der In (14C-Restaktivität oder Prüfsubstanzkonzentration) bezogen auf den zeitlichen Verlauf zu schätzen. Insbesondere bei 14C-Messungen sind ausschließlich Daten aus dem anfänglichen linearen Bereich der Kurve im Anschluss an die beendete "Lag"-Phase zu verwenden; dabei sollten vorzugsweise eher einige wenige repräsentative Daten als umfangreichere, aber unsicherere Daten ausgewählt werden. Unsicherheitsfaktoren sind unter anderem auch die der empfohlenen direkten Messung der 14C-Restaktivität inhärenten Fehler (siehe folgende Erläuterungen). Unter Umständen kann die Berechnung von zwei unterschiedlichen Konstanten für die Abbaurate erforderlich sein, wenn der Abbau in zwei Phasen erfolgt. In diesem Fall werden zwei getrennte Phasen der Abbaukurve definiert. Für die Kolben mit den einzelnen Wiederholungen sollten die Konstante der Abbaurate (k) und die Halbwertszeit (t, = ln2jk) berechnet werden, wenn aus einem Kolben mehrere Teilproben entnommen werden; alternativ können die Durchschnittswerte verwendet werden, wenn jeweils der vollständige Inhalt eines Kolbens als Probe entnommen wird (siehe Abschnitt 1.8.9.2 letzter Absatz). Beim erstgenannten Verfahren sollten die Konstante der Abbaurate und die Halbwertszeit für alle Kolben mit den einzelnen Wiederholungen sowie ein Durchschnittswert mit einer Standardabweichung protokolliert werden. Bei hohen Prüfsubstanzkonzentrationen kann die Abbaukurve beträchtlich von einer Gerade abweichen (semilogarithmische Darstellung), und die Kinetik erster Ordnung ist unter Umständen nicht gültig. Die Definition einer Halbwertszeit wäre daher nicht sinnvoll. Für einen begrenzten Datenbereich kann jedoch die Pseudo-Kinetik erster Ordnung angenommen und die Halbwertszeit des Abbaus DT50 (Dauer bis zu einem Abbau von 50 %) geschätzt werden. Dabei ist jedoch zu berücksichtigen, dass der zeitliche Verlauf des Abbaus über den ausgewählten Datenbereich hinaus nicht mit DT50 prognostiziert werden kann, da dieser Parameter ausschließlich einen vorgegebenen Datensatz beschreibt. Analyse-Tools zur einfacheren statistischen Berechnung und zur Kurvenanpassung sind allgemein zugänglich; der Einsatz dieser Software wird empfohlen.
Wenn spezifische chemische Analysen durchgeführt werden, sind die Konstanten der Abbaurate und die Halbwertszeiten des primären Abbaus wie oben beschrieben für die Gesamtmineralisation zu schätzen. Gelegentlich können Datenpunkte aus der gesamten Abbauphase verwendet werden, wenn der Prozess durch den primären Abbau eingeschränkt wird. (Im Gegensatz zu Messungen der 14C-Aktivität werden diese Messungen direkt durchgeführt.)
Wenn mit 14C markierte Substanzen verwendet werden, sollte eine Massenbilanz ausgedrückt als Prozentanteil der eingesetzten Ausgangskonzentration zumindest am Ende des Tests protokolliert werden.
2.1.2 Restaktivität
Beim biologischen Abbau des mit 14C markierten Anteils einer organischen Substanz wird das 14C größtenteils in 14CO2 umgewandelt; ein weiterer Anteil des 14C wird beim Wachstum der Biomasse und/oder bei der Synthese extrazellulärer Metaboliten verbraucht. Entsprechend wird der Kohlenstoff auch bei biologischem "Endabbau" einer Substanz nicht zu 100 % in 14CO2 umgewandelt. Das in den durch Biosynthese gebildeten Produkten enthaltene 14C wird anschließend infolge "sekundärer Mineralisation" langsam als 14CO2 freigesetzt. Aus diesen Gründen weisen grafische Darstellungen der organischen 14C-Restaktivität (gemessen nach der Abtrennung des CO2) oder des erzeugten 14CO2 bezogen auf den zeitlichen Verlauf auch nach Abschluss des Abbaus noch "Tailings" auf. Dies erschwert die Auswertung der Daten unter kinetischen Gesichtspunkten; daher sollte im Allgemeinen nur der erste Abschnitt der Kurve (nach dem Ende der "Lag"-Phase und vor Erreichen eines Abbaus von etwa 50 %) für die Schätzung der Abbaurate-Konstante berücksichtigt werden. Beim Abbau der Prüfsubstanz ist die gesamte 14C-Restaktivität immer höher als die 14C-Aktivität in Verbindung mit der verbliebenen noch unveränderten Prüfsubstanz. Wird die Prüfsubstanz durch eine Reaktion erster Ordnung abgebaut und erfolgt eine Mineralisation einer konstanten Fraktion a in CO2, beträgt die anfängliche Steigung der 14C-Abbaukurve (Gesamtanteil an organischem 14C bezogen auf den zeitlichen Verlauft das afache der Steigung der entsprechenden Kurve der Prüfsubstanzkonzentration (bzw. genauer gesagt des mit 14C markierten Anteils der Prüfsubstanz). Die Konstante der Abbaurate wird ausgehend von unkorrigierten Messungen der 14C-Gesamtaktivität daher eher mit einem Sicherheitszuschlag berechnet. Verfahren zum Schätzen der Prüfsubstanzkonzentrationen aus der gemessenen radiochemischen Aktivität aufgrund verschiedener vereinfachender Annahmen wurden in der Literatur beschrieben (2)(9)(10)(11). Diese Verfahren sind bei rasch abbaubaren Substanzen am leichtesten anzuwenden.
2.2 Auswertung der Ergebnisse
Wenn festgestellt wird, dass k unabhängig von der hinzugegebenen Konzentration ist (d. h. wenn die berechnete Konstante k bei unterschiedlichen Konzentrationen der Prüfsubstanz etwa gleich ist), kann angenommen werden, dass die Konstante des Abbaus erster Ordnung repräsentativ für die betreffenden Testbedingungen sowie für die Wasserprobe und die Testtemperatur ist. In welchem Umfang die Ergebnisse verallgemeinert oder auf andere Systeme extrapoliert werden können, ist von Fachleuten zu beurteilen. Wenn eine hohe Prüfsubstanzkonzentration eingesetzt wird und der Abbau daher nicht nach der Kinetik erster Ordnung verläuft, können die Daten nicht zur direkten Schätzung einer Rate für den Abbau erster Ordnung oder einer entsprechenden Halbwertszeit verwendet werden. Aus einem Test mit einer hohen Prüfsubstanzkonzentration abgeleitete Daten können jedoch trotzdem für eine Schätzung des Umfangs der Gesamtmineralisation und/oder für den Nachweis und die Quantifizierung von Transformationsprodukten geeignet sein.
Wenn statt des biologischen Abbaus die Raten sonstiger Verlustprozesse bekannt sind (z.B. Hydrolyse- oder Verflüchtigungsdaten), können diese Daten von der im Test festgestellten Nettoverlustrate abgezogen werden, um die Rate des biologischen Abbaus zu schätzen. Hydrolysedaten können z.B. aus sterilen Kontrollen oder aus Paralleltests mit höheren Prüfsubstanzkonzentrationen ermittelt werden.
Die indirekte und die direkte Bestimmung von 14CO2 (Abschnitt 1.8.9.4 und Anhang 3) kommt ausschließlich für die Messung des Umfangs der Mineralisation der Prüfsubstanz in CO2 in Betracht. Die Analyse der Konzentrationen von mit 14C markierten Prüfsubstanzen und der Bildung wichtigerer Transformationsprodukte kann durch Radiochromatographie (RAD-TLC) oder HPLC erfolgen (Abschnitt 1.8.9.4 Absatz 3). Voraussetzung für eine direkte Schätzung der Halbwertszeit ist, dass keine wichtigeren Transformationsprodukte (definiert als >_ 10 % der eingesetzten Menge der Prüfsubstanz) enthalten sind. Wenn wichtigere Transformationsprodukte in einem größeren Umfang als im vorstehenden Satz genannt vorkommen, ist eine detaillierte Auswertung der Daten vorzunehmen. Die detaillierte Auswertung kann Testwiederholungen und/oder Bestimmungen der Transformationsprodukte beinhalten (siehe Abschnitt 1.8.9.5 Absatz 1), wenn das Verhalten der Transformationsprodukte nicht aufgrund von Erfahrungswerten (z.B. Informationen zu den Abbauwegen) mit angemessener Zuverlässigkeit beurteilt werden kann. Da das Verhältnis des in der Prüfsubstanz enthaltenen und in CO2 umgewandelten Kohlenstoffs nicht immer gleich ist (weitgehend von der Konzentration der Prüfsubstanz und sonstiger vorhandener Substrate sowie von den Testbedingungen und den jeweiligen Mikroorganismen abhängig), lässt dieser Test (im Gegensatz zu einem DOC-Die-Away-Test) eine direkte Schätzung des biologischen Endabbaus nicht zu; das Ergebnis ist jedoch ähnlich dem Ergebnis einer Respirometer-Untersuchung. Der Umfang der Mineralisation wird entsprechend kleiner oder gleich dem Mindestumfang des biologischen Endabbaus sein. Um ein umfassenderes Bild vom biologischen Endabbau (Mineralisation und Aufnahme in die vorhandene Biomasse) zu erhalten, sollte am Ende des Tests die Phasenverteilung des 14C analysiert werden (siehe Anhani 1). Das in den vorhandenen Feststoffen enthaltene 14C beinhaltet das in die Bakterienmasse aufgenommene 4C sowie das in organische Partikel sorbierte 14C.
2.3 Validität des Tests
Wenn die Referenzsubstanz binnen des erwarteten Zeitraums nicht abgebaut wird (bei Anilin und Natriumbenzoat gewöhnlich unter zwei Wochen), ist die Validität des Tests zu bezweifeln und weiter zu überprüfen; alternativ kann der Test auch mit einer frischen Wasserprobe wiederholt werden. Bei einem ISO-Ringtest der Methode, an dem sieben europäische Labors beteiligt waren, lagen die angepassten Abbaurate-Konstanten zwischen 0,3 und 1,7 d-1 bei durchschnittlich 0,8 d-1, einer Temperatur von 20 °C und einer Standardabweichung von ± 0,4 d-1 (t1/2 = 0,9 Tage). Typisch waren "Lag`-Phasen von 1 bis 7 Tagen. Für das untersuchte Wasser wurde eine bakterielle Biomasse von 103 - 104 KBE (koloniebildenden Einheiten) pro ml protokolliert. Die Abbauraten in nährstoffreichen mitteleuropäischen Gewässern waren höher als in nordischen oligotrophen Gewässern; dies könnte auf die unterschiedlichen Trophiezustände oder auf eine vorherige Belastung mit chemischen Stoffen zurückzuführen sein.
Die Gesamtwiederfindung (Massenbilanz) am Ende des Versuchs sollte zwischen 90 % und 110 % bei radioaktiv markierten Substanzen liegen; bei nicht radioaktiv markierten Substanzen sollte die anfängliche Rückgewinnung am Anfang des Versuchs zwischen 70 % und 110 % betragen. Die angegebenen Bereiche sind als Idealbereiche zu verstehen und sollten nicht als Kriterien für die Annehmbarkeit eines Tests betrachtet werden.
3. Prüfbericht
Der Typ der Studie (z.B. pelagischer Test oder Test mit suspendiertem Sediment) ist im Prüfbericht klar anzugeben; außerdem enthält der Bericht mindestens folgende Informationen:
Prüfsubstanz und Referenzsubstanz(en):
Oberflächenwasser:
Für Wasserproben sind mindestens die folgenden Informationen anzugeben:
fakultativ:
Bei Tests mit suspendierten Sedimenten sollten außerdem die folgenden Informationen zu den Sedimenten angegeben werden:
Testbedingungen:
wenn der Test nicht im Dunkeln durchgeführt werden muss, Informationen zur Qualität des "diffusen Lichts";
Ergebnisse:
4. Literatur
1. OECD TG 309 (2004) Aerobic Mineralisation in surface water - Simulation Biodegradation Test.
2. ISO/DIS 14592-1 (1999) Water quality - Evaluation of the aerobic biodegradability of organic compounds at low concentrations - Part 1: Shake flask batch test with surface water or surface waterisediment suspensions.
3. Prüfmethode C.23. Aerobic and anaerobic transformation in soil.
4. Prüfmethode C.24. Aerobic and anaerobic transformation in aquatic sediments.
5. OECD (1993). Leitlinien für die Prüfung von Chemikalien OECD, Paris.
6. ISO 8245 (1999). Water quality - Guidelines an the determination of total organic carbon (TOC) and dissolved organic carbon (DOC).
7. ISO 10634 (1995). Water quality - Guidance for the preparation and treatment of poorly watersoluble organic compounds for the subsequent evaluation of their biodegradability in an aqueous medium.
8. OECD-Entwurf (2000). Guidance Document an aquatic toxicity testing of difficult substances and mixtures. Environmental Health and Safety Publications. Series an Testing and Assessment. No 22.
9. Simkins, S. und Alexander, M. (1984). Models for mineralization kinetics with the variables of substrate concentration and population density. Appl. Environ. Microbiol. 47, S. 394-401.
10. Ingerslev, F. und N. Nyholm. (2000). Shakeflask test for determination of biodegradation rates of 14C-labeled chemicals at low concentrations in surface water systems. Ecotoxicol. Environ. Saf. 45, S. 274-283.
11. ISO/CD 14592-1 (1999). Ring test report: Water Quality - Evaluation of the aerobic biodegradability of organic compounds at low concentrations part 1-report of 19981999 ringtest. Shake flask batch test with surface water or surface waterisediment suspensions.
| Phasenverteilung 14C | Anhang 1 |
Um das Verfahren zu überprüfen, sollten die Routinemessungen der 14C-Gesamtaktivität von organischem Kohlenstoff (TOA) um Massenbilanzmessungen unter direkter Bestimmung des entwickelten 14CO2 nach Bindung in einem Absorber ergänzt werden (siehe Anhang 3). Eine positive 14CO2 Bildung ist an sich schon als direkter Nachweis für einen biologischen Abbau zu betrachten (im Gegensatz zum abiotischen Abbau oder zu sonstigen Verlustprozessen wie z.B. Verflüchtigung oder Sorption). Weitere hilfreiche Informationen zur Beschreibung des biologischen Abbauverhaltens sind aus Messungen der TOA-Verteilung zwischen dem gelösten Stadium (14C-Aktivität von gelöstem organischem Kohlenstoff, DOA) und dem Partikelstadium (14C-Aktivität von als Feststoff vorliegendem organischem Kohlenstoff, POA) nach Abtrennung der Feststoffe durch Membranfiltration oder Zentrifugierung zu entnehmen. Die POA besteht aus der in der mikrobiologischen Biomasse und auf sonstigen Feststoffen sorbierten Prüfsubstanz sowie dem in der Prüfsubstanz enthaltenen Kohlenstoff, der zur Synthese neuen Zellmaterials verwendet und dadurch in die Feststoff-Biomasse aufgenommen wurde. Die Bildung gelösten organischen 14C-Materials kann als DOA am Ende des biologischen Abbaus (Plateau der Abbau-/Zeit-Kurve) geschätzt werden.
Die Phasenverteilung des 14C-Restanteils, der auf den ausgewählten Proben nach Filtration auf einem Membranfilter (etwa aus Polycarbonat) mit einer Porengröße von 0,22 µm oder 0,45 µm verblieben ist, wird geschätzt. Wenn die Sorption der Prüfsubstanz auf dem Filter nicht vernachlässigbar gering ist (vor der Durchführung des Versuchs festzustellen), kann anstelle der Filtration eine Zentrifugierung mit hoher Geschwindigkeit (2.000 g; 10 min) erfolgen.
Anschließend ist das Filtrat bm das Zentrifugat wie in Anhang 3 für ungefilterte Proben beschrieben zu behandeln. Membranfilter sind in einer geeigneten Szintillationsflüssigkeit zu lösen; die Zählung erfolgt dann in der üblichen Weise; im Allgemeinen werden Korrekturen der jeweiligen Querempfindlichkeit unter Anwendung eines externen Standardverhältnisverfahrens vorgenommen; alternativ kann auch ein Oxydationsmittel verwendet werden. Wenn eine Zentrifugierung erforderlich ist, muss das aus der Partikelfraktion gebildete Pellet in 1-2 ml destilliertem Wasser aufgelöst und in ein Szintillationsfläschchen gegeben werden. Anschließend werden zwei Spülungen mit 1 ml destilliertem Wasser vorgenommen und das Spülwasser in das Fläschchen gebracht. Wenn erforderlich, kann die Suspension zur Flüssigkeitsszintillationszählung in ein Gel eingebettet werden.
| Semikontinuierliches Verfahren | Anhang 2 |
Eine Inkubation über maximal mehrere Monate kann erforderlich sein, um einen hinreichenden Abbau von persistenten Substanzen zu erzielen. Die Testdauer sollte im Allgemeinen höchstens 60 Tage betragen, wenn die Merkmale der ursprünglichen Wasserprobe nicht auch nach einer Erneuerung der Testsuspension unverändert erhalten bleiben. Hat allerdings binnen der ersten 60 Tage der Abbau der Prüfsubstanz noch immer nicht begonnen, kann die Testdauer auf maximal 90 Tage ohne Erneuerung der Testsuspension ausgedehnt werden.
Während der Inkubation über längere Zeiträume kann die Diversität des mikrobiologischen Materials durch verschiedene Verlustprozesse sowie durch den möglichen Verbrauch wesentlicher Nährstoffe und primärer Kohlenstoffsubstrate aus der Wasserprobe reduziert werden. Daher wird empfohlen, mit einem semikontinuierlichen Test in angemessener Weise die Abbaurate langsam abbauender Substanzen zu bestimmen. Der Test sollte mit dem semikontinuierlichen Verfahren begonnen werden, wenn erfahrungsgemäß eine Inkubationsdauer von drei Monaten erforderlich ist, um die Substanz zu 20 % abzubauen. Alternativ kann der normale Batch-Test in einen seminkontinuierlichen Test abgewandelt werden, wenn während der Prüfung im Batch-Test binnen etwa 60 Tagen kein Abbau der Prüfsubstanz erfolgt ist. Das semikontinuierliche Verfahren kann ausgesetzt und der Test als Batch-Test fortgesetzt werden, wenn ein erheblicher Abbau verzeichnet wurde (z.B. > 20 %).
Beim semikontinuierlichen Test wird alle zwei Wochen etwa ein Drittel des Volumens der Testsuspension durch frisch entnommenes Wasser ersetzt, zu dem die Prüfsubstanz in der Ausgangskonzentration hinzugegeben wurde: Sedimentmaterial wird ebenso in der Ausgangskonzentration (zwischen 0,01 und 1 g/l) zum zur Erneuerung zu verwendenden Wasser hinzugegeben, wenn der fakultative Test mit dem suspendierten Sediment durchgeführt wird. Bei der Durchführung des Tests mit den suspendierten Sedimentpartikeln muss gewährleistet sein, dass auch während der Erneuerung des Wassers ein System mit vollständiger Suspension besteht und dass die Verweildauer für Feststoffe und Wasser gleich ist; ansonsten kann die erwünschte Ähnlichkeit mit einem homogenen wässerigen System ohne feste Phasen beeinträchtigt werden kann. Aus diesen Gründen wird für das semikontinuierliche Verfahren eine Ausgangskonzentration der suspendierten Sedimente im unteren Segment des vorgegebenen Konzentrationsbereichs bevorzugt.
Die vorgesehene Zugabe der Prüfsubstanz setzt voraus, dass die Ausgangskonzentration der Prüfsubstanz nicht durch eine teilweise Erneuerung der Testsuspension überschritten und entsprechend - wie häufig bei hohen Prüfsubstanzkonzentrationen festzustellen - eine Anpassung vermieden wird. Da das Verfahren sowohl eine Neubeimpfung als auch einen Ausgleich für abgebaute Nährstoffe und Primärsubstrate beinhaltet, wird die ursprüngliche mikrobiologische Diversität wiederhergestellt, und die Testdauer kann theoretisch unendlich ausgedehnt werden. Beim semikontinuierlichen Verfahren ist darauf zu achten, dass die Restkonzentration der Prüfsubstanz unter Berücksichtigung der jeweiligen Mengen der hinzugegebenen und der entnommenen Prüfsubstanz korrigiert wird. Die Gesamtkonzentration und die Konzentration der gelösten Prüfsubstanz sind für Verbindungen mit schwachem Sorptionsverhalten beliebig austauschbar. Die Sorption ist unter den vorgegebenen Bedingungen (0,1-1 g Feststoffel) bei Substanzen mit log Kow < 3 (für neutrale, lipophile Verbindungen) unerheblich (< 5 %). Dies wird aus dem folgenden Berechnungsbeispiel deutlich: 0,1 g/l Feststoffe entsprechen etwa 10 mg Kohlenstoff pro Liter (Kohlenstofffraktion, fc = 0,01). Wenn angenommen wird, dass
Log Kow(der Prüfsubstanz) = 3,
Koc = 0,42 x Kow und
der Verteilungskoeffizient Kd = fc x Koc ist,
beträgt die gelöste Fraktion der Gesamtkonzentration (C-Wasser (Cw)/C-Gesamt (Ct):
Cw/Ct = 1/(1 + Kd x SS) = 1(1 + Koc x fc x SS) = 1/(1 + 0,42 x 103 x 0,01 x 0,1 x 10-3) = 0,999
| Bestimmung des 14CO2-Anteils | Anhang 3 |
Indirekte 14CO2-Bestimmung
Bei Routinemessungen ist die indirekte Methode im Allgemeinen am wenigsten zeitaufwändig und am präzisesten, wenn die Prüfsubstanz nicht flüchtig ist und wenn aus der Prüfsubstanz keine flüchtigen Transformationsprodukte gebildet werden. Dazu sind einfach ungefilterte Proben (z.B. 5 ml in Szintillationsfläschchen) zu geben. Für die Tests geeignet ist eine anfängliche Probenaktivität von 5000 dpm-10.000 dpm (80-170 Bq) die änfängliche Aktivität sollte mindestens etwa 1000 dpm betragen. Das CO2 sollte nach dem Ansäuern auf einen pH-Wert von 2-3 mit 1-2 Tropfen konzentriertem H3 PO4 oder HCl erfolgen. Die CO2-Abtrennung kann durch Sprudeln mit Luft über etwa 1/2-1 Stunde erfolgen. Alternativ können die Fläschchen auch 1-2 Stunden heftig geschüttelt werden (z.B. auf einem Mikroplatten-Schüttler) oder vorsichtiger über Nacht geschüttelt werden. Die Wirksamkeit der CO2-Abtrennung ist zu überprüfen (durch Verlängerung der Belüftung oder der Dauer des Schüttelns). Anschließend sollte eine zur Zählung wässeriger Proben geeignete geeignete Szintillationsflüssigkeit hinzugegeben, die Probe in einem Wirbelmischer homogenisiert und die Radioaktivität mit einem Flüssigkeitsszintillationszähler bestimmt werden; dabei ist die in den Blindproben festgestellte Hintergrundaktivität (FB abzuziehen. Wenn das im Test verwendete Wasser stark gefärbt ist oder hohe Feststoffkonzentrationen enthält, weisen die Proben im Allgemeinen eine einheitliche Querempfindlichkeit auf, in diesem Fall sind Querempfindlichkeitskorrekturen mit einem externen Standard hinreichend. Ist das im Test verwendete Wasser stark gefärbt, müssen die Querempfindlichkeitskorrekturen unter Umständen durch Zugabe eines internen Standards vorgenommen werden. Bei hoher Feststoffkonzentration kann unter Umständen keine homogene Lösung bzw. kein homogenes Gel hergestellt werden, oder die Proben weisen sehr unterschiedliche Querempfindlichkeiten auf. In diesem Fall kann die im Folgenden beschriebene Methode zum Zählen von Testschlämmen verwendet werden. Wenn der Test mit einem suspendierten Sediment durchgeführt wird, sollte die 14CO2-Messung indirekt durch Entnahme einer homogenen 10-ml-Probe des im Test verwendeten Wassers bzw. der im Test verwendeten Suspension und anschließende Trennung der Phasen durch Zentrifugieren bei geeigneter Geschwindigkeit (z.B. 40.000 m/s2, 15 min) erfolgen. Danach sollte die wässerige Phase wie oben beschrieben behandelt werden. Die 14C-Aktivität von als Feststoff vorliegendem organischem Kohlenstoff (POA) sollte durch erneute Suspendierung des Sediments in einer kleinen Menge destillierten Wassers, Einbringung in Szintillationsfläschchen und Zugabe einer Szintillationsflüssigkeit zur Bildung eines Gels ermittelt werden. (Für diesen Zweck geeignete Szintillationsflüssigkeiten sind verfügbar.) Je nach Beschaffenheit der Feststoffe (z.B. Anteil an organischem Material) kann die Probe unter Umständen über Nacht mit einem Gewebelöser verarbeitet und dann in einem Wirbelmischer homogenisiert werden, bevor die Szintillationsflüssigkeit hinzugegeben wird. Alternativ kann die POA durch Verbrennung des überschüssigen Sauerstoffs mit einem Oxydationsmittel bestimmt werden. Beim Zählen sollten grundsätzlich interne Standards einbezogen werden; unter Umständen müssen Querempfmdlichkeitskorrekturen unter Zugabe eines internen Standards zu den einzelnen Proben vorgenommen werden.
Direkte 14CO2 Bestimmung
Wenn das entstandene 14CO2 direkt gemessen werden soll, sind bei Beginn des Tests zusätzliche Kolben vorzubereiten. Bei der Probenahme wird jeweils der gesamte Inhalt der Kolben entnommen. Die Kolben werden auf einen pH-Wert von 2-3 angesäuert und das 14CO2 in einem internen (bereits bei Beginn des Tests in die Testkolben gebrachten) Absorber oder einem externen Absorber gebunden. Als Absorptionsmedium kann wahlweise Alkali (z.B. 1 N NaOH-Lösung oder ein NaOHPellet), Ethanolamin oder ein im Handel erhältlicher Absorber auf Ethanolaminbasis verwendet werden. Für direkte Messungen des 14CO2-Gehalts sollten die Kolben z.B. mit Butylkautschuk-Septen verschlossen werden.
Abbildung 1a Beispiel einer arithmetischen Darstellung der Daten (Restaktivität/Zeit)
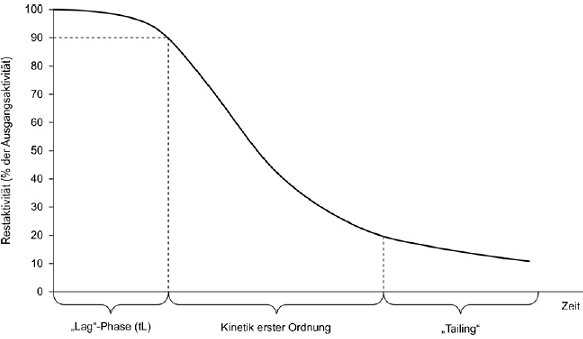
Abbildung 1b Beispiel einer semilogarithmischen Darstellung der Daten (In Restaktivität/Zeit)
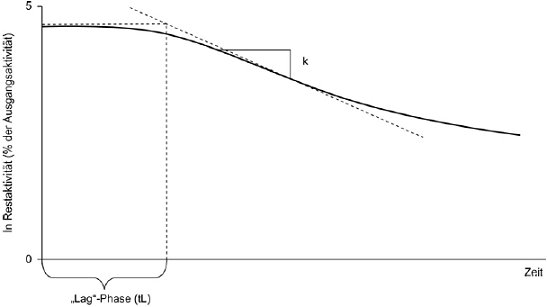
| Anhang VI |
C.26 Lemna SP. Wachstumsinhibiptionstest
1. Methode
Diese Methode entspricht der Prüfrichtlinie OECD TG 221 (2006) (1). Unter den Behörden der EU bestand breiter Konsens darüber, dass der Lemna-Test bei stark kolorierten Substanzen eine geeignete Alternative zur Prüfung an einer Alge ist (2)(3).
1.1 Einleitung
Mit dieser Prüfmethode soll die Toxizität von Substanzen für Süßwasserpflanzen der Gattung Lemna (Wasserlinse) beurteilt werden. Die Methode beruht auf bestehenden Leitlinien (4)(5)(6)(7)(8)(9), sieht aber Änderungen der entsprechenden Methoden vor, die neueren Forschungsergebnissen und Anhörungen bezüglich einer Reihe wesentlicher Aspekte Rechnung tragen. Die vorgeschlagene Methode wurde durch einen internationalen Ringtest validiert (10).
Diese Prüfmethode beschreibt Toxizitätstests mit Lemna gibba und Lemna minor, die beide umfassend untersucht wurden und Gegenstand der genannten Standards waren. Die Taxonomie von Lemna spp. ist schwierig; problematisch sind die zahlreichen Phänotypen. Bei Lemna kann die Reaktion auf Giftstoffe zu genetischer Variabilität führen; zurzeit sind jedoch keine hinreichenden Daten zu den Ursachen dieser Variabilität verfügbar, welche die Empfehlung eines spezifischen Klons für diese Prüfmethode rechtfertigen würden. Es wird darauf hingewiesen, dass der Test nicht axenisch durchgeführt wird; in verschiedenen Stadien während der Durchführung des Tests wird jedoch mit geeigneten Maßnahmen versucht, Verunreinigungen durch andere Organismen auf ein Minimum zu begrenzen.
Die Durchführung von Tests sowohl unter Erneuerung der Testlösung (semistatische Tests und Durchflusstests) als auch ohne Erneuerung der Testlösung (statische Tests) wird detailliert beschrieben. Abhängig von den Zielsetzungen der Tests sowie von rechtlichen Anforderungen wird empfohlen, den Einsatz von semistatischen Methoden sowie von Durchflussmethoden zu prüfen (z.B. für Substanzen, die durch Verflüchtigung, Photodegradation, Ausfällung oder biologischen Abbau rasch verloren gehen). Weitere Informationen sind Quelle (11) zu entnehmen.
1.2 Begriffsbestimmungen
Im Zusammenhang mit dieser Prüfmethode werden die folgenden Begriffsbestimmungen zugrunde gelegt und folgende Abkürzungen verwendet:
Biomasse: Trockengewicht des in einer Population enthaltenen lebenden Materials; bei diesem Test werden typischerweise Surrogate für die betreffende Biomasse (z.B. Frondzahl oder Frondfläche) gemessen; entsprechend bezieht sich der Begriff "Biomasse" auch auf diese Surrogatparameter.
Chlorose: Gelbfärbung des Blattmaterials.
KIon: ein Organismus oder eine Zelle, der bzw. die durch geschlechtslose Reproduktion aus einem einzelnen Organismus gewonnen wurde; aus jeweils demselben Klon gewonnene Organismen sind entsprechend genetisch identisch.
Kolonie: Gesamtheit der miteinander verbundenen Mutter- und Tochter-Fronds (gewöhnlich 2 bis 4); gelegentlich auch als Pflanze bezeichnet.
ECx: Konzentration der im Prüfmedium aufgelösten Prüfsubstanz, bei der sich binnen einer festgelegten Expositionsdauer eine Reduzierung des Wachstums von Lemna um x % (z.B. 50 %) ergibt. (Die Expositionsdauer ist ausdrücklich zu nennen, wenn die Dauer von der vollständigen oder normalen Testdauer abweicht.) Um einen von der Wachstumsrate oder vom Zellertrag abweichenden EC-Wert eindeutig zu kennzeichnen, wird die Bezeichnung "Er C" für die Wachstumsrate und "Er C" für den Zellertrag jeweils gefolgt von der verwendeten Messvariablen (z.B. Er C (Frondzahl)) verwendet.
Durchflusstest: ein Test, bei dem die Testlösungen kontinuierlich ersetzt werden.
Frond: eine separateleinzelne "blattartige" Struktur einer Wasserlinsen-Pflanze; kleinste reproduktionsfähige Einheit (d. h. einzelner Organismus).
Aufwölbungen: Fronds mit einer Wölbung oder Schwellung.
Wachstum: Zunahme der Messvariablen, z.B. Frondzahl, Trockengewicht, Feuchtgewicht oder Frondfläche während der Testdauer.
Wachstumsrate (durchschnittliche spezifische Wachstumsrate): logarithmische Zunahme der Biomasse während der Expositionsdauer.
Niedrigste Konzentration mit beobachteter Wirkung (LOEC): niedrigste geprüfte Konzentration, bei der beobachtet wurde, dass die Substanz binnen einer bestimmten Expositionsdauer gegenüber der Kontrollprobe eine statistisch signifikante Wachstumsreduzierung bewirkt (bei p < 0,05); allerdings müssen sämtliche Testkonzentrationen über der LOEC schädliche Folgen haben, die mindestens den bei der LOEC beobachteten schädlichen Folgen gleichwertig sind. Wenn diese beiden Bedingungen nicht erfüllt werden können, ist umfassend darzulegen, warum die LOEC (und entsprechend die NOEC) gewählt wurde.
Messvariablen: alle Variablentypen, die gemessen werden, um mit mindestens einer Reaktionsvariablen den Endpunkt des Tests zu beschreiben; Messvariablen bei dieser Methode sind Frondzahl, Frondfläche, Frischgewicht und Trockengewicht.
Monokukur: Kultur mit einer Pflanzenart.
Nekrose: totes (d. h. weißes oder mit Wasser durchfeuchtetes) Blattmaterial.
Höchste geprüfte Konzentration ohne beobachtete schädliche Wirkung (NOEC): Testkonzentration unmittelbar unterhalb der LOEC.
Phänotyp: zu beobachtende Merkmale eines Organismus, die durch Interaktion der Gene dieses Organismus mit seiner Umgebung bestimmt werden.
Reaktionsvariablen: Variablen für die geschätzte Toxizität, abgeleitet aus beliebigen gemessenen Variablen zur Beschreibung der Biomasse durch verschiedene Berechnungsmethoden; bei dieser Methode sind die Wachstumsraten und der Zellertrag Reaktionsvariablen, die aus Messvariablen wie z.B. Frondzahl, Frondfläche, Frischgewicht oder Trockengewicht abgeleitet werden.
Semistatischer (Erneuerungs-)Test: Test, bei dem die Testlösung während der Testdauer regelmäßig oder in bestimmten Intervallen ersetzt wird.
Statischer Test: Testmethode, bei der die Testlösung während der Testdauer nicht erneuert wird.
Endpunkt des Tests: beschreibt den allgemeinen Faktor, der bezogen auf die Kontrolle als Testziel durch die Prüfchemikalie verändert wird; bei dieser Methode wird als Endpunkt des Tests die Wachstumshemmung angenommen; diese kann durch verschiedene Reaktionsvariablen ausgedrückt werden, die jeweils auf mindestens einer Messvariablen beruhen.
Prüfmedium: gesamtes synthetisches Nährmedium, in dem die zu prüfenden Pflanzen wachsen, wenn sie der Prüfsubstanz ausgesetzt werden; die Prüfsubstanz wird im Allgemeinen im Prüfmedium aufgelöst.
Zellertrag: Wert einer Messvariablen zur Beschreibung der Biomasse am Ende der Expositionsdauer abzüglich der Messvariablen am Anfang der Expositionsdauer.
1.3 Prinzip der Prüfmethode
Exponentiell wachsende Pflanzenkulturen der Gattung Lemna werden über einen Zeitraum von sieben Tagen als Monokulturen in unterschiedlichen Konzentrationen der Prüfsubstanz kultiviert. Ziel des Tests ist die Quantifizierung von durch die Substanz bedingten Auswirkungen auf das Pflanzenwachstum während dieses Zeitraums, ausgehend von Bewertungen der ausgewählten Messvariablen. Die Frondzahl ist die primäre Messvariable. Mindestens eine weitere Messvariable (gesamte Frondfläche, Trockengewicht oder Frischgewicht) wird ebenfalls gemessen, da sich einige Substanzen erheblich stärker als die Frondzahlen auf die Messvariablen auswirken können. Um die substanzabhängigen Auswirkungen zu quantifizieren, wird das Wachstum der Testlösungen mit dem Wachstum der Kontrolllösungen verglichen, bei dem eine spezifizierte Wachstumshemmung von x % (z.B. 50 %) festgestellt wird, die dann als ECx (z.B. EC50) anzugeben ist.
Der Endpunkt des Tests ist die Wachstumshemmung ausgedrückt als logarithmische Zunahme der Messvariablen (durchschnittliche spezifische Wachstumsrate) während der Expositionsdauer. Aus den in einer Reihe von Testlösungen erfassten durchschnittlichen spezifischen Wachstumsraten wird die Konzentration bestimmt, bei der sich eine spezifizierte Hemmung der Wachstumsrate von x % (z.B. 50 %) ergibt; diese Konzentration wird als ErCx bezeichnet (z.B. ErC50).
Bei dieser Prüfmethode wird als weitere Reaktionsvariable der Zellertrag verwendet; diese Variable kann erforderlich sein, damit in manchen Ländern bestimmte maßgebliche Rechtsvorschriften erfüllt werden. Die Variable ergibt sich aus den Messvariablen am Ende der Expositionsdauer abzüglich der Messvariablen am Beginn der Expositionsdauer. Aus dem in einer Reihe von Testlösungen ermittelten Zellertrag werden die Konzentrationen berechnet, bei denen sich eine festgelegte Zellertrag-Hemmung von x % (z.B. 50 %) ergibt; diese Konzentration wird als EyCx bezeichnet (z.B. EyC50).
Außerdem können die niedrigste Konzentration mit beobachteter Wirkung (LOEC) und die höchste geprüfte Konzentration ohne beobachtete schädliche Wirkung (NOEC) statistisch bestimmt werden.
1.4 Informationen zur Prüfsubstanz
Eine Analysemethode mit geeigneter Empfmdlichkeit für die Quantifizierung der im Prüfmedium enthaltenen Substanz sollte verfügbar sein.
Zur Festlegung der Testbedingungen hilfreiche Informationen zur Prüfsubstanz sind die Strukturformel, die Wasserlöslichkeit, die Stabilität in Wasser, die Lichtbeständigkeit, pKa, Kow, der Dampfdruck und die biologische Abbaubarkeit. Aus der Wasserlöslichkeit und dem Dampfdruck kann die Henry-Konstante berechnet werden, aus der zu entnehmen ist, ob während der Testdauer erhebliche Verluste der Prüfsubstanz zu erwarten sind. Die Konstante gibt Aufschluss darüber, ob bestimmte Maßnahmen zur Überwachung dieser Verluste durchgeführt werden sollten. Wenn Informationen zur Löslichkeit und zur Stabilität der Prüfsubstanz nicht zuverlässig sind, sollten die Substanzen unter den Testbedingungen (d. h. Nährmedium, Temperatur und Beleuchtung) untersucht werden.
Wenn der Steuerung des pH-Wertes des Prüfmediums besondere Bedeutung zukommt (z.B. beim Testen von Metallen oder sonstigen hydrolytisch instabilen Substanzen), wird die Zugabe einer Pufferlösung zum Nährmedium empfohlen (siehe Abschnitt 1.7.4 Absatz 1). Weitere Hinweise zu Prüfsubstanzen mit physikalisch-chemischen Merkmalen, welche die Durchführung des Tests erschweren, sind Quelle (11) zu entnehmen.
1.5 Referenzsubstanz
Um das Prüfverfahren zu testen, können Referenzsubstanzen wie z.B. das im internationalen Ringtest (10) verwendete 3,5-Dichlorphenol geprüft werden. Die Prüfsubstanzen sollten mindestens zweimal jährlich bzw., wenn die Tests seltener durchgeführt werden, gleichzeitig mit der Bestimmung der Toxizität der Prüfsubstanzen getestet werden.
1.6 Validität des Tests
Die Tests sind nur dann gültig, wenn die Zeit bis zur Verdopplung der Frondzahl der Kontrolle weniger als 2,5 Tage (60 h) und damit etwa einer Erhöhung um das Siebenfache binnen sieben Tagen bei einer durchschnittlichen spezifischen Wachstumsrate von 0,275 d-1 entspricht. Für die Medien und Testbedingungen gemäß dieser Methode kann dieses Kriterium mit dem Prüfprotokoll des statischen Tests (8) erfüllt werden. Außerdem wird angenommen, dass dieses Kriterium auch bei den Bedingungen des semistatischen Tests und des Durchflusstests erfüllt wird. Wie die Verdopplungszeit zu berechnen ist, wird in Abschnitt 2.1 beschrieben.
1.7 Beschreibung der Methode
1.7.1 Apparatur
Sämtliche Geräte, die mit dem Prüfmedium in Berührung kommen, sollten aus Glas oder einem sonstigen chemisch inerten Material bestehen. Die zur Kultivierung und für die Tests verwendeten Glasgeräte sollten steril sein und von chemischen Verunreinigungen befreit werden, die in das Prüfmedium gelangen könnten. Die Prüfgefäße sollten so groß sein, dass die Fronds der verschiedenen Kolonien in den Kontrollgefäßen wachsen können, ohne sich am Ende der Testdauer zu überlagern. Dass die Wurzeln gegebenenfalls den Boden der Prüfgefäße berühren, ist unerheblich; in jedem Fall ist jedoch eine Mindesttiefe von 20 mm und ein Mindestvolumen von 100 ml pro Prüfgefäß zu empfehlen. Die Art der Prüfgefäße ist nicht entscheidend, sofern diese Anforderungen erfüllt sind. Bewährt haben sich Bechergläser, Kristallisierungsschalen und Petrischalen mit geeigneten Abmessungen. Die Prüfgefäße werden abgedeckt, um die Verdampfung und zufällige Verunreinigungen zu minimieren und trotzdem den erforderlichen Luftaustausch zu ermöglichen. Die Prüfgefäße und insbesondere die verwendeten Abdeckungen dürfen keine Schatten erzeugen und keine Änderungen des Lichtspektrums bewirken.
Kulturen und Prüfgefäße sollten nicht zusammen gelagert werden. Um diese Anforderung zu erfüllen, werden am besten getrennte Wachstumskammern bzw. getrennte Inkubatoren oder Räume verwendet. Beleuchtung und Temperatur müssen kontrolliert werden können, und für Beleuchtung und Temperatur müssen gleichbleibende Werte aufrechterhalten werden können (siehe Abschnitt 1.7.8).
1.7.2 Testorganismus
Für diesen Test wird entweder Lemna gibba oder Lemna minor verwendet. In Anlage 1 sind Kurzbeschreibungen von in Toxizitätstests verwendeten Wasserlinsenarten zusammengestellt. Das Pflanzenmaterial kann aus einer Kultursammlung oder aus einem anderen Labor bezogen oder aus der Natur entnommen werden. Bei Entnahme aus der Natur sollten die Pflanzen mindestens acht Wochen vor der Verwendung in dem Medium kultiviert werden, das auch für die Tests verwendet wird. Orte in der freien Natur, aus denen die Ausgangskulturen entnommen werden, dürfen keinen offensichtlichen Quellen von Verunreinigungen ausgesetzt sein. Wenn die Kulturen aus einem anderen Labor oder aus einer Kultursammlung bezogen werden, sollten sie ebenfalls mindestens drei Wochen unter ähnlichen Bedingungen kultiviert werden. Angegeben werden sollten auch die Herkunft des Pflanzenmaterials, der Arten und des Klons (wenn bekannt), die für die Tests verwendet werden.
Für die Tests sollten Monokulturen verwendet werden, die keine sichtbaren Verunreinigungen durch andere Organismen als Algen und Protozoen aufweisen. Gesunde Pflanzen der Art L. minor bestehen aus Kolonien mit zwei bis fünf Fronds; gesunde L. gibba-Kolonien können bis zu sieben Fronds umfassen.
Qualität und Einheitlichkeit der für die Tests verwendeten Pflanzen haben erhebliche Auswirkungen auf das Ergebnis der Tests; entsprechend sorgfältig sollten die Pflanzen ausgewählt werden. Nach Möglichkeit sollten junge, rasch wachsende Pflanzen ohne sichtbare Läsionen oder Verfärbungen (Chlorose) verwendet werden. Hochwertige Kulturen weisen einen hohen Anteil an Kolonien mit mindestens zwei Fronds auf. Zahlreiche einzelne Fronds deuten auf Umweltstress hin (z.B. auf Nährstoffmangel); Pflanzenmaterial aus diesen Kulturen sollte für die Tests nicht verwendet werden.
1.7.3 Kultivierung
Um den Kultivierungsaufwand zu reduzieren (z.B. wenn über einen bestimmten Zeitraum keine Tests mit Lemna vorgesehen sind), können die Kulturen bei verringerter Beleuchtung und niedrigerer Temperatur (4-10 °C) gelagert werden. Nähere Informationen zur Kultivierung sind Anlage 2 zu entnehmen. Bei offensichtlichen Anzeichen für Verunreinigungen durch Algen oder sonstige Organismen muss eine Oberflächensterilisation einer Teilprobe der Lemna-Fronds vorgenommen werden und anschließend eine Übertragung in ein frisches Medium erfolgen (siehe Anhang 2). In diesem Fall sollte die verbleibende verunreinigte Kultur verworfen werden.
Mindestens sieben Tage vor Durchführung der Tests wird eine hinreichende Anzahl an Kolonien keimfrei in ein frisches steriles Medium gebracht und 7-10 Tage bei Testbedingungen kultiviert.
1.7.4 Prüfmedium
Für Lemna minor und Lemna gibba werden jeweils die im Folgenden genannten Medien empfohlen. Wenn angenommen wird, dass das Medium mit der Prüfsubstanz reagieren und die Toxizitätswirkung beeinflussen könnte, ist sorgfältig zu prüfen, ob eine pH-Pufferlösung zum Prüfmedium gegeben werden sollte (beim L. minor-Medium MOPS (4-Morpholinpropan-Sulfonsäure, CAS-Nr. 1132-61-2; EINECS-Nr. 214-478-5) und beim L. gibba-Medium NaHCO3). Das Steinberg-Medium (12) ist ebenfalls annehmbar, wenn die Validitätskriterien erfüllt werden.
Zur Kultivierung von L. minor und für Tests mit L. minor wird eine Modifizierung des SIS-Lemna-Nährmediums (SIS = schwedisches Standardmedium) empfohlen. Die Zusammensetzung dieses Mediums ist Anlage 3 zu entnehmen.
Zur Kultivierung von L. gibba und für Tests mit L. gibba wird das Nährmedium 20X - AAP (siehe Anlage 3) empfohlen.
Das in Anlage 3 beschriebene Steinberg-Medium ist für L. minor ebenfalls geeignet, kann aber auch für L. gibba verwendet werden, wenn die Validitätskriterien erfüllt sind.
1.7.5 Testlösungen
Die Testlösungen werden gewöhnlich durch Verdünnung einer Stammlösung hergestellt. Zum Herstellen von Stammlösungen der Prüfsubstanz wird die Substanz im Allgemeinen im Nährmedium gelöst.
Die höchste getestete Konzentration der Prüfsubstanz sollte in der Regel die Wasserlöslichkeit der Substanz bei den jeweiligen Testbedingungen nicht überschreiten. Allerdings wird darauf hingewiesen, dass Lemna spp. auf der Oberfläche treiben und Substanzen ausgesetzt werden könnten, die sich am Übergang zwischen Wasser und Umgebungsluft sammeln (z.B. schlecht wasserlösliche oder hydrophobe Substanzen oder oberflächenaktive Wirkstoffe). Unter diesen Umständen besteht eine Belastung durch nicht in der Lösung enthaltene Materialien, und die Testkonzentrationen können je nach den Merkmalen der Prüfsubstanz die Wasserlöslichkeit überschreiten. Bei Prüfsubstanzen mit geringerer Wasserlöslichkeit muss unter Umständen mit einem organischen Lösungsmittel oder einem Dispergiermittel eine konzentrierte Stammlösung oder eine Dispersion der Substanz hergestellt werden, damit leichter die exakten Mengen der Prüfsubstanz zum Prüfmedium hinzugegeben werden können und damit die Dispergierung und die Auflösung der Substanz begünstigt wird. Es sollte unbedingt versucht werden, die Verwendung dieser Materialien zu vermeiden. Infolge der Verwendung unterstützender Lösungsmittel oder Dispergiermittel sollte keine Phytotoxizität entstehen. Häufig verwendete Lösungsmittel, die bei Konzentrationen bis zu 100 µl×l-1 keine phototoxische Wirkung haben, sind z.B. Aceton und Dimethylformamid. Wenn ein Lösungsmittel oder ein Dispergiermittel verwendet wird, sollte die Endkonzentration protokolliert und auf ein Minimum (≤100 µl×l-1) beschränkt werden; alle behandelten Proben und die Kontrollproben sollten das Lösungsmittel bzw. das Dispergiermittel in derselben Konzentration enthalten. Weitere Informationen zur Verwendung von Dispergiermitteln sind Quelle (11) zu entnehmen.
1.7.6 Test- und Kontrollgruppen
Die vorherige Kenntnis der Toxizität der Prüfsubstanz für Lemna (z.B. aufgrund eines Tests zur Bereichsermittlung) erleichtert die Auswahl geeigneter Testkonzentrationen. Beim definitiven Toxizitätstest sollten in der Regel mindestens fünf Testkonzentrationen in geometrischen Reihen angeordnet sein. Der Trennfaktor zwischen den Testkonzentrationen sollte höchstens 3,2 betragen; bei flachen Konzentrations-Reaktionskurven kommen jedoch auch höhere Werte in Betracht. Die Verwendung von weniger als fünf Konzentrationen sollte begründet werden. Für jede Testkonzentration sollten mindestens drei Wiederholungen verwendet werden.
Beim Festlegen des Testkonzentrationsbereichs (zur Bereichsermittlung und/oder für den definitiven Toxizitätstest) sind folgende Punkte zu berücksichtigen:
Die Tests sollten jeweils Kontrollen beinhalten, bei denen dasselbe Nährmedium, dieselbe Anzahl an Fronds und Kolonien und die gleichen Umgebungsbedingungen und Verfahren wie in den Prüfgefäßen gegeben sind und nur die Prüfsubstanz fehlt. Wenn ein zusätzliches Lösungsmittel oder Dispergiermittel verwendet wird, sollte eine zusätzliche Kontrollbehandlung mit dem Lösungsmittel/Dispergiermittel erfolgen, das in derselben Konzentration wie in den Gefäßen mit der Prüfsubstanz vorliegt. Die Anzahl der Kontrollgefäße zur Durchführung von Wiederholungen (sowie ggf. der Lösungsmittelgefäße) sollte mindestens identisch mit der Anzahl der für die verschiedenen Testkonzentrationen verwendeten Gefäße sein; im Idealfall sollten sogar doppelt so viele Gefäße verwendet werden.
Wenn die NOEC nicht bestimmt werden muss, kann das Prüfprotokoll geändert werden, indem die Anzahl der Konzentrationen erhöht und die Anzahl der Wiederholungen verringert wird. Zur Kontrolle sollten mindestens drei Wiederholungen durchgeführt werden.
1.7.7 Exposition
Kolonien mit zwei bis vier sichtbaren Fronds werden unter keimfreien Bedingungen aus der Impfkultur übertragen und zufällig den Prüfgefäßen zugewiesen. Die Prüfgefäße sollten jeweils insgesamt neun bis zwölf Fronds enthalten. Die Anzahl der Fronds und Kolonien sollte in allen Prüfgefäßen jeweils identisch sein. Erfahrungen mit dieser Methode sowie Daten aus Ringtests haben gezeigt, dass drei Wiederholungen pro Behandlung und pro Wiederholung anfänglich neun bis zwölf Fronds hinreichend sind, um Unterschiede hinsichtlich der Wachstumshemmungen in der Größenordnung von ca. 4 bis 7 % aufgrund der Wachstumsrate (pro Zellertrag 10 bis 15 % berechnet) zwischen den Behandlungen feststellen zu können (10).
Die Prüfgefäße müssen randomisiert im Inkubator angeordnet werden, um die Auswirkungen räumlich unterschiedlicher Lichtintensitäten und Temperaturen zu minimieren. Außerdem sind eine blockweise Anordnung oder ein zufälliges Einsetzen der Gefäße während der Durchführung der Messungen (oder noch häufiger) erforderlich.
Wenn aufgrund eines vorläufigen Stabilitätstests anzunehmen ist, dass die Prüfsubstanzkonzentration während der Testdauer (7 Tage) nicht aufrechterhalten werden kann (d. h. wenn die gemessene Konzentration unter 80 % der gemessenen Ausgangskonzentration fällt), wird ein semistatischer Test empfohlen. In diesem Fall sollten die Kolonien während der Testdauer mindestens zweimal (z.B. an den Tagen 3 und 5) frisch hergestellten Test- und Kontrolllösungen ausgesetzt werden. Wie häufig die Lösungen dem frischen Medium ausgesetzt werden, hängt von der Stabilität der Prüfsubstanz ab. Bei sehr instabilen oder flüchtigen Substanzen ist unter Umständen eine häufigere Exposition erforderlich, um die Konzentrationen annähernd konstant zu halten. Unter gewissen Umständen kann auch die Durchführung eines Durchflusstests erforderlich sein (11) (13).
Das Expositionsszenario beim Besprühen wird in dieser Prüfmethode nicht berücksichtigt; diesbezüglich wird auf Quelle (14) verwiesen.
1.7.8 Inkubationsbedingungen
Durch kontinuierliche fluoreszierende Beleuchtung mit warmem oder kaltweißem Licht sollte eine Lichtintensität hergestellt werden, die bei Messung unter photosynthetisch aktiver Strahlung (400-700 nm) an Punkten jeweils in demselben Abstand von der Lichtquelle wie die Lemna-Fronds im Bereich 85 - 135 µE×m-2.s-1(entsprechend etwa 6.500 - 10.000 lx) liegt. Abweichungen von der gewählten Lichtintensität dürfen im Testbereich höchstens ± 15 % betragen. Dabei ist zu beachten, dass die Messwerte von der Methode zur Feststellung und zur Messung der Lichtintensität (insbesondere vom Sensortyp) abhängen. Kugelförmige Sensoren (die auf Licht aus allen Winkeln über und unter der Messebene reagieren) sowie "Kosinus"-Sensoren (die auf Licht aus allen Winkeln über der Messebene ansprechen) sind gegenüber unidirektionalen Sensoren zu bevorzugen, da diese Sensoren bei Mehrpunkt-Lichtquellen des hier beschriebenen Typs höhere Messwerte ergeben.
Die Temperatur der Prüfgefäße sollte 24 ± 2 °C betragen. Der pH-Wert des Kontrollmediums darf während des Tests höchstens um 1,5 Einheiten ansteigen. Auch bei Abweichungen von mehr als 1,5 Einheiten sind Testergebnisse jedoch dann nicht ungültig, wenn nachgewiesen werden kann, dass die Validitätskriterien erfüllt sind. Erhöhte Sorgfalt ist bei der Beurteilung von Verschiebungen des pH-Wertes in Sonderfällen geboten (z.B. beim Testen instabiler Substanzen oder beim Testen von Metallen). Weitere Informationen in diesem Zusammenhang sind Quelle (11) zu entnehmen.
1.7.9 Dauer
Der Test wird sieben Tage nach Einbringung der Pflanzen in die Prüfgefäße beendet.
1.7.10 Messungen und analytische Bestimmungen
Bei Beginn des Tests wird die Anzahl der in den Prüfgefäßen enthaltenen Fronds ermittelt und protokolliert; zu zählen sind alle herausragenden, deutlich erkennbaren Fronds. Die Anzahl der normal oder anomal aussehenden Fronds ist bei Beginn des Tests, alle drei Tage während der Expositionsdauer (d. h. binnen des Zeitraums von sieben Tagen mindestens zweimal) und am Ende des Tests zu bestimmen. Änderungen in der Entwicklung der Pflanzen (z.B. Änderungen der Größe oder des Aussehens der Fronds, Anzeichen für eine Nekrose, Chlorose oder Aufwölbungen, das Aufbrechen von Kolonien oder der Verlust der Schwimmfähigkeit sowie Veränderungen der Wurzellänge oder der sonstigen Beschaffenheit der Wurzeln) sind zu protokollieren. Wesentliche Merkmale des Prüfmediums (z.B. Vorliegen nicht gelösten Materials oder Algenwachstum im Prüfgefäß) sollten ebenfalls verzeichnet werden.
Ergänzend zur Ermittlung der Frondzahl während des Tests sind auch die Auswirkungen der Prüfsubstanz auf eine (oder mehrere) der folgenden Messvariablen zu bewerten:
Die Gesamtfläche der Fronds hat den Vorteil, dass sie für jedes einzelne Prüfgefäß und für jedes einzelne Kontrollgefäß jeweils bei Beginn des Tests, während der Durchführung des Tests und am Ende des Tests bestimmt werden kann. Das Trockengewicht und das Frischgewicht sollten bei Beginn des Tests an einer Probe der Impfkultur ermittelt werden, die typisch für das bei Beginn des Tests verwendete Material ist; eine weitere Feststellung sollte am Ende des Tests anhand des Pflanzenmaterials jeweils aus den Prüfgefäßen und aus den Kontrollgefäßen erfolgen. Wenn die Frondfläche nicht gemessen wird, sollte eher das Trockengewicht als das Frischgewicht ermittelt werden.
Die Gesamtfläche der Fronds, das Trockengewicht und das Frischgewicht können wie folgt bestimmt werden:
1.7.10.1 Häufigkeit der Messungen und der analytischen Bestimmungen
Bei statischen Tests sollte jeweils bei Beginn des Tests und am Ende des Tests der pH-Wert der behandelten Lösungen gemessen werden. Bei semistatischen Tests sollte für alle Batches der "frischen" Testlösung jeweils vor den Erneuerungen der pH-Wert ermittelt werden; außerdem sollte der pH-Wert der "verbrauchten" Lösungen bestimmt werden.
Die Lichtintensität sollte in der Wachstumskammer, im Inkubator oder im jeweiligen Raum in dem Abstand von der Lichtquelle gemessen werden, der auch für die Lemna-Fronds gegeben ist. Während des Tests sollte mindestens eine Messung vorgenommen werden. Die Temperatur des Mediums in einem Surrogatgefäß, das Bedingungen ausgesetzt wird, die in der Wachstumskammer bzw. im Inkubator oder im jeweiligen Raum gegeben sind, sollte mindestens täglich protokolliert werden.
Während des Tests wird die Konzentration der Prüfsubstanz in geeigneten Intervallen bestimmt. Bei statischen Tests ist die Konzentration mindestens bei Beginn des Tests und am Ende des Tests zu ermitteln.
Bei semistatischen Tests, bei denen davon ausgegangen wird, dass die Konzentration der Prüfsubstanz nicht im Bereich von 20 % der Nennkonzentration aufrechterhalten werden kann, müssen alle frisch hergestellten Testlösungen sowie alle Lösungen jeweils nach einer Erneuerung analysiert werden (siehe Abschnitt 1.7.7 Absatz 3). Bei Tests, bei denen die gemessene Ausgangskonzentration der Prüfsubstanz zwar nicht im Bereich von 20 % der Nennkonzentration liegt, bei denen jedoch hinreichend nachgewiesen werden kann, dass die Ausgangskonzentrationen wiederholbar und stabil sind (d. h. dass die Konzentrationen im Bereich von 80-120 % der Ausgangskonzentration liegen), sind chemische Bestimmungen nur bei der höchsten und bei der niedrigsten Konzentration erforderlich. In jedem Fall brauchen die Prüfsubstanzkonzentrationen für alle Testkonzentrationen vor der Erneuerung jeweils nur bei einer einzigen Wiederholung (bzw. bei Gefäßen mit zusammengefassten Wiederholungen jeweils bei nur einem Gefäß) erneut bestimmt zu werden.
Bei Durchflusstests ist ähnlich zu verfahren, wie bei den semistatischen Tests, d. h. Analysen sind jeweils bei Beginn des Tests, während des Tests und am Ende des Tests durchzuführen; Messungen der "verbrauchten" Lösungen sind jedoch nicht erforderlich. Bei diesem Testtyp sollten der Durchfluss des Verdünnungsmittels und der Prüfsubstanz-Stammlösung täglich geprüft werden.
Wenn nachgewiesen wird, dass die Konzentration der Prüfsubstanz während der gesamten Testdauer in befriedigender Weise im Bereich von 20 % der Nennkonzentration oder der gemessenen Ausgangskonzentration aufrechterhalten werden konnte, können die Ergebnisse auch ausgehend von den Nennwerten bzw. von den gemessenen Ausgangswerten durchgeführt werden. Beträgt die Abweichung von der Nennkonzentration oder von der gemessenen Ausgangskonzentration mehr als 20 %, sollte bei der Analyse der Ergebnisse von der geometrischen mittleren Konzentration während der Expositionsdauer oder von Modellen ausgegangen werden, die den Rückgang der Prüfsubstanzkonzentration beschreiben (11).
1.7.11 Limit-Test
Unter gewissen Umständen, z.B. wenn ein vorläufiger Test darauf hindeutet, dass die Prüfsubstanz bei Konzentrationen bis zu 100 mg ×l-1 bm bis zur Löslichkeitsgrenze im Prüfmedium (maßgeblich ist die jeweils niedrigere Konzentration) keine toxische Wirkung hat, kann ein Limit-Test durchgeführt werden, in dem die Reaktionen einer Kontrollgruppe und einer Behandlungsgruppe (100 mg×l-1 bm eine mit der Löslichkeitsgrenze identische Konzentration) verglichen werden. Es wird nachdrücklich empfohlen, diese Tests durch Analysen der Expositionskonzentration zu verifizieren. Alle oben beschriebenen Testbedingungen und Validitätskriterien beziehen sich auf einen Limit-Test; allerdings sollte die Anzahl der behandelten Wiederholungen mindestens doppelt so hoch sein. Das Wachstum der Kontrollgruppe und der Behandlungsgruppe kann mit einem statistischen Test zum Vergleich der Mittelwerte analysiert werden (z.B. mit einem Student-Test).
2. Daten und Abschlussbericht
2.1 Verdopplungszeit
Um die Dauer bis zur Verdopplung der Frondzahl (Td) zu bestimmen und um sicherzustellen, dass dieses Validitätskriterium von der Studie erfüllt wird (siehe Abschnitt 1.6), ist mit den Daten der Kontrollgefäße die folgende Gleichung zu berechnen:
Td = ln 2/µ
Dabei steht µ für die durchschnittliche spezifische Wachstumsrate, die bestimmt wurde, wie in Abschnitt 2.2.1 in Absatz 2 beschrieben.
2.2 Reaktionsvariablen
Mit der Prüfung sollen die Auswirkungen der Prüfsubstanz auf das vegetative Wachstum von Lemna bestimmt werden. Diese Prüfmethode beschreibt zwei Reaktionsvariablen, da in den Mitgliedsländern unterschiedliche Präferenzen und rechtliche Anforderungen bestehen. Damit die Testergebnisse in allen Mitgliedsländern anerkannt werden können, sollten die Auswirkungen mit Hilfe der beiden im Folgenden beschriebenen Reaktionsvariablen a) und b) beurteilt werden.
Es wird darauf hingewiesen, dass die mit diesen beiden Reaktionsvariablen berechneten Toxizitätswerte nicht vergleichbar sind; der entsprechende Unterschied muss bei der Verwendung der Testergebnisse berücksichtigt werden. Die mit der durchschnittlichen spezifischen Wachstumsrate (Er Cx berechneten Werte für ECx werden im Allgemeinen höher sein als die anhand des Zellertrags (EyCx) ermittelten Werte, wenn die für diese Testmethode vorgesehenen Bedingungen eingehalten werden; dies ist auf die unterschiedliche mathematische Grundlage der beiden Berechnungsverfahren zurückzuführen. Die auftretenden Unterschiede sollten jedoch nicht als Anzeichen für eine unterschiedliche Empfindlichkeit der beiden Reaktionsvariablen betrachtet werden; beide Werte sind einfach mathematisch verschieden. Das Konzept der durchschnittlichen spezifischen Wachstumsrate beruht auf dem im Allgemeinen exponentiellen Verlauf des Wasserlinsenwachstums bei nicht beschränkten Kulturen, bei denen die Toxizität aufgrund der Auswirkungen auf die Wachstumsrate geschätzt wird, ohne jedoch von der absoluten Höhe der jeweiligen Wachstumsrate der Kontrollprobe, von der Steigung der Konzentration-Reaktionskurve oder von der Testdauer abhängig zu sein. Auf der Reaktionsvariable Zellertrag beruhende Ergebnisse hingegen hängen von allen übrigen genannten Variablen ab. EyCx ist von der spezifischen Wachstumsrate der in den einzelnen Tests verwendeten Wasserlinsenarten sowie von der maximalen spezifischen Wachstumsrate abhängig, die je nach Art sowie sogar zwischen den einzelnen Klonen unterschiedlich sein kann. Diese Reaktionsvariable sollte nicht verwendet werden, um die Empfindlichkeit von Algenarten oder auch nur verschiedener Klone gegenüber Giftstoffen zu vergleichen. Die Verwendung der durchschnittlichen spezifischen Wachstumsrate zur Schätzung der Toxizität wird in der Wissenschaft bevorzugt; bei dieser Prüfmethode werden jedoch auch Toxizitätsschätzungen aufgrund des Zellertrags berücksichtigt, um den derzeitigen rechtlichen Anforderungen einiger Länder Rechnung zu tragen.
Toxizitätsschätzungen sollten auf der Frondzahl sowie auf einer weiteren Messvariablen (gesamte Frondfläche, Trockengewicht oder Frischgewicht) beruhen, da sich manche Substanzen erheblich stärker auf andere Messvariablen auswirken können als die Frondzahl. Diese Auswirkungen wurden nicht festgestellt, wenn ausschließlich die Frondzahl berechnet wurde.
Die Anzahl der Fronds sowie alle sonstigen protokollierten Messvariablen (gesamte Frondfläche, Trockengewicht oder Frischgewicht) werden zusammen mit den Konzentrationen der Prüfsubstanz für jede Messung tabellarisch zusammengestellt. Anschließende Datenanalysen z.B. zur Schätzung von LOEC-, NOEC- oder ECx-Werten sollten auf den Werten der einzelnen Wiederholungen, nicht aber auf berechneten Mittelwerten der einzelnen Behandlungsgruppen beruhen.
2.2.1 Durchschnittliche spezifische Wachstumsrate
Die durchschnittliche spezifische Wachstumsrate in einem bestimmten Zeitraum wird als logarithmische Zunahme der Wachstumsvariablen (d. h. der Frondzahl und einer sonstigen Messvariablen (gesamte Frondfläche, Trockengewicht oder Frischgewicht)) mit der nachstehenden Formel für die einzelnen Wiederholungen der Kontrolllösungen und der behandelten Lösungen berechnet.
| µi-j = | ln (Nj) - ln (Ni) |
|
| |
| t |
wobei
Für jede Behandlungsgruppe und für jede Kontrollgruppe sind die mittlere Wachstumsrate sowie die Varianzschätzungen zu berechnen.
Die durchschnittliche spezifische Wachstumsrate sollte für die gesamte Testdauer berechnet werden. (In der vorstehenden Formel bezeichnet "i" den Beginn des Tests und der Zeitpunkt "j" das Ende des Tests.) Für alle Konzentrationen der Testlösungen und der Kontrolllösungen sind ein Mittelwert für die durchschnittliche spezifische Wachstumsrate zu berechnen und die entsprechenden Varianzschätzungen vorzunehmen. Außerdem sollte die abschnittsbezogene Wachstumsrate bestimmt werden, um die Auswirkungen der Prüfsubstanz während der Expositionsdauer beurteilen zu können (z.B. durch Prüfung der logarithmisch transformierten Wachstumskurven). Erhebliche Unterschiede zwischen der abschnittsbezogenen Wachstumsrate und der durchschnittlichen Wachstumsrate deuten auf Abweichungen vom konstanten exponentiellen Wachstum hin und erfordern eine genaue Überprüfung der Wachstumskurven. In diesem Fall wurde ein vorsichtigerer Ansatz in einem Vergleich der spezifischen Wachstumsraten der behandelten Kulturen während der Dauer der maximalen Hemmung mit den spezifischen Wachstumsraten der Kontrolllösungen im selben Zeitraum bestehen.
Die Hemmung der Wachstumsrate in Prozent (Ir) kann anschließend für jede Testkonzentration (Behandlungsgruppe) nach der folgenden Formel berechnet werden:
| %Ir = | (µc- µr) | x 100 |
|
| ||
| µc |
wobei
2.2.2 Zellertrag
Die Auswirkungen auf den Zellertrag werden ausgehend von zwei Messvariablen, d. h. der Frondzahl und einer sonstigen Messvariablen (der gesamten Frondfläche, dem Trockengewicht oder dem Frischgewicht) der jeweiligen Prüfgefäße am Anfang und am Ende des Tests bestimmt. Für das Trockengewicht und das Frischgewicht wird die Ausgangsbiomasse ausgehend von einer Frond-Probe aus dem betreffenden Batch bestimmt, aus der die Prüfgefäße geimpft wurden (siehe Abschnitt 1.7.3 Absatz 2). Für die Testkonzentrationen und für die
Kontrolllösungen ist jeweils ein mittlerer Zellertrag zu berechnen; die Varianzen sind jeweils zu schätzen. Die prozentuale Hemmung des Zellertrags (% Iy) kann für die Behandlungsgruppen wie folgt berechnet werden:
| %Iy = | (bc- bT) | x 100 |
|
| ||
| bc |
wobei
2.2.3 Darstellung der Konzentration-Reaktionskurven
Die Konzentrations-Reaktionskurven der mittleren Hemmung der Reaktionsvariablen in Prozent (Ir bzw. Iy berechnet gemäß der Anweisung im letzten Absatz in Abschnitt 2.2.1 bzw. gemäß der Anweisung in Abschnitt 2.2.2) und die logarithmische Konzentration der Prüfsubstanz sollten grafisch dargestellt werden.
2.2.4 Schätzung von ECx
Schätzungen der ECx Werte (z.B. EC50) sollten sowohl auf der durchschnittlichen spezifischen Wachstumsrate (EyCx) als auch auf dem Zellertrag (EyCx) beruhen, und beide Werte sollten ihrerseits von der Frondzahl und von einer weiteren Messvariablen (gesamte Frondfläche, Trockengewicht oder Frischgewicht) ausgehen, weil sich manche Prüfsubstanzen in besonderer Weise auf die Frondzahl und sonstige Messvariablen auswirken. Die gewünschten Toxizitätsparameter bestehen entsprechend aus vier ECx-Werten für alle berechneten Hemmkonzentrationen x: EyCx(Frondzahl), EyCx(gesamte Frondfläche, Trockengewicht oder Frischgewicht), EyCx(Frondzahl), und EyCx(gesamte Frondfläche, Trockengewicht oder Frischgewicht).
2.3 Statistische Verfahren
Ziel ist die Ermittlung einer quantitativen Konzentrations-Reaktionsbeziehung durch Regressionsanalyse. Im Anschluss an eine linearisierte Transformation der Reaktionsdaten (z.B. in Einheiten nach dem Probit-, Logitoder Weibull-Modell) (16) kann eine gewichtete lineare Regression vorgenommen werden; nichtlineare Regressionsverfahren, mit denen die unvermeidlichen Unregelmäßigkeiten der Daten und Abweichungen von gleichförmigen Verteilungen besser verarbeitet werden können, werden jedoch bevorzugt. Gegen null bzw. gegen die vollständige Hemmung können diese Unregelmäßigkeiten durch die Transformation vergrößert werden und die Analyse beeinträchtigen (16). Es wird darauf hingewiesen, dass Standard-Analysmethoden mit Probit-, Logitoder Weibull-Transformationen für quantitative Daten (z.B. Mortalität oder Überlebensraten) vorgesehen sind und zur Verwendung in Verbindung mit Wachstums- oder Zellertragsdaten entsprechend modifiziert werden müssen. Spezifische Verfahren zur Bestimmung von ECx-Werten aus kontinuierlichen Daten sind den Quellen (17), (18) und (19) zu entnehmen.
Für jede zu analysierende Reaktionsvariable sind aufgrund der Konzentrations-Reaktionsbeziehung Punktschätzungen für die EC, Werte zu ermitteln. Nach Möglichkeit sollten für jede Schätzung die 95%-Konfidenzintervalle bestimmt werden. Die Qualität der Übereinstimmung der Reaktionsdaten mit dem Regressionsmodell sollte grafisch oder statistisch bewertet werden. Die Regressionsanalyse sollte mit den Reaktionen der einzelnen Wiederholungen (und nicht mit den Mittelwerten der Behandlungsgruppe) durchgeführt werden.
Schätzwerte für EC50 und für die Konfidenzintervalle können auch durch lineare Interpolation mit einem Bootstrapping-Algorithmus (20) erzielt werden, wenn die verfügbaren Regressionsmodelleimethoden für die betreffenden Daten nicht geeignet sind.
Für eine Schätzung der LOEC und entsprechend auch der NOEC müssen die Mittelwerte der behandelten Lösungen durch Varianzanalyseverfahren (ANOVA) verglichen werden. Der Mittelwert der einzelnen Konzentrationen ist dann mit einer geeigneten Methode zur Durchführung von Mehrfachvergleichen bzw. zur Durchführung von Trendtests mit dem Mittelwert der Kontrollgruppe zu vergleichen. Dunnett- und Williams-Tests können hilfreich sein (21)(22) (2 3)(24). Die ANOVA-Annahme der Varianzhomogenität muss einer Überprüfung unterzogen werden. Die entsprechende Bewertung kann anhand einer grafischen Darstellung oder aufgrund eines formalen Tests vorgenommen werden (25). Geeignet sind Levene- und Bartlett-Tests. Wenn die Annahme der Varianzhomogenität nicht erfüllt ist, kann gelegentlich eine Korrektur durch logarithmische Datentransformation erfolgen. Bei außerordentlicher Varianzheterogenität, die durch Transformation nicht korrigiert werden kann, sollten Analysen durch Methoden wie z.B. Jonckheere-Trendtests (Stepdown) erwogen werden. Weitere Hinweise zur Bestimmung von NOEC-Werten sind Quelle (19) zu entnehmen.
Aufgrund neuer Forschungsergebnisse wird empfohlen, das Konzept der NOEC aufzugeben und durch Punktschätzungen von ECx-Werten zu ersetzen, die durch Regression ermittelt wurden. Für diesen Lemna-Test wurde noch kein geeigneter Wert für x definiert. Ein Bereich von 10 bis 20 % scheint jedoch geeignet (abhängig von der auswählten Reaktionsvariablen); vorzugsweise sollten sowohl EC10 als auch EC20 protokolliert werden.
3. Abschlussbericht
3.1 Prüfbericht
Der Prüfbericht muss folgende Informationen enthalten:
Prüfsubstanz:
Im Test verwendete Art:
Prüfbedingungen:
Ergebnisse:
4. Literatur
1. OECD TG 221 (2006) Lemna Sp. Growth Inhibition Test.
2. Die Nutzung von Lemna-Studien für kolorierte Substanzen ist in Abschnitt 13.5.3 des EU-Handbuchs für Entscheidungen/Beschlüsse vom Juli 2006 ausgeführt: http://ecb.jrc.ec.europa.euinewchemicals.
3. Guidance an information requirements and chemical safety assessment - Kapitel R.7b: Endpoint specific guidance; Tabelle 7.8.3: Summary of dfficult substance testing issues, verfügbar unter http://guidance.echa.europa.euldocs/guidance_document/information_requirements_en.htm?time= 1234958685#A
4. ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). S. 733-742. Annual Book of ASTM Standards, Vol. 11.05 Biological Effects and Environmental Fate; Biotechnology; Pesticides, ASTM, West Conshohocken, PA.
5. USEPA - United States Environmental Protection Agency. (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., "Public draft". EPA 712-C-96-156. 8pp.
6. AFNOR - Association Francaise de Normalisation. (1996). XP T 90-337: Determination de Pinhibition de la croissance de Lemna minor. 10pp.
7. SSI - Swedish Standards Institute. (1995). Water quality- Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 S. (Schwedisch).
8. Environment Canada. (1999). Biological Test Method: Test for Measuring the Inhibition of Growth Using the Freshwater Macrophyte, Lemna minor. EPS 1/RM/37-120 S.
9. Environment Canada. (1993) Proposed Guidelines for Registration of Chemical Pesticides: Non-Target Plant Testing and Evaluation. Canadian Wildlife Service, Technical Report Series No. 145.
10. Sims I., Whitehouse P., and Lacey R. (1999) The OECD Lemna Growth Inhibition Test. Development and Ringtesting of draft OECD Test Guideline. R&D Technical Report EMA 003. WRc plc - Environment Agency.
11. OECD (2000). Guidance Document an Aquatic Toxicity Testing of Difficult Substances and Mixtures. OECD Environmental Health and Safety Publications, Series an Testing and Assessment No. 23.
12. ISO DIS 20079. Water Quality - Determination of the Toxic Effect of Water Constituents and Waste Water to Duckweed (Lemna minor) - Duckweed Growth Inhibition Test.
13. Walbridge C. T. (1977). A flowthrough testing procedure with duckweed (Lemna minor L.). Environmental Research Laboratory - Duluth, Minnesota 55804. US EPA Report No. EPA-6003-77.108. September 1977.
14. Lockhart W. L., Billeck B. N. and Baron C. L. (1989). Bioassays with a floating plant (Lemna minor) for effects of sprayed and dissolved glyphosate. Hydrobiologia 118119, S. 353-359.
15. Huebert, D.B. and Shay J.M. (1993) Considerations in the assessment of toxicityusing duckweeds. Environmental Toxicology and Chemistry 12, S. 481-483.
16. Christensen, E.R., Nyholm, N. (1984): Ecotoxicological Assays with Algae: Weibull Dose-Response Curves. Env. Sci. Technol. 19, S. 713-718.
17. Nyholm, N. Serensen, P.S., Kusk, K.O. and Christensen, E.R. (1992): Statistical treatment of data from microbial toxicity tests. Environ. Toxicol. Chem. 11, S. 157-167.
18. Bruce R.D. and Versteeg D.J. (1992) A statistical procedure for modelling continuous toxicity data. Environmental Toxicology and Chemistry 11, S. 1485-1494.
19. OECD. (2004). Guidance Document an Statistical Analysis of Ecotoxicity Data.
20. Norberg-King T.J. (1988) An interpolation estimate for chronic toxicity: The ICp approach. National Effluent Toxicity Assessment center Technical Report 05-88. USEPA, Duluth, MN.
21. Dunnett, C.W. (1955) A multiple comparisons procedure for comparing several treatments with a control. J. Amer. Statist. Assoc. 50, S. 1096-1121.
22. Dunnett, C.W. (1964) New tables for multiple comparisons with a control. Biometrics, 20, 482-491.
23. Williams, D.A. (1971) A test for differences between treatment means when several dose levels are compared with a zero dose control. Biometrics 27, S. 103-117.
24. Williams, D.A. (1972) The comparison of several dose levels with a zero dose control. Biometrics 28, S. 510-531.
25. Brain P. and Cousens R. (1989). An equation to describe doseresponses wherethereis stimulation of growth at low doses. Weed Research 29, S. 93-96.
| Beschreibung Lemna spp. | Anlage 1 |
Die gemeinsprachlich als Wasserlinse (Lemna spp.) bezeichnete Wasserpflanze zählt zur Familie der Lemnaceae, die weltweit in vier Gattungen mit verschiedenen Arten vorkommt. Das jeweilige Aussehen und die Taxonomie wurden umfassend dokumentiert (1)(2). Lemna gibba und L. minor sind für gemäßigte Regionen typische Arten; diese Arten werden häufig in Toxizitätstests verwendet. Beide Arten besitzen einen treibenden oder auch untergetauchten Stängel (Frond); von der Mitte der Unterseite der Fronds geht jeweils eine Wurzel aus. Lemna spp. bilden selten Blüten aus; die Pflanzen vermehren sich durch Austrieb neuer Fronds (3). Im Vergleich zu älteren Pflanzen sind jüngere Pflanzen eher blasser, besitzen kürzere Wurzeln und bestehen aus zwei bis drei Fronds unterschiedlicher Größe. Dank der geringen Größe, des einfachen Aufbaus, der geschlechtslosen Reproduktion und der kurzen Generationsdauer ist Lemna für Labortests in besonderer Weise geeignet (4)(5).
Da von unterschiedlichen Empfindlichkeiten der verschiedenen Arten auszugehen ist, sind ausschließlich Vergleiche der Empfindlichkeit jeweils einer einzigen Art annehmbar.
Beispiele für Lenm-Arten, die in Tests verwendet wurden: Artenreferenz
Lemna aequinoctialis: Eklund, B. (1996). The use of the red alga Ceramium strictum and the duckweed Lemna aequinoctialis in aquatic ecotoxicological bioassays. Licentiate in Philosophy Thesis 1996:2. Dep. of Systems Ecology, Stockholm University.
Lemna major. Clark, N. A. (1925). The rate of reproduction of Lemna major as a function of intensity and duration of light. J. phys. Chem. 29, S. 935-941.
Lemna minor. United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., "Public draft". EPA 712-C-96-156. 8pp.
Association Francaise de Normalisation (AFNOR). (1996). XP T 90-337: Determination de Pinhibition de la croissance de Lemna minor. 10 S.
Swedish Standards Institute (SIS). (1995). Water quality - Determination of growth inhibition (7-d) Lemna minor, duckweed. SS 02 82 13. 15 S. (Schwedisch).
Lemna gibba: ASTM International. (2003). Standard Guide for Conducting Static Toxicity Test With Lemna gibba G3. E 1415-91 (Reapproved 1998). S. 733-742.
United States Environmental Protection Agency (USEPA). (1996). OPPTS 850.4400 Aquatic Plant Toxicity Test Using Lemna spp., "Public draft". EPA 712-C-96-156. 8 S.
Lemna paucicostata: Nasu, Y., Kugimoto, M. (1981). Lemna (duckweed) as an indicator of water pollution. I. The sensitivity of Lemna paucicostata to heavy metals. Arch. Environ. Contam. Toxicol. 10, S. 1959-1969.
Lemna perpusilla: Clark, J. R. et al. (1981). Accumulation an d depuration of metals by duckweed (Lemna perpusilla). Ecotoxicol. Environ. Saf. 5, S. 87-96.
Lemna trisulca: Huebert, D. B., Shay, J. M. (1993). Considerations in the assessment of toxicity using duckweeds. Environ. Toxicol. and Chem. 12, S. 481-483.
Lemna valdiviana: Hutchinson, T.C., Czyrska, H. (1975). Heavy metal toxicityand synergism to floating aquatic weeds. Verh: Int. Ver. Limnol. 19, S. 2102-2111.
Bezugsquellen für Lemna-Arten
University of Toronto Culture Collection of Algae and Cyanobacteria
Department of Botany, University of Toronto
Toronto, Ontario, Canada, M5S 3 B2
Tel: +1-416-978-3641
Telefax:+ l-416- 9 7 8- 5 8 7 8
E-Mail: jacreman@botany.utoronto.ca
http://www.botany.utoronto.calutcc
North Carolina State University
Forestry Dept
Duckweed Culture Collection
Campus Box 8002
Raleigh, NC 27695-8002
United States
Telefon 001 (919) 515-7572
astomp@unity.ncsu.edu
Institute of Applied Environmental Research (UM) Stockholm University
SE -106 91 Stockholm
Schweden
Tel: +46 8.674 7240
+46 8.674 7636
Umweltbundesamt (UBA)
FG 111 3.4
Schichauweg 58
12307 Berlin
Deutschland
E-Mail: Lemna@uba.de
http://www.umweltbundesamt.delcontact.htm
Literatur
1. Hillman, W.S. (1961). The Lemnaceae or duckweeds: A review of the descriptive and experimental Literature. The Botanical Review 27 S. 221-287.
2. Landolt, E. (1986). Biosystematic investigations in the family of duckweed (Lemnaceae). Vol. 2. Geobotanischen Inst. ETH, Stiftung Rubel, Zürich, Switzerland.
3. Björndahl, G. (1982). Growth performance, nutrient uptake and human utilization of duckweeds (Lemnaceae family). ISBN 82-991150-0-0. The Agricultural Research Council of Norway, University of Oslo.
4. Wang, W. (1986). Toxicity tests of aquatic pollutants by using common duckweed. Environmental Pollution, Ser B 11, S. 1-14.
5. Wang, W. (1990). Literature review an duckweed toxicity testing. Environmental Research, 52, S. 7-22.
| Haltung der Stammkultur | Anlage 2 |
Die Stammkulturen können über längere Zeiträume bei niedrigeren Temperaturen (4-10 °C) gebrauchsfähig gelagert werden. Das Lemna-Nährmedium kann identisch mit dem für die Tests verwendeten Nährmedium sein; für Stammkulturen können jedoch auch andere nährstoffreiche Medien verwendet werden.
Regelmäßig wird eine gewisse Anzahl junger, hellgrüner Pflanzen entnommen und mit einem keimfreien Verfahren in neue Kulturgefäße mit einem frischen Medium gebracht. Unter den hier empfohlenen kühleren Bedingungen können in Intervallen von bis zu drei Monaten Teilkulturen hergestellt werden.
In den Prüfungen sollten chemische reine (mit Säure gereinigte) und sterile gläserne Kulturgefäße verwendet werden; die Handhabung sollte keimfrei erfolgen. Bei einer Verunreinigung der Stammkultur (z.B. durch Algen oder Pilze) sind entsprechende Schritte zur Entfernung der verunreinigenden Organismen erforderlich. Algen und die meisten sonstigen verunreinigenden Organismen können durch eine Oberflächensterilisation entfernt werden. Von dem verunreinigten Pflanzenmaterial wird eine Probe genommen; von dem Material werden die Wurzeln abgeschnitten. Anschließend wird das Material heftig in sauberem Wasser geschüttelt und dann 30 Sekunden bis 5 Minuten in eine 0,5%ige Natriumhypochloridlösung (viv) getaucht. Danach wird das Pflanzenmaterial mit sterilem Wasser gespült und in mehreren Schritten in Kulturgefäße jeweils mit frischem Nährmedium gebracht. Bei dieser Behandlung werden viele Fronds sterben, besonders bei längerer Expositionsdauer, einige der überlebenden Fronds sollten jedoch frei von Verunreinigungen sein. Diese Fronds können dann zur Impfung neuer Kulturen verwendet werden.
| Medien | Anlage 3 |
Für L. minor und L. gibba werden unterschiedliche Nährmedien empfohlen. Für L. minor sollte ein modifiziertes schwedisches Standardmedium (SIS) verwendet werden; für L. gibba ist das Medium 20X AAP zu empfehlen. Die Zusammensetzungen beider Medien werden im Folgenden beschrieben. Bei der Herstellung dieser Medien sollten chemische Stoffe in Reagenzien- oder Analysequalität und entionisiertes Wasser verwendet werden.
Schwedisches Standard-Lemna-Nälirmedium (SIS)
| Stammlösung Nr. | Substanz | Konzentration in der Stammlösung (g× l-1) | Konzentration im hergestellten Medium (mg×l-1) | Hergestelltes Medium | |
| Element | Konzentration (mg×l-1) | ||||
| I | NaNO3 | 8,50 | 85 | Na; N | 32; 14 |
| KH2PO4 | 1,34 | 13,4 | K; P | 6,0; 2,4 | |
| II | MgSO4×7H2O | 15 | 75 | Mg; S | 7,4; 9,8 |
| III | CaCl2×2H2O | 7,2 | 36 | Ca; Cl | 9,8; 17,5 |
| IV | Na2CO3 | 4,0 | 20 | C | 2,3 |
| V | H3BO3 | 1,0 | 1,00 | B | 0,17 |
| MnCl2×4H2O | 0,20 | 0,20 | Mn | 0,056 | |
| Na2Mo04×2H2O | 0,010 | 0,010 | Mo | 0,0040 | |
| ZnS04. 7 H2O | 0,050 | 0,050 | Zn | 0,011 | |
| CuSO4×5H2O | 0,0050 | 0,0050 | Cu | 0,0013 | |
| Co(N03)2×6H2O | 0,010 | 0,010 | Co | 0,0020 | |
| VI | FeCl3×6H2O | 0,17 | 0,84 | Fe | 0,17 |
| Na2-EDTA2×H2O | 0,28 | 1,4 | - | - | |
| VII | MOPS (Puffer) | 490 | 490 | - | - |
Hinweis: Bei bestimmten Prüfsubstanzen kann eine weitere Stammlösung VII (MOPS-Pufferlösung) erforderlich sein (siehe Abschnitt 1.4 letzter Absatz).
Nährmedium 20X AAP
Die Stammlösungen werden in sterilem destilliertem oder entionisiertem Wasser hergestellt.
Sterile Stammlösungen sollten kühl und dunkel gelagert werden. Unter diesen Bedingungen beträgt die Lagerfähigkeit der Stammlösungen mindestens 6-8 Wochen.
Für das Medium 20X AAP werden fünf Stamm-Nährlösungen (A1, A2, A3, B und C) hergestellt; dabei sind chemische Stoffe mit Reagenzienqualität zu verwenden. Jeweils 20 ml der Stamm-Nährlösungen werden zu etwa 850 ml entionisiertem Wasser hinzugegeben, um das Nährmedium herzustellen. Der pH-Wert wird wahlweise mit 0,1 oder 1 mol HCl oder NaOH auf 7,5 ± 0,1 eingestellt; durch Hinzugabe von entionisiertem Wasser wird ein Volumen von einem Liter hergestellt. Anschließend wird das Medium durch einen Membranfilter mit einer Porengröße von (etwa) 0,2 µm in einen sterilen Behälter gefiltert.
Das für die Prüfung vorgesehene Nährmedium sollte 1-2 Tage vor der Verwendung hergestellt werden, damit sich der pH-Wert stabilisieren kann. Der pH-Wert des Nährmediums sollte vor der Verwendung geprüft und ggf. durch Zugabe von 0,1 oder 1 M NaOH oder HCl wie oben beschrieben korrigiert werden.
| Stammlösung Nr. | Substanz | Konzentration in der Stammlösung (g×l-1)* | Konzentration im hergestellten Medium (mg×l-1)* | Hergestelltes Medium | |
| Element | Konzentration (mg×l-1)* | ||||
| A1 | NaNO3 | 26 | 510 | Na; N | 190; 84 |
| MgCl2×6H2O | 12 | 240 | Mg | 58,08 | |
| CaCl2×2H2O | 4,4 | 90 | Ca | 24.04 | |
| A2 | MgSO4×7H2O | 15 | 290 | S | 38,22 |
| A3 | KZHPO4×3H2O | 1,4 | 30 | K; P | 9,4;3,7 |
| B | H3BO3 | 0,19 | 3,7 | B | 0,65 |
| MnCl2×4H2O | 0,42 | 8,3 | Mn | 2,3 | |
| FeCl3×6H2O | 0,16 | 3,2 | Fe | 0,66 | |
| Na2EDTA×2H2O | 0,30 | 6,0 | - | - | |
| ZnCl2 | 3,3 mg×l-1 | 66 µg×l-1 | Zn | 31 µg×l-1 | |
| CoCl2×6H2O | 1,4 mg×l-1 | 29 µg×l-1 | Co | 7,1 µg×l-1 | |
| Na2 MoO4×2H2O | 7,3 mg×l-1 | 145 µg×l-1 | Mo | 58 µg×l-1 | |
| CuCl22×H2O | 0,012 mg×l-1 | 0,24 µg×l-1 | Cu | 0,080 µg×l-1 | |
| C | NaHCO3 | 15 | 300 | Na; C | 220; 43 |
| *) Wenn nicht anders angegeben.
Fußnote:
Die theoretisch geeignete Bicarbonat-Endkonzentration | |||||
STEINBERG-Medium (nach ISO 20079)
Konzentrationen und Stammlösungen
Tabelle 1: STEINBERG-Medium mit stabilisiertem pH-Wert (modifiziert nach Altenburger)
| Substanz | Nährmedium | ||
| Makroelemente | Molmasse | mg/l | mmol/l |
| KNO3 | 101,12 | 350,00 | 3,46 |
| Ca(NO3)2×4H2O | 236,15 | 295,00 | 1,25 |
| KH2PO4 | 136,09 | 90,00 | 0,66 |
| K2HPO4 | 174,18 | 12,60 | 0,072 |
| MgSO4×7H2O | 246,37 | 100,00 | 0,41 |
| Mikroelemente | Molmasse | µg/l | µmol/l |
| H3BO3 | 61,83 | 120,00 | 1,94 |
| ZnSO4×7H2O | 287,43 | 180,00 | 0,63 |
| Na2MoO4×2H2O | 241,92 | 44,00 | 0,18 |
| MnCl2×4H2O | 197,84 | 180,00 | 0,91 |
| FeCl3×6H2O | 270,21 | 760,00 | 2,81 |
| EDTA-Dinatrium-Dihydrat | 372,24 | 1500,00 | 4,03 |
Tabelle 2: Stammlösungen (Makroelemente)
| 1. Makroelemente (50fach konzentriert) | g/l |
| Stammlösung 1: | |
| KNO3 | 17,50 |
| KH2PO4 | 4,5 |
| K2HPO4 | 0,63 |
| Stammlösung 2: MgSO4×7H2O | 5,00 |
| Stammlösung 3: Ca(NO3)2×4H2O | 14,75 |
Tabelle 3: Stammlösungen (Mikroelemente)
| 2. Mikroelemente (1000fach konzentriert) | mg/l |
| Stammlösung 4: H3BO3 | 120,0 |
| Stammlösung 5: ZnSO4×7H2O | 180,0 |
| Stammlösung 6: Na2 MoO4×2H2O | 44,0 |
| Stammlösung 7: MnCl2×4H2O | 180,0 |
| Stammlösung 8: | |
| FeCl3×6H2O | 760,00 |
| EDTA-Dinatrium-Dihydrat | 1.500,00 |
Herstellung der Endkonzentration des STEINBERG-Mediums (modifiziert)
Herstellung des 10fach konzentrierten STEINBERG-Mediums (modifiziert) zur Zwischenlagerung
 |
ENDE