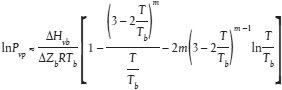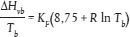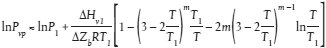umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (2)
| zurück |
|
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode oder sonstige anzuwendende Prüfmethode für den betreffenden Endpunkt sind in Teil 0 Tabelle 1 aufgeführt.
| 1. Methode
Diese Methode entspricht der Prüfrichtlinie OECD TG 104 (2004). 1.1 Einleitung Diese geänderte Fassung von Methode A.4 (1) beinhaltet als zusätzliche Methode die "Effusionsmethode: isotherme Thermogravimetrie"; diese Methode wurde entwickelt für Substanzen mit sehr niedrigen Drücken (bis zu einem Mindestdruck von 10-10 Pa). Angesichts der Verfahrenserfordernisse, insbesondere bei der Ermittlung des Dampfdrucks für Substanzen mit niedrigem Dampfdruck, werden auch andere Verfahren zur Anwendung dieser Methode im Hinblick auf sonstige Einsatzbereiche neu bewertet. Bei thermodynamischem Gleichgewicht hängt der Dampfdruck einer reinen Substanz ausschließlich von der Temperatur ab. Die zugrunde liegenden Prinzipien werden an anderer Stelle erläutert (2)(3). Kein einzelnes Messverfahren ist für sämtliche Dampfdrucke von unter 10-10 Pa bis zu 105 Pa geeignet. Entsprechend umfasst diese Beschreibung acht Methoden zur Messung des Dampfdrucks, die in verschiedenen Dampfdruckbereichen eingesetzt werden können. Die vorgesehenen Einsatzmöglichkeiten und Messbereiche der einzelnen Methoden sind in Tabelle 1 zusammengestellt. Die Methoden können nur im Zusammenhang mit Verbindungen eingesetzt werden, bei denen unter den Testbedingungen kein Abbau erfolgt. In Fällen, in denen die Versuchsmethoden aus technischen Gründen nicht eingesetzt werden können, kann der Dampfdruck auch geschätzt werden; eine empfohlene Schätzmethode wird in der Anlage beschrieben. 1.2 Begriffsbestimmungen und Einheiten Als Dampfdruck einer Substanz wird der Sättigungsdruck über einer festen oder flüssigen Substanz bezeichnet. In den Messungen sollte die Einheit Pascal (Pa) als SI-Einheit für Druckwerte verwendet werden. Im Folgenden sind weitere, früher verwendete Einheiten jeweils mit den entsprechenden Umrechnungsfaktoren zusammengestellt:
Die SI-Einheit der Temperatur ist Kelvin (K). Angaben in Grad Celsius werden mit folgender Formel in Kelvin umgerechnet: T= t + 273,15 wobei T = Kelvin oder thermodynamische Temperatur und t = Temperatur in Grad Celsius
1.3 Prinzip der Prüfmethode Im Allgemeinen wird der Dampfdruck bei verschiedenen Temperaturen gemessen. In einem begrenzten Temperaturbereich ist der Logarithmus des Dampfdrucks einer reinen Substanz eine lineare Umkehrfunktion der thermodynamischen Temperatur gemäß der vereinfachten Clapeyron-Clausius-Gleichung:
wobei
1.4 Referenzsubstanzen Referenzsubstanzen brauchen nicht unbedingt verwendet zu werden. Sie dienen in erster Linie zur gelegentlichen Überprüfung der Leistungsfähigkeit einer Methode und sollen Vergleiche der mit unterschiedlichen Methoden erzielten Ergebnisse ermöglichen. 1.5 Beschreibung der Methode 1.5.1 Dynamische Methode (Cottrell-Verfahren) 1.5.1.1 Prinzip Der Dampfdruck wird durch die Messung der Siedetemperatur einer Substanz bei verschiedenen vorgegebenen Drücken zwischen etwa 103 und 105 Pa gemessen. Diese Methode wird auch für die Bestimmung der Siedetemperatur empfohlen. Für diesen Zweck kann die Methode bei Temperaturen bis zu 600 K eingesetzt werden. Wegen des hydrostatischen Drucks der Flüssigkeitssäule liegen die Siedetemperaturen von Flüssigkeiten bei einer Tiefe von 3 bis 4 cm etwa 0,1 °C höher als an der Oberfläche. Beim Cottrell-Verfahren (4) wird das Thermometer über der Flüssigkeit in den Dampf gebracht und die siedende Flüssigkeit kontinuierlich über die Thermometerkugel gepumpt. Eine dünne Flüssigkeitsschicht, die sich bei Atmosphärendruck im Gleichgewicht mit dem Dampf befindet, bedeckt die Kugel. Das Thermometer gibt dann den echten Siedepunkt an; Fehler durch Überhitzung oder hydrostatischen Druck werden ausgeschlossen. Die ursprünglich von Cottrell verwendete Pumpe ist in Abbildung 1 dargestellt. Rohr A enthält die siedende Flüssigkeit. Ein Platindraht B, der in den Boden eingesiegelt wurde, begünstigt ein gleichmäßiges Siedeverhalten. Das seitliche Rohr C führt zu einem Kondensator, und der Spritzschutz D verhindert, dass das kalte Kondensat in das Thermometer E gelangt. Wenn die Flüssigkeit in A siedet, werden die entstehenden Blasen und die Flüssigkeit mit dem Trichter abgetrennt und über die beiden Arme der Pumpe F über die Thermometerkugel gegossen.
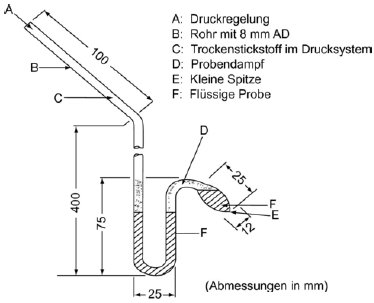
Cottrell-Pumpe (4) A: Thermoelement 1.5.1.2 Apparatur In Abbildung 2 ist eine sehr genaue Apparatur dargestellt, die auf dem Cottrell-Prinzip beruht. Die Apparatur besteht aus einem Rohr mit einem Siedebereich im unteren Teil, einem Kühler im mittleren Bereich und einem Auslass und einem Flansch im oberen Bereich. Die Cottrell-Pumpe befindet sich im Siedebereich, der mit einer elektrischen Patrone beheizt wird. Die Temperatur wird mit einem ummantelten Thermoelement oder einem Widerstandsthermomenter gemessen, das über den oben befindlichen Flansch in das Gerät geführt wird. Der Auslass ist mit einem Druckregelsystem verbunden. Letzteres besteht aus einer Vakuumpumpe, einem Puffervolumen, einem Druckwächter zur Druckregulierung unter Stickstoffeinleitung und einem Druckmesser. 1.5.1.3 Verfahren Die Substanz wird in den Siedebereich gebracht. Bei nicht pulverigen Feststoffen können Probleme auftreten, die sich gelegentlich aber durch Beheizung des Kühlmantels beheben lassen. Die Apparatur ist am Flansch versiegelt, und die Prüfsubstanz wird entgast. Schäumende Substanzen können mit dieser Methode nicht gemessen werden. In der Apparatur wird der niedrigste gewünschte Druck eingestellt und die Heizung eingeschaltet. Dann wird der Temperatursensor mit einem Aufzeichnungsgerät verbunden. Das Gleichgewicht ist dann erreicht, wenn eine gleichbleibende Siedetemperatur bei konstantem Druck aufgezeichnet wird. Insbesondere ist darauf zu achten, dass während des Siedevorgangs nicht gegen die Apparatur gestoßen wird. Außerdem muss auf dem Kühler eine vollständige Kondensation erfolgen. Bei der Bestimmung des Dampfdrucks von niedrigschmelzenden Feststoffen ist darauf zu achten, dass der Kondensator nicht verblockt. Nach der Aufzeichnung dieses Gleichgewichtspunkts wird ein höherer Druck eingestellt. Der Prozess wird auf diese Weise fortgesetzt, bis ein Druck von 105 Pa erreicht ist (insgesamt etwa 5 bis 10 Messpunkte). Zur Kontrolle sind die Gleichgewichtspunkte bei abnehmenden Drücken zu reproduzieren. 1.5.2 Statische Methode 1.5.2.1 Prinzip Bei der statischen Methode (5) wird der Dampfdruck bei thermodynamischem Gleichgewicht und einer gegebenen Temperatur bestimmt. Diese Methode eignet sich für Substanzen und für aus mehreren Bestandteilen bestehende Flüssigkeiten und Feststoffe im Druckbereich von 10-1 bis 105 Pa sowie bei entsprechend sorgfältiger Vorgehensweise auch im Bereich von 1 bis 10 Pa. 1.5.2.2 Apparatur Die Apparatur besteht aus einem Bad mit konstanter Temperatur (Genauigkeit ± 0,2 K), einem mit einer Unterdruckleitung verbundenen Probenbehälter, einem Druckmesser und einem System zur Druckregelung. Die Probenkammer (Abbildung 3a) ist über ein Ventil und einen Differenzdruckmesser (ein U-Rohr mit einer geeigneten Manometerflüssigkeit) verbunden, das als Nullpunktanzeige dient. Im Differenzdruckmesser können Quecksilber, Silikone und Phthalate verwendet werden; maßgeblich sind der jeweilige Druckbereich und das chemische Verhalten der Prüfsubstanz. Aus Gründen des Umweltschutzes sollte nach Möglichkeit jedoch auf Quecksilber verzichtet werden. Die Prüfsubstanz darf sich nicht merklich in der im U-Rohr enthaltenen Flüssigkeit auflösen oder mit dieser reagieren. Statt eines U-Rohrs kann auch ein Druckmesser verwendet werden (Abbildung 3b). Für den Druckmesser kann Quecksilber im Bereich des Atmosphärendrucks bis zu einem Mindestdruck von 102 Pa eingesetzt werden; Silikon-Flüssigkeiten und Phthalate sind für Drücke unter 102 Pa bis zu 10 Pa geeignet. Unter 102 Pa können sonstife Druckmesser eingesetzt werden; Kapazitätsmanometer mit Heizmembran können sogar bei Drücken unter 10-1 Pa verwendet werden. Die Temperatur wird an der Außenwand des Probenbehälters oder im Behälter selbst gemessen. 1.5.2.3 Verfahren In der in Abbildung 3a beschriebenen Apparatur wird das U-Rohr mit der gewählten Flüssigkeit gefüllt; vor Durchführung der Messungen ist die Flüssigkeit bei höherer Temperatur zu entgasen. Die Prüfsubstanz wird in die Apparatur gebracht und bei niedrigerer Temperatur entgast. Bei aus mehreren Bestandteilen bestehenden Proben sollte die Temperatur so niedrig sein, dass die Zusammensetzung des jeweiligen Materials erhalten bleibt. Durch Rühren kann das erforderliche Gleichgewicht schneller herbeigeführt werden. Die Probe kann dann mit flüssigem Stickstoff oder mit Trockeneis abgekühlt werden. Dabei ist allerdings sicherzustellen, dass die Luft oder die Pumpflüssigkeit nicht kondensieren. Bei geöffnetem Ventil über der Probe wird über mehrere Minuten ein Sog ausgeübt, um die eingeschlossene Luft zu entfernen. Wenn erforderlich, wird die Entgasung mehrmals wiederholt.
Wenn die Probe bei geschlossenem Ventil beheizt wird, erhöht sich der Dampfdruck. Dadurch ändert sich das Gleichgewicht der Flüssigkeit im U-Rohr. Um die Änderung auszugleichen, wird Stickstoff oder Luft in die Apparatur geleitet, bis die Differenzdruckanzeige wieder auf null steht. Der dazu erforderliche Druck kann an einem Druckmesser oder an einem genaueren Messgerät abgelesen werden. Dieser Druck entspricht dem Dampfdruck der Substanz bei der Messtemperatur. Bei der in Abbildung 3b dargestellten Apparatur wird der Dampfdruck direkt abgelesen. Der Dampfdruck wird in geeigneten geringen Temperaturintervallen (insgesamt etwa 5 bis 10 Messpunkte) bis zur gewünschten Höchsttemperatur gemessen. Zur Kontrolle sind Messungen bei niedrigen Temperaturen durchzuführen. Wenn die in den mehrfachen Messungen ermittelten Werte nicht auf der Kurve liegen, die sich bei den höheren Temperaturen ergibt, kann dies auf eine der folgenden Ursachen zurückzuführen sein:
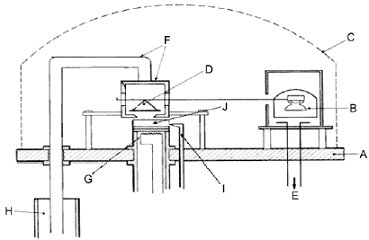
1.5.3 Isoteniskopmethode 1.5.3.1 Prinzip Das Isoteniskop (6) beruht auf dem Prinzip der statischen Methode. Bei dieser Methode wird eine Probe in einen Kolben gebracht, in dem eine gleichbleibende Temperatur besteht, und der mit einem Druckmesser und einer Vakuumpumpe verbunden ist. Verunreinigungen mit höherer Flüchtigkeit als die Prüfsubstanz werden durch Entgasen bei reduziertem Druck entfernt. Der Dampfdruck der Probe bei bestimmten Temperaturen wird durch einen mit einem inerten Gas ausgeübten bekannten Druck ausgeglichen. Das Isoteniskop wurde entwickelt, um den Dampfdruck bestimmter flüssiger Kohlenwasserstoffe zu messen, kann aber auch zur Untersuchung von Feststoffen eingesetzt werden. Für Systeme mit mehreren Bestandteilen ist diese Methode im Allgemeinen nicht geeignet. Die Ergebnisse weisen nur leichte Fehler bei Proben mit nicht flüchtigen Verunreinigungen auf. Die Methode wird für den Bereich 102 bis 105 Pa empfohlen. 1.5.3.2 Apparatur In Abbildung 4 wird ein Messsystem dargestellt. Eine vollständige Beschreibung ist ASTM D 2879-86 (6) zu entnehmen. 1.5.3.3 Verfahren Bei Flüssigkeiten dient die Substanz selbst als Flüssigkeit im Differenzdruckmesser. In das Isoteniskop wird so viel Flüssigkeit eingebracht, dass der Kolben und der kurze Fuß des Druckmessers gefüllt sind. Das Isoteniskop wird mit einem Vakuumsystem verbunden und mit Hilfe des Vakuumsystems vollständig entleert. Anschließend wird das Isoteniskop mit Stickstoff gefüllt. Die Absaugung und die Reinigung des Systems wird zweimal wiederholt, um den verbliebenen Sauerstoff zu entfernen. Das befüllte Isoteniskop wird in horizontal ausgerichtet, damit sich die Probe in Kolben und Druckmesser als dünne Schicht ausbreitet. Der Systemdruck wird auf 133 Pa reduziert, und die Probe wird allmählich erwärmt, bis eben der Siedepunkt erreicht ist (Abtrennung gelöster Gase). Danach wird das Isoteniskop so ausgerichtet, dass die Probe wieder in den Kolben zurückfließt und den kurzen Fuß des Druckmessers ausfüllt. Dabei wird ein Druck von 133 Pa aufrechterhalten. Die vorgezogene Spitze des Probenkolbens wird mit kleiner Flamme erwärmt, bis sich der freigesetzte Probendampf hinreichend ausgedehnt hat, um einen Teil der Probe aus dem oberen Bereich des Kolbens und des Druckmesserarms in den Druckmesser hinein zu verdrängen und somit einen stickstofffreien Raum mit Dampf gefüllt hat. Darauf wird das Isoteniskop in ein Bad mit konstanter Temperatur gebracht, und Druck und Stickstoffzufuhr werden so eingestellt, dass sie mit dem Druck und dem Stickstoffanteil der Probe übereinstimmen. Wenn das erwünschte Gleichgewicht erreicht ist, entspricht der Stickstoffdruck dem Dampfdruck der Prüfsubstanz. Bei Feststoffen sowie je nach Druck- und Temperaturbereich werden Manometerflüssigkeiten wie z.B. Silikon-Flüssigkeiten oder Phthalate verwendet. Die entgaste Manometerflüssigkeit wird in eine dafür vorgesehene Rufwölbung am langen Arm des Isoteniskops gebracht. Anschließend wird der zu prüfende Feststoff in den Probenkolben gegeben und bei einer höheren Temperatur entgast. Darauf wird das Isoteniskop so geneigt, dass die Manometerflüssigkeit in das U-Rohr fliegen kann. 1.5.4 Effusionsmethode: Dampfdruckgleichgewicht (7) 1.5.4.1 Prinzip Eine Probe der Prüfsubstanz wird in einem kleinen Ofen erhitzt und in eine Gasglocke gebracht, in der ein Unterdruck hergestellt wurde. Der Ofen wird mit einem Deckel verschlossen, in dem sich kleine Löcher mit einem bestimmten Durchmesser befinden. Der aus der Substanz erzeugte Dampf entweicht durch eines dieser Löcher und wird unmittelbar auf die Schale einer hoch empfindlichen Waage geleitet, die sich ebenfalls im Vakuum der Glasglocke befindet. Bei manchen Ausführungen ist die Waagschale von einem Kühlgefäß umgeben, über das die Wärme thermisch abgeleitet und unter Abstrahlung der Wärme so gekühlt wird, dass sich der entweichende Dampf darauf niederschlägt. Die Energie des Dampfstrahls wirkt als Kraft auf die Waage. Der Dampfdruck kann auf zweierlei Weise ermittelt werden: entweder direkt aus der auf die Waagschale wirkenden Kraft oder mit Hilfe der Hertz-Knudsen-Gleichung aufgrund der Verdampfungsgeschwindigkeit (2): p = G ((2πRT x 103)/M)0,5 wobei G = Verdampfungsgeschwindigkeit (kg s-1 m-2) M = Molmasse (g mol-1) T = Temperatur (K) R = Universale Gaskonstante (J mol-1 K-1) P = Dampfdruck (Pa) Der empfohlene Bereich liegt zwischen 10-3 und 1 Pa. 1.5.4.2 Apparatur Abbildung 5 veranschaulicht das Grundprinzip der Apparatur:
1.5.5 Effusionsmethode: Knudsen-Zelle 1.5.5.1 Prinzip Die Methode beruht auf der Schätzung der Masse der Prüfsubstanz, die im Ultravakuum pro Zeiteinheit aus einer Knudsen-Zelle (8) als Dampf durch eine Mikrobohrung strömt. Die Masse des ausströmenden Dampfs kann entweder aufgrund des Masseverlustes der Zelle oder durch Kondensation des Dampfs bei niedriger Temperatur und anschließende chromatographische Messung der Menge der verflüchtigten Substanz bestimmt werden. Der Dampfdruck wird mit Hilfe der Hertz-Knudsen-Gleichung (siehe Abschnitt 1.5.4.1) unter Berücksichtigung von Korrekturfaktoren berechnet, die von den Parametern der jeweiligen Apparatur abhängen (9). Empfohlen wird diese Methode für den Bereich 10-10 bis 1 Pa (10)(11)(12)(13)(14). 1.5.5.2 Apparatur Abbildung 6 veranschaulicht das Grundprinzip der Apparatur:
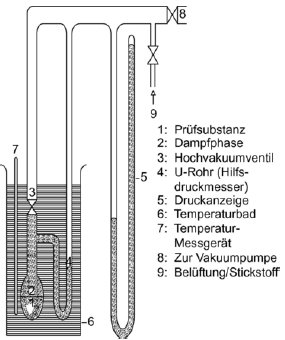
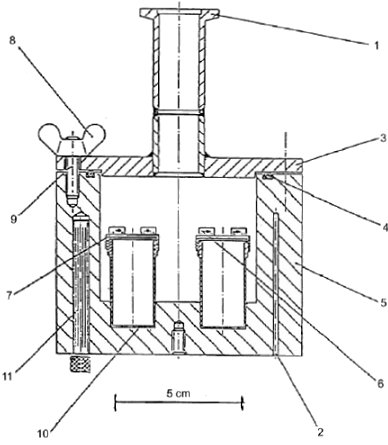
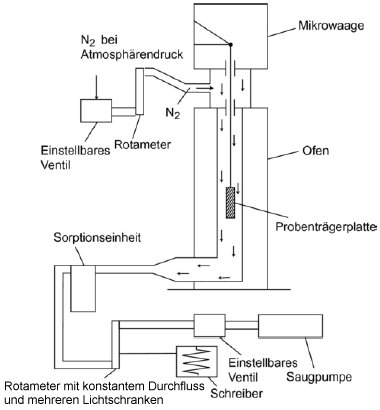
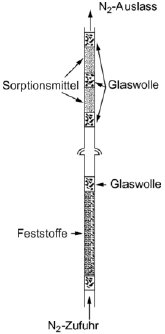
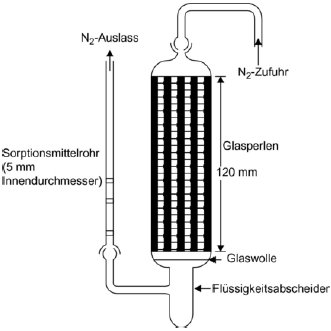
1.5.6 Effusionsmethode: isotherme Thermogravimetrie 1.5.6.1 Prinzip Die Methode beruht auf der Bestimmung der Geschwindigkeit einer beschleunigten Verdampfung der Prüfsubstanz bei höheren Temperaturen und bei Umgebungsdruck durch Thermogravimetrie (10)(15)(16)(17)(18)(19)(20). Die Verdampfungsgeschwindigkeiten vT ergeben sich daraus, dass die jeweils ausgewählte Verbindung einer Atmosphäre mit einem langsam strömenden inerten Gas ausgesetzt wird; bei gegebenen isothermen Temperaturen T (ausgedrückt in Kelvin) wird dann über bestimmte Zeitspannen der Gewichtsverlust überwacht. Die Dampfdrücke pT werden aus den Werten für vT aufgrund der linearen Beziehung zwischen dem Logarithmus des Dampfdrucks und dem Logarithmus der Verdampfungsgeschwindigkeit bestimmt. Wenn erforderlich, kann durch eine Regressionsanalyse von log pT bezogen auf 1/T eine Extrapoherung auf Temperaturen von 20 und 25 °C erfolgen. Diese Methode kommt für Substanzen mit Mindestdampfdrücken bis zu 10-10 Pa (10-12 mbar) in Betracht; um Fehlinterpretationen der gemessenen Gewichtsverluste zu vermeiden, sollte die Reinheit annähernd 100 % betragen. 1.5.6.2 Apparatur In Abbildung 7 wird das allgemeine Prinzip der Apparatur dargestellt: Die in einer Kammer mit Temperaturüberwachung an einer Mikrowaage aufgehängte Probenträgerplatte wird von trockenem Stickstoffgas umströmt, in dem die verdampften Moleküle der Prüfsubstanz geführt werden. Nach dem Verlassen der Kammer wird der Gasstrom durch eine Sorptionseinheit gereinigt. 1.5.6.3 Verfahren Die Prüfsubstanz wird als homogene Schicht auf eine aufgeraute Glasplatte aufgebracht. Bei Feststoffen wird die Platte gleichförmig mit einer Lösung befeuchtet, in der die Prüfsubstanz in einem geeigneten Lösungsmittel gelöst wurde; anschließend wird die Lösung in einer inerten Atmosphäre getrocknet. Für die Messung wird die beschichtete Platte in den Thermogravimetrie-Analysator gehängt, dort wird der Gewichtsverlust kontinuierlich als zeitabhängige Funktion bestimmt. Aus dem Gewichtsverlust Am der Probenplatte wird die Verdampfungsgeschwindigkeit vT bei einer bestimmten Temperatur mit der folgenden Formel berechnet: vT = Δm/F×t (gcm-2 h-1) wobei F = Oberfläche der beschichteten Prüfsubstanzen (im Allgemeinen die Oberfläche der Probenplatte) und t = Dauer des Gewichtsverlusts Δm Der Dampfdruck pT wird ausgehend von der Funktion der Verdampfungsgeschwindigkeit vT wie folgt berechnet: Log pT = C + D log vT wobei C und D spezifische Konstanten für die jeweilige Apparatur sind und vom Durchmesser der Messkammer sowie vom Gasstrom abhängen. Diese Konstanten sind einmal zu bestimmen, indem eine Reihe von Verbindungen mit bekanntem Dampfdruck gemessen und dann durch Regressionsanalyse log pT gegenüber log vT ermittelt wird (11)(21)(22). Die Beziehung zwischen dem Dampfdruck pT und der Temperatur T in Kelvin wird wie folgt ausgedrückt: Log pT = A + B 1/T Dabei sind A und B Konstanten, die sich aus der Regressionsanalyse von log pT gegenüber 1/T ergeben. Mit dieser Formel kann der Dampfdruck für jede beliebige Temperatur extrapoliert werden. 1.5.7 Gassättigungsmethode (23) 1.5.7.1 Prinzip Das inerte Gas wird bei Raumtemperatur und mit einer bekannten Durchflussgeschwindigkeit so langsam durch bm über eine Probe der Prüfsubstanz geleitet, dass eine Sättigung eintreten kann. Die Herbeiführung der Sättigung in der Gasphase ist von entscheidender Bedeutung. Die mitgeführte Substanz wird eingeschlossen (im Allgemeinen mit einem Sorptionsmittel); anschließend wird die Menge der abgetrennten Substanz bestimmt. Alternativ zum Dampfeinschluss mit anschließender Analyse kommen in den Gasstrom integrierte Analyseverfahren wie z.B. die Gaschromatographie zur quantitativen Bestimmung des mitgeführten Materials in Betracht. Der Dampfdruck wird ausgehend von der Annahme berechnet, dass sich der Gasstrom entsprechend dem idealen Gasgesetz verhält und dass der Gesamtdruck eines Gasgemischs mit der Summe der Drücke der einzelnen enthaltenen Gase übereinstimmt. Der Teildruck der Prüfsubstanz, d. h. der Dampfdruck, wird aus dem bekannten Volumen des gesamten Gasstroms und aus dem Gewicht des mitgeführten Materials berechnet. Das Gassättigungsverfahren ist für feste und flüssige Substanzen geeignet. Es kann bei Mindestdampfdrücken bis zu 10-10 Pa genutzt werden (10)(11)(12)(13)(14). Bei Dampfdrücken unter 103 Pa ist diese Methode die zuverlässigste Methode. Über 103 Pa werden die Dampfdrücke im Allgemeinen überschätzt; dies ist wahrscheinlich auf die Bildung von Aerosolen zurückzuführen. Da die Dampfdruckmessungen bei Raumtemperatur erfolgen, brauchen keine Extrapolierungen hoher Temperaturen vorgenommen zu werden; entsprechend entfallen die häufig mit der Extrapolierung hoher Temperaturen verbundenen schwerwiegenden Fehler. 1.5.7.2 Apparatur Für das Verfahren wird ein Behälter mit konstanter Temperatur benötigt. In Abbildung 8 ist ein Behälter mit Haltern für jeweils drei feste und flüssige Proben skizziert, in dem jeweils drei wahlweise feste oder flüssige Proben analysiert werden können. Die Temperatur wird mit einer Genauigkeit von mindestens ± 0,5 °C überwacht. Im Allgemeinen wird Stickstoff als inertes Trägergas verwendet; gelegentlich können aber auch sonstige Gase erforderlich sein (24). Das Trägergas muss trocken sein. Der Gasstrom wird in sechs Teilströme getrennt, die jeweils durch Kegelventile (mit Bohrungen von ca. 0,79 mm) geregelt und über ein Kupferrohr mit einem Innendurchmesser von 3,8 mm in den Behälter geleitet werden. Nach dem Temperaturausgleich strömt das Gas durch die Probe und verlässt den Behälter durch die Adsorptionsfalle. Feste Proben werden in ein mit Glaswollestopfen verschlossenes Glasrohr mit einem Innendurchmesser von 5 mm gegeben (siehe Abbildung 9). In Abbildung 10 sind ein Halter für feste Proben und ein Sorptionssystem dargestellt. Die am besten reproduzierbare Methode zur Messung des Dampfdrucks von Flüssigkeiten besteht darin, die zu prüfende Flüssigkeit auf Glasperlen oder auf ein inertes Sorptionsmittel wie z.B. Kieselerde aufzubringen und den Halter mit diesen Perlen zu packen. Alternativ kann das Trägergas auch über eine grobe Fritte und eine Blase durch die Säule mit der flüssigen Prüfsubstanz geleitet werden.
Das Sorptionssystem enthält einen vorderen und einen hinteren Sorptionsabschnitt. Bei sehr niedrigen Dampfdrücken werden nur geringe Anteile vom Sorptionsmittel abgetrennt; dabei kann die Adsorption auf der Glaswolle und im Glasrohr zwischen der Probe und dem Sorptionsmittel ein ernsthaftes Problem sein. Mit festem CO2 gekühlte Abscheider stellen eine weitere wirksame Möglichkeit zur Aufnahme des verdampften Materials dar. Diese Abscheider verursachen keinerlei Gegendruck auf die Sättigungssäule; außerdem lässt sich das abgetrennte Material leicht quantitativ bestimmen. 1.5.7.3 Verfahren Die Durchflussgeschwindigkeit des ausströmenden Trägergases wird bei Raumtemperatur gemessen. Während der Messung wird die Durchflussgeschwindigkeit häufig geprüft, um sicherzustellen, dass das Gesamtvolumen des Trägergases zuverlässig ermittelt wird. Vorzugsweise erfolgt eine kontinuierliche Überwachung mit einem Mengendurchflussmesser. Die Sättigung der Gasphase kann eine beträchtliche Kontaktzeit und entsprechend verhältnismäßig geringe Gasdurchflüsse erfordern (25). Im Anschluss an die Messungen werden der vordere und der hintere Sorptionsabschnitt getrennt analysiert. Die Verbindungen in den einzelnen Abschnitten werden durch Zugabe eines Lösungsmittels desorbiert. Die entstehenden Lösungen werden einer quantitativen Analyse unterzogen, um die jeweils aus den Abschnitten desorbierten Gewichte zu bestimmen. Die Wahl der Analysemethode (sowie die Wahl des Sorptionsmittels und des desorbierenden Lösungsmittels) hängt von der Beschaffenheit des zu prüfenden Materials ab. Die Desorptionsleistung wird bestimmt, indem eine bekannte Probenmenge auf das Sorptionsmittel gespritzt, die Substanz desorbiert und anschließend die zurückgewonnene Menge analysiert wird. Die Desorptionsleistung muss mit der gleichen oder zumindest annähernd gleichen Probenkonzentration wie in der eigentlichen Prüfung bestimmt werden. Um sicherzustellen, dass das Trägergas mit der Prüfsubstanz gesättigt ist, werden drei verschiedene Durchflussgeschwindigkeiten eingestellt. Wenn sich der berechnete Dampfdruck auch bei unterschiedlichen Durchflüssen nicht ändert, wird angenommen, dass das Gas gesättigt ist. Der Dampfdruck wird mit folgender Gleichung berechnet: p = W / V x RT / M wobei p = Dampfdruck (Pa) W = Gewicht der verdampften Prüfsubstanz (g) V = Volumen des gesättigten Gases (m3) R = universale Gaskonstante 8,314 0 mol-1 K-1) T = Temperatur (K) M = Molmasse der Prüfsubstanz (g mol-1) Die gemessenen Volumina sind unter Berücksichtigung von Druck- und Temperaturunterschieden zwischen Durchflussmesser und Sättigungssäule zu korrigieren. 1.5.8 Rotationsmethode 1.5.8.1 Prinzip Bei dieser Methode wird ein Rotationsviskosimeter eingesetzt, bei dem das Messelement aus einer kleinen Stahlkugel besteht, die in einem Magnetfeld aufgehängt durch Drehfelder in Drehungen versetzt wird (26)(27)(28). Aufnehmerspulen ermöglichen eine Messung der Drehzahl. Wenn die Kugel eine bestimmte Drehzahl erreicht hat (in der Regel etwa 400 Umdrehungen pro Sekunde), wird die Erregung unterbrochen, und infolge der Gasreibung erfolgt eine Verzögerung. Der Rückgang der Drehzahl wird zeitabhängig gemessen. Aus der druckabhängigen Verlangsamung der Stahlkugel wird der Dampfdruck abgeleitet. Diese Methode wird für den Bereich von 10-4 bis 0, 5 Pa empfohlen. 1.5.8.2 Apparatur In Abbildung 11 ist die Apparatur schematisch dargestellt. Der Messfühler wird in ein Gehäuse mit konstanter Temperatur gebracht, in dem die Temperatur mit einer Genauigkeit von 0,1 °C geregelt wird. Der Probenbehälter wird in ein getrenntes Gehäuse gesetzt, dessen Temperatur ebenfalls mit einer Genauigkeit von 0,1 °C geregelt wird. Alle übrigen Teile der Apparatur werden auf einer höheren Temperatur gehalten, um eine Kondensatbildung auszuschließen. Die gesamte Apparatur ist mit einem Hochvakuumsystem verbunden. 2. Daten und Abschlussbericht 2.1 Daten Der Dampfdruck sollte mit den genannten Methoden jeweils bei mindestens zwei Temperaturen gemessen werden. Im Bereich 0 bis 50 °C werden Messungen vorzugsweise bei drei Temperaturen durchgeführt, um zu prüfen, ob die Dampfdruckkurve linear verläuft. Bei der Effusionsmethode (Knudsen-Zelle und isotherme Thermogravimetrie) und bei der Gassättigungsmethode wird statt des üblichen Bereichs von 0 bis 50 °C ein Temperaturbereich von 120 bis 150 °C empfohlen. 2.2 Prüfbericht Der Prüfbericht muss folgende Informationen enthalten:
Wenn ein Übergang (Änderung des Aggregatzustandes oder Zersetzung) beobachtet wird, sollten folgende Informationen vermerkt werden:
Alle für die Auswertung der Ergebnisse maggeblichen Informationen und Bemerkungen sind zu protokollieren; dies gilt insbesondere für Verunreinigungen und für die physikalische Beschaffenheit der Prüfsubstanz. 3. LITERATUR 1. Amtsblatt der Europäischen Gemeinschaften L 383 A, S. 26-47 (1992). 2. Ambrose, D. (1975). Experimental Thermodynamics, Vol. II, Le Nein dre, B., an d Vodar, B., Eds., Butterworths, London. 3. Weissberger R., ed. (1959). Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3' ed., Vol I, Part I. Chapter IX, Interscience Publ., New York. 4. Glasstone, S. (1946). Textbook of Physical Chemistry, 2nd ed., Van Nostrand Company, New York. 5. NF T 20-048 AFNOR (September 1985). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within a range from 10-1 to 105 Pa - Static method. 6. ASTM D 2879-86, Standard test method for vapour pressure - temperature relationship and initial decomposition temperature of liquids by isoteniscope. 7. NF T 20-047 AFNOR (September 1985). Chemical products for industrial use - Determination of vapour pressure of solids and liquids within range from 10-3 to 1 Pa - Vapour pressure balance method. B. Knudsen, M. (1909). Ann. Phys. Lpz., 29, 1979; (1911), 34, 593. 9. Ambrose, D., Lawrenson, I.J., Sprake, C.H.S. (1975). J. Chem. Thermodynamics 7, S. 1173. 10. Schmuckler, M.E., Barefoot, A.C., Kleier, D.A., Cobranchi, D.P. (2000), Vapor pressures of sulfonylurea herbicides; Pest Management Science 56, S. 521-532. 11. Tomlin, C.D.S. (ed.), The Pesticide Manual, Twelfth Edition (2000). 12. Friedrich, K., Stammbach, K., Gas chromatographic determination of small vapour pressures determination of the vapour pressures of some triazine herbicides. J. Chromatog. 16 (1964), S. 22-28. 13. Grayson, B.T., Fosbraey, L.A., Pesticide Science 16 (1982), S. 269-278. 14. Rordorf, B.F., Prediction of vapor pressures, boiling points and enthalpies of fusion for twentynine halogenated dibenzo-pdioxins, Thermochimia Acta 112 Issue 1 (1987), S. 117-122. 15. Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection; Pesticide Science 4 (1973), S. 137-147. 16. Gückel, W., Synnatschke, G., Ritttig, R., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection II. Application to Formulated Products; Pesticide Science 5 (1974), S. 393-400. 17. Gückel, W., Kaestel, R., Lewerenz, J., Synnatschke, G., A Method for Determining the Volatility of Active Ingredients Used in Plant Protection. Part III: The Temperature Relationship between Vapour Pressure and Evaporation Rate; Pesticide Science 13 (1982), S. 161-168. 18. Gückel, W., Kaestel, R., Kroehl, T., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part IV: An Improved Thermogravimetric Determination Based an Evaporation Rate; Pesticide Science 45 (1995), S. 27-31. 19. Kroehl, T., Kaestel, R., Koenig, W., Ziegler, H., Koehle, H., Parg, A., Methods for Determining the Vapour Pressure of Active Ingredients Used in Crop Protection. Part V: Thermogravimetry Combined with Solid Phase MicroExtraction (SPME); Pesticide Science 53 (1998), S. 300-310. 20. Tesconi, M., Yalkowsky, S.H., A Novel Thermogravimetric Method for Estimating the Saturated Vapor Pressure of Low-Volatility Compounds; Journal of Pharmaceutical Science 87(12) (1998), S. 1512-20. 21. Lide, D.R. (ed.), CRC Handbook of Chemistry and Physics, 81th ed.(2000), Vapour Pressure in the Range - 25 °C to 150 °C. 22. Meister, R.T. (ed.), Farm Chemicals Handbook, Vol. 88 (2002). 23. 40 CFR, 796. (1993). S. 148-153, Office of the Federal Register, Washington DC. 24. Rordorf B.F. (1985). Thermochimica Acta 85, S. 435. 25. Westcott et al. (1981). Environ. Sci. Technol. 15, S. 1375. 26. Messer G., Röhl, P., Grosse G., and Jitschin W. (1987). J. Vac. Sci. Technol. (A), 5(4), S. 2440. 27. Comsa G., Fremerey J.K., and Lindenau, B. (1980). J. Vac. Sci. Technol. 17(2), S. 642. 28. Fremerey, J.K. (1985). J. Vac. Sci. Technol. (A), 3(3), S. 1715.
Einleitung Geschätzte Dampfdrücke können verwendet werden,
Schätzmethode Der Dampfdruck von Flüssigkeiten und Feststoffen kann aufgrund der modifizierten Watson-Korrelation geschätzt werden (a). In diesem Fall braucht als Prüfwert nur der normale Siedepunkt bestimmt zu werden. Diese Methode kommt im Druckbereich 105 Pa bis 10-5 Pa in Betracht. Detaillierte Informationen zu dieser Methode sind dem "Handbook of Chemical Property Estimation Methods" zu entnehmen (b). Außerdem wird auf die OECD Environmental Monograph No. 67 verwiesen (c). Berechnungsverfahren Der Dampfdruck wird wie folgt berechnet: Tb wobei
Außerdem ist folgende Berechnung vorzunehmen: wobei KF ein empirischer Faktor ist, der die Polarität der Substanz berücksichtigt; im Anhang sind die Faktoren KF für verschiedene Verbindungen zusammengestellt. Verhältnismäßig häufig sind Daten verfügbar, bei denen ein Siedepunkt bei reduziertem Druck angegeben wird. In diesen Fällen wird der Dampfdruck wie folgt berechnet: wobei Tl = Siedepunkt bei reduziertem Druck Pl Bericht Bei der Schätzmethode sollte die vorgenommene Berechnung im Bericht umfassend dokumentiert werden. Literatur a) Watson, K.M. (1943). Ind. Eng. Chem, 35, 398. b) Lyman, W.J., Reehl, W.F., Rosenblatt, D. H. (1982). Handbook of Chemical Property Estimation Methods, McGraw Hill. c) OECD Environmental Monograph No. 67. Application of Structure-Activity Relationships to the Estimation of Properties Important in Exposure Assessment (1993). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A.5 Oberflächenspannung
1. Methoden
Den meisten der hier beschriebenen Methoden liegt die OECD-Prüfrichtlinie (1) zugrunde. Die Grundprinzipien sind in (2) angegeben.
1.1 Einleitung
Die hier beschriebenen Methoden sind zur Messung der Oberflächenspannung wässriger Lösungen anzuwenden.
Zweckdienlich ist, dass vor der Durchführung dieser Prüfungen Vorabinformationen über die Wasserlöslichkeit, die Struktur, die Hydrolyseeigenschaften und die kritische Konzentration für Mizellbildung des Stoffes vorliegen.
Die nachstehenden Methoden können für die meisten chemischen Substanzen ohne Einschränkung in Bezug auf ihren Reinheitsgrad angewendet werden.
Die Messung der Oberflächenspannung nach der Ringmethode beschränkt sich auf wässrige Lösungen mit einer dynamischen Viskosität unter ca. 200 mPa s.
1.2 Definition und Einheiten
Die freie Oberflächenenthalpie pro Oberflächeneinheit bezeichnet man als Oberflächenspannung.
Die Oberflächenspannung wird in folgenden Einheiten angegeben:
N/m (SI-Einheit) oder
mN/m (SI-Untereinheit)
1 N/m = 103 dyn/cm
1 mN/m = 1 dyn/cm im veralteten CGS-System
1.3 Referenzsubstanzen
Referenzsubstanzen müssen nicht in allen Fällen verwendet werden, in denen eine neue Prüfsubstanz untersucht wird. Die Referenzsubstanzen sollten in erster Linie dazu dienen, die Methode von Zeit zu Zeit zu überprüfen und einen Vergleich mit den Ergebnissen aus anderen Methoden zu ermöglichen.
Referenzsubstanzen, die einen weiten Bereich von Oberflächenspannungen abdecken, sind in der Literatur (1) (3) aufgeführt.
1.4 Prinzip der Methoden
Gemessen wird die maximale Kraft, die in vertikaler Richtung auf einen Bügel oder einen Ring ausgeübt werden muss, um diesen aus seinem Kontakt mit der Oberfläche der in ein Messgerät gefüllten Prüfflüssigkeit zu ziehen, bzw. die auf eine Platte ausgeübt werden muss, deren einer Rand in Kontakt mit der Oberfläche steht, um den gebildeten Film hochzuziehen.
Stoffe, die mindestens in einer Konzentration von 1 mg/l in Wasser löslich sind, werden in wässriger Lösung in einer einzigen Konzentration geprüft.
1.5 Qualitätskriterien
Die Genauigkeit dieser Methoden überschreitet wahrscheinlich alle Kontrollerfordernisse des Umweltschutzes.
1.6 Beschreibung der Methoden
Eine Lösung der Prüfsubstanz wird in destilliertem Wasser zubereitet. Die Konzentration dieser Lösung sollte bei 90 % der Sättigungslöslichkeit der Substanz in Wasser liegen; wenn diese Konzentration höher liegt als 1 g/l, wird für die Prüfung eine Konzentration von 1 g/l verwendet. Substanzen mit einer Wasserlöslichkeit unter 1 mg/l brauchen nicht geprüft zu werden.
1.6.1 Plattenmethode
Siehe ISO 304 und NF T 73-060 (Surface active agents - determination of surface tension by drawing up liquid films).
1.6.2 Bügelmethode
Siehe ISO 304 und NF T 73-060 (Surface active agents - determination of surface tension by drawing up liquid films).
1.6.3 Ringmethode
Siehe ISO 304 und NF T 73-060 (Surface active agents - determination of surface tension by drawing up liquid films).
1.6.4 OECD-Ringmethode
1.6.4.1 Apparatur
Zur Ausführung der in Betracht kommenden Messungen eignen sich handelsübliche Tensiometer. Sie bestehen aus folgenden Teilen:
1.6.4.1.1 Beweglicher Probentisch
Der bewegliche Probentisch dient als Untersatz für das thermostatisierte Messgefäß, in welchem sich die zu untersuchende Flüssigkeit befindet. Er ist zusammen mit dem Kraftmesssystem auf ein Stativ montiert.
1.6.4.1.2 Kraftmesssystem
Das Kraftmesssystem (siehe Abbildung) befindet sich über dem Probentisch. Der Fehler der Kraftmessung sollte einen Wert von ± 10-6 N nicht übersteigen, was einer Fehlergrenze von ± 0,1 mg bei der Massenbestimmung entspricht. In den meisten Fällen erfolgt die Einteilung der Messskala handelsüblicher Tensiometer in mN/m, so dass die Oberflächenspannung direkt in mN/m mit einer Genauigkeit von 0,1 mN/m abgelesen werden kann.
1.6.4.1.3 Messkörper (Ring)
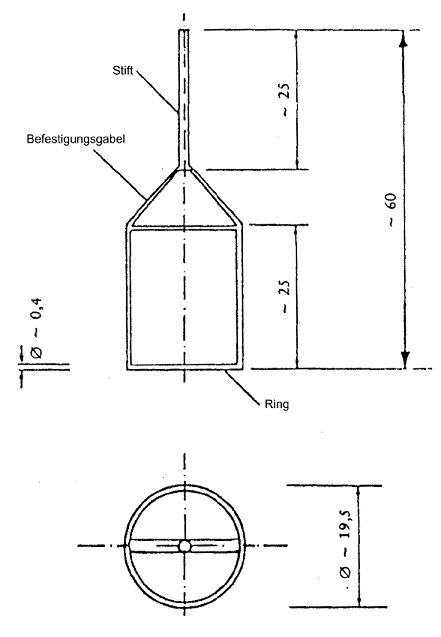
Üblicherweise wird der Ring aus Platin-Iridium-Draht mit einer Stärke von etwa 0,4 mm und einem mittleren Umfang von 60 mm hergestellt. Der Drahtring ist horizontal mittels einer Befestigungsgabel aus Draht und einem Metallstift aufgehängt, welche die Verbindung zum Kraftmesssystem darstellen (siehe Abbildung).
Abbildung Messkörper (alle Abmessungen in mm)
1.6.4.1.4 Messgefäß
Zur Aufnahme der Prüflösung bei den Messungen sollte ein thermostatisiertes Glasgefäß benutzt werden. Die Anordnung sollte so ausgelegt werden, dass während der Messung die Temperatur sowohl der Prüflösung wie auch die der sich über deren Oberfläche befindlichen Gasphase konstant bleiben und die Probe nicht verdampfen kann. Hierfür sollten zylindrische Glasgefäße mit einem Innendurchmesser von nicht weniger als 45 mm zur Anwendung kommen.
1.6.4.2 Vorbereitung der Apparatur
1.6.4.2.1 Reinigung
Die Glasgefäße müssen sorgfältig gereinigt werden. Falls notwendig, sollten sie mit heißer Chromschwefelsäure und anschließend mit sirupartiger Phosphorsäure (83 bis 98 Gew.- % H3 PO4) gewaschen, sorgfältig mit Leitungswasser gespült und schließlich nochmals mit doppelt destilliertem Wasser ausgewaschen werden, bis man eine neutrale Reaktion erhält. Daraufhin trocknet man das Gefäß oder spült es mit der zu untersuchenden Probenlösung aus.
Der Ring sollte zunächst sorgfältig mit Wasser abgewaschen werden, um alle wasserlöslichen Substanzen zu entfernen. Anschließend wird er kurzzeitig in Chromschwefelsäure getaucht, in doppelt destilliertem Wasser bis zur neutralen Reaktion gespült und schließlich kurz über einer Methanolflamme erhitzt.
Anmerkung
Verunreinigungen durch Substanzen, die weder durch Chromschwefelsäure noch Phosphorsäure gelöst oder zersetzt werden, wie beispielsweise Silikone, sind mittels geeigneter organischer Lösungsmittel zu entfernen.
1.6.4.2.2 Eichung der Apparatur
Die Validierung der Apparatur besteht in einer Überprüfung des Nullpunktes. Dieser sollte so eingestellt werden, dass die Instrumentenanzeige eine zuverlässige Bestimmung in mN/m zulässt.
Aufstellung
Das Gerät muss waagerecht aufgestellt werden, was sich beispielsweise unter Zuhilfenahme einer Wasserwaage, die man auf die Grundplatte des Tensiometers legt, und entsprechender Einstellungen mit den dort vorgesehenen Stellschrauben erzielen lässt.
Nullpunkteinstellung
Nach der Befestigung des Rings an der Apparatur und vor dem Eintauchen in die Flüssigkeit sind der Nullpunkt der Tensiometeranzeige einzustellen und die Parallelität des Rings zur Flüssigkeitsoberfläche zu überprüfen. Dazu kann man die Flüssigkeitsoberfläche als Spiegel benutzen.
Eichen
Das eigentliche Eichen vor den Untersuchungen lässt sich auf zweierlei Weise durchführen:
| Φa = σr/σa | (1) |
Hierbei ist:
| σr | = mg/2b (mN/m) |
| m | = Masse des Reiters |
| g | = Erdbeschleunigung (981 cm s-2 in Meereshöhe) |
| b | = mittlerer Umfang des Rings (cm) |
| σa | = abgelesener Wert am Tensiometer nach dem Anbringen des Reiters auf dem Ring (mN/m) |
| Φb = σo/σg | (2) |
Hierbei ist:
σo = angegebener Literaturwert für die Oberflächenspannung von Wasser (mN/m)
σg = gemessener Wert der Oberflächenspannung von Wasser (mN/m) beide bei der gleichen Temperatur.
1.6.4.3 Vorbereitung der Proben
Von den zu untersuchenden Substanzen sind wässrige Lösungen in den erforderlichen Konzentrationen herzustellen. Die Lösungen dürfen keine ungelösten Bestandteile enthalten.
Die Lösung ist bei konstanter Temperatur zu halten (± 0,5 °C). Da sich die Oberflächenspannung der im Messbehälter befindlichen Lösung im Verlauf der Zeit verändert, sollten Messungen zu verschiedenen Zeitpunkten vorgenommen und entsprechend eine Kurve erstellt werden, die die Oberflächenspannung in Abhängigkeit von der Zeit darstellt. Ein Gleichgewichtszustand ist erreicht, sobald keine weiteren Änderungen auftreten.
Verschmutzung durch Staub oder gasförmige Substanzen beeinträchtigt die Messung. Aus diesem Grunde sollten die Arbeiten unter einer Schutzhaube vorgenommen werden.
1.6.5 Prüfbedingungen
Die Messungen sind bei etwa 20 °C auszuführen, und die Temperaturkonstanz sollte mit ± 0,5 °C eingehalten werden.
1.6.6 Durchführung der Prüfung
Die zu messenden Lösungen werden in das sorgfältig gereinigte Messgefäß gefüllt, wobei darauf geachtet werden sollte, Schaumbildung zu vermeiden. Anschließend wird das Messgefäß auf den Tisch der Testapparatur gestellt. Das Tischoberteil mit dem Messgefäß wird nun so weit hochgeschraubt, bis der Ring unter die Oberfläche der zu messenden Lösung taucht. Daraufhin wird das Tischoberteil langsam und gleichmäßig abgesenkt (mit einer Geschwindigkeit von ca. 0,5 cm/min), um den Ring aus der Oberfläche herauszuziehen, bis ein maximaler Wert der Kraft erreicht ist. Der am Ring haftende Flüssigkeitsfilm darf nicht von ihm abreißen. Nach Beendigung der Messung wird der Ring wieder unter die Oberfläche getaucht und der Vorgang wiederholt, bis ein konstanter Wert der Oberflächenspannung erreicht ist. Bei jeder Bestimmung sollte die Zeitmessung mit dem Einfüllen der Lösung in das Messgefäß beginnen. Die Ablesung erfolgt jeweils zu dem Zeitpunkt, bei dem die Maximalkraft beim Herausziehen des Rings aus der Flüssigkeitsoberfläche erreicht ist.
2. Daten
Zur Berechnung der Oberflächenspannung wird zunächst der in mN/m an der Apparatur abgelesene Wert mit dem Eichfaktor Φa oder Φb (je nach dem verwendeten Eichverfahren) multipliziert. Man erhält einen Wert, der jedoch nur annähernd gilt und infolgedessen einer Korrektur bedarf.
Harkins und Jordan (4) haben empirische Korrekturfaktoren für Oberflächenspannungswerte bestimmt, die mit der Ringmethode gemessen wurden. Diese Faktoren sind von den Ringdimensionen, der Dichte der Flüssigkeit und ihrer Oberflächenspannung abhängig.
Da es umständlich ist, für jede einzelne Messung den Korrekturfaktor aus den Tabellen von Harkins und Jordan zu bestimmen, um die Oberflächenspannung wässriger Lösungen zu berechnen, kann eine vereinfachte Methode angewandt werden, die darin besteht, die korrigierten Werte für die Oberflächenspannung direkt aus der nachstehenden Tabelle abzulesen. (Für Ablesewerte, die zwischen den Tabellenwerten liegen, ist eine Interpolation möglich.)
Tabelle Korrektur der gemessenen Oberflächenspannungswerte
Nur für wässrige Lösungen, ρ = 1 g/cm3
| R | = 9,55 mm (mittlerer Ringradius) |
| r | = 0,185 mm (Radius des Ringdrahtes) |
| Experimenteller Wert (mN/m) | Korrigierter Wert (mN/m) | |
| Eichung mit Gewichten (vgl. 1.6.4.2.2 a) | Eichung mit Wasser (vgl. 1.6.4.2.2 b) | |
| 20 | 16,9 | 18,1 |
| 22 | 18,7 | 20,1 |
| 24 | 20,6 | 22,1 |
| 26 | 22,4 | 24,1 |
| 28 | 24,3 | 26,1 |
| 30 | 26,2 | 28,1 |
| 32 | 28,1 | 30,1 |
| 34 | 29,9 | 32,1 |
| 36 | 31,8 | 34,1 |
| 38 | 33,7 | 36,1 |
| 40 | 35,6 | 38,2 |
| 42 | 37,6 | 40,3 |
| 44 | 39,5 | 42,3 |
| 46 | 41,4 | 44,4 |
| 48 | 43,4 | 46,5 |
| 50 | 45,3 | 48,6 |
| 52 | 47,3 | 50,7 |
| 54 | 49,3 | 52,8 |
| 56 | 51,2 | 54,9 |
| 58 | 53,2 | 57,0 |
| 60 | 55,2 | 59,1 |
| 62 | 57,2 | 61,3 |
| 64 | 59,2 | 63,4 |
| 66 | 61,2 | 65,5 |
| 68 | 63,2 | 67,7 |
| 70 | 65,2 | 69,9 |
| 72 | 67,2 | 72,0 |
| 74 | 69,2 | - |
| 76 | 71,2 | - |
| 78 | 73,2 | - |
Die Zusammenstellung dieser Tabelle erfolgte auf der Grundlage der Harkins-Jordan-Korrekturen und entsprechend der DIN-Norm (DIN 53914) für Wasser und wässrige Lösungen (Dichte ρ = 1 g/cm3). Sie gilt für einen handelsüblichen Ring mit folgenden Abmessungen: R = 9,55 mm (mittlerer Ringradius) und r = 0,185 mm (Radius des Ringdrahtes). Die Tabelle enthält korrigierte Werte für Oberflächenspannungsmessungen nach einer Eichung entweder mit Gewichten oder mit Wasser.
Alternativ lässt sich die Oberflächenspannung ohne vorhergehende Eichung nach der folgenden Gleichung berechnen:
σ= (f x F)/4πR
Hierbei ist:
| F | = die vom Kraftmesssystem angegebene Kraft beim Abreißen des Films |
| R | = der Ringradius |
| f | = der Korrekturfaktor (1) |
3. Abschlussbericht
3.1 Prüfbericht
Im Prüfbericht ist, wenn möglich, Folgendes anzugeben:
3.2 Interpretation der Ergebnisse
Ausgehend davon, dass destilliertes Wasser bei 20 °C eine Oberflächenspannung von 72,75 mN/m hat, sollten Stoffe mit einer Oberflächenspannung unter 60 mN/m unter den Bedingungen dieses Verfahrens als oberflächenaktiv betrachtet werden.
4. Literatur
(1) OECD, Paris, 1981, Test Guideline 115, Decision of the Council C(81) 30 final.
(2) R. Weissberger (Hrsg.), Technique of Organic Chemistry, Physical Methods of Organic Chemistry, 3rd ed., Interscience Publ., New York, 1959, Vol. I, Part I, Chapter XIV.
(3) Pure Appl. Chem., 1976, Vol. 48, 511.
(4) Harkins, W.D., Jordan, H.F., J. Amer. Chem. Soc, 1930, Vol. 52, 1751.
| weiter . | |