

| zurück | |
| Methoden zur Bestimmung der physikalisch-chemischen Eigenschaften
A.19 Niedermolekularer Anteil von Polymeren | Anhang V zur RL 67/548/EWG |
A.19.1 Methode
Diese gelpermeationschromatographische Methode entspricht der OECD TG 119 (1996). Die wichtigsten Grundsätze und weitere technische Informationen werden in den Literaturhinweisen (1) genannt.
A.19.1.1 Einleitung
Da die Eigenschaften von Polymeren so unterschiedlich sind, ist es unmöglich, nur eine einzige Methode zu nennen, die alle Bedingungen für die Trennung und Auswertung erfüllt und somit sämtliche Eventualitäten und Besonderheiten bei der Trennung von Polymeren berücksichtigt. Insbesondere für komplexe polymere Systeme ist die Gelpermeationschromatographie (GPC) häufig nicht geeignet. Wenn die GPC nicht anwendbar ist, kann die Molmasse mit Hilfe anderer Methoden bestimmt werden (siehe Anhang). In solchen Fällen muß die verwendete Methode in allen Einzelheiten beschrieben und die Gründe für deren Verwendung genannt werden.
Die beschriebene Methode beruht auf DIN-Norm 55672 (1). Diese Norm enthält ausführliche Informationen darüber, wie die Versuche durchgeführt und wie die Daten ausgewertet werden müssen. Wenn Änderungen der Versuchsbedingungen notwendig sein sollten, müssen diese Änderungen begründet werden. Es können andere Normen herangezogen werden, diese müssen jedoch belegt werden. Im beschriebenen Verfahren werden Polystyrolproben bekannter Polydispersität zur Kalibrierung verwendet; es kann jedoch vorkommen, daß das Verfahren für bestimmte Polymere, z.B. wasserlösliche und langkettige, verzweigte Polymere, angepaßt werden muß.
A.19.1.2Definitionen und Einheiten
Der niedermolekulare Anteil wird willkürlich auf unter 1000 Dalton festgelegt.
Die zahlengemittelte Molmasse Mn und die gewichtsgemittelte Molmasse Mw werden anhand folgender Gleichungen bestimmt:
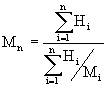
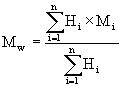
wobei,
| Hi | die Höhe des Detektorsignals von der Grundlinie für das Retentionsvolumen Vi, |
| Mi | die Molmasse der Polymerfraktion bei dem Retentionsvolumen Vi und |
| n | die Zahl der Datenpunkte bezeichnet. |
Die Breite der Molmassenverteilung, die ein Maß für die Dispersität des Systems ist, wird durch das Verhältnis Mw/Mn ausgedrückt.
A.19.1.3Referenzsubstanzen
Da es sich bei der GPC um eine relative Methode handelt, muß eine Kalibrierung vorgenommen werden. Hierzu werden in der Regel eng verteilte, linear aufgebaute Polystyrolstandards mit bekannten mittleren Molmassen Mn und Mw und einer bekannten Molmassenverteilung verwendet. Die Eichkurve kann für die Bestimmung der Molmassen unbekannter Proben nur herangezogen werden, wenn die Bedingungen für die Trennung der Probe und der Standards identisch sind.
Ein fester Bezug zwischen der Molmasse und dem Elutionsvolumen ist nur unter den spezifischen Bedingungen des betreffenden Versuchs zulässig. Diese Bedingungen umfassen vor allem die Temperatur, das Lösungsmittel (oder die Lösungsmittelmischung), die chromatographischen Bedingungen und die Trennsäule bzw. das Trennsäulensystem.
Bei den auf diese Weise ermittelten Molmassen der Probe handelt es sich um relative Werte, die als "polystyrol-äquivalente Molmasse" bezeichnet werden. Das bedeutet, daß - in Abhängigkeit von den strukturellen und chemischen Unterschieden zwischen der Probe und den Standards - die Molmassen mehr oder weniger von den absoluten Werten abweichen können. Werden andere Standards verwendet, z.B. Polyethylenglykol, Polyethylenoxid, Polymethylmethacrylat, Polyacrylsäure, so muß dies begründet werden.
A.19.1.4Prinzip der Prüfmethode
Sowohl die Molmassenverteilung der Probe als auch die mittleren Molmassen (Mn, Mw) können mit Hilfe der GPC bestimmt werden. Bei der GPC handelt es sich um eine besondere Form der Flüssigchromatographie, bei der die Probe nach den hydrodynamischen Volumina der einzelnen Bestandteile (2) aufgetrennt wird.
Die Trennung erfolgt, indem die Probe durch eine Säule läuft, die mit einem porösen Material, in der Regel einem organischen Gel, gefüllt ist. Kleine Moleküle durchdringen die Poren, während große Moleküle ausgeschlossen werden. Der Weg der großen Moleküle ist daher kürzer, und folglich werden diese zuerst eluiert. Die Moleküle mittlerer Größe durchdringen einige der Poren und werden zu einem späteren Zeitpunkt eluiert. Die kleinsten Moleküle, mit einem durchschnittlichen hydrodynamischen Radius, der kleiner ist als die Poren des Gels, können alle Poren durchdringen. Diese werden zuletzt eluiert.
Im Idealfall erfolgt die Trennung ausschließlich über die Größe der Moleküle, doch ist es in der Praxis schwierig, gewisse störende Absorptionseffekte zu vermeiden. Ungleichmäßige Säulenfüllungen und Totvolumen können zur weiteren Verschlechterung der Trennung führen (2).
Die Detektion erfolgt beispielsweise über den Brechungsindex oder die UV-Absorption und ergibt eine einfache Verteilungskurve. Um tatsächliche Molmassenwerte für die Kurve zu erhalten, ist es notwendig, die Säule zu kalibrieren, indem Polymere mit bekannter Molmasse sowie idealerweise auch mit im großen und ganzen vergleichbarer Struktur, z.B. verschiedene Polystyrolstandards, auf diese Säule aufgegeben werden. In der Regel ergibt sich eine Gauß'sche Kurve, die gelegentlich durch einen kleinen Schwanz in Richtung der niedrigen Molmassen verzerrt ist; die vertikale Achse zeigt die Häufigkeit der verschiedenen eluierten Molmassenfraktionen, die horizontale Achse log Molmasse.
Der niedermolekulare Anteil wird aus dieser Kurve abgeleitet. Die Berechnung kann nur dann genau sein, wenn die niedermolekularen Fraktionen in Bezug auf die Masse äquivalent zum Polymer als ganzes sind.
A.19.1.5 Qualitätskriterien
Die Wiederholbarkeit (Relative Standardabweichung: RSA) für den Wert des Elutionsvolumens sollte besser als 0,3 % sein. Die geforderte Wiederholbarkeit der Analyse muß durch Korrektur mittels eines internen Standards gewährleistet sein, wenn ein Chromatogramm zeitabhängig ausgewertet wird und nicht dem obengenannten Kriterium (1) entspricht. Die Polydispersitäten sind von den Molmassen der Standards abhängig. Für die Polystyrolstandards sind folgende Werte charakteristisch:
| Mp < 2000 | Mw / Mn < 1,20 |
| 2000 ≤ Mp ≤ 106 | Mw / Mn < 1,05 |
| Mp > 106 | Mw / Mn < 1,20 |
| (Mp bezeichnet die Molmasse des Standards am Peakmaximum) | |
A.19.1.6Beschreibung der Prüfmethode
A.19.1.6.1Vorbereitung der Standardpolystyrollösungen
Die Polystyrolstandards werden vorsichtig im gewählten Elutionsmittel gelöst. Die Empfehlungen des Herstellers müssen bei der Vorbereitung der Lösungen berücksichtigt werden.
Die Konzentrationen der gewählten Standards sind von verschiedenen Faktoren abhängig, z.B. Injektionsvolumen, Viskosität der Lösung und Empfindlichkeit des analytischen Detektors. Das maximale Injektionsvolumen muß der Länge der Säule angepaßt werden, um eine überbeladung zu vermeiden. Normalerweise liegen die Injektionsvolumina für analytische Trennungen mittels GPC durch eine Säule von 30 cm x 7,8 mm zwischen 40 und 100 µl. Größere Volumen sind möglich, doch sollten 250 µl nicht überschritten werden. Das optimale Verhältnis zwischen Injektionsvolumen und Konzentration muß vor der eigentlichen Kalibrierung der Säule bestimmt werden.
A.19.1.6.2 Vorbereitung der Probelösung
Im Prinzip gelten die zuvor genannten Anforderungen auch für die Vorbereitung der Probelösungen. Die Probe wird in einem geeigneten Lösungsmittel, z.B. Tetrahydrofuran (THF), durch vorsichtiges Schütteln gelöst. Die Lösung sollte unter keinen Umständen mittels Ultraschallbad gelöst werden. Wenn nötig, wird die Probelösung mit Hilfe eines Membranfilters mit einer Porengröße von 0,2 bis 2 µm gereinigt.
Die Anwesenheit ungelöster Partikel muß im Abschlußbericht dokumentiert werden, da diese auf hohe Molmassenfraktionen zurückzuführen sein könnte. Es sollte ein geeignetes Verfahren verwendet werden, um die Gewichtsanteile der ungelösten Partikel zu bestimmen. Die Lösung sollte innerhalb von 24 Stunden verbraucht werden.
A.19.1.6.3Berichtigungen aufgrund von Verunreinigungen und Zusatzstoffen
Die Korrektur des Gehalts an Fraktionen mit M < 1000 aufgrund bestimmter vorhandener nichtpolymerer Komponenten (z.B. Verunreinigungen und/oder Zusatzstoffe) ist in der Regel notwendig, sofern der gemessene Gehalt nicht bereits < 1 % ist. Dies wird durch die direkte Analyse der Polymerlösung oder des GPC-Eluats erreicht.
Wenn das Eluat nach Durchlaufen der Säule für eine weitere Analyse zu verdünnt ist, muß es konzentriert werden. Es kann u. U. erforderlich sein, das Eluat bis zur Trocknung einzudampfen und den Rückstand neu aufzulösen. Die Konzentrierung des Eluats muß unter Bedingungen erfolgen, die sicherstellen, daß im Eluat keine Veränderungen auftreten. Die Behandlung des Eluats nach der GPC ist abhängig davon, welches analytische Verfahren für die quantitative Bestimmung eingesetzt wird.
A.19.1.6.4 Apparatur
Die GPC-Apparatur besteht aus folgenden Komponenten:
Es muß sichergestellt sein, daß das GPC-System gegenüber dem verwendeten Lösungsmittel inert ist (z.B. durch die Verwendung von Stahlkapillaren für das Lösungsmittel THF).
A.19.1.6.5Injektion und Lösungsmittelzugabesystem
Auf die Säule wird eine bestimmte Menge der Probelösung, entweder automatisch oder manuell in einer scharf begrenzten Zone aufgegeben. Ein zu schnelles Zurückziehen oder Drücken des Spritzen-kolbens (bei manueller Ausführung) kann Veränderungen in der beobachteten Molmassenverteilung zur Folge haben. Die Lösungsmittelzugabe sollte möglichst pulsationafrei sein, wobei idealerweise ein Pulsationsdämpfer eingesetzt wird. Die Durchflußgeschwindigkeit liegt in der Größenordnung von 1 ml/min.
A.19.1.6.6 Säule
Je nach Art der Probe wird das Polymer durch Verwendung einer einfachen oder mehrerer in Reihe geschalteter Säulen charakterisiert. Im Handel ist eine Reihe poröser Säulenmaterialien mit definierten Eigenschaften (z.B. Porengröße, Ausschlußgrenzen) erhältlich. Die Wahl des Trenngels oder der Länge der Säule ist sowohl von den Eigenschaften der Probe (hydrodynamisches Volumen, Molmassenverteilung) als auch von den spezifischen Bedingungen für die Trennung wie z.B. Lösungsmittel, Temperatur und Durchflußgeschwindigkeit (1) (2) (3) abhängig.
A.19.1.6.7Theoretische Böden
Die für die Trennung verwendete Säule bzw. Säulenkombination muß durch die Anzahl der theoretischen Böden charakterisiert sein. Dies umfaßt (wenn THF als Elutionsmittel verwendet wird) die Aufgabe einer Lösung von Ethylenbenzol oder einer anderen geeigneten nichtpolaren Substanz auf die Säule. Die Zahl der theoretischen Böden ergibt sich aus folgender Gleichung:
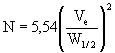 oder
oder 
| wobei, | N | die Zahl der theoretischen Böden, |
| Ve | das Elutionsvolumen am Peakmaximum, | |
| W | die Peakbreite an der Grundlinie, | |
| W1/2 | die Peakbreite in halber Höhe bezeichnet. |
A.19.1.6.8 Trennleistung
Außer der Zahl der theoretischen Böden, die für die Bestimmung der Bandbreite notwendig ist, spielt auch die Trennleistung eine Rolle, die sich aus der Steilheit der Eichkurve ergibt. Die Trennleistung einer Säule wird aus folgender Beziehung abgeleitet:

wobei,
| Ve,Mx | das Elutionsvolumen für Polystyrol mit der Molmasse Mx, |
| Ve,(10Mx) | das Elutionsvolumen für Polystyrol mit einer zehnmal größeren Molmasse bezeichnet. |
Die Auflösung (R) des Systems wird allgemein wie folgt definiert:

wobei,
| Ve1, Ve2 | die Elutionsvolumen der beiden Polystyrolstandards am Peakmaximum, |
| W1, W2 | die Peakbreite an der Grundlinie, |
| M1, M2 | die Molmassen am Peakmaximum (sollten um den Faktor 10 differieren) bezeichnet. |
Der R-Wert für das Säulensystem sollte größer als 1,7 (4) sein.
A.19.1.6.9 Lösungsmittel
Alle Lösungsmittel müssen von höchster Reinheit sein (THF wird in einer Reinheit von 99,5 % verwendet). Die Größe des Lösungsmittelreservoirs (gegebenenfalls in einer Inertgasatmosphäre) muß für die Kalibrierung der Säule und mehrere Probenanalysen ausreichend sein. Das Lösungsmittel muß entgast werden, bevor es mit Hilfe der Pumpe auf die Säule aufgegeben wird.
A.19.1.6.10 Temperaturkontrolle
Die Temperatur von Injektionsschleife Säulen, Detektor und Säulenmaterial sollte konstant und auf das gewählte Lösungsmittel abgestimmt sein.
A.19.1.6.11 Detektor
Der Detektor dient zur mengenmäßigen Erfassung der Konzentration der aus der Säule eluierten Probe. Um eine unnötige Verbreiterung der Peaks zu vermeiden, muß das Kuvettenvolumen der Detektorzelle so klein wie möglich gehalten werden. Außer bei Lichtstreuungs- und Viskositätsdetektoren sollte es nicht mehr als 10 µl betragen. Für die Detektion wird in der Regel die Differentialrefraktometrie eingesetzt. Wenn es die spezifischen Eigenschaften der Probe oder des Elutionsmittels erfordern, können auch andere Detektortypen verwendet werden, z.B. UV/VIS-, IR-, Viskositätsdetektoren etc.
A.19.2 Daten und Berichterstattung
A.19.2.1 Daten
Im Hinblick auf die detaillierten Auswertungskriterien wie auch für die Anforderungen bezüglich Datenerfassung und -verarbeitung sollte die DIN-Norm (1) angewendet werden.
Für jede Probe müssen zwei unabhängige Versuche durchgeführt werden, die getrennt analysiert werden. Ferner ist es absolut unerläßlich, auch Daten aus Blindproben zu ermitteln, die unter den gleichen Bedingungen getestet werden wie die Probe.
Es ist ausdrücklich darauf hinzuweisen, daß es sich bei den gemessenen Werten um Relativwerte handelt, die der Molmasse des verwendeten Standards äquivalent sind.
Nach der Bestimmung der Retentionsvolumina oder der Retentionszeiten (u. U. mit Hilfe eines internen Standards korrigiert) werden die log Mp Werte (wobei Mp das Peakmaximum des Eichstandards ist) gegen eine dieser Größen aufgetragen. Mindestens zwei Eichpunkte sind pro Molmassendekade notwendig, und mindestens fünf Meßpunkte sind für die Gesamtkurve erforderlich, durch die die geschätzte Molmasse der Probe erfaßt werden soll. Der niedermolekulare Endpunkt der Eichkurve wird durch n-Hexylbenzol oder eine andere geeignete nichtpolare Substanz definiert. Zahlenmittel und Gewichtsmittel der Molmasse werden im allgemeinen mittels elektronischer Datenverarbeitung auf der Grundlage der in Abschnitt 1.2 genannten Formeln ermittelt. Bei manueller Auswertung kann die ASTM D 3536-91 herangezogen werden (3).
Wenn unlösliche Polymeranteile in der Säule zurückgehalten werden, ist ihre Molmasse wahrscheinlich höher als die der löslichen Fraktion. Wird dies nicht berücksichtigt, kann der niedermolekulare Anteil zu hoch eingeschätzt werden; im Anhang ist beschrieben, wie der unlösliche Polymeranteil berücksichtigt werden kann.
Die Verteilungskurve muß in Form einer Tabelle oder als Zahl (differentielle Häufigkeit oder Summenprozent gegen log M) dargestellt werden. Bei der graphischen Darstellung sollte eine Molmassendekade in der Regel 4 cm breit sein, und das Peakmaximum sollte etwa 8 cm sein. Bei integralen Verteilungskurven sollte der Abstand auf der Ordinate zwischen 0 und 100 % ca. 10 cm betragen.
A.19.2.2 Prüfbericht
Der Prüfbericht muß folgende Informationen enthalten:
A.19.2.2.1 Prüfsubstanz
A.19.2.2.2 Instrumentierung
A.19.2.2.3 Systemkalibrierung
A.19.2.2.4 Angaben zum niedermolekularen Anteil
A.19.2.2.5 Auswertung
A.19.3 Literaturhinweise
(1) DIN 55672 (1995) Gelpermeationschromatographie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1.
(2) Yau, W.W., Kirkland, J.J., and Bly, D.D. eds (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons.
(3) ASTM D 3536-91, (1991). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography - GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania.
(4) ASTM D 5296-92, (1992). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania.
Anhang
zur RL 67/548/EWG Anhang V A.19.
Korrektur des niedermolekularen Anteils um unlösliche Polymerfraktionen
Sind unlösliche Polymeranteile in einer Probe vorhanden, so führt dies zu Masseverlusten während der GPC-Analyse. Das unlösliche Polymer kann an der Säule bzw. im Probenfilter zurückgehalten werden, während der lösliche Teil der Probe die Säule durchläuft. Wenn das Brechungsindexinkrement (dn/dc) des Polymers geschätzt oder gemessen werden kann, kann auch der Masseverlust der Probe in der Säule abgeschätzt werden. In diesem Fall wird eine Korrektur anhand einer externen Kalibrierung mit Standardmaterialien bekannter Konzentration und bekanntem dn/dc zur Eichung des Refraktometers vorgenommen. In dem folgenden Beispiel wird ein Polymethylmethacrylat (pMMA)-Standard verwendet.
Bei der externen Kalibrierung zur Analyse von Acrylpolymeren wird ein pMMA-Standard bekannter Konzentration in Tetrahydrofuran mittels GPC untersucht; die sich daraus ergebenden Daten dienen der Ermittlung der Refraktometerkonstanten mit folgender Gleichung:
K = R / (C x V x dn/ dc )
wobei
| K | die Refraktometerkonstante (in Mikrovoltsekunde/ml), |
| R | die Meßgröße für den pMMA-Standard (in Mikrovoltsekunde), |
| C | die Konzentration des pMMA-Standards (in mg/ml), |
| V | das Injektionsvolumen (in ml) und |
| dn/dc | das Brechungsindexinkrement für pMMA in Tetrahydrofuran (in ml/mg) bezeichnet. |
Die folgenden Daten sind für einen pMMA-Standard charakteristisch:
| R | = 2 937 891 |
| C | = 1,07 mg/ml |
| V | = 0,1 ml |
| dn/dc | = 9 x 10-5 ml/mg. |
Der sich daraus ergebende Wert K = 3,05 ξ 1011 wird dann zur Berechnung des theoretischen Detektorsignals herangezogen, wenn 100 % des injizierten Polymers den Detektor passiert haben.
| weiter . | |