
umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (5)
 | zurück |
|
A.18 Zahlengemittelte Molmasse und Molmassenverteilung von Polymeren
1. Methode
Diese gelpermeationschromatografische Methode entspricht der OECD TG 118 (1996). Die wichtigsten Grundsätze und weitere technische Informationen werden in den Literaturhinweisen (1) genannt.
1.1 Einleitung
Da die Eigenschaften von Polymeren so unterschiedlich sind, ist es unmöglich, nur eine einzige Methode zu nennen, die alle Bedingungen für die Trennung und Auswertung erfüllt und somit sämtliche Eventualitäten und Besonderheiten bei der Trennung von Polymeren berücksichtigt. Insbesondere für komplexe polymere Systeme ist die Gelpermeationschromatografie (GPC) häufig nicht geeignet. Wenn die GPC nicht anwendbar ist, kann die Molmasse mit Hilfe anderer Methoden bestimmt werden (siehe Anlage). In solchen Fällen muss die verwendete Methode in allen Einzelheiten beschrieben und die Gründe für deren Verwendung genannt werden.
Die beschriebene Methode beruht auf DIN-Norm 55672 (1). Diese Norm enthält ausführliche Informationen darüber, wie die Versuche durchgeführt und wie die Daten ausgewertet werden müssen. Wenn Änderungen der Versuchsbedingungen notwendig sein sollten, müssen diese Änderungen begründet werden. Es können andere Normen herangezogen werden, diese müssen jedoch belegt werden. Im beschriebenen Verfahren werden Polystyrolproben bekannter Polydispersität zur Kalibrierung verwendet; es kann jedoch vorkommen, dass das Verfahren für bestimmte Polymere, z.B. wasserlösliche und langkettige, verzweigte Polymere, angepasst werden muss.
1.2 Definitionen und Einheiten
Die zahlengemittelte Molmasse Mn und die gewichtsgemittelte Molmasse werden anhand folgender Gleichungen bestimmt:

| Dabei ist: | |
| Hi = | die Höhe des Detektorsignals von der Grundlinie für das Retentionsvolumen Vi |
| Mi = | die Molmasse der Polymerfraktion bei dem Retentionsvolumen Vi und |
| n = | die Zahl der Datenpunkte |
Die Breite der Molmassenverteilung, die ein Maß für die Dispersität des Systems ist, wird durch das Verhältnis Mw/Mn ausgedrückt.
1.3 Referenzsubstanzen
Da es sich bei der GPC um eine relative Methode handelt, muss eine Kalibrierung vorgenommen werden. Hierzu werden in der Regel eng verteilte, linear aufgebaute Polystyrolstandards mit bekannten mittleren Molmassen Mn und Mw und einer bekannten Molmassenverteilung verwendet. Die Eichkurve kann für die Bestimmung der Molmassen unbekannter Proben nur herangezogen werden, wenn die Bedingungen für die Trennung der Probe und der Standards identisch sind.
Ein fester Bezug zwischen der Molmasse und dem Elutionsvolumen ist nur unter den spezifischen Bedingungen des betreffenden Versuchs zulässig. Diese Bedingungen umfassen vor allem die Temperatur, das Lösungsmittel (oder die Lösungsmittelmischung), die chromatografischen Bedingungen und die Trennsäule bzw. das Trennsäulensystem.
Bei den auf diese Weise ermittelten Molmassen der Probe handelt es sich um relative Werte, die als "polystyro äquivalente" Molmasse bezeichnet werden. Das bedeutet, dass - in Abhängigkeit von den strukturellen und chemischen Unterschieden zwischen der Probe und den Standards - die Molmassen mehr oder weniger von den absoluten Werten abweichen können. Werden andere Standards verwendet, z.B. Polyethylenglykol, Polyethylenoxid, Polymethylmethacrylat, Polyacrylsäure, so muss dies begründet werden.
1.4 Prinzip der Prüfmethode
Sowohl die Molmassenverteilung der Probe als auch die mittleren Molmassen (Mn, Mw) können mit Hilfe der GPC bestimmt werden. Bei der GPC handelt es sich um eine besondere Form der Flüssigchromatografie, bei der die Probe nach den hydrodynamischen Volumina der einzelnen Bestandteile (2) aufgetrennt wird.
Die Trennung erfolgt, indem die Probe durch eine Säule läuft, die mit einem porösen Material, in der Regel einem organischen Gel, gefüllt ist. Kleine Moleküle durchdringen die Poren, während große Moleküle ausgeschlossen werden. Der Weg der großen Moleküle ist daher kürzer, und folglich werden diese zuerst eluiert. Die Moleküle mittlerer Größe durchdringen einige der Poren und werden zu einem späteren Zeitpunkt eluiert. Die kleinsten Moleküle, mit einem durchschnittlichen hydrodynamischen Radius, der kleiner ist als die Poren des Gels, können alle Poren durchdringen. Diese werden zuletzt eluiert.
Im Idealfall erfolgt die Trennung ausschließlich über die Größe der Moleküle, doch ist es in der Praxis schwierig, gewisse störende Absorptionseffekte zu vermeiden. Ungleichmäßige Säulenfüllungen und Totvolumen können zur weiteren Verschlechterung der Trennung führen (2).
Die Detektion erfolgt beispielsweise über den Brechungsindex oder die UV-Absorption und ergibt eine einfache Verteilungskurve. Um tatsächliche Molmassenwerte für die Kurve zu erhalten, ist es notwendig, die Säule zu kalibrieren, indem Polymere mit bekannter Molmasse und idealerweise auch mit im großen und ganzen vergleichbarer Struktur, z.B. verschiedene Polystyrolstandards, auf diese Säule aufgegeben werden. In der Regel ergibt sich eine Gauß'sche Kurve, die manchmal durch einen kleinen Schwanz in Richtung der niedrigen Molmassen verzerrt ist; die vertikale Achse zeigt die Häufigkeit der verschiedenen eluierten Molmassenfraktionen, die horizontale Achse log Molmasse.
1.5 Qualitätskriterien
Die Wiederholbarkeit (Relative Standardabweichung:
RSA) für den Wert des Elutionsvolumens sollte besser als 0,3 % sein.
Die geforderte Wiederholbarkeit der Analyse muss durch Korrektur mittels eines internen Standards gewährleistet sein, wenn ein Chromatogramm zeitabhängig ausgewertet wird und nicht dem oben genannten Kriterium (1) entspricht.
Die Polydispersitäten sind von den Molmassen der Standards abhängig.
Für die Polystyrolstandards sind folgende Werte charakteristisch:
|
Mp < 2.000 | Mw/Mn < 1,20 |
|
2.000 ≤ Mp ≤ 106 | Mw/Mn < 1,05 |
|
Mp > 106 | Mw//Mn < 1,20 |
| (Mp bezeichnet die Molmasse des Standards am Peakmaximum.) | |
1.6 Beschreibung der Testmethode
1.6.1 Vorbereitung der Standardpolystyrollösungen
Die Polystyrolstandards werden vorsichtig im gewählten Elutionsmittel gelöst. Die Empfehlungen des Herstellers müssen bei der Vorbereitung der Lösungen berücksichtigt werden.
Die Konzentrationen der gewählten Standards sind von verschiedenen Faktoren abhängig, z.B. Injektionsvolumen, Viskosität der Lösung und Empfindlichkeit des analytischen Detektors. Das maximale Injektionsvolumen muss der Länge der Säule angepasst werden, um eine Überladung zu vermeiden. Normalerweise liegen die Injektionsvolumina für analytische Trennungen mittels GPC durch eine Säule von 30 cm x 7,8 mm zwischen 40 und 100 µl. Größere Volumen sind möglich, doch sollten 250 µl nicht überschritten werden. Das optimale Verhältnis zwischen Injektionsvolumen und Konzentration muss vor der eigentlichen Kalibrierung der Säule bestimmt werden.
1.6.2 Vorbereitung der Probelösung
Im Prinzip gelten die zuvor genannten Anforderungen auch für die Vorbereitung der Probelösungen. Die Probe wird in einem geeigneten Lösungsmittel, z.B. Tetrahydrofuran (THF), durch vorsichtiges Schütteln gelöst. Das Polymer sollte unter keinen Umständen mittels Ultraschallbad gelöst werden. Wenn nötig, wird die Probelösung mit Hilfe eines Membranfilters mit einer Porengröße von 0,2 bis 2 µm gereinigt.
Die Anwesenheit ungelöster Partikel muss im Abschlußbericht dokumentiert werden, da diese auf hohe Molmassenfraktionen zurückzuführen sein könnte. Es sollte ein geeignetes Verfahren verwendet werden, um die Gewichtsanteile der ungelösten Partikel zu bestimmen. Die Lösung sollte innerhalb von 24 Stunden verbraucht werden.
1.6.3 Apparatur
- Lösungsmittelvorratsgefäß
- Vorrichtung zum Entgasen (gegebenenfalls)
- Pumpe
- Pulsationsdämpfer (gegebenenfalls)
- Injektionssystem
- Chromatografiesäulen
- Detektor
- Durchflussmesser (gegebenenfalls)
- Datenaufzeichnungs-/-verarbeitungsgerät
- Abfallbehältnis
Es muss sichergestellt sein, dass das GPC-System gegenüber dem verwendeten Lösungsmittel inert ist (z.B. durch die Verwendung von Stahlkapillaren für das Lösungsmittel THF).
1.6.4 Injektion und Lösungsmittelzugabesystem
Auf die Säule wird eine bestimmte Menge der Probelösung, entweder automatisch oder manuell in einer scharf begrenzten Zone aufgegeben. Ein zu schnelles Zurückziehen oder Drücken des Spritzenkolbens (bei manueller Ausführung) kann Veränderungen in der beobachteten Molmassenverteilung zur Folge haben. Die Lösungsmittelzugabe sollte möglichst pulsationsfrei erfolgen, wobei idealerweise ein Pulsationsdämpfer eingesetzt wird. Die Durchflussgeschwindigkeit liegt in der Größenordnung von 1 ml/min.
1.6.5 Säule
Je nach Art der Probe wird das Polymer durch Verwendung einer einfachen oder mehrerer in Reihe geschalteter Säulen charakterisiert. Im Handel ist eine Reihe poröser Säulenmaterialien mit definierten Eigenschaften (z.B. Porengröße, Ausschlussgrenzen) erhältlich. Die Wahl des Trenngels oder der Länge der Säule ist sowohl von den Eigenschaften der Probe (hydrodynamisches Volumen, Molmassenverteilung) als auch von den spezifischen Bedingungen für die Trennung wie z.B. Lösungsmittel, Temperatur und Durchflussgeschwindigkeit (1) (2) (3) abhängig.
1.6.6 Theoretische Böden
Die für die Trennung verwendete Säule bzw. Säulenkombination muss durch die Anzahl der theoretischen Böden charakterisiert sein. Dies umfasst (wenn THF als Elutionsmittel verwendet wird) die Aufgabe einer Lösung von Ethylenbenzol oder einer anderen geeigneten nichtpolaren Substanz auf die Säule. Die Zahl der theoretischen Böden ergibt sich aus folgender Gleichung:

| Dabei ist: | |
| N = | die Zahl der theoretischen Böden |
| Ve = | das Elutionsvolumen am Peakmaximum |
| W = | die Peakbreite an der Grundlinie |
| W1/2 = | die Peakbreite in halber Höhe |
1.6.7 Trennleistung
Außer der Zahl der theoretischen Böden, die für die Bestimmung der Bandbreite notwendig ist, spielt auch die Trennleistung eine Rolle, die sich aus der Steilheit der Eichkurve ergibt. Die Trennleistung einer Säule wird aus folgender Beziehung abgeleitet:

Dabei ist:
| Ve, Mx | = das Elutionsvolumen für Polystyrol mit der Molmasse Mx |
| Ve,(10.Mx) | = das Elutionsvolumen für Polystyrol mit einer zehnmal größeren Molmasse |
Die Auflösung (R) des Systems wird allgemein wie folgt definiert:
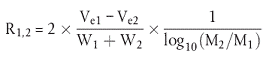
Dabei ist:
| Ve1, Ve2 | = die Elutionsvolumen der beiden Polystyrolstandards am Peakmaximum |
| W1, W2 | = die Peakbreite an der Grundlinie |
| M1, M2 | = die Molmassen am Peakmaximum (sollten um den Faktor 10 differieren) |
Der R-Wert für das Säulensystem sollte größer als 1,7 (4) sein.
1.6.8 Lösungsmittel
Alle Lösungsmittel müssen von höchster Reinheit sein (T'HF wird in einer Reinheit von 99,5 % verwendet). Die Größe des Lösungsmittelreservoirs (gegebenenfalls in einer Inertgasatmosphäre) muss für die Kalibrierung der Säule und mehrere Probenanalysen ausreichend sein. Das Lösungsmittel muss entgast werden, bevor es mit Hilfe der Pumpe auf die Säule aufgegeben wird.
1.6.9 Temperaturkontrolle
Die Temperatur von Injektionsschleife, Säulen, Detektor und Säulenmaterial sollte konstant und auf das gewählte Lösungsmittel abgestimmt sein.
1.6.10 Detektor
Der Detektor dient zur mengenmäßigen Erfassung der Konzentration der aus der Säule eluierten Probe. Um eine unnötige Verbreiterung der Peaks zu vermeiden, muss das Kuvettenvolumen der Detektorzelle so klein wie möglich gehalten werden. Außer bei Lichtstreuungs- und Viskositätsdetektoren sollte es nicht mehr als 10 Il betragen. Für die Detektion wird in der Regel die Differentialrefraktometrie eingesetzt. Wenn es die spezifischen Eigenschaften der Probe oder des Elutionsmittels erfordern, können auch andere Detektortypen verwendet werden, z.B. UV/VIS-, IR-, Viskositätsdetektoren etc.
2. Daten und Berichterstattung
2.1 Daten
Im Hinblick auf die detaillierten Auswertungskriterien wie auch für die Anforderungen bezüglich Datenerfassung und -Verarbeitung sollte die DIN-Norm (1) angewendet werden.
Für jede Probe müssen zwei unabhängige Versuche durchgeführt werden, die getrennt analysiert werden.
Mn, Mw, Mw/Mn und Mp müssen für jede der Messungen bekannt sein. Es ist ausdrücklich darauf hinzuweisen, dass es sich bei den gemessenen Werten um Relativwerte handelt, die der Molmasse des verwendeten Standards äquivalent sind.
Nach der Bestimmung der Retentionsvolumina oder der Retentionszeiten (u. U. mit Hilfe eines internen Standards korrigiert) werden die log Mp Werte (wobei Mp das Peakmaximum des Eichstandards ist) gegen eine dieser Größen aufgetragen. Mindestens zwei Eichpunkte sind pro Molmassendekade notwendig, und mindestens fünf Messpunkte sind für die Gesamtkurve erforderlich, durch die die geschätzte Molmasse der Probe erfasst werden soll. Der niedermolekulare Endpunkt der Eichkurve wird durch n-Hexylbenzol oder eine andere geeignete nichtpolare Substanz definiert. Zahlenmittel und Gewichtsmittel der Molmasse werden im Allgemeinen mittels elektronischer Datenverarbeitung auf der Grundlage der in Abschnitt 1.2 genannten Formeln ermittelt. Bei manueller Auswertung kann die ASTM D 3536-91 herangezogen werden (3).
Die Verteilungskurve muss in Form einer Tabelle oder als Abbildung (differentielle Häufigkeit oder Summenprozent gegen log M) dargestellt werden. Bei einer grafischen Darstellung sollte eine Molmassendekade in der Regel 4 cm breit sein, und das Peakmaximum sollte etwa 8 cm sein. Bei integralen Verteilungskurven sollte der Abstand auf der Ordinate zwischen 0 und 100 % bei ca. 10 cm liegen.
2.2 Prüfbericht
Der Prüfbericht muss folgende Informationen enthalten:
2.2.1 Prüfsubstanz
- verfügbare Informationen über die Prüfsubstanz (Identität, Zusatzstoffe, Verunreinigungen)
- Beschreibung der Probenbehandlung, Beobachtungen, Probleme
2.2.2 Instrumentierung
- Reservoir des Elutionsmittels, Inertgas, Entgasung des Elutionsmittels, Zusammensetzung des Elutionsmittels, Verunreinigungen
- Pumpe, Pulsationsdämpfer, Injektionssystem
- Trennsäulen (Hersteller, alle Angaben zu den Säuleneigenschaften, z.B. Porengröße, Art des Trennmaterials etc., Zahl, Länge und Anordnung der verwendeten Säulen)
- Zahl der theoretischen Böden der Säule (oder Säulenkombination), Trennleistung (Auflösungsvermögen des Systems)
- Angaben über die Peaksymmetrie
- Säulentemperatur, Art der Temperaturkontrolle
- Detektor (Messprinzip, Typ, Kuvettenvolumen)
- gegebenenfalls Durchflussmesser (Hersteller, Messprinzip)
- Datenaufzeichnungs- und -Verarbeitungssystem (Hardware und Software)
2.2.3 Systemkalibrierung
- detaillierte Beschreibung des für die Erstellung der Eichkurve verwendeten Verfahrens
- Angaben zu Qualitätskriterien dieses Verfahrens (z.B. Korrelationskoeffizient, Quadratsummenfehler usw.)
- Angaben über alle Extrapolationen und Näherungen während des Versuchsablaufs sowie in der Auswertung und Verarbeitung der Daten
- alle Messungen zur Erstellung der Eichkurve müssen in einer Tabelle dokumentiert sein, die für jeden Eichpunkt folgende Angaben enthält:
- Name der Probe,
- Hersteller der Probe,
- charakteristische Werte der Standards Mp, Mn, Mw, Mw/Mn, wie sie vom Hersteller genannt oder aus Messungen abgeleitet wurden, sowie alle Einzelheiten zur Bestimmungsmethode,
- Injektionsvolumen und Injektionskonzentration,
- für die Kalibrierung verwendeter Mp-Wert,
- am Peakmaximum gemessenes Elutionsvolumen oder korrigierte Retentionszeit,
- Mp, berechnet am Peakmaximum,
- prozentualer Fehler von berechneten Mp und Kalibrierwert Mp.
2.2.4 Auswertung
- Auswertung über die Zeit: verwendete Verfahren zur Gewährleistung der geforderten Reproduzierbarkeit (Korrekturverfahren, interner Standard etc.)
- Angaben darüber, ob die Bewertung auf der Grundlage des Elutionsvolumens oder der Retentionszeit vorgenommen wurde
- Angaben zu den Grenzen der Auswertung, wenn ein Peak nicht vollständig analysiert wurde
- Beschreibung der Glättungsmethoden, falls verwendet
- Vorbereitung und Vorbehandlung der Probe
- Angaben zur Anwesenheit ungelöster Partikel, falls vorhanden
- Injektionsvolumen (l) und Injektionskonzentration (mg/ml)
- Beobachtungen von Effekten, die zu Abweichungen vom idealen GPC-Profil führen
- ausführliche Beschreibung aller Änderungen im Prüfverfahren
- Einzelheiten zu den Fehlerbereichen
- alle weiteren Angaben und Beobachtungen, die für die Auswertung der Ergebnisse relevant sind.
3. Literaturhinweise
(1) DIN 55672 (1995). Gelpermeationschromatografie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1.
(2) Yau, W.W., Kirkland, J.J., and Bly, D.D. eds, (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons.
(3) ASTM D 3536-91, (1991). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography-GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania.
(4) ASTM D 5296-92 (1992). Standard Test Method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania.
| Beispiele anderer Methoden zur Bestimmung der zahlengemittelten Molmasse (Mn) von Polymeren | Anlage |
Die Gelpermeationschromatografie (GPC) ist die bevorzugte Methode zur Bestimmung von Mn, insbesondere wenn eine Reihe von Standards zur Verfügung steht, deren Struktur mit der des Polymers vergleichbar ist. Wo jedoch praktische Schwierigkeiten beim Einsatz der GPC auftreten oder wo zu erwarten ist, dass kein korrekter Wert für Mn erhalten wird (und was bestätigt werden soll), stehen Alternativen zur Verfügung, wie z.B.:
1. Nutzung kolligativer Eigenschaften
1.1 Ebullioskopie/Kryoskopie
besteht in der Messung der Siedepunkterhöhung (Ebullioskopie) oder Gefrierpunkterniedrigung (Kryoskopie) eines Lösungsmittels, wenn das Polymer zugefügt wird. Die Methode beruht auf der Tatsache, dass der Siede-/ Gefrierpunkt des Lösungsmittels von der Molmasse des gelösten Polymers abhängig ist (1) (2).
Anwendungsbereich: Mn < 20.000.
1.2 Dampfdruckerniedrigung
besteht in der Messung des Dampfdrucks einer gewählten Bezugsflüssigkeit vor und nach der Zugabe einer bekannten Menge des Polymers (1) (2).
Anwendungsbereich: Mn < 20.000 (theoretisch; in der Praxis jedoch nur von eingeschränktem Bedeutungswert).
1.3 Membranosmometrie
beruht auf dem Prinzip der Osmose, d. h. der natürlichen Tendenz von Lösungsmittelmolekülen, eine semipermeable Membran von einer verdünnten in Richtung einer konzentrierten Lösung zu durchdringen, um ein Gleichgewicht zu erreichen. In dem Test hat die verdünnte Lösung die Konzentration Null, während die konzentrierte Lösung das Polymer enthält. Die Wanderung des Lösungsmittels durch die Membran verursacht eine Druckdifferenz, die von der Konzentration und der Molmasse des Polymers (1) (3) (4) abhängig ist.
Anwendungsbereich: Mn < 20.000-200.000.
1.4 Dampfphasen-Osmometrie
besteht im Vergleich der Verdunstungsgeschwindigkeit eines reinen Lösungsmittelaerosols mit mindestens drei Aerosolen, die das Polymer in unterschiedlichen Konzentrationen (1) (5) (6) enthalten.
Anwendungsbereich: Mn < 20.000.
2. Endgruppen-Analyse
Um diese Methode verwenden zu können, muss sowohl die Gesamtstruktur des Polymermoleküls als auch die Art der Endgruppe der Kette bekannt sein (die beispielsweise mittels NMR oder Titration/Derivatisierung vom Hauptgerüst unterschieden werden muss). Über die Endgruppenzahl kann die Molmasse des Polymers errechnet werden (7) (8) (9).
Anwendungsbereich: Mn bis zu 50.000 (mit abnehmender Zuverlässigkeit).
Literaturhinweise
(1) Billmeyer, F.W. Jr., (1984). Textbook of Polymer Science, 3rd ed., John Wiley, New York.
(2) Glover, C.A., (1975). Absolute Colligative Property Methods. Kapitel 4. In: Polymer Molecular Weights, Teil I, P.E. Slade, Jr. ed., Marcel Dekker, New York.
(3) ASTM D 3750-79, (1979). Standard Practice for Determination of Number-Average Molecular Weight of Polymers by Membrane Osmometry. American Society for Testing and Materials, Philadelphia, Pennsylvania.
(4) Coll, H. (1989). Membrane Osmometry. In: Determination of Molecular Weight, A.R. Cooper ed., J. Wiley and Sons, 25-52.
(5) ASTM 3592-77, (1977). Standard Recommended Practice for Determination of Molecular Weight by Vapour Pressure. American Society for Testing and Materials, Philadelphia, Pennsylvania.
(6) Morris, C.E.M., (1989). Vapour Pressure Osmometry. In: Determination of Molecular Weight, A.R. Cooper ed., John Wiley and Sons.
(7) Schröder, E., Müller, G, und Arndt, K.-F., (1989). Polymer Characterisation, Carl Hanser Verlag, München.
(8) Garmon, R.G., (1975). End-Group Determinations, Kapitel 3. In: Polymer Molecular Weights, Teil I, P.E. Slade, Jr. ed. Marcel Dekker, New York.
(9) Amiya, S., et al. (1990). Pure and Applied Chemistry, 62, 2139-2146.
A.19 Niedermolekularer Anteil von Polymeren
1. Methode
Diese gelpermeationschromatografische Methode entspricht der OECD TG 119 (1996). Die wichtigsten Grundsätze und weitere technische Informationen werden in den Literaturhinweisen (1) genannt.
1.1 Einleitung
Da die Eigenschaften von Polymeren so unterschiedlich sind, ist es unmöglich, nur eine einzige Methode zu nennen, die alle Bedingungen für die Trennung und Auswertung erfüllt und somit sämtliche Eventualitäten und Besonderheiten bei der Trennung von Polymeren berücksichtigt. Insbesondere für komplexe polymere Systeme ist die Gelpermeationschromatografie (GPC) häufig nicht geeignet. Wenn die GPC nicht anwendbar ist, kann die Molmasse mit Hilfe anderer Methoden bestimmt werden (siehe Anlage). In solchen Fällen muss die verwendete Methode in allen Einzelheiten beschrieben und die Gründe für deren Verwendung genannt werden.
Die beschriebene Methode beruht auf DIN-Norm 55672 (1). Diese Norm enthält ausführliche Informationen darüber, wie die Versuche durchgeführt und wie die Daten ausgewertet werden müssen. Wenn Änderungen der Versuchsbedingungen notwendig sein sollten, müssen diese Änderungen begründet werden. Es können andere Normen herangezogen werden, diese müssen jedoch belegt werden. Im beschriebenen Verfahren werden Polystyrolproben bekannter Polydispersität zur Kalibrierung verwendet; es kann jedoch vorkommen, dass das Verfahren für bestimmte Polymere, z.B. wasserlösliche und langkettige, verzweigte Polymere, angepasst werden muss.
1.2 Definitionen und Einheiten
Der niedermolekulare Anteil wird willkürlich auf unter 1.000 Dalton festgelegt.
Die zahlengemittelte Molmasse Mn und die gewichtsgemittelte Molmasse Mw werden anhand folgender Gleichungen bestimmt:

| Hi | = die Höhe des Detektorsignals von der Grundlinie für das Retentionsvolumen Vi |
| Mi, | = die Molmasse der Polymerfraktion bei dem Retentionsvolumen Vi |
| n | = die Zahl der Datenpunkte |
Die Breite der Molmassenverteilung, die ein Maß für die Dispersität des Systems ist, wird durch das Verhältnis Mw/Mn ausgedrückt.
1.3 Referenzsubstanzen
Da es sich bei der GPC um eine relative Methode handelt, muss eine Kalibrierung vorgenommen werden. Hierzu werden in der Regel eng verteilte, linear aufgebaute Polystyrolstandards mit bekannten mittleren Molmassen Mn und Mw und einer bekannten Molmassenverteilung verwendet. Die Eichkurve kann für die Bestimmung der Molmassen unbekannter Proben nur herangezogen werden, wenn die Bedingungen für die Trennung der Probe und der Standards identisch sind.
Ein fester Bezug zwischen der Molmasse und dem Elutionsvolumen ist nur unter den spezifischen Bedingungen des betreffenden Versuchs zulässig. Diese Bedingungen umfassen vor allem die Temperatur, das Lösungsmittel (oder die Lösungsmittelmischung), die chromatografischen Bedingungen und die Trennsäule bzw. das Trennsäulensystem.
Bei den auf diese Weise ermittelten Molmassen der Probe handelt es sich um relative Werte, die als "polystyro äquivalente Molmasse" bezeichnet werden. Das bedeutet, dass - in Abhängigkeit von den strukturellen und chemischen Unterschieden zwischen der Probe und den Standards - die Molmassen mehr oder weniger von den absoluten Werten abweichen können. Werden andere Standards verwendet, z.B. Polyethylenglykol, Polyethylenoxid, Polymethylmethacrylat, Polyacrylsäure, so muss dies begründet werden.
1.4 Prinzip der Prüfmethode
Sowohl die Molmassenverteilung der Probe als auch die mittleren Molmassen (Mn, Mw) können mit Hilfe der GPC bestimmt werden. Bei der GPC handelt es sich um eine besondere Form der Flüssigchromatografie, bei der die Probe nach den hydrodynamischen Volumina der einzelnen Bestandteile (2) aufgetrennt wird.
Die Trennung erfolgt, indem die Probe durch eine Säule läuft, die mit einem porösen Material, in der Regel einem organischen Gel, gefüllt ist. Kleine Moleküle durchdringen die Poren, während große Moleküle ausgeschlossen werden. Der Weg der großen Moleküle ist daher kürzer, und folglich werden diese zuerst eluiert. Die Moleküle mittlerer Größe durchdringen einige der Poren und werden zu einem späteren Zeitpunkt eluiert. Die kleinsten Moleküle, mit einem durchschnittlichen hydrodynamischen Radius, der kleiner ist als die Poren des Gels, können alle Poren durchdringen. Diese werden zuletzt eluiert.
Im Idealfall erfolgt die Trennung ausschließlich über die Größe der Moleküle, doch ist es in der Praxis schwierig, gewisse störende Absorptionseffekte zu vermeiden. Ungleichmäßige Säulenfüllungen und Totvolumen können zur weiteren Verschlechterung der Trennung führen (2).
Die Detektion erfolgt beispielsweise über den Brechungsindex oder die UV-Absorption und ergibt eine einfache Verteilungskurve. Um tatsächliche Molmassenwerte für die Kurve zu erhalten, ist es notwendig, die Säule zu kalibrieren, indem Polymere mit bekannter Molmasse sowie idealerweise auch mit im Großen und Ganzen vergleichbarer Struktur, z.B. verschiedene Polystyrolstandards, auf diese Säule aufgegeben werden. In der Regel ergibt sich eine Gaußsche Kurve, die gelegentlich durch einen kleinen Schwanz in Richtung der niedrigen Molmassen verzerrt ist; die vertikale Achse zeigt die Häufigkeit der verschiedenen eluierten Molmassenfraktionen, die horizontale Achse log Molmasse.
Der niedermolekulare Anteil wird aus dieser Kurve abgeleitet. Die Berechnung kann nur dann genau sein, wenn die niedermolekularen Fraktionen in Bezug auf die Masse äquivalent zum Polymer als Ganzes sind.
1.5 Qualitätskriterien
Die Wiederholbarkeit (Relative Standardabweichung:
RSA) für den Wert des Elutionsvolumens sollte besser als 0,3 % sein.
Die geforderte Wiederholbarkeit der Analyse muss durch Korrektur mittels eines internen Standards gewährleistet sein, wenn ein Chromatogramm zeitabhängig ausgewertet wird und nicht dem oben genannten Kriterium (1) entspricht.
Die Polydispersitäten sind von den Molmassen der Standards abhängig.
Für die Polystyrolstandards sind folgende Werte charakteristisch:
| Mp < 2.000 | Mw/Mn < 1,20 |
| 2.000 ≤ Mp ≤ 106 | Mw/Mn < 1,05 |
| Mp > 106 | Mw/Mn < 1,20 |
(Mp bezeichnet die Molmasse des Standards am Peakmaximum)
1.6 Beschreibung der Prüfmethode
1.6.1 Vorbereitung der Standardpolystyrollösungen
Die Polystyrolstandards werden vorsichtig im gewählten Elutionsmittel gelöst. Die Empfehlungen des Herstellers müssen bei der Vorbereitung der Lösungen berücksichtigt werden.
Die Konzentrationen der gewählten Standards sind von verschiedenen Faktoren abhängig, z.B. Injektionsvolumen, Viskosität der Lösung und Empfindlichkeit des analytischen Detektors. Das maximale Injektionsvolumen muss der Länge der Säule angepasst werden, um eine Überbeladung zu vermeiden. Normalerweise liegen die Injektionsvolumina für analytische Trennungen mittels GPC durch eine Säule von 30 cm x 7,8 mm zwischen 40 und 100 µl. Größere Volumen sind möglich, doch sollten 250 µl nicht überschritten werden. Das optimale Verhältnis zwischen Injektionsvolumen und Konzentration muss vor der eigentlichen Kalibrierung der Säule bestimmt werden.
1.6.2 Vorbereitung der Probelösung
Im Prinzip gelten die zuvor genannten Anforderungen auch für die Vorbereitung der Probelösungen. Die Probe wird in einem geeigneten Lösungsmittel, z.B. Tetrahydrofuran (THF), durch vorsichtiges Schütteln gelöst. Die Lösung sollte unter keinen Umständen mittels Ultraschallbad gelöst werden. Wenn nötig, wird die Probelösung mit Hilfe eines Membranfilters mit einer Porengröße von 0,2 bis 2 µm gereinigt.
Die Anwesenheit ungelöster Partikel muss im Abschlussbericht dokumentiert werden, da diese auf hohe Molmassenfraktionen zurückzuführen sein könnte. Es sollte ein geeignetes Verfahren verwendet werden, um die Gewichtsanteile der ungelösten Partikel zu bestimmen. Die Lösung sollte innerhalb von 24 Stunden verbraucht werden.
1.6.3 Berichtigungen aufgrund von Verunreinigungen und Zusatzstoffen
Die Korrektur des Gehalts an Fraktionen mit M < 1.000 aufgrund bestimmter vorhandener nichtpolymerer Komponenten (z.B. Verunreinigungen und/oder Zusatzstoffe) ist in der Regel notwendig, sofern der gemessene Gehalt nicht bereits < 1 % ist. Dies wird durch die direkte Analyse der Polymerlösung oder des GPC-Eluats erreicht.
Wenn das Eluat nach Durchlaufen der Säule für eine weitere Analyse zu verdünnt ist, muss es konzentriert werden. Es kann u. U. erforderlich sein, das Eluat bis zur Trocknung einzudampfen und den Rückstand neu aufzulösen. Die Konzentrierung des Eluats muss unter Bedingungen erfolgen, die sicherstellen, dass im Eluat keine Veränderungen auftreten. Die Behandlung des Eluats nach der GPC ist abhängig davon, welches analytische Verfahren für die quantitative Bestimmung eingesetzt wird.
1.6.4 Apparatur
Die GPC-Apparatur besteht aus folgenden Komponenten:
- Lösungsmittelvorratsgefäß,
- Vorrichtung zum Entgasen (gegebenenfalls),
- Pumpe,
- Pulsationsdämpfer (gegebenenfalls),
- Injektionssystem,
- Chromatografiesäulen,
- Detektor,
- Durchflussmesser (gegebenenfalls),
- Datenaufzeichnungs-/-verarbeitungsgerät,
- Abfallbehältnis.
Es muss sichergestellt sein, dass das GPC-System gegenüber dem verwendeten Lösungsmittel inert ist (z.B. durch die Verwendung von Stahlkapillaren für das Lösungsmittel THF).
1.6.5 Injektion und Lösungsmittelzugabesystem
Auf die Säule wird eine bestimmte Menge der Probelösung, entweder automatisch oder manuell in einer scharf begrenzten Zone aufgegeben. Ein zu schnelles Zurückziehen oder Drücken des Spritzenkolbens (bei manueller Ausführung) kann Veränderungen in der beobachteten Molmassenverteilung zur Folge haben. Die Lösungsmittelzugabe sollte möglichst pulsationsfrei sein, wobei idealerweise ein Pulsationsdämpfer eingesetzt wird. Die Durchflussgeschwindigkeit liegt in der Größenordnung von 1 ml/min.
1.6.6 Säule
Je nach Art der Probe wird das Polymer durch Verwendung einer einfachen oder mehrerer in Reihe geschalteter Säulen charakterisiert. Im Handel ist eine Reihe poröser Säulenmaterialien mit definierten Eigenschaften (z.B. Porengröße, Ausschlussgrenzen) erhältlich. Die Wahl des Trenngels oder der Länge der Säule ist sowohl von den Eigenschaften der Probe (hydrodynamisches Volumen, Molmassenverteilung) als auch von den spezifischen Bedingungen für die Trennung wie z.B. Lösungsmittel, Temperatur und Durchflussgeschwindigkeit (1) (2) (3) abhängig.
1.6.7 Theoretische Böden
Die für die Trennung verwendete Säule bzw. Säulenkombination muss durch die Anzahl der theoretischen Böden charakterisiert sein. Dies umfasst (wenn THF als Elutionsmittel verwendet wird) die Aufgabe einer Lösung von Ethylenbenzol oder einer anderen geeigneten nichtpolaren Substanz auf die Säule. Die Zahl der theoretischen Böden ergibt sich aus folgender Gleichung:
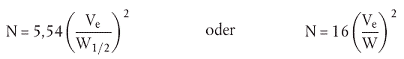
Dabei ist:
| N | = die Zahl der theoretischen Böden |
| Ve | = das Elutionsvolumen am Peakmaximum |
| W | = die Peakbreite an der Grundlinie |
| W1/2 | = die Peakbreite in halber Höhe |
1.6.8 Trennleistung
Außer der Zahl der theoretischen Böden, die für die Bestimmung der Bandbreite notwendig ist, spielt auch die Trennleistung eine Rolle, die sich aus der Steilheit der Eichkurve ergibt. Die Trennleistung einer Säule wird aus folgender Beziehung abgeleitet:
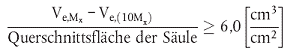
Dabei ist:
| Ve, Mx | = das Elutionsvolumen für Polystyrol mit der Molmasse Mx |
| Ve,(10.Mx) | = das Elutionsvolumen für Polystyrol mit einer zehnmal größeren Molmasse |
Die Auflösung (R) des Systems wird allgemein wie folgt definiert:
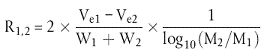
Dabei ist:
| Ve1, Ve2 | = die Elutionsvolumen der beiden Polystyrolstandards am Peakmaximum |
| W1, W2 | = die Peakbreite an der Grundlinie |
| M1, M2 | = die Molmassen am Peakmaximum (sollten um den Faktor 10 differieren) |
Der R-Wert für das Säulensystem sollte größer als 1,7 (4) sein.
1.6.9 Lösungsmittel
Alle Lösungsmittel müssen von höchster Reinheit sein (THF wird in einer Reinheit von 99,5 % verwendet). Die Größe des Lösungsmittelreservoirs (gegebenenfalls in einer Inertgasatmosphäre) muss für die Kalibrierung der Säule und mehrere Probenanalysen ausreichend sein. Das Lösungsmittel muss entgast werden, bevor es mit Hilfe der Pumpe auf die Säule aufgegeben wird.
1.6.10 Temperaturkontrolle
Die Temperatur von Injektionsschleife, Säulen, Detektor und Säulenmaterial sollte konstant und auf das gewählte Lösungsmittel abgestimmt sein.
1.6.11 Detektor
Der Detektor dient zur mengenmäßigen Erfassung der Konzentration der aus der Säule eluierten Probe. Um eine unnötige Verbreiterung der Peaks zu vermeiden, muss das Kuvettenvolumen der Detektorzelle so klein wie möglich gehalten werden. Außer bei Lichtstreuungs- und Viskositätsdetektoren sollte es nicht mehr als 10 µl betragen. Für die Detektion wird in der Regel die Differentialrefraktometrie eingesetzt. Wenn es die spezifischen Eigenschaften der Probe oder des Elutionsmittels erfordern, können auch andere Detektortypen verwendet werden, z.B. UV/VIS-, IR-, Viskositätsdetektoren usw.
2. Daten und Berichterstattung
2.1 Daten
Im Hinblick auf die detaillierten Auswertungskriterien wie auch für die Anforderungen bezüglich Datenerfassung und -Verarbeitung sollte die DIN-Norm (1) angewendet werden.
Für jede Probe müssen zwei unabhängige Versuche durchgeführt werden, die getrennt analysiert werden. Ferner ist es absolut unerlässlich, auch Daten aus Blindproben zu ermitteln, die unter den gleichen Bedingungen getestet werden wie die Probe.
Es ist ausdrücklich darauf hinzuweisen, dass es sich bei den gemessenen Werten um Relativwerte handelt, die der Molmasse des verwendeten Standards äquivalent sind.
Nach der Bestimmung der Retentionsvolumina oder der Retentionszeiten (u. U. mit Hilfe eines internen Standards korrigiert) werden die log Mp Werte (wobei Mp das Peakmaximum des Eichstandards ist) gegen eine dieser Größen aufgetragen. Mindestens zwei Eichpunkte sind pro Molmassendekade notwendig, und mindestens fünf Messpunkte sind für die Gesamtkurve erforderlich, durch die die geschätzte Molmasse der Probe erfasst werden soll. Der niedermolekulare Endpunkt der Eichkurve wird durch n-Hexylbenzol oder eine andere geeignete nichtpolare Substanz definiert. Zahlenmittel und Gewichtsmittel der Molmasse werden im Allgemeinen mittels elektronischer Datenverarbeitung auf der Grundlage der in Abschnitt 1.2 genannten Formeln ermittelt. Bei manueller Auswertung kann die ASTM D 3536-91 herangezogen werden (3).
Wenn unlösliche Polymeranteile in der Säule zurückgehalten werden, ist ihre Molmasse wahrscheinlich höher als die der löslichen Fraktion. Wird dies nicht berücksichtigt, kann der niedermolekulare Anteil zu hoch eingeschätzt werden; in der Anlage ist beschrieben, wie der unlösliche Polymeranteil berücksichtigt werden kann.
Die Verteilungskurve muss in Form einer Tabelle oder als Zahl (differentielle Häufigkeit oder Summenprozent gegen log M) dargestellt werden. Bei der grafischen Darstellung sollte eine Molmassendekade in der Regel 4 cm breit sein, und das Peakmaximum sollte etwa 8 cm sein. Bei integralen Verteilungskurven sollte der Abstand auf der Ordinate zwischen 0 und 100 % ca. 10 cm betragen.
2.2 Prüfbericht
Der Prüfbericht muss folgende Informationen enthalten:
2.2.1 Prüfsubstanz
- Verfügbare Informationen über die Prüfsubstanz (Identität, Zusatzstoffe, Verunreinigungen)
- Beschreibung der Probenbehandlung, Beobachtungen, Probleme
2.2.2 Instrumentierung
- Reservoir des Elutionsmittels, Inertgas, Entgasung des Elutionsmittels, Zusammensetzung des Elutionsmittels, Verunreinigungen
- Pumpe, Pulsationsdämpfer, Injektionssystem
- Trennsäulen (Hersteller, alle Angaben zu den Säuleneigenschaften, z.B. Porengröße, Art des Trennmaterials etc., Zahl, Länge und Anordnung der verwendeten Säulen)
- Zahl der theoretischen Böden der Säule (oder Säulenkombination), Trennleistung (Auflösungsvermögen des Systems)
- Angaben über die Peaksymmetrie
- Säulentemperatur, Art der Temperaturkontrolle
- Detektor (Messprinzip, Typ, Kuvettenvolumen)
- gegebenenfalls Durchflussmesser (Hersteller, Messprinzip)
- Datenaufzeichnungs- und -Verarbeitungssystem (Hardware und Software)
2.2.3 Systemkalibrierung
- Detaillierte Beschreibung des für die Erstellung der Eichkurve verwendeten Verfahrens
- Angaben zu Qualitätskriterien dieses Verfahrens (z.B. Korrelationskoeffizient, Quadratsummenfehler usw.)
- Angaben über alle Extrapolationen und Näherungen während des Versuchsablaufs sowie in der Auswertung und Verarbeitung der Daten
- Alle Messungen zur Erstellung der Eichkurve müssen in einer Tabelle dokumentiert sein, die für jeden Eichpunkt folgende Angaben enthält:
- Name der Probe,
- Hersteller der Probe,
- charakteristische Werte der Standards Mp, Mn, Mw, Mw/Mn, wie sie vom Hersteller genannt oder aus Messungen abgeleitet wurden, sowie alle Einzelheiten zur Bestimmungsmethode,
- Injektionsvolumen und Injektionskonzentration,
- für die Kalibrierung verwendeter Mp-Wert,
- am Peakmaximum gemessenes Elutionsvolumen oder korrigierte Retentionszeit,
- Mp, berechnet am Peakmaximum,
- prozentualer Fehler vom berechneten Mp und Kalibrierwert Mp.
2.2.4 Angaben zum niedermolekularen Anteil
- Beschreibung der für die Analyse verwendeten Methoden sowie der Art und Weise, wie die Versuche durchgeführt wurden
- Angaben zu dem prozentuellen Anteil der niedermolekularen Fraktionen (w/w) im Verhältnis zur Gesamtprobe
- Angaben zu Verunreinigungen, Zusatzstoffen und anderen nichtpolymeren Fraktionen (% w/w) im Verhältnis zur Gesamtprobe
2.2.5 Auswertung
- Auswertung über die Zeit: verwendete Verfahren zur Gewährleistung der geforderten Reproduzierbarkeit (Berichtigungsverfahren, interner Standard usw.)
- Angaben darüber, ob die Bewertung auf der Grundlage des Elutionsvolumens oder der Retentionszeit vorgenommen wurde
- Angaben zu den Grenzen der Auswertung, wenn ein Peak nicht vollständig analysiert wurde
- Beschreibung der Glättungsmethoden, falls verwendet
- Vorbereitung und Vorbehandlung der Probe
- Angaben zur Anwesenheit ungelöster Partikel, falls vorhanden
- Injektionsvolumen (µl) und Injektionskonzentration (mg/ml)
- Beobachtungen von Effekten, die zu Abweichungen vom idealen GPC-Profil führen
- Ausführliche Beschreibung aller Änderungen im Prüfverfahren
- Einzelheiten zu den Fehlerbereichen
- Alle weiteren Angaben und Beobachtungen, die für die Auswertung der Ergebnisse relevant sind
3. Literaturhinweise
(1) DIN 55672 (1995) Gelpermeationschromatografie (GPC) mit Tetrahydrofuran (THF) als Elutionsmittel, Teil 1.
(2) Yau, W.W., Kirkland, J.J., and Bly, D.D. eds (1979). Modern Size Exclusion Liquid Chromatography, J. Wiley and Sons.
(3) ASTM D 3536-91, (1991). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution by Liquid Exclusion Chromatography (Gel Permeation Chromatography - GPC). American Society for Testing and Materials, Philadelphia, Pennsylvania.
(4) ASTM D 5296-92, (1992). Standard Test method for Molecular Weight Averages and Molecular Weight Distribution of Polystyrene by High Performance Size-Exclusion Chromatography. American Society for Testing and Materials, Philadelphia, Pennsylvania.
| Korrektur des niedermolekularen Anteils um unlösliche Polymerfraktionen | Anlage |
Sind unlösliche Polymeranteile in einer Probe vorhanden, so führt dies zu Masseverlusten während der GPC-Analyse.
Das unlösliche Polymer kann an der Säule bzw. im Probenfilter zurückgehalten werden, während der lösliche Teil der Probe die Säule durchläuft. Wenn das Brechungsindexinkrement (dn/dc) des Polymers geschätzt oder gemessen werden kann, kann auch der Masseverlust der Probe in der Säule abgeschätzt werden.
In diesem Fall wird eine Korrektur anhand einer externen Kalibrierung mit Standardmaterialien bekannter Konzentration und bekanntem dn/dc zur Eichung des Refraktometers vorgenommen.
In dem folgenden Beispiel wird ein Polymethylmethacrylat (pMMA)-Standard verwendet.
Bei der externen Kalibrierung zur Analyse von Acrylpolymeren wird ein pMMA-Standard bekannter Konzentration in Tetrahydrofuran mittels GPC untersucht; die sich daraus ergebenden Daten dienen der Ermittlung der Refraktometerkonstanten mit folgender Gleichung:
K = R/(C x V x dn/dc)
Dabei ist:
| K | = die Refraktometerkonstante (in Mikrovoltsekunde/ml) |
| R | = die Messgröße für den pMMA-Standard (in Mikrovoltsekunde) |
| C | = die Konzentration des pMMA-Standards (in mg/ml) |
| V | = das Injektionsvolumen (in ml) |
| dn/dc | = das Brechungsindexinkrement für pMMA in Tetrahydrofuran (in ml/mg) |
Die folgenden Daten sind für einen pMMA-Standard charakteristisch:
| R | = 2.937.891 |
| C | = 1,07 mg/ml |
| V | = 0,1 ml |
| dn/ac | = 9 x 10-5 ml/mg. |
Der sich daraus ergebende Wert K = 3,05 x 1011 wird dann zur Berechnung des theoretischen Detektorsignals herangezogen, wenn 100 % des injizierten Polymers den Detektor passiert haben.
A.20 Lösungs-/Extraktionsverhalten von Polymeren in Wasser 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode oder sonstige anzuwendende Prüfmethode für den betreffenden Endpunkt sind in Teil 0 Tabelle 1 aufgeführt.
1. Methode
Die beschriebene Methode entspricht der geänderten Fassung der OECD TG 120 (1997). Weitere technische Informationen werden in den Literaturhinweisen (1) gegeben.
1.1 Einleitung
Bestimmte Polymere, wie z.B. Emulsionspolymere, müssen eventuell vorbehandelt werden, bevor die nachstehend beschriebene Methode verwendet werden kann. Die Methode ist nicht anwendbar für flüssige Polymere und Polymere, die unter den Testbedingungen mit Wasser reagieren.
Wenn die Methode nicht praktikabel oder nicht möglich ist, sollte das Lösungs-/Extraktionsverhalten mittels anderer Methoden untersucht werden. In diesem Fall muss die verwendete Methode in allen Einzelheiten beschrieben und ihre Verwendung begründet werden.
1.2 Referenzsubstanzen
Keine.
1.3 Prinzip der Prüfmethode
Das Lösungs-/Extraktionsverhalten von Polymeren in einem wässrigen Medium wird mit Hilfe der Kolbenmethode ermittelt (siehe A.6 Wasserlöslichkeit, Kolbenmethode), wobei die unten beschriebenen Änderungen vorgenommen wurden.
1.4 Qualitätskriterien
Keine.
1.5 Beschreibung der Prüfmethode
1.5.1 Ausstattung
Für die Durchführung der Methode ist folgende Ausstattung erforderlich:
- Zerkleinerungsgerät, z.B. Mühle zur Herstellung von Partikeln bekannter Größe,
- Schüttelgerät mit der Möglichkeit zur Temperaturkontrolle,
- Membranfiltersystem,
- geeignete Analysegeräte,
- genormte Siebe.
1.5.2 Probenvorbereitung
Eine repräsentative Probe muss zunächst mit Hilfe geeigneter Siebe auf eine Partikelgröße zwischen 0,125 und 0,25 mm reduziert werden. Für die Stabilität der Probe oder für den Zerkleinerungsprozess kann dazu u. U. eine Kühlung erforderlich sein. Gummiartige Materialien können bei der Temperatur von Flüssigstickstoff (1) zerkleinert werden.
Wenn die erforderliche Partikelgrößenfraktion nicht erreicht werden kann, sollten Maßnahmen ergriffen werden, um die Partikelgröße so weit wie möglich zu reduzieren; die Ergebnisse sollten dokumentiert werden. Im Bericht muss festgehalten werden, wie die zerkleinerte Probe vor dem Test aufbewahrt wurde.
1.5.3 Verfahren
Je 10 g Prüfsubstanz werden in drei mit einem Glasstopfen versehene Gefäße gegeben; jedes Gefäß wird mit 1.000 ml Wasser aufgefüllt. Wenn sich eine Polymermenge von 10 g als unpraktikabel erweist, sollte die nächstgrößere Menge, die verarbeitet werden kann, eingesetzt und mit Wasser entsprechend aufgefüllt werden.
Die Gefäße werden fest verschlossen und dann bei 20 °C geschüttelt. Es sollte ein Schüttel- oder Rührgerät verwendet werden, das bei einer konstanten Temperatur arbeitet. Nach 24 Stunden wird der Inhalt eines jeden Gefäßes zentrifugiert oder filtriert und die Polymerkonzentration in der klaren wässrigen Phase mit Hilfe eines geeigneten analytischen Verfahrens bestimmt. Sollten keine geeigneten analytischen Verfahren für die wässrige Phase zur Verfügung stehen, kann die Gesamtlöslichkeit/-extrahierbarkeit anhand der Trockenmasse des Filterrückstands oder des zentrifugierten Niederschlags abgeschätzt werden.
Es ist in der Regel notwendig, quantitativ zwischen Verunreinigungen und Zusatzstoffen einerseits und den niedermolekularen Fraktionen andererseits zu differenzieren. Im Fall einer gravimetrischen Bestimmung ist es ferner wichtig, eine Blindprobe durchzuführen, in der keine Prüfsubstanz eingesetzt wird, um Rückstände aus dem Versuchsverfahren zu berücksichtigen.
Das Lösungs-/Extraktionsverhalten von Polymeren in Wasser bei 37 °C bei pH-Werten von 2 und 9 kann auf gleiche Weise bestimmt werden wie für die Untersuchung bei 20 °C beschrieben. Die pH-Werte können entweder durch Zugabe einer geeigneten Pufferlösung oder entsprechender Säuren bzw. Basen wie z.B. Salzsäure, Essigsäure, Natrium- oder Kaliumhydroxid oder NH3 p. a. erreicht werden.
In Abhängigkeit von der eingesetzten Analysemethode sollten ein oder zwei Tests durchgeführt werden. Wenn hinreichend genaue Methoden zur direkten Analyse der wässrigen Phase der Polymerkomponente zur Verfügung stehen, sollte ein Test (wie oben beschrieben) ausreichen. Wenn solche Methoden jedoch nicht verfügbar sind und die Bestimmung des Lösungs-/Extraktionsverhaltens des Polymers auf indirekte Analysen beschränkt ist, bei denen lediglich der gesamte organische Kohlenstoff (TOC) des wässrigen Extrakts bestimmt wird, sollte ein zusätzlicher Test durchgeführt werden. Dieser zusätzliche Test sollte ebenfalls dreimal durchgeführt werden, wobei zehnmal kleinere Polymerproben und die gleichen Mengen Wasser wie im ersten Test verwendet werden.
1.5.4 Analyse
1.5.4.1 Test mit einer Probengröße
Es ist möglich, dass Methoden für die direkte Analyse von Polymerkomponenten in der wässrigen Phase zur Verfügung stehen. Alternativ können auch indirekte Analysen der gelösten/extrahierten Polymerkomponenten durchgeführt werden, in denen der Gesamtgehalt der löslichen Anteile bestimmt und eine Berichtigung um nichtpolymerspezifische Bestandteile vorgenommen wird.
Eine Analyse der wässrigen Phase für das gesamte Polymer ist möglich entweder durch ein hinreichend empfindliches Verfahren, wie z.B.
- TOC unter Verwendung eines Peroxosulfat- oder Dichromataufschlusses zur Darstellung von CO2 und einer IR-Analyse oder chemischen Analyse,
- Atomabsorptionsspektrometrie (AAS) oder das ICP-Emissionsäquivalent (Inductively Coupled Plasma) für silizium- oder metallhaltige Polymere,
- UV-Absorption der Spektrofluorimetrie für Arylpolymere,
- LC-MS für Proben mit geringer niedriger Molmasse,
oder durch Eindampfen des wässrigen Extrakts im Vakuum und Analyse des Rückstands mit Hilfe der Spektroskopie (IR, UV usw.) oder AAS/ICP.
Wenn eine Analyse der wässrigen Phase als solche nicht praktikabel ist, sollte der wässrige Extrakt mittels eines nicht mit Wasser mischbaren organischen Lösungsmittels, z.B. einem chlorierten Kohlenwasserstoff, extrahiert werden. Das Lösungsmittel wird anschließend abgezogen, der Rückstand wird (wie oben für den Polymergehalt beschrieben) analysiert. Alle Bestandteile dieses Rückstands, die als Verunreinigungen oder Zusatzstoffe identifiziert werden, müssen für die Bestimmung des Lösungs-/Extraktionsgrades des Polymers subtrahiert werden.
Wenn relativ große Mengen solcher Stoffe vorhanden sind, kann es u. U. notwendig sein, den Rückstand beispielsweise einer HPLC- oder GC-Analyse zu unterwerfen, um die Verunreinigungen von den vorhandenen Monomeren bzw. Monomerderivaten zu unterscheiden, um deren tatsächlichen Gehalt zu bestimmen.
In einigen Fällen ist es u. U. ausreichend, das organische Lösungsmittel abzuziehen und den trockenen Rückstand auszuwiegen.
1.5.4.2 Test mit zwei unterschiedlichen Probengrößen
Alle wässrigen Extrakte werden auf ihren TOC analysiert.
An dem nichtgelösten/nichtextrahierten Teil einer Probe wird eine gravimetrische Analyse durchgeführt. Wenn nach der Zentrifugation oder Filtration noch Polymerablagerungen an den Wänden des Gefäßes zu finden sind, sollte das Gefäß so lange mit dem Filtrat gespült werden, bis es frei von allen sichtbaren Rückständen ist. Im Anschluss wird das Filtrat erneut zentrifugiert oder filtriert. Die auf dem Filter oder im Zentrifugenglas verbliebenen Rückstände werden bei 40 °C im Vakuum getrocknet und gewogen. Die Trocknung wird fortgesetzt, bis ein konstantes Gewicht erzielt wurde.
2. Daten
2.1 Test mit einer Probengrösse
Die einzelnen Ergebnisse für die drei Kolben und die Durchschnittswerte sollten in Masseeinheiten pro Lösungsvolumen (mg/l) bzw. Masseeinheiten pro Masse der Polymerprobe (mg/g) angegeben werden. Außerdem sollte der Gewichtsverlust der Probe (berechnet als Quotient aus der Masse des eluierten Anteils und der Masse der ursprünglichen Probe) angegeben werden. Die relativen Standardabweichungen (RSA) sollten berechnet werden. Die Zahlen sollten sowohl für die gesamte Substanz (Polymer + Additive usw.) als auch für das Polymer allein (d. h. nach Abzug der Zusatzstoffe) genannt werden.
2.2 Test mit zwei unterschiedlichen Probengrössen
Die einzelnen TOC-Werte der wässrigen Extrakte der beiden Dreifachversuche sowie der Durchschnittswert für jeden Versuch sollten sowohl in Masseeinheiten pro Lösungsvolumen (normalerweise mg C/l) als auch in Maßeinheiten pro Gewicht der ursprünglichen Probe (normalerweise mg C/g) ausgedrückt werden.
Wenn es keinen Unterschied zwischen den Ergebnissen mit hohem bzw. niedrigem Probe-Wasser-Verhältnis gibt, deutet dies darauf hin, dass alle extrahierbaren Komponenten auch tatsächlich extrahiert worden sind. In diesem Fall ist eine direkte Analyse in der Regel nicht erforderlich.
Die Massen der einzelnen Rückstände sollten als prozentualer Anteil der Ausgangsmasse der Proben angegeben werden. Die Durchschnittswerte sollten ermittelt werden. Die Differenz zwischen 100 und den gefundenen Prozentsätzen stellt den Prozentgehalt des löslichen und extrahierbaren Materials der ursprünglichen Probe dar.
3. Abschlussbericht
3.1 Testbericht
Der Testbericht muss folgende Angaben enthalten:
3.1.1 Prüfsubstanz
- Verfügbare Angaben zur Prüfsubstanz (Identität, Zusatzstoffe, Verunreinigungen, Gehalt der niedermolekularen Spezies)
3.1.2 Versuchsbedingungen
- Beschreibung der verwendeten Verfahren und Versuchsbedingungen
- Beschreibung der Analyse- und Nachweismethoden
3.1.3 Ergebnisse
- Ergebnisse der Löslichkeit/Extrahierbarkeit in mg/l; Einzel- und Durchschnittswerte für die Extraktionstests in den verschiedenen Lösungen, aufgeschlüsselt nach Polymergehalt und Verunreinigungen, Zusatzstoffen usw.
- Ergebnisse der Löslichkeit/Extrahierbarkeit in mg/g Polymer
- TOC-Werte der wässrigen Extrakte, Masse des eluierten Teils und errechnete Prozentsätze (gegebenenfalls)
- pH-Wert der einzelnen Proben
- Angaben zu den Werten der Blindproben
- Gegebenenfalls Hinweise auf die chemische Instabilität der Prüfsubstanz sowohl während der Prüfung als auch während des Analyseverfahrens
- Alle Informationen, die für die Auswertung der Ergebnisse von Bedeutung sind
4. Hinweise
(1) DIN 53733 (1976) Zerkleinerung von Kunststofferzeugnissen für Prüfzwecke.
A.21 Brandfördernde Eigenschaften (Flüssige Stoffe) 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode oder sonstige anzuwendende Prüfmethode für den betreffenden Endpunkt sind in Teil 0 Tabelle 1 aufgeführt.
| 1. Verfahren
1.1 Einleitung Mit diesem Prüfverfahren soll festgestellt werden, inwieweit ein flüssiger Stoff die Verbrennungsgeschwindigkeit oder die Verbrennungsintensität eines brennbaren Stoffes erhöhen kann oder ein Gemisch mit einem brennbaren Stoff bilden kann, welches sich spontan entzündet, wenn beide sorgfältig gemischt werden. Es beruht auf dem UN-Test auf brandfördernde (oxidierende) Eigenschaften für flüssige Stoffe (1) und ist ihm gleichwertig. Da dieses Verfahren A.21 jedoch in erster Linie für die Verordnung (EG) Nr. 1907/2006 eingeführt wurde, ist lediglich der Vergleich mit einem Referenzstoff vorgeschrieben. Weitere Tests und der Vergleich mit zusätzlichen Referenzstoffen können erforderlich sein, wenn die Testergebnisse für andere Zwecke verwendet werden sollen 1. Dieser Test muss nicht durchgeführt werden, wenn anhand der Strukturformel hinreichend nachgewiesen wurde, dass der Stoff mit anderen brennbaren Stoffen nicht exotherm reagieren kann. Es ist nützlich, Vorausinformationen über die potenziellen explosiven Eigenschaften der Stoffe zu haben, bevor dieser Test durchgeführt wird. Dieser Test ist nicht auf feste Stoffe, Gase, explosive oder leichtentzündliche Stoffe oder auf organische Peroxide anwendbar. Dieser Test muss nicht durchgeführt werden, wenn bereits Ergebnisse für den getesteten Stoff aus dem UN-Test auf brandfördernde (oxidierende) Eigenschaften für flüssige Stoffe (1) vorliegen. 1.2 Definitionen und Einheiten Die durchschnittliche Druckanstiegszeit ist der Durchschnitt der gemessenen Zeiten, die vergehen, bis ein Gemisch bei einem Test einen Druckanstieg von 690 kPa auf 2.070 kPa über atmosphärischem Druck erzeugt. 1.3 Referenzstoff Als Referenzstoff ist 65 % (w/w) Salpetersäure in wässriger Lösung (analysenrein) erforderlich 2. Wenn der Experimentator davon ausgeht, dass die Ergebnisse dieses Tests auch für andere Zwecke verwendet werden sollen (1), kann die Prüfung zusätzlicher Referenzstoffe sinnvoll sein 3. 1.4 Prinzip des Prüfverfahrens Die Testflüssigkeit wird in einem Massenverhältnis von 1:1 mit Fasercellulose gemischt und in ein Druckgefäß gefüllt. Kommt es beim Mischen oder beim Einfüllen spontan zur Entzündung, sind keine weiteren Tests mehr nötig. Kommt es nicht spontan zur Entzündung, so wird der gesamte Test durchgeführt. Das Gemisch wird in einem Druckgefäß erhitzt, und die Zeit, die im Durchschnitt vergeht, bis der Druck von 690 kPa auf 2.070 kPa über atmosphärischen Druck angestiegen ist, wird ermittelt. Diese Zeit wird mit der durchschnittlichen Druckanstiegszeit für das 1:1-Gemisch des/der Referenzstoffe(s) und der Cellulose verglichen. 1.5 Qualitätskriterien In fünf hintereinander erfolgten Prüfungen mit ein und demselben Stoff sollten die Ergebnisse um nicht mehr als 30 % vom arithmetischen Mittel abweichen. Ergebnisse, die um mehr als 30 % abweichen, sind nicht zu berücksichtigen. Misch- und Einfüllverfahren sollten verbessert und die Testreihe sollte wiederholt werden. 1.6 Beschreibung des Verfahrens 1.6.1 Vorbereitung 1.6.1.1 Brennbarer Stoff Getrocknete Fasercellulose mit einer Faserlänge von 50 bis 250m und einem durchschnittlichen Durchmesser von 25 μm 4 wird als brennbares Material verwendet. Sie wird in einer Schicht von nicht mehr als 25 mm Dicke bei 105 °C 4 Stunden lang bis zur Gewichtskonstanz getrocknet und in einem Exsikkator mit Trocknungsmittel aufbewahrt, bis sie vollkommen abgekühlt ist und benötigt wird. Der Wassergehalt der getrockneten Cellulose sollte weniger als 0,5 % der Trockenmasse 5 betragen. Gegebenenfalls sollte die Trocknungszeit verlängert werden 6. Während des gesamten Tests ist Cellulose aus derselben Vorbereitungsprozedur zu verwenden, 1.6.1.2 Apparatur 1.6.1.2.1 Druckgefäß Ein Druckgefäß ist erforderlich. Das Gefäß besteht aus einem zylindrischen Druckgefäß aus Stahl von einer Länge von 89 mm und einem Außendurchmesser von 60 mm (siehe Abbildung 1). Seitlich ist der Kolben an zwei gegenüberliegenden Seilen abgeflacht (wo sich der Durchmesser des Gefäßes auf 50 mm verringert), um die Handhabung bei der Einführung von Zündstopfen und Entlüftungsstopfen zu erleichtern. Das Gefäß, das eine Bohrung von 20 mm im Durchmesser hat, ist an einem Ende in einer Tiefe von 19 mm vergrößert und mit einem Gewinde versehen, so dass ein 1" British Standard Pipe (BSP) oder eine metrische Entsprechung eingeführt werden kann. Ein Druckablassarm wird in die nicht abgeflachte Seite des Druckgefäßes 35 mm von einem Ende und im Winkel von 90° zu den abgeflachten Seiten eingeschraubt. Dazu ist eine Bohrung von 12 mm Tiefe vorgesehen, die mit einem Gewinde versehen ist, in das das 1/2"-BSP-Gewinde (oder metrische Entsprechung) am unteren Ende des Seitenarms eingeschraubt werden kann. Gegebenenfalls wird eine Dichtung aus inertem Material angebracht, um den Arm gasundurchlässig zu machen. Der Seitenarm ragt 55 mm aus dem Druckgefäß heraus und hat eine Bohrung von 6 mm. Das Ende des Seitenarms ist vergrößert und mit einem Gewinde versehen, so dass ein Membrandruckaufnehmer eingeschraubt werden kann. Jedes beliebige Druckmessgerät kann verwendet werden, sofern es gegen die heißen Gase oder Spaltprodukte beständig ist und auf eine Druckanstiegsgeschwindigkeit von 690 bis 2.070 kPa in höchstens 5 ms anspricht. Das weiter vom Seitenarm entfernte Ende wird mit einem Zündstopfen verschlossen, an den zwei Elektroden angebracht sind. Die eine ist vom Stopfen isoliert, die andere ist über diesen geerdet. Das andere Ende des Druckgefäßes wird mit einer Berstscheibe (Berstdruck rund 2.200 kPa) verschlossen, die von einem Stopfen mit einer Bohrung von 20 mm gehalten wird. Gegebenenfalls wird am Zündstopfen eine Dichtung aus inertem Material verwendet, um Gasundurchlässigkeit zu gewährleisten. Ein Ständer (Schaubild 2) hält die Vorrichtung beim Gebrauch in der richtigen Position. Er besteht in der Regel aus einer Weichstahl-Grundplatte mit der Abmessung 235 mm x 184 mm x 6 mm und einem 185 mm langen quadratischen Hohlkörper mir der Abmessung 70 mm x 70 mm x 4 mm. Von zwei gegenüberliegenden Seiten des Hohlkörpers wird an einem Längsende jeweils ein Seitenteil abgeschnitten, so dass ein Gestell mit zwei flachwandigen Beinen und einem 86 mm langen ganzen Kasten darauf entsteht. Die Enden dieser flachwandigen Beine werden in einem Winkel von 60° zur Horizontalen abgeschnitten und an die Grundplatte angeschweißt. Ein 22 mm weiter und 46 mm tiefer Spalt wird in eine Seite am oberen Ende des Kastens geschnitten, so dass bei der Einführung der Druckgefäßvorrichtung mit dem Zündstopfen voran in den Kastenteil der Vorrichtung der Seitenarm in den Spalt passt. Ein Stahlstück von 30 mm Länge und 6 mm Dicke wird als Zwischenstück unten an der Innenseite des Kastens angeschweißt. Zwei Flügelschrauben von 7 mm sind an der gegenüberliegenden Seite eingeschraubt und halten das Druckgefäß. Zwei 12 mm breite Streifen von 6 mm dickem Stahl, die an die Seitenteile am Boden des Kastens angeschweißt sind, halten das Druckgefäß von unten. 1.6.1.2.2 Zündvorrichtung Die Zündvorrichtung besteht aus einem 25 cm langem Ni/Cr-Draht mit einem Durchmesser von 0,6 mm und einem Widerstand von 3,85 Ohm/m. Der Draht wird mit Hilfe eines Stabes von 5 mm Durchmesser zu einer Wendel gedreht und wird an den am Zündstopfen befindlichen Elektroden befestigt. Die Wendel sollte einer der Darstellungen in Abbildung 3 entsprechen. Die Unterseite der Zündwendel sollte 20 mm vom Boden des Gefäßes entfernt sein. Wenn die Elektroden nicht nachstellbar sind, sollten die Enden des Zünddrahtes zwischen der Wendel und dem Boden des Gefäßes mit einer Keramikumhüllung isoliert werden. Der Draht wird durch konstante Stromversorgung von mindestens 10 A erhitzt. 1.6.2 Durchführung des Tests 7 Die Apparatur, die komplett mit Druckaufnehmer und Heizsystem montiert ist, bei der jedoch die Berstscheibe nicht eingeführt ist, wird mit dem Zündstopfen nach unten auf dem Ständer befestigt. 2,5 g der zu testenden Flüssigkeit werden mit 2,5 g getrockneter Cellulose in einem Becherglas mit einem Rührstab aus Glas gemischt 8. Aus Sicherheitsgründen sollte der Mischvorgang mit einem Schutzschirm zwischen Experimentator und Gemisch durchgeführt werden. Entzündet sich das Gemisch beim Mischen oder beim Einfüllen, sind keine weiteren Tests erforderlich. Das Gemisch wird in kleinen Portionen mit leichtem Klopfen in das Druckgefäß gefüllt, wobei darauf geachtet werden muss, dass das Gemisch die Zündwendel ausreichend umhüllt und damit ein guter Kontakt gewährleistet ist. Es ist wichtig, dass sich die Wendel während des Füllens nicht verformt, da das zu falschen Ergebnissen führen kann 9. Die Berstscheibe wird in die vorgesehene Druckgefäßöffnung eingelegt und mit dem Halterungsstopfen fest eingeschraubt. Das gefüllte Gefäß wird auf den Ständer montiert, wobei das Ende mit der Berstscheibe nach oben zeigt. Der Ständer sollte sich in einem geeigneten gepanzerten Abzugsschrank oder in einer Brennkammer befinden. Das Stromkabel ist an den äußeren Anschlusssteckern am Zündstopfen angeschlossen. Die Stromstärke beträgt 10 A. Zwischen dem Beginn des Mischens und dem Anschalten des Stroms sollten nicht mehr als 10 Minuten vergehen. Das vom Druckaufnehmer erzeugte Signal wird durch ein geeignetes Messdatenerfassungssystem aufgezeichnet, das sowohl die Messung als auch die Aufzeichnung eines Zeit-Druck-Profils ermöglicht (z.B. ein Transientenrecorder in Verbindung mit einem grafischen Drucker). Das Gemisch wird mindestens 60 s lang oder so lange, bis die Berstscheibe aufreißt, erhitzt. Reißt die Scheibe nicht auf, so sollte man das Gemisch abkühlen lassen, bevor die Apparatur vorsichtig abgebaut werden kann, wobei Vorkehrungsmaßnahmen gegen einen eventuellen Druckaufbau getroffen werden sollten. Es werden fünf Prüfgänge mit dem Prüfstoff und dem/ den Referenzstoff(en) durchgeführt. Es wird festgehalten, wie viel Zeit vergeht, bis der Druck von 690 kPa auf 2.070 kPa über atmosphärischem Druck steigt. Die durchschnittliche Druckanstiegszeit wird berechnet. In manchen Fällen können Stoffe einen (zu hohen oder zu niedrigen) Druckanstieg erzeugen, der nicht auf die brandfördernden Eigenschaften des Stoffes zurückzuführen ist. In diesen Fällen muss der Test gegebenenfalls mit einem inerten Stoff, z.B. Diatomit (Kieselgur), anstelle der Cellulose wiederholt werden, um die Art der Reaktion festzustellen. 2. Daten Druckanstiegszeiten für die Testsubstanz und den/die Referenzstoff(e), Druckanstiegszeiten für die Tests mit einem inerten Stoff, soweit durchgeführt. 2.1 Ergebnisverarbeitung Sowohl für die Testsubstanz als auch für den/die Referenzstoff(e) werden die durchschnittlichen Druckanstiegszeiten berechnet. Die durchschnittliche Druckanstiegszeit wird für die Tests mit einem inerten Stoff berechnet (sofern durchgeführt). In Tabelle 1 sind einige Ergebnisbeispiele aufgeführt. Tabelle 1: Ergebnisbeispiele a
3. Bericht 3.1 Prüfbericht Der Prüfbericht sollte folgende Angaben enthalten:
Bei der Bewertung der Prüfergebnisse ist Folgendes zu beachten:
Ein flüssiger Stoff wird als brandfördernd beurteilt, wenn
Um falsche positive Ergebnisse zu vermeiden, sollten die beim Test des Stoffes mit einem inerten Stoff erhaltenen Ergebnisse in die Analyse der Ergebnisse mit einbezogen werden. 4. Bezugsdokumente (1) UN-Empfehlungen für die Beförderung gefährlicher Güter, Test- und Kriterienhandbuch, 3. geänderte Ausgabe. UN-Veröffentlichungsnummer: ST/SG/AC.10/11/Rev. 3, 1999, S. 342. Test O.2: Test auf oxidierende Eigenschaften für flüssige Stoffe. Abbildung 1 Druckgefäß Abbildung 2 Ständer Abbildung 3 Zündvorrichtung Anmerkung: Eines dieser Systeme kann verwendet werden. ... 2) Die Säure ist vor dem Test zu titrieren, um die genaue Konzentration zu überprüfen. 3) Beispiel: 50 % (w/w) Perchlorsäure und 40 % (w/w) Natriumchlorat werden in Bezugsdokument 1 verwendet. 4) Zum Beispiel Cellulosestaub aus der Whatman-Säulen-Chromatografie CF 11, Katalognr. 4021 050. 5) Was beispielsweise durch Karl-Fischer-Titration zu bestätigen ist. 6) Als Alternative kann der Wassergehalt beispielsweise auch durch Vakuumerhitzen auf 105 °C während 24 Stunden erreicht werden. 7) Gemische von Oxidationsmitteln und Cellulose sind als potenziell explosiv zu behandeln und mit Vorsicht zu handhaben. 8) Dazu kann auch ein Gemisch der zu testenden Flüssigkeit und der Cellulose im Verhältnis 1:1 in größerer Menge zubereitet werden, wovon dann 5 ± 0,1 g in das Druckgefäß gefüllt werden. Das Gemisch ist für jeden Prüfgang frisch zuzubereiten. 9) Insbesondere muss vermieden werden, dass nebeneinander liegende Schleifen der Wendel miteinander in Kontakt kommen. 10) Siehe Bezugsdokument 1 zur Analyse der Ergebnisse nach den Beförderungsbestimmungen der UN bei Verwendung verschiedener Referenzstoffe. | ||||||||||||||||||||||||||||||||||
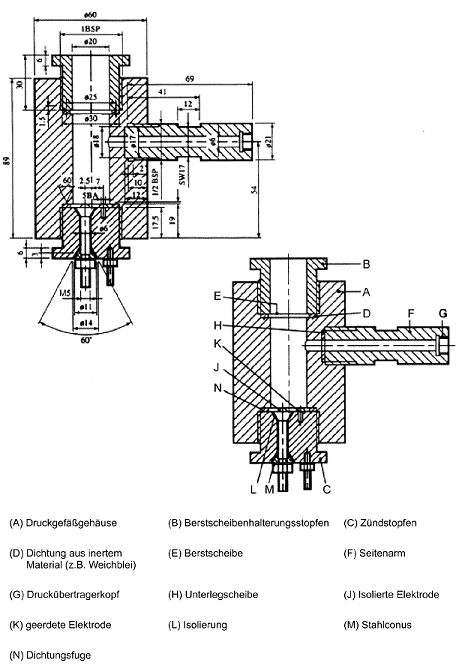
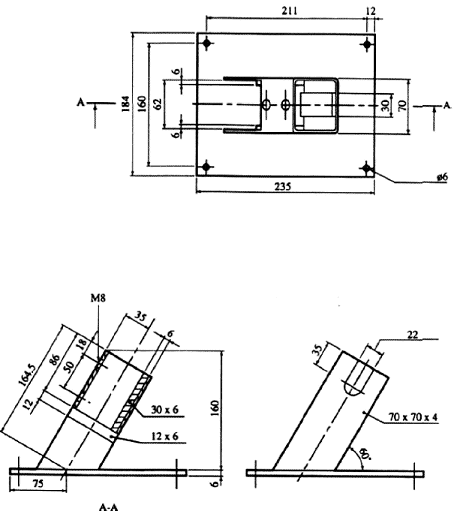
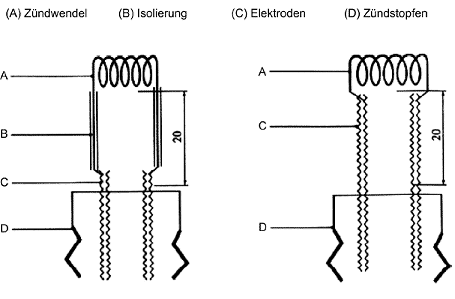
A.22 Längengewichteter mittlerer geometrischer Durchmesser von Fasern
1. Methode
1.1 Einleitung
Mit dieser Testmethode wird ein Verfahren zur Messung des längengewichteten mittleren geometrischen Durchmessers (GWGMD - Length Weighted Geometric Mean Diameter) von künstlichen Mineralfasern (MMMF - Man Made Mineral Fibres) beschrieben. Da der LWGMD der Population mit einer Wahrscheinlichkeit von 95 % zwischen den 95 %-Vertrauensintervallen (LWGMD ± 2 Standardfehler) der Probe liegt, entspricht der im Bericht angegebene Wert (der Testwert) der unteren 95 %-Vertrauensgrenze der Probe (d. h. LWGMD - 2 Standardfehler). Diese Methode basiert auf einer aktualisierten Fassung Quni 1994) des Entwurfs einer HSEIndustrieverfahrensweisung, die am 26. September 1993 in Chester zwischen ECHA und HSE vereinbart und für und aus einem zweiten laborinternen Versuch entwickelt wurde (1, 2). Diese Messmethode kann zur Kennzeichnung des Faserdurchmessers von Schüttgutstoffen oder Produkten verwendet werden, die MMMF enthalten, z.B. feuerfeste Keramikfasern (PCF - Refractory Ceramic Fibres), künstliche Glasfasern (MMVF - Man-Made Vitreous Fibres), kristalline und polykristalline Fasern.
Die Längengewichtung dient zur Kompensation der Auswirkungen auf die Durchmesserverteilung, zu denen es durch den Bruch langer Fasern bei der Probenahme oder beim Umgang mit dem Material kommt. Die Größenverteilung der Durchmesser der MMMF wird durch geometrische statistische Verfahren (geometrisches Mittel) gemessen, da diese Durchmesser normalerweise Größenverteilungen aufweisen, die näherungsweise dem Lognormal entsprechen.
Die Messung der Länge und des Durchmessers ist ein mühsamer, zeitaufwändiger Prozess, werden jedoch nur jene Fasern gemessen, die eine unendlich dünne Linie in einem REM-Sichtfeld berühren, so ist die Wahrscheinlichkeit, eine bestimmte Faser auszuwählen, proportional zu deren Länge. Da damit die Länge in den Berechnungen der Längengewichtung berücksichtigt wird, muss lediglich der Durchmesser gewichtet werden; der LWGMD - 2SF kann dann auf die beschriebene Weise berechnet werden.
1.2 Begriffsbestimmungen
Partikel: Ein Objekt mit einem Länge-Breite-Verhältnis von weniger als 3:1.
Faser: Ein Objekt mit einem Länge-Breite-Verhältnis (Seitenverhältnis) von mindestens 3:1.
1.3 Umfang und Einschränkungen
Durch diese Methode sollen die Durchmesserverteilungen untersucht werden, deren mittlerer Durchmesser zwischen 0,5 µm und 6 µm liegt. Größere Durchmesser können mithilfe geringerer REM-Vergrößerungsfaktoren gemessen werden, allerdings stößt diese Methode bei feineren Faserverteilungen zunehmend an seine Grenzen; bei mittleren Durchmessern unter 0,5 µm wird die Messung mit Transmissions-Elektronenmikroskopen (TEM) empfohlen.
1.4 Prinzip der Testmethode
Aus der Fasermatte oder aus losen Fasern wird eine bestimmte Anzahl repräsentativer Kernproben entnommen. Die Länge der losen Fasern wird durch Brechen verringert und es wird eine repräsentative Teilprobe in Wasser dispergiert. Aliquote Teile werden extrahiert und durch ein Polycarbonatfilter mit einer Porengröße von 0,2 µm gefiltert und zur Untersuchung unter einem Rasterelektronenmikroskop (SEM) vorbereitet. Die Faserdurchmesser werden mit einem Rastervergrößerungsfaktor von x 10.000 oder mehr (1) nach einem Line-Intercept-Verfahren gemessen, das eine unverfälschte Schätzung des mittleren Durchmessers ergibt. Das untere 95 %-Vertrauensintervall (auf der Basis eines einseitigen Tests) wird so berechnet, dass ein Schätzwert für den niedrigsten Wert des mittleren geometrischen Faserdurchmessers des Materials entsteht.
(1) Dieser Vergrößerungsfaktor bezieht sich auf 3µm-Fasem; bei 6µm-Fasern ist ein Vergrößerungsfaktor von x 5.000 möglicherweise geeigneter.
1.5 Beschreibung der Testmethode
1.5.1 Sicherheits-Vorsichtsmaßnahmen
Die Belastung des menschlichen Organismus durch Schwebfasern ist zu minimieren; daher ist bei der Arbeit mit den trockenen Fasern ein Abzugsschrank oder eine Glove-Box zu verwenden. In periodischen Abständen ist die Belastung des menschlichen Organismus zu überwachen, um die Wirkung der Schutzverfahren zu überprüfen. Bei der Handhabung von MMMF sind Einweghandschuhe zu tragen, um Hautreizungen zu verringern und eine Querkontaminierung zu vermeiden.
1.5.2 Geräte/Apparatur
- Press- und Formwerkzeug (für 10 MPa).
- Polycarbonat-Kapillarporenfilter mit 0,2 µm Porengröße (Durchmesser 25 mm).
- Zelluloseestermembranfilter mit 5 prn Porengröße zur Verwendung als Zusatzfilter.
- Glasfiltervorrichtung (oder Einwegfiltersysteme) für die Aufnahme von Filtern mit 25 mm Durchmesser (z.B. Millipore-Glasmikroanalyse-Filtersatz, Typ XX10.025 00).
- Frisch destilliertes Wasser, das zur Ausfilterung von Mikroorganismen durch einen Filter mit 0,2 µm Porengröße gefiltert wurde.
- Sputter-Beschichtungsvorrichtung mit Gold- oder Gold/Palladium-Target.
- Rasterelektronenmikroskop mit einem Auflösevermögen bis 10 nm und Vergrößerungsfaktor bis x 10000.
- Diverses: Spatel, Skalpellmesser Typ 24, Pinzette, REM-Röhrchen, Kohlenstoffkleber oder Kohlenstoffklebeband, Kolloidalsilber ("Silver dag").
- Ultraschallsonde oder Tisch-Ultraschallbad.
- Kernbohrer oder Korkbohrer für die Entnahme von Kernproben aus MMMF-Fasermatten.
1.5.3 Testverfahren
1.5.3.1 Probenahme
Bei Fasermatten und Platten wird ein 25 mm-Kernbohrer oder Korkbohrer zur Entnahme von Proben aus dem Querschnitt verwendet. Diese Proben sind gleichmäßig über die Breite a der Schmalseite der Matte anzuordnen oder, wenn lange Abschnitte der Matte zur Verfügung stehen, aus beliebigen Stellen zu entnehmen. Die gleichen Hilfsmittel können auch für die Entnahme von Zufallsproben aus losen Fasern verwendet werden. Es sind sechs Stichproben möglichst so zu entnehmen, das räumliche Schwankungen im losen Material damit wiedergegeben werden.
Die sechs Kernproben sind in einem Stempel mit 50 mm Durchmesser unter 10 MPa zu zerkleinern. Das Material wird mit einem Spachtel durchmischt und bei 10 MPa erneut gepresst. Anschließend wird das Material aus dem Stempel entnommen und in einer verschlossenen Glasflasche gelagert.
1.5.3.2 Vorbereitung der Probe
Falls erforderlich, können organische Bindemittel entzogen werden, indem die Faser ca. eine Stunde lang bei 450 °C in einen Ofen eingelegt wird.
Die Probe nach dem Coneand-Quarter-Verfahren in vier gleiche Teile unterteilen (dieser Schritt sollte in einem Staubschrank erfolgen).
Mit einem Spatel eine geringe Menge (< 0,5 g) der Probe in 100 ml frisch destilliertes Wasser geben, das durch einen 0,2 µm-Membranfilter gefiltert wurde (andere Beschaffungsquellen von hochreinem Wasser sind ebenfalls zulässig, wenn sie nachgewiesenermagen von ausreichender Qualität sind). Die Probe mithilfe einer mit 100 W betriebenen Ultraschallsonde, die so eingestellt ist, dass eine Kavitation eintritt, gründlich dispergieren (Steht keine Sonde zur Verfügung, ist wie folgt vorzugehen: 30 Sekunden lang mehrmals schütteln und umkehren; fünfminütige Ultraschallbehandlung in einem Tisch-Ultraschallbad; dann wieder 30 Sekunden lang mehrmals schütteln und umkehren).
Sofort nach der Dispersion der Faser mehrere aliquote Teile (z.B. drei aliquote Teile mit 3, 6 und 10 ml) mit einer breiten Pipette (Aufnahmevermögen 2-5 ml) entnehmen.
Jeder aliquote Teil wird durch einen 0,2 µm-Polycarbonatfilter mit MEC-Zusatzfilter mit 5 µm-Poren unter Verwendung eines 25-mm-Glasfiltertrichters mit zylindrischem Vorratsbehälter vakuumgefiltert. Rund 5 ml des gefilterten destillierten Wassers wird in den Trichter gegeben und den aliquoten Teil langsam in das Wasser gegeben, wobei die Pipettenspitze unterhalb des Meniskus gehalten wird. Pipette und Vorratsbehälter müssen nach dem Pipettieren gründlich gespült werden, da dünne Fasern dazu neigen, sich eher an der Oberfläche anzusammeln.
Filter vorsichtig entnehmen und vom Zusatzfilter trennen, bevor er zum Trocknen in einen Behälter eingelegt wird.
Einen Viertel- oder halben Filterquerschnitt der Filterrückstände mit ruckartigen Bewegungen mit einem Skalpell Typ 24 ausschneiden. Den ausgeschnittenen Querschnitt mit Kohlenstoffklebeband oder Kohlenstoffkleber vorsichtig auf dem Träger (Stub') des REM befestigen. An mindestens drei Stellen ist Kolloidalsilber zur Verbesserung des elektrischen Kontakts der Filterränder und des "Stub" aufzubringen. Wenn der Klebstoff bzw. das Kolloidalsilber getrocknet ist, durch Sputter-Beschichtung ca. 50 nm Gold oder Gold/Palladium auf die Oberfläche des Filterrückstands aufbringen.
1.5.3.3 Kalibrierung und Betrieb des REM
1.5.3.3.1 Kalibrierung
Die Kalibrierung des REM ist mindestens einmal wöchentlich (idealerweise täglich) anhand eines freigegebenen Kalibriergitters zu kontrollieren. Die Kalibrierung ist anhand eines freigegebenen Normals zu kontrollieren; stimmt der Messwert (REM) nicht auf ± 2 % mit dem zertifizierten Wert überein, muss die Kalibrierung des REM nachjustiert und erneut kontrolliert werden.
Das REM muss bei Verwendung einer Probenmatrix mindestens die Auflösung eines sichtbaren Mindestdurchmessers von 0,2 µm bei einem Vergrößerungsfaktor x 2000 ermöglichen.
1.5.3.3.2 Betrieb
Das REM ist mit einem Vergrößerungsfaktor von 10.000 1 unter Bedingungen zu betrieben, die eine gute Auflösung und eine akzeptable Bildqualität bei geringer Abtastgeschwindigkeit von beispielsweise 5 Sekunden je Aufnahme ergeben. Da die Betriebsvoraussetzungen unterschiedlicher REM sich voneinander unterscheiden können, sind bei Materialien mit relativ geringer Atommasse Beschleunigungsspannungen von 5-10 keV zu verwenden, um eine möglichst gute Sichtbarkeit und Auflösung zu erreichen; dabei ist eine geringe Punktgröße und ein kurzer Arbeitsabstand einzustellen. Wird ein Linear-Trverse-Verfahren angewandt, ist eine Neigung von 0° zu verwenden, um Refokussierung auf ein Minimum zu beschränken; weist das REM eine euzentrische Stufe auf, ist der euzentrische Arbeitsabstand zu verwenden. Ein geringerer Vergrößerungsfaktor kann verwendet werden, wenn das Material keine kleinen Fasern (mit geringem Durchmesser) enthält und große Faserdurchmesser (> 5 µm) vorliegen.
___________________
1) Siehe vorherige Fußnote zu 3 µm-Fasern.
1.5.3.4 Größenbestimmung
1.5.3.4.1 Bewertung der Probe bei geringer Vergrößerung
Zunächst ist die Probe mit einem geringen Vergrößerungsfaktor auf Anzeichen von Klumpenbildung größerer Fasern zu untersuchen und die Faserndichte zu ermitteln. Bei übermäßiger Klumpenbildung wird empfohlen, eine neue Probe herzustellen.
Aus Gründen der statistischen Genauigkeit muss eine bestimmte Mindestzahl Fasern gemessen werden, eine hohe Faserndichte ist dabei erstrebenswert, da die Untersuchung leerer Felder zeitaufwändig ist und keinen Beitrag zum Analyseergebnis liefert. Ist der Filter jedoch überladen, ist die Messung aller messbaren Fasern erschwert und es besteht die Gefahr, dass kleinere Fasern durch größere Fasern überdeckt und dadurch übersehen werden.
Eine Verfälschung in Richtung einer zu hoch geschätzten LWGMD kann dann eintreten, wenn Faserdichten von mehr als 150 Fasern je Millimeter Linear-Traverse vorliegen. Durch geringe Faserkonzentrationen nimmt andererseits die Dauer der Analyse zu, weshalb es oft kostengünstiger ist, eine Probe herzustellen, deren Faserdichte näher am Optimum liegt, statt ständig die Faserzahlen in Filtern geringer Konzentrationen zu zählen. Die optimale Faserdichte soll durchschnittlich ca. 1 oder 2 zählbare Fasern je Sichtfeld bei einer 5000-fachen Vergrößerung ergeben. Allerdings ist die optimale Dichte von der Größe (Durchmesser) der Fasern abhängig, also muss der Bediener mit entsprechendem Sachverstand entscheiden, ob die Faserdichte dem Optimum nahekommt oder nicht.
1.5.3.4.2 Längengewichtung der Faserdurchmesser
Es werden nur diejenigen Fasern gezählt, die eine (unendlich) dünne Linie auf dem Raster des REM berühren (oder kreuzen). Hierzu wird eine waagerechte (oder vertikale) Linie durch die Rastermitte gezogen.
Alternativ dazu wird ein einzelner Punkt in der Mitte des Rasters angeordnet und ein kontinuierlicher Abtastvorgang in einer Richtung über den Filter hinweg gestartet. Der Durchmesser jeder Faser mit einem Seitenverhältnis von mehr als 3:1, die diesen Punkt berührt oder kreuzt, wird gemessen und aufgezeichnet.
1.5.3.4.3 Bestimmung der Fasergröße
Es wird empfohlen, mindestens 300 Fasern zu messen. Jede Faser wird nur ein einziges Mal am Schnittpunkt mit der auf dem Bild gezeichneten Linie oder Punkt (oder nahe dem Schnittpunkt, wenn die Faserkanten verdeckt sind) gemessen. Werden Fasern mit uneinheitlichen Querschnitten festgestellt, ist eine Messung am durchschnittlichen Faserdurchmesser zugrundezulegen. Bei der Festlegung des Randes und der Messung des geringsten Abstands zwischen den Faserrändern ist vorsichtig vorzugehen. Die Größenbestimmung kann online oder offline an gespeicherten Bildern oder Fotoaufnahmen erfolgen. Die Verwendung von halbautomatischen Bildmesssystemen, bei denen die Daten direkt in eine Tabellenkalkulation geladen werden, ist zu empfehlen, da diese Verfahren Zeit sparen, Abschriftfehler vermeiden und eine automatische Berechnung ermöglichen.
Die Enden langer Fasern sind bei geringer Vergrößerung zu prüfen, damit sichergestellt ist, dass sie sich nicht in den Sichtbereich des Messfeldes rollen und nur einmal gemessen werden.
2. Daten
2.1 Behandlung der Ergebnisse
Die Faserdurchmesser weisen normalerweise keine Normalverteilung auf. Mit Hilfe einer Log-Transformation kann jedoch eine Verteilung ermittelt werden, die näherungsweise der Normalverteilung entspricht.
Das arithmetische Mittel (mittlerer 1nD) und die Standardabweichung (SDlnD) der 1nD-Werte (log to base e) der n Faserdurchmesser (D) wird berechnet.
mittl.1nD = ΣlnD / n (1)
SDlnD = {[Σ(lnD - mittl. lnD)]2/ (n-1)}0,5 (2)
Die Standardabweichung wird durch die Quadratwurzel der Anzahl Messungen (n) dividiert und daraus der Standardfehler (SE,J ermittelt.
SElnd = SD / n0,5 (3)
Das Zweifache des Standardfehlers wird vom Mittelwert abgezogen und der Exponentialwert dieses Wertes (Mittelwert minus dem Zweifachen des Standardfehlers) berechnet; dies ergibt den geometrischen Mittelwert minus zwei geometrischen Standardfehlern.
LWGMD - 2SE = e(mitt1.1ndD-2SElnd) (4)
3. Berichtserstellung
Testbericht
Der Testbericht muss mindestens folgende Angaben enthalten:
- den Wert für LWGMD-2SE,
- etwaige Abweichungen, vor allem jene, die sich auf Präzision oder Genauigkeit der Ergebnisse auswirken (mit entsprechenden Begründungen).
4. LITERATUR
1) B. Tylee SOP MF 240. Health and Safety Executive. February 1999.
2) G. Burdett und G. Revell. Development of a standard method to measure the lengthweigthed geometric mean fibre diameter. Results of the Second interlaboratory exchange. IR/LIMF/94107. Project 1142.75 HPD. Health and Safety Executive. Research and Laboratory Services Division, 1994.
A.23 1-Octanol/Wasser-Verteilungskoeffizient: Methode zur Prüfung unter langsamem Rühren 14
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 123 (2006). Die POW-Werte (POW = 1-Octanol/ Wasser-Verteilungskoeffizient) konnten mit der Methode zur Prüfung unter langsamem Rühren bis zu log POW 8,2 genau bestimmt werden (1). Entsprechend kommt diese Methode für die direkte Bestimmung der POW-Werte stark hydrophober Substanzen in Betracht.
2. Weitere Methoden zur Bestimmung des 1-Octanol/Wasser-Verteilungskoeffizienten (POW) sind die 'Schüttelmethode' (2) und die Bestimmung des POW aufgrund des Retentionsverhaltens bei der HPLC mit Phasenumkehr (3). Die 'Schüttelmethode' ist wegen der Übertragung von Octanol-Mikrotröpfchen in die wässrige Phase jedoch fehleranfällig. Mit steigenden POW-Werten führt das Vorhandensein dieser Tröpfchen in der wässrigen Phase zu einer zunehmenden Überschätzung der Konzentration der Prüfsubstanz im Wasser. Daher ist diese Methode nur bei Substanzen mit log POW < 4 geeignet. Die zweite Methode beruht auf direkt bestimmten POW-Werten, die zur Kalibrierung der Beziehung zwischen dem Retentionsverhalten in der HPLC und den gemessenen POW- Werten verwendet werden. Es gab einen Entwurf einer OECD-Richtlinie zur Bestimmung des 1-Octanol/Wasser-Verteilungskoeffizienten ionisierbarer Substanzen (4), der aber nicht mehr verwendet werden soll.
3. Diese Prüfmethode wurde in den Niederlanden entwickelt. Die Genauigkeit der in dieser Prüfmethode beschriebenen Verfahren wurde in einer Ringtest-Validierungsstudie unter Beteiligung von 15 Labors validiert und optimiert (5).
Bedeutung und Anwendung
4. Bei inerten organischen Substanzen wurden hoch signifikante Beziehungen zwischen den 1-Octanol/Wasser- Verteilungskoeffizienten (POW) und der jeweiligen Bioakkumulation in Fischen festgestellt. Außerdem wurde eine Korrelation zwischen den POW-Werten und der Toxizität für Fische sowie zwischen den POW-Werten und der Sorption chemischer Stoffe in Feststoffen wie z.B. in Böden und in Sedimenten nachgewiesen. Das Literaturverzeichnis enthält eine umfassende Übersicht über die verschiedenen Zusammenhänge (6).
5. Zwischen dem 1-Octanol/Wasser-Verteilungskoeffizienten und sonstigen für die Umwelttoxizität und für das chemische Verhalten erheblichen Merkmalen von Substanzen wurden vielfältige Beziehungen festgestellt. Entsprechend hat sich der 1-Octanol/Wasser-Verteilungskoeffizient zum Schlüsselparameter für die Bewertung des mit chemischen Stoffen verbundenen Umweltrisikos sowie zur Prognose der Persistenz chemischer Stoffe in der Umwelt entwickelt.
6. Bei der Methode zur Prüfung unter langsamem Rühren soll die Bildung von 1-Octanol-Mikrotröpfchen in der wässrigen Phase verringert werden. Entsprechend ist ausgeschlossen, dass die Konzentration in der wässrigen Phase wegen der ansonsten mit diesen Tröpfchen verbundenen Moleküle der Prüfsubstanz überschätzt wird. Daher eignet sich die Methode zur Prüfung unter langsamem Rühren insbesondere zur Bestimmung der POW-Werte von Substanzen, bei denen log POW-Werte im Bereich von mindestens 5 zu erwarten sind und bei denen die Schüttelmethode (2) eher fehleranfällig wäre.
7. Der Verteilungskoeffizient einer Substanz für Wasser und ein lipophiles Lösungsmittel (1-Octanol) beschreibt die Gleichgewichtsverteilung des jeweiligen chemischen Stoffs zwischen den beiden Phasen. Der Verteilungskoeffizient für Wasser und 1-Octanol (POW) wird definiert als Verhältnis der Gleichgewichtskonzentrationen der Prüfsubstanz in mit Wasser gesättigtem 1-Octanol (CO) und in mit 1-Octanol gesättigtem Wasser (CW).
POW = CO/CW
Für das Konzentrationsverhältnis wird keine Einheit angegeben. Meist wird das Verhältnis als Zehnerlogarithmus (log POW) ausgedrückt. POW ist temperaturabhängig; entsprechend sollte die Messtemperatur berücksichtigt werden.
8. Um den Verteilungskoeffizienten zu bestimmen, werden Wasser, 1-Octanol und die Prüfsubstanz bei einer konstanten Temperatur in ein Gleichgewicht gebracht. Anschließend werden die Konzentrationen der Prüfsubstanz in den beiden Phasen bestimmt.
9. Die bei der Prüfung mit der Schüttelmethode auftretenden Schwierigkeiten infolge der Entstehung von Mikrotröpfchen können mit der hier vorgeschlagenen Methode unter langsamem Rühren verringert werden. Bei der Prüfung unter langsamem Rühren werden Wasser, 1-Octanol und die Prüfsubstanz in einem thermostatgeregelten Rührbehälter in ein Gleichgewicht gebracht. Durch das Rühren wird der Austausch zwischen den Phasen beschleunigt. Beim Rühren entstehen begrenzte Turbulenzen, welche den Austausch zwischen 1-Octanol und Wasser begünstigen, ohne dass Mikrotröpfchen entstehen können (1).
10. Da sich das Vorhandensein anderer Substanzen auf den Aktivitätskoeffizienten der Prüfsubstanz auswirken kann, sollte die Prüfsubstanz als reine Substanz untersucht werden. Daher sollte die jeweilige Substanz für die Messung der 1-Octanol/Wasser-Verteilung in der höchsten auf dem Markt verfügbaren Reinheit verwendet werden.
11. Diese Methode ist anzuwenden bei reinen Substanzen, bei denen weder eine Dissoziation noch eine Assoziation erfolgt und die keine erhebliche Grenzflächenaktivität aufweisen. Die Methode kann zur Bestimmung der 1- Octanol/Wasser-Verteilung dieser Substanzen und von Gemischen eingesetzt werden. Wenn die Methode für Gemische eingesetzt wird, hängt die 1-Octanol/Wasser-Verteilung von der chemischen Zusammensetzung des zu prüfenden Gemischs sowie von der Zusammensetzung des als wässrige Phase genutzten Elektrolyten ab. Wenn die Methode um verschiedene Schritte ergänzt wird, kann sie auch für dissoziierende und assoziierende Verbindungen eingesetzt werden (siehe Nummer 12).
12. Aufgrund der unterschiedlichen Gleichgewichte in Wasser und 1-Octanol, die sich bei der 1-Octanol-/Wasser- Verteilung dissoziierender Substanzen wie z.B. organischer Säuren und Phenole, organischer Basen und organometallischer Substanzen ergeben, ist das Verteilungsverhältnis für 1-Octanol/Wasser ebenfalls in hohem Maße von der Zusammensetzung des Elektrolyten abhängig (7)(8). Die Bestimmung des Verteilungsverhältnisses für 1- Octanol/Wasser setzt voraus, dass pH-Wert und Elektrolytzusammensetzung während der Messung überwacht und protokolliert werden. Die Bewertung der Verteilungsverhältnisse muss durch Fachleute erfolgen. Unter Berücksichtigung der Dissoziationskonstante(n) müssen geeignete pH-Werte so ausgewählt werden, dass für jedes Ionisierungsstadium ein Verteilungsverhältnis bestimmt werden kann. Zur Prüfung organometallischer Verbindungen müssen Pufferlösungen eingesetzt werden, die nicht als Komplexbildner wirken (8). Unter Berücksichtigung des aktuellen Kenntnisstandes bezüglich der Chemie der wässrigen Phase (Komplexbildungskonstanten, Dissoziationskonstanten) sollten die Versuchsbedingungen so gewählt werden, dass die Speziation der Prüfsubstanz in der wässrigen Phase bestimmt werden kann. Die Ionenkonzentration sollte in allen Prüfungen gleich sein; um dies zu gewährleisten, sollte ein Hintergrundelektrolyt eingesetzt werden.
13. Schwierigkeiten können sich bei der Prüfung in Verbindung mit der Untersuchung von Substanzen mit geringer Wasserlöslichkeit oder mit hohem POW ergeben, weil die Konzentrationen im Wasser sehr gering werden und eine genaue Bestimmung entsprechend problematisch ist. Diese Prüfmethode erläutert, wie diesem Problem begegnet werden kann.
Informationen zur Prüfsubstanz
14. Die chemischen Reagenzien sollten mindestens Analysequalität besitzen. Es wird empfohlen, nicht markierte Prüfsubstanzen mit bekannter chemischer Zusammensetzung und einer Reinheit von mindestens 99 % oder radioaktiv markierte Prüfsubstanzen mit bekannter chemischer Zusammensetzung und radiochemischer Reinheit einzusetzen. Beim Einsatz von Indikatorsubstanzen mit kurzer Halbwertszeit sollten Korrekturen unter Berücksichtigung des Zerfallsverhaltens vorgenommen werden. Wenn die Prüfsubstanzen radioaktiv markiert wurden, sollte eine speziell für den jeweiligen chemischen Stoff vorgesehene Analysemethode eingesetzt werden, um sicherzustellen, dass die gemessene Radioaktivität unmittelbar auf die Prüfsubstanz zurückzuführen ist.
15. Log POW kann mit einer im Handel erhältlichen und für diesen Zweck vorgesehenen Software geschätzt werden; alternativ kann die Schätzung auch aufgrund des Verhältnisses der Löslichkeiten in beiden Lösungsmitteln erfolgen.
16. Bevor POW durch eine Prüfung unter langsamem Rühren bestimmt wird, sollten die folgenden Informationen zur Prüfsubstanz bekannt sein:
- die Strukturformel,
- geeignete Analysemethoden zur Bestimmung der Konzentration der Substanz in Wasser und in 1-Octanol,
- die Dissoziationskonstante(n) ionisierbarer Substanzen (OECD-Richtlinie 112) (9),
- die Wasserlöslichkeit (OECD-Richtlinie 105) (10),
- Informationen zur abiotischen Hydrolyse (OECD-Richtlinie 111) (11),
- die leichte biologische Abbaubarkeit (OECD-Richtlinie 301) (12),
- der Dampfdruck (OECD-Richtlinie 104) (13).
Geräte und Apparatur
17. Für die Prüfungen werden Standardlaborgeräte benötigt; insbesondere sind folgende Geräte erforderlich:
- Magnetrührer sowie Magnetrührstäbe mit Teflonbeschichtung zum Umrühren der wässrigen Phase,
- geeignete Analyseinstrumente zur Bestimmung der Konzentration der Prüfsubstanz in den erwarteten Konzentrationen,
- Rührgefäße mit einem Absperrhahn unten am Gefäß. Abhängig vom geschätzten Wert für log POW und von der Nachweisgrenze (LOD) der zu prüfenden Verbindung ist der Einsatz eines Reaktionsbehälters mit identischer Geometrie, aber mit einem Volumen von mehr als einem Liter in Betracht zu ziehen, damit hinreichend Wasser für die chemische Extraktion und für die durchzuführende Analyse entnommen werden kann. Dies führt zu höheren Konzentrationen im Wasserextrakt und ermöglicht entsprechend zuverlässigere Analysen. Anlage 1 enthält eine Tabelle mit Schätzwerten für die erforderlichen Mindestvolumina, der Nachweisgrenze der Verbindung, dem geschätzten log POW und der Löslichkeit in Wasser. Die Tabelle beruht auf der Beziehung zwischen log POW und dem Verhältnis zwischen der Löslichkeit in Octanol und in Wasser gemäß Pinsuwan u. a. (14):
Log POW = 0,88 log SR + 0,41
Dabei ist
SR = SOCt/SW (Molarität)
sowie auf der von Lyman (15) definierten Beziehung für die Prognose der Löslichkeit in Wasser. Die mit der in Anlage 1 genannten Formel bestimmten Wasserlöslichkeiten sind als erste Schätzung zu betrachten. Der Prüfende kann die Wasserlöslichkeit auch aufgrund einer sonstigen Beziehung schätzen, die er für besser geeignet hält, Auskunft über die Beziehung zwischen Hydrophobizität und Löslichkeit zu geben. Bei festen Verbindungen wird z.B. die Berücksichtigung des Schmelzpunktes bei der Prognose der Löslichkeit empfohlen. Wenn eine modifizierte Formel verwendet wird, sollte sichergestellt werden, dass die Gleichung zur Berechnung der Löslichkeit in Octanol noch gültig ist. In Anlage 2 ist ein Rührgefäß mit Glasmantel und einem Inhalt von etwa einem Liter dargestellt. Die Proportionen des in Anlage 2 dargestellten Gefäßes haben sich als günstig erwiesen und sollten auch dann beibehalten werden, wenn eine anders dimensionierte Apparatur verwendet wird,
- entscheidend ist eine Vorrichtung, mit der während der Prüfung unter langsamem Rühren eine konstante Temperatur aufrechterhalten werden kann.
18. Die Gefäße sollten aus einem inerten Material bestehen, damit die Adsorption durch die Oberfläche der Gefäße vernachlässigt werden kann.
Herstellung der Prüflösungen
19. Der Wert für POW sollte mit 1-Octanol der höchsten im Handel erhältlichen Reinheit (mindestens + 99 %) bestimmt werden. Die Reinigung von 1-Octanol durch die Extraktion mit einer Säure, einer Base und Wasser und eine anschließende Trocknung werden empfohlen. Außerdem kann 1-Octanol auch durch Destillation gereinigt werden. Zur Herstellung der Standardlösungen der Prüfsubstanzen ist gereinigtes 1-Octanol zu verwenden. Das für die Bestimmung von POW zu verwendende Wasser sollte durch Glas oder Quarz destilliert oder mit einem Reinigungssystem hergestellt worden sein; alternativ kann auch HPLC-Wasser verwendet werden. Destilliertes Wasser sollte durch ein 0,22-µm-Filter gefiltert werden; mit Blindproben sollte sichergestellt werden, dass die konzentrierten Extrakte keine Verunreinigungen enthalten, welche die Prüfsubstanz verändern könnten. Wenn ein Glasfaserfilter verwendet wird, sollte das Filter durch mindestens dreistündige Erhitzung auf 400 °C gereinigt werden.
20. Beide Lösungsmittel werden vor Durchführung der Prüfung wechselseitig gesättigt, indem sie in einem hinreichend großen Gefäß in ein Gleichgewicht gebracht werden. Dazu wird das aus zwei Phasen bestehende System zwei Tage lang langsam gerührt.
21. Eine geeignete Konzentration der Prüfsubstanz wird ausgewählt und in 1-Octanol (mit Wasser gesättigt) gelöst. Der 1-Octanol/Wasser-Verteilungskoeffizient ist in verdünnten Lösungen mit 1-Octanol und Wasser zu bestimmen. Daher sollte die Konzentration der Prüfsubstanz höchstens 70 % ihrer Löslichkeit bei einer Höchstkonzentration von 0,1 M in beiden Phasen betragen (1). Die für die Prüfungen verwendeten 1-Octanol-Lösungen dürfen keine suspendierten Feststoffe aus der Prüfsubstanz enthalten.
22. Eine geeignete Menge der Prüfsubstanz wird in 1-Octanol (mit Wasser gesättigt) gelöst. Wenn die Schätzung für log POW einen Wert über fünf ergibt, ist besonders darauf zu achten, dass die für die Prüfung verwendeten 1-Octanol-Lösungen keine suspendierten Feststoffe aus der Prüfsubstanz enthalten. Dazu ist bei chemischen Stoffen mit einem geschätzten log POW > 5 wie folgt zu verfahren:
- Die Prüfsubstanz wird in 1-Octanol (mit Wasser gesättigt) gelöst.
- Es wird hinreichend Zeit gelassen, damit sich suspendierte Feststoffe absetzen können. Während des Absetzens wird die Konzentration der Prüfsubstanz überwacht.
- Nachdem sich die gemessenen Konzentrationen der 1-Octanol-Lösung stabilisiert haben, wird die Stammlösung mit einer geeigneten Menge 1-Octanol verdünnt.
- Anschließend wird die Konzentration der verdünnten Stammlösung gemessen. Wenn die gemessene Konzentration mit der Verdünnung übereinstimmt, kann die verdünnte Stammlösung zur Prüfung unter langsamem Rühren verwendet werden.
Extraktion und Analyse der Proben
23. Für die Untersuchung der Prüfsubstanz sollte eine validierte Analysemethode verwendet werden. Der Prüfende muss nachweisen, dass die Konzentrationen in dem mit Wasser gesättigten 1-Octanol sowie in der mit 1-Octanol gesättigten wässrigen Phase während der Prüfung über der bei dem eingesetzten Analyseverfahren für die Methode festgesetzten Quantifizierungsgrenze liegen. In Fällen, in denen Extraktionsmethoden erforderlich sind, muss die analytische Wiederfindung der Prüfsubstanzen aus der wässrigen Phase und aus der 1-Octanol-Phase vor der Prüfung erfolgen. Die Anforderungen an die Analysesignale sind aufgrund von Blindwerten zu korrigieren; dabei ist darauf zu achten, dass keine Analytübertragungen zwischen den Proben vorkommen können.
24. Bei hydrophoben Prüfsubstanzen sind wegen der eher niedrigen Konzentrationen der Prüfsubstanzen in der wässrigen Phase vor der Analyse wahrscheinlich eine Extraktion der wässrigen Phase mit einem organischen Lösungsmittel und eine Vorkonzentration des Extrakts vorzunehmen. Aus demselben Grund müssen die eventuell verwendeten Blindprobenkonzentrationen reduziert werden. Dazu sind hochreine und vorzugsweise für Rückstandsanalysen vorgesehene Lösungsmittel zu verwenden. Die Verwendung sorgfältig vorgereinigter Glasgeräte (z.B. durch Spülen mit einem Lösungsmittel oder unter starker Erwärmung) kann zusätzlich helfen, Kreuzkontamination zu vermeiden.
25. Log POW kann mit einem geeigneten Programm ermittelt oder von Fachleuten mit entsprechender Erfahrung geschätzt werden. Bei Werten über 6 müssen Blindwert-Korrekturen und Analyteintragungen sorgfältig überwacht werden. Wenn der geschätzte Wert für log POW über 6 liegt, muss ein Surrogatstandard für die Wiederfindungskorrektur verwendet werden, damit die erforderlichen hohen Vorkonzentrationsfaktoren erreicht werden können. Auf dem Markt werden verschiedene Computer-Programme zur Schätzung von log POW angeboten 1 (z.B. Clog P(16), KOWWIN(17), ProLogP(18) und ACD log P(19)). Das Literaturverzeichnis enthält Beschreibungen der verschiedenen Schätzverfahren (20-22).
26. Die Quantifizierungsgrenzen (LOQ) für die Bestimmung der Prüfsubstanz in 1-Octanol und Wasser werden mit anerkannten Methoden bestimmt. Als Faustregel kann die Konzentration in Wasser oder 1-Octanol, bei der sich ein Signal-/Rauschverhältnis von 10 ergibt, als Quantifizierungsgrenze für die betreffende Methode angenommen werden. Entsprechend sollten eine geeignete Methode zur Extraktion und zur Vorkonzentration ausgewählt und Verfahren für die analytische Wiederfindung spezifiziert werden. Der Vorkonzentrationsfaktor ist so auszuwählen, dass sich bei der Analyse ein Signal der geforderten Stärke ergibt.
27. Ausgehend von den Parametern der Analysemethode und von den erwarteten Konzentrationen wird die Probengröße bestimmt, die für eine genaue Bestimmung der Konzentration der jeweiligen Verbindung ungefähr erforderlich ist. Wasserproben, die so klein sind, dass kein hinreichendes Analysesignal festgestellt werden kann, sollten nicht verwendet werden. Ebenso sollte die Verwendung zu großer Wasserproben vermieden werden, weil ansonsten möglicherweise zu wenig Wasser für die erforderlichen Analysen (n = 5) verbleibt. In Anlage 1 wird das Proben-Mindestvolumen abhängig vom Gefäßvolumen, von der Nachweisgrenze der Prüfsubstanz und von der Löslichkeit der Prüfsubstanz angegeben.
28. Die Quantifizierung der Prüfsubstanzen erfolgt durch den Vergleich mit Kalibrierungskurven der entsprechenden Verbindung. Die Konzentrationen in den analysierten Proben müssen innerhalb der Bandbreite der Standardkonzentrationen liegen.
29. Bei Prüfsubstanzen mit log POW-Werten über 6 muss der Wasserprobe vor der Extraktion ein Surrogatstandard zugesetzt werden, um die bei der Extraktion und bei der Vorkonzentration der Wasserprobe auftretenden Verluste zu erfassen. Für exakte Wiederfindungskorrekturen müssen die Surrogate Merkmale aufweisen, die denen der Prüfsubstanz möglichst ähnlich sind oder vollständig mit deren Merkmalen übereinstimmen. Dazu werden vorzugsweise mit einem (stabilen) Isotop markierte, den Prüfsubstanzen analoge Verbindungen verwendet (z.B. deuterierte oder mit 13C markierte Verbindungen). Wenn mit einem stabilen Isotop (d. h. mit 13C oder 2H) markierte Verbindungen nicht verwendet werden können, sollte anhand zuverlässiger Daten in der Fachliteratur nachgewiesen werden, dass die physikalisch-chemischen Merkmale der Surrogate den Merkmalen der Prüfsubstanzen sehr nahekommen. Bei der Flüssigflüssig-Extraktion der wässrigen Phase können Emulsionen entstehen. Diese Emulsionen können durch die Zugabe von Salz und Ausfällen der Emulsion über Nacht verringert werden. Die Methoden zur Extraktion und zur Vorkonzentration der Proben sind zu protokollieren.
30. Aus der 1-Octanol-Phase gezogene Proben können vor der Analyse erforderlichenfalls mit einem geeigneten Lösungsmittel verdünnt werden. Die Verwendung von Surrogatstandards zur Wiederfindungskorrektur wird für Substanzen empfohlen, bei denen in Wiederfindungsprüfungen starke Schwankungen zu verzeichnen waren (relative Standardabweichung > 10 %).
31. Die Analysemethode ist detailliert zu protokollieren. Zu erfassen sind unter anderem die Extraktionsmethode, Vorkonzentrations- und Verdünnungsfaktoren, Geräteparameter, die Kalibrierungsroutine, der Kalibrierungsbereich, Angaben zur analytischen Wiederfindung der Prüfsubstanzen aus dem Wasser, Zugaben von Surrogatstandards zur Wiederfindungskorrektur, Blindwerte, Nachweisgrenzen und Quantifizierungsgrenzen.
Durchführung der Prüfung
Optimale 1-Octanol-/Wasser-Verhältnisse
32. Bei der Auswahl der Volumina von Wasser und 1-Octanol sollten die folgenden Punkte berücksichtigt werden: die Quantifizierungsgrenze (LOQ) in 1-Octanol und Wasser, die Vorkonzentrationsfaktoren der Wasserproben, das Volumen der aus 1-Octanol und aus Wasser genommenen Proben und die zu erwartenden Konzentrationen. Aus durchführungspraktischen Gründen sollte das 1-Octanol-Volumen bei dem System zur Prüfung unter langsamem Rühren so gewählt werden, dass die 1-Octanol-Schicht hinreichend stark (> 0,5 cm) ist, damit die Proben ohne Beeinträchtigung aus der 1-Octanol-Phase entnommen werden können.
33. Typische Phasenverhältnisse für die Bestimmung von Verbindungen mit log POW-Werten von mindestens 4,5 sind 20 bis 50 ml 1-Octanol und 950 bis 980 ml Wasser in einem 1-l-Gefäß.
Prüfbedingungen
34. Während der Prüfung wird das Reaktionsgefäß mit einem Thermostaten so geregelt, dass die Temperaturschwankungen unter 1 °C liegen. Die Untersuchung sollte bei einer Temperatur von 25 °C durchgeführt werden.
35. Das Prüfsystem sollte vor Tageslichteinfall geschützt werden, indem die Prüfungen entweder im Dunkeln durchgeführt werden oder das Reaktionsgefäß mit einer Aluminiumfolie bedeckt wird.
36. Außerdem sollte die Prüfung in (möglichst) staubfreier Umgebung erfolgen.
37. Das 1-Octanol-Wasser-System wird gerührt, bis das erforderliche Gleichgewicht hergestellt ist. In einem Pilottest unter langsamem Rühren bei regelmäßiger Entnahme von Proben aus der wässrigen Phase und aus der 1-Octanol-Phase wird die Zeitspanne bis zur Herstellung des Gleichgewichts ermittelt. Zwischen den verschiedenen Probenahmen sollten jeweils mindestens 5 Stunden liegen.
38. Die POW-Werte sind in mindestens drei unabhängig voneinander durchzuführenden Prüfungen unter langsamem Rühren zu bestimmen.
Bestimmung der zur Herstellung des Gleichgewichts erforderlichen Zeitspanne
39. Es wird angenommen, dass das Gleichgewicht dann erreicht ist, wenn das Konzentrationsverhältnis für 1-Octanol und Wasser in einem Zeitraum mit vier Messzeitpunkten bei einem p-Wert von 0,05 so weit zurückgegangen ist, dass der Endwert nicht mehr signifikant von 0 abweicht. Die Zeitspanne zur Herstellung des Gleichgewichts beträgt mindestens einen Tag. Erst dann kann mit der Probenahme begonnen werden. Als Faustregel kann davon ausgegangen werden, dass bei Substanzen, bei denen log POW auf unter 5 geschätzt wird, die Probenahme am zweiten und am dritten Tag erfolgen kann. Bei stärker hydrophoben Verbindungen dauert die Herstellung des Gleichgewichts unter Umständen länger. Bei einer Verbindung mit log POW 8,23 (Decachlorbiphenyl) wurde das Gleichgewicht nach 144 Stunden erreicht. Ob das Gleichgewicht hergestellt ist, wird anhand mehrfacher Probenahmen aus einem einzigen Gefäß beurteilt.
Beginn der Prüfung
40. Zu Beginn der Prüfung wird das Reaktionsgefäß Wasser befüllt, das mit 1-Octanol gesättigt wurde. Dabei sollte genügend Zeit zum Erreichen der thermostatgeregelten Temperatur gelassen werden.
41. Die gewünschte Menge der Prüfsubstanz (im erforderlichen Volumen des mit Wasser gesättigten 1-Octanol) wird vorsichtig in das Reaktionsgefäß gegeben. Dies ist ein entscheidender Schritt bei der Prüfung, da bei der Mischung der beiden Phasen Verwirbelungen vermieden werden müssen. Dazu kann die 1-Octanol-Phase langsam mit einer Pipette dicht über der Wasseroberfläche gegen die Wand des Prüfgefäßes getropft werden. Die 1- Octanol-Phase fließt dann an der Glaswand hinab und bildet einen Film auf der wässrigen Phase. Eine direkte Dekantierung von 1-Octanol in den Kolben sollte in jedem Fall vermieden werden; unter keinen Umständen sollten 1-Octanol-Tropfen direkt auf die Wasseroberfläche fallen.
42. Nach dem Beginn des Rührvorgangs sollte die Rührgeschwindigkeit langsam gesteigert werden. Wenn die Rührmotoren nicht in geeigneter Weise eingestellt werden können, sollte der Einsatz eines Transformators in Erwägung gezogen werden. Die Rührgeschwindigkeit sollte so eingestellt werden, dass am Übergang zwischen Wasser und 1-Octanol ein Wirbel mit einer Tiefe von 0,5 cm bis höchstens 2,5 cm entsteht. Die Rührgeschwindigkeit ist zu verringern, wenn der Wirbel tiefer als 2,5 cm wird; andernfalls können 1-Octanol-Mikrotröpfchen in der wässrigen Phase entstehen und dazu führen, dass die Konzentration der Prüfsubstanz im Wasser überschätzt wird. Die Rührgeschwindigkeit, bei der ein Wirbel mit einer Tiefe von höchstens 2,5 cm entsteht, wird ausgehend von den Ergebnissen der Ringtest-Validierungsstudie empfohlen (5). Diese Rührgeschwindigkeit gewährleistet einen Kompromiss zwischen der möglichst raschen Herstellung des erforderlichen Gleichgewichts und der Vermeidung von 1-Octanol-Mikrotröpfchen.
Probenahme und Behandlung der Proben
43. Vor der Probenahme sollte der Rührmotor ausgeschaltet und gewartet werden, bis die Flüssigkeiten zur Ruhe gekommen sind. Nach der Probenahme wird der Rührmotor bei geringer Rührgeschwindigkeit wieder eingeschaltet und die Rührgeschwindigkeit wie oben beschrieben langsam gesteigert.
44. Die Probenahme aus der wässrigen Phase erfolgt aus einem Absperrhahn unten am Reaktionsgefäß. Dabei ist das Totvolumen des im Hahn enthaltenen Wassers (in dem in Anlage 2 abgebildeten Gefäß etwa 5 ml) zu verwerfen. Das im Hahn enthaltene Wasser wurde nicht umgerührt und befindet sich daher nicht im Gleichgewicht mit der übrigen Flüssigkeit. Das Volumen der Wasserproben wird protokolliert; außerdem ist die Menge der im verworfenen Wasservolumen enthaltenen Prüfsubstanz bei der Berechnung der Massenbilanz zu berücksichtigen. Verdampfungsverluste sollten vermieden werden; dazu sollte dem Wasser Gelegenheit gegeben werden, ruhig in den Abscheidetrichter abzufließen, damit die Wasser-1-Octanol-Schicht nicht gestört wird.
45. Die Probenahme aus der 1-Octanol-Phase erfolgt, indem eine kleine Aliquote (ca. 100 µl) mit einer 100- Mikroliter-Glas-Metall-Spritze aus der 1-Octanol-Schicht gezogen wird. Dabei ist darauf zu achten, dass der Übergangsbereich nicht berührt wird. Anschließend wird das Volumen der als Probe entnommenen Flüssigkeit protokolliert. Eine kleine Aliquote ist ausreichend, da die 1-Octanol-Probe verdünnt wird.
46. Bei der Handhabung der Proben sollten unnötige Übertragungsschritte vermieden werden. Daher sollte das Probenvolumen gravimetrisch bestimmt werden. Bei Wasserproben kann dies geschehen, indem die Wasserproben in einem Abscheidetrichter gesammelt werden, der bereits die erforderliche Lösungsmittelmenge erhält.
47. Bei der hier beschriebenen Prüfmethode wird POW aufgrund von drei Prüfungen der zu untersuchenden Verbindung (drei Prüfeinheiten) unter langsamem Rühren bestimmt; dabei müssen jeweils identische Bedingungen gegeben sein. Die zum Nachweis des hergestellten Gleichgewichts vorgenommene Regression sollte auf den Ergebnissen von mindestens vier CO/CW-Werten zu aufeinanderfolgenden Zeitpunkten beruhen. Anhand dieser Ergebnisse kann eine Varianz als Maß für die Unsicherheit des pro Prüfeinheit ermittelten Durchschnittswerts berechnet werden.
48. POW kann über die Varianz der in den einzelnen Prüfeinheiten ermittelten Daten beschrieben werden. Aufgrund dieser Information wird POW nämlich als gewichteter Durchschnitt der Ergebnisse der einzelnen Prüfeinheiten berechnet. Dazu wird der Kehrwert der Varianz der Ergebnisse der Prüfeinheiten als Gewichtung angenommen. Dies hat zur Folge, dass sich Daten mit großen Schwankungen (ausgedrückt in einer hohen Varianz) und entsprechend geringerer Zuverlässigkeit weniger auf das Ergebnis auswirken als Daten mit niedriger Varianz.
49. In entsprechender Weise wird die gewichtete Standardabweichung berechnet. Die Standardabweichung beschreibt die Wiederholbarkeit der POW-Messung. Eine niedrige gewichtete Standardabweichung ist Ausdruck einer sehr hohen Wiederholbarkeit der POW-Bestimmung in ein und demselben Labor. Im Folgenden wird die formale statistische Behandlung der Daten beschrieben.
Auswertung der Ergebnisse
Nachweis der Herstellung des Gleichgewichts
50. Für jeden Probenahmezeitpunkt wird der Logarithmus des Verhältnisses der Konzentration der Prüfsubstanz in 1-Octanol und Wasser (log (CO/CW)) berechnet. Die Herstellung des chemischen Gleichgewichts wird nachgewiesen, indem dieses Verhältnis bezogen auf die betreffende Zeit dargestellt wird. Ein Plateau in der aus Messungen zu mindestens vier aufeinanderfolgenden Zeitpunkten erstellten Kurve zeigt, dass ein Gleichgewicht erreicht und die Verbindung tatsächlich in 1-Octanol gelöst ist. Andernfalls sind die Prüfungen fortzusetzen, bis sich aus den Werten von vier aufeinanderfolgenden Zeitpunkten eine Kurve ergibt, deren Steigung sich bei einem p-Wert von 0,05 nicht mehr signifikant von null unterscheidet, und die entsprechend zeigt, dass log CO/CW nicht mehr zeitabhängig ist.
Berechnung von Log POW
51. Log POW der Versuchseinheit wird als gewichteter Durchschnitt von log Co/Cw für den Teil der Kurve zur Darstellung des Verhältnisses von log Co/Cw zur Zeitspanne berechnet, in dem das Gleichgewicht nachgewiesen wurde. Zur Berechnung des gewichteten Durchschnitts werden die Daten mit dem Kehrwert der Varianz so berechnet, dass der Einfluss der Daten auf das Endergebnis umgekehrt proportional zur Unsicherheit der Daten ist.
Durchschnittlicher Wert für log POW
52. Der durchschnittliche Wert für log POW bei verschiedenen Versuchseinheiten wird als Durchschnitt der Ergebnisse der einzelnen mit der jeweiligen Varianz gewichteten Versuchseinheiten berechnet.
Die Berechnung erfolgt nach der nachstehenden Formel:
Log POW,Av = (Σwi x log POW,i) x (Σwi)-1
Dabei ist
Log POW,i = log POW der jeweiligen Versuchseinheit i,
log POW, Av = gewichteter Durchschnitt der jeweils ermittelten Werte für log POW,
wi = statistische Gewichtung von log POW der Versuchseinheit i.
Der Kehrwert der Varianz von log POW,i wird als wi eingesetzt (wi = var(log POW,i)-1).
53. Der Fehler des Durchschnitts von log POW wird geschätzt als die in den einzelnen Versuchseinheiten während der Gleichgewichtsphase ermittelte Wiederholbarkeit von log CO/CW und als gewichtete Standardabweichung von log POW, Av (σlog POW, Av) ausgedrückt, die wiederum ein Maß für den mit log POW, Av verbundenen Fehler ist. Die gewichtete Standardabweichung kann wie folgt aus der gewichteten Varianz (varlog POW, Av) berechnet werden:
varlog Pow, Av = (Σwi x (log POW,i - log POW, Av)2) x (Σwi x (n - 1))-1
σlog Pow, Av = (varlog Pow, Av)0,5
Dabei steht n für die Anzahl der Versuchseinheiten.
54. Der Prüfbericht sollte folgende Informationen enthalten:
Prüfsubstanz:
- Handelsname (Common name), chemischer Name, CAS-Nummer, Strukturformel (mit Angabe der Lage der Markierung bei Verwendung radioaktiv markierten Materials) und relevante physikalisch-chemische Eigenschaften (siehe Nummer 17),
- Reinheit (Verunreinigungen) der Prüfsubstanz,
- Reinheit der Markierung von markierten Chemikalien und molare Aktivität (soweit zutreffend),
- vorläufige Schätzung von log POW sowie Methode zur Ableitung des Wertes.
Prüfbedingungen:
- Daten der Durchführung der Untersuchungen,
- Temperatur während der Prüfung,
- Volumina an 1-Octanol und Wasser bei Beginn der Prüfung,
- Volumina der entnommenen 1-Octanol- und Wasserproben,
- in den Prüfgefäßen verbliebene Volumina an 1-Octanol und Wasser,
- Beschreibung der Prüfgefäße und der Rührbedingungen (Geometrie von Rührstab und Prüfgefäß, Wirbeltiefe in mm und - wenn bekannt - Rührgeschwindigkeit),
- Analysemethoden zur Bestimmung der Prüfsubstanz und Methode zur Bestimmung der Quantifizierungsgrenze,
- Probenahmezeiten,
- pH-Wert der wässrigen Phase und verwendete Pufferlösungen, wenn der pH-Wert unter Berücksichtigung der ionisierbaren Moleküle korrigiert wurde,
- Anzahl der Wiederholungen.
Ergebnisse:
- Wiederholbarkeit und Empfindlichkeit der verwendeten Analysemethoden,
- bestimmte Konzentrationen der Prüfsubstanz in 1-Octanol und in Wasser abhängig von der Zeit,
- Berechnung der Massenbilanz,
- Temperatur und Standardabweichung oder Temperaturbereich während der Prüfung,
- zeitabhängige Regression des Konzentrationsverhältnisses,
- durchschnittlicher Wert log POW, Av und Standardfehler,
- Diskussion und Auswertung der Ergebnisse,
- beispielhafte Rohdaten repräsentativer Analysen (sämtliche Rohdaten sind gemäß den GLP-Standards zu speichern); u. a. Daten zur Wiederfindung von Surrogaten, Anzahl der bei der Kalibrierung verwendeten Konzentrationen (sowie Kriterien für den Korrelationskoeffizienten der Kalibrierungskurve) und Ergebnisse der Qualitätssicherung/Qualitätskontrolle (QA/QC),
- wenn verfügbar: Validierungsbericht zum Untersuchungsverfahren (im Literaturverzeichnis anzugeben).
1. De Bruijn JHM, Busser F, Seinen W, Hermens J. (1989). Determination of octanol/water partition coefficients with the 'slow-stirring' method. Environ. Toxicol. Chem. 8: 499-512.
2. Kapitel A.8 dieses Anhangs, Verteilungskoeffizient.
3. Kapitel A.8 dieses Anhangs, Verteilungskoeffizient.
4. OECD (2000). OECD Draft Guideline for the Testing of Chemicals: 122 Partition Coefficient (n-Octanol/Water): pH-Metric Method for Ionisable Substances. Paris.
5. Tolls J (2002). Partition Coefficient 1-Octanol/Water (Pow) Slow-Stirring Method for Highly Hydrophobic Chemicals, Validation Report. RIVM contract-Nrs 602730 M/602700/01.
6. Boethling RS, Mackay D (eds.) (2000). Handbook of property estimation methods for chemicals. Lewis Publishers Boca Raton, FL, USA.
7. Schwarzenbach RP, Gschwend PM, Imboden DM (1993). Environmental Organic Chemistry. Wiley, New York, NY.
8. Arnold CG, Widenhaupt A, David MM, Müller SR, Haderlein SB, Schwarzenbach RP (1997). Aqueous speciation and 1-octanol-water partitioning of tributyl- and triphenyltin: effect of pH and ion composition. Environ. Sci. Technol. 31: 2596-2602.
9. OECD (1981) OECD Guidelines for the Testing of Chemicals: 112 Dissociation Constants in Water. Paris.
10. Kapitel A.6 dieses Anhangs, Wasserlöslichkeit.
11. Kapitel C.7 dieses Anhangs, Abbaubarkeit - abiotischer Abbau: Hydrolyse in Abhängigkeit vom pH-Wert.
12. Kapitel C.4 - Teile II - VII (Methoden A bis F) dieses Anhangs -Bestimmung der 'leichten' biologischen Abbaubarkeit.
13. Kapitel A.4 dieses Anhangs, Dampfdruck.
14. Pinsuwan S, Li A and Yalkowsky S.H. (1995). Correlation of octanol/water solubility ratios and partition coefficients, J. Chem. Eng. Data. 40: 623-626.
15. Lyman WJ (1990). Solubility in water. In: Handbook of Chemical Property Estimation Methods: Environmental Behavior of Organic Compounds, Lyman WJ, Reehl WF, Rosenblatt DH, Eds. American Chemical Society, Washington, DC, 2-1 to 2-52.
16. Leo A, Weininger D (1989). Medchem Software Manual. Daylight Chemical Information Systems, Irvine, CA.
17. Meylan W (1993). SRC-LOGKOW for Windows. SRC, Syracuse, N.Y.
18. Compudrug L (1992). ProLogP. Compudrug, Ltd, Budapest.
19. ACD. ACD logP; Advanced Chemistry Development: Toronto, Ontario M5H 3V9, Canada, 2001.
20. Lyman WJ (1990). Octanol/water partition coefficient. In Lyman WJ, Reehl WF, Rosenblatt DH, eds, Handbook of chemical property estimation, American Chemical Society, Washington, D.C.
21. Rekker RF, de Kort HM (1979). The hydrophobic fragmental constant: An extension to a 1.000 data point set. Eur. J. Med. Chem. Chim. Ther. 14: 479-488.
22. Jübermann O (1958). Houben-Weyl, ed, Methoden der Organischen Chemie: 386-390.
_____
1) Die Angaben dienen nur zur Information. Wenn mit anderen Programmen nachweislich dieselben Ergebnisse ermittelt werden, können auch diese Programme verwendet werden.
| Tabelle zur Berechnung der für den Nachweis von prüßubstanzen mit unterschiedlichen logPOW-Werten in der wässrigen Phase mindestens erforderlichen Volumina | Anlage 1 |
Voraussetzungen:
- Maximales Volumen der einzelnen Aliquoten = 10 % des Gesamtvolumens; 5 Aliquoten = 50 % des Gesamtvolumens.
- Konzentration der Prüfsubstanzen = 0,7 x Löslichkeit in beiden Phasen; bei niedrigeren Konzentrationen sind größere Volumina erforderlich.
- Zur Bestimmung der Nachweisgrenze verwendetes Volumen = 100 ml.
- log POW vs. Log SW und log POW vs. SR (SOCT/SW) sind sinnvolle Darstellungen der Beziehungen zwischen den Prüfsubstanzen.
Geschätzter Wert für Sw
| log POW | Formel | log Sw | Sw (mg/l) |
| 4 | (-)0,922 x log POW + 4,184 | 0,496 | 3,133E+00 |
| 4,5 | (-)0,922 x log POW + 4,184 | 0,035 | 1,084E+00 |
| 5 | (-)0,922 x log POW + 4,184 | - 0,426 | 3,750E-01 |
| 5,5 | (-)0,922 x log POW + 4,184 | - 0,887 | 1,297E-01 |
| 6 | (-)0,922 x log POW + 4,184 | - 1,348 | 4,487E-02 |
| 6,5 | (-)0,922 x log POW + 4,184 | - 1,809 | 1,552E-02 |
| 7 | (-)0,922 x log POW + 4,184 | - 2,270 | 5,370E-03 |
| 7,5 | (-)0,922 x log POW + 4,184 | - 2,731 | 1,858E-03 |
| 8 | (-)0,922 x log POW + 4,184 | - 3,192 | 6,427E-04 |
Geschätzter Wert für Soct
| log POW | Formel | Soct (mg/l) |
| 4 | log POW = 0,88log SR + 0,41 | 3,763E+04 |
| 4,5 | log POW = 0,88log SR + 0,42 | 4,816E+04 |
| 5 | log POW = 0,88log SR + 0,43 | 6,165E+04 |
| 5,5 | log POW = 0,88log SR + 0,44 | 7,890E+04 |
| 6 | log POW = 0,88log SR + 0,45 | 1,010E+05 |
| 6,5 | log POW = 0,88log SR + 0,46 | 1,293E+05 |
| 7 | log POW = 0,88log SR + 0,47 | 1,654E+05 |
| 7,5 | log POW = 0,88log SR + 0,48 | 2,117E+05 |
| 8 | log POW = 0,88log SR + 0,49 | 2,710E+05 |
| Gesamtmasse der Prüfsubstanz (mg) | MasseOct/MasseWasser | MasseH2O (mg) | KonzH2O (mg/l) | Masseoct (mg) | Konzoct (mg/l) |
| 1.319 | 526 | 2,5017 | 2,6333 | 1.317 | 26.333 |
| 1.686 | 1.664 | 1,0127 | 1,0660 | 1.685 | 33.709 |
| 2.158 | 5.263 | 0,4099 | 0,4315 | 2.157 | 43.149 |
| 2.762 | 16.644 | 0,1659 | 0,1747 | 2.762 | 55.230 |
| 3.535 | 52.632 | 0,0672 | 0,0707 | 3.535 | 70.691 |
| 4.524 | 166.436 | 0,0272 | 0,0286 | 4.524 | 90.480 |
| 5.790 | 526.316 | 0,0110 | 0,0116 | 5.790 | 115.807 |
| 7.411 | 1.664.357 | 0,0045 | 0,0047 | 7.411 | 148.223 |
| 9.486 | 5.263.158 | 0,0018 | 0,0019 | 9.486 | 189.713 |
Berechnung der Volumina
Erforderliches Mindestvolumen der H2O-Phase bei den verschiedenen LOD-Konzentrationen
| log Kow | LOD (¼g/l)' | 0,001 | 0,01 | 0,10 | 1,00 | 10 |
| 4 | 0,04 | 0,38 | 3,80 | 38 | 380 | |
| 4,5 | 0,09 | 0,94 | 9,38 | 94 | 938 | |
| 5 | 0,23 | 2,32 | 23,18 | 232 | 2.318 | |
| 5,5 | 0,57 | 5,73 | 57,26 | 573 | 5.726 | |
| 6 | 1,41 | 14,15 | 141 | 1.415 | 14.146 | |
| 6,5 | 3,50 | 34,95 | 350 | 3.495 | 34.950 | |
| 7 | 8,64 | 86,35 | 864 | 8.635 | 86.351 | |
| 7,5 | 21,33 | 213 | 2.133 | 21.335 | 213.346 | |
| 8 | 52,71 | 527 | 5.271 | 52.711 | 527.111 | |
| Volumen für Nachweisgrenze LOD (l) | 0,1 |
Erläuterungen
< 10 % des Gesamtvolumens der wässrigen Phase, 1-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 2-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 5-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 10-l-Ausgleichsgefäß.
Mehr als 10 % des Volumens eines 10-l-Ausgleichsgefäßes.
Übersicht über die benötigten Volumina abhängig von der Wasserlöslichkeit und von log POW
Erforderliches Mindestvolumen der H2O-Phase bei den verschiedenen LOD-Konzentrationen (ml)
| log POW | Sw (mg/l) | LOD (μg/l) -> | 0,001 | 0,01 | 0,10 | 1,00 | 10 |
| 4 | 10 | 0,01 | 0,12 | 1,19 | 11,90 | 118,99 | |
| 5 | 0,02 | 0,24 | 2,38 | 23,80 | 237,97 | ||
| 3 | 0,04 | 0,40 | 3,97 | 39,66 | 396,62 | ||
| 1 | 0,12 | 1,19 | 11,90 | 118,99 | 1.189,86 | ||
| 4,5 | 5 | 0,02 | 0,20 | 2,03 | 20,34 | 203,37 | |
| 2 | 0,05 | 0,51 | 5,08 | 50,84 | 508,42 | ||
| 1 | 0,10 | 1,02 | 10,17 | 101,68 | 1.016,83 | ||
| 0,5 | 0,20 | 2,03 | 20,34 | 203,37 | 2.033,67 | ||
| 5 | 1 | 0,09 | 0,87 | 8,69 | 86,90 | 869,01 | |
| 0,5 | 0,17 | 1,74 | 17,38 | 173,80 | 1.738,02 | ||
| 0,375 | 0,23 | 2,32 | 23,18 | 231,75 | 2.317,53 | ||
| 0,2 | 0,43 | 4,35 | 43,45 | 434,51 | 4.345,05 | ||
| 5,5 | 0,4 | 0,19 | 1,86 | 18,57 | 185,68 | 1.856,79 | |
| 0,2 | 0,37 | 3,71 | 37,14 | 371,36 | 3.713,59 | ||
| 0,1 | 0,74 | 7,43 | 74,27 | 742,72 | 7.427,17 | ||
| 0,05 | 1,49 | 14,85 | 148,54 | 1.485,43 | 14.854,35 | ||
| 6 | 0,1 | 0,63 | 6,35 | 63,48 | 634,80 | 6.347,95 | |
| 0,05 | 1,27 | 12,70 | 126,96 | 1.269,59 | 12.695,91 | ||
| 0,025 | 2,54 | 25,39 | 253,92 | 2.539,18 | 25.391,82 | ||
| 0,0125 | 5,08 | 50,78 | 507,84 | 5.078,36 | 50.783,64 | ||
| 6,5 | 0,025 | 2,17 | 21,70 | 217,02 | 2.170,25 | 21.702,46 | |
| 0,0125 | 4,34 | 43,40 | 434,05 | 4.340,49 | 43.404,93 | ||
| 0,006 | 9,04 | 90,43 | 904,27 | 9.042,69 | 90.426,93 | ||
| 0,003 | 18,09 | 180,85 | 1.808,54 | 18.085,39 | 180.853,86 | ||
| 7 | 0,006 | 7,73 | 77,29 | 772,89 | 7.728,85 | 77.288,50 | |
| 0,003 | 15,46 | 154,58 | 1.545,77 | 15.457,70 | 154.577,01 | ||
| 0,0015 | 23,19 | 231,87 | 2.318,66 | 23.186,55 | 231.865,51 | ||
| 0,001 | 46,37 | 463,73 | 4.637,31 | 46.373,10 | 463.731,03 | ||
| 7,5 | 0,002 | 19,82 | 198,18 | 1.981,77 | 19.817,73 | 198.177,33 | |
| 0,001 | 39,64 | 396,35 | 3.963,55 | 39.635,47 | 396.354,66 | ||
| 0,0005 | 79,27 | 792,71 | 7.927,09 | 79.270,93 | 792.709,32 | ||
| 0,00025 | 158,54 | 1.585,42 | 15.854,19 | 158.541,86 | 1.585.418,63 | ||
| 8 | 0,001 | 33,88 | 338,77 | 3.387,68 | 33.876,77 | 338.767,72 | |
| 0,0005 | 67,75 | 677,54 | 6.775,35 | 67.753,54 | 677.535,44 | ||
| 0,00025 | 135,51 | 1.355,07 | 13.550,71 | 135.507,09 | 1.355.070,89 | ||
| 0,000125 | 271,01 | 2.710,14 | 27.101,42 | 271.014,18 | 2.710.141,77 | ||
| Volumen für Nachweisgrenze LOD (l) | 0,1 | ||||||
| Beispiel eines Prüfgefäßes mit Glasmantel zur Bestimmung von POW unter langsamem Rühren | Anlage 2 |
A.24. Verteilungskoeffizient (N-Octanol/Wasser), Hochleistungs-Flüssigkeitschromatographie (HPLC-Methode) 16
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 117 (2004).
1. Als Verteilungskoeffizient (P) bezeichnet man das Verhältnis der Gleichgewichtskonzentrationen eines gelösten Stoffs in einem Zweiphasensystem aus zwei weitgehend unmischbaren Lösungsmitteln. Für n-Octanol und Wasser gilt:
![]()
Der Verteilungskoeffizient (P) ist der Quotient zweier Konzentrationen. Er wird ohne Maßeinheit gewöhnlich in Form seines Zehnerlogarithmus angegeben.
2. Der Pow-Wert ist ein Schlüsselparameter in Studien zur Persistenz chemischer Stoffe in der Umwelt. Eine hoch signifikante Beziehung zwischen dem Pow-Wert nicht ionisierter Stoffe und ihrer Bioakkumulation in Fischen wurde nachgewiesen. Außerdem wurde nachgewiesen, dass der Pow-Wert ein hilfreicher Parameter zur Prognose der Adsorption im Boden und in Sedimenten und zur Feststellung quantitativer Struktur-Wirkungs- Beziehungen bei vielfältigen biologischen Wirkungen ist.
3. Der ursprüngliche Vorschlag für diese Prüfmethode beruhte auf einem Artikel von C. V. Eadsforth und P. Moser (1). 1986 hat das deutsche Umweltbundesamt die Entwicklung der Prüfmethode und einen OECD-Ringversuch koordiniert (2).
4. log Pow-Werte von - 2 bis 4 (gelegentlich bis zu 5 und höher) 1 können experimentell mit der Schüttelmethode bestimmt werden (Kapitel A.8 dieses Anhangs, OECD-Prüfrichtlinie 107). Mit der HPLC- Methode können log Pow-Werte von 0 bis 6 ermittelt werden (1)(2)(3)(4)(5). Bei dieser Methode muss unter Umständen der Pow-Wert geschätzt werden, um geeignete Referenzstoffe zuordnen und Schlussfolgerungen aus den in der Prüfung generierten Daten ziehen zu können. Die Berechnungsmethoden werden in der Anlage zu dieser Prüfmethode kurz erläutert. Die HPLC wird mit isokratischer Elution durchgeführt.
5. Die Pow-Werte hängen von den Umgebungsbedingungen (Temperatur, pH-Wert, Ionenkonzentration usw.) ab. Diese sollten im Versuch definiert werden, damit die Pow-Werte richtig interpretiert werden können. Für ionisierbare Stoffe könnte in Zukunft eine andere Methode zur Verfügung stehen (z.B. der Entwurf der OECD- Prüfrichtlinie zur pH-Messung ionisierter Stoffe (6)), die als alternative Methode angewandt werden sollte. Nach diesem Entwurf der OECD-Richtlinie kann zwar unter Umständen der Pow-Wert dieser ionisierbaren Stoffe ermittelt werden; manchmal ist jedoch eher die HPLC-Methode bei einem für die jeweiligen Umgebungsbedingungen relevanten pH-Wert zu empfehlen (siehe Nummer 9).
6. Analysen durch Umkehrphasen-HPLC werden mit Analysensäulen mit handelsüblichen Festphasen durchgeführt, die chemisch an Siliciumdioxid gebundene langkettige Kohlenwasserstoffe (z.B. C8 oder C18) enthalten.
7. Eine in eine solche Säule injizierte Chemikalie partitioniert zwischen der mobilen Lösungsmittelphase und der stationären Kohlenwasserstoffphase, während sie mit der mobilen Phase durch die Säule eluiert wird. Die Stoffe werden proportional entsprechend dem jeweiligen Kohlenwasserstoff/Wasser-Verteilungskoeffizienten abgetrennt; dabei werden zunächst die hydrophilen Stoffe und zuletzt die lipophilen Stoffe eluiert. Die Retentionszeit wird als Kapazitätsfaktor k ausgedrückt und mit folgender Formel bestimmt:
![]()
Dabei ist tR die Retentionszeit des Prüfstoffs und t0 die Totzeit, d. h. die Zeit, die ein Lösungsmittelmolekül durchschnittlich zum Wandern durch die Säule benötigt. Quantitative Analysemethoden sind nicht erforderlich, es müssen lediglich die Retentionszeiten bestimmt werden.
8. Der Octanol/Wasser-Verteilungskoeffizient eines Prüfstoffs kann berechnet werden, indem der Kapazitätsfaktor k des Prüfstoffs bestimmt und der so ermittelte Faktor k in die folgende Gleichung eingesetzt wird:
![]()
Dabei sind:
a, b = lineare Regressionskoeffizienten.
Die vorstehende Gleichung kann durch lineare Regression des Logarithmus der Octanol/Wasser-Verteilungskoeffizienten von Referenzsubstanzen gegen den Logarithmus der Kapazitätsfaktoren der Referenzsubstanzen ermittelt werden.
9. Durch Umkehrphasen-HPLC können Verteilungskoeffizienten im log Pow-Bereich von 0 bis 6 bestimmt werden; in Ausnahmefällen sind Berechnungen aber auch im log Pow-Bereich von 6 bis 10 möglich. Dazu muss unter Umständen allerdings die mobile Phase modifiziert werden (3). Für starke Säuren und Basen sowie für Metallkomplexe, Stoffe, die mit dem Elutionsmittel reagieren, und für grenzflächenaktive Stoffe ist diese Methode nicht geeignet. Messungen an ionisierbaren Stoffen können nur an deren nicht ionisierter Form (freie Säure oder freie Base) durch Verwendung eines geeigneten Puffers mit einem pH-Wert unter (freie Säure) bzw. über (freie Base) dem pKa-Wert durchgeführt werden. Alternativ könnte in Zukunft eine pH-Messmethode zur Prüfung ionisierbarer Stoffe zur Verfügung stehen, die als Alternativmethode (6) angewendet werden könnte. Wenn der log Pow-Wert für eine Einstufung oder eine Bewertung des Umweltrisikos ermittelt wird, ist die Prüfung bei einem für die jeweilige natürliche Umgebung typischen pH-Bereich vorzunehmen (d. h. im pH- Bereich von 5,0 bis 9).
10. Manchmal können Verunreinigungen die Interpretation der Ergebnisse erschweren, weil die Zuordnung der Peaks nicht eindeutig ist. Für Gemische, die ein nicht aufgelöstes Band ergeben, sollten jeweils die obere und die untere Grenze des log Pow-Peaks und die prozentuale Fläche jedes log Pow-Peaks angegeben werden. Für Gemische, die aus einer Gruppe von Homologen bestehen, sollte auch der gewichtete durchschnittliche log Pow- Wert angegeben werden (7); dieser ist ausgehend von den einzelnen Pow-Werten und den entsprechenden prozentualen Flächen zu berechnen (8). Alle Peaks, die mit einer Fläche von mindestens 5 % zur gesamten Peak-Fläche beitragen, sind in der Berechnung zu berücksichtigen (9):
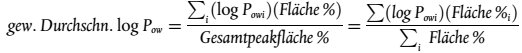
Der gewichtete durchschnittliche log Pow-Wert gilt nur für aus Homologen (z.B. einer Reihe von Alkanen) bestehende Stoffe oder Gemische (z.B. für Tallöle). Die Messung von Gemischen kann zu aussagekräftigen Ergebnissen führen, wenn die Empfindlichkeit des verwendeten analytischen Detektors bei allen in dem Gemisch enthaltenen Stoffen gleich ist und eine angemessene Auflösung erreicht werden kann.
11. Die Dissoziationskonstante, die Strukturformel und die Löslichkeit in der mobilen Phase sollten bekannt sein, bevor diese Methode angewendet wird. Außerdem sind Informationen zur Hydrolyse hilfreich.
12. Um die Zuverlässigkeit der Messung zu erhöhen, sind Doppelbestimmungen durchzuführen.
- Wiederholbarkeit: Der aus wiederholten Messungen unter identischen Bedingungen und mit derselben Gruppe von Referenzstoffen ermittelte log Pow-Wert sollte im Bereich von ± 0,1 log-Einheiten liegen.
- Reproduzierbarkeit: Wenn die Messungen mit einer anderen Reihe von Referenzstoffen wiederholt werden, können andere Ergebnisse ermittelt werden. In der Regel liegt der Korrelationskoeffizient R der Beziehung zwischen log k und log Pow bei einer Reihe von Prüfstoffen etwa bei 0,9; dies entspricht einem Octanol-/Wasser-Verteilungskoeffizienten von log Pow ± 0,5 log-Einheiten.
13. Der Ringversuch hat gezeigt, dass mit der HPLC-Methode log Pow-Werte im Bereich von ± 0,5 Einheiten der mit der Schüttelmethode bestimmten Werte ermittelt werden können (2). Weitere Vergleiche sind der Literatur zu entnehmen (4)(5)(10)(11)(12). Die höchste Genauigkeit wird mit Korrelationsdiagrammen strukturverwandter Referenzstoffe erzielt (13).
14. Um den gemessenen Kapazitätsfaktor k eines Stoffs mit seinem Pow zu korrelieren, muss eine Kalibrierkurve mit mindestens sechs Datenpunkten erstellt werden (siehe Nummer 24). Die Wahl der geeigneten Referenzstoffe obliegt dem Benutzer. Die log Pow-Werte der Referenzstoffe sollten in der Regel den log Pow-Wert des Prüfstoffs einschließen; d. h. mindestens ein Referenzstoff sollte einen Pow-Wert über dem des Prüfstoffs und ein anderer einen Pow-Wert unter dem des Prüfstoffs haben. Extrapolationen sollten nur ausnahmsweise vorgenommen werden. Die Referenzstoffe sollten vorzugsweise mit dem Prüfstoff strukturverwandt sein. Die log- Pow-Werte der für die Kalibrierung verwendeten Referenzstoffe müssen auf zuverlässigen Versuchsdaten beruhen. Bei Stoffen mit hohem log Pow (normalerweise über 4) können berechnete Werte verwendet werden, wenn keine zuverlässigen experimentellen Daten verfügbar sind. Wenn extrapolierte Werte verwendet werden, ist ein Grenzwert anzugeben.
15. Für viele Gruppen von Chemikalien liegen umfangreiche Listen mit log Pow-Werten vor (14)(15). Wenn keine Daten zu den Verteilungskoeffizienten strukturverwandter Stoffe verfügbar sind, kann eine mit sonstigen Referenzstoffen ermittelte allgemeinere Kalibrierung verwendet werden. In Tabelle 1 sind empfohlene Referenzstoffe und die jeweiligen Pow-Werte zusammengestellt. Bei ionisierbaren Stoffen beziehen sich die angegebenen Werte auf die nicht ionisierte Form. Im Ringversuch wurden die Werte einer Plausibilitäts- und Qualitätsprüfung unterzogen.
Tabelle 1: Empfohlene Referenzstoffe
| CAS-Nummer | Referenzstoff | log Pow | pKa | |
| 1 | 78-93-3 | 2-Butanon (Methylethylketon) | 0,3 | |
| 2 | 1122-54-9 | 4-Acetylpyridin | 0,5 | |
| 3 | 62-53-3 | Anilin | 0,9 | |
| 4 | 103-84-4 | Acetanilid | 1,0 | |
| 5 | 100-51-6 | Benzylalkohol | 1,1 | |
| 6 | 150-76-5 | 4-Methoxyphenol | 1,3 | pKa = 10,26 |
| 7 | 122-59-8 | Phenoxyessigsäure | 1,4 | pKa = 3,12 |
| 8 | 108-95-2 | Phenol | 1,5 | pKa = 9,92 |
| 9 | 51-28-5 | 2,4-Dinitrophenol | 1,5 | pKa = 3,96 |
| 10 | 100-47-0 | Benzonitril | 1,6 | |
| 11 | 140-29-4 | Phenylacetonitril | 1,6 | |
| 12 | 589-18-4 | 4-Methylbenzylalkohol | 1,6 | |
| 13 | 98-86-2 | Acetophenon | 1,7 | |
| 14 | 88-75-5 | 2-Nitrophenol | 1,8 | pKa = 7,17 |
| 15 | 121-92-6 | 3-Nitrobenzoesäure | 1,8 | pKa = 3,47 |
| 16 | 106-47-8 | 4-Chloranilin | 1,8 | pKa = 4,15 |
| 17 | 98-95-3 | Nitrobenzol | 1,9 | |
| 18 | 104-54-1 | Zinnamylalkohol (Zimtalkohol) | 1,9 | |
| 19 | 65-85-0 | Benzoesäure | 1,9 | pKa = 4,19 |
| 20 | 106-44-5 | p-Cresol | 1,9 | pKa = 10,17 |
| 21 | 140-10-3 (trans) | Zimtsäure | 2,1 | pKa = 3,89 (cis) 4,44 (trans) |
| 22 | 100-66-3 | Anisol | 2,1 | |
| 23 | 93-58-3 | Methylbenzoat | 2,1 | |
| 24 | 71-43-2 | Benzol | 2,1 | |
| 25 | 99-04-7 | 3-Methylbenzoesäure | 2,4 | pKa = 4,27 |
| 26 | 106-48-9 | 4-Chlorphenol | 2,4 | pKa = 9,1 |
| 27 | 79-01-6 | Trichlorethylen | 2,4 | |
| 28 | 1912-24-9 | Atrazin | 2,6 | |
| 29 | 93-89-0 | Ethylbenzoat | 2,6 | |
| 30 | 1194-65-6 | 2,6-Dichlorbenzonitril | 2,6 | |
| 31 | 535-80-8 | 3-Chlorbenzoesäure | 2,7 | pKa = 3,82 |
| 32 | 108-88-3 | Toluol | 2,7 | |
| 33 | 90-15-3 | 1-Naphthol | 2,7 | pKa = 9,34 |
| 34 | 608-27-5 | 2,3-Dichloranilin | 2,8 | |
| 35 | 108-90-7 | Chlorbenzol | 2,8 | |
| 36 | 1746-13-0 | Allyl-Phenylether | 2,9 | |
| 37 | 108-86-1 | Brombenzol | 3,0 | |
| 38 | 100-41-4 | Ethylbenzol | 3,2 | |
| 39 | 119-61-9 | Benzophenon | 3,2 | |
| 40 | 92-69-3 | 4-Phenylphenol | 3,2 | pKa = 9,54 |
| 41 | 89-83-8 | Thymol | 3,3 | |
| 42 | 106-46-7 | 1,4-Dichlorbenzol | 3,4 | |
| 43 | 122-39-4 | Diphenylamin | 3,4 | pKa = 0,79 |
| 44 | 91-20-3 | Naphthalin | 3,6 | |
| 45 | 93-99-2 | Phenylbenzoat | 3,6 | |
| 46 | 98-82-8 | Isopropylbenzol | 3,7 | |
| 47 | 88-06-2 | 2,4,6-Trichlorphenol | 3,7 | pKa = 6 |
| 48 | 92-52-4 | Biphenyl | 4,0 | |
| 49 | 120-51-4 | Benzylbenzoat | 4,0 | |
| 50 | 88-85-7 | 2,4-Dinitro-6-secbutylphenol | 4,1 | |
| 51 | 120-82-1 | 1,2,4-Trichlorbenzol | 4,2 | |
| 52 | 143-07-7 | Dodecansäure | 4,2 | pKa = 5,3 |
| 53 | 101-84-8 | Diphenylether | 4,2 | |
| 54 | 85-01-8 | Phenanthren | 4,5 | |
| 55 | 104-51-8 | n-Butylbenzol | 4,6 | |
| 56 | 103-29-7 | Dibenzyl | 4,8 | |
| 57 | 3558-69-8 | 2,6-Diphenylpyridin | 4,9 | |
| 58 | 206-44-0 | Fluoranthen | 5,1 | |
| 59 | 603-34-9 | Triphenylamin | 5,7 | |
| 60 | 50-29-3 | DDT | 6,5 |
Vorabschätzung des Verteilungskoeffizienten
16. Erforderlichenfalls kann der Verteilungskoeffizient der Prüfsubstanz geschätzt werden; die Schätzung sollte vorzugsweise mit einer Berechnungsmethode (siehe Anlage) bzw. gegebenenfalls auch aufgrund des Verhältnisses der Löslichkeit der Prüfsubstanz in den reinen Lösungsmitteln erfolgen.
17. Benötigt werden ein Flüssigphasenchromatograph mit einer Pumpe mit niedriger Förderleistung und ein geeignetes Detektionssystem. Angesichts der Vielzahl chemischer Gruppen kommen ein UV-Detektor mit einer Wellenlänge von 210 nm oder ein RI-Detektor in Betracht. Die Leistung der HPLC-Säule kann durch polare Gruppen in der stationären Phase erheblich beeinträchtigt werden. Deshalb sollten die stationären Phasen so wenig polare Gruppen wie möglich enthalten (16). Es können handelsübliche Mikroteilchenfüllungen für die Umkehrphasenchromatografie oder Fertigsäulen verwendet werden. Zwischen das Dosiersystem und die Analysensäule kann eine Vorsäule gesetzt werden.
18. Zur Zubereitung des Elutionsmittels werden für die HPLC-Methode ausreichend reines Methanol und destilliertes oder entionisiertes Wasser verwendet; das Elutionsmittel wird vor seiner Verwendung entgast. Es ist das Verfahren der isokratischen Elution anzuwenden. Dabei werden Methanol-Wasser-Verhältnisse mit einem Wassergehalt von mindestens 25 % empfohlen. Im Normalfall ist eine Methanol-Wasser-Mischung im Volumenverhältnis 3:1 für die Eluierung von Verbindungen mit einem log P-Wert von 6 bei einer Elutionszeit von einer Stunde und einer Durchflussrate von 1 ml/min ausreichend. Für Verbindungen mit einem log P-Wert von über 6 kann eine Verkürzung der Elutionszeit (auch der der Referenzstoffe) durch Senkung der Polarität der mobilen Phase oder Kürzung der Säulenlänge erforderlich sein.
19. Der Prüfstoff und die Referenzstoffe müssen in der mobilen Phase in ausreichender Konzentration löslich sein, um nachgewiesen werden zu können. Nur in Ausnahmefällen dürfen in der Methanol-Wasser-Mischung Zusatzstoffe verwendet werden, da diese die Eigenschaften der Säule verändern. In diesen Fällen muss sichergestellt werden, dass keine Auswirkungen auf die Retentionszeit der Prüfstoffe und der Referenzstoffe gegeben sind. Wenn die Methanol-Wasser-Mischung ungeeignet ist, können andere Mischungen aus einem organischen Lösungsmittel und Wasser verwendet werden, beispielsweise Ethanol-Wasser, Acetonitril-Wasser oder 2-Propanol-Wasser.
20. Der pH-Wert des Lösungsmittels ist für ionisierbare Stoffe kritisch. Er sollte innerhalb des pH-Bereichs der Säule liegen, der sich im Allgemeinen zwischen 2 und 8 bewegt. Die Anwendung eines Puffers ist zu empfehlen. Dabei muss darauf geachtet werden, dass kein Salz ausfällt und es nicht zur Beschädigung der Säule kommt, was bei einer Reihe von Mischungen von organischer Phase und Puffer möglich ist. HPLC-Messungen mit einer an Siliciumdioxid gebundenen stationären Phase und einem pH-Wert über 8 sind nicht empfehlenswert, da die Leistung der Säule bei Verwendung einer alkalischen mobilen Phase rapide nachlassen kann.
21. Der Prüfstoff und die Referenzstoffe müssen ausreichend rein sein, um die Peaks der Chromatogramme den jeweiligen Stoffen zuordnen zu können. Die für Prüf- oder Kalibrierungszwecke verwendeten Stoffe werden möglichst in der mobilen Phase gelöst. Wird für die Lösung des Prüfstoffs und der Referenzstoffe ein anderes Lösungsmittel als die mobile Phase verwendet, so ist die mobile Phase für die letzte Verdünnung vor der Injektion zu verwenden.
22. Die Temperatur darf während der Messungen um höchstens ± 1 °C schwanken.
23. Die Totzeit t0 lässt sich durch Verwendung nicht chromatografisch verzögerter organischer Verbindungen (z.B. Thioharnstoff oder Formamid) bestimmen. Ein genauerer Wert für die Totzeit ergibt sich aus den gemessenen Retentionszeiten oder aus etwa sieben Komponenten einer homologen Reihe (z.B. n-Alkyl-Methyl-Ketone) (17). Die Retentionszeiten tR (nC + 1) werden gegen tR (nC) aufgetragen; nC ist die Anzahl der Kohlenstoffatome. Es ergibt sich eine Gerade, tR (nC + 1) = A tR (nC) + (1 - A)t0; A ergibt sich aus k(nC + 1)/k(nC) und ist konstant. Die Totzeit t0 wird aus dem Schnittpunkt (1 - A) t0 und der Steigung A bestimmt.
24. Der nächste Schritt besteht in der Erstellung einer Korrelationskurve log k/log P für geeignete Referenzstoffe mit log P-Werten etwa im Bereich des für den Prüfstoff zu erwartenden Wertes. In der Praxis werden dazu 6 bis 10 Referenzstoffe gleichzeitig eingespritzt. Die Retentionszeiten werden am besten mithilfe eines mit dem Detektionssystem gekoppelten registrierenden Integrators bestimmt. Die Logarithmen der entsprechenden Kapazitätsfaktoren (log k) werden als Funktion von log P aufgezeichnet. Die Regressionsgleichung wird in regelmäßigen Abständen (mindestens einmal täglich) vorgenommen, damit eventuelle Leistungsveränderungen der Säule berücksichtigt werden können.
Bestimmung des Pow-Wertes des Prüfstoffs
25. Der Prüfstoff wird in den geringsten noch nachweisbaren Mengen eingespritzt. Die Retentionszeit wird doppelt bestimmt. Der Verteilungskoeffizient des Prüfstoffs kann aus dem berechneten Kapazitätsfaktor auf der Kalibrierkurve interpoliert werden. Bei sehr niedrigen und sehr hohen Verteilungskoeffizienten ist eine Extrapolation erforderlich. In diesen Fällen ist besonders auf die Vertrauensgrenzen der Regressionsgeraden zu achten. Wenn die Retentionszeit der Probe außerhalb des Bereichs der für die Standards ermittelten Retentionszeiten liegt, ist ein Grenzwert anzugeben.
26. Der Bericht muss folgende Angaben enthalten:
- wenn bestimmt, die Vorabschätzung des Verteilungskoeffizienten, die geschätzten Werte und die verwendete Methode; wenn eine Berechnungsmethode verwendet wurde, die vollständige Beschreibung der Methode einschließlich der Datenbasis und detaillierter Informationen zur Auswahl von Substrukturen;
- Prüf- und Referenzstoffe: Reinheit, Strukturformel und CAS-Nummer,
- Beschreibung der Ausrüstung und der Betriebsbedingungen: Analysensäule, Vorsäule
- mobile Phase, Detektionsverfahren, Temperaturbereich, pH-Wert;
- Elutionsprofile (Chromatogramme);
- Totzeit und entsprechendes Messverfahren;
- Retentionswerte und Pow-Werte aus der Literatur für zur Kalibrierung verwendete Referenzstoffe;
- nähere Angaben zur Anpassung der Regressionsgeraden (log k/log Pow) und zum Korrelationskoeffizienten der Geraden einschließlich Konfidenzintervallen;
- durchschnittliche Retentionswerte und interpolierter log Pow-Wert für den Prüfstoff;
- bei Mischungen: Elutionsprofil-Chromatogramm mit Abgrenzungswerten;
- log Pow-Werte im Verhältnis zur prozentualen Fläche des log Pow-Peaks;
- Berechnung mit einer Regressionsgeraden;
- gegebenenfalls berechnete durchschnittliche log Pow-Werte.
(1) C.V. Eadsforth und P. Moser. (1983). Assessment of Reverse Phase Chromatographic Methods for Determining Partition Coefficients. Chemosphere. 12, 1459.
(2) W. Klein, W. Kördel, M. Weiss und H.J. Poremski. (1988). Updating of the OECD Test Guideline 107 Partition Coefficient n-Octanol-Water, OECD Laboratory Intercomparison Test on the HPLC Method. Chemosphere. 17, 361.
(3) C.V. Eadsforth. (1986). Application of Reverse H.P.L.C. for the Determination of Partition Coefficient. Pesticide Science. 17, 311.
(4) H. Ellgehausen, C. D'Hondt und R. Fuerer (1981). Reversedphase chromatography as a general method for determining octan-1-ol/water partition coefficients. Pesticide. Science. 12, 219.
(5) B. McDuffie (1981). Estimation of Octanol Water Partition Coefficients for Organic Pollutants Using Reverse Phase High Pressure Liquid Chromatography. Chemosphere. 10, 73.
(6) OECD (2000). Guideline for Testing of Chemicals - Partition Coefficient (noctanol/water): pH-metric Method for Ionisable Substances. Draft Guideline, November 2000.
(7) OSPAR (1995). , Harmonised Offshore Chemicals Notification Format (HOCFN) 1995", Oslo and Paris Conventions for the Prevention of Marine Pollution Programmes and Measures Committee (PRAM), Annex 10, Oviedo, 20.-24. Februar 1995.
(8) M. Thatcher, M. Robinson, L. R. Henriquez und C. C. Karman. (1999). An User Guide for the Evaluation of Chemicals Used and Discharged Offshore, A CIN Revised CHARM III Report 1999. Version 1.0, 3. August.
(9) E. A. Vik, S. Bakke und K. Bansal. (1998). Partitioning of Chemicals. Important Factors in Exposure Assessment of Offshore Discharges. Environmental Modelling & Software Vol. 13, S. 529-537.
(10) L.O. Renberg, S.G. Sundstroem und K. Sundh-Nygård. (1980). Partition coefficients of organic chemicals derived from reversedphase thinlayer chromatography. Evaluation of methods and application on phosphate esters, polychlorinated paraffins and some PCB-substitutes. Chemosphere. 9, 683.
(11) W.E. Hammers, G.J. Meurs und C.L. De-Ligny. (1982). Correlations between liquid chromatographic capacity ratio data on Lichrosorb RP-18 and partition coefficients in the octanolwater system. J. Chromatography 247, 1.
(12) J.E. Haky und A.M. Young. (1984). Evaluation of a simple HPLC correlation method for the estimation of the octanolwater partition coefficients of organic compounds. J. Liq. Chromatography. 7, 675.
(13) S. Fujisawa und E. Masuhara. (1981). Determination of Partition Coefficients of Acrylates Methacrylates and Vinyl Monomers Using High Performance Liquid Chromatography. Journal of Biomedical Materials Research. 15, 787.
(14) C. Hansch und A. J. Leo. (1979). Substituent Constants for Correlation Analysis in Chemistry and Biology. John Willey, New York.
(15) C. Hansch, Vorsitz; A. J. Leo, Dir. (1982). Log P and Parameter Database: A tool for the quantitative prediction of bioactivity - Available from Pomona College Medical Chemistry Project, Pomona College, Claremont, California 91711.
(16) R. F. Rekker, H. M. de Kort. (1979). The hydrophobic fragmental constant: An extension to a 1.000 data point set. Eur. J. Med. Chem. - Chim. Ther. 14, 479.
(17) G.E. Berendsen, P.J. Schoenmakers, L. de Galan, G. Vigh, Z. Varga-Puchony, und J. Inczédy. (1980). On determination of holdup time in reversedphase liquid chromatography. J. Liq. Chromato. 3, 1669.
| Methoden zur Berechnung von Pow-Werten | Anlage |
1. Diese Anlage enthält eine kurze Einführung in die Berechnung von Pow-Werten. Weitere Informationen sind der Literatur zu entnehmen (1)(2).
2. Die berechneten Pow-Werte werden zu folgenden Zwecken verwendet:
- Auswahl der anzuwendenden Versuchsmethode: Schüttelmethode bei log Pow zwischen - 2 und 4 und HPLC-Methode bei log Pow zwischen 0 und 6;
- Festlegung der Bedingungen der HPLC (Referenzstoffe, Methanol-Wasser-Verhältnis);
- Plausibilitätsprüfung der durch Versuchsverfahren ermittelten Werte;
- Abgabe einer Einschätzung, wenn Versuchsverfahren nicht verwendet werden können.
Prinzip der Berechnungsverfahren
3. Die hier vorgeschlagenen Berechnungsverfahren beruhen auf der theoretischen Aufspaltung des Moleküls in geeignete Substrukturen, für die zuverlässige log Pow-Inkremente bekannt sind. Der log Pow-Wert wird als Summe seiner entsprechenden Teilwerte und der Korrekturglieder für intramolekulare Wechselwirkungen berechnet. Aufstellungen über Konstanten von Substrukturen und Korrekturglieder liegen vor (1)(2)(3)(4)(5)(6). Einige davon werden regelmäßig aktualisiert (3).
Zuverlässigkeit berechneter Werte
4. Im Allgemeinen nimmt die Zuverlässigkeit der Berechnungsverfahren in dem Maße ab, in dem die Komplexität des Prüfstoffs zunimmt. Bei einfachen Stoffen mit niedrigem Molekulargewicht und einer oder zwei funktionellen Gruppen ist mit einer Abweichung von 0,1 bis 0,3 log Pow-Einheiten von den Ergebnissen der verschiedenen Fragmentmethoden gegenüber den Messwerten zu rechnen. Die Fehlerspanne hängt von der Zuverlässigkeit der verwendeten Konstanten für die Substrukturen, der Fähigkeit der Erkennung intramolekularer Wechselwirkungen (z.B. Wasserstoffbindungen) und der richtigen Anwendung der Korrekturglieder ab. Bei ionisierbaren Stoffen ist die richtige Berücksichtigung der Ladung und des Ionisierungsgrades wichtig (10).
Fujita-Hansch'sche π-Methode
5. Die ursprünglich von Fujita et al. (7) für hydrophobe Substituenten eingeführte Konstante π wird wie folgt definiert:
![]()
wobei PhX ein aromatischer Abkömmling und PhH der Ausgangsstoff ist.

Die π-Methode ist vorwiegend bei aromatischen Stoffen von Bedeutung. π-Werte liegen für zahlreiche Substituenten vor (4)(5).
Rekker-Methode
6. Mit der Rekker-Methode (8) wird log Pow wie folgt berechnet:
![]()
wobei ai für die Häufigkeit steht, mit der eine bestimmte Substruktur im Molekül vorkommt, und fi das log Pow-Inkrement der Substruktur ist. Die Glieder für die Wechselwirkungen lassen sich als ein ganzes Vielfaches einer einzigen Konstante Cm (der so genannten magischen Konstante) angeben. Die Substrukturkonstanten fi und Cm wurden aus einer Liste von 1.054 experimentell ermittelten Pow-Werten (825 Verbindungen) mithilfe der mehrfachen Regressionsanalyse bestimmt (6)(8). Die Bestimmung der Glieder für die Wechselwirkungen erfolgt nach den in der Literatur angegebenen Regeln (6)(8)(9).
Hansch-Leo-Methode
7. Nach Hansch und Leo (4) wird der log Pow-Wert wie folgt berechnet:
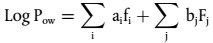
wobei fi eine Substrukturkonstante und Fj ein Korrekturglied (, Faktor") ist und ai und bj für die entsprechende Häufigkeit des Vorkommens stehen. Listen der Substrukturwerte für einzelne Atome und Gruppen und für Korrekturglieder Fj wurden durch die Trialand-Error-Methode aus experimentell bestimmten Pow-Werten abgeleitet. Die Korrekturglieder sind in unterschiedliche Kategorien eingeordnet worden (1)(4). Um sämtliche Regeln und Korrekturglieder zu berücksichtigen, wurden geeignete Software-Pakete entwickelt (3).
8. Die Berechnung der log Pow-Werte komplexer Moleküle kann beträchtlich verbessert werden, wenn das Molekül in größere Substrukturen zerlegt wird, für die zuverlässige log Pow-Werte vorliegen, sei es aus Tabellen (3) (4), sei es aus eigenen Messungen. Solche Substrukturen (z.B. Heterocyclen, Anthrachinon, Azobenzol) können dann mit den Hansch'schen π-Werten oder mit den Substrukturkonstanten nach Rekker oder Leo kombiniert werden.
Bemerkungen
- Die Berechnungsmethoden können auf teilweise oder vollständig ionisierte Stoffe nur dann angewendet werden, wenn die erforderlichen Korrekturfaktoren berücksichtigt werden.
- Wenn von intramolekularen Wasserstoffbindungen ausgegangen werden kann, müssen die entsprechenden Korrekturglieder (etwa + 0,6 bis + 1,0 log Pow-Einheiten) addiert werden (1). Hinweise auf das Vorliegen solcher Bindungen sind Stereo-Modellen oder spektroskopischen Daten des Stoffs zu entnehmen.
- Wenn mehrere tautomere Formen möglich sind, ist die wahrscheinlichste Form als Berechnungsgrundlage anzunehmen.
- Die Überarbeitungen der Listen der Substrukturkonstanten sind sorgfältig zu verfolgen.
Literatur zu Berechnungsmethoden
(1) W.J. Lyman, W.F. Reehl und D.H. Rosenblatt (Hrsg.). Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York (1982).
(2) W.J. Dunn, J.H. Block und R.S. Pearlman (Hrsg.). Partition Coefficient, Determination and Estimation, Pergamon Press, Elmsford (New York) und Oxford (1986).
(3) Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3).
(4) C. Hansch und A.J. Leo. Substituent Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York (1979).
(5) Leo, C. Hansch und D. Elkins. (1971) Partition coefficients and their uses. Chemical Reviews. 71, 525.
(6) R. F. Rekker, H. M. de Kort. (1979). The hydrophobic fragmental constant: An extension to a 1.000 data point set. Eur. J. Med. Chem. - Chim. Ther. 14, 479.
(7) Toshio Fujita, Junkichi Iwasa & Corwin Hansch (1964). A New Substituent Constant, À, Derived from Partition Coefficients. J. Amer. Chem. Soc. 86, 5175.
(8) R.F. Rekker. The Hydrophobic Fragmental Constant, Pharmacochemistry Library, Vol. 1, Elsevier, New York (1977).
(9) C.V. Eadsforth und P. Moser. (1983). Assessment of Reverse Phase Chromatographic Methods for Determining Partition Coefficients. Chemosphere. 12, 1459.
(10) R.A. Scherrer. ACS - Symposium Series 255, S. 225, American Chemical Society, Washington, D.C. (1984).
A.25 Dissoziationskonstanten in Wasser (Titrationsverfahren - Spektrofotometrisches Verfahren - Konduktometrisches Verfahren) 17
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 112 (1981).
Voraussetzungen
- Geeignete Analysemethode
- Wasserlöslichkeit
Leitlinien
- Strukturformel
- Elektrische Leitfähigkeit für konduktometrisches Verfahren
Einschränkende Aussagen
- Alle Prüfmethoden können an reinen Stoffen oder Stoffen handelsüblicher Qualität durchgeführt werden. Die potenziellen Auswirkungen von Verunreinigungen auf die Ergebnisse sind zu berücksichtigen.
- Das Titrationsverfahren ist für Stoffe mit geringer Löslichkeit nicht geeignet (siehe Prüflösungen unten).
- Das spektrofotometrische Verfahren ist nur bei Stoffen anwendbar, deren UV/VIS-Absorptionsspektren sich in der dissoziierten und undissoziierten Form jeweils deutlich unterscheiden. Dieses Verfahren eignet sich möglicherweise auch für Stoffe mit geringer Löslichkeit und nicht-saure/basische Dissoziationsformen (z.B. Komplexbildung).
- In Fällen, in denen die Onsagersche Gleichung anwendbar ist, kann das konduktometrische Verfahren angewendet werden, selbst bei mäßig niedrigen Konzentrationen und auch in Fällen von Nichtsäure-/Basen-Gleichgewichten.
Standarddokumente
Diese Prüfmethode beruht auf den Verfahren, auf die im Abschnitt "Literaturhinweise" verweisen wird, sowie auf dem Leitfaden der EPA "Preliminary Draft Guidance for Premanufacture Notification" vom 18. August 1978.
Methode - Einleitung, Zweck, Anwendungsbereich, Relevanz, Anwendung und Versuchsgrenzen
Die Dissoziation eines Stoffes in Wasser ist wichtig für die Beurteilung seiner Auswirkungen auf die Umwelt. Sie bestimmt die Form des Stoffes, die wiederum für das Verhalten und den Transport des Stoffes maßgeblich ist. Sie beeinflusst möglicherweise die Anlagerung der Chemikalie an Böden und Sedimente und ihr Eindringen in biologische Zellen (Absorption).
Definitionen und Einheiten
Unter Dissoziation versteht man den reversiblen Vorgang der Spaltung einer chemischen Spezies in zwei oder mehrere chemische Stoffe, die auch in ionischer Form vorliegen können. Dieser Vorgang wird im Allgemeinen wie folgt angegeben:
Die der Reaktion zugrunde liegende Gleichgewichtskonstante der Konzentration ist
K = [R+][X-] / [RX]
Handelt es sich bei R beispielsweise um Wasserstoff (eine Säure), ist die Konstante
Kα = [H+] ⋅ ([X-] / [HX])
oder
pKα = pH - log ([X-] / [HX])
Die folgenden Referenzstoffe müssen nicht in allen Fällen verwendet werden, in denen eine neue Substanz untersucht wird.
Sie werden hier vor allem angegeben, damit die Methode gelegentlich kalibriert werden kann und um die Ergebnisse mit denen anderer Methoden vergleichen zu können.
| pKa 1 | Temp. in °C | |
| p-Nitrophenol | 7,15 | 25 1 |
| Benzoesäure | 4,12 | 20 |
| p-Chloroanilin | 3,93 | 20 |
| 1) Für 20 °C ist kein Wert verfügbar, aber es kann davon ausgegangen werden, dass die Variabilität der Messergebnisse höher ist als die zu erwartende Temperaturabhängigkeit. | ||
Sinnvoll wäre ein Stoff mit mehreren pKs (siehe "Prinzip der Prüfmethode"), z.B.
| Zitronensäure | pKa (8) | Temp. in °C |
| 1) 3,14 | 20 | |
| 2) 4,77 | 20 | |
| 3) 6,39 | 20 |
Prinzip der Prüfmethode
Das beschriebene chemische Verfahren ist im umweltrelevanten Temperaturbereich in der Regel nur wenig temperaturabhängig. Zur Bestimmung der Dissoziationskonstante ist eine Messung der Konzentrationen des chemischen Stoffes in seiner dissoziierten und undissoziierten Form erforderlich. Aus dem stöchiometrischen Verhältnis der Dissoziationsreaktion (siehe "Definitionen und Einheiten") lässt sich die jeweilige Konstante bestimmen. In dem bei dieser Prüfmethode beschriebenen besonderen Fall verhält sich der Stoff wie eine Säure oder Base, und die Bestimmung erfolgt am besten mittels Bestimmung der relativen Konzentrationen der ionisierten und nichtionisierten Formen des Stoffes und des pH-Werts der Lösung. Welche Beziehung zwischen diesen Begriffen besteht, ist der Gleichung für pKa in "Definitionen und Einheiten" zu entnehmen. Für einige Stoffe ergeben sich mehrere Dissoziationskonstanten, und es lassen sich ähnliche Gleichungen aufstellen. Einige der hier beschriebenen Methoden eignen sich auch für nicht-saure/basische Dissoziationen.
Wiederholbarkeit
Die Dissoziationskonstante sollte mit einer Toleranz von ± 0,1 log-Einheiten repliziert werden (mindestens dreifache Bestimmung).
Beschreibung der Prüfverfahren
Zur Bestimmung des pKa-Wertes stehen zwei grundlegende Ansätze zur Verfügung - Titration einer bekannten Menge des Stoffes mit einer Standardsäure bzw. Standardbase und Bestimmung der relativen Konzentration der ionisierten und nichtionisierten Formen und ihrer pH-Abhängigkeit.
Die auf diesen Grundsätzen basierenden Methoden können als Titrationsverfahren, spektrofotometrische Verfahren und konduktometrische Verfahren klassifiziert werden.
Beim Titrier- und konduktometrischen Verfahren sollte die chemische Substanz in destilliertem Wasser gelöst werden. Beim spektrofotometrischen und bei anderen Verfahren werden Pufferlösungen verwendet. Die Konzentration des Prüfstoffs sollte den niedrigeren der beiden Werte - 0,01 M bzw. die Hälfte der Sättigungskonzentration - nicht überschreiten, und bei der Herstellung der Lösungen sollte die reinste verfügbare Form der Substanz verwendet werden. Sofern der Stoff nur schwer löslich ist, kann er in einer kleinen Menge wassermischbaren Lösungsmittels gelöst werden, bevor er den oben genannten Konzentrationen zugegeben wird.
Die Lösungen sollten mithilfe eines Tyndall-Strahls auf Emulsionen überprüft werden, insbesondere wenn ein Zusatzlösungsmittel zur Verbesserung der Löslichkeit verwendet wurde. Sofern Pufferlösungen verwendet werden, sollte die Konzentration 0,05 M nicht überschreiten.
Temperaturkonstanz sollte mit einer Toleranz von ± 1 °C gewährleistet sein. Die Bestimmung sollte am besten bei 201 °C erfolgen.
Wenn eine erhebliche Temperaturabhängigkeit vermutet wird, sollte die Bestimmung bei mindestens zwei weiteren Temperaturwerten erfolgen. Die Temperaturintervalle sollten im vorliegenden Fall 101 °C betragen, und Temperaturkonstanz sollte mit einer Toleranz von ± 0,11 °C gewährleistet sein.
Die Methode richtet sich nach der Art des Prüfstoffs. Er muss ausreichend empfindlich sein, damit eine Bestimmung der unterschiedlichen Spezies bei jeder Konzentration der Testlösung möglich ist.
Die Bestimmung der Prüflösung erfolgt durch Titration mit der Standardsäure- bzw. Standardbasenlösung, wobei nach jeder Zugabe des Titranten der pH-Wert gemessen wird. Vor Erreichen des Äquivalenzpunktes sollten mindestens 10 inkrementelle Zugaben erfolgen. Wenn ein Gleichgewicht relativ schnell erreicht wird, kann ein Kompensationschreiber (Potenziometer) verwendet werden. Für dieses Verfahren müssen sowohl die Gesamtmenge des Stoffes als auch seine Konzentration genau bekannt sein. Es müssen Vorkehrungen zum Ausschluss von Kohlendioxid getroffen werden. In den Standardtests sind Verfahren, Vorsichtsmaßnahmen und Berechnungsmethoden im Einzelnen beschrieben (vgl. Literaturhinweise (1), (2), (3), (4)).
Spektrofotometrisches Verfahren
Es wird eine Wellenlänge ermittelt, bei der sich die Extinktionskoeffizienten der ionisierten und nichtionisierten Formen des Stoffes deutlich unterscheiden. Das UV/VIS-Absorptionsspektrum wird aus Lösungen mit konstanter Konzentration bei einem pH-Wert, bei dem der Stoff im Wesentlichen nichtionisiert und bei dem sie vollständig ionisiert vorliegt, sowie bei mehreren pH-Zwischenwerten ermittelt. Dies geschieht entweder durch Zugabe von Inkrementen konzentrierter Säure (Base) zu einem relativ großen Volumen einer Lösung der Substanz in einem Mehrkomponentenpuffer bei einem anfangs hohen (niedrigen) pH-Wert (Literaturhinweis 5) oder durch Zugabe gleicher Mengen einer Stammlösung des Stoffes z.B. in Wasser oder Methanol zu konstanten Volumina verschiedener Pufferlösungen, die den gewünschten pH-Bereich abdecken. Aus den pH- und Absorptionswerten bei der gewählten Wellenlänge wird eine ausreichende Anzahl von Werten für den pKa-Wert berechnet; dabei werden Daten aus mindestens 5 pH-Bereichen verwendet, in denen die Substanz zu mindestens 10 Prozent und zu weniger als 90 Prozent ionisiert ist. Für weitere Einzelheiten zum Versuch und zur Berechnungsmethode siehe Literaturhinweis (1).
Anhand einer Zelle mit niedriger, bekannter Zellkonstante wird die Leitfähigkeit einer ca. 0,1 M-Lösung der Substanz in Leitfähigkeitswasser gemessen. Ferner werden die Leitfähigkeiten einiger akkurat hergestellter Verdünnungen dieser Lösung gemessen. Die Konzentration wird jedes Mal halbiert, und die Reihen sollten verschiedene Konzentrationen in abgestufter Reihenfolge umfassen. Die Grenzleitfähigkeit bei unendlicher Verdünnung lässt sich ermitteln, indem ein ähnlicher Versuch mit Na-Salz durchgeführt und extrapoliert wird. Der Dissoziationsgrad lässt sich sodann aus der Leitfähigkeit der jeweiligen Lösung nach der Onsagerschen Gleichung ermitteln, und die Dissoziationskonstante kann anschließend nach dem Ostwaldschen Verdünnungsgesetz als K = α2C/(1 - α) berechnet werden, wobei C der Konzentration in Mol pro Liter und α dem dissoziierten Teil entspricht. Es mÌssen Vorkehrungen zum Ausschluss von CO2 getroffen werden. Für weitere Einzelheiten zum Versuch und zur Berechnungsmethode siehe Standardtexte und Literaturhinweise (1), (6) und (7).
Der pKa-Wert wird für 10 Messpunkte auf der Titrationskurve gemessen. Es werden die Mittelwerte und Standardabweichungen dieser pKa-Werte errechnet. Es sollte eine Korrelationskurve pH-Wert/Volumen der Standardbase oder -säure in tabellarischer Form erstellt werden.
Spektrofotometrisches Verfahren
Das Absorptionsmaß und der pH-Wert werden für jedes Spektrum tabellarisch erfasst. Aus den Datenpunkten der Zwischenspektren werden mindestens fünf Werte für den pKa berechnet, ebenso wie die Mittelwerte und Standardabweichungen dieser Ergebnisse.
Die Äquivalentleitfähigkeit Λwird für jede Säurekonzentration und für jede Konzentration eines Gemisches aus einem Säureäquivalent plus einem 0,98 Äquivalent karbonatfreier Natronlauge ermittelt. Die Säure liegt im Überschuss vor, um einen hydrolysebedingten Überschuss an OH-zu vermeiden. 1/Λ wird gegen Ö_C aufgetragen, und Λo des Salzes lässt sich durch Extrapolation zur Nullkonzentration ermitteln.
Λo der Säure lässt sich anhand von Literaturwerten für H+ und Na+ berechnen. Der pKa lässt sich für jede Konzentration aus α = Λi /Λo und Ka = α2C/(1 - α) ermitteln. Man erhält bessere Werte für Ka, indem Korrekturen für Mobilität und Aktivität vorgenommen werden. Es sollten die Mittelwerte und Standardabweichungen dieser pKa-Werte berechnet werden.
Anzugeben sind sämtliche Rohdaten, die berechneten pKa-Werte und die Berechnungsmethode (am besten in tabellarischer Form, wie in Literaturhinweis (1) empfohlen), ebenso wie die oben beschriebenen statistischen Parameter. Bei Titrationsverfahren sind nähere Angaben zur Standardisierung der Titranten zu machen.
Beim spektrofotometrischen Verfahren sind alle Spektren anzugeben. Beim konduktometrischen Verfahren sollten nähere Angaben zur Bestimmung der Zellkonstante gemacht werden. Zudem sind Angaben zum angewandten Verfahren, zur Analysemethode und zur Art des verwendeten Puffers zu machen.
Die Prüftemperatur(en) sollte(n) festgehalten werden.
(1) Albert, A. & Sergeant, E.P., Ionization Constants of Acids and Bases, Wiley, Inc., New York, 1962.
(2) Nelson, N.H. & Faust, S.D., Acidic dissociation constants of selected aquatic herbicides, Env. Sci. Tech. 3, II, S. 1186-1188 (1969).
(3) ASTM D 1293 - Annual ASTM Standards, Philadelphia, 1974.
(4) Standard Method 242. APHA/AWWA/WPCF, Standard Methods for the Examination of Water and Waste Water, 14. Ausgabe, American Public Health Association, Washington, D.C., 1976.
(5) Clark, J. & Cunliffe, A.E., Rapid spectrophotometric measurement of ionisation constants in aqueous solution. Chem. Ind. (London) 281, (März 1973).
(6) ASTM D 1125 - Annual ASTM Standards, Philadelphia, 1974.
(7) Standard Method 205 - APHA/AWWA/NPCF (siehe (4)).
(8) Handbook of Chemistry and Physics, 60th ed. CRC-Press, Boca Raton, Florida, 33431 (1980).
| weiter . |  |
...
X
⍂
↑
↓