Bundesministerium für Gesundheit Bonn, den 31. Juli 2009
An den
Präsidenten des Bundesrates
Herrn Ministerpräsidenten
Peter Müller
Sehr geehrter Herr Präsident,
mit dem Gewebegesetz vom 20. Juli 2007, das am 1. August 2007 in Kraft getreten ist, sind in Umsetzung europäischen Rechts Qualitäts- und Sicherheitsstandards für menschliche Gewebe und Zellen im bestehenden Rechtsrahmen geregelt worden. In der Folge wurde auch die Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) mit der ersten Änderungsverordnung an das Gewebegesetz angepasst sowie das Transplantationsgesetz durch die Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung (TPG-GewV) konkretisiert.
Der Bundesrat hat anlässlich seiner Beschlüsse zum Gewebegesetz, zur ersten Änderungsverordnung zur AMWHV und zur TPG-GewV die Bundesregierung in Entschließungen gebeten, spätestens zwei Jahre nach Inkrafttreten des Gewebegesetzes über die dann vorliegenden Erfahrungen zu berichten - Bundesratsdrucksachen 385/07(B) 
 , 938/07(B) und 939/07(B) .
, 938/07(B) und 939/07(B) .
Als Anlage übersende ich Ihnen den erbetenen Erfahrungsbericht der Bundesregierung, der unter Mitwirkung der Länder, der Bundesressorts, der zuständigen Bundesoberbehörde sowie der Verbände und Fachgesellschaften erstellt wurde.
Mit freundlichen Grüßen
Ulla Schmidt
Erfahrungsbericht der Bundesregierung an den Bundesrat zum Gesetz über Qualität und Sicherheit von menschlichen Geweben und Zellen (Gewebegesetz)
Abkürzungsverzeichnis
| AMG | Arzneimittelgesetz |
| AMRadV | Verordnung über radioaktive oder mit ionisierenden Strahlen behandelte Arzneimittel |
| AMWHV | Arzneimittel- und Wirkstoffherstellungsverordnung |
| BVerwGE | Sammlung der wichtigsten Entscheidungen des Bundesverwaltungsgerichts |
| EG | Europäische Gemeinschaft |
| EGV | Vertrag zur Gründung der Europäischen Gemeinschaft |
| EMEA | Europäische Arzneimittelagentur |
| EU | Europäische Union |
| EWR | Europäischer Wirtschaftsraum |
| G-BA | Gemeinsamer Bundesausschusses |
| GKV | Gesetzliche Krankenversicherung |
| PEI | Paul-Ehrlich-Institut |
| TFG | Transfusionsgesetz |
| TPG | Transplantationsgesetz |
| TPG-GewV | Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung nach dem Transplantationsgesetz |
| ZLG | Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten |
Abbildungs- und Tabellenverzeichnis
| Abb. 3.2.1 | Übersicht zur Abgrenzung von bekannten Gewebezubereitungen zu anderen Arzneimitteln menschlicher oder tierischer Herkunft ( § 20c AMG) |
| Abb. 3.4.1 | Übersicht zu den Dokumentations- und Meldepflichten für Gewebe und Gewebezubereitungen ( § 63c AMG) |
| Abb. 3.6.1a | Übersicht zu den Erlaubnispflichten für die Gewinnung und die Be- oder Verarbeitung von Stammzellen des blutbildenden Systems (§§ 20b, 20c und 13 AMG) |
| Abb. 3.6.1b | Übersicht zur Genehmigungs- und Zulassungspflicht von Zubereitungen mit Stammzellen des blutbildenden Systems nach dem Arzneimittelgesetz (§§ 21a und 21 AMG) |
| Tab. 3.2.1 | Indizien für die Abgrenzung eines nicht industriellen Verfahrens zu einem industriellen Verfahren (§ § 20c AMG) |
| Tab. 3.3.2 | Antragssituation beim PEI zu Gewebezubereitungen und Blutstammzellzubereitungen mit Stand vom 30. Juni 2009 unter Berücksichtigung der Stichtage nach § 142 Absatz 2 AMG (§§ 21a und 21 AMG) |
1 Berichtsauftrag
Der Bundesrat hat am 6. Juli 2007 anlässlich seines Beschlusses zum Gesetz über Qualität und Sicherheit von menschlichen Geweben und Zellen (Gewebegesetz) eine Entschließung angenommen (Anlage 1). Darin begrüßt er, dass zum Gewebegesetz ein vertretbarer Kompromiss zustande gekommen sei.
Dieser beinhalte die Unterteilung in bekannte und neuartige Gewebe beziehungsweise Gewebezubereitungen mit der Folge, dass die Bestimmungen bezüglich der Be- und Verarbeitung von bekannten Geweben sowie deren Konservierung, Lagerung und Inverkehrbringen vereinfacht wurden. Die Bundesregierung wird aufgefordert, sobald als möglich, jedoch spätestens zwei Jahre nach Inkrafttreten des Gewebegesetzes, dem Bundesrat über die dann vorliegenden Erfahrungen zu berichten.
Am 15. Februar 2008 hat der Bundesrat ferner Entschließungen anlässlich seiner Beschlüsse zur Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) und zur Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung nach dem Transplantationsgesetz (TPG-GewV) angenommen (Anlagen 2 und 3). Danach soll der vorliegende Erfahrungsbericht ausdrücklich auch die Konsequenzen beinhalten, die sich durch die geänderte AMWHV sowie die TPG-GewV für die Gewebeeinrichtungen ergeben. Besonders berücksichtigt werden soll ob die getroffenen Regelungen zur Gewebeentnahme sowohl in der AMWHV als auch der TPG-GewV rechtlich notwendig, fachlich sinnvoll und praktisch umsetzbar sind.
2 Vorbereitung und Themen des Berichts
Zur Vorbereitung des Berichts wurden die Länder (Gesundheits- und Wissenschaftsbereich), das Paul-Ehrlich-Institut (PEI) als die zuständige Bundesoberbehörde, die Verbände und Fachgesellschaften sowie die Bundesressorts gebeten, ihre Erfahrungen und Einschätzungen hinsichtlich der neuen Regelungen nach dem Gewebegesetz (siehe im Einzelnen nachfolgend) zu schildern. Für die Berichterstattung gingen zahlreiche Stellungnahmen ein. Sie wurden berücksichtigt, soweit sie im Zusammenhang mit dem Berichtsauftrag standen.
Zur AMWHV wurde erfragt, ob sich die im Rahmen der 1. Änderungsverordnung zur AMWHV eingefügten Begriffsbestimmungen, insbesondere zur Gewebe- und Entnahmeeinrichtung, in der Praxis bewährt haben.
Es wurde um Einschätzung gebeten, wie sich die Sondervorschriften für Entnahme- und Gewebeeinrichtungen sowie für Gewebespenderlabore nach der AMWHV in der Praxis generell ausgewirkt haben und ob sich im Gesetzesvollzug oder bei der Überwachung besondere Probleme oder Schwierigkeiten ergeben hatten. Sofern ein Handlungsbedarf für eine Änderung der AMWHV im Hinblick auf die Entnahme-oder Gewebeeinrichtungen gesehen wurde, sollte dies ebenfalls mitgeteilt werden.
Zur TPG-GewV ist abfragt worden, ob sich die Regelungen insbesondere im Hinblick auf ihre Auswirkungen auf die Gewebeeinrichtungen in der Durchführung durch die zuständigen Behörden bewährt haben und ob Handlungsbedarf für Änderungen in der TPG-GewV gesehen wird.
Der Bericht enthält insbesondere Aussagen zu folgenden Themenkomplexen:
- - Erlaubnis für Entnahmeeinrichtungen und Labore für die Gewinnung von Gewebe und für die Laboruntersuchungen ( § 20b AMG)
- - Erlaubnis für die Be- oder Verarbeitung, Konservierung, Prüfung, Lagerung oder das Inverkehrbringen von Gewebe oder Gewebezubereitungen ( § 20c AMG), auch in Abgrenzung zur Herstellungserlaubnis nach § 13 AMG
- - Genehmigung von Gewebezubereitungen sowie Bescheinigung für das erstmalige Verbringen von Gewebezubereitungen in den Geltungsbereich des Arzneimittelgesetzes ( § 21a AMG), auch in Abgrenzung zur Zulassung nach § 21 AMG und zur zentralen Zulassung nach der Verordnung (EG) Nr. 1394/20071
- - besondere Dokumentations- und Meldepflichten bei Blut- und Gewebezubereitungen ( § 63c AMG)
- - Einfuhrerlaubnis und Qualitätszertifikate für Gewebe und bestimmte Gewebezubereitungen ( § 72b AMG)
- - Zubereitungen mit Stammzellen des blutbildenden Systems
- - besondere Bestimmungen der AMWHV für Gewebe- und Entnahmeeinrichtungen sowie Gewebespenderlabore und
- - besondere Bestimmungen der TPG-GewV für Gewebeeinrichtungen.
Aufgrund der zum Teil berichteten Schwierigkeiten beim Vollzug, aber auch der bisher noch begrenzten Erfahrungen mit den Vorschriften des Gewebegesetzes, der AMWHV und der TPG-GewV sowie aus Gründen der Vereinfachung und besseren Verständlichkeit wird den einzelnen Themenkomplexen zunächst eine Darstellung der jeweiligen Rechtslage vorangestellt. Diese beschränkt sich auf die grundlegenden Anforderungen und erhebt keinen Anspruch auf Vollständigkeit oder Erfassung aller Einzelfalle.
Daran schließt sich eine allgemeine Einschätzung der Bundesregierung sowie die Behandlung wesentlicher Einzelfragen zu den jeweiligen Stellungnahmen mit einer Bewertung durch die Bundesregierung an.
Bei den Einzelfragen werden insbesondere Aspekte erörtert, die in den Stellungnahmen eine besondere Rolle gespielt haben oder Anregungen für eine Fortentwicklung des Gewebegesetzes, der AMWHV oder der TPG-GewV enthalten.
Eine besondere Bedeutung nahmen in den Stellungnahmen Zubereitungen mit Stammzellen des blutbildenden Systems ein. Da sich die Stellungnahmen einerseits auf Zubereitungen aus dem Knochenmark und damit auf das Gewebegesetz, andererseits auf Zubereitungen der Blutstammzellen aus dem peripheren Blut und dem Nabelschnurblut und damit auf Regelungen des Arzneimittelgesetzes und des Transfusionsgesetzes bezogen und sich darüber hinaus auf alle im Erfahrungsbericht behandelten Themenkomplexe erstrecken werden diese als gesonderter Themenkomplex behandelt (siehe Abschnitt 3.6).
Der vorliegende Erfahrungsbericht berücksichtigt des Weiteren die Änderungen durch das Gesetz zur Änderung arzneimittelrechtlicher und anderer Vorschriften vom 17. Juli 2009 (BGBl. I S. 1990, nachfolgend 15. AMG-Novelle), soweit diese Gewebe und Gewebezubereitungen betrifft. Diese gehen auf den Beschluss der Task Force "Pharma" vom 22. Januar 2008 zum Zukunftsprojekt "Individualisierte Arzneimitteltherapie und Tissue Engineering" (Dritter Bericht der Task Force "Pharma" vom März 2009, 3. Handlungsempfehlung zum Tissue Engineering, S. 20) und einen Vorschlag des Bundesrates zur Schaffung einer Regelung für externe Prüfeinrichtungen für Gewebe oder Gewebezubereitungen (Beschluss des Bundesrates zur AMWHV, Bundesrats-Drucksache 938/07 (PDF) Nummer 2, vgl. Anlage 2) zurück.
Nicht berücksichtigt wurden Stellungnahmen zur Versorgung der Bevölkerung mit Geweben und Gewebezubereitungen in Deutschland. Diese sind Gegenstand des Berichts der Bundesregierung nach Artikel 7a des Gewebegesetzes.
Der vorliegende Bericht erstreckt sich grundsätzlich auf die Erfahrungen im Zeitraum seit Inkrafttreten des Gewebegesetzes am 1. August 2007 bis zum 31. Dezember 2008. Die Erfahrungen und statistischen Daten des PEI zu § 21a AMG konnten bis 30. Juni 2009 berücksichtigt werden.
Mit dem Gewebegesetz vom 20. Juli 2007 (BGBl. I S. 1574), das am 1. August 2007 in Kraft getreten ist, sind in Umsetzung europäischen Rechts2 Qualitäts- und Sicherheitsstandards für menschliche Gewebe und Zellen im bereits bestehenden Rechtsrahmen verankert worden, vor allem im Transplantationsgesetz (TPG), Arzneimittelgesetz (AMG) und Transfusionsgesetz (TFG).
Im Transplantationsgesetz betreffen diese Standards im Wesentlichen Aspekte der Erweiterung des Anwendungsbereichs auf Knochenmark, embryonale und fötale Organe und Gewebe und menschliche Zellen sowie der bereichsspezifischen Regelungen der Entnahme und Untersuchung sowie der Dokumentation und Rückverfolgung der Gewebe durch Gewebeeinrichtungen und Einrichtungen der medizinischen Versorgung. Im Arzneimittelgesetz sind insbesondere die Erlaubnis für die Gewinnung von Gewebe und die Laboruntersuchungen und für die Be- oder Verarbeitung von Gewebe oder Gewebezubereitungen, die Genehmigung von Gewebezubereitungen, besondere Dokumentations- und Meldepflichten für Gewebezubereitungen sowie die Einfuhrerlaubnis und Qualitätszertifikate für die Einfuhr von Gewebe und bestimmten Gewebezubereitungen aus Drittländern betroffen. Die Änderungen im Transfusionsgesetz betreffen insbesondere die Anforderungen an die Qualifikation der ärztlichen Person der Spendeeinrichtung sowie die Kompetenz der Bundesärztekammer zum Erlass von konkretisierenden Richtlinien zum Stand der Erkenntnisse der medizinischen Wissenschaft und Technik zur Gewinnung von Blut und Blutbestandteilen.
3.1 Erlaubnis für die Gewinnung von Gewebe und die Laboruntersuchungen ( § 20b AMG)
3.1.1 Rechtslage
Alle Einrichtungen, die menschliches Gewebe im Sinne von § 1a Nummer 4 TPG zur Verwendung bei Menschen gewinnen sowie die für die Gewinnung erforderlichen Laboruntersuchungen durchführen (nachfolgend Gewebespenderlabore), bedürfen unabhängig von der weiteren Be- oder Verarbeitung des Gewebes grundsätzlich einer Erlaubnis der zuständigen Behörde nach § 20b Absatz 1 AMG. Die Gewinnung umfasst neben der direkten Entnahme des Gewebes vom lebenden oder toten Spender auch extrakorporale Entnahmen wie z.B. die Entnahme von Herzklappen oder Leberzellen aus nicht vermittlungsfähigen Organen oder von Geweben aus Plazenta. Einrichtungen, die ausschließlich Gewebe gewinnen (z.B. Krankenhäuser) oder nur die für die Gewinnung erforderlichen Laboruntersuchungen durchführen, steht neben dem Erlaubnisverfahren nach § 20b Absatz 1 AMG das vereinfachte Erlaubnisverfahren nach § 20b Absatz 2 AMG zur Verfügung. Sie bedürfen danach keiner eigenen Erlaubnis nach § 20b Absatz 1 AMG, wenn sie mit einem Hersteller oder Be- oder Verarbeiter von Gewebe oder Gewebezubereitungen vertraglich kooperieren.
3.1.2 Allgemeine Einschätzung
Nach der überwiegenden Anzahl der eingereichten Stellungnahmen hat sich das vereinfachte Verfahren nach § 20b AMG, insbesondere das Verfahren nach § 20b Absatz 2 AMG, bewährt. Für Entnahmeeinrichtungen, aber auch für die zuständigen Behörden stellt das Verfahren eine erhebliche Vereinfachung dar.
Positiv bewertet wird zudem der in § 20b Absatz 1 Satz 4 AMG eingeräumte Ermessensspielraum der zuständigen Behörde für die Besichtigung der Entnahmeeinrichtung. Demgegenuber ist aus Sicht einzelner Fachgesellschaften das Erlaubnisverfahren nach § 20b AMG mit einem großen Aufwand beziehungsweise einem hohen Maß an Bürokratie und steigenden Kosten verbunden. Länder, Fachgesellschaften, Antragsteller sowie ein Verband berichten von einer Reihe noch bestehender Schwierigkeiten beim Vollzug des § 20b AMG und fordern eine Vereinheitlichung der Vollzugspraxis der zuständigen Behörden. Schwierigkeiten bestünden unter anderem bei der praktischen Durchführung des Verfahrens nach § 20b Absatz 2 AMG. Zudem erfolge die Erteilung der Erlaubnisse nach § 20b AMG mit zum Teil erheblichen zeitlichen Verzögerungen. Des Weiteren wird darauf hingewiesen, dass die vertragliche Ausgestaltung nach § 20b Absatz 2 AMG häufig schwierig und zeitaufwändig sei und insbesondere kleineren Einrichtungen Schwierigkeiten bereite. Die Bundesregierung vertritt die Auffassung, dass die Umsetzung neuer Vorschriften erfahrungsgemäß eine gewisse Anlaufzeit benötigt und es immer eines Erfahrungsaustausches der am Verfahren Beteiligten bedarf. Die von der Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) zwischenzeitlich erarbeitete Verfahrensanweisung zu §§ 20b, 20c und 72b AMG (einschließlich Checklisten, Antragsformulare)3 konnte bereits zu einer Vereinheitlichung der Vollzugspraxis beitragen und wird zukünftig auch zu einer Verkürzung der Bearbeitungszeit führen.
3.1.3 Einzelfragen
Aus den Stellungnahmen ergibt sich, dass in der Praxis Schwierigkeiten beim Vollzug des vereinfachten Verfahrens nach § 20b Absatz 2 AMG aufgetreten sind. Nach § 20b Absatz 2 AMG hat der Gesetzgeber das nachfolgend beschriebene Verfahren vorgesehen:
Der im Umgang mit Behörden zumeist erfahrene Hersteller oder Be- oder Verarbeiter, der bereits über eine Erlaubnis nach § 13 oder § 20c AMG verfügt, zeigt die mit ihm kooperierende Entnahmeeinrichtung oder das Gewebespenderlabor bei der für diese örtlich zuständigen Behörde an.
Die für die Entnahmeeinrichtung oder das Gewebespenderlabor zuständige Behörde prüft die personellen, räumlichen und technischen Anforderungen (§ 20b Absatz 1 Satz 3 Nummer 1 bis 4 AMG in Verbindung mit den Vorschriften von TPG, AMWHV, TPG-GewV) wie z.B. den Vertrag zwischen dem Hersteller oder Be- oder Verarbeiter und der Entnahmeeinrichtung oder dem Gewebespenderlabor, Entnahme-, Test- und Transportanweisungen und andere qualitätsrelevante Arbeitsanweisungen. Hierbei auftretende Fragen sind von der zuständigen Behörde unmittelbar mit dem Anzeigenden (z.B. .verantwortlicher Person") und nicht, wie sich aus den Stellungnahmen ergibt mit der Entnahmeeinrichtung zu klaren. Die zuständige Behörde kann der Anzeige innerhalb einer Frist von einem Monat widersprechen.
Sofern die zuständige Behörde innerhalb dieser Frist nicht widerspricht, erteilt sie gemäß § 20b Absatz 2 Satz 7 in Verbindung mit Absatz 1 Satz 5 AMG dem Hersteller oder Be- oder Verarbeiter die Erlaubnis nach § 20b AMG. Dies sollte, auch wenn es gesetzlich nicht ausdrücklich vorgesehen ist innerhalb der Frist von einem Monat erfolgen. Im Fall eines Widerspruchs der zuständigen Behörde (z.B. Mängelrüge) erfolgt eine weitere fachliche und rechtliche Prüfung durch die für die Entnahmeeinrichtung oder das Gewebespenderlabor zuständige Behörde. Nach Abschluss der Prüfung erteilt sie dem Hersteller oder Be- oder Verarbeiter die Erlaubnis oder versagt diese, wenn eine oder mehrere Voraussetzungen des § 20b Absatz 1 Satz 3 AMG vorliegen.
Der Hersteller oder Be- oder Verarbeiter hat die Entnahmeeinrichtung oder das Gewebespenderlabor nach Ablauf der Ein-Monats-Frist bei der für ihn zuständigen Behörde anzuzeigen. Diese Anzeige ist nach der gesetzgeberischen Intention lediglich deklaratorischer Natur. Eine Erweiterung der bereits bestehenden Erlaubnis des Herstellers nach § 13 AMG oder des Be- oder Verarbeiters nach § 20c AMG um die Entnahmeeinrichtung (Betriebsstätte) ist nach § 20b Absatz 2 AMG nicht vorgesehen (siehe auch § 14 Absatz 4 Nummer 4 AMG).
Eine erneute Prüfung der Entnahmeeinrichtung durch die für den Hersteller oder Be- oder Verarbeiter zuständige Behörde, wie sie in verschiedenen Stellungnahmen vorgetragen wurde, soll deshalb nicht stattfinden. Insoweit entspricht die in der Verfahrensanweisung der ZLG beschriebene Prüfung der Unterlagen im Verfahren nach § 20b Absatz 2 AMG (Abschnitt 3.4.2) nicht dem in § 20b Absatz 2 AMG vorgesehenen Verfahren. Die Bundesregierung regt deshalb eine Anpassung der Verfahrensanweisung der ZLG an die geltende Rechtslage an.
Vor dem Hintergrund der zum Teil erheblich verzögerten Erlaubniserteilung fordern verschiedene Antragsteller sowie eine Fachgesellschaft und ein Verband für § 20b Absatz 1 AMG eine Fristenregelung.
Vorgeschlagen werden verschiedene Fristenmodelle: Zum einen wird eine § 20c Absatz 5 AMG entsprechende Drei-Monats-Frist für die Erlaubniserteilung angeregt, zum anderen wird auf eine § 20b Absatz 2 AMG entsprechende Ein-Monats-Frist mit Verlängerungsmöglichkeit um weitere zwei Monate (auch im Sinne eines fiktiven Verwaltungsaktes) abgestellt. Daneben wird für § 20b Absatz 2 AMG eine unverzügliche Erlaubniserteilung nach Anzeigeneingang vorgeschlagen. Die Bundesregierung wird die verschiedenen Vorschläge für eine Fristenregelung prüfen und gegebenenfalls im Rahmen einer Gesetzesänderung berücksichtigen.
Aus verschiedenen Stellungnahmen ergibt sich, dass Verunsicherung besteht, wenn sich die für die Entnahmeeinrichtung oder das Gewebespenderlabor zuständige Behörde zum Antrag überhaupt nicht beziehungsweise nicht innerhalb der Ein-Monats-Frist äußert. Wille des Gesetzgebers war es, dass Entnahmeeinrichtungen und Gewebespenderlabore ihre Tätigkeit unmittelbar nach Ablauf der Ein-Monats-Frist beginnen können. Die Bundesregierung wird prüfen, ob diesem Anliegen gesetzlich noch besser nachgekommen werden kann.
Mehrere Länder halten darüber hinaus die Aufnahme einer Verpflichtung des Erlaubnisinhabers zur Änderungsanzeige in § 20b AMG für erforderlich, wie dies in § 20c Absatz 6 AMG oder auch in § 20 AMG für die Herstellungserlaubnis nach § 13 AMG vorgesehen ist. Die Bundesregierung halt die Forderung Für erwägenswert und wird eine Gesetzesänderung prüfen.
Mehrere Länder regen an, dass die für die Entnahmeeinrichtung oder das Gewebespenderlabor zuständige Behörde die für den Hersteller oder den Be- oder Verarbeiter zuständige Behörde über den Status der Anzeige (Erlaubniserteilung oder Widerspruch) informieren muss. Nach dem Wortlaut der Vorschrift wird die für den Hersteller oder den Be- oder Verarbeiter zuständige Behörde durch Anzeige des Herstellers oder des Be- oder Verarbeiters über den Abschluss des Verfahrens in Kenntnis gesetzt. Aus Sicht der Bundesregierung wird die ordnungsgemäße Durchführung des vereinfachten Verfahrens auf diese Weise hinreichend gewährleistet, so dass für eine ergänzende Regelung in § 20b Absatz 2 AMG kein Bedarf gesehen wird.
Ein Land regt an zu prüfen, ob eine Zuverlässigkeitsprüfung der .verantwortlichen Person" auch im Rahmen von §§ 20b und 20c AMG angezeigt ist (vgl. § 14 Absatz 1 Nummer 3 AMG). Die Richtlinie 2004/23/EG enthält eine solche Bestimmung nicht. Die Mitgliedstaaten können zwar gemäß Artikel 4 Absatz 2 der Richtlinie 2004/23/EG strengere Schutzmaßnahmen einführen. Aus Sicht der Bundesregierung besteht aber keine zwingende Notwendigkeit, über den Regelungsrahmen der Richtlinie hinauszugehen.
In einigen Stellungnahmen von Fachgesellschaften und Antragstellern wird zunächst allgemein eine weitere Vereinfachung und transparentere Gestaltung des Verfahrens gefordert. Konkret vorgeschlagen wird, klinisch tätige Ärztinnen und Ärzte sowie akkreditierte infektionsdiagnostische Labore von der Erlaubnis nach § 20b AMG freizustellen. Die Bundesregierung sieht vor dem Hintergrund von Artikel 5 der Richtlinie 2004/23/EG keinen Spielraum, die Anforderungen des § 20b AMG herabzusetzen.
Darüber hinaus wird von zwei Fachgesellschaften und einem Antragsteller thematisiert, dass Einrichtungen, die die für die Gewinnung von Gewebe erforderlichen Laboruntersuchungen durchführen, eine Erlaubnis nach § 20b AMG benötigen. Im Fall der parallelen Organ- und Gewebeentnahme mussten diese zusätzlich zu den für die Organspende nach dem Transplantationsgesetz notwendigen Untersuchungen durchgeführt werden, was zu Kostensteigerungen führe. Die Bundesregierung sieht auf Grund von Artikel 5 Absatz 2 der Richtlinie 2004/23/EG keine Möglichkeit, von einer Erlaubnis für die für die Gewinnung von Geweben erforderlichen Laboruntersuchungen nach § 20b AMG abzusehen. Labore, die die für die Gewinnung von Gewebe erforderlichen Laboruntersuchungen durchführen, bedürfen nach § 20b Absatz 1 AMG einer Erlaubnis der zuständigen Behörde. Als Alternative zu einer eigenen Erlaubnis können Labore auch mit einer Gewebe be- oder verarbeitenden Gewebeeinrichtung nach § 20b Absatz 2 AMG als deren Betriebsstätte kooperieren. In Deutschland verfügen genügend Labore über eine Erlaubnis nach § 13 AMG beziehungsweise nach § 20b AMG. Es ist zu erwarten, dass die Zahl der über eine entsprechende Erlaubnis nach § 20b AMG verfügenden Labore in Zukunft weiter zunehmen wird, so dass auch im Fall der parallelen Organ- und Gewebeentnahme die Anforderungen des Arzneimittelgesetzes erfüllt werden können. Es ist empfehlenswert, von vornherein mit einem Labor mit AMG-Erlaubnis zu kooperieren, wenn die Möglichkeit besteht, dass dem Organspender auch Gewebe entnommen wird, oder das Organ später auch als Ausgangsmaterial für die Be- oder Verarbeitung zu Gewebezubereitungen in Betracht kommt.
In Stellungnahmen mehrerer Länder und Fachgesellschaften wird eine unzureichende Regelung der Entnahme von Gewebe durch mobile Teams einer Entnahmeeinrichtung thematisiert. Die Bundesregierung vertritt die Auffassung, dass die Tätigkeit mobiler Teams von der Erlaubnis der sie jeweils entsendenden Entnahmeeinrichtung nach § 20b AMG umfasst sein kann. Dies folgt aus dem Wortlaut der Vorschrift und ihrer Entstehungsgeschichte (vgl. Beschlussempfehlung und Bericht des Ausschusses für Gesundheit, Bundestags-Drucksache 016/5443, S. 57). Der insoweit offen formulierte § 20b Absatz 1 Satz 1 AMG umfasst neben stationären Entnahmeeinrichtungen auch mobile Teams, d.h. von einer Entnahmeeinrichtung entsandtes Personal, das die Gewebeentnahme außerhalb der von der Erlaubnis nach § 20b AMG erfassten Raume durchführt (§ 34 Absatz 2 und 4 AMWHV). Dies entspricht der seit langem üblichen und allgemein anerkannten Praxis im Blutspendewesen (vgl. "Richtlinie für die Überwachung der Herstellung und des Verkehrs mit Blutzubereitungen", 1996) und gilt gleichermaßen für den Bereich der Gewebeentnahme.
Das mobile Team trägt die Verantwortung dafür, dass die Entnahmestelle geeignet ist, und sollte, wie bereits in verschiedenen Bundesländern praktiziert, die Entnahmestelle vorab der zuständigen Behörde anzeigen. Die Bundesregierung sieht insoweit keinen weiteren Handlungsbedarf.
In verschiedenen Stellungnahmen wird gefordert, das vereinfachte Verfahren nach § 20b Absatz 2 AMG
Für Antragsteller aus EU-Mitgliedstaaten/EWR-Vertragsstaaten zu öffnen. Dies wird zum Teil mit Hinweis auf Artikel 28 und 30 EGV begründet. Die Bundesregierung vertritt die Auffassung, dass dieses Verfahren Antragstellern aus EU-Mitgliedstaaten/EWR-Vertragsstaaten nicht zur Verfügung stehen kann. § 20b Absatz 2 AMG knüpft an eine Erlaubnis nach § 13 AMG oder § 20c AMG an, die nur Herstellern oder Be- oder Verarbeitern mit Sitz im Geltungsbereich des Arzneimittelgesetzes erteilt werden kann.
Antragsteller aus EU-Mitgliedstaaten/EWR-Vertragsstaaten können aber grundsätzlich einen Anspruch auf Erteilung einer Erlaubnis nach § 20b Absatz 1 AMG haben. Voraussetzung ist, dass der Antragsteller im Geltungsbereich des Arzneimittelgesetzes eine Entnahmeeinrichtung im Sinne von § 20b Absatz 1 AMG oder ein Gewebespenderlabor (vgl. § 2 Nummer 13 AMWHV) als eigene Einrichtung oder aufgrund vertraglicher Vereinbarung begründet und die dafür notwendigen Nachweise erbringt. Einen Sitz in der Bundesrepublik Deutschland muss der Antragsteller in diesem Fall nicht begründen (vgl. Urteil des EuGH vom 28. Februar 1984, Rs. C-247/81). Der Verfahrensaufwand für deutsche und in einem EU-Mitgliedstaat / EWR-Vertragsstaat ansässige Hersteller oder Be- oder Verarbeiter ist für Anträge nach § 20b Absatz 1 AMG und § 20b Absatz 2 AMG nahezu vergleichbar. Die Bundesregierung ist der Ansicht, dass die Anwendung von § 20b Absatz 1 AMG auf Hersteller in EU-Mitgliedstaaten/EWR-Vertragsstaaten keine unvereinbare Maßnahme gleicher Wirkung im Sinne von Artikel 28, 30 EG beziehungsweise von Artikel 11, 13 des Abkommens über den Europäischen Wirtschaftsraum darstellt.
Schlieslich regen mehrere Länder eine Verfahrensvereinfachung für die Entnahme einer geringen Menge autologen Blutes für die Herstellung biotechnologisch bearbeiteter Gewebeprodukte an beziehungsweise begrüßen die im Rahmen der 15. AMG-Novelle vorgesehene Gesetzesänderung in den § 13 Absatz 1a Nummer 2 und § 20b Absatz 4 AMG. Damit ist zukünftig für die Entnahme des autologen Gewebes und des autologen Blutes für die Herstellung biotechnologisch bearbeiteter Gewebeprodukte gleichermaßen eine Erlaubnis nach § 20b AMG erforderlich; die Erlaubnis nach § 13 AMG entfällt.
3.2 Erlaubnis für die Be- oder Verarbeitung, Konservierung, Prüfung, Lagerung oder das Inverkehrbringen von Gewebe oder Gewebezubereitungen ( § 20c AMG)
3.2.1 Rechtslage
Einrichtungen, die Gewebe oder Gewebezubereitungen im Sinne des § 20c AMG be- oder verarbeiten, konservieren prüfen, lagern oder in Verkehr bringen, bedürfen einer Erlaubnis der zuständigen Behörde nach dieser Vorschrift. § 20c AMG ist von § 13 AMG abzugrenzen. Nach den eingereichten Stellungnahmen liegen bislang nur wenige oder keine Erfahrungen vor. In den Stellungnahmen wird gleichwohl eine klarere Abgrenzung beider Vorschriften, eine Konkretisierung beziehungsweise gesetzliche Definition der maßgeblichen Begriffe gefordert.
Eine Abgrenzung lässt sich aus dem Arzneimittelgesetz und dessen Systematik, aus den Gesetzesmaterialien und den begrenzt vorliegenden Erfahrungen aus der Praxis vornehmen. § 20c AMG4 findet auf sogenannte bekannte (klassische) Gewebe und Gewebezubereitungen Anwendung (z.B. muskuloskeletale Gewebe, Augenhornhäute und Herzklappen). Davon sind bestimmte Gewebe abzugrenzen, die nicht von den Vorschriften des Arzneimittelgesetzes erfasst sind wie z.B. Gewebe, die innerhalb eines Behandlungsvorgangs einer Person entnommen und rückübertragen werden (z.B. Schadelkalotte oder Hauttransplantate), vgl. § 4a Absatz 1 Nummer 3 AMG.
Von den genannten Ausnahmen abgesehen findet das Arzneimittelgesetz auf Gewebe und Gewebezubereitungen Anwendung. Im Hinblick auf die in § 4 Absatz 30 Satz 2 AMG genannten Zellen und Gewebe gilt dies nur eingeschränkt (unter anderem §§ 20b, 20c und 64 ff. AMG). Hierunter fallen:
- - Samen- und Eizellen einschließlich imprägnierter Eizellen sowie
- - Hoden- und Nebenhodengewebe, solange diese Gewebe ausschließlich im Sinne der Reproduktionsmedizin zur Gewinnung von Samenzellen verwendet werden.
Von den bekannten Geweben und Gewebezubereitungen sind des Weiteren Arzneimittel und Gewebezubereitungen abzugrenzen die zwar von den Vorschriften des Arzneimittelgesetzes, nicht aber von der Vorschrift des § 20c AMG erfasst werden. Ausgangspunkt für die Abgrenzung ist die Begriffsbestimmung in § 4 Absatz 30 AMG. Danach sind Gewebezubereitungen Arzneimittel, die Gewebe im Sinne von § 1a Nummer 4 TPG sind oder aus solchen Geweben hergestellt worden sind, d.h. Gewebe und Zellen menschlichen Ursprungs (vgl. § 1 TPG sowie Artikel 2 Absatz 1 der Richtlinie 2004/23/EG). Keine Gewebezubereitungen im Sinne von § 4 Absatz 30 AMG sind somit Gewebezubereitungen tierischen Ursprungs (z.B. xenogene Arzneimittel, xenogene somatische Zelltherapeutika) sowie andere Arzneimittelzubereitungen aus Stoffen menschlicher Herkunft (z.B. Blutzubereitungen). Ihre Herstellung unterliegt der Erlaubnispflicht nach § 13 AMG. Gewebezubereitungen im Sinne des § 4 Absatz 30 AMG sind neben bekannten Gewebezubereitungen auch Arzneimittel für neuartige Therapien nach § 4 Absatz 9 AMG in Verbindung mit der Verordnung (EG) Nr. 1394/2007 (d.h. Gentherapeutika, somatische Zelltherapeutika und biotechnologisch bearbeitete Gewebeprodukte), soweit diese menschlichen Ursprungs sind. Für die Herstellung von Arzneimitteln für neuartige Therapien menschlichen Ursprungs bedarf es ebenfalls einer Erlaubnis nach § 13 AMG. Für die (verbleibenden) Gewebezubereitungen kommt es für die Anwendung des § 20c AMG in Abgrenzung zu § 13 AMG auf die Unterscheidung zwischen nicht industriellem und industriellem Verfahren (Arzneimittel im Sinne der Richtlinie 2001/83/EG5), dem Einsatz hinreichend bekannter oder vergleichbarer Be- oder Verarbeitungsverfahren und nicht hinreichend bekannter Be- oder Verarbeitungsverfahren an. Die nachfolgende Übersicht veranschaulicht die beschriebenen Abgrenzungen:
Abb. 3.2.1 Übersicht zur Abgrenzung von bekannten Gewebezubereitungen zu anderen Arzneimitteln menschlicher oder tierischer Herkunft ( § 20c AMG)
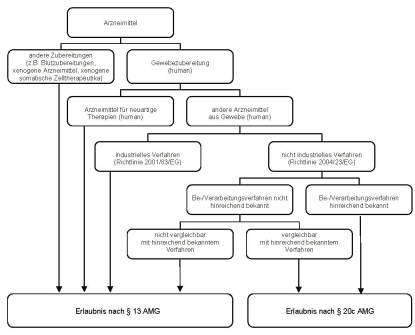
Für § 20c Absatz 1 AMG kommt es somit im Regelfall auf die Begriffe "nicht industrielles Verfahren" und "hinreichend bekanntes Be- oder Verarbeitungsverfahren" an. Die Begriffe sind gesetzlich nicht definiert, werden jedoch in den Gesetzesmaterialien (Beschlussempfehlung und Bericht des Ausschusses für Gesundheit, Bundestags-Drucksache 016/5443, S. 57) konkretisiert. Danach liegt eine Herstellung durch ein industrielles Verfahren vor, wenn "bei der Be- oder Verarbeitung von Gewebe anspruchsvolle technische oder aufwändige Verfahren eingesetzt" werden. Darüber hinaus enthält die Antwort der Bundesregierung auf die Kleine Anfrage der Abgeordneten Dr. Harald Terpe, Birgitt Bender, Elisabeth Scharfenberg, weiterer Abgeordneter und der Fraktion BÜNDNIS 90/DIE GRÜNEN (Bundestags-Drucksache 016/9988, S. 5) nahere Ausführungen zur Auslegung des Begriffes "industrielles Verfahren". Hiernach sind weitere Indizien die Herstellung in größerem Umfang oder auf Vorrat für einen nicht bekannten Abnehmerkreis.
Bekannte Gewebezubereitungen werden in der Regel in nicht industriellen Verfahren be- und verarbeitet. Auf die Einrichtung (z.B. Krankenhaus, pharmazeutisches Unternehmen), die die in § 20c AMG genannten Tätigkeiten durchführt, kommt es nach Ansicht der Bundesregierung bei der Abgrenzung von § 20c zu § 13 AMG nicht an. Ob ein .nicht industrielles Verfahren" vorliegt, ist vielmehr im konkreten Einzelfall auf der Grundlage der vorliegenden Indizien zu beurteilen. Im Einzelnen können folgende Indizien, auch in Kombination, für die Beurteilung herangezogen werden:
Tab. 3.2.1 Indizien für die Abgrenzung eines nicht industriellen Verfahrens zu einem industriellen Verfahren ( § 20c AMG)
| nicht industrielles Verfahren | industrielles Verfahren |
|---|
| Einsatz einfacher Verfahren | Einsatz automatisierter, anspruchsvoller technischer oder aufwändiger maschineller Verfahren |
| Einsatz einfacher Verfahrensschritte | Einsatz technologisch hochwertiger oder komplizierter Verfahrensschritte |
| Be- oder Verarbeitung in geringem Umfang | Be- oder Verarbeitung im größeren Umfang |
| Be- oder Verarbeitung erfolgt im Einzelfall für überschaubaren Abnehmerkreis | Be- oder Verarbeitung erfolgt auf Vorrat für einen nicht bekannten Abnehmerkreis |
Ein charakteristisches Beispiel für die Abgrenzung zwischen industriellem und nicht industriellem Verfahren ist die Be- oder Verarbeitung von Augenhornhaut, für die abhängig von der späteren Indikation zwei Herstellungsverfahren zur Verfügung stehen. Bei der Gewebezubereitung "Humane Augenhornhaut, organkultiviert"
Für den Ersatz der kompletten Augenhornhaut (perforierende Keratoplastik) kommt kein aufwändiges Be- oder Verarbeitungsverfahren zum Einsatz: Die Augenhornhaut wird nach der Entnahme zunächst mit Kochsalzlösung gespült und anschließend in ein Kulturmedium mit Antibiotika und Antimykotika eingebracht. Sowohl die Augenhornhaut als auch das Kulturmedium werden regelmäßig kontrolliert. In einem Brutschrank kann die Augenhornhaut bei etwa 34°C bis zu vier Wochen aufbewahrt werden. Die beschriebenen Be- oder Verarbeitungsschritte und die Lagerung unterliegen der Erlaubnis nach § 20c AMG. Im Gegensatz dazu werden bei der Gewebezubereitung "Lamellar präparierte organkultivierte humane Augenhornhaut" für den Ersatz von einer Schicht der Augenhornhaut (lamellare Keratoplastik) mittels eines neuartigen Mikrokeratoms endotheliale Lamellen hergestellt. Bei einem Mikrokeratom handelt es sich um ein äußerst präzises elektronisch gesteuertes Schneideinstrument zur Herstellung von Gewebescheiben definierter Dicke und Große. Dieses Verfahren wurde vom PEI als technisch aufwändig und somit als industrielles Verfahren eingestuft.
Ein hinreichend bekanntes Be- oder Verarbeitungsverfahren liegt nach den Materialien zum Gewebegesetz (Beschlussempfehlung und Bericht des Ausschusses für Gesundheit, Bundestags-Drucksache 016/5443, S. 57) vor, wenn "die Be- oder Verarbeitungsverfahren der Gewebe und Gewebezubereitungen bereits seit zehn Jahren oder länger in der Europäischen Union bekannt sind. Auch bei erst seit wenigen Jahren bekannten Verfahren kann ein hinreichender Bekanntheitsgrad erreicht sein, falls die Verfahren mit bekannten Verfahren vergleichbar sind oder das Gefährdungspotenzial möglicher Auswirkungen sicher einschätzbar ist." Erst wenn die wesentlichen Verfahrensschritte so neu sind, dass die Auswirkungen auf die Gewebezubereitung nicht hinreichend bekannt sind und das Gefährdungspotenzial möglicher Auswirkungen daher nicht einschätzbar ist, liegt kein hinreichender Bekanntheitsgrad vor. Danach lage im oben genannten Beispiel der Augenhornhaut ein hinreichend bekanntes Be- oder Verarbeitungsverfahren vor wenn die Herstellung der lamellaren Hornhaut mittels Skalpell während der Operation erfolgen wurde.
Ein neues Verfahren, das mit einem bekannten Verfahren vergleichbar ware, konnte z.B. bei der Herstellung mittels Trepan (Rundmesser zum kreisförmigen Ausschneiden der Augenhornhaut) vorliegen.
Ein weiteres charakteristisches Beispiel für ein nicht industrielles, hinreichend bekanntes Verfahren ist die Thermodesinfektion von Femurköpfen (Oberschenkelköpfen) im automatisierten Verfahren. Der im Rahmen einer endoprothetischen Hüftgelenkoperation zur Implantation eines künstlichen Gelenks entnommene Femurkopf wird zunächst entknorpelt und für maximal vier Wochen gefroren zwischengelagert. Die Thermodesinfektion zur Inaktivierung von HIV / Hepatitis-B- und Hepatitis-C-Erregern erfolgt entweder unmittelbar nach der Entnahme oder nach einer Zwischenlagerung. Zum Einsatz kommt ein automatisiertes Verfahren, das sogenannte Marburger Knochenbank-System zur thermischen Desinfektion allogener Hüftköpfe von Lebendspendern: Der Femurkopf wird dabei in einer Kochsalzlösung ohne weitere Zusatzstoffe (Ringer-Lösung) in einem geschlossenen Behältnis über einen bestimmten Zeitraum auf mindestens 82,5°C Kerntemperatur erhitzt und anschließend bei - -35°C bis zu zwei Jahren gefrierkonserviert.
Nach dem Auftauen werden Teile des Femurkopfes an den jeweiligen Defekt der Patientin oder des Patienten angepasst und transplantiert. Die Thermodesinfektion kommt seit mehr als zehn Jahren zum Einsatz und wurde vom PEI als nicht industrielles, hinreichend bekanntes Verfahren eingestuft.
Die Beurteilung des Bekanntheitsgrades eines Verfahrens ist ein dynamischer Prozess, der durch den aktuellen Stand von Wissenschaft und Technik bestimmt wird. Ein Be- oder Verarbeitungsverfahren, das heute als neu und damit nach § 13 AMG als erlaubnispflichtig eingestuft wird, kann sich nach einer entsprechenden Erfahrungszeit zu einem hinreichend bekannten Verfahren entwickeln. Zustandig für die Beurteilung der Be- und Verarbeitungsverfahren im Rahmen der §§ 20c und 13 AMG ist die jeweils zuständige Behörde. Sie wird dabei vom PEI unterstützt, das über das herzustellende Benehmen in das Erlaubnisverfahren eingebunden ist. Die Unterstützung durch das PEI wird von mehreren Ländern positiv bewertet. Nach Ansicht der Bundesregierung wurde eine gesetzliche Definition der verschiedenen unbestimmten Rechtsbegriffe des § 20c AMG dem schnellen wissenschaftlichen und technischen Fortschritt in diesem Bereich nicht hinreichend Rechnung tragen. Die fachliche Konkretisierung und praktikable Auslegung der unbestimmten Rechtsbegriffe in § 20c AMG sind aus Sicht der Bundesregierung durch das beschriebene Verfahren gewährleistet.
3.2.2 Allgemeine Einschätzung
Nach einem erheblichen Teil der zu § 20c AMG eingereichten Stellungnahmen liegen noch keine oder nur begrenzte Erfahrungen vor. Soweit bereits Erfahrungen vorliegen, wird das Verfahren überwiegend positiv bewertet.
Länder, Fachgesellschaften und Antragsteller berichten im Hinblick auf § 20c AMG zudem von Schwierigkeiten bei der Durchführung der Vorschriften und fordern eine bundesweite Angleichung der Verwaltungspraxis.
Mehrere Länder berichten von einem hohen Beratungsbedarf im Hinblick auf das in § 20c Absatz 2 Nummer 5 AMG in Verbindung mit der AMWHV geforderte Qualitätsmanagementsystem. Die Verfahrensanweisung der ZLG zu §§ 20b, 20c und 72b AMG (einschließlich Checklisten und Antragsformularen)6 hat zur Vereinheitlichung der Verwaltungspraxis beigetragen. Darüber hinaus ist das PEI bestrebt, durch ständigen Austausch mit Vertretern der Landesbehörden, durch Begleitung bei Besichtigungen und im Rahmen gemeinsamer Veranstaltungen eine weitere Angleichung der §§ 20c und 21a AMG zu erzielen. Die Bundesregierung unterstützt im Rahmen ihrer Möglichkeiten die Bestrebungen der Beteiligten zur Verfahrensvereinheitlichung.
3.2.3 Einzelfragen
In zwei Stellungnahmen wird thematisiert, dass für das Inverkehrbringen einerseits eine Erlaubnis nach § 20c AMG, andererseits eine Genehmigung nach § 21a AMG erforderlich sei, da beide Vorschriften an den Begriff des Inverkehrbringens anknüpfen. Aus Sicht der Bundesregierung besteht kein Klarstellungsbedarf.
§ 20c AMG regelt die Erlaubnispflicht für Personen (Einrichtungen), die bestimmte Tätigkeiten ausüben. Damit ist die Vorschrift mit den arzneimittelrechtlichen personenbezogenen Erlaubnissen für das Herstellen oder den Handel (Inverkehrbringen) vergleichbar. Bei solchen Erlaubnissen werden insbesondere die Anforderungen in Bezug auf die Eignung von Personen, Raumen und Einrichtungen sowie Herstellungs- oder Prüfverfahren geprüft. Die Vorschrift erfasst auch Falle, in denen eine Einrichtung Gewebe oder Gewebezubereitungen an andere abgibt als Variante des Inverkehrbringens, ohne diese selbst zu be- oder verarbeiten oder zu prüfen. Anknupfungspunkt für die Erlaubnis ist also eine Tätigkeit der Einrichtung in Bezug auf das Gewebe oder die Gewebezubereitung. Demgegenuber knüpft die Genehmigung nach § 21a AMG an die Gewebezubereitung als solche und damit an die Qualität, Unbedenklichkeit und Funktionalität des Arzneimittels an. Diese ist mit der produktbezogenen Zulassung nach § 21 AMG vergleichbar.
In mehreren Stellungnahmen wird angeregt, in § 20c AMG die Tätigkeit der Prüfung von Geweben und Gewebezubereitungen zu ergänzen. Eine entsprechende redaktionelle Anpassung ist mit der 15. AMGNovelle erfolgt.
Des Weiteren wird eine gesetzliche Regelung für den Transport von Geweben angeregt. Aus Sicht der Bundesregierung besteht kein Regelungsbedarf. Die Erlaubnis des § 20c AMG umfasst die Be- und Verarbeitung, Konservierung, Prüfung, Lagerung und das Inverkehrbringen. Aus Sicht der Bundesregierung ist hiervon der Transport durch die Gewebeeinrichtung begriffsnotwendig umfasst. Nahere Bestimmungen zur Durchführung des Transports enthalten zudem die §§ 7, 35 und 39 AMWHV.
Ein Land hat gebeten zu prüfen, ob eine Zuverlässigkeitsprüfung der verantwortlichen Person im Rahmen der §§ 20b und 20c AMG angezeigt ist (vgl. § 14 Absatz 1 Nummer 3 AMG). Die Richtlinie 2004/23/EG enthält eine solche Anforderung nicht. Zwar können die Mitgliedstaaten gemäß Artikel 4 Absatz 2 der Richtlinie 2004/23/EG strengere Schutzmaßnahmen einführen, aus Sicht der Bundesregierung besteht jedoch kein Grund, über den Regelungsrahmen der Richtlinie hinauszugehen.
Ein anderes Land halt es für erforderlich, die Verantwortlichkeiten eines Informationsbeauftragten auf die verantwortliche Person nach § 20c Absatz 2 AMG zu übertragen. Ein Informationsbeauftragter wird nach § 74a Absatz 1 Satz 1 AMG von einem pharmazeutischen Unternehmer, der Fertigarzneimittel in den Verkehr bringt, beauftragt und ist für die wissenschaftliche Information über die Arzneimittel zuständig.
In der Richtlinie 2004/23/EG ist dies für die Erlaubnis nach § 20c AMG nicht vorgesehen. Aus Sicht der Bundesregierung ist es nicht erforderlich, über den Regelungsrahmen der Richtlinie hinauszugehen.
Von einem Verband wird die Vorsorgeverpflichtung des Be- oder Verarbeiters nach § 20c Absatz 7 Satz 4 und 5 AMG für den Fall, dass er die Be- oder Verarbeitung von Geweben oder Gewebezubereitungen einstellt beanstandet. Die Bundesregierung verweist auf die entsprechende Vorgabe in Artikel 21 Absatz 5 der Richtlinie 2004/23/EG.
Des Weiteren regt ein Land eine Klarstellung des § 64 Absatz 3 Satz 3 AMG an, wonach vor Erteilung einer Erlaubnis nach § 20c AMG verpflichtend eine Besichtigung durch die zuständige Behörde durchzuführen ist. Eine entsprechende Klarstellung ist im Rahmen der 15. AMG-Novelle erfolgt. Nach Erteilung der Erlaubnis sind Gewebeeinrichtungen in der Regel alle zwei Jahre zu besichtigen (vgl. § 64 Absatz 3 Satz 2 AMG).
In zwei Stellungnahmen wird angeregt, die Umwandlung einer Erlaubnis nach § 13 AMG in eine Erlaubnis nach § 20c AMG zu ermöglichen. Dem Inhaber einer Erlaubnis nach § 13 AMG steht es offen, eine Erlaubnis nach § 20c AMG zu beantragen. Die zuständige Behörde wird nach den vorgelegten Unterlagen prüfen ob die Voraussetzungen nach § 20c Absatz 1 AMG (nicht industrielles Verfahren, hinreichend bekanntes Be- oder Verarbeitungsverfahren etc.) und § 20c Absatz 2 AMG in Verbindung mit der AMWHV vorliegen und entsprechend bescheiden. Ein Regelungsbedarf besteht aus Sicht der Bundesregierung nicht.
In einer weiteren Stellungnahme wird um Klarstellung gebeten, dass für Forschungszwecke be- oder verarbeitetes Gewebe, das nicht im oder am Menschen angewendet wird, nicht der Erlaubnis nach § 20c AMG unterliegt. Nach Ansicht der Bundesregierung ist eine solche Klarstellung nicht erforderlich. § 2 Absatz 1 und § 4 Absatz 30 AMG stellen hinreichend klar, dass der Anwendungsbereich des Arzneimittelgesetzes nur dann eröffnet ist, wenn die Gewebezubereitungen zur Anwendung im oder am menschlichen Körper bestimmt sind. Zudem wird auf Erwägungsgrund (11) der Richtlinie 2004/23/EG verwiesen wonach die Richtlinie nicht für die forschungsbedingte Nutzung menschlicher Gewebe und Zellen gilt, es sei denn, die Gewebe und Zellen werden in klinischen Versuchen im oder am menschlichen Körper eingesetzt. Dem trägt das Arzneimittelgesetz Rechnung.
Aus der Praxis wird zudem berichtet, dass die Einbeziehung von reproduktionsmedizinischen Einrichtungen in den Geltungsbereich des Gewebegesetzes wenig Akzeptanz findet. Die Bundesregierung sieht keinen Handlungsspielraum, Keimzellen im Sinne des § 4 Absatz 30 AMG von einer gesetzlichen Regelung auszunehmen. Nach Artikel 2 Absatz 1 der Richtlinie 2004/23/EG ist die Richtlinie auf menschliche Zellen und Geweben anzuwenden, was nach Erwägungsgrund (7) der Richtlinie auch Keimzellen (Eizellen, Samenzellen) einschließt.
Anlässlich seiner Entschließung zur AMWHV hatte der Bundesrat die Bundesregierung gebeten zu klaren, wie externe Prüfeinrichtungen, die z.B. mikrobiologische Prüfungen von Geweben oder Gewebezubereitungen vornehmen hinsichtlich ihrer Erlaubnispflicht behandelt werden. Im Rahmen der 15. AMGNovelle wurde in Anlehnung an § 14 Absatz 4 AMG eine Vorschrift für externe Prüfeinrichtungen in § 20c Absatz 2 Satz 2 AMG vorgeschlagen, was in mehreren Stellungnahmen begrüßt wurde. Ein Land weist in diesem Zusammenhang darauf hin, dass auch Fallkonstellationen berücksichtigt werden sollten, bei denen sich die externe Prüfeinrichtung nicht im Zuständigkeitsbereich der für die Erlaubnis nach § 20c AMG zuständigen Behörde befindet. Nach § 20c Absatz 4 Satz 3 AMG wird die Erlaubnis für eine bestimmte Betriebsstätte erteilt, im Falle des § 20c Absatz 2 Satz 2 AMG auch für eine bestimmte Betriebsstätte des beauftragten oder des anderen Betriebes (zur vergleichbaren Regelung in § 14 Absatz 4 AMG siehe dementsprechend § 16 AMG). Im Übrigen verweist die Bundesregierung auf die Zuständigkeit der Länder Für die verfahrensrechtliche und organisatorische Ausgestaltung.
3.3 Genehmigung von Gewebezubereitungen ( § 21a AMG)
3.3.1 Rechtslage
Bis zum Inkrafttreten des Gewebegesetzes war für das Inverkehrbringen von Gewebezubereitungen grundsätzlich eine Zulassung nach § 21 AMG erforderlich. Mit dem Gewebegesetz wurde für das Inverkehrbringen von bekannten Gewebezubereitungen ein gesondertes Genehmigungsverfahren in § 21a AMG eingeführt, das gegenüber dem Zulassungsverfahren nach § 21 AMG vereinfacht ist.
Zur Abgrenzung des vereinfachten Genehmigungsverfahrens gegenüber dem Zulassungsverfahren nach § 21 AMG kommt es - wie auch bei § 20c AMG - auf die Unterscheidung zwischen nicht industriellem und industriellem Verfahren, dem Einsatz hinreichend bekannter oder vergleichbarer Be- oder Verarbeitungsverfahren und nicht hinreichend bekannter (neuer) Be- oder Verarbeitungsverfahren an. Auf die Erläuterungen zu § 20c AMG wird verwiesen (siehe Abschnitt 3.2.1). Für die Abgrenzung ist nach der Gesetzesbegründung zu § 21a AMG maßgeblich, dass die wesentlichen Be- oder Verarbeitungsschritte und -verfahren bekannt sind. Unwesentliche Neuerungen wie z.B. die Verwendung eines anderen Hilfsstoffes oder Stabilisators führen nicht zu einer Neuartigkeit der Be- oder Verarbeitungsverfahren und damit auch nicht zu einer Zulassungspflicht nach § 21 AMG. Erst wenn die wesentlichen Be- oder Verarbeitungsschritte so neuartig sind, dass die Auswirkungen auf die Gewebezubereitung und ihre Anwendung im oder am Menschen nicht hinreichend bekannt sind, muss aus Sicherheitsgründen ein Zulassungsverfahren nach § 21 AMG für das Inverkehrbringen beantragt werden (Beschlussempfehlung und Bericht des Ausschusses Für Gesundheit, Bundestags-Drucksache 016/5443, S. 58).
Vom Zulassungsverfahren nach § 21 AMG unterscheidet sich das Genehmigungsverfahren nach § 21a AMG dadurch, dass die Genehmigungsbehörde neben der Prüfung des Arzneimittels als solches auf Funktionalität, Unbedenklichkeit und Qualität insbesondere die Be- oder Verarbeitungsverfahren bewertet, mit denen die Gewebezubereitung hergestellt wird. Diese müssen so gestaltet sein, dass die Gewebezubereitung ihre Funktion erfüllt und nicht schädlich für die Patientin oder den Patienten ist. Nach § 21a Absatz 1 Satz 1 AMG müssen sich die Wirkungen und Nebenwirkungen der Gewebezubereitung aus dem wissenschaftlichen Erkenntnismaterial ergeben. Für den Nachweis der Anwendungsgebiete sowie der Art und Dauer der Anwendung können Unterlagen aus ärztlichem Erfahrungsmaterial oder eigenen Studien oder Veröffentlichungen ausreichen (§ 21a Absatz 3 AMG). Von umfangreichen pharmakologischtoxikologischen und klinischen Studien kann regelmäßig abgesehen werden (Beschlussempfehlung und Bericht des Ausschusses für Gesundheit, Bundestags-Drucksache 016/5443, S. 58).
Auf bestrahlte Gewebezubereitungen findet hingegen aufgrund § 7 Absatz 1 AMG in Verbindung mit der Verordnung über radioaktive oder mit ionisierenden Strahlen behandelte Arzneimittel (AMRadV) das Zulassungsverfahren nach § 21 AMG Anwendung;
Ausnahmen davon werden in § 1 AMRadV definiert.
Grundsätzlich ist es nach § 7 Absatz 1 AMG verboten, Arzneimittel in den Verkehr zu bringen, bei deren Herstellung ionisierende Strahlen (Elektronen-, Gamma oder Röntgenstrahlen) verwendet worden sind.
Das Verkehrsverbot des § 7 Absatz 1 AMG gilt aufgrund der AMRadV jedoch nicht für Arzneimittel, bei deren Herstellung diese Strahlen zur Verminderung der Keimzahl oder zur Inaktivierung von Blutbestandteilen oder Tumormaterial oder zur Modifizierung von Bestandteilen verwendet worden sind, sofern die Arzneimittel durch das PEI im Hinblick auf die Behandlung mit ionisierenden Strahlen zur Verminderung der Keimzahl oder zur Inaktivierung von Blutbestandteilen oder Tumormaterial oder zur Modifizierung von Bestandteilen nach § 25 Absatz 1 AMG zugelassen worden sind oder nach § 21 Absatz 2 Nummer 1a, 1b, 1c, 2, 5 oder 6 AMG ohne Zulassung in den Verkehr gebracht werden dürfen (siehe § 1 Absatz 2 Nummer 4 AMRadV). Daraus folgt, dass bestrahlte Gewebezubereitungen, auch wenn es sich um hinreichend bekannte Arzneimittel handelt, einer Zulassung nach § 21 AMG bedürfen, wenn sie in Verkehr gebracht werden sollen.
Soll eine bekannte Gewebezubereitung aus einem EU-Mitgliedstaat/EWR-Vertragsstaat in den Geltungsbereich des Arzneimittelgesetzes erstmalig verbracht werden, ist eine Bescheinigung nach § 21a Absatz 9 AMG erforderlich. Die Bescheinigung soll sicherstellen, dass nur gleichwertige Produkte in den Geltungsbereich des Arzneimittelgesetzes verbracht werden. Das Bescheinigungsverfahren ist im Vergleich zur Einfuhrerlaubnis nach § 72b AMG vereinfacht und auf die erstmalige Einfuhr beschränkt. Eine Genehmigung nach § 21a Absatz 1 AMG muss aber beantragt werden, wenn eine Gleichwertigkeit der Anforderungen in den EU-Mitgliedstaaten/EWR-Vertragsstaaten verneint wird oder die Richtlinie 2004/23/EG und ihre Durchführungsrichtlinien in dem EU-Mitgliedstaat/EWR-Vertragsstaat noch nicht umgesetzt wurden.
Für bekannte Gewebezubereitungen, die aus einem Drittland eingeführt und im Geltungsbereich des Arzneimittelgesetzes in Verkehr gebracht werden sollen, ist eine Genehmigung für das Inverkehrbringen nach § 21a Absatz 1 AMG des PEI und eine Einfuhrerlaubnis nach § 72b Absatz 1 AMG der zuständigen Behörde erforderlich (siehe dazu Abschnitt 3.5).
3.3.2 Allgemeine Einschätzung
Nach der überwiegenden Anzahl der eingereichten Stellungnahmen konnte zu § 21a AMG aufgrund bisher fehlender Erfahrungen (noch) keine Bewertung abgegeben werden. Soweit bereits Erfahrungen in der Praxis gewonnen werden konnten, wird § 21a AMG überwiegend positiv bewertet. Betont wird, dass die Regelungen im Arzneimittelgesetz, auch wenn sie personal- und zeitintensive Auswirkungen haben, zu einer signifikanten Verbesserung der Qualität von Gewebezubereitungen führen werden. Aus Sicht der Forschung wird die Vereinheitlichung von Qualitätsstandards auf hohem Niveau auch unter dem Gesichtspunkt einer möglichen Akquirierung zusätzlicher finanzieller Mittel positiv eingeschätzt.
Nach Ansicht des PEI haben sich die Vorschriften und Anforderungen an das Genehmigungsverfahren nach § 21a AMG für bekannte Gewebezubereitungen als vorteilhaft gegenüber dem kostenintensiveren und aufwändigeren Zulassungsverfahren nach § 21 AMG erwiesen. Allerdings ist der Berichtszeitraum für eine aussagekräftige Einschätzung des Genehmigungsverfahrens zu kurz und stellt damit nur eine vorläufige Bestandsaufnahme dar.
Das Genehmigungsverfahren nach § 21a AMG hat alle Beteiligten vor neue Anforderungen gestellt. Zu fachlichen und rechtlichen Fragestellungen des § 21a AMG steht das PEI den Antragstellern und zuständigen Behörden beratend zur Seite. Auf der Homepage des PEI wurden umfangreiche Informationen einschließlich Antragsformulare eingestellt und spezielle Service-Telefonnummern und E-Mail-Adressen eingerichtet. Darüber hinaus hat das PEI in einer Vielzahl von Fachveranstaltungen über das Genehmigungsverfahren nach § 21a AMG informiert.
Zu dem in § 142 Absatz 2 AMG für die Genehmigung nach § 21a AMG festgelegten Stichtag (1. Februar 2008) gingen beim PEI 560 Anträge ein (341 für Gewebezubereitungen und 219 für Blutstammzellzubereitungen).
Diese enthielten zum Teil Sammelanträge oder wurden zunächst vorsorglich zur Fristwahrung gestellt und später teilweise wieder zurückgenommen. Nach Prüfung und Rücksprache mit den Antragstellern reduzierte sich die Zahl der Anträge auf 374 (181 für Gewebezubereitungen und 193 für Blutstammzellzubereitungen).
Bis auf wenige Ausnahmen enthielten die bis zum Stichtag eingereichten Anträge nach § 21a AMG nicht die für eine Beurteilung ausreichenden Unterlagen, insbesondere waren die Unterlagen zum Nachweis der Qualität, Sicherheit und Funktionalität der Gewebezubereitungen unzureichend.
Das PEI unterstützte die Antragsteller im Rahmen seiner Möglichkeiten beratend und durch organisatorische Maßnahmen. Durch eine Gruppierung der bekannten Gewebezubereitungen und die Bereitstellung von Musteranträgen (z.B. für vergleichbare Augenhornhäute oder muskuloskeletale Gewebezubereitungen) wurde das Verfahren der Genehmigung nach § 21a AMG weiter vereinfacht und der Aufwand Für die Erstellung, Bearbeitung und Prüfung identischer oder ähnlicher Gewebezubereitungen begrenzt.
Zu dem in § 142 Absatz 2 AMG für die Zulassung nach § 21 AMG festgelegten Stichtag (30. September 2008) wurden 8 Anträge eingereicht.
Bis zum 30. Juni 2009 konnte das PEI eine Genehmigung nach § 21a Absatz 1 AMG für eine muskuloskeletale Gewebezubereitung erteilen. Daneben liegen 28 Alt-Zulassungen nach § 21 AMG für Haut und muskuloskeletale Gewebezubereitungen vor, die bereits vor Inkrafttreten des Gewebegesetzes erteilt wurden (Stand vom 8. März 2009, siehe Bundesanzeiger Nummer 67 vom 6. Mai 2009, S. 1609).
Bis zum 30. Juni 2009 wurden zwei Anträge auf Bescheinigung nach § 21a Absatz 9 AMG gestellt, weitere 17 Anträge von Antragstellern aus EU-Mitgliedstaaten/EWR-Vertragsstaaten wurden nach § 21a Absatz 1 AMG gestellt. Anzahl und Status der Anträge sind nachfolgend in tabellarischer Form zusammengefasst:
Tab. 3.3.2 Antragssituation beim PEI zu Gewebezubereitungen und Blutstammzellzubereitungen mit Stand vom 30. Juni 2009 unter Berücksichtigung der Stichtage nach § 142 Absatz 2 AMG (§§ 21a und 21 AMG)
| Verfahrensart | Anträge zum Stichtag 1.2.2008 | Anträge zum Stichtag 30.9.2008 | Anträge mit Stand 16.6.2009 | Bescheidung mit Stand 30.6.2009 |
|---|
Inverkehrbringen von Gewebezubereitungen:
- Genehmigung von Gewebezubereitungen nach § 21a Absatz 1 AMG | 341 | Stichtag nicht relevant | 181 | 1 |
| - Genehmigung von Blutstammzellzubereitungen nach § 21a AMG | 219 | Stichtag nicht relevant | 193 | 0 |
| - Zulassung von Gewebezubereitungen nach § 21 Absatz 1 AMG | Stichtag nicht relevant | 8 | 9 | 28 (alt) |
Einfuhr von Gewebezubereitungen:
- Bescheinigung nach § 21a Absatz 9 AMG (erstmaliges Verbringen aus einem EU-Mitgliedstaat / EWR-Vertragsstaat) | Stichtag nicht relevant | Stichtag nicht relevant | 2 | 0 |
| - Genehmigung nach § 21a Absatz 1 AMG (Antragsteller aus einem EU-Mitgliedstaat/ EWR-Vertragsstaat) | Stichtag nicht relevant | Stichtag nicht relevant | 17 | 0 |
3.3.3 Einzelfragen
Nach einem Großteil der Stellungnahmen ist der in § 21a Absatz 1 AMG verwendete Begriff des Inverkehrbringens klarstellungsbedürftig insbesondere im Hinblick auf lokale Gewebebanken in Krankenhäusern.
Nach den Stellungnahmen lässt sich folgendes Meinungsspektrum, was unter einer Abgabe an andere nicht zu verstehen ist, festhalten:
- - herstellende und anwendende Person müssen personenidentisch sein
- - herstellende und anwendende Person müssen innerhalb einer Abteilung eines Krankenhauses tätig sein, es ist keine Personenidentität, aber Verfügungsberechtigung einer Person erforderlich
- - herstellende und anwendende Person können verschiedenen Abteilungen eines Krankenhauses angehören, solange die Verfügungsberechtigung einer Person gewährleistet ist
- - herstellende und anwendende Person können verschiedenen Krankenhäusern angehören, solange es sich um denselben Krankenhausträger oder dieselbe Institution handelt.
Nach Ansicht der Bundesregierung stellt sich die Rechtslage wie folgt dar:
Der in § 21a AMG verwendete Begriff des Inverkehrbringens ist in § 4 Absatz 17 AMG gesetzlich definiert.
Danach ist als Inverkehrbringen das Vorrätighalten zum Verkauf oder zu sonstiger Abgabe, das Feilhalten, das Feilbieten und die Abgabe an andere zu verstehen. Maßgeblich kommt es auf den Begriff der Abgabe an andere an. Die Definition der Abgabe in § 13 Absatz 1 Satz 3 AMG (Personenidentität) in der bis zur 15. AMG-Novelle geltenden Fassung kann dabei nicht zugrunde gelegt werden, weil diese einen anderen Sachzusammenhang als das Inverkehrbringen nach § 4 Absatz 17 AMG betrifft (Herstellung).
Eine Abgabe an andere im Sinne des § 4 Absatz 17 AMG liegt vor, wenn die tatsächliche Verfügungsgewalt über eine Gewebezubereitung wechselt (BVerwGE 131, 1 ff.; Kloesel/Cyran, Arzneimittelrecht, Loseblattsammlung, Stand 1. August 2008, § 4 AMG Anmerkung 57; Rehmann, Arzneimittelgesetz, 2. Auflage 2003, § 4 Rn. 19). Mit der Verfügungsgewalt ist die Verfügungsberechtigung gemeint (Kloesel/Cyran, Arzneimittelrecht, § 4 AMG Anmerkung 57 mit weiteren Nachweisen). Maßgeblich für die Beurteilung, wer die Verfügungsgewalt inne hat, ist die Organisation der Herstellung und Anwendung der Gewebezubereitung im Krankenhaus und die Ausgestaltung der Erlaubnis durch die zuständige Behörde. Wird z.B. die Erlaubnis für eine bestimmte Betriebsstätte innerhalb des Krankenhauses erteilt, so liegt es nahe, dass mit dem Wechsel der Gewebezubereitung von der herstellenden Betriebsstätte in die anwendende Abteilung des Krankenhauses auch ein Wechsel in der Verfügungsgewalt vorliegt. Die Verfügungsgewalt kann demgegenüber noch gegeben sein, wenn der Leiter der Abteilung eines Krankenhauses unmittelbar für die Herstellung der Gewebezubereitung in seiner Abteilung zuständig ist und sie auch in seiner Abteilung anwendet wenn auch unter Zuhilfenahme der ihm unterstellten Ärzte. Eine Abgabe an andere liegt dann nicht vor und eine Genehmigung nach § 21a AMG ist in diesem Fall nicht erforderlich. Wechselt jedoch die Gewebezubereitung von der herstellenden Abteilung in eine andere Abteilung eines Krankenhauses oder sogar in ein anderes Krankenhaus desselben Trägers, durfte regelmäßig ein Wechsel in der Verfügungsgewalt vorliegen und eine Genehmigung nach § 21a AMG erforderlich sein. Aus Sicht der Bundesregierung besteht kein Handlungsbedarf.
Darüber hinaus wird in den Stellungnahmen der Länder die Abgrenzung von bekannten Gewebezubereitungen zu Arzneimitteln für neuartige Therapien thematisiert. Die Abgrenzung sei im Einzelfall sowohl fachlich als auch verfahrenstechnisch (Zuständigkeit der Europäischen Arzneimittelagentur EMEA) schwierig und sollte deshalb transparenter und einfacher gestaltet werden. Als Beispiele werden die Anwendung des Kollagenaseverdaus (sogenannter enzymatischer Verdau) auf Gewebe genannt, bei denen dieses Verfahren bislang keine Anwendung gefunden hat, sowie die Herstellung von Stammzellen aus dem Knochenmark zur Anwendung am Herzen von Herzinfarktpatienten. Es wird zudem angeregt, bereits erfolgte Einstufungen zu veröffentlichen. Die Bundesregierung ist der Auffassung, dass sich bekannte Gewebezubereitungen von Arzneimitteln für neuartige Therapien nur durch eine wissenschaftlich fundierte fachliche Bewertung im Einzelfall abgrenzen lassen, und sieht keinen Handlungsbedarf. Arzneimittel für neuartige Therapien unterliegen seit dem 30. Dezember 2008 nach der Verordnung (EG) Nr. 1394/2007 grundsätzlich der zentralen Zulassung durch die Europäische Kommission. Das Arzneimittelgesetz wurde im Rahmen der 15. AMG-Novelle an die Verordnung (EG) Nr. 1394/2007 angepasst (vgl. insbesondere § 4 Absatz 9, §§ 4b und 21 Absatz 1 AMG). Die Abgrenzung der bekannten Gewebezubereitungen von den Arzneimitteln für neuartige Therapien ist von den zuständigen Behörden, dem PEI und der EMEA im Rahmen der jeweiligen Zuständigkeiten vorzunehmen. Die zuständigen Behörden können bei Abgrenzungsschwierigkeiten entsprechend § 21 Absatz 4 AMG beziehungsweise zukünftig nach § 4b Absatz 3 in Verbindung mit § 21 Absatz 4 AMG die Zulassungs- oder Genehmigungspflicht auf Antrag durch das PEI feststellen lassen. Die Feststellung, ob ein Arzneimittel für neuartige Therapien nicht routinemäßig hergestellt wird und damit nur genehmigungspflichtig ist (vgl. § 4b AMG) oder schon der Zulassungspflicht durch die Europäische Kommission unterliegt, ist zukünftig von den zuständigen Behörden im Benehmen mit dem PEI zu treffen (vgl. § 4b Absatz 4 AMG).
Im Falle der beiden vorstehend genannten Beispiele (Verfahren des enzymatischen Verdaus, Knochenstammzellen zur Anwendung am Herzen) vertritt die Bundesregierung die Auffassung, dass es sich um Arzneimittel für neuartige Therapien handelt. Die Herstellungsverfahren werden entweder bei neuen (anderen) Geweben oder im Zusammenhang mit einer neuartigen Therapie verwendet. In beiden Fällen ist es erforderlich sorgfältig zu prüfen, ob die Herstellungsverfahren die Wirksamkeit und Unbedenklichkeit des neuartigen Arzneimittels gewährleisten.
Eine Veröffentlichung der Einstufungen von Arzneimitteln für neuartige Therapien ist nicht vorgesehen.
Die EMEA veröffentlicht jedoch auf der Grundlage des Artikel 13 Absatz 3 der Verordnung (EG) Nr. 726/2004 nach Streichung aller vertraulichen Angaben geschäftlicher Art den vom Ausschuss für Humanarzneimittel erstellten Bericht über die Beurteilung des Humanarzneimittels und die Gründe für das Gutachten zugunsten der Erteilung der zentralen Zulassung (siehe im Internet unter http://www.emea.europa.eu/htms/human/epar/eparintro.htm ). Die zentrale Zulassung wird gemäß Artikel 13 Absatz 2 der Verordnung (EG) Nr. 726/2004 im Amtsblatt der Europäischen Union veröffentlicht.
In einer weiteren Stellungnahme wird thematisiert, dass der rechtliche Status von sogenannten komplexen Geweben wie z.B. Gesicht und Gliedmaßen ungeklärt sei. Es wird eine gesetzliche Regelung im Transplantationsgesetz angeregt. Aus Sicht der Bundesregierung unterfallen auch komplexe Gewebe den Qualitäts- und Sicherheitsanforderungen der Richtlinie 2004/23/EG. Die Bundesregierung wird prüfen, ob im Hinblick auf die Besonderheiten dieser Gewebe Änderungen sinnvoll und sachgerecht sind.
Die beschriebene Regelungssituation für bestrahlte Gewebezubereitungen (siehe Abschnitt 3.3.1) wird von zwei Fachgesellschaften und im Hinblick auf die Zulassungspflicht nach § 21 AMG vom PEI thematisiert.
Es wird vorgeschlagen, auf ein Zulassungsverfahren zu verzichten und stattdessen auch für bestrahlte Gewebezubereitungen eine Genehmigung nach § 21a AMG zu erteilen. Dieser Vorschlag bedarf aus Sicht der Bundesregierung einer eingehenden differenzierten fachlichen Bewertung.
Des Weiteren wird seitens des PEI und weiterer Befragter angeregt, entsprechend § 22 Absatz 4 AMG eine Vorlage der Erlaubnis nach § 20c AMG für das Genehmigungsverfahren nach § 21a AMG vorzusehen.
Im Rahmen der 15. AMG-Novelle wurde eine entsprechende Vorschrift in § 21a Absatz 2 Satz 2 AMG zur Vorlage der Erlaubnis nach § 20c AMG ergänzt.
Ein Verband regt eine Klarstellung der Begrifflichkeiten in § 21a Absatz 2 Nummer 6, 7 und 8 AMG sowie eine Erweiterung des § 21a Absatz 3 AMG auf andere Aspekte an; es ist zu vermuten, dass sich dieser Vorschlag auf Aspekte der Herstellung bezieht. Aus Sicht der Bundesregierung besteht kein Änderungsbedarf.
Die Begrifflichkeiten des § 21a Absatz 2 AMG gehen auf Artikel 6 Absatz 2 der Richtlinie 2004/23/EG in Verbindung mit Anhang II der Richtlinie 2006/86/EG7 zurück. Im Übrigen kann medizinisches Erkenntnismaterial im Sinne des § 21a Absatz 3 AMG nur sinnvoll auf § 21a Absatz 2 Nummer 3 AMG, also auf die Anwendungsgebiete sowie die Art und Dauer der Anwendung, angewendet werden.
In einigen Stellungnahmen wird ausgeführt, dass die Genehmigungspraxis nach § 21a AMG mit einem erheblichen bürokratischen Aufwand und entsprechenden Kosten verbunden sei. Die Anforderungen der Genehmigungsanträge, die das PEI Antragstellern auf seiner Homepage zur Verfügung stellt, seien zu umfangreich. Eine Gewebebank etwa, die verschiedenste Gewebe verarbeiten und in Verkehr bringen möchte habe trotz der vorgeschlagene Kostenverordnung eine erhebliche Gebührenlast zu tragen, da für jede Gewebeart eine Genehmigung erforderlich sei. Gleichzeitig wird begrüßt, dass bei im Wesentlichen gleichen Herstellungsverfahren nach der Kosten-Verordnung für Amtshandlungen des PEI ermäßigte Gebühren berechnet werden können. Die Bundesregierung sieht keinen Handlungsbedarf. Das PEI steht Antragstellern und zuständigen Behörden stets beratend zur Seite und stellt zahlreiche Informationen zur Verfügung, um den bürokratischen Aufwand im Interesse aller Beteiligten gering zu halten (vgl. Abschnitt 3.3.2). Die Genehmigungspraxis des PEI wird zudem erweisen, inwieweit Anträge oder Unterlagen von verschiedenen Anträgen bei gleichartigen Geweben oder Herstellungsverfahren zusammengefasst eingereicht und entsprechend die Kosten gesenkt werden können. So können z.B. bereits jetzt Antragsteller verschiedene Arten der Be- und Verarbeitung von Blutstammzellpräparaten (aus Knochenmark, peripherem Blut oder Nabelschnurblut) in einem Antrag zusammenfassen, ohne dass für jede Art der Herstellung ein getrenntes Antragsverfahren erforderlich wird beziehungsweise für jede Art der Herstellung getrennte Kosten erhoben werden.
In einer Stellungnahme wird angemerkt, dass durch das Verfahren nach § 21a Absatz 9 AMG für das Verbringen aus EU-Mitgliedstaaten/EWR-Vertragsstaaten bürokratische Hürden geschaffen wurden, denen Importeure bei der Einfuhr in andere EU-Mitgliedstaaten nicht unterlagen. Zur Vereinfachung des Verfahrens wird vorgeschlagen, Zertifikate europäischer und internationaler Organisationen anzuerkennen.
Die Bundesregierung kann diesem Vorschlag nicht folgen. Die Verbringungsregelung in § 21a Absatz 9 AMG berücksichtigt insbesondere die Möglichkeit der Mitgliedstaaten, höhere Anforderungen festzulegen als in den EG-Richtlinien vorgesehen sind (vgl. Artikel 4 Absatz 2 der Richtlinie 2004/23/EG). Sie soll sicherstellen, dass nur gleichwertige Gewebezubereitungen aus Mitgliedstaaten der Europäischen Union in den Geltungsbereich des Arzneimittelgesetzes verbracht werden. Damit soll der hohe Gesundheitsschutz der Bevölkerung gewahrt werden, insbesondere im Hinblick auf den in Deutschland erreichten hohen Standard der verwendeten Testverfahren. Durch das beschriebene vereinfachte Verfahren der Bescheinigung wird für das Verbringen kein unverhältnismäßiger Aufwand verursacht. Zu den Einzelheiten des Verfahrens wird auf Abschnitt 3.3.1 verwiesen.
Soweit im Hinblick auf § 21a AMG ausgeführt wird, es fehle an einer europarechtlichen Grundlage, verweist die Bundesregierung insbesondere auf Artikel 6 der Richtlinie 2004/23/EG.
3.4 Besondere Dokumentations- und Meldepflichten bei Blut- und Gewebezubereitungen ( § 63c AMG)
3.4.1 Rechtslage
§ 63c AMG normiert spezielle Dokumentations- und Meldepflichten für Blut- und Gewebezubereitungen sowie deren Ausgangsmaterialien (Blut, Blutbestandteile und Gewebe), die sogenannte Hämo- und Gewebevigilanz, und zwar Für:
- - genehmigungspflichtige Gewebezubereitungen einschließlich Zubereitungen aus dem Knochenmark (siehe Abschnitte 3.3.1 und 3.6.1)
- - genehmigungspflichtige Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut, die zur autologen oder gerichteten, für eine bestimmte Person vorgesehene Anwendung bestimmt sind (siehe Abschnitt 3.6.1)
- - zulassungspflichtige Blutzubereitungen im Sinne der Richtlinie 2002/98/EG8 (z.B. gefrorenes Frischplasma, Thrombozyten oder Erythrozyten)
- - nicht zulassungs- und genehmigungspflichtige Blut- und Gewebezubereitungen sowie
- - Blut, Blutbestandteile und Gewebe.
Abzugrenzen von den genannten Blut- und Gewebezubereitungen sind solche, die zulassungspflichtige Arzneimittel im Sinne der Richtlinie 2001/83/EG sind (z.B. Blutgerinnungsfaktoren, Immunglobuline, Albumine oder industriell hergestellte Gewebezubereitungen). Dies betrifft auch Arzneimittel, die der zentralen Zulassung durch die Europäische Kommission unterliegen wie z.B. Arzneimittel für neuartige Therapien (vgl. Verordnung (EG) Nr. 1394/2007) beziehungsweise nicht routinemäßig hergestellte Arzneimittel für neuartige Therapien, die mit der 15. AMG-Novelle einer Genehmigungspflicht nach § 4b Absatz 3 AMG unterstellt werden. Für diese Arzneimittel gelten die Dokumentations- und Meldepflichten nach § 63b AMG (siehe auch Beschlussempfehlung und Bericht des Ausschusses für Gesundheit, Bundestags-Drucksache 016/5443, S. 58). § 63c Absatz 1 und 2 AMG verpflichtet zunächst den Inhaber einer Zulassung beziehungsweise den Inhaber einer Genehmigung für Blut- oder Gewebezubereitungen (§§ 21, 21a AMG), alle Verdachtsfälle von schwerwiegenden Zwischenfällen oder schwerwiegenden unerwünschten Reaktionen, die innerhalb und außerhalb von Deutschland aufgetreten sind und einen Einfluss auf die Qualität und Sicherheit der Blut- und Gewebezubereitungen haben oder auf sie zurückgeführt werden können, sowie die Anzahl der Rückrufe zu dokumentieren. Gemas § 63c Absatz 2 AMG muss er jeden Verdacht eines schwerwiegenden Zwischenfalls oder einer schwerwiegenden unerwünschten Reaktion dokumentieren und unverzüglich (spätestens innerhalb von 15 Tagen) dem PEI melden. § 63c Absatz 3 AMG verpflichtet zudem Gewebeeinrichtungen sowie Blut- und Plasmaspendeeinrichtungen, jeden Verdacht eines schwerwiegenden Zwischenfalls oder einer schwerwiegenden unerwünschten Reaktion im Zusammenhang mit nicht zulassungs-oder genehmigungspflichtigen Blut- oder Gewebezubereitungen sowie - unabhängig von einer Zulassungs- oder Genehmigungspflicht - mit Blut, Blutbestandteilen oder Gewebe unverzüglich der zuständigen Behörde zu melden. Diese leitet die Meldung an das PEI weiter. Die Dokumentations- und Meldepflichten des § 63c AMG beschränken sich auf schwerwiegende Zwischenfälle oder schwerwiegende unerwünschte Reaktionen, die Einfluss auf die Qualität und Sicherheit der Blut- und Gewebezubereitungen haben oder auf sie zurückgeführt werden können. Nicht erfasst von den Dokumentations- und Meldepflichten werden somit z.B. Anwendungs- oder Verwechslungsfehler von Ärztinnen oder Ärzten.
Die dargestellten Dokumentations- und Meldepflichten nach § 63c AMG werden ergänzt durch Dokumentations-und Meldepflichten nach § 40 AMWHV für Entnahmeeinrichtungen und Gewebespenderlabore sowie nach § 13b TPG und §§ 8 und 9 TPG-GewV für Einrichtungen der medizinischen Versorgung (Definition vgl. § 1a Nummer 9 TPG, z.B. Krankenhäuser). Die nachfolgende Übersicht stellt die wesentlichen Inhalte der Dokumentations- und Meldepflichten für den Bereich der Gewebe und Gewebezubereitungen schematisch dar:
Abb. 3.4.1 Übersicht zu den Dokumentations- und Meldepflichten für Gewebe und Gewebezubereitungen ( § 63c AMG)
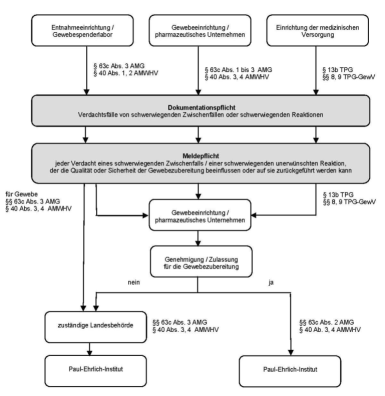
Der Umfang der meldepflichtigen Daten ist in § 63c Absatz 2 Satz 2 und Absatz 3 Satz 2 AMG festgelegt.
Der Inhaber einer Zulassung oder einer Genehmigung für Blut- oder Gewebezubereitungen ist nach § 63c Absatz 4 AMG zudem verpflichtet, auf Anforderung, mindestens jedoch jährlich dem PEI einen aktualisierten Bericht über die Unbedenklichkeit der Arzneimittel vorzulegen (Periodic Safety Update Report - PSUR).
3.4.2 Allgemeine Einschätzung
Soweit bereits Erfahrungen zu den besonderen Dokumentations- und Meldepflichten des § 63c AMG vorliegen, hat sich die Vorschrift nach der Mehrzahl dieser Stellungnahmen bewährt. Grostenteils liegen jedoch noch keine Erfahrungen zu § 63c AMG vor. Nach Auskunft des PEI sind bis 30. Juni 2009 nur wenige Meldungen eingegangen. Es liegt die Vermutung nahe, dass die Meldepflichten bei den Entnahme-und Gewebeeinrichtungen, Gewebespenderlaboren sowie den Ärztinnen und Ärzten noch nicht hinreichend bekannt sind beziehungsweise von ihnen noch nicht hinreichend beachtet werden. Auf die Busgeldbewehrung in § 97 Absatz 2 Nummer 7 und 24e AMG (Geldbuße bis 25 000 Euro) sowie mögliche straf- und zivilrechtliche Konsequenzen wird hingewiesen.
In einzelnen Stellungnahmen wird darauf hingewiesen, dass die Meldepflicht zu umfangreich und in der Praxis daher kaum zu bewältigen sei. Vereinzelt wird zudem von Durchführungsschwierigkeiten berichtet.
Die Bundesregierung weist darauf hin, dass die Durchführung neuer Regelungen erfahrungsgemäß Zeit beansprucht und daher zunächst mehr Erfahrungen gesammelt werden sollten, bevor mögliche Änderungen erwogen werden. Durch Meldeformulare des PEI für schwerwiegende Zwischenfälle und unerwünschte Reaktionen nach § 63c AMG soll das Meldeverfahren standardisiert und damit für die Meldepflichtigen vereinfacht werden. Das PEI hat in einer Arbeitsgemeinschaft mit Mitgliedern von Gewebe- sowie Blutspendeeinrichtungen entsprechende Meldeformulare erarbeitet und stimmt diese derzeit mit den Ländern ab. Im Anschluss daran werden die Meldeformulare auf der Homepage des PEI zur Verfügung gestellt.
3.4.3 Einzelfragen
Das PEI und einzelne Fachgesellschaften teilen mit, dass nach ihrer Auffassung die Begriffe "schwerwiegender
Zwischenfall", "schwerwiegende unerwünschte Reaktion" und "Verdacht" nicht eindeutig definiert seien. Die Begriffe "schwerwiegender Zwischenfall" und "schwerwiegende unerwünschte Reaktion" sind in § 63c Absatz 6 und 7 AMG umfassend definiert. Während schwerwiegende Zwischenfälle typischerweise produktassoziiert sind (z.B. mikrobielle oder virale Kontamination oder anderweitig mangelhafte Gewebezubereitung, die z.B. aus Mängeln im Hinblick auf Gewinnung, Transport, Lagerung, Verpackung oder Kennzeichnung herrühren), sind schwerwiegende unerwünschte Reaktionen in der Regel spender- oder empfängerassoziiert (z.B. Infektionen beim Spender oder Empfänger). Der Begriff des "Verdachts" ist in § 63c AMG nicht definiert. Ein Verdachtsfall bei einer schwerwiegenden unerwünschten Reaktion liegt vor bei Hinweisen auf einen kausalen (auch zeitlichen) Zusammenhang zwischen der Anwendung der Blut- oder Gewebezubereitung und der schwerwiegenden unerwünschten Reaktion. Die Meldeverpflichtung betrifft nur Verdachtsfälle mit schwerwiegenden unerwünschten Reaktionen (u.a. lebensbedrohliche Situation, Behinderung, Krankenhausaufenthalt). Das PEI beabsichtigt, zur Unterstützung der Fachkreise auf seiner Homepage Falldefinitionen zur Verfügung zu stellen. Dies soll zunächst im Bereich der Hämovigilanz erfolgen. Nach einer entsprechenden Erfahrungszeit konnten die Falldefinitionen um den Bereich der Gewebevigilanz ergänzt werden. Im Ergebnis ist aus Sicht der Bundesregierung eine Klarstellung der Begrifflichkeiten nicht erforderlich.
Zudem wird angemerkt, dass der Begriff "schwerwiegender Zwischenfall" zu weit gefasst sei, da er alle kausalen Zwischenfälle von der Gewinnung bis zur Abgabe erfasse. Insgesamt sei von einer geringen Meldebereitschaft der Ärztinnen und Ärzte auszugehen, weil viele befürchteten, dass unerwünschte Reaktionen trotz fehlender Kausalität als ärztlicher Fehler angesehen wurden. Nach § 13b TPG und § 8 TPG-GewV sind für Einrichtungen der medizinischen Versorgung alle Verdachtsfälle schwerwiegender Zwischenfälle meldepflichtig die auf die Entnahme, Untersuchung, Aufbereitung, Be- oder Verarbeitung, Konservierung, Aufbewahrung oder Abgabe einschließlich Transport des verwendeten Gewebes zurückgeführt werden können, nicht aber Anwendungsfehler. Nach Ansicht der Bundesregierung ist es nicht möglich den Begriff des "schwerwiegenden Zwischenfalls" enger zu fassen. Die Definition beruht auf Artikel 3 Buchstabe g der Richtlinie 2002/98/EG und Artikel 3 Buchstabe m der Richtlinie 2004/23/EG. Die Mitgliedstaaten können keine im Verhältnis zum Regelungsrahmen der Richtlinien weniger strengen Regelungen einführen. Jedoch ist in § 63c AMG klargestellt, dass nur solche Zwischenfälle gemeldet werden müssen die einen Einfluss auf das Gewebeprodukt haben können, z.B. defekte Behälter oder Fehler beim Transport oder der Lagerung. Aus Sicht der Bundesregierung besteht kein Änderungsbedarf.
Des Weiteren wird berichtet, dass die Verwendung der unterschiedlichen Begriffe "schwerwiegende Nebenwirkung" in § 63b AMG und "schwerwiegende unerwünschte Reaktion" in § 63c AMG schwer verständlich sei, weil sich die Meldepflicht der Ärztinnen und Ärzte bislang immer auf Nebenwirkungen erstreckt hatte. Es wird daher eine Angleichung der Begrifflichkeiten angeregt. Die Bundesregierung vertritt die Auffassung, dass sich die Begriffe auf unterschiedliche Sachverhalte beziehen und sich deshalb inhaltlich unterscheiden (vgl. Gesetzentwurf der Bundesregierung, Bundesrats-Drucksache 543/06 (PDF) , S. 96). Der Begriff "Nebenwirkung" ist in § 4 Absatz 13 AMG definiert. Danach sind schwerwiegende Nebenwirkungen schädliche unbeabsichtigte Reaktionen, die beim bestimmungsgemäßen Gebrauch eines Arzneimittels auftreten. Der Begriff der schwerwiegenden unerwünschten Reaktion nach § 63c Absatz 7 AGM umfasst hingegen unbeabsichtigte Reaktionen sowohl beim Spender als auch beim Empfänger, die im Zusammenhang mit der Gewinnung oder Übertragung von Gewebe- oder Blutzubereitungen auftreten, und zwar im Inland oder in Drittländern. Diese Definition beruht auf der Richtlinie 2002/98/EG und der Richtlinie 2004/23/EG. Eine begriffliche Angleichung von § 63c AMG an § 63b AMG ist daher nicht möglich.
Das PEI regt an, die Dokumentationspflicht des § 63c Absatz 1 AMG auf nicht schwerwiegende Zwischenfälle und Reaktionen zu erweitern. Nach der derzeitigen Regelungslage sei eine umfassende Beurteilung der Risiken der Blut- und Gewebezubereitungen nicht möglich. Es bestehe die Gefahr, dass eine unverhältnismäßige Häufung nicht schwerwiegender Zwischenfälle und Reaktionen, die ebenfalls ein Risiko darstellen, übersehen werde. Eine entsprechende Dokumentationspflicht für alle Verdachtsfälle von Nebenwirkungen eines Arzneimittels enthält § 63b Absatz 1 AMG in Verbindung mit § 4 Absatz 13 AMG.
Nach Artikel 11 Absatz 1 der Richtlinie 2004/23/EG und Artikel 15 Absatz 1 der Richtlinie 2002/98/EG besteht für Blut- und Gewebezubereitungen hingegen eine Dokumentationspflicht nur für schwerwiegende Zwischenfälle und schwerwiegende unerwünschte Reaktionen. Ob darüber hinaus Dokumentations- und Berichtspflichten erforderlich sind, wird von der Bundesregierung geprüft.
Zwei Länder schlagen vor, dass alle Verdachtsfälle bei nicht zulassungs- oder genehmigungspflichtigen Blut- oder Gewebezubereitungen sowie Blut und Gewebe nicht nur der zuständigen Behörde, sondern auch dem PEI unverzüglich gemeldet und von letzterem bewertet werden. § 63c Absatz 3 AMG sieht Für den genannten Bereich eine Meldung an die zuständige Behörde vor, die diese an das PEI weiterleitet.
Verdachtsfälle bei zulassungs- oder genehmigungspflichtigen Blut- oder Gewebezubereitungen werden nach § 63c Absatz 2 AMG hingegen dem PEI gemeldet. Die Bundesregierung vertritt die Auffassung, dass der unterschiedliche Meldeablauf in § 63c Absatz 2 und 3 AMG aufgrund des unterschiedlichen Status der Arzneimittel (zulassungs- oder genehmigungspflichtiges Arzneimittel / nicht zulassungs- oder genehmigungspflichtiges Arzneimittel) gerechtfertigt ist. Die Meldung bei nicht zulassungs- oder genehmigungspflichtigen Arzneimitteln erfolgt bei der zuständigen Behörde, bei der in der Regel bereits die Unterlagen zum Herstellungsverfahren vorliegen. Bei der produktspezifischen Bewertung im Rahmen des § 63c AMG wird die zuständige Behörde durch das PEI unterstützt. Damit wird eine sachgerechte Bewertung der Meldung gewährleistet. Ein Änderungsbedarf wird aus Sicht der Bundesregierung nicht gesehen.
Eine Fachgesellschaft trägt vor, dass es auf gesetzlicher Ebene an einer Umsetzung der Anforderungen von Artikel 5 Absatz 1 Buchstabe a und Artikel 6 Absatz 1 Buchstabe a der Richtlinie 2006/86/EG an die Meldepflicht von Entnahmeeinrichtungen fehle. Die Meldepflicht ergebe sich lediglich aus § 40 Absatz 1 und Absatz 2 AMWHV. Zudem sei die Aufspaltung der Meldepflichten auf Gesetzes- und Verordnungsebene unübersichtlich. Hierzu ist aus Sicht der Bundesregierung zu sagen, dass nach der Systematik der Dokumentations- und Meldepflichten zwischen solchen für die Entnahmeeinrichtungen (einschließlich Gewebespenderlaboren), Gewebeeinrichtungen beziehungsweise pharmazeutischen Unternehmen und Einrichtungen der medizinischen Versorgung zu unterscheiden ist (siehe die Übersicht in Abschnitt 3.4.1). Diese gehen zurück auf die Richtlinie 2002/98/EG und deren Durchführungsrichtlinie 2005/61/EG9 sowie die Richtlinie 2004/23/EG und deren Durchführungsrichtlinie 2006/86/EG. Nach Artikel 249 Absatz 3 EGV hat der Gesetzgeber die Wahl, in welcher Form und durch welche Mittel er Richtlinien umsetzt. Durchführungsrichtlinien der Europäischen Kommission enthalten konkretisierende Regelungen, die im nationalen Recht in der Regel durch Rechtsverordnung umgesetzt werden. Die konkrete Umsetzung in verschiedenen Rechtsverordnungen ist bedingt durch die gesetzessystematisch unterschiedlich betroffenen Bereiche der Gewinnung und Laboruntersuchung, der Be- oder Verarbeitung beziehungsweise Herstellung sowie der Anwendung. Artikel 5 Absatz 1 Buchstabe a und Artikel 6 Absatz 1
Buchstabe a der Richtlinie 2006/86/EG wurden in § 63c Absatz 3 AMG und § 40 Absatz 1 und 2 AMWHV umgesetzt zum Begriff der Gewebeeinrichtung in § 63c Absatz 3 AMG wird auf § 1a Nummer 8 TPG und § 2 Nummer 10 AMWHV verwiesen. Er erfasst auch Entnahmeeinrichtungen. Nach Ansicht der Bundesregierung besteht kein Änderungsbedarf.
Ein Land schlägt vor, dass zum Zwecke der Angleichung auch die nach § 21 AMG zugelassenen Gewebezubereitungen (siehe Abschnitt 3.4.1) dem Anwendungsbereich des § 63c AMG unterstellt werden. In der Richtlinie 2001/83/EG ist dies nicht vorgesehen. Nach Einschätzung der Bundesregierung ist es deshalb nicht möglich, diese Arzneimittelgruppen von dem weitergehenden Regelungsrahmen der Richtlinie 2001/83/EG und damit des § 63b AMG auszunehmen.
Ein Land schlägt zudem vor, dass die nach § 63c AMG erhobenen Daten in Form einer Jahresstatistik allen Herstellern von Blut- und Gewebezubereitungen sowie den Ärztinnen und Ärzten zur Verfügung gestellt werden sollten. Aus Sicht der Bundesregierung besteht insoweit kein Handlungsbedarf. Das PEI wertet die bei ihm nach § 63c AMG eingegangenen Meldungen statistisch aus und stellt die Auswertung der interessierten Öffentlichkeit z.B. auf Fachtagungen vor. Für den Bereich der Blutzubereitungen wurde zudem eine Fachpublikation mit den statistischen Daten aus dem Meldezeitraum 1997 bis 2007 erstellt ( Transfusion Medicine, 2009); eine weitere Fachpublikation mit den statistischen Daten aus dem Jahr 2008 ist bereits in Vorbereitung. Für den Bereich der Gewebezubereitungen bedarf es zunächst einer ausreichenden Datenlage, um eine statistische Auswertung der Meldungen vornehmen zu können.
3.5 Einfuhrerlaubnis und Zertifikate für Gewebe und bestimmte Gewebezubereitungen ( § 72b AMG)
3.5.1 Rechtslage
Für die Einfuhr von Gewebe im Sinne von § 1a Nummer 4 TPG oder Gewebezubereitungen im Sinne von § 20c AMG (vgl. Abschnitt 3.2.1) aus einem Drittland ist eine Einfuhrerlaubnis nach § 72b Absatz 1 AMG erforderlich. Sie wird von der zuständigen Behörde erteilt, die ihre Entscheidung im Benehmen mit dem PEI trifft (vgl. § 72b Absatz 1 Satz 2 in Verbindung mit § 20c Absatz 1 Satz 3 AMG). Voraussetzung für die qualitätsgerechte Einfuhr von Gewebe und Gewebezubereitungen ist darüber hinaus nach § 72b Absatz 2 AMG die Vorlage eines/r
- - Qualitätszertifikats der Behörde des Herkunftslandes (Gleichwertigkeit und gegenseitige Anerkennung) oder
- - Qualitätsbescheinigung der zuständigen Behörde (Gleichwertigkeit und Besichtigung) oder
- - Bescheinigung der zuständigen Behörde, dass die Einfuhr im öffentlichen Interesse liegt.
§ 72b Absatz 2 Satz 1 Nummer 2 AMG sieht grundsätzlich vor, dass die zuständige Behörde die Entnahme-oder Gewebeeinrichtung oder das Gewebespenderlabor im Herkunftsland vor Erteilung der Qualitätsbescheinigung besichtigt. Es steht jedoch nach § 72b Absatz 2 Satz 2 AMG in ihrem Ermessen, ob sie eine Besichtigung vornimmt oder nicht, wenn die eingereichten Unterlagen keinen Anlass zur Beanstandung geben oder ihr die Einrichtung oder Betriebsstätte sowie das Qualitätssicherungssystem bekannt sind.
Für die Einfuhr von Gewebe und Gewebezubereitungen zur unmittelbaren Anwendung bei Menschen steht über die Verweisung des § 72b Absatz 1 Satz 3 AMG auf § 72 Absatz 2 AMG ein vereinfachtes Erlaubnisverfahren zur Verfügung. Die Anforderungen dieses Erlaubnisverfahrens wurden mit der 15. AMGNovelle weiter präzisiert. Danach darf die Erlaubnis nur dann versagt werden, wenn nicht nachgewiesen wird dass für die Beurteilung der Qualität und Sicherheit der Arzneimittel und für die gegebenenfalls erforderliche Überführung der Arzneimittel in ihre anwendungsfähige Form nach dem Stand von Wissenschaft und Technik qualifiziertes Personal und geeignete Raume vorhanden sind. Es wurde im Rahmen der 15. AMG-Novelle klargestellt, dass die Erlaubnisvorschrift des § 72 Absatz 2 AMG auch dann in Anspruch genommen werden kann, wenn geringfügige Arbeitsschritte durchgeführt werden, die für die Anwendungsfähigkeit des Arzneimittels erforderlich sind. Für die qualitätsgerechte Einfuhr von Gewebe und Gewebezubereitungen zur unmittelbaren Anwendung gelten im Übrigen die dargestellten Voraussetzungen des § 72b Absatz 2 AMG.
Neben der Einfuhrerlaubnis nach § 72b Absatz 1 AMG ist für das Inverkehrbringen von Gewebezubereitungen grundsätzlich eine Genehmigung nach § 21a Absatz 1 AMG erforderlich; es wird auf die Einzelheiten im Abschnitt 3.3 verwiesen.
3.5.2 Allgemeine Einschätzung
Nach der überwiegenden Anzahl der eingereichten Stellungnahmen konnte zu § 72b AMG aufgrund der fehlenden Erfahrungen noch keine Bewertung abgegeben werden. Das PEI wurde bisher erst in einem Fall, wie in § 72b Absatz 1 Satz 2 in Verbindung mit § 20c Absatz 1 Satz 3 AMG vorgesehen, durch eine zuständige Behörde ins Benehmen gesetzt. Teilweise wird auch von Durchführungsschwierigkeiten berichtet.
Soweit zu § 72b AMG bereits Erfahrungen vorliegen, wird in den Stellungnahmen hervorgehoben, dass das Erlaubnisverfahren nach § 72b Absatz 1 AMG zu einer Vereinfachung geführt hat. Dies wird insbesondere vor dem Hintergrund der Dringlichkeit der Einfuhr von Gewebe und Gewebezubereitungen begrüßt.
Positiv bewertet wird zudem, dass die zuständige Behörde auf eine Besichtigung der Einrichtung oder der Betriebsstätte im Drittland unter den in § 72b Absatz 2 Satz 2 AMG genannten Voraussetzungen verzichten kann, was für die in diesem Bereich notwendige Flexibilität sorgt.
3.5.3 Einzelfragen
Vereinzelt wird in den Stellungnahmen ausgeführt, dass durch das Erlaubnisverfahren des § 72b AMG für die Einfuhr aus Drittländern bürokratische Hürden geschaffen wurden, denen Importeure bei der Einfuhr in andere EU-Mitgliedstaaten nicht unterlagen. Zur Vereinfachung des Verfahrens wird vorgeschlagen, Zertifikate europäischer und internationaler Organisationen anzuerkennen. Die Bundesregierung kann diesem Vorschlag nicht folgen. Die Einfuhr von Gewebe oder Gewebezubereitungen unterliegt nach Artikel 9 der Richtlinie 2004/23/EG der Erlaubnispflicht. Durch Zertifikate europäischer und internationaler Organisationen kann nicht hinreichend gewährleistet werden, dass die Gewinnung oder Be- oder Verarbeitung sowie die Laboruntersuchungen nach Standards durchgeführt wurden, die den Standards der Guten fachlichen Praxis der EG-Richtlinien für Gewebe mindestens gleichwertig sind. Diese stellen den bestehenden hohen Gesundheitsschutz in der Europäischen Union sicher.
Zudem wird darauf hingewiesen, dass die Feststellung der Gleichwertigkeit der Standards für Gewinnung, Be- oder Verarbeitung sowie Laboruntersuchungen mit denjenigen der Guten fachlichen Praxis durch die zuständige Behörde sehr aufwändig sei, insbesondere im Hinblick auf die Prüfung von fremdsprachigen Unterlagen sowie die Besichtigung im Drittland. Die Bundesregierung ist der Ansicht, dass die Prüfung der Unterlagen sowie die grundsätzlich vorgesehene Besichtigung im Drittland notwendig sind, um einen hohen Gesundheitsschutz der Bevölkerung zu gewährleisten. Um einen zügigen Ablauf des Erlaubnisverfahrens zu ermöglichen, werden die zuständigen Behörden vom PEI unterstützt, vgl. § 72b Absatz 1 Satz 2 in Verbindung mit § 20c Absatz 1 Satz 3 AMG. Auf das Ermessen der zuständigen Behörde, von einer Besichtigung im Drittland nach § 72b Absatz 2 Satz 2 AMG abzusehen, wird hingewiesen.
In weiteren Stellungnahmen wird angeregt, dass für die Einfuhr von Lymphozyten und Lymphozytenzubereitungen aus Drittländern § 72b AMG Anwendung finden solle. Die Bundesregierung ist der Ansicht, dass kein Änderungsbedarf besteht. Lymphozyten und Lymphozytenzubereitungen sind Blutzubereitungen im Sinne des § 4 Absatz 2 AMG und unterfallen daher den Regelungen der §§ 72 und 72a AMG. Für die Einfuhr von Lymphozyten und Lymphozytenzubereitungen zur unmittelbaren Anwendung steht das vereinfachte Erlaubnisverfahren nach § 72 Absatz 2 AMG zur Verfügung (vgl. Abschnitt 3.5.1).
Ein Qualitätszertifikat oder eine Qualitätsbescheinigung ist in diesem Fall nach § 72a Absatz 1a Nummer 2 AMG nicht erforderlich. Für Lymphozyten und Lymphozytenzubereitungen, die nicht zur unmittelbaren Anwendung bestimmt sind, ist eine Einfuhrerlaubnis nach § 72 Absatz 1 AMG erforderlich. Diese Regelung entspricht Artikel 2 Absatz 3 der Richtlinie 2005/62/EG10 und ist zur Sicherung eines hohen Gesundheitsschutzes notwendig.
3.6 Sonderthema: Zubereitungen mit Stammzellen des blutbildenden Systems
3.6.1 Rechtslage
Zubereitungen mit Stammzellen des blutbildenden Systems sind Arzneimittel nach § 2 Absatz 1 AMG.
Nach der Systematik des Arzneimittelgesetzes ist nach der Herkunft der Stammzellen des blutbildenden Systems zu differenzieren: Zubereitungen, die aus dem Knochenmark hergestellt werden, sind nach § 4 Absatz 30 AMG in Verbindung mit § 1a Nummer 4 TPG als Gewebezubereitung zu qualifizieren. Blutstammzellzubereitungen, die aus dem peripheren Blut oder dem Nabelschnurblut hergestellt werden, sind hingegen nach § 4 Absatz 2 AMG Blutzubereitungen. Diese Differenzierung ist bei den Anforderungen an die Gewinnung, die Be- oder Verarbeitung, das Inverkehrbringen, das erstmalige Verbringen und die Einfuhr zu beachten.
Soweit es sich bei den Zubereitungen aus dem Knochenmark oder den Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut um Arzneimittel im Sinne der Richtlinie 2001/83/EG oder um Arzneimittel für neuartige Therapien im Sinne des § 4 Absatz 9 AMG handelt, finden die speziellen Vorschriften des Arzneimittelgesetzes für Gewebe und Gewebezubereitungen überwiegend keine Anwendung (zu weiteren Einzelheiten siehe Abschnitt 3.2.1). Es gelten insbesondere die Vorschriften zur Herstellungserlaubnis (§§ 13 ff. AMG), zur Zulassung (§§ 21, 22 ff. AMG), zu den Dokumentations- und Meldepflichten ( § 63b AMG) sowie zur Einfuhrerlaubnis und den Zertifikaten (§§ 72 und 72a AMG).
Die Anforderungen des Gewebegesetzes an Zubereitungen mit Stammzellen des blutbildenden Systems sowie an ihre Ausgangsbestandteile werden nachfolgend dargestellt.
Gewinnung, Be- oder Verarbeitung Die Gewinnung von Knochenmark und die Durchführung der dafür erforderlichen Laboruntersuchungen unterliegen den Anforderungen des § 20b AMG. Daneben sind die Vorschriften des Transplantationsgesetzes zu beachten, insbesondere §§ 8d und 8e TPG. Die Be- oder Verarbeitung, Konservierung, Lagerung, Prüfung oder das Inverkehrbringen des Knochenmarks beziehungsweise der Zubereitungen aus dem Knochenmark richten sich dementsprechend nach § 20c AMG.
Die Gewinnung von Blutstammzellen aus dem peripheren Blut oder aus Nabelschnurblut und die nachfolgende Herstellung von Blutstammzellzubereitungen unterliegen den Anforderungen des § 13 AMG. Für die Gewinnung der Blutstammzellen sind zusätzlich die Vorschriften des Transfusionsgesetzes zu beachten. Die nachfolgende Übersicht bildet diese grundsätzliche Abgrenzung graphisch nach:
Abb. 3.6.1a Übersicht zu den Erlaubnispflichten für die Gewinnung und die Be- oder Verarbeitung von Stammzellen des blutbildenden Systems (§§ 20b, 20c und 13 AMG)
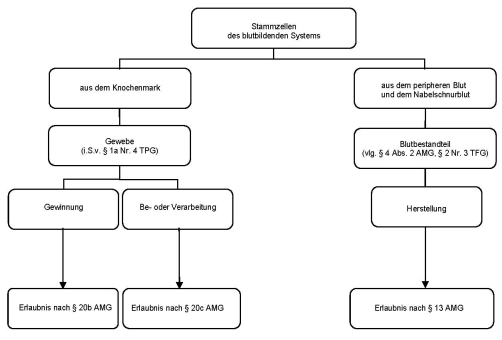
Inverkehrbringen
Zubereitungen aus dem Knochenmark unterfallen der Genehmigungspflicht des § 21a AMG (vgl. zur Abgrenzung zu § 21 AMG Abschnitt 3.3.1). Gemas § 21a Absatz 1 Satz 3 AMG unterliegen außerdem Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut, die zur autologen oder gerichteten für eine bestimmte Person vorgesehenen Anwendung bestimmt sind, der Genehmigungspflicht des § 21a AMG.
Soweit die Voraussetzungen des § 21a Absatz 1 AMG nicht vorliegen, werden Zubereitungen aus dem Knochenmark sowie Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut nach § 21 Absatz 1 AMG oder zentral nach der Verordnung (EG) Nr. 726/0411 in Verbindung mit der Verordnung (EG) Nr. 1394/2007 über Arzneimittel für neuartige Therapien zugelassen. Werden Arzneimittel für neuartige Therapien auf einer nicht routinemäßigen Basis hergestellt und ausschließlich in Deutschland verschrieben, hergestellt und angewendet, sind sie zukünftig nach § 4b AMG zu genehmigen.
Die nachfolgende Übersicht bildet diese grundsätzliche Abgrenzung für das Arzneimittelgesetz graphisch nach:
Abb. 3.6.1b Übersicht zur Genehmigungs- und Zulassungspflicht von Zubereitungen mit Stammzellen des blutbildenden Systems nach dem Arzneimittelgesetz (§§ 21a und 21 AMG)
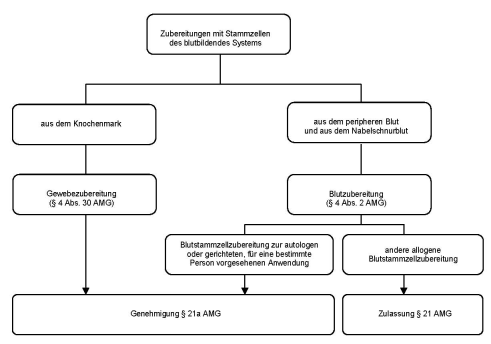
Gewebe- und Hämovigilanz
Zubereitungen aus dem Knochenmark sowie Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut sowie Blut(-bestandteile) und Gewebe unterliegen den besonderen Dokumentations-und Meldepflichten des § 63c AMG. Auf die Ausführungen im Abschnitt 3.4 wird verwiesen.
Sind sie Arzneimittel im Sinne der Richtlinie 2001/83/EG, richtet sich die Pharmakovigilanz nach § 63b AMG.
Erstmaliges Verbringen, Einfuhr
Für das erstmalige Verbringen von Zubereitungen aus dem Knochenmark aus einem EU-Mitgliedstaat/ EWR-Vertragsstaat in den Geltungsbereich des Arzneimittelgesetzes bedarf es einer Bescheinigung nach § 21a Absatz 9 AMG. Für die Einfuhr von Knochenmark und Zubereitungen aus dem Knochenmark aus Drittländern bedarf es einer Einfuhrerlaubnis nach § 72b AMG (siehe Abschnitt 3.5.1). Neben der Einfuhrerlaubnis nach § 72b Absatz 1 AMG ist für das Inverkehrbringen von Zubereitungen aus dem Knochenmark grundsätzlich eine Genehmigung nach § 21a Absatz 1 AMG erforderlich (siehe naher Abschnitt 3.3.1). Darüber hinaus bedarf es für die Einfuhr aus Drittländern nach § 72b Absatz 2 AMG eines Qualitätszertifikats des Herkunftslandes oder einer Qualitätsbescheinigung der für den Einführer zuständigen Behörde oder einer Bescheinigung dieser Landesbehörde, dass die Einfuhr im öffentlichen Interesse liegt.
Für die Einfuhr von Blutstammzellzubereitungen aus dem peripheren Blut oder aus Nabelschnurblut aus Drittländern bedarf es einer Erlaubnis nach § 72 Absatz 1 AMG. Darüber hinaus bedarf es grundsätzlich eines Zertifikats oder einer Bescheinigung nach § 72a Absatz 1 AMG. Letzteres soll nach § 72a Absatz 1a Nummer 2 AMG zukünftig nicht für solche Blutstammzellzubereitungen gelten, die zur gerichteten, Für eine bestimmte Person vorgesehene Anwendung bestimmt sind. Die mit der 15. AMG-Novelle eingeführte Erleichterung ermöglicht es, für einzelne Patientinnen oder Patienten bestimmte Blutstammzellzubereitungen einzuführen die zumeist in Notfällen für Anwendungen in lebensbedrohlichen Situationen benötigt werden.
Für die Einfuhr von Zubereitungen aus dem Knochenmark sowie Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut zur unmittelbaren Anwendung bei Menschen besteht die Möglichkeit, eine vereinfachte Erlaubnis nach § 72 Absatz 2 AMG zu beantragen (vgl. im Einzelnen Abschnitt 3.5.1). Für Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut zur unmittelbaren Anwendung bei Menschen ist nach § 72a Absatz 1a Nummer 2 AMG kein Qualitätszertifikat und keine Qualitätsbescheinigung erforderlich.
Nach Ansicht der Bundesregierung können auf der Grundlage der §§ 72, 72a und 72b AMG in der durch die 15. AMG-Novelle geänderten Fassung zukünftig angemessene Lösungen für die Einfuhr von Knochenmark, Zubereitungen aus dem Knochenmark sowie Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut in Eil- und Notfällen gefunden werden.
3.6.2 Allgemeine Einschätzung
Soweit in den Stellungnahmen Aussagen zum Bereich der Zubereitungen aus dem Knochenmark und/oder Blutstammzellzubereitungen aus dem peripheren Blut oder dem Nabelschnurblut getroffen werden, wird überwiegend thematisiert, dass es keine einheitliche Regelung gebe und ein sachlicher Grund Für die Differenzierung nicht ersichtlich sei. Die unterschiedlichen Regelungen führten zu einem zusätzlichen bürokratischen Aufwand und konnten die Wahl der optimalen Therapie negativ beeinflussen. Zudem mussten die Qualitätsanforderungen für alle genannten Zubereitungen identisch sein. Vereinzelt wird vorgeschlagen, die Vorschriften für diese Zubereitungen insgesamt den Regelungen für Blutzubereitungen oder insgesamt denjenigen für Gewebezubereitungen zu unterstellen oder aber einheitliche Sondervorschriften zu schaffen.
Die Bundesregierung ist der Auffassung, dass eine Angleichung der Regelungen sachlich nicht gerechtfertigt ist. Die unterschiedlichen Vorschriften im Transfusionsgesetz und Transplantationsgesetz für die Gewinnung von Knochenmark und Blutstammzellen aus dem peripheren Blut oder dem Nabelschnurblut gehen auf die unterschiedlichen Verfahren zur Gewinnung von Stammzellen des blutbildenden Systems zurück. Die verschiedenartigen Risiken und Belastungen für die Spender gebieten differenzierte Anforderungen.
So ist z.B. für die Gewinnung von Blutstammzellen aus dem peripheren Blut eine Vorbehandlung des Spenders zur Mobilisierung der Stammzellen notwendig. Dem Spender werden hierzu hormonähnliche Substanzen (Wachstumsfaktoren) verabreicht, die Auswirkungen auf verschiedene Zellarten haben und die Ausschüttung der Blutstammzellen in das periphere Blut bewirken. Dabei können akute Nebenwirkungen beim Spender auftreten. Die Anforderungen an diese Vorbehandlung sind in § 9 in Verbindung mit § 8 Absatz 2 bis 4 TFG geregelt. Insbesondere sind eine Begutachtung des Verfahrens zur Mobilisierung der Stammzellen und ein entsprechendes Votum der Ethik-Kommission erforderlich. Diese Begutachtung ist zur Gewährleistung der einwandfreien Durchführung der Spendervorbehandlung nach dem Stand der medizinischen Wissenschaft sowie auch der Einhaltung ethischer Anforderungen erforderlich.
Bei der Gewinnung von Knochenmark wird hingegen ein operativer Eingriff vorgenommen, der andere Belastungen für den Spender hat. Mit dem Gewebegesetz wurde die Differenzierung der Vorschriften für die Gewinnung von Blutstammzellen aus dem peripheren Blut und dem Nabelschnurblut im Transfusionsgesetz einerseits und von Knochenmark im Transplantationsgesetz andererseits grundsätzlich beibehalten.
Diese Differenzierung steht auch im Einklang mit den EG-Vorschriften. Zwar werden in Artikel 2 Absatz 1 der Richtlinie 2004/23/EG Stammzellen des blutbildenden Systems unabhängig von ihrer Herkunft dem Anwendungsbereich dieser Richtlinie unterstellt. Es ist jedoch gemäß Artikel 249 Absatz 3 EGV den Mitgliedstaaten überlassen, wie sie eine Richtlinie umsetzen, so dass in Deutschland die bisherige Systematik beibehalten werden konnte.
In verschiedenen Stellungnahmen wird zudem darauf hingewiesen, dass die Regelungen für das Inverkehrbringen und die Einfuhr für Knochenmark, Zubereitungen aus Knochenmark sowie Blutstammzellen und Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut nicht praktikabel seien und zu Durchführungsschwierigkeiten führten. Teilweise konnten potentielle Schaden für Patienten nur mit großem logistischen und finanziellen Aufwand abgewendet werden. Vorgeschlagen wird daher, die genannten Zubereitungen und ihre Ausgangsstoffe von den Vorschriften für das Inverkehrbringen und die Einfuhr im Arzneimittelgesetz auszunehmen. Nach Auffassung der Bundesregierung kann diesem Vorschlag nicht gefolgt werden. Die oben dargestellten Regelungen für das Inverkehrbringen und die Einfuhr sind auch für Knochenmark, Zubereitungen aus Knochenmark sowie Blutstammzellen und Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut erforderlich, um dem Sicherheitsbedürfnis der Patientinnen und Patienten gerecht zu werden. Sie entsprechen den Anforderungen der Artikel 6 Absatz 1 und Artikel 9 Absatz 1 der Richtlinie 2004/23/EG und berücksichtigen die in Artikel 9 Absatz 3 Buchstabe a und b der Richtlinie 2004/23/EG vorgesehenen Erleichterungen für die Einfuhr von den genannten Zubereitungen und ihren Ausgangsstoffen (zu weiteren Einzelheiten siehe Abschnitt 3.6.1).
3.6.3 Einzelfragen
Einzelne Verbände weisen darauf hin, dass im Hinblick auf die Ausnahmevorschrift in § 21 Absatz 2 Nummer 1d AMG Rechtsunsicherheit bestehe. Danach sind genehmigungspflichtige Gewebezubereitungen im Sinne von § 21a AMG von der Zulassungspflicht nach § 21 Absatz 1 AMG ausgenommen. Vorgeschlagen wird § 21 Absatz 2 Nummer 1d AMG um "Blutstammzellzubereitungen, die zur autologen oder gerichteten für eine bestimmte Person vorgesehenen Anwendung bestimmt sind" entsprechend der Regelung des § 21a AMG zu ergänzen. Die Bundesregierung sieht keinen Änderungsbedarf. Blutstammzellzubereitungen aus dem peripheren Blut und dem Nabelschnurblut, die zur autologen oder gerichteten, für eine bestimmte Person vorgesehenen Anwendung bestimmt sind, werden bereits von § 21 Absatz 2 Nummer 1a AMG umfasst, sie bedürfen folglich keiner Zulassung nach § 21 Absatz 1 AMG.
In zwei Stellungnahmen wird erläutert, dass die Verbringungsregelung des § 21a Absatz 9 AMG für individuell hergestellte Zubereitungen aus dem Knochenmark nicht umsetzbar sei. In der kurzen Zeit, die Für die Therapie zur Verfügung stehe, sei es kaum möglich, die Gleichwertigkeitsprüfung durchzuführen.
Vorgeschlagen wird, international anerkannte Standards und Akkreditierungsverfahren als Indikatoren einer Gleichwertigkeit der Anforderungen heranzuziehen. Nach Ansicht der Bundesregierung kann dem Vorschlag nicht gefolgt werden (zu Begründung und Verfahrenserleichterungen siehe Abschnitte 3.3.1 und 3.3.3). Im Verfahren nach § 21a Absatz 9 AMG kann auf eilbedürftige Einzelfalle Rücksicht genommen werden. In einzelnen Notfällen kommt ein Verbringen aus Gründen des rechtfertigenden Notstandes in Betracht.
In einer Stellungnahme wird angemerkt, dass sich die Regelung des § 72b Absatz 2 Satz 2 AMG für die Einfuhr von Zubereitungen aus dem Knochenmark zur gerichteten Anwendung nicht bewährt habe. Die Prüfung der Unterlagen sei sehr zeitaufwändig und teilweise stelle sich aufgrund der Schwierigkeit, einen passenden Spender zu finden, erst kurzfristig heraus, von welcher Entnahmeeinrichtung Knochenmark oder Zubereitungen aus dem Knochenmark bezogen werden können. Auch der Fall, dass der zuständigen Behörde eine Einrichtung im Drittland bereits bekannt ist, werde kaum eintreten. Es gebe unzählige potentielle Entnahmeeinrichtungen, die bisher nicht von den Landesbehörden besichtigt worden seien.
Die Bundesregierung ist der Ansicht, dass kein Änderungsbedarf besteht. Gemas § 72b Absatz 2 Satz 2 AMG kann die zuständige Behörde von einer Besichtigung absehen, wenn ihr die eingereichten Unterlagen keinen Anlass zur Beanstandung geben oder wenn die Entnahmeeinrichtung ihr bekannt ist. Im Eilfall, etwa wenn die Einfuhr aus Gründen des Gesundheitsschutzes eines Patienten dringend notwendig ist kann die zuständige Behörde gemäß § 72b Absatz 2 Satz 1 Nummer 3 AMG bescheinigen, dass die Einfuhr im öffentlichen Interesse liegt. Auch kann die zuständige Behörde entscheiden, den Betrieb des Einführers im Drittland und die einzelnen Entnahmeeinrichtungen, mit denen der Einführer im Drittland kooperiert stichprobenartig zu besichtigen.
3.7 Sonstiges
Im Zusammenhang mit dem Gewebegesetz wird § 4a Satz 1 Nummer 3 AMG thematisiert. Die Vorschrift werde in der Praxis regional unterschiedlich ausgelegt, so dass Klarstellungsbedarf bestehe. Aus Sicht der Bundesregierung besteht kein Klarstellungsbedarf mehr. Im Rahmen der 15. AMG-Novelle wurde die Vorschrift gestrichen. Stattdessen wird der neue § 20d AMG für Gewebe und Gewebezubereitungen eine Ausnahme von der Erlaubnispflicht nach § 20b Absatz 1 und § 20c Absatz 1 AMG vorsehen. Danach soll es keiner Erlaubnis bedürfen, wenn die in den Vorschriften genannten Tätigkeiten, mit Ausnahme des Inverkehrbringens, von einer Person, die Ärztin oder Arzt oder sonst zur Ausübung der Heilkunde bei Menschen befugt ist, ausgeübt werden, um die Gewebe oder Gewebezubereitung persönlich bei ihren Patienten anzuwenden; dies wird nicht für die klinische Prüfung gelten.
Darüber hinaus wird von zwei Ländern § 4a Satz 1 Nummer 4 AMG (jetzt § 4a Satz 1 Nummer 3 AMG) thematisiert es wird eine Konkretisierung dessen, was unter .innerhalb eines Behandlungsvorgangs" zu verstehen ist, angeregt. Aus Sicht der Bundesregierung besteht auch insoweit kein Handlungsbedarf mehr. Im Rahmen der 15. AMG-Novelle wurde § 4a Satz 1 Nummer 3 AMG präzisiert. Danach findet das Arzneimittelgesetz keine Anwendung, wenn Gewebe innerhalb eines Behandlungsvorgangs einer Person entnommen werden, um auf diese .ohne Änderung ihrer stofflichen Beschaffenheit" rückübertragen zu werden. Damit wurde verdeutlicht, dass geringfügige Arbeitsschritte, die im Hinblick auf die Anwendungsfähigkeit des Gewebes erforderlich sein können (z.B. das Säubern und Spulen oder Dehnen des autologen Gewebes, das Glätten seiner Schutzränder oder seine sachgerechte Aufbewahrung bis zur Anwendung) und nicht die Beschaffenheit des Gewebes verändern, nicht vom Anwendungsbereich des Arzneimittelgesetzes erfasst werden.
Darüber hinaus wurde vorgetragen, dass im Hinblick auf die Übergangsvorschriften nach § 142 Absatz 2 AMG Unsicherheit bestehe, insbesondere wie lange diese gelten. Die Übergangsvorschriften gelten Für die Anträge auf Erlaubnis nach §§ 13, 20b und 20c AMG mit Stichtag 1. Oktober 2007 und für die Genehmigung beziehungsweise Zulassung nach §§ 21a, 21 AMG mit Stichtag 1. Februar 2008 beziehungsweise
30. September 2008. Wer bis zu den genannten Stichtagen einen entsprechenden Antrag bei der zuständigen Behörde gestellt hat, darf weiter diese Tätigkeiten bis zur Entscheidung der zuständigen Behörde über die Anträge ausüben. Der Umfang der Antragsprüfung richtet sich nach dem konkreten Antrag, also zum Beispiel nach § 20b oder § 20c AMG einerseits und § 13 AMG andererseits. Eine Erlaubnis nach § 13 AMG oder § 20c AMG wird erst nach einer Besichtigung der Betriebsstätte erteilt (§ 64 Absatz 3 Satz 3 AMG). Bei einer Erlaubnis nach § 20b AMG kann von der Besichtigung abgesehen werden (§ 20b Absatz 1 Satz 4 und § 64 Absatz 3 Satz 3 AMG).
Die Arzneimittel- und Wirkstoffherstellungsverordnung (AMWHV) wurde als Artikel 1 der Verordnung zur Ablösung der Betriebsverordnung für pharmazeutische Unternehmer am 3. November 2006 erlassen (BGBl. I S. 2523). Sie wurde zwischenzeitlich einmal durch Artikel 1 der Verordnung vom 26. März 2008 (BGBl. I S. 521) geändert. Die AMWHV vom 3. November 2006 verfolgte das Ziel, die bis dahin geltende Betriebsverordnung für pharmazeutische Unternehmer abzulösen und die im Rahmen der 14. AMGNovelle erfolgten Änderungen umzusetzen.
Die erste Änderungsverordnung betrifft speziell Entnahme- und Gewebeeinrichtungen und Gewebespenderlabore.
Mit ihr wurden neben den erforderlichen Anpassungen an die durch das Gewebegesetz geänderte Rechtslage insbesondere mehrere europäische Durchführungsrichtlinien zur Richtlinie 2004/23/EG sowie aus dem Blutbereich umgesetzt.
4.1 Rechtslage
Im Einzelnen enthält die AMWHV ergänzende Anforderungen zum Qualitätsmanagementsystem (§ 32 AMWHV), zur Feststellung der Spendereignung und der für die Gewinnung erforderlichen Gewebespenderlabore (§ 33 AMWHV), zur Gewinnung von Gewebe durch die Entnahmeeinrichtungen (§ 34 AMWHV), zum Transport zur Be- oder Verarbeitung und Entgegennahme in der Gewebeeinrichtung ( § 35 AMWHV), zur Be- oder Verarbeitung und Lagerung durch diese ( § 36 AMWHV), zur Prüfung von Gewebe und Gewebezubereitungen (§ 37 AMWHV), zu Freigabe, Inverkehrbringen, Einfuhr und Transport durch die Gewebeeinrichtung (§§ 38 und 39 AMWHV), zur Meldung schwerwiegender unerwünschter Reaktionen und schwerwiegender Zwischenfälle und Rückrufe (§ 40 AMWHV) sowie zur Aufbewahrung der Dokumentation (§ 41 AMWHV).
4.2 Allgemeine Einschätzung
In den Stellungnahmen wurden keine wesentlichen Probleme von den beim Vollzug der AMWHV Beteiligten mitgeteilt. Dabei ist zu berücksichtigen, dass sich mangels eines ausreichenden Erfahrungszeitraums seit dem Inkrafttreten der ersten Änderungsverordnung zur AMWHV am 5. April 2008 nur wenige Stellungnahmen speziell mit der AMWHV befasst haben.
Insgesamt wird die Umsetzung der EG-rechtlichen Vorgaben in deutsches Recht sowie die Schaffung eines Sonderabschnittes in der AMWHV für Entnahme- und Gewebeeinrichtungen sowie für Gewebespenderlabore befürwortet. Insbesondere wird begrüßt, dass eine klare Unterscheidung zwischen Gewebeeinrichtungen und Entnahmeeinrichtungen vorgenommen worden ist, die auch der Systematik der EG-Richtlinien Für Gewebe entspricht.
Bemängelt werden die Schnittstellen und Querverweise in der AMWHV und in der TPG-GewV (§ 33 Absatz 1, § 34 Absatz 3 und 7, § 35 Absatz 1 AMWHV, § 5 Absatz 2 TPG-GewV). Dies erschwere in der Praxis die Rechtsanwendung.
Die Umsetzung der europäischen Durchführungsrichtlinien durch zwei verschiedene Verordnungen folgt der Entscheidung des Gesetzgebers, die europäischen Vorgaben je nach Inhalt im Transplantationsgesetz und im Arzneimittelgesetz zu regeln. Eine Zusammenführung beider Verordnungen erscheint insoweit nicht sinnvoll. Zudem ist zu berücksichtigen, dass neue Regelwerke mit zum Teil neuen Begriffsdefinitionen wie die AMWHV einen gewissen Zeitraum benötigen, bis die Anwendung der neuen Vorgaben reibungslos und auch einheitlich erfolgen kann. Vor diesem Hintergrund sieht die Bundesregierung derzeit keinen grundsätzlichen Änderungsbedarf der AMWHV.
4.3 Einzelfragen
In den Stellungnahmen wird mehrfach thematisiert, dass die Begriffsbestimmungen für die Gewebe- und Entnahmeeinrichtungen im Arzneimittel- und im Transplantationsrecht nicht deckungsgleich seien. Es wird angeregt, § 2 Absatz 10 und 11 AMWHV zu Einzelaspekten (z.B. Transport) weiter zu präzisieren.
Zum Teil wird auch dargelegt, dass der Begriff des Gewebespenderlabors nicht ausreichend sei.
Daneben wird insbesondere die Notwendigkeit der "Doppelregelung" zum Entnahmebericht in § 34 Absatz 7 Satz 5 AMWHV und in § 5 Absatz 2 der TPG-GewV in Frage gestellt. Auch der Bundesrat hat sich in seiner Entschließung zur AMWHV für eine Überführung der Regelung des § 5 Absatz 2 TPG-GewV in die AMWHV ausgesprochen (Empfehlung des federführenden Gesundheitsausschusses, Bundesrats-Drucksache 938/07 (PDF) , S. 8 f.).
Bei Stammzellpräparaten wird darauf hingewiesen, dass die in § 36 Absatz 8 AMWHV geforderten Angaben zur Kennzeichnung nicht auf ein Kryoetikett passten. Zudem lagen diese Angaben nicht bereits vor der Kryokonservierung vor.
Im Hinblick auf den Gesetzesvollzug durch die Überwachungsbehörden wird zum Teil angemerkt, dass die Auslegung und die Anwendung der Vorschriften durch die Behörden nicht einheitlich seien.
Die Bundesregierung ist der Ansicht, dass die in der AMWHV verwendeten Begriffsbestimmungen in Übereinstimmung mit der im Arzneimittelgesetz verwendeten Terminologie stehen. Die AMWHV dient als Rechtsverordnung zur Konkretisierung der gesetzlichen Vorgaben des Arzneimittelgesetzes sowie zur Umsetzung verbindlicher EG-rechtlicher Vorgaben. Die Bundesregierung wird jedoch den Wunsch nach einer klareren Abgrenzung von Begriffsdefinitionen prüfen. Gleiches gilt für Einzelaspekte der Verordnung, Für die der Wunsch nach einer Präzisierung beziehungsweise Konkretisierung geäußert worden ist.
5 Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung nach dem Transplantationsgesetz
Die auf Grund des § 16a Satz 1 und 2 TPG erlassene TPG-GewV vom 26. März 2008 (BGBl. I S. 512) regelt die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung, sofern dies der Abwehr von Gefahren für die menschliche Gesundheit und zur Risikovorsorge dient. In der Verordnung werden die durch das Gewebegesetz in §§ 8d ff. TPG verankerten Pflichten der Gewebeeinrichtungen sowie der Einrichtungen der medizinischen Versorgung, die Gewebe übertragen, konkretisiert.
Gleichzeitig wurden die entsprechenden Vorgaben der Richtlinie 2006/17/EG12 und der Richtlinie 2006/86/EG umgesetzt.
5.1. Rechtslage
Im Einzelnen regelt die Verordnung die Anforderungen an die Entnahme von Geweben ( § 2 TPG-GewV), an die ärztliche Beurteilung der medizinischen Eignung des Spenders (§ 3 TPG-GewV), an die Laboruntersuchungen und die Untersuchungsverfahren ( § 4 TPG-GewV), an die Spenderakte und den Entnahmebericht (§ 5 TPG-GewV) und die Voraussetzungen für die Verwendung von Keimzellen im Rahmen von Maßnahmen einer medizinisch unterstützten Befruchtung ( § 6 TPG-GewV) sowie die Anforderungen an die Dokumentation von übertragenen Geweben in Einrichtungen der medizinischen Versorgung (§ 7 TPG-GewV) und Meldung schwerwiegender Zwischenfälle und schwerwiegender unerwünschter Reaktionen durch Einrichtungen der medizinischen Versorgung (§§ 8 und 9 TPG-GewV).
5.2 Allgemeine Einschätzung
Die Befragung hat ergeben, dass ein Teil der Befragten auf noch unzureichende Erfahrungen innerhalb des Berichtszeitraums verweisen. Vielfach wurde jedoch festgestellt, dass sich die Regelungen der TPG-GewV grundsätzlich bewährt haben. Die Vorgaben der TPG-GewV wurden als gut umsetzbar bezeichnet.
Ein Handlungsbedarf für eine gesetzliche Änderung wird daher grundsätzlich nicht gesehen.
Ein Verband stellt fest, dass sich der bürokratische Aufwand allgemein erhöht habe. Vereinzelt werden Auslegungsfragen angesprochen wie z.B. der Verbleib der Spenderakte. Nach Auffassung der Bundesregierung bedarf es weiterer Erfahrungen in der Praxis, um prüfen zu können, ob gegebenenfalls konkreter gesetzgeberischer Handlungsbedarf besteht.
5.3 Einzelfragen
Im besonderen Fokus der Stellungnahmen stehen die Regelungen über die erforderlichen Laboruntersuchungen bei der Verwendung von Keimzellen bei einer Partnerspende im Rahmen von Maßnahmen einer künstlichen Befruchtung. Die vorgesehenen Möglichkeiten, innerhalb einer Partnerspende von der Durchführung biologischer Laboruntersuchungen abzusehen, werden vor allem von Reproduktionsmedizinern als unzureichend gesehen. Nach Auffassung eines Verbandes seien die in Anlage 4 Nummer 1 und 3 TPG-GewV gestellten Anforderungen überzogen und im homologen System insbesondere aus medizinischen Gründen nicht notwendig. Nach Aussage einer Fachgesellschaft gabe es in der Praxis zudem unterschiedliche Auslegungen der zuständigen Behörden einerseits und der Reproduktionsmediziner andererseits im Hinblick auf Anforderungen an validierte Verfahren im Zusammenhang mit der Exposition des Personals nach Anlage 4 Nummer 1 Buchstabe a TPG-GewV.
Nach Ansicht der Bundesregierung stellt sich die Rechtslage wie folgt dar:
Die erforderlichen Laboruntersuchungen für die Verwendung von Keimzellen sind in § 6 Absatz 1 in Verbindung mit Anlage 4 TPG-GewV festgelegt. Nach Anlage 4 Nummer 1 Buchstabe a TPG-GewV kann bei der Verwendung von Samenzellen, die zur intrauterinen Samenübertragung aufbereitet und nicht gelagert werden von der Durchführung der vorgesehenen Laboruntersuchungen bei der Partnerspende abgesehen werden sofern die Gewebeeinrichtung nachweisen kann, dass dem Risiko der Kreuzkontamination und der Exposition des Personals durch die Anwendung validierter Verfahren begegnet wurde. Nach Aussage der Europäischen Kommission beschränkt sich diese Ausnahme, die auf Anhang III Nummer 2.2 der Richtlinie 2006/17/EG beruht, ausschließlich auf die intrauterine Samenspende. Andere Verfahren der Fortpflanzungsmedizin sind von dieser Ausnahme nicht erfasst. Im Hinblick auf den Zeitpunkt der Probennahme regelt Anlage 4 Nummer 3 Buchstabe a in Verbindung mit Anhang II Nummer 2 Buchstabe e Doppelbuchstabe aa TPG-GewV, dass eine Blutprobe zum Zeitpunkt der Spende zu entnehmen ist oder, falls dies nicht möglich ist, innerhalb von sieben Tagen vor oder nach der Spende. Mit dieser Regelung werden die Anforderungen des Anhangs III Nummer 4.2 in Verbindung Anhang II Nummer 2.5 Buchstabe a der Richtlinie 2006/17/EG umgesetzt.
Ein Fachverband verweist auf die derzeit geltenden Richtlinien über künstliche Befruchtung des Gemeinsamen Bundesausschusses (G-BA), in denen die vorgesehenen biologischen Untersuchungen im Reproduktionsfall, d.h. von Behandlungsbeginn bis zum Eintritt einer Schwangerschaft, zu Lasten der GKV zu veranlassen sind. Die biologischen Untersuchungen bei der Partnerspende, die unter den oben genannten Voraussetzungen nicht zur intrauterinen Insemination verwendet werden, sind jeweils erneut im unmittelbaren Zusammenhang mit der Keimzellgewinnung durchzuführen - dies bedeutet z.B. bis zu 3 zusätzliche HIV-Tests pro Partner - und seien eine nicht unerhebliche finanzielle Zusatzbelastung für die GKV und die Patientinnen und Patienten. Die Richtlinien des G-BA werden zurzeit den Vorgaben der TPG-GewV angepasst. Dabei haben sich auch Schwierigkeiten im Hinblick auf die Anforderungen gemäß Anlage 4 Nummer 1 Buchstabe d und e TPG-GewV ergeben, da die schon in der Richtlinie 2006/17/EG festgelegten biologischen Untersuchungen (HTLV-I-Antikörper, RhD, Malaria u.a.) nicht ausreichend konkret ausgelegt werden, um daraus im Rahmen der Richtlinie des G-BA Leistungsansprüche abzuleiten.
Aus Sicht der Bundesregierung sollte die Notwendigkeit für Laboruntersuchungen bei der Partnerspende von Keimzellen überprüft werden. Die Bundesregierung wird diesbezüglich die von medizinischer Seite vorgebrachten Einwände gegen die geltende Richtlinie 2006/17/EG der Europäischen Kommission vortragen und eine sachgerechte Lösung auf europäischer Ebene anstreben.
6 Zusammenfassung und Schlussbemerkung
Die für den Erfahrungsbericht der Bundesregierung zum Gewebegesetz, zur AMWHV und zur TPG-GewV eingereichten Stellungnahmen zeigen, dass erst begrenzte Erfahrungen zum neuen Geweberecht vorliegen.
Soweit bereits Erfahrungen vorliegen, werden diese überwiegend positiv bewertet. Dies betrifft insbesondere die Vorschriften zur Erlaubnis für die Gewinnung von Geweben und Laboruntersuchungen ( § 20b AMG), die Vorschriften zur Erlaubnis für die Be- oder Verarbeitung, Konservierung, Prüfung, Lagerung oder das Inverkehrbringen von Gewebe oder Gewebezubereitungen ( § 20c AMG) sowie die Vorschriften zur Genehmigung von Gewebezubereitungen (§ 21a AMG), insbesondere die im Vergleich zu den allgemeinen Vorschriften enthaltenen Verfahrensvereinfachungen nach §§ 20b und 21a AMG. In Teilbereichen sind abschließende Beurteilungen noch nicht möglich. Dies betrifft neben dem Genehmigungsverfahren Für Gewebezubereitungen insbesondere die Vorschriften zu den besonderen Dokumentations-und Meldepflichten bei Blut- und Gewebezubereitungen ( § 63c AMG) sowie die Vorschriften zur Einfuhrerlaubnis und zu den Qualitätszertifikaten für Gewebe und bestimmte Gewebezubereitungen (§ 72b AMG). Mangels eines ausreichenden Erfahrungszeitraums ist auch eine abschließende Bewertung zur AMWHV und zur TPG-GewV noch nicht möglich.
Auf der Grundlage der in den Stellungnahmen übermittelten Erfahrungen und Anregungen für eine Fortentwicklung des Gewebegesetz können aus Sicht der Bundesregierung folgende Änderungen der bestehenden Regelungen in Betracht gezogen werden:
- - Ergänzung einer Fristenregelung in § 20b Absatz 1 AMG sowie
- - Ergänzung einer Verpflichtung zur Änderungsanzeige für nachträgliche Änderungen bei der Gewinnung von Gewebe und den erforderlichen Laboruntersuchungen in § 20b AMG.
Auf der Grundlage der eingereichten Stellungnahmen wird die Bundesregierung insbesondere folgende Sachverhalte beziehungsweise Vorschläge prüfen:
- - bestehende Zulassungspflicht für bestrahlte Gewebezubereitungen nach § 21 AMG
- - rechtliche Bewertung von sogenannten komplexen Geweben sowie
- - Erweiterung der Dokumentations- und Berichtspflichten nach § 63c AMG auf nicht schwerwiegende Zwischenfälle und Reaktionen zur Erkennung einer unverhältnismäßigen Häufung solcher Zwischenfälle und Reaktionen.
Die Durchführung neuer Regelungen beansprucht erfahrungsgemäß einige Zeit. Nach Auffassung der Bundesregierung sollten zunächst mehr Erfahrungen gesammelt werden, bevor darüber hinaus gehende Änderungen erwogen werden. Insgesamt sieht die Bundesregierung keinen Anlass für grundlegende strukturelle Änderungen am Gewebegesetz, an der AMWHV und der TGP-GewV. Zur Frage der Notwendigkeit von Laboruntersuchungen bei der Partnerspende von Keimzellen wird die Bundesregierung die diesbezüglich vorgebrachten Einwände gegen die geltende Richtlinie 2006/17/EG der Europäischen Kommission vortragen und eine sachgerechte Lösung auf europäischer Ebene anstreben.
Nach den aus der Praxis übermittelten Einschätzungen haben die Regelungen des Gewebegesetzes zu einer deutlichen Vereinheitlichung der Qualitäts- und Sicherheitsstandards und damit zu einer signifikanten Verbesserung der Qualität von Gewebezubereitungen geführt. Nach Einschätzung der Bundesregierung gewährleisten die Vorschriften des Gewebegesetzes in angemessener Weise die Sicherheit von Gewebe und Gewebezubereitungen und somit den Schutz der Patientinnen und Patienten. Sie setzen Standards, die zunehmend weltweit eingeführt werden.
Anlagen
Anlage 1
Bundesrats-Drucksache 385/07(B) 06.07.07
Beschluss des Bundesrates
Gesetz über Qualität und Sicherheit von menschlichen Geweben und Zellen (Gewebegesetz)
Der Bundesrat hat in seiner 835. Sitzung am 6. Juli 2007 beschlossen, zu dem vom Deutschen Bundestag am 24. Mai 2007 verabschiedeten Gesetz einen Antrag gemäß Artikel 77 Absatz 2 des Grundgesetzes nicht zu stellen.
Der Bundesrat hat ferner die nachstehende Entschließung gefasst:
Der Bundesrat begrüßt, dass zum Gewebegesetz letztendlich ein vertretbarer Kompromiss zwischen den Ländern und der Bundesregierung zustande gekommen ist.
Inhalt dieses Kompromisses ist unter anderem die nunmehr vorgesehene Unterteilung in bekannte und neuartige Gewebe bzw. Gewebezubereitungen. Danach werden die Bestimmungen bezüglich der Be- und Verarbeitung von bekannten Geweben sowie deren Konservierung, Lagerung und Inverkehrbringen vereinfacht.
Um zu gewährleisten, dass auf diesem Weg tatsächlich das ursprüngliche Ziel einer praktikablen, unbürokratischen und trotzdem sicheren Regelung für bekannte Gewebe erreicht wurde, wird die Bundesregierung gebeten sobald als möglich, jedoch spätestens zwei Jahre nach Inkrafttreten des Gewebegesetzes, dem Bundesrat über die dann vorliegenden Erfahrungen zu berichten.
Anlage 2
Bundesrat Drucksache 938/07(B) 15.02.08
Beschluss des Bundesrates
Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung
Der Bundesrat hat in seiner 841. Sitzung am 15. Februar 2008 beschlossen, der Verordnung gemäß Artikel 80 Absatz 2 des Grundgesetzes nach Maßgabe der sich aus der Anlage ergebenden Änderungen zuzustimmen.
Der Bundesrat hat ferner die nachstehende Entschließung gefasst:
- 1. Aus Anlass der Behandlung der Verordnung zur Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung sowie der TPG-Gewebeverordnung (vgl. BR-Drucksache 939/07 (PDF) ) betont der Bundesrat nochmals seine bereits mit Beschluss vom 6. Juli 2007 (vgl. BR-Drucksache 385/07 (PDF) Beschluss -) zum Ausdruck gebrachte Position, dass für Gewebe zwar sichere, aber gleichzeitig auch praktikable und unbürokratische Regelungen erforderlich sind. Aus diesem Grund soll der mit Beschluss vom 6. Juli 2007 (vgl. BR-Drucksache 385/07(B) ) erbetene Erfahrungsbericht der Bundesregierung ganz ausdrücklich auch die Konsequenzen, die sich durch die geänderte Arzneimittel- und Wirkstoffherstellungsverordnung sowie die TPG-Gewebeverordnung für die Gewebeeinrichtungen ergeben, mit beinhalten. Dabei soll besonders berücksichtigt werden, ob die gegenwärtig getroffenen Regelungen zur Gewebeentnahme sowohl in der Arzneimittel- und Wirkstoffherstellungsverordnung als auch der TPG-Gewebeverordnung rechtlich notwendig, fachlich sinnvoll und praktisch umsetzbar sind.
- 2. Der Bundesrat bittet die Bundesregierung, im Rahmen der 15. Novelle des Arzneimittelgesetzes zu klaren, wie externe Prüfeinrichtungen, die zum Beispiel mikrobiologische Prüfungen von Geweben oder Gewebezubereitungen vornehmen, hinsichtlich ihrer Erlaubnispflicht behandelt werden.
Begründung:
§ 20c Arzneimittelgesetz sieht für Gewebe oder Gewebezubereitungen, die nicht mit industriellen Verfahren be- oder verarbeitet, konserviert, gelagert oder in Verkehr gebracht wurden und deren wesentliche Be- oder Verarbeitungsverfahren in der Europäischen Union hinreichend bekannt sind abweichend von § 13 Arzneimittelgesetz eine erleichterte Erlaubnis für die Be- oder Verarbeitung, Konservierung, Lagerung und das Inverkehrbringen vor.
Es ist zu überlegen, ob eine § 14 Absatz 4 Arzneimittelgesetz entsprechende Regelung, nämlich die Möglichkeit, externe Prüfeinrichtungen in die Erlaubnis nach § 20c Arzneimittelgesetz aufnehmen zu können, in § 20c aufgenommen werden soll.
Eine solche Regelung wurde vermeiden, dass alle externen Prüfeinrichtungen von Betrieben mit einer Erlaubnis nach § 20c Arzneimittelgesetz einer eigenständigen Erlaubnis bedürfen.
Anlage 3
Bundesrat Drucksache 939/07(B) 15.02.08
Beschluss des Bundesrates
Verordnung über die Anforderungen an Qualität und Sicherheit der Entnahme von Geweben und deren Übertragung nach dem Transplantationsgesetz (TPG-Gewebeverordnung - TPG-GewV)
Der Bundesrat hat in seiner 841. Sitzung am 15. Februar 2008 beschlossen, der Verordnung gemäß Artikel 80 Absatz 2 des Grundgesetzes zuzustimmen.
Der Bundesrat hat ferner die nachstehende Entschließung gefasst:
Aus Anlass der Behandlung der TPG-Gewebeverordnung sowie der Änderung der Arzneimittel- und Wirkstoffherstellungsverordnung (vgl. BR-Drucksache 938/07 (PDF) ) betont der Bundesrat nochmals seine bereits mit Beschluss vom 6. Juli 2007 (vgl. BR-Drucksache 385/07(B) ) zum Ausdruck gebrachte Position, dass für Gewebe zwar sichere, aber gleichzeitig auch praktikable und unbürokratische Regelungen erforderlich sind. Aus diesem Grund soll der mit Beschluss vom 6. Juli 2007 (vgl. BR-Drucksache 385/07 (PDF) -Beschluss -) erbetene Erfahrungsbericht der Bundesregierung ganz ausdrücklich auch die Konsequenzen, die sich durch die geänderte Arzneimittel- und Wirkstoffherstellungsverordnung sowie die TPG-Gewebeverordnung Für die Gewebeeinrichtungen ergeben, mit beinhalten. Dabei soll besonders berücksichtigt werden ob die gegenwärtig getroffenen Regelungen zur Gewebeentnahme sowohl in der Arzneimittel-und Wirkstoffherstellungsverordnung als auch der TPG-Gewebeverordnung rechtlich notwendig, fachlich sinnvoll und praktisch umsetzbar sind.
- 1 Verordnung (EG) Nr. 1394/2007 vom 13. November 2007 über Arzneimittel für neuartige Therapien und zur Änderung der Richtlinie 2001/83/EG und der Verordnung (EG) Nr. 726/2004, ABl. L 324 vom 12. Dezember 2007, S. 121.
- 2 Richtlinie 2004/23/EG des Europäischen Parlamentes und des Rates vom 31. März 2004 zur Festlegung von Qualitäts- und Sicherheitsstandards für die Spende, Beschaffung, Testung, Verarbeitung, Konservierung, Lagerung und Verteilung von menschlichen Geweben und Zellen, ABl. L 102 vom 31. März 2004, S. 48.
- 3 Verfahrensanweisung 15111601 vom 23. Juli 2008, Qualitätssicherungshandbuch der ZLG, Kapitel 15.
- 4 § 20c ist an § 22 Absatz 3 Nummer 1 AMG angelehnt.
- 5 Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel, ABl. L 311 vom 28. November 2001, S. 67, zuletzt geändert durch Richtlinie 2008/29/EG vom 11. März 2008, ABl. L 81 vom 20. März 2008, S. 51.
- 6 Verfahrensanweisung 15111601, Qualitätssicherungshandbuch der ZLG, Kapitel 15.
- 7 Richtlinie 2006/86/EG der Kommission vom 24. Oktober 2006 zur Umsetzung der Richtlinie 2004/23/EG des Europäischen Parlaments und des Rates hinsichtlich der Anforderungen an die Rückverfolgbarkeit, der Meldung schwerwiegender Zwischenfälle und unerwünschter Reaktionen sowie bestimmter technischer Anforderungen an die Kodierung, Verarbeitung, Konservierung, Lagerung und Verteilung von menschlichen Geweben und Zellen, ABl. L 294 vom 25. Oktober 2006, S. 32.
- 8 Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates vom 27. Januar 2003 zur Festlegung von Qualitäts- und Sicherheitsstandards für die Gewinnung, Testung, Verarbeitung, Lagerung und Verteilung von menschlichem Blut und Blutbestandteilen und zur Änderung der Richtlinie 2001/83/EG, ABl. L 33 vom 8. Februar 2003, S. 30.
- 9 Richtlinie 2005/61/EG der Kommission vom 30. September 2005 zur Durchführung der Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates in Bezug auf die Anforderungen an die Rückverfolgbarkeit und die Meldung ernster Zwischenfälle und ernster unerwünschter Reaktionen, ABl. L 256 vom 1. Oktober 2005, S. 32.
- 10 Richtlinie 2005/62/EG der Kommission vom 30. September 2005 zur Durchführung der Richtlinie 2002/98/EG des Europäischen Parlaments und des Rates in Bezug auf gemeinschaftliche Standards und Spezifikationen für ein Qualitätssystem für Blutspendeeinrichtungen, ABl. L 256 vom 1. Oktober 2005, S. 41.
- S. 1, zuletzt geändert durch Verordnung (EG) Nr. 1394/2007 vom 13. November 2007, ABl. L 324 vom 10. Dezember 2007, S. 130.
- 11 Verordnung (EG) Nr. 726/2004 des Europäischen Parlaments und des Rates vom 31. März 2004 zur Festlegung von Gemeinschaftsverfahren für die Genehmigung und Überwachung von Human- und Tierarzneimitteln und zur Errichtung einer Europäischen Arzneimittel-Agentur, ABl. L 136 vom 30. April 2004,
- 12 Richtlinie 2006/17/EG der Kommission vom 8. Februar 2006 zur Durchführung der Richtlinie 2004/23/EG des Europäischen Parlaments und des Rates hinsichtlich technischer Vorschriften für die Spende, Beschaffung und Testung von menschlichen Geweben und Zellen hinsichtlich der Anforderungen an Gewebeeinrichtungen, die Gewebe entnehmen oder untersuchen, ABl. L 38 vom 9. Februar 2006, S. 40.