
umwelt-online: Richtlinie 2000/45/EG zur Festlegunggemeinschaftlicher Analysemethoden für die Bestimmung von Vitamin A,Vitamin E und Tryptophan in Futtermitteln (2)
 | zurück |  |
5.4. Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (von 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben (4.2.4) pipettiert. Das Lösungsmittel wird unter reduziertem Druck und bei einer Badtemperatur von höchstens 40 °C am Rotationsverdampfer (4.1) fast bis zur Trockne eingedampft. Darauf wird unter Stickstoff (3.9) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom (3.9) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10-100 ml) Methanol (3.3) aufgenommen (die Konzentration von DL-a-Tocopherol sollte 5 µg/ml bis 30 µg/ml betragen).
5.5. Bestimmung durch HPLC
Die Abtrennung von Vitamin E erfolgt mit Hilfe einer Umkehrphasen-C18-Säule (4.5.1), und die Konzentration wird mit Hilfe eines Fluoreszenzdetektors (Anregung: 295 nm, Emission: 330 nm) oder eines UV-Detektors (292 nm) (4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z.B. 20 µl) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase (3.8) eluiert. Die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen derselben Probenlösung werden ermittelt. In der gleichen Weise werden die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen (5.6.2) bestimmt.
HPLC-Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| Flüssigkeitschromatographiesäule (4.5.1): | 250 mm × 4 mm, C18, 5 oder 10 µm Korngröße oder gleichwertiges Material |
| Mobile Phase (3.8): | Mischung von Methanol (3.3) und Wasser z.B. 980 + 20 (v + v) |
| Durchflussrate: | 1-2 ml/Min. |
| Detektor (4.5.2): | Fluoreszenzdetektor (Anregung: 295 nm Emission: 330 nm) oder UV-Detektor (292 nm) |
5.6. Eichung (DL-α-Tocopherol-Acetat oder DL-α-Tocopherol)
5.6.1. DL-α-Tocopherol-Acetat-Standard
5.6.1.1. Herstellen der Arbeits-Standardlösung
25 ml der DL-α-Tocopherol-Acetat-Stammlösung (3.10.1) in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) pipettieren und wie unter Nummer 5.2 beschrieben hydrolysieren. Anschließend mit Petrolether (3.2) gemäß Nummer 5.3 extrahieren und mit Petrolether auf 500 ml auffüllen. 25 ml dieses Extrakts werden am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft, das verbleibende Lösungsmittel wird mit einem Stickstoffstrom (3.9) abgeblasen, und der Rückstand wird in 25,0 ml Methanol (3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 45,5 µg DL-α-Tocopherol je ml; das entspricht 50 µg DL-α-Tocopherol-Acetat je ml. Die Arbeits-Standardlösung muss vor Gebrauch frisch zubereitet werden.
5.6.1.2. Herstellen der Kalibrierlösungen und Erstellung der Kalibrierkurve
1,0 ml, 2,0 ml, 4,0 ml und 10,0 ml der Arbeits-Standardlösung werden in eine Reihe von 20-ml-Messkolben pipettiert; bis zur Marke mit Methanol (3.3) auffüllen und mischen. Der Sollgehalt dieser Lösungen beträgt 2,5 µg/ml, 5,0 µg/ml, 10,0 µg/ml und 25,0 µg/ml DL-α-Tocopherol-Acetat, d. h. 2,28 µg/ml, 4,55 µg/ml, 9,10 µg/ml und 22,8 µg/ml DL-α-Tocopherol.
20 µl von jeder Kalibrierlösung werden mehrmals eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Unter Verwendung der mittleren Peakhöhen (-flächen) wird eine Kalibrierkurve erstellt.
5.6.1.3. UV-Kontrolle der DL-α-Tocopherol-Acetat-Stammlösung (3.10.1)
5,0 ml der DL-α-Tocopherol-Acetat-Stammlösung (3.10.1) werden mit Ethanol auf 25,0 ml aufgefüllt, das UV-Spektrum dieser Lösung wird gegen Ethanol (3.1) im Spektralphotometer (4.6) zwischen 250 nm und 320 nm gemessen.
Das Extinktionsmaximum soll bei 284 nm liegen.
| (E | 1 % | = 43,6 bei 284 nm in Ethanol |
| 1 m |
Bei der vorliegenden Verdünnung sollte eine Extinktion von 0,84 bis 0,88 erzielt werden.
5.6.2. DL-α-Tocopherol-Standard
5.6.2.1. Herstellen der Arbeits-Standardlösung
2,0 ml der DL-α-Tocopherol-Stammlösung (3.11.1) werden in einen 50-ml-Messkolben pipettiert, in Methanol (3.3) gelöst und bis zur Marke mit Methanol aufgefüllt. Der Sollgehalt dieser Lösung beträgt 40 µg DL-α-Tocopherol je ml; das entspricht 44,0 µg DL-α-Tocopherol-Acetat je ml. Die Arbeits-Standardlösung muss vor Gebrauch frisch zubereitet werden.
5.6.2.2. Herstellen der Kalibrierlösungen und Erstellung der Kalibrierkurve
1,0 ml, 2,0 ml, 4,0 ml und 10,0 ml der Arbeits-Standardlösung werden in eine Reihe von 20-ml-Messkolben pipettiert; bis zur Marke mit Methanol (3.3) auffüllen und mischen. Der Sollgehalt dieser Lösungen beträgt 2,0 µg/ml, 4,0 µg/ml, 8,0 µg/ml und 20,0 µg/ml DL-α-Tocopherol, d. h. 2,20 µg/ml, 4,40 µg/ml, 8,79 µg/ml und 22,0 µg/ml DL-α-Tocopherol-Acetat.
20 µl von jeder Kalibrierlösung werden mehrmals eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Unter Verwendung der mittleren Peakhöhen (-flächen) wird eine Kalibrierkurve erstellt.
5.6.2.3. UV-Kontrolle der DL-α-Tocopherol-Stammlösung (3.11.1)
2,0 ml der DL-α-Tocopherol-Stammlösung (3.11.1) werden mit Ethanol auf 25,0 ml aufgefüllt, das UV-Spektrum dieser Lösung wird gegen Ethanol (3.1) im Spektralphotometer (4.6) zwischen 250 nm und 320 nm gemessen. Das Extinktionsmaximum soll bei 292 nm liegen.
| (E | 1 % | = 75,8 bei 292 nm in Ethanol |
| 1 m |
Bei der vorliegenden Verdünnung sollte eine Extinktion von 0,6 erzielt werden.
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-E-Peaks der Probenlösung wird anhand der Kalibrierkurve (5.6.1.2 oder 5.6.2.2) die Konzentration der Probenlösung in gg/ml (berechnet als α-Tocopherol-Acetat) bestimmt.
Der Vitamin-E-Gehalt w in mg/kg der Probe wird nach folgender Formel berechnet:
| w = | 500 · β · V2 | [mg/kg] |
|
| ||
| V1 · m |
Dabei ist:
β = Vitamin-E-Konzentration der Probenlösung (5.4) in µg/ml
V1 = Volumen der Probenlösung (5.4) in ml
V2 = Volumen des bei 5.4 verwendeten aliquoten Teils in ml
m = Probeneinwaage in g
7. Bemerkungen
7.1. Bei Proben mit niedrigen Vitamin-E-Gehalten kann es zweckmäßig sein, die Petroletherextrakte von zwei Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen.
7.2. Die Einwaage für die Analyse sollte höchstens 2 g Fett enthalten.
7.3. Bei schlechter Phasentrennung können etwa 10 ml Ethanol (3.1) zur Zerstörung der Emulsion zugegeben werden.
7.4. Nach der spektralphotometrischen Vermessung der DL-α-Tocopherol-Acetat- oder DL-α-Tocopherol-Lösung nach 5.6.1.3 bzw. 5.6.2.3 empfiehlt es sich, der Lösung (3.10.1 bzw. 3.10.2) etwa 10 mg BHT (3.12) zuzusetzen und die Lösung im Kühlschrank aufzubewahren (Haltbarkeitsdauer höchstens vier Wochen).
7.5. Statt BHT kann Hydrochinon verwendet werden.
7.6. Bei Verwendung einer Normalphasensäule ist die Trennung von α-, β-, χ- und δ-Tocopherol möglich.
7.7. Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden.
7.8. Statt Natriumsulfid-Lösung können etwa 50 mg EDTA verwendet werden.
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen bei ein und derselben Probe darf 15 % des höheren Resultats nicht überschreiten.
9. Ergebnisse eines Ringversuchs 1
| Vormischung | Vormischung | Mineralfutter | Eiweißkonzentrat | Ferkelaufzuchtfutter | |
| L | 12 | 12 | 12 | 12 | 12 |
| n | 48 | 48 | 48 | 48 | 48 |
| Mittelwert [mg/kg] | 17.380 | 1.187 | 926 | 315 | 61,3 |
| sr [mg/kg] | 384 | 45,3 | 25,2 | 13,0 | 2,3 |
| r [mg/kg] | 1.075 | 126,8 | 70,6 | 36,4 | 6,4 |
| CVr [%] | 2,2 | 3,8 | 2,7 | 4,1 | 3,8 |
| sR [mg/kg] | 830 | 65,0 | 55,5 | 18,9 | 7,8 |
| R [mg/kg] | 2.324 | 182,0 | 155,4 | 52,9 | 21,8 |
| CVR [%] | 4,8 | 5,5 | 6,0 | 6,0 | 12,7 |
| L: Zahl der Labors n: Zahl der Einzelwerte sr: Wiederholdstandardabweichung sR: Vergleichstandardabweichung r: Wiederholbarkeit R: Vergleichbarkeit CVr: Wiederholvariationskoeffizient CVR: Vergleichsvariationskoeffizient 1) Durchgeführt von der Fachgruppe Futtermittel des Verbands Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA). | |||||
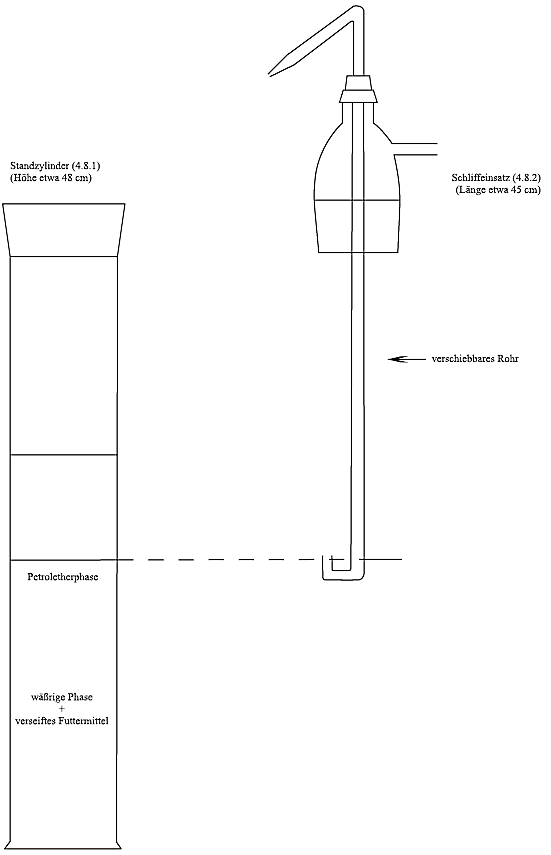
Abbildung 1: Extraktionsapparat (4.8)
Teil C
Bestimmung von Tryptophan
1. Zweck und Anwendungsbereich
Die Methode dient zur Bestimmung des Gesamtgehalts an Tryptophan und des Gehalts an freiem Tryptophan in Futtermitteln. Sie dient nicht zur Unterscheidung zwischen D- und L-Formen.
2. Prinzip
Zur Bestimmung des Gesamtgehalts an Tryptophan wird die Probe unter alkalischen Bedingungen mit gesättigter Bariumhydroxid-Lösung hydrolysiert und 20 Std. auf 110 °C erhitzt. Nach der Hydrolyse wird ein interner Standard zugegeben.
Zur Bestimmung des Gehalts an freiem Tryptophan wird die Probe unter leicht sauren Bedingungen in Gegenwart eines internen Standards extrahiert.
Tryptophan und interner Standard im Hydrolysat bzw. im Extrakt werden durch HPLC mit Fluoreszenzdetektor bestimmt.
3. Reagenzien
3.1. Es ist bidestilliertes Wasser oder Wasser gleichwertiger Qualität zu verwenden (Leitfähigkeit < 10 µS/cm).
3.2. Standardsubstanz: Tryptophan (Reinheit/Gehalt > 99 %), im Vakuum über Phosphorpentoxid getrocknet
3.3. Interner Standard: a-Methyl-Tryptophan (Reinheit/Gehalt > 99 %), im Vakuum über Phosphorpentoxid getrocknet
3.4. Bariumhydroxid-Octahydrat (das Ba(OH)2 · 8 H2O darf nicht übermäßig der Luft ausgesetzt werden, um die Bildung von BaCO3 zu vermeiden, das die Bestimmung stören könnte (siehe Bemerkung unter 9.3).
3.5. Natriumhydroxid
3.6. Orthophosphorsäure, w = 85 %
3.7. Salzsäure, ρ20, 1,19 g/ml
3.8. Methanol, HPLC-Qualität
3.9. Petrolether, Siedeintervall 40 bis 60 °C
3.10. Natriumhydroxid-Lösung, c = 1 mol/l: 40,0 g NaOH (3.5) in Wasser lösen und mit Wasser (3.1) auf 1 Liter auffüllen.
3.11. Salzsäure, c = 6 mol/l: 492 ml HCl (3.7) mit Wasser auf 1 Liter auffüllen.
3.12. Salzsäure, c = 1 mol/l: 82 ml HCl (3.7) mit Wasser auf 1 Liter auffüllen.
3.13. Salzsäure, c = 0,1 mol/l: 8,2 ml HCl (3.7) mit Wasser auf 1 Liter auffüllen.
3.14. Orthophosphorsäure, c = 0,5 mol/l: 34 ml Orthophosphorsäure (3.6) mit Wasser (3.1) auf 1 Liter auffüllen.
3.15. Konzentrierte Tryptophan-Lösung (3.2), c = 2,50 µmol/ml: 0,2553 g Tryptophan (3.2) in einem 500-ml-Messkolben in Salzsäure (3.13) lösen und mit Salzsäure bis zur Marke auffüllen. Bei - 18 °C höchstens vier Wochen aufbewahren.
3.16. Konzentrierte interne Standardlösung, c = 2,50 µmol/ml: 0,2728 g α-Methyl-Tryptophan (3.3) in einem 500-ml-Messkolben in Salzsäure (3.13) lösen und mit Salzsäure (3.13) bis zur Marke auffüllen. Bei - 18 °C höchstens vier Wochen aufbewahren.
3.17. Eichstandardlösung von Tryptophan und internem Standard:
2,00 ml konzentrierte Tryptophan-Lösung (3.15) und 2,00 ml konzentrierte interne Standardlösung (α-Methyl-Tryptophan) (3.16) mit Wasser (3.1) und Methanol (3.8) auf etwa dasselbe Volumen und etwa dieselbe Methanolkonzentration (10-30 %) wie das fertige Hydrolysat verdünnen.
Diese Lösung muss vor Gebrauch frisch hergestellt werden. Bei der Zubereitung vor direktem Sonnenlicht schützen.
3.18. Essigsäure
3.19. 1,1,1-Trichlor-2-methyl-2-propanol
3.20. Ethanolamin > 98 %
3.21. Lösung von 1 g 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in 100 ml Methanol (3.8)
3.22. Mobile Phase für HPLC: 3,00 g Essigsäure (3.18) + 900 ml Wasser (3.1) + 50,0 ml Lösung (3.21) von 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in Methanol (3.8) (1 g/100 ml). Der pH-Wert wird mit Ethanolamin (3.20) auf 5,00 eingestellt. Mit Wasser (3.1) auf 1.000 ml auffüllen.
4. Geräte
4.1. HPLC-Einrichtung mit Fluoreszenzdetektor
4.2. Flüssigkeitschromatographiesäule, 125 mm × 4 mm, C18, Korngröße 3 µm, oder gleichwertiges Material
4.3. pH-Meter
4.4. Polypropylen-Kolben, 125 ml, Weithals mit Schraubkappe
4.5. Membranfilter, 0,45 µm
4.6. Autoklav, 110 (± 2) °C, 1,4 (± 0,1) bar
4.7. Mechanisches Schüttelgerät oder Magnetrührer
4.8. Vortex-Schüttelgerät
5. Durchführung
5.1. Vorbereitung der Proben
Die Probe wird so fein vermahlen, dass sie ein Sieb mit 0,5 mm Maschenweite passieren kann. Proben mit hohem Feuchtigkeitsgehalt müssen vor dem Vermahlen entweder bei einer Temperatur von höchstens 50 °C luftgetrocknet oder gefriergetrocknet werden. Proben mit hohem Fettgehalt sollten vor dem Vermahlen mit Petrolether (3.9) extrahiert werden.
5.2. Bestimmung von freiem Tryptophan (Extrakt)
Eine geeignete Menge (1-5 g) der vorbereiteten Probe (5.1) auf 1 mg genau in einen Erlenmeyerkolben einwiegen. 100,0 ml Salzsäure, c = 0,1 mol/l, (3.13) und 5,00 ml konzentrierte interne Standardlösung (3.16) zugeben. 60 Min. mit einem mechanischen Schüttelgerät oder einem Magnetrührer (4.7) schütteln bzw. rühren. Absetzen lassen und 10,0 ml des Überstands in ein Becherglas pipettieren. 5 ml Orthophosphorsäure, c = 0,5 mol/l, (3.14) zugeben. Der pH-Wert wird mit Natriumhydroxid-Lösung, c = 1,0 mol/l, (3.10) auf 3,0 eingestellt. Ausreichend Methanol (3.8) für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatographie notwendige Volumen verdünnen (etwa dasselbe Volumen wie die Kalibrierstandardlösung (3.17)).
Einige ml der Lösung werden durch ein 0,45-pm-Membranfilter (4.5) filtriert, bevor ein Aliquot auf die HPLC-Säule gegeben wird. Die Chromatographie gemäß Nummer 5.4 durchführen.
Standardlösung und Extrakte sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Extrakte am selben Tag zu analysieren, können sie bei 5 °C maximal drei Tage aufbewahrt werden.
5.3. Bestimmung des Gesamttryptophans (Hydrolysat)
0,1 bis 1 g der vorbereiteten Probe (5.1) werden auf 0,2 mg genau in den Polypropylenkolben (4.4) eingewogen. Der eingewogene Teil der Probe sollte einen Stickstoffgehalt von etwa 10 mg haben. 8,4 g Bariumhydroxid-Octahydrat (3.4) und 10 ml Wasser zugeben. Mit Hilfe eines Vortex-Schüttelgeräts (4.8) oder Magnetrührgeräts (4.7) mischen. Den teflonbeschichteten Magneten in der Mischung lassen. Die Gefäßwände mit 4 ml Wasser abspülen. Schraubkappe aufsetzen und Kolben locker verschließen. In einem Autoklaven (4.6) 30-60 Minuten mit kochendem Wasser erhitzen. Den Autoklaven schließen und 20 Stunden bei 110 (± 2) °C autoklavieren.
Vor dem Öffnen des Autoklaven ist die Temperatur auf knapp unter 100 °C zu senken. Um Kristallisation des Ba(OH)2 · 8 HO2 zu vermeiden, sind der warmen Mischung 30 ml Wasser mit Zimmertemperatur zuzugeben. Leicht schütteln oder rühren. 2,00 ml konzentrierte interne Standardlösung (-Methyl-Tryptophan) (3.16) zugeben. Die Gefäße im Wasser-/Eisbad 15 Min. kühlen.
Anschließend 5 ml Orthophosphorsäure, c = 0,5 mol/l, (3.14) zugeben. Das Gefäß im Kühlbad belassen und unter Rühren mit HCl, c = 6 mol/l, (3.11) neutralisieren; der pH-Wert wird mit HCl, c = 1 mol/l, (3.12) auf 3,0 eingestellt. Ausreichend Methanol für eine Methanolkonzentration von 10 bis 30 % im Endvolumen zugeben. In einen Messkolben mit ausreichendem Volumen überführen und mit Wasser auf das für die Chromatographie notwendige Volumen verdünnen (z.B. 100 ml). Die Zugabe von Methanol sollte keine Präzipitation verursachen.
Einige ml der Lösung werden durch ein 0,45-pm-Membranfilter (4.5) filtriert, bevor ein Aliquot auf die HPLC-Säule gegeben wird. Die Chromatographie gemäß Nummer 5.4 durchführen.
Standardlösung und Hydrolysate sind vor direktem Sonnenlicht zu schützen. Falls es nicht möglich ist, die Hydrolysate am selben Tag zu analysieren, können sie bei 5 °C maximal drei Tage aufbewahrt werden.
5.4. HPLC-Bestimmung
Die folgenden Angaben für eine isokratische Trennung sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen (siehe Bemerkungen 9.1 und 9.2):
| Flüssigkeitschromatographiesäule (4.2): | 125 mm x 4 mm, C18, 3 µm Korngröße oder gleichwertiges Material |
| Säulentemperatur: | Zimmertemperatur |
| Mobile Phase (3.22): | 3,00 g Essigsäure (3.18) + 900 ml Wasser |
| Durchflussrate: | (3.1) + 50,0 ml Lösung (3.21) von 1,1,1-Trichlor-2-methyl-2-propanol (3.19) in Methanol (3.8) (1 g/100 ml).
Mit Ethanolamin (3.20) auf einen pH-Wert von 5,00 einstellen. Mit Wasser (3.1) auf 1.000 ml auffüllen 1 ml/Min. |
| Gesamtlaufzeit: | etwa 34 Min. |
| Detektionswellenlänge: | Anregung: 280 nm/Emission: 356 nm |
| Injektionsvolumen: | 20 µl |
6. Berechnung der Ergebnisse
| A x B x C x D x E x MW | = g Tryptophan je 100 g Probe |
|
| |
| F x G x H x 10.000 x W |
A = Peakfläche von internem Standard; Kalibrierstandardlösung (3.17)
B = Peakfläche von Tryptophan, Extrakt (5.2) oder Hydrolysat (5.3)
C = Volumen der konzentrierten Tryptophanlösung (3.15), das der Kalibrierlösung (3.17) zugegeben wurde, in ml (2 ml)
D = Konzentration der konzentrierten Tryptophanlösung (3.15), die der Kalibrierlösung (3.17) zugegeben wurde, in µmol/ml (= 2,50)
E = Volumen der konzentrierten internen Standardlösung (3.16), das bei der Extraktion (5.2) (= 5,00 ml) bzw. bei der Hydrolyse (5.3) (2,00 ml) zugegeben wurde, in ml
F = Peakfläche von internem Standard, Extrakt (5.2) oder Hydrolysat (5.3)
G = Peakfläche von Tryptophan, Kalibrierstandardlösung (3.17)
H = Volumen der konzentrierten internen Standardlösung (3.16), das der Kalibrierstandardlösung (3.17) zugegeben wurde, in ml (2,00 ml)
W = Probengewicht in g (korrigiert auf Originalgewicht, falls getrocknet und/oder entfettet) MW = Molekulargewicht von Tryptophan (= 204,23)
7. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen bei ein und derselben Probe darf 10 % des höheren Resultats nicht überschreiten.
8. Ergebnisse eines Ringversuchs
Es wurde ein EG-Ringversuch (4. Vergleichstest) durchgeführt, bei dem drei Proben von bis zu 12 Labors analysiert wurden, um die Hydrolysemethode zu zertifizieren.
Jede Probe wurde mehrfach (fünfmal) analysiert.
Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
| Probe 1 Schweinefutter | Probe 2 Schweinefutter mit Zusatz von L-Tryptophan | Probe 3 Futterkonzentrat für Schweine | |
| L | 12 | 12 | 12 |
| n | 50 | 55 | 50 |
| Mittelwert [g/kg] | 2,42 | 3,40 | 4,22 |
| sr [g/kg] | 0,05 | 0,05 | 0,08 |
| r [g/kg] | 0,14 | 0,14 | 0,22 |
| CVr [%] | 1,9 | 1,6 | 1,9 |
| sR [g/kg] | 0,15 | 0,20 | 0,09 |
| R [g/kg] | 0,42 | 0,56 | 0,25 |
| CVR [%] | 6,3 | 6,0 | 2,2 |
| L: Zahl der Labors, die Ergebnisse eingereicht haben n: Zahl der Einzelergebnisse ohen Ausreißer (Ausreißertest von Cochran, Dixon) sr: Wiederholdstandardabweichung sR: Vergleichstandardabweichung r: Wiederholbarkeit R: Vergleichbarkeit CVr: Wiederholvariationskoeffizient CVR: Vergleichsvariationskoeffizient | |||
Es wurde ein weiterer EG-Ringversuch (3. Vergleichstest) durchgeführt, bei dem zwei Proben von bis zu 13 Labors analysiert wurden, um die Methode zur Extraktion von freiem Tryptophan zu zertifizieren.
Jede Probe wurde mehrfach (fünfmal) analysiert.
Die Ergebnisse sind in nachstehender Tabelle zusammengefasst:
| Probe 4 Mischung aus Weizen und Soja | Probe 5 Mischung aus Weizen und Soja (= Probe 4) mit Tryptophan-Zusatz (0,457 g/kgl) | |
| L | 12 | 12 |
| n | 55 | 60 |
| Mittelwert [g/kg] | 0,391 | 0,931 |
| sr [g/kg] | 0,005 | 0,012 |
| r [g/kg] | 0,014 | 0,034 |
| CVr [%] | 1,34 | 1,34 |
| sR [g/kg] | 0,018 | 0,048 |
| R [g/kg] | 0,050 | 0,134 |
| CVR [%] | 4,71 | 5,11 |
| L: Zahl der Labors, die Ergebnisse eingereicht haben n: Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon) sr: Wiederholstandardabweichung sR: Vergleichstandardabweichung r: Wiederholbarkeit R: Vergleichbarkeit CVr: Wiederholvariationskoeffizient CVR: Vergleichsvariationskoeffizient | ||
Es wurde ein weiterer EG-Vergleichstest durchgeführt, bei dem vier Proben von bis zu sieben Labors analysiert wurden, um die Tryptophan-Hydrolyse zu zertifizieren.
Die Ergebnisse sind in nachstehender Tabelle zusammengefasst.
Jede Probe wurde mehrfach (fünfmal) analysiert.
| Probe 1 Schweinemischfutter (CRM 117) | Probe 2 Fischmehl mit geringem Fettgehalt (CRM 118) | Probe 3 Sojamehl (CRM 119) | Probe 4 Magermilchpulver (CRM 120) | |
| L | 7 | 7 | 7 | 7 |
| n | 25 | 30 | 30 | 30 |
| Mittelwert [g/kg] | 2,064 | 8,801 | 6,882 | 5,236 |
| sr [g/kg] | 0,021 | 0,101 | 0,089 | 0,040 |
| r [g/kg] | 0,059 | 0,283 | 0,249 | 0,112 |
| CVr [%] | 1,04 | 1,15 | 1,30 | 0,76 |
| sR [g/kg] | 0,031 | 0,413 | 0,283 | 0,221 |
| R [g/kg] | 0,087 | 1,156 | 0,792 | 0,619 |
| CVR [%] | 1,48 | 4,69 | 4,11 | 4,22 |
| L: Zahl der Labors, die Ergebnisse eingereicht haben n: Zahl der Einzelergebnisse ohne Ausreißer (Ausreißertest von Cochran, Dixon) sr: Wiederholstandardabweichung sR: Vergleichstandardabweichung r: Wiederholbarkeit R: Vergleichbarkeit CVr: Wiederholvariationskoeffizient CVR: Vergleichsvariationskoeffizient | ||||
9. Bemerkungen
9.1. Unter besonderen Chromatographiebedingungen kann eine bessere Trennung von Tryptophan und α-Methyl-Tryptophan erreicht werden.
Isokratische Elution gefolgt von Gradientensäulenreinigung:
| Flüssigkeitschromatographiesäule: | 125 mm × 4 mm, C18, 5 µm Korngröße oder gleichwertiges Material | ||
| Säulentemperatur: | 32 °C | ||
| Mobile Phase: | A: 0,01 mol/l KH2PO4/Methanol, 95 + 5 (V + V). B : Methanol | ||
| Gradientenprogramm: | 0 Min. | 100 % A | 0 % B |
| 15 Min. | 100 % A | 0 % B | |
| 17 Min. | 60 % A | 40 % B | |
| 19 Min. | 60 % A | 40 % B | |
| 21 Min. | 100 % A | 0 % B | |
| 33 Min. | 100 % A | 0 % B | |
| Durchflussrate: | 1,2 ml/Min. | ||
| Gesamtlaufzeit: | etwa 33 Min. | ||
9.2. Die Durchführung der Chromatographie variiert je nach Art der HPLC und der verwendeten Säulenpackung. Das System muss so gewählt werden, dass eine Grundlinientrennung zwischen Tryptophan und dem internen Standard möglich ist. Die Abbauprodukte müssen deutlich von Tryptophan und internem Standard getrennt werden. Es sollten Hydrolysate ohne internen Standard analysiert werden, um die Grundlinie unter dem internen Standard auf Verunreinigungen zu analysieren. Die Laufzeit muss lang genug sein, dass alle Abbauprodukte eluiert werden, andernfalls können späte Elutionspeaks anschließende Chromatographiedurchgänge beeinträchtigen.
Im Betriebsbereich sollte das Chromatographiesystem eine lineare Reaktion ergeben. Die lineare Reaktion sollte bei einer konstanten (der normalen) Konzentration des internen Standards und unterschiedlichen Tryptophankonzentrationen gemessen werden. Die Größe des Tryptophan- und des internen Standardpeaks müssen sich innerhalb des linearen Bereichs des HPLC/Fluoreszenzsystems befinden. Sollten der Tryptophan- und/oder der interne Standardpeak zu klein oder zu hoch sein, ist die Analyse mit einer anderen Probengröße und/oder einem anderen Endvolumen zu wiederholen.
9.3. Bariumhydroxid
Älteres Bariumhydroxid ist schwerer löslich. Dies kann dazu führen, dass die Lösung für die HPLC-Bestimmung nicht klar ist und zu niedrigen Tryptophan-Werten führen kann.
_________
1) ABl. Nr. L 170 vom 03.08.1970 S. 2.
2) ABl. C 241 vom 29.08.1994 S. 1.
3) ABl. Nr. L 270 vom 14.12.1970 S. 1.
4) ABl. Nr. L 297 vom 18.11.1999 S. 8.
5) ABl. Nr. L 86 vom 06.04.1979 S. 30.
6) ABl. Nr. L 105 vom 03.05.2000 S. 36.
7) ABl. Nr. L 237 vom 22.09.1993 S. 23.
8) ABl. Nr. L 125 vom 23.05.1996 S. 35.
| ENDE | |
...
X
⍂
↑
↓