
 Für einen individuellen Ausdruck passen Sie bitte die Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. ▢ Regelwerk, EU 2000, Lebensmittel - Futtermittel |  |
Richtlinie 2000/45/EG der Kommission vom 6. Juli 2000 zur Festlegung gemeinschaftlicher Analysemethoden für die Bestimmung von Vitamin A, Vitamin E und Tryptophan in Futtermitteln
(Text von Bedeutung für den EWR)
(ABl. Nr. L 174 vom 13.07.2000 S. 32)
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Richtlinie 70/373/EWG des Rates vom 20. Juli 1970 über die Einführung gemeinschaftlicher Probenahmeverfahren und Analysemethoden für die amtliche Untersuchung von Futtermitteln 1, zuletzt geändert durch die Akte über den Beitritt Österreichs, Finnlands und Schwedens 2, insbesondere auf Artikel 2,
in Erwägung nachstehender Gründe:
(1) Gemäß der Richtlinie 70/373/EWG werden die amtlichen Untersuchungen von Futtermitteln zur Feststellung, ob die aufgrund der Rechts- und Verwaltungsvorschriften festgelegten Anforderungen an Beschaffenheit und Zusammensetzung der Futtermittel erfüllt sind, nach gemeinschaftlichen Probenahmeverfahren und Analysemethoden durchgeführt.
(2) Gemäß der Richtlinie 70/524/EWG des Rates vom 23. November 1970 über Zusatzstoffe in der Tierernährung 3, zuletzt geändert durch die Verordnung (EG) Nr. 2439/1999 der Kommission 4, muss der Gehalt an Vitamin A und Vitamin E bei der Etikettierung angegeben werden, wenn diese Stoffe Vormischungen oder Futtermitteln zugesetzt werden.
(3) Gemäß der Richtlinie 79/373/EWG des Rates vom 2. April 1979 über den Verkehr mit Mischfuttermitteln 5, zuletzt geändert durch die Richtlinie 2000/16/EG 6, und der Richtlinie 93/74/EWG des Rates vom 13. September 1993 über Futtermittel für besondere Ernährungszwecke 7, zuletzt geändert durch die Richtlinie 96/25/EG 8, müssen Aminosäuren auf dem Etikett von Futtermitteln angegeben werden.
(4) Es sind gemeinschaftliche Analysemethoden für die Kontrolle dieser Stoffe festzulegen.
(5) Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Futtermittelausschusses -
hat folgende Richtlinie erlassen:
Die Mitgliedstaaten schreiben vor, dass die Analysen für die amtliche Untersuchung von Futtermitteln und Vormischungen auf ihren Gehalt an Vitamin A, Vitamin E und Tryptophan nach den im Anhang beschriebenen Methoden durchgeführt werden.
Die Mitgliedstaaten setzen bis spätestens 31. August 2000 die erforderlichen Rechts- und Verwaltungsvorschriften in Kraft, um dieser Richtlinie nachzukommen. Sie setzen die Kommission davon unverzüglich in Kenntnis.
Sie wenden die Maßnahmen ab 1. September 2000 an.
Wenn die Mitgliedstaaten diese Vorschriften erlassen, nehmen sie in den Vorschriften selbst oder durch einen Hinweis bei der amtlichen Veröffentlichung auf diese Richtlinie Bezug. Die Mitgliedstaaten regeln die Einzelheiten dieser Bezugnahme.
Diese Richtlinie tritt am 20. Tag nach ihrer Veröffentlichung im
Amtsblatt der Europäischen Gemeinschaften in Kraft.
Diese Richtlinie ist an die Mitgliedstaaten gerichtet.
| Anhang |
Teil A
Bestimmung von Vitamin A
1. Zweck und Anwendungsbereich
Die Methode dient zur Bestimmung von Vitamin A (Retinol) in Futtermitteln und Vormischungen. Unter Vitamin A wird der nach dieser Methode ermittelte Gehalt an alltrans-Vitamin-A-Alkohol und seinen cis-Isomeren verstanden. Der Gehalt wird in Internationalen Einheiten (IE) je kg angegeben. Eine IE entspricht der Aktivität von 0,300 µg alltrans-Vitamin-A-Alkohol oder 0,344 µg alltrans-Vitamin-A-Acetat oder 0,550 µg alltrans-Vitamin-A-Palmitat.
Die untere Grenze der Bestimmbarkeit beträgt 2 000 IE Vitamin A/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxid-Lösung hydrolysiert, und das Vitamin A wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft, der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-A-Gehalt wird mittels Umkehrphasen-Hochleistungsflüssigkeits-Chromatographie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt. Die Arbeitsbedingungen werden so gewählt, dass keine Ruftrennung zwischen alltrans-Vitamin-A-Alkohol und seinen cis-Isomeren erfolgt.
3. Reagenzien
3.1. Ethanol, σ = 96 %
3.2. Petrolether, Siedeintervall 40 bis 60 °C
3.3. Methanol
3.4. Kaliumhydroxid-Lösung, β = 50 g/100 ml
3.5. Natriumascorbat-Lösung, β = 10 g/100 ml (siehe Bemerkungen 7.7)
3.6. Natriumsulfid, Na2S · x H2O (x = 7-9)
3.6.1. Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (bei x = 9) (siehe Bemerkungen unter 7.8)
3.7. Phenolphthalein-Lösung, β = 2 g/100 ml in Ethanol (3.1)
3.8. 2-Propanol
3.9. Mobile Phase für HPLC: Mischung von Methanol (3.3) und Wasser, z.B. 980 + 20 (v + v). Das Mischungsverhältnis muss der jeweils verwendeten Säule angepasst werden.
3.10. Stickstoff, sauerstoffrei
3.11. Alltrans-Vitamin-A-Acetat, reinst, mit zertifizierter Aktivität, z.B. 2,80 × 106 IE/g
3.11.1. Stammlösung von alltrans-Vitamin-A-Acetat: 50 mg Vitamin-A-Acetat (3.11) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und bis zur Marke mit demselben Lösungsmittel auffüllen. Der Sollgehalt dieser Lösung beträgt 1.400 IE Vitamin A je ml. Der genaue Gehalt ist nach Nummer 5.6.3.1 zu bestimmen.
3.12. Alltrans-Vitamin-A-Palmitat, reinst, mit zertifizierter Aktivität, z.B. 1,80 × 106 IE/g
3.12.1. Stammlösung von alltrans-Vitamin-A-Palmitat: 80 mg Vitamin-A-Palmitat (3.12) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In 2-Propanol (3.8) lösen und bis zur Marke mit demselben Lösungsmittel auffüllen. Der Sollgehalt dieser Lösung beträgt 1.400 IE Vitamin A je ml. Der genaue Gehalt ist nach Nummer 5.6.3.2 zu bestimmen.
3.13. 2,6-Ditertbutyl-4-methylphenol (BHT) (siehe Bemerkungen unter 7.5)
4. Geräte
4.1. Vakuum-Rotationsverdampfer
4.2. Braunglasgeräte
4.2.1. Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse
4.2.2. Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml
4.2.3. Scheidetrichter, konische Form, 1.000 ml, mit Schliffstopfen
4.2.4. Spitzkolben, 250 ml, mit Schliffhülsen
4.3. Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung
4.4. Phasentrennungsfaltenfilter, Durchmesser 185 mm (z.B. Schleicher & Schuell 597 Hy 1/2)
4.5. HPLC-Einrichtung mit Injektionssystem
4.5.1. Flüssigkeitschromatographiesäule, 250 mm × 4 mm, C18, 5 oder 10 µm Korngröße oder gleichwertiges Material (Leistungskriterium: nur ein Peak für alle Retinol-Isomeren unter diesen HPLC-Bedingungen)
4.5.2. UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung
4.6. Spektralphotometer mit Quarz-Küvetten von 10 mm Schichtdicke
4.7. Wasserbad mit Magnetrührer
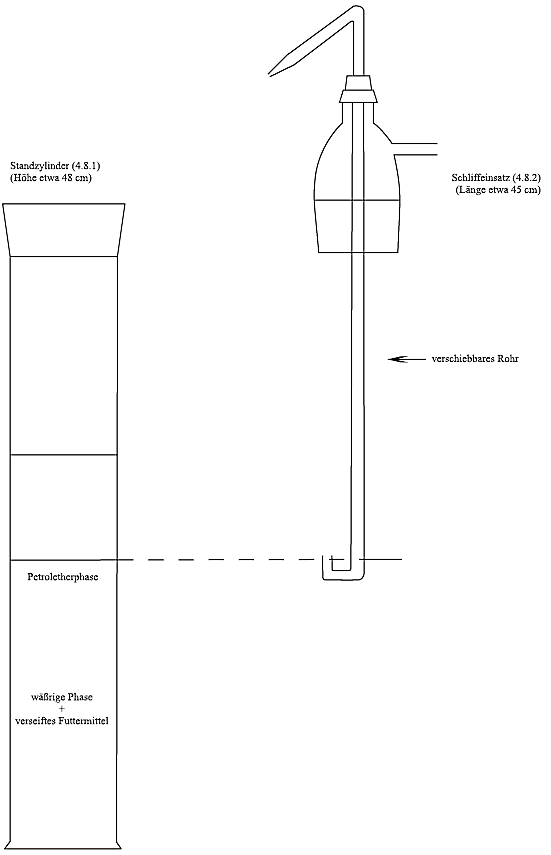
4.8. Extraktionsapparat (Abbildung 1) bestehend aus:
4.8.1. 1-Liter-Standzylinder mit Schliff und Schliffstopfen
4.8.2. Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr sollte ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann.
5. Durchführung
Anmerkung: Vitamin A ist empfindlich gegenüber Licht (UV Strahlung und Oxidationsmitteln. Es muss deshalb unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder mit Aluminiumfolie abgedeckte Glasgeräte) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion sollte das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen rechtzeitig lüften).
5.1. Vorbereitung der Probe
Die Probe unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-A-Verluste auftreten können.
5.2. Verseifung
Je nach Vitamin-A-Gehalt 2 bis 25 g der Probe auf 0,01 g genau in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) einwiegen. Nacheinander unter Umschütteln 130 ml Ethanol (3.1), etwa 100 mg BHT (3.13), 2 ml Natriumascorbat-Lösung (3.5) und 2 ml Natriumsulfid-Lösung (3.6) zusetzen. Den Kolben mit einem Rückflusskühler (4.3) verbinden und in ein Wasserbad mit Magnetrührer (4.7) geben. Bis zum Sieden erhitzen und 5 Min. am Rückflusskühler kochen. Anschließend werden 25 ml Kaliumhydroxid-Lösung (3.4) durch den Rückflusskühler (4.3) zugegeben, und es wird 25 Min. weiter gekocht unter ständigem Rühren und schwachem Stickstoffstrom. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3. Extraktion
Die Verseifungslösung wird mit 250 ml Wasser quantitativ in einen 1 000-ml-Scheidetrichter (4.2.3) oder in den Extraktionsapparat (4.8) überführt. Der Verseifungskolben wird mit 25 ml Ethanol (3.1) und anschließend mit 100 ml Petrolether (3.2) nachgewaschen, und die Flüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen sollte etwa 2:1 betragen. 2 Min. gründlich schütteln und dann 2 Min. stehenlassen.
5.3.1. Extraktion mit Hilfe eines Scheidetrichters (4.2.3)
Nach Phasentrennung (siehe Bemerkung unter 7.3) wird die Petroletherphase in einen anderen Scheidetrichter (4.2.3) überführt. Diese Extraktion noch zweimal mit je 100 ml Petrolether (3.2) und zweimal mit je 50 ml Petrolether (3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter durch zweimaliges Schwenken (Vermeidung von Emulsionsbildung) mit Portionen von jeweils 100 ml Wasser und durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung (3.7) farblos bleibt (viermaliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch ein trockenes Phasentrennungsfilter (4.4) in einen 500-ml-Messkolben (4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether (3.2) nachwaschen, mit Petrolether bis zur Marke (3.2) auffüllen und gut umschütteln.
5.3.2. Extraktion mit Hilfe eines Extraktionsapparats (4.8)
Nach Phasentrennung (siehe Bemerkung unter 7.3) wird der Stopfen des Standzylinders (4.8.1) am Schliffeinsatz (4.8.2) wieder aufgesetzt, und das verschiebbare Rohr wird so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1.000-ml-Scheidetrichter (4.2.3) überführt. 100 ml Petrolether (3.2) in den Standzylinder geben, mit Stopfen verschließen und gründlich schütteln. Nach der Phasentrennung die obere Phase wie zuvor in den Scheidetrichter überführen. Diese Extraktion noch zweimal mit je 100 ml Petrolether (3.2) und zweimal mit je 50 ml Petrolether (3.2) wiederholen und die Petroletherphasen in den Scheidetrichter überführen.
Die vereinigten Petroletherextrakte wie unter Nummer 5.3.1 erläutert waschen und wie dort beschrieben weiter verfahren.
5.4. Vorbereitung der Probenlösung für die HPLC
Ein aliquoter Teil der Petrolether-Lösung (von 5.3.1 oder 5.3.2) wird in einen 250-ml-Spitzkolben (4.2.4) pipettiert. Das Lösungsmittel wird unter reduziertem Druck und bei einer Badtemperatur von höchstens 40 °C am Rotationsverdampfer (4.1) fast bis zur Trockne eingedampft. Darauf wird unter Stickstoff (3.10) Druckausgleich geschaffen und der Kolben vom Rotationsverdampfer abgenommen. Das restliche Lösungsmittel wird im Stickstoffstrom (3.10) abgeblasen, und der Rückstand wird sofort in einer definierten Menge (10-100 ml) Methanol (3.3) aufgenommen (die Konzentration von Vitamin A sollte 5 IE/ml bis 30 IE/ml betragen).
5.5. Bestimmung durch HPLC
Die Abtrennung von Vitamin A erfolgt mit Hilfe einer Umkehrphasen-C18-Säule (4.5.1), und die Konzentration wird mit Hilfe eines UV-Detektors (325 nm) oder eines Fluoreszenzdetektors (Anregung: 325 nm/Emission: 475 nm) (4.5.2) gemessen.
Hierzu wird ein aliquoter Teil (z.B. 20 µl) der nach 5.4 erhaltenen methanolischen Lösung auf die Trennsäule gegeben und mit der mobilen Phase (3.9) eluiert. Die mittlere Peakhöhe (-fläche) von mehreren Einspritzungen derselben Probenlösung wird ermittelt. In der gleichen Weise werden die mittleren Peakhöhen (-flächen) von mehreren Einspritzungen der Kalibrierlösungen (5.6.2) bestimmt.
HPLC-Parameter
Die folgenden Angaben sind Richtwerte; andere Parameter können verwendet werden, sofern sie zu vergleichbaren Ergebnissen führen:
| Flüssigkeitschromatographiesäule (4.5.1): | 250 mm × 4 mm, C18, 5 oder 10 µm Korngröße oder gleichwertiges Material |
| Mobile Phase (3.9): | Mischung von Methanol (3.3) und Wasser z.B. 980 + 20 (v + v) |
| Durchflussrate: | 1-2 ml/Min. |
| Detektor (4.5.2): | UV-Detektor (325 nm) oder Fluoreszenzdetektor (Anregung: 325 nm/Emission: 475 nm) |
5.6. Kalibrierung
5.6.1. Herstellen der Arbeits-Standardlösungen
20 ml der Vitamin-A-Acetat-Stammlösung (3.11.1) oder 20 ml der Vitamin-A-Palmitat-Stammlösung (3.12.1) in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) pipettieren und wie unter Nummer 5.2 beschrieben, aber ohne Zugabe von BHT, hydrolysieren. Anschließend mit Petrolether (3.2) gemäß Nummer 5.3 extrahieren und mit Petrolether (3.2) auf 500 ml auffüllen. 100 ml dieses Extrakts werden am Rotationsverdampfer (siehe 5.4) fast bis zur Trockne eingedampft, das verbleibende Lösungsmittel wird im Stickstoffstrom (3.10) abgeblasen, und der Rückstand wird in 10,0 ml Methanol (3.3) aufgenommen. Der Sollgehalt dieser Lösung beträgt 560 IE Vitamin A je ml. Der genaue Gehalt ist nach Nummer 5.6.3.3 zu bestimmen. Die Arbeits-Standardlösung muss vor Gebrauch frisch bereitet werden.
2,0 ml dieser Arbeits-Standardlösung werden in einen 20-ml-Messkolben pipettiert, es wird bis zur Marke mit Methanol (3.3) aufgefüllt und gemischt. Der Sollgehalt dieser verdünnten Arbeits-Standardlösung beträgt 56 IE Vitamin A je ml.
5.6.2. Herstellen der Kalibrierlösungen und Erstellung der Kalibrierkurve
1,0 ml, 2,0 ml, 5,0 ml und 10,0 ml der verdünnten Arbeits-Standardlösung werden in eine Reihe von 20-ml-Messkolben pipettiert; bis zur Marke mit Methanol (3.3) auffüllen und mischen. Der Sollgehalt dieser Lösungen beträgt 2,8 IE, 5,6 IE, 14,0 IE und 28,0 IE Vitamin A je ml.
20 µl von jeder Kalibrierlösung werden mehrmals eingespritzt, und die mittleren Peakhöhen (-flächen) werden gemessen. Die erhaltenen Peakhöhen (-flächen) dienen zur Erstellung der Kalibrierkurve unter Berücksichtigung der Ergebnisse der UV-Kontrolle (5.6.3.3).
5.6.3. UV-Kontrolle der Standardlösungen
5.6.3.1. Vitamin-A-Acetat-Stammlösung
2,0 ml der Vitamin-A-Acetat-Stammlösung (3.11.1) werden in einen 50-ml-Messkolben (4.2.2) pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin A je ml. 3,0 ml dieser verdünnten Vitamin-A-Acetat-Lösung werden in einen 25-ml-Messkolben pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralphotometer (4.6) zwischen 300 nm und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 nm und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
| (E | 1 % | für Vitamin-A-Acetat = 1.530 bei 326 nm in 2-Propanol) |
| 1 cm |
5.6.3.2. Vitamin-A-Palmitat-Stammlösung
2,0 ml der Vitamin-A-Palmitat-Stammlösung (3.12.1) werden in einen 50-ml-Messkolben (4.2.2) pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 56 IE Vitamin A je ml. 3,0 ml dieser verdünnten Vitamin-A-Palmitat-Lösung werden in einen 25-ml-Messkolben pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralphotometer (4.6) zwischen 300 nm und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 nm und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E326 × 19,0
| (E | 1 % | für Vitamin-A-Palmitat = 957 bei 326 nm in 2-Propanol) |
| 1 cm |
5.6.3.3. Vitamin-A-Arbeits-Standardlösung
3,0 ml der gemäß 5.6.1 hergestellten, unverdünnten Vitamin-A-Arbeits-Standardlösung werden in einen 50-ml-Messkolben (4.2.2) pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. 5,0 ml dieser Lösung werden in einen 25-ml-Messkolben pipettiert, es wird bis zur Marke mit 2-Propanol (3.8) aufgefüllt. Der Sollgehalt dieser Lösung beträgt 6,72 IE Vitamin A je ml. Das UV-Spektrum dieser Lösung wird gegen 2-Propanol (3.8) im Spektralphotometer (4.6) zwischen 300 nm und 400 nm gemessen. Das Extinktionsmaximum muss zwischen 325 nm und 327 nm liegen.
Berechnung des Vitamin-A-Gehalts:
IE Vitamin A/ml = E325 × 18,3
| (E | 1 % | für Vitamin-A-Alkohol = 1.821 bei 325 nm in 2-Propanol) |
| 1 cm |
6. Berechnung der Ergebnisse
Aus der mittleren Höhe (Fläche) der Vitamin-A-Peaks der Probenlösung wird anhand der Kalibrierkurve (5.6.2) die Konzentration der Probenlösung in IE/ml bestimmt.
Der Vitamin-A-Gehalt w in IE/kg der Probe wird nach folgender Formel berechnet:
| w = | 500 · β · V2 · 1.000 | [IE/kg] |
|
| ||
| V1 · m |
Dabei ist:
β = Vitamin-A-Konzentration der Probenlösung (5.4) in IE/ml
V1 = Volumen der Probenlösung (5.4) in ml
V2 = Volumen des bei 5.4 verwendeten aliquoten Teils in ml
m = Probeneinwaage in g
7. Bemerkungen
7.1. Bei Proben mit niedrigen Vitamin-A-Gehalten kann es zweckmäßig sein, die Petroletherextrakte von zwei Verseifungsansätzen (Einwaage 25 g) zu einer Probenlösung für die HPLC-Bestimmung zu vereinigen.
7.2. Die Einwaage für die Analyse sollte höchstens 2 g Fett enthalten.
7.3. Bei schlechter Phasentrennung können etwa 10 ml Ethanol (3.1) zur Zerstörung der Emulsion zugegeben werden.
7.4. Bei Lebertran oder anderen reinen Fetten wird empfohlen, die Verseifungsdauer auf 45 bis 60 Min. zu erhöhen.
7.5. Statt BHT kann Hydrochinon verwendet werden.
7.6. Bei Verwendung einer Normalphasensäule ist die Trennung der Retinolisomeren möglich.
7.7. Statt Natriumascorbat-Lösung können etwa 150 mg Ascorbinsäure verwendet werden.
7.8. Statt Natriumsulfid-Lösung können etwa 50 mg EDTA verwendet werden.
8. Wiederholbarkeit
Die Differenz zwischen den Ergebnissen zweier paralleler Bestimmungen bei ein und derselben Probe darf 15 % des höheren Resultats nicht überschreiten.
9. Ergebnisse eines Ringversuchs 1
| Vormischung | Vormischung | Mineralfutter | Eiweißkonzentrat | Ferkelaufzuchtfutter | |||
| L | 13 | 12 | 13 | 12 | 13 | ||
| n | 48 | 45 | 47 | 46 | 49 | ||
| Mittelwert [IE/kg] | 17,02 x 106 | 1,21 x 106 | 537.100 | 151.800 | 18.070 | ||
| sr [IE/kg] | 0,51 x 106 | 0,039 x 106 | 22.080 | 12.280 | 682 | ||
| r [IE/kg] | 1,43 x 106 | 0,109 x 106 | 61.824 | 34.384 | 1.910 | ||
| CVr [%] | 3,0 | 3,5 | 4,1 | 8,1 | 3,8 | ||
| sR [IE/kg] | 1,36 x 106 | 0,069 x 106 | 46.300 | 23.060 | 3.614 | ||
| R [IE/kg] | 3,81 x 106 | 0,193 x 106 | 129.640 | 64.568 | 10.119 | ||
| CVR [%] | 8,0 | 6,2 | 8,6 | 15 | 20 | ||
| L: Zahl der Labors n: Zahl der Einzelwerte sr: Wiederholstandardabweichung sR: Vergleichstandardabweichung r: Wiederholbarkeit R: Vergleichbarkeit CVr: Wiederholvariationskoeffizient CVR: Vergleichsvariationskoeffizient 1) Durchgeführt von der Fachgruppe Futtermittel des Verbands Deutscher Landwirtschaftlicher Untersuchungs- und Forschungsanstalten (VDLUFA). | |||||||
Abbildung 1: Extraktionsapparat (4.8)
Teil B
Bestimmung von Vitamin E
1. Zweck und Anwendungsbereich
Die Methode dient zur Bestimmung von Vitamin E in Futtermitteln und Vormischungen. Der Vitamin-E-Gehalt wird in mg DL-α-Tocopherol-Acetat je kg angegeben. 1 mg DL-α-Tocopherol-Acetat entspricht 0,91 mg DL-α-Tocopherol (Vitamin E).
Die untere Grenze der Bestimmbarkeit beträgt 2 mg Vitamin E/kg.
2. Prinzip
Die Probe wird mit ethanolischer Kaliumhydroxid-Lösung hydrolysiert, und das Vitamin E wird mit Petrolether extrahiert. Das Lösungsmittel wird eingedampft, der Rückstand wird in Methanol gelöst und, falls notwendig, auf die erforderliche Konzentration verdünnt. Der Vitamin-E-Gehalt wird durch Umkehrphasen-Hochleistungsflüssigkeits-Chromatographie (RP-HPLC) unter Verwendung eines UV- oder Fluoreszenzdetektors bestimmt.
3. Reagenzien
3.1. Ethanol, α = 96 %
3.2. Petrolether, Siedeintervall 40 bis 60 °C
3.3. Methanol
3.4. Kaliumhydroxid-Lösung, β = 50 g/100 ml
3.5. Natriumascorbat-Lösung, β = 10 g/100 ml (siehe Bemerkungen unter 7.7)
3.6. Natriumsulfid, Na2S · x H2O (x = 7-9)
3.6.1. Natriumsulfid-Lösung, c = 0,5 mol/l in Glycerin, β = 120 g/l (bei x = 9) (siehe Bemerkungen unter 7.8)
3.7. Phenolphthalein-Lösung, β = 2 g/100 ml in Ethanol (3.1)
3.8. Mobile Phase für HPLC: Mischung von Methanol (3.3) und Wasser, z.B. 980 + 20 (v + v). Das Mischungsverhältnis muss der jeweils verwendeten Säule angepasst werden.
3.9. Stickstoff, sauerstofffrei
3.10. DL-α-Tocopherol-Acetat, reinst, mit zertifizierter Aktivität
3.10.1. DL-α-Tocopherol-Acetat-Stammlösung: 100 mg DL-α-Tocopherol-Acetat (3.10) auf 0,1 mg genau in einen 100-ml-Messkolben einwiegen. In Ethanol (3.1) lösen und bis zur Marke mit demselben Lösungsmittel auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol-Acetat. (UV-Kontrolle siehe 5.6.1.3; Stabilisierung siehe Bemerkungen unter 7.4).
3.11. DL-α-Tocopherol, reinst, mit zertifizierter Aktivität
3.11.1. 100 mg DL-α-Tocopherol (3.10) auf 0,1 mg genau in einen 100-ml Messkolben einwiegen. In Ethanol (3.1) lösen und bis zur Marke mit demselben Lösungsmittel auffüllen. 1 ml dieser Lösung enthält 1 mg DL-α-Tocopherol. (UV-Kontrolle siehe 5.6.2.3; Stabilisierung siehe Bemerkungen unter 7.4).
3.12. 2,6-Ditertbutyl-4-methylphenol (BHT) (siehe Bemerkungen unter 7.5)
4. Geräte
4.1. Vakuum-Rotationsverdampfer
4.2. Braunglasgeräte
4.2.1. Stehkolben oder Erlenmeyerkolben, 500 ml, mit Schliffhülse
4.2.2. Messkolben mit Schliffstopfen, enghalsig, 10, 25, 100 und 500 ml
4.2.3. Scheidetrichter, konische Form, 1.000 ml, mit Schliffstopfen
4.2.4. Spitzkolben, 250 ml, mit Schliffhülsen
4.3. Allihn-Rückflusskühler, Mantellänge 300 mm, Kernschliff mit Adapter für Gaseinleitung
4.4. Phasentrennungsfaltenfilter, Durchmesser 185 mm (z.B. Schleicher & Schuell 597 Hy 1/2)
4.5. HPLC-Einrichtung mit Injektionssystem
4.5.1. Flüssigkeitschromatographiesäule, 250 mm × 4 mm, C18, Korngröße 5 oder 10 µm oder gleichwertiges Material
4.5.2. UV- oder Fluoreszenzdetektor mit variabler Wellenlängeneinstellung
4.6. Spektralphotometer mit Quarz-Küvetten von 10 mm Schichtdicke
4.7. Wasserbad mit Magnetrührer
4.8. Extraktionsapparat (Abbildung 1) bestehend aus:
4.8.1. 1-Liter-Standzylinder mit Schliff und Schliffstopfen
4.8.2. Schliffeinsatz mit Seitenarm und einem in der Höhe verschiebbaren Rohr in der Mitte. Das verschiebbare Rohr sollte ein U-förmiges unteres Ende und eine Düse am entgegengesetzten Ende haben, so dass die obere Flüssigkeitsphase aus dem Zylinder in den Scheidetrichter überführt werden kann.
5. Durchführung
Anmerkung: Vitamin E ist empfindlich gegenüber Licht (UV-Strahlung und Oxidationsmitteln. Es muss deshalb unter Ausschluss von Licht (Verwendung von Braunglasgeräten oder mit Aluminiumfolie abgedeckte Glasgeräte) und Sauerstoff (Stickstoffspülung) gearbeitet werden. Während der Extraktion sollte das Luftpolster über der Flüssigkeit durch Stickstoff ersetzt werden (zur Vermeidung von Überdruck Stopfen rechtzeitig lüften).
5.1. Vorbereitung der Probe
Die Probe unter Vermeidung von Erwärmung so fein vermahlen, dass sie ein Sieb mit 1 mm Maschenweite passieren kann. Die Zerkleinerung darf erst unmittelbar vor dem Einwiegen und Verseifen erfolgen, da sonst Vitamin-E-Verluste auftreten können.
5.2. Verseifung
Je nach Vitamin-E-Gehalt 2 bis 25 g der Probe auf 0,01 g genau in einen 500-ml-Steh- oder Erlenmeyerkolben (4.2.1) einwiegen. Nacheinander unter Umschütteln 130 ml Ethanol (3.1), etwa 100 mg BHT (3.12), 2 ml Natriumascorbat-Lösung (3.5) und 2 ml Natriumsulfid-Lösung (3.6) zusetzen. Den Kolben mit dem Rückflusskühler (4.3) verbinden und in ein Wasserbad mit Magnetrührer (4.7) geben. Bis zum Sieden erhitzen und 5 Min. am Rückflusskühler kochen. Anschließend werden 25 ml Kaliumhydroxid-Lösung (3.4) durch den Rückflusskühler (4.3) zugegeben, und es wird 25 Min. weiter gekocht unter ständigem Rühren und schwachem Stickstoffstrom. Den Kühler mit etwa 20 ml Wasser ausspülen und den Kolbeninhalt auf Zimmertemperatur abkühlen.
5.3. Extraktion
Die Verseifungslösung wird mit 250 ml Wasser quantitativ in einen 1 000-ml-Scheidetrichter (4.2.3) oder in den Extraktionsapparat (4.8) überführt. Der Verseifungskolben wird mit 25 ml Ethanol (3.1) und anschließend mit 100 ml Petrolether (3.2) nachgewaschen, und die Flüssigkeiten werden ebenfalls in den Scheidetrichter oder den Extraktionsapparat überführt. Das Wasser/Ethanol-Verhältnis in den vereinigten Lösungen sollte etwa 2:1 betragen. 2 Min. gründlich schütteln und dann 2 Min. stehenlassen.
5.3.1. Extraktion mit Hilfe eines Scheidetrichters (4.2.3)
Nach Phasentrennung (siehe Bemerkung unter 7.3) wird die Petroletherphase in einen anderen Scheidetrichter (4.2.3) überführt. Diese Extraktion noch zweimal mit je 100 ml Petrolether (3.2) und zweimal mit je 50 ml Petrolether (3.2) wiederholen.
Die vereinigten Extrakte werden im Scheidetrichter durch zweimaliges Schwenken (Vermeidung von Emulsionsbildung) mit Portionen von jeweils 100 ml Wasser und durch mehrmaliges Schütteln mit weiteren Portionen von 100 ml Wasser so oft gewaschen, bis das Waschwasser bei Zugabe von Phenolphthalein-Lösung (3.7) farblos bleibt (viermaliges Waschen ist normalerweise ausreichend). Zur Entfernung eventuell suspendierten Wassers wird der gewaschene Extrakt durch ein trockenes Phasentrennungsfilter (4.4) in einen 500-ml-Messkolben (4.2.2) filtriert. Scheidetrichter und Filter mit 50 ml Petrolether (3.2) nachwaschen, mit Petrolether (3.2) bis zur Marke auffüllen und gut umschütteln.
5.3.2. Extraktion mit Hilfe eines Extraktionsapparats (4.8)
Nach Phasentrennung (siehe Bemerkung unter 7.3) wird der Stopfen des Standzylinders (4.8.1) am Schliffeinsatz (4.8.2) wieder aufgesetzt und das verschiebbare Rohr wird so eingestellt, dass sich das U-förmige untere Ende gerade über dem Niveau der Grenzfläche befindet. Durch Druckausübung von einer am Seitenarm angebrachten Stickstoffleitung wird die obere Petroletherphase in einen 1.000-ml-Scheidetrichter (4.2.3) überführt. 100 ml Petrolether (3.2) in den Standzylinder geben, mit Stopfen verschließen und gründlich schütteln. Nach der Phasentrennung die obere Phase wie zuvor in den Scheidetrichter überführen. Diese Extraktion noch zweimal mit je 100 ml Petrolether (3.2) und zweimal mit je 50 ml Petrolether (3.2) wiederholen und die Petroletherphasen in den Scheidetrichter überführen.
Die vereinigten Petroletherextrakte wie unter Nummer 5.3.1 erläutert waschen und wie dort beschrieben weiter verfahren.
 | weiter . | |
...
X
⍂
↑
↓