
 Für einen individuellen Ausdruck passen Sie bitte die Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. ▢ Regelwerk, EU 2006 |  |
Richtlinie 2006/56/EG der Kommission vom 12. Juni 2006 zur Änderung der Anhänge der Richtlinie 93/85/EWG des Rates zur Bekämpfung der bakteriellen Ringfäule der Kartoffel
(ABl. Nr. L 182 vom 04.07.2006 S. 1)
Die Kommission der Europäischen Gemeinschaften -
gestützt auf den Vertrag zur Gründung der Europäischen Gemeinschaft,
gestützt auf die Richtlinie 93/85/EWG des Rates vom 4. Oktober 1993 zur Bekämpfung der bakteriellen Ringfäule der Kartoffel 1, insbesondere auf Artikel 12,
in Erwägung nachstehender Grunde:
(1) Ein für die Kartoffel gefährlicher Schadorganismus ist Clavibacter michiganensis (Smith) Davis et al. ssp. sepedonicus (Spieckermann et Kotthoff Davis et al., der Erreger der bakteriellen Ringfäule (im Folgenden "Schadorganismus" genannt).
(2) In einigen Teilen der Gemeinschaft tritt der Schadorganismus noch immer auf.
(3) Mit der Richtlinie 93/85 /EWG wurde im Einzelnen festgelegt, welche Maßnahmen die Mitgliedstaaten treffen müssen, um den Schadorganismus zu lokalisieren und das Ausmaß seiner Verbreitung zu ermitteln, sein Auftreten und seine Verschleppung zu verhindern und ihn, soweit er tatsächlich festgestellt wird, im Interesse der Tilgung wirksam zu bekämpfen und seine Ausbreitung zu verhüten.
(4) Seither hat es in Bezug auf die Biologie und die Verfahren zum Nachweis und zur Identifizierung des Schadorganismus bedeutende neue Entwicklungen gegeben. Darüber hinaus machen die bisherigen Erfahrungen mit der Bekämpfung des Schadorganismus die Überarbeitung bestimmter technischer Aspekte der Bekämpfungsmaßnahmen erforderlich.
(5) Angesichts der genannten Entwicklungen sollten die in den Anhängen der Richtlinie 93/85/EWG vorgesehenen Maßnahmen überarbeitet und aktualisiert werden.
Hinsichtlich der Nachweis- und Identifizierungsverfahren werden die Richtlinienbestimmungen um neue Methoden wie die Fluoreszenz-in-situ-Hybridisierung (FISH) und die Polymerase-Kettenreaktion (PCR) ergänzt und in bestimmten technischen Punkten verbessert.
(6) Auch die Bekämpfungsmaßnahmen werden unter bestimmten technischen Gesichtspunkten verbessert, namentlich - zur Rückverfolgbarkeit des Schadorganismus - der Art und Weise der Konservierung getesteter Proben, der zur Feststellung des Ausmaßes der wahrscheinlichen Kontamination erforderlichen Daten, der Einzelheiten der Mitteilung eines bestätigten Auftretens des Schadorganismus und der betreffenden Befallszone sowie der Maßnahmen, die an als kontaminiert ausgewiesenen Produktionsorten und innerhalb der abgegrenzten Sicherheitszonen durchzuführen sind.
(7) Die in dieser Richtlinie vorgesehenen Maßnahmen entsprechen der Stellungnahme des Ständigen Ausschusses für Pflanzenschutz -
- hat folgende Richtlinie erlassen:
Die Anhänge der Richtlinie 93/85/EWG erhalten die Fassung des Anhangs der vorliegenden Richtlinie.
(1) Die Mitgliedstaaten erlassen und veröffentlichen bis spätestens 31. März 2007 die erforderlichen Rechts- und Verwaltungsvorschriften, um dieser Richtlinie nachzukommen. Sie teilen der Kommission unverzüglich den Wortlaut dieser Rechtsvorschriften mit und fügen eine Entsprechungstabelle dieser Rechtsvorschriften und der vorliegenden Richtlinie bei.
Sie wenden diese Vorschriften ab 1. April 2007 an.
Bei Erlass dieser Vorschriften nehmen die Mitgliedstaaten in den Vorschriften selbst oder durch einen Hinweis bei der amtlichen Veröffentlichung auf die vorliegende Richtlinie Bezug. Die Mitgliedstaaten regeln die Einzelheiten dieser Bezugnahme.
(2) Die Mitgliedstaaten teilen der Kommission unverzüglich den Wortlaut der wichtigsten innerstaatlichen Vorschriften mit, die sie auf dem unter die vorliegende Richtlinie fallenden Gebiet erlassen.
Diese Richtlinie tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Richtlinie ist an die Mitgliedstaaten gerichtet.
Brüssel, den 12. Juni 2006
| Testschema für die Diagnose, den Nachweis und die Identifizierung des Erregers der Bakteriellen Ringfäule der Kartoffel, Clavibacter Michiganensis (Smith) Davis et al. ssp. Sepedonicus (Spieckermann et Kotthof) Davis et al. | Anhang I |
Anwendungsbereich des Testschemas
Das folgende Testschema beschreibt die verschiedenen Verfahren
- zur Diagnose der bakteriellen Ringfäule in Kartoffelknollen und Kartoffelpflanzen,
- zum Nachweis von Clavibaau michiganensis ssp. sepedonicus in Proben von Kartoffelknollen und Kartoffelpflanzen,
- zur Identifizierung von Clavibacter michiganensis ssp. sepedonicus (C. m. subsp. sepedonicus).
Grundregeln
Optimierte Protokolle für die verschiedenen Methoden, validierte Reagenzien sowie die Einzelheiten für die Vorbereitung der Test- und Kontrollmaterialien sind in den Anlagen festgelegt. Anlage 1 enthält eine Liste der an der Optimierung und Validierung von Protokollen beteiligten Laboratorien.
Da die Protokolle dem Nachweis eines Quarantäneschadorganismus dienen und als Kontrollmaterialen lebensfähige Kulturen von C. m. subsp. sepedonicus (Cms) voraussetzen, müssen die Testverfahren in geeigneten Quarantäneeinrichtungen durchgeführt werden, die über angemessene Abfallentsorgungsanlagen und über eine entsprechende Zulassung durch die für Pflanzenquarantäne zuständige Behörde verfügen.
Die Testparameter müssen den eindeutigen und reproduzierbaren Nachweis von C. m. subsp. sepedonicus auf den für die gewählten Methoden vorgegebenen Schwellenwerten gewährleisten.
Die präzise Vorbereitung von Positivkontrollen ist unerlässlich.
Das Testen auf der Grundlage vorgegebener Schwellenwerte erfordert auch eine korrekte Einstellung, Wartung und Eichung der Testgeräte, den sorgfältigen Umgang mit und die Aufbewahrung von Reagenzien sowie Vorkehrungen zur Verhütung der Kontamination zwischen Proben, beispielsweise durch Trennung der Positivkontrollen von Testproben. Zur Vermeidung administrativer und sonstiger Fehler, insbesondere bei der Etikettierung und Dokumentierung, sind Qualitätskontrollen durchzuführen.
Ein Verdachtsfall im Sinne von Artikel 4 Absatz 2 der Richtlinie 93/85/EWG setzt ein positives Ergebnis der Diagnose- oder Screeningtests an einer Probe nach den Vorgaben der Flussdiagramme voraus.
Fällt der erste Screeningtest (IF- oder PCR-/FISH-Test) positiv aus, so besteht Verdacht einer C. m.-subsp.-sepedonicus-Kontamination, und es muss ein zweiter Screeningtest durchgeführt werden. Fällt dieser zweite Test ebenfalls positiv aus, so gilt der Verdacht als bestätigt (Befallsverdacht) und die im Flussdiagramm vorgegebenen Tests müssen fortgesetzt werden. Fällt der zweite Screeningtest negativ aus, so gilt die Probe als nicht kontaminiert.
Ein positiver IF-Test im Sinne von Artikel 4 Absatz 2 wird daher definiert als ein durch einen zweiten Screeningtest (PCRIFISH) bestätigter positiver IF-Befund.
Ein bestätigter Verdacht im Sinne von Artikel 5 Absatz 1 der Richtlinie 93/85/EWG erfordert die Isolierung und Identifizierung einer C. m.-subsp.-sepedonicus-Reinkultur und die Bestätigung der Pathogenität.
1. Flussdiagramme
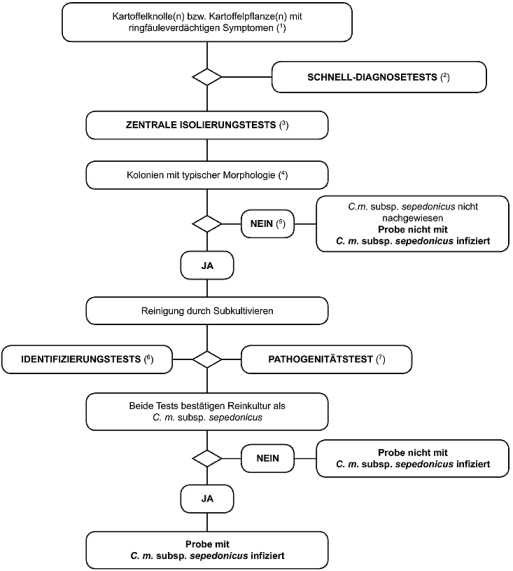
1.1. Nachweisverfahren zur Diagnose der bakteriellen Ringfäule in Kartoffelknollen und Kartoffelpflanzen mit Ringfäulesymptomen
Das Testverfahren ist für Kartoffelknollen und Kartoffelpflanzen mit ringfäuletypischen oder ringfäuleverdächtigen Symptomen geeignet. Es umfasst einen Schnell-Screeningtest, die Isolierung des Erregers aus infiziertem Gefäßgewebe auf Diagnosemedien und - bei positivem Ergebnis - die Identifizierung der Kultur als C. m. subsp. sepedonicus.
______
1) Beschreibung der Symptome in Abschnitt 2.
2) Geeignete Tests:
- IF-Test (Abschnitt 4).
- PCR-Test (Abschnitt 6).
- FISH-Test (Abschnitt 5).
3) Obgleich sich der Erregerdurch Verdünnungsausstrich relativeinfach aus Pflanzenmaterial mittypischen Symptomen isolieren lässt, kann das Kultivieren in fortgeschrittenem Infektionsstadium scheitern. Auf krankem Gewebe wachsende saprophytische Bakterien können den Erreger auf dem Isolierungsmedium überwuchern oder sein Wachstum hemmen. Daher wird empfohlen, sowohl nichtselektive als auch selektive Medien, vorzugsweise MTNA (Abschnitt 8) oder den Biotest (Abschnitt 7) zu verwenden.
4) Beschreibung einer typischen Kolonienmorphologie in Abschnitt B.
5) Fällt der Isolierungstest negativ aus, liegen aber typische Krankheitssymptome vor, so ist die Isolierung zu wiederholen.
6) Eine Reinkultur von C. m. subsp. sepedonicus wird durch die Tests gemäß Abschnitt 9 verlässlich identifiziert.
7) Beschreibung des Pathogenitätstests in Abschnitt 10.
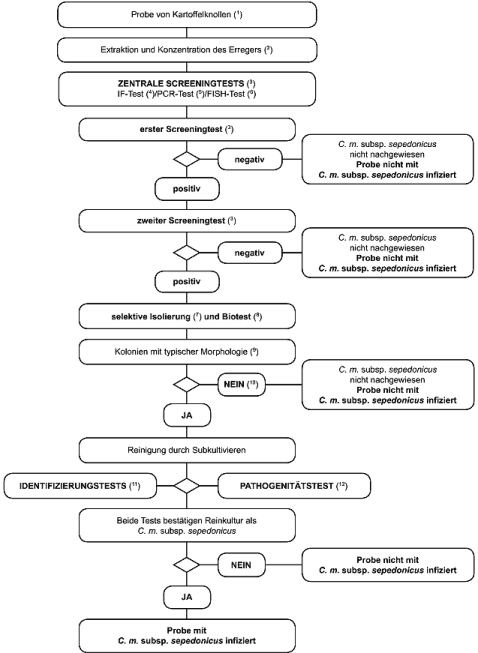
1.2. Verfahren zum Nachweis und zur Identifizierung von C. m. subsp. sepedonicus in Proben symptomfreier Kartoffelknollen
Grundsatz
Das Testverfahren dient dem Nachweis latenter Infektionen von Kartoffelknollen. Ein positives Ergebnis bei mindestens zwei Screeningtests, die auf unterschiedlichen biologischen Grundsätzen beruhen, ist durch Isolierung des Erregers zu bestätigen. Bei Isolierung typischer Kolonien ist eine Reinkultur als C. m. subsp. sepedonicus zu bestätigen. Ein positiver Screeningtest allein reicht nicht aus, um die Probe als verdächtig einzustufen.
Screeningtests und Isolierungstests müssen eine Nachweisgrenze von 103 bis 104 Zellen/ml resuspendiertes Pellet gewährleisten, die als Positivkontrollen in jede Testreihe einzubeziehen sind.
_________
1) Eine Standardprobe besteht aus 200 Knollen; das Verfahren ist aber auch für kleinere Proben geeignet, wenn keine 200 Knollen zur Verfügung stehen.
2) Beschreibung der Methoden der Erregerextraktion und -konzentration in Abschnitt 3.1.
3) Fallen mindestens zwei Tests, die auf unterschiedlichen biologischen Grundsätzen beruhen, positiv aus, so muss der Erreger isoliert und bestätigt werden. Es ist mindestens ein Screeningtest durchzuführen. Fällt dieser Test negativ aus, so gilt die Probe als negativ. Fällt der Test positiv aus, so sind zur Abklärung des ersten positiven Ergebnisses und nach unterschiedlichen biologischen Grundsätzen ein zweiter oder weitere Screeningtests durchzuführen. Fallen der zweite oder andere Tests negativ aus, so gilt die Probe als negativ. Weitere Tests sind in diesem Falle nicht erforderlich.
4) Immunfluoreszenztest (IF-Test).
Fürden IF-Test stets polyklonale Antikörper verwenden; zusätzlich können monoklonale Antikörper eine größere Spezifität gewährleisten (siehe Abschnitt 4).
5) PCR-Test.
Validierte PCR-Reagenzien und -Protokolle verwenden (siehe Abschnitt 6).
6) Fish-Test.
Validierte Reagenzien und Protokolle verwenden (siehe Abschnitt 5).
7) Selektive Isolierung.
Mittels MTNA-Medium oder NCP-88-Medium und einer 1/100-Verdünnung des resuspendierten Pellets; in den meisten Fällen ist diese Methode für die direkte Isolierung von C. m. subsp. sepedonicus geeignet.
Typische Kolonien bilden sich 3-10 Tage nach dem Ausstrich.
Der Erreger kann sodann gereinigt und identifiziert werden.
Zur vollen Ausschöpfung des Testpotenzials sind die Nabelendstücke sorgfältig vorzubereiten, um Sekundärbakterien aus den Knollen, die auf dem Medium mit C. m. subsp. sepedonicus konkurrieren und den Erreger überwuchern könnten, zu vermeiden.
Scheitert der Ausstrichtest, so ist der Erreger aus Pflanzen zu isolieren, die für den Biotest (siehe Abschnitt 8) verwendet werden.
8) Der Biotest wird verwendet für die Isolierung von C. m. subsp. sepedonicus aus Kartoffelextraktpellets durch selektive Anreicherung in Auberginen (Solanum melongena). Der Biotest setzt optimale Inkubationsbedingungen, wie im Testverfahren beschrieben, voraus. Bakterien, die das Wachstum von C. m. subsp. sepedonicus auf dem MTNA-Medium oder dem NCP-88-Medium hemmen, dürften diesen Test nicht beeinträchtigen (siehe Abschnitt 7).
9) Beschreibung einer typischen Kolonienmorphologie in Abschnitt B.
10) Das Kultivieren oder Biotest können aufgrund konkurrierender oder hemmender saprophytischer Bakterien scheitern. Fallen Screeningtests positiv, die Isolierungstests jedoch negativ aus, so sind die Isolierungstests aus demselben Pellet oder durch Entnahme zusätzlichen Gefäßgewebes am Nabelende durchschnittener Knollen derselben Probe zu wiederholen; erforderlichenfalls sind weitere Proben zu testen.
11) Eine Reinkultur von C.-m.-subsp.-sepedonicus-verdächtigen Kulturen wird durch die Tests gemäß Abschnitt 9 verlässlich identifiziert.
12) Beschreibung des Pathogenitätstests in Abschnitt 10.
Verfahren zum Nachweis und zur Identifizierung von Clavfacter michiganensis ssp. sepedmicus in Proben symptomfreier Kartoffelpflanzen

___________
1) Empfohlene Probengrößen in Abschnitt 3.2.
2) Beschreibung von Methoden zur Erregerextraktion und -konzentration in Abschnitt 3.2.
3) Fallen mindestens zwei Tests, die auf unterschiedlichen biologischen Grundsätzen beruhen, positiv aus, so muss der Erreger isoliert und bestätigt werden.
Es ist mindestens ein Screeningtest durchzuführen.
Fällt dieser Test negativ aus, so gilt die Probe als negativ.
Fällt der Test positiv aus, so sind zur Abklärung des ersten positiven Ergebnisses und nach unterschiedlichen biologischen Grundsätzen ein zweiter oder weitere Screeningtests durchzuführen.
Fallen der zweite oder andere Tests negativ aus, so gilt die Probe als negativ.
Weitere Tests sind in diesem Falle nicht erforderlich.
4) Beschreibung des selektiven Isolierungstests und einer typischen Kolonienmorphologie in Abschnitt B.
5) Beschreibung des IF-Tests in Abschnitt 4.
6) Beschreibung des PCR-Tests in Abschnitt 6.
7) Beschreibung des FISH-Tests in Abschnitt 5.
8) Beschreibung des Biotests in Abschnitt 7.
9) Das Kultivieren oder Biotests können aufgrund konkurrierender oder hemmender saprophytischer Bakterien scheitern. Fallen Screeningtests positiv, die Isolierungstests jedoch negativ aus, so sind die Isolierungstests zu wiederholen; erforderlichenfalls sind weitere Proben zu testen.
10) Eine Reinkultur von C.-m.-subsp.sepedonicus-verdächtigen Kulturen wird durch die Tests gemäß Abschnitt 9 verlässlich identifiziert.
11) Beschreibung des Pathogenitätstests in Abschnitt 10.
2. Visuelle prüfung auf Ringfäulesymptome
2.1. Kartoffelpflanzen
Unter europäischen Klimabedingungen zeigen sich Symptome in Feldbeständen nur selten und häufig nur gegen Ende der Saison. Die Symptome sind außerdem oft verschleiert oder werden mit anderen Krankheiten, Alterungsprozessen oder mechanischen Schäden verwechselt. Daher können Ringfäulesymptome bei Feldbesichtigungen leicht übersehen werden. Welkesymptome unterscheiden sich stark von denen der Schleimkrankheit; der Welkeprozess ist gewöhnlich langsam und zunächst auf die Blattränder beschränkt. Infizierte junge Blätter wachsen oft weiter, allerdings in infizierten Zonen weniger. Dadurch entstehen ungewöhnliche Blattformen. An Blättern, deren Gefäßgewebe weiter unten am Stängel blockiert ist, entwickeln sich häufig chlorotische, gelb bis schwach-bräunliche Verfärbungen zwischen den Blattadern. Infizierte Blättchen, Blätter und selbst Stängel können absterben. Oft bleiben Blätter und Knollen auch einfach kleiner. Mitunter bleiben die Pflanzen in ihrem Wachstum gestaucht. Farbabbildungen verschiedener Ringfäulesymptome können über folgende Internet-Website abgerufen werden: http://forum.europa.eu.int/Pubfic/irc/sanco/Home/main
2.2. Kartoffelknollen
Erste Symptome sind eine leichte Glasigkeit oder Durchsichtigkeit des Gewebes insbesondere nah am Nabelende ohne Aufweichen des Gewebes um das Gefäßsystem. Der Gefäßbündelring am Nabelende kann leicht dunkler gefärbt sein als üblich. Die ersten erkennbaren Symptome sind eine Gelbfärbung des Gefäßbündelrings und, wenn auf die Knolle ein leichter Druck ausgeübt wird, eine aus den Gefäßen austretende käsige Substanz. Diese Absonderung enthält Millionen von Bakterien. In diesem Stadium kann sich eine Braunfärbung des Gefäßgewebes entwickeln, und die Knollensyrnptome sind mit denen der durch Ralstonia solanacearum verursachten Schleimkrankheit vergleichbar. Die Symptome können zunächst auf einen Teil des Rings, der sich nicht unbedingt in der Nähe des Nabelendes befinden muss, beschränkt sein und sich dann allmählich auf den gesamten Ring ausdehnen. Mit fortschreitender Infektion wird das Gefäßgewebe zerstört, der äußerlich dem Gefäßbündelgefäß angrenzende Gewebebereich kann sich ablösen. In fortgeschrittenen Stadien der Infektion erscheinen auf der Oberfläche der Knolle Risse, die an den Rändern oft eine gelblich-braune Färbung aufweisen. In Europa sind kürzlich verschiedene Fälle aufgetreten, in denen das Knolleninnere zur gleichen Zeit verfault wie der Gefäßbündelring, wodurch eine sekundäre Ausbreitung erfolgt, die dazu führt, dass das Knolleninnere zerfällt und Nekrose eintritt. Sekundärer Pilz- oder Bakterienbefall kann die Symptome verschleiern, und es kann schwierig, wenn nicht sogar unmöglich werden, die Symptome einer fortgeschrittenen Ringfäule von anderen Knollenfäulen zu unterscheiden. Es können auch atypische Symptome auftreten. Farbabbildungen der verschiedenen Symptome können über folgende Internet-Website abgerufen werden: http://forum.europa.eu.int/Public/irc/sanco/Home/main
3. Probenaufbereitung
3.1. Kartoffelknollen
Hinweis:
- Die Standardprobe umfasst 200 Knollen je Test. Für umfangreichere Testreihen müssen mehr Proben dieser Größe untersucht werden. Mehr als 200 Knollen in der Probe führen zu Hemmreaktionen oder erschweren die Ergebnisauswertung. Das Verfahren ist aber auch für Proben mit weniger als 200 Knollen geeignet, wenn nur wenige Knollen zur Verfügung stehen.
- Die Validienmg aller im Folgenden beschriebenen Nachweismethoden beruht auf der Untersuchung von Proben einer Größe von 200 Knollen.
- Der im Folgenden beschriebene Kartoffelextrakt kann auch zum Nachweis des Erregers der Schleimkrankheit der Kartoffel, Ralstonia solanacearum, verwendet werden.
Fakultative Vorbehandlung vor der Probenaufbereitung:
Knollen waschen. Zwischen den einzelnen Proben geeignete Desinfektionsmittel (Chlorverbindungen im Falle des PCR-Tests, um etwa vorhandene pathogene DNA zu entfernen) und Detergenzien verwenden. Knollen an der Luft trocknen lassen. Dieses Waschverfahren ist besonders für Proben mit viel überschüssiger Erde und in Fällen hilfreich (jedoch nicht vorgeschrieben), in denen ein PCR-Test oder ein direktes Isolierungsverfahren durchgeführt werden sollte.
3.1.1. Mit einem sauberen, sterilen Skalpell oder Gemüsemesser die Schale am Nabelende der Knolle entfernen, so dass das Gefäßbündelgewebe sichtbar wird. Am Nabelende jeder Knolle sorgfältig ein kleines, kegelförmiges Gewebe-stück aus dem Gefäßbereich herausschneiden. Dabei darauf achten, dass möglichst wenig nicht vaskuläres Gewebe erfasst wird (siehe Internet-Website: http://forum.europa.eu.int/Public/irc/sanco/Home/main).
Hinweis:
Knollen mit ringfäuleverdächtigen Symptomen (sichtbarer Fäulnis) sind auszusondern und separat zu testen.
Werden beim Entfernen des kegelförmigen Gewebestückchens ringfäuleverdächtige Symptome festgestellt, so sollte diese Knolle visuell geprüft werden, nachdem sie am Nabelende durchgeschnitten wurde. Durchschnittene Knollen mit verdächtigen Symptomen sollten zur Wundenverkorkung 2 Tage bei Raumtemperatur aufbewahrt und anschließend unter Quarantänebedingungen (bei 4-10 °C) gelagert werden, bis alle Tests abgeschlossen sind. Alle Knollen der Probe, einschließlich solcher mit Verdachtssymptomen, sind gemäß Anhang II aufzubewahren.
3.1.2. Die kegelförmigen Nabelendstückchen in unbenutzten verschließbaren und/oder verplombbaren Einwegbehältnissen sammeln (werden Behältnisse wieder verwendet, so sind sie mit Chlorverbindungen gründlich zu reinigen und zu desinfizieren). Die Gewebestückchen sind möglichst sofort zu verarbeiten bzw. - soweit dies nicht möglich ist - in ihrem Behältnis ohne Zugabe von Puffer für maximal 72 Stunden gekühlt bzw. für maximal 24 Stunden bei Raumtemperatur aufzubewahren. Ein Austrocknen und Verkorken der Gewebestückchen und Saprophyten-Bewuchs während der Lagerung können den Nachweis des Ringfäulebakteriums verhindern.
3.1.3. Die Gewebestückchen nach einem der folgenden Verfahren verarbeiten: Entweder
- die Stückchen mit einer ausreichenden Menge (ungefähr 40 ml) Extraktionspuffer (Anlage 3) bedecken und auf einem Schüttler (50-100 rpm) für 4 Stunden bei einer Temperatur von weniger als 24 °C bzw. 16-24 Stunden bei Kühlschranktemperatur schütteln;
oder - die Stückchen mit einer ausreichenden Menge (ungefähr 40 ml) Extraktionspuffer (Anlage 3) entweder in einem Waring- oder Ultra-Thurax-Mixer oder durch Zerkleinerung in einem robusten verschlossenen Einweg-Mazerationsbeutel (z.B. Stomacher- oder Bioreba-Plastikbeutel, 150 mm X 250 mm; strahlensterilisiert) mit einem Gummihammer oder einem geeigneten Zerkleinerungsgerät (z.B. Homex) homogenisieren.
Hinweis:
Das Risiko der Kreuzkontamination von Proben ist groß, wenn letztere mit einem Mixer homogenisiert werden.
Es sind Vorkehrungen zu treffen, um jedes Versprühen oder Verschütten während des Extraktionsprozesses zu vermeiden.
Ferner ist sicherzustellen, dass für jede Probe frisch sterilisierte Mixerblätter und Gefäße verwendet werden.
Beim PCR-Test ist eine DNA-Übertragung auf Behälter oder Zerkleinerungsgeräte zu vermeiden.
Die Zerkleinerung sollte in Einwegbeuteln und Einwegröhrchen erfolgen, wenn der PCR-Test angewendet werden soll.
3.1.4. Den Überstand umfüllen. Wenn er sehr trübe ist, entweder durch langsames Zentrifugieren (bei nicht mehr als 180 g für 10 Minuten zwischen 4-10 °C) oder durch Vakuumfiltration (40-100 pm) klären und den Filter mit zusätzlichem (10 ml) Extraktionspuffer (Anlage 3) waschen.
3.1.5. Die Bakterien durch 15-minütiges Zentrifugieren des erhaltenen Überstandes bei 7 000 g (oder für 10 Minuten bei 10 000 g) bei einer Temperatur zwischen 4-10 °C konzentrieren und den Überstand verwerfen, ohne das Pellet aufzurühren.
3.1.6. Das Pellet in 1,5 ml Pelletpuffer (Anlage 3) resuspendieren. 500 μ l für das Testen auf C. m. subsp. sepedonicus, 500 μl für Ralstonia solanacearum und 500 µl für Referenzzwecke verwenden. Zum Referenzaliquot von 500 µl und den restlichen Testaliquots steriles Glyzerin zugeben, um eine Endkonzentration von 10-25 % (v/v) zu erreichen, vortexen und bei - 16 bis - 24 °C (Wochen) oder bei - 68 bis - 86 °C (Monate) lagern. Die Testaliquots während der Testung bei 4-10 °C aufbewahren.
Von wiederholtem Einfrieren und Auftauen wird abgeraten.
Muss der Extrakt befördert werden, so ist sicherzustellen, dass der Transport innerhalb von 24 bis 48 Stunden erfolgt und eine Kühlbox verwendet wird.
3.1.7. Zur Vermeidung von Kontaminationen müssen alle C. m. subsp. sepedonicus Positivkontrollen und Proben unbedingt separat behandelt werden. Dies gilt für IF-Objektträger und für sämtliche Tests.
3.2. Kartoffelpflanzen
Hinweis:
Zum Nachweis latenter C. m. subsp. sepedonicus Populationen wird empfohlen, Mischproben zu testen. Das Verfahren ist für Mischproben von bis zu 200 Stängelstücken geeignet. (Werden Erhebungen durchgeführt, so sollten diese an einer statistisch repräsentativen Probe der zu untersuchenden Pflanzenpopulation ausgerichtet werden.)
3.2.1. Mit einem sauberen, sterilen Messer oder einer Gartenschere am Stängelansatz genau über der Erde ein 1-2 cm großes Stängelstück herausschneiden.
Die Stängelstücke kurz mit 70 %igem Ethanol desinfizieren und sofort auf Papiertüchern trocken tupfen. Die Stängelstücke in einem verschlossenen sterilen Behältnis sammeln und wie folgt verfahren:
3.2.2. Die Stängelstücke einer der folgenden Behandlungen unterziehen: Entweder
- die Stücke mit einer ausreichenden Menge (ungefähr 40 ml) Extraktionspuffer (Anlage 3) bedecken und auf einem Schüttler (bei 50-100 rpm) für 4 Stunden bei einer Temperatur von weniger als 24 °C bzw. für 16-24 Stunden bei Kühlschranktemperatur schütteln;
oder - die Stücke mit einer ausreichenden Menge (ungefähr 40 ml) Extraktionspuffer (Anlage 3) durch Zerkleinern in einem robusten Mazerationsbeutel (z.B. Stomacher- oder Bioreba-Beutel) mit einem Gummihammer oder einem geeigneten Zerkleinerungsgerät (z.B. Homex) unverzüglich verarbeiten. Soweit dies nicht möglich ist, die Stängelstücke für maximal 72 Stunden bei Kühlschranktemperatur und höchstens 24 Stunden bei Raumtemperatur lagern.
3.2.3. Den Überstand umfüllen, nachdem das Mazerat 15 Minuten gestanden hat.
3.2.4. Eine weitere Klärung des Extrakts oder Konzentration der Bakterien ist in der Regel nicht erforderlich, kann jedoch durch Filtrieren und/oder Zentrifugieren (siehe Abschnitte 3.1.4-3.1.6) erreicht werden.
3.2.5. Den unverdünnten bzw. konzentrierten Probenextrakt in zwei gleiche Teile teilen. Eine Hälfte während der Testung bei 4-10 °C aufbewahren, die andere Hälfte mit 10-25 %igem (v/v) sterilem Glyzerin bei - 16 bis - 24 °C (Wochen) oder bei - 68 bis - 86 °C (Monate) lagern für den Fall, dass weitere Tests erforderlich werden.
4. IF-TEST
Grundsatz
Der IF-Test wird als Hauptscreeningtest empfohlen, weil er nachweislich stabil genug ist, um die vorgeschriebenen Schwellenwerte zu erreichen.
Wird der IF-Test als Hauptscreeningtest angewandt und fällt der IF-Befund positiv aus, so muss als zweiter Screeningtest der PCR- oder der FISH-Test durchgeführt werden. Wird der IF-Test als zweiter Screeningtest angewandt und fällt der IF-Befund positiv aus, so sind zum Abschluss der Analyse weitere Tests (siehe Flussdiagramm) erforderlich.
Hinweis:
Stets einen polyklonalen Antikörper verwenden, wenn der IF-Test als Hauptscreeningtest angewandt wird. Bei positivem IF-Befund mit polyklonalem Antikörper kann ein weiterer Test der Proben mit einem monoklonalen Antikörper unter Umständen spezifischer sein, wenn auch weniger empfindlich.
Antikörper gegen einen Referenzstamm von C. m. subsp. sepedonicus verwenden. Es wird empfohlen, für jede neue Antikörpercharge den Titer zu bestimmen. Der Titer wird definiert als die höchste Verdünnung, bei der eine optimale Reaktion eintritt, wenn eine Suspension aus 105 bis 106 Zellen/ml des homologen Stammes von C. m. subsp. sepedonicus mit einem geeigneten Fluorescein-Isothiocyanat-(FITC)-Konjugat nach Herstellerspezifikationen getestet wird. Das polyklonale oder monoklonale Rohserum sollte einen IF-Titer von mindestens 1:2000 ergeben. Beim Test sollten die Antikörper in Arbeitsverdünnung(en) (AV) sehr nahe am oder auf dem Titerwert verwendet werden. Validierte Antikörper verwenden (siehe Website: http://fonun.europa.eu.int/Pubfic/irc/sanco/Home/main).
Der Test ist an frisch vorbereitenen Probenextrakten durchzuführen. Er kann erforderlichenfalls auch an Extrakten vorgenommen werden, die bei - 68 bis - 86 °C unter Glyzerinzugabe gelagert wurden. Das Glyzerin kann durch Zugabe von 1 ml Pelletpuffer (Anlage 4), 15-minütiges Rezentrifugieren bei 7 000 g und Resuspendieren in gleicher Menge Pelletpuffer entfernt werden. Dieser Arbeitsschritt ist häufig nicht erforderlich, vor allem, wenn die Proben durch Abflammen am Objektträger fixiert werden (siehe Abschnitt 2.2).
Separate Objektträger mit Positivkontrollen des homologen Stammes oder eines anderen Referenzstammes von C. m. subsp. sepedonicus, nach Anlage 2 in Kartoffelextrakt und fakultativ in Puffer suspendiert, herstellen.
Als ähnliche Kontrolle auf demselben Objektträger nach Möglichkeit natürlich infiziertes Gewebe (durch Lyophilisierung oder Einfrieren bei - 16 bis - 24 °C gelagert) verwenden.
Als Negativkontrollen Aliquots des Probenextrakts verwenden, deren vorheriges Testergebnis negativ war. Multiwell-Objektträger mit vorzugsweise 10 Sichtfeldern von mindestens 6 mm Durchmesser verwenden. Das Kontrollmaterial nach demselben Verfahren testen wie die Probe(n).
4.1. Die Objektträger nach einem der folgenden Verfahren vorbereiten:
- Pellets mit relativ wenig Stärkesediment:
Ein abgemessenes Standardvolumen (15 µl reichen für ein Feld von 6 mm Durchmesser aus - bei größeren Feldern ein entsprechend größeres Volumen verwenden) des 1/100 verdünnten resuspendierten Kartoffelpellets auf das erste Feld pipettieren. Dieselbe Menge unverdünntes Pellet (1/1) auf die restlichen Felder der Reihe pipettieren. Die zweite Reihe kann, wie in Abbildung 1 dargestellt, als Duplikat oder für eine zweite Probe verwendet werden.
- Andere Pellets:
Dezimalverdünnungen (1/0 und 1/100) des resuspendierten Pellets in Pelletpuffer herstellen. Ein abgemessenes Standardvolumen (15 µl reichen für ein Feld von 6 mm Durchmesser aus - bei größeren Feldern ein entsprechend größeres Volumen verwenden) des resuspendierten Pellets und jeder Verdünnung auf eine Felderreihe pipettieren. Die zweite Reihe kann, wie in Abbildung 2 dargestellt, als Duplikat oder für eine zweite Probe verwendet werden.
4.2. Tropfen bei Umgebungstemperatur oder durch Erwärmen auf 40 bis 45 °C trocknen lassen. Bakterienzellen entweder durch 15-minütiges Erhitzen bei 60 °C, Abflammen, mit 95 %igem Ethanol oder nach genauen Anweisungen des Antikörper-Lieferanten am Objektträger fixieren.
Soweit erforderlich können fixierte Objektträger vor weiteren Tests eingefroren in einem trockenen Behältnis für kurze Zeit (jedoch höchstens drei Monate) gelagert werden.
4.3. IF-Verfahren:
- Bei Objektträgervorbereitung nach 4.1 i:
Einen Satz Zweifachverdünnungen des Antikörpers in IF-Puffer herstellen. Im ersten Feld sollte 112 des Titers (TJ2), in den anderen Feldern 114 des Titers (TJ4), 112 des Titers (TJ2), der Titer (T) und das Doppelte des Titers (2T) enthalten sein.
- Bei Objektträgervorbereitung nach 4.1 ii:
Herstellung der Arbeitsverdünnung (AV) des Antikörpers in IF-Puffer. Die Arbeitsverdünnung beeinflusst die Spezifität.
Abb. 1: Vorbereitung des Objektträgers nach 4.1 i und 4.3 i
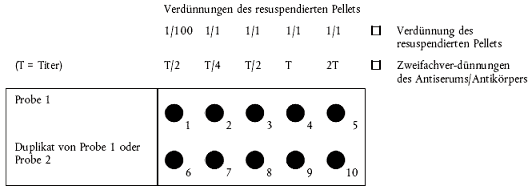
Abb. 2: Vorbereitung des Objektträgers nach 4.1 ii und 4.3 ii

4.3.1. Die Objektträger auf feuchten Papiertüchern auslegen. Alle Testfelder mit Antikörperverdünnung(en) bedecken. Das auf die Felder aufgetragene Antikörpervolumen muss mindestens dem Volumen des aufgetragenen Extraktes entsprechen.
Liegen keine spezifischen Anweisungen des Antikörper-Lieferanten vor, so ist folgendes Verfahren anzuwenden:
4.3.2. Objektträger abgedeckt für 30 Minuten bei Raumtemperatur (18-25 °C) auf feuchten Papiertüchern inkubieren.
4.3.3. Antikörpertropfen vom Objektträger abschütteln, und die Objektträger sorgfältig mit IF-Puffer spulen. 5 Minuten mit IF-Puffer-Tween (Anlage 3) und anschließend 5 Minuten in IF-Puffer waschen. Kreuzkontaminationen durch Sprüh- oder Tropfenbildung vermeiden. Überschüssige Feuchtigkeit durch Abtupfen sorgfältig entfernen.
4.3.4. Objektträger auf feuchten Papiertüchern auslegen. Testfelder mit der zur Titer-Bestimmung verwendeten Verdünnung des FITC-Konjugats bedecken. Das auf die Felder aufgetragene Konjugatvolumen muss dem Volumen des aufgetragenen Antikörpers entsprechen.
4.3.5. Objektträger abgedeckt für 30 Minuten bei Raumtemperatur (18-25 °C) auf feuchten Papiertüchern inkubieren. 4.3.6. Tropfen vom Objektträger abschütteln. Wie unter 4.3.3 angegeben waschen und spulen. Überschüssige Feuchtigkeit sorgfältig entfernen.
4.3.7. Auf jedes Feld 5-10 µl 0,1 M phosphatgepuffertes Glyzerin (Anlage 3) oder eine handelsübliche Antifading-Deckflüssigkeit auftragen und Deckglas auflegen.
4.4. IF-Test-Befund:
4.4.1. Test-Objektträger auf einem Epifluoreszenz-Mikroskop mit Filtern, die für FITC-Anregung geeignet sind, unter Öl- oder Wasserimmersion bei einer Vergrößerung von 500-1 000 untersuchen. Jedes Feld in zwei rechtwinklig zueinander stehenden Durchmessern sowie entlang des Feldrandes mikroskopisch untersuchen. Bei Proben, die keine oder nur wenige Zellen zeigen, mindestens 40 Mikroskopfelder untersuchen.
Zunächst den Objektträger mit der Positivkontrolle prüfen. Die Zellen müssen stark fluoreszieren und bei dem festgelegten Antikörpertiter oder der festgelegten Arbeitsverdünnung vollständig gefärbt sein. Bei abweichender Färbung zum Normalzustand ist der IF-Test (Abschnitt 4) zu wiederholen.
4.4.2. Auf Anwesenheit hell fluoreszierender Zellen mit charakteristischer C. m. subsp. sepedonicus Morphologie in den Testfeldern auf den Testobjektträgern prüfen (siehe Website: http://forum.europa.eu.int/Pubfic/irc/sanco/Home/main). Die Intensität der Fluoreszenz muss der des positiven Kontrollstammes bei gleicher Antikörperverdünnung zumindest entsprechen oder besser sein. Unvollständig gefärbte oder nur schwach fluoreszierende Zellen sind nicht zu berücksichtigen.
Bei jeglichem Verdacht auf eine Kontamination ist der Test zu wiederholen. Dies kann der Fall sein, wenn alle Objektträger in einer Charge wegen Pufferkontamination positive Zellen zeigen oder wenn auf der Objektträgerbeschichtung (außerhalb der Felder) positive Zellen gefunden werden.
4.4.3. Der Immunofluoreszenztest hat in Bezug auf die Spezifität verschiedene Probleme. Es ist damit zu rechnen, dass in Pellets aus Kartoffelnabelenden und Stängelstücken Hintergrundpopulationen mit fluoreszierenden Zellen und atypischer Morphologie sowie kreuzreagierende saprophytische Bakterien mit C. m. sepedonicus ähnlicher Größe und Morphologie auftreten.
4.4.4. Nur fluoreszierende Zellen von typischer Größe und Morphologie bei Verwendung des Titers oder der Arbeitsverdünnung des Antikörpers (siehe 4.3) berücksichtigen.
4.4.5. Auswertung des IF-Befundes:
- Für jede Probe, bei der stark fluoreszierende Zellen mit charakteristischer Morphologie gefunden werden, sind die mittlere Anzahl typischer Zellen je Mikroskopfeld zu schätzen und die Zahl typischer Zellen je ml resuspendiertes Pellet zu berechnen (Anlage 4).
Der IF-Befund gilt bei Proben mit mindestens 5 x 103 typischen Zellen je ml resuspendiertes Pellet als positiv. Die Probe gilt in diesem Falle als potenziell kontaminiert, und es sind weitere Tests erforderlich. - Der IF-Befund gilt bei Proben mit weniger als 5 x103 Zellen je ml resuspendiertes Pellet als negativ. Weitere Tests sind in diesem Falle nicht erforderlich.
5. FISH-TEST
Grundsatz
Wird der FISH-Test als Hauptscreeningtest angewandt und fällt dieser Test positiv aus, so muss obligatorisch als zweiter Screeningtest der IF-Test durchführt werden. Wird der FISH-Test als zweiter Screeningtest durchgeführt und fällt der Test positiv aus, so sind zur Sicherung der Diagnose weitere Tests nach dem Flussdiagramm erforderlich.
Hinweis:
Validierte C. m. subsp. sepedonicus spezifische Oligo-Sonden (Anlage 7) verwenden. Vorausgehende Tests mit diesem Verfahren müssen gewährleisten, mindestens 103-104 Zellen von C. m. subsp. sepedonicus je ml, die Probenextrakten mit zuvor negativem Testergebnis zugefügt wurden, reproduzierbar nachzuweisen.
Das im Folgenden beschriebene Verfahren ist vorzugsweise an frisch vorbereitetem Probenextrakt durchzuführen, kann aber auch bei Probenextrakten angewendet werden, die bei -16 bis - 24 °C oder bei - 68 bis - 86 °C unter Glyzerinzugabe gelagert wurden.
Als Negativkontrollen sind Aliquots von Probenextrakten zu verwenden, die zuvor mit negativem Testergebnis auf C. m. subsp. sepedonicus untersucht wurden.
Als Positivkontrollen aus einer 3-5 Tage alten Kultur Suspensionen mit 105 bis 106 C. m. subsp. sepedonicus Zellen je ml (z.B. Stamm NCPPB 4053 oder PD 406) in 0,01M Phosphatpuffer (PB) herstellen (siehe Anlage 2). Separate Objektträger mit Positivkontrollen des homologen Stammes oder eines anderen Referenzstammes von C. m. subsp. sepedonicus, in Kartoffelextrakt suspendiert, herstellen (siehe Anlage 2).
Die FITC-markierte eubakterielle Oligo-Sonde bietet insofern eine Kontrolle für den Hybridisierungsprozess, als sie alle Eubakterien anfärbt, die in der Probe vorhanden sind.
Das Kontrollmaterial nach demselben Verfahren testen wie die Probe(n).
5.1. Fixierung des Kartoffelextrakts
Das folgende Protokoll basiert auf Wullings et al., (1998):
5.1.1. Fixierlösung herstellen (siehe Anlage 7).
5.1.2. 100 µl von jedem Probenextrakt in ein Eppendorf-Röhrchen pipettieren und für 8 Minuten bei 7 000 g zentrifugieren.
5.1.3. Überstand verwerfen und das Pellet in 500 µl Fixiermittel, das < 24 Stunden zuvor angesetzt wurde, auflösen. Vortexen und über Nacht bei 4 °C inkubieren.
Als alternatives Fixiermittel kann 96 %iges Ethanol verwendet werden. Dazu das Pellet (Punkt 5.1.2) in 50 µl 0,01M PB und 50 µl 96 %igem Ethanol auflösen. Vortexen und für 30-60 Minuten bei 4 °C inkubieren.
5.1.4. Für 8 Minuten bei 7 000 g zentrifugieren. Überstand verwerfen und das Pellet in 75 µl 0,01M PB resuspendieren (siehe Anlage 3).
5.1.5. 16 µl der fixierten Suspensionen auf einen sauberen Multitest-Objektträger (siehe Abb. 3) auftragen. Zwei verschiedene Proben je Objektträger auftragen, unverdünnt, und 10 µl zur Herstellung einer 1 100 Verdünnung (in 0,01M PB) verwenden. Die restliche Probenlösung (49 µl) kann nach Zugabe von 1 Volumen 96 %igem Ethanol bei - 20 °C gelagert werden. Für den Fall, dass der FISH-Test wiederholt werden muss, das Ethanol abzentrifugieren und dasselbe Volumen von 0,01M PB zugeben (durch Vortexen vermischen).
Abb. 3: Layout des FISH-Objektträgers

5.1.6. Objektträger an der Luft (oder bei 37 °C auf einer Wärmebank) trocknen lassen und anschließend durch Abflammen fixieren.
In dieser Phase des Verfahrens kann die Hybridisierung unterbrochen und am nächsten Tag fortgesetzt werden. Objektträger sind staubfrei und trocken bei Raumtemperatur zu lagern.
5.2. Vorhybridisierung und Hybridisierung
5.2.1. Eine Lysozymlösung mit 10 mg Lysozym (Sigma L-6876) in 10 ml Puffer (100 mM Tris-HCI, 50 mM EDTA, pH 8,0) herstellen. Diese Lösung kann gelagert, sollte jedoch höchstens einmal aus dem Frost aufgetaut werden. Jede Probe sorgfältig mit ungefähr 50 μl Lysozymlösung bedecken und für 10 Minuten bei Raumtemperatur inkubieren. Objektträger anschließend nur einmal in entmineralisiertes Wasser tauchen und mit Filterpapier trocknen.
Alternativ können anstelle von Lysozym 50 μl von 40-400 μg ml-1 Proteinase K in Puffer (20 mM Tris-HCl, 2 mM CaCl2, pH 7,4) auf jedes Feld gegeben und bei 37 °C für 30 Minuten inkubiert werden.
5.2.2. Die Zellen in aufsteigender Ethanolreihe von 50 %, 80 % und 96 % für jeweils 1 Minute dehydratisieren. Objektträger in einem Objektträgerhalter lufttrocknen lassen.
5.2.3. Durch Auslegen des Bodens einer luftdichten Box mit Zellstoff- oder Filterpapier, das in lx Hybmix (Anlage 7) eingeweicht wurde, eine feuchte Inkubationskammer vorbereiten. Die Box im Hybridisierungsofen bei 55 °C für mindestens 10 Minuten vorinkubieren.
5.2.4. Die Hybridisierungslösung (Anlage 7) herstellen (45 μl je Objektträger) und für 5 Minuten bei 55 °C vorinkubieren.
5.2.5. Objektträger auf eine heiße (45 °C) Platte legen und 10 µl Hybridisierungslösung in jede der 4 Felder auf dem (den) Objektträger(n) geben.
5.2.6. Jeden Objektträger ohne Lufteinschluss mit zwei Deckgläsern (24 x 24 mm) abdecken. Objektträger in die vorgewärmte feuchte Kammer stellen und im Hybridisierungsofen über Nacht bei 55 °C im Dunkeln inkubieren.
5.2.7. Drei Becher vorbereiten mit 1 l Reinstwasser (UPW), 1 l 1x Hybmix (334 ml 3x Hybmix und 666 ml UPW) und 1 l 1/2x Hybmix (167 ml 3x Hybmix und 833 ml UPW) und jeweils im Wasserbad bei 55 °C einzeln vorinkubieren.
5.2.8. Deckgläser abnehmen und Objektträger in einen Objektträgerhalter stellen.
5.2.9. Auswaschen der überschüssigen Sonde durch 15-minütige Inkubation im Becher mit lx Hybmix bei 55 °C.
5.2.10. Objektträgerhalter in 1/2 Hybmix-Waschlösung stellen und für weitere 15 Minuten inkubieren.
5.2.11. Objektträger kurz in den Becher mit Reinstwasser (UPW) tauchen und auf Filterpapier setzen. Überschüssige Feuchtigkeit durch leichtes Abdecken der Oberfläche mit Filterpapier aufsaugen. 5-10 μl Antifading-Deckflüssigkeit (z.B. Vectashield, Vecta Laboratories, CA, USA o. Ä.) auf jedes Feld geben und den gesamten Objektträger mit einem großen Deckglas (24 x 60 mm) abdecken.
5.3. FISH-Test-Befund
5.3.1. Den Objektträger unverzüglich unter einem Epifluoreszenz-Mikroskop bei 630- oder 1 000facher Vergrößerung unter Ölimmersion untersuchen. Mit einem für die Fluorescein-Isothiocyanat-Markierung (FITC) geeigneten Filter werden eubakterielle Zellen (einschließlich der meisten Gram-negativen Zellen) in der Probe leuchtend grün angefärbt. Mit einem Filter für Tetramethylrhodamin-5-Isothiocyanat leuchten Cy3-angefärbte Zellen von C. m. subsp. sepedonicus fluoreszierend rot. Die Zeltmorphologie mit der der Positivkontrollen vergleichen. Die Zellen müssen stark fluoreszieren und vollständig gefärbt sein. Der FISH-Test (Abschnitt 9.4) ist bei abweichender Färbung zum Normalzustand zu wiederholen. Jedes Sichtfeld in zwei rechtwinklig zueinander stehenden Durchmessern sowie entlang des Feldrandes mikroskopisch untersuchen. Bei Proben, die keine oder nur wenige Zellen zeigen, mindestens 40 Mikroskopfelder untersuchen.
5.3.2. Auf stark fluoreszierende Zellen mit charakteristischer Morphologie von C. m. subsp. sepedonicus in den Testfeldern auf den Testobjektträgern untersuchen (siehe Website: http://forum.europa.euint/Public/irc/sanco/Home/main). Die Intensität der Fluoreszenz muss der des positiven Kontrollstammes entsprechen oder stärker sein. Unvollständig gefärbte oder schwach fluoreszierende Zellen sind nicht zu berücksichtigen.
5.3.3. Bei jeglichem Verdacht auf eine Kontamination ist der Test zu wiederholen. Dies kann der Fall sein, wenn alle Objektträger in einer Charge wegen Pufferkontamination positive Zellen zeigen oder wenn positive Zellen auf der Objektträgerbeschichtung (außerhalb der Felder) gefunden werden.
5.3.4. Der FISH-Test hat in Bezug auf die Spezifität verschiedene Probleme. Es ist damit zu rechnen, dass in Pellets aus Kartoffelnabelenden und Stängelstücken Hintergrundpopulationen fluoreszierender Zellen mit atypischer Morphologie und kreuzreagierende saprophytische Bakterien mit C. m. subsp. sepedonicus ähnlicher Größe und Morphologie auftreten, wenn auch weniger häufig als beim IF-Test.
5.3.5. Es sind nur fluoreszierende Zellen typischer Größe und Morphologie (siehe 5.3.2) zu berücksichtigen.
5.3.6. Auswertung des FISH-Test-Befundes:
- Gültig sind FISH-Test-Ergebnisse, wenn mit Hilfe des FITC-Filters grün fluoreszierende Zellen mit C. m. subsp. sepedonicus typischer Größe und Morphologie und mit Hilfe des Rhodaminfilters rot fluoreszierende Zellen in allen Positivkontrollen, jedoch in keiner einzigen Negativkontrolle festgestellt werden. Werden hell fluoreszierende Zellen einer charakteristischen Morphologie festgestellt, für jedes mikroskopische Feld die durchschnittliche Anzahl typischer Zellen schätzen und die Zahl der typischen Zellen je ml resuspendiertes Pellet (Anlage 4) berechnen. Proben mit mindestens 5 X 103 typischen Zellen je ml resuspendiertes Pellet gelten als potenziell kontaminiert, und weitere Tests sind erforderlich. Proben mit weniger als 5 X 103 typischen Zellen je ml resuspendiertes Pellet gelten als negativ.
- Der FISH-Test ist negativ, wenn mit dem Rhodaminfilter keine rot fluoreszierenden Zellen mit C. m. subsp. sepedonicus typischer Größe und Morphologie festgestellt werden, vorausgesetzt allerdings, dass sich in den Positivkontrollen bei Verwendung des Rhodaminfilters typische rot fluoreszierende Zellen nachweisen lassen.
 | weiter . | |
_________________________
1) ABl. Nr. L 259 vom 18.10.1993 S. 2.
...
X
⍂
↑
↓