
umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (3)
 | zurück |
|
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 105 (1995). Sie ist eine überarbeitete Fassung der ursprünglichen TG 105, die 1981 angenommen wurde. Es gibt keine inhaltlichen Unterschiede zwischen der derzeitigen Fassung und der von 1981. Geändert wurde hauptsächlich das Format. Die Überarbeitung stützte sich auf die EU-Prüfmethode 'Wasserlöslichkeit' (1).
2. Die Wasserlöslichkeit eines Stoffs kann durch Verunreinigungen erheblich beeinflusst werden. Diese Prüfmethode behandelt die Bestimmung der Wasserlöslichkeit von im Wesentlichen reinen Substanzen, die in Wasser stabil und nicht flüchtig sind. Vor Bestimmung der Wasserlöslichkeit sollten Vorinformationen über die Strukturformel, den Dampfdruck, die Dissoziationskonstante und das Hydrolyseverhalten (als Funktion des pH-Wertes) des Stoffes vorliegen.
3. Die beiden nachstehend beschriebenen Methoden, d. h. die Säulen-Elutions-Methode und die Kolbenmethode, decken Löslichkeiten von unter bzw. über 10-2 g/l ab. Ein einfacher Vorversuch wird ebenfalls beschrieben. Er ermöglicht die Bestimmung der bei der eigentlichen Prüfung zu verwendenden ungefähren Probenmenge und die zum Erreichen der Sättigungskonzentration notwendige Zeit.
4. Die Wasserlöslichkeit einer Substanz wird durch ihre Massen-Sättigungskonzentration in Wasser bei einer bestimmten Temperatur angegeben.
5. Die Wasserlöslichkeit wird als Masse des gelösten Stoffs je Lösungsvolumen ausgedrückt. Die SI-Einheit ist kg/m3, aber g/l kann auch verwendet werden.
6. Bei der Untersuchung der Prüfsubstanz brauchen keine Referenzsubstanzen verwendet zu werden.
7. Die Prüfung wird vorzugsweise bei 20 °C ± 0,5 °C durchgeführt. Die gewählte Temperatur ist in allen wichtigen Teilen der Apparatur konstant zu halten.
8. Etwa 0,1 g der Probe (feste Prüfsubstanzen müssen pulverisiert sein) werden in einen mit Glasstopfen verschließbaren 10-ml-Messzylinder gegeben. Dann werden portionsweise zunehmende Volumen Wasser von Raumtemperatur zugesetzt. Nach jedem Zusatz einer Wassermenge wird die Mischung 10 Minuten geschüttelt und mit bloßem Auge auf ungelöste Teilchen der Probe untersucht. Wenn nach Zusatz von 10 ml Wasser die Probe oder Teile von ihr ungelöst bleiben, ist der Versuch in einem 100-ml-Messzylinder zu wiederholen. Die ungefähre Löslichkeit ist in Tabelle 1 unter demjenigen Volumen Wasser angegeben, bei dem die Probe vollständig gelöst wird. Bei geringer Löslichkeit kann es lange (bis zu 24 Stunden) dauern, bis die Prüfsubstanz gelöst ist. Ist die Prüfsubstanz nach 24 Stunden noch nicht gelöst, sollte der Versuch verlängert werden (maximal 96 Stunden) oder es sollte weiter verdünnt werden, um festzustellen, ob die Säulen-Elutions- oder die Kolben-Methode zu benutzen ist.
| ml Wasser auf 0,1 g lösliche Substanz | 0,1 | 0,5 | 1 | 2 | 10 | 100 | > 100 |
| Ungefähre Löslichkeit in g/l | > 1.000 | 1.000-200 | 200-100 | 100-50 | 50-10 | 10-1 | < 1 |
Säulen-Elutions-Methode
Prinzip
9. Diese Methode basiert auf der Elution einer Prüfsubstanz mit Wasser aus einer Mikrosäule, die mit einem inerten Trägermaterial gefüllt ist, welches mit einem Überschuss an Prüfsubstanz beschichtet ist (2). Die Wasserlöslichkeit wird durch die Massenkonzentration des Eluats ausgedrückt, wenn dieses ein Plateau in Abhängigkeit von der Zeit erreicht hat.
Apparatur
10. Die Apparatur besteht aus einer Mikrosäule (Abbildung 1), die auf konstanter Temperatur gehalten wird. Sie ist entweder mit einer Umwälzpumpe (Abbildung 2) oder mit einem Niveaugefäß (Abbildung 3) verbunden. Die Mikrosäule enthält ein inertes Trägermaterial, das durch einen kleinen Pfropfen aus Glaswolle in der Säule gehalten wird, der gleichzeitig zum Herausfiltern von Partikeln dient. Als Trägermaterial können Glaskugeln, Diatomeenerde oder andere inerte Stoffe verwendet werden.
11. Die Mikrosäule in Abbildung 1 eignet sich für die Apparatur mit Umwälzpumpe. Sie hat einen Kopfraum für fünf Säulenbett-Volumina (werden zu Beginn des Versuchs verworfen) und das Volumen von fünf Proben (werden während des Versuchs entnommen). Der Kopfraum kann kleiner gehalten werden, wenn während des Versuchs Wasser zugegeben werden kann, um die anfänglich mit Verunreinigungen entnommenen fünf Säulenbett-Volumina zu ersetzen. Die Säule ist durch einen Schlauch aus inertem Material mit der Umwälzpumpe verbunden, die mit einem Fluss von etwa 25 ml/h fördern kann. Die Umwälzpumpe kann z.B. eine Schlauch- oder eine Membranpumpe sein. Dabei ist darauf zu achten, dass es nicht zu einer Verunreinigung und/oder Absorption durch das Schlauchmaterial kommt.
12. Abbildung 3 zeigt die schematische Darstellung mit Verwendung eines Niveaugefäßes. In dieser Darstellung ist die Mikrosäule mit einem Einweghahn versehen. Sie wird mit einem Glasschliff-Verbindungsstück und einem Schlauch aus inertem Material an das Niveaugefäß angeschlossen. Die Durchflussrate vom Niveaugefäß sollte etwa 25 ml/h betragen.

Abmessungen in mm
A. Anschluss für Glasschliff-Stopfen
B. Kopfraum
C. Innenmaß 5
D. Außenmaß 19
E. Glaswollpfropfen
F. Absperrhahn

A. atmosphärischer Druckausgleich
B. Durchflussmesser
C. Mikrosäule
D. thermostatgeregelte Pumpe
E. Umwälzpumpe
F. 2-Wege-Hahn zur Probenentnahme
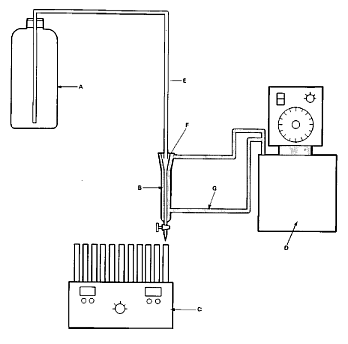
A. Niveaugefäß (z.B. 2,5-Liter-Kolben)
B. Säule
C. Fraktionssammler
D. Thermostat
E. Teflonschlauch
F. Glasschliff-Stopfen
G. Wasserschlauch (zwischen Thermostat und Säule, Innendurchmesser ungefähr 8 mm)
13. Etwa 600 mg Trägermaterial werden in einen 50-ml-Rundkolben eingefüllt. Eine geeignete Menge Prüfsubstanz wird in einem flüchtigen Lösungsmittel von Analysenqualität gelöst, und eine ausreichende Menge dieser Lösung wird zum Trägermaterial hinzugefügt. Das Lösungsmittel muss vollständig abgezogen werden, z.B. in einem Rotationsverdampfer, da sonst während der Elutionsphase wegen Verteilungseffekten auf der Oberfläche keine vollständige Sättigung dieses Materials mit Wasser erzielt wird. Das beladene Trägermaterial lässt man etwa 2 Stunden lang in etwa 5 ml Wasser quellen. Dann wird die Suspension in die Mikrosäule gefüllt. Es ist auch möglich, das trockene, beladene Trägermaterial in die mit Wasser gefüllte Mikrosäule zu geben. Auch hier wird der Quellvorgang von etwa 2 Stunden abgewartet.
14. Das Aufbringen der Prüfsubstanz auf das Trägermaterial kann problematisch werden und zu fehlerhaften Ergebnissen führen, z.B. wenn sich die Prüfsubstanz ölartig auf dem Träger niederschlägt. Diese Probleme sollten untersucht und Einzelheiten dazu dokumentiert werden.
Verfahren mit Umwälzpumpe
15. Der Säulenfluss wird in Gang gesetzt. Eine Durchflussleistung von etwa 25 ml/h wird empfohlen (etwa zehn Säulenbett-Volumina/h bei der beschriebenen Säule). Mindestens die ersten fünf Säulenbett-Volumina werden verworfen, um wasserlösliche Verunreinigungen zu entfernen. Danach lässt man die Umwälzpumpe bis zur Einstellung des Gleichgewichts laufen. Das Gleichgewicht ist erreicht, wenn bei fünf aufeinander folgenden Proben die Konzentrationen um nicht mehr als ± 30 % streuen. Diese Proben sollten in solchen zeitlichen Abständen genommen werden, in denen mindestens zehn Säulenbett-Volumina die Säule durchlaufen haben. Je nach verwendeter Analysemethode kann es ratsam sein, eine Konzentrations-Zeit-Kurve zu erstellen, um zu zeigen, dass das Gleichgewicht erreicht ist.
Verfahren mit Niveaugefäß
16. Aufeinander folgende Eluatfraktionen werden gesammelt und ihre Konzentrationen mit der gewählten Analysemethode bestimmt. Fraktionen des mittleren Eluatbereichs, bei denen die Konzentrationen in mindestens fünf aufeinander folgenden Proben konstant bleiben (± 30 %), werden zur Bestimmung der Wasserlöslichkeit benutzt.
17. Das bevorzugte Elutionsmittel ist bidestilliertes Wasser. Es kann auch deionisiertes Wasser mit einem spezifischen Widerstand von mehr als 10 Megaohm/cm und einem Gesamtgehalt an organischem Kohlenstoff unter 0,01 % verwendet werden.
18. Bei beiden Verfahren wird ein zweiter Durchlauf mit halber Durchflussrate durchgeführt. Stimmen die Ergebnisse der beiden Versuche überein, wird das Prüfergebnis als zufriedenstellend betrachtet. Ist die gemessene Löslichkeit bei dem niedrigeren Durchfluss höher, muss die Durchflussleistung so lange weiter halbiert werden, bis zwei aufeinander folgende Versuchsdurchläufe die gleiche Löslichkeit ergeben.
19. Bei beiden Verfahren sollten die Fraktionen durch Prüfung des Tyndall-Effekts auf kolloidale Substanzpartikel untersucht werden. Wenn solche Substanzpartikel vorkommen, ist das Prüfergebnis unbrauchbar. Die Prüfung sollte dann wiederholt werden, nachdem die Filterfunktion der Säule verbessert wurde.
20. Der pH-Wert jeder Probe sollte vorzugsweise mit speziellen Indikatorstäbchen bestimmt werden.
Kolben-Methode
Prinzip
21. Die Prüfsubstanz (Feststoffe müssen pulverisiert werden) wird bei einer Temperatur in Wasser aufgelöst, die leicht über der Prüftemperatur liegt. Wenn die Sättigung erreicht ist, wird die Lösung abgekühlt und auf der Prüftemperatur gehalten. Alternativ kann die Messung direkt bei der Prüftemperatur durchgeführt werden, wenn durch entsprechende Probenahme gesichert ist, dass das Sättigungsgleichgewicht erreicht ist. Dann wird die Massenkonzentration der Prüfsubstanz in der wässrigen Lösung, die keine ungelösten Substanzpartikel enthalten darf, mit einer geeigneten Analysemethode (3) bestimmt.
Geräte
22. Folgende Geräte werden benötigt:
- übliche Laborglasgeräte und -instrumente,
- eine Vorrichtung zum Schütteln der Lösungen bei konstanter Temperatur,
- eine Zentrifuge (möglichst thermostatisiert), falls diese bei Emulsionen erforderlich wird, und
- Analysegeräte.
Verfahren
23. Die zur Sättigung des vorgegebenen Wasservolumens erforderliche Prüfsubstanzmenge wird anhand der Ergebnisse des Vorversuches abgeschätzt. Etwa das Fünffache dieser Menge wird jeweils in drei mit Glasstopfen versehene Glasgefäße eingewogen (z.B. Zentrifugenröhrchen oder Kolben). Jedem Gefäß wird ein je nach Analysemethode und Löslichkeitsbereich gewähltes Wasservolumen zugesetzt. Die Gefäße werden fest verschlossen und dann bei 30 °C geschüttelt. Hierzu sollte ein Schüttel- oder Rührgerät verwendet werden, das bei einer konstanten Temperatur arbeitet, z.B. Magnetrührstäbe in einem thermostatisierten Wasserbad. Nach einem Tag wird eines der Gefäße 24 Stunden unter gelegentlichem Schütteln bei Prüftemperatur stehen gelassen, bis sich das Gleichgewicht eingestellt hat. Dann wird der Inhalt des Gefäßes bei Prüftemperatur zentrifugiert und die Konzentration der Prüfsubstanz in der klaren wässrigen Phase mit einem geeigneten Analyseverfahren bestimmt. Mit den beiden anderen Kolben wird nach zwei bzw. drei Tagen genauso verfahren, nachdem zuvor das Sättigungsgleichgewicht bei 30 °C eingestellt wurde. Weichen die gemessenen Konzentrationen bei mindestens den letzten beiden Gefäßen um nicht mehr als 15 % voneinander ab, ist die Prüfung als zufriedenstellend anzusehen. Wenn die Prüfergebnisse der Gefäße 1, 2 und 3 eine steigende Tendenz aufweisen, sollte die gesamte Prüfung unter Verlängerung der Zeiten für die Gleichgewichtseinstellung wiederholt werden.
24. Die Prüfung kann auch ohne Präinkubation bei 30 °C durchgeführt werden. Um den Grad des erreichten Sättigungsgleichgewichts zu bestimmen, werden so lange Proben entnommen, bis die gemessenen Konzentrationen nicht länger von der Rührzeit beeinflusst werden.
25. Der pH-Wert jeder Probe sollte vorzugsweise mit speziellen Indikatorstäbchen bestimmt werden.
Analytische Bestimmungen
26. Eine substanzspezifische Methode ist vorzuziehen, da bereits kleine Mengen von löslichen Verunreinigungen große Fehler bei der Bestimmung der Löslichkeit verursachen können. Beispiele für solche Analysemethoden sind: Gas- oder Flüssigchromatographie, Titrierverfahren, fotometrische Methoden, voltametrische Verfahren.
Daten
Säulen-Elutions-Methode
27. Für jeden Durchlauf werden der Mittelwert und die Standardabweichung von mindestens fünf aufeinander folgenden Proben aus dem Bereich des Sättigungsplateaus berechnet. Die für zwei Prüfungen mit unterschiedlichen Durchflussleistungen berechneten Mittelwerte sollten nicht um mehr als 30 % voneinander abweichen.
Kolben-Methode
28. Die einzelnen Ergebnisse für jeden der drei Kolben, die nicht um mehr als 15 % voneinander abweichen sollten, werden gemittelt.
Prüfbericht
Säulen-Elutions-Methode
29. Der Prüfbericht muss folgende Informationen enthalten:
- die Ergebnisse des Vorversuchs,
- Angaben zur Prüfsubstanz (Identität und Verunreinigungen), gegebenenfalls Vorreinigung,
- die Konzentrationen, Durchflussraten und pH-Werte jeder Probe,
- die Mittelwerte und Standardabweichungen von mindestens fünf Proben aus dem Bereich des Sättigungsplateaus eines jeden Durchlaufs,
- den Durchschnitt von mindestens zwei aufeinanderfolgenden Durchläufen,
- die Wassertemperatur während des Sättigungsvorgangs,
- die Analysemethode,
- die Art des verwendeten Trägermaterials,
- die Beladung des Trägermaterials,
- das verwendete Lösungsmittel,
- gegebenenfalls Hinweise auf eine chemische Instabilität der Prüfsubstanz während des Prüfungsvorgangs,
- alle für die Auswertung der Ergebnisse sachdienlichen Informationen und Bemerkungen, insbesondere in Bezug auf Verunreinigungen und den Aggregatzustand der Prüfsubstanz.
Kolben-Methode
30. Der Prüfbericht muss folgende Informationen enthalten:
- die Ergebnisse des Vorversuchs,
- Angaben zur Prüfsubstanz (Identität und Verunreinigungen), gegebenenfalls Vorreinigung,
- die einzelnen Analysenergebnisse und die Durchschnittswerte, wenn pro Kolben mehr als ein Wert bestimmt wurde,
- den pH-Wert jeder Probe,
- den Mittelwert derjenigen Kolben, deren Ergebnisse übereinstimmen,
- die Prüftemperatur,
- die Analysemethode,
- gegebenenfalls Hinweise auf eine chemische Instabilität der Prüfsubstanz während des Prüfungsvorgangs,
- alle für die Auswertung der Ergebnisse sachdienlichen Informationen und Bemerkungen, insbesondere in Bezug auf Verunreinigungen und den Aggregatzustand der Prüfsubstanz.
1. Richtlinie 92/69/EWG der Kommission vom 31. Juli 1992 zur siebzehnten Anpassung der Richtlinie 67/548/EWG des Rates zur Angleichung der Rechts- und Verwaltungsvorschriften für die Einstufung, Verpackung und Kennzeichnung gefährlicher Stoffe an den technischen Fortschritt, ABl.L 383 vom 29.12.1992 S. 113.
2. NF T 20-045 (AFNOR) (September 1985). Chemical products for industrial use - Determination of water solubility of solids and liquids with low solubility - Column elution method.
3. NF T 20-046 (AFNOR) (September 1985). Chemical products for industrial use - Determination of water solubility of solids and liquids with high solubility - Flask method.
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode oder sonstige anzuwendende Prüfmethode für den betreffenden Endpunkt sind in Teil 0 Tabelle 1 aufgeführt.
| 1. Methode
Der hier beschriebenen Schüttelmethode liegt die OECD-Prüfrichtlinie (1) zugrunde. 1.1 Einleitung Zur Durchführung der Prüfung ist es nützlich, Vorinformationen über die Strukturformel, die Dissoziationskonstante, die Wasserlöslichkeit, das Hydrolyseverhalten, die n-Oktanol-Löslichkeit und die Oberflächenspannung des Stoffes in wässriger Lösung zu haben. Messungen von ionischen Substanzen sollten nur an deren nicht ionisierter Form (freie Säure oder freie Base) durch Verwendung eines geeigneten Puffers mit einem pH-Wert von mindestens einer pH-Einheit unter (freie Säure) oder über (freie Base) dem pK-Wert durchgeführt werden. Diese Prüfmethode beinhaltet zwei getrennte Verfahren: die Schüttelmethode und die Hochleistungs-Flüssigkeitschromatografie (HPLC). Die erste findet dann Anwendung, wenn der log-Pow-Wert (Definitionen siehe unten) im Bereich - 2 bis 4 liegt, die letzte dann, wenn dieser Wert im Bereich 0 bis 6 liegt. Vor der Messung mit einer der beiden Methoden sollte eine Vorab-Schätzung des Verteilungskoeffizienten durchgeführt werden. Die Schüttelmethode gilt nur für im Wesentlichen reine Substanzen, die in Wasser und n-Oktanol löslich sind. Sie ist nicht auf oberflächenaktive Stoffe anwendbar (für diese sollte ein berechneter oder ein geschätzter Wert auf der Grundlage der einzelnen Löslichkeiten in n-Oktanol und Wasser vorgelegt werden). Die HPLC-Methode ist nicht für starke Säuren und Basen, Metallkomplexe, oberflächenaktive Stoffe oder für Substanzen anwendbar, die mit dem Eluenten reagieren. Für diese Stoffe sollte ein berechneter oder ein geschätzter Wert auf der Grundlage der einzelnen Löslichkeiten in n-Oktanol und Wasser vorgelegt werden. Die HPLC-Methode ist bezüglich Verunreinigungen in der Prüfsubstanz weniger empfindlich als die Schüttelmethode. Dennoch kann die Interpretation der Ergebnisse in einigen Fällen durch das Vorliegen von Verunreinigungen erschwert werden, weil die Zuordnung der Peaks nicht eindeutig ist. Für Mischungen, die ein nicht aufgelöstes Band ergeben, sollten die obere und die untere Grenze des Zehnerlogarithmus (log P) angegeben werden. 1.2 Definitionen und Einheiten Als Verteilungskoeffizient (P) bezeichnet man das Verhältnis der Gleichgewichtskonzentrationen (ci) einer gelösten Substanz in einem Zweiphasensystem aus zwei weitgehend unmischbaren Lösungsmitteln. Im Falle von n-Oktanol und Wasser ergibt sich: pow = (cn- Oktanol)/cWasser Der Verteilungskoeffizient (P) ist somit der Quotient zweier Konzentrationen. Er wird gewöhnlich in Form seines Zehnerlogarithmus (log P) angegeben. 1.3 Referenzsubstanzen Schüttelmethode Referenzsubstanzen müssen nicht in allen Fällen verwendet werden, in denen eine neue Prüfsubstanz untersucht wird. Die Referenzsubstanzen sollten in erster Linie dazu dienen, die Methode von Zeit zu Zeit zu überprüfen und einen Vergleich mit den Ergebnissen aus anderen Methoden zu ermöglichen. HPLC-Methode Um die HPLC-Messdaten einer Substanz mit deren P-Wert zu korrelieren, ist eine Eichkurve log P/ chromatografische Daten unter Verwendung von mindestens sechs Bezugspunkten aufzustellen. Die Wahl der geeigneten Referenzsubstanzen obliegt dem Benutzer. Soweit möglich, sollte mindestens eine Referenzsubstanz einen Pow-Wert über dem der Prüfsubstanz und eine andere einen Pow-Wert unter dem der Prüfsubstanz haben. Für log-P-Werte unter 4 kann bei der Eichung von Daten ausgegangen werden, die mit Hilfe der Schüttelmethode erhalten worden sind. Für log-P-Werte über 4 kann man sich bei der Eichung auf kalibrierte Literaturwerte stützen, sofern diese mit den berechneten Werten übereinstimmen. Aus Gründen einer größeren Genauigkeit sollten vorzugsweise Referenzsubstanzen verwendet werden, die strukturell mit der Prüfsubstanz verwandt sind. Es liegen umfangreiche Listen mit log-Pow-Werten für zahlreiche Gruppen von Chemikalien vor (2) (3). Wenn keine Verteilungskoeffizienten zu strukturell verwandten Verbindungen vorhanden sind, kann eine allgemeinere Eichung auf der Grundlage anderer Referenzsubstanzen vorgenommen werden. Eine Liste der empfohlenen Referenzsubstanzen und deren Pow-Werten ist in Anlage 2 enthalten. 1.4 Prinzip der Methode 1.4.1 Schüttelmethode Zur Bestimmung des Verteilungskoeffizienten müssen nach Einstellung des Gleichgewichts zwischen allen wechselwirkenden Komponenten des Verteilungssystems die Konzentrationen der in beiden Phasen gelösten Substanz ermittelt werden. Die einschlägige Literatur zeigt, dass hierfür verschiedene Techniken vorhanden sind, wie z.B. die gründliche Mischung der beiden Phasen mit anschließender Phasentrennung zur Bestimmung der Gleichgewichtskonzentration der untersuchten Substanz. 1.4.2 HPLC-Methode Die HPLC-Methode wird an Analysensäulen durchgeführt, die mit einer handelsüblichen festen Phase mit langen, chemisch an Siliziumdioxid gebundenen Kohlenwasserstoffketten (z.B. C8, C18) gefüllt sind. Chemikalien, die in eine solche Säule eingespritzt werden, bewegen sich darin wegen der unterschiedlichen Verteilungsgrade zwischen der mobilen und der stationären (Kohlenwasserstoffe) Phase mit unterschiedlicher Geschwindigkeit. Substanzgemische werden entsprechend dem hydrophoben Charakter der Bestandteile eluiert - zuerst die wasserlöslichen und zuletzt die öllöslichen; dabei erfolgt die Elution proportional zum jeweiligen Kohlenwasserstoff-Wasser-Verteilungskoeffizienten. Dadurch kann die Beziehung zwischen der Retentionszeit an einer solchen (Phasenumkehr-)Säule und dem Verteilungskoeffizienten für n-Oktanol/Wasser aufgestellt werden. Der Verteilungskoeffizient wird vom Kapazitätsfaktor k über die Formel k = (tr- to)/to abgeleitet, wobei tR die Retentionszeit der Prüfsubstanz und to die durchschnittliche Zeit ist, die ein Lösungsmittelmolekül für die Wanderung durch die Säule benötigt (Totzeit). Quantitative Analysenmethoden sind nicht erforderlich; es müssen lediglich die Elutionszeiten bestimmt werden. 1.5 Qualitätskriterien 1.5.1 Wiederholbarkeit Schüttelmethode Um die Genauigkeit des Verteilungskoeffizienten zu gewährleisten, sind Doppelbestimmungen bei drei verschiedenen Prüfbedingungen durchzuführen. Dazu sollen sowohl die eingesetzte Menge der untersuchten Substanz als auch das Verhältnis der Lösungsmittelvolumina verändert werden. Die so ermittelten Werte des Verteilungskoeffizienten, angegeben als deren Zehnerlogarithmus, sollen in einem Bereich von ± 0,3 log-Einheiten liegen. HPLC-Methode Um die Zuverlässigkeit der Messung zu erhöhen, sind Doppelbestimmungen durchzuführen. Die aus den Einzelmessungen abgeleiteten log-P-Werte sollen in einem Bereich von ± 0,1 log-Einheiten liegen. 1.5.2 Empfindlichkeit Schüttelmethode Der Messbereich der Methode wird durch die Nachweisgrenze des Analysenverfahrens festgelegt. Dieses sollte die Bestimmung von log-Pow-Werten innerhalb eines Bereichs von - 2 bis 4 erlauben (sofern es die Bedingungen zulassen, kann dieser Bereich gelegentlich auf Pow-Werte bis 5 erweitert werden, wenn die Konzentration der gelösten Substanz in keiner Phase größer als 0,01 Mol/l ist). HPLC-Methode Die HPLC-Methode erlaubt die Bestimmung von Verteilungskoeffizienten innerhalb eines Pow-Bereichs von 0 bis 6. Normalerweise lässt sich der Verteilungskoeffizient einer Verbindung innerhalb eines Bereichs von ± 1 log-Einheit des bei der Schüttelmethode gewonnenen Wertes bestimmen. Typische Korrelationen sind in der Literatur angegeben (4) (5) (6) (7) (8). Eine höhere Genauigkeit ist gewöhnlich zu erreichen, wenn die Korrelationskurven von strukturell verwandten Referenzsubstanzen ausgehen (9). 1.5.3 Anwendbarkeit Schüttelmethode Das Nernst'sche Verteilungsgesetz gilt nur für verdünnte Lösungen bei konstanter Temperatur, konstantem Druck und pH-Wert. Es gilt streng nur für eine reine Substanz, die zwischen zwei reinen Lösungsmitteln verteilt ist. Wenn mehrere gelöste Stoffe in einer oder beiden Phasen gleichzeitig vorkommen, kann dadurch das Ergebnis beeinflusst werden. Dissoziation oder Assoziation gelöster Moleküle führen zu Abweichungen vom Nernst'schen Verteilungsgesetz. Solche Abweichungen zeigen sich darin, dass der Verteilungskoeffizient von der Konzentration der Lösung abhängig wird. Wegen der auftretenden multiplen Verteilungsgleichgewichte sollte diese Prüfmethode für ionische Verbindungen nicht ohne entsprechende Korrekturen angewendet werden. Für derartige Verbindungen sollte die Benutzung von Pufferlösungen anstelle von Wasser erwogen werden; dabei sollte der pH-Wert des Puffers mindestens 1 pH-Einheit vom pKa-Wert der Substanz entfernt sein und die Bedeutung dieses pH-Wertes für die Umwelt berücksichtigt werden. 1.6 Beschreibung der Methode 1.6.1 Abschätzung des Verteilungskoeffizienten Der Verteilungskoeffizient wird vorzugsweise durch ein Berechnungsverfahren abgeschätzt (siehe Anlage 1); wo möglich, kann er aus dem Löslichkeitsverhältnis der Prüfsubstanz in den reinen Lösungsmitteln abgeschätzt werden (10). 1.6.2 Schüttelmethode 1.6.2.1 Vorbereitung n-Oktanol: Die Bestimmung des Verteilungskoeffizienten soll mit sehr reinem Reagens durchgeführt werden. Wasser: Es soll in Glas- oder Quarzgefäßen destilliertes bzw. doppelt destilliertes Wasser verwendet werden. Für ionische Verbindungen sollten, wenn begründbar, anstelle von Wasser Pufferlösungen verwendet werden. Anmerkung Direkt aus einem Ionenaustauscher entnommenes Wasser soll nicht benutzt werden. 1.6.2.1.1 Vorsättigung der Lösungsmittel Vor der Bestimmung des Verteilungskoeffizienten werden die Phasen des Lösungsmittelsystems durch Schütteln bei Prüftemperatur gegenseitig gesättigt. Dazu ist es zweckmäßig, zwei große Vorratsflaschen gefüllt mit sehr reinem n-Oktanol bzw. Wasser mit jeweils einer ausreichenden Menge des anderen Lösungsmittels zu versetzen, mit einem mechanischen Schüttelapparat 24 Stunden zu schütteln und dann so lange stehen zu lassen, bis sich die Phasen getrennt haben und der Sättigungszustand erreicht ist. 1.6.2.1.2 Vorbereitung der Prüfung Das Gesamtvolumen des Zweiphasensystems soll das Prüfgefäß nahezu ausfüllen. Dadurch können Materialverluste aufgrund von Verdampfung verhindert werden. Das Volumenverhältnis und die einzusetzenden Mengen der Substanz werden durch die folgenden Angaben festgelegt:
Es sind drei Prüfungen durchzuführen. Bei der ersten wird das berechnete Volumenverhältnis n-Oktanol/Wasser eingesetzt, bei der zweiten wird dieses Verhältnis halbiert, bei der dritten verdoppelt (z.B. 1:1, 1:2, 2:1). 1.6.2.1.3 Prüfsubstanz Es wird eine Vorratslösung in mit Wasser vorgesättigtem n-Oktanol hergestellt. Die Konzentration dieser Vorratslösung soll vor deren Gebrauch zur Bestimmung des Verteilungskoeffizienten exakt bestimmt werden. Diese Lösung soll so gelagert werden, dass ihre Stabilität gewährleistet ist. 1.6.2.2 Prüfbedingungen Die Prüftemperatur sollte zwischen 20 und 25 °C liegen und konstant (± 1 °C) gehalten werden. 1.6.2.3 Messverfahren 1.6.2.3.1 Einstellen des Verteilungsgleichgewichts Für jede der Prüfbedingungen sollen zwei Prüfgefäße vorbereitet werden, die jeweils die erforderlichen, genau abgemessenen Mengen der beiden Lösungsmittel sowie die erforderliche Menge an Vorratslösung enthalten. Die n-Oktanol-Phasen sollten volumetrisch bestimmt werden. Die Prüfgefäße sollten entweder mit einem geeigneten Schüttelapparat oder von Hand geschüttelt werden. Bei Verwendung eines Zentrifugenglases besteht ein empfohlenes Verfahren darin, das Glas rasch um 180 °C um seine Querachse zu drehen, so dass eventuell eingeschlossene Luft durch beide Phasen aufsteigt. Erfahrungsgemäß reichen im Allgemeinen 50 solcher Umdrehungen zur Einstellung des Verteilungsgleichgewichts aus. Zur Sicherheit werden 100 Umdrehungen in 5 Minuten empfohlen. 1.6.2.3.2 Phasentrennung Zur Trennung der Phasen sollte die Mischung, sofern erforderlich, in einer Laborzentrifuge bei Raumtemperatur zentrifugiert werden. Wenn eine Zentrifuge ohne Thermostat benutzt wird, sollten die Zentrifugengläser vor der Analyse mindestens 1 Stunde bei Prüftemperatur aufbewahrt werden, damit sich das Gleichgewicht einstellt. 1.6.2.4 Analyse Zur Ermittlung des Verteilungskoeffizienten müssen die Konzentrationen der Prüfsubstanz in beiden Phasen analysiert werden. Dies kann dadurch geschehen, dass von jeder der beiden Phasen aus jedem Glas und für jede Prüfbedingung ein aliquoter Teil entnommen und mit dem gewählten Verfahren analysiert wird. Die in den beiden Phasen vorhandene Gesamtmenge der Substanz ist zu berechnen und mit der eingesetzten Menge zu vergleichen. Die Probenahme aus der wässrigen Phase sollte so erfolgen, dass die Gefahr des Einschlusses von Spuren an n-Oktanol möglichst weitgehend vermindert wird; z.B. kann eine Glasspritze mit auswechselbarer Nadel zur Probenahme verwendet werden. Zuerst sollte die Spritze teilweise mit Luft gefüllt werden. Diese Luft sollte vorsichtig herausgedrückt werden, während die Nadel durch die n-Oktanol-Schicht hindurchgeführt wird. Ein ausreichendes Volumen an wässriger Phase wird in die Spritze gezogen. Die Spritze wird schnell aus der Lösung entfernt und die Nadel abgenommen. Der Inhalt der Spritze kann dann als wässrige Probe weiterverwendet werden. Die Konzentration in den beiden voneinander getrennten Phasen sollte am besten mit einem substanzspezifischen Verfahren ermittelt werden. Beispiele für möglicherweise geeignete Analysenverfahren sind:
1.6.3 HPLC-Methode 1.6.3.1 Vorbereitung Apparatur Erforderlich ist ein mit einer pulsfreien Pumpe und einem geeigneten Detektor ausgestatteter Flüssigkeitschromatograf. Dabei wird die Verwendung eines Einspritzventils mit Dosierschleife empfohlen. Die Leistung der HPLC-Säule kann durch das Vorhandensein polarer Gruppen in der stationären Phase ernsthaft beeinträchtigt werden. Deshalb sollten die stationären Phasen ein Minimum an polaren Gruppen haben (11). Es können handelsübliche Mikroteilchenfüllungen für die Umkehrphasenchromatografie oder Fertigsäulen verwendet werden. Zwischen dem Dosiersystem und der Analysensäule kann eine Vorsäule angebracht werden. Mobile Phase Zur Zubereitung des Elutionsmittels werden für die HPLC-Methode ausreichend reines Methanol und Wasser verwendet; das Elutionsmittel wird vor seiner Verwendung entgast. Es sollte das Verfahren der isokratischen Elution angewendet werden. Dabei werden Methanol-Wasser-Verhältnisse mit einem Mindestgehalt an Wasser von 25 % empfohlen. Im Normalfall ist eine Methanol-Wasser-Mischung im Volumenverhältnis 3:1 für die Eluierung von Verbindungen mit einem log-P-Wert von 6 bei einer Elutionszeit von einer Stunde ausreichend (Durchflussrate: 1 ml/min). Für Verbindungen mit einem hohen log-P-Wert kann eine Verkürzung der Elutionszeit (auch der der Referenzsubstanzen) durch Senkung der Polarität der mobilen Phase oder Kürzung der Säulenlänge erforderlich sein. Stoffe mit einer sehr geringen Löslichkeit in n-Oktanol ergeben bei der HPLC-Methode häufig anormal niedriger log-Pow-Werte; die Peaks dieser Stoffe begleiten mitunter die Lösungsmittelfront. Dies liegt wahrscheinlich daran, dass der Verteilungsprozess zu langsam ist, um innerhalb der normalerweise für eine HPLC-Trennung benötigten Zeit den Gleichgewichtszustand zu erreichen. In solchen Fällen kann die Verminderung der Durchflussrate und/oder des Methanol-Wasser-Verhältnisses ein wirksames Verfahren sein, um zu einem zuverlässigen Wert zu gelangen. Prüf- und Referenzsubstanz sollten in der mobilen Phase in ausreichender Konzentration lösbar sein, um nachgewiesen werden zu können. Nur in Ausnahmefällen dürfen in der Methanol-Wasser-Mischung Zusatzstoffe verwendet werden, da diese Zusatzstoffe die Eigenschaften der Säule verändern. Für Chromatogramme, die mit Zusatzstoffen erhalten wurden, ist der Einsatz einer weiteren Säule desselben Typs zwingend vorgeschrieben. Wenn die Methanol-Wasser-Mischung ungeeignet ist, können andere Mischungen aus einem organischen Lösungsmittel und Wasser verwendet werden, so z.B. Ethanol-Wasser oder Acetonitril-Wasser. Der pH-Wert des Lösungsmittels ist für ionische Verbindungen kritisch. Er sollte innerhalb des pH-Betriebsbereichs der Säule liegen, der sich im Allgemeinen zwischen 2 und 8 bewegt. Die Anwendung eines Puffers ist ratsam. Dabei muss darauf geachtet werden, dass kein Salz ausfällt und es nicht zur Beschädigung der Säule kommt, was bei einer Reihe von Mischungen von organischer Phase und Puffer möglich ist. HPLC-Messungen mit an Siliziumdioxid gebundener stationärer Phase und einem pH-Wert über 8 sind nicht empfehlenswert, da die Verwendung einer alkalischen mobilen Phase zu einem rapiden Nachlassen der Leistung der Säule führen kann. Referenz-/Prüfsubstanzen Die Referenzsubstanzen sollten den höchstmöglichen Reinheitsgrad haben. Das für Prüf- oder Eichzwecke zu verwendende Substanzgemisch wird, wenn möglich, in der mobilen Phase gelöst. Prüfbedingungen Die Temperatur sollte im Verlauf der Messungen um nicht mehr als ± 2 K schwanken. 1.6.3.2 Messung Berechnung der Totzeit to Die Totzeit lässt sich entweder durch Verwendung einer homologen Reihe (z.B. n-Alkyl-Methyl-Ketone) oder durch nicht chromatografisch verzögerte organische Verbindungen (z.B. Thioharnstoff oder Formamid) bestimmen. Zur Berechnung der Totzeit to mit Hilfe einer homologen Reihe werden mindestens 7 Komponenten einer homologen Reihe eingespritzt und die jeweiligen Retentionszeiten gemessen. Die Retentionszeiten tr(nc + 1) werden in Abhängigkeit von tr(nc) aufgetragen und anschließend der Schnittpunkt a und die Steigung b der Regressionsgleichung: tr(nc + 1) = a + b tr(nc) bestimmt (nc = Anzahl der Kohlenstoffatome). Die Totzeit to ergibt sich dann aus: to = a/(1 - b) Eichkurve Der nächste Schritt besteht in der Aufstellung einer Korrelationskurve log k/log P für geeignete Referenzsubstanzen. In der Praxis werden dazu zwischen 5 und 10 Standard-Referenzsubstanzen, deren log-P-Wert in der Nähe des erwarteten Bereichs liegt, gleichzeitig eingespritzt und die Retentionszeiten am besten mit Hilfe eines mit dem Nachweissystem gekoppelten registrierenden Integrators bestimmt. Die Logarithmen der entsprechenden Kapazitätsfaktoren (log k) werden berechnet und gegen die mittels der Schüttelmethode bestimmten log-P-Werte aufgezeichnet. Die Eichung wird in regelmäßigen Abständen, mindestens einmal täglich, vorgenommen, so dass eventuelle Veränderungen in der Leistung der Säule berücksichtigt werden können. Bestimmung des Kapazitätsfaktors der Prüfsubstanz Die Prüfsubstanz wird in möglichst geringer Menge der mobilen Phase eingespritzt. Die Retentionszeit wird (doppelt) bestimmt zur Berechnung des Kapazitätsfaktors k. Aus der Korrelationskurve der Referenzsubstanzen kann der Verteilungskoeffizient der Prüfsubstanz interpoliert werden. Bei sehr niedrigen und sehr hohen Verteilungskoeffizienten ist eine Extrapolation erforderlich. In diesen Fällen ist besonders auf die Vertrauensgrenzen der Regressionsgeraden zu achten. 2. Daten Schüttelmethode Die Zuverlässigkeit der ermittelten P-Werte kann durch Vergleich der Mittelwerte der Doppelbestimmungen mit dem Gesamtmittelwert geprüft werden. 3. Abschlussbericht Im Prüfbericht ist, wenn möglich, Folgendes anzugeben
Für die Schüttelmethode:
Für die HPLC-Methode:
4. Literatur (1) OECD, Paris, 1981, Test Guideline 107, Decision of the Council C(81) 30 final. (2) C. Hansch und A.J. Leo, Substitution Constants for Correlation Analysis in Chemistry and Biology, John Wiley, New York, 1979. (3) Log P and Parameter Database, A tool for the quantitative prediction of bioactivity (C. Hansch, chairman; A.J. Leo, dir.) - Erhältlich bei Pomona College Medicinal Chemistry Project, 1982, Pomona College, Claremont, California, 91711. (4) L. Renberg, G. Sundström und K. Sundh-Nygärd, Chemosphere, 1980, vol. 80, 683. (5) H. Ellgehausen, C. D'Hondt und R. Fuerer, Pesric. Sei., 1981, vol. 12, 219. (6) B. McDuffie, Chemosphere, 1981, vol. 10, 73. (7) W.E. Hammers et al., J. Chromatog., 1982, vol. 247, 1. (8) J.E. Haky und A.M. Young, J. Liq. Chromat., 1984, vol. 7, 675. (9) S. Fujisawa und E. Masuhara, J. Biomed. Mat. Res., 1981, vol. 15, 787. (10) O. Jubermann, Verteilen und Extrahieren, in: Methoden der Organischen Chemie (Houben Weyl), Allgemeine Laboratoriumspraxis (herausgegeben von E. Müller), Georg Thieme Verlag, Stuttgart, 1958, Band I/l, 223-339. (11) R.F. Rekker und H.M. de Kort, Euro. J. Med. Chem., 1979, vol. 14, 479. (12) A. Leo, C. Hansch und D. Elkins, Partition coefficients and their uses. Chem. Rev., 1971, vol. 71, 525. (13) R.F. Rekker, The Hydrophobie Fragmental Constant, Elsevier, Amsterdam, 1977. (14) NF T 20-043 AFNOR (1985). Chemical products for industrial use - Determination of partition coefficient - Flask shaking method. (15) C.V. Eadsforth und P. Moser, Chemosphere, 1983, vol. 12, 1 459. (16) A. Leo, C. Hansch und D. Elkins, Chem. Rev., 1971, vol. 71, 525. (17) C. Hansch, A. Leo, S.H. Unger, K.H. Kim, D. Nikaitani und E.J. Lien, J. Med. Chem., 1973, vol. 16, 1 207. (18) W.B. Neely, D.R. Branson und G.E. Blau, Environ. Sei. Technol., 1974, vol. 8, 1 113. (19) D.S. Brown und E.W. Flagg, J. Environ. Qual., 1981, vol. 10, 382. (20) J.K. Seydel und K.J. Schaper, Chemische Struktur und biologische Aktivität von Wirkstoffen, Verlag Chemie, Weinheim, New York, 1979. (21) R. Franke, Theoretical Drug Design Methods, Elsevier, Amsterdam, 1984. (22) Y.C. Martin, Quantitative Drug Design, Marcel Dekker, New York, Basel, 1978. (23) N.S. Nirrlees, S.J. Noulton, CT. Murphy und P.J. Taylor, J. Med. Chem., 1976, vol. 19, 615.
Eine allgemeine Einführung in die Berechnungsverfahren, Daten und Beispiele werden im Handbook of Chemical Property Estimation Methods (a) gegeben. Berechnete Pow-Werte können verwendet werden:
Abschätzverfahren Vorläufige Abschätzung des Verteilungskoeffizienten Der Wert des Verteilungskoeffizienten kann durch Verwendung der Löslichkeitswerte der Prüfsubstanz in den reinen Lösungsmitteln abgeschätzt werden: Dafür gilt: PSchätzwert = Sättigung cn-Oktand/Sättigung cWasser Berechnungsverfahren Prinzip der Berechnungsverfahren Sämtliche Berechnungsverfahren beruhen auf der formalen Aufspaltung des Moleküls in geeignete Substrukturen, für die zuverlässige log-Pow-Inkremente bekannt sind. Der log-Pow-Wert des gesamten Moleküls wird danach als Summe seiner entsprechenden Teilwerte plus Summe der Korrekturglieder für intramolekulare Wechselwirkungen berechnet. Aufstellungen über die Konstanten von Substrukturen und den Korrekturgliedern liegen vor (b) (c) (d) (e). Einige davon werden regelmäßig aktualisiert (b). Qualitätskriterien Im Allgemeinen nimmt die Zuverlässigkeit des Berechnungverfahrens in dem Maße ab, in dem die Komplexität der Prüfsubstanz zunimmt. Bei einfachen Substanzen mit niedrigem Molekulargewicht und einer oder zwei funktioneller Gruppen ist mit einer Abweichung von 0,1 bis 0,3 log-Pow-Einheiten von den Ergebnissen der verschiedenen Fragmentmethoden gegenüber dem Messwert zu rechnen. Bei komplexeren Substanzen kann die Fehlerspanne größer sein. Dies hängt von der Zuverlässigkeit und der Verfügbarkeit der Konstanten für die Substrukturen sowie von der Fähigkeit der Erkennung intramolekularer Wechselwirkungen (z.B. Wasserstoffbindungen) und der richtigen Anwendung der Korrekturglieder ab (was mit dem Computer-Programm CLOGP-3 ein geringeres Problem ist) (b). Bei ionischen Substanzen ist die richtige Berücksichtigung der Ladung oder des Ionisierungsgrades wichtig. Berechnungsverfahren Hansch'sche π-Methode Die ursprünglich für hydrophobe Substituenten verwendete Konstante π, eingeführt von Fujita et al. (f), , wird wie folgt definiert: πx = log Pow (PhX) - log Pow (PhH) wobei Pow (PhX) der Verteilungskoeffizient eines aromatischen Abkömmlings und Pow (PhH) derjenige der Ausgangssubstanz ist: (z.B. πCl = log Pow (C6 H5 Cl) - log Pow (C6 H6) = 2,84 - 2,13 = 0,71). Nach seiner Definition ist π-Methode vorwiegend bei der aromatischen Substitution anwendbar. Die-Werte liegen für eine große Anzahl von Substituenten tabelliert vor (b) (c) (d). Sie werden für die Berechnung der log-Pow-Werte für aromatische Moleküle oder Substrukturen verwendet. Rekker-Methode Nach Rekker (g) wird der log-Pow-Wert wie folgt berechnet: (Wechselwirkungsglieder) wobei fi die verschiedenen Konstanten der Substrukturen und ai die Häufigkeit ihres Vorkommens in der Prüfsubstanz darstellen. Die Korrekturglieder lassen sich als ein ganzes Vielfaches einer einzigen Konstante Cm (der so genannten "magischen Konstante") angeben. Die Substrukturkonstanten fi und Cm wurden aus einer Liste von 1.054 experimentell ermittelten Pow-Werten (825 Verbindungen) mit Hilfe der mehrfachen Regressionsanalyse bestimmt (c) (h). Die Bestimmung der Glieder für die Wechselwirkungen erfolgt auf der Grundlage der in der Literatur angegebenen Regeln (e) (h) (i). Hansch-Leo-Methode Nach Hansch und Leo (c) wird der log-Pow-Wert aus der Beziehung errechnet, wobei fi die verschiedenen Konstanten der Substrukturen, Fj die Korrekturglieder und ai, bj die entsprechenden Vorkommenshäufigkeiten sind. Eine Liste der Substrukturwerte für einzelne Atome und Gruppen, abgeleitet aus experimentell bestimmten Pow-Werten, und eine Liste der Korrekturglieder Fj (so genannte "Faktoren") wurden durch die Trialanderror-Methode erhalten. Die Korrekturglieder sind in mehrere unterschiedliche Kategorien eingeordnet worden (a) (c). Es ist relativ kompliziert und zeitraubend, alle Regeln und Korrekturglieder zu berücksichtigen. Software-Pakete sind entwickelt worden (b). Kombinierte Methode Die Berechnung der log-Pow-Werte komplexer Substanzen kann beträchtlich verbessert werden, wenn das Molekül in größere Substrukturen zerlegt wird, für die zuverlässige log-Pow-Werte vorliegen, sei es aus Tabellen (b) (c), sei es aus eigenen Messungen. Solche Substrukturen (z.B. Heterozyklen, Anthrakinon, Azobenzen) können dann mit den Hansch'schen π -Werten oder mit den Substrukturkonstanten nach Rekker oder Leo kombiniert werden. Anmerkungen
Abschlussbericht Bei der Verwendung der Berechnungs-/Abschätzmethoden sollte der Prüfbericht, wenn möglich, Folgendes anzugeben:
Literatur (a) WJ. Lyman, W.F. Reehl und D.H. Rosenblatt (Hrsg.), Handbook of Chemical Property Estimation Methods, McGraw-Hill, New York, 1983. (b) Pomona College, Medicinal Chemistry Project, Claremont, California 91711, USA, Log P Database and Med. Chem. Software (Program CLOGP-3). (c) C. Hansch und A.J. Leo, Substituent Constants for Correlation Analysis in Chemistry and Biology. John Wiley, New York, 1979. (d) A. Leo und C. Hansch, D. Elkins, Chem. Rev. 1971, vol. 71, 525. (e) R.F. Rekker und H.M. de Kort, Eur. J. Med. Chem. - Chim. Ther., 1979, vol. 14, 479. (f) T. Fujita, J. Iwasa und C. Hansch, J. Amer. Chem. Soc, 1964, vol. 86, 5175. (g) R.F. Rekker, The Hydrophopic Fragmental Constant, Pharmacochemistry Library, vol. 1, Elsevier, New York, 1977. (h) C.V. Eadsforth und P. Moser, Chemosphere, 1983, vol. 12, 1459. (i) R.A. Scherrer, ACS, American Chemical Society, Washington D.C., 1984, Symposium Series 255, 225.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

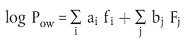
| weiter . |  |
...
X
⍂
↑
↓