
umwelt-online: Verordnung (EU) 2017/735 zur Änderung - zwecks Anpassung an den technischen Fortschritt - des Anhangs der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (3)
 | zurück |  |
B.61 Fluorescein-Leckage-Prüfmethode zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 460 (2012). Bei der Fluorescein-Leckage-Prüfmethode handelt es sich um eine In-vitro-Prüfmethode, die unter bestimmten Umständen und mit bestimmten Einschränkungen zur Einstufung von Chemikalien (Stoffen und Gemischen) als Stoffe mit augenverätzender und stark augenreizender Wirkung gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (Kategorie 1), in der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 (CLP) (Kategorie 1) und durch die US-Umweltschutzbehörde (EPA) (Kategorie I) eingesetzt werden kann (1) (2). Im Sinne dieser Prüfmethode sind Stoffe mit stark augenreizender Wirkung als Chemikalien definiert, die nach Applikation eine Gewebeschädigung im Auge verursachen, die nicht innerhalb von 21 Tagen ausheilt, oder eine massive Verschlechterung des Sehvermögens auslösen, während Stoffe mit augenverätzender Wirkung als Chemikalien definiert sind, die eine irreversible Gewebeschädigung im Auge verursachen. Diese Chemikalien werden in UN-GHS-Kategorie 1, EU-CLP-Kategorie 1 oder US-EPA-Kategorie I eingestuft.
Obwohl die FL-Prüfmethode den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird sie als Teil einer gestuften Prüfstrategie zu gesetzgeberischen Einstufungs- und Kennzeichnungszwecken empfohlen. Somit wird die FL-Prüfmethode als erster Schritt innerhalb eines Top-Down-Ansatzes zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung, insbesondere bei bestimmten Arten von Chemikalien (z.B. wasserlösliche Stoffe und Gemische), empfohlen (3) (4).
Gegenwärtig herrscht allgemeines Einvernehmen darüber, dass der In-vivo-Augentest (TM B.5 (5)) in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Reizungen für verschiedene Chemikalienklassen vorherzusagen. Allerdings kann der In-vivo-Augentest eventuell durch strategische Kombination verschiedener alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie ersetzt werden (4). Der Top-Down-Ansatz (4) sollte zum Einsatz kommen, wenn die vorliegenden Informationen darauf schließen lassen, dass eine Chemikalie ein hohes Reizungspotenzial besitzt.
Basierend auf dem Vorhersagemodell unter Nummer 35 können durch die FL-Prüfmethode Chemikalien innerhalb eines begrenzten Anwendungsbereichs ohne weitere Testung als Stoffe mit augenverätzender oder stark augenreizender Wirkung (UN-GHS Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) identifiziert werden. Gleiches wird für Gemische angenommen, obwohl Gemische bei der Validierung nicht verwendet wurden. Daher kann mithilfe der FL-Prüfmethode die augenverätzende/-reizende Wirkung von Chemikalien entsprechend der sequenziellen Prüfstrategie TM B.5 bestimmt werden (5). Jedoch müsste eine Chemikalie, die nach der FL-Prüfmethode nicht als Stoff mit augenverätzender oder stark augenreizender Wirkung vorhergesagt wurde, mit einer oder mehreren weiteren Prüfmethoden (in vitro und/oder in vivo) geprüft werden, mit denen folgende Chemikalien identifiziert werden können: i) Chemikalien, die nach der FL-Prüfmethode in vitro falsch-negative Stoffe mit augenverätzender/-reizender Wirkung sind (UN-GHS Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I); ii) Chemikalien, die nicht als augenverätzend/-reizend eingestuft sind (UN-GHS "Keine Einstufung", EU-CLP "Keine Einstufung", US-EPA-Kategorie IV); und/oder iii) Chemikalien, die als Stoffe mit leichter/mäßiger augenreizender Wirkung eingestuft sind (UN-GHS-Kategorien 2A und 2B, EU-CLP-Kategorie 2, US-EPA-Kategorien II und III).
Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung der potenziellen augenverätzenden oder -reizenden Wirkung einer Prüfchemikalie, gemessen als ihre Fähigkeit, an einem impermeablen, konfluenten epithelialen Monolayer eine Schädigung hervorzurufen. Die Integrität der transepithelialen Permeabilität ist eine wichtige Funktion eines Epithels, das beispielsweise in der Bindehaut und Hornhaut zu finden ist. Die transepitheliale Permeabilität wird durch verschiedene undurchlässige Verbindungen (Tight Junctions) kontrolliert. Es wurde nachgewiesen, dass eine Zunahme der Permeabilität des Hornhautepithels in vivo mit der Entzündungsaktivität und Oberflächenschädigung, die mit fortschreitender Augenreizung zu beobachten sind, im Zusammenhang steht.
In der FL-Prüfmethode werden die toxischen Wirkungen nach kurzer Exposition gegenüber der Prüfchemikalie anhand einer Zunahme der Permeabilität für Fluorescein-Natrium durch das epitheliale Monolayer von Madin-Darby Canine Kidney (MDCK)-Zellen, die auf permeablen Inserts kultiviert werden, bestimmt. Die auftretende Fluorescein-Leckage ist proportional zu der von der Chemikalie induzierten Schädigung an den Tight Junctions, desmosomalen Verbindungen und Zellmembranen. Anhand der Leckagemenge kann das augentoxische Potenzial einer Prüfchemikalie geschätzt werden. Anlage 1 zeigt ein Diagramm von auf einer Insertmembran kultivierten Zellen für die FL-Prüfmethode.
Definitionen sind Anlage 2 zu entnehmen.
Vorbemerkungen und Einsatzgrenzen
Diese Prüfmethode basiert auf dem INVITTOX-Protokoll Nr. 71 (6), das in einer internationalen Validierungsstudie des Europäischen Zentrums zur Validierung alternativer Methoden (ECVAM) in Zusammenarbeit mit dem US-amerikanischen Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) und dem Japanese center for the Validation of Alternative Methods (JaCVAM) bewertet wurde.
Die FL-Prüfmethode wird weder zur Identifizierung von Chemikalien empfohlen, die als Chemikalien mit leichter/mäßiger Reizwirkung eingestuft werden sollten, noch von Chemikalien, die nicht als augenreizend eingestuft werden sollten (Stoffe und Gemische) (d. h. GHS-Kat. 2A/2B, "keine Einstufung"; EU-CLP-Kat. 2, "keine Einstufung"; US-EPA-Kat. II/III/IV), wie durch die Validierungsstudie nachgewiesen (3) (7).
Die Prüfmethode ist nur an wasserlöslichen Chemikalien (Stoffen und Gemischen) anwendbar. Durch die FL-Prüfmethode wird das stark augenreizende Potenzial von Chemikalien, die wasserlöslich sind und/oder bei denen die toxische Wirkung nicht durch Verdünnung verändert wird, in der Regel exakt vorhergesagt (7). Um eine Chemikalie als wasserlöslich unter Versuchsbedingungen einzustufen, sollte sie in einer sterilen calciumhaltigen (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien Hanks-Salzlösung (Hanks' Balanced Salt Solution, HBSS) mit einer Konzentration von ≥ 250 mg/ml löslich sein (eine Dosis über der Schwelle von 100 mg/ml). Ist die Prüfchemikalie jedoch unter der Konzentration 100 mg/ml löslich, bewirkt aber bereits eine FL-Induktion von 20 % bei dieser Konzentration (was bedeutet: FL20 < 100 mg/ml), kann sie dennoch in GHS-Kat. 1 oder EPA-Kat. I eingestuft werden.
Aufgrund der anerkannten Einsatzgrenzen dieser Prüfmethode sind starke Säuren und Basen, Zellfixierlösungen und stark flüchtige Chemikalien aus dem Anwendungsbereich ausgeschlossen. Diese Chemikalien weisen Mechanismen auf, die von der FL-Prüfmethode nicht gemessen werden, z.B. übermäßige Koagulation, Verseifung oder spezifische chemische Reaktionen. Andere anerkannte Einsatzgrenzen dieser Methode beruhen auf den Ergebnissen der Vorhersagefähigkeit bei gefärbten und viskosen Prüfchemikalien (7). Es ist zu beachten, dass beide Arten von Chemikalien nach kurzer Expositionsdauer schwer aus dem Monolayer zu entfernen sind und dass die Vorhersagefähigkeit der Prüfmethode verbessert werden könnte, wenn eine größere Anzahl von Spülschritten verwendet wird. Feste, in Flüssigkeit suspendierte Chemikalien neigen zu Ausfällung und die Endkonzentration, der die Zellen ausgesetzt sind, kann schwer zu bestimmen sein. Durch Ausschluss von Chemikalien dieser chemischen und physikalischen Klassen aus der Datenbank wird die Genauigkeit der FL-Prüfmethode bei den EU-, EPA- und GHS-Klassifizierungssystemen erheblich verbessert (7).
Basierend auf dem Zweck dieser Prüfmethode (Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung) sind Falsch-Negativ-Raten (siehe Nummer 13) unkritisch, da solche Chemikalien anschließend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequenziellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz (weight-of-evidence approach) (5) im Rahmen anderer angemessen validierter In-vitro-Tests oder an Kaninchen geprüft werden (siehe auch Nummern 3 und 4).
Andere anerkannte Einsatzgrenzen der FL-Prüfmethode beruhen auf Falsch-Negativ- und Falsch-Positiv-Raten. Bei Einsatz als erster Schritt innerhalb eines Top-Down-Ansatzes zur Identifizierung von wasserlöslichen Stoffen und Gemischen mit augenverätzender oder stark augenreizender Wirkung (UN-GHS-Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) lag die Falsch-Positiv-Rate der FL-Prüfmethode im Bereich von 7 % (7/103; UN-GHS und EU-CLP) bis 9 % (9/99; US-EPA) und die Falsch-Negativ-Rate im Bereich von 54 % (15/28; US-EPA) bis 56 % (27/48; UN-GHS und EU-CLP) im Vergleich zu den In-vivo-Ergebnissen. Chemikaliengruppen, die bei der FL-Prüfmethode zu Falsch-Positiv- und/oder Falsch-Negativ-Ergebnissen führen, werden hier nicht definiert.
Einige technische Einsatzgrenzen sind für die MDCK-Zellkultur spezifisch. Die Tight Junctions, die die Passage des Fluorescein-Natrium-Farbstoffs durch den Monolayer hemmen, werden mit steigender Zellpassagenzahl zunehmend beschädigt. Die unvollständige Ausbildung der Tight Junctions führt zu einer verstärkten Fluorescein-Leckage in der unbehandelten Kontrolle. Daher muss eine zulässige maximale Leckage in den unbehandelten Kontrollen festgelegt werden (siehe Nummer 38: 0 % Leckage). Wie bei allen In-vitro-Tests besteht die potenzielle Möglichkeit, dass die Zellen im Laufe der Zeit eine Transformation durchlaufen, sodass die Passagenzahlbereiche für die Tests unbedingt angegeben werden müssen.
Der gegenwärtige Anwendungsbereich könnte in einigen Fällen ausgedehnt werden, aber erst nach Analyse eines erweiterten Datenbestands von untersuchten Prüfchemikalien, der vorzugsweise durch Testung zusammengetragen wurde (3). Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen.
Laboratorien, die diese Prüfmethode erstmals anwenden, sollen die in Anlage 3 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung der FL-Prüfmethode nachzuweisen, bevor sie FL-Testdaten zum Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen.
Testprinzip
Die FL-Prüfmethode ist ein zellbasierter In-vitro-Zytotoxizitätstest an einem konfluenten Monolayer von tubulären MDCK CB997-Epithelzellen, die auf semipermeablen Inserts kultiviert wurden und den nicht proliferierenden Zustand des In-vivo-Hornhautepithels abbilden. Die MDCK-Zelllinie ist gründlich erprobt und bildet Tight Junctions und desmosomale Verbindungen, die mit denjenigen auf der apikalen Seite des Bindehaut- und Hornhautepithels vergleichbar sind. Die Tight Junctions und desmosomalen Verbindungen verhindern in vivo, dass gelöste Stoffe und Fremdstoffe das Hornhautepithel durchdringen. Der Verlust der transepithelialen Impermeabilität aufgrund beschädigter Tight Junctions und desmosomaler Verbindungen ist eines der ersten Anzeichen einer chemikalieninduzierten Augenreizung.
Die Prüfchemikalie wird auf den konfluenten Zellenlayer auf der apikalen Seite des Inserts appliziert. Eine kurze Expositionsdauer von einer Minute wird routinemäßig angewandt, um die normale Ausscheidungsrate bei Exposition am Menschen widerzuspiegeln. Ein Vorteil der kurzen Expositionsdauer ist, dass wasserbasierte Stoffe und Gemische unverdünnt getestet werden können, falls sie nach der Expositionsdauer einfach zu entfernen sind. Dies ermöglicht einen direkteren Vergleich der Ergebnisse mit den Wirkungen der Chemikalie beim Menschen. Die Prüfchemikalie wird dann entfernt und der nicht toxische, stark fluoreszierende Fluorescein-Natrium-Farbstoff wird 30 Minuten lang auf die apikale Seite des Monolayers appliziert. Die von der Prüfchemikalie hervorgerufene Schädigung an den Tight Junctions wird anhand der Fluorescein-Menge bestimmt, die während einer festgelegten Zeitdauer durch die Zellschicht dringt.
Die Menge an Fluorescein-Natrium-Farbstoff, die durch den Monolayer und die Insertmembran in eine festgelegte Lösungsmenge in der Mulde (in die der Fluorescein-Natrium-Farbstoff durchtritt) gelangt, wird durch spektrofluorometrische Messung der Fluorescein-Konzentration in der Mulde bestimmt. Die Menge der Fluorescein-Leckage (FL) wird bezogen auf die gemessene Fluoreszenzintensitität (FI) aus zwei Kontrollen berechnet: einer Blindkontrolle und einer Maximal-Leckage-Kontrolle. Der Leckageprozentsatz und somit das Ausmaß der Schädigung an den Tight Junctions wird bezogen auf diese Kontrollen für jede der festgelegten Konzentrationen der Prüfchemikalie bestimmt. Dann wird der FL20-Wert (d. h. die Konzentration, bei der es zu 20 % FL relativ zu dem erfassten Wert für den unbehandelten konfluenten Monolayer und Inserts ohne Zellen kommt) berechnet. Der FL20-Wert (mg/ml) wird im Vorhersagemodell zur Identifizierung von Stoffen mit augenverätzender und schwer augenreizender Wirkung verwendet (siehe Nummer 35).
Der Erholungsfaktor ist ein wichtiger Teil des Toxizitätsprofils einer Prüfchemikalie, der auch im In-vivo-Augenreizungstest bewertet wird. Vorläufige Analysen haben ergeben, dass Daten zur Erholung (bis zu 72 h nach Exposition gegenüber der Chemikalie) die Vorhersagekapazität des INVITTOX-Protokolls 71 potenziell verbessern könnten, jedoch ist eine weitergehende Bewertung erforderlich, bei der zusätzliche, vorzugsweise aus weiteren Prüfungen ermittelte Daten von Vorteil wären (6). Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen.
Verfahren
Vorbereitung des Zellmonolayers
Das MDCK CB997-Zellmonolayer wird unter Verwendung von subkonfluenten Zellen hergestellt, die in Zellkulturflaschen in DMEM/F12-Nährstoffmischung (1x-Konzentrat mit L-Glutamin, 15 mM HEPES, Calcium (mit einer Konzentration von 1,0-1,8 mM) und 10 % hitzeinaktiviertem FCS/FBS) kultiviert wurden. Wichtig ist, dass alle Medien/Lösungen, die im FL-Test verwendet werden, Calcium mit einer Konzentration zwischen 1,8 mM (200 mg/l) und 1,0 mM (111 mg/l) enthalten, um die Bildung und Integrität der Tight Junctions sicherzustellen. Die Anzahl der Zellpassagen sollte Gegenstand einer Kontrolle sein, damit eine gleichmäßige und reproduzierbare Bildung der Tight Junctions gewährleistet ist. Vorzugsweise sollten Zellen verwendet werden, die nach 3-30 Passagen ab dem Auftauen gewonnen wurden, da Zellen in diesem Bereich eine ähnliche Funktionalität aufweisen, was zur Reproduzierbarkeit der Testergebnisse beiträgt.
Vor Durchführung der FL-Prüfmethode werden die Zellen durch Trypsinisation aus der Flasche entnommen und zentrifugiert. Eine geeignete Menge an Zellen wird in die Inserts von 24-Mulden-Platten ausgesät (siehe Anlage 1). Zum Aussäen der Zellen sollten Inserts (12 mm Durchmesser) mit einer Zellulosemischester-Membran, einer Dicke von 80-150 µm und einer PorengrÆße von 0,45 µm verwendet werden. In der Validierungsstudie wurden Millicell-HA-Inserts (12 mm) verwendet. Die Eigenschaften des Insert- und Membrantyps sind wichtig, da sich diese auf das Zellwachstum und die Bindung der Chemikalie auswirken. Bestimmte Arten von Chemikalien können eine Bindung mit der Millicell-HA-Insertmembran eingehen, was die Interpretation der Ergebnisse beeinflussen könnte. Es sollten Leistungschemikalien (siehe Anlage 3) verwendet werden, um die Gleichwertigkeit bei Verwendung anderer Membrane nachzuweisen.
Die Bindung der Chemikalie an die Insertmembran ist bei kationischen Chemikalien, wie beispielsweise Benzalkoniumchlorid, die von der geladenen Membran angezogen werden, häufiger anzutreffen (7). Durch die Bindung der Chemikalie an die Insertmembran kann sich die Dauer der Exposition gegenüber der Chemikalie verlängern, was zu einer Überschätzung des toxischen Potenzials der Chemikalie führt; andererseits kann sich aber auch die Leckage von Fluorescein durch das Insert physikalisch bedingt verringern, da das Färbemittel an die kationische, an die Insertmembran gebundene Chemikalie gebunden wird, was wiederum zu einer Unterschätzung des toxischen Potenzials der Chemikalie führt. Dies lässt sich überwachen, indem nur die Membran der höchsten Konzentration der geprüften Chemikalie ausgesetzt und dann Fluorescein-Natrium-Farbstoff bei normaler Konzentration während der Standarddauer beigegeben wird (ohne Zellkontrolle). Wenn der Fluorescein-Natrium-Farbstoff gebunden wird, hat die Insertmembran nach dem Abspülen des Prüfmaterials die Farbe Gelb. Daher müssen die Bindungseigenschaften der Prüfchemikalie unbedingt bekannt sein, damit die Wirkung der Chemikalie auf die Zellen ausgewertet werden kann.
Durch die Beimpfung der Inserts mit den Zellen sollte zum Zeitpunkt der Exposition gegenüber der Chemikalie ein konfluenter Monolayer gewonnen worden sein. 1,6 x 105 Zellen sollten pro Insert beigegeben werden (400 µl einer Zellsuspension mit einer Dichte von 4 x 105 Zellen/ml). Unter diesen Umständen wird ein konfluenter Monolayer in der Regel nach 96 Stunden in Kultur gewonnen. Die Inserts sollten vor der Beimpfung sichtgeprüft werden, um sicherzustellen, dass etwaige Schäden, die bei der unter Nummer 30 beschriebenen visuellen Kontrolle erfasst werden, der Handhabung zuzuschreiben sind.
Die MDCK-Zellkulturen sollten in Inkubatoren in einer befeuchteten Atmosphäre bei 5 % ± 1 % CO2 und 37 ± 1 °C kultiviert werden. Die Zellen sollten nicht durch Bakterien, Viren, Mycoplasmen oder Pilze kontaminiert sein.
Applikation der Prüf- und Kontrollchemikalien
Für jede Versuchsdurchführung sollte eine frische Stammlösung der Prüfchemikalie hergestellt und nach der Zubereitung innerhalb von 30 Minuten verwendet werden. Die Prüfchemikalien sollten in einer calciumhaltigem (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien Hanks-Salzlösung (HBBS) vorbereitet werden, um die Bindung von Serumproteinen zu vermeiden. Die Löslichkeit der Chemikalie bei 250 mg/ml in HBSS sollte vor der Prüfung bewertet werden. Wenn die Chemikalie bei dieser Konzentration 30 Minuten lang eine stabile Suspension oder Emulsion bildet (d. h. gleichförmig bleibt und sich nicht absetzt oder in mehrere Phasen trennt), kann HBSS als Lösungsmittel verwendet werden. Stellt sich jedoch heraus, dass die Chemikalie bei dieser Konzentration nicht in HBSS löslich ist, sollte die Verwendung anderer Prüfmethoden anstelle des FL-Tests in Erwägung gezogen werden. Der Einsatz von leichtem Mineralöl als Lösungsmittel in Fällen, in denen die Chemikalie nachweislich nicht in HBSS löslich ist, sollte mit Bedacht geprüft werden, da keine ausreichenden Daten vorliegen, die eine Schlussfolgerung hinsichtlich der Leistungsfähigkeit des FL-Tests unter solchen Bedingungen zulassen.
Alle zu prüfenden Chemikalien werden in einer sterilen, calciumhaltigen (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien HBSS aus der Stammlösung bei fünf festgelegten Konzentrationen, die auf Gewichts-/Volumenbasis verdünnt werden, hergestellt: 1, 25, 100, 250 mg/ml sowie eine unverdünnte oder gesättigte Lösung. Beim Prüfen einer festen Chemikalie sollte eine sehr hohe Konzentration von 750 mg/ml eingeschlossen werden. Bei dieser Konzentration muss für die Applikation auf die Zellen möglicherweise eine Direktverdrängerpipette verwendet werden. Wenn die ermittelte Toxizität zwischen 25 und 100 mg/ml liegt, sollten die folgenden zusätzlichen Konzentrationen zweimal geprüft werden: 1, 25, 50, 75, 100 mg/ml. Der FL20-Wert sollte aus diesen Konzentrationen abgeleitet werden, sofern die Akzeptanzkriterien erfüllt waren.
Die Prüfchemikalien werden nach Entfernung des Zellkulturmediums und zweimaliger Spülung mit steriler, warmer (37 °C), calciumhaltiger (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreier HBSS auf den konfluenten Zellmonolayer appliziert. Zuvor wurden die Filter visuell auf bereits vorhandene Schäden kontrolliert, die fälschlicherweise potenziellen Unverträglichkeiten gegenüber Prüfchemikalien zugeschrieben werden könnten. Es sollten mindestens drei Replikate für jede Konzentration der Prüfchemikalie sowie für die Kontrollen bei der Durchführung verwendet werden. Nach einer Minute Exposition bei Zimmertemperatur sollte die Prüfchemikalie vorsichtig abgesaugt werden, der Monolayer zweimal mit steriler, warmer (37 °C), calciumhaltiger (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreier HBSS abgespült und die Fluorescein-Leckage sofort bestimmt werden.
Bei jeder Durchführung sollten gleichzeitige Negativ- und Positivkontrollen verwendet werden, um nachzuweisen, dass die Monolayer-Integrität (Negativkontrolle) und die Empfindlichkeit der Zellen (Positivkontrolle) innerhalb eines festgelegten historischen Akzeptanzbereichs liegen. Als Positivkontrolle wird Brij 35 (CAS Nr. 9002-92-0) mit einer Konzentration von 100 mg/ml vorgeschlagen. Diese Konzentration sollte eine Fluorescein-Leckage von ungefähr 30 % bewirken (akzeptabler Bereich 20-40 % Fluorescein-Leckage, d. h. Beschädigung der Zellschicht). Die vorgeschlagene Negativkontrolle ist calciumhaltige (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreie HBSS (unbehandelt, Blindkontrolle). Ferner sollte eine Maximal-Leckage-Kontrolle bei jeder Durchführung eingeschlossen sein, um die Berechnung von FL20-Werten zu ermöglichen. Die Maximal-Leckage wird über ein Kontroll-Insert ohne Zellen bestimmt.
Bestimmung der Fluorescein-Permeabilität
Unmittelbar nach Entfernung der Prüf- und Kontrollchemikalien werden 400 µl einer 0,1-mg/ml-Fluorescein-Natrium-Lösung (0,01 % (w/v) in calciumaltiger [mit einer Konzentration von 1,0-1,8 mM], phenolrotfreier HBSS) den Inserts beigegeben (z.B. Millicell-HA). Die Kulturen werden 30 Minuten bei Zimmertemperatur gehalten. Am Ende der Inkubation mit Fluorescein werden die Inserts vorsichtig aus den Mulden entnommen. Jeder Filter wird visuell kontrolliert und etwaige Schäden, die eventuell während der Handhabung aufgetreten sind, werden protokolliert.
Die Menge an Fluorescein, die durch Monolayer und Insert gedrungen ist, wird in der Lösung bestimmt, die nach Entfernen der Inserts in den Mulden verblieben ist. Die Messungen werden in einem Spektrofluorometer bei einer Anregungs- bzw. Emissionswellenlänge von 485 nm bzw. 530 nm durchgeführt. Die Empfindlichkeit des Spektrofluorometers sollte so eingestellt werden, dass die numerische Differenz zwischen der maximalen FL (Insert ohne Zellen) und der minimalen FL (Insert mit konfluentem Monolayer, behandelt mit Negativkontrolle) am größten ist. Aufgrund der Unterschiede bei dem verwendeten Spektrofluorometer wird die Verwendung einer Empfindlichkeit vorgeschlagen, die eine Fluoreszenzintensitität > 4.000 bei der maximalen Fluorescein-Leckage-Kontrolle ergibt. Der maximale FL-Wert sollte nicht größer als 9.999 sein. Die Fluoreszenzintensitität bei maximaler Leckage sollte im linearen Bereich des verwendeten Spektrofluorometers liegen.
Interpretation der Ergebnisse und Vorhersagemodell
Die FL-Menge ist proportional zu der durch die Chemikalie hervorgerufenen Schädigung an den Tight Junctions. Der FL-Prozentsatz für jede geprüfte Konzentration der Chemikalie wird aus den FL-Werten berechnet, die für die Prüfchemikalie mit Bezug auf die FL-Werte aus der Negativkontrolle (Messwert aus dem konfluenten Monolayer der mit der Negativkontrolle behandelten Zellen) und einer maximalen Leckage-Kontrolle (Messwert für die FL durch ein Insert ohne Zellen) ermittelt wurden.
Mittlere Fluoreszenzintensität bei maximaler Leckage = x
Mittlere Fluoreszenzintensität bei 0 % Leckage (Negativkontrolle) = y
Die mittlere 100 % Leckage wird durch Subtraktion der mittleren 0 % Leckage von der mittleren maximalen Leckage ermittelt,
d. h. x - y = z
Die prozentuale Leckage für jede festgelegte Dosis wird durch Subtraktion der 0 % Leckage von der mittleren Fluoreszenzintensität der drei Replikatmesswerte (m) und Division dieses Werts durch die 100 % Leckage bestimmt, d. h. % FL = [(m-y)/z] x 100 %. Dabei sind:
m = mittlere Fluoreszenzintensität der drei Replikatmessungen für die verwendete Konzentration
% FL = Prozentsatz des Fluorescein, das durch die Zellschicht dringt
Für die Berechnung der Chemikalienkonzentration, die 20 % FL bewirkt, sollte folgende Gleichung herangezogen werden:
FLD = [(A-B)/(C-B)] x (MC - MB) + MB
Dabei sind:
D = prozentuale Hemmung
A = prozentuale Schädigung (20 % Fluorescein-Leckage)
B = prozentuale Fluorescein-Leckage < A
C = prozentuale Fluorescein-Leckage > A
MC = Konzentration (mg/ml) von C
MB = Konzentration (mg/ml) von B
Der FL20-Grenzwert für die Vorhersage von Chemikalien als Stoffe mit augenverätzender/schwer augenreizender Wirkung ist nachfolgend angegeben:
| FL20 (mg/ml) | UN-GHS E&K | EU-CLP E&K | US-EPA E&K |
| ≤ 100 | Kategorie 1 | Kategorie 1 | Kategorie I |
| E&K: Einstufung und Kennzeichnung | |||
Die FL-Prüfmethode wird nur für die Identifizierung von wasserlöslichen Stoffen mit augenverätzender oder schwer augenreizender Wirkung empfohlen (UN-GHS-Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) (siehe Nummern 1 und 10).
Zur Identifizierung von wasserlöslichen Chemikalien (Stoffen und Gemischen) (3) (6) (7) als Stoff mit schwer augenschädigender Wirkung (UN-GHS/EU-CLP-Kategorie 1) oder als Stoff mit augenverätzender oder stark augenreizender Wirkung (US-EPA-Kategorie I) sollte die Prüfchemikalie einen FL20-Wert von ≤ 100 mg/ml induzieren.
Akzeptanz der Ergebnisse
Der mittlere maximale Fluorescein-Leckage-Wert (x) sollte größer 4.000 (siehe Nummer 31) sein, der mittlere 0 % Leckage-Wert (y) kleiner-gleich 300 sein und der mittlere 100 %-Leckage-Wert (z) zwischen 3.700 und 6.000 liegen.
Eine Prüfung gilt als akzeptabel, wenn die Positivkontrolle 20 % bis 40 % Schädigung an der Zellschicht verursacht hat (gemessen als prozentuale Fluorescein-Leckage).
Daten und Berichterstattung
Daten
Je Versuchsdurchführung sollten die Daten von einzelnen Replikat-Mulden (z.B. Fluoreszenzintensitätswerte und berechnete prozentuale FL-Werte für jede Prüfchemikalie, einschließlich Einstufung) in tabellarischer Form angegeben werden. Darüber hinaus sollten die Mittelwerte ± Standardabweichung von einzelnen Replikatmessungen angegeben werden.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalien und Kontrollchemikalien
- chemische Bezeichnung(en) wie der vom Chemical Abstracts Service (CAS) benutzte strukturelle Name, gefolgt von anderen Bezeichnungen, soweit bekannt;
- CAS-Nummer der Chemikalie, soweit bekannt;
- Reinheit und Zusammensetzung des Stoffs oder Gemischs (in Gewichtsprozent), soweit diesbezügliche Informationen vorliegen;
- physikalisch-chemische Eigenschaften, soweit sie für die Studie relevant sind (z.B. physikalischer Zustand, Flüchtigkeit, pH-Wert, Stabilität, Wasserlöslichkeit, Chemikalienklasse);
- Behandlung der Prüf-/Kontrollchemikalien vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- Lagerbedingungen:
Rechtfertigung der Prüfmethode und des verwendeten Protokolls
- Dies sollte Überlegungen in Bezug auf Anwendungsbereich und Einsatzgrenzen der Prüfmethode umfassen.
Prüfbedingungen
- Beschreibung des verwendeten Zellsystems, einschließlich Echtheitszeugnis und Mycoplasma-Status der Zelllinie;
- Details des angewandten Prüfverfahrens;
- verwendete Konzentration(en) der Prüfchemikalie;
- Dauer der Exposition gegenüber der Prüfchemikalie;
- Dauer der Inkubation mit Fluorescein;
- Beschreibung etwaiger Änderungen am Testverfahren;
- Beschreibung der angewandten Bewertungskriterien;
- Verweis auf historische Daten des Modells (z.B. Negativ- und Positivkontrollen, Referenzchemikalien, soweit zutreffend);
- Informationen über die nachgewiesene technische Kompetenz des Labors.
Ergebnisse
- tabellarische Darstellung der Daten von einzelnen Prüfchemikalien und Kontrollen für jede Versuchsdurchführung und Replikatmessung (einschließlich individueller Ergebnisse, Mittelwerte und Standardabweichungen);
- abgeleitete Einstufung(en) mit Bezug auf das Vorhersagemodell und/oder angewandte Entscheidungskriterien;
- Beschreibung anderer beobachteter Wirkungen;
Erörterung der Ergebnisse
- Dies sollte Überlegungen bezüglich eines nicht aussagekräftigen Ergebnisses (Nummer 35: FL20 > 100 mg/ml) und weitere Prüfungen umfassen.
Schlussfolgerungen
Literaturhinweise
(1) UN (2009), Globally Harmonized System of Classification and Labelling of Chemicals (GHS), dritte überarbeitete Auflage, New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Verfügbar unter: [http://www.unece.org/trans/danger/publi/ghs/ghs_rev03/03files_e.html]
(2) U.S. EPA (1996), Label Review Manual: 2nd Edition, EPA737-B-96-001, Washington, DC: U.S., Environmental Protection Agency.
(3) EC-ECVAM (2009), Statement on the scientific validity of cytotoxicity/cell-function based in vitro assays for eye irritation testing.
(4) Scott, L. et al. (2010), A proposed eye irritation testing strategy to reduce and replace in vivo studies using Bottom-Up and Top-Down approaches, Toxicol. In Vitro 24, 1-9
(5) Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung.
(6) EC-ECVAM (1999), INVITOX Protocol 71: Fluorescein Leakage Test, Ispra, Italien: European Centre for the Validation of Alternative Methods (ECVAM). Verfügbar unter: [http://ecvam-dbalm.jrc.ec.europa.eu
(7) EC-ECVAM (2008), Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing.
(8) OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, OECD Series on Testing and Assessment No. 34. OECD, Paris.
_____
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
.
| Diagramm von MDCK-Zellen, die auf einer Insertmembran für die FL-Prüfmethode kultiviert wurden | Anlage 1 |
Eine konfluente Schicht von MDCK-Zellen wird auf der semipermeablen Membran eines Inserts kultiviert. Die Inserts werden in die Mulden von 24-Mulden-Platten eingesetzt.
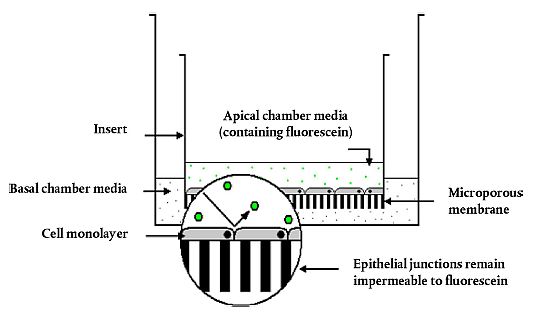
Abbildung entnommen aus: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Thesis, University of Nottingham, UK.
.
| Definitionen | Anlage 2 |
Definitionen
Chemikalie: Stoff oder Gemisch.
Empfindlichkeit: Anteil aller positiven/aktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (8).
EPA-Kategorie I: Chemikalien mit verätzender Wirkung (irreversible Schädigung des Augengewebes) oder Hornhautkomplikation oder -reizwirkung, die länger als 21 Tage anhält (2).
Ersatzprüfung: Eine Prüfung, die eine routinemäßig angewandte Prüfung zur Identifikation von Gefahren und/oder zur Risikobewertung ersetzen soll und die im Vergleich zur akzeptierten Prüfung nachweislich in allen denkbaren Prüfsituationen und mit allen Chemikalien einen gleichwertigen oder besseren Schutz der Gesundheit von Mensch oder Tier oder der Umwelt gewährleistet, je nachdem, was zutrifft.
EU CLP (Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen): Umsetzung des UN-GHS-Systems zur Einstufung von Chemikalien (Stoffen und Gemischen) in der Europäischen Union (EU).
Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Chemikalie entscheiden zu können und diese Entscheidung zu untermauern.
Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
FL20: Kann durch Bestimmung der Konzentration geschätzt werden, bei der die geprüfte Chemikalie 20 % Fluorescein-Leckage durch die Zellschicht bewirkt.
Fluorescein-Leckage: Fluorescein-Menge, die durch die Zellschicht dringt, spektrofluorometrisch gemessen.
Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch: Wird im UN-GHS verwendet und bezeichnet ein Gemisch oder eine Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren.
Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode.
Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann.
GHS (Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN)): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten.
GHS-Kategorie 1: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind.
Leistungschemikalien: Teilmenge der Referenzchemikalien, die von einem Labor verwendet werden können, um seine Kompetenz hinsichtlich der validierten Referenzprüfmethode nachzuweisen.
Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert.
Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert.
Nicht eingestuft: Chemikalien, die nicht als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorien 1, 2A oder 2B, der EU-CLP-Kategorien 1 oder 2 oder der US-EPA-Kategorien I, II oder III eingestuft sind.
Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Die Relevanz beinhaltet die Prüfung der Genauigkeit (Übereinstimmung) einer Prüfmethode (8).
Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind.
Spezifität: Anteil aller negativen/inaktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode.
Stoff: Wird im UN-GHS verwendet und bezeichnet chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können.
Stoff mit augenreizender Wirkung: a) Chemikalie, die nach Applikation auf die Vorderfläche des Auges eine reversible Augenschädigung verursacht; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorien 2A oder 2B, der EU-CLP-Kategorie 2 oder der US-EPA-Kategorien II oder III eingestuft sind.
Stoff mit augenverätzender Wirkung: a) Chemikalie, die das Augengewebe irreversibel schädigt; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorie 1, der EU-CLP-Kategorie 1 oder der US-EPA-Kategorie I eingestuft sind.
Stoff mit stark augenreizender Wirkung: a) Chemikalie, die nach Applikation auf die Vorderfläche des Auges das Augengewebe derart schädigt, dass die Schädigung nicht innerhalb von 21 Tagen nach der Applikation des Stoffs zurückgeht oder dass das Sehvermögen schwer beeinträchtigt ist; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorie 1, der EU-CLP-Kategorie 1 oder der US-EPA-Kategorie I eingestuft sind.
Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden (8).
Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
.
| Leistungschemikalien für die FL-Prüfmethode | Anlage 3 |
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Laboratorien ihre technische Kompetenz nachweisen, indem sie die in Tabelle 1 empfohlenen acht Chemikalien in die richtige Augenschädigungsklasse einstufen. Diese Chemikalien wurden so ausgewählt, dass sie die Bandbreite der lokalen Augenreizungen/-verätzungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405, TM B.5 (5)) (d. h. den Kategorien 1, 2A, 2B oder "Keine Einstufung" gemäß UN-GHS) basieren. Angesicht der validierten Zweckmäßigkeit des FL-Tests (d. h. zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung) lässt sich die Leistungsfähigkeit lediglich anhand von zwei Testergebnissen zu Einstufungszwecken (verätzend/stark reizend oder nicht verätzend/nicht stark reizend) nachweisen. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger Daten aus der FL-Prüfmethode. Aus diesem Grund wurden die Leistungschemikalien aus der Publikation Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing (8) ausgewählt, die zur retrospektiven Validierung der FL-Prüfmethode herangezogen wurde.
Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des FL-Tests
| Chemikalie | CAS-Nr. | Chemikalienklasse 1 | Physikalischer Zustand | In-vivo- Klassifizierung 2 | In-vitro- Klassifizierung 3 |
| Benzalkoniumchlorid (5 %) | 8001-54-5 | Oniumverbindung | flüssig | Kategorie 1 | Verätzend/stark reizend |
| Promethazin-Hydrochlorid | 58-33-3 | Amin/Amidin, heterocyclisch, organische Schwefelverbindung | fest | Kategorie 1 | Verätzend/stark reizend |
| Natriumhydroxid (10 %) | 1310-73-2 | Alkali | flüssig | Kategorie 1 | Verätzend/stark reizend |
| Natrium-laurylsulfat (15 %) | 151-21-3 | Carbonsäure (Salz) | flüssig | Kategorie 1 | Verätzend/stark reizend |
| 4-Carboxy-Benzaldehyd | 619-66-9 | Carbonsäure, Aldehyd | fest | Kategorie 2(A) | Nicht verätzend/nicht stark reizend |
| Ammoniumnitrat | 6484-52-2 | Anorganisches Salz | fest | Kategorie 2(A) | Nicht verätzend/nicht stark reizend |
| Ethyl-2-methylaceto-acetat | 609-14-3 | Ketone, Ester | flüssig | Kategorie 2(B) | Nicht verätzend/nicht stark reizend |
| Glyzerin | 56-81-5 | Alkohol | flüssig | Keine Einstufung | Nicht verätzend/nicht stark reizend |
| Abkürzungen:
CAS-Nr. = Registernummer des Chemical Abstracts Service
1) Jede Prüfchemikalie wurde anhand einer Standard-Klassifizierungsregelung auf Basis des Klassifizierungssystems der National Library of Medicine Medical Subject Headings (MeSH) einer Chemikalienklasse zugeordnet (abrufbar über http//www.nlm.nih.gov/mesh). 2) Gestützt auf Ergebnisse aus dem In-vivo-Kaninchenaugentest (OECD TG 405, TM B.5) unter Verwendung von UN-GHS und EU-CLP. 3) Gestützt auf Ergebnisse aus dem FL-Test (INVITTOX-Protokoll Nr. 71 (6)) | |||||
B.62 Alkalischer In-vivo-Comet-Assay an Säugetierzellen
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 489 (2016). Der alkalische In-vivo-Comet-Assay (Einzelzell-Gelelektrophorese (Single Cell Gel Electrophoresis, SCGE)) (im Folgenden "Comet-Assay") wird für die Erkennung von DNA-Strangbrüchen in Zellen oder Zellkernen eingesetzt, die aus Geweben von Tieren, gewöhnlich Nagern, isoliert wurden, die potenziell toxischen Stoffen ausgesetzt wurden. Der Comet-Assay wurde überprüft, und verschiedene Expertengruppen haben Empfehlungen abgegeben (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (11).
Der Comet-Assay dient zum Nachweis von Chemikalien, die DNA-Schäden auslösen. Durch den Comet-Assay können unter alkalischen Bedingungen (> pH 13) Einzel- und Doppelstrangbrüche nachgewiesen werden, die beispielsweise auf direkte Interaktionen mit der DNA, alkali-labile Stellen oder transiente DNA-Strangbrüche aufgrund einer DNA-Exzisionsreparatur zurückzuführen sind. Diese Strangbrüche können repariert werden, ohne dass eine anhaltende Wirkung eintritt, können tödlich für die Zelle sein oder können zu einer Mutation führen, die eine dauerhafte lebensfähige Veränderung bewirkt. Zudem können sie Chromosomenschäden hervorrufen, die mit vielen Erkrankungen des Menschen, darunter auch Krebs, im Zusammenhang stehen.
Eine formale Validierungsstudie des In-vivo-Comet-Assays an Nagetieren wurde 2006-2012 unter Koordinierung des Japanischen Zentrums zur Validierung alternativer Methoden (JaCVAM) in Zusammenarbeit mit dem Europäischen Zentrum zur Validierung alternativer Methoden (ECVAM) und den US-amerikanischen Validierungszentren Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) und NTP Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM) durchgeführt (12). Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des Comet-Assays und basiert auf dem bei der Validierungsstudie angewandten Schlussprotokoll (12) sowie auf weiteren einschlägigen veröffentlichten und unveröffentlichten (laboreigenen) Daten.
Die Definitionen der Schlüsselbegriffe sind Anlage 1 zu entnehmen. Es ist zu beachten, dass für diesen Assay viele verschiedene Plattformen eingesetzt werden können (Objektträger, Gelproben, 96-Mulden-Platten usw.). Zur Vereinfachung wird im Folgenden der Begriff "Objektträger" verwendet, wobei aber alle anderen Plattformen darin eingeschlossen sind.
Vorbemerkungen und Einsatzgrenzen
Der Comet-Assay ist eine Methode zur Bestimmung von DNA-Strangbrüchen in eukaryotischen Zellen. In Agarose eingebettete Einzelzellen/Zellkerne auf einem Objektträger werden mit Detergenzien und hoher Salzkonzentration lysiert. Durch diesen Lyseschritt werden die Zell- und Zellkernmembrane zerstört, sodass die spiralisierten DNA-Schleifen (allgemein als Nukleoide bezeichnet) und die DNA-Fragmente freigelegt werden können. Die Elektrophorese bei hohem pH-Wert führt zu kometartigen Strukturen, die bei Verwendung geeigneter Farbstoffe unter dem Fluoreszenzmikroskop beobachtet werden können; die DNA-Fragmente wandern je nach Größe vom "Kopf" in den "Schweif" und die Intensität des Kometenschweifs relativ zur Gesamtintensität (Kopf plus Schweif) entspricht dem Grad des DNA-Bruchs (13) (14) (15).
Der alkalische In-vivo-Comet-Assay ist insbesondere für die Bewertung der genotoxischen Wirkung relevant, da die Reaktionen im Rahmen des Assays von der In-vivo-ADME (Absorption, distribution, metabolism and excretion, Resorption, Verteilung, Metabolisierung und Ausscheidung) sowie von DNA-Reparaturprozessen abhängig sind. Diese unterscheiden sich je nach Tierart, Gewebetyp und Art der DNA-Schädigung.
Zur Einhaltung der Tierschutzanforderungen, insbesondere zur Verringerung der Verwendung von Versuchstieren (3R-Prinzip: Replace, Reduce, Refine - Vermeiden, Verringern, Verbessern) kann dieser Test auch mit anderen toxikologischen Studien, z.B. Toxizitätsstudien mit wiederholter Verabreichung (10) (16) (17), integriert werden oder der Endpunkt kann mit anderen genotoxischen Endpunkten, wie beispielsweise dem In-vivo-Erythrozyten-Mikrokerntest bei Säugern, kombiniert werden (18) (19) (20). Der Comet-Assay wird überwiegend an Nagetieren durchgeführt, wurde aber auch bei anderen Säugetier- und Nichtsäugetierarten angewandt. Die Verwendung von Nichtnagetierarten sollte im Einzelfall wissenschaftlich und ethisch begründet werden. Es wird dringend empfohlen, den Comet-Assay an anderen Arten als Nagetieren nur im Rahmen einer anderen Toxizitätsstudie und nicht als eigenständigen Test durchzuführen.
Expositionspfad und zu untersuchende Gewebe sollten basierend auf allen verfügbaren/vorliegenden Informationen über die Prüfchemikalien, z.B. beabsichtigter/erwarteter Expositionspfad beim Menschen, Metabolisierung und Verteilung, Potenzial für Wirkungen an Kontaktstellen, strukturelle Warnungen, andere Daten zur Genotoxizität oder Toxizität und Zweck der Studie, gewählt werden. Das genotoxische Potenzial der Prüfchemikalien kann daher gegebenenfalls im Zielgewebe bzw. in den Zielgeweben karzinogener und/oder anderer toxischer Wirkungen untersucht werden. Der Test gilt ferner als hilfreich für weitere Untersuchungen zur Genotoxizität, die in einem In-vitro-System nachgewiesen wurde. Es ist zweckmäßig, einen In-vivo-Comet-Assay an einem bestimmten Gewebe durchzuführen, wenn begründeterweise von einer entsprechenden Exposition des Gewebes ausgegangen werden kann.
Der Test wurde am ausführlichsten im Rahmen von Kooperationsstudien wie beispielsweise der JaCVAM-Studie (12) und in Rothfuss et al., 2010 (10) am somatischen Gewebe männlicher Ratten validiert. In der internationalen Validierungsstudie von JaCVAM wurden Leber und Magen verwendet. Die Leber, weil sie das aktivste Organ in der Metabolisierung von Chemikalien und häufig ein Zielorgan für Karzinogenität ist. Der Magen, weil er gewöhnlich die erste Kontaktstelle für Chemikalien nach oraler Aufnahme ist, obwohl andere Bereiche des Magen-Darm-Trakts wie Zwölffingerdarm und Dünndarm ebenfalls als Kontaktstellengewebe untersucht werden sollten und beim Menschen unter Umständen relevanter sind als der Drüsenmagen von Nagetieren. Es muss unbedingt darauf geachtet werden, dass solches Gewebe nicht übermäßig hohen Prüfchemikalienkonzentrationen ausgesetzt wird (21). Das Verfahren ist grundsätzlich bei allen Geweben anwendbar, von denen analysierbare Einzelzell-/Zellkernsuspensionen gewonnen werden können. Proprietäre Daten verschiedener Labors belegen die erfolgreiche Anwendung bei verschiedenen Geweben, und es liegen zahlreiche Publikationen vor, in denen die Anwendbarkeit des Verfahrens bei anderen Organen oder Geweben als Leber und Magen, z.B. Dünndarm (22), Niere (23) (24), Haut (25) (26) oder Harnblase (27) (28), Lunge und bronchoalveoläre Lavage-Zellen (relevant bei Untersuchungen zu eingeatmeten Chemikalien) (29) (30), aufgezeigt wurde. Ferner wurden Tests an mehreren Organen gleichzeitig durchgeführt (31) (32).
Obwohl die genotoxischen Wirkungen in Keimzellen durchaus von Interesse sein mögen, ist zu beachten, dass der standardmäßige alkalische Comet-Assay, der in der Prüfmethode beschrieben wird, für die Messung von DNA-Strangbrüchen in reifen Keimzellen als ungeeignet angesehen wird. Da im Rahmen einer Auswertung der Fachliteratur zum Einsatz des Comet-Assays bei der Untersuchung der Genotoxizität in Keimzellen von hohen und variablen Background-Werten der DNA-Schädigung berichtet wurde (33), werden Änderungen des Protokolls sowie verbesserte Standardisierungs- und Validierungsstudien für notwendig erachtet, bevor der Comet-Assay an reifen Keimzellen (z.B. Spermien) in die Prüfmethode aufgenommen werden kann. Darüber hinaus ist das Expositionsschema, das in dieser Prüfmethode beschrieben wird, nicht optimal. Für eine aussagekräftige Analyse der DNA-Strangbrüche in reifen Spermien wären längere Expositions- oder Probenahmezeiten erforderlich. Genotoxische Wirkungen, die durch den Comet-Assay in Hodenzellen in verschiedenen Differenzierungsstadien gemessen wurden, wurden in der Fachliteratur beschrieben (34) (35). Allerdings sollte beachtet werden, dass Gonaden eine Mischung aus somatischen und Keimzellen enthalten. Aus diesem Grund spiegeln positive Ergebnisse an einer Gonade (Hoden) nicht unbedingt eine Keimzellenschädigung wider, deuten aber darauf hin, dass die geprüfte(n) Chemikalie(n) und/oder ihre Metaboliten die Gonade erreicht haben.
Vernetzungen können mit den Standard-Versuchsbedingungen des Comet-Assays nicht zuverlässig identifiziert werden. Unter bestimmten geänderten Versuchsbedingungen könnten DNA-DNA- und DNA-Protein-Vernetzungen sowie sonstige Basenveränderungen wie beispielsweise oxidierte Basen erkannt werden (23) (36) (37) (38) (39). Jedoch wären weitere Arbeiten notwendig, um die notwendigen Protokolländerungen angemessen zu charakterisieren. Daher ist der Nachweis von Vernetzungsmitteln nicht der primäre Zweck des hierin beschriebenen Tests. Der Test eignet sich, auch mit Änderungen, nicht zum Nachweis von Aneugenen.
Nach aktuellem Kenntnisstand hat der In-vivo-Comet-Assay verschiedene zusätzliche Einsatzgrenzen (siehe Anlage 3). Es wird davon ausgegangen, dass die Prüfmethode in Zukunft überprüft und gegebenenfalls angesichts der gewonnenen Erkenntnisse überarbeitet werden wird.
Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Diese Überlegungen erübrigen sich, wenn die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist.
Prinzip der Prüfmethode
Die Prüfchemikalie wird den Tieren über einen geeigneten Applikationsweg verabreicht. Dosierung und Probenahme sind unter den Nummern 36-40 ausführlich beschrieben. Zu den ausgewählten Probenahmezeitpunkten werden die untersuchten Gewebe seziert und Einzelzell-/Zellkernsuspensionen hergestellt (In-situ-Perfusion kann vorgenommen werden, sofern sie als sinnvoll angesehen wird, z.B. Leber) und in Softagar eingebettet, um sie auf Objektträgern zu immobilisieren. Die Zellen/Zellkerne werden mit Lysepuffer behandelt, um die Zell- und/oder Zellkernmembran zu entfernen, und einer starken Lauge, z.B. pH ≥ 13, ausgesetzt, um das "Entwinden" der DNA und die Freisetzung der relaxierten DNA-Schleifen und -Fragmente zu ermöglichen. Die nukleare DNA im Agar wird dann der Elektrophorese unterzogen. Normale nichtfragmentierte DNA-Moleküle bleiben an der Stelle, an der sich die nukleare DNA im Agar befand, während fragmentierte DNA und relaxierte DNA-Schleifen zur Anode wandern. Nach der Elektrophorese wird die DNA durch einen geeigneten fluoreszierenden Farbstoff sichtbar gemacht. Die Präparate sollten mikroskopisch und mittels voll- oder halbautomatischer Bildanalysesysteme untersucht werden. Der Umfang der DNA-Wanderung bei der Elektrophorese und der Migrationsabstand entsprechen der Menge und Größe der DNA-Fragmente. Beim Comet-Assay gibt es verschiedene Endpunkte. Es wurde empfohlen, den DNA-Inhalt im Schweif (% DNA im Schweif oder % Schweif-Intensität) zur Bewertung der DNA-Schädigung heranzuziehen (12) (40) (41) (42). Nach der Analyse einer ausreichenden Anzahl von Zellkernen werden die Testergebnisse mit geeigneten Analysemethoden analysiert.
Es ist zu beachten, dass Änderungen verschiedener Aspekte der Methodik, einschließlich Vorbereitung der Proben, Elektrophoresebedingungen, visuelle Analyseparameter (z.B. Farbstoffintensität, Lichtintensität der Mikroskoplampe und Einsatz von Mikroskopfiltern und Kameradynamik), und der Umgebungsbedingungen (z.B. Hintergrundbeleuchtung), untersucht wurden und sich auf die DNA-Wanderung auswirken können (43) (44) (45) (46).
Überprüfung der Eignung des Labors
Jedes Labor sollte seine experimentelle Kompetenz in Bezug auf den Comet-Assay belegen, indem es seine Fähigkeit zur Herstellung von Einzelzell- oder Zellkernsuspensionen von ausreichender Qualität für jedes Zielgewebe und jede verwendete Tierart nachweist. Die Qualität der Zubereitungen wird in erster Linie anhand des prozentualen DNA-Anteils im Schweif bei mit Vehikel behandelten Tieren bewertet, deren Werte in einen reproduzierbaren niedrigen Bereich fallen. Die aktuellen Daten deuten darauf hin, dass der prozentuale DNA-Anteil im Schweif im Gruppenmittel (basierend auf dem Mittelwert der Mediane - Erläuterungen zu diesen Begriffen unter Nummer 57) in der Rattenleber vorzugsweise 6 % nicht überschreiten sollte, was mit den Werten aus der JaCVAM-Validierungsstudie (12) sowie aus anderen veröffentlichten und proprietären Daten im Einklang stehen würde. Zum gegenwärtigen Zeitpunkt liegen nicht genügend Daten vor, um Empfehlungen für optimale oder akzeptable Bereiche bei anderen Geweben abzugeben. Dies schließt die Verwendung anderer Gewebe, sofern gerechtfertigt, nicht aus. Der Prüfbericht sollte eine Beurteilung der Leistung des Comet-Assays bei diesen Geweben unter Bezugnahme auf die veröffentliche Literatur oder proprietäre Daten beinhalten. Erstens ist ein niedriger prozentualer DNA-Anteil im Schweif in den Kontrollen wünschenswert, um eine hinreichend dynamische Bandbreite für den Nachweis einer positiven Wirkung zu gewährleisten. Zweitens sollte jedes Labor die erwarteten Reaktionen für direkte Mutagene und Promutagene für verschiedene Wirkungsweisen gemäß den Beispielen in Tabelle 1 (Nummer 29) reproduzieren können.
Beispielsweise können positive Stoffe aus der JaCVAM-Validierungsstudie (12) oder anderen veröffentlichten Daten (siehe Nummer 9), gegebenenfalls mit Begründung, ausgewählt werden, wobei die eindeutigen positiven Wirkungen in den untersuchten Geweben nachgewiesen werden. Zudem sollte die Fähigkeit zum Nachweis von schwachen Wirkungen bekannter Mutagene, wie z.B. niedrig dosiertem EMS, nachgewiesen werden, indem beispielsweise Dosis-Wirkungs-Beziehungen an einer hinreichenden Anzahl von Dosierungen mit entsprechendem Abstand aufgestellt werden. Anfänglich sollte sich das Labor auf den Nachweis der Kompetenz bei den am häufigsten verwendeten Geweben (z.B. Rattenleber) konzentrieren, wobei ein Vergleich zwischen vorliegenden Daten und erwarteten Ergebnissen angestellt werden könnte (12). Die Daten von anderen Geweben wie z.B. Magen, Zwölffingerdarm, Dünndarm, Blut usw. könnten gleichzeitig erfasst werden. Das Labor muss seine Kompetenz für sämtliche Gewebe aller Tierarten, die untersucht werden sollen, nachweisen und aufzeigen, dass eine akzeptable positive Reaktion bei einem bekannten Mutagen (z.B. EMS) im jeweiligen Gewebe erreicht werden kann.
Vehikel-/Negativkontrolldaten sollten erfasst werden, um die Reproduzierbarkeit negativer Reaktionen nachzuweisen und sicherzustellen, dass die technischen Aspekte des Assays vorschriftsmäßig kontrolliert wurden, oder darauf hinzuweisen, dass historische Kontrollbereiche erneut festgelegt werden sollten (siehe Nummer 22).
Es ist zu beachten, dass das Labor, da mehrere Gewebe bei der Sektion gewonnen und für die Comet-Analyse aufgearbeitet werden können, in der Lage sein muss, mehrere Gewebe von einem einzelnen Tier zu gewinnen und somit sicherzustellen, dass keine potenzielle DNA-Läsion verloren geht und die Comet-Analyse nicht beeinträchtigt wird. Die Zeitdauer zwischen der Tötung des Tiers und der Gewinnung der Gewebe zur Aufbereitung kann kritisch sein (siehe Nummer 44).
Da bei der Entwicklung der Kompetenz für diesen Test Tierschutzbelange berücksichtigt werden müssen, können Gewebe von in anderen Tests verwendeten Tieren zur Entwicklung der Kompetenz bei den verschiedenen Aspekten des Tests eingesetzt werden. Darüber hinaus muss während der verschiedenen Phasen der Etablierung einer neuen Prüfmethode in einem Labor nicht unbedingt eine vollständige Studie durchgeführt werden. Ferner können weniger Tiere oder Prüfkonzentrationen bei der Entwicklung der notwendigen Fähigkeiten verwendet werden.
Historische Kontrolldaten
Im Verlauf der Untersuchungen zum Nachweis der Kompetenz sollte das Labor eine Datenbank mit historischen Daten aufbauen, um Positiv- und Negativkontrollbereiche und -verteilungen für die betreffenden Gewebe und Tierarten zu erfassen. Empfehlungen zur Erstellung und Verwendung historischer Daten (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus der Datenbank und die Akzeptanzkriterien für einen bestimmten Versuch) sind der Literatur zu entnehmen (47). Verschiedene Gewebe und Tierarten sowie verschiedene Vehikel und Verabreichungswege können unterschiedliche prozentuale Schweif-DNA-Werte in den Negativkontrollen ergeben. Daher müssen unbedingt Negativkontrollbereiche für jedes Gewebe und jede Tierart erfasst werden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder Xbar-Karten (48)), um zu ermitteln, wie variabel ihre Daten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist. Die Auswahl der für Positivkontrollen geeigneten Stoffe, der Dosisbereiche und der Versuchsbedingungen (z.B. Elektrophoresebedingungen) muss zum Nachweis schwacher Wirkungen vielleicht ebenfalls optimiert werden (siehe Nummer 17).
Sämtliche Änderungen am Versuchsprotokoll sind auf ihre Übereinstimmung mit den bereits vorhandenen Datenbanken historischer Kontrolldaten des Labors zu prüfen. Bei größeren Inkonsistenzen sollte eine neue Datenbank historischer Kontrolldaten erstellt werden.
Beschreibung der Methode
Vorbereitungen
Auswahl der Tierart
In der Regel werden junge gesunde und geschlechtsreife Nagetiere üblicher Laborstämme verwendet (die bei Behandlungsbeginn 6 bis 10 Wochen alt sind, wenngleich etwas ältere Tiere auch zulässig sind). Die Auswahl der Nagetierarten sollte basieren auf i) Arten, die in anderen Toxizitätsstudien verwendet werden (um eine Korrelation zwischen den Daten herstellen zu können und integrierte Studien zu ermöglichen), ii) Arten, die in einer Kanzerogenitätsstudie Tumore entwickelt haben (bei der Untersuchung des Mechanismus der Karzinogenese) oder iii) Arten, deren Metabolismus dem des Menschen am ähnlichsten ist, soweit bekannt. In diesem Test werden routinemäßig Ratten verwendet, aber es können auch andere Arten zum Einsatz kommen, sofern dies ethisch und wissenschaftlich begründet ist.
Haltungs- und Fütterungsbedingungen
Bei Nagern soll die Temperatur im Versuchstierraum 22 1 °C (± 3 1 °C) betragen. Die relative Luftfeuchte sollte bei 50 bis 60 % liegen, wenigstens 30 % betragen und außer bei Reinigung des Raumes 70 % vorzugsweise nicht übersteigen. Der Raum sollte künstlich beleuchtet sein, wobei die Beleuchtung im 12-Stunden-Rhythmus ein- und ausgeschaltet werden sollte. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung einer Prüfchemikalie gewährleistet werden muss, wenn diese über das Futter verabreicht wird. Nagetiere sollten in kleinen gleichgeschlechtlichen Gruppen (maximal fünf pro Käfig) untergebracht werden, sofern kein aggressives Verhalten zu erwarten ist. Die Tiere sind nur dann einzeln zu halten, wenn dies wissenschaftlich begründet ist. Es sind möglichst feste Böden zu verwenden, da Maschendrahtböden Verletzungen verursachen können (49). Es muss für eine entsprechende Ausgestaltung des Lebensumfelds gesorgt werden.
Vorbereitung der Versuchstiere
Die Tiere werden den Kontroll- und Behandlungsgruppen nach dem Zufallsprinzip zugeordnet. Sie werden individuell gekennzeichnet und über einen Zeitraum von mindestens fünf Tagen vor Behandlungsbeginn unter Laborbedingungen eingewöhnt. Zur individuellen Kennzeichnung der Tiere muss eine minimalinvasive Methode gewählt werden. Geeignete Methoden sind Anbringen von Ringen, Marken oder Mikrochips oder eine biometrische Identifizierung. Kupieren der Ohren oder Zehen ist bei diesen Tests wissenschaftlich nicht gerechtfertigt. Die Käfige sind so anzuordnen, dass sich ihre Position möglichst wenig auswirkt. Zu Beginn des Versuchs sollte die Abweichung des Körpergewichts der Tiere möglichst gering sein und nicht mehr als ± 20 % betragen.
Vorbereitung der Dosierung
Feste Prüfchemikalien sollten vor der Verabreichung an die Tiere in geeigneten Vehikeln gelöst oder suspendiert oder dem Futter oder Trinkwasser beigemischt werden. Flüssige Prüfchemikalien können direkt verabreicht oder zuvor verdünnt werden. Bei Exposition durch Inhalation können die Prüfchemikalien je nach ihren physikalisch-chemischen Eigenschaften als Gas, Dampf oder festes/flüssiges Aerosol verabreicht werden (50) (51).
Es sind frische Zubereitungen der Prüfchemikalie zu verwenden, es sei denn, die Stabilität der Chemikalie bei Lagerung wird nachgewiesen und die entsprechenden Lagerbedingungen werden definiert.
Prüfbedingungen
Vehikel
Das Vehikel sollte bei den gewählten Dosierungen keine toxischen Wirkungen hervorrufen und nicht im Verdacht stehen, mit den Prüfchemikalien eine chemische Reaktion einzugehen. Werden keine allgemein bekannten Vehikel verwendet, so sind Daten zu deren Kompatibilität in Bezug auf Versuchstiere, Verabreichungsweg und Endpunkt beizubringen. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. Es ist zu beachten, dass einige Vehikel (insbesondere zähflüssige Vehikel) Entzündungen hervorrufen und die Background-Werte der DNA-Strangbrüche an der Kontaktstelle, insbesondere bei mehrfachen Verabreichungen, erhöhen können.
Kontrollen
Positivkontrollen
Zum gegenwärtigen Zeitpunkt sollte jeder Versuch eine Gruppe von mindestens drei analysierbaren Tieren eines Geschlechts oder je Geschlecht, sofern beide Geschlechter verwendet werden (siehe Nummer 32), die mit einem Positivkontrollstoff behandelt wurden, umfassen. Möglicherweise kann die Eignung künftig adäquat nachgewiesen werden, sodass weniger Positivkontrollen erforderlich sind. Bei Verwendung mehrerer Probenahmezeitpunkte (z.B. bei Protokoll mit Einzelverabreichung) müssen nur bei einer Probenahme Positivkontrollen einbezogen werden. In einem solchen Fall sollte für einen ausgewogenen Versuchsplan gesorgt werden (siehe Nummer 48). Die gleichzeitig als Positivikontrollen verwendeten Stoffe müssen nicht auf demselben Weg verabreicht werden wie die Prüfchemikalie. Allerdings ist es wichtig, dass bei der Messung der Wirkungen an Kontaktstellen derselbe Verabreichungsweg angewandt wird. Die Positivkontrollstoffe sollten nachweislich DNA-Strangbrüche in allen für die Prüfchemikalie untersuchten Geweben induzieren. EMS ist wahrscheinlich der am besten geeignete Positivkontrollstoff, da er in allen untersuchten Geweben DNA-Strangbrüche hervorgerufen hat. Die Dosierungen der Positivkontrollstoffe sollten jeweils so gewählt werden, dass mäßige Wirkungen für eine kritische Bewertung der Leistungsfähigkeit und Empfindlichkeit des Tests erreicht werden. Sie könnten auf Dosis-Wirkungs-Kurven beruhen, die das Labor während des Nachweises seiner Kompetenz aufgestellt hat. Der prozentuale DNA-Anteil im Schweif bei Tieren, die gleichzeitig mit einer Positivkontrolle behandelt wurden, sollte mit dem vorab ermittelten Laborbereich für jedes Gewebe und jede Probenahmezeit für diese Tierart übereinstimmen (siehe Nummer 16). Beispiele für Positivkontrollstoffe und einige ihrer Zielgewebe (bei Nagern) sind Tabelle 1 zu entnehmen. In wissenschaftlich begründeten Fällen können auch andere als die in Tabelle 1 genannten Stoffe ausgewählt werden.
Tabelle 1 Beispiele für Positivkontrollstoffe und einige der jeweils zutreffenden Zielgewebe
| Stoffe und CAS-Nr. |
| Ethylmethansulfonat (CAS-Nr. 62-50-0) für sämtliche Gewebe |
| Ethylnitrosoharnstoff (CAS-Nr. 759-73-9) für Leber und Magen, Zwölffingerdarm oder Dünndarm |
| Methylmethansulfonat (CAS-Nr. 66-27-3) für Leber, Magen, Zwölffingerdarm oder Dünndarm, Lunge oder bronchoalveoläre Lavage-Zellen, Niere, Harnblase, Lunge, Hoden und Knochenmark/Blut |
| N-Methyl-N2-nitro-N-nitrosoguanidin (CAS-Nr. 70-25-7) für Magen, Zwölffingerdarm oder Dünndarm |
| 1,2-Dimethylhydrazin-2HCl (CAS-Nr. 306-37-6) für Leber und Darm |
| N-methyl-N-nitrosoharnstoff (CAS-Nr. 684-93-5) für Leber, Knochenmark, Blut, Niere, Magen, Dünndarm und Hirn. |
Negativkontrollen
Jeder Versuch sollte für jede Probenahmezeit und jedes Gewebe eine Gruppe von Negativkontrolltieren umfassen, die nur mit dem Vehikel und ansonsten auf dieselbe Weise wie die Behandlungsgruppen behandelt wurden. Der prozentuale DNA-Anteil im Schweif in den Negativkontrolltieren sollte innerhalb des vorab ermittelten Background-Bereichs des Labors für jedes Gewebe und jede Probenahmezeit für diese Tierart liegen (siehe Nummer 16). Liegen keine historischen oder veröffentlichten Kontrolldaten vor, aus denen hervorgeht, dass weder das gewählte Vehikel noch die Zahl der Verabreichungen oder der Verabreichungsweg schädliche oder genotoxische Wirkungen hervorruft, sollten Vorversuche vor der Komplettstudie durchgeführt werden, um die Akzeptanz der Vehikelkontrolle nachzuweisen.
Verfahren
Anzahl und Geschlecht der Tiere
Obwohl nur wenige Daten über weibliche Tiere vorliegen, die bei einem Vergleich zwischen den Geschlechtern in Bezug auf den Comet-Assay herangezogen werden können, reagieren männliche und weibliche Tiere bei anderen In-vivo-Genotoxizitätstest in der Regel ähnlich, sodass die meisten Studien an beiden Geschlechtern durchgeführt werden könnten. Daten, aus denen nennenswerte Unterschiede zwischen männlichen und weiblichen Tieren hervorgehen (z.B. Unterschiede in der systemischen Toxizität, im Stoffwechsel, in der Bioverfügbarkeit usw., einschließlich unter anderem eine Dosisfindungsstudie) legen die Verwendung von Tieren beider Geschlechter nahe. In diesem Fall kann es angebracht sein, eine Studie an männlichen und weiblichen Tieren durchzuführen, z.B. als Teil einer Toxizitätsstudie mit wiederholter Verabreichung. Gegebenenfalls ist die Verwendung eines faktoriellen Versuchsplans geeignet, wenn beide Geschlechter einbezogen werden. Einzelheiten zur Analyse der Daten bei Verwendung eines solchen Versuchsplans sind in Anlage 2 enthalten.
Zu Beginn der Studie (und während des Kompetenznachweises) sollten die Gruppengrößen so festgelegt werden, dass man pro Gruppe mindestens fünf analysierbare Tiere eines Geschlechts - oder je Geschlecht, sofern beide Geschlechter einbezogen werden - erhält (weniger in der gleichzeitigen Positivkontrollgruppe - siehe Nummer 29). Sollte es sich beim Menschen um eine geschlechtsspezifische Exposition handeln, z.B. bei bestimmten Pharmazeutika, ist der Versuch an Tieren des betreffenden Geschlechts durchzuführen. Bei einer gemäß den unter Nummer 33 festgelegten Parametern durchgeführten Studie mit drei Dosisgruppen und gleichzeitigen Negativ- und Positivkontrollgruppen (wobei jede Gruppe aus fünf Tieren desselben Geschlechts besteht) kann als Richtwert für die üblicherweise erforderliche Höchstmenge an Tieren eine Anzahl von 25 bis 35 Versuchstieren angegeben werden.
Behandlungsplan
Die Tiere sollten täglich über einen Zeitraum von mindestens zwei Tagen behandelt werden (d. h. zwei oder mehr Behandlungen in Abständen von ungefähr24 Stunden), und die Probenahme sollte einmal 2 bis 6 Stunden (oder zum Zeitpunkt Tmax) nach der letzten Behandlung erfolgen (12). Proben aus verlängerten Verabreichungsschemata (z.B. tägliche Dosierung über28 Tage) sind zulässig. Es wurde nachgewiesen, dass der Comet-Assay erfolgreich mit dem Erythrozyten-Mikrokerntest kombiniert werden kann (10) (19). Jedoch sollte die Logistik im Zusammenhang mit Gewebeproben für die Comet-Analyse unter Berücksichtigung der Anforderungen an Gewebeproben für andere Arten von toxikologischen Bewertungen sorgfältig geplant werden. Die Probenahme 24 Stunden nach der letzten Verabreichung, was bei einer allgemeinen Toxizitätsstudie üblicherweise der Fall ist, ist in den meisten Fällen ungeeignet (siehe Nummer 40 zum Probenahmezeitpunkt). Die Anwendung anderer Behandlungs- und Probenahmepläne sollte begründet werden (siehe Anlage 3). Beispielsweise könnte eine Einzelbehandlung mit mehreren Probenahmen erfolgen, jedoch sollte in einem solchen Fall beachtet werden, dass bei einer Studie mit Einzelverabreichung mehrere Tiere benötigt werden, da mehrere Probenahmezeitpunkte erforderlich sind. Allerdings ist dies gelegentlich vorzuziehen, wenn beispielsweise die Prüfchemikalie eine übermäßige Toxizität nach wiederholter Verabreichung induziert.
Jede Art der Testdurchführung ist akzeptabel, solange die Prüfchemikalie eine positive Wirkung hervorruft oder im Falle einer Negativstudie die Exposition der Zielgewebe oder die Toxizität für diese Zielgewebe durch direkte oder indirekte Hinweise nachgewiesen wurde oder die Limit-Dosis erreicht wird (siehe Nummer 36).
Die Prüfchemikalien können auch in Form von zwei Teilmengen am selben Tag im Abstand von nicht mehr als 2 bis 3 Stunden verabreicht werden, wenn es sich um ein großes Volumen handelt. In diesen Fällen sollte der Zeitpunkt der Probenahme ausgehend vom Zeitpunkt der letzten Verabreichung geplant werden (siehe Nummer 40).
Dosierungen
Wenn zunächst eine Dosisfindungsstudie durchgeführt wird, da keine geeigneten Daten aus anderen einschlägigen Studien zur Dosierungswahl vorliegen, sollte diese im selben Labor unter Verwendung von Tieren derselben Art und Rasse und desselben Geschlechts sowie nach demselben Behandlungsverfahren stattfinden, die nach den gegenwärtigen Ansätzen für die Durchführung von Dosisfindungsstudien in der Hauptstudie zu verwenden sind. Ziel der Studie sollte sein, die maximal verträgliche Dosis (MTD) zu ermitteln, die als die Dosis definiert ist, die für die Dauer der Studie zu leichten toxischen Wirkungen (z.B. eindeutige klinische Anzeichen wie Abweichungen in Verhalten oder Reaktionen, geringer Rückgang des Körpergewichts oder zytotoxische Wirkung auf ein Zielgewebe) führt, jedoch weder zum Tod führt noch Anzeichen von Schmerzen, Leiden oder Ängsten hervorruft, die eine Tötung erforderlich machen würden. Bei einer nicht toxischen Prüfchemikalie beträgt die Höchstdosis (Limit-Dosis) bei einem Behandlungszeitraum von mindestens 14 Tagen 1.000 mg/kg Körpergewicht/Tag. Bei Behandlungszeiträumen von weniger als 14 Tagen beträgt die Höchstdosis (Limit-Dosis) 2.000 mg/kg Körpergewicht/Tag. Bei bestimmten Arten von Prüfchemikalien (z.B. Humanarzneimittel), die spezifischen Vorschriften unterliegen, können andere Grenzwerte gelten.
Chemikalien, die eine Sättigung der toxikokinetischen Eigenschaften aufweisen oder Entgiftungsprozesse einleiten, die nach einer Langzeitgabe möglicherweise zu einem Rückgang der Exposition führen, entsprechen möglicherweise nicht den Dosierungskriterien und sollten anhand einer Einzelfallprüfung bewertet werden.
Bei der akuten und subakuten Version des Comet-Assays sollten neben der Höchstdosis (MTD, maximal mögliche Dosis, maximale Expositionsdosis oder Limit-Dosis) eine absteigende Folge von mindestens zwei zusätzlichen Dosierungen mit geeignetem Abstand (vorzugsweise weniger als 10) für jeden Probenahmezeitpunkt gewählt werden, um dosisbezogene Wirkungen nachzuweisen. Jedoch sollten die verwendeten Dosierungen vorzugsweise einen Bereich vom Höchstwert bis zu einer Dosierung, die wenig oder keine Toxizität erzeugt, abdecken. Werden bei allen untersuchten Dosierungen toxische Wirkungen im Zielgewebe beobachtet, sind weitere Untersuchungen bei nichttoxischen Dosierungen anzuraten (siehe Nummern 54 und 55). Studien, in denen der Verlauf der Dosis-Wirkungs-Kurve umfassender untersucht werden soll, erfordern gegebenenfalls weitere Dosisgruppen.
Verabreichung
Bei der Versuchsplanung ist der zu erwartende Expositionsweg beim Menschen zu berücksichtigen. Daher können mit entsprechender Begründung Verabreichungswege wie etwa eine Aufnahme über die Nahrung, über das Trinkwasser, eine topische, subkutane, intravenöse, orale (über eine Magensonde), intratracheale Verabreichung, durch Inhalation oder Implantation, gewählt werden. In jedem Fall sollte der Verabreichungsweg so gewählt werden, dass eine angemessene Exposition des/der Zielgewebe(s) sichergestellt ist. Eine intraperitoneale Injektion wird in der Regel nicht empfohlen, da sie keinen typischen Expositionsweg beim Menschen darstellt; sie sollte nur mit spezieller Begründung angewandt werden (z.B. bestimmte Stoffe, die als Positivkontrollen verwendet werden, zu Untersuchungszwecken oder für einige intraperitoneal verabreichte Arzneimittel). Die Höchstmenge an Flüssigkeit, die je Gabe über eine Magensonde oder eine Injektion verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen können aber auch 2 ml/100 g Körpergewicht in Betracht gezogen werden. Werden größere Volumina verwendet (sofern durch Tierschutzvorschriften erlaubt), ist dies zu begründen. Soweit möglich sollten verschiedene Dosierungen durch Anpassung der Konzentration der Dosisformulierung erreicht werden, um bei allen Dosen ein konstantes Volumen im Verhältnis zum Körpergewicht sicherzustellen.
Probenahmezeitpunkt
Der Probenahmezeitpunkt ist eine kritische Variable, da er von dem Zeitraum abhängt, der erforderlich ist, bis die Prüfchemikalien ihre maximale Konzentration im Zielgewebe erreichen und DNA-Strangbrüche induziert werden; er muss jedoch vor dem Zeitpunkt liegen, zu dem diese Brüche entfernt oder repariert werden oder zum Zelltod führen. Die Persistenz einiger Läsionen, die zu den im Comet-Assay nachgewiesenen DNA-Strangbrüchen führen, kann sehr kurz sein, zumindest bei einigen in vitro geprüften Chemikalien (52) (53). Wenn solche transienten DNA-Läsionen vermutet werden, sollten Maßnahmen ergriffen werden, um deren Verlust zu verringern, indem sichergestellt wird, dass die Proben der Gewebe ausreichend früh, möglichst vor den nachfolgend angegebenen Standardzeiten, genommen werden. Die optimalen Probenahmezeitpunkte können chemikalien- oder pfadspezifisch sein, was z.B. zu einer raschen Gewebeexposition bei intravenöser Verabreichung oder bei Exposition durch Inhalation führt. Die Probenahmezeitpunkte sollten, sofern vorhanden, anhand von kinetischen Daten (z.B. Zeitpunkt (Tmax), zu dem die höchste Plasma- oder Gewebekonzentration (Cmax) erreicht wird oder im stabilen Zustand bei mehrfacher Verabreichung) bestimmt werden. Liegen keine kinetischen Daten vor, besteht ein akzeptabler Kompromiss für die Messung der Genotoxizität in der Probenahme 2 bis 6 Stunden nach der letzten Behandlung im Falle von zwei oder mehr Behandlungen oder 2 bis 6 und 16 bis 26 Stunden nach einer Einzelverabreichung. In einem solchen Fall ist jedoch darauf zu achten, dass alle Tiere gleichzeitig nach der letzten (oder einzigen) Dosis seziert werden. Ferner können Informationen über das Auftreten toxischer Wirkungen in Zielorganen (falls verfügbar) bei der Wahl geeigneter Probenahmezeitpunkte als Grundlage dienen.
Beobachtungen
Allgemeine klinische Beobachtungen in Bezug auf die Gesundheit der Tiere sollten mindestens einmal täglich, vorzugsweise zum gleichen Zeitpunkt und unter Berücksichtigung des Zeitraums, in dem der Wirkungsgipfel nach Verabreichung der Dosis zu erwarten ist, vorgenommen und protokolliert werden (54). Mindestens zweimal täglich sind alle Tiere auf Morbidität und Mortalität zu überprüfen. Bei länger andauernden Studien sollten alle Tiere mindestens einmal wöchentlich und am Ende des Prüfzeitraums gewogen werden. Die Futteraufnahme sollte bei jedem Futterwechsel und mindestens einmal wöchentlich gemessen werden. Wenn die Prüfchemikalie über das Trinkwasser verabreicht wird, sollte auch die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen werden. Tiere, die nicht letale Anzeichen übermäßiger Toxizität aufweisen, sollten vor Ende des Prüfzeitraums getötet werden und werden in der Regel nicht für die Comet-Analyse verwendet.
Gewebegewinnung
Da die Induktion von DNA-Strangbrüchen (Comets) praktisch in jedem Gewebe untersucht werden kann, sollte die Begründung der Auswahl der zu gewinnenden Gewebe eindeutig definiert werden und auf dem Grund für die Durchführung der Studie sowie vorliegenden Daten zu ADME, Genotoxizität, Karzinogenität oder sonstiger Toxizität für die untersuchte Prüfchemikalie beruhen. Wichtige zu berücksichtigende Faktoren sind der Verabreichungsweg (basierend auf dem bzw. den wahrscheinlichen Expositionspfaden beim Menschen), die vorhergesagte Resorption und Verteilung im Gewebe, die Rolle der Metabolisierung und der mögliche Wirkmechanismus der Prüfchemikalien. Die Leber ist das am häufigsten untersuchte Gewebe, für das auch die meisten Daten vorliegen. Wenn keine Hintergrundinformationen vorliegen und keine spezifischen zu untersuchenden Gewebe identifiziert werden, wäre daher eine Probenahme an der Leber gerechtfertigt, da sie ein primärer Ort des Metabolismus xenobiotischer Stoffe und häufig sowohl der/den ursprünglichen Chemikalie(n) als auch Metaboliten ausgesetzt ist. In einigen Fällen kann jedoch die Untersuchung einer direkten Kontaktstelle (z.B. bei oral verabreichten Chemikalien der Drüsenmagen oder Zwölffingerdarm/Dünndarm oder bei inhalierten Chemikalien die Lunge) am zweckmäßigsten sein. Zusätzliche oder alternative Gewebe sollten basierend auf den jeweiligen Gründen für die Durchführung des Versuchs gewählt werden. Allerdings kann es sinnvoll sein, mehrere Gewebe derselben Tiere zu untersuchen, vorausgesetzt, das Labor hat seine Kompetenz bei solchen Geweben sowie im Umgang mit mehreren Geweben gleichzeitig nachgewiesen.
Vorbereitung der Proben
Für die nachstehend (Nummern 44-49) beschriebenen Prozesse ist es wichtig, dass alle Lösungen oder stabilen Suspensionen vor ihrem Verfallsdatum verwendet oder bei Bedarf frisch zubereitet werden. In der nachfolgenden Beschreibung gelten die Zeiträume, um i) die einzelnen Gewebe nach der Sektion zu entnehmen, ii) die Gewebe in Zell-/Zellkernsuspensionen aufzuarbeiten und iii) die Suspension aufzuarbeiten und die Objektträger vorzubereiten, alle als kritische Variablen (siehe Definitionen in Anlage 1). Für jeden dieser Schritte sollte bei der Festlegung der Methode und beim Kompetenznachweis eine akzeptable Zeitdauer festgelegt worden sein.
Die Tiere werden im Einklang mit den einschlägigen Tierschutzvorschriften und nach dem 3R-Prinzip zu den entsprechenden Zeitpunkten nach der letzten Behandlung mit einer Prüfchemikalie getötet. Die ausgewählten Gewebe werden entfernt und seziert und ein Teil wird für den Comet-Assay gewonnen. Gleichzeitig sollte eine Sektion aus demselben Gewebeteil herausgeschnitten und für eine mögliche histopathologische Analyse (siehe Nummer 55) nach Standardmethoden (12) in eine Formaldehydlösung oder eine geeignete Fixierlösung gelegt werden. Das Gewebe für den Comet-Assay wird in einen Zerkleinerungspuffer gelegt, ausreichend mit kaltem Zerkleinerungspuffer gespült, um Restblut zu beseitigen, und bis zur Verarbeitung in eiskaltem Zerkleinerungspuffer aufbewahrt. Eine In-situ-Perfusion kann ebenfalls durchgeführt werden, z.B. bei Leber, Niere.
Es wurden zahlreiche Methoden für die Zell-/Zellkernisolierung veröffentlicht. Dazu gehören das Zerkleinern von Geweben wie beispielsweise Leber und Niere, das Abschaben der Schleimhautoberflächen des Magen-Darm-Trakts, die Homogenisierung und die enzymatische Verdauung. In der JaCVAM-Validierungsstudie wurden lediglich isolierte Zellen untersucht. Um die Methode festzulegen und um zwecks Nachweis der Kompetenz auf die JaCVAM-Versuchsdaten Bezug nehmen zu können, werden isolierte Zellen bevorzugt. Jedoch wurde gezeigt, dass sich das Testergebnis bei Verwendung von isolierten Zellen oder von Zellkernen nicht wesentlich unterschied (8). Ferner führten unterschiedliche Methoden für die Isolierung der Zellen (z.B. Homogenisierung, Zerkleinerung, enzymatische Verdauung und Siebfiltration) zu vergleichbaren Ergebnissen (55). Folglich können entweder isolierte Zellen oder Zellkerne verwendet werden. Das Labor sollte die gewebespezifischen Methoden zur Einzelzell-/Zellkernisolierung sorgfältig prüfen und validieren. Wie bereits unter Nummer 40 erwähnt, kann die Persistenz einiger Läsionen, die zu den vom Comet-Assay nachgewiesenen DNA-Strangbrüchen führen, sehr kurz sein (52) (53). Daher ist es ungeachtet der Methode für die Zubereitung der Einzelzell-/Zellkernsuspensionen wichtig, dass die Gewebe möglichst bald nach Tötung der Tiere aufbereitet und unter Bedingungen aufbewahrt werden, die den Verlust der Läsionen verringern (z.B. durch Lagerung bei niedriger Temperatur). Die Zellsuspensionen sollten bis zur Verwendung eiskalt aufbewahrt werden, sodass die Abweichungen zwischen Proben minimal sind und angemessene Reaktionen bei den Positiv- und Negativkontrollen nachgewiesen werden können.
Vorbereitung der Objektträger
Die Objektträger sollten schnellstmöglich (im Idealfall innerhalb einer Stunde) nach Zubereitung der Einzelzell-/Zellkernsuspensionen vorbereitet werden. Die Temperatur und die Zeit zwischen der Tötung der Tiere und der Vorbereitung der Objektträger sollten eng kontrolliert und unter Laborbedingungen validiert werden. Durch das Volumen der Zellsuspension, das der Agarose mit niedrigem Schmelzpunkt (gewöhnlich 0,5-1,0 %) zur Vorbereitung der Objektträger beigegeben wird, sollte der Anteil der Agarose mit niedrigem Schmelzpunkt nicht auf weniger als 0,45 % reduziert werden. Die optimale Zelldichte wird durch das Bildanalysesystem bestimmt, das zur Auswertung der Comets verwendet wird.
Lyse
Die Lysebedingungen stellen ebenfalls eine kritische Variable dar und können sich auf die Strangbrüche, die aus spezifischen Arten von DNA-Veränderungen resultieren (bestimmte Alkylierungen und Bildung von Addukten zu Basen der DNA), auswirken. Daher empfiehlt es sich, die Lysebedingungen bei allen Objektträgern innerhalb eines Versuchs so konstant wie möglich zu halten. Nach der Vorbereitung sollten die Objektträger mindestens eine Stunde (oder über Nacht) bei ungefähr 2-8 1 °C bei gedämpften Lichtverhältnissen (z.B. gelbes Licht (oder vor Licht geschützt)) zur Vermeidung einer Exposition gegenüber weißem Licht, das UV-Komponenten enthalten kann, in gekühlte Lyselösung eingelegt werden. Nach dieser Inkubationszeit sollten die Objektträger abgespült werden, um vor der alkalischen Entwindung Detergenz- und Salzreste zu beseitigen. Dies kann unter Verwendung von gereinigtem Wasser, neutralisierender Pufferlösung oder Phosphat-Pufferlösung erfolgen. Elektrophorese-Pufferlösung kann ebenfalls verwendet werden. Dies würde die alkalischen Bedingungen in der Elektrophoresekammer erhalten.
Entwindung und Elektrophorese
Die Objektträger sollten nach dem Zufallsprinzip auf die Plattform einer Unterwasser-Elektrophoreseapparatur gesetzt werden, die so viel Elektrophoreselösung enthält, dass die Oberflächen der Objektträger vollständig bedeckt sind (die Höhe der Bedeckung sollte bei allen Versuchsdurchführungen gleich sein). Bei anderen Arten von Comet-Assay-Elektrophoreseapparaturen, z.B. mit aktiver Kühlung, Umwälzung und Hochleistungsnetzteil, führt eine höhere Bedeckung durch das Lösungsmittel zu einer höheren Stromstärke bei konstanter Spannung. Die Objektträger sollten nach einem ausgewogenen Versuchsplan in den Elektrophoresetank eingelegt werden, damit die Wirkungen von Trends oder Randeffekte im Tank abgeschwächt und Schwankungen zwischen den Chargen möglichst gering gehalten werden. Dies bedeutet, dass jede Elektrophorese-Versuchsdurchführung die gleiche Anzahl von Objektträgern von jedem in der Studie eingeschlossenen Tier und Proben aus den verschiedenen Dosisgruppen, Negativ- und Positivkontrollen umfassen sollte. Die Objektträger sollten mindestens 20 Minuten im Elektrophoresetank belassen werden, damit die DNA sich entwinden kann, und dann unter kontrollierten Bedingungen elektrophoretisch untersucht werden. Diese Bedingungen sollten eine optimale Empfindlichkeit und dynamische Bandbreite des Tests garantieren (d. h. zu akzeptablen Werten des prozentualen DNA-Anteil im Schweif bei Negativ- und Positivkontrollen zur Optimierung der Empfindlichkeit führen). Das Ausmaß der DNA-Wanderung steht im linearen Verhältnis zur Dauer der Elektrophorese sowie zum Potenzial (V/cm). Auf der Grundlage der JaCVAM-Studie könnte dies 0,7 V/cm bei mindestens 20 Minuten sein. Die Dauer der Elektrophorese wird als kritische Variable betrachtet, und die Elektrophoresezeit sollte so eingestellt werden, dass die dynamische Bandbreite optimiert wird. Längere Elektrophoresezeiten (z.B. 30 oder 40 Minuten zur Maximierung der Empfindlichkeit) bewirken bei bekannten Mutagenen in der Regel stärkere Positivreaktionen. Allerdings können längere Elektrophoresezeiten auch zu einer übermäßigen Wanderung in den Kontrollproben führen. Die Spannung sollte in jedem Versuch konstant und die Variabilität der anderen Parameter in einem schmalen, festgelegten Bereich gehalten werden. In der JaCVAM-Studie ergab beispielsweise ein Wert von 0,7 V/cm eine anfängliche Stromstärke von 300 mA. Die Tiefe der Pufferlösung sollte so eingestellt werden, dass die erforderlichen Bedingungen erreicht und während des gesamten Versuchs eingehalten werden. Die Stromstärke am Anfang und Ende der Elektrophoresezeit sollte protokolliert werden. Daher sollten die optimalen Bedingungen während des anfänglichen Nachweises der Kompetenz im jeweiligen Labor bei jedem untersuchten Gewebe ermittelt werden. Die Temperatur der Elektrophoreselösung sollte während der Entwindung und Elektrophorese niedrig gehalten werden (in der Regel 2-10 1 °C) (10). Die Temperatur der Elektrophoreselösung am Anfang der Entwindung sowie am Anfang und Ende der Elektrophorese ist jeweils zu protokollieren.
Nach der Elektrophorese sollten die Objektträger mindestens 5 Minuten lang in die neutralisierende Pufferlösung eingelegt bzw. darin abgespült werden. Gele können angefärbt und "frisch" (z.B. innerhalb von 1-2 Tagen) ausgewertet oder zur späteren Auswertung (z.B. innerhalb von 1-2 Wochen nach Anfärbung) dehydriert werden (56). Allerdings sollten die Bedingungen während des Nachweises der Kompetenz validiert und historische Daten für jede dieser Bedingungen gesondert erfasst und protokolliert werden. Im letzteren Fall sollten die Objektträger zur Dehydration mindestens 5 Minuten lang in reines Ethanol eingelegt, an der Luft getrocknet und bis zur Auswertung entweder bei Zimmertemperatur oder in einem Behälter in einem Kühlschrank aufbewahrt werden.
Messmethoden
Comets sollten mithilfe eines automatischen oder halbautomatischen Bildanalysesystems quantitativ ausgewertet werden. Die Objektträger werden mit einem geeigneten fluoreszierenden Farbstoff, z.B. SYBR Gold, Green I, Propidiumjodid oder Ethidiumbromid, angefärbt und mit geeigneter Vergrößerung (z.B. 200x) unter einem Mikroskop mit Epi-Fluoreszenz- und entsprechenden Detektoren oder mit einer Digitalkamera (z.B. CCD-Kamera) gemessen.
Die Zellen lassen sich, wie im Atlas of Comet Assay Images (57) beschrieben, in drei Kategorien einteilen, nämlich in auswertbar, nicht auswertbar und Hedgehog (weitere Ausführungen unter Nummer 56). Nur auswertbare Zellen (eindeutig definierter Kopf und Schweif, keine Interferenzen mit Nachbarzellen) sollten für den prozentualen DNA-Anteil im Schweif ausgewertet werden, um Artefakte zu vermeiden. Die Häufigkeit von nicht auswertbaren Zellen muss nicht angegeben werden. Die Häufigkeit von Hedgehogs sollte durch visuelle Auswertung (da das Fehlen eines klar definierten Kopfes bedeutet, dass sie mittels Bildanalyse nicht leicht erkannt werden) von mindestens 150 Zellen pro Probe (näheren Erläuterungen unter Nummer 56) bestimmt und gesondert dokumentiert werden.
Alle zu analysierenden Objektträger, einschließlich der Positiv- und Negativkontrollen, sollten vor der Analyse unabhängig kodiert und "blind" ausgewertet werden, damit die Auswertung ohne Kenntnis der Behandlungsbedingungen erfolgt. Bei jeder Probe (pro Gewebe pro Tier) sollten mindestens 150 Zellen (ohne Hedgehogs - siehe Nummer 56) analysiert werden. Die Auswertung von 150 Zellen pro Tier bei mindestens fünf Tieren pro Dosis (weniger in der gleichzeitigen Positivkontrolle - siehe Nummer 29) gewährleistet entsprechend der Analyse von Smith et al., 2008, (5) eine hinreichende statistische Aussagekraft. Bei Verwendung von Objektträgern könnten dies zwei oder drei ausgewertete Objektträger pro Probe sein, wenn fünf Tiere pro Gruppe zum Einsatz kommen. Es sollten mehrere Bereiche des Objektträgers in einer solchen Dichte untersucht werden, dass eine Überschneidung von Schweifen ausgeschlossen ist. Die Auswertung am Rand der Objektträger ist zu vermeiden.
DNA-Strangbrüche im Comet-Assay können anhand von unabhängigen Endpunkten wie z.B. dem prozentualen DNA-Anteil im Schweif, der Schweiflänge und dem Schweifmoment gemessen werden. Sofern ein geeignetes Bildanalysesystem verwendet wird, können alle drei Messungen durchgeführt werden. Jedoch wird der prozentuale DNA-Anteil im Schweif (auch als prozentuale Schweif-Intensität bezeichnet) für die Bewertung und Interpretation der Ergebnisse empfohlen (12) (40) (41) (42). Dieser Anteil wird anhand der DNA-Fragmentintensität im Schweif, ausgedrückt als Prozentsatz der Gesamtintensität der Zelle, bestimmt (13).
Gewebeschädigung und Zytotoxizität
Positive Ergebnisse im Comet-Assay sind möglicherweise nicht allein auf Genotoxizität zurückzuführen sein. Auch toxische Wirkungen im Zielgewebe können eine Zunahme der DNA-Wanderung hervorrufen (12) (41). Hingegen ist bei bekannten Genotoxinen häufig eine geringe oder mäßige Zytotoxizität zu beobachten (12), was zeigt, dass der Comet-Assay alleine keine Unterscheidung zwischen DNA-Wanderung, die durch Genotoxizität ausgelöst wird, und durch Zytotoxizität hervorgerufener DNA-Wanderung erlaubt. Wenn jedoch eine Zunahme der DNA-Wanderung beobachtet wird, wird empfohlen, eine Untersuchung auf einen oder mehrere Indikatoren für Zytotoxizität durchzuführen, da dies bei der Interpretation der Ergebnisse hilfreich sein kann. Eine Zunahme der DNA-Wanderung ist bei Vorliegen eindeutiger Anzeichen von Zytotoxizität mit Vorsicht zu interpretieren.
Es wurden zahlreiche Messgrößen für die Zytotoxität vorgeschlagen, wobei histopathologische Veränderungen als relevante Messgröße für Gewebetoxizität gelten. Beobachtungen wie Entzündung, Zellinfiltration, apoptotische oder nekrotische Veränderungen wurden mit einer Zunahme der DNA-Wanderung in Verbindung gebracht. Wie die JaCVAM-Validierungsstudie (12) gezeigt hat, gibt es jedoch keine endgültige Liste der mit einer Zunahme der DNA-Wanderung stets einhergehenden, histopathologischen Veränderungen. Änderungen bestimmter biologischer Parameter (z.B. AST, ALT) können ebenfalls nützliche Informationen über die Gewebeschädigung liefern, und ferner können zusätzliche Indikatoren wie z.B. Caspase-Aktivierung, TUNEL-Färbung, Annexin-V-Färbung usw. in Betracht gezogen werden. Allerdings wurden über die Verwendung dieser Indikatoren in In-vivo-Studien nur wenig Daten veröffentlicht, und nicht alle sind gleich zuverlässig.
Hedgehogs (oder Clouds, Geisterzellen) sind Zellen, deren mikroskopisches Bild einen kleinen oder nicht existenten Kopf und lange, diffuse Schweife zeigt und die als schwer beschädigte Zellen gelten, obwohl die Genese der Hedgehogs unsicher ist (siehe Anlage 3). Aufgrund ihres Aussehens sind Messungen des prozentualen Anteils der Schweif-DNA mittels Bildanalyse unzuverlässig, weshalb Hedgehogs gesondert zu bewerten sind. Das Auftreten von Hedgehogs sollte vermerkt und angegeben werden, und jede relevante Zunahme, bei der davon ausgegangen wird, dass sie auf die Prüfchemikalie zurückzuführen ist, ist zu untersuchen und mit Vorsicht zu interpretieren. Bei solchen Überlegungen kann die Kenntnis der potenziellen Wirkungsweise der Prüfchemikalien von Nutzen sein.
Daten und Berichterstattung
Behandlung der Ergebnisse
Die Versuchseinheit ist das Tier. Daher sollten sowohl die Daten für die einzelnen Tiere als auch die zusammengefassten Ergebnisse in tabellarischer Form dargestellt werden. Aufgrund der hierarchischen Art der Daten wird empfohlen, für jeden Objektträger den Median des prozentualen Anteils der Schweif-DNA zu bestimmen und für jedes Tier den Mittelwert der Mediane zu berechnen (12). Anschließend wird der Mittelwert aus den Mittelwerten für die einzelnen Tiere bestimmt, um den Gruppenmittelwert zu erhalten. Alle diese Werte sind in den Bericht aufzunehmen. Alternative Ansätze (siehe Nummer 53) können verwendet werden, sofern dies wissenschaftlich und statistisch begründet ist. Statistische Analysen können nach verschiedenen Ansätzen durchgeführt werden (58) (59) (60) (61). Bei der Auswahl der anzuwendenden statistischen Methoden sollte die Notwendigkeit einer Transformation (z.B. Logarithmus oder Quadratwurzel) der Daten und/oder der Addition einer kleinen Zahl (z.B. 0,001) zu allen Werten (auch Werten ungleich Null), wie in den obigen Referenzdokumenten erörtert, berücksichtigt werden, um die Wirkungen von Null-Zellwerten abzuschwächen. Nähere Informationen zur Analyse von Wechselwirkungen zwischen Behandlung und Geschlecht bei Verwendung beider Geschlechter sowie zur nachfolgenden Analyse der Daten, wenn Unterschiede bestehen oder keine Unterschiede festgestellt werden, sind Anlage 2 zu entnehmen. Daten zur Toxizität bei Tieren und klinische Anzeichen sind ebenfalls anzugeben.
Gültigkeitskriterien
Die Akzeptanz eines Versuchs beruht auf folgenden Kriterien:
- Die Daten der gleichzeitigen Negativkontrolle gelten als zulässig für die Aufnahme in die Datenbank des Labors mit historischen Negativkontrolldaten (siehe Nummer 16).
- Gleichzeitige Positivkontrollen (siehe Nummer 29) sollten Reaktionen hervorrufen, die mit denen kompatibel sind, die in der Datenbank mit historischen Positivkontrollen generiert werden und gegenüber den gleichzeitigen Negativkontrollen eine statistisch signifikante Zunahme aufweisen.
- Eine angemessene Zahl an Zellen und Dosierungen wurde analysiert (Nummer 52 und Nummern 36, 37 und 38).
- Die Kriterien für die Wahl der höchsten Dosierung stimmen mit den unter Nummer 36 beschriebenen überein.
Bewertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn
- mindestens eine der Versuchsdosierungen eine statistisch signifikante Zunahme gegenüber der gleichzeitigen Negativkontrolle aufweist,
- ein geeigneter Trendtest zeigt, dass die Zunahme dosisabhängig ist,
- Ergebnisse außerhalb der Verteilung der historischen Negativkontrolldaten für eine bestimmte Tierart, ein Vehikel, einen Verabreichungsweg, ein Gewebe und eine bestimmte Anzahl von Verabreichungen liegen.
Sind alle diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie DNA-Strangbrüche in den in diesem Versuchssystem untersuchten Geweben auslösen kann. Sind nur ein oder zwei dieser Kriterien erfüllt, siehe Nummer 62.
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn
- keine der Versuchskonzentrationen eine statistisch signifikante Zunahme gegenüber der gleichzeitigen Negativkontrolle aufweist,
- ein geeigneter Trendtest zeigt, dass es keine dosisabhängige Zunahme gibt,
- alle Ergebnisse innerhalb der Verteilung der historischen Negativkontrolldaten für eine bestimmte Tierart, ein Vehikel, einen Verabreichungsweg, ein Gewebe und eine bestimmte Anzahl von Verabreichungen liegen,
- eine Exposition der Zielgewebe oder die Toxizität für die Zielgewebe direkt oder indirekt nachgewiesen wurde.
Es wird dann davon ausgegangen, dass die Prüfchemikalie keine DNA-Strangbrüche in den in diesem Versuchssystem untersuchten Geweben auslösen kann.
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
In den Fällen, in denen die Reaktion, weder eindeutig negativ noch eindeutig positiv ist (d. h. nicht alle unter Nummer 59 oder 60 genannten Kriterien sind erfüllt), und um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung bewertet und/oder weitere Untersuchungen durchgeführt werden, wenn dies wissenschaftlich begründet ist. Die Auswertung weiterer Zellen (soweit dies angebracht ist) oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter optimierten Versuchsbedingungen (z.B. Abstände der Dosierungen, andere Verabreichungswege, andere Probenahmezeitpunkte oder andere Gewebe), könnte hilfreich sein.
In seltenen Fällen erlaubt der Datensatz selbst nach weiteren Untersuchungen keine definitive Aussage zu positiven oder negativen Ergebnissen, so dass die Reaktion als nicht eindeutig eingestuft wird.
Um die biologische Relevanz eines positiven oder mehrdeutigen Ergebnisses zu bewerten, sind Informationen zur Zytotoxizität für das Zielgewebe erforderlich (siehe Nummern 54 und 55). Wenn lediglich im Falle von eindeutigen Anzeichen für zytotoxische Wirkungen positive oder mehrdeutige Ergebnisse beobachtet werden, würde die Studie als nicht eindeutig im Hinblick auf die Genotoxizität abgeschlossen werden, außer wenn genügend Informationen vorliegen, die eine endgültige Schlussfolgerung zulassen. Bei einem negativen Studienergebnis mit Anzeichen für Toxizität bei allen untersuchten Dosierungen sind weitere Untersuchungen bei nichttoxischen Dosierungen anzuraten.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalie:
- Herkunft, Partienummer, sofern vorhanden;
- Stabilität der Prüfchemikalie, letztes Verwendungsdatum oder Datum für erneute Analyse, soweit bekannt.
Einkomponentige Substanz:
- Aussehen, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
- chemische Bezeichnung, wie z.B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer , SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, falls zutreffend und praktisch durchführbar usw.
Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
- so weit wie möglich charakterisiert durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
Lösungsmittel/Vehikel:
- Begründung der Auswahl des Lösungsmittels/Vehikels;
- Löslichkeit und Stabilität der Prüfchemikalie im Lösungsmittel/Vehikel, falls bekannt;
- Zubereitung der Dosisformulierungen;
- analytische Bestimmungen der Formulierungen (z.B. Stabilität, Homogenität, nominale Konzentrationen).
Versuchstiere:
- verwendete Tierart/Rasse, wissenschaftliche und ethische Begründung für deren Wahl;
- Anzahl, Alter und Geschlecht der Tiere;
- Herkunft der Tiere, Haltungsbedingungen, Futter, Ausgestaltung der Käfige usw.;
- individuelles Gewicht der Tiere am Anfang und am Ende des Versuchs, einschließlich Bereich, Mittelwert und Standardabweichung des Körpergewichts für jede Gruppe.
Prüfbedingungen:
- Positiv- und Negativ-(Vehikel-/Lösungsmittel-)Kontrollen;
- Ergebnisse einer Dosisfindungsstudie (falls durchgeführt);
- Begründung der gewählten Dosisstufen;
- Angaben zur Zubereitung der Prüfchemikalie;
- Angaben zur Verabreichung der Prüfchemikalie;
- Begründung des Verabreichungswegs;
- Injektionsstelle (bei Studien mit subkutaner oder intravenöser Verabreichung);
- Methoden für die Aufbereitung der Proben, falls verfügbar, histopathologische Analysen, insbesondere bei einer Chemikalie mit positiver Reaktion im Comet-Assay;
- Begründung der Gewebeauswahl;
- Methoden zur Überprüfung, ob die Prüfchemikalie ins Zielgewebe oder in den allgemeinen Kreislauf gelangt ist, im Falle negativer Ergebnisse;
- tatsächliche Dosis (mg/kg Körpergewicht/Tag), berechnet aus der Konzentration der Prüfchemikalie im Futter/Wasser (ppm) und der Futter- bzw. Wasseraufnahme, falls zutreffend;
- Angaben zu Futter- und Wasserqualität;
- nähere Angaben zu Behandlungs- und Stichprobenentnahmeplänen und Begründungen der Wahl (z.B. toxikokinetische Daten, falls verfügbar);
- Schmerzlinderungs-, Analgesiemethode;
- Angaben zur Methode der humanen Tötung;
- Verfahren zur Isolierung und Konservierung der Gewebe;
- Methoden zur Zubereitung von Einzelzell-/Zellkernsuspensionen;
- Herkunft und Partienummern aller Reagenzien (soweit möglich);
- Methoden zur Bewertung der Zytotoxizität;
- Elektrophoresebedingungen;
- angewandte Anfärbemethoden und
- Methoden zur Auswertung und Messung von Comets.
Ergebnisse:
- gegebenenfalls allgemeine klinische Beobachtungen vor und während des Testzeitraums bei jedem Tier;
- Nachweise für Zytotoxizität, falls untersucht;
- bei Studien von mehr als einer Woche: individuelles Gewicht der Tiere während der Studie, einschließlich Bereich, Mittelwert und Standardabweichung des Körpergewichts für jede Gruppe; Futteraufnahme;
- gegebenenfalls Dosis-Wirkungs-Beziehung;
- für jedes Gewebe/Tier prozentualer DNA-Anteil im Schweif (oder andere Messwerte, falls ausgewählt) und Mediane pro Objektträger, Mittelwerte pro Tier und Mittelwerte pro Gruppe;
- gleichzeitige und historische Negativkontrolldaten mit Bereichen, Mittelwerten/Medianen und Standardabweichungen für jedes bewertete Gewebe;
- gleichzeitige und historische Positivkontrolldaten;
- für andere Gewebe als die Leber Dosis-Wirkungs-Kurve, einschließlich Positivkontrolle. Diese kann auf den während des Nachweises der Kompetenz erfassten Daten basieren (siehe Nummern 16 und 17). Ferner sollte mit Verweisen auf die aktuelle Literatur belegt werden, dass Größenordnung und Streuung der Reaktionen auf die Kontrollen bei diesem Gewebe angemessen sind;
- statistische Analysen und angewandte Methoden; Kriterien für die Einstufung einer Wirkung als positiv, negativ oder nicht eindeutig;
- Häufigkeit von Hedgehogs in jeder Gruppe und pro Tier.
Erörterung der Ergebnisse
Schlussfolgerung
Referenzdokumente
Literaturhinweise
(1) Kirkland, D., G. Speit (2008), Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens III. Appropriate follow-up testing in vivo, Mutation Research, Vol. 654/2, 114-32.
(2) Brendler-Schwaab, S. et al. (2005), The in vivo Comet assay: use and status in genotoxicity testing, Mutagenesis, Vol. 20/4, 245-54.
(3) Burlinson, B. et al. (2007), Fourth International Workgroup on Genotoxicity Testing: result of the in vivo Comet assay workgroup, Mutation Research, Vol. 627/1, 31-5.
(4) Burlinson, B. (2012), The in vitro and in vivo Comet assays, Methods in Molecular Biology, Vol. 817, 143-63.
(5) Smith, C.C. et al. (2008), Recommendations for design of the rat Comet assay, Mutagenesis, Vol. 23/3, 233-40.
(6) Hartmann, A. et al. (2003), Recommendations for conducting the in vivo alkaline Comet assay, Mutagenesis, Vol. 18/1, 45-51.
(7) McKelvey-Martin, V.J. et al. (1993), The single cell gel electrophoresis assay (Comet assay): a European review, Mutation Research, Vol. 288/1, 47-63.
(8) Tice, R.R. et al. (2000), Single cell gel/Comet assay: guidelines for in vitro and in vivo genetic toxicology testing, Environmental and Molecular Mutagenesis, Vol. 35/3, 206-21.
(9) Singh, N.P. et al. (1988), A simple technique for quantitation of low levels of DNA damage in individual cells, Experimental Cell Research, Vol. 175/1, 184-91.
(10) Rothfuss, A. et al. (2010), Collaborative study on fifteen compounds in the rat-liver Comet assay integrated into 2- and 4-week repeat-dose studies, Mutation Research, Vol. 702/1, 40-69.
(11) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(12) OECD (2014), Reports of the JaCVAM initiative international pre-validation and validation studies of the in vivo rodent alkaline comet assay for the detection of genotoxic carcinogens, Series on Testing and Assessment, Nos. 195 and 196, OECD Publishing, Paris.
(13) Olive, P.L., J.P. Banath, R.E. Durand (1990), Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells using the "Comet" assay, Radiation Research, Vol. 122/1, 86-94.
(14) Tice, R.R., D.L. Strauss (1995), The single cell gel electrophoresis/Comet assay: a potential tool for detecting radiation-induced DNA damage in humans, Stem Cells, Vol. 13/1, 207-14.
(15) Collins, A.R (2004), The Comet assay for DNA damage and repair: principles, applications, and limitations, Molecular Biotechnology, Vol. 26/3, 249-61.
(16) Rothfuss, A. et al. (2011), Improvement of in vivo genotoxicity assessment: combination of acute tests and integration into standard toxicity testing, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 723/2, 108-20.
(17) Kushwaha, S. et al. (2010), Evaluation of multi-organ DNA damage by Comet assay from 28 days repeated dose oral toxicity test in mice: A practical approach for test integration in regulatory toxicity testing, Regulatory Toxicology and Pharmacology, Vol. 58/1, 145-54.
(18) Vasquez, M.Z. (2010), Combining the in vivo Comet and micronucleus assays: a practical approach to genotoxicity testing and data interpretation, Mutagenesis, Vol. 25/2, 187-99.
(19) Bowen, D.E. (2011), Evaluation of a multi-endpoint assay in rats, combining the bone-marrow micronucleus test, the Comet assay and the flow-cytometric peripheral blood micronucleus test, Mutation Research, Vol. 722/1, 7-19.
(20) Recio, L. et al. (2010), Dose-response assessment of four genotoxic chemicals in a combined mouse and rat micronucleus (MN) and Comet assay protocol, The Journal of Toxicological Science, Vol. 35/2, 149-62.
(21) O'Donovan, M., B. Burlinson (2013), Maximum dose levels for the rodent comet assay to examine damage at the site of contact or to the gastrointestinal tract, Mutagenesis, Vol. 28/6, 621-3.
(22) Hartmann, A. (2004), Use of the alkaline in vivo Comet assay for mechanistic genotoxicity investigations, Mutagenesis, Vol. 19/1, 51-9.
(23) Nesslany, F. (2007), In vivo Comet assay on isolated kidney cells to distinguish genotoxic carcinogens from epigenetic carcinogens or cytotoxic compounds, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 630/1, 28-41.
(24) Brendler-Schwaab, S.Y., B.A. Herbold (1997), A new method for the enrichment of single renal proximal tubular cells and their first use in the Comet assay, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 393/1-2, 175-8.
(25) Toyoizumi, T. et al. (2011), Use of the in vivo skin Comet assay to evaluate the DNA-damaging potential of chemicals applied to the skin, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 726/2, 175-80.
(26) Struwe, M. et al. (2008), Detection of photogenotoxicity in skin and eye in rat with the photo Comet assay, Photochemical and Photobiological Sciences, Vol. 7/2, 240-9.
(27) Wada, K. et al. (2012), A comparison of cell-collecting methods for the Comet assay in urinary bladders of rats, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 742/1-2, 26-30.
(28) Wang, A. et al. (2007), Measurement of DNA damage in rat urinary bladder transitional cells: improved selective harvest of transitional cells and detailed Comet assay protocols, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 634/ 1-2, 51-9.
(29) Burlinson, B. et al. (2007), In Vivo Comet Assay Workgroup, part of the Fourth International Workgroup on Genotoxicity Testing. Fourth International Workgroup on Genotoxicity testing: results of the in vivo Comet assay workgroup, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 627/1, 31-5.
(30) Jackson, P. et al. (2012), Pulmonary exposure to carbon black by inhalation or instillation in pregnant mice: effects on liver DNA strand breaks in dams and offspring, Nanotoxicology, Vol. 6/5, 486-500.
(31) Sasaki, Y.F. et al. (2000), The comet assay with multiple mouse organs: comparison of Comet assay results and carcinogenicity with 208 chemicals selected from the IARC monographs and U.S. NTP Carcinogenicity Database, Critical Reviews in Toxicology, Vol. 30/6, 629-799.
(32) Sekihashi, K. et al. (2002), Comparative investigations of multiple organs of mice and rats in the Comet assay, Mutation Research, Vol. 517/1-2, 53-74.
(33) Speit, G, M. Vasquez, A. Hartmann (2009), The comet assay as an indicator test for germ cell genotoxicity, Mutation Research, Vol. 681/1, 3-12.
(34) Zheng, H., P.L. Olive (1997), Influence of oxygen on radiation-induced DNA damage in testicular cells of C3H mice, International Journal of Radiation Biology, Vol. 71/3, 275-282.
(35) Cordelli, E. et al. (2003), Evaluation of DNA damage in different stages of mouse spermatogenesis after testicular X irradiation, Journal of Radiation Research, Vol. 160/4, 443-451.
(36) Merk, O., G. Speit (1999), Detection of crosslinks with the Comet assay in relationship to genotoxicity and cytotoxicity, Environmental and Molecular Mutagenesis, Vol. 33/2, 167-72.
(37) Pfuhler, S., H.U. Wolf (1996), Detection of DNA-crosslinking agents with the alkaline Comet assay, Environmental and Molecular Mutagenesis, Vol. 27/3, 196-201.
(38) Wu, J.H., N.J. Jones (2012), Assessment of DNA interstrand crosslinks using the modified alkaline Comet assay, Methods in Molecular Biology, Vol. 817, 165-81.
(39) Spanswick, V.J., J.M. Hartley, J.A. Hartley (2010), Measurement of DNA interstrand crosslinking in individual cells using the Single Cell Gel Electrophoresis (Comet) assay, Methods in Molecular Biology, Vol. 613, 267-282.
(40) Kumaravel, T.S., A.N. Jha (2006), Reliable Comet assay measurements for detecting DNA damage induced by ionizing radiation and chemicals, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 605(1-2), 7-16.
(41) Burlinson, B. et al. (2007), Fourth International Workgroup on Genotoxicity Testing: result of the in vivo Comet assay workgroup, Mutation Research, Vol. 627/1, 31-5.
(42) Kumaravel, T.S. et al. (2009), Comet Assay measurements: a perspective, Cell Biology and Toxicology, Vol. 25/1, 53-64.
(43) Ersson, C., L. Möller (2011), The effects on DNA migration of altering parameters in the Comet assay protocol such as agarose density, electrophoresis conditions and durations of the enzyme or the alkaline treatments, Mutagenesis, Vol. 26/6, 689-95.
(44) Møller, P. et al. (2010), Assessment and reduction of Comet assay variation in relation to DNA damage: studies from the European Comet Assay Validation Group, Mutagenesis, Vol. 25/2, 109-11.
(45) Forchhammer, L. et al. (2010), Variation in the measurement of DNA damage by Comet assay measured by the ECVAG inter-laboratory validation trial, Mutagenesis, Vol. 25/2, 113-23.
(46) Azqueta, A. et al. (2011), Towards a more reliable comet assay: Optimising agarose concentration, unwinding time and electrophoresis conditions, Mutation Research, Vol. 724/1-2, 41-45.
(47) Hayashi, M. et al. (2011), Compilation and use of genetic toxicity historical control data, Mutation Research, Vol. 723/2, 87-90.
(48) Ryan, T. P. (2000), Statistical Methods for Quality Improvement, John Wiley and Sons, New York2nd ed.
(49) Appendix A of the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (ETS No. 123)
(50) Kapitel B.8 dieses Anhangs: Prüfung auf subakute Toxizität nach Inhalation: 28-Tage-Test.
(51) Kapitel B.29 dieses Anhangs: Prüfung auf subchronische Toxizität nach Inhalation: 90-Tage-Test.
(52) Blakey, D.H., G.R. Douglas (1984), Transient DNA lesions induced by benzo[a]pyrene in Chinese hamster ovary cells, Mutation Research, Vol. 140/2-3, 141-45.
(53) Blakey, D.H., G.R. Douglas (1990), The role of excision repair in the removal of transient benzo[a]pyrene-induced DNA lesions in Chinese hamster ovary cells, Mutation Research, Vol. 236/1, 35-41.
(54) OECD (2002), "Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation", OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, No. 19, OECD Publishing, Paris.
(55) Nakajima, M. (2012), Tissue sample preparation for in vivo rodent alkaline Comet assay, Genes and Environment, Vol. 34/1, 50-4.
(56) Hartmann, A. et al. (2003), Recommendations for conducting the in vivo alkaline Comet assay, Mutagenesis, Vol.18/1, 45-51.
(57) Atlas of Comet Assay Images, Scientist Press Co., Ltd., Tokio, Japan.
(58) Lovell, D.P., G. Thomas, R. Dubow (1999), Issues related to the experimental design and subsequent statistical analysis of in vivo and in vitro Comet studies, Teratogenesis Carcinogenesis Mutagenesis, Vol. 19/2, 109-19.
(59) Wiklund, S.J., E. Agurell (2003), Aspects of design and statistical analysis in the Comet assay, Mutagenesis, Vol. 18/2, 167-75.
(60) Bright, J. et al. (2011), Recommendations on the statistical analysis of the Comet assay, Pharmaceutical Statistics, Vol. 10/6, 485-93.
(61) Lovell, D.P., T. Omori (2008), Statistical issues in the use of the Comet assay, Mutagenesis, Vol. 23/3, 171-82.
.
| Definitionen | Anlage 1 |
Alkalische Einzelzell-Gelelektrophorese: Empfindliches Verfahren zum Nachweis primärer DNA-Schädigungen auf Einzelzell-/Zellkernebene.
Chemikalie: Stoff oder Gemisch.
Comet: Kometähnliche Form, die Nucleoide nach Einwirkung eines elektrophoretischen Felds annehmen: Der Kopf ist der Zellkern, und der Schweif wird durch die DNA gebildet, die aufgrund des elektrischen Felds aus dem Zellkern gewandert ist.
Kritische Variable/kritischer Parameter: Dies ist eine Protokollvariable, bei der sich eine kleine Änderung erheblich auf die Schlussfolgerung des Tests auswirken kann. Kritische Variablen können gewebespezifisch sein. Kritische Variablen sollten (insbesondere innerhalb eines Tests) nicht geändert werden, ohne zu prüfen, wie sich die Änderung auf die Assay-Reaktion auswirkt, wie beispielsweise durch die Größenordnung und Variabilität bei den Positiv- und Negativkontrollen angedeutet. Im Prüfbericht sollten Änderungen an kritischen Variablen, die während des Tests oder gegenüber dem Standardprotokoll des Labors vorgenommen wurden, angegeben und einzeln begründet werden.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Schweif-Intensität oder % DNA im Schweif: Dies entspricht der Intensität des Comet-Schweifs bezogen auf die Gesamtintensität (Kopf plus Schweif) und spiegelt den als Prozentsatz ausgedrückten Anteil der DNA-Schädigung wider.
UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
.
| Faktorieller Versuchsplan zur Ermittlung geschlechtsspezifischer Unterschiede beim In-vivo-Comet-Assay | Anlage 2 |
Faktorieller Versuchsplan und Analyse
Bei diesem Versuchsplan werden mindestens fünf männliche und fünf weibliche Tiere je Konzentration getestet. Dies ergibt einen Versuch, bei dem mindestens 40 Tiere verwendet werden (20 männliche und 20 weibliche zzgl. entsprechender Positivkontrollen).
Der Versuchsaufbau, der zu den einfacheren faktoriellen Versuchsplänen zählt, entspricht einer zweifaktoriellen Varianzanalyse, wobei Geschlecht und Konzentration die Hauptfaktoren sind. Die Daten können anhand vieler Standard-Statistiksoftwareanwendungen wie SPSS, SAS, STATA oder Genstat oder auch mit R analysiert werden.
Bei der Analyse wird die Varianz im Datensatz in die zwischen den Geschlechtern, die zwischen den Konzentrationen und die in Zusammenhang mit den Interaktionen zwischen den Geschlechtern und Konzentrationen partinioniert. Jeder der Terme wird anhand eines Schätzwerts der Varianz zwischen den Replikatversuchstieren innerhalb der gleichgeschlechtlichen Tiergruppen überprüft, die die gleiche Konzentration erhalten haben. Die zugrundeliegende Methodik ist in vielen Standardwerken zur Statistik (siehe Referenzdokumente) und in den in Statistiksoftware-Pakten mitgelieferten Hilfe-Funktionen im Einzelnen beschrieben.
Die Analyse erfolgt durch Prüfung des Terms der Interaktion "Geschlecht x Konzentration" in der ANOVA-Tabelle 1. Liegt kein signifikanter Term für die Interaktion vor, ergeben die kombinierten Werte für beide Geschlechter oder mehrere Konzentrationen eine zulässige Grundlage für statistische Tests zwischen den Konzentrationen aufgrund des Terms der ANOVA für die Varianz der innerhalb eines solchen Gruppe zusammengefassten Werte.
Die Analyse wird fortgesetzt mit der Partitionierung der geschätzten Varianz zwischen den Konzentrationen in kontrastierende Klassen, die die Grundlage bilden für einen Test auf lineare und quadratische Abhängigkeit der Reaktionen bei den verschiedenen Konzentrationen. Liegt dagegen eine signifikante Interaktion "Geschlecht x Konzentration" vor, so kann dieser Term auch in Gegenüberstellung der Interaktion "linear x Geschlecht" und "quadratisch x Geschlecht" partitioniert werden. Anhand dieser Terme kann geprüft werden, ob die Reaktionen auf die Konzentrationen bei beiden Geschlechtern parallel verlaufen oder ob die Geschlechter unterschiedlich reagieren.
Der Schätzwert für die Varianz der innerhalb der Gruppen zusammengefassten Werte kann als Grundlage für paarweise Tests zu Abweichungen zwischen den Mittelwerten dienen. Diese Vergleiche könnten zwischen den Mittelwerten für die beiden Geschlechter und zwischen den Mittelwerten für die verschiedenen Konzentrationen durchgeführt werden, beispielsweise um einen Vergleich mit den Negativkontrollen vorzunehmen. Bei signifikanter Interaktion können die Mittelwerte verschiedener Konzentrationen innerhalb eines Geschlechts oder die Mittelwerte beider Geschlechter bei derselben Konzentration verglichen werden.
Referenzdokumente
Es sind viele Werke zur Statistik erhältlich, in denen die Theorie, der Aufbau, die Methodik, die Analyse und die Interpretation faktorieller Versuchspläne erörtert werden, von einfachen Zweifaktorenanalysen bis hin zu komplexeren Formen, wie sie in der "Design of Experiment"-Methode verwendet werden. Die folgende Auflistung ist nicht erschöpfend. Einige Bücher enthalten Beispiele für die Anwendung vergleichbarer Versuchspläne, in einigen Fällen auch mit einem Code zur Durchführung der Analysen unter Verwendung verschiedener Softwarepakete.
(1) Box, G.E.P, Hunter, W.G. and Hunter, J.S. (1978). Statistics for Experimenters. An Introduction to Design, Data Analysis, and Model Building. New York: John Wiley & Sons.
(2) Box G.E.P. & Draper, N.R. (1987) Empirical model-building and response surfaces. John Wiley & Sons Inc.
(3) Doncaster, C.P. & Davey, A.J.H. (2007) Analysis of Variance and Covariance: How to choose and Construct Models for the Life Sciences. Cambridge University Press.
(4) Mead, R. (1990) The Design of Experiments. Statistical principles for practical application. Cambridge University Press.
(5) Montgomery D.C. (1997) Design and Analysis of Experiments. John Wiley & Sons Inc.
(6) Winer, B.J. (1971) Statistical Principles in Experimental Design. McGraw Hill.
(7) Wu, C.F.J & Hamada, M.S. (2009) Experiments: Planning, Analysis and Optimization. John Wiley & Sons Inc.
_____
1) Statistiker, die mit einem Modellierungsansatz wie dem Ansatz der allgemeinen linearen Modelle (GLM) arbeiten, folgen bei der Analyse möglicherweise einem anderen, wenn auch vergleichbarem Ansatz, werden jedoch nicht notwendigerweise eine Herleitung der herkömmlichen Anova-Tabelle vornehmen, die auf algorithmische Lösungswege für statistische Berechnungen aus dem Vor-Computerzeitalter zurückgeht.
.
| Gegenwärtige Einsatzgrenzen des Tests | Anlage 3 |
Nach aktuellem Kenntnisstand sind mit dem In-vivo-Comet-Assay verschiedene Einsatzgrenzen verbunden. Es wird davon ausgegangen, dass diese Einsatzgrenzen in dem Maße verringert oder enger definiert werden, wie zunehmende Erfahrungen mit der Anwendung des Tests zur Klärung von Sicherheitsfragen in einem regulatorischen Umfeld beitragen.
1. Einige Arten von DNA-Schädigungen können kurzlebig sein, d. h. zu rasch repariert werden, um 24 Stunden oder noch später nach der letzten Dosis beobachtet werden zu können. Es liegt weder eine Liste der Arten von kurzlebigen Schädigungen noch der Chemikalien vor, die diese Art von Schädigung wahrscheinlich auslösen, und es ist zudem nicht bekannt, über welchen Zeitraum diese Art von Schädigung nachweisbar ist. Die optimalen Probenahmezeitpunkte können außerdem spezifisch für die jeweilige Chemikalie oder den Verabreichungsweg sein und sollten auf kinetischen Daten (z.B. Zeitpunkt Tmax, zu dem die höchste Plasma- oder Gewebekonzentration erreicht wird) basieren, falls solche Daten vorliegen. In den meisten Validierungsstudien, auf die sich diese Prüfmethode stützt, wurde eine Sektion 2 oder 3 Stunden nach Verabreichung der letzten Dosis angegeben. In der veröffentlichten Literatur wird beschrieben, dass bei den meisten Studien die letzte Dosis 2 bis 6 Stunden vor der Tötung verabreicht wurde. Daher wurden diese Versuche als Grundlage für die Empfehlung in der Prüfmethode verwendet, wonach in Ermangelung von Daten, die eine andere Vorgehensweise nahelegen, die letzte Dosis zu einem festgelegten Zeitpunkt zwischen 2 und 6 Stunden vor der Sektion verabreicht werden sollte.
2. Es gibt keine validierten Daten zur Empfindlichkeit des Versuchs in Bezug auf den Nachweis kurzlebiger DNA-Schädigungen nach Verabreichung im Futter oder Trinkwasser im Vergleich zur Verabreichung über eine Magensonde. DNA-Schädigungen nach Verabreichung im Futter oder Trinkwasser wurden zwar nachgewiesen, aber es gibt nur wenige solcher Berichte verglichen mit der weitaus größeren Erfahrung mit Magensonden und intraperitonealer Verabreichung. Somit kann die Empfindlichkeit des Tests bei Chemikalien, die bei Verabreichung im Futter oder Trinkwasser kurzlebige Schädigungen auslösen, reduziert sein.
3. Da keine Interlaborstudien an anderen Geweben als Leber und Magen durchgeführt wurden, gibt es keine Empfehlung dazu, wie eine empfindliche und reproduzierbare Reaktion in anderen Geweben als Leber, wie z.B. zu erwartende Bereiche bei Positiv- und Negativkontrollen, zu erreichen ist. Für die Leber konnte ferner keine Einigung über die Festlegung einer niedrigeren Grenze für den Negativkontrollwert erzielt werden.
4. Obwohl in verschiedenen Veröffentlichungen die unklare zytotoxische Wirkung in vitro aufgezeigt wurde, wurden nur sehr wenige In-vivo-Daten veröffentlicht, sodass keine einzelne Messgröße für die Zytotoxizität empfohlen werden konnte. Histopathologische Veränderungen wie Entzündung, Zellinfiltration, apoptotische oder nekrotische Veränderungen wurden mit einer Zunahme der DNA-Wanderung in Verbindung gebracht. Wie die JaCVAM-Validierungsstudie (OECD, 2014) gezeigt hat, führen diese Veränderungen nicht immer zu positiven Comet-Ergebnissen, sodass keine endgültige Liste der stets mit einer Zunahme der DNA-Wanderung einhergehenden, histopathologischen Veränderungen vorliegt. Es wurde vorgeschlagen, Hedgehogs (oder Clouds, Geisterzellen) als Indikator für Zytotoxizität zu verwenden, doch die Genese der Hedgehogs ist unklar. Es liegen Daten vor, die darauf hindeuten, dass sie durch Zytotoxizität der Chemikalien, mechanisch/enzyminduzierte Schäden, die während der Aufbereitung der Proben hervorgerufen werden (Guerard et al., 2014), und/oder eine extremere Wirkung der Genotoxizität der Prüfchemikalie verursacht werden können. Andere Daten scheinen darauf hinzudeuten, dass sie auf umfangreiche, aber vielleicht reparierbare DNA-Schädigungen zurückzuführen sind (Lorenzo et al., 2013).
5. Gewebe oder Zellkerne wurden erfolgreich für eine spätere Analyse eingefroren. Dies führt gewöhnlich zu einem messbaren Effekt bei der Reaktion auf die Vehikel- und Positivkontrolle (Recio et al., 2010; Recio et al., 2012; Jackson et al., 2013). Wenn das Labor dieses Verfahren anwendet, sollte es seine Kompetenz bei Einfriermethoden nachweisen und belegen, dass die Bereiche der prozentualen DNA-Anteile im Schweif in den Zielgeweben bei den mit dem Vehikel behandelten Tieren ausreichend niedrig sind und positive Reaktionen noch nachweisbar sind. In der Literatur wurden verschiedene Methoden für das Einfrieren von Gewebe beschrieben. Allerdings besteht kein Einvernehmen darüber, wie Gewebe am besten einzufrieren und aufzutauen sind und wie zu bewerten ist, ob eine potenziell geänderte Wirkung die Empfindlichkeit des Tests beeinträchtigen kann.
6. Neuere Arbeiten zeigen, dass davon auszugehen ist, dass die Liste der kritischen Variablen weiter verkürzt wird und die Parameter für kritische Variablen näher definiert werden (Guerard et al., 2014).
Referenzdokumente
(1) Guerard, M., C. Marchand, U. Plappert-Helbig (2014), Influence of Experimental Conditions on Data Variability in the Liver Comet Assay, Environmental and Molecular Mutagenesis, Vol. 55/2, pp. 114-21.
(2) Jackson, P. et al. (2013), Validation of use of frozen tissues in high-throughput comet assay with fully-automatic scoring, Mutagenesis, Vol. 28/6, pp. 699-707.
(3) Lorenzo, Y. et al. (2013), The comet assay, DNA damage, DNA repair and cytotoxicity: hedgehogs are not always dead, Mutagenesis, Vol. 28/4, pp. 427-32.
(4) OECD (2014), Reports of the JaCVAM initiative international pre-validation and validation studies of the in vivo rodent alkaline comet assay for the detection of genotoxic carcinogens, Series on Testing and Assessment, Nos. 195 and 196, OECD Publishing, Paris.
(5) Recio L, Hobbs C, Caspary W, Witt KL, (2010), Dose-response assessment of four genotoxic chemicals in a combined mouse and rat micronucleus (MN) and Comet assay protocol, J. Toxicol. Sci. 35:149-62.
(6) Recio, L. et al. (2012), Comparison of Comet assay dose-response for ethyl methanesulfonate using freshly prepared versus cryopreserved tissues, Environmental and Molecular Mutagenesis, Vol. 53/2, pp. 101-13."
(16) In Teil C wird Kapitel C.13 durch Folgendes ersetzt:
"C.13 Bioakkumulationsprüfung am Fisch mit aquatischer Exposition und Exposition über das Futter
Einleitung
Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie 305 (2012). Mit ihrer Überarbeitung werden im Wesentlichen zwei Ziele verfolgt. Erstens soll ein Test auf Bioakkumulation infolge der Aufnahme über das Futter 1 einbezogen werden, der für die Bestimmung des Bioakkumulationspotenzials von Stoffen mit sehr niedriger Wasserlöslichkeit geeignet ist. Zweitens soll eine Prüfmethode bereitgestellt werden, bei der aus Tierschutzgründen gegebenenfalls weniger Fische verwendet werden und die somit kostengünstiger ist.
In den Jahren seit der Verabschiedung der konsolidierten Prüfmethode C.13 (1) wurden zahlreiche Stoffe geprüft und von Laboratorien und Regulierungsbehörden umfangreiche Erfahrungen gesammelt. Dies hat zu der Überzeugung geführt, dass die Komplexität des Versuchs verringert werden kann, wenn bestimmte Kriterien erfüllt sind (siehe Nummer 88), und dass ein gestufter Ansatz möglich ist. Die Erfahrung hat gezeigt, dass biologische Faktoren wie Wachstum und Lipidgehalt von Fischen die Ergebnisse erheblich beeinflussen können und berücksichtigt werden sollten. Darüber hinaus wurde erkannt, dass die Prüfung von Stoffen mit sehr geringer Wasserlöslichkeit technisch nicht durchführbar ist. Zudem kann bei Stoffen mit sehr geringer Wasserlöslichkeit im aquatischen Milieu die aquatische Exposition im Vergleich zur Exposition über das Futter geringfügiger sein. Dies hat zur Entwicklung einer Prüfmethode geführt, bei der die Fische über das Futter exponiert werden (siehe Nummern 7-14 und Nummer 97 ff.). Die Prüfung mit Exposition über das Futter wurde 2010 validiert (Ringtest) (51).
Die wichtigsten Änderungen beinhalten Folgendes:
- Die Prüfung nur einer Prüfkonzentration gilt als ausreichend, wenn davon ausgegangen werden kann, dass der Biokonzentrationsfaktor (bioconcentration factor, BCF) von der Prüfkonzentration unabhängig ist.
- Es kann ein Versuch mit minimaler aquatischer Exposition und weniger Probenahmezeitpunkten ins Auge gefasst werden, sofern bestimmte Kriterien erfüllt sind.
- Der Lipidgehalt der Fische sollte gemessen werden, damit der BCF auf Basis eines Lipidgehalts von 5 % ausgedrückt werden kann.
- Neben der Schätzung des BCF im stationären Zustand (steady state) wird der Schwerpunkt verstärkt auf die Schätzung des kinetischen BCF (soweit möglich) gelegt.
- Für bestimmte Stoffgruppen wird die Prüfung mit Exposition über das Futter vorgeschlagen, wenn diese Art der Prüfung im Vergleich zur Prüfung mit aquatischer Exposition als geeigneter gilt.
- Das Fischgewicht sollte bestimmt werden, damit der BCFk um den Effekt der Verdünnung durch Wachstum korrigiert werden kann.
Vor der Durchführung eines Bioakkumulationstests sollte Folgendes über die Prüfchemikalie bekannt sein:
- Empfindlichkeit der Analysetechnik zwecks Messung der Gewebe- sowie der Wasser- und Futterkonzentrationen sowohl für den Prüfstoff als auch für mögliche Metaboliten (siehe Nummer 65).
- Löslichkeit in Wasser [PM A.6; (2)]; diese sollte nach einer Methode bestimmt werden, die für den (geschätzten) Löslichkeitsbereich geeignet ist, um einen zuverlässigen Wert zu erhalten. Bei hydrophoben Stoffen ist dies im Allgemeinen die Säulen-Elutions-Methode.
- n-Octanol/Wasser-Verteilungskoeffizient, KOW 2 [PM A.8 (4), A.24 (5), A.23 (6)]; oder andere geeignete Informationen über das Verteilungsverhalten (z.B. Sorption an Lipiden, KOC); dieses sollte nach einer Methode bestimmt werden, die für den Bereich von KOW (Schätzwert) geeignet ist, um einen zuverlässigen Wert zu erhalten. Bei hydrophoben Stoffen ist dies im Allgemeinen die Prüfung unter langsamem Rühren [TM A.23 (6)];
- Stabilität des Stoffs in Wasser (Hydrolyse [TM C.7 (7)]);
- Stabilität des Stoffs im Futter (insbesondere wenn ein Prüfungsansatz mit Exposition über das Futter gewählt wird);
- Informationen über die Phototransformation, die für die Bestrahlungsbedingungen der Prüfung relevant sind (8);
- Oberflächenspannung (z.B. bei Stoffen, bei denen log KOW nicht bestimmt werden kann) [TM A.5 (9)];
- Dampfdruck [TM A.4 (10)];
- Etwaige Informationen über den biotischen oder abiotischen Abbau in Wasser, z.B. über leichte biologische Abbaubarkeit [PM C.4 Teile II bis VII (11), PM C.29 (12)], soweit zutreffend;
- Informationen über Metaboliten: Struktur, log KOW, Bildung und Abbaubarkeit, falls zutreffend;
- Säuredissoziationskonstante (pKa) für Stoffe, die ionisieren könnten. Der pH-Wert des Prüfwassers sollte angepasst werden, um sicherzustellen, dass der Stoff in ionisierter Form verwendet wird, sofern dies mit der verwendeten Fischart vereinbar ist.
Diese Prüfmethode beschreibt ein Verfahren zur Charakterisierung des Potenzials verschiedener Stoffe zur Bioakkumulation in Fischen, unabhängig von der gewählten Expositionsmethode oder dem gewählten Probenahmeschema. Obwohl Durchflusstests empfohlen werden, sind - sofern die Validitätskriterien erfüllt sind (siehe Nummern 24 und 113) - auch semistatische Methoden zulässig. Für die Exposition über das Futter ist zwar kein Durchflusssystem notwendig, um wässrige Konzentrationen des Prüfstoffs aufrechtzuerhalten, es trägt jedoch dazu bei, angemessene Konzentrationen gelösten Sauerstoffs aufrechtzuerhalten, das Wasser sauber zu halten und Einflussfaktoren wie Ausscheidungsprodukte zu beseitigen.
Ungeachtet der gewählten Methode enthält diese Prüfmethode genügend Einzelheiten für die Durchführung des Tests, räumt jedoch gleichzeitig genügend Spielraum für die Anpassung des Versuchsplans an die jeweiligen Laborgegebenheiten ein, auch für den Fall, dass die Prüfstoffe unterschiedliche Eigenschaften aufweisen. Die Prüfung bei aquatischer Exposition eignet sich am besten für stabile organische Stoffen mit log KOW-Werten zwischen 1,5 und 6,0 (13), kann aber auch bei stark hydrophoben Stoffen (mit log KOW > 6,0) angewendet werden, wenn eine stabile und vollständig gelöste Konzentration des Prüfstoffs in Wasser nachgewiesen werden kann. Kann keine stabile Konzentration des Prüfstoffs in Wasser nachgewiesen werden, wäre ein Versuch mit aquatischer Exposition ungeeignet und die Exposition müsste über das Fischfutter erfolgen (wenngleich Interpretation und Verwendung der Ergebnisse des futterbasierten Tests auch vom Rechtsrahmen abhängen können). Vorläufige Schätzwerte für den Biokonzentrationsfaktor (BCF, manchmal auch als KB bezeichnet) für organische Stoffe mit log KOW-Werten von bis zu 9,0 können nach der Gleichung von Bintein et al. (14) bestimmt werden. Die vorläufigen Schätzwerte für den Biokonzentrationsfaktor für derart stark hydrophoben Stoffe können höher sein als in Laborversuchen erwartete Steady-state-Biokonzentrationsfaktor (BCFSS), insbesondere, wenn für die vorläufige Schätzung ein einfaches lineares Modell angewendet wird. Zu den Parametern, die das Bioakkumulationspotenzial charakterisieren, gehören die Aufnahmekonstante (k1), die Ausscheidungskonstante (k2), der Steady-state-Biokonzentrationsfaktor (BCFSS), der kinetische Biokonzentrationsfaktor (BCFK) und der futterbezogene Biomagnifikationsfaktor (BMF) 1.
Radioaktiv markierte Prüfstoffe können die Analyse von Wasser-, Futter- und Fischproben erleichtern und für die Entscheidung, ob die Metaboliten identifiziert und quantifiziert werden sollten, herangezogen werden. Wenn nur der Gesamtgehalt an radioaktiven Rückständen gemessen wird (z.B. durch Verbrennung oder Solubilisierung von Gewebe), wird der BCF oder der BMF anhand des Gesamtwerts des Ausgangsstoffes, etwa verbliebener Stoffwechselprodukte und auch des assimilierten Kohlenstoffs bestimmt. BCF- oder BMF-Werte, die auf Basis des Gesamtgehalts an radioaktiven Rückständen ermittelt werden, sind daher nicht direkt mit einem BCF oder BMF vergleichbar, der nur aus der spezifischen chemischen Analyse des Ausgangsstoffes abgeleitet wurde. Trennungsverfahren wie TLC, HPLC oder GC 3 können vor der Analyse in Studien mit radioaktiver Markierung angewendet werden, um den BCF oder BMF anhand des Ausgangsstoffes zu bestimmen. Bei Anwendung von Trennungsverfahren sollten der Ausgangsstoff und die relevanten Stoffwechselprodukte identifiziert und quantifiziert werden 4 (siehe Nummer 65), wenn der BCF oder BMF anhand der Konzentration des Ausgangstoffes in den Fischen und nicht Gesamtgehalts an radioaktiv markierten Rückständen berechnet werden soll. Auch ist es aufgrund der Analyse und Identifizierung der Rückstände in den Geweben möglich, Untersuchungen des Fischstoffwechsels oder In-vivo-Verteilungsstudien mit einer Bioakkumulationsstudie zu kombinieren. Die Möglichkeit einer Metabolismus-Studie lässt sich mithilfe geeigneter Instrumente (z.B. der OECD QSAR Toolbox (15) und QSAR-spezifischer Programme) vorherbestimmen.
Die Entscheidung, ob und mit welchem Versuchsaufbau eine Prüfung mit aquatischer Exposition oder mit Exposition über das Futter durchgeführt werden soll, sollte unter Berücksichtigung der Faktoren gemäß Nummer Punkt 3 und der geltenden Rechtvorschriften getroffen werden. Für Stoffe beispielsweise, die einen hohen log KOW aufweisen, bei denen aufgrund der Empfindlichkeit verfügbarer Analyseverfahren aber dennoch eine gute Wasserlöslichkeit nachweisbar ist, sollte in erster Linie eine Prüfung mit aquatischer Exposition in Erwägung gezogen werden. Es kann allerdings sein, dass die Informationen über die Wasserlöslichkeit für diese hydrophoben Arten von Stoffen nicht endgültig sind, weshalb vor der Entscheidung über die anzuwendende Methode untersucht werden sollte, ob stabile und messbare wasserlösliche Konzentrationen (stabile Emulsionen sind nicht zulässig), die für eine Prüfung mit aquatischer Exposition geeignet sind, hergestellt werden können (16). Auf der Grundlage der Ausschlusskriterien "Wasserlöslichkeit" und "Octanol/Wasser-Verteilungskoeffizient" lässt sich keine genauen Anweisung für die anzuwendende Methode geben, da andere Faktoren (wie Analyseverfahren, Abbau, Adsorption usw.) aus den oben genannten Gründen einen erheblichen Einfluss auf die Anwendbarkeit der Methode haben können. Bei Stoffen mit einem log KOW-Wert von über 5 und einer Wasserlöslichkeit von unter ~0,01-0,1 mg/l wird die Prüfung mit aquatischer Exposition jedoch möglicherweise immer schwieriger.
Auch andere Faktoren, die die Wahl der Prüfmethode beeinflussen können, sollten geprüft werden, wie das Potenzial des Stoffes zur Anlagerung an Prüfgefäße und Apparaturen, seine Stabilität in wässriger Lösung im Vergleich zur Stabilität im Futter (17) (18) usw.
Informationen über diese praktischen Aspekte lassen sich möglicherweise auch aus anderen abgeschlossenen Studien im Wassermilieu herleiten. Weitere Informationen über die Prüfung von Aspekten im Zusammenhang mit der Durchführung von Bioakkumulationsstudien sind in der Fachliteratur verfügbar (z.B. (19)).
Bei Stoffen, bei denen die Löslichkeit oder die Aufrechterhaltung der wässrigen Konzentration sowie die Analyse dieser Konzentrationen keine Einschränkungen für die Durchführung der Methode mit aquatischer Exposition darstellen, ist diese Methode für die Bestimmung des Biokonzentrationspotenzials des Stoffes zu bevorzugen. Es sollte in jedem Fall sichergestellt werden, dass die zu verwendende(n) Expositions-konzentration(en) den Kriterien für die Wasserlöslichkeit in den Prüfmedien genügt (genügen). Für die Aufrechterhaltung stabiler Konzentrationen des gelösten Prüfstoffes sind verschiedene Methoden denkbar, z.B. die Verwendung von Stammlösungen oder passive Dosierung (z.B. nach der Säulen-Elutions-Methode), sofern nachgewiesen werden kann, dass stabile Konzentrationen aufrechterhalten werden können und die Prüfmedien nicht von den Empfehlungen unter Nummer 27 abweichen.
Bei stark hydrophoben Stoffen (log KOW > 5 und Löslichkeit unter ~ 0,01-0,1 mg/l) kann sich die Prüfung mit aquatischer Exposition als zunehmend schwierig erweisen. Gründe hierfür können sein, dass die wässrige Konzentration nicht auf einem Wert gehalten werden kann, der als hinreichend konstant erachtet wird (z.B. aufgrund der Sorption am Glas der Prüfgefäße oder rascher Aufnahme durch die Fische), oder dass die zu verwendenden wässrigen Konzentrationen so gering sind, dass sie innerhalb der Größenordnung der analytischen Quantifizierungsgrenze oder darunter liegen 5. Für diese stark hydrophoben Stoffe wird die Prüfung mit Exposition über das Futter empfohlen, vorausgesetzt, sie entspricht den einschlägigen Rahmenvorschriften und dem Erfordernis der Risikobewertung.
Bei Tensiden sollte geprüft werden, ob - angesichts der Eigenschaften des Stoffes - der Biokonzentrationstest im aquatischen Milieu durchführbar ist; ansonsten wäre die Prüfung mit Exposition über das Futter geeigneter. Tenside sind oberflächenaktive Stoffe, die die Grenzflächenspannung zwischen zwei Flüssigkeiten verringern. Aufgrund ihres amphiphilen Charakters (d. h. sie enthalten sowohl einen hydrophilen als auch einen hydrophoben Teil) akkumulieren sie an Grenzflächen wie der Wasser/Luft-Grenzfläche, der Wasser/Futter-Grenzfläche und Glaswänden, was die Bestimmung ihrer Konzentration im Wassermilieu behindert.
Bei der Prüfung mit Exposition über das Futter können einige der bei komplexen Gemischen mit Komponenten unterschiedlicher Wasserlöslichkeitsgrenzen auftretenden Expositionsprobleme insoweit umgangen werden, als eine vergleichbare Exposition aller Komponenten des Gemischs über das Futter wahrscheinlicher ist als über das Wasser (siehe (20)).
Es ist zu beachten, dass die Methode mit Exposition über das Futter einen futterbezogenen Biomagnifikationsfaktor (BMF) und nicht etwa einen Biokonzentrationsfaktor (BCF) 1 ergibt. Es existieren Ansätze zur Schätzung eines kinetischen Biokonzentrationsfaktors (BCFK) anhand von Daten aus dem Versuch mit Exposition über das Futter (siehe Anlage 8), jedoch sollten diese mit Vorsicht angewendet werden, denn sie setzen im Allgemeinen eine Kinetik erster Ordnung voraus und sind nur auf bestimmte Gruppen von Verbindungen anwendbar. Auf Tenside lassen sie sich wahrscheinlich nicht anwenden (siehe Nummer 12).
Ein Versuch mit minimaler aquatischer Exposition und weniger Probenahmezeitpunkten, der es gestattet, die Zahl der erforderlichen Versuchstiere und/oder Ressourcen (siehe Nummer 83 ff.) zu begrenzen, sollte nur bei Stoffen angewendet werden, bei denen mit gutem Grund davon ausgegangen werden kann, dass Aufnahme und Ausscheidung ungefähr nach dem Schema der Kinetik erster Ordnung ablaufen (also eher bei nicht ionisierten organischen Stoffen, siehe Nummer 88).
_____
1) Für Definitionen und Einheiten siehe Anlage 1.
2) Manchmal als POW bezeichnet; wird nach einer Schüttelmethode gemäß PM A.8 (4), einer HPLC-Methode gemäß PM A.24 (5) und einer Methode mit langsamem Rühren gemäß PM A.23 (6) bestimmt. Gelegentlich findet die Generatorsäulenmethode (Generator-Column-Methode) zur Bestimmung von log KOW Anwendung. Es gibt eine begrenzte Anzahl von Studien, bei denen diese Methode angewandt wird, überwiegend für chlorierte Biphenyle und Dibenzodioxine (z.B. Li und Doucette, 1993) (3). Bei Stoffen, die ionisieren könnten, sollte sich log KOW auf die nicht ionisierte Form beziehen.
3) TLC: Thin Layer Chromatography (Dünnschichtchromatographie); HPLC: High Pressure Liquid Chromatography (Hochdruckflüssigkeitschromatografie); GC: Gas Chromatography (Gaschromatografie)
4) Bestimmte Rahmenvorschriften geben die Analyse von Metaboliten möglicherweise vor, wenn bestimmte Bedingungen vorliegen (siehe Nummer 65).
5) Im Allgemeinen sollten die während der Aufnahmephase in Wasser gemessenen Konzentrationen mindestens eine Zehnerpotenz über der Quantifizierungsgrenze liegen, sodass in der Ausscheidungsphase der Studie mehr als eine Halbwertzeit der Körperbelastung gemessen werden kann.
C.13 - I: Biokonzentrationsprüfung am Fisch mit aquatischer Exposition
Testprinzip
Der Test besteht aus zwei Phasen: der Expositionsphase (oder Aufnahmephase) und der Post-Expositionsphase (oder Ausscheidungsphase). Während der Aufnahmephase wird eine Gruppe von Fischen ein und derselben Spezies je nach Eigenschaften des Prüfstoffes einer oder mehreren ausgewählten Prüfstoffkonzentrationen ausgesetzt (siehe Nummer 49). Sie werden anschließend für die Ausscheidungsphase in ein Medium ohne Prüfstoff eingesetzt. Eine Ausscheidungsphase ist immer erforderlich, es sei denn, die Aufnahme des Stoffes während der Aufnahmephase war unbedeutend. Die Konzentration des Prüfstoffes in/auf den Fischen (oder bestimmten Gewebeteilen von Fischen) wird in beiden Testphasen beobachtet. Zusätzlich zu der behandelten Gruppe wird eine Kontrollgruppe von Fischen unter - abgesehen vom fehlenden Prüfstoff - gleichen Bedingungen gehalten, um eventuelle schädigende Wirkungen, die während der Biokonzentrationsprüfung beobachtet werden, mit einer Kontrollgruppe vergleichen zu können und um Hintergrundkonzentrationen des Prüfstoffs zu erfahren 1.
Bei der Prüfung mit aquatischer Exposition dauert die Aufnahmephase in der Regel 28 Tage. Sie kann ggf. verlängert (siehe Nummer 18) oder verkürzt werden, wenn nachgewiesen wird, dass schon zu einem früheren Zeitpunkt ein stationärer Zustand (steady state) erreicht wird (für Definitionen und Einheiten siehe Anlage 1). Die Dauer der Aufnahmephase und die Zeit bis zum Erreichen des stationären Zustands kann anhand der Gleichungen in Anlage 5 vorausgeschätzt werden. Die Ausscheidungsphase beginnt, wenn die Fische dem Prüfstoff nicht länger ausgesetzt sind, d. h. mit der Umsetzung der Tiere in ein sauberes Gefäß, das ein bis auf den Prüfstoff identisches Medium enthält. Wenn möglich, sollte der Biokonzentrationsfaktor sowohl als Quotient der Konzentration in den Fischen (Cf) und im Wasser (Cw) bei stationärem Zustand (BCFSS; für die Definition siehe Anlage 1) berechnet werden, als auch als kinetischer Biokonzentrationsfaktor BCFK; für Definitionen und Einheiten siehe Anlage 1), der geschätzt wird als Quotient der Aufnahme- (k1) und der Ausscheidungskonstante (k2), wobei eine Kinetik erster Ordnung vorausgesetzt wird 2.
Wird innerhalb von 28 Tagen kein stationärer Zustand (steady state) erreicht, wird der BCF entweder nach dem kinetischen Ansatz (siehe Nummer 38) berechnet oder die Aufnahmephase kann verlängert werden. Dauert die Aufnahmephase bis zum Erreichen des stationären Zustands unangemessen lange (siehe Nummern 37 und 38, Anlage 5), ist der kinetische Ansatz zu bevorzugen. Alternativ sollte bei stark hydrophoben Stoffen eine Studie mit Exposition über das Futter in Betracht gezogen werden 3, vorausgesetzt, eine solche Studie läuft den einschlägigen Rahmenvorschriften nicht zuwider.
Die Aufnahmekonstante, die Ausscheidungskonstante (oder -konstanten, soweit komplexere Modelle verwendet werden), der Biokonzentrationsfaktor (steady-state und/oder kinetisch) und, wenn möglich, die Konfidenzgrenzen jedes einzelnen dieser Parameter werden nach dem Modell berechnet, das die gemessenen Konzentrationen des Prüfstoffs in den Fischen und im Wasser am besten beschreibt (siehe Anlage 5).
Die Zunahme der Fischmasse während der Prüfung führt zu einer Verringerung der Prüfstoffkonzentration in den wachsenden Fischen (sogenannte Verdünnung durch Wachstum), weshalb der kinetische BCF zu niedrig geschätzt wird, wenn er nicht um das Wachstum korrigiert wird (siehe Nummern 72 und 73).
Der BCF basiert auf der Gesamtkonzentration in den Fischen (d. h. auf dem Gesamt-nassgewicht der Fische). Für besondere Zwecke können jedoch auch bestimmte Gewebe oder Organe (z.B. Muskeln, Leber) verwendet werden, sofern die Fische groß genug sind, oder die Fische können in essbare (Filets) und nicht-essbare (Viszera) Fraktionen unterteilt werden. Da bei vielen organischen Stoffen ein eindeutiger Zusammenhang zwischen Biokonzentrationspotenzial und Hydrophobie besteht, gibt es entsprechend auch einen eindeutigen Zusammenhang zwischen dem Lipidgehalt der Versuchsfische und der beobachteten Biokonzentration derartiger Stoffe. Um derart variable Testergebnisse bei stark lipophilen Stoffen (d. h. Stoffen mit log KOW > 3) zu begrenzen, sollte die Biokonzentration zusätzlich zu dem aus der Studie direkt abgeleiteten Wert auf einen Fischlipidgehalt von 5 % (basierend auf dem Ganzkörpernassgewicht) standardisiert werden. Dies ist notwendig, um eine Grundlage für den Vergleich zwischen verschiedenen Stoffen und/oder Versuchsspezies zu ermöglichen. Ein Lipidgehalt von 5 % ist ein gängiger Wert, denn er entspricht dem durchschnittlichen Lipidgehalt von Fischen, die bei dieser Prüfmethode normalerweise verwendet werden (21).
Angaben zum Prüfstoff
Zusätzlich zu den in der Einleitung genannten Prüfstoffeigenschaften (Nummer 3) müssen auch Informationen über die toxische Wirkung auf die Versuchsspezies vorliegen - vorzugsweise der asymptotische (d. h. zeitunabhängige) LC50-Wert - und/oder Schätzwerte zur Toxizität aus Langzeitversuchen an Fischen (z.B. TM C.47 (22), C.15 (23), C.14 (24)).
Eine geeignete Analysemethode von bekannter Zuverlässigkeit, Genauigkeit und Empfindlichkeit sowie Einzelheiten der Probenvorbereitung und -aufbewahrung sollten für die Quantifizierung des Prüfstoffs in den Testlösungen und im biologischen Material verfügbar sein. Ebenso sollten die analytischen Nachweisgrenzen des Prüfstoffs in Wasser sowie in den Fischgeweben bekannt sein. Wird ein radioaktiv markierter Prüfstoff verwendet, sollte dieser von höchstmöglicher Reinheit (von möglichst > 98 %) sein; auch der Prozentanteil der auf Verunreinigungen zurückzuführenden Radioaktivität sollte bekannt sein.
Gültigkeit des Tests
Ein Test wird als gültig betrachtet, wenn folgende Bedingungen erfüllt sind:
Die Temperaturschwankung des Wassers beträgt weniger als ± 21 °C, denn große Schwankungen können sich auf die biologischen Parameter, die für die Aufnahme und Ausscheidung relevant sind, auswirken und bei den Tieren Stress auslösen;
die Konzentration an gelöstem Sauerstoff fällt nicht unter 60 % Sättigung;
die Konzentration des Prüfstoffs in den Prüfgefäßen wird auf dem während der Aufnahmephase gemessenen Durchschnittswert ± 20 % gehalten;
die Konzentration des Prüfstoffs liegt unter der Grenze seiner Löslichkeit in Wasser, wobei die potenzielle Wirkung des Prüfwassers auf die tatsächliche Löslichkeit zu berücksichtigen ist 4;
Mortalität oder andere Schadwirkungen/Krankheiten bei Kontroll- und Versuchsfischen betragen bei Prüfungsende weniger als 10 %; erstreckt sich die Prüfung über mehrere Wochen oder Monate, sollten Mortalität oder andere Schadwirkungen bei beiden Fischgruppen weniger als 5 % pro Monat betragen und insgesamt 30 % nicht überschreiten. Signifikante Unterschiede im durchschnittlichen Wachstum zwischen den Versuchs- und Kontrollgruppen könnten auf eine toxische Wirkung des Prüfstoffs hinweisen.
Referenzstoffe
Die Verwendung von Referenzstoffen mit bekanntem Biokonzentrationspotential und schwachem Metabolismus wäre zur Kontrolle des Versuchsablaufs möglicherweise sinnvoll (z.B. wenn ein Labor keine vorherige Prüfungserfahrung hat oder die Versuchsbedingungen geändert wurden).
Beschreibung der Prüfmethode
Apparatur
Für alle Teile der Apparatur sollten Materialien, die sich auflösen, sorbieren oder auslaugen und die Fische schädigen können, möglichst vermieden werden. Verwendet werden können rechteckige oder zylindrische Behälter aus chemisch inertem Material und mit besatzgerechtem Fassungsvermögen (siehe Nummer 43). Die Verwendung von Rohren aus Weichkunststoff sollte auf ein Minimum beschränkt werden. Rohre aus Polytetrafluorethylen, Edelstahl und/oder Glas sind zu bevorzugen. Die Erfahrung hat gezeigt, dass bei Prüfstoffen mit hohem Adsorptionskoeffizient, wie synthetischen Pyrethroiden, die Verwendung von silanisiertem Glas nötig sein kann. In solchen Fällen sollte die Apparatur nach der Benutzung entsorgt werden. Die Versuchssysteme sollten den in der Studie zu verwendenden Prüfstoffkonzentrationen möglichst so lange ausgesetzt werden, bis die Expositionskonzentrationen nachweislich stabil sind; erst dann sollten die Prüforganismen eingesetzt werden.
Wasser
Allgemein wird für den Test natürliches Wasser verwendet, das aus einer unverschmutzten Quelle gleichbleibend guter Qualität stammt. Rekonstituiertes Wasser (d. h. entmineralisiertes Wasser, dem spezifische Nährstoffe in bekannten Mengen zugegeben wurden) ist jedoch möglicherweise besser geeignet, um im Zeitverlauf gleichbleibend gute Qualität zu gewährleisten. Das Verdünnungswasser (d. h. das Wasser, das vor dem Einfüllen in das Prüfgefäß mit dem Prüfstoff gemischt wird; siehe Nummer 30) muss von einer Qualität sein, die ein Überleben der gewählten Versuchsspezies für die Dauer der Akklimatisations- und Prüfperiode ermöglicht, ohne dass diese ein abnormes Erscheinungsbild oder Verhalten zeigen. Im Idealfall sollte nachgewiesen werden, dass die Prüfspezies im Verdünnungswasser (z.B. in einer Laborkultur oder in einem Lebenszyklus-Toxizitätstest) überleben, wachsen und sich vermehren. Über das Verdünnungswasser sollten zumindest Angaben über pH-Wert, Härte, Gesamtfeststoffgehalt, gesamten organischen Kohlenstoff (TOC 5) und möglichst auch für Ammonium, Nitrit und Alkalität bzw. - für die Meerwasserspezies -Salinität vorliegen. Nicht alle Parameter, die für einen optimalen Schutz der Fische wichtig sind, sind bekannt, doch werden in Anlage 2 für eine Reihe von Parametern die empfohlenen Höchstkonzentrationen für Süß- und Meeresprüfwässer genannt.
Während der Prüfungsdauer sollte das Verdünnungswasser von gleichbleibender Qualität sein. Der pH-Wert sollte bei Prüfungsbeginn zwischen 6,0 und 8,5 liegen, doch während des Prüfungsablaufs sollte er um nicht mehr als ± 0,5 pH-Einheiten schwanken. Um sicherzugehen, dass das Verdünnungswasser das Prüfungsergebnis nicht übermäßig stark beeinflusst (beispielsweise durch Komplexierung des Prüfstoffs) oder schädigende Wirkungen auf den Fischbestand hat, sollten in gewissen Abständen, zumindest am Anfang und am Ende der Prüfung, Proben zur Analyse entnommen werden. Soweit das Verdünnungswasser bekanntermaßen von relativ konstanter Qualität ist, sollten alle drei Monate der Gehalt an Schwermetallen (z.B. Cu, Pb, Zn, Hg, Cd und Ni), an Hauptanionen und -kationen (z.B. Ca2+, Mg2+, Na+, K+, Cl- und SO42-), an Pestiziden (z.B. der Gesamtgehalt an phosphororganischen und chlororganischen Pestiziden), der TOC und die Schwebstoffe bestimmt werden. Hat sich die Wasserqualität über mindestens ein Jahr als konstant erwiesen, können diese Untersuchungen reduziert und in größeren Zeitabständen (z.B. alle sechs Monate) durchgeführt werden.
Der natürliche Partikelgehalt und der gesamte organische Kohlenstoff des Verdünnungswassers sollten möglichst niedrig sein, um Adsorption des Prüfstoffs an organische Stoffe zu vermeiden, weil deren Bioverfügbarkeit dadurch reduziert und somit der BCF zu niedrig geschätzt werden könnte. Der maximal zulässige Wert beträgt 5 mg/l für Partikel (Trockenstoff, der einen Filter von 0,45 µm nicht passiert) und 2 mg/l für den gesamten organischen Kohlenstoff (siehe Anlage 2). Gegebenenfalls sollte das Wasser vor der Verwendung gefiltert werden. Der Anteil von Fischausscheidungen und Futterresten am organischen Kohlenstoffgehalt des Wassers sollte so gering wie möglich sein (siehe Nummer 46).
Prüflösungen
Eine Stammlösung des Prüfstoffs wird in entsprechender Konzentration vorbereitet, vorzugsweise durch einfaches Mischen oder Einrühren des Prüfstoffs in das Verdünnungswasser. Als mögliche Alternative, die sich in bestimmten Fällen anbietet, kann ein Dosierungssystem mit Festphasen-Desorption verwendet werden. Die Verwendung von Lösungs- oder Dispergiermitteln (Lösungsvermittlern) wird im Allgemeinen nicht empfohlen (siehe (25)), auch wenn dies in bestimmten Fällen nötig sein sollte, um eine entsprechend konzentrierte Stammlösung herzustellen; doch sollte die Verwendung solcher Materialien auf ein Minimum beschränkt und deren kritische Mizellenkonzentration nicht überschritten werden (falls relevant). In Frage kommende Lösungsmittel sind Ethanol, Methanol, Dimethylformamid und Triethylenglykol. Geeignete Dispergiermittel sind Tween 80, Methylzellulose 0,01 % und HCO-40. Die Lösungsmittelkonzentration im endgültigen Prüfmedium sollte bei allen Behandlungen (ungeachtet der Prüfstoffkonzentration) identisch sein und die entsprechenden Toxizitätsschwellenwerte, die für das Lösungsmittel unter den Prüfungsbedingungen festgelegt wurden, sollten nicht überschritten werden. Die Höchstkonzentration beträgt 100 mg/l (oder 0,1 ml/l). Es ist unwahrscheinlich, dass eine Lösungsmittelkonzentration von 100 mg/l die gelöste Höchstkonzentration des Prüfstoffs, die in dem Medium erreicht werden kann, erheblich beeinflussen wird (25). Der Beitrag des Lösungsmittels (zusammen mit dem Prüfstoff) zum Gesamtgehalt des Prüfwasser an organischem Kohlenstoff sollte bekannt sein. Während der gesamten Prüfungsdauer sollte die Konzentration des gesamten organischen Kohlenstoffs in den Prüfgefäßen die Konzentration des organischen Kohlenstoffs aus dem Prüfstoff und, falls verwendet, dem Lösungsmittel oder Lösungsvermittler 6 um nicht mehr als 10 mg/l (± 20 %) überschreiten. Der Gehalt an organischen Stoffen kann bei Durchflussprüfungen an Fischen die Menge an frei gelöstem Prüfstoff erheblich beeinflussen, insbesondere bei lipophilen Stoffen. Festphasen-Mikroextraktion (siehe Nummer 60) kann wichtige Informationen über das Verhältnis zwischen gebundenen und frei gelösten Verbindungen liefern, wobei bei Letzteren davon ausgegangen wird, dass sie dem bioverfügbaren Teil entsprechen. Die Prüfstoffkonzentration sollte trotz Verwendung eines Lösungsmittels oder Lösungsvermittlers unter der Löslichkeitsgrenze des Prüfstoffs im Prüfmedium liegen. Bei Verwendung biologisch leicht abbaubarer Stoffe ist Vorsicht geboten, da diese im Durchflusstest Probleme in Form von Bakterienwachstum verursachen können. Falls eine Stammlösung nicht ohne Lösungsvermittler hergestellt werden kann, sollte die Eignung einer Prüfung mit aquatischer Exposition zugunsten einer Prüfung mit Exposition über das Futter überdacht werden.
Für Durchflussprüfungen ist ein System erforderlich, das kontinuierlich eine Stammlösung des Prüfstoffs abgibt und verdünnt (z.B. Dosierpumpe, Proportionalverdünner, Sättigungsvorrichtung), oder ein Dosierungssystem mit Festphasen-Desorption, um die Prüfkonzentrationen den Prüfkammern zuzuführen. Die Inhalte der Prüfkammern sollten täglich mindestens fünf Mal ausgetauscht werden, wobei das Durchflussverfahren zu bevorzugen ist. Ist dies nicht möglich (wenn z.B. schädigende Wirkungen auf die Prüforganismen zu erwarten sind), kann ein semistatisches Verfahren angewendet werden, sofern die Validitätskriterien erfüllt sind (siehe Nummer 24). Die Durchflussgeschwindigkeit der Stammlösungen und des Verdünnungswassers sollten 48 Stunden vor sowie einmal täglich während der Prüfung kontrolliert werden. Dabei sollte auch die Durchflussgeschwindigkeit für jede Prüfkammer bestimmt und sichergestellt werden, dass sie innerhalb oder zwischen den Kammern um nicht mehr als 20 % abweicht.
Auswahl der Spezies
Wichtig für die Auswahl der Spezies ist, dass die Tiere leicht und in geeigneter Größe erhältlich sind und problemlos im Labor gehalten werden können. Andere Kriterien zur Auswahl der Fischspezies sind u. a. ihr Freizeit-, Handels- und ökologischer Wert sowie eine vergleichbare Empfindlichkeit, ein erfolgreicher Einsatz in früheren Prüfungen usw. Die empfohlenen Versuchsspezies sind in Anlage 3 aufgeführt. Es können auch andere Spezies verwendet werden, doch muss das Prüfverfahren dann u. U. angepasst werden, um geeignete Prüfungsbedingungen zu schaffen. Die Gründe für die Auswahl der Spezies und der Versuchsmethode sollten in diesem Fall genau dokumentiert werden. Im Allgemeinen verkürzt sich durch die Verwendung kleinerer Fischspezies die Zeit bis zum Erreichen des stationären Zustands, jedoch werden mehr Fische (Proben) benötigt, um den Lipidgehalt und die Prüfstoffkonzentrationen in den Fischen angemessen zu analysieren. Außerdem können Unterschiede bei Atmungsrate und Metabolismus zwischen jungen und älteren Fischen Ergebnisvergleiche zwischen verschiedenen Prüfungen und Versuchsspezies erschweren. Es ist zu beachten, dass bei Fischarten, die in einer (juvenilen) wachstumsintensiven Lebensphase geprüft werden, die Interpretation der Daten schwierig sein kann.
Haltung der Fische (relevant bei aquatischer Exposition und Exposition über das Futter)
Die Stammpopulation der Fische sollte mindestens zwei Wochen lang im Wasser (siehe Nummer 28) bei Prüftemperatur eingewöhnt und ausreichend gefüttert werden (siehe Nummer 45). Es sollten Wasser und Futter derselben Art verwendet werden wie während der Prüfung.
Nach 48-stündiger Eingewöhnung werden die Mortalitäten erfasst; dabei wird wie folgt vorgegangen:
- bei einer Mortalität von mehr als 10 % der Population innerhalb von sieben Tagen: Austausch des gesamten Besatzes;
- bei einer Mortalität zwischen 5 und 10 % der Population innerhalb von sieben Tagen: weitere sieben Tage Akklimatisation - bei einer Mortalität innerhalb der folgenden sieben Tage von über 5 %: Austausch des gesamten Besatzes;
- bei einer Mortalität unter 5 % der Population innerhalb von sieben Tagen: Akzeptanz des gesamten Besatzes.
Die Versuchsfische sollten keine sichtbaren Erkrankungen oder Anormalitäten aufweisen. Alle kranken Fische sind zu entfernen. Die Fische sollten zwei Wochen vor und während der Prüfung nicht gegen Krankheiten behandelt werden.
Prüfungsablauf
Vorversuch
Es kann sinnvoll sein, einen Vorversuch durchzuführen, um die Bedingungen für den Hauptversuch zu optimieren, z.B. in Bezug auf die Wahl der Prüfstoffkonzentration(en), die Dauer der Aufnahme- und Ausscheidungsphase, oder um zu bestimmen, ob die Prüfung vollständig durchgeführt werden muss. Der Vorversuch sollte so aufgebaut sein, dass die erforderlichen Informationen ermittelt werden können. Es kann geprüft werden, ob ein Versuch unter minimalen Bedingungen für die Ableitung eines BCF ausreicht oder ob ein vollständiger Versuch notwendig ist (siehe Nummern 83-95 zum Versuch unter minimalen Bedingungen).
Expositionsbedingungen
Dauer der Aufnahmephase
Die Dauer der Aufnahmephase lässt sich anhand praktischer Erfahrungen (z.B. aus einer früheren Untersuchung oder einer Akkumulationsstudie an einem strukturell verwandten Stoff) oder aus bestimmten empirischen Beziehungen, wobei auf Wissen über die Wasserlöslichkeit oder den Octanol/Wasser-Verteilungskoeffizienten des Prüfstoffs (vorausgesetzt, die Aufnahme folgt der Kinetik erster Ordnung; siehe Anlage 5) zurückgegriffen wird, vorabschätzen.
Die Aufnahmephase sollte 28 Tage dauern, sofern ein stationärer Zustand nachweislich nicht bereits zu einem früheren Zeitpunkt erreicht wurde (siehe Anlage 1, Definitionen und Einheiten). Ein "steady state" oder stationärer Zustand gilt in der graphischen Darstellung des gegen die Zeit aufgetragenen Prüfstoffes in Fischen (Cf) als erreicht, wenn die Kurve parallel zur Zeitachse verläuft und wenn drei aufeinanderfolgende Cf-Analysen von Proben, die im Abstand von mindestens zwei Tagen entnommen wurden, um nicht mehr als ± 20 % voneinander abweichen bzw. wenn es in der Zeit zwischen der ersten und der letzten der nacheinander durchgeführten Analysen keinen bedeutenden Anstieg von Cf gibt. Werden gepoolte Proben analysiert, sind mindestens vier aufeinanderfolgende Analysen erforderlich. Für Prüfstoffe, die nur langsam aufgenommen werden, wäre ein zeitlicher Abstand zwischen den Probenahmen von sieben Tagen geeigneter. Stellt sich innerhalb von 28 Tagen kein stationärer Zustand ein, so wird der BCF entweder nach dem kinetischen Ansatz berechnet (bei dem kein stationärer Zustand erreicht werden muss) oder die Aufnahmephase kann um weitere Messungen verlängert werden, bis ein stationärer Zustand erreicht ist, oder um 60 Tage, je nach dem, welcher Termin als erster eintritt. Außerdem muss die Prüfstoffkonzentration in den Fischen am Ende der Aufnahmephase hinreichend hoch sein, um eine zuverlässige Schätzung von k2 aus der Ausscheidungsphase zu gewährleisten. Wenn sich nach 28 Tagen keine signifikante Aufnahme gezeigt hat, kann der Versuch gestoppt werden.
Dauer der Ausscheidungsphase
Bei Stoffen, die der Kinetik erster Ordnung folgen, reicht eine halbe Aufnahmephase in der Regel für eine angemessene Reduzierung (z.B. 95 %) der stoffbedingten Körperbelastung aus (zur Erläuterung dieser Schätzung siehe Anlage 5). Ist die Zeit für eine 95 %ige Ausscheidung jedoch unangemessen lang und wird die normale Dauer der Aufnahmephase beispielsweise um mehr als das Doppelte überschritten (d. h. mehr als 56 Tage), kann die Phase entsprechend verkürzt werden (z.B. bis die Prüfstoffkonzentration weniger als 10 % der Konzentration bei stationärem Zustand beträgt). Bei Stoffen mit komplexeren Aufnahme- und Ausscheidungsmustern, als sie durch ein Einkompartiment-Fischmodell dargestellt werden können (bei einer Kinetik erster Ordnung), sind jedoch unter Umständen längere Ausscheidungsphasen notwendig. Wenn solche komplexen Muster beobachtet und/oder erwartet werden, empfiehlt es sich, den Rat eines Biostatistikers und/oder Pharmakokinetikers einzuholen, um einen vorschriftsmäßigen Versuchsaufbau sicherzustellen. Da die Ausscheidungsphase verlängert wird, kann die Anzahl der erforderlichen Fischproben an Grenzen stoßen und können Wachstumsunterschiede zwischen den Fischen die Ergebnisse beeinflussen. Die Phase richtet sich auch nach dem Zeitraum, über den die Konzentration des Prüfstoffs in den Fischen oberhalb der analytischen Quantifizierungsgrenze bleibt.
Zahl der Versuchsfische
Die Zahl der Fische je Prüfkonzentration sollte so gewählt werden, dass bei jeder Probenahme mindestens vier Fische je Probe zur Verfügung stehen. Die Fische sollten nur gepoolt werden, wenn eine Analyse einzelner Fische nicht möglich ist. Wird bei der Kurvenanpassung (und den abgeleiteten Parametern) eine höhere Genauigkeit angestrebt oder sind Metabolismusuntersuchungen erforderlich (z.B. zur Unterscheidung zwischen Metaboliten und Ausgangsstoff bei Verwendung radioaktiv markierter Prüfstoffe) sind mehr Fische je Probe erforderlich. Der Lipidgehalt sollte an demselben biologischen Material bestimmt werden, das auch für die Bestimmung der Konzentration des Prüfstoffs verwendet wird. Sollte dies nicht möglich sein, sind eventuell zusätzliche Fische erforderlich (siehe Nummer 56 und 57).
Werden ausgewachsene (d. h. geschlechtsreife) Fische verwendet, sollten sie nicht in der Laichphase sein oder kürzlich erst gelaicht haben (entweder vor noch während des Versuchs). Zudem sollte angegeben werden, ob es sich um männliche oder weibliche Tiere handelt bzw. ob beide Geschlechter für den Versuch eingesetzt werden. Werden beide Geschlechter verwendet, sollten Unterschiede bei Wachstum und Lipidgehalt zwischen den Geschlechtern vor Beginn der Exposition als nicht signifikant dokumentiert werden, insbesondere wenn ein Pooling der männlichen und weiblichen Fische voraussichtlich notwendig sein wird, um nachweisbare Stoffkonzentrationen und/oder Lipidgehalte sicherzustellen.
Für jede Prüfung werden Fische von vergleichbarem Gewicht gewählt, sodass die kleinsten nicht weniger als zwei Drittel des Gewichts der größten Tiere aufweisen. Alle sollten derselben Altersgruppe angehören und dieselbe Herkunft haben. Da Gewicht und Alter eines Fisches einen bedeutenden Einfluss auf die BCF-Werte (12) haben können, sollten diese Angaben sorgfältig dokumentiert werden. Das Wiegen einer Teilprobe der Fischpopulation vor Durchführung der Prüfung wird empfohlen, damit das Durchschnittsgewicht geschätzt werden kann (siehe Nummer 61).
Besatz
Es werden hohe Wasser/Fisch-Verhältnisse gewählt, damit die durch den Einsatz der Fische zu Beginn des Versuchs verursachte Verringerung der Konzentration der Prüfverbindung im Wasser auf ein Mindestmaß zu begrenzen und auch eine Verringerung der Konzentration des gelösten Sauerstoffs zu vermeiden. Wichtig ist, dass die Besatzrate für die jeweils verwendete Prüfspezies geeignet ist. In der Regel wird eine Besatzrate von 0,1-1,0 g Fisch (Nassgewicht) pro Liter Wasser und Tag empfohlen. Höhere Besatzraten sind möglich, wenn nachgewiesen werden kann, dass die erforderliche Prüfstoffkonzentration mit einer Toleranzmarge von ± 20 % aufrechterhalten werden kann und die Sättigungskonzentration des gelösten Sauerstoffs nicht unter 60 % sinkt (siehe Nummer 24).
Bei der Entscheidung über die geeignete Besatzrate ist dem normalen Lebensraum der Fischspezies Rechnung zu tragen. So erfordern auf dem Meeresboden lebende Fische im Aquarium bei gleicher Wassermenge möglicherweise eine größere Bodenfläche als pelagische Fischspezies.
Fütterung
Während der Akklimatisations- und Prüfphase sind die Fische mit geeignetem Futter von bekanntem Lipid- und Gesamtproteingehalt in einer Menge zu füttern, die gewährleistet, dass die Tiere gesund bleiben und ihr Gewicht halten (geringfügiges Wachstum ist zulässig). Die Fische werden während der gesamten Dauer der Akklimatisations- und Prüfphase täglich mit einer der Art, den Versuchsbedingungen und dem Brennwert des Futters (bei Regenbogenforellen beispielsweise ca. 1 bis 2 % Körpergewicht/Tag) angepassten Menge Futter gefüttert. Die Fütterungsrate sollte so gewählt werden, dass schnelles Wachstum und eine starke Zunahme des Lipidgehalts vermieden werden. Die Futtermenge sollte ggf. (zum Beispiel einmal pro Woche) neu berechnet werden, um die Fütterungsrate beibehalten zu können. Dazu kann das Gewicht der Fische in den einzelnen Prüfkammern anhand des Gewichts der Fische in der Kammer geschätzt werden, die zuletzt beprobt wurde. Die restlichen Fische in der Kammer sollten nicht gewogen werden.
Nicht gefressenes Futter und Exkremente sollten täglich aus den Prüfkammern entfernt werden, und zwar kurz nach der Fütterung (innerhalb von 30 Minuten bis 1 Stunde). Die Kammern sollten während der gesamten Prüfungsdauer so sauber wie möglich gehalten werden, um die Konzentration organischer Stoffe möglichst gering zu halten (siehe Nummer 29), da das Vorhandensein von organischem Kohlenstoff die Bioverfügbarkeit des Prüfstoffs u. U. beeinträchtigen kann (12).
Da viele Futtersorten aus Fischmehl gewonnen werden, sollte sichergestellt werden, dass das Futter, beispielsweise aufgrund seines etwaigen Gehalts an Spuren von Pestiziden, Schwermetallen bzw. des Prüfstoffs als solchem, die Prüfungsergebnisse nicht beinflusst oder Schadwirkungen auslöst.
Licht und Temperatur
Es wird eine Photoperiode von 12 bis 16 Stunden empfohlen, und die Temperatur (± 21 °C) sollte der Prüfspezies (siehe Anlage 3) angepasst sein. Art und Eigenschaften der Beleuchtung sollten bekannt sein. Eine mögliche Phototransformation des Prüfstoffs unter den Bestrahlungsbedingungen der Prüfung sollte berücksichtigt werden. Die Beleuchtung sollte so gewählt werden, dass eine Exposition der Fische gegenüber unnatürlichen Photoprodukten vermieden wird. In einigen Fällen mag die Verwendung eines Filters angebracht sein, um UV-Strahlungen unter290 mm herauszufiltern.
Prüfkonzentrationen
Die Prüfung wurde ursprünglich für unpolare organische Stoffe entwickelt. Bei dieser Art von Stoffen wird davon ausgegangen, dass die Exposition der Fische gegenüber einer einzigen Konzentration ausreicht, da keine konzentrationsabhängigen Wirkungen erwartet werden, obwohl nach den geltenden Rahmenvorschriften möglicherweise zwei Konzentrationen erforderlich sind. Wenn Stoffe außerhalb dieses Bereichs geprüft werden oder andere Anzeichen einer möglichen Konzentrationsabhängigkeit bekannt sind, sollte die Prüfung mit zwei oder mehr Konzentrationen durchgeführt werden. Wird nur eine Konzentration geprüft, ist die Verwendung dieser einzigen Konzentration zu begründen (siehe Nummer 79). Zudem sollte die Prüfkonzentration so niedrig wie praktisch oder technisch möglich sein (d. h. nicht nahe der Löslichkeitsgrenze liegen).
In bestimmten Fällen kann davon ausgegangen werden, dass die Biokonzentration eines Stoffs von der Wasserkonzentration abhängt (z.B. bei Metallen, deren Aufnahme durch die Fische sich zumindest teilweise kontrollieren lässt). In solchen Fällen müssen mindestens zwei, vorzugsweise aber mehr Konzentrationen geprüft werden (siehe Nummer 49), die unter Umweltgesichtspunkten relevant sind. Auch für Stoffe, bei denen die Prüfkonzentrationen aus praktischen Gründen nahe der Löslichkeitsgrenze liegen müssen, wird die Prüfung mit mindestens zwei Konzentrationen empfohlen, da dies einen Einblick in die Zuverlässigkeit der Expositionskonzentrationen gibt. Die gewählten Prüfkonzentrationen sollten die ökologisch realistische Konzentration sowie die Konzentration umfassen, die für die Zwecke der jeweiligen Bewertung relevant ist.
Die Prüfstoffkonzentration(en) sollte(n) so gewählt werden, dass sie unter ihrem chronischen Wirkungsniveau oder unter 1 % ihres akuten asymptotischen LC50-Werts, innerhalb eines unter Umweltgesichtspunkten relevanten Bereichs sowie mindestens eine Zehnerpotenz über ihrer für die angewandte Analysemethode maßgeblichen Quantifizierungsgrenze in Wasser liegen. Die höchstzulässige Prüfkonzentration kann auch ermittelt werden, indem der akute 96-h-LC50-Wert durch ein angemessenes Akut/Chronisch-Verhältnis dividiert wird (z.B. liegt der Quotient bei bestimmten Stoffen bei 3, bei einigen wenigen jedoch über 100). Wird eine zweite Konzentration verwendet, sollte sie um Faktor 10 von der nächsthöheren Konzentration abweichen. Sollte dies aufgrund des Toxizitätskriteriums (das die obere Prüfkonzentration begrenzt) und der analytischen Grenze (die die untere Testkonzentration begrenzt) nicht möglich sein, kann ein Faktor unter 10 gewählt und die Verwendung eines radioaktiv markierten Stoffes (von höchster Reinheit; z. B vorzugsweise > 98 %) sollte in Erwägung gezogen werden. Es ist darauf zu achten, dass keine der verwendeten Konzentrationen die Löslichkeitsgrenze des Prüfstoffs in den Prüfmedien überschreitet.
Kontrollen
Zusätzlich zu den Versuchsreihen sollte eine Verdünnungswasserkontrolle bzw. (siehe Nummern 30 und 31) eine Kontrolle mit dem Lösungsmittel angelegt werden.
Häufigkeit der Wasserqualitätsmessungen
Während der Prüfung sollten der gelöste Sauerstoff, der TOC, der pH-Wert und die Temperatur in allen Prüf- und Kontrollgefäßen gemessen werden. Die Gesamthärte und (ggf.) die Salinität sollten in der (den) Kontrolle(n) und in einem Prüfgefäß gemessen werden. Werden zwei oder mehr Konzentrationen geprüft, sind diese Parameter bei der höheren (oder höchsten) Konzentration zu messen. Der gelöste Sauerstoff und (ggf.) die Salinität sollten mindestens dreimal - zu Beginn, ungefähr in der Mitte und am Ende der Aufnahmephase - sowie einmal pro Woche in der Ausscheidungsphase gemessen werden. Der TOC sollte zu Beginn der Prüfung (24 bzw. 48 Std. vor Beginn der Prüfung oder der Aufnahmephase), bevor die Fische eingesetzt werden, und sowohl während der Aufnahme- als auch der Ausscheidungsphase mindestens einmal pro Woche gemessen werden. Die Temperatur sollte täglich gemessen und protokolliert werden, der pH-Wert zu Beginn und am Ende jeder Prüfungsphase und die Härte einmal pro Prüfung. Die Temperatur sollte in mindestens einem Gefäß möglichst kontinuierlich überprüft werden.
Beprobung von Fischen und Wasser
Probenahmeplan für Fische und Wasser
Vor dem Einsatz der Fische sowie während der Aufnahme- und Ausscheidungsphase ist den Prüfkammern Wasser zur Bestimmung der Prüfstoffkonzentration zu entnehmen. Die Wasserproben sollten gleichzeitig mit den Fischproben vor deren Fütterung entnommen werden. Eine häufigere Probenahme kann sinnvoll sein, um stabile Konzentrationen nach dem Einsetzen der Fische zu gewährleisten. Während der Aufnahmephase sollten die Prüfstoffkonzentrationen bestimmt werden, um Übereinstimmung mit den Validitätskriterien zu gewährleisten (Nummer 24). Wenn in Wasserproben zu Beginn der Ausscheidungsphase kein Prüfstoff nachzuweisen ist, kann dies rechtfertigen, dass in der restlichen Ausscheidungsphase kein Prüf- und Kontrollwasser auf Prüfstoff gemessen wird.
Während der Aufnahmephase sollten mindestens fünf, während der Ausscheidungsphase mindestens vier Fischproben für die Untersuchung auf Prüfstoff entnommen werden. Da es in manchen Fällen schwierig sein wird, anhand dieser Probenzahl eine einigermaßen genaue Schätzung des BCF vorzunehmen (insbesondere wenn es sich nicht um eine reine Aufnahme- und Ausscheidungskinetik erster Ordnung handelt), kann es ratsam sein, in beiden Phasen häufiger Proben zu entnehmen (siehe Anlage 4).
Der Lipidgehalt sollte zumindest zu Anfang und am Ende der Aufnahmephase sowie am Ende der Ausscheidungsphase anhand desselben biologischen Materials bestimmt werden, das auch zur Bestimmung der Prüfstoffkonzentration verwendet wird. Ist dies nicht machbar, sollte der Lipidgehalt bei drei separaten Fischen an jedem der drei Endpunkte bestimmt werden. Die Anzahl Fische pro Behälter sollte zu Beginn der Prüfung entsprechend angepasst werden 7. Alternativ können, wenn in Kontrollfischen (d. h. Fischen aus der Stammpopulation) keine erheblichen Prüfstoffmengen nachgewiesen werden, die Kontrollfische der Prüfung ausschließlich auf Lipidgehalt untersucht werden, und die Prüfstoffanalyse in der (den) Prüfgruppe(n) (und die zugehörigen Werte der Aufnahmekonstante, der Ausscheidungskonstante und des BCF) können um Veränderungen des Lipidgehalts der Kontrollgruppe während der Prüfung korrigiert werden 8.
Tote oder kranke Fische sollten nicht auf Prüfstoffkonzentration oder Lipidgehalt untersucht werden.
Ein Beispiel für einen denkbaren Probenahmeplan wird in Anlage 4 gegeben. Mithilfe anderer hypothetischer KOW-Werte lassen sich leicht andere Beprobungspläne festlegen, um die für eine Aufnahme von 95 % erforderliche Expositionsdauer zu berechnen (für Berechnungen siehe Anlage 5).
Die Beprobung sollte während der Aufnahmephase so lange fortgesetzt werden, bis der stationäre Zustand erreicht ist (für Definitionen und Einheiten siehe Anlage 1) oder die Ausscheidungsphase auf andere Weise beendet wird (nach 28 oder 60 Tagen, siehe Nummern 37 und 38). Vor Beginn der Ausscheidungsphase sollten die Fische in saubere Gefäße umgesetzt werden.
Probenahme und Probenvorbereitung
Wasserproben für die Analyse sollten z.B. durch Absaugen durch eine inerte Schlauchleitung an einem zentralen Punkt der Prüfkammer gezogen werden. Der nicht-bioverfügbare Teil des Prüfstoffs kann offensichtlich nicht immer durch Filtrieren oder Zentrifugieren vom bioverfügbaren Teil getrennt werden. Bei Anwendung einer Trennungstechnik sollte diese angesichts der genannten Probleme stets im Prüfbericht begründet oder validiert werden (25). Insbesondere bei stark hydrophoben Stoffen (d. h. Stoffen mit einem log KOW > 5) (12) (26), bei denen es zu Adsorption an die Filtermatrix oder an die Zentrifugationsgefäße kommen könnte, sollten die Proben diesen Verfahren nicht unterzogen werden. Stattdessen sollten Vorkehrungen getroffen werden, um die Behälter so sauber wie möglich zu halten (siehe Nummer 46), und der Gesamtgehalt an organischem Kohlenstoff sollte sowohl während der Aufnahme- als auch während der Ausscheidungsphase kontrolliert werden (siehe Nummer 53). Zur Vermeidung möglicher Probleme aufgrund geringerer Bioverfügbarkeit kann die Probenahme bei schwer löslichen und stark hydrophoben Stoffen durch Festphasen-Mikroextraktion erfolgen.
Die beprobten Fische sollten unverzüglich auf geeignete und humane Weise getötet werden (bei Messungen ganzer Fische sollten diese nur mit Wasser abgespült (siehe Nummer 28) und trocken getupft werden). Die Fische sollten gewogen und ihre Gesamtlänge sollte gemessen werden 9. Bei jedem einzelnen Fisch sollten die Gewichts- und Längenmessungen der gemessenen Stoffkonzentration (und ggf. dem Lipidgehalt) zugeordnet werden, z.B. mithilfe eines individuellen Kenncodes für jeden beprobten Fisch.
Die Analyse der Fisch- und Wasserproben sollte vorzugsweise direkt nach der Probenahme erfolgen, um Degradation oder sonstige Verluste zu vermeiden und während der Prüfung die ungefähren Aufnahme- und Ausscheidungskonstanten berechnen zu können. Eine unverzügliche Analyse verhindert auch, dass ein Plateau (stationärer Zustand) erst spät bestimmt wird.
Kann keine sofortige Analyse erfolgen, sollten die Proben auf geeignete Weise gelagert werden. Informationen zur der für den betreffenden Prüfstoff geeigneten Lagerungsmethode - beispielsweise Tiefkühlung, Lagerung bei 41 °C, Extraktion usw. - sollten vor Beginn der Prüfung eingeholt werden. Die Dauer der Lagerung sollte gewährleisten, dass sich der Stoff während der Lagerung nicht verschlechtert.
Qualität der Analysemethode
Da das gesamte Verfahren im Wesentlichen von der Zuverlässigkeit, Genauigkeit und Empfindlichkeit der auf den Prüfstoff angewandten Analysemethoden abhängt, muss in Versuchen überprüft werden, ob die betreffende Methode in Bezug auf Zuverlässigkeit, Genauigkeit und Reproduzierbarkeit der chemischen Analyse sowie Wiederfindung des Prüfstoffs in Wasser und Fischen geeignet ist. Dies sollte im Rahmen von Vorversuchen geschehen. Zudem muss sichergestellt werden, dass der Prüfstoff im verwendeten Verdünnungswasser nicht nachweisbar ist. Erforderlichenfalls müssen die Prüfstoffkonzentrationen in Wasser und in den Fischen um die Wiederfindungs- und Hintergrundwerte der Kontrollen berichtigt werden. Die Fisch- und Wasserproben sollten stets so behandelt werden, dass Verunreinigungen und Verluste (z.B. infolge von Adsorption an das Probenahmegerät) auf ein Mindestmaß begrenzt sind.
Analyse der Fischproben
Werden für den Test radioaktiv markierte Materialien verwendet, können die Proben entweder auf ihre gesamte radioaktive Markierung analysiert werden (d. h. Ausgangsstoff und Metaboliten) oder aber gereinigt werden, damit der Ausgangsstoff separat untersucht werden kann. Soll der BCF auf dem Ausgangsstoff basieren, sollten die Hauptmetaboliten zumindest am Ende der Aufnahmephase bestimmt werden (siehe Nummer 6). Hauptmetaboliten sind jene, die mindestens 10 % der Gesamtrückstände in Fischgeweben entsprechen, die mindestens 5 % an zwei aufeinanderfolgenden Probenahmezeitpunkten entsprechen, die steigende Werte während der Aufnahmephase zeigen und die bekanntermaßen toxikologisch bedenklich sind. Wenn der BCF für den ganzen Fisch bezogen auf die gesamten radioaktiv markierten Rückstände ≥ 500 beträgt, ist es u. U. ratsam - und bei gewissen Kategorien von Stoffen wie Pestiziden unbedingt empfehlenswert - die Hauptmetaboliten zu identifizieren und zu quantifizieren. Die Quantifizierung solcher Metabolite wird von bestimmten Regulierungsbehörden möglicherweise gefordert. Wenn die Abbauprodukte, die ≥ 10 % des gesamten radioaktiv markierten Rückstands in den Fischgeweben ausmachen, identifiziert und quantifiziert werden, dann sollten auch die Abbauprodukte im Prüfwasser identifiziert und quantifiziert werden. Ist dies nicht machbar, sollte dies im Prüfbericht erläutert werden.
Die Konzentration des Prüfstoffes wird normalerweise für jeden gewogenen Fisch einzeln bestimmt. Ist dies nicht möglich, können die Proben jedes Probenahmevorgangs gepoolt werden, doch beschränkt dies die statistischen Verfahren, die auf die Daten angewendet werden können, sodass im Test eine Anzahl Fische eingesetzt werden muss, die dem Pooling, dem statistischen Verfahren und der gewünschten Aussagekraft Genüge leistet. Die Referenzdokumente (27) und (28) können als Einführung in relevante Pooling-Verfahren konsultiert werden.
Der BCF sollte auf einen Fisch mit 5 % Lipidgehalt (basierend auf dem Nassgewicht) standardisiert und nicht nur bezogen auf das aus der Studie direkt abgeleitete Körpergewicht ausgedrückt werden (siehe Nummer 21), es sei denn, es kann argumentiert werden, dass der Prüfstoff nicht überwiegend in Lipiden akkumuliert. Der Lipidgehalt der Fische sollte möglichst bei jeder Probenahme bestimmt werden, vorzugsweise an dem Extrakt, das für die Untersuchung des Prüfstoffs hergestellt wurde, da die Lipide häufig erst aus dem Extrakt entfernt werden müssen, bevor dieses chromatographisch analysiert werden kann. Jedoch erfordert die Analyse der Prüfstoffe häufig spezifische Extraktionsverfahren, die den Prüfmethoden zur Bestimmung des Lipidgehalts zuwiderlaufen können. In diesem Fall (bis geeignete zerstörungsfreie instrumentelle Methoden verfügbar sind) wird die Anwendung einer anderen Strategie zur Bestimmung des Lipidgehalts von Fischen empfohlen (siehe Nummer 56). Zur Bestimmung des Lipidgehalts sollten geeignete Methoden angewendet werden (20). Das Chloroform/Methanol-Extraktionsverfahren (29) bietet sich als Standardmethode an (30), als alternatives Verfahren ist die Smedes-Methode (31) geeignet. Letztere zeichnet sich durch vergleichbare Extraktionseffizienz, hohe Genauigkeit, den Einsatz weniger toxisch wirkender organischer Lösungsmittel und einfache Durchführung aus. Andere Methoden, deren Genauigkeit im Vergleich zu den empfohlenen Methoden gut ist, könnten ebenfalls angewendet werden, sofern dies entsprechend begründet wird. Wichtig ist, dass die angewandte Methode präzisiert wird.
Messung des Fischwachstums
Bei Prüfungsbeginn sind 5-10 Fische aus der Stammpopulation einzeln zu wiegen und ihre Gesamtlänge ist zu messen. Dies können dieselben Fische sein, die für die Bestimmung des Lipidgehalts verwendet werden (siehe Nummer 56). Das Gewicht und die Länge von Fischen, die je Probenahme aus den Prüf- und Kontrollgruppen entnommen werden, sollten vor der chemischen oder der Lipidanalyse gemessen werden. Anhand der Messwerte aus diesen Fischproben lassen sich Gewicht und Länge der in den Prüf- und Kontrollbehältern verbleibenden Fische abschätzen (siehe Nummer 45).
Daten und Berichterstattung
Auswertung der Ergebnisse
Die Aufnahmekurve für den Prüfstoff ergibt sich, indem seine Konzentration in/auf den Fischen (oder bestimmten Geweben) in der Aufnahmephase auf eine arithmetischen Skala gegen die Zeit auftragen wird. Hat die Kurve ein Plateau erreicht, d. h. ist sie nahezu asymptotisch zur Zeitachse, sollte der BCF im stationären Zustand (BCFSS) nach folgender Gleichung berechnet werden:
Cfbei stationärem Zustand(Mittelwert) / Cwbei stationärem Zustand(Mittelwert)
Die Entwicklung von Cf kann durch das Fischwachstum beeinflusst werden (Nummern 72 und 73). Die mittlere Expositionskonzentration (Cw) unterliegt Schwankungen im Zeitverlauf. Es kann davon ausgegangen werden, dass eine zeitgewichtete durchschnittliche Konzentration bei Bioakkumulationsstudien relevanter und genauer ist, selbst wenn die Schwankung innerhalb des entsprechenden Validitätsbereichs liegt (siehe Nummer 24). Für die Berechnung einer zeitgewichteten durchschnittlichen Wasserkonzentration siehe Anlage 5 Abschnitt 1.
Der kinetische Biokonzentrationsfaktor (BCFK) sollte als Quotient k1/k2, den beiden kinetischen Konstanten erster Ordnung, ermittelt werden. Die Konstanten k1 und k2 sowie der BCFK können durch gleichzeitiges Anpassen der Aufnahme- und der Ausscheidungsphase abgeleitet werden. Alternativ können k1 und k2 nacheinander bestimmt werden (siehe Anlage 5 (für Beschreibung und Vergleich dieser Methoden siehe Anlage 5). Die Ausscheidungskonstante (k2) muss ggf. um die Verdünnung durch Wachstum korrigiert werden (siehe Nummern 72 und 73). Wenn die Aufnahme- und/oder die Ausscheidungskurve eindeutig keiner Kinetik erster Ordnung folgen, sollten komplexere Modelle verwendet (siehe Referenzen in Anlage 5) und der Rat eines Biostatistikers eingeholt werden.
Daten zu Fischgewicht/-länge
Während der Aufnahmephase und der Ausscheidungsphase werden zu jedem Probenahmezeitpunkt die Nassgewichte und Gesamtlängen der einzelnen Fische (einschließlich der Stammpopulation zu Beginn der Aufnahme), aufgeschlüsselt nach Test- und Kontrollgruppen, tabellarisch erfasst. Bei jedem einzelnen Fisch sollten die Gewichts- und die Längenmessung der analysierten Stoffkonzentration zugeordnet werden, z.B. mithilfe eines individuellen Kenncodes für jeden untersuchten Fisch. Das Gewicht ist der bevorzugte Wachstumsparameter zur Korrektur der kinetischen BCF-Werte um die Verdünnung durch Wachstum (für die Methode zur Korrektur der Daten um die Verdünnung durch Wachstum siehe Nummer 73 und Anlage 5).
Korrektur um die Verdünnung durch Wachstum und Lipidstandardisierung
Das Fischwachstum während der Ausscheidungsphase kann die gemessenen Stoffkonzentrationen in den Fischen verringern, sodass die Gesamtausscheidungskonstante (k2) größer ist als sie bei den Ausscheidungsprozessen (z.B. Atmung, Metabolismus, Egestion) alleine wäre. Die kinetischen Biokonzentrationsfaktoren sollten um die Verdünnung durch Wachstum korrigiert werden. Ein BCFSS wird ebenfalls durch das Wachstum beeinflusst, doch existiert kein anerkanntes Verfahren für die Korrektur eines BCFSS um das Wachstum. In Fällen erheblichen Wachstums sollte der BCFK, der um das Wachstum korrigiert wurde (BCFKg), ebenfalls abgeleitet werden, da dies ein aussagekräftigeres Maß für den Biokonzentrationsfaktor sein kann. Der Lipidgehalt von Versuchsfischen (der eng mit der Bioakkumulation von hydrophoben Stoffen) assoziiert wird, kann in der Praxis variieren, sodass eine Standardsisierung auf einen bestimmten Lipidgehalt (5 % w/w) erforderlich ist, um sowohl den kinetischen Biokonzentrationsfaktor als auch den Biokonzentrationsfaktor bei stationärem Zustand auf sinnvolle Weise darzustellen, es sei denn, es kann argumentiert werden, dass der Prüfstoff nicht hauptsächlich in Lipiden akkumuliert (z.B. können sich bestimmte perfluorierte Stoffe an Proteine binden). Für Gleichungen und Beispiele für diese Berechnungen siehe Anlage 5.
Um einen kinetischen BCF um die Verdünnung durch Wachstum zu korrigieren, sollte die Ausscheidungskonstante um das Wachstum korrigiert werden. Diese wachstumskorrigierte Ausscheidungskonstante (k2g) wird berechnet durch Subtraktion der Wachstumskonstante (kg, wie aus den gemessenen Gewichtsdaten bestimmt) von der Gesamtausscheidungskonstante (k2). Anschließend wird der wachstumskorrigierte kinetische Biokonzentrationsfaktor durch Division der Aufnahmekonstante (k1) durch die wachstumskorrigierte Ausscheidungskonstante (k2g) berechnet (siehe Anlage 5). In bestimmten Fällen funktioniert dieser Ansatz nur bedingt. Beispielsweise kann bei Prüfstoffen, die sehr langsam ausgeschieden werden, die abgeleitete Konstante k2g bei wachsenden Fischen sehr klein sein, weshalb der Fehler bei den beiden Konstanten, die zur Ableitung verwendet wurden, kritisch wird und kg-Schätzungen in bestimmten Fällen möglicherweise größer als k2 sind. Ein alternativer Ansatz, bei dem die Notwendigkeit einer Korrektur um die Verdünnung durch Wachstum umgangen wird, besteht darin, anstelle der üblichen Daten über die Prüfstoffmasse pro Fischmasseneinheit (Konzentration) Daten über die Prüfstoffmasse pro Fischausscheidung (basierend auf ganzen Fischen) zu verwenden. Dies lässt sich einfach bewerkstelligen, da bei Versuchen, die nach dieser Prüfmethode durchgeführt werden, protokollierte Gewebekonzentrationen einzelnen Fischgewichten zugeordnet werden sollten. Dieses einfache Verfahren wird in Anlage 5 beschrieben. Es ist zu beachten, dass k2 auch bei Anwendung dieses alternativen Ansatzes angegeben werden sollte.
Der kinetische Biokonzentrationsfaktor und der Biokonzentrationsfaktor bei stationärem Zustand sollten ebenfalls bezogen auf einen Standard-Lipidgehalt von Fischen von 5 % (w/w) angegeben werden, es sei denn, es kann argumentiert werden, dass der Prüfstoff nicht vorwiegend in Lipiden akkumuliert. Daten über die Konzentration im Fisch oder der BCF sind entsprechend dem Verhältnis zwischen 5 % und dem tatsächlichen durchschnittlichen Lipidgehalt eines Fisches (in % Nassgewicht) zu standardisieren (siehe Anlage 5).
Wurden die chemische Analyse und die Lipidanalyse an ein und demselben Fisch durchgeführt, sollten diese lipidstandardisierten Daten für einzelne Fische bei der Berechnung eines lipidstandardisierten BCF berücksichtigt werden. Alternativ ist es möglich, soweit enn das Wachstum bei Kontroll- und Versuchsfischen vergleichbar ist, nur den Lipidgehalt von Kontrollfischen für die Lipidkorrektur zu verwenden (siehe Nummer 56). Eine Methode zur Berechnung eines lipidstandardisierten BCF ist in Anlage 5 beschrieben.
Interpretation der Ergebnisse
Wenn die gemessenen Prüflösungskonzentrationen an der Nachweisgrenze der Analysemethode liegen, sollten die Ergebnisse mit Vorsicht interpretiert werden.
Das Durchschnittswachstum der Prüf- und Kontrollgruppen sollte grundsätzlich nicht allzu stark variieren, um toxische Wirkungen auszuschließen. Die Wachstumskonstanten oder die Wachstumskurven der beiden Gruppen sollten nach einem geeigneten Verfahren miteinander verglichen werden 10.
Gut definierte Aufnahme- und Ausscheidungskurven weisen auf eine gute Qualität der Biokonzentrationsdaten hin. Bei den Konstanten sollte ein χ2 Goodness-of-Fit-Test eine gute Anpassung (d. h. einen kleinen Messfehlerprozentsatz (32)) für das Bioakkumulationsmodell ergeben, damit die Konstanten als zuverlässig angesehen werden können (siehe Anlage 5). Werden mehrere Prüfkonzentrationen verwendet, sollte die Schwankung der Aufnahme-/Ausscheidungskonstanten zwischen den Testkonzentrationen weniger als 20 % 11 betragen. Ist dies nicht der Fall, könnte dies auf eine Abhängigkeit von der Konzentration hindeuten. Werden erhebliche Unterschiede bei den Aufnahme-/Ausscheidungskonstanten zwischen den verwendeten Prüfkonzentrationen beobachtet, sollten diese dokumentiert und möglichst begründet werden. Bei einem guten Versuchsplan liegt die 95 %-Konfidenzgrenze des BCF in der Regel bei ungefähr ± 20 % des errechneten BCF.
Werden zwei oder mehr Konzentrationen geprüft, wird anhand der Ergebnisse der beiden oder aller Konzentrationen kontrolliert, ob die Ergebnisse übereinstimmend sind und eine Konzentrationsabhängigkeit besteht. Wird nur eine einzige Konzentration geprüft, um die Verwendung von Tieren und/oder Ressourcen zu verringern, sollte die Verwendung dieser einzigen Konzentration begründet werden.
Der resultierende BCFSS ist zweifelhaft, wenn der BCFK erheblich größer ist als der BCFSS, da dies darauf hindeuten kann, dass kein stationärer Zustand erreicht wurde oder die Verdünnung durch Wachstum sowie Ausscheidungsprozesse nicht berücksichtigt wurden. In Fällen, in denen der BCFSS wesentlich größer ist als der BCFK, sollte die Ableitung der Aufnahme- und Ausscheidungskonstanten auf Fehler überprüft und neu evaluiert werden. Die Schätzung des BCFK lässt sich möglicherweise durch ein anderes Anpassungsverfahren verbessern (siehe Anlage 5).
Prüfbericht
Abgesehen von den Angaben zum Prüfstoff unter Nummer 3 muss der Prüfbericht folgende Informationen enthalten:
Prüfstoff:
Physikalischer Zustand und, soweit relevant, physikalisch-chemische Eigenschaften;
- chemische Bezeichnung, wie z.B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer , SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar usw. (einschließlich des Gehalts an organischem Kohlenstoff, falls zutreffend);
- bei mehrkomponentigen Stoffen und UVCB-Stoffen (chemische Stoffe unbekannter oder variabler Zusammensetzung, komplexe Reaktionsprodukte und biologische Materialien): Beschreibung, soweit möglich, der chemischen Identität der Einzelkomponenten und jeweils des Prozentsatzes der Gesamtmasse des Stoffs. Es sollte zusammengefasst werden, inwieweit die bei der Prüfung verwendete Analysemethode ein Maß für die Konzentration des Stoffes ist; alle Analysemethoden sollten mit Angaben zu Genauigkeit, Nachweisgrenze und Quantifizierungsgrenze beschrieben werden;
- im Falle der radioaktiven Markierung: die exakte Position der markierten Atome und der prozentuale Anteil der auf Verunreinigungen zurückgeführten Radioaktivität;
- Angaben zur Toxizität des Prüfstoffs in Fischen (idealerweise die Prüfspezies). Die Toxizität sollte als akuter 96-h-LC50-Wert sowie als NOAEC- und LOAEC-Wert aus einer Langzeitstudie (d. h. einem Test im frühen Entwicklungsstadium oder einem Test, der den gesamten Lebenszyklus abdeckt, falls verfügbar) angegeben werden;
- Lagerbedingungen und Stabilität der Prüfchemikalie oder des Prüfstoffs unter Lagerbedingungen, falls diese vor der Verwendung gelagert werden.
Prüfspezies:
Wissenschaftlicher Name, Stamm, Herkunft, etwaige Vorbehandlungen, Akklimatisierung, Alter, Geschlecht (falls relevant), Größenbereich (Gewicht und Länge) usw.;
Prüfbedingungen:
- angewandtes Prüfverfahren (z.B. Durchflussverfahren, semistatisches Verfahren); vollständiger oder minimierter Versuch (einschließlich Begründung und Rechtfertigung);
- Art und Eigenschaften der verwendeten Beleuchtung und Photoperiode(n);
- Versuchsplan (z.B. Zahl und Größe der Prüfkammern, Wasservolumen-Austauschrate, Besatzrate, Zahl der Replikate, Anzahl Fische pro Replikat, Zahl der Prüfkonzentrationen, Dauer der Aufnahme- und Ausscheidungsphase, Häufigkeit der Entnahme von Fisch- und Wasserproben);
- Methode der Vorbereitung der Stammlösungen und Erneuerungshäufigkeit (falls verwendet, müssen Angaben zum Lösungsmittel, seiner Konzentration und seinem Beitrag zum organischen Kohlenstoffgehalt des Prüfwassers gemacht werden) oder Beschreibung des alternativen Dosierungssystems;
- die nominellen Prüfkonzentrationen, der Durchschnitt der gemessenen Werte sowie deren Standardabweichungen in den Prüfbehältern sowie das Verfahren, nach dem diese ermittelt wurden;
- Herkunft des Verdünnungswassers, Beschreibung etwaiger Vorbehandlungen, Ergebnisse etwaiger Nachweise, dass die Versuchsfische in dem Wasser überleben können, sowie Wassereigenschaften: pH-Wert, Härte, Temperatur, Konzentration des gelösten Sauerstoffs, Restchlor (falls gemessen), TOC, suspendierte Feststoffe, Salinität des Prüfmediums (gegebenenfalls) sowie die Ergebnisse aller anderen durchgeführten Messungen;
- Wasserqualität innerhalb der Prüfbehälter, pH-Wert, Härte, TOC, Temperatur und Konzentration des gelösten Sauerstoffs; Messverfahren und -häufigkeit;
- ausführliche Angaben zur Fütterung, z.B. Art des Futters, Herkunft, Zusammensetzung (zumindest der Lipid- und Proteingehalt, falls möglich), gewählte Fütterungsrate, Futtermenge und Häufigkeit;
- Angaben zur Behandlung der Fisch- und Wasserproben, einschließlich Einzelheiten zu Vorbereitung, Lagerung, Extraktion und Analyseverfahren (und -genauigkeit) für den Prüfstoff und den Lipidgehalt;
- angewandte Methoden zur Randomisierung der Behandlung und Zuordnung der Fische zu den Prüfbehältern;
- Datum des Einsatzes der Prüforganismen in die Prüflösungen und Prüfungsdauer;
- Beschreibung der Dosisfindungstests und der Ergebnisse, falls verfügbar.
Ergebnisse:
- Ergebnisse etwaiger Vorversuche;
- Mortalität der Kontrollfische und der Fische in den einzelnen Expositionskammern sowie etwa beobachtete Verhaltensanomalien;
- Beschreibung etwa festgestellter Schadwirkungen;
- vollständige Beschreibung aller angewandten chemischen Analyseverfahren, einschließlich Nachweis- und Quantifizierungsgrenzen, Variabilität und Wiederfindung;
- Lipidgehalt der Fische, einschließlich der angewandten Methode, und Lipidstandardisierungsfaktor (Ln, Faktor zur Angabe der Ergebnisse bezogen auf einen Lipidgehalt von 5 %), falls abgeleitet;
- tabellarisch dargestellte Daten zu Fischgewicht (und -länge), bezogen auf die Chemikalienkonzentrationen in den einzelnen Fischen (und den Lipidgehalt, falls zutreffend), sowohl für die Kontroll- als auch die Expositionsgruppen (z.B. unter Verwendung von individuellen Kenncodes für jede Fischprobe) und Berechnungen der abgeleiteten Wachstumskonstante(n);
- tabellarisch dargestellte Daten zur Prüfcstoffkonzentration in den Fischen (Cf, bezogen auf einzelnen Fische) und im Wasser (Cw) (gegebenenfalls mit Mittelwerten für Prüf- und Kontrollgruppe, Standardabweichung und Abweichungsbereich) für alle Probenahmen (wobei Cf in mg/kg Nassgewicht des Ganzkörpers oder bestimmter Gewebe, z.B. Lipid, und Cw in mg/l ausgedrückt wird). Auch die Cw-Werte für die Kontrollreihe (Hintergrund) sollten dokumentiert werden;
- Kurven (einschließlich aller gemessenen Daten), aus denen Folgendes hervorgeht (falls zutreffend, können die Konzentrationen bezogen auf den Ganzkörper und den Lipidgehalt normalisiert auf 5 % des Fischs oder bestimmter Fischgewebe ausgedrückt werden):
- Wachstum, d. h. Fischgewicht im Verhältnis zur Zeit oder durch natürliches Logarithmieren transformiertes Gewicht im Verhältnis zur Zeit (einschließlich der abgeleiteten Wachstumskonstante, kg);
- Aufnahme und Ausscheidung des Prüfstoffs durch die Fische (in einem Diagramm);
- Zeit zum stationären Zustand (falls erreicht);
- durch natürliches Logarithmieren transformierte Konzentration im Verhältnis zur Dauer der Aufnahmephase (einschließlich der abgeleiteten Aufnahmekonstanten k1);
- durch natürliches Logarithmieren transformierte Konzentration (ln(Konzentration)) im Verhältnis zur Dauer der Ausscheidungsphase (einschließlich der abgeleiteten Ausscheidungskonstanten k2); und
- Aufnahme- und Ausscheidungskurve, aus denen sowohl die Daten als auch das angepasste Modell hervorgehen.
- Werden bei einer visuellen Überprüfung einer grafischen Darstellung "Ausreißer" festgestellt, kann ein statistisch gültiger Ausreißertest durchgeführt werden, um falsche Datenpunkte zu entfernen; die Gründe für die Auslassung solcher Punkte sind zu dokumentieren.
- der Biokonzentrationsfaktor bei stationärem Zustand (BCFSS), d. h. wenn der stationäre Zustand (fast) erreicht ist;
- kinetischer Konzentrationsfaktor (BCFK) sowie die abgeleiteten Aufnahme- und Ausscheidungskonstanten k1 und k2, zusammen mit den Varianzen in k2 (Steigung und Achsenabschnitt) bei sequenzieller Anpassung;
- Konfidenzgrenzen, Standardabweichung (soweit verfügbar) sowie Berechnungs-/Datenanalyseverfahren für jeden Parameter bei jeder Konzentration des verwendeten Prüfstoffs;
- etwaige Angaben zu radioaktiv markierten Prüfstoffmetaboliten und deren Akkumulation;
- Wachstumskonstante(n) (einschließlich 95 %-Konfidenzintervall(e)) und berechnete, um das Wachstum korrigierte Ausscheidungskonstante (k2g), Halbwertszeit und BCF-Werte (BCFKg);
- etwaige Besonderheiten im Zusammenhang mit dem Test, Abweichungen von den genannten Verfahren und andere relevante Informationen;
- nachstehende Übersichtstabelle mit wichtigen gemessenen und berechneten Daten:
Stoffbezogene Aufnahme- und Ausscheidungskonstanten und Biokonzentrationsfaktoren (BCF) kg (Wachstumskonstante, Tag-1): Wert eintragen (95 % CI) 1 k1 (Gesamtaufnahmekonstante; l kg-1 Tag-1): Wert eintragen (95 % CI) 1 k2 (Gesamtausscheidungskonstante, Tag-1): Wert eintragen (95 % CI) 1 k2g (wachstumskorrigierte Ausscheidungskonstante, Tag-1): Wert eintragen (95 % CI) 1 Cf (Stoffkonzentration im Fisch bei stationärem Zustand; mg kg-1): Wert eintragen ± SD 2 Cw (Stoffkonzentration im Wasser; mg l-1): Wert eintragen ± SD 2 Ln (Lipidnormalisierungsfaktor): Wert eintragen 3 BCFSS (BCF bei stationärem Zustand; l kg-1) Wert eintragen ± SD 2 BCFSSL (lipidnormalisierter BCF bei stationärem Zustand; l kg-1) Wert eintragen 3 ± SD 2 BCFK (kinetischer BCF; l kg-1) Wert eintragen (95 % CI) 1 BCFK (wachstumskorrigierter kinetischer BCF; l kg-1) Wert eintragen (95 % CI) 1 t1/2g (wachstumskorrigierte Halbwertzeit; Tag): Wert eintragen (95 % CI) 1 BCFKL (lipidnormalisierter kinetischer BCF; l kg-1): Wert eintragen BCFKLG (lipidnormalisierter, wachstumskorrigierter kinetischer BCF; l kg-1): Wert eintragen 1) CI: Konfidenzintervall (falls schätzbar) 2) SD: Standardabweichung (falls schätzbar)
Ergebnisse der Art "an der Nachweis-/Quantifizierungsgrenze nicht nachweisbar/quantifizierbar" sollten durch Vorsuche und einen entsprechenden Versuchsplan vermieden werden, da sie für die Berechnung der Konstanten nicht berücksichtigt werden können.
_____
1) Für die meisten Prüfstoffe gilt, dass sie im Kontrollwasser möglichst nicht nachweisbar sein sollten.
Hintergrundkonzentrationen sollten nur für natürlich vorkommende Stoffen (z.B. bestimmte Metalle) und bei in der Umwelt allgemein vorkommende Stoffe relevant sein.
2) Folgen die Messdaten offensichtlich nicht der Kinetik erster Ordnung, sollten komplexere Modelle verwendet (siehe Referenzdokumente in Anlage 5) und der Rat eines Biostatistikers eingeholt werden.
3) Die Aufnahme kann bei der Biokonzentrationsprüfung aufgrund niedriger Expositionskonzentrationen wegen geringer Wasserlöslichkeit begrenzt sein, wohingegen bei der Prüfung mit Exposition über das Futter weitaus höhere Expositionskonzentrationen erreicht werden können.
4) Bei mehrkomponentigen Stoffen, UVCB-Stoffen und Gemischen sollte zur Bestimmung der geeigneten Expositionskonzentrationen die Wasserlöslichkeit jeder relevanten Komponente berücksichtigt werden.
5) TOC umfasst organischen Kohlenstoff aus Partikeln und gelöstem organischen Kohlenstoff, d. h. TOC = POC + DOC.
6) Wird ein Lösungsmittel oder Lösungsvermittler verwendet (obwohl dies in der Regel nicht empfohlen wird), sollte der daraus resultierende organische Kohlenstoff dem organischen Kohlenstoff aus dem Prüfstoff hinzugerechnet werden, damit die Konzentration des organischen Kohlenstoffs in den Prüfgefäßen bewertet werden kann.
7) Wird der Lipidgehalt nicht bei denselben Fischen bestimmt, wie dies beim Prüfstoff der Fall ist, sollten die Fische zumindest ein ähnliches Gewicht besitzen und (ggf.) vom gleichen Geschlecht sein.
8) Diese Alternative gilt nur, wenn die Fische in allen Prüfgruppen in vergleichbarer Größenordnung gehalten, nach demselben Schema entfernt und auf dieselbe Weise gefüttert werden. Dies gewährleistet, dass das Fischwachstum in allen Gruppen vergleichbar ist, wenn die geprüfte Konzentration unterhalb des toxischen Bereiches liegt. Es wird davon ausgegangen, dass bei vergleichbarem Wachstum auch der Lipidgehalt ähnlich ist. Wachstumsunterschiede in der Kontrolle würden auf eine stoffbedingte Wirkung hindeuten und die Studie invalidieren.
9) Zusätzlich zum Gewicht sollte auch die Gesamtlänge protokolliert werden, da ein Vergleich des Längenwachstums während des Versuchs ein geeigneter Indikator einer Schadwirkung ist.
10) Es kann ein t-Test an Wachstumskonstanten durchgeführt werden, um festzustellen, ob es zwischen Kontroll- und Prüfgruppen Wachstumsunterschiede gibt, oder aber ein F-Test im Falle einer Varianzanalyse. Ein F-Test oder Likelihood-Ratio-Test kann die Auswahl des geeigneten Wachstumsmodells vereinfachen (OECD Monograph 54 (32)).
11) Diese Prozentsätze setzen voraus, dass die Analysemethoden zuverlässig sind und die Halbwertzeit < 14 Tage beträgt. Wenn die Analysemethoden weniger zuverlässig sind oder die Halbwertzeit (erheblich) verlängert ist, werden diese Zahlen größer.
C.13 - II: Fischtest - Minimale aquatische Exposition
Einleitung
Die zunehmende Erfahrung von Laboratorien und Regulierungsbehörden mit der Durchführung und Interpretation des vollständigen Tests zeigt, dass die Kinetik erster Ordnung - mit wenigen Ausnahmen - für die Schätzung der Aufnahme- und Ausscheidungskonstanten geeignet ist, d. h. Aufnahme- und Ausscheidungskonstanten können mit einem Minimum an Probenahme geschätzt und der kinetische BCF kann berechnet werden.
Der anfängliche Zweck der Prüfung alternativer Versuchspläne für BCF-Studien bestand darin, einen weniger komplexen Test zu entwickeln, der in einem Prüfzwischenschritt eingesetzt werden könnte, um BCF-Schätzwerte basierend auf KOW und QSAR zu widerlegen oder zu bestätigen, und somit die Notwendigkeit einer vollständigen Prüfung für zahlreiche Stoffen zu vermeiden und die Kosten sowie den Einsatz von Tieren durch die Verringerung der Probenahmen und der Analysesequenzen auf ein Minimum zu beschränken. Wenngleich die wichtigsten Elemente des Versuchsplans der bisherigen Prüfmethode übernommen wurden, um die Einbeziehung der Prüfergebnisse in die vorhandenen BCF-Daten zu ermöglichen und die Prüfung sowie Auslegung der Daten zu erleichtern, bestand das Ziel darin, BCF-Schätzwerte von ausreichender Genauigkeit und Präzision für Risikobewertungsentscheidungen bereitzustellen. Es gelten vielfach dieselben Kriterien wie für die vollständige Prüfung, z.B. die Validitätskriterien (siehe Nummer 24) und Prüfungsabbruch, wenn am Ende der Aufnahmephase eine unerhebliche Aufnahme festgestellt wird (siehe Nummern 16 und 38).
Für einen Versuchsplan mit minimaler Exposition kämen generell Stoffe in Frage, für diese Prüfmethode grundsätzlich entwickelt wurde, d. h. unpolare organische Stoffe (siehe Nummer 49). Gibt es Anhaltspunkte dafür, dass der Prüfstoff ein abweichendes Verhalten zeigen könnte (z.B. eindeutige Abweichung von der Kinetik erster Ordnung), sollte zu regulatorischen Zwecke eine vollständige Prüfung durchgeführt werden.
Der minimierte Test dauert in der Regel ebenso lang wie der BCF-Standardtest durchgeführt, erfordert jedoch weniger Fischprobenahmen (für die Begründung siehe Anlage 6). Jedoch kann die Ausscheidungsphase bei schnell ausgeschiedenen Stoffen verkürzt werden, um zu vermeiden, dass die Konzentrationen in den Fischen vor Prüfungsende unter die Nachweis-/Quantifizierungsgrenze sinken. Mit einem Fischtest mit minimaler Exposition kann anhand einer einzigen Konzentration festgestellt werden, ob eine vollständige Prüfung durchgeführt werden muss oder nicht und ob die resultierenden Daten für die Berechnung der Konstanten und BCF robust genug sind (siehe Nummer 93). Auf eine vollständige Prüfung kann verzichtet werden, wenn die ermittelten BCF unter regulatorischen Gesichtspunkten in keiner Weise bedenklich sind.
In bestimmten Fällen kann es von Vorteil sein, den minimierten Test mit mehr als einer Prüfkonzentration als Vorversuch durchzuführen, um zu bestimmen, ob die BCF-Schätzwerte für einen bestimmten Stoff konzentrationsabhängig sind. Sollten die BCF-Schätzwerte aus dem minimierten Test eine Konzentrationsabhängigkeit zeigen, muss eine vollständige Prüfung durchgeführt werden. Ergibt der minimierte Test, dass die BCF-Schätzwerte nicht konzentrationsabhängig sind, die Ergebnisse jedoch nicht als schlüssig angesehen werden, könnte anschließend eine vollständige Prüfung mit einer Einzelkonzentration durchgeführt und die Zahl der benötigten Tiere im Vergleich zu einer vollständigen Prüfung mit zwei (oder mehr) Konzentrationen auf diese Weise verringert werden.
Stoffe, die potenziell für den minimierten Test in Frage kommen, sollten
- der Aufnahme- und Ausscheidungskinetik erster Ordnung möglichst folgen, z.B. abgeleitet aus Analogien mit ähnlichen Stoffen;
- einen log KOW < 6 aufweisen, es sei denn, es wird eine schnelle Metabolisierung erwartet 1;
- im Hinblick auf das Analyseverfahren hinreichend wasserlöslich sein (siehe Nummer 24);
- in den Fischen und im Wasser eindeutig quantifizierbar sein (d. h. die Konzentrationen sollten mindestens eine Zehnerpotenz über der Quantifizierungsgrenze liegen), wobei eine radioaktive Markierung empfohlen wird (siehe Nummer 23); und
- eine Ausscheidungsphase aufweisen, die länger als die prognostizierte Halbwertzeit ist (für Berechnungen siehe Anlage 5), oder die Dauer der Ausscheidungsphase sollte entsprechend angepasst werden (siehe Nummer 91). Eine Abweichung von dieser Regel ist zulässig, wenn von einer schnellen Metabolisierung ausgegangen wird.
Probenahmeplan für minimierte Versuche
Beprobung der Fische
Die Entnahme von Fischproben wird auf vier Zeitpunkte reduziert:
- in der Mitte und am Ende der Aufnahmephase (letztere ist gleichzeitig auch der Beginn der Ausscheidungsphase), z.B. nach 14 und 28 Tagen (33);
- in der Mitte der Ausscheidungsphase und bei Versuchsende (soweit die Stoffkonzentration < 10 % der Höchstkonzentration beträgt, oder zumindest deutlich nach einer Halbwertzeit des Prüfstoffs), z.B. nach 7 und 14 Tagen der Ausscheidung (33). Wird eine schnelle Ausscheidung erwartet oder beobachtet, sollte die Ausscheidungsphase verkürzt werden, um zu vermeiden, dass die Konzentrationen in den Fischen unter die Quantifizierungsgrenze fallen;
- Lipidmessung wie bei der umfassenden Prüfung;
- Wachstumskorrektur wie bei der umfassenden Prüfung;
- der BCF wird als kinetischer BCF berechnet.
Wasserbeprobung
Beim minimierten Versuchsplan werden die Wasserproben wie bei der vollständigen Prüfung (siehe Nummer 54), mindestens jedoch fünf Mal in gleichen Zeitabständen über die Aufnahmephase verteilt, sowie in der Ausscheidungsphase wöchentlich entnommen.
Änderungen des Versuchsplans
Je nach Eigenschaften des Prüfstoffs, gültigen QSAR-Prognosen und des konkreten Versuchszwecks können Änderungen des Versuchsplans in Betracht gezogen werden:
- Ist mehr Präzision erforderlich, könnten mehr Fische (6 oder 8 anstatt 4) für die Probe am Ende der Aufnahmephase verwendet werden.
- Einbeziehung einer "Extragruppe" von Fischen, wenn eine Ausscheidungsphase von 14 Tagen (oder das prognostizierte Ende der Ausscheidungsphase) für eine adäquate Ausscheidung (d. h. > 50 %) umzulänglich war. Wenn die prognostizierte Dauer der Ausscheidungsphase kürzer oder länger als 14 Tage ist, sollte der Probenahmeplan angepasst werden (d. h. eine Gruppe sollte am prognostizierten Ende der Ausscheidungsphase und eine zweite Gruppe nach der Halbwertzeit beprobt werden).
- Verwendung von zwei Prüfkonzentrationen, um eine mögliche Konzentrationsabhängigkeit zu untersuchen. Sollten die Ergebnisse des minimierten Versuchs, der mit zwei Prüfkonzentrationen durchgeführt wurde, zeigen, dass der BCF nicht konzentrationsabhängig ist (d. h. weniger als 20 % abweicht), kann eine einzige Prüfkonzentration für die vollständige Prüfung als ausreichend angesehen werden (falls diese durchgeführt wird).
- Es kann davon ausgegangen werden, dass Modelle für Bioakkumulationsprozesse wie die von Arnot et al. (35) vorgeschlagenen Prozesse die Planung der Dauer der Aufnahme- und Ausscheidungsphase erleichtern (siehe auch Anlage 5).
Berechnungen
Grund für diesen Ansatz ist die Tatsache, dass der Biokonzentrationsfaktor bei einer vollständigen Prüfung entweder als Biokonzentrationsfaktor bei stationärem Zustand (BCFSS) - durch Berechnung des Quotienten der Prüfstoffkonzentration im Fischgewebe und der Prüfstoffkonzentration im Wasser - oder als kinetischer Biokonzentrationsfaktor (BCFK) durch Berechnung des Quotienten der Aufnahmekonstanten k1 und der Ausscheidungskonstanten k2 - bestimmt werden kann. Der BCFK ist auch dann gültig, wenn während der Aufnahme keine Stoffkonzentration im stationären Zustand erreicht wird, vorausgesetzt, die Aufnahme und Ausscheidung folgen in etwa Prozessen der Kinetik erster Ordnung. Als absolutes Minimum werden zwei Datenpunkte für die Schätzung der Aufnahme- und der Ausscheidungskonstanten benötigt - einer am Ende der Aufnahmephase (d. h. zu Beginn der Ausscheidungsphase) und einer am Ende (oder nach einem signifikanten Teil) der Ausscheidungsphase. Der zwischengeschaltete Probenahmezeitpunkt wird zur Kontrolle der Aufnahme- und Ausscheidungskinetik empfohlen 2. Für Berechnungen siehe Anlagen 5 und 6.
Auswertung der Ergebnisse
Zur Bewertung der Gültigkeit und Aussagekraft des Versuchs sollte überprüft werden, ob die Dauer der Ausscheidungsphase eine Halbwertzeit überschreitet. Ebenso sollte der BCFKm (aus einem minimierten Versuch abgeleiteter kinetischer BCF) mit dem minimierten BCFSS-Wert (BCFSS, der am Ende der Aufnahmephase in der Annahme berechnet wird, dass ein Fließgewicht erreicht wird. Dies kann nur angenommen werden, da die Probenahmezeitpunkte für einen entsprechenden Nachweis nicht ausreichen) verglichen werden. Ist BCFKm < als der minimierte BCFSS, sollte letzterer als Wert der Wahl herangezogen werden. Ist der BCFKm kleiner als 70 % des minimierten BCFSS, sind die Ergebnisse unschlüssig, und es sollte ein vollständiger Versuch durchgeführt werden.
Wenn der minimierte Versuch einen BCFKm im Bereich eines aus regulatorischer Sicht bedenklichen Wertes ergibt, sollte eine vollständige Prüfung durchgeführt werden. Ist das Ergebnis weit von einem aus regulatorischer Sicht bedenklichen Wert entfernt (liegt es erheblich darüber oder darunter), so ist eine vollständige Prüfung u. U. nicht erforderlich oder es kann eine vollständige Prüfung mit nur einer Konzentration durchgeführt werden, sofern dies in maßgeblichen Rahmenvorschriften vorgeschrieben ist.
Wird nach einem minimierten Versuch mit nur einer Konzentration eine vollständige Prüfung als notwendig erachtet, kann diese mit einer zweiten Konzentration durchgeführt werden. Bei übereinstimmenden Ergebnissen kann auf eine weitere vollständige Prüfung mit einer weiteren Konzentration verzichtet werden, da die Biokonzentration des Stoffs voraussichtlich nicht konzentrationsabhängig ist. Wurde der minimierte Versuch mit zwei Konzentrationen durchgeführt und erweisen sich die Ergebnisse nicht als konzentrationsabhängig, so braucht die vollständige Prüfung mit nur einer Konzentration durchgeführt zu werden (siehe Nummer 87).
Prüfbericht
Der Prüfbericht für den minimierten Test sollte alle für die vollständige Prüfung erforderlichen Informationen enthalten (siehe Nummer 81), außer solche, die nicht generiert werden können (wie eine Kurve zur Anzeige der Zeit bis zum Erreichen des stationären Zustands und des Biokonzentrationsfaktors bei stationärem Zustand; für letzteren sollte stattdessen der minimierte BCFss angegeben werden). Zudem sollten die Gründe für die Anwendung des minimierten Tests und der resultierende BCFKm angegeben werden.
_____
1) Kann tatsächlich von einer schnellen Metabolisierung ausgegangen werden, so kann dies de facto durch den minimierten Test nachgewiesen werden.
2) Werden nur zwei Datenpunkte gemessen, können die Konfidenzgrenzen für den BCFKm nach Bootstrapping- Methoden geschätzt werden. Wenn Datenzwischenpunkte verfügbar sind, können die Konfidenzgrenzen für den BCFKm wie bei der vollständigen Prüfung berechnet werden.
C.13 - III: Bioakkumulationstest an Fischen - Exposition über das Futter
Einleitung
Die in diesem Abschnitt beschriebene Methode betrifft Stoffe, für die die Methode mit aquatischer Exposition nicht geeignet ist (beispielsweise weil keine stabilen, messbaren Wasserkonzentrationen aufrechterhalten werden können oder weil innerhalb einer Expositionsdauer von 60 Tagen keine angemessene Körperbelastung erreicht werden kann; siehe vorangegangenen Abschnitt zur Methode mit aquatischer Exposition). Es ist zu beachten, dass der Endpunkt bei diesem Test ein futterbezogener Biomagnifikationsfaktor (BMF) und kein Biokonzentrationsfaktor (BCF) 1 ist.
Im Mai 2001 wurde auf der SETAC Europe-Konferenz in Madrid eine neue Methode zur Prüfung der Bioakkumulation von Stoffen mit sehr geringer Wasserlöslichkeit vorgestellt (36). Diese Arbeiten basierten auf verschiedenen Bioakkumulationsstudien (über die in der Fachliteratur berichtet wurde), bei denen eine Dosierungsmethode mit dotiertem Futter (z.B. (37)) zum Einsatz kam. Anfang 2004 wurde der Entwurf eines Protokolls (38) zur Messung des Bioakkumulationspotenzials von Stoffen mit geringer Wasserlöslichkeit, bei denen der Standard-Bioakkumulationstest mit aquatischer Exposition nicht durchführbar war, vorgelegt, zusammen mit einem unterstützenden Beweisdokument (39) einer PBT-Arbeitsgruppe der EU. Als weiterer Rechtsfertigungsgrund für die Anwendung der Methode wurde angegeben, dass die potenzielle Umweltbelastung durch diese schwer wasserlöslichen Stoffe (d. h. Stoffe mit log KOW > 5) weitestgehend über das Futter erfolgt (vgl. (40) (41) (42) (43) (44)). Aus diesem Grund wird in einigen veröffentlichten Chemikalienverordnungen auf Prüfungen mit Exposition über das Futter verwiesen 2. Es ist jedoch zu beachten, dass bei der hier beschriebenen Methode die Exposition über die wässrige Phase sorgfältig vermieden wird und ein BMF-Wert aus dieser Prüfmethode somit nicht direkt mit einem BMF-Wert aus einer Feldstudie (bei der die aquatische Exposition und die Exposition über das Futter miteinander kombiniert werden kann) verglichen werden kann.
Dieser Abschnitt der vorliegenden Prüfmethode basiert auf diesem Protokoll (38) und entspricht einer neuen Methode, die in der vorherigen Fassung der Prüfmethode C.13 nicht enthalten war. Bei dieser alternativen Prüfung kann die Exposition über das Futter unter kontrollierten Laborbedingungen direkt untersucht werden.
Um zu entscheiden, wann die Prüfung mit Exposition über das Futter der Prüfung mit aquatischer Exposition vorzuziehen ist, sollten potenzielle Prüfer die Nummern 1 bis 14 dieser Prüfmethode konsultieren, die Informationen über verschiedene stoffliche Kriterien enthalten, die vor der Durchführung einer Prüfung berücksichtigt werden sollten.
Für radioaktiv markierte Stoffe gelten ähnliche Überlegungen wie bei der Methode mit aquatischer Exposition (siehe Nummern 6 und 65).
Die futterbasierte Methode gestattet es, mehrere Stoffe in einem einzigen Versuch zu prüfen, so lange bestimmte Kriterien erfüllt sind; siehe Nummer 112. Der Einfachheit halber wird hier eine Prüfung beschrieben, bei der nur ein Prüfstoff zum Einsatz kommt.
Der futterbasierte Test ähnelt der Methode mit aquatischer Exposition in vielerlei Hinsicht - bis auf den Expositionspfad. Daher überschneiden sich viele Aspekte der hier beschriebenen Methode mit der im vorstehende beschriebenen Methode mit aquatischer Exposition. Es wird, soweit möglich, auf die entsprechenden Punkte im vorangegangenen Abschnitt verwiesen, jedoch ist im Interesse der Leserfreundlichkeit und des guten Verständnisses ein gewisses Maß an Duplizierung unvermeidbar.
Prinzip der Prüfmethode
Durchfluss- oder semistatische Prüfbedingungen sind möglich (siehe Nummer 4); Durchflussbedingungen werden allerdings empfohlen, um die potenzielle aquatische Exposition gegenüber dem Prüfstoff infolge einer Desorption aus dotiertem Futter oder Exkrementen zu begrenzen. Die Prüfung umfasst zwei Phasen: Aufnahme (mit Prüfstoff dotiertes Futter) und Ausscheidung (reines, unbehandeltes Futter) (siehe Nummer 16). In der Aufnahmephase wird eine "Prüfgruppe" Fische täglich mit einem handelsüblichen prüfstoffdotierten Futter bekannter Zusammensetzung gefüttert. Die Fische sollten im Idealfall das gesamte angebotene Futter aufnehmen (siehe Nummer 141) und erhalten anschließend während der Ausscheidungsphase unbehandeltes handelsübliches Futter. Wie bei der Methode mit aquatischer Exposition können ggf. mehrere Prüfgruppen unterschiedlich stark dotierten Prüfstoffkonzentrationen ausgesetzt werden, jedoch reicht für die meisten stark hydrophoben organischen Prüfstoffe eine Prüfgruppe aus (siehe Nummern 49 und 107). Unter semistatischen Bedingungen sollten die Fische am Ende der Aufnahmephase in ein neues Medium und/oder eine neue Prüfkammer umgesetzt werden (falls das in der Aufnahmephase verwendete Medium und/oder die verwendete Apparatur durch Auslaufen mit dem Prüfstoff kontaminiert sind). Die Prüfstoffkonzentrationen in den Fischen werden in beiden Prüfungsphasen gemessen. Zusätzlich zu der mit dotiertem Futter gefütterten Fischgruppe (Prüfgruppe) wird eine Kontrollgruppe unter identischen Bedingungen gehalten und auf identische Weise gefüttert, außer dass die aus handelsüblichem Fischfutter bestehende Nahrung nicht mit dem Prüfstoff dotiert wurde. Diese Kontrollgruppe ermöglicht die Quantifizierung der Hintergrundwerte des Prüfstoffs in nicht exponierten Fischen und dient als Vergleich für behandlungsbedingte Schadwirkungen, die bei der bzw. den Prüfgruppe(n) festgestellt wurden 3. Sie ermöglicht zudem den Vergleich der Wachstumskonstanten zwischen den Gruppen und somit die Kontrolle, ob ähnliche Mengen des angebotenen Futters aufgenommen wurden (potenzielle Unterschiede in der Schmackhaftigkeit zwischen Futterarten sollten bei der Begründung unterschiedlicher Wachstumskonstanten ebenfalls berücksichtigt werden; siehe Nummer 138). Es ist wichtig, dass die Prüf- und die Kontrollgruppe während der Aufnahme- und Ausscheidungsphase gleichwertiges Futter erhalten.
Nach den Erfahrungen der Prüfmethodenentwickler reicht eine Aufnahmephase von 7-14 Tagen im Allgemeinen aus (38) (39). Auf diese Weise lassen sich die Kosten für die Durchführung der Prüfung begrenzen und für die meisten Stoffe gleichzeitig eine ausreichende Exposition sicherstellen. In bestimmten Fällen kann die Aufnahmephase jedoch verlängert werden (siehe Nummer 127). Während dieser Phase erreicht die Prüfstoffkonzentration in den Fischen möglicherweise keinen stationären Zustand, weshalb die Datenauswertung und Ergebnisse dieser Methode in der Regel auf einer kinetischen Analyse von Geweberückständen beruhen. (Hinweis: Wie schon bei der Prüfung mit aquatischer Exposition kann die bis zum Erreichen des stationären Zustands erforderliche Zeit anhand von Gleichungen geschätzt werden - siehe Anlage 5). Die Ausscheidungsphase beginnt, wenn die Fische erstmals nicht dotiertes Futter erhalten, und dauert bis zu 28 Tage oder bis der Prüfstoff nicht mehr im ganzen Fisch quantifizierbar ist, je nachdem, welcher Zeitpunkt zuerst eintritt. Die Ausscheidungsphase kann je nach gemessenen Prüfstoffkonzentrationen und Fischgröße verkürzt oder über28 Tage hinaus verlängert werden.
Diese Methode ermöglicht es, für den Prüfstoff im Fisch die stoffspezifische Halbwertzeit (t1/2, anhand der Ausscheidungskonstanten k2), die Assimilationseffizienz (Absorption im Darm, a), den kinetischen futterbezogenen Biomagnifikationsfaktor (BMFK), den wachstumskorrigierten kinetischen futterbezogenen Biomagnifikationsfaktor (BMFKg) und den lipidkorrigierten 4 kinetischen futterbezogenen Biomagnifikationsfaktor (BMFKL) (und/oder den wachstums- und lipidkorrigierten kinetischen futterbezogenen Biomagnifikationsfaktor, BMFKgL) zu bestimmen. Wie bei der Methode mit aquatischer Exposition führt die Zunahme der Fischmasse während der Prüfung zu einer Verdünnung der Prüfstoffkonzentration in wachsenden Fischen, was dazu führt, dass der (kinetische) BMF zu niedrig geschätzt wird, wenn er nicht um das Wachstum korrigiert wird (siehe Nummern 162 und 163). Wird ferner davon ausgegangen, dass in der Aufnahmephase ein stationärer Zustand erreicht wurde, kann ein indikativer BMF bei stationärem Zustand berechnet werden. Es gibt Ansätze, die die Schätzung eines kinetischen Biokonzentrationsfaktors (BCFK) auf Basis der in der futterbasierten Studie gewonnenen Daten ermöglichen (z.B. (44) (45) (46) (47) (48)). Pros und Kontras solcher Ansätze werden in Anlage 8 erörtert.
Der Test wurde primär für unpolare organische Stoffe mit geringer Löslichkeit entwickelt, die in Fischen ungefähr der Aufnahme- und Ausscheidungskinetik erster Ordnung folgen. Wird ein Stoff geprüft, der nicht der Aufnahme- und Ausscheidungskinetik erster Ordnung folgt, sollten komplexere Modelle verwendet (siehe Referenzdokumente in Anlage 5) und Biostatistiker und/oder Pharmakokinetiker konsultiert werden.
Der BMF wird normalerweise bestimmt, indem ganze Fische einer Prüfstoffanalyse unterzogen werden (basierend auf dem Nassgewicht). Sofern für die Ziele der Studie relevant, können Proben bestimmter Gewebe (z.B. Muskel, Leber) entnommen werden, wenn der Fisch in genießbare und ungenießbare Teile zerlegt wird (siehe Nummer 21). Zudem kann der Magen-Darm-Trakt entfernt und separat analysiert werden, um dessen Anteil an den Prüfstoffkonzentrationen für Probenahmepunkte am Ende der Aufnahmephase und kurz vor Beginn der Ausscheidungsphase oder im Rahmen eines Massenbilanzansatzes zu bestimmen.
Der Lipidgehalt der Ganzfischproben sollte gemessen werden, damit Konzentrationen zur Berücksichtigung des Lipidgehalts sowohl des Futters als auch und der Fische korrigiert werden können (siehe Nummern 56 und 57 und Anlage 7).
Zur Berechnung des während der Prüfung eventuell eingetretenen Wachstums sollte das Gewicht der beprobten Fische gemessen und protokolliert und der für die einzelnen Fische bestimmten Stoffkonzentration zugeordnet werden (z.B. über einen individuellen Kenncode für jeden beprobten Fisch). Die Gesamtlänge der einzelnen Fische sollte, soweit möglich, ebenfalls gemessen werden 5. Gewichtsdaten sind auch für die Schätzung des BCF anhand der Ausscheidungsdaten aus dem futterbasierten Test unerlässlich.
Angaben zum Prüfstoff
Es sollten die unter den Nummern 3 und 22 genannten Prüfstoffangaben vorliegen. Eine Methode zur Analyse der Prüfstoffkonzentrationen in Wasser ist in der Regel nicht notwendig; ausreichend empfindliche Methoden für die Messung der Konzentrationen in Fischfutter und Fischgewebe sind hingegen erforderlich.
Mit der Methode lassen sich im Rahmen eines einzigen Tests mehrere Stoffe untersuchen. Allerdings sollten die Prüfstoffe so kompatibel sein, dass sie nicht interagieren oder ihre chemische Identität bei der Dotierung ins Fischfutter verändern. Bezweckt wird, dass die Messergebnisse für jeden der zusammen getesteten Prüfstoffe nicht allzu stark von den Werten abweichen, die sich ergeben hätten, wenn jeder Prüfstoff einzeln getestet worden wäre. Mit Voranalysen sollten belegen, dass jeder Stoff in einer mehrfach dotierten Futter- und Fischgewebeprobe i) mit hoher Rate (z.B. > 85 % des Nennwerts) und ii) der für die Prüfung erforderlichen Empfindlichkeit wiedergefunden werden kann. Die Gesamtdosis zusammen geprüfter Stoffe sollte unter der kombinierten Konzentration liegen, die toxische Wirkungen hervorrufen könnte (siehe Nummer 51). Zudem sollten bei der Versuchsplanung mögliche Schadwirkungen in den Fischen und potenzielle interaktive Wirkungen (z.B. metabolische Wirkungen), die mit der gleichzeitigen Prüfung mehrerer Stoffe assoziiert werden, berücksichtigt werden. Die simultane Prüfung ionisierbarer Stoffe sollte vermieden werden. Unter Expositionsgesichtspunkten ist die Methode auch für komplexe Gemische (siehe Nummer 13, obwohl die gleichen analytischen Grenzen gelten wie für jede andere Methode) geeignet.
Validität der Prüfung
Ein Test wird als gültig betrachtet, wenn folgende Bedingungen erfüllt sind (siehe Nummer 24):
- Die Wassertemperatur schwankt bei den Behandlungs- oder Kontrollgruppen um weniger als ± 2 °C.
- Die Konzentration an gelöstem Sauerstoff fällt nicht unter einen Luftsättigungswert von 60 %.
- Die Prüfstoffkonzentration im Fischfutter vor und am Ende der Aufnahmephase liegt im Bereich von ± 20 % (basierend auf mindestens drei Probenahmen zu jedem der beiden Zeitpunkte).
- Bei Voranalysen des dotierten Futters sollte ein hohes Maß an Homogenität des Stoffs im Futter nachgewiesen werden; mindestens drei Stoffkonzentrationen, die in den zu Prüfungsbeginn entnommenen Proben gemessen wurden, sollten um nicht mehr als ± 15 % vom Mittelwert abweichen.
- Prüfstoffkonzentrationen werden nicht nachgewiesen oder sind im Vergleich zu behandelten Proben nur in typischen Spurenwerten in nicht dotiertem Futter oder in Kontrollfischgewebe vorhanden.
- Mortalität oder andere Schadwirkungen/Krankheiten bei Fischen der Kontroll- und der Prüfgruppe liegen am Ende der Prüfung unter 10 %; wird der Test aus einem bestimmten Grund verlängert, sollten Schadwirkungen bei beiden Fischgruppen weniger als 5 % pro Monat betragen und insgesamt 30 % nicht überschreiten. Signifikante Unterschiede im durchschnittlichen Wachstum bei beprobten Fischen der Prüf- und Kontrollgruppen könnten auf eine toxische Wirkung des Prüfstoffs hindeuten.
Referenzstoffe
Führt ein Labor die Prüfung zum ersten Mal durch oder wurden wesentliche Änderungen vorgenommen (beispielsweise Änderungen des Fischstammes oder der Bezugsquelle, wesentliche Änderung der Fischgröße, des Fischfutters oder des Dotierungsverfahrens usw.), sollte unter Verwendung eines Referenzstoffs eine technische Eignungsprüfung durchgeführt werden. Der Referenzstoff wird hauptsächlich verwendet, um nachzuweisen, ob das Verfahren der Futterdotierung maximale Homogenität und die Bioverfügbarkeit des Prüfstoffs gewährleistet. Ein Referenzstoff, der beispielsweise bereits bei unpolaren hydrophoben Stoffen verwendet wurde, ist Hexachlorbenzol (HCB), doch sollten aufgrund der Gefahrstoffeigenschaft von HCB auch andere Stoffe, für die zuverlässige Daten zu Aufnahme und Biomagnifikation vorliegen, erwogen werden 6. Falls HCB verwendet wird, sollte der Prüfbericht, wie auch für Prüfstoffe, alle wesentliche Referenzstoffangaben wie Name, Reinheit, CAS-Nummer , Struktur, Toxizitätsdaten (falls verfügbar) enthalten (siehe Nummern 3 und 22).
Beschreibung der Prüfmethode
Apparatur
Materialien und Apparatur sollten, wie bereits für die Methode mit aquatischer Exposition beschrieben, verwendet werden (siehe Nummer 26). Es sollte ein Durchflussverfahren oder ein statisches Erneuerungsverfahren angewandt werden, das Verdünnungswasser in ausreichender Menge zu den Prüfbehälter führt. Die Durchflussraten sollten protokolliert werden.
Wasser
Das Prüfwasser sollte, wie bereits für die Methode mit aquatischer Exposition beschrieben, verwendet werden (siehe Nummern 27-29). Das Prüfmedium sollte wie beschrieben charakterisiert werden und während der Prüfung von gleichbleibender Quaklität sein. Der natürliche Partikelgehalt und der gesamte organische Kohlenstoff (TOC) sollten vor Testbeginn möglichst gering sein (≤ 5 mg/l Partikel; ≤ 2 mg/l TOC). Der TOC muss lediglich vor der Prüfung als Teil der Charakterisierung des Prüfwassers gemessen werden (siehe Nummer 53).
Futter
Empfohlen wird handelsübliches Fischfutter (schwimmfähiges und/oder langsam absinkendes pelletiertes Futter), das zumindest in Bezug auf den Protein- und Fettgehalt charakterisiert ist. Das Futter sollte eine einheitliche Pelletgröße aufweisen, um die Exposition über das Futter zu optimieren, d. h. die Fische nehmen auf diese Weise mehr Futter auf und fressen nicht nur die größeren Stücke. Die Pellets sollten zu Prüfungsbeginn eine der Größe der Fische angepasste Größe aufweisen (z.B. sind Pelletdurchmesser von ungefähr 0,6-0,85 mm für Fische einer Gesamtlänge von 3 bis 7 cm und Pelletdurchmesser von ungefähr 0,85-1,2 mm für Fische einer Gesamtlänge von 6 bis 12 cm geeignet). Die Pelletgröße kann zu Beginn der Ausscheidungsphase dem Fischwachstum angepasst werden. Ein Beispiel einer geeigneten Zusammensetzung (handelsüblichen) Fischfutters findet sich in Anlage 7. Bei der Entwicklung dieser Methode wurde routinemäßig Prüffutter mit einem Gesamtlipidgehalt von 15 bis 20 % (w/w) verwendet. Fischfutter mit einer derart hohen Lipidkonzentration ist in bestimmten Regionen möglicherweise nicht erhältlich. In diesem Fall könnten die Versuche mit einer niedrigeren Lipidkonzentration im Futter durchgeführt werden und die Fütterungsrate könnte (auf Basis von Vorversuchen) angepasst werden, um die Fische gesund zu halten. Der Gesamtlipidgehalt des Futters der Prüf- und Kontrollgruppe muss vor Beginn der Prüfung und am Ende der Aufnahmephase gemessen und protokolliert werden. Der Prüfbericht sollte Herstellerangaben zu Nährstoffgehalt, Feuchtigkeit, Faser- und Aschegehalt und, falls möglich, Mineralien und Pestizidrückstände (z.B."standardmäßige" prioritäre Schadstoffe) des handelsüblichen Futters enthalten.
Bei der Dotierung des Futters mit dem Prüfstoff sollte sichergestellt werden, dass das Prüffutter homogen ist. Die Prüfstoffkonzentration im Futter der Prüfgruppe sollte unter Berücksichtigung der Empfindlichkeit des Analyseverfahrens, der Toxizität des Prüfstoffs (NOEC, soweit bekannt) und der relevanten physikalisch-chemischen Daten gewählt werden. Der Referenzstoff, falls verwendet, sollte vorbehaltlich der Analyseempfindlichkeit möglichst in einer Konzentration von ungefähr 10 % der Prüfstoffkonzentration (in jedem Fall so niedrig wie möglich sein) einbezogen werden (Beispiel: bei Hexachlorbenzol wurde eine Konzentration im Futter von 1-100 µg/g als akzeptabel erachtet; siehe (47) für weitere Informationen über Assimilationseffizienzen von HCB).
Das Fischfutter kann je nach physikalischen Eigenschaften und Löslichkeit auf verschiedene Weise mit dem Prüfstoff dotiert werden (für nähere Informationen über Dotierungsmethoden siehe Anlage 7):
- Ist der Stoff in Triglyceriden löslich und stabil, sollte er vor seiner Beimischung zum Fischfutter in einer geringen Menge Fischöl oder pflanzlichem Speiseöl gelöst werden. In diesem Fall sollte unter Berücksichtigung des natürlichen Lipidgehalts des dotierten Futters die Herstellung von Rationen mit zu hohem Lipidgehalt unbedingt vermieden werden, indem die bekannte Mindestmenge Öl beigemischt wird, die erforderlich ist, um eine gute Verteilung und Homogenität des Prüfstoffs im Futter zu erreichen, oder
- das Futter sollte mithilfe eines geeigneten organischen Lösungsmittels dotiert werden, soweit dies die Homogenität und Bioverfügbarkeit nicht beeinträchtigt (im Futter können sich infolge der Verdunstung des Lösungsmittels (Mikro-)Kristalle des Prüfstoffs bilden, und es lässt sich nicht einfach nachweisen, dass dies nicht der Fall ist; siehe (49)), oder
- nicht zähflüssige Flüssigkeiten sollten dem Fischfutter direkt beigegeben, jedoch gut vermischt werden, um Homogenität und gute Assimilation zu gewährleisten. Gutes Mischen dürfte die Homogenität des dotierten Futters gewährleisten.
In bestimmten Fällen, z.B. bei weniger hydrophoben Prüfstoffen, die mit größerer Wahrscheinlichkeit aus dem Futter desorbieren, müssen vorbereitete Fischpellets möglicherweise mit einer geringen Menge Maiskeim- oder Fischöl beschichtet werden (siehe Nummer 142). In diesen Fällen sollten das Kontrollfutter gleich behandelt und das fertig zubereitete Futter für die Lipidmessung verwendet werden.
Die Ergebnisse für den Referenzstoff, falls verwendet, sollten mit den Daten einer in der Fachliteratur beschriebenen, unter ähnlichen Bedingungen und mit einer vergleichbarer Fütterungsrate durchgeführten Studie komparabel sein (siehe Nummer 45), und die spezifischen Parameter des Referenzstoffs sollten die relevanten Kriterien unter Nummer 113 (Unterpunkte 3, 4 und 5) erfüllen.
Wenn als Vehikel für den Prüfstoff Öl oder ein Trägerlösungsmittel verwendet wird, sollte eine entsprechende Menge desselben Vehikels (Prüfstoff ausgenommen) mit dem Kontrolfutter vermischt werden, um ein dem dotierten Futter gleichwertiges Futter zu erhalten. Dabei ist wichtig, dass die Prüf- und Kontrollgruppen während der Aufnahme- und der Ausscheidungsphase gleichwertiges Futter erhalten.
Das dotierte Futter sollte unter Bedingungen gelagert werden, die die Stabilität des Prüfstoffs innerhalb der Futtermischung garantieren (z.B. durch Kühlung); diese Bedingungen sind anzugeben.
Auswahl der Fischspezies
Es können die gleichen Fischspezies verwendet werden, die auch für die aquatische Exposition in Frage kommen (siehe Nummer 32 und Anlage 3). Vor Veröffentlichung dieser Prüfmethode wurden für futterbasierte Bioakkumulationsstudien mit organischen Stoffen Regenbogenforellen (Oncorhynchus mykiss), Karpfen (Cyprinus carpio) und Dickkopfelritzen (Pimephales promelas) verwendet. Die Prüfspezies sollten ein Fressverhalten aufweisen, das den raschen Verzehr der verabreichten Futterration gewährleistet, um Faktoren, die die Konzentration des Prüfstoffs im Futter beeinflussen könnten (wie Auslaugen ins Wasser und Möglichkeit aquatischer Exposition), auf ein Mindestmaß zu begrenzen. Es sollten Fische innerhalb des empfohlenen Größen-/Gewichtsbereichs (siehe Anlage 3) verwendet werden. Sie sollten nicht so klein sein, dass einzelne Fische nur mit Schwierigkeiten analysiert werden können. Bei Spezies, die in einer Lebensphase schnellen Wachstums untersucht werden, kann die Datenauswertung schwierig sein, und hohe Wachstumsraten können die Berechnung der Assimilationseffizienz beeinflussen 7.
Haltung der Fische
Die Kriterien bezüglich Akklimatisierung, Mortalität und Erkrankung vor Prüfungsbeginn sind dieselben wie für die Methode mit aquatischer Exposition (siehe Nummern 33-35).
Durchführung des Tests
Vorversuch und Dosisfindungstest
Ein analytischer Vorversuch ist notwendig, um Wiederfindung des Prüfstoffs im dotierten Fischfutter/Fischgewebe nachzuweisen. Ein Dosisfindungstest zur Entscheidung über die geeignete Prüfstoffkonzentration im Futter ist nicht immer erforderlich. Um nachzuweisen, dass keine Schadwirkungen auftreten, und die Schmackhaftigkeit des dotierten Futters, die Empfindlichkeit der Methode für die Analyse des Fischgewebes und des Futters und die Wahl der geeigneten Fütterungsrate sowie die Zeitabstände für die Probenahmen während der Ausscheidungsphase usw. zu bewerten, können Fütterungsvorversuche durchgeführt werden, sind aber nicht erforderlich. Eine Vorstudie kann hilfreich sein, um abzuschätzen, wie viele Fische während der Ausscheidungsphase beprobt werden müssen. Dies kann zu einer erheblichen Verringerung der Zahl der Versuchsfische führen, insbesondere bei Prüfstoffen, die metabolisierungsanfällig sind.
Expositionsbedingungen
Dauer der Aufnahmephase
Eine Aufnahmephase von 7-14 Tagen, in der eine Gruppe Fische täglich das Kontrollfutter und eine zweite Gruppe Fische täglich eine auf die Prüfspezies und Versuchsbedingungen zugeschnittene Ration Prüffutter (z.B. 1-2 % des Körpergewichts (Nassgewicht) bei Regenbogenforellen) erhält, reicht in der Regel aus. Die Fütterungsrate sollte so gewählt werden, dass schnelles Wachstum und eine starke Zunahme des Lipidgehalts vermieden werden. Die Aufnahmephase kann erforderlichenfalls auf Basis praktischer Erfahrungen aus früheren Versuchen oder von Informationen über das Aufnahme-/Ausscheidungsverhalten des Prüfstoffs (oder eines analogen Stoffs) bei Fischen verlängert werden. Der Prüfungsbeginn ist definiert als der Zeitpunkt der ersten Fütterung mit dotiertem Futter. Ein Versuchstag reicht vom Zeitpunkt der Fütterung bis kurz vor den Zeitpunkt der nächsten Fütterung (z.B. eine Stunde). Somit beginnt der erste Versuchstag (Aufnahmephase) zum Zeitpunkt der ersten Fütterung mit dotiertem Futter und endet kurz vor der zweiten Fütterung mit dotiertem Futter. In der Praxis endet die Aufnahmephase kurz vor (z.B. eine Stunde vor) der ersten Fütterung mit undotiertem Prüfstoff, da die Fische das dotierte Futter in den dazwischenliegenden 24 Stunden weiterhin verdauen und den Prüfstoff absorbieren. Im Interesse der Analysemethode muss sichergestellt werden, dass eine ausreichend hohe (nicht toxische) Körperbelastung durch den Prüfstoff erreicht wird, damit in der Ausscheidungsphase zumindest ein Rückgang um eine Zehnerpotenz gemessen werden kann. In besonderen Fällen kann eine (auf bis zu 28 Tage) verlängerte Aufnahmephase mit weiteren Probenahmen beschlossen werden, um Informationen über die Aufnahmekinetik zu erhalten. Während der Aufnahme erreicht die Konzentration möglicherweise keinen stationären Zustand. Wie auch bei der Prüfung mit aquatischer Exposition können Gleichungen zur Schätzung der Zeit bis zum Erreichen des stationären Zustands (als Anhaltspunkt für die wahrscheinlich erforderliche Zeit bis zum Erreichen nennenswerter Konzentrationen in den Fischen) verwendet werden (siehe Anlage 5).
In bestimmten Fällen ist möglicherweise bekannt, dass die Aufnahme des Prüfstoffs durch die Fische über einen Zeitraum von 7-14 Tagen aufgrund der unzulänglichen Empfindlichkeit der Methode oder einer schlechten Assimilationseffizienz nicht ausreichen wird, um mit der verwendeten Futterkonzentration eine Prüfstoffkonzentration in den Fischen zu erreichen, die hoch genug ist, um während der Ausscheidung zumindest einen Rückgang um eine Zehnerpotenz messen zu können. In solchen Fällen kann es von Vorteil sein, die anfängliche Fütterungsphase über 14 Tage hinaus zu verlängern, oder es sollte eine höhere Prüfstoffkonzentration im Futter in Betracht gezogen werden, insbesondere bei stark metabolisierenden Stoffen. Es sollte jedoch beachtet werden, dass die Körperbelastung während der Aufnahme unterhalb der (geschätzten) chronischen Konzentration bleibt, bei der keine statistisch signifikante Wirkung beobachtet wird (NOEC) (siehe Nummer 138).
Dauer der Ausscheidungsphase
Die Ausscheidung dauert in der Regel bis zu 28 Tage und beginnt, wenn die Fische der Prüfgruppe im Anschluss an die Aufnahmephase reines, unbehandeltes Futter Nahrung erhalten. Die Ausscheidung beginnt mit der ersten Fütterung mit "undotiertem" Futter und nicht etwa direkt nach der letzten Fütterung mit "dotiertem" Futter, da die Fische das Futter in den dazwischenliegenden 24 Stunden weiterhin verdauen und den Prüfstoff absorbieren, wie unter Nummer Punkt 126 beschrieben. Somit wird die erste Probe in der Ausscheidungsphase kurz vor der zweiten Fütterung mit undotiertem Futter entnommen. Mit dieser Ausscheidungsphase sollen Stoffe mit einer potenziellen Halbwertzeit von bis zu 14 Tagen erfasst werden, was mit der Halbwertzeit bioakkumulativer Stoffe übereinstimmt 8, sodass 28 Versuchstage zwei Halbwertzeiten solcher Stoffe umfassen. Bei sehr stark bioakkumulierenden Stoffen kann es von Vorteil sein, die Ausscheidungsphase zu verlängern (falls aufgrund von Vorversuchen indiziert).
Wird ein Stoff so langsam ausgeschieden, dass in der Ausscheidungsphase keine exakte Halbwertzeit bestimmt werden kann, reichen die erhaltenen Informationen möglicherweise dennoch aus, um ein hohes Bioakkumulationsniveau anzuzeigen und Bewertungen vorzunehmen. Wird ein Stoff hingegen so schnell ausgeschieden, dass eine zuverlässige Konzentration zum Zeitpunkt Null (Konzentration am Ende der Aufnahme/Anfang der Ausscheidung, C0,d) und k2 nicht abgeleitet werden können, kann eine konservative Schätzung von k2 vorgenommen werden (siehe Anlage 7).
Wenn frühere Analysen der (z.B. nach 7 oder 14 Tagen) beprobten Fische zeigen, dass der Stoff vor Ablauf der vollen 28 Tage unterhalb des Quantifizierungsniveaus ausgeschieden wurde, können nachfolgende Probenahmen eingestellt und die Prüfung beendet werden.
In bestimmten Fällen lässt sich am Ende der Aufnahmephase (oder bei der zweiten Ausscheidungsprobe) keine messbare Aufnahme des Prüfstoffs feststellen. Wenn nachgewiesen werden kann, dass i) die Validitätskriterien gemäß Nummer 113 erfüllt sind und ii) eine mangelnde Aufnahme nicht auf andere Prüfungsmängel (z.B. zu kurze Aufnahmephase, mangelhafte Futterdotierung mit entsprechend schlechter Bioverfügbarkeit, mangelnde Empfindlichkeit des Analyseverfahrens, kein Futterverzehr durch die Fische usw.) zurückzuführen ist, kann die Prüfung beendet werden, ohne dass sie mit einer längeren Aufnahmephase erneut durchgeführt werden muss. Hat sich im Vorversuch gezeigt, dass dies der Fall sein kann, kann im Rahmen eines Massenbilanzansatzes soweit möglich eine Analyse der Exkremente auf unverdauten Prüfstoff ratsam sein.
Anzahl Versuchsfische
Ähnlich wie bei der Prüfung mit aquatischer Exposition sollten Fische von vergleichbarem Gewicht und vergleichbarer Länge gewählt werden, wobei der kleinste Fisch nicht weniger als zwei Drittel des Gewichts des größten Fisches haben sollte (siehe Nummern 40-42).
Die Gesamtzahl der für die Studie verwendeten Fische sollte sich nach dem Probenahmezeitplan richten (mindestens eine Probenahme am Ende der Aufnahmephase und 4-6 Probenahmen während der Ausscheidungsphase, je nach Dauer der Phasen), wobei die Empfindlichkeit des Analyseverfahrens, die am Ende Aufnahmephase wahrscheinliche Konzentration (auf Basis früherer Erkenntnisse) und die Ausscheidungsphase (falls frühere Erkenntnisse eine Schätzung zulassen) berücksichtigt werden sollten. Bei jeder Probenahme sollten 5-10 Fische entnommen werden, wobei die Wachstumsparameter (Gewicht und Gesamtlänge) vor der chemischen Analyse oder der Lipidanalyse gemessen werden.
Aufgrund der unvermeidbaren Größen-, Wachstums- und physiologischen Unterschiede zwischen den Fischen und der wahrscheinlich unterschiedlichen Futtermenge, die jeder Fisch verzehrt, sollten zu jedem Probenahmezeitpunkt mindestens 5 Fische aus der Prüfgruppe und 5 Fische aus der Kontrollgruppe entnommen werden, um die Durchschnittskonzentration und ihre Variabilität bestimmen zu können. Die Variabilität der Fischparameter trägt wahrscheinlich stärker zur unkontrollierten Gesamtvariabilität der Prüfung bei als die inhärente Variabilität der angewandten Analysemethoden und rechtfertigt somit in bestimmten Fällen die Verwendung von bis zu 10 Fischen je Probenahme. Sind die Hintergrundkonzentrationen des Prüfstoffs in den Kontrollfischen zu Beginn der Ausscheidung jedoch nicht messbar, reicht eine chemische Analyse von 2-3 Kontrollfischen bei der letzten Probenahme nur dann aus, wenn die übrigen Kontrollfische bei jeder Probenahme weiterhin zur Erfassung von Gewicht und Gesamtlänge beprobt werden (um zwar so, dass zur Wachstumsbestimmung jedes Mal dieselbe Anzahl Fische aus der Prüf- und der Kontrollgruppe untersucht wird). Die Fische sollten gelagert und einzeln gewogen (selbst wenn es sich als notwendig erweisen sollte, die Probenergebnisse anschließend zu kombinieren) und ihre Gesamtlänge sollte gemessen werden.
So erfordert ein Standardtest mit einer beispielsweise 28 Tage dauernden Ausscheidungsphase und fünf Ausscheidungsproben insgesamt 59-120 Fische aus der Prüf- und 50-110 Fische aus der Kontrollgruppe, wobei davon ausgegangen wird, dass es das Analyseverfahren für den Stoff erlaubt, den Lipidgehalt am selben Fisch zu analysieren. Können die Lipidanalyse und die chemische Analyse nicht am selben Fisch durchgeführt werden und ist die Verwendung von Kontrollfischen ausschließlich zum Zwecke der Lipidanalyse nicht möglich (siehe Nummer 56), wären weitere 15 Fische erforderlich (drei aus der Stammpopulation zu Prüfungsbeginn, jeweils drei aus der Kontroll- und der Prüfgruppe zu Beginn der Ausscheidung und jeweils drei aus der Kontroll- und Prüfgruppe am Ende des Versuchs). Ein Beispiel eines Probenahmeplans mit den entsprechenden Fischzahlen findet sich in Anlage 4.
Besatz
Es empfiehlt sich ein ebenso großes Wasser/Fisch-Verhältnis wie bei der Methode mit aquatischer Exposition (siehe Nummern 43 und 44). Obwohl sich die Fisch/Wasser-Besatzraten nicht auf die Expositionskonzentrationen in diesem Versuch auswirken, wird eine Besatzrate von 0,1-1,0 g Fisch (Nassgewicht) pro Liter Wasser und Tag empfohlen, um angemessene Konzentrationen an gelöstem Sauerstoff aufrechtzuerhalten und die Stressbelastung der Prüforganismen möglichst gering zu halten.
Prüffutter und Fütterung
Während der Akklimatisierungsphase sollten die Fische, wie oben beschrieben, geeignetes Futter erhalten (Nummer 117). Wird der Test unter Durchflussbedingungen durchgeführt, sollte der Durchfluss während der Fütterung unterbrochen werden.
Während der Prüfung sollte die Prüfgruppe nur das vorstehend beschriebene Futter erhalten (Nummern 116-121). Neben stoffspezifischen Faktoren, der Empfindlichkeit der Analysemethode, der erwarteten Prüfstoffkonzentration im Futter unter bestimmten Umweltbedingungen und der chronischen Toxizitätswerte/Körperbelastung sollte zur Festlegung der Zielkonzentration des Prüfstoffs im Futter auch dessen Schmackhaftigkeit berücksichtigt werden (damit die Fische das Futter nicht ablehnen). Die nominelle Dotierungskonzentration sollte im Bericht festgehalten werden. Erfahrungsgemäß sind Dotierungskonzentrationen von 1-1.000 µg/g für Versuche mit Prüfstoffen geeignet, die keinen spezifischen toxischen Mechanismus aufweisen. Bei Stoffen, die über einen unspezifischen Mechanismus wirken, sollten die Geweberückstände 5 µmol/g Lipid nicht überschreiten, da Rückstände über diesem Niveau wahrscheinlich chronische Wirkungen hervorrufen (19) (48) (50) 9. Bei anderen Stoffen sollte darauf geachtet werden, dass aufgrund der akkumulierten Exposition keine Schadwirkungen auftreten (siehe Nummer 127). Dies gilt insbesondere, wenn mehrere Stoffe gleichzeitig geprüft werden (siehe Nummer 112).
Das Fischfutter kann auf drei verschiedene Arten mit der geeigneten Menge Prüfstoff dotiert werden (siehe Nummer 119 und Anlage 7). Die Methoden und Verfahren für die Futterdotierung sollten im Bericht festgehalten werden. Die Kontrollfische erhalten unbehandeltes Futter, das eine gleichwertige Menge an undotiertem Öl enthält, soweit Öl als Vehikel in der Aufnahmephase im dotierten Futter verwendet wird, oder das mit "reinem" Lösungsmittel behandelt wurde, wenn ein Lösungsmittel bei der Zubereitung des Futters für die Prüfgruppe als Vehikel verwendet wurde. Die Prüfstoffkonzentration im behandelten und unbehandelten Futter sollte vor Beginn und am Ende der Aufnahmephase mindestens drei Mal analysiert werden. Nach der Exposition gegenüber dem behandelten Futter (Aufnahmephase) erhalten die Fische (beide Gruppen) unbehandeltes Futter (Ausscheidungsphase).
Die Fische erhalten eine bestimmte Ration (je nach Spezies; z.B. ca. 1-2 % des Nasskörpergewichts pro Tag im Falle der Regenbogenforelle). Die Fütterungsrate sollte gewährleisten, dass schnelles Wachstum und eine starke Zunahme des Lipidgehalts vermieden werden. Die exakte Fütterungsrate, die während des Versuchs festgelegt wurde, sollte protokolliert werden. Die anfängliche Fütterung sollte auf der Grundlage der Gewichtsmessungen der Stammpopulation unmittelbar vor Prüfungsbeginn erfolgen. Die Futtermenge sollte je nach Nassgewicht der bei jeder Probenahme entnommenen Fische angepasst werden, um das Wachstum während des Versuchs zu berücksichtigen. Die Gewichte und Längen der Fische in den Prüf- und Kontrollbehältern können auf Basis der Gewichte und Gesamtlängen der für die einzelnen Probenahmen verwendeten Fische geschätzt werden; die übrigen Fische in den Prüf- oder Kontrollbehältern sollten nicht gewogen oder gemessen werden. Es ist wichtig, dass die vorgegebene Fütterungsrate während des gesamten Versuchs aufrechterhalten wird.
Die Fütterung sollte beobachtet werden, um sicherzustellen, dass die Fische das gesamte verabreichte Futter verzehren und die für die Berechnungen verwendeten Ingestionsraten korrekt sind. Bei der Festlegung einer Fütterungsrate, die gewährleistet, dass bei einer einmal täglichen Fütterung das gesamte Futter aufgenommen wird, sollten Fütterungsvorversuche oder vorherige Erfahrungswerte berücksichtigt werden. Wird systematisch ein Teils des Futters nicht verzehrt, empfiehlt es sich, die Dosis mit einer zweiten Fütterung über den Versuchstag zu verteilen (d. h. eine Tagesration - zwei Fütterungen). Falls erforderlich, sollte die zweite Fütterung zu einem bestimmten Zeitpunkt erfolgen und zeitlich so festgelegt werden, dass vor der Beprobung der Fische so viel Zeit wie möglich vergeht (diese zweite Fütterung erfolgt beispielsweise in der ersten Hälfte eines Versuchstags).
Obwohl die Fische das Futter in der Regel rasch aufnehmen, muss unbedingt sichergestellt werden, dass der Prüfstoff an das Futter adsorbiert bleibt. Es sollte vermieden werden, dass sich der Prüfstoff aus dem Futter ins Wasser verteilt und von den Fischen zusätzlich zum Futter auch in wässrigen Konzentrationen aufgenommen werden. Dies wird erreicht, indem nicht verzehrtes Futter (und Exkremente) innerhalb einer Stunde, jedoch vorzugsweise innerhalb von 30 Minuten, nach der Fütterung aus den Prüf- und Kontrollbehälten entfernt wird (werden). Zudem kann ein System angewendet werden, bei dem das Wasser kontinuierlich über einen Aktivkohlefilter gereinigt wird und "gelöste" Verunreinigungen absorbiert werden. Durchflusssysteme können dazu beitragen, Partikel und gelöste Stoffe schnell wegzuspülen 10. In bestimmten Fällen lässt sich das Problem durch Modifikation des Verfahrens für die Zubereitung des dotierten Futters abschwächen (siehe Nummer 119).
Licht und Temperatur
Wie bei der Methode mit aquatischer Exposition (siehe Nummer 48) wird eine Photoperiode von 12-16 Stunden und eine für die verwendete Prüfspezies geeignete Temperatur (± 2 °C) empfohlen (siehe Anlage 3). Art und Merkmale der Beleuchtung sollten bekannt sein und dokumentiert werden.
Kontrollen
Es sollte eine Kontrollgruppe aus Fischen gebildet werden, die dieselbe Futterration wie die Prüfgruppe erhalten, jedoch ohne Prüfstoff. Wurde in der Prüfgruppe Öl oder ein Lösungsmittel als Vehikel für die Futterdotierung verwendet, sollte das Futter der Kontrollgruppe auf exakt dieselbe Weise, jedoch ohne Prüfstoff, verabreicht werden, d. h. Prüf- und Kontrollgruppe sollten dasselbe Futter erhalten (siehe Nummer 121 und 139).
Häufigkeit der Wasserqualitätsmessungen
Die Bedingungen, die für die Methode mit aquatischer Exposition beschrieben wurden, gelten analog, außer dass der TOC nur vor der Prüfung als Teil der Prüfwassercharakterisierung gemessen werden muss (siehe Nummer 53).
Beprobung und Analyse von Fischen und Futter
Analyse der Futterproben
Proben des Futters der Prüf- und Kontrollgruppe sollten zumindest vor Beginn und am Ende der Aufnahmephase und zumindest dreifach auf Prüfstoff und Lipidgehalt untersucht werden. Die Analysemethoden sowie die Verfahren, mit denen die Homogenität des Futters gewährleistet werden soll, sind im Bericht anzugeben.
Die Proben sollten nach der vorgegebenen und validierten Methode auf Prüfstoff analysiert werden. Es sollte ein Vorversuch durchgeführt werden, um für die vorgesehene Probenmatrix die Quantifizierungsgrenze, die Wiederfindungsrate (in Prozent), etwaige Interferenzen und die Variabilität der Analyse zu bestimmen. Wird radioaktiv markiertes Material geprüft, sollten dieselben Aspekte geprüft werden wie bei der Methode mit aquatischer Exposition, wobei die Futteranalyse die Wasseranalyse ersetzt (siehe Nummer 65).
Analyse der Fischproben
Für jede Beprobung werden 5-10 einzelne Fische aus den Prüf- und Kontrollgruppen entnommen (in bestimmten Fällen kann die Zahl der Kontrollfische verringert werden; siehe Nummer 134).
Die Proben sollten an jedem Versuchstag zum selben Zeitpunkt (je nach Fütterungstermin) entnommen werden, und zwar möglichst zu einem Zeitpunkt, der die Wahrscheinlichkeit, dass Futter während der Aufnahmephase und zu Beginn der Ausscheidungsphase im Darm verbleibt, auf ein Minimum begrenzt, um unerwünschte Verfälschungen der Gesamtprüfstoffkonzentrationen zu vermeiden (d. h. zu beprobende Fische sollten gegen Ende eines Versuchstags entnommen werden, wobei zu beachten ist, dass ein Versuchstag mit dem Zeitpunkt der Fütterung beginnt und mit dem Zeitpunkt der nächsten Fütterung, also ungefähr 24 Stunden später, endet. Die Ausscheidung beginnt mit der ersten Fütterung des undotierten Futters; siehe Nummer 128). Die erste Probenahme in der Ausscheidungsphase (kurz vor der zweiten Fütterung mit undotiertem Futter) ist wichtig, denn sie dient der Extrapolation der Konzentration zum Zeitpunkt Null, einen Tag vor dieser Messung (C0,d, die Konzentration in den Fischen am Ende der Aufnahme/Anfang der Ausscheidung). Es besteht auch die Möglichkeit, den Magen-Darm-Trakt der Fische zu entfernen und am Ende der Aufnahme sowie an den Tagen 1 und 3 der Ausscheidung separat zu analysieren.
Bei jeder Probenahme sollten aus beiden Prüfbehältern Fische entnommen und auf dieselbe Weise behandelt werden wie bei der Methode mit aquatischer Exposition beschrieben (siehe Nummer 61-63).
Die Prüfstoffkonzentrationen im Ganzfisch (Nassgewicht) werden mindestens am Ende der Aufnahmephase und während der Ausscheidungsphase und zwar sowohl für die Kontroll- als auch für die Prüfgruppe gemessen. Für die Ausscheidungsphase werden 4-6 Probenahmen empfohlen (z.B. an den Tagen 1, 3, 7, 14 und 28). Optional kann eine zusätzliche Probenahme nach 1-3 Tagen der Aufnahme hinzugezogen werden, um die Assimilationseffizienz ab der linearen Aufnahmephase zu schätzen, wenn die Fische sich noch am Anfang der Expositionsperiode befinden. Zwei Abweichungen vom Versuchsplan sind möglich: i) wenn die Aufnahmephase zur Untersuchung der Aufnahmekinetik verlängert wird, sind für diese Phase weitere Probenahmezeitpunkte vorzusehen, um mehr Fische untersuchen zu können (siehe Nummer 126); ii) wenn am Ende der Aufnahmephase keine Aufnahme messbar ist, muss der Versuch beendet werden (siehe Nummer 131). Jeder entnommene Fisch sollte gewogen (und seine Gesamtlänge gemessen) werden, um die Wachstumskonstanten bestimmen zu können. Die Stoffkonzentrationen in bestimmten Fischgeweben (genießbare und ungenießbare Teile) können auch am Ende der Aufnahmephase und zu ausgewählten Zeitpunkten während der Ausscheidung gemessen werden. Wird radioaktiv markiertes Material geprüft, sollten ähnliche Aspekte wie bei der Methode mit aquatischer Exposition geprüft werden, wobei die Futteranalyse die Wasseranalyse ersetzt (siehe Nummer 65).
Bei regelmäßiger Verwendung eines Referenzstoffs (siehe Nummer 25) sollten die Konzentrationen in der Prüfgruppe möglichst am Ende der Aufnahme und zu allen für den Prüfstoff festgelegten Ausscheidungszeitpunkten gemessen werden (Ganzfische); in der Kontrollgruppe müssen die Konzentrationen nur am Ende der Aufnahme analysiert werden (Ganzfische). Unter bestimmten Umständen (beispielsweise, wenn die Analyseverfahren für die Prüf- und die Referenzsubstanz inkompatibel sind, sodass mehr Fische notwendig werden, um den Probenahmeplan einzuhalten) kann ein anderer Ansatz gewählt werden, um die Zahl der zusätzlichen Fische auf ein Mindestmaß zu begrenzen. Die Referenzstoffkonzentrationen werden während der Ausscheidung nur an den Tagen 1 und 3 sowie zwei weiteren Probenahmezeitpunkten gemessen, die so gewählt werden, dass die Konzentration zum Zeitpunkt Null (C0,d) und k2 für den Referenzstoff zuverlässig geschätzt werden kann.
Soweit möglich, sollte der Lipidgehalt der einzelnen Fische bei jeder Probename oder zumindest am Anfang und Ende der Aufnahmephase und am Ende der Ausscheidungsphase bestimmt werden. (siehe Nummern 56 und 67). Je nach Analysemethode (siehe Nummer 67 und Anlage 4) ist es u. U. möglich, zur Bestimmung des Lipidgehalts und der Prüfstoffkonzentration dieselben Fische zu verwenden, was sich im Interesse der Begrenzung der Versuchstierzahl empfiehlt. Sollte dies jedoch nicht möglich sein, kann derselbe Ansatz wie bei der Methode mit aquatischer Exposition gewählt werden (siehe Nummer 56 für alternative Möglichkeiten der Lipidgehaltmessung). Die für die Quantifizierung des Lipidgehalts angewandte Methode sollte im Bericht dokumentiert werden.
Qualität der Analysemethode
Es sollten Versuchskontrollen durchgeführt werden, um die Spezifität, Genauigkeit, Präzision und Reproduzierbarkeit des stoffspezifischen Analyseverfahrens sowie die Wiederfindung des Prüfstoffs im Futter und in den Fischen zu gewährleisten.
Messung des Fischwachstums
Zu Beginn der Prüfung muss eine Probe Fische aus der Stammpopulation gewogen (und ihre Gesamtlänge gemessen) werden. Diese Fische sollten kurz vor der ersten Fütterung mit dotiertem Futter (z.B. 1 Stunde davor) entnommen und dem Versuchstag 0 zugeordnet werden. Die Zahl der Fische in dieser Probe sollte der für die Probenahmen während der Prüfung vorgesehenen Zahl zumindest entsprechen. Dabei kann sich um dieselben Fische handeln, die vor Beginn der Aufnahmephase für die Lipidanalyse verwendet wurden (siehe Nummer 153). Zu jedem Probenahmezeitpunkt werden die Fische zunächst gewogen und längenvermessen. Für jeden Fisch sollten Messgewicht (und Messlänge) der analysierten Stoffkonzentration (und ggf. dem Lipidgehalt) zugeordnet werden, z.B. anhand eines individuellen Kenncodes für jeden untersuchten Fisch. Anhand der Messwerte für diese Fischproben können Gewicht (und Länge) der in den Prüf- und Kontrollbehältern verbleibenden Fische geschätzt werden.
Evaluierung des Versuchs
Die Mortalität der Fische sollte täglich protokolliert werden. Es sollte außerdem auf Schadwirkungen wie Verhaltensanomalien oder Pigmentierung geachtet werden; entsprechende Vorkommnisse sind aufzuzeichnen. Fische gelten als tot, wenn keine Atmung mehr stattfindet und es bei leichter mechanischer Stimulierung zu keiner Reaktion kommt. Tote oder eindeutig moribunde Fische sollten entfernt werden.
Daten und Berichterstattung
Auswertung der Ergebnisse
Die Prüfungsergebnisse werden verwendet, um anhand des Gesamtnassgewichts der Fische die Ausscheidungskonstante (k2) abzuleiten. Die Wachstumskonstante, kg, wird anhand der mittleren Zunahme des Fischgewichts berechnet und ggf. zur Bestimmung der wachstumskorrigierten Ausscheidungskonstante, k2g, verwendet. Zudem sollten die Assimilationseffizienz (a; Absorption aus dem Darm), der kinetische Biomagnifikationsfaktor (BMFK) (ggf. der wachstumskorrigierte Biomagnifikationsfaktor, BMFKg), der lipidkorrigierte Wert (BMFKL oder BMFKgL, falls um die Verdünnung durch Wachstum korrigiert) und die Fütterungsrate dokumentiert werden. Soweit die Zeit bis zum Erreichen des stationären Zustands in der Aufnahmephase geschätzt werden kann (z.B. 95 % des stationären Zustands oder t95 = 3,0/k2), kann auch der BMF im stationären Zustand (BMFSS) einbezogen werden (siehe Nummern 105 und 106 und Anlage 5), falls der t95-Wert darauf hindeutet, dass ein stationären Zustand erreicht zu sein scheint. Auf diesen BMFSS sollte dieselbe Lipidgehaltkorrektur angewendet werden wie auf den kinetisch abgeleiteten BMF (BMFK), um einen lipidkorrigierten Wert, BMFSSL, zu erhalten (dabei ist zu beachten, dass ein anerkanntes Verfahren für die Korrektur eines BMF im stationären Zustand um den Effekt der Verdünnung durch Wachstum existiert). Formeln und Berechnungsbeispiele finden sich in Anlage 7. Es existieren verschiedene Ansätze zur Schätzung eines kinetischen Biokonzentrationsfaktors (BCFK) anhand von Daten aus der futterbasierten Studie. Sie sind Gegenstand von Anlage 8.
Daten zu Fischgewicht und Fischlänge
Die Nassgewichte und Längen der einzelnen Fische werden, aufgeschlüsselt nach Prüf- und Kontrollgruppen, für alle Probenahmezeitpunkte in der Aufnahmephase (Stammpopulation zu Beginn der Aufnahme; Kontroll- und Prüfgruppe am Ende der Aufnahme) und ggf. in der Anfangsphase (z.B. Tag 1-3 der Aufnahme) und in der Ausscheidungsphase (z.B. Tage 1, 2, 4, 7, 14, 28 für die Kontroll- und die Prüfgruppe) tabellarisch dargestellt. Das Gewicht ist der bevorzugte Wachstumsparameter für die Korrektur um den Effekt der Verdünnung durch das Wachstum. Angaben zu der (den) angewandten Methode(n) für die Korrektur der Daten um den Effekt der Verdünnung durch das Wachstum siehe Nummer 162 und 163 sowie Anlage 5.
Daten zur Prüfstoffkonzentration in den Fischen
Die Messungen der Prüfstoffrückstände in einzelnen Fischen (oder gepoolten Fischproben, falls Messungen an einzelnen Fischen nicht möglich sind), werden, ausgedrückt als Nassgewichtskonzentration (w/w), für die Prüf- und die Kontrollfische, aufgeschlüsselt nach Probenahmezeitpunkten, tabellarisch dargestellt. Wurde die Lipidanalyse bei jedem entnommenen Fisch durchgeführt, können einzelne lipidkorrigierte Konzentrationen als Lipidkonzentration (w/w Lipid) abgeleitet und tabellarisch dargestellt werden.
- Die Messungen der Prüfstoffrückstände in einzelnen Fischen (oder gepoolten Fischproben, falls Messungen an einzelnen Fischen nicht möglich sind, siehe Nummer 66) für die Ausscheidungsphase werden in ihre natürlichen Logarithmen umgewandelt und gegen die Zeit (Tag) aufgetragen. Werden bei einer visuellen Überprüfung der graphischen Darstellung "Ausreißer" festgestellt, kann ein statistisch gültiger Ausreißertest angewendet werden, um falsche Datenpunkte zu entfernen; die Gründe dafür sind zu dokumentieren.
- Für ln(Konzentration) gegen Ausscheidung (Tag) wird eine lineare Korrelation der kleinsten Quadrate berechnet. Steigung und Achsenabschnitt der Geraden werden als Gesamtausscheidungskonstante (k2) und natürlicher Logarithmus der abgeleiteten Konzentration zum Zeitpunkt Null (C0,d) angegeben (für nähere Informationen siehe Anlagen 5 und 7). Sollte dies nicht möglich sein, da die Konzentrationen unter die Quantifizierungsgrenze für die zweite Ausscheidungsprobe fallen, kann eine konservative Schätzung von k2 durchgeführt werden (siehe Anlage 7).
- Die Abweichungen in der Steigung und im Achsenabschnitt der Geraden werden nach statistischen Standardverfahren berechnet, und die 90 %- (oder 95 %)-Konfidenzintervalle um diese Ergebnisse werden bewertet und dargestellt.
- Ferner wird die gemessene mittlere Konzentration in den Fischen am letzten Tag der Aufnahme (gemessene Konzentration zum Zeitpunkt Null, C0,m) berechnet und mit dem abgeleiteten Wert C0,d verglichen. Ist der abgeleitete Wert geringer als der gemessene Wert, kann die Differenz auf das Vorhandensein von unverdautem dotiertem Futter im Darm hindeuten. Ist der abgeleitete Wert wesentlich höher als der gemessene Wert, kann dies ein Anzeichen dafür sein, dass der abgeleitete Wert aus der linearen Regression der Ausscheidungsdaten falsch ist und neu bewertet werden muss (siehe Anlage 7).
Ausscheidungsrate und Biomagnifikationsfaktor
Zur Errechnung des Biomagnifikationsfaktors aus den Daten sollte zunächst die Assimilationseffizienz (Absorption des Prüfstoffs im Darm, ±) bestimmt werden. Hierzu sollte die Gleichung A7.1 in Anlage 7 verwendet werden, wobei die abgeleitete Konzentration in den Fischen zum Zeitpunkt Null der Ausscheidungsphase (C0,d), die (Gesamt-)Ausscheidungskonstante (k2), die Konzentration im Futter (CFutter), die Futteringestionskonstante (I) und die Dauer der Aufnahmephase (t) bekannt sein müssen. Die Steigung und der Achsenabschnitt der linearen Beziehung zwischen dem natürlichen Logarithmus der Konzentration und dem Ausscheidungszeitpunkt werden als Gesamtausscheidungskonstante (k2 = Steigung) und Konzentration zum Zeitpunkt Null (C0,d = eAchsenabschnitt) wie oben angegeben. Die abgeleiteten Werte sollten auf biologische Plausibilität geprüft werden (z.B. Assimilationseffizienz ist als Bruchteil nicht größer als 1). (I) wird durch Division der Masse des Futters durch die Masse des täglich gefütterten Fisches (wird dieser im Wert von 2 % seines Körpergewichts gefüttert, entspricht (I) 0,02) berechnet. Jedoch sollte die für die Berechnung verwendete Fütterungsrate um das Fischwachstum korrigiert werden (eventuell unter Verwendung der bekannten Wachstumskonstanten zur Schätzung des Fischgewichts zu jedem Zeitpunkt während der Aufnahmephase; siehe Anlage 7). In Fällen, in denen k2 und C0,d nicht berechnet werden können, weil bei der zweiten Ausscheidungsprobe die Konzentrationen beispielsweise unter die Nachweisgrenze gefallen sind, kann eine konservative Schätzung von k2 und eines "oberen" BMFk vorgenommen werden (siehe Anlage 7).
Nachdem die Assimilationseffizienz (α) bestimmt wurde, kann der Biomagnifikationsfaktor durch Multiplikation von α mit der Ingestionskonstante (I) und Division durch die (Gesamt-)Ausscheidungskonstante (k2) berechnet werden. Der wachstumskorrigierte Biomagnifikationsfaktor wird auf dieselbe Weise berechnet, jedoch anhand der wachstumskorrigierten Ausscheidungskonstanten (k2g; siehe Nummern 162 und 163). Eine alternative Schätzung der Assimilationseffizienz kann abgeleitet werden, wenn die Gewebeanalyse an den in der frühen, linearen Phase der Aufnahmephase entnommenen Fischen durchgeführt wurde; siehe Nummer 151 und Anlage 7. Dieser Wert entspricht einer unabhängigen Schätzung der Assimilationseffizienz bei einem im Wesentlichen nicht exponierten Organismus (d. h. Fische ganz am Anfang der Aufnahmephase). Die anhand der Ausscheidungsdaten geschätzte Assimilationseffizienz wird in der Regel zur Berechnung des BMF herangezogen.
Lipidkorrektur und Korrektur um den Effekt der Verdünnung durch Wachstum
Das Fischwachstum während der Ausscheidungsphase kann die gemessenen Stoffkonzentrationen in den Fischen verringern, wodurch die Gesamtausscheidungskonstante (k2) größer ist als sie es bei den Ausscheidungsprozessen (z.B. Atmung, Metabolismus, Egestion) alleine wäre (siehe Nummer 72). Der Lipidgehalt der Prüffische (der eng mit der Bioakkumulation hydrophober Stoffe in Zusammenhang steht) und der Lipidgehalt des Futters können in der Praxis derart variieren, sodass eine Korrektur notwendig ist, um sinnvolle Biomagnifikationsfaktoren zu erhalten. Der Biomagnifikationsfaktor sollte um den Effekt der Verdünnung durch Wachstum (wie der kinetische BCF bei der Methode mit aquatischer Exposition) und den Lipidgehalt des Futters im Vergleich zu dem des Fisches (Lipidkorrekturfaktor) korrigiert werden. Gleichungen und Beispiele für diese Berechnungen finden sich in Anlage 5 bzw. Anlage 7.
Zwecks Korrektur um den Effekt der Verdünnung durch Wachstum sollte die wachstumskorrigierte Ausscheidungskonstante (k2g) berechnet werden (siehe Anlage 5 für Gleichungen). Diese wachstumskorrigierte Ausscheidungskonstante (k2g) wird verwendet, um den wachstumskorrigierten Biomagnifikationsfaktor zu berechnen (siehe Nummer 73). In bestimmten Fällen ist dieser Ansatz nicht möglich. Ein alternativer Ansatz, der die Notwendigkeit einer Korrektur um den Effekt der Verdünnung durch Wachstum umgeht, besteht in der Verwendung von Daten über die Prüfstoffmasse pro Fisch (Ganzfische) bei der Ausscheidung anstelle der üblichen Daten zur Prüfstoffmasse pro Fischmasseneinheit (Konzentration). Diese Berechnung ist einfach, da Prüfungen nach dieser Methode die protokollierten Gewebekonzentrationen mit dem Gewicht der einzelnen Tiere korreliert werden. Dieses einfache Verfahren wird in Anlage 5 beschrieben. Es ist zu beachten, dass k2 auch bei Anwendung dieses alternativen Ansatzes geschätzt und angegeben werden sollte.
Zwecks Korrektur um den Lipidgehalt des Futters und der Fische, wenn die Lipidanalyse nicht bei allen beprobten Fischen durchgeführt wurde, werden die mittleren Lipidfraktionen (w/w) in den Fischen und im Futter berechnet 11. Der Lipidkorrekturfaktor (Lc) wird sodann durch Division der mittleren Lipidfraktion der Fische durch die mittlere Lipidfraktion des Futters berechnet. Der Biomagnifikationsfaktor (ggf. wachstumskorrigiert) wird durch den Lipidkorrekturfaktor dividiert, um den lipidkorrigierten Biomagnifikationsfaktor zu berechnen.
Wurde die Stoff- und Lipidanalyse an jedem Probenahmezeitpunkt an ein und denselben Fischen durchgeführt, können die lipidkorrigierten Gewebedaten für einzelne Fische zur direkten Berechnung eines lipidkorrigierten BMF verwendet werden (siehe (37)). Die Kurve der Daten über die lipidkorrigierte Konzentration ergibt C0,d (bezogen auf die Lipide) und k2. Anschließend kann die mathematische Analyse nach den Gleichungen in Anlage 7 erfolgen, die Assimilationseffizienz (a) wird jedoch anhand der lipidstandardisierten Futteringestionskonstante (ILipid) und der auf die Lipide bezogenen Konzentration im Futter (CFutter-Lipid) berechnet. Die lipidkorrigierten Parameter werden sodann in ähnlicher Weise verwendet, um den BMF zu berechnen (es ist zu beachten, dass die Korrektur um die Wachstumskonstante auch auf die Lipidfraktion und nicht auf das Nassgewicht der Fische angewendet werden sollte, um den lipid- und wachstumskorrigierten BMFKgL zu berechnen).
Interpretation der Ergebnisse
Das durchschnittliche Wachstum sollte bei Prüf- und Kontrollgruppen grundsätzlich nicht allzu stark variieren, um toxische Wirkungen auszuschließen. Die Wachstumskonstanten oder die Wachstumskurven der beiden Gruppen sollten nach einem geeigneten Verfahren miteinander verglichen werden 12).
Prüfbericht
Nach Abschluss des Versuchs ist ein Schlussbericht mit Angaben zu Prüfstoff, Prüfspezies und Prüfbedingungen (siehe Nummer 81 und Methode mit aquatischer Exposition) zu erstellen. Daneben sind folgende Angaben erforderlich:
Prüfstoff:
- Angaben zur Stabilität des Prüfstoffs im zubereiteten Futter;
Prüfbedingungen:
- Nominale Stoffkonzentration im Futter, Dotierungsverfahren, Menge des für die Futterdotierung verwendeten (Lipid-)Vehikels, Messungen der Prüfstoffkonzentration im dotierten Futter bei jeder Analyse (mindestens dreifach vor Versuchsbeginn sowie am Ende der Aufnahme) und Mittelwerte;
- Art und Qualität des Trägeröls oder -lösungsmittels (Güte, Lieferant usw.), das für die Futterdotierung verwendet wird;
- verwendete Futterart (Sofortanalyse 13, Güte oder Qualität, Lieferant usw.), Fütterungsrate in der Aufnahmephase, verabreichte Futtermenge und Häufigkeit der Fütterung (einschließlich Anpassungen je nach Gewicht der beprobten Fische);
- Zeitpunkt, an dem die Fische für die Stoffanalyse entnommen und getötet wurden (z.B. 1 Stunde vor der Fütterung am folgenden Tag), nach Probenahmezeitpunkten;
Ergebnisse:
- Ergebnisse aus etwaigen Vorversuchen;
- Beschreibung etwaiger festgestellter Schadwirkungen;
- vollständige Beschreibung aller angewandten chemischen Analyseverfahren, einschließlich Nachweis- und Quantifizierungsgrenzen, Variabilität und Wiederfindung;
- gemessene Lipidkonzentrationen im Futter (dotiertes Futter und Kontrollfutter), individuelle Werte, Mittelwerte und Standardabweichungen;
- tabellarisch dargestellte Daten zu Fischgewicht (und Fischlänge), bezogen auf einzelne Fische, sowohl für die Kontroll- als auch die Prüfgruppe (z.B. mittels individuellen Kenncodes für die einzelnen Fische) und Berechnungen, abgeleitete Wachstumskonstante(n) und 95 %-Konfidenzintervalle;
- tabellarisch dargestellte Daten zur Prüfstoffkonzentration in den Fischen, mittlere gemessene Konzentration am Ende der Aufnahme (C0,m) und abgeleitete (Gesamt-)Ausscheidungskonstante (k2) und Konzentration in den Fischen zu Beginn der Ausscheidungsphase (C0,d), zusammen mit den Varianzen in diesen Werten (Steigung und Achsenabschnitt);
- tabellarisch dargestellte Daten zum Lipidgehalt der Fische (ggf. aufgelistet nach spezifischen Stoffkonzentrationen), Mittelwerte für Prüfgruppe und Kontrolle zu Prüfungsbeginn, am Ende der Aufnahmephase und am Ende der Ausscheidungsphase;
- Kurven (einschließlich aller gemessenen Daten), aus denen Folgendes hervorgeht (falls zutreffend, können die Konzentrationen bezogen auf den Ganzkörper des Tiers oder bestimmte Gewebe ausgedrückt werden):
- Wachstum (d. h. Fischgewicht (und Fischlänge) im Verhältnis zur Zeit) oder durch natürliches Logarithmieren transformiertes Gewicht im Verhältnis zur Zeit;
- Ausscheidung des Prüfstoffs durch die Fische; und
- durch natürliches Logarithmieren transformierte Konzentration (ln(Konzentration)) im Verhältnis zur Dauer der Ausscheidungsphase (einschließlich der abgeleiteten Ausscheidungskonstanten k2, durch natürliches Logarithmieren abgeleitete Konzentration in den Fischen zu Beginn der Ausscheidungsphase, C0,d);
- werden bei einer visuellen Überprüfung einer graphischen Darstellung "Ausreißer" festgestellt, kann ein statistisch gültiger Ausreißertest durchgeführt werden, um falsche Datenpunkte zu entfernen; die Gründe hierfür sind zu dokumentieren;
- berechnete wachstumskorrigierte Ausscheidungskonstante und wachstumskorrigierte Halbwertszeit;
- berechnete Assimilationseffizienz (±);
- Brutto-BMF (futterbezogen), lipid- und wachstumskorrigierter kinetischer BMF (brutto und lipidkorrigiert auf Basis des Ganzfischnassgewichts), ggf. gewebespezifischer BMF;
- etwaige Angaben zu Metaboliten radioaktiv markierter Prüfstoffe und deren Akkumulation;
- etwaige Besonderheiten im Zusammenhang mit der Prüfung, etwaige Abweichungen von den genannten Verfahren sowie etwaige anderen relevanten Informationen;
- Übersichtstabelle relevanter Mess- und Berechnungsdaten (wie nachfolgend):
Stoffspezifische Ausscheidungskonstanten und Biomagnifikationsfaktoren (BMFK) kg (Wachstumskonstante, Tag-1): Wert eintragen (95 % CI) 1 k2 (Gesamtausscheidungskonstante, Tag-1): Wert eintragen (95 % CI) k2g (wachstumskorrigierte Ausscheidungskonstante, Tag-1): Wert eintragen (95 % CI) 1 C0,m (gemessene Konzentration zum Zeitpunkt Null, Konzentration in den Fischen am Ende der Aufnahmephase) (µg/g): Wert eintragen ± SD 2 C0,d (abgeleitete Konzentration zum Zeitpunkt Null der Ausscheidungsphase; µg/g): Wert eintragen ± SD 2 I (festgelegte Futteringestionsrate; g Futter/g Fisch/Tag): Wert eintragen Ig (effektive Fütterungsrate, wachstumskorrigiert; g Futter/g Fisch/Tag) 2: Wert eintragen ± SD 2 CFutter (Stoffkonzentration im Futter; µg/g): Wert eintragen ± SD 2 α (stoffspezifische Assimilationseffizienz): Wert eintragen ± SD 2 BMFK (kinetischer nahrungsbezogener BMF): Wert eintragen (95 % CI) 1 BMFK (wachstumskorrigierter kinetischer nahrungsbezogener BMF): Wert eintragen (95 % CI) 1 t1/2g (wachstumskorrigierte Halbwertzeit, in Tagen): Wert eintragen ± SD 2 Lc (Lipidkorrekturfaktor): Wert eintragen BMFKgL (lipidkorrigierter wachstumskorrigierter kinetischer BMF): Wert eintragen BMFSS-L (indikativer lipidkorrigierter BMF bei stationärem Zustand) 2: Wert eintragen ± SD 2 1) CI: Konfidenzintervall (falls schätzbar) 2) SD: Standardabweichung (falls schätzbar)
Literaturhinweise
(1) Kapitel C.13 dieses Anhangs, Biokonzentration: Durchfluss-Fischtest.
(2) Kapitel A.6 dieses Anhangs, Löslichkeit in Wasser
(3) Li A, Doucette W.J. (1993), The effect of cosolutes on the aqueous solubilities and octanol/water partition coefficients of selected polychlorinated biphenyl congeners. Environ Toxicol Chem 12: 2031-2035
(4) Kapitel A.8 dieses Anhangs, Partition Coefficient (n-octanol/water): Shake Flask Method.
(5) Kapitel A.24 dieses Anhangs, Verteilungskoeffizient n-Oktanol/Wasser, HPLC-Methode.
(6) Kapitel A.23 dieses Anhangs, Verteilungskoeffizient (1-Oktanol/Wasser): Methode zur Prüfung unter langsamem Rühren.
(7) Kapitel C.7 dieses Anhangs, Hydrolyse als Funktion des pH.
(8) (OECD (1997), OECD Environmental Health and Safety Publications Series on Testing and Assessment Number 7: Guidance Document on Direct Phototransformation of Chemicals in Water OCDE/GD(97)21. Organisation for Economic Co-operation and Development (OECD), Paris, Frankreich.
(9) Kapitel A.5 dieses Anhangs, Oberflächenspannung wässriger Lösungen.
(10) Kapitel A.4 dieses Anhangs, Dampfdruck.
(11) Kapitel C.4 dieses Anhangs, "Leichte" biologische Abbaubarkeit.
(12) Kapitel C.29 dieses Anhangs, "Leichte" biologische Abbaubarkeit - C.29, Leichte biologische Abbaubarkeit -
(13) Connell D.W. (1988), Bioaccumulation behaviour of persistent chemicals with aquatic organisms. Rev. Environ. Contam. Toxicol. 102: 117-156.
(14) Bintein S., Devillers J. and Karcher W. (1993), Nonlinear dependence of fish bioconcentration on n-octanol/water partition coefficient. SAR QSAR Environ. Res. 1: 29-39.
(15) OECD (2011), QSAR Toolbox 2.1. February 2011. Abrufbar unter: http://www.oecd.org/document/54/0,3746,en_2649_34379_42923638_1_1_1_1,00.html.
(16) Brown R.S., Akhtar P., Åkerman J., Hampel L., Kozin I.S., Villerius L.A. and Klamer H.J.C. (2001), Partition controlled delivery of hydrophobic substances in toxicity tests using poly(dimethylsiloxane) (PDMS) films. Environ. Sci. Technol. 35: 4097-4102.
(17) Fernandez J.D., Denny J.S. and Tietge J.E. (1998), A simple apparatus for administering 2,3,7,8-tetrachlorodibenzo-p-dioxin to commercially available pelletized fish food. Environ. Toxicol. Chem. 17: 2058-2062.
(18) Nichols J.W., Fitzsimmons P.N., Whiteman F.W. and Dawson T.D. (2004), A physiologically based toxicokinetic model for dietary uptake of hydrophobic organic compounds by fish: I. Feeding studies with 2,22, 5,52-tetrachlorobiphenyl. Toxicol. Sci. 77: 206-218.
(19) Parkerton T.F., Arnot J.A., Weisbrod A.V., Russom C., Hoke R.A., Woodburn K., Traas T., Bonnell M., Burkhard L.P. and Lampi M.A. (2008), Guidance for evaluating in vivo fish bioaccumulation data. Integr. Environ. Assess. Manag. 4: 139-155.
(20) Verbruggen E.M.J., Beek M., Pijnenburg J. and Traas T.P. (2008). Ecotoxicological environmental risk limits for total petroleum hydrocarbons on the basis of internal lipid concentrations. Environ. Toxicol. Chem. 27: 2436-2448.
(21) Schlechtriem C., Fliedner A. and Schäfers C. (2012), Determination of lipid content in fish samples from bioaccumulation studies: Contributions to the revision of OECD Test Guideline 305. Environmental Sciences Europe 2012, 24:13. published: 3 April 2012.
(22) Kapitel C.47 dieses Anhangs, Toxozitätsprüfung an Fischen im frühen Entwicklungsstadium.
(23) Kapitel C.15 dieses Anhangs, Fish, Kurzfristige Toxizitätsprüfung an Embryonen und Jungfischen mit Dottersack.
(24) Kapitel C.14 dieses Anhangs, Fische, Wachstumstest an Jungfischen.
(25) OECD (2000), OECD Environmental Health and Safety Publications Series on Testing and Assessment No. 23: Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures ENV/JM/MONO(2000)6. Organisation for Economic Co-operation and Development (OECD), Paris, France.
(26) US-EPA (1994), Great Lake water quality initiative technical support document for the procedure to determine bioaccumulation factors 822-R-94-002. US EPA, Office of Water, Office of Science and Technology, Washington, DC, USA.
(27) US-FDA (1999), Pesticide analytical manual (PAM). Vol.1. US Food and Drug Administration, Rockville, MD, USA.
(28) US-EPA (1974), Section 5, A (1) Analysis of Human or Animal Adipose Tissue, in Analysis of Pesticide Residues in Human and Environmental Samples, Thompson, J.F., Editor. US-EPA, Research Triangle Park, NC, USA
(29) Bligh E.G. and Dyer W.J. (1959), A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37: 911-917.
(30) Gardner W.S., Frez W.A., Cichocki E.A. and Parrish C.C. (1985), Micromethod for lipids in aquatic invertebrates. Limnol. Oceanogr. 30: 1099-1105.
(31) Smedes F. (1999), Determination of total lipid using non-chlorinated solvents. Analyst. 124: 1711-1718.
(32) OECD (2006), OECD Environmental Health and Safety Publications Series on Testing and Assessment No. 54: Current approaches in the statistical analysis of ecotoxicity data: a guidance to application. ENV/JM/MONO(2006)18. Organisation for Economic Co-operation and Development (OECD), Paris, Frankreich.
(33) Springer T.A., Guiney P.D., Krueger H.O. and Jaber M.J. (2008), Assessment of an approach to estimating aquatic bioconcentration factors using reduced sampling. Environ. Toxicol. Chem. 27: 2271-2280.
(34) Springer T.A. (2009), Statistical Research Related to Update of OECD Guideline 305. Wildlife International, Ltd, Easton, MD, USA.
(35) Arnot J.A., Meylan W., Tunkel J., Howard P.H., Mackay D., Bonnell M. and Boethling R.S. (2009), A quantitative structure-activity relationship for predicting metabolic biotransformation rates for organic chemicals in fish. Environ. Toxicol. Chem. 28: 1168-1177.
(36) Parkerton T., Letinski D., Febbo E., Davi R., Dzambia C., Connelly M., Christensen K. and Peterson D. (2001), A practical testing approach for assessing bioaccumulation potential of poorly water soluble organic chemicals (presentation). in SETAC Europe 12th Annual Meeting: Madrid, Spain.
(37) Fisk A.T., Cymbalisty C.D., Bergman Ã. and Muir D.C.G. (1996), Dietary accumulation of C12- and C16-chlorinated alkanes by juvenile rainbow trout (Oncorhynchus mykiss). Environ. Toxicol. Chem. 15: 1775-1782.
(38) Anonymous (2004), Fish, dietary bioaccumulation study - Basic protocol, document submitted to the TC-NES WG on PBT.
(39) Anonymous (2004), Background document to the fish dietary study protocol, document submitted to the TC-NES WG on PBT.
(40) Bruggeman W.A., Opperhuizen A., Wijbenga A. and Hutzinger O. (1984), Bioaccumulation of super-lipophilic chemicals in fish, Toxicol. Environ. Chem. 7: 173-189.
(41) Muir D.C.G., Marshall W.K. and Webster G.R.B. (1985), Bioconcentration of PCDDs by fish: effects of molecular structure and water chemistry. Chemosphere. 14: 829-833.
(42) Thomann R.V. (1989), Bioaccumulation model of organic chemical distribution in aquatic food chains. Environ. Sci. Technol. 23: 699-707.
(43) Nichols J.W., Fitzsimmons P.N. and Whiteman F.W. (2004), A physiologically based toxicokinetic model for dietary uptake of hydrophobic organic compounds by fish: II. Stimulation of chronic exposure scenarios. Toxicol. Sci. 77: 219-229.
(44) Gobas F.A.P.C., de Wolf W., Burkhard L.P., Verbruggen E. and Plotzke K. (2009), Revisiting bioaccumulation criteria for POPs and PBT assessments. Integr. Environ. Assess. Manag. 5: 624-637.
(45) Sijm D.T.H.M. and van der Linde A. (1995), Size-dependent bioconcentration kinetics of hydrophobic organic chemicals in fish based on diffusive mass transfer and allometric relationships. Environ. Sci. Technol. 29: 2769-2777.
(46) Sijm D.T.H.M., Verberne M.E., de Jonge W.J., Pärt P. and Opperhuizen A. (1995), Allometry in the uptake of hydrophobic chemicals determined in vivo and in isolated perfused gills. Toxicol. Appl. Pharmacol. 131: 130-135.
(47) Fisk A.T., Norstrom R.J., Cymbalisty C.D. and Muir D.G.G. (1998), Dietary accumulation and depuration of hydrophobic organochlorines: Bioaccumulation parameters and their relationship with the octanol/water partition coefficient. Environ. Toxicol. Chem. 17: 951-961.
(48) McGrath J.A., Parkerton T.F. and Di Toro D.M. (2004), Application of the narcosis target lipid model to algal toxicity and deriving predicted-no-effect concentrations. Environ. Toxicol. Chem. 23: 2503-2517.
(49) Poppendieck D.G. (2002), Polycyclic Aromatic Hydrocarbon Desorption Mechanisms from Manufactured Gas Plant Site Samples. Dissertation. Department of Civil, Architectural and Environmental Engineering, University of Texas, Austin, Texas, USA.
(50) McCarty L.S. and Mackay D. (1993), Enhancing ecotoxicological modelling and assessment: body residues and modes of toxic action. Environ. Sci. Technol. 27: 1718-1728.
(51) OECD (2012), OECD Environmental Health and Safety Publications Series on Testing and Assessment No. 175: Part I - Validation Report of a ring test for the OECD TG 305 dietary exposure bioaccumulation fish test. Part II - Additional Report including comparative analysis of trout and carp results ENV/JM/MONO(2012)20. Organisation for Economic Co-operation and Development (OECD), Paris, Frankreich.
____
1) Vgl. Anlage 1 für Definitionen und Einheiten.
2) Zum Zwecke der Verordnung (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (ABl. Nr. L 396 vom 30.12.2006 S. 1) wurde dieser Aspekt in den Leitlinien zu Informationsanforderungen und Stoffsicherheitsbeurteilung, Kapitel R.7c, R.7.10.3.1, R.7.10.4.1 und Abbildung R7.10-2, Sgeregelt.
3) Bei den meisten Prüfstoffen sollten sich im Idealfall keine Nachweise im Kontrollwasser finden. Hintergrundkonzentrationen sollten nur bei natürlich vorkommenden Stoffen (z.B. bestimmten Metallen) und in der Umwelt allgemein vorkommenden Stoffen relevant sein.
4) Da der BMF als Verhältnis der Konzentration eines Stoffs in einem Organismus zur Konzentration des Stoffs in der Nahrung des Organismus bei stationärem Zustand definiert ist, werden Lipide durch Korrektur um den Lipidgehalt des Organismus und des Futters berücksichtigt und folglich - exakter - als "Korrektur" beschrieben. Dieser Ansatz unterscheidet sich von der Festlegung eines "standardisierten" Lipidgehalts für den Organismus, wie dies beim Biokonzentrationstest mit aquatischer Exposition der Fall ist.
5) Die Gesamtlänge sollte auch während der Prüfung protokolliert werden, denn sie ist ein guter Indikator für das Auftreten einer Schadwirkung.
6) HCB ist in den Anhängen A und C des Stockholmer Übereinkommens sowie in den Anhängen I und III der Verordnung (EG) Nr. 850/2004 über persistente organische Schadstoffe (ABl. Nr. L 158 vom 30.04.2004 S. 7) aufgeführt.
7) Bei schnellem Wachstum während der Aufnahmephase sinkt die eigentliche Fütterungsrate gegenüber der zu Expositionsbeginn festgelegten Rate.
8) Bei einem Versuch mit aquatischer Exposition entspräche eine Halbwertzeit von 14 Tagen einem BCF von etwa 10.000 L/kg bei Fischen von 1 g mit einer entsprechenden Aufnahmerate von etwa 500 L/kg/Tag (nach der Gleichung nach Sijm et al (46)).
9) Da die tatsächlichen internen Konzentrationen erst nach abgeschlossener Prüfung bestimmt werden können, muss die erwartete interne Konzentration im Futter geschätzt werden (z.B. auf der Grundlage des erwarteten BMF und der Konzentration im Futter; siehe Gleichung A5.8 in Anlage 5).
10) Das Vorhandensein von Prüfstoff im Prüfmedium infolge der Exkretion durch die Fische oder des Auslaugens aus dem Futter lässt sich u. U. nicht vollständig vermeiden. Folglich besteht eine Möglichkeit darin, die Stoffkonzentration im Wasser am Ende der Aufnahmephase zu messen, insbesondere wenn ein semistatisches Systems angewendet wird, um festzustellen, ob es zu einer aquatischen Exposition gekommen ist.
11) Dieser Ansatz gilt speziell für die futterbasierte Studie und unterscheidet sich von dem bei der Methode mit aquatischer Exposition verwendeten Ansatz. Aus diesem Grunde wurde anstelle von "Standardisierung" der Begriff "Korrektur" verwendet, um Irreführung zu vermeiden - siehe auch Fußnote in Nummer 106.
12) Es kann ein t-Test auf Wachstumskonstanten durchgeführt werden, um festzustellen, ob es zwischen Kontroll- und Prüfgruppen Wachstumsunterschiede gibt, oder ein F-Test im Falle einer Varianzanalyse. Ein F-Test oder Likelihood-Ratio-Test kann die Auswahl des geeigneten Wachstumsmodells vereinfachen (OECD Monograph 54 (32)).
13) Verfahren zu Analyse des Protein-, Lipid-, Rohfaser- und Aschegehalts von Futtermitteln; diese Angaben sind normalerweise beim Futtermittellieferant erhältlich.
| weiter . | |
...
X
⍂
↑
↓