
umwelt-online: Verordnung (EU) 2017/735 zur Änderung - zwecks Anpassung an den technischen Fortschritt - des Anhangs der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (2)
 | zurück |  |
.
| Vorhersagefähigkeit der BCOP-Prüfmethode | Anlage 2 |
Tabelle 1 Vorhersagefähigkeit der BCOP-Prüfmethode für die Ermittlung von Chemikalien, die schwere Augenschäden verursachen ([UN-GHS/EU-CLP Kategorie 1 vs. nicht Kategorie 1 (Kategorie 2 + Keine Einstufung); US-EPA-Kategorie I vs. nicht Kategorie I (Kategorie II + Kategorie III + Kategorie IV)]
| Klassifizierungs- system | Nr. | Genauigkeit | Empfindlichkeit | Falsche Negative | Spezifität | Falsche Positive | |||||
| % | Nr. | % | Nr. | % | Nr. | % | Nr. | % | Nr. | ||
| UN-GHS
EU-CLP | 191 | 78,53 | 150/191 | 86,15 | 56/65 | 13,85 | 9/65 | 74,60 | 94/126 | 25,40 | 32/126 |
| US-EPA | 190 | 78,95 | 150/190 | 85,71 | 54/63 | 14,29 | 9/63 | 75,59 | 96/127 | 24,41 | 31/127 |
Tabelle 2 Vorhersagefähigkeit der BCOP-Prüfmethode für die Ermittlung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern ("Stoffe ohne Reizwirkung") [UN-GHS/EU-CLP Keine Einstufung vs. nicht Keine Einstufung (Kategorie 1 + Kategorie 2); US-EPA-Kategorie IV vs. nicht Kategorie IV (Kategorie I + Kategorie II + Kategorie III)]
| Klassifizierungs- system | Nr. | Genauigkeit | Empfindlichkeit | Falsche Negative | Spezifität | Falsche Positive | |||||
| % | Nr. | % | Nr. | % | Nr. | % | Nr. | % | Nr. | ||
| UN-GHS
EU-CLP | 196 | 68,88 | 135/196 | 100 | 107/107 | 0 | 0/107 | 31,46 | 28/89 | 68,54 | 61/89 |
| US-EPA | 190 | 82,11 | 156/190 | 93,15 | 136/146 | 6,85 | 10/146 | 45,45 | 20/44 | 54,55 | 24/44 |
.
| Leistungschemikalien für die BCOP-Prüfmethode | Anlage 3 |
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Laboratorien ihre technische Kompetenz nachweisen, indem sie die 13 in Tabelle 1 empfohlenen Chemikalien in die richtige Augengefährdungsklasse einstufen. Die Chemikalien wurden so ausgewählt, dass sie die Bandbreite von Augengefährdungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405) (17) und dem UN-GHS-Klassifizierungssystem (d. h. den Kategorien 1, 2A, 2B oder "Nicht eingestuft") basieren (4). Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger In-vitro-Daten aus der BCOP-Prüfmethode. Referenzdaten können aus dem Streamlined Summary Document (3) und den Background Review Documents des ICCVAM für die BCOP-Prüfmethode bezogen werden (2) (18).
Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des BCOP-Tests
| Chemikalie | CAS-Nr. | Chemikalien- klasse 1 | Physikalischer Zustand | In Vivo- Klassifizierung 2 | BCOP- Klassifizierung |
| Benzalkoniumchlorid (5 %) | 8001-54-5 | Oniumverbindung | flüssig | Kategorie 1 | Kategorie 1 |
| Chlorhexidin | 55-56-1 | Amin, Amidin | fest | Kategorie 1 | Kategorie 1 |
| Dibenzoyl-D-Weinsäure | 2743-38-6 | Carbonsäure, Ester | fest | Kategorie 1 | Kategorie 1 |
| Imidazol | 288-32-4 | heterocyclisch | fest | Kategorie 1 | Kategorie 1 |
| Trichloressigsäure (30 %) | 76-03-9 | Carbonsäure | flüssig | Kategorie 1 | Kategorie 1 |
| 2,6-Dichlorbenzoyl-chlorid | 4659-45-4 | Acylhalogenid | flüssig | Kategorie 2A | Keine genaue/zuverlässige Vorhersage möglich |
| Ethyl-2-methylaceto-acetat | 609-14-3 | Ketone, Ester | flüssig | Kategorie 2B | Keine genaue/zuverlässige Vorhersage möglich |
| Ammoniumnitrat | 6484-52-2 | Anorganisches Salz | fest | Kategorie 2 3 | Keine genaue/zuverlässige Vorhersage möglich |
| EDTA (Ethylendiamintetraessigsäure), Di-Kaliumsalz | 25102-12-9 | Amin, Carbonsäure (Salz) | fest | Nicht eingestuft | Nicht eingestuft |
| Tween 20 | 9005-64-5 | Ester, Polyether | flüssig | Nicht eingestuft | Nicht eingestuft |
| 2-Mercaptopyrimidin | 1450-85-7 | Acylhalogenid | fest | Nicht eingestuft | Nicht eingestuft |
| Phenylbutazon | 50-33-9 | heterocyclisch | fest | Nicht eingestuft | Nicht eingestuft |
| Polyoxyethylen-23-laurylether (BRIJ-35) (10 %) | 9002-92-0 | Alkohol | flüssig | Nicht eingestuft | Nicht eingestuft |
| Abkürzungen:
CAS-Nr. = Registernummer des Chemical Abstracts Service
1) Jede Prüfchemikalie wurde anhand einer Standard-Klassifizierungsregelung auf Basis des Klassifizierungssystems der National Library of Medicine Medical Subject Headings (MeSH) einer Chemikalienklasse zugeordnet (verfügbar unter http//www.nlm.nih.gov/mesh). 2) Gestützt auf Ergebnisse aus dem In-vivo-Kaninchenaugentest (OECD TG 405) (17) unter Verwendung des UN-GHS (4). 3) Die Einstufung in die Kategorie 2A oder 2B ist von der Auswertung der Kriterien des UN-GHS zur Unterscheidung zwischen diesen beiden Kategorien abhängig, d. h. für eine Einstufung in die Kategorie 2A müssen an 1 von 3 gegenüber 2 von 3 Tieren Wirkungen an Tag 7 beobachtet werden. Die In-vivo-Studie umfasste drei Tiere. Alle Endpunkte mit Ausnahme einer Bindehautrötung bei einem Tier, gingen bis Tag 7 oder früher auf einen Wert von null zurück. Das eine Tier, das sich bis Tag 7 nicht vollständig regeneriert hatte, wies (an Tag 7) einen Bindehautrötungswert von 1 auf, der an Tag 10 ganz zurückging. | |||||
.
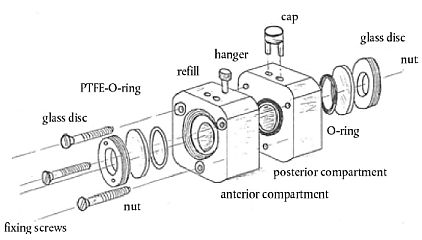
| BCOP-Hornhauthalter | Anlage 4 |
BCOP-Hornhauthalter sind aus einem trägen Material (z.B. Polypropylen) gefertigt. Sie bestehen aus zwei Hälften (einer Vorder- und einer Hinterkammer) sowie zwei identischen zylindrischen Innenkammern. Jede Kammer hat ein Fassungsvermögen von ca. 5 ml und schließt mit einer Glasscheibe ab, durch die die Trübungsmesswerte abgelesen werden. Jede Innenkammer hat einen Durchmesser von 1,7 cm und ist 2,2 cm tief 1. Ein auf der Hinterkammer positionierter Dichtungsring verhindert Leckagen. Die Hornhäute werden mit der Endothel-Seite nach unten auf den Dichtungsring der Hinterkammern platziert, während die Vorderkammern auf die Epithel-Seite der Hornhäute gesetzt werden. Die Kammern werden mit drei Schrauben aus rostfreiem Stahl an den Außenkanten der Kammer fixiert. Jede Kammer schließt mit einer Glasscheibe ab, die für den leichten Zugang zur Hornhaut abgenommen werden kann. Um Leckagen zu verhindern, befindet sich zwischen Glasscheibe und Kammer ein weiterer Dichtungsring. Zwei Öffnungen auf der Oberseite jeder Kammer gestatten den Ein- und Auslass des Mediums und der Prüfchemikalien. Sie werden während der Behandlung und der Inkubation mit Gummistöpseln verschlossen. Die Lichtübertragung durch die Hornhauthalter kann sich potenziell verändern, da Lichtstreuung oder Reflexion durch Abnutzung oder Ablagerung bestimmter chemischer Rückstände an den Bohrungen der Innenkammer oder an der Glasscheibe beeinflusst werden können. Dies könnte höhere oder niedrigere Referenzwerte für die Lichtübertragung durch die Hornhauthalter (und umgekehrt niedrigere oder höhere Referenztrübungswerte) zur Folge haben und sich als deutliche Veränderungen bei den erwarteten ersten Referenzmessungen der Hornhauttrübung in den einzelnen Kammern bemerkbar machen (d. h. die anfänglichen Hornhauttrübungswerte bei spezifischen einzelnen Hornhauthaltern können regelmäßig um mehr als zwei oder drei Trübungseinheiten von den erwarten Referenzwerten abweichen). Jedes Labor sollte in Erwägung ziehen, ein Programm zur Evaluierung der Veränderungen der Lichtübertragung durch die Hornhauthalter einzurichten, das die Art der geprüften Chemikalien und die Häufigkeit des Gebrauchs der Kammern berücksichtigt. Zur Festlegung von Referenzwerten können die Hornhauthalter vor dem regelmäßigen Einsatz durch Messung der Referenztrübungswerte (oder Lichtübertragung) der vollständig mit dem Medium gefüllten Kammern, ohne Hornhäute, geprüft werden. Anschließend werden die Hornhauthalter während ihres Einsatzes in regelmäßigen Abständen auf Veränderungen der Lichtübertragung kontrolliert. Jedes Labor kann die Häufigkeit der Kontrollen der Hornhauthalter abhängig von der Art der geprüften Chemikalien, der Häufigkeit des Gebrauchs und der beobachteten Veränderungen der Referenzwerte für die Hornhauttrübung festlegen. Werden deutliche Veränderungen in der Lichtübertragung durch die Hornhauthalter festgestellt, sind eine Reinigung und/oder Polierung der Innenfläche der Hornhauthalter mit geeigneten Verfahren oder ein Austausch in Betracht zu ziehen.
Hornhauthalter: Explosionsdarstellung
_____
1) Es handelt sich um Abmessungen einer Halterung für Hornhäute von Kühen im Alter von 12 bis 60 Monaten.
Soweit Tiere im Alter von sechs bis 12 Monaten verwendet werden, müsste die Halterung so konzipiert sein, dass jede Kammer 4 ml fasst und jede Innenkammer einen Durchmesser von 1,5 cm aufweist und 2,2 cm tief ist. Bei jedem neuen Halterungsmodell ist ausschlaggebend, dass das Verhältnis zwischen der exponierten Hornhautoberfläche und dem Fassungsvermögen der Hinterkammer dem jeweiligen Verhältniswert bei herkömmlichen Hornhauthaltern entspricht.
Dadurch wird sichergestellt, dass die Durchlässigkeitswerte zwecks Berechnung des IVIS-Wertes nach der vorgeschlagenen Formel korrekt bestimmt werden.
.
| Opazimeter | Anlage 5 |
Opazimeter
Der Opazimeter ist ein Gerät zur Messung der Lichtübertragung. Bei dem zur Validierung des BCOP-Tests verwendeten Gerät OP-KIT von Electro Design (Riom, Frankreich) wird beispielsweise Licht aus einer Halogenleuchte durch eine Kontrollkammer (leere Kammer ohne Fenster oder Flüssigkeit) zu einer Fotozelle geleitet und mit dem Lichtstrahl verglichen, der durch die Versuchskammer, welche die Kammer mit der Hornhaut enthält, zu einer Fotozelle geleitet wird. Der Unterschied bei der Lichtübertragung aus den Fotozellen wird errechnet und als numerischer Trübungswert digital angezeigt. Die Trübungseinheiten sind vorgegeben. Andere Arten von Opazimetern mit einem anderen Aufbau (bei denen z.B. keine parallelen Messungen der Kontrollkammer und Versuchskammer erforderlich sind) können verwendet werden, wenn sie nachweislich ähnliche Ergebnisse wie das validierte Gerät liefern.
Der Opazimeter sollte eine lineare Reaktion innerhalb eines Bereichs von Trübungsmesswerten ergeben, der den Schwellenwerten für die vom Vorhersagemodell beschriebenen unterschiedlichen Klassifizierungen (d. h. bis zu dem Schwellenwert, der die verätzende/stark reizende Wirkung bestimmt) Rechnung trägt. Um lineare und akkurate Messwerte bis zu 75-80 Trübungseinheiten zu gewährleisten, muss der Opazimeter mit einer Reihe von Kalibratoren geeicht werden. Die Kalibratoren werden in die Kalibrierkammer (eine für die Aufnahme der Kalibratoren konzipierte Hornhautkammer) platziert und auf dem Opazimeter abgelesen. Die Kalibrierkammer ist derart konzipiert, dass die Kalibratoren in ungefähr demselben Abstand zu Lichtquelle und Fotozelle gehalten werden, in dem sich die Hornhäute bei der Trübungsmessung befinden würden. Die Referenzwerte und der anfängliche Sollwert sind von der Art des verwendeten Geräts abhängig. Die Linearität der Trübungsmessungen sollte durch geeignete (instrumentspezifische) Verfahren sichergestellt werden. Beim Gerät OP-KIT von Electro Design (Riom, Frankreich) wird der Opazimeter beispielsweise zunächst auf null Trübungseinheiten geeicht, wobei die Kalibrierkammer ohne Kalibrator verwendet wird. Anschließend werden nacheinander drei unterschiedliche Kalibratoren in die Kalibrierkammer gesetzt, und die Trübungswerte werden gelesen. Die Kalibratoren 1, 2 und 3 sollten Trübungswerte ergeben, die ihren eingestellten Werten von 75, 150 bzw. 225 Trübungseinheiten ± 5 % entsprechen."
(13) In Teil B erhält Kapitel B.48 folgende Fassung:
"B.48 Test am isolierten Hühnerauge zur Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 438 (2013). Der Test am isolierten Hühnerauge (Isolated Chicken Eye, ICE) wurde vom Organisationsübergreifenden Koordinationsausschuss zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods, ECVAM) und des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese Centre for the Validation of Alternative Methods, JaCVAM) in den Jahren 2006 und 2010 evaluiert (1) (2) (3). Bei der ersten Evaluierung wurde der ICE-Test als wissenschaftlich fundierte Prüfmethode für den Einsatz als Screening-Test zur Identifizierung von Chemikalien (Stoffen und Gemischen), die schwere Augenschäden (Kategorie 1) gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN) (1) (2) (4) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 verursachen, anerkannt. Bei der zweiten Evaluierung wurde der ICE-Test im Hinblick auf seine Eignung als Screening-Test zur Identifizierung von Chemikalien, die nicht als augenreizend oder schwer augenschädigend gemäß UN-GHS einzustufen sind, beurteilt (3) (4). Die Ergebnisse der Validierungsstudie und die Empfehlungen der Peer-Review-Gruppe bestätigten die ursprüngliche Empfehlung, den ICE-Test für die Einstufung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) zu verwenden, da sich die verfügbare Datenbank seit der ursprünglichen Validierung des ICCVAM nicht geändert hat. In jener Phase wurden keine weiteren Empfehlungen für eine Erweiterung des Anwendungsbereichs des ICE-Tests auch auf andere Kategorien ausgesprochen. Es wurde eine Neubewertung des in der Validierungsstudie verwendeten In-vitro- und In-vivo-Datensatzes durchgeführt. Der Schwerpunkt lag dabei auf der Evaluierung der Zweckmäßigkeit des ICE-Tests für die Identifizierung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend (5) erfordern. Die Neubewertung führte zu dem Ergebnis, dass der ICE-Test auch verwendet werden kann, um Chemikalien zu identifizieren, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS (4) (5) erfordern. Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des ICE-Tests auf Grundlage dieser Evaluierungen. Die Hauptunterschiede zwischen der ursprünglichen Fassung der OECD-Prüflinie aus dem Jahr2009 und der aktualisierten Fassung von 2013 sind u. a. die Verwendung des ICE-Tests zur Identifizierung von Chemikalien, die keine Einstufung nach dem UN-GHS-Klassifizierungssystem erfordern, eine Aktualisierung der Elemente des Prüfberichts, eine Aktualisierung der Definitionen in Anlage 1 sowie eine Aktualisierung der Leistungschemikalien in Anlage 2.
Gegenwärtig herrscht allgemeiner Konsens darüber, dass der In-vivo-Draize-Augentest in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Augenreizungen für verschiedene Chemikalienklassen vorherzusagen. Unter Umständen ist es jedoch möglich, den Draize-Augentest durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie zu ersetzen (6). Der Top-Down-Ansatz (7) wird verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie ein hohes Reizpotenzial aufweist. Umgekehrt wird der Bottom-Up-Ansatz (7) verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie keine ausreichende Augenreizung für eine Einstufung hervorruft. Der ICE-Test ist eine In-vitro-Prüfmethode, die unter bestimmten Bedingungen und mit bestimmten Grenzen, wie unter den Nummern 8 bis 10 erläutert, zur Einstufung und Kennzeichnung von Chemikalien mit augengefährdender Wirkung angewendet werden kann. Auch wenn der Test den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird der ICE-Test als erster Schritt im Rahmen einer Prüfstrategie wie des von Scott et al. vorgeschlagenen Top-Down-Ansatzes (7) empfohlen, um ohne weitere Tests (4) schwer augenschädigende Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, zu identifizieren. Der ICE-Test wird auch für die Identifizierung von Chemikalien empfohlen, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS (Keine Einstufung (No Category), NC) (4) erfordern, und kann daher als erster Schritt im Rahmen einer Prüfstrategie mit Bottom-up-Ansatz verwendet werden (7). Im Falle einer Chemikalie, bei der mit dem ICE-Test nicht vorhergesagt werden kann, dass sie schwere Augenschäden verursacht oder keine Einstufung als augenreizend/schwer augenschädigend erfordert, müssten jedoch weitere Tests (in vitro und/oder in vivo) durchgeführt werden, um eine eindeutige Einstufung festzulegen. Außerdem sollten die zuständigen Regulierungsbehörden konsultiert werden, bevor der ICE-Test in einem Bottom-Up-Ansatz unter einer anderen Klassifizierungsregelung als dem UN-GHS angewendet wird.
Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie, gemessen als ihre Fähigkeit, im isolierten Hühnerauge eine toxische Wirkung hervorzurufen oder nicht. Toxische Wirkungen auf die Hornhaut werden gemessen durch i) qualitative Bewertung der Trübung, ii) qualitative Bewertung der Schädigung der Epithel-Zellschicht durch Applikation von Fluorescein auf das Auge (Fluorescein-Verfärbung), iii) quantitative Messung verstärkter Dicke (Schwellung) und iv) qualitative Beurteilung makroskopischer morphologischer Oberflächenschädigungen. Hornhauttrübungen, Hornhautschwellungen und Hornhautschädigungen nach Applikation einer Prüfchemikalie werden zunächst einzeln bewertet und anschließend zwecks Klassifizierung des Augenreizwertes (Eye Irritancy Classification) kombiniert.
Definitionen sind Anlage 1 zu entnehmen.
Vorbemerkungen und Einsatzgrenzen
Diese Prüfmethode basiert auf dem im OECD Guidance Document 160 (8) empfohlenen Protokoll, das im Anschluss an eine internationale Validierungsstudie des ICCVAM (1) (3) (9) und unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods), des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese center for the Validation of Alternative Methods) und der Abteilung Toxikologie und angewandte Pharmakologie des niederländischen Forschungsinstituts TNO Quality of Life entwickelt wurde. Das Protokoll beruht auf Informationen aus veröffentlichten Protokollen und auf dem von TNO derzeit verwendeten Protokoll (10) (11) (12) (13) (14).
Bei der dieser Prüfmethode zugrunde liegenden Validierung wurde ein breites Spektrum von Chemikalien getestet. Die empirische Datenbank der Validierungsstudie umfasste 152 Chemikalien (72 Stoffe und 80 Gemische) (5). Die Prüfmethode ist auf Feststoffe, Flüssigkeiten, Emulsionen und Gele anwendbar. Die Flüssigkeiten können wässrig oder nichtwässrig, die Feststoffe wasserlöslich oder wasserunlöslich sein. Gase und Aerosole wurden in einer Validierungsstudie bisher noch nicht bewertet.
Der ICE-Test kann zur Identifizierung von schwer augenschädigenden Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 (4) einzustufen sind, angewendet werden. In diesem Anwendungsbereich beruhen die anerkannten Einsatzgrenzen des ICE-Tests auf den hohen Falsch-Positiv-Raten für Alkohole und den hohen Falsch-Negativ-Raten für Feststoffe und Tenside (1) (3) (9). Die Falsch-Negativ-Raten sind in diesem Kontext (UN-GHS-Kategorie 1 wird als nicht UN-GHS-Kategorie 1 erkannt) jedoch nicht maßgeblich, da alle Prüfchemikalien mit negativem Ergebnis anschließend im Rahmen anderer angemessen validierter In-vitro-Tests oder als letzte Möglichkeit - je nach Vorschriften - an Kaninchen anhand einer sequenziellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz (weight-of-evidence approach) geprüft werden. Es ist zu beachten, dass Feststoffe beim In-vivo-Draize-Augenreizungstest zu variablen und extremen Expositionsbedingungen führen können, aus denen sich möglicherweise irrelevante Vorhersagen über das tatsächliche Reizungspotenzial ableiten (15). Prüfer können diese Prüfmethode jedoch für alle Arten von Chemikalien einsetzen und ein Positivergebnis als Indikator für eine schwer augenschädigende Wirkung, d. h. eine Einstufung in UN-GHS-Kategorie 1, ohne weitere Tests akzeptieren. Positivergebnisse bei Verwendung von Alkohol sollten angesichts des Risikos von Falsch-Positiv-Prognosen (over-prediction) jedoch mit einer gewissen Zurückhaltung interpretiert werden.
Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist der ICE-Test hinsichtlich der Identifizierung von Chemikalien mit schwer augenschädigender Wirkung (UN-GHS-Kategorie 1) eine allgemeine Genauigkeit von 86 % (120/140), eine Falsch-Positiv-Rate von 6 % (7/113) und eine Falsch-Negativ-Rate von 48 % (13/27) auf (4) (5).
Der ICE-Test kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN-GHS-Klassifizierungssystem erfordern (4). Bevor der ICE-Test in einem Bottom-Up-Ansatz unter einer anderen Klassifizierungsregelung angewendet wird, sollten die zuständigen Regulierungsbehörden konsultiert werden. Diese Prüfmethode kann für alle Arten von Chemikalien eingesetzt werden, wobei ein Negativergebnis als Indikator für die Nichteinstufung einer Chemikalie als augenreizend oder schwer augenschädigend akzeptiert werden könnte. Auf der Basis eines Ergebnisses aus der Validierungsdatenbank besteht bei Bewuchsschutzfarben mit organischen Lösungsmitteln jedoch das Risiko von Falsch-Negativ-Prognosen (under-prediction) (5).
Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist der ICE-Test hinsichtlich der Identifizierung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern, eine allgemeine Genauigkeit von 82 % (125/152), eine Falsch-Positiv-Rate von 33 % (26/79) und eine Falsch-Negativ-Rate von 1 % (1/73) auf (4) (5). Werden Stoffe einer bestimmten Chemikalienklasse (d. h. Bewuchsschutzfarben mit organischen Lösungsmitteln) aus der Datenbank ausgeschlossen, so liegt die Genauigkeit des ICE-Tests gemessen am UN-GHS-Klassifizierungssystem bei 83 % (123/149), die Falsch-Positiv-Rate bei 33 % (26/78) und die Falsch-Negativ-Rate bei 0 % (0/71) (4) (5).
Aufgrund der beträchtlichen Anzahl von Chemikalien der UN-GHS-Kategorie 1, die zu niedrig in die UN-GHS-Kategorien 2, 2A oder 2B eingestuft sind, und von Chemikalien, die keine Einstufung erfordern, jedoch zu hoch in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, wird der ICE-Test nicht für die Identifizierung von Prüfchemikalien empfohlen, die als augenreizend (d. h. UN-GHS-Kategorie 2 oder Kategorie 2A) oder leicht augenreizend (UN-GHS-Kategorie 2B) eingestuft werden sollten. Diesbezüglich können weitere Tests mit einer anderen geeigneten Methode erforderlich sein.
Bei allen Verfahren, die Hühneraugen involvieren, sind die geltenden Regeln und Verfahrensvorschriften der Prüfanstalt für den Umgang mit Human- bzw. Tiermaterial (u. a. Gewebe und Gewebeflüssigkeiten) einzuhalten. Universelle Laborregeln sollten beachtet werden (16).
Die im Kaninchenaugentest evaluierten Bindehaut- und Irisverletzungen werden beim ICE-Test zwar außer Acht gelassen, doch werden bei der Prüfmethode Auswirkungen auf die Hornhaut berücksichtigt, die wesentliche Einflussfaktoren für die In-vivo-Klassifizierung nach dem UN-GHS-Klassifizierungssystem bilden. Auch wurde - obwohl sich die Reversibilität von Hornhautläsionen per se mit dem ICE-Test nicht beurteilen lässt - ausgehend von Kaninchenaugenstudien vorgeschlagen, dass eine Bewertung der anfänglichen Tiefe der Hornhautverletzung herangezogen werden kann, um einige Arten irreversibler Wirkungen zu identifizieren (17). Insbesondere sind weitere wissenschaftliche Erkenntnisse erforderlich, um zu verstehen, wie irreversible Wirkungen auftreten, die nicht mit einer anfänglichen starken Verletzung im Zusammenhang stehen. Schließlich sei auch erwähnt, dass das Potenzial für eine mit der Augenexposition verbundene systemische Toxizität mit der ICE-Methode nicht bewertet werden kann.
Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Beispielsweise können histopathologische Befunde potenziell nützlich sein, wenn eine umfassendere Charakterisierung der Hornhautschädigung erforderlich ist. Zur Evaluierung dieser Möglichkeit sollten Anwender die Augen aufbewahren und histopathologische Proben zubereiten, die zum Aufbau einer Datenbank und zur Entwicklung von Entscheidungskriterien herangezogen werden können, mit denen sich die Genauigkeit dieser Prüfmethode weiter verbessern lässt. Die OECD hat einen Leitfaden (Guidance Document) für die Anwendung von Prüfmethoden zur Untersuchung der okularen Toxizität erarbeitet. Dieser enthält ausführliche Verfahrensanweisungen für die Entnahme histopathologischer Proben und Angaben darüber, wo die Proben und/oder histopathologischen Daten einzureichen sind (8).
Laboratorien, die diese Prüfmethode erstmals anwenden, sollten die in Anlage 2 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung des ICE-Tests nachzuweisen, bevor sie ICE-Testdaten zum Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen.
Testprinzip
Die ICE-Prüfmethode ist ein organtypisches Modell, das die Zellstruktur des Hühnerauges in vitro kurzfristig funktionsfähig hält. Bei dieser Prüfmethode werden durch die Prüfchemikalie hervorgerufene Schäden als Hornhautschwellungen, Hornhauttrübung und korneale Fluorescein-Verfärbung festgestellt. Während die beiden letztgenannten Parameter eine qualitative Bewertung erfordern, muss die Hornhautschwellung quantitativ bestimmt werden. Jedes Messergebnis wird entweder in einen quantitativen Wert umgerechnet, der seinerseits für die Berechnung eines Gesamtreizindexes (Overall Irritancy Index) zugrunde gelegt wird, oder einer Qualitätskategorie zugeordnet, die wiederum für die Einordnung in Klassen für Chemikalien mit in vitro augengefährdender Wirkung (UN-GHS-Kategorie 1 oder keine Einstufung nach UN-GHS) herangezogen wird. Jedes dieser Ergebnisse kann dann verwendet werden, um das Potenzial einer Prüfchemikalie, in vivo schwere Augenschäden hervorzurufen oder keine Einstufung als augengefährdend zu erfordern, vorherzusagen (siehe Entscheidungskriterien). Allerdings ist keine Einstufung bei Chemikalien möglich, bei denen mit dem ICE-Test nicht vorhergesagt wird, dass sie schwere Augenschäden verursachen oder nicht einzustufen sind (siehe Nummer 11).
Bezugsquelle und Alter der Hühneraugen
Traditionell werden für diesen Test Augen von Hühnern verwendet, die in einer Schlächterei für den menschlichen Verzehr geschlachtet wurden; auf diese Weise wird der Einsatz von Versuchstieren vermieden. Nur Augen von gesunden Tieren, die als für die Nahrungskette geeignet angesehen werden, dürfen verwendet werden.
Es wurde keine kontrollierte Studie zur Bestimmung des optimalen Alters der Hühner durchgeführt, doch werden für diesen Test bisher Hühner verwendet, bei denen es sich nach Alter und Gewicht um Junghühner handelt (d. h. Hühner im Alter von ungefähr sieben Wochen mit einem Gewicht von 1,5 bis 2,5 kg), die in der Regel in einer Geflügelschlächterei geschlachtet werden.
Gewinnung und Beförderung der Augen zum Labor
Die Köpfe sollten unmittelbar nach dem Betäuben der Tiere, normalerweise durch Elektroschock, und nach dem Entbluten durch Nackenstich abgesetzt werden. Sie sollten aus einer in Nähe des Labors gelegenen Quelle bezogen werden, um die Köpfe möglichst schnell vom Schlachthof zum Labor befördern zu können, damit sich ihr Zustand nicht verschlechtert und/oder Bakterienkontaminationen auf ein Mindestmaß begrenzt werden. Der Zeitabstand zwischen der Gewinnung der Hühnerköpfe und ihrem Einsetzen in die Superfusionskammer nach der Ausschälung muss möglichst gering sein (d. h. höchstens zwei Stunden betragen), damit die Akzeptanzkriterien des Tests erfüllt werden. Alle für die Prüfung verwendeten Augen sollten aus einer an ein und demselben Tag gewonnenen Partie Augen stammen.
Da die Augen im Labor seziert werden, sind die intakten Köpfe in Kunststoffbehältnissen, die mit Tüchern, welche in isotonischer Kochsalzlösung getränkt wurden, ausgeschlagen sind, und bei Umgebungstemperatur (normalerweise zwischen 18 1 °C und 25 1 °C) vom Schlachthof zum Labor zu befördern.
Auswahlkriterien und Anzahl der Augen, die im ICE-Test eingesetzt werden
Augen mit starker Ausgangs-Fluorescein-Verfärbung (d. h. > 0,5) oder mit starker Hornhauttrübung (d. h. > 0,5) nach der Ausschälung werden verworfen.
Jede Behandlungsgruppe und die gleichzeitige Positivkontrolle umfassen mindestens drei Augen. Die Negativkontrollen bzw. die Lösungsmittelkontrolle (wenn ein anderes Lösungsmittel als Kochsalzlösung verwendet wird) bestehen aus mindestens einem Auge.
Im Fall von Feststoffen mit dem GHS-Ergebnis "Keine Einstufung" (No Category, NC) wird ein zweiter Durchlauf mit drei Augen empfohlen, um das Negativergebnis zu bestätigen oder zu verwerfen.
Verfahren
Vorbereitung der Augen
Die Augenlider werden sorgfältig weggeschnitten, wobei darauf zu achten ist, dass die Hornhaut nicht beschädigt wird. Die Unversehrtheit der Hornhaut wird mithilfe eines Tropfens von 2 %igem (w/v) Natrium-Fluorescein, der für einige Sekunden auf die Hornhautoberfläche appliziert und anschließend mit isotonischer Kochsalzlösung abgespült wird, schnell überprüft. Fluorescein-behandelte Augen werden sodann mit einem Spaltlampenmikroskop auf Hornhautschädigung untersucht (d. h. Fluorescein-Verfärbung und Hornhauttrübungswerte müssen < 0,5 betragen).
Liegt keine Schädigung vor, wird das Auge weiter freigelegt, wobei dafür Sorge zu tragen ist, dass die Hornhaut nicht beschädigt wird. Der Augapfel wird aus der Augenhöhle herausgezogen, indem die Nickhaut mithilfe einer chirurgischen Zange festgehalten und die Augenmuskulatur mit einer gebogenen, stumpfendigen Schere durchtrennt wird. Dieser Schritt ist wichtig, um zu vermeiden, dass die Hornhaut durch übermäßigen Druck geschädigt wird (Kompressionsartefakte).
Beim Herauslösen des Auges aus der Augenhöhle sollte ein sichtbarer Teil des Sehnervs noch anhaften. Das Auge wird anschließend auf eine saugfähige Unterlage gesetzt, und Nickhaut sowie anderes Bindegewebe werden weggeschnitten.
Das auf diese Weise ausgeschälte Auge wird in einer Klemme aus rostfreiem Stahl fixiert, wobei die Hornhaut vertikal positioniert sein muss. Die Klemme wird sodann in eine Kammer des Superfusionsgeräts gesetzt (18). Im Superfusionsgerät sind die Klemmen so zu positionieren, dass die gesamte Hornhaut mit der Kochsalzinfusion (3-4 Tropfen pro Minute bzw. 0,1-0,15 ml/min) versorgt wird. Die Temperatur in den Kammern des Superfusionsgeräts sollte auf 32 ± 1,5 °C gehalten werden. Anlage 3 zeigt ein Schaubild eines typischen Superfusionsgeräts mit Augenklemmen; das Gerät ist im Handel fertig oder als Bausatz erhältlich. Es kann den Bedürfnissen des jeweiligen Labors angepasst werden (z.B. um Augen in verschiedener Anzahl aufzunehmen).
Nach dem Einsetzen ins Superfusionsgerät werden die Augen erneut mit einem Spaltlampenmikroskop untersucht, um sicherzustellen, dass sie während des Sezierens nicht beschädigt wurden. Bei dieser Gelegenheit sollte mit dem Pachometeraufsatz des Spaltlampenmikroskops auch die Hornhautdicke im Apex gemessen werden. Augen mit i) einer Fluorescein-Verfärbung von > 0,5, ii) einer Hornhauttrübung von > 0,5 oder iii) etwaigen anderen Anzeichen einer Schädigung sind zu ersetzen. Einzelne Augen, auf die keines der genannten Kriterien zutrifft, werden verworfen, wenn sie eine Hornhautdicke aufweisen, die mehr als 10 % vom mittleren Dickenwert aller Augen zusammengerechnet abweicht. Anwender sollten sich darüber im Klaren sein, dass Spaltlampenmikroskope mit unterschiedlicher Spaltbreiteneinstellung unterschiedliche Dickenmesswerte ergeben können. Die Spaltbreite sollte auf 0,095 mm eingestellt sein.
Sobald alle Augen untersucht und für einwandfrei befunden wurden, werden sie zwecks Äquilibrierung des Testsystems vor Applikation der Prüfchemikalie für ungefähr 45 bis 60 Minuten inkubiert. Nach der Äquilibrierung werden Referenzmessungen von Hornhautdicke und Hornhauttrübung zum Zeitpunkt Null vorgenommen, welche als Referenzszenario dienen (d. h. Zeitpunkt = 0). Der beim Sezieren ermittelte Fluorescein-Wert wird als Referenzmesswert für diesen Endpunkt verwendet.
Applikation der Prüfchemikalie
Unmittelbar nach den Referenzmessungen zum Null-Zeitpunkt wird das Auge (in seiner Halterung) aus dem Superfusionsgerät entnommen und zur Applikation der Prüfchemikalie auf die Hornhaut horizontal positioniert.
Flüssige Prüfchemikalien werden in der Regel unverdünnt getestet, können jedoch verdünnt werden, sofern dies für notwendig gehalten wird (z.B. als Teil des Studienkonzepts). Bevorzugtes Lösungsmittel für verdünnte Prüfchemikalien ist physiologische Kochsalzlösung. Unter kontrollierten Bedingungen können auch alternative Lösungsmittel verwendet werden, deren Eignung jedoch nachzuweisen ist.
Flüssige Prüfchemikalien werden so auf die Hornhaut appliziert, dass die gesamte Hornhautoberfläche gleichmäßig von der Prüfchemikalie bedeckt ist; das Standardvolumen beträgt 0,03 ml.
Feste Prüfchemikalien sollten wenn möglich im Mörser oder mit einem vergleichbaren Zerkleinerungsgerät so fein wie möglich gemahlen werden. Das Pulver wird so auf die Hornhaut appliziert, dass die Oberfläche von der Prüfchemikalie gleichmäßig bedeckt ist; die Standardmenge beträgt 0,03 g.
Die Prüfchemikalie (flüssig oder fest) wird für 10 Sekunden appliziert und anschließend mit isotonischer Kochsalzlösung (ungefähr 20 ml) bei Umgebungstemperatur vom Auge abgespült. Das Auge (in seiner Halterung) wird anschließend wieder in aufrechter Ausgangsstellung in das Superfusionsgerät eingesetzt. Bei Bedarf können nach der 10-sekündlichen Applikation und zu späteren Zeitpunkten (z.B. bei der Feststellung von Rückständen der Prüfchemikalie auf der Hornhaut) weitere Spülungen vorgenommen werden. Im Allgemeinen ist die Menge an Kochsalzlösung, die zusätzlich für die Spülungen verwendet wird, nicht maßgeblich; wichtig ist jedoch die Beobachtung von Anhaftungen der Chemikalie an der Hornhaut.
Kontrollchemikalien
Jeder Versuch sollte gleichzeitige Negativ- oder Lösungsmittel-/Vehikelkontrollen und gleichzeitige Positivkontrollen umfassen.
Beim Prüfen unverdünnter (100 %iger) Flüssigkeiten oder Feststoffe wird als gleichzeitige Negativkontrolle im ICE-Test physiologische Kochsalzlösung verwendet, um unspezifische Veränderungen im Testsystem festzustellen und um sicherzustellen, dass die Testbedingungen keine ungerechtfertigten Reizwirkungen hervorrufen.
Zum Testen verdünnter Flüssigkeiten umfasst der Test auch eine Gruppe gleichzeitiger Lösungsmittel-/Vehikelkontrollen, um unspezifische Veränderungen im Testsystem nachzuweisen und um sicherzustellen, dass die Testbedingungen keine ungerechtfertigten Reizwirkungen hervorrufen. Wie unter Nummer 31 erwähnt, sind nur Lösungsmittel/Vehikel zulässig, die das Testsystem nachweislich nicht beeinträchtigen.
Jeder Versuch umfasst als gleichzeitige Positivkontrolle einen bekannten Augenreizstoff, damit überprüft werden kann, ob eine adäquate Wirkung eintritt. Da der ICE-Test bei dieser Prüfmethode eingesetzt wird, um verätzende oder stark reizende Stoffe zu identifizieren, sollte es sich bei der Positivkontrolle um eine Referenzchemikalie handeln, die bei dieser Prüfmethode eine starke Wirkung hervorruft. Um zu gewährleisten, dass die Veränderlichkeit der Reaktion der Positivkontrolle im Zeitverlauf bewertet werden kann, sollte die Reaktionsstärke jedoch nicht allzu heftig sein. Es sollten genügend In-vitro-Daten für die Positivkontrolle generiert werden, damit eine statistisch vorgegebene vertretbare Spanne für die Positivkontrolle berechnet werden kann. Liegen für eine bestimmte Positivkontrolle keine angemessenen historischen Daten zur ICE-Prüfmethode vor, so können Studien erforderlich werden, um die notwendigen Daten zu generieren.
Beispiele für Positivkontrollen für flüssige Prüfchemikalien sind 10 %ige Essigsäure oder 5 %iges Benzalkoniumchlorid, während für Positivkontrollen für feste Prüfchemikalien Natriumhydroxid oder Imidazol in Frage kommen.
Referenzchemikalien sind nützlich für die Evaluierung des Augenreizpotenzials unbekannter Chemikalien einer bestimmten Chemikalien- oder Produktklasse oder zur Evaluierung des relativen Reizpotenzials eines Augenreizstoffes innerhalb einer spezifischen Spanne von Reizwirkungen.
Gemessene Endpunkte
Behandelte Hornhäute werden vor der Behandlung evaluiert sowie in Abständen von 30, 75, 120, 180 und 240 Minuten (± 5 Minuten) nach dem Abspülen im Anschluss an die Behandlung. Diese Zeitreihe gestattet eine angemessene Anzahl von Messungen während des vierstündigen Behandlungszeitraums und lässt genügend Zeit zwischen den Messungen, um alle Augen vorschriftsgemäß beobachten zu können.
Die evaluierten Endpunkte sind Hornhauttrübung, Hornhautschwellung, Fluorescein-Verfärbung und morphologische Effekte (z.B. durchlöchertes oder abgelöstes Epithel). Alle Endpunkte mit Ausnahme der Fluorescein-Verfärbung (die ausschließlich vor der Behandlung sowie 30 Minuten nach Applikation der Prüfchemikalie ermittelt wird) werden zu jedem der vorgenannten Zeitpunkte bestimmt.
Hornhauttrübung, Fluorescein-Verfärbung, morphologische Effekte und, sofern sie vorliegen, histopathologische Befunde sollten fotografiert werden.
Nach der abschließenden Untersuchung am Ende des vierstündigen Behandlungszeitraums sind die Augen in einer geeigneten Fixierlösung (z.B. neutrales gepuffertes Formalin) aufzubewahren, damit sie gegebenenfalls histopathologisch untersucht werden können (Einzelheiten siehe Nummer 14 und Literaturhinweis (8)).
Hornhautschwellungen werden durch Hornhautdickenmessungen bestimmt, die mit dem optischen Pachometeraufsatz eines Spaltlampenmikroskops vorgenommen werden. Der Schwellungsmesswert wird als Prozentsatz ausgedrückt und auf Basis der Hornhautdickenmesswerte nach folgender Formel berechnet:

Für alle getesteten Augen wird die mittlere Hornhautschwellung (in Prozent) für sämtliche Beobachtungszeitpunkte berechnet. Auf Basis des höchsten Mittelwertes für die Hornhautschwellung (beliebiger Beobachtungszeitpunkt) wird anschließend jeder Prüfchemikalie ein Gesamtwert für die Kategorie (overall category score) zugeordnet (siehe Nummer 51).
Die Hornhauttrübung wird anhand der Hornhautfläche bewertet, die am stärksten getrübt ist (siehe Tabelle 1). Für alle getesteten Augen wird der mittlere Trübungswert für jeden Beobachtungszeitpunkt berechnet. Auf Basis des höchsten Mittelwertes für die Hornhauttrübung (beliebiger Beobachtungszeitpunkt) wird anschließend jeder Prüfchemikalie ein Gesamtwert für die Kategorie (overall category score) zugeordnet (siehe Nummer 51).
Tabelle 1 Hornhauttrübungswerte
| Wert | Beobachtung |
| 0 | keine Trübung |
| 0,5 | sehr schwache Trübung |
| 1 | gestreute oder diffuse Strukturen; Iris deutlich sichtbar |
| 2 | leicht erkennbare lichtdurchlässige Struktur; Iris weniger deutlich sichtbar |
| 3 | starke Hornhauttrübung; Einzelheiten der Iris nicht sichtbar; Pupillengröße kaum erkennbar |
| 4 | vollständige Trübung/Iris unsichtbar |
Die Fluorescein-Verfärbung wird nur für den 30-minütigen Beobachtungszeitpunkt bewertet (siehe Tabelle 2). Anschließend wird für alle getesteten Augen die mittlere Fluorescein-Verfärbung für den 30-minütigen Beobachtungszeitpunkt berechnet und zur Ermittlung des jeder Prüfchemikalie zugeordneten Gesamtwertes für die Kategorie herangezogen (siehe Nummer 51).
Tabelle 2 Fluorescein-Verfärbungswerte
| Wert | Beobachtung |
| 0 | keine Fluorescein-Verfärbung |
| 0,5 | sehr geringfügige Verfärbung einzelner Zellen |
| 1 | Verfärbung einzelner Zellen auf der gesamten behandelten Fläche der Hornhaut |
| 2 | Punktuelle oder konfluierende dichte Zellverfärbung |
| 3 | konfluierende großflächige Fluorescein-Verfärbung der Hornhaut |
Morphologische Effekte umfassen "durchlöcherte" Hornhaut-Epithelzellen, "abgelöste"Epithelzellen, "aufgeraute" Hornhautoberfläche und "Verklebung" der Hornhaut mit der Prüfchemikalie. Diese Befunde können unterschiedlich stark ausgeprägt sein und gleichzeitig auftreten. Ihre Einstufung erfolgt subjektiv je nach Auswertung des Prüfers.
Daten und Berichterstattung
Datenauswertung
Die Ergebnisse für die Hornhauttrübung, die Hornhautschwellung und die Fluorescein-Verfärbung sind einzeln zu evaluieren, um für jeden Endpunkt eine ICE-Klasse zu generieren. Kombiniert ergeben die ICE-Klassen für die einzelnen Endpunkte eine Reizklasse (Irritancy Classification) für die einzelnen Prüfchemikalien.
Entscheidungskriterien
Nach der Evaluierung jedes Endpunktes können die ICE-Klassen entsprechend einer vorgegebenen Spanne zugeordnet werden. Die Auswertung der Hornhautschwellung (Tabelle 3), der Hornhauttrübung (Tabelle 4) und der Fluorescein-Verfärbung (Tabelle 5) und ihre Einstufung in vier ICE-Klassen erfolgen anhand der nachstehenden Bewertungsskalen. Die Werte für die Hornhautschwellung in Tabelle 3 sind nur gültig, wenn die Dicke mit einem Spaltlampenmikroskop (z.B. Haag-Streit BP900) mit Tiefenmessgerät Nr. 1 und einer Spaltbreiteneinstellung von 9,5 (= 0,095 mm) gemessen wird. Anwender sollten sich darüber im Klaren sein, dass Spaltlampenmikroskope mit unterschiedlicher Spaltbreiteneinstellung unterschiedliche Dickenmesswerte ergeben können.
Tabelle 3 Kriterien für die Einstufung in ICE-Klassen - Hornhautschwellung
| Mittlere Hornhautschwellung (in %) * | ICE-Klasse |
| 0 bis 5 | I |
| > 5 bis 12 | II |
| > 12 bis 18 (> 75 Minuten nach der Behandlung) | II |
| > 12 bis 18 (d 75 Minuten nach der Behandlung) | III |
| > 18 bis 26 | III |
| > 26 bis 32 (> 75 Minuten nach der Behandlung) | III |
| > 26 bis 32 (d 75 Minuten nach der Behandlung) | IV |
| > 32 | IV |
| *) Höchster Mittelwert für die Hornhauttrübung an einem beliebigen Beobachtungszeitpunkt | |
Tabelle 4 Kriterien für die Einstufung in ICE-Klassen - Hornhauttrübung
| Höchster Mittelwert für die Hornhauttrübung * | ICE-Klasse |
| 0,0-0,5 | I |
| 0,6-1,5 | II |
| 1,6-2,5 | III |
| 2,6-4,0 | IV |
| *) Höchster Mittelwert an einem beliebigen Beobachtungszeitpunkt (auf Basis der Trübungswerte gemäß Tabelle 1). | |
Tabelle 5 Kriterien für die Einstufung in ICE-Klassen - mittlere Fluorescein-Verfärbung
| Mittlere Verfärbung 30 Minuten nach der Behandlung * | ICE-Klasse |
| 0,0-0,5 | I |
| 0,6-1,5 | II |
| 1,6-2,5 | III |
| 2,6-3,0 | IV |
| *) Auf Basis der Werte gemäß Tabelle 2. | |
Die In-vitro-Klassifizierung einer Prüfchemikalie richtet sich nach der GHS-Klasse, die der Kombination von Kategorien entspricht, die für die Hornhautschwellung, die Hornhauttrübung und die Fluorescein-Verfärbung ermittelt wurden, und wird gemäß Tabelle 6 vorgenommen.
Tabelle 6 In-vitro-Gesamtklassifizierung
| UN-GHS-Klassifizierung | Kombinationen der drei Endpunkte |
| Keine Einstufung | 3 x I
2 x I, 1 x II |
| Keine Vorhersage möglich | Andere Kombinationen |
| Kategorie 1 | 3 x IV
2 x IV, 1 x III 2 x IV, 1 x II * 2 x IV, 1 x I * Hornhauttrübung > 3 nach 30 Min. (bei mindestens zwei Augen) |
| *) weniger gängige Kombinationen. | |
Studienakzeptanzkriterien
Ein Test gilt als akzeptabel, wenn die gleichzeitigen Negativ- oder Vehikel-/Lösungsmittelkontrollen sowie die gleichzeitigen Positivkontrollen "Keine Einstufung" nach GHS bzw. GHS-Kategorie 1 ergeben.
Prüfbericht
Der Prüfbericht sollte die folgenden Informationen umfassen, soweit sie für die Studie relevant sind:
Prüfchemikalien und Kontrollchemikalien
- chemische Bezeichnung(en) wie der vom Chemical Abstracts Service (CAS) benutzte strukturelle Name, gefolgt von anderen Bezeichnungen, soweit bekannt;
- CAS-Registrierungsnummer (CAS-Nr.), soweit bekannt;
- Reinheit und Zusammensetzung der Prüfchemikalien/Kontrollchemikalien (in Gewichtsprozent), soweit diesbezügliche Informationen vorliegen;
- physikalisch-chemische Eigenschaften wie physikalischer Zustand, Flüchtigkeit, pH-Wert, Stabilität, Chemikalienklasse, Wasserlöslichkeit, soweit sie für die Studie relevant sind;
- Behandlung der Prüfchemikalien/Kontrollchemikalien vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- Stabilität, soweit bekannt.
Informationen zu Auftraggeber und Prüfanstalt
- Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
- Angaben zur Identifizierung der Bezugsquelle der Augen (z.B. Einrichtung, in der sie gewonnen wurden);
Testbedingungen
- Beschreibung des angewandten Testsystems;
- verwendetes Spaltlampenmikroskop (z.B. Modell) und Einstellungen des verwendeten Spaltlampenmikroskops;
- Verweis auf historische Ergebnisse von Negativ- und Positivkontrollen und gegebenenfalls historische Daten, die akzeptable Reihen gleichzeitiger Referenzkontrollen dokumentieren;
- das zur Gewährleistung der Integrität (d. h. Genauigkeit und Zuverlässigkeit) der Prüfmethode im Zeitverlauf angewandte Verfahren (z.B. regelmäßige Testung von Leistungschemikalien).
Gewinnung und Vorbereitung der Augen
- Alter und Gewicht des Spendertieres und, soweit verfügbar, andere spezifische Angaben zu den Tieren, von denen die Augen gewonnen wurden (z.B. Geschlecht, Stamm);
- Lager- und Transportbedingungen der Augen (z.B. Datum und Uhrzeit der Gewinnung der Augen, Zeitabstand zwischen der Gewinnung der Hühnerköpfe und dem Einsetzen der ausgeschälten Augen in die Superfusionskammer);
- Vorbereitung und Einsetzen der Augen, einschließlich Angaben zu ihrer Qualität, zur Temperatur der Augenkammern und zu den Kriterien für die Auswahl der bei den Tests verwendeten Augen.
Testverfahren
- Anzahl der verwendeten Replikate;
- Identität der verwendeten Negativ- und Positivkontrollen (soweit zutreffend, auch der Lösungsmittel- und Referenzkontrollen);
- verwendete Dosis, Applikations- und Expositionsdauer der Prüfchemikalie;
- Beobachtungszeitpunkte (vor und nach der Behandlung);
- Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
- Beschreibung der angewandten Studienakzeptanzkriterien;
- Beschreibung etwaiger Änderungen am Testverfahren.
Ergebnisse
- tabellarische Aufstellung der Hornhautschwellungs-, Hornhauttrübungs- und Fluorescein-Verfärbungswerte für jedes einzelne Auge und an jedem Beobachtungszeitpunkt, einschließlich der Mittelwerte aller getesteten Augen an jedem Beobachtungszeitpunkt;
- höchster Mittelwert für die Hornhautschwellung, die Hornhauttrübung und die Fluorescein-Verfärbung an einem beliebigen Beobachtungszeitpunkt und die zugehörige ICE-Klasse;
- Beschreibung sonstiger festgestellter Wirkungen;
- die abgeleitete In-vitro-Klassifizierung nach GHS;
- gegebenenfalls Aufnahmen vom Auge.
Erörterung der Ergebnisse
Schlussfolgerung
Literaturhinweise
(1) ICCVAM (2007). Test Method Evaluation Report - In Vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm.
(2) ESAC (2007). Statement on the conclusion of the ICCVAM retrospective study on organotypic in vitro assays as screening tests to identify potential ocular corrosives and severe eye irritants. Verfügbar unter: http://ecvam.jrc.it/index.htm.
(3) ICCVAM (2010). ICCVAM Test Method Evaluation Report - Current Status of in vitro Test Methods for Identifying Mild/Moderate Ocular Irritants: The Isolated Chicken Eye (ICE) Test Method. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 10-7553A. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm.
(4) Vereinte Nationen (UN) (2011). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), vierte überarbeitete Ausgabe, UN New York und Genf, 2011. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev04/04files_e.html.
(5) Streamlined Summary Document Supporting OECD Test Guideline 438 on the Isolated Chicken Eye for Eye Irritation/Corrosion. Series on Testing and Assessment Nr. 188 (Part 1 und Part 2), OECD, Paris.
(6) Kapitel B.5 dieser Anlage, Akute Augenreizung/-verätzung.
(7) Scott, L., Eskes, C., Hoffman, S., Adriaens, E., Alepee, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Hamernik, K., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., Mcnamee, P., Osborn, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielmann, H., Stokes, W., Trouba, K., Vassallo, M., Van den Berghe, C., Van Goethem, F., Vinardell, P., Zuang, V (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-up and Top-down Approaches. Toxicology in Vitro, 24, 1-9.
(8) OECD (2011). Guidance Document on The Bovine Corneal Opacity and Permeability (BCOP) and Isolated Chicken Eye (ICE) Test Methods: Collection of Tissues for Histological Evaluation and Collection of Data on Non-Severe Irritants. Series on Testing and Assessment, Nr. 160, OECD, Paris.
(9) ICCVAM (2006). Background review document: Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Isolated Chicken Eye Test Method. NIH-Veröffentlichung Nr. 06-4513. Research Triangle Park: National Toxicology Program. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_brd_ice.htm.
(10) Prinsen, M.K. und Koëter, B.W.M. (1993). Justification of the enucleated eye test with eyes of slaughterhouse animals as an alternative to the Draize eye irritation test with rabbits. Fd. Chem. Toxicol. 31:69-76.
(11) DB-ALM (INVITTOX) (2009). Protokoll Nr. 80: Chicken enucleated eye test (CEET) / Isolated Chicken Eye Test, 13pp. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/.
(12) Balls, M., Botham, P.A., Bruner, L.H. und Spielmann H. (1995). The EC/HO international validation study on alternatives to the Draize eye irritation test. Toxicol. in Vitro, 9:871-929.
(13) Prinsen, M.K. (1996). The chicken enucleated eye test (CEET): A practical (pre)screen for the assessment of eye irritation/corrosion potential of test materials. Food Chem. Toxicol. 34:291-296.
(14) Chamberlain, M., Gad, S.C., Gautheron, P. und Prinsen, M.K. (1997). IRAG-Arbeitsgruppe I: Organotypic models for the assessment/prediction of ocular irritation. Food Chem. Toxicol. 35:23-37.
(15) Prinsen, M.K. (2006). The Draize Eye Test and in vitro alternatives; a left-handed marriage? Toxicology in Vitro, 20:78-81.
(16) Siegel, J.D., Rhinehart, E., Jackson, M., Chiarello, L. und das Healthcare Infection Control Practices Advisory Committee (2007), Guideline for Isolation Precautions: Preventing Transmission of Infectious Agents in Healthcare Settings. Verfügbar unter: http://www.cdc.gov/ncidod/dhqp/pdf/isolation2007.pdf.
(17) Maurer, J.K., Parker, R.D. und Jester, J.V. (2002). Extent of corneal injury as the mechanistic basis for ocular irritation: key findings and recommendations for the development of alternative assays. Reg. Tox. Pharmacol., 36:106-117.
(18) Burton, A.B.G., M. York und R.S. Lawrence (1981). The in vitro assessment of severe irritants. Fd. Cosmet.- Toxicol., 19:471-480.
_____
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
.
| Definitionen | Anlage 1 |
Augenreizung: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind.
Austauschbar mit "Reversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 2" (4).
Bottom-Up-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie keine Einstufung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zuerst Chemikalien, die keine Einstufung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden.
Chemikalie: Stoff oder Gemisch.
Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Chemikalie entscheiden zu können und diese Entscheidung zu untermauern.
Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
Fluorescein-Verfärbung: Subjektiver Messwert im Rahmen des ICE-Tests für die Fluorescein-Natrium-Verfärbung der Epithel-Zellen in der Hornhaut nach Applikation einer Prüfchemikalie. Das Ausmaß der Fluorescein-Verfärbung ist ein Indikator der Schädigung des Hornhautepithels.
Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (4).
Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode.
Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann.
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (4).
Hornhaut: Der Iris und Pupille überdeckende transparente vordere Teil des Augapfels, über den Licht ins Augeninnere übertragen wird.
Hornhautschwellung: Objektiver Messwert im Rahmen des ICE-Tests für die Umfangszunahme der Hornhaut nach Applikation einer Prüfchemikalie. Der Wert wird als Prozentsatz ausgedrückt und auf Basis von Referenz-Hornhautdickenmessungen (Messung vor der Applikation) und der in regelmäßigen Abständen nach der Applikation der Prüfchemikalie im ICE-Test aufgezeichneten Dicke berechnet. Das Ausmaß der Hornhautschwellung ist ein Indikator der Hornhautschädigung.
Hornhauttrübung: Messwert für die Undurchsichtigkeit der Hornhaut nach Applikation einer Prüfchemikalie. Eine verstärkte Hornhauttrübung ist ein Indikator für die Schädigung der Hornhaut.
Irreversible Wirkungen am Auge: siehe "Schwere Augenschädigung" und "UN-GHS-Kategorie 1".
Keine Einstufung nach UN-GHS: Stoffe, die die Voraussetzungen für eine Einstufung in die UN-GHS-Kategorien 1 oder2 (2A oder 2B) nicht erfüllen. Austauschbar mit "Nicht eingestuft".
Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert.
Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert.
Nicht eingestuft: Stoffe, die nicht als augenreizend (UN-GHS-Kategorie 2) oder schwer augenschädigend (UN-GHS-Kategorie 1) eingestuft sind. Austauschbar mit der UN-GHS-Klasse "Keine Einstufung".
Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Referenzchemikalie: Eine zum Vergleich mit einer Prüfchemikalie verwendete Bezugsgröße. Eine Referenzchemikalie sollte die folgenden Eigenschaften aufweisen: i) beständige und zuverlässige Quelle(n); ii) strukturelle und funktionelle Ähnlichkeit zur Klasse der geprüften Chemikalien; iii) bekannte physikalische/chemische Eigenschaften; iv) unterstützende Daten zu bekannten Wirkungen und v) bekannte Potenz innerhalb der gewünschten Wirkungsspanne.
Reversible Wirkungen am Auge: siehe "Augenreizung" und "UN-GHS-Kategorie 2".
Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Austauschbar mit "Irreversible Wirkungen am Auge" und mit "UN-GHS-Kategorie 1" (4).
Spaltlampenmikroskop: Ein Instrument zur direkten Untersuchung des Auges, vergrößert mit einem binokularen Aufrechtbild-Stereomikroskop. Beim ICE-Test wird dieses Instrument eingesetzt, um die Vorderkammern des Hühnerauges visuell zu untersuchen und um die Hornhautdicke anhand eines Pachometer-Aufsatzes objektiv zu messen.
Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (4).
Tensid: Auch als oberflächenaktiver Stoff bezeichnet. Hierbei handelt es sich um Stoffe, wie waschaktive Substanzen (Detergenzien), die die Oberflächenspannung einer Flüssigkeit herabsetzen und so die Bildung von Schaum oder das Eindringen in feste Stoffe ermöglichen; auch bekannt als Netzmittel.
Top-Down-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zuerst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden.
UN-GHS-Kategorie 1: siehe "Schwere Augenschädigung" und/oder "Irreversible Wirkungen am Auge".
UN-GHS-Kategorie 2: siehe "Augenreizung" und/oder "Reversible Wirkungen am Auge".
Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden.
Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
.
| Leistungschemikalien für den ICE | Anlage 2 |
Vor der routinemäßigen Anwendung eines Tests, der den Anforderungen der vorliegenden Prüfmethode genügt, haben Laboratorien ihre technische Kompetenz nachzuweisen, indem sie die 13 in Tabelle 1 empfohlenen Chemikalien in die richtige Augengefährdungsklasse einstufen. Die Chemikalien wurden so ausgewählt, dass sie die Bandbreite von Augengefährdungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405) und dem UN-GHS-Klassifizierungssystem (d. h. den UN-GHS-Kategorien 1, 2A, 2B oder "Keine Einstufung") basieren (4) (6). Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger Daten aus dem ICE-In-vitro-Test. Referenzdaten können aus dem SSD (5) und den Background Review Documents des ICCVAM für den ICE-Test bezogen werden (9).
Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des ICE-Tests
| Chemikalie | CAS-Nr. | Chemikalienklasse 1 | Physikalischer Zustand | In-Vivo- Klassifizierung 2 | In-Vitro- Klassifizierung 3 |
| Benzalkonium-chlorid (5 %) | 8001-54-5 | Onium-verbindung | flüssig | Kategorie 1 | Kategorie 1 |
| Chlorhexidin | 55-56-1 | Amin, Amidin | fest | Kategorie 1 | Kategorie 1 |
| Dibenzoyl-D-Weinsäure | 2743-38-6 | Carbonsäure, Ester | fest | Kategorie 1 | Kategorie 1 |
| Imidazol | 288-32-4 | heterocyclisch | fest | Kategorie 1 | Kategorie 1 |
| Trichloressig-säure (30 %) | 76-03-9 | Carbonsäure | flüssig | Kategorie 1 | Kategorie 1 |
| 2,6-Dichlorbenzoyl-chlorid | 4659-45-4 | Acylhalogenid | flüssig | Kategorie 2 A | Keine Vorhersage möglich 4 |
| Ammonium-nitrat | 6484-52-2 | Anorganisches Salz | fest | Kategorie 2A 5 | Keine Vorhersage möglich 4 |
| Ethyl-2-methylaceto-acetat | 609-14-3 | Ketone, Ester | flüssig | Kategorie 2B | Keine Vorhersage möglich 4 |
| Dimethyl-sulfoxid | 67-68-5 | Organische Schwefel-verbindung | flüssig | Nicht eingestuft | Nicht eingestuft |
| Glyzerin | 56-81-5 | Alkohol | flüssig | Nicht eingestuft | Nicht eingestuft (Grenzfall) |
| Methylcyclo-pentan | 96-37-7 | Kohlenwasser-stoff (cyclisch) | flüssig | Nicht eingestuft | Nicht eingestuft |
| n-Hexan | 110-54-3 | Kohlenwasser-stoff (acyclisch) | flüssig | Nicht eingestuft | Nicht eingestuft |
| Triacetin | 102-76-1 | Lipid | flüssig | Nicht eingestuft | Nicht eingestuft |
| Abkürzungen:
CAS-Nr. = Registernummer des Chemical Abstracts Service
1) Jede Prüfchemikalie wurde anhand einer Standard-Klassifizierungsregelung auf Basis des Klassifizierungssystems der National Library of Medicine Medical Subject Headings (MeSH) einer Chemikalienklasse zugeordnet (abrufbar über http//www.nlm.nih.gov/mesh). 2) gestützt auf Ergebnisse aus dem In-vivo-Kaninchenaugentest (OECD TG 405) unter Verwendung des UN-GHS-Klassifizierungssystems (4)(6). 3) gestützt auf Ergebnisse des ICE-Tests gemäß Tabelle 6. 4) Kombination anderer ICE-Werte als der in Tabelle 6 angegebenen Werte zur Identifizierung der GHS-Kategorie "Keine Einstufung" und der GHS-Kategorie 1 (siehe Tabelle 6). 5) Die Einstufung in die Kategorie 2A oder 2B ist von der Auswertung der Kriterien des UN-GHS zur Unterscheidung zwischen diesen beiden Kategorien abhängig, d. h. für eine Einstufung in die Kategorie 2A müssen an 1 von 3 gegenüber 2 von 3 Tieren Wirkungen an Tag 7 beobachtet werden. Die In-vivo-Studie umfasste drei Tiere. Alle Endpunkte mit Ausnahme einer Bindehautrötung bei einem Tier, gingen bis Tag 7 oder früher auf einen Wert von null zurück. Das eine Tier, das sich bis Tag 7 nicht vollständig regeneriert hatte, wies (an Tag 7) einen Bindehautrötungswert von 1 auf, der an Tag 10 ganz zurückging. | |||||
.
| Schaubilder des ICE-Superfusionsgeräts und der ICE-Augenklemmen | Anlage 3 |
(Siehe Burton et al. (18) für weitere generische Beschreibungen von Superfusionsgeräten und Augenklemmen)
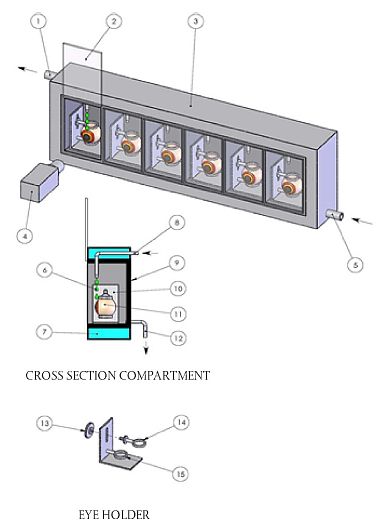
| Nr. | Beschreibung | Nr. | Beschreibung |
| 1 | Warmwasserauslass | 9 | Kammer |
| 2 | Schiebefenster | 10 | Augenhalter |
| 3 | Superfusionsgerät | 11 | Hühnerauge |
| 4 | Optisches Messinstrument | 12 | Auslass Kochsalzlösung |
| 5 | Warmwassereinlass | 13 | Feststellschraube |
| 6 | Kochsalzlösung | 14 | Oberer justierbarer Fixierarm |
| 7 | Warmes Wasser | 15 | Unterer unbeweglicher Fixierarm" |
| 8 | Einlass Kochsalzlösung | ||
(14) In Teil B erhält Kapitel B.49 folgende Fassung:
"B.49 In-vitro-Mikronukleustest an Säugetierzellen
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 487 (2016) und ist Teil einer Reihe von Prüfmethoden zur genetischen Toxikologie. Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (1).
Der In-vitro-Mikronukleustest (MNvit) ist ein Genotoxizitätstest zum Nachweis von Mikronuklei im Zytoplasma von Interphasezellen. Mikronuklei oder Mikrokerne können aus azentrischen Chromosomenfragmenten (d. h. Chromosomen, denen ein Zentromer fehlt) oder aus ganzen Chromosomen entstehen, die während der Anaphase der Zellteilung nicht zu den Polen wandern können. Der MNvit-Test ist daher eine In-vitro-Methode, die eine breite Basis für die In-vitro-Erforschung potenzieller Chromosomenschädigungen bildet, da sowohl Aneugene als auch Klastogene (2) (3) in Zellen nachgewiesen werden können, die während oder nach Kontakt mit der Prüfchemikalie eine Zellteilung durchlaufen haben (weitere Einzelheiten siehe Nummer 13). Mikrokerne stellen auf Tochterzellen übertragene Schäden dar, während in Metaphasezellen festgestellte Chromosomenaberrationen unter Umständen nicht übertragen werden. In beiden Fällen sind die Veränderungen möglicherweise nicht mit dem Überleben der Zellen kompatibel.
Die vorliegende Prüfmethode gestattet die Verwendung von Protokollen mit und ohne den Aktin-Polymerisationsinhibitor Cytochalasin B (cytoB). Durch Beigabe von cytoB vor der Mitose entstehen Zellen mit zwei Kernen. Dies ermöglicht die Identifizierung und selektive Analyse von Mikrokernen in Zellen, die eine Mitose durchlaufen haben (4) (5). Diese Prüfmethode gestattet auch die Verwendung von Protokollen ohne Zytokinese-Block, vorausgesetzt, es gibt Beweise dafür, dass die analysierte Zellpopulation eine Mitose vollzogen hat.
Zusätzlich zum MNvit-Test zur Identifizierung von Chemikalien, die Mikrokerne erzeugen, können auch die immunchemische Markierung von Kinetochoren oder die Hybridisierung mit Zentromer- bzw. Telomer-Sonden (Fluoreszenz-in-situ-Hybridisierung (FISH)) zusätzliche Informationen über die Mechanismen der Chromosomenschädigung und der Bildung von Mikrokernen liefern (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Diese Markierungs- und Hybridisierungsverfahren können angewandt werden, wenn eine verstärkte Mikrokernbildung festgestellt wird und der Prüfer erkennen will, ob diese Zunahme das Ergebnis klastogener und/oder aneugener Ereignisse ist.
Da Mikrokerne in Zellen im Stadium der Interphase mit relativer Objektivität bewertet werden können, muss das Laborpersonal nur bestimmen, wie viele Zellen zwei Kerne (bei Hinzufügung von cytoB) bzw. (in allen anderen Fällen) wie viele Zellen einen Mikrokern enthalten. Infolgedessen können die Objektträger relativ schnell bewertet werden, und die Analyse lässt sich automatisieren. Dies macht es praktisch möglich, Tausende statt Hunderte von Zellen pro Behandlung zu bewerten und so die Aussagekraft des Tests zu erhöhen. Da schließlich die Mikrokerne von verzögert transportierten Chromosomen herrühren können, besteht die Möglichkeit, Aneuploidie induzierende Agenzien nachzuweisen, deren Untersuchung in konventionellen Chromosomenaberrationstests nur schwer möglich ist, z.B. Kapitel B.10 dieses Anhangs (18). Allerdings gestattet der in dieser Prüfmethode beschriebene MNvit-Test die Differenzierung zwischen Veränderungen der Chromosomenzahl und/oder Polyploidie induzierenden Chemikalien und Klastogenizität verursachenden Chemikalien nur, wenn besondere Techniken wie die unter Nummer 4 genannte FISH eingesetzt werden.
Der MNvit-Test ist zuverlässig und kann bei einer Vielfalt von Zelltypen, mit oder ohne Zusatz von cytoB, durchgeführt werden. Umfassende Daten belegen die Validität des MNvit-Tests bei Verwendung unterschiedlicher Zelltypen (kultivierter Zelllinien oder primärer Zellkulturen) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Hierzu zählen insbesondere die internationalen Validierungsstudien, koordiniert durch die Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23), und die Berichte des International Workshop on Genotoxicity Testing (5) (17). Die verfügbaren Daten wurden außerdem vom Europäischen Zentrum zur Validierung von Alternativmethoden (ECVAM) der Europäischen Kommission in einer retrospektiven Validierungsstudie nach dem Weight-of-Evidence-Ansatz neu bewertet, und die Prüfmethode wurde vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM als wissenschaftlich validiert anerkannt (37) (38) (39).
Beim MNvit-Test an Säugetierzellen können kultivierte Zelllinien oder primäre Zellkulturen menschlichen Ursprungs oder von Nagetieren zum Einsatz kommen. Da die Hintergrundfrequenz von Mikrokernen die Empfindlichkeit des Tests beeinflusst, empfiehlt es sich, Zelltypen mit einer stabilen und festgelegten Hintergrundfrequenz der Mikrokernbildung zu verwenden. Die verwendeten Zellen werden unter dem Gesichtspunkt der Wachstumsfähigkeit in Kultur, der Karyotypstabilität (einschließlich Chromosomenzahl) und der spontanen Häufigkeit von Mikrokernen ausgewählt (40). Zum gegenwärtigen Zeitpunkt lassen die verfügbaren Daten keine konkreten Empfehlungen zu, deuten jedoch darauf hin, dass es bei der Bewertung chemischer Gefahren wichtig ist, den p53-Status, die genetische (Karyotyp-)Stabilität, die DNA-Reparaturfähigkeit und die Herkunft (Nagetier oder Mensch) der für die Tests ausgewählten Zellen zu berücksichtigen. Anwendern dieser Prüfmethode wird daher empfohlen, den Einfluss dieser und anderer Zelleigenschaften auf die Leistung einer Zelllinie bei der Erkennung der Mikrokerninduktion zu berücksichtigen, da sich die Erkenntnisse auf diesem Gebiet ständig weiterentwickeln.
Es gelten die Definitionen gemäß Anlage 1.
Vorbemerkungen und Einsatzgrenzen
In vitro durchgeführte Tests erfordern in der Regel den Zusatz eines exogenen Stoffwechselaktivierungssystems, sofern die Zellen nicht im Hinblick auf die Prüfchemikalien metabolisch kompetent sind. Mit diesem exogenen Stoffwechselaktivierungssystem lassen sich die In-vivo-Bedingungen jedoch nicht gänzlich nachvollziehen. Es sind auch Bedingungen zu vermeiden, die zu künstlich positiven Ergebnissen führen, die nicht die Genotoxizität der Prüfchemikalie widerspiegeln. Dazu gehören unter anderem Veränderungen des pH-Wertes (41) (42) (43) bzw. der Osmolalität, Wechselwirkung mit dem Zellkulturmedium (44) (45) oder hochgradige Zytotoxizität (siehe Nummer 29).
Zur Analyse der Mikrokerninduktion ist es wesentlich, dass sowohl die behandelten als auch die unbehandelten Kulturen eine Mitose durchlaufen haben. Das informativste Stadium für die Auswertung von Mikrokernen liegt in Zellen vor, die eine Mitose während oder nach der Behandlung mit der Prüfchemikalie vollzogen haben. Bei hergestellten Nanomaterialien sind besondere Anpassungen dieser Prüfmethode erforderlich, die hier jedoch nicht beschrieben werden.
Bevor die Prüfmethode auf ein Gemisch angewendet wird, um zu Regulierungszwecken Daten zu gewinnen, sollte geprüft werden, ob, und falls ja, warum, sie für diesen Zweck geeignete Ergebnisse liefern kann. Diese Überlegungen erübrigen sich, sofern die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist.
Testprinzip
Zellkulturen menschlichen Ursprungs oder von Säugetieren werden sowohl mit als auch ohne exogene Stoffwechselaktivierung mit der Prüfchemikalie in Kontakt gebracht, außer wenn Zellen mit eigener adäquater Metabolisierungskapazität verwendet werden (siehe Nummer 19).
Während oder nach dem Kontakt mit der Prüfchemikalie werden die Zellen so lange kultiviert, dass Chromosomenschäden oder andere Wirkungen auf den Zellzyklus/die Zellteilung zur Bildung von Mikrokernen in Interphasezellen führen können. Zur Induktion einer Aneuploidie sollte die Prüfchemikalie während der Mitose vorhanden sein. Gewonnene und gefärbte Interphasezellen werden auf das Vorhandensein von Mikrokernen untersucht. Im Idealfall sollten Mikrokerne nur in Zellen ausgewertet werden, die während des Kontakts mit der Prüfchemikalie oder ggf. während der Zeit nach der Behandlung eine Mitose durchlaufen haben. In Kulturen, die mit einem Zytokinese-Blocker behandelt wurden, ist dies ohne Weiteres möglich, indem nur zweikernige Zellen ausgewertet werden. Wenn kein Zytokinese-Blocker verwendet wurde, ist es wichtig nachzuweisen, dass die analysierten Zellen aufgrund einer Zunahme der Zellpopulation wahrscheinlich während oder nach dem Kontakt mit der Prüfchemikalie eine Zellteilung durchlaufen haben. Bei allen Protokollen ist es wichtig nachzuweisen, dass sowohl in den Kontrollkulturen als auch in den behandelten Kulturen eine Zellproliferation stattgefunden hat. Das Ausmaß der von der Prüfchemikalie induzierten Zytotoxizität oder Zytostase sollte in allen Kulturen bewertet werden, die auf das Vorhandensein von Mikrokernen untersucht werden.
Beschreibung der Methode
Zellen
Es können kultivierte primäre Lymphozyten aus dem peripheren Blut von Menschen oder anderen Säugetieren (7) (20) (46) (47) sowie verschiedene Zelllinien von Nagetieren wie CHO, V79, CHL/IU und L5178Y oder menschliche Zelllinien wie TK6 verwendet werden (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (siehe Nummer 6). Andere Zelllinien wie HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2-Zellen (52) (53), A549 und primäre Embryonalzellen des Syrischen Hamsters (54) wurden in Mikronukleustests verwendet, sind zu diesem Zeitpunkt jedoch nicht umfassend validiert worden. Die Verwendung dieser Zelllinien und -typen sollte daher anhand ihrer nachgewiesenen Leistung im Test begründet werden, wie im Abschnitt "Akzeptanzkriterien" beschrieben. CytoB kann Berichten zufolge das Wachstum von L5178Y-Zellen beeinträchtigen und wird daher bei dieser Zelllinie nicht empfohlen (23). Wenn aus Gründen des Tierschutzes primäre Zellen verwendet werden, sollten, soweit möglich, Zellen menschlichen Ursprungs in Betracht gezogen und nach humanethischen Grundsätzen und Vorschriften beprobt werden.
Lymphozyten aus dem peripheren Blut von Menschen sollten jungen (ca. 18- bis 35-jährigen) nicht rauchenden Personen ohne bekannte Erkrankung entnommen werden, die bekanntermaßen in letzter Zeit nicht mit genotoxischen Einwirkungen (z.B. in Form von Chemikalien oder ionisierender Strahlung) in einer Intensität in Kontakt gekommen sind, die zu einem Anstieg der Hintergrundrate von Mikrokernzellen führen würde. Dies stellt eine niedrige und gleichmäßige Hintergrundrate von Mikrokernzellen sicher. Die Hintergrundrate von Mikrokernzellen nimmt mit fortschreitendem Alter zu. Dieser Trend ist bei Frauen ausgeprägter als bei Männern (55). Wenn Zellen von mehreren Spendern zur Verwendung gepoolt werden, ist die Anzahl der Spender anzugeben. Es muss nachgewiesen werden, dass sich die Zellen seit dem Beginn der Behandlung mit der Prüfchemikalie bis zur Zellentnahme geteilt haben. Zellkulturen werden in einer Phase des exponentiellen Wachstums gehalten (Zelllinien) oder zur Teilung angeregt (primäre Lymphozytenkulturen), damit die Zellen in unterschiedlichen Stadien des Zellzyklus exponiert werden, da die Empfindlichkeit der Zellstadien gegenüber den Prüfchemikalien möglicherweise nicht bekannt ist. Die Primärzellen, die mit mitogenen Stoffen zur Teilung stimuliert werden müssen, werden in der Regel während der Behandlung mit der Prüfchemikalie nicht weiter synchronisiert (z.B. humane Lymphozyten nach einer 48-stündigen mitogenen Stimulierung). Die Verwendung synchronisierter Zellen während der Behandlung mit der Prüfchemikalie wird nicht empfohlen, kann jedoch zulässig sein, sofern sie begründet wird.
Kulturmedien und Inkubationsbedingungen
Zur Kultivierung sind geeignete Kulturmedien und Inkubationsbedingungen (Kulturgefäße, ggf. befeuchtete Atmosphäre mit einer CO2-Konzentration von 5 %, Temperatur von 371 °C) zu verwenden. Zelllinien sind routinemäßig auf Stabilität der modalen Chromosomenzahl und Mykoplasmaverunreinigung zu überprüfen, und Zellen sollten bei Verunreinigung oder bei veränderter modaler Chromosomenzahl nicht herangezogen werden. Die normale Dauer des Zellzyklus bei den gewählten Zelllinien oder der im Prüflabor verwendeten primären Kulturen sollte bekannt sein und mit den veröffentlichten Zelleigenschaften übereinstimmen.
Vorbereitung der Kulturen
Zelllinien: Die Zellen werden aus Stammkulturen gewonnen und im Kulturmedium in einer solchen Dichte überimpft, dass die Zellen in Suspensions- oder Monolayerkultur bis zum Zeitpunkt der Gewinnung weiterhin exponentiell wachsen (z.B. sollte eine Konfluenz bei in Monolayerkultur gezüchteten Zellen vermieden werden).
Lymphozyten: Mit einem Antikoagulans (z.B. Heparin) behandeltes Vollblut oder separierte Lymphozyten werden einem Kulturmedium beigegeben (z.B. bei humanen Lymphozyten für die Dauer von 48 Stunden), das ein Mitogen (z.B. Phytohämagglutinin (PHA) im Fall von humanen Lymphozyten) enthält, um eine Zellteilung vor der Behandlung mit der Prüfchemikalie und cytoB herbeizuführen.
Stoffwechselaktivierung
Bei Zellen mit ungeeigneter Stoffwechselkapazität sollten exogene metabolisierende Systeme eingesetzt werden. Das am häufigsten verwendete, standardmäßig empfohlene System, sofern nicht anders begründet, ist eine durch Ko-Faktoren ergänzte post-mitochondriale Fraktion (S9) aus der Leber von Nagetieren (in der Regel Ratten), die mit enzyminduzierenden Agenzien wie Aroclor 1254 (56) (57) oder einem Gemisch aus Phenobarbital und b-Naphtoflavon (58) (59) (69) vorbehandelt wurde. Das letztgenannte Gemisch verstößt nicht gegen das Stockholmer Übereinkommen über persistente organische Schadstoffe (61) und hat sich bei der Induktion von Mischfunktionsoxidasen als ebenso wirksam wie Aroclor 1254 erwiesen (58) (59) (60). Die S9-Fraktion wird im Endmedium in der Regel in Konzentrationen von 1 bis 2 % v/v verwendet, kann jedoch auf 10 % v/v erhöht werden. Die Verwendung von Produkten, die den Mitoseindex senken, insbesondere Komplexbildnern für Calcium (62), sollte während der Behandlung vermieden werden. Die Wahl der Art und Konzentration des exogenen Metabolisierungssystems oder metabolischen Agens ist möglicherweise von der geprüften chemischen Klasse abhängig.
Vorbereitung der Prüfchemikalie
Feste Prüfchemikalien sollten vor der Zellbehandlung in geeigneten Lösungsmitteln gelöst und ggf. verdünnt werden. Flüssige Prüfchemikalien können dem Versuchssystem vor der Behandlung direkt beigegeben und/oder verdünnt werden. Gasförmige oder flüchtige Prüfchemikalien sind mithilfe geeigneter Änderungen an den Standardprotokollen, wie z.B. durch Behandlung in hermetisch verschlossenen Gefäßen (63) (64) (65), zu prüfen. Zubereitungen der Prüfchemikalie sollten kurz vor der Behandlung hergestellt werden, es sei denn, die Stabilität der Chemikalie bei Lagerung wird nachgewiesen.
Prüfbedingungen
Lösungsmittel
Das Lösungsmittel sollte so gewählt werden, dass eine optimale Löslichkeit der Prüfchemikalie gewährleistet ist, ohne dass die Durchführung des Versuchs negativ beeinträchtigt wird, z.B. durch Änderung des Zellwachstums, Beeinträchtigung der Integrität der Prüfchemikalie, Reaktion mit Kulturgefäßen, Behinderung des Stoffwechselaktivierungssystems. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels (oder Kulturmediums) in Erwägung zu ziehen. Gründlich erprobte Lösungsmittel sind Wasser oder Dimethylsulfoxid (DMSO). In der Regel sollen organische Lösungsmittel 1 % v/v nicht übersteigen. Wenn cytoB in DMSO gelöst wird, darf die für die Prüfchemikalie und cytoB verwendete Gesamtmenge an organischen Lösungsmitteln nicht mehr als 1 % v/v betragen; anderenfalls sind unbehandelte Kontrollen zu verwenden, um sicherzustellen, dass der Anteil an organischen Lösungsmitteln keine schädliche Wirkung hat. Wässrige Lösungsmittel (Kochsalzlösung oder Wasser) sollen 10 % v/v im Endmedium nicht übersteigen. Kommen weniger gründlich erprobte Lösungsmittel zur Verwendung (z. B Ethanol oder Aceton), ist deren Verwendung durch Daten zu stützen, die ihre Verträglichkeit mit der Prüfchemikalie, mit dem Versuchssystem sowie ihre mangelnde Genotoxizität in der verwendeten Konzentration belegen. Liegen keine Daten vor, die dies belegen, ist es wichtig, unbehandelte Kontrollen (siehe Anlage 1) sowie Lösungsmittelkontrollen einzubeziehen, um nachzuweisen, dass durch die gewählten Lösungsmittel keine schädlichen oder chromosomalen Wirkungen (z.B. Aneuploidie oder Klastogenizität) ausgelöst werden.
Verwendung von cytoB als Zytokinese-Blocker
Eine der wichtigsten Überlegungen bei der Durchführung des MNvit-Tests gilt der Vergewisserung, dass die bewerteten Zellen während der Behandlung oder ggf. während der Inkubationszeit nach der Behandlung eine Mitose durchlaufen haben. Die Auswertung von Mikrokernen soll daher auf Zellen beschränkt werden, die während oder nach der Behandlung eine Mitose vollzogen haben. CytoB ist das am häufigsten verwendete Agens zur Zytokinese-Blockierung, da es den Aktinaufbau hemmt und somit die Trennung der Tochterzellen nach der Mitose verhindert, was wiederum zur Bildung zweikerniger Zellen führt (6) (66) (67). Zugleich kann bei Verwendung von cytoB die Auswirkung der Prüfchemikalie auf die Zellproliferationskinetik gemessen werden. CytoB sollte bei der Verwendung menschlicher Lymphozyten als Zytokinese-Blocker eingesetzt werden, da die Zellzyklendauer zwischen einzelnen Spendern variiert und nicht alle Lymphozyten auf Phytohämagglutinin reagieren. Bei anderen Zelltypen ist cytoB nicht zwingend erforderlich, sofern nachgewiesen werden kann, dass diese eine Teilung durchlaufen haben, wie unter Nummer 27 beschrieben. Außerdem wird cytoB im Allgemeinen nicht verwendet, wenn die Proben mithilfe durchflusszytometrischer Methoden auf Mikrokerne untersucht werden.
Für jeden Zelltyp sollte das Labor die geeignete cytoB-Konzentration festlegen, um in den Lösungsmittelkontrollkulturen eine optimale Frequenz der zweikernigen Zellen zu erreichen. Ferner sollte die Konzentration nachweislich eine gute Ausbeute an zweikernigen Zellen zur Auswertung ergeben. Die geeignete cytoB-Konzentration liegt in der Regel zwischen 3 und 6 µ/ml (19).
Messung von Zellproliferation und Zytotoxizität und Wahl der Behandlungskonzentrationen
Bei der Festlegung der höchsten zu testenden Konzentration der Prüfchemikalie sind Konzentrationen zu vermeiden, die zu künstlich positiven Reaktionen führen können, wie z.B. zu übermäßiger Zytotoxizität (siehe Nummer 29), Ausfällungen im Kulturmedium (siehe Nummer 30) oder ausgeprägten Änderungen des pH-Werts oder der Osmolalität (siehe Nummer 9). Sofern die Prüfchemikalie zum Zeitpunkt der Zugabe den pH-Wert des Mediums erheblich verändert, lässt sich der pH-Wert auch durch Anwendung eines Puffers im Endmedium einstellen, damit künstlich positive Reaktionen vermieden und geeignete Kulturbedingungen aufrechterhalten werden.
Es werden Messungen der Zellproliferation vorgenommen, um sicherzustellen, dass genügend behandelte Zellen während des Tests eine Mitose durchlaufen haben und dass die Behandlungen bei geeigneten Zytotoxizitätsniveaus durchgeführt werden (siehe Nummer 29). Die Zytotoxizität sollte im Hauptversuch mit und ohne Stoffwechselaktivierung und unter Verwendung eines geeigneten Indikators für Zelltod und -wachstum bestimmt werden (siehe Nummern 26 und 27). Wenngleich die Bewertung der Zytotoxizität im Rahmen eines ersten Vorversuchs nützlich sein kann, um die im Hauptversuch verwendeten Konzentrationen besser bestimmen zu können, ist ein Vorversuch nicht zwingend erforderlich. Wird er durchgeführt, ersetzt er nicht die Messung der Zytotoxizität im Hauptversuch.
Die Behandlung von Kulturen mit cytoB und die Messung der relativen Häufigkeit von einkernigen, zweikernigen und mehrkernigen Zellen in der Kultur sind ein genaues Verfahren zur Quantifizierung des Effekts auf die Zellproliferation und die zytotoxische oder zytostatische Wirkung einer Behandlung (6) und stellen sicher, dass nur Zellen mikroskopisch bewertet werden, die während oder nach der Behandlung eine Teilung vollzogen haben. Es wird empfohlen, die zytotoxische und zytostatische Wirkung einer Behandlung mittels des Zytokinese-Block-Proliferationsindex (CBPI) (6) (27) (68) oder des Replikationsindex (RI) bei mindestens 500 Zellen pro Kultur abzuleiten (Formeln siehe Anlage 2). Hierzu werden die Werte in behandelten Kulturen und Kontrollkulturen miteinander verglichen. Die Bewertung anderer Zytotoxizitäts-Indikatoren (z.B. Zellintegrität, Apoptose, Nekrose, Metaphasezählung, Zellzyklus) könnte ebenfalls nützliche Informationen liefern, darf jedoch nicht als Ersatz für den CBPI oder RI verwendet werden.
In Studien ohne cytoB muss nachgewiesen werden, dass sich die Zellen in der Kultur geteilt haben, sodass ein erheblicher Teil der bewerteten Zellen während oder nach Behandlung mit der Prüfchemikalie eine Teilung durchlaufen hat. Anderenfalls kann es zu falsch negativen Reaktionen kommen. Die Messung der relativen Populationsverdopplung (RPD) oder der relativen Erhöhung der Zellzahl (RICC) sind empfohlene Verfahren zur Schätzung der zytostatischen Wirkung eines Behandlung (17) (68) (69) (70) (71) (Formeln siehe Anlage 2). Bei längeren Probenahmephasen (z.B. Behandlung über einen Zeitraum von 1,5 bis 2 normalen Zellzykluslängen und Gewinnung nach weiteren 1,5 bis 2 normalen Zellzykluslängen, woraus sich eine Probenahmezeit von insgesamt mehr als 3 bis 4 normalen Zellzykluslängen ergibt, wie unter Nummern 38 und 39 beschrieben) könnte es bei der RPD zu einer Unterschätzung der Toxizität kommen (71). Unter diesen Umständen ist die RICC möglicherweise ein besseres Verfahren. Anderenfalls erhält man anhand einer Bewertung der Zytotoxizität nach 1,5 bis 2 normalen Zellzykluslängen einen hilfreichen Schätzwert. Die Bewertung anderer Marker für Zytotoxizität oder Zytostase (z.B. Zellintegrität, Apoptose, Nekrose, Metaphasezählung, Proliferationsindex (PI), Zellzyklus, nukleoplasmische Brücken oder Kernknospen) könnten nützliche Informationen liefern, dürfen jedoch nicht als Ersatz für die RPD oder die RICC verwendet werden.
Es sollten mindestens drei Versuchskonzentrationen (ausgenommen das Lösungsmittel und Positivkontrollen), die die Akzeptanzkriterien erfüllen (geeignete Zytotoxizität, Anzahl der Zellen usw.), ausgewertet werden. Unabhängig von der Art der Zellen (Zelllinien oder Primärkulturen von Lymphozyten) können für jede überprüfte Konzentration Replikat- oder Einfachkulturen herangezogen werden. Wenngleich die Verwendung von Zweifachkulturen ratsam ist, sind Einfachkulturen auch zulässig, vorausgesetzt, es wird für Einfach- oder Zweifachkulturen jeweils die gleiche Gesamt-Zellpopulation ausgewertet. Die Verwendung von Einzelkulturen ist insbesondere dann relevant, wenn mehr als drei Konzentrationen bewertet werden (siehe Nummern 44 und 45). Die Ergebnisse aus den unabhängigen Replikatkulturen bei einer festgelegten Konzentration können zu Datenanalysezwecken gepoolt werden. Bei Prüfchemikalien mit geringer oder keiner Zytotoxizität sind in der Regel Konzentrationsintervalle mit zwei- bis dreifacher Konzentration geeignet. Wenn Zytotoxizität auftritt, sollten die Prüfkonzentrationen einen Bereich ausgehend vom Wert, bei dem Zytotoxizität auftritt (siehe Beschreibung unter Nummer 29), bis zu Konzentrationen mit mäßiger oder geringer oder nicht vorhandener Toxizität umfassen. Viele Prüfchemikalien zeigen steile Konzentrations-Wirkungs-Kurven. Um Daten zu mäßiger oder geringer Toxizität zu erhalten oder um die Dosis-Wirkungs-Beziehungen im Einzelnen auszuwerten, wird es daher erforderlich sein, Konzentrationen mit kleineren Abständen und/oder mehr als drei Konzentrationen zu verwenden (Einfach- oder Replikatkulturen), insbesondere in Fällen, in denen ein Wiederholungsversuch erforderlich ist (siehe Nummer 60).
Beruht die höchste Konzentration auf der Zytotoxizität, sollte die Höchstkonzentration darauf abzielen, unter Verwendung der empfohlenen Zytotoxizitätsparameter (d. h. Verringerung der RICC und RPD bei Zelllinien, wenn cytoB nicht verwendet wird, und Verringerung des CBPI oder RI, wenn cytoB verwendet wird, auf 45 ± 5 % der gleichzeitigen Negativkontrollen) eine Zytotoxizität von 55 ± 5 % zu erreichen (72). Ausschließlich am oberen Ende dieses Zytotoxizitätsbereichs von 55 ± 5 % anzutreffende positive Ergebnisse sollten mit Vorsicht interpretiert werden (71).
Im Falle schwer löslicher Chemikalien, die bei Konzentrationen unterhalb der niedrigsten unlöslichen Konzentration nicht zytotoxisch sind, sollte die höchste analysierte Konzentration am Ende der Behandlung mit der Prüfchemikalie eine Trübung oder eine mit bloßem Auge oder mithilfe eines Inversmikroskops erkennbare Ausfällung bewirken. Auch wenn Zytotoxizität oberhalb der niedrigsten unlöslichen Konzentration auftritt, ist es ratsam, nur eine Konzentration zu testen, die eine Trübung oder sichtbare Ausfällung hervorruft, da artefaktische Wirkungen eine Folge dieser Ausfällung sein könnten. Bei der Konzentration, bei der es zu einer Ausfällung kommt, ist unbedingt sicherzustellen, dass die Ausfällung nicht die Durchführung des Versuchs beeinträchtigt (z.B. Färbung oder Auswertung). Es ist möglicherweise sinnvoll, die Löslichkeit im Kulturmedium vor dem Versuch zu bestimmen.
Wenn keine Ausfällung bzw. keine begrenzende Zytotoxizität festgestellt wird, sollte die Höchstkonzentration bei Versuchen 10 mM, 2 mg/ml oder 2 µl/ml betragen, je nachdem, welcher Wert am niedrigsten ist (73) (74) (75). Sofern die Zusammensetzung der Prüfchemikalie nicht genau definiert ist, z.B. ein Stoff mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien (UVCB) (76), ein Umweltextrakt usw., muss die höchste Konzentration möglicherweise höher angesetzt werden (z.B. bei 5 mg/ml), sofern keine ausreichende Zytotoxizität vorhanden ist, um die Konzentration der einzelnen Bestandteile zu erhöhen. Es sei jedoch darauf hingewiesen, dass diese Anforderungen sich von denen für Humanpharmazeutika unterscheiden können (93).
Kontrollen
Bei jedem Gewinnungszeitpunkt sind parallel laufende Negativkontrollen (siehe Nummer 21) anzulegen, bei denen das Behandlungsmedium lediglich Lösungsmittel enthält und die auf die gleiche Weise wie die Behandlungskulturen bearbeitet werden.
Gleichzeitige Positivkontrollen müssen durchgeführt werden, um die Eignung des Labors zum Nachweis von Klastogenen und Aneugenen unter den Bedingungen des verwendeten Prüfprotokolls sowie ggf. die Wirksamkeit des exogenen Stoffwechselaktivierungssystems nachzuweisen. Beispiele für Positivkontrollen sind nachstehender Tabelle 1 zu entnehmen. Es können andere geeignete Positivkontrollchemikalien verwendet werden, sofern dies begründet ist.
Zum gegenwärtigen Zeitpunkt sind keine Aneugene bekannt, die zu ihrer genotoxischen Wirkung eine Stoffwechselaktivierung benötigen (17). Da In-vitro-Tests auf Genotoxizität in Säugetierzellen für die gleichzeitigen Kurzzeitbehandlungen mit und ohne Zusatz eines Stoffwechselaktivierungssystems über die gleiche Behandlungsdauer ausreichend standardisiert sind, kann sich die Hinzuziehung von Positivkontrollen auf ein Klastogen beschränken, das eine Stoffwechselaktivierung erfordert. In diesem Fall wird durch die Reaktion einer einzelnen Positivkontrolle sowohl die Aktivität des Stoffwechselaktivierungssystems als auch die Reaktionsfähigkeit des Testsystems nachgewiesen. Im Falle einer Langzeitbehandlung (ohne S9) sollte jedoch eine gesonderte Positivkontrolle erfolgen, da die Behandlungsdauer beim Test mit Stoffwechselaktivierung eine andere ist. Wird als einzige Positivkontrolle für die Kurzzeitbehandlung mit und ohne Stoffwechselaktivierung ein Klastogen ausgewählt, sollte für die Langzeitbehandlung ohne Stoffwechselaktivierung ein Aneugen ausgewählt werden. Positivkontrollen für Klastogenizität und Aneugenizität sollten in metabolisch kompetenten Zellen verwendet werden, die kein S9 benötigen.
Jede Positivkontrolle sollte bei einer oder mehreren Konzentrationen durchgeführt werden, die voraussichtlich eine reproduzierbare und erkennbare Zunahme gegenüber dem Hintergrund ergeben, womit sich die Empfindlichkeit des Versuchssystems nachweisen lässt (d. h. die Wirkungen sind eindeutig, lassen aber beim Ablesen nicht sofort die Identität der kodierten Objektträger erkennen), und die Wirkung sollte nicht durch einen Zytotoxizitätswert beeinträchtigt werden, der die in dieser Prüfmethode vorgegebenen Grenzen überschreitet.
Tabelle 1 Zur Beurteilung der Eignung des Labors und zur Wahl der Positivkontrollen empfohlene Referenzchemikalien
| Kategorie | Chemikalie | CAS-Nr. |
| 1. Klastogene, die ohne Stoffwechselaktivierung wirken | ||
| Methylmethansulfonat | 66-27-3 | |
| Mitomycin C | 50-07-7 | |
| 4-Nitroquinolin-N-oxid | 56-57-5 | |
| Cytosinarabinosid | 147-94-4 | |
| 2. Klastogene, die eine Stoffwechselaktivierung erfordern | ||
| Benzo[a]pyren | 50-32-8 | |
| Cyclophosphamid | 50-18-0 | |
| 3. Aneugene | ||
| Colchicin | 64-86-8 | |
| Vinblastin | 143-67-9 | |
Verfahren
Behandlungsplan
Um die Wahrscheinlichkeit zu maximieren, dass ein Aneugen oder Klastogen in einem bestimmten Stadium des Zellzyklus erkannt wird, muss eine ausreichende Anzahl an Zellen während aller Stadien ihrer Zellzyklen mit der Prüfchemikalie behandelt werden. Alle Behandlungen sollten während des exponentiellen Wachstums der Zellen beginnen und enden, und die Zellen sollten bis zum Zeitpunkt der Probenahme weiter wachsen. Der Behandlungsplan für Zelllinien und primäre Zellkulturen kann daher in gewisser Weise von dem für Lymphozyten abweichen, die für den Beginn ihres Zellzyklus einer mitogenen Stimulation bedürfen (17). Bei Lymphozyten ist es am wirksamsten, die Behandlung mit der Prüfchemikalie 44 bis 48 Stunden nach der PHA-Stimulation zu beginnen, wenn sich die Zellen asynchron teilen (6).
Veröffentlichte Daten (19) legen nahe, dass die meisten Aneugene und Klastogene entdeckt werden, wenn eine kurze Behandlung von 3 bis 6 Stunden mit oder ohne Vorhandensein von S9 erfolgt, woraufhin die Prüfchemikalie entfernt wird und die Zellen in einem Zeitraum beprobt werden, der etwa der 1,5-fachen bis 2-fachen Dauer des normalen Zellzyklus nach Behandlungsbeginn entspricht (7).
Für eine gründliche Bewertung, die erforderlich wäre, um auf ein negatives Ergebnis zu schließen, sollten bei Durchführung einer Kurzzeitbehandlung mit und ohne Stoffwechselaktivierung und einer Langzeitbehandlung ohne Stoffwechselaktivierung alle drei der folgenden Versuchsbedingungen angewandt werden, und zwar eine (siehe Nummern 56, 57 und 58):
- Die Zellen sollten 3 bis 6 Stunden lang ohne Zusatz eines Stoffwechselaktivierungssystems mit der Prüfchemikalie behandelt werden und in einem Zeitraum beprobt werden, der etwa der 1,5-fachen bis 2-fachen Dauer des normalen Zellzyklus nach Behandlungsbeginn entspricht (19).
- Die Zellen sollten 3 bis 6 Stunden lang mit Zusatz eines Stoffwechselaktivierungssystems mit der Prüfchemikalie behandelt werden und in einem Zeitraum beprobt werden, der etwa der 1,5-fachen bis 2-fachen Dauer des normalen Zellzyklus nach Behandlungsbeginn entspricht (19).
- Die Zellen sollten kontinuierlich ohne Stoffwechselaktivierung behandelt werden, wobei eine Beprobung nach Ablauf eines Zeitraums erfolgt, der etwa der 1,5-fachen bis 2-fachen Dauer des normalen Zellzyklus entspricht.
Führt eine der oben genannten Versuchsbedingungen zu einem positiven Befund, ist es möglicherweise nicht erforderlich, Untersuchungen anhand der anderen Behandlungsverfahren durchzuführen.
Wenn bekannt ist oder der Verdacht besteht, dass die Prüfchemikalie die Dauer des Zellzyklus beeinflusst (z.B. bei der Prüfung von Nucleosidanalogen), was insbesondere für p53-kompetente Zellen gilt (35) (36) (77), können die Zeiträume für die Probenahme oder Regenerierung um bis zu eine weitere 1,5-fache bis 2-fache Dauer des normalen Zellzyklus (d. h. insgesamt die 3-fache bis 4-fache Dauer des Zellzyklus nach Beginn der Kurzzeit- und Langzeitbehandlungen) ausgedehnt werden. Diese Optionen beziehen sich auf Situationen, in denen mögliche Wechselwirkungen zwischen der Prüfchemikalie und cytoB befürchtet werden. Bei verlängerten Probenahmezeiten (d. h. einer Kulturzeit von insgesamt der 3-fachen bis 4-fachen Zellzyklusdauer) ist unbedingt sicherzustellen, dass sich die Zellen noch aktiv teilen. Bei Lymphozyten beispielsweise kann das exponentielle Wachstum 96 Stunden nach der Stimulation nachlassen, und Monolayerkulturen können konfluent werden.
Die empfohlenen Pläne für die Zellbehandlung sind in Tabelle 2 zusammengefasst. Diese allgemeinen Behandlungspläne können je nach Stabilität oder Reaktivität der Prüfchemikalie oder den besonderen Wachstumseigenschaften der verwendeten Zellen (mit entsprechender Begründung) modifiziert werden.
Tabelle 2 Zellbehandlung und Entnahmezeitpunkte beim MNvit-Test
| Lymphozyten, Primärzellen und Zelllinien, die mit cytoB behandelt wurden | + S9 Kurzzeitbehandlung | 3-6 Stunden Behandlung in Anwesenheit von S9; Entfernung von S9 und Behandlungsmedium; Zugabe von neuem Medium und cytoB; Zellentnahme nach 1,5-2,0 normalen Zellzyklen nach Behandlungsbeginn. |
| - S9 Kurzzeitbehandlung | 3-6 Stunden Behandlung; Entfernung des Behandlungsmediums; Zugabe von neuem Medium und cytoB; Zellentnahme nach 1,5-2,0 normalen Zellzyklen nach Behandlungsbeginn. | |
| - S9 Verlängerte Behandlung | Behandlung über 1,5-2,0 normale Zellzyklen in Anwesenheit von cytoB; Zellentnahme am Ende der Behandlungsdauer. | |
| Ohne cytoB behandelte Zelllinien (Identisch mit den obigen Behandlungsplänen mit der Ausnahme, dass kein cytoB zugegeben wird) | ||
Bei Monolayerkulturen können am Ende der 3- bis 6-stündigen Behandlung mitotische Zellen vorhanden sein (erkennbar daran, dass sie rund sind und sich von der Oberfläche ablösen). Da diese mitotischen Zellen sich leicht lösen, können sie bei Entfernung des Mediums mit der Prüfchemikalie verloren gehen. Kann nachgewiesen werden, dass die Zahl der mitotischen Zellen gegenüber den Kontrollen erheblich zugenommen hat, was mit hoher Wahrscheinlichkeit auf einen mitotischen Arrest hinweist, sollten die Zellen durch Zentrifugieren gewonnen und anschließend der Kultur wieder zugeführt werden, um zu vermeiden, dass Zellen verlorengehen, die sich in der Mitose befinden und zum Zeitpunkt der Gewinnung dem Risiko einer Mikrokernbildung/Chromosomenaberration ausgesetzt sind.
Zellgewinnung und Präparation des Objektträgers
Jede Kultur wird gesondert gewonnen und aufbereitet. Die Zellpräparation kann eine Behandlung mit hypotoner Lösung beinhalten, dieser Schritt ist jedoch nicht notwendig, wenn eine angemessene Ausbreitung der Zellen anderweitig sichergestellt wird. Bei der Präparation der Objektträger können verschiedene Techniken angewandt werden, vorausgesetzt, es werden Zellpräparate hoher Qualität zur Auswertung bereitgestellt. Dabei sollten Zellen mit intakter Zellmembran und intaktem Zytoplasma zurückgehalten werden, um die Erkennung von Mikrokernen sowie (bei der Zytokinese-Block-Methode) die zuverlässige Identifizierung zweikerniger Zellen zu ermöglichen.
Die Objektträger können mit verschiedenen Verfahren angefärbt werden, beispielsweise mit Giemsa oder fluoreszierenden DNA-spezifischen Farbstoffen. Durch Verwendung eines DNA-spezifischen Farbstoffs (z.B. Acridin Orange (78) oder Hoechst 33258 Plus Pyronin-Y (79)) lassen sich bestimmte Artefakte vermeiden, die bei der Verwendung eines nicht DNA-spezifischen Farbstoffs auftreten. Zur Identifizierung des Inhalts (ganze Chromosomen werden eingefärbt, azentrische Chromosomenfragmente nicht) von Mikrokernen können Anti-Kinetochor-Antikörper, FISH mit panzentromerischen DNA-Sonden oder eine präparierte In-situ-Markierung mit panzentromer-spezifischen Primern, zusammen mit der geeigneten DNA-Gegenfärbung, verwendet werden, wenn mechanistische Informationen zu ihrer Bildung benötigt werden (16) (17). Andere Methoden zur Unterscheidung zwischen Klastogenen und Aneugenen können ebenfalls angewandt werden, wenn sie sich als wirksam erwiesen haben. Beispielsweise könnten bei bestimmten Zelllinien die Messungen von sub-2N-Kernen als hypodiploide Ereignisse mittels Techniken wie Bildanalyse, Laser-Scan-Zytometrie oder Durchflusszytometrie ebenfalls nützliche Informationen liefern (80) (81) (82). Morphologische Beobachtungen von Kernen könnten ebenfalls auf eine mögliche Aneuploidie hinweisen. Ein Test auf Chromosomenaberrationen in der Metaphase, vorzugsweise am gleichen Zelltyp und unter Verwendung eines Protokolls mit vergleichbarer Empfindlichkeit, könnte ebenfalls eine nützliche Methode sein, um festzustellen, ob die Mikrokerne auf einen Chromosomenbruch zurückzuführen sind (in dem Wissen, dass ein Chromosomenverlust mit dem Test auf Chromosomenaberrationen nicht erkannt würde).
Analyse
Alle Objektträger, auch die für das Lösungsmittel und die unbehandelten Kontrollen (sofern verwendet) und die Positivkontrollen, sind vor der mikroskopischen Untersuchung der Mikrokernfrequenzen von unabhängiger Seite zu kodieren. Bei der Verwendung eines automatisierten Auswertungssystems, wie Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse, sollten geeignete Techniken zum Ausgleich systematischer Messfehler verwendet werden. Unabhängig davon, welche automatisierte Plattform zur Zählung der Mikrokerne verwendet wird, sind CBPI, RI, RPD oder RICC parallel auszuwerten.
In Kulturen, die mit cytoB behandelt wurden, sind die Mikrokernfrequenzen von mindestens 2.000 zweikernigen Zellen pro Konzentration und Kontrolle (83) zu analysieren, die, sofern Replikate verwendet werden, gleichmäßig auf die Replikate verteilt sein sollten. Bei der Verwendung von Einfachkulturen pro Dosis (siehe Nummer 28) sollten in einer solchen Kultur mindestens 2.000 zweikernige Zellen pro Kultur (83) ausgewertet werden. Wenn wesentlich weniger als 1.000 zweikernige Zellen pro Kultur (bei Zweifachkulturen) bzw. 2.000 (bei Einfachkulturen) in jeder Konzentration für die Auswertung zur Verfügung stehen und wenn keine signifikante Zunahme an Mikrokernen festgestellt wird, ist der Test mit einer höheren Anzahl an Zellen oder mit weniger zytotoxischen Konzentrationen zu wiederholen, je nachdem, was angemessen ist. Es ist darauf zu achten, dass keine zweikernigen Zellen ausgewertet werden, die unregelmäßige Formen aufweisen oder deren zwei Kerne in der Größe beträchtlich voneinander abweichen. Außerdem dürfen zweikernige Zellen nicht mit schlecht auf dem Träger verteilten mehrkernigen Zellen verwechselt werden. Zellen mit mehr als zwei Hauptkernen werden nicht auf Mikrokerne untersucht, da in diesen Zellen die zugrunde liegende Mikrokernfrequenz höher sein könnte (84). Die Auswertung einkerniger Zellen ist akzeptabel, wenn die Prüfchemikalie die Wirkung von cytoB beeinträchtigt. In diesen Fällen könnte ein Wiederholungstest ohne cytoB sinnvoll sein. Die Auswertung einkerniger Zellen zusätzlich zu den zweikernigen Zellen könnte nützliche Informationen liefern (85) (86), ist jedoch nicht zwingend erforderlich.
In Zelllinien, die ohne cytoB-Behandlung getestet wurden, sind die Mikrokerne in mindestens 2.000 Zellen pro Prüfkonzentration und Kontrolle (83) zu analysieren, die, sofern Replikate verwendet werden, gleichmäßig auf die Replikate verteilt sein sollten. Bei der Verwendung von Einzelkulturen je Konzentration (siehe Nummer 28) sind in dieser Kultur mindestens 2.000 Zellen auszuwerten. Wenn wesentlich weniger als 1.000 Zellen pro Kultur (bei Zweifachkulturen) bzw. 2.000 Zellen (bei Einfachkulturen) in jeder Konzentration für die Auswertung zur Verfügung stehen und wenn keine signifikante Zunahme an Mikrokernen festgestellt wird, ist der Test mit einer höheren Anzahl an Zellen oder mit weniger zytotoxischen Konzentrationen zu wiederholen, je nachdem, was angemessen ist.
Wenn cytoB verwendet wird, ist von mindestens 500 Zellen pro Kultur ein CBPI oder RI zur Bewertung der Zellproliferation (siehe Anlage 2) festzulegen. Werden die Behandlungen ohne cytoB durchgeführt, muss unbedingt nachgewiesen werden, dass die Zellen in der Kultur eine Zellteilung vollzogen haben, wie unter den Nummern 24 bis 28 erläutert.
Kompetenz des Labors
Um ausreichende Erfahrungen mit der Prüfung zu sammeln, bevor routinemäßige Tests erfolgen, sollte das Labor eine Reihe von Versuchen mit positiven Referenzchemikalien durchgeführt haben, die sich unterschiedlicher Mechanismen (mindestens einer mit und einer ohne Stoffwechselaktivierung sowie einer mit einem aneugenen Wirkmechanismus, ausgewählt aus den in Tabelle 1 aufgeführten Chemikalien) und verschiedener Negativkontrollen (einschließlich unbehandelter Kulturen und verschiedener Lösungsmittel/Vehikel) bedienen. Die Reaktionen dieser Positiv- und Negativkontrollen sollten mit der Literatur im Einklang stehen. Dies gilt nicht für erfahrene Labors, d. h. für Labors, die über eine Datenbank mit historischen Daten gemäß der Definition unter den Nummern 49 bis 52 verfügen.
Eine Auswahl von Positivkontrollchemikalien (siehe Tabelle 1) sollte anhand von Kurz- und Langzeitbehandlungen ohne Stoffwechselaktivierung und darüber hinaus anhand einer Kurzzeitbehandlung mit Stoffwechselaktivierung untersucht werden, um die Fähigkeit des Labors zum Nachweis klastogener und aneugener Chemikalien, zur Bestimmung der Wirksamkeit des Stoffwechselaktivierungssystems und des Nachweises der Eignung der Auswertungsverfahren (visuelle mikroskopische Untersuchung, Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse) zu belegen. Zum Nachweis der Empfindlichkeit und dynamischen Bandbreite des Versuchssystems sollte eine Spanne der Konzentrationen der ausgewählten Chemikalien festgelegt werden, um reproduzierbare und konzentrationsbezogene Zunahmen gegenüber dem Hintergrund zu erhalten.
Historische Kontrolldaten
Das Labor sollte Folgendes bestimmen:
- Bereich und Verteilung historischer Positivkontrollen
- Bereich und Verteilung historischer Negativkontrollen (unbehandelt, Lösungsmittel)
Bei der erstmaligen Erfassung von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen veröffentlichten Negativkontrolldaten entsprechen, sofern solche vorhanden sind. Werden weitere Versuchsdaten zur Verteilung der Kontrollen hinzugefügt, sollten gleichzeitige Negativkontrollen idealerweise innerhalb von 95 % der Kontrollgrenzen der gewählten Verteilung liegen (87) (88). Die Datenbank des Labors zu historischen Negativkontrollen sollte zunächst mit mindestens 10 Versuchen angelegt werden. Vorzugsweise sollte sie jedoch aus mindestens 20 Versuchen bestehen, die unter vergleichbaren Versuchsbedingungen durchgeführt wurden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder X-Bar-Karten (88)), um zu ermitteln, wie variabel ihre Positiv- und Negativkontrolldaten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist (83). Weitere Empfehlungen zum Aufbau und zur Verwendung von Sammlungen historischer Daten (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus historischen Datensätzen und die Gültigkeitskriterien für einen bestimmten Versuch) sind den Literaturangaben zu entnehmen (87).
Sämtliche Änderungen am Versuchsprotokoll sind auf Übereinstimmung mit den bereits vorhandenen historischen Kontrolldaten zu prüfen. Bei größeren Inkonsistenzen sollte eine neue historische Kontrolldatenbank erstellt werden.
Negativkontrolldaten sollten das Auftreten von Zellen mit Mikrokernen aus einer Einzelkultur oder aus einer Summe von Replikatkulturen erfassen (vgl. Beschreibung unter Nummer 28). Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der Kontrollgrenzen von 95 % der gewählten Verteilung in der Datenbank des Labors zu historischen Negativkontrollen liegen (87) (88). Sofern gleichzeitige Negativkontrolldaten außerhalb der Kontrollgrenzen von 95 % liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um "extreme Ausreißer" handelt und nachgewiesen werden kann, dass das Testsystem "unter Kontrolle" ist (siehe Nummer 50) und nachweislich kein technisches oder menschliches Versagen vorliegt.
Daten und Berichterstattung
Interpretation der Ergebnisse
Bei Verwendung des Zytokinese-Block-Verfahrens werden nur die Frequenzen zweikerniger Zellen mit Mikrokernen (unabhängig von der Anzahl der Mikrokerne pro Zelle) zur Bewertung der Mikrokern-Induktion herangezogen. Die Auswertung der Anzahl an Zellen mit einem, zwei oder mehr Mikrokernen kann separat ausgewiesen werden und könnte nützliche Informationen liefern, ist jedoch nicht zwingend notwendig.
Festzulegen sind auch Maßnahmen, die gleichzeitig zur Bestimmung der Zytotoxizität und/oder Zytostase aller behandelten Kulturen und aller Negativ- und Positivkontrollkulturen durchgeführt werden (16). Der CBPI oder der RI ist für alle behandelten Kulturen und Kontrollkulturen zu berechnen, da sich bei Anwendung der Zytokinese-Block-Methode die Messungen des Zellzyklus verzögern. Kommt cytoB nicht zum Einsatz, sollte die relative Populationsverdopplung (RPD) oder die relative Erhöhung der Zellzahl (RICC) herangezogen werden (siehe Anlage 2).
Es sind die Daten für die einzelnen Kulturen zu dokumentieren. Zusätzlich sollten alle Daten in tabellarischer Form zusammengefasst werden.
Gültigkeitskriterien
Die Akzeptanz eines Versuchs beruht auf folgenden Kriterien:
- Die Aufnahme der gleichzeitigen Negativkontrolle in die Datenbank des Labors zu historischen Negativkontrollen ist zulässig, wenn sie der Beschreibung unter Nummer 50 entspricht.
- Gleichzeitige Positivkontrollen (siehe Nummer 50) sollten Reaktionen hervorrufen, die mit den Reaktionen kompatibel sind, die in der Datenbank zu historischen Positivkontrollen erzeugt werden, und eine statistisch signifikante Zunahme gegenüber den gleichzeitigen Negativkontrollen aufweisen.
- Die Kriterien für die Zellproliferation in der Lösungsmittelkontrolle müssen erfüllt sein (Nummern 25, 26 und 27).
- Es wurden alle drei Versuchsbedingungen getestet, es sei denn, eine führte zu positiven Ergebnissen (Nummern 36-40).
- Eine angemessene Zahl an Zellen und Konzentrationen ist analysierbar (Nummer 28 sowie Nummern 44, 45 und 46).
- Die Kriterien für die Auswahl der höchsten Konzentration entsprechen den Kriterien unter den Nummern 24-31.
Bewertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn unter den geprüften Versuchsbedingungen (siehe Nummern 36-39)
- mindestens eine der Versuchskonzentrationen eine statistisch signifikante Zunahme gegenüber der gleichzeitigen Negativkontrolle aufweist (89),
- die Zunahme unter mindestens einer Versuchsbedingung bei der Bewertung anhand eines geeigneten Trendtests dosisabhängig ist (siehe Nummer 28),
- eines der Ergebnisse außerhalb der Verteilung der historischen Negativkontrolldaten liegt (z.B. auf einer Poisson-Verteilung beruhende Kontrollgrenzen von 95 %, siehe Nummer 52).
Sind alle diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie in diesem Testsystem Chromosomenbrüche und/oder Gewinn bzw. Verlust von Chromosomenmaterial auslösen kann. Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (90) (91) (92).
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn unter allen geprüften Versuchsbedingungen (siehe Nummern 36-39)
- keine der Versuchskonzentrationen eine statistisch signifikante Zunahme gegenüber der gleichzeitigen Negativkontrolle aufweist,
- die Bewertung anhand eines geeigneten Trendtests keine dosisabhängige Zunahme ergibt,
- alle Ergebnisse innerhalb der Verteilung der historischen Negativkontrolldaten liegen (z.B. auf einer Poisson-Verteilung beruhende Kontrollgrenzen von 95 %, siehe Nummer 52).
In diesem Fall wird davon ausgegangen, dass die Prüfchemikalie keine Chromosomenbrüche und/oder Gewinn bzw. Verlust von Chromosomenmaterial in diesem Testsystem auslösen kann. Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (90) (91) (92).
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
In den Fällen, in denen die Reaktion, wie oben beschrieben, weder eindeutig negativ noch eindeutig positiv ist, und/oder um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen bewertet werden. Die Auswertung weiterer Zellen oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter veränderten Versuchsbedingungen (z.B. Abstände der Konzentrationen, andere Stoffwechselaktivierungsbedingungen (d. h. Konzentration oder Herkunft von S9), könnte gegebenenfalls hilfreich sein.
In seltenen Fällen erlaubt der Datensatz selbst nach weiteren Untersuchungen keine definitive Aussage zu positiven oder negativen Ergebnissen, und es wird daher die Schlussfolgerung gezogen, dass die Reaktion nicht eindeutig ist.
Wenn Prüfchemikalien im MNvit-Test Mikrokerne induzieren, kann dies darauf zurückzuführen sein, dass sie Chromosomenbrüche, Chromosomenverlust oder eine Kombination aus beidem verursachen. Deshalb können weitere Analysen unter Einsatz von Anti-Kinetochor-Antikörpern, zentromer-spezifischen In-situ-Sonden oder mithilfe sonstiger Methoden vorgenommen werden, um festzustellen, ob der Mechanismus der Mikrokern-Induktion von einer klastogenen und/oder aneugenen Wirkung herrührt.
Testbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalie:
- Herkunft, Partienummer, ggf. begrenztes Verwendungsdatum;
- Stabilität der Prüfchemikalie, falls bekannt;
- Reaktivität der Prüfchemikalien mit dem Lösungsmittel/Vehikel oder dem Zellkulturmedium;
- Löslichkeit und Stabilität der Prüfchemikalie im Lösungsmittel, falls bekannt;
- Messung des pH-Werts, der Osmolalität und ggf. des Niederschlag im Kulturmedium, dem die Prüfchemikalie zugegeben wurde.
Einkomponentige Substanz:
- Aussehen, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
- chemische Bezeichnung, wie z.B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, falls zutreffend und praktisch durchführbar usw.
Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
- So weit wie möglich charakterisiert durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten.
Lösungsmittel:
- Begründung der Auswahl des Lösungsmittels;
- Anteil des Lösungsmittels im endgültigen Kulturmedium.
Zellen:
- Typ und Herkunft der verwendeten Zellen;
- Eignung des verwendeten Zelltyps;
- bei Zelllinien: Nichtvorhandensein von Mykoplasma;
- bei Zelllinien: Angaben über die Dauer des Zellzyklus oder den Proliferationsindex;
- bei Verwendung von Lymphozyten: Geschlecht und Alter der Blutspender, sachdienliche Informationen zum Spender, Vollblut oder separierte Lymphozyten, verwendetes Mitogen;
- normale Zellzyklusdauer (Negativkontrolle);
- bei Zelllinien: ggf. Passagenanzahl;
- bei Zelllinien: zum Erhalt der Zellkultur verwendete Verfahren;
- bei Zelllinien: Modalwert der Chromosomen.
Prüfbedingungen:
- Bezeichnung des Zytokinese-Block-Stoffs (z.B. cytoB), falls verwendet, dessen Konzentration und Dauer der Zellexposition;
- Konzentration der Prüfchemikalie, ausgedrückt als Endkonzentration im Kulturmedium (z.B. µg oder mg/ml oder mM des Kulturmediums);
- Begründung für die Auswahl der Konzentrationen und die Anzahl der Kulturen, darunter z.B. Angaben zur Zytotoxizität und Löslichkeitsgrenze;
- Medienzusammensetzung, ggf. CO2-Konzentration, Feuchtigkeit;
- Konzentration (und/oder Volumen) des Lösungsmittels und der dem Kulturmedium beigegebenen Prüfchemikalie;
- Inkubationstemperatur und -dauer;
- Behandlungsdauer;
- Zeitpunkt der Gewinnung nach der Behandlung;
- ggf. Zelldichte bei der Beimpfung;
- Art und Zusammensetzung des Stoffwechselaktivierungssystems (Herkunft von S9, Zubereitungsmethode des S9-Gemisches, Konzentration oder Volumen des S9-Gemisches und von S9 im Endmedium), Qualitätskontrollen von S9 (z.B. enzymatische Aktivität, Sterilität, Stoffwechselfähigkeit);
- Positiv- und Negativkontrollchemikalien, Endkonzentrationen, Bedingungen sowie Behandlungsdauer und Regenerationszeit;
- Methoden zur Präparation des Objektträgers und verwendete Färbetechnik;
- Kriterien für die Auswertung von Mikrokernzellen (Auswahl der analysierbaren Zellen und Identifizierung von Mikrokernen);
- Anzahl der analysierten Zellen;
- Methoden zur Bestimmung der Zytotoxizität;
- evtl. zusätzliche Angaben zur Zytotoxizität und zum verwendeten Verfahren;
- Kriterien zur Einstufung der Studien als positiv, negativ oder nicht eindeutig;
- verwendete statistische Analysemethode(n);
- Methoden, z.B. die Verwendung von Kinetochor-Antikörpern oder Pan-Zentromer-Sonden, um ggf. zu beschreiben, ob Mikrokerne ganze oder fragmentierte Chromosomen enthalten;
- Methoden zur Bestimmung von pH-Wert, Osmolalität und Ausfällung.
Ergebnisse:
- Definition geeigneter Zellen für die Analyse;
- bei Zelllinien: für jede Kultur die Anzahl der behandelten Zellen und Anzahl der gewonnenen Zellen, wenn kein cytoB verwendet wird;
- verwendete Messung der Zytotoxizität, z.B. CBPI oder RI bei der Zytokinese-Block-Methode; RICC oder RPD, wenn keine Zytokinese-Block-Verfahren verwendet werden; sonstige Beobachtungen bei Bedarf (z.B. Zell-Konfluenz, Apoptose, Nekrose, Metaphasezählung, Frequenz zweikerniger Zellen);
- Ausfällungszeichen und Bestimmungszeit;
- Angaben zum pH-Wert und zur Osmolalität des Behandlungsmediums, falls ermittelt;
- Verteilung einkerniger, zweikerniger und mehrkerniger Zellen, falls eine Zytokinese-Block-Methode verwendet wird;
- Anzahl der Zellen mit Mikrokernen, gesondert für jede behandelte Kultur und Kontrollkultur, und Angabe, ob diese von zweikernigen oder einkernigen Zellen stammen, sofern zutreffend;
- nach Möglichkeit Dosis-Wirkungs-Verhältnis;
- Daten zu gleichzeitigen Negativ-(Lösungsmittel-)Kontrollen und Positivkontrollen (Konzentrationen und Lösungsmittel);
- Daten zu historischen Negativ-(Lösungsmittel-)Kontrollen und Positivkontrollen mit Bereichen, Mittelwerten und Standardabweichungen und 95 %-Grenzen für die Verteilung sowie Anzahl der Datensätze;
- statistische Analysen, ggf. p-Werte.
Diskussion der Ergebnisse:
Schlussfolgerungen
Literaturhinweise
(1) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(2) Kirsch-Volders, M. (1997). Towards a validation of the micronucleus test. Mutation Research, Vol. 392/1-2, 1-4.
(3) Parry, J.M., A. Sors (1993). The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project. Mutation Research, Vol. 287/1, 3-15.
(4) Fenech, M., A.A. Morley (1985). Solutions to the kinetic problem in the micronucleus assay. Cytobios, Vol. 43/172-173, 233-246.
(5) Kirsch-Volders, M. et al. (2000). Report from the In Vitro Micronucleus Assay Working Group. Environmental and Molecular Mutagenesis, Vol. 35/3, 167-172.
(6) Fenech, M. (2007). Cytokinesis-block micronucleus cytome assay. Nature Protocols, Vol. 2/5, 1084-1104.
(7) Fenech, M., A.A. Morley (1986). Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation. Mutation Research, Vol. 161/2, 193-198.
(8) Eastmond, D.A., J.D. Tucker (1989). Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody. Environmental and Molecular Mutagenesis, Vol. 13/1, 34-43.
(9) Eastmond, D.A., D. Pinkel (1990). Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probe. Mutation Research, Vol. 234/5, 9-20.
(10) Miller, B.M. et al. (1991). Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA. Mutation Research, Vol. 6/4, 297-302.
(11) Farooqi, Z., F. Darroudi, A. T. Natarajan (1993). The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes. Mutation Research, Vol. 8/4, 329-334.
(12) Henderson, L. et al. (1993). Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe. Mutation Research, Vol. 319/3, 205-213.
(13) Norppa, H., L. Renzi, C. Lindholm (1993). Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization. Mutation Research, Vol. 8/6, 519-525.
(14) Eastmond, D.A, D.S. Rupa, L.S. Hasegawa (1994). Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes. Mutation Research, Vol. 322/1, 9-20.
(15) Marshall, R.R. et al. (1996). Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy. Mutation Research, Vol. 372/2, 233-245.
(16) Zijno, P. et al. (1996). Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes. Mutation Research, Vol. 372/2, 211-219.
(17) Kirsch-Volders et al. (2003). Report from the in vitro micronucleus assay working group. Mutation Research, Vol. 540/2, 153-163.
(18) Kapitel B.10: In-vitro-Test auf Chromosomenaberrationen in Säugetierzellen.
(19) Lorge, E. et al. (2006). SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study. Mutation Research, Vol. 607/1, 13-36.
(20) Clare, G. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes. Mutation Research, Vol. 607/1, 37-60.
(21) Aardema, M.J. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells. Mutation Research, Vol. 607/1, 61-87.
(22) Wakata, A. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells. Mutation Research, Vol. 607/1, 88-124.
(23) Oliver, J. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells. Mutation Research, Vol. 607/1, 125-152.
(24) Albertini, S. et al. (1997). Detailed data on in vitro MNT and in vitro CA: industrial experience. Mutation Research, Vol. 392/1-2, 187-208.
(25) Miller, B. et al. (1997). Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience. Mutation Research, Vol. 392/1-2, 45-59.
(26) Miller, B. et al. (1998). Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft für Umwelt-Mutationsforschung, Mutation Research, Vol. 410, 81-116.
(27) Kalweit, S. et al. 1999. Chemically induced micronucleus formation in V79 cells - comparison of three different test approaches. Mutation Research, Vol. 439/2, 183-190.
(28) Kersten, B. et al. 1999. The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity. Mutation Research, Vol. 445/1, 55-71.
(29) von der Hude, W. et al. (2000). In vitro micronucleus assay with Chinese hamster V79 cells - results of a collaborative study with in situ exposure to 26 chemical substances. Mutation Research, Vol. 468/2, 137-163.
(30) Garriott, M.L., J.B. Phelps, W.P. Hoffman (2002). A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity. Mutation Research, Vol. 517/1-2, 123-134.
(31) Matsushima, T. et al. 1999. Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU). Mutation Research, Vol. 14/6, 569-580.
(32) Elhajouji, A., E. Lorge (2006). Sonderausgabe: SFTG International collaborative study on in vitro micronucleus test. Mutation Research, Vol. 607/1, 1-152.
(33) Kirkland, D. (2010). Evaluation of different cytotoxic and cytostatic measures for the in vitromicronucleus test (MNVit): Introduction to the collaborative trial. Mutation Research, Vol. 702/2, 139-147.
(34) Hashimoto K. et al. (2011). Comparison of four different treatment conditions of extended exposure in the in vitro micronucleus assay using TK6 lymphoblastoid cells. Regulatory Toxicology and Pharmacology, Vol. 59/1, 28-36.
(35) Honma, M., M. Hayashi (2011). Comparison of in vitro micronucleus and gene mutation assay results for p53-competent versus p53-deficient human lymphoblastoid cells. Environmental and Molecular Mutagenesis, Vol. 52/5, 373-384.
(36) Zhang, L.S. et al. (1995). A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test. Mutation Research Letters, Vol. 347/3-4, 105-115.
(37) ECVAM (2006). Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. 25. Sitzung des ESAC, 16./17. November2006, verfügbar unter: http://ecvam.jrc.it/index.htm.
(38) ESAC (2006). Peer-Review des Wissenschaftlich Beratenden Ausschusses (ESAC) des ECVAM, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel. Verfügbar unter: http://ecvam.jrc.it/index.htm.
(39) Corvi, R. et al. (2008). ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT). Mutagenesis, Vol. 23/4, 271-283.
(40) Publikation des ILSI (Entwurf). Lorge, E., M.M. Moore, J. Clements, M. O'Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, A. Sutter, V. Thybaud, B. Gollapudi, M. Aardema, J. Young-Tannir. Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. Mutation Research.
(41) Scott, D. et al. (1991). International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9. Mutation Research, Vol. 257/2, 147-205.
(42) Morita, T. et al. (1992). Clastogenicity of low pH to various cultured mammalian cells. Mutation Research, Vol. 268/2, 297-305.
(43) Brusick, D. (1986). Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations. Environmental Mutagenesis, Vol. 8/6, 789-886.
(44) Long, L.H. et al. (2007). Different cytotoxic and clastogenic effects of epigallocatechin gallate in various cell-culture media due to variable rates of its oxidation in the culture medium. Mutation Research, Vol. 634/1-2, 177-183.
(45) Nesslany, F. et al. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environmental and Molecular Mutation, Vol. 49, 439-452.
(46) Fenech, M., A.A. Morley (1985). Measurement of micronuclei in lymphocytes. Mutation Research, Vol. 147/1-2, 29-36.
(47) Fenech, M. (1997). The advantages and disadvantages of cytokinesis-blood micronucleus method. Mutation Research, Vol. 392, 11-18.
(48) Payne, C.M. et al. (2010). Hydrophobic bile acid-induced micronuclei formation, mitotic perturbations, and decreases in spindle checkpoint proteins: relevance to genomic instability in colon carcinogenesis. Nutrition and Cancer, Vol. 62/6, 825-840.
(49) Bazin, E. et al. (2010). Genotoxicity of a Freshwater Cyanotoxin, Cylindrospermopsin, in Two Human Cell Lines: Caco-2 and HepaRG. Environmental and Molecular Mutagenesis, Vol. 51/3, 251-259.
(50) Le Hegarat, L. et al. (2010). Assessment of the genotoxic potential of indirect chemical mutagens in HepaRG cellsby the comet and the cytokinesis-block micronucleus assays. Mutagenesis, Vol. 25/6, 555-560.
(51) Josse, R. et al. (2012). An adaptation of the human HepaRG cells to the in vitro micronucleus assay. Mutagenesis, Vol. 27/3, 295-304.
(52) Ehrlich, V. et al. (2002). Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells. Mutation Research, Vol. 17/3, 257-260.
(53) Knasmüller, S. et al. (2004). Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge. Toxicology, Vol. 198/1-3, 315-328.
(54) Gibson, D.P. et al. (1997). Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals. Mutation Research, Vol. 392/1-2, 61-70.
(55) Bonassi, S. et al. (2001). HUman Micro Nucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei. Environmental and Molecular Mutagenesis, Vol. 37/1, 31-45.
(56) Maron, D.M., B.N. Ames (1983). Revised methods for the Salmonella mutagenicity test. Mutation Research, Vol. 113/3-4, 173-215.
(57) Ong, T.-m. et al. (1980). Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver. Journal of Environmental Pathology and Toxicology, Vol. 4/1, 55-65.
(58) Elliott, B.M. et al. (1992). Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, Vol. 7, 175-177.
(59) Matsushima, T. et al. (1976)."A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems", in In Vitro Metabolic Activation in Mutagenesis Testing, de Serres, F.J. et al. (eds), Elsevier, North-Holland, 85-88.
(60) Johnson, T.E., D. R. Umbenhauer, S.M. Galloway (1996). Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9. Environmental and Molecular Mutagenesis, Vol. 28, 51-59.
(61) UNEP (2001). Stockholmer Übereinkommen über persistente organische Schadstoffe, Umweltprogramm der Vereinten Nationen (UNEP). Verfügbar unter: http://www.pops.int/
(62) Tucker, J.D., M.L. Christensen (1987). Effects of anticoagulants upon sister-chromatid exchanges, cell-cycle kinetics, and mitotic index in human peripheral lymphocytes. Mutation Research, Vol. 190/3, 225-8.
(63) Krahn, D.F., F.C. Barsky, K.T. McCooey (1982)."CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids", in Genotoxic Effects of Airborne Agents. Tice, R.R., D.L. Costa, K.M. Schaich (eds.), Plenum, New York, 91-103.
(64) Zamora, P.O. et al. (1983). Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay. Environmental Mutagenesis, Vol. 5/6, 795-801.
(65) Asakura, M. et al. (2008). An improved system for exposure of cultured mammalian cells to gaseous compounds in the chromosomal aberration assay. Mutation Research, Vol. 652/2, 122-130.
(66) Fenech, M. (1993). The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations. Mutation Research, Vol. 285/1, 35-44.
(67) Phelps, J.B., M.L. Garriott, W.P. Hoffman (2002). A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity. Mutation Research, Vol. 521/1-2, 103-112.
(68) Kirsch-Volders, M. et al. (2004). Corrigendum to "Report from the in vitro micronucleus assay working group" . Mutation Research, 564, 97-100.
(69) Lorge, E. et al. (2008). Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects. Mutation Research, Vol. 655/1-2, 1-3.
(70) Surralles, J. et al. (1995). Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures. Mutation Research, Vol. 341/3, 169-184.
(71) Honma, M. (2011). Cytotoxicity measurement in in vitro chromosome aberration test and micronucleus test. Mutation Research, Vol. 724, 86-87.
(72) Pfuhler, S. et al. (2011). In vitro genotoxicity test approaches with better predictivity: Summary of an IWGT workshop. Mutation Research, Vol. 723/2, 101-107.
(73) OECD (2014). Document supporting the WNT decision to implement revised criteria for the selection of the top concentration in the in vitro mammalian cell assays on genotoxicity (Test Guidelines 473, 476 and 487). ENV/JM/TG(2014)17. Auf Anfrage erhältlich.
(74) Morita T., M. Honma, K. Morikawa (2012). Effect of reducing the top concentration used in the in vitro chromosomal aberration test in CHL cells on the evaluation of industrial chemical genotoxicity. Mutation Research, Vol. 741, 32-56.
(75) Brookmire, L., J.J. Chen, D.D. Levy (2013). Evaluation of the Highest Concentrations Used in the in vitro Chromosome Aberrations Assay. Environmental and Molecular Mutagenesis, Vol. 54/1, 36-43.
(76) EPA, Office of Chemical Safety and Pollution Prevention (2011). Chemical Substances of Unknown or Variable Composition, Complex Reaction Products and Biological Materials: UVCB Substances. http://www.epa.gov/opptintr/newchems/pubs/uvcb.txt.
(77) Sobol, Z. et al. (2012). Development and validation of an in vitro micronucleus assay platform in TK6 cells. Mutation Research, Vol. 746/1, 29-34.
(78) Hayashi, M., T. Sofuni, M. Jr. Ishidate (1983). An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test. Mutation Research, Vol. 120/4, 241-247.
(79) MacGregor, J. T., C.M. Wehr, R.G. Langlois (1983). A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y. Mutation Research, Vol. 120/4, 269-275.
(80) Bryce, S.M. et al. (2011). Miniaturized flow cytometry-based CHO-K1 micronucleus assay discriminates aneugenic and clastogenic modes of action. Environmental and Molecular Mutagenesis, Vol. 52/4, 280-286.
(81) Nicolette, J. et al. (2011). in vitro micronucleus screening of pharmaceutical candidates by flow cytometry in Chinese hamster V79 cells. Environmental and Molecular Mutagenesis, Vol. 52/5, 355-362.
(82) Shi, J., R. Bezabhie, A. Szkudlinska (2010). Further evaluation of a flow cytometric in vitro micronucleus assay in CHO-K1 cells: a reliable platform that detects micronuclei and discriminates apoptotic bodies. Mutagenesis, Vol. 25/1, 33-40.
(83) OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment Nr. 198, OECD Publishing, Paris.
(84) Fenech, M. et al. (2003). HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutation Research, Vol. 534/1-2, 65-75.
(85) Elhajouji, A., M. Cunha, M. Kirsch-Volders (1998). Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis-block assay. Mutagenesis, Vol. 13/2, 193-8.
(86) Kirsch-Volders, M. et al. (2011). The in vitro MN assay in 2011: origin and fate, biological significance, protocols, high throughput methodologies and toxicological relevance. Archives of Toxicology, Vol. 85/8, 873-99.
(87) Hayashi, M. et al. (2010). Compilation and use of genetic toxicity historical control Data. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 723/2, 87-90.
(88) Ryan, T. P. (2000). Statistical Methods for Quality Improvement. 2. Auflage, John Wiley and Sons, New York
(89) Hoffman, W.P., M.L. Garriott, C. Lee (2003)."In vitro micronucleus test", in Encyclopedia of Biopharmaceutical Statistics, 2. Auflage. Chow, S. (Hrsg.), Marcel Dekker, Inc. New York, 463-467.
(90) Fleiss, J. L., B. Levin, M.C. Paik (2003). Statistical Methods for Rates and Proportions, 3. Auflage, John Wiley & Sons, New York.
(91) Galloway, S.M. et al. (1987). Chromosome aberration and sister chromatid exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals. Environmental and Molecular Mutagenesis, Vol. 10/suppl. 10, 1-175.
(92) Richardson, C. et al. (1989). Analysis of Data from in vitro Cytogenetic Assays, in Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Hrsg.), Cambridge University Press, Cambridge, 141-154.
(93) International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use.
.
| Definitionen | Anlage 1 |
Aneugen: Chemikalie oder Prozess, die/der durch Wechselwirkung mit den Komponenten des mitotischen und meiotischen Zellteilungszyklus-Apparats zu Aneuploidie in Zellen oder Organismen führt.
Aneuploidie: Abweichung von der normalen diploiden (oder haploiden) Chromosomenzahl durch ein einziges Chromosom oder mehr, nicht aber durch einen ganzen (oder mehrere) Chromosomensatz/-sätze (Polyploidie).
Apoptose: Programmierter Zelltod, der durch eine Reihe von Schritten charakterisiert ist, an deren Ende eine Desintegration von Zellen in membranumschlossene Partikel steht, die schließlich durch Phagozytose oder Shedding abgebaut werden.
Chemikalie: Stoff oder Gemisch.
Genotoxisch: Allgemeine Bezeichnung für alle Arten von DNA- oder Chromosomenschäden, einschließlich Brüchen, Deletionen, Addukten, Nukleotidmodifikationen und -verknüpfungen, Reunionen, Genmutationen, Chromosomenaberrationen und Aneuploidie. Nicht alle genotoxischen Effekte führen zu Mutationen oder stabilen Chromosomenschäden.
Kinetochor: Proteinhaltige Struktur, die sich am Zentromer eines Chromosoms sammelt, an der während der Zellteilung die Spindelfasern anhaften, wodurch die ordnungsgemäße Beförderung der Tochterchromosomen zu den Polen der Tochterzellen ermöglicht wird.
Klastogen: Chemikalie oder Ereignis, die/das strukturelle Chromosomenaberrationen in Zellpopulationen oder eukaryontischen Organismen auslöst.
Konzentrationen: Beziehen sich auf Endkonzentrationen der Prüfchemikalie im Kulturmedium.
Interphasezellen: Zellen, die sich nicht im Stadium der Mitose befinden.
Lösungsmittelkontrolle: allgemeiner Begriff zur Bezeichnung der Kontrollkulturen, die nur mit dem Lösungsmittel behandelt werden, in dem die Prüfchemikalie gelöst wird.
Mikronuclei/Mikrokerne: Kleine Kerne zusätzlich zu den Hauptkernen der Zellen und von diesen getrennt, die während der Telophase der Mitose oder Meiose durch zurückgebliebene Chromosomenteile oder ganze Chromosomen gebildet werden.
Mitose: Teilung des Zellkerns, die in der Regel in Prophase, Prometaphase, Metaphase, Anaphase und Telophase gegliedert ist.
Mitoseindex: Anteil der Zellen einer Zellpopulation, die sich zum Beobachtungszeitpunkt in Metaphase befinden; ein Hinweis auf den Grad der Zellproliferation dieser Population.
Mutagen: Auslöser einer Erbgutveränderung der DNA-Basenpaarsequenz(en) in Genen oder in der Chromosomenstruktur (Chromosomenaberrationen).
Non-Disjunktion: Unvermögen der paarweise angeordneten Chromatiden, sich zu trennen und sich auf die in Entwicklung begriffenen Tochterzellen aufzuteilen. Dabei entstehen Tochterzellen mit abweichenden Chromosomenzahlen.
p53-Status: Das p53-Protein ist an der Regulierung des Zellzyklus, der Apoptose und der DNA-Reparatur beteiligt. Zellen, denen ein funktionales p53-Protein fehlt und die nicht in der Lage sind, den Zellzyklus aufzuhalten oder beschädigte Zellen über Apoptose oder andere Mechanismen (z.B. Einleitung einer DNA-Reparatur) im Zusammenhang mit Aufgaben des p53-Proteins als Reaktion auf DNA-Schäden zu beseitigen, sollten theoretisch eher zu Genmutationen oder Chromosomenaberrationen neigen.
Polyploidie: Zahlenmäßige Chromosomenaberrationen in Zellen oder Organismen, von denen ein oder mehrere ganze Chromosomensätze betroffen sind, im Gegensatz zur Aneuploidie, bei der nur ein oder mehrere einzelne Chromosomen betroffen sind.
Proliferationsindex (PI): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2).
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Relative Erhöhung der Zellzahl (RICC): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2).
Relative Populationsverdopplung (RPD): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2).
Replikationsindex (RI): Anteil der abgeschlossenen Zellteilungszyklen in einer behandelten Kultur im Verhältnis zur nicht behandelten Kontrollgruppe während der Expositions- und Regenerationsphase (Formel siehe Anlage 2).
S9-Gemisch: Gemisch aus der S9-Leberfraktion und den für die metabolische Enzymaktivität notwendigen Ko-Faktoren.
S9-Leberfraktion: Überstand des Leberhomogenats nach Zentrifugieren bei 9.000 g, d. h. Rohleberextrakt.
Unbehandelte Kontrollen: Kulturen, die nicht behandelt werden (d. h. weder mit einer Prüfchemikalie noch mit Lösungsmittel), jedoch gleichzeitig in gleicher Weise aufbereitet werden wie die Kulturen, die mit der Prüfchemikalie behandelt werden.
Zellproliferation: Zunahme der Anzahl von Zellen als Ergebnis der mitotischen Zellteilung.
Zentromer: DNA-Bereich eines Chromosoms, an dem die beiden Chromatiden zusammengehalten werden und an dem beide Kinetochoren Seite an Seite angeordnet sind.
Zytokinese: Prozess der Zellteilung unmittelbar nach der Mitose, bei dem zwei Tochterzellen, jeweils mit einem einzigen Kern, gebildet werden.
Zytokinese-Block-Proliferationsindex (CBPI): Anteil der Zellen aus zweiter Teilung in der behandelten Population im Verhältnis zur nicht behandelten Kontrollgruppe (Formel siehe Anlage 2).
Zytostase: Hemmung des Zellwachstums (Formel siehe Anlage 2).
Zytotoxizität: Bei den unter diese Prüfmethode fallenden Versuchen, die mit Zusatz von Cytochalasin B durchgeführt werden, bezeichnet Zytotoxizität eine Verringerung des Zytokinese-Block-Proliferationsindex (CBPI) oder Replikationsindex (RI) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 26 und Anlage 2).
Bei den unter diese Prüfmethode fallenden Versuche, die ohne Zusatz von Cytochalasin B durchgeführt werden, bezeichnet Zytotoxizität eine Verringerung der relativen Populationsverdopplung (RPD) bzw. der relativen Erhöhung der Zellzahl (RICC) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 27 und Anlage 2).
.
| Formeln zur Bewertung der Zytotoxizität | Anlage 2 |
Wenn cytoB zum Einsatz kommt, sollte die Bewertung der Zytotoxizität auf dem Zytokinese-Block-Proliferationsindex (CBPI) oder dem Replikationsindex (RI) beruhen (17) (69). Der CBPI gibt die mittlere Zahl der Kerne pro Zelle an und kann zur Berechnung der Zellproliferation eingesetzt werden. Der RI gibt die relative Zahl der Zellzyklen pro Zelle während der Exposition gegenüber cytoB in behandelten Kulturen im Vergleich zu Kontrollkulturen an und kann zur Berechnung der Zytostase in % verwendet werden:
% Zytostase = 100 -100{(CBPIT -1) ÷ (CBPIC -1)}
und:
T = mit der Prüfchemikalie behandelte Kultur
C = Vehikel-Kontrollkultur
Dabei gilt:
| ((Anzahl einkerniger Zellen) + (2 x Anzahl zweikerniger Zellen) + (3 x Anzahl mehrkerniger Zellen)) | |
| CBPI = |
|
| (Gesamtzellpopulation) |
Damit entspricht ein CBPI von 1 (alle Zellen sind einkernig) einer Zytostase von 100 %.
Zytostase = 100 - RI
| ((Anzahl zweikerniger Zellen) + (2 x Anzahl mehrkerniger Zellen))/(Gesamtzellpopulation)T | ||
| RI = |
| x 100 |
| ((Anzahl zweikerniger Zellen) + (2 x Anzahl mehrkerniger Zellen))/(Gesamtzellpopulation)C |
T = behandelte Kulturen
C = Kontrollkulturen
Ein RI von 53 % bedeutet somit, dass sich in der behandelten Kultur gegenüber der Anzahl der Zellen in der Kontrollkultur, die sich geteilt und zwei- und mehrkernige Zellen gebildet haben, nur 53 % geteilt haben, was eine Zytostase von 47 % bedeutet.
Wenn kein cytoB zum Einsatz kommt, wird eine Bewertung der Zytotoxizität auf der Basis der relativen Erhöhung der Zellzahl (RICC) oder der relativen Populationsverdopplung (RPD) empfohlen (69), da bei beiden der Anteil der Zellpopulation berücksichtigt wird, der eine Teilung durchlaufen hat.
| (zahlenmäßige Zunahme von Zellen in behandelten Kulturen(Ende-Beginn)) | ||
| RICC(%) = |
| x 100 |
| (zahlenmäßige Zunahme von Zellen in Kontrollkulturen(Ende-Beginn)) | ||
| (Anzahl von Populationsverdoppelungen in behandelten Kulturen) | ||
| RPD(%) = |
| x 100 |
| (Anzahl von Populationsverdoppelungen in Kontrollkulturen) |
Dabei gilt:
Populationsverdopplung = [log ((Zellzahl nach Behandlung ÷ Anfängliche Zellzahl)] ÷ log 2
Somit zeigt eine RICC oder eine RPD von 53 % eine Zytotoxizität/Zytostase von 47 % an.
Bei Verwendung eines Proliferationsindex (PI) kann die Zytotoxizität durch das Zählen aller Klone bewertet werden, die aus 1 Zelle (cl1), 2 Zellen (cl2), 3 bis 4 Zellen (cl4) sowie 5 bis 8 Zellen (cl8) bestehen.
| ((1 x cl1) + (2 x cl2) + (3 x cl4) + (4 x cl8) | |
| PI = |
|
| (cl1 + cl2 + cl4 + cl8) |
Der PI wurde als aussagefähiger und zuverlässiger Parameter für die Zytotoxizität auch bei Zelllinien verwendet, die in vitro ohne cytoB kultiviert wurden (35) (36) (37) (38) und kann als nützlicher zusätzlicher Parameter angesehen werden.
In jedem Fall sollen die behandelten Kulturen und die Negativkontrollkulturen vor der Behandlung die gleiche Zellzahl aufweisen.
Wenngleich der RCC-Wert (d. h. Zellzahl in behandelten Kulturen/Zellzahl in Kontrollkulturen) in der Vergangenheit als Bestimmungsgröße für die Zytotoxizität herangezogen wurde, wird er nicht mehr empfohlen, da er zu einer Unterschätzung der Toxizität führen kann.
Bei der Verwendung automatisierter Auswertungssysteme, wie Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse, kann die Zellzahl in der Formel durch die Anzahl der Kerne ersetzt werden.
In den Negativkontrollkulturen sollte die Populationsverdopplung bzw. der Replikationsindex mit den Anforderungen an die Probenahme von Zellen nach der Behandlung zu einem Zeitpunkt, der etwa der 1,5-bis 2-fachen Dauer des normalen Zellzyklus entspricht, übereinstimmen."
(15) In Teil B werden die folgenden Kapitel angefügt:
"B.59 In-chemico-Hautsensibilisierung: Direkt-Peptidreaktivitätstest (DPRA)
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 442C (2015). Ein Hautallergen ist gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP) 1 ein Stoff, der bei Hautkontakt eine allergische Reaktion auslöst. Diese Prüfmethode ist ein In-chemico-Verfahren (Direkt-Peptidreaktivitätstest - DPRA) zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren gemäß UN-GHS und CLP.
Es besteht allgemeines Einvernehmen über die der Hautsensibilisierung zugrunde liegenden biologischen Vorgänge. Das vorhandene Wissen über die chemischen und biologischen Mechanismen im Zusammenhang mit der Hautsensibilisierung wurde in Form eines Adverse Outcome Pathway (AOP) (2), ausgehend von dem auslösenden molekularen Ereignis über die Zwischenvorgänge bis hin zur schädlichen Auswirkung auf die Gesundheit, nämlich die allergische Kontaktdermatitis beim Menschen oder die Kontakt-Überempfindlichkeit bei Nagetieren, zusammengefasst. Innerhalb des Hautsensibilisierungs-AOP ist das auslösende molekulare Ereignis die kovalente Bindung von elektrophilen Stoffen an nukleophile Zentren in Hautproteinen.
Die Bewertung der Hautsensibilisierung erfolgte normalerweise an Labortieren. Bei den klassischen Methoden an Meerschweinchen, dem Maximierungstest an Meerschweinchen (Guinea Pig Maximisation Test, GMPT) nach Magnusson/Kligman und dem Bühler-Test (Kapitel B.6 (3)), werden die Induktions- und die Auslösephase der Hautsensibilisierung untersucht. Ein Test an der Maus, der Lokale Lymphknotentest (LLNA, Kapitel B.42 (4)) und die beiden nicht radioaktiven Abwandlungen dieses Tests, LLNA: DA (Kapitel B.50 (5)) und LLNA: BrdU-ELISA (Kapitel B.51 (6)), bei denen jeweils nur die Induktionsreaktion bewertet wird, haben ebenfalls an Akzeptanz gewonnen, da sie sowohl in Bezug auf den Tierschutz als auch die objektive Messung der Induktionsphase der Hautsensibilisierung Vorteile gegenüber Tests am Meerschweinchen bieten.
Vor kurzem wurden mechanistisch basierte In-chemico- und In-vitro-Prüfmethoden für die Bewertung der Gefahr einer Hautsensibilisierung durch Chemikalien als wissenschaftlich fundiert befunden. Allerdings sind Methoden ohne Tierversuche (in silico, in chemico, in vitro) im Rahmen von integrierten Test- und Bewertungsansätzen (Integrated Approaches to Testing and Assessment, IATA) erforderlich, um die gegenwärtig angewandten Tierversuche angesichts der eingeschränkten mechanistischen AOP-Abdeckung der gegenwärtig verfügbaren Prüfmethoden ohne Tierversuche zu ersetzen (2) (7).
Der DPRA wird für das auslösende molekulare Ereignis im AOP der Hautsensibilisierung, nämlich die Proteinreaktivität, vorgeschlagen, indem die Reaktivität von Prüfchemikalien gegenüber synthetischen lysin- oder cysteinhaltigen Modellpeptiden quantifiziert wird (8). Anhand der prozentualen Peptid-Depletionswerte für Cystein und Lysin wird ein Stoff dann in eine von vier Reaktivitätsklassen eingestuft, um die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren zu unterstützen (9).
Der DPRA wurde in einer Validierungsstudie unter der Leitung eines Europäischen Referenzlabors für Alternativen zu Tierversuchen (European Union Reference Laboratory for Alternatives to Animal Testing, EURL ECVAM) und in einem anschließenden unabhängigen Peer-Review des Wissenschaftlich Beratenden Ausschusses (ESAC) des EURL ECVAM bewertet und aus wissenschaftlicher Sicht für zulässig befunden (10), um im Rahmen eines IATA die Unterscheidung zwischen Hautallergenen und Nichtsensibilatoren im Hinblick auf die Gefahreneinstufung und -kennzeichnung zu unterstützen. Beispiele für die Verwendung von DPRA-Daten in Kombination mit anderen Informationen werden in der Literatur beschrieben (11) (12) (13) (14).
Definitionen sind Anlage I zu entnehmen.
Vorbemerkungen, Anwendbarkeit und Einsatzgrenzen
Die Korrelation zwischen Proteinreaktivität und Hautsensibilisierungspotenzial ist gut nachgewiesen (15) (16) (17). Da die Proteinbindung jedoch nur einen einzigen Schlüsselvorgang, wenn auch das auslösende molekulare Ereignis für den AOP der Hautsensibilisierung, darstellt, reichen die mit Prüfmethoden und Nicht-Prüfmethoden ermittelten Informationen alleine möglicherweise nicht aus, um zu dem Schluss zu gelangen, dass Chemikalien kein Hautsensibilisierungspotenzial besitzen. Daher sollten die mit dieser Prüfmethode ermittelten Daten im Rahmen integrierter Ansätze, wie z.B. IATA, betrachtet und mit anderen ergänzenden Informationen, die beispielsweise aus In-vitro-Tests in Bezug auf andere Schlüsselvorgänge des Hautsensibilisierungs-AOP abgeleitet werden, sowie mit anderen Nicht-Prüfmethoden, einschließlich der Übertragung von Informationen (read across) zu chemischen Analoga, kombiniert werden.
Diese Prüfmethode kann zusammen mit anderen ergänzenden Informationen zur Unterstützung der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kategorie 1) und Nichtsensibilisatoren im Rahmen eines IATA eingesetzt werden. Diese Prüfmethode kann alleine weder zur Einstufung von Hautallergenen in die Unterkategorien 1A und 1B gemäß Definition in UN-GHS/CLP noch zur Vorhersage des Potenzials im Rahmen von Sicherheitsbewertungsentscheidungen verwendet werden. Jedoch kann ein positives Ergebnis mit dem DPRA je nach Rechtsrahmen alleine zur Einstufung einer Chemikalie in die UN-GHS/CLP-Kategorie 1 herangezogen werden.
Es hat sich gezeigt, dass die DPRA-Prüfmethode Labors mit Erfahrung auf dem Gebiet der Hochleistungsflüssigchromatographie (HPLC) übertragen werden kann. Der Grad der Reproduzierbarkeit bei Vorhersagen, der bei der Prüfmethode erwartet werden kann, liegt in der Größenordnung von 85 % innerhalb von Labors und von 80 % zwischen Labors (10). Die Ergebnisse der Validierungsstudie (18) und veröffentlichter Studien (19) weisen insgesamt darauf hin, dass die Genauigkeit des DPRA bei der Unterscheidung von Sensibilatoren (d. h. UN-GHS-/CLP-Kategorie 1) gegenüber Nichtsensibilisatoren 80 % (N = 157) bei einer Empfindlichkeit von 80 % (88/109) und einer Spezifität von 77 % (37/48) im Vergleich zu den Ergebnissen des LLNA beträgt. Beim DPRA ist die Wahrscheinlichkeit einer Unterschätzung bei Chemikalien mit geringem bis mittlerem Hautsensibilisierungspotenzial (d. h. UN-GHS/CLP-Unterkategorie 1B) größer als bei Chemikalien mit hohem Hautsensibilisierungspotenzial (d. h. UN-GHS/CLP-Unterkategorie 1A) (18) (19). Jedoch sind die Genauigkeitswerte, die hier für den DPRA als eigenständige Testmethode angegeben werden, lediglich als Anhaltspunkte zu betrachten, da die Prüfmethode in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 9 betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA-Test sowie andere Tierversuche die Situation bei den relevanten Arten, d. h. Menschen, nicht vollständig widerspiegeln. Auf der Grundlage der verfügbaren Gesamtdaten über den DPRA wurde nachgewiesen, dass der Test bei Prüfchemikalien, die eine Vielzahl an organischen funktionellen Gruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzialen (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (8) (9) (10) (19). Insgesamt deuten diese Informationen auf die Zweckmäßigkeit des DPRA für die Erkennung der Gefahr einer Hautsensibilisierung hin.
Der Begriff "Prüfchemikalie" bezeichnet bei dieser Prüfmethode das, was getestet wird, und bezieht sich nicht auf die Anwendbarkeit des DPRA für die Prüfung von Stoffen und/oder Gemischen. Diese Prüfmethode ist nicht für die Untersuchung von Metallverbindungen geeignet, da diese bekanntermaßen mit Proteinen reagieren, die über andere Mechanismen als die kovalente Bindung wirken. Eine Prüfchemikalie sollte in einem geeigneten Lösungsmittel in einer Endkonzentration von 100 mM löslich sein (siehe Nummer 18). Prüfchemikalien, die in dieser Konzentration nicht löslich sind, können unter Umständen jedoch trotzdem in geringeren löslichen Konzentrationen getestet werden. In einem solchen Fall könnte ein positives Ergebnis dennoch zur Identifizierung der Prüfchemikalie als Hautallergen unterstützend herangezogen werden, während bei einem negativen Ergebnis keine verbindlichen Rückschlüsse auf die fehlende Reaktivität gezogen werden dürfen. Gegenwärtig stehen nur begrenzte Informationen über die Anwendbarkeit des DPRA auf Gemische mit bekannter Zusammensetzung zur Verfügung (18) (19). Dennoch gilt der DPRA für die Prüfung von mehrkomponentigen Substanzen und Gemischen mit bekannter Zusammensetzung als technisch geeignet (siehe Nummer 18). Bevor diese Prüfmethode auf ein Gemisch angewendet wird, um zu Regulierungszwecken Daten zu gewinnen, sollte geprüft werden, ob, und falls ja, warum, sie für diesen Zweck geeignete Ergebnisse liefert. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs gesetzlich vorgeschrieben ist. Das gegenwärtige Vorhersagemodell kann aufgrund des festgelegten Molverhältnisses von Prüfchemikalie und Peptid nicht bei komplexen Gemischen mit unbekannter Zusammensetzung oder bei Stoffen mit unbekannter oder variabler Zusammensetzung, komplexen Reaktionsprodukten oder biologischen Materialien (z.B. UVCB-Stoffen) eingesetzt werden. Für diesen Zweck wird es erforderlich sein, ein neues Vorhersagemodell auf der Grundlage eines gravimetrischen Ansatzes zu entwickeln. Wenn nachgewiesen werden kann, dass die Prüfmethode bei anderen spezifischen Chemikalienkategorien nicht anwendbar ist, sollte sie bei diesen nicht verwendet werden.
Diese Prüfmethode ist eine In-chemico-Methode, die kein Stoffwechselsystem beinhaltet. Chemikalien, die ihr Hautsensibilisierungspotenzial erst durch eine enzymatische Bioaktivierung erlangen (d. h. Prohaptene), können mit dieser Prüfmethode nicht erkannt werden. Chemikalien, die erst nach einer abiotischen Umwandlung zu Sensibilisatoren werden (d. h. Prähaptene), werden Berichten zufolge in einigen Fällen mit der Prüfmethode richtig erkannt (18). Angesichts der vorstehenden Ausführungen sollten mit der Prüfmethode erhaltene negative Ergebnisse im Kontext der angegebenen Grenzen und in Verbindung mit anderen Informationsquellen im Rahmen eines IATA interpretiert werden. Prüfchemikalien, die nicht kovalent an das Peptid binden, sondern seine Oxidation (d. h. Cystein-Dimerisierung) fördern, könnten zu einer potenziellen Überschätzung der Peptid-Depletion führen, mit der Folge möglicher falscher Positiv-Vorhersagen und/oder einer Einstufung in eine höhere Reaktivitätsklasse (siehe Nummern 29 und 30).
Der DPRA unterstützt, wie bereits erwähnt, die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren. Er kann jedoch bei Verwendung im Rahmen von integrierten Ansätzen wie IATA möglicherweise auch zur Bewertung der Sensibilisierungspotenz beitragen (11). Es sind allerdings weitere Untersuchungen notwendig, vorzugsweise auf der Grundlage von Humandaten, um herauszufinden, inwieweit die Ergebnisse des DPRA möglicherweise zur Potenzbewertung herangezogen werden können.
Testprinzip
Der DPRA ist eine In-chemico-Methode, bei der die Restkonzentration eines cystein- oder lysinhaltigen Peptids nach 24-stündiger Inkubation mit der Prüfchemikalie bei 25 ± 2,5 °C quantifiziert wird. Die synthetischen Peptide enthalten Phenylalanin als Hilfsmittel zum Nachweis. Die relative Peptidkonzentration wird mittels Hochleistungsflüssigchromatographie (HPLC) mit Gradientenelution und UV-Detektion bei 220 nm gemessen. Anschließend werden die prozentualen Peptid-Depletionswerte für Cystein und Lysin berechnet, die als Eingangsparameter in ein Vorhersagemodell (siehe Nummer 29) zur Einstufung der Prüfchemikalie in eine von vier Reaktivitätsklassen einfließen, die zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilatoren verwendet werden.
Vor der routinemäßigen Verwendung des unter dieser Prüfmethode beschriebenen Verfahrens sollten Laboratorien ihre technische Kompetenz anhand der zehn in Anlage 2 aufgeführten Leistungsstoffe nachweisen.
Verfahren
Diese Prüfmethode basiert auf dem DPRA DB-ALM-Protokoll Nr. 154 (20), das für die vom EURL ECVAM koordinierte Validierungsstudie verwendet wurde. Es wird empfohlen, dieses Protokoll bei der Durchführung und Anwendung der Methode im Labor zugrunde zu legen. Nachfolgend werden die wichtigsten Komponenten und Verfahren des DPRA beschrieben. Bei der Verwendung eines anderen HPLC-Aufbaus ist dessen Gleichwertigkeit mit dem im DB-ALM-Protokoll beschriebenen validierten Aufbau nachzuweisen (z.B. durch Prüfung der Leistungsstoffe in Anlage 2).
Vorbereitung der cystein- oder lysinhaltigen Peptide
Die Stammlösungen von cysteinhaltigen (Ac-RFAACAA-COOH) und lysinhaltigen (Ac-RFAAKAA-COOH) synthetischen Peptiden mit einer Reinheit von mehr als 85 % (vorzugsweise im Bereich von 90-95 %) sollten kurz vor ihrer Inkubation mit der Prüfchemikalie frisch zubereitet werden. Die Endkonzentration des Cysteinpeptids sollte 0,667 mM in einem Phosphatpuffer mit pH 7,5 und die Endkonzentration des Lysinpeptids 0,667 mM in einem Ammoniumacetat-Puffer mit pH 10,2 betragen. Die HPLC-Sequenz ist so einzustellen, dass die HPLC-Analyse in weniger als 30 Stunden abgeschlossen ist. Bei dem in der Validierungsstudie verwendeten und in dieser Prüfmethode beschriebenen HPLC-Aufbau können in einem einzigen HPLC-Durchlauf bis zu 26 Analyseproben (bestehend aus der Prüfchemikalie, der Positivkontrolle und einer entsprechenden Anzahl an Lösungsmittelkontrollen, abhängig von der Anzahl der einzelnen im Test verwendeten Lösungsmittel, jeweils dreifach getestet) abgearbeitet werden. Alle im gleichen Durchlauf analysierten Replikate sollten die gleichen Cystein- und Lysinpeptid-Stammlösungen verwenden. Es wird empfohlen, vor der Verwendung der einzelnen Peptidchargen ihre Löslichkeit entsprechend nachzuweisen.
Vorbereitung der Prüfchemikalie
Vor der Durchführung des Tests sollte die Löslichkeit der Prüfchemikalie in einem geeigneten Lösungsmittel gemäß dem im DPRA DB-ALM-Protokoll (20) beschriebenen Solubilisierungsverfahren bewertet werden. Ein geeignetes Lösungsmittel löst die Prüfchemikalie vollständig auf. Da die Prüfchemikalie beim DPRA mit hohem Überschuss mit den Cystein- oder den Lysinpeptiden inkubiert wird, gilt die visuelle Prüfung auf Bildung einer klaren Lösung als ausreichend, um festzustellen, dass die Prüfchemikalie (und - bei Testung einer mehrkomponentigen Substanz oder eines Gemischs - alle Bestandteile) aufgelöst wurde(n). Geeignete Lösungsmittel sind Acetonitril, Wasser, ein 1:1-Gemisch aus Wasser und Acetonitril, Isopropanol, Aceton oder ein 1:1-Gemisch aus Aceton und Acetonitril. Andere Lösungsmittel können verwendet werden, solange sie die Stabilität des Peptids nicht beeinträchtigen. Dies wird mithilfe von Referenzkontrollen C (d. h. Proben, bei denen das Peptid alleine im jeweiligen Lösungsmittel aufgelöst ist; siehe Anlage 3) überwacht. Ist die Prüfchemikalie in keinem dieser Lösungsmittel löslich, sollte als letzte Möglichkeit versucht werden, sie in 300 µL DMSO zu lösen und die erhaltene Lösung mit 2.700 µL Acetonitril zu verdünnen. Ist die Prüfchemikalie auch in diesem Gemisch unlöslich, sollte versucht werden, sie in gleicher Menge in 1..500 µL DMSO zu lösen und die erhaltene Lösung mit 1.500 µL Acetonitril zu verdünnen. Zum Ansetzen einer 100-mM-Lösung wird die Prüfchemikalie vorgewogen in Glasgefäße gegeben und unmittelbar vor dem Test in einem geeigneten Lösungsmittel aufgelöst. Bei Gemischen und mehrkomponentigen Substanzen mit bekannter Zusammensetzung wird aus der Summe der Anteile der einzelnen Bestandteile (ausgenommen Wasser) ein einziger Wert für die Reinheit und anhand der Molekulargewichte der einzelnen Bestandteile im Gemisch (ausgenommen Wasser) und ihres jeweiligen Anteils ein einziges scheinbares Molekulargewicht bestimmt. Aus den erhaltenen Werten für die Reinheit und das scheinbare Molekulargewicht wird dann das erforderliche Gewicht der Prüfchemikalie für die Zubereitung einer 100-mM-Lösung berechnet. Bei Polymeren, für die sich kein vorherrschendes Molekulargewicht bestimmen lässt, kann das Molekulargewicht des Monomers (oder das scheinbare Molekulargewicht der verschiedenen Monomere, aus denen sich das Polymer zusammensetzt) für die Zubereitung einer 100-mM-Lösung herangezogen werden. Bei der Prüfung von Gemischen, mehrkomponentigen Substanzen oder Polymeren mit bekannter Zusammensetzung sollte jedoch auch in Erwägung gezogen werden, die unverdünnte Chemikalie zu testen. Bei Flüssigkeiten wird die unverdünnte Chemikalie so, wie sie ist, ohne vorherige Verdünnung getestet und in einem molaren Verhältnis von 1:10 und 1:50 mit den Cystein- bzw. Lysinpeptiden inkubiert. Bei festen Stoffen wird die Prüfchemikalie auf ihre höchste lösliche Konzentration in dem gleichen Lösungsmittel aufgelöst, das zum Ansetzen der scheinbaren 100-mM-Lösung verwendet wurde. Anschließend wird sie so, wie sie ist, ohne weitere Verdünnung getestet und in einem molaren Verhältnis von 1:10 und 1:50 mit den Cystein- bzw. Lysinpeptiden inkubiert. Übereinstimmende Ergebnisse (reaktiv oder nicht reaktiv) zwischen der scheinbaren 100-mM-Lösung und der unverdünnten Chemikalie sollten eine verbindliche Schlussfolgerung bezüglich des Ergebnisses zulassen.
Vorbereitung der Positivkontrolle, Referenzkontrollen und Koelutionskontrollen
Als Posivivkontrolle (PC) sollte Zimtaldehyd (CAS 104-55-2; > 95 % von lebensmitteltauglicher Reinheit) mit einer Konzentration von 100 mM in Acetonitril verwendet werden. Andere geeignete Positivkontrollen, die vorzugsweise Depletionswerte im mittleren Bereich ergeben, können verwendet werden, sofern historische Daten für die Ableitung vergleichbarer Akzeptanzkriterien für einen Testdurchlauf zur Verfügung stehen. Außerdem sollte die HPLC-Sequenz auch Referenzkontrollen (d. h. Proben, die nur das im entsprechenden Lösungsmittel gelöste Peptid enthalten) umfassen. Diese dienen dazu, die Eignung des HPLC-Systems vor der Analyse (Referenzkontrollen A) und die Stabilität der Referenzkontrollen im Zeitverlauf (Referenzkontrollen B) zu verifizieren und zu bestätigen, dass das zur Lösung der Prüfchemikalie verwendete Lösungsmittel die prozentuale Peptid-Depletion nicht beeinflusst (Referenzkontrollen C) (siehe Anlage 3). Mithilfe der geeigneten Referenzkontrolle für jede Chemikalie wird die prozentuale Peptid-Depletion für diese Chemikalie berechnet (siehe Nummer 26). Darüber hinaus sollte für jede analysierte Prüfchemikalie eine Koelutionskontrolle, bestehend aus der Prüfchemikalie alleine, in die Ablaufsequenz aufgenommen werden, um eine mögliche Koelution der Prüfchemikalie mit dem Lysinpeptid bzw. dem Cysteinpeptid zu erkennen.
Inkubation der Prüfchemikalie mit den Cystein- und Lysinpeptid-Lösungen
Die Cystein- und Lysinpeptid-Lösungen sollten in Autosampler-Glasgefäßen in einem Verhältnis von 1:10 bzw. 1:50 mit der Prüfchemikalie inkubiert werden. Wenn es unmittelbar nach der Zugabe der Prüfchemikalie zur Peptidlösung aufgrund der geringen Wasserlöslichkeit der Prüfchemikalie zu einer Ausfällung kommt, kann nicht mit Sicherheit festgestellt werden, welche Menge der Prüfchemikalie in der Lösung verblieben ist, um mit dem Peptid zu reagieren. In diesem Fall könnte ein positives Ergebnis trotzdem verwendet werden. Ein negatives Ergebnis ist hingegen unsicher und sollte entsprechend vorsichtig interpretiert werden (siehe auch die Vorschriften unter Nummer 11 für die Prüfung von Chemikalien, die bis zu einer Konzentration von 100 mM unlöslich sind). Die Reaktionslösung wird bei 25 ± 2,5 °C 24 ± 2 Stunden vor der Durchführung der HPLC-Analyse im Dunkeln gelassen. Jede Prüfchemikalie wird für beide Peptide in dreifacher Ausfertigung analysiert. Die Proben sind vor der HPLC einer Sichtprüfung zu unterziehen. Bei festgestellter Ausfällung oder Phasentrennung können die Proben als Vorsichtsmaßnahme bei geringer Geschwindigkeit (100-400 x g) zentrifugiert werden, damit die Ausfällung auf den Boden des Gefäßes sinkt, da große Ausfällungsmengen die Leitungen oder Säulen der HPLC-Apparatur verstopfen können. Wird nach der Inkubationszeit eine Ausfällung oder Phasentrennung festgestellt, kann die Peptid-Depletion unterschätzt werden. In diesem Fall kann bei einem negativen Ergebnis nicht mit hinreichender Sicherheit auf das Fehlen von Reaktivität geschlossen werden.
Erstellung der HPLC-Standardkalibrierungskurve
Sowohl für die Cystein- als auch die Lysinpeptide sollte eine Standardkalibrierungskurve erstellt werden. Die Peptidstandards werden in einer Lösung mit 20 % oder 25 % Acetonitril/Puffer unter Verwendung eines Phosphatpuffers (pH 7,5) für das Cysteinpeptid und eines Ammoniumacetat-Puffers (pH 10,2) für das Lysinpeptid zubereitet. Aus Verdünnungsreihen-Standards der Peptid-Stammlösung (0,667 mM) werden sechs Kalibrierungslösungen hergestellt, die den Bereich von 0,534 bis 0,0167 mM abdecken. Eine Blindkontrolle des Verdünnungspuffers sollte ebenfalls in die Standardkalibrierungskurve aufgenommen werden. Geeignete Kalibrierungskurven sollten einen Wert von r2 > 0,99 aufweisen.
Vorbereitung des HPLC-Systems und Analyse
Vor der Durchführung der Analyse ist die Eignung des HPLC-Systems zu verifizieren. Die Peptid-Depletion wird durch ein mit einem UV-Detektor (Fotodiodenarray-Detektor oder Festwellenlängen-Absorptionsdektektor mit 220-nm-Signal) verbundenes HPLC-Gerät überwacht. Die entsprechende Säule wird in das HPLC-System eingebaut. Bei dem im validierten Protokoll beschriebenen HPLC-Aufbau wird das Modell Zorbax SB-C-18 2,1 mm x 100 mm x 3,5 µm als bevorzugte Säule verwendet. Bei dieser Säule für die Umkehrphasen-HPLC sollte das gesamte System mindestens 2 Stunden vor dem Betrieb bei 301 °C mit 50 % Phase A (0,1 % v/v Trichloressigsäure in Wasser) und 50 % Phase B (0,085 % v/v Trichloressigsäure in Acetonitril) äquilibriert werden. Die HPLC-Analyse erfolgt mit einer Flussrate von 0,35 ml/min und einem linearen Gradienten von 10 % bis 25 % Acetonitril über einen Zeitraum von 10 Minuten, gefolgt von einem schnellen Anstieg auf 90 % Acetonitril, um andere Materialien zu entfernen. Standard, Probe und Kontrolle werden jeweils in gleicher Menge injiziert. Zwischen den Injektionen wird die Säule unter den Ausgangsbedingungen 7 Minuten lang neu äquilibriert. Bei Verwendung einer anderen Säule für die Umkehrphasen-HPLC müssen die oben genannten Einstellparameter möglicherweise angepasst werden, um eine angemessene Elution und Integration der Cystein- und Lysinpeptide sicherzustellen. Dies gilt auch für die Injektionsmenge, die je nach verwendetem System variieren kann (normalerweise im Bereich von 3-10 µl). Wichtig ist auch, dass bei der Verwendung eines anderen Aufbaus der HPLC-Apparatur dessen Gleichwertigkeit mit dem vorstehend beschriebenen validierten Aufbau nachgewiesen wird (z.B. durch Prüfung der Leistungsstoffe in Anlage 2). Die Extinktion wird bei 220 nm überwacht. Bei Verwendung eines Fotodiodenarray-Detektors sollte die Extinktion bei 258 nm ebenfalls aufgezeichnet werden. Es wird darauf hingewiesen, dass einige Acetonitril-Chargen die Peptidstabilität beeinträchtigen können. Dies muss bei der Verwendung einer neuen Acetonitril-Charge bewertet werden. Als Indikator für eine Koelution kann das Verhältnis der Peakfläche bei 220 nm und der Peakfläche bei 258 nm verwendet werden. Beispielsweise wäre bei jeder Probe ein Verhältnis im Bereich von 90 % < mittleres 2 Flächenverhältnis der Kontrollproben < 100 % ein guter Anhaltspunkt dafür, dass keine Koelution eingetreten ist.
Es gibt unter Umständen Prüfchemikalien, die die Oxidation des Cysteinpeptids fördern könnten. Der Peak des dimerisierten Cysteinpeptids kann visuell überwacht werden. Scheint eine Dimerisierung erfolgt zu sein, sollte dies vermerkt werden, da die prozentuale Peptid-Depletion überschätzt werden kann, was zu falschen Positiv-Vorhersagen und/oder einer Einstufung in eine höhere Reaktivitätsklasse führen kann (siehe Nummer 29 und 30).
Die HPLC-Analyse der Cystein- und Lysinpeptide kann gleichzeitig (wenn zwei HPLC-Systeme zur Verfügung stehen) oder an verschiedenen Tagen durchgeführt werden. Erfolgt die Analyse an verschiedenen Tagen, sind alle Prüfchemikalienlösungen für beide Tests am jeweiligen Tag frisch herzustellen. Die Analyse wird zeitlich so angesetzt, dass die Injektion der ersten Probe 22 bis 26 Stunden nach Mischung der Prüfchemikalie mit der Peptidlösung stattfindet. Die HPLC-Sequenz ist so einzustellen, dass die HPLC-Analyse in weniger als 30 Stunden abgeschlossen ist. Bei dem in der Validierungsstudie verwendeten und bei dieser Prüfmethode beschriebenen HPLC-Aufbau können in einem einzigen HPLC-Durchlauf bis zu 26 Analyseproben abgearbeitet werden (siehe auch Nummer 17). Ein Beispiel für eine HPLC-Analysesequenz ist Anlage 3 zu entnehmen.
Daten und Berichterstattung
Datenauswertung
Die Konzentration des Cystein- oder Lysinpeptids der einzelnen Proben wird bei 220 nm fotometrisch bestimmt. Hierzu wird die Peakfläche (Fläche unter der Kurve (Area Under the Curve, AUC) der entsprechenden Peaks gemessen, und anhand der aus den Standards abgeleiteten linearen Kalibrierungskurve wird die Peptidkonzentration berechnet.
Die prozentuale Peptid-Depletion der einzelnen Proben erhält man nach folgender Formel durch Messung der Peakfläche und deren Division durch die mittlere Peakfläche der jeweiligen Referenzkontrollen C (siehe Anlage 3)

Akzeptanzkriterien
Ein Testdurchlauf gilt als gültig, wenn die folgenden Kriterien erfüllt sind:
- Die Standardkalibrierungskurve weist einen Wert von r2 > 0,99 auf;
- der Mittelwert der prozentualen Peptid-Depletion der drei Replikate für die Positivkontrolle mit Zimtaldehyd liegt zwischen 60,8 % und 100 % beim Cysteinpeptid und zwischen 40,2 % und 69,0 % beim Lysinpeptid, und die maximale Standardabweichung (SA) für die Replikate der Positivkontrolle beträgt < 14,9 % für die prozentuale Cystein-Depletion und < 11,6 % für die prozentuale Lysin-Depletion; und
- die mittlere Peptidkonzentration der Referenzkontrollen A beträgt 0,50 ± 0,05 mM, und der Variationskoeffizient (VK) der Peptid-Peakflächen für die neun Referenzkontrollen B und C in Acetonitril beträgt < 15,0 %.
Wenn eines oder mehrere dieser Kriterien nicht erfüllt sind, ist der Testdurchlauf zu wiederholen.
Die Ergebnisse für eine Prüfchemikalie gelten als gültig, wenn die folgenden Kriterien erfüllt sind:
- Die maximale Standardabweichung bei den Replikaten der Prüfchemikalie beträgt < 14,9 % für die prozentuale Cystein-Depletion und < 11,6 % für die prozentuale Lysin-Depletion;
- die mittlere Peptidkonzentration der drei Referenzkontrollen C im entsprechenden Lösungsmittel beträgt 0,50 ± 0,05 mM. Wenn diese Kriterien nicht erfüllt sind, werden die Daten verworfen, und der Testdurchlauf für diese spezifische Prüfchemikalie ist zu wiederholen.
Vorhersagemodell
Für jede Prüfchemikalie wird der Mittelwert der prozentualen Cystein- und Lysin-Depletion berechnet. Eine negative Depletion wird bei der Berechnung des Mittelwerts mit "0" angenommen. Unter Verwendung des Vorhersagemodells Cystein 1:10/Lysin 1:50 in Tabelle 1 wird eine durchschnittliche Peptid-Depletion von 6,38 % als Schwelle zur Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren im Rahmen eines IATA herangezogen. Die Anwendung des Vorhersagemodells für die Einstufung einer Prüfchemikalie in eine Reaktivitätsklasse (d. h. geringe, mittlere und hohe Reaktivität) kann ggf. als nützliche Information für die Potenzbewertung im Rahmen eines IATA dienen.
Tabelle 1 Vorhersagemodell Cystein 1:10/Lysin 1:50 1
| Mittelwert der prozentualen Cystein- und Lysin-Depletion | Reaktivitätsklasse | DPRA-Vorhersage 2 |
| 0 % < Mittelwert der prozentualen Depletion < 6,38 % | keine oder minimale Reaktivität | negativ |
| 6,38 % < Mittelwert der prozentualen Depletion < 22,62 % | geringe Reaktivität | positiv |
| 22,62 % < Mittelwert der prozentualen Depletion < 42,47 % | mittlere Reaktivität | |
| 42,47 % < Mittelwert der prozentualen Depletion < 100 % | hohe Reaktivität | |
| 1) Die Werte beziehen sich auf statistisch generierte Schwellenwerte und nicht auf die Genauigkeit der Messung.
2) Eine DPRA-Vorhersage ist im Rahmen eines IATA und gemäß den Bestimmungen unter den Nummern 9 und 12 zu betrachten. | ||
Es könnte Fälle geben, in denen die Prüfchemikalie (der Stoff oder einer oder mehrere Bestandteile einer mehrkomponentigen Substanz oder eines Gemischs) eine starke Extinktion bei 220 nm aufweist und die gleiche Retentionszeit wie das Peptid besitzt (Koelution). Eine Koelution kann vermieden werden, indem der HPLC-Aufbau so verändert wird, dass die Elutionszeit der Prüfchemikalie und des Peptids weiter auseinander liegen. Wird ein anderer HPLC-Aufbau verwendet, um eine Koelution aufzulösen, so ist seine Gleichwertigkeit mit dem validierten Aufbau nachzuweisen (z.B. durch Prüfung der Leistungsstoffe in Anlage 2). Bei Vorliegen einer Koelution kann der Peak des Peptids nicht integriert und die prozentuale Peptid-Depletion nicht berechnet werden. Koeluieren solche Prüfchemikalien sowohl mit den Cystein- als auch den Lysinpeptiden, ist die Analyse als "nicht aussagekräftig" anzugeben. Liegt eine Koelution nur mit dem Lysinpeptid vor, kann das Vorhersagemodell Cystein 1:10 in Tabelle 2 verwendet werden.
Tabelle 2 Vorhersagemodell Cystein 1:10 1
| Prozentuale Cystein-Depletion | Reaktivitätsklasse | DPRA-Vorhersage 2 |
| 0 % < prozentuale Cystein-Depletion < 13,89 % | keine oder minimale Reaktivität | negativ |
| 13,89 % < prozentuale Cystein-Depletion < 23,09 % | geringe Reaktivität | positiv |
| 23,09 % < prozentuale Cystein-Depletion < 98,24 % | mittlere Reaktivität | |
| 98,24 % < prozentuale Cystein-Depletion < 100 % | hohe Reaktivität | |
| 1) Die Werte beziehen sich auf statistisch generierte Schwellenwerte und nicht auf die Genauigkeit der Messung.
2) Eine DPRA-Vorhersage ist im Rahmen eines IATA und gemäß den Bestimmungen unter den Nummern 9 und 12 zu betrachten. | ||
Es könnte andere Fälle geben, in denen sich die Retentionszeiten der Prüfchemikalie und eines der beiden Peptide nicht vollständig überlappen. In solchen Fällen können die Peptid-Depletionswerte geschätzt und im Vorhersagemodell Cystein 1:10/Lysin 1:50 verwendet werden. Eine genaue Einstufung der Prüfchemikalie in eine Reaktivitätsklasse ist jedoch nicht möglich.
Bei einem eindeutigen Ergebnis ist eine einzige HPLC-Analyse sowohl für das Cystein- als auch das Lysinpeptid pro Prüfchemikalie ausreichend. Liegen die Ergebnisse jedoch nahe an der Schwelle zur Unterscheidung zwischen positivem und negativem Ergebnis (d. h. handelt es sich um grenzwertige Ergebnisse), sind möglicherweise weitere Tests erforderlich. Fällt der Mittelwert der prozentualen Depletion in den Bereich von 3 % bis 10 % (Vorhersagemodell Cystin 1:10/Lysin 1:50) bzw. die prozentuale Cystein-Depletion in den Bereich von 9 % bis 17 % (Vorhersagemodell Cystin 1:10), sind ein zweiter Testdurchlauf sowie ein dritter Durchlauf (bei abweichenden Ergebnissen zwischen den ersten beiden Durchläufen) in Erwägung zu ziehen.
Prüfbericht
Der Prüfbericht sollte folgende Angaben enthalten:
Prüfchemikalie
- Einkomponentige Substanz
- chemische Bezeichnung, wie z.B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer (n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
- Aussehen, Wasserlöslichkeit, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, soweit verfügbar;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar.
- Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
- Charakterisierung, so weit wie möglich, z.B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Komponenten, soweit verfügbar;
- Aussehen, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften, soweit verfügbar;
- Molekulargewicht oder scheinbares Molekulargewicht im Fall von Gemischen/Polymeren mit bekannter Zusammensetzung oder andere für die Durchführung der Studie relevante Informationen;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar.
Kontrollen
- Positivkontrolle
- Chemische Bezeichnung, wie z.B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer (n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
- Aussehen, Wasserlöslichkeit, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, soweit verfügbar;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar;
- ggf. Verweis auf historische Ergebnisse von Positivkontrollen, die geeignete Akzeptanzkriterien für einen Testdurchlauf dokumentieren.
- Lösungsmittel/Vehikel
- Verwendetes Lösungsmittel/Vehikel und ggf. das Verhältnis ihrer Bestandteile;
- chemische Bezeichnung(en), wie z.B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer (n) und/oder andere Kennungen;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- Aussehen, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern andere Lösungsmittel/Vehikel als die in der Prüfmethode genannten verwendet werden und soweit verfügbar;
- Lagerbedingungen und Stabilität, soweit verfügbar;
- Begründung der Auswahl des Lösungsmittels für jede Prüfchemikalie;
- bei Acetonitril die Ergebnisse des Tests der Auswirkung auf die Peptidstabilität.
Vorbereitung von Peptiden, Positivkontrolle und Prüfchemikalie
- Charakterisierung der Peptidlösungen (Lieferant, Los, genaues Peptidgewicht, hinzugefügte Menge für die Stammlösung);
- Charakterisierung der Positivkontrolllösung (genaues Gewicht des für die Positivkontrolle verwendeten Stoffs, hinzugefügte Menge für die Testlösung);
- Charakterisierung der Prüfchemikalienlösungen (genaues Gewicht der Prüfchemikalien, hinzugefügte Menge für die Testlösung);
Einstellung des HPLC-Geräts und Analyse
- Typ des HPLC-Geräts, HPLC und Vorsäulen, Detektor, Autosampler;
- für die HPLC-Analyse relevante Parameter, wie Temperatur der Säule, Injektionsvolumen, Flussrate und Gradient.
Eignung des Systems
- Peptid-Peakfläche bei 220 nm jedes Replikats der Standard- und Referenzkontrolle A;
- grafische Darstellung der linearen Kalibrierungskurve und Angabe des r2-Wertes;
- Peptidkonzentration jedes Replikats der Referenzkontrolle A;
- mittlere Peptidkonzentration (mM) der drei Referenzkontrollen A, Standardabweichung und Variationskoeffizient;
- Peptidkonzentration der Referenzkontrollen A und C.
Ablauf der Analyse
- Für Referenzkontrollen:
- Peptid-Peakfläche bei 220 nm jedes B- und C-Replikats;
- mittlere Peptid-Peakfläche bei 220 nm der neun Referenzkontrollen B und C in Acetonitril, Standardabweichung und Variationskoeffzient (für die Stabilität der Referenzkontrollen über die Analysezeit);
- für jedes verwendete Lösungsmittel: mittlere Peptid-Peakfläche bei 220 nm der drei entsprechenden Referenzkontrollen C (zur Berechnung der prozentualen Peptid-Depletion);
- für jedes verwendete Lösungsmittel: die Peptidkonzentration (mM) der drei entsprechenden Referenzkontrollen C;
- für jedes verwendete Lösungsmittel: die mittlere Peptidkonzentration (mM) der drei entsprechenden Referenzkontrollen C, Standardabweichung und Variationskoeffizient.
- Für die Positivkontrolle:
- Peptid-Peakfläche bei 220 nm jedes Replikats;
- prozentuale Peptid-Depletion jedes Replikats;
- Mittelwert der prozentualen Peptid-Depletion der drei Replikate, Standardabweichung und Variationskoeffizient.
- Für jede Prüfchemikalie:
- Aussehen der Ausfällung im Reaktionsgemisch am Ende der Inkubationszeit, sofern beobachtet. Angabe, ob die Ausfällung wieder löslich gemacht oder zentrifugiert wurde;
- Auftreten von Koelution;
- ggf. Beschreibung anderer relevanter Beobachtungen;
- Peptid-Peakfläche bei 220 nm jedes Replikats;
- prozentuale Peptid-Depletion jedes Replikats;
- Mittelwert der prozentualen Peptid-Depletion der drei Replikate, Standardabweichung und Variationskoeffizient;
- Mittelwert der prozentualen Cystein- und Lysin-Depletion;
- verwendetes Vorhersagemodell und DPRA-Vorhersage.
Leistungstests
- Ggf. das angewandte Verfahren zum Nachweis der Kompetenz des Labors bei der Durchführung der Prüfmethode (z.B. durch Prüfung der Leistungsstoffe) oder zum Nachweis der reproduzierbaren Leistung der Prüfmethode im Zeitverlauf.
Erörterung der Ergebnisse
- Erörterung der anhand der DPRA-Prüfmethode erhaltenen Ergebnisse;
- Erörterung der Ergebnisse der Prüfmethode im Rahmen eines IATA, sofern andere sachdienliche Informationen verfügbar sind.
Schlussfolgerung
Literaturhinweise
(1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). 5. überarbeitete Auflage, UN New York und Genf, 2013. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html
(2) OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment, Nr. 168, OECD, Paris.
(3) Kapitel B.6: Sensibilisierung der Haut
(4) Kapitel B.42: Lokaler Lymphknotentest
(5) Kapitel B.50: Hautsensibilisierung: Lokaler Lymphknotentest: DA.
(6) Kapitel B.51: Hautsensibilisierung: Lokaler Lymphknotentest BrdU-ELISA
(7) Adler et al. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Archives of Toxicology, 85:367-485.
(8) Gerberick et al. (2004). Development of a peptide reactivity assay for screening contact allergens. Toxicological Sciences, 81:332-343.
(9) Gerberick et al. (2007). Quantification of chemical peptide reactivity for screening contact allergens: A classification tree model approach. Toxicological Sciences, 97:417-427.
(10) EC EURL-ECVAM (2013). Recommendation on the Direct Peptide Reactivity Assay (DPRA) for skin sensitisation testing. Verfügbar unter: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-direct-peptide-reactivity-assay-dpra
(11) Jaworska et al. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: from theory to practice. Journal of Applied Toxicology, Online-Veröffentlichung, 14. Mai 2013, DOI: 10.1002/jat.2869.
(12) Bauch et al. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potential. Regulatory Toxicology and Pharmacology, 63: 489-504.
(13) Nukada et al. (2013). Data integration of non-animal tests for the development of a test battery to predict the skin sensitizing potential and potency of chemicals. Toxicology in Vitro, 27:609 618.
(14) Ball et al (2011). Evaluating the sensitization potential of surfactants: integrating data from the local lymph node assay, guinea pig maximization test, and in vitro methods in a weight-of-evidence approach. Regulatory Toxicology and Pharmacology, 60:389-400.
(15) Landsteiner und Jacobs (1936). Studies on the sensitization of animals with simple chemical compounds. Journal of Experimental Medicine, 64:625-639.
(16) Dupuis and Benezra (1982). Allergic contact dermatitis to simple chemicals: a molecular approach. New York und Basel: Marcel Dekker Inc.
(17) Lepoittevin et al. (1998). Allergic contact dermatitis: the molecular basis. Springer, Berlin.
(18) EC EURL ECVAM (2012). Direct Peptide Reactivity Assay (DPRA) Validation Study Report, 74pp. Verfügbar unter: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-direct-peptide-reactivity-assay-dpra
(19) Natsch et al. (2013). A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. Journal of Applied Toxicology, Online-Veröffentlichung, 9. April 2013, DOI:10.1002/jat.2868.
(20) DB-ALM (INVITTOX) Protokoll 154. Direct Peptide Reactivity assay (DPRA) for skin sensitisation testing, 17pp. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/
(21) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Series on Testing and Assessment, Nr. 34. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris, Frankreich.
(22) FDA (Food and Drug Administration) (2001). Guidance for Industry: Bioanalytical Method Validation, 22pp. Verfügbar unter: www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidance/ucm070107.pdf -138
(23) ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology and Toxicology of Chemicals (Technical Report Nr. 87).
_____
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006. ABl. Nr. L 353 vom 31.12.2008 S. 1.
2) Unter dem Begriff "Mittelwert" oder "mittlere/r/s" ist im gesamten Dokument das arithmetische Mittel zu verstehen.
.
| Definitionen | Anlage 1 |
AOP (Adverse Outcome Pathway): Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den auslösenden molekularen Vorgang bis in zu einem In-vivo-Ergebnis von Interesse (2).
Auslösender molekularer Vorgang: Chemisch induzierte Störung eines biologischen Systems auf molekularer Ebene, die als Ausgangspunkt des Adverse Outcome Pathway identifiziert wird.
Chemikalie: Stoff oder Gemisch.
Eignung des Systems: Feststellung der Leistung (z.B. Empfindlichkeit) eines Instruments durch Analyse eines Referenzstandards vor dem Durchlauf der zu analysierenden Charge (22).
Einkomponentige Substanz: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist.
Empfindlichkeit: Der Anteil aller positiven/wirkenden Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Empfindlichkeit ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (21).
Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (1).
Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (21).
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeitnehmer, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1).
Gültige Prüfmethode: Eine Prüfmethode, die eine ausreichende Relevanz und Zuverlässigkeit für einen bestimmten Zweck aufweist und auf wissenschaftlich fundierten Grundsätzen beruht. Eine Prüfmethode ist nie im absoluten Sinn, sondern nur in Bezug auf einen definierten Zweck (21) gültig.
IATA (Integrated Approach to Testing and Assessment - Integrierter Test- und Bewertungsansatz): Strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbewertung (Potenzial/Potenz und Exposition) einer Chemikalie oder Chemikaliengruppe, bei dem alle maßgeblichen Daten strategisch integriert und gewichtet werden, um als Grundlage für fundierte regulatorische Entscheidungen über potenzielle Gefahren und/oder Risiken und/oder die Notwendigkeit weiterer gezielter und somit minimaler Testungen herangezogen zu werden.
Kalibrierungskurve: Beziehung zwischen dem im Versuch ermittelten Reaktionswert und der Analysekonzentration (auch als Standardkurve bezeichnet) eines bekannten Stoffs.
Mehrkomponentige Substanz: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens > 10 % w/w und < 80 % w/w vorhanden sind. Eine mehrkomponentige Substanz ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einer mehrkomponentigen Substanz besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Eine mehrkomponentige Substanz wird durch eine chemische Reaktion gebildet.
Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie: Der Begriff "Prüfchemikalie" bezeichnet das, was getestet wird.
Referenzkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert.
Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Sie berücksichtigt auch die Genauigkeit (Übereinstimmung) einer Prüfmethode (21).
Reproduzierbarkeit: Übereinstimmung der Ergebnisse von Tests, die an der gleichen Chemikalie bei einheitlichem Prüfprotokoll durchgeführt werden (siehe Zuverlässigkeit) (21).
Spezifität: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (21).
Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (1).
UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Variationskoeffizient: Kennwert der Varianz. Er wird für eine Gruppe von Replikatdaten mittels Division der Standardabweichung durch den Mittelwert berechnet. Multipliziert mit 100 ergibt sich ein Prozentwert.
Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Labors über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (21).
.
| Anlage 2 |
Leistungsstoffe
In-chemico-Hautsensibilisierung: Direkt-Peptidreaktivitätstest
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Labors ihre technische Kompetenz nachweisen, indem sie die erwartete DPRA-Vorhersage für die zehn in Tabelle 1 empfohlenen Leistungsstoffe richtig treffen und bei acht von zehn Leistungsstoffen für jedes Peptid Werte für die Cystein- und Lysin-Depletion erhalten, die im jeweiligen Referenzbereich liegen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Stoffe im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger In-vitro-Daten aus dem DPRA und dass diese in der vom EURL ECVAM koordinierten Validierungsstudie verwendet wurden, um die erfolgreiche Durchführung der Prüfmethode in den an der Studie beteiligten Labors nachzuweisen.
Tabelle 1 Empfohlene Leistungsstoffe für den Nachweis der technischen Kompetenz zur Durchführung des Direkt-Peptidreaktivitätstests
| Leistungsstoffe | CAS-Nr. | Aggregat- zustand | In-vivo- Vorhersage 1 | DPRA- Vorhersage 2 | Bandbreite 3 der prozentualen Cysteinpeptid- Depletion | Bandbreite 3 der prozentualen Lysinpeptid- Depletion |
| 2,4-Dinitrochlorbenzol | 97-00-7 | fest | Sensibilisator (extrem) | positiv | 90-100 | 15-45 |
| Oxazolon | 15646-46-5 | fest | Sensibilisator (extrem) | positiv | 60-80 | 10-55 |
| Formaldehyd | 50-00-0 | flüssig | Sensibilisator (stark) | positiv | 30-60 | 0-24 |
| Benzylidenaceton | 122-57-6 | fest | Sensibilisator (mäßig) | positiv | 80-100 | 0-7 |
| Farnesal | 19317-11-4 | flüssig | Sensibilisator (schwach) | positiv | 15-55 | 0-25 |
| 2,3-Butandion | 431-03-8 | flüssig | Sensibilisator (schwach) | positiv | 60-100 | 10-45 |
| 1-Butanol | 71-36-3 | flüssig | Nicht-sensibilisator | negativ | 0-7 | 0-5,5 |
| 6-Methylcoumarin | 92-48-8 | fest | Nicht-sensibilisator | negativ | 0-7 | 0-5,5 |
| Milchsäure | 50-21-5 | flüssig | Nicht-sensibilisator | negativ | 0-7 | 0-5,5 |
| 4-Methoxyacetophenon | 100-06-1 | fest | Nicht-sensibilisator | negativ | 0-7 | 0-5,5 |
| 1) Die In-vivo-Gefahrenidentifizierung und (Potenz-)Vorhersagen basieren auf Daten des LLNA (19). Die In-vivo-Potenz wird anhand der vom ECETOC (23) vorgeschlagenen Kriterien abgeleitet.
2) Eine DPRA-Vorhersage ist im Rahmen eines IATA und gemäß den Bestimmungen unter den Nummern 9 und 11 zu betrachten. 3) Bestimmung der Bandbreiten auf der Grundlage von mindestens zehn Depletionswerten von sechs unabhängigen Labors. | ||||||
.
| Beispiele für die Analysesequenz | Anlage 3 |
| Kalibrierungsstandards und Referenzkontrollen | STD1 STD2 STD3 STD4 STD5 STD6 Verdünnungspuffer Referenzkontrolle A, Rep. 1 Referenzkontrolle A, Rep. 2 Referenzkontrolle A, Rep. 3 |
| Koelutionskontrollen | Koelutionskontrolle 1 für Prüfchemikalie 1 Koelutionskontrolle 2 für Prüfchemikalie 2 |
| Referenzkontrollen | Referenzkontrolle B, Rep. 1 Referenzkontrolle B, Rep. 2 Referenzkontrolle B, Rep. 3 |
| Erster Replikatsatz | Referenzkontrolle C, Rep. 1 Zimtaldehyd, Rep. 1 Probe 1, Rep. 1 Probe 2, Rep. 1 |
| Zweiter Replikatsatz | Referenzkontrolle C, Rep. 2 Zimtaldehyd, Rep. 2 Probe 1, Rep. 2 Probe 2, Rep. 2 |
| Dritter Replikatsatz | Referenzkontrolle C, Rep. 3 Zimtaldehyd, Rep. 3 Probe 1, Rep. 3 Probe 2, Rep. 3 |
| Referenzkontrollen | Referenzkontrolle B, Rep. 4 Referenzkontrolle B, Rep. 5 Referenzkontrolle B, Rep. 6 |
| Die Analysesequenz umfasst drei Sätze von Referenzkontrollen (d. h. Proben, die nur aus dem im geeigneten Lösungsmittel gelösten Peptid bestehen):
Referenzkontrolle A: Zur Verifizierung der Eignung des HPLC-Systems. Referenzkontrolle B: Wird zu Beginn und am Ende der Analysesequenz aufgenommen, um die Stabilität der Referenzkontrollen im Zeitverlauf zu verifizieren. Referenzkontrolle C: Wird in die Analysesequenz aufgenommen, um zu verifizieren, dass das zur Lösung der Prüfchemikalie verwendete Lösungsmittel keinen Einfluss auf die prozentuale Peptid-Depletion hat. | |
B.60 In-vitro-Hautsensibilisierung: ARE-Nrf2 Luciferase-Prüfmethode
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 442D (2015). Ein Hautallergen ist gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 (CLP) ein Stoff, der bei Hautkontakt eine allergische Reaktion auslöst. Diese Prüfmethode ist ein In-vitro-Verfahren (ARE-Nrf2 Luciferase-Test) zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren gemäß UN-GHS (1) und CLP.
Es besteht allgemeines Einvernehmen über die der Hautsensibilisierung zugrunde liegenden biologischen Schlüsselvorgänge. Das vorhandene Wissen über die chemischen und biologischen Mechanismen im Zusammenhang mit der Hautsensibilisierung wurde in Form eines Adverse Outcome Pathway (AOP) (2) - vom molekularen auslösenden Vorgang über die dazwischen liegenden Vorgänge bis hin zur schädlichen Auswirkung auf die Gesundheit, z.B. allergische Kontaktdermatitis beim Menschen oder Kontakt-Überempfindlichkeit bei Nagetieren, - zusammengefasst (2) (3). Der molekulare auslösende Vorgang ist die kovalente Bindung von elektrophilen Stoffen an nukleophile Zentren in Hautproteinen. Der zweite Schlüsselvorgang in diesem AOP findet in den Keratinozyten statt und umfasst entzündliche Reaktionen sowie Genexpression in Verbindung mit spezifischen Zellsignalketten wie beispielsweise ARE (Antioxidant/Electrophile Response Element)-abhängigen Ketten. Der dritte Schlüsselvorgang ist die Aktivierung von dendritischen Zellen, die normalerweise durch Expression von spezifischen Zelloberflächenmarkern, Chemokinen und Cytokinen bewertet werden. Der vierte Schlüsselvorgang ist die T-Zellproliferation, die indirekt im Lokalen Lymphknotentest (LLNA) an der Maus (4) bewertet wird.
Die Bewertung der Hautsensibilisierung erfolgte normalerweise an Labortieren. Bei den klassischen Methoden an Meerschweinchen, dem Maximierungstest an Meerschweinchen (GMPT) nach Magnusson/Kligman und dem Bühler-Test (TM B.6 (5)), werden die Induktions- und die Auslösephase der Hautsensibilisierung untersucht. Ein Test an der Maus, der Lokale Lymphknotentest (LLNA) (TM B.42 (4)) sowie die beiden nicht radioaktiven Abwandlungen dieses Tests, LLNA: DA (TM B.50 (6)) und LLNA: BrdU-ELISA (TM B.51 (7)), bei denen jeweils nur die Induktionsreaktion bewertet wird, haben ebenfalls an Akzeptanz gewonnen, da sie sowohl in Bezug auf den Tierschutz als auch in Bezug auf die objektive Messung der Induktionsphase der Hautsensibilisierung Vorteile gegenüber Tests am Meerschweinchen bieten.
Vor Kurzem wurden mechanistisch basierte In-chemico- und In-vitro-Prüfmethoden für die Bewertung der Gefahr einer Hautsensibilisierung durch Chemikalien als wissenschaftlich fundiert befunden. Allerdings sind Methodenkombinationen ohne Tierversuche (in silico, in chemico, in vitro) im Rahmen von Integrierten Test- und Bewertungsansätzen (Integrated Approaches to Testing and Assessment, IATA) erforderlich, um die gegenwärtig verwendeten Tierversuche angesichts der eingeschränkten mechanistischen AOP-Abdeckung der gegenwärtig verfügbaren Prüfmethoden ohne Tierversuche vollständig zu ersetzen (2) (3).
Diese Prüfmethode (ARE-Nrf2 Luciferase-Test) wird für den unter Nummer 2 erläuterten zweiten Schlüsselvorgang vorgeschlagen. Es wurde berichtet, dass Hautallergene Gene induzieren können, die durch das Antioxidant Response Element (ARE) reguliert werden (8) (9). Kleinmolekulare elektrophile Stoffe wie beispielsweise Hautallergene können auf das Sensorprotein Keap1 (Kelch-like ECH-associated protein 1) einwirken, beispielsweise durch kovalente Modifizierung seines Cysteinrests, was zu einer Dissoziation vom Transkriptionsfaktor Nrf2 (nuclear factor-erythroid 2-related factor2) führt. Das dissoziierte Nrf2 kann dann ARE-abhängige Gene wie beispielsweise jene, die Phase-II-Entgiftungsenzyme codieren, aktivieren (8) (10) (11).
Gegenwärtig ist der einzige In-vitro-ARE-Nrf2-Luciferase-Test, der durch diese Prüfmethode abgedeckt wird, der KeratinoSensTM-Test, für den Validierungsstudien abgeschlossen wurden (9) (12) (13), denen ein unabhängiger Peer-Review des Europäischen Referenzlabors für Alternativen zu Tierversuchen (European Union Reference Laboratory for Alternatives to Animal Testing, EURL ECVAM) folgte 14. Der KeratinoSensTM-Test wurde als aus wissenschaftlicher Sicht für die Anwendung als Teil eines IATA zulässig befunden, um die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren im Hinblick auf die Gefahreneinstufung und -kennzeichnung zu unterstützen (14). Labors, die die Prüfmethode durchführen möchten, können die im KeratinoSensTM-Test verwendete rekombinante Zelllinie durch Abschluss einer Lizenzvereinbarung mit dem Entwickler der Prüfmethode erhalten (15).
Definitionen sind Anlage 1 zu entnehmen.
Vorbemerkungen, Anwendbarkeit und Einsatzgrenzen
Da sich die Aktivierung der Keap1-Nrf2-ARE-Kette nur auf den zweiten Schlüsselvorgang des Hautsensibilisierungs-AOP bezieht, reichen Informationen aus Prüfmethoden auf der Grundlage der Aktivierung dieser Kette wahrscheinlich alleine nicht aus, um Schlussfolgerungen über das Hautsensibilisierungspotenzial von Chemikalien zu ziehen. Daher sollten die mit der gegenwärtigen Prüfmethode ermittelten Daten im Rahmen integrierter Ansätze, wie z.B. IATA, betrachtet und mit anderen ergänzenden Informationen, die beispielsweise aus In-vitro-Tests in Bezug auf andere Schlüsselvorgänge des Hautsensibilisierungs-AOP abgeleitet werden, sowie anderen Nicht-Prüfmethoden, einschließlich chemischer Analogien, kombiniert werden. Beispiele für die Verwendung der ARE-Nrf2-Luciferase-Prüfmethode in Kombination mit anderen Informationen werden in der Literatur beschrieben (13) (16) (17) (18) (19).
Diese Prüfmethode kann zur Unterstützung der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kategorie 1) und Nichtsensibilisatoren im Rahmen eines IATA eingesetzt werden. Diese Prüfmethode kann alleine weder zur Einstufung von Hautallergenen in die Unterkategorien 1A und 1B gemäß Definition in UN-GHS/CLP noch zur Vorhersage der Potenz im Rahmen von Sicherheitsbewertungsentscheidungen verwendet werden. Jedoch kann ein positives Ergebnis je nach Rechtsrahmen alleine zur Einstufung einer Chemikalie in die UN-GHS/CLP-Kategorie 1 herangezogen werden.
Die Daten aus der Validierungsstudie und den internen Prüfungen im Rahmen des unabhängigen Peer-Review der Prüfmethode haben ergeben, dass der KeratinoSensTM-Test an Labors mit Erfahrung auf dem Gebiet der Zellkultur übertragen werden kann. Der Grad der Reproduzierbarkeit, der bei der Prüfmethode erwartet werden kann, liegt in der Größenordnung von 85 % innerhalb und zwischen Labors (14). Die Genauigkeit (77 % -155/201), Empfindlichkeit (78 % - 71/91) und Spezifität (76 % - 84/110) des KeratinoSensTM-Tests für die Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kat. 1) und Nichtsensibilisatoren wurden unter Berücksichtigung aller Daten, die vom EURL ECVAM für die Bewertung und das Peer-Review der Prüfmethode vorgelegt wurden, im Vergleich zu den LLNA-Ergebnissen ermittelt (14). Diese Zahlen sind vergleichbar mit den Zahlen, die kürzlich basierend auf internen Prüfungen von ungefähr 145 Stoffen veröffentlicht wurden (Genauigkeit 77 %, Empfindlichkeit 79 %, Spezifität 72 %) (13). Beim KeratinoSensTM-Test ist die Wahrscheinlichkeit einer Unterschätzung bei Chemikalien mit geringer bis mittlerer Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1B) größer als bei Chemikalien mit hoher Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1A) (13) (14). Insgesamt deuten diese Informationen auf die Zweckmäßigkeit des KeratinoSensTM-Tests für die Erkennung der Gefahr einer Hautsensibilisierung hin. Jedoch sind die Genauigkeitswerte, die hier für den KeratinoSensTM-Test als eigenständiger Test angegeben werden, lediglich als Anhaltspunkte zu betrachten, da die Prüfmethode in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 9 oben betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA sowie andere Tierversuche die Situation bei der untersuchten Spezies, d. h. Menschen, nicht vollständig widerspiegeln.
Der Begriff "Prüfchemikalie" bezeichnet bei dieser Prüfmethode das, was getestet wird, und bezieht sich nicht auf die Anwendbarkeit der ARE-Nrf2-Luciferase-Prüfmethode hinsichtlich der Prüfung von Stoffen und/oder Gemischen. Auf der Grundlage der gegenwärtig verfügbaren Daten über den KeratinoSensTM-Test wurde nachgewiesen, dass der Test bei Prüfchemikalien, die eine Vielzahl an organischen Funktionsgruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzen (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (9) (12) (13) (14). Es wurden zwar hauptsächlich einkomponentige Stoffe getestet, jedoch liegen auch begrenzte Daten über die Prüfung von Gemischen vor (20). Die Prüfmethode ist dennoch für die Prüfung von mehrkomponentigen Stoffen und Gemischen technisch geeignet. Bevor diese Prüfmethode jedoch für die Generierung von Daten für einen bestimmten Regelungszweck verwendet wird, sollte geprüft werden, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefert, und wenn dem so ist, warum. Diese Überlegungen erübrigen sich, sofern die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist. Zudem sollte bei mehrkomponentigen Stoffen oder Gemischen die mögliche Interferenz der zytotoxischen Komponenten mit den beobachteten Reaktionen beachtet werden. Die Prüfmethode ist bei löslichen Prüfchemikalien oder solchen Prüfchemikalien anwendbar, die eine stabile Dispersion (d. h. ein Kolloid oder eine Suspension, worin sich die Prüfchemikalie nicht absetzen oder in anderen Phasen vom Lösungsmittel trennen kann) in Wasser oder DMSO (einschließlich aller technischen Komponenten im Falle der Prüfung eines mehrkomponentigen Stoffs oder Gemischs) bilden. Prüfchemikalien, die diese Bedingungen bei der höchsten erforderlichen Konzentration von 2.000 µM (siehe Nummer 22) nicht erfüllen, können trotzdem bei niedrigeren Konzentrationen geprüft werden. In einem solchen Fall könnten Ergebnisse, die die unter Nummer 39 beschriebenen Bedingungen für die Positivität erfüllen, dennoch unterstützend zur Identifizierung der Prüfchemikalie als Hautallergen herangezogen werden, während ein negatives Ergebnis, das bei Konzentrationen < 1.000 µM erzielt wird, als nicht aussagekräftig zu betrachten ist (siehe Vorhersagemodell unter Nummer 39). Im Allgemeinen wurden Stoffe mit einem LogP-Wert bis 5 erfolgreich geprüft, während extrem hydrophobe Stoffe mit LogP > 7 außerhalb der bekannten Anwendbarkeit der Prüfmethode liegen (14). Für Stoffe mit einem LogP-Wert zwischen 5 und 7 liegen nur beschränkte Informationen vor.
Negative Ergebnisse sind mit Vorsicht auszulegen, da Stoffe mit ausschließlicher Reaktivität gegenüber Lysinresten durch die Prüfmethode als negativ erkannt werden können. Außerdem können Prohaptene (d. h. Chemikalien, die beispielsweise über P450-Enzyme aktiviert werden müssen) und Prähaptene (d. h. Chemikalien, die durch Selbstoxidation aktiviert werden) aufgrund der begrenzten Stoffwechselfähigkeit der verwendeten Zelllinie (21) sowie der Versuchsbedingungen, insbesondere bei geringer Oxidationsgeschwindigkeit, zu negativen Ergebnissen führen. Andererseits können Prüfchemikalien, die zwar nicht als Allergene, aber dennoch als chemische Stressoren wirken, zu falschen positiven Ergebnissen führen (14). Zudem lassen sich hochgradig zytotoxische Prüfchemikalien nicht immer zuverlässig bewerten. Schließlich können Prüfchemikalien, die mit dem Luciferase-Enzym interferieren, die Aktivität der Luciferase in zellbasierten Tests stören, was entweder zu scheinbarer Hemmung oder verstärkter Lumineszenz führt (22). Beispielweise wurde berichtet, dass Phytoöstrogenkonzentrationen > 1 µM die Lumineszenzsignale in anderen auf Luciferase basierenden Reporter-Gen-Tests aufgrund der Überaktivierung des Luciferase-Reporter-Gens stören (23). Infolgedessen muss die Luciferase-Expression, die bei hohen Konzentrationen von Phytoöstrogenen oder ähnlichen Chemikalien, die vermutlich eine mit Phytoöstrogen vergleichbare Überaktivierung des Luciferase-Reporter-Gens bewirken, sorgfältig untersucht werden (23). In Fällen, in denen die Nichtanwendbarkeit der Prüfmethode bei anderen spezifischen Kategorien von Prüfchemikalien nachgewiesen werden kann, sollte die Prüfmethode bei diesen spezifischen Kategorien nicht verwendet werden.
Abgesehen von der Unterstützung bei der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren liefert der KeratinoSensTM-Test auch Konzentrations-/Wirkungs-Informationen, die potenziell zur Bewertung der Sensibilisierungspotenz bei Verwendung in integrierten Ansätzen wie beispielsweise IATA beitragen können (19). Darüber hinaus sind weitere Untersuchungen, vorzugsweise auf der Grundlage verlässlicher Humandaten, notwendig, um herauszufinden, inwieweit die Ergebnisse des KeratinoSensTM-Tests zur Potenzbewertung (24) und Einstufung von Allergenen in Unterkategorien gemäß UN-GHS/CLP beitragen können.
Testprinzip
Die ARE-Nrf2-Luciferase-Prüfmethode basiert auf einer immortalisierten, adhärenten Zelllinie, die aus humanen und mit einem wählbaren Plasmid stabil transfizierten HaCaT-Keratinozyten abgeleitet wurde. Die Zelllinie enthält das Luciferase-Gen unter der transkriptionalen Kontrolle eines konstitutiven Promoters, verschmolzen mit einem ARE-Element aus einem Gen, das bekanntermaßen durch Kontaktsensibilisatoren hochreguliert wird (25) (26). Das Luciferase-Signal spiegelt die Aktivierung durch Sensibilisatoren der endogenen Nrf2-abhängigen Gene wider, wobei die Abhängigkeit des Luciferase-Signals in der rekombinanten Zelllinie von Nrf2 nachgewiesen wurde (27). Dies ermöglicht die quantitative Messung (durch Lumineszenzerkennung) der Luciferase-Geninduktion unter Verwendung von bekannten lichterzeugenden Luciferase-Substraten als Indikator für die Aktivität des Nrf2-Transkriptionsfaktors in Zellen nach Exposition gegenüber elektrophilen Stoffen.
Prüfchemikalien werden im KeratinoSensTM-Test als positiv angesehen, wenn sie eine statistisch signifikante Induktion der Luciferase-Aktivität oberhalb einer gegebenen Schwelle (d. h. > 1,5-facher Wert oder Anstieg um 50 %) bewirken, und zwar unterhalb einer festgelegten Konzentration, die sich nicht signifikant auf die Zellviabilität auswirkt (d. h. unter 1.000 µM und bei einer Konzentration, bei der die Zellviabilität mehr als 70 % beträgt (9) (12)). Zu diesem Zweck wird die maximal-fache Induktion der Luciferase-Aktivität über eine (Negativ-)Lösungsmittel-Kontrolle (Imax) bestimmt. Da die Zellen ferner verschiedenen Konzentrationen der Prüfchemikalien ausgesetzt werden, sollte die erforderliche Konzentration für eine statistisch signifikante Induktion der Luciferase-Aktivität oberhalb der Schwelle (d. h. EC1.5-Wert) aus der Dosis-Wirkungs-Kurve interpoliert werden (für Berechnungen siehe Nummer 32). Schließlich sollten parallele zytotoxische Messungen durchgeführt werden, um zu bewerten, ob die Induktionswerte der Luciferase-Aktivität bei subzytotoxischen Konzentrationen auftreten.
Vor der routinemäßigen Anwendung des ARE-Nrf2-Luciferase-Tests, der den Anforderungen der vorliegenden Prüfmethode genügt, sollte die technische Leistungsfähigkeit der Labors anhand der in Anlage 2 aufgeführten zehn Leistungsstoffe nachgewiesen werden.
Es stehen Leistungsnormen (28) zur Verfügung, die die Validierung neuer oder geänderter In-vitro-ARE-Nrf2-Luciferase-Prüfmethoden ähnlich dem KeratinoSensTM-Test sowie die rechtzeitige Anpassung dieser Prüfmethoden für deren Einbeziehung ermöglichen. Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird nur für Prüfmethoden garantiert, die gemäß diesen Leistungsnormen validiert wurden, sofern diese Prüfmethoden von der OECD überprüft und in die entsprechende Prüfrichtlinie aufgenommen wurden.
Verfahren
Gegenwärtig ist die einzige unter diese Prüfmethode fallende Methode der wissenschaftlich validierte KeratinoSensTM-Test (9) (12) (13) (14). Es liegen Standardarbeitsanweisungen für den KeratinoSensTM-Test vor, die bei der Umsetzung und Verwendung dieser Prüfmethode im Labor angewendet werden sollten (15). Labors, die die Prüfmethode durchführen möchten, können die im KeratinoSensTM-Test verwendete rekombinante Zelllinie durch Abschluss einer Lizenzvereinbarung mit dem Entwickler der Prüfmethode erhalten. Nachfolgend werden die Hauptkomponenten und Verfahren der ARE-Nrf2-Luciferase-Prüfmethode beschrieben.
Vorbereitung der Keratinozytenkulturen
Es sollte eine transgene Zelllinie mit einer stabilen Insertion des Luciferase-Reporter-Gens unter der Kontrolle des ARE-Elements verwendet werden (z.B. KeratinoSensTM-Zelllinie). Bei Erhalt werden die Zellen propagiert (z.B. 2 bis 4 Passagen) und als homogener Stamm eingefroren aufbewahrt. Zellen aus diesem ursprünglichen Stamm können bis zu einer Höchstzahl von Passagen (z.B. 25 im Fall von KeratinoSensTM) propagiert und für routinemäßige Tests unter Verwendung des geeigneten Trägermediums eingesetzt werden (im Fall von KeratinoSensTM ist dies serum- und Geneticin-haltiges DMEM).
Für die Prüfung sollten die Zellen zu 80 bis 90 % konfluent sein, und es sollte darauf geachtet werden, dass die Zellen nicht zu vollständiger Konfluenz zusammengewachsen sind. Einen Tag vor der Prüfung werden die Zellen gewonnen und in 96-Mulden-Platten (10.000 Zellen/Mulde im Fall von KeratinoSensTM) verteilt. Eine Sedimentation der Zellen bei der Beimpfung ist zu vermeiden, um eine homogene quantitative Verteilung der Zellen in den Mulden zu gewährleisten. Ist dies nicht der Fall, kann dieser Schritt zu einer hohen Variabilität zwischen den Mulden führen. Bei jeder Wiederholung werden drei Replikate für die Messungen der Luciferase-Aktivität und ein paralleles Replikat für den Zellviabilitätstest verwendet.
Vorbereitung der Prüfchemikalie und Kontrollstoffe
Die Prüfchemikalie sowie die Kontrollstoffe werden am Tag der Prüfung vorbereitet. Für den KeratinoSensTM-Test werden die Prüfchemikalien in Dimethylsulfoxid (DMSO) bis zur gewünschten Endkonzentration gelöst (z.B. 200 mM). Die DMSO-Lösungen können als selbststerilisierend angesehen werden, sodass keine sterile Filtration erforderlich ist. Prüfchemikalien, die nicht in DMSO löslich sind, werden in sterilem Wasser oder Kulturmedium gelöst und die Lösungen beispielsweise durch Filtration sterilisiert. Bei einer Prüfchemikalie ohne festgelegtes Molekulargewicht wird im KeratinoSensTM-Test eine Stammlösung mit einer Standardkonzentration hergestellt (40 mg/ml oder 4 % (w/v)). Falls andere Lösungsmittel als DMSO, Wasser oder Kulturmedium zum Einsatz kommen, sollten ausreichende wissenschaftliche Gründe vorgelegt werden.
Basierend auf den DMSO-Stammlösungen der Prüfchemikalie werden Verdünnungsreihen unter Verwendung von DMSO hergestellt, um 12 Hauptkonzentrationen der zu prüfenden Chemikalie zu erhalten (von 0,098 bis 200 mM im KeratinoSensTM-Test). Bei einer nicht in DMSO löslichen Prüfchemikalie werden die Verdünnungen zum Erhalt der Hauptkonzentrationen unter Verwendung von sterilem Wasser oder Kulturmedium hergestellt. Unabhängig vom verwendeten Lösungsmittel werden die Hauptkonzentrationen dann weiter 25-fach im serumhaltigen Kulturmedium verdünnt und schließlich für die Behandlung mit einem weiteren 4-fachen Verdünnungsfaktor verwendet, sodass die Endkonzentrationen der geprüften Chemikalie im KeratinoSensTM-Test zwischen 0,98 und 2.000 µM liegen. Andere Konzentrationen kÆnnen verwendet werden, sofern dies gerechtfertigt ist (z.B. bei Zytotoxizität oder schlechter Löslichkeit).
Die Negativ-(Lösungsmittel-)Kontrolle, die im KeratinoSensTM-Test verwendet wird, ist DMSO (CAS Nr. 67-68-5, > 99 % Reinheit), wobei sechs Mulden pro Platte vorbereitet werden. Dabei werden die gleichen Verdünnungen wie für die Hauptkonzentrationen unter Nummer 22 hergestellt, sodass die Endkonzentration der Negativ-(Lösungsmittel-)Kontrolle 1 % beträgt. Diese Konzentration wirkt sich bekanntermaßen nicht auf die Zellviabilität aus und entspricht der DMSO-Konzentration in der geprüften Chemikalie und der Positivkontrolle. Bei einer nicht in DSMO löslichen Chemikalie, bei der Verdünnungen in Wasser hergestellt wurden, muss der DMSO-Wert in allen Mulden der endgültigen Prüflösung wie bei den anderen Prüfchemikalien und Kontrollstoffen an 1 % angepasst werden.
Die Positivkontrolle, die im Fall des KeratinoSensTM-Tests verwendet ist, ist Zimtaldehyd (CAS Nr. 14371-10-9, > 98 % Reinheit), wobei eine Reihe von 5 Hauptkonzentrationen zwischen 0,4 und 6,4 mM in DMSO (ausgehend von einer Stammlösung mit 6,4 mM) hergestellt und wie für die Hauptkonzentrationen unter Nummer 22 beschrieben verdünnt wird, sodass die Endkonzentrationen der Positivkontrolle zwischen 4 und 64 µM liegen. Andere geeignete Positivkontrollen, vorzugsweise mit EC1,5-Werten im mittleren Bereich, kÆnnen verwendet werden, sofern historische Daten vorliegen, aus denen vergleichbare Versuchsakzeptanzkriterien abgeleitet werden können.
Applikation der Prüfchemikalie und Kontrollstoffe
Für jede Prüfchemikalie und jeden Positivkontrollstoff ist ein Versuch erforderlich, um eine Vorhersage (positiv oder negativ), bestehend aus mindestens zwei unabhängigen Wiederholungen mit je drei Replikaten (d. h. n = 6), abzuleiten. Weichen die Ergebnisse zwischen den beiden unabhängigen Wiederholungen ab, sollte eine dritte Wiederholung mit drei Replikaten (d. h. n = 9) durchgeführt werden. Jede unabhängige Wiederholung wird an einem anderen Tag mit einer frischen Stammlösung der Prüfchemikalie und unabhängig voneinander gewonnenen Zellen vorgenommen. Die Zellen können jedoch aus derselben Passage stammen.
Nach der Beimpfung wie unter Nummer 20 beschrieben lässt man die Zellen 24 Stunden in den 96-Mulden-Mikrotiterplatten wachsen. Das Medium wird dann entfernt und durch frisches Kulturmedium (150 µl serumhaltiges Kulturmedium, jedoch ohne Geneticin im Fall von KeratinoSensTM) ersetzt, dem 50 µl der25-fach verdünnten Prüfchemikalie und Kontrollstoffe zugegeben werden. Mindestens eine Mulde pro Platte sollte zur Bewertung der Background-Werte leer gelassen werden (ohne Zellen und Behandlung).
Die behandelten Platten werden im KeratinoSensTM-Test ungefähr 48 Stunden bei 37±1 1 °C in der Gegenwart von 5 % CO2 inkubiert. Es ist darauf zu achten, dass eine Verdunstung der flüchtigen Prüfchemikalien sowie eine Kreuzkontamination zwischen Mulden durch die Prüfchemikalien vermieden wird, indem die Platten beispielsweise vor der Inkubation mit den Prüfchemikalien mit einer Folie abgedeckt werden.
Messungen der Luciferase-Aktivität
Zur Gewährleistung angemessener Lumineszenz-Messwerte sind drei Faktoren entscheidend:
- Wahl eines empfindlichen Luminometers
- Verwendung eines Plattenformats mit ausreichender Höhe zur Vermeidung einer Kreuzkontamination
- Verwendung eines Luciferase-Substrats mit ausreichender Lichtausbeute zur Gewährleistung einer hinreichenden Empfindlichkeit und geringen Variabilität.
Vor der Prüfung sollte ein Kontrollversuch wie in Anlage 3 beschrieben durchgeführt werden, um sicherzustellen, dass die drei genannten Bedingungen erfüllt sind.
Nach einer Expositionsdauer von 48 Stunden mit der Prüfchemikalie und den Kontrollstoffen im KeratinoSensTM-Test werden die Zellen mit einer phosphatgepufferten Kochsalzlösung gewaschen. Ferner wird jeder Mulde der relevante Lysepuffer für Lumineszenzmessungen 20 Minuten bei Zimmertemperatur hinzugefügt.
Die Platten mit dem Zelllysat werden dann zur Messung in das Luminometer gestellt, das im KeratinoSensTM-Test wie folgt programmiert wird: i) Hinzufügen des Luciferase-Substrats zu jeder Mulde (d. h. 50 µl), ii) Abwarten von 1 Sekunde und iii) Integration der Luciferase-Aktivit´t für2 Sekunden. Bei Verwendung anderer Einstellungen, z.B. je nach verwendetem Luminometermodell, sollten diese begründet werden. Darüber hinaus kann auch ein Glimmsubstrat verwendet werden, sofern der in Anlage 3 beschriebene Qualitätssicherungsversuch erfolgreich durchgeführt wird.
Bewertung der Zytotoxizität
Für den KeratinoSensTM-Zellviabilitätstest wird das Medium nach der Expositionsdauer von 48 Stunden durch frisches Medium ersetzt. Dieses Medium enthält MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolylblau Tetrazoliumbromid; CAS Nr. 298-93-1) und Zellen, die 4 Stunden bei 37 1 °C in der Gegenwart von 5 % CO2 inkubiert wurden. Das MTT-Medium wird dann entfernt und die Zellen werden über Nacht lysiert (z.B. durch Hinzufügen von 10 %iger SDS-Lösung zu jeder Mulde). Nach dem Schütteln wird die Absorption bei 600 nm mit einem Photometer gemessen.
Daten und Berichterstattung
Datenauswertung
Die folgenden Parameter werden im KeratinoSensTM-Test berechnet:
- durchschnittliche maximal-fache Induktion der Luciferase-Aktivität (Imax), die bei jeder Konzentration der geprüften Chemikalie und Positivkontrolle beobachtet wurde;
- EC1,5-Wert entsprechend der Konzentration, bei der die Induktion der Luciferase-Aktivität über der 1,5-fachen Schwelle liegt (d. h. um 50 % verstärkte Luciferase-Aktivität wurde erzielt); und
- IC50- und IC30-Konzentrationswerte für eine Verringerung der Zellviabilität um 50 % und 30 %.
- Die n-fache Induktion der Luciferase-Aktivität wird durch Gleichung 1 und die allgemeine maximal-fache Induktion (Imax) als Durchschnitt der einzelnen Wiederholungen berechnet.
Gleichung 1:
| (LProbe - LBlindversuch) | |
| n-fache Induktion = |
|
| (LLösungsmittel - LBlindversuch) |
Dabei sind:
LProbe: Lumineszenz-Messwert in der Prüfchemikalien-Mulde
LLeer: Lumineszenz-Messwert in der leeren Mulde (ohne Zellen und Behandlung)
LLösungsmittel: durchschnittlicher Lumineszenz-Messwert in den Mulden, die Zellen und (Negativ-)Lösungsmittel-Kontrolle enthalten
EC1,5 wird durch lineare Interpolation anhand von Gleichung 2 und der EC1,5-Gesamtwert als geometrisches Mittel der einzelnen Wiederholungen berechnet.
Gleichung 2:
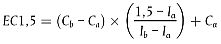
Dabei sind:
Ca: niedrigste Konzentration in µM bei > 1,5-facher Induktion
Cb: höchste Konzentration in µM bei < 1,5-facher Induktion
Ia: n-fache Induktion, gemessen bei der niedrigsten Konzentration bei > 1,5-facher Induktion (Mittel von drei Replikat-Mulden)
Ib: n-fache Induktion, gemessen bei der höchsten Konzentration bei < 1,5-facher Induktion (Mittel von drei Replikat-Mulden)
Die Viabilität wird durch Gleichung 3 berechnet:
Gleichung 3:
| (VProbe - VBlindversuch) | ||
| Viabilität = |
| x 100 |
| VLösungsmittel - VBlindversuch) |
Dabei sind:
VProbe: MTT-Absorptions-Messwert in der Prüfchemikalien-Mulde
Vleer: MTT-Absorptions-Messwert in der leeren Mulde (ohne Zellen und Behandlung)
VLösungsmittel: durchschnittlicher MTT-Absorptions-Messwert in den Mulden, die Zellen und (Negativ-)Lösungsmittel-Kontrolle enthalten
IC50 und IC30 werden durch lineare Interpolation anhand von Gleichung 4 und die IC50- und IC30-Gesamtwerte als geometrisches Mittel der einzelnen Wiederholungen berechnet.
Gleichung 4:
Dabei sind:
X: prozentuale Verringerung bei der zu berechnenden Konzentration (50 und 30 bei IC50 und IC30)
Ca: niedrigste Konzentration in µM bei > x % Verringerung der Viabilität
Cb: höchste Konzentration in µM bei < x % Verringerung der Viabilität
Va: prozentuale Viabilität bei der niedrigsten Konzentration bei > x % Verringerung der Viabilität
Vb: prozentuale Viabilität bei der höchsten Konzentration bei < x % Verringerung der Viabilität
Für jede Konzentration, bei der die Induktion der Luciferase-Aktivität mehr als das 1,5-fache beträgt, wird die statistische Signifikanz berechnet (z.B. durch einen zweiseitigen Student-t-Test), wobei die Lumineszenzwerte für die drei Replikate mit den Lumineszenzwerten in den Mulden der (Negativ-)Lösungsmittel-Kontrolle verglichen werden, um zu ermitteln, ob die Induktion der Luciferase-Aktivität statistisch signifikant ist (p < 0,05). Die niedrigste Konzentration, bei der die Induktion der Luciferase-Aktivität mehr als das 1,5-fache beträgt, ist der Wert zur Bestimmung des EC1,5-Werts. Es wird in jedem Fall geprüft, ob dieser Wert unter dem IC30-Wert liegt, was darauf hindeutet, dass die Verringerung der Zellviabilität bei der bestimmenden Konzentration für den EC1,5-Wert weniger als 30 % beträgt.
Es wird empfohlen, die Daten anhand von Diagrammen visuell zu überprüfen. Wenn keine eindeutige Dosis-Wirkungs-Kurve beobachtet wird oder wenn die ermittelte Dosis-Wirkungs-Kurve pfadspezifisch ist (d. h. die Schwelle von 1,5 zweimal überschritten wird), sollte der Versuch wiederholt werden, um zu prüfen, ob dies für die Prüfchemikalie spezifisch oder auf einen Versuchsartefakt zurückzuführen ist. Falls die biphasige Wirkung in einem unabhängigen Versuch reproduziert werden kann, ist der niedrigere EC1,5-Wert (Konzentration, bei der die Schwelle von 1,5 erstmalig überschritten wird) anzugeben.
In den seltenen Fällen, in denen eine statistisch nicht signifikante Induktion über dem 1,5-fachen gefolgt von einer höheren Konzentration mit einer statistisch signifikanten Induktion beobachtet wird, werden die Ergebnisse aus dieser Wiederholung nur dann als gültig und positiv angesehen, wenn die statistisch signifikante Induktion über der Schwelle von 1,5 bei einer nicht zytotoxischen Konzentration ermittelt wurde.
Bei Prüfchemikalien, die bereits bei der niedrigsten Testkonzentration von 0,98 µM eine 1,5-fache oder höhere Induktion erzeugen, wird der EC1,5-Wert < 0,98 basierend auf der visuellen Überprüfung der Dosis-Wirkungs-Kurve festgelegt.
Akzeptanzkriterien
Bei der Verwendung des KeratinoSensTM-Tests sollten die folgenden Akzeptanzkriterien erfüllt werden. Erstens sollte die Induktion der Luciferase-Aktivität, die bei der Positivkontrolle (Zimtaldehyd) erzeugt wird, bei mindestens einer der geprüften Konzentrationen (4 bis 64 µM) statistisch signifikant über der Schwelle von 1,5 (z.B. bei Verwendung eines T-Tests) liegen.
Zweitens sollte der EC1,5-Wert innerhalb von zwei Standardabweichungen vom historischen Mittelwert der Prüfanstalt liegen (z.B. zwischen 7 µM und 30 µM basierend auf dem Validierungsdatensatz), der regelmäßig aktualisiert werden sollte. Darüber hinaus sollte die durchschnittliche Induktion in den drei Replikaten für Zimtaldehyd bei 64 µM zwischen 2 und 8 liegen. Ist letzteres Kriterium nicht erfüllt, sollte die Dosis-Wirkung von Zimtaldehyd sorgfältig überprüft werden. Tests sind nur dann akzeptabel, wenn die Dosis-Wirkungs-Beziehung eindeutig ist, wobei die Induktion der Luciferase-Aktivität bei zunehmenden Konzentrationen in der Positivkontrolle ansteigt.
Schließlich sollte der durchschnittliche Variationskoeffizient des Lumineszenz-Messwerts für die Negativ-(Lösungsmittel-)Kontrolle (DMSO) bei jeder Wiederholung, bestehend aus 6 dreifach geprüften Mulden, unter 20 % liegen. Liegt die Variabilität über diesem Wert, sollten die Ergebnisse ignoriert werden.
Interpretation der Ergebnisse und Vorhersagemodell
Eine KeratinoSensTM-Vorhersage wird als positiv betrachtet, wenn die folgenden vier Bedingungen alle bei 2 von 2 oder bei denselben 2 von 3 Wiederholungen erfüllt sind. Andernfalls wird die KeratinoSensTM-Vorhersage als negativ erachtet (Abbildung 1):
- Imax ist größer als (>) der 1,5-fache Wert und statistisch signifikant abweichend im Vergleich zur (Negativ-)Lösungsmittel-Kontrolle (wie durch einen zweiseitigen, unpaarigen Student-T-Test bestimmt);
- die Zellviabilität ist größer als (>) 70 % der niedrigsten Konzentration mit Induktion von Luciferase-Aktivität über dem 1,5-fachen Wert (d. h. bei bestimmender Konzentration für den EC1,5-Wert);
- der EC1,5-Wert ist kleiner als (<) 1.000 µM (oder < 200 µg/ml bei Prüfchemikalien ohne festgelegtes Molekulargewicht);
- es besteht eine augenscheinliche allgemeine Dosis-Wirkungs-Beziehung für die Induktion der Luciferase-Aktivität (oder eine biphasige Wirkung wie unter Nummer 33 erwähnt).
Wenn bei einer gegebenen Wiederholung die ersten drei Bedingungen erfüllt sind, jedoch keine eindeutige Dosis-Wirkungs-Beziehung für die Induktion der Luciferase-Aktivität beobachtet werden kann, ist das Ergebnis dieser Wiederholung als nicht aussagekräftig anzusehen, sodass eventuell weitere Prüfungen erforderlich sind (Abbildung 1). Zudem ist ein negatives Ergebnis bei Konzentrationen < 1.000 µM (oder < 200 µg/ml bei Prüfchemikalien ohne festgelegtes Molekulargewicht) ebenfalls als nicht aussagekräftig zu betrachten (siehe Nummer 11).
Abbildung 1 Vorhersagemodell des KeratinoSensTM-Tests. Eine KeratinoSensTM-Vorhersage sollte im Rahmen eines IATA sowie gemäß den Nummern 9 und 11 in Betracht gezogen werden
In seltenen Fällen können Prüfchemikalien, bei denen die Induktion der Luciferase-Aktivität sehr nahe bei zytotoxischen Werten erfolgt, in einigen Wiederholungen bei nicht zytotoxischen Werten (d. h. bestimmende Konzentration für den EC1,5-Wert unter (<) IC30) und in anderen Wiederholungen nur bei zytotoxischen Werten (d. h. bestimmende Konzentration für den EC1,5-Wert größer als (>) IC30) positiv sein. Solche Prüfchemikalien sind erneut im Rahmen einer sehr genauen Dosis-Wirkungs-Analyse unter Verwendung eines geringeren Verdünnungsfaktors (z.B. 1,33 oder Ö2 (= 1,41)-fache Verdünnung zwischen Mulden) zu prüfen, um festzustellen, ob die Induktion bei zytotoxischen Werten aufgetreten ist oder nicht (9).
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalie
- Einkomponentiger Stoff:
- chemische Bezeichnung, wie z.B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer (n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
- physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar.
- Mehrkomponentiger Stoff, UVCB-Stoff und Gemisch:
- Charakterisierung, so weit wie möglich, z.B. durch die chemische Zusammensetzung (siehe oben), Reinheit, das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften (siehe oben) der einzelnen Komponenten, soweit verfügbar;
- physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar;
- Molekulargewicht oder scheinbares Molekulargewicht im Fall von Gemischen/Polymeren mit bekannter Zusammensetzung oder andere für die Durchführung der Studie relevante Informationen;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar.
Kontrollen
- Positivkontrolle:
- Chemische Bezeichnung, wie z.B. IUPAC- oder CAS Bezeichnung(en), CAS-Nummer (n), SMILES- oder InChI-Code, Strukturformel und/oder andere Kennungen;
- physikalisches Erscheinungsbild, Löslichkeit in Wasser, Löslichkeit in DMSO, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, sofern verfügbar und falls zutreffend;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- Behandlung vor der Testung, soweit zutreffend (z.B. Erwärmung, Zerkleinerung);
- geprüfte Konzentration(en);
- Lagerbedingungen und Stabilität, soweit verfügbar;
- ggf. Verweis auf historische Ergebnisse von Positivkontrollen, die geeignete Akzeptanzkriterien für einen Testdurchlauf dokumentieren.
- Negativ-(Vehikel-)Kontrolle:
- Chemische Kennung, wie beispielsweise IUPAC- oder CAS-Bezeichnung(en), CAS-Nummer (n) und/oder sonstige Kennungen;
- Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- physikalisches Erscheinungsbild, Molekulargewicht und weitere relevante physikalisch-chemische Eigenschaften, falls andere als die in dieser Prüfmethode genannten Negativkontrollen/Vehikel verwendet werden, und sofern verfügbar;
- Lagerbedingungen und Stabilität, soweit verfügbar;
- Begründung der Auswahl des Lösungsmittels für jede Prüfchemikalie.
Prüfbedingungen
- Name und Anschrift des Auftraggebers, der Prüfanstalt und des Studienleiters;
- Beschreibung der angewandten Prüfmethode;
- verwendete Zelllinie, zugehörige Lagerbedingungen und Quelle (z.B. Einrichtung, von der sie gewonnen wurden);
- Passagenzahl und Konfluenzwert der geprüften Zellen;
- angewandte Zellzählungsmethode bei der Beimpfung vor der Prüfung und ergriffene Maßnahmen zur Gewährleistung einer homogenen quantitativen Verteilung der Zellen (siehe Nummer 20);
- verwendetes Luminometer (z.B. Modell), einschließlich Geräteeinstellungen, verwendetes Luciferase-Substrat und Nachweis geeigneter Lumineszenzmessungen basierend auf der in Anlage 3 beschriebenen Kontrollprüfung;
- angewandtes Verfahren zum Nachweis der Leistungsfähigkeit des Labors hinsichtlich der Durchführung der Prüfmethode (z.B. durch Prüfen von Leistungsstoffen) oder zum Nachweis der reproduzierbaren Durchführung der Prüfmethode im Zeitverlauf.
Prüfverfahren
- Anzahl der Wiederholungen und verwendeten Replikate
- Konzentrationen der Prüfchemikalie, Applikationsverfahren und angewandte Expositionsdauer (falls von den Empfehlungen abweichend)
- Beschreibung der angewandten Bewertungs- und Entscheidungskriterien;
- Beschreibung der angewandten Studienakzeptanzkriterien;
- Beschreibung etwaiger Änderungen am Testverfahren.
Ergebnisse
- Tabellarische Darstellung der Imax-, EC1,5- und Viabilitätswerte (d. h. IC50, IC30), die für die Prüfchemikalie und die Positivkontrolle bei jeder Wiederholung ermittelt wurden, sowie der Mittelwerte (Imax: Durchschnitt; EC1,5- und Viabilitätswerte: geometrisches Mittel) und Standardabweichung, die anhand der Daten aus allen einzelnen Wiederholungen berechnet wurden, und Angabe der Einstufung der Prüfchemikalie gemäß dem Vorhersagemodell;
- ermittelter Variationskoeffizient bei den Lumineszenz-Messwerten für die Negativ-Kontrolle bei jedem Versuch;
- Diagramm zur Darstellung der Dosis-Wirkungs-Kurven für die Induktion der Luciferase-Aktivität und Viabilität;
- Beschreibung etwaiger sonstiger relevanter Beobachtungen, falls zutreffend.
Erörterung der Ergebnisse
- Erörterung der Ergebnisse aus dem KeratinoSensTM-Test;
- Analyse der Prüfergebnisse im Rahmen eines IATA, sofern sonstige relevante Informationen vorliegen.
Schlussfolgerung
Literaturhinweise
(1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), fünfte überarbeitete Ausgabe, UN New York und Genf, 2013. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html.
(2) OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 168. OECD, Paris.
(3) Adler S., Basketter D., Creton S., Pelkonen O., van Benthem J., Zuang V., Andersen K.E., Angers-Loustau A., Aptula A., Bal-Price A., Benfenati E., Bernauer U., Bessems J., Bois F.Y., Boobis A., Brandon E., Bremer S., Broschard T., Casati S., Coecke S., Corvi R., Cronin M., Daston G., Dekant W., Felter S., Grignard E., Gundert-Remy U., Heinonen T., Kimber I., Kleinjans J., Komulainen H., Kreiling R., Kreysa J., Leite S.B., Loizou G., Maxwell G., Mazzatorta P., Munn S., Pfuhler S., Phrakonkham P., Piersma A., Poth A., Prieto P., Repetto G., Rogiers V., Schoeters G., Schwarz M., Serafimova R., Tähti H., Testai E., van Delft J., van Loveren H., Vinken M., Worth A., Zaldivar J.M. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Archives of Toxicology 85, 367-485.
(4) Kapitel B.42 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest.
(5) Kapitel B.6 dieses Anhangs: Sensibilisierung der Haut.
(6) Kapitel B.50 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest: DA.
(7) Kapitel B.51 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest: BrdU-ELISA.
(8) Natsch A. (2010). The Nrf2-Keap1-ARE Toxicity Pathway as a Cellular Sensor for Skin Sensitizers-Functional Relevance and Hypothesis on Innate Reactions to Skin Sensitizers. Toxicological Sciences 113, 284-292.
(9) Emter R., Ellis G., Natsch A. (2010). Performance of a novel keratinocyte-based reporter cell line to screen skin sensitizers in vitro. Toxicology and Applied Pharmacology 245, 281-290.
(10) Dinkova-Kostova A.T., Holtzclaw W.D., Kensler T.W. (2005). The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 18, 1779-1791.
(11) Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. (2013). The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 1(1), 45-49.
(12) Natsch A., Bauch C., Foertsch L., Gerberick F., Normann K., Hilberer A., Inglis H., Landsiedel R., Onken S., Reuter H., Schepky A., Emter R. (2011). The intra- and inter-laboratory reproducibility and predictivity of the KeratinoSens assay to predict skin sensitizers in vitro: results of a ring-study in five laboratories. Toxicol. In Vitro 25, 733-744.
(13) Natsch A., Ryan C.A., Foertsch L., Emter R., Jaworska J., Gerberick G.F., Kern P. (2013). A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. Journal of Applied Toxicology, 33, 1337-1352.
(14) EURL-ECVAM (2014). Recommendation on the KeratinoSensTM assay for skin sensitisation testing, 42 ff. Verfügbar unter: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations/recommendation-keratinosens-skin-sensitisation.
(15) DB-ALM (INVITTOX) (2013) Protocol 155: KeratinoSensTM., 17 ff. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methods AndProtocols/index
(16) Natsch A., Emter R., Ellis G. (2009). Filling the concept with data: integrating data from different in vitro and in silico assays on skin sensitizers to explore the battery approach for animal-free skin sensitization testing. Toxicol. Sci. 107, 106-121.
(17) Ball N., Cagen S., Carrillo J.C., Certa H., Eigler D., Emter R., Faulhammer F., Garcia C., Graham C., Haux C., Kolle S.N., Kreiling R., Natsch A., Mehling A. (2011). Evaluating the sensitization potential of surfactants: integrating data from the local lymph node assay, guinea pig maximization test, and in vitro methods in a weight-of-evidence approach. Regul. Toxicol. Pharmacol., 60, 389-400.
(18) Bauch C., Kolle S.N., Ramirez T., Eltze T., Fabian E., Mehling A., Teubner W., van Ravenzwaay B., Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potentials. Regul. Toxicol. Pharmacol., 63, 489-504.
(19) Jaworska J., Dancik Y., Kern P., Gerberick F., Natsch A. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: from theory to practice. J Appl Toxicol. 33, 1353-1364.
(20) Andres E., Sa-Rocha V.M., Barrichello C., Haupt T., Ellis G., Natsch A. (2013). The sensitivity of the KeratinoSensTM assay to evaluate plant extracts: A pilot study. in Toxicology in Vitro, 27, 1220-1225.
(21) Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1683-1969.
(22) Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology. Chemistry and Biology 17, 646-657.
(23) OECD (2012). BG1Luc Estrogen Receptor Transactivation Test Method for Identifying Estrogen Receptor Agonists and Antagonists. OECD Guidelines for Chemical Testing No. 457. OECD, Paris.
(24) ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology and Toxicology of Chemicals (Technical Report Nr. 87).
(25) Gildea L.A., Ryan C.A., Foertsch L.M., Kennedy J.M., Dearman R.J., Kimber I., Gerberick G.F. (2006). Identification of gene expression changes induced by chemical allergens in dendritic cells: opportunities for skin sensitization testing. J. Invest. Dermatol., 126, 1813-1822.
(26) Ryan C.A., Gildea L.A., Hulette B.C., Dearman R.J., Kimber I., Gerberick G.F. (2004). Gene expressing changes in peripheral blood-derived dendritic cells following exposure to a contact allergen. Toxicol. Lett. 150, 301-316.
(27) Emter R., van der Veen J.W., Adamson G., Ezendam J., van Loveren H., Natsch A. (2013). Gene expression changes induced by skin sensitizers in the KeratinoSensTM cell line: Discriminating Nrf2-dependent and Nrf2-independent events. Toxicol. in vitro 27, 2225-2232.
(28) OECD (2015). Performance Standards for assessment of proposed similar or modified in vitro skin sensitisation ARE-Nrf2 luciferase test methods. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 213, OECD, Paris.
(29) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 34. OECD, Paris, Frankreich.
(30) NAFTA (North American Free Trade Agreement) (2012). Technical Working Group on Pesticides - (Quantitative) Structure Activity Relationship ((Q)SAR) Guidance Document. 186 S. http://www.epa.gov/oppfead1/international/naftatwg/guidance/qsar-guidance.pdf
_____
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
.
| Anlage 1 |
Definitionen
AOP (Adverse Outcome Pathway): Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den molekularen auslösenden Vorgang bis hin zu einem In-vivo-Ergebnis von Interesse (2).
ARE: Antioxidant Response Element (auch als EpRE (elektrophile Response Element) bezeichnet), ist ein Response-Element, das in der vorgelagerten Promoterregion vieler zytoprotektiven und Phase-II-Gene anzutreffen ist. Bei Aktivierung durch Nfr2 steuert es die transkriptionelle Induktion dieser Gene.
Chemikalie: Stoff oder Gemisch.
EC1,5: Interpolierte Konzentration bei einer 1,5-fachen Induktion der Luciferase-Aktivität.
Einkomponentiger Stoff: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist.
Empfindlichkeit: Anteil aller positiven/aktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (29).
Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (1).
Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (29).
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1).
Gültige Prüfmethode: Eine Prüfmethode, die eine ausreichende Relevanz und Zuverlässigkeit für einen bestimmten Zweck aufweist und auf wissenschaftlich fundierten Grundsätzen beruht. Eine Prüfmethode ist niemals gültig im absoluten Sinn, sondern nur in Bezug auf einen festgelegten Zweck (29).
IATA (Integrated Approach to Testing and Assessment - Integrierter Test- und Bewertungsansatz): Strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbewertung (Potenzial/Potenz und Exposition) einer Chemikalie oder Chemikaliengruppe, bei dem alle maßgeblichen Daten strategisch integriert und gewichtet werden, um als Grundlage für fundierte regulatorische Entscheidungen über potenzielle Gefahren und/oder Risiken und/oder die Notwendigkeit weiterer gezielter und somit minimaler Testungen herangezogen zu werden.
IC30: Konzentration, die eine Verringerung der Zellviabilität um 30 % bewirkt.
IC50: Konzentration, die eine Verringerung der Zellviabilität um 50 % bewirkt.
Imax: Maximaler Induktionsfaktor der Luciferase-Aktivität im Vergleich zur (Negativ-)Lösungsmittel-Kontrolle bei jeder Prüfchemikalienkonzentration.
Keap1: Kelch-like ECH-associated protein 1 ist ein Sensorprotein, durch das die Nrf2-Aktivität reguliert werden kann. Unter nicht induzierten Bedingungen wirkt das Keap1-Sensorprotein auf den Transkriptionsfaktor Nrf2 für eine Ubiquitinylierung und den proteolytischen Abbau im Proteasom ein. Die kovalente Modifizierung der reaktiven Cysteinreste von Keap1 durch kleine Moleküle kann zur Nrf2-Dissoziation von Keap1 führen (8) (10) (11)).
Lösungsmittel-/Vehikelkontrolle: Replikat, das alle Komponenten eines Testsystems, einschließlich des verwendeten Lösungsmittels, jedoch außer der Prüfchemikalie, enthält. Dies wird verwendet, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel aufgelöst wurden, zu bestimmen.
Mehrkomponentiger Stoff: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens > 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet.
Nrf2: Nuclear factor (erythroid-derived 2)-like 2 ist ein Transkriptionsfaktor, der am Antioxidant Response Pathway beteiligt ist. Wenn Nrf2 nicht ubiquitinyliert ist, baut es sich im Zytoplasma auf und transloziert in den Kern, wo es sich mit dem ARE in der vorgelagerten Promoterregion vieler zytoprotektiven Gene vereint, wobei deren Transkription initiiert wird (8) (10) (11).
Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie: Der Begriff "Prüfchemikalie" bezeichnet das, was getestet wird.
Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Sie beinhaltet die Prüfung der Genauigkeit (Übereinstimmung) einer Prüfmethode (29).
Reproduzierbarkeit: Übereinstimmung zwischen den Ergebnissen, die bei Prüfungen der gleichen Chemikalie bei einheitlichem Protokoll erzielt werden (siehe Zuverlässigkeit) (29).
Spezifität: Anteil aller negativen/inaktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (29).
Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (1).
UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
Variationskoeffizient: Kennwert der Varianz. Er wird für eine Gruppe replizierter Daten mittels Division der Standardabweichung durch den Mittelwert berechnet. Multipliziert mit 100 ergibt sich ein Prozentwert.
Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (29).
.
| Anlage 2 |
Leistungsstoffe
In-vitro-Hautsensibilisierung: ARE-Nrf2 Luciferase-Prüfmethode
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Labors ihre technische Leistungsfähigkeit demonstrieren, indem sie die erwartete KeratinoSensTM-Vorhersage für die in Tabelle 1 empfohlenen 10 Leistungsstoffe korrekt ermitteln und die EC1,5- und IC50-Werte, die in den betreffenden Referenzbereich fallen, bei mindestens 8 der 10 Leistungsstoffe bestimmen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien waren kommerzielle Verfügbarkeit, Verfügbarkeit hochwertiger In-vivo-Referenzdaten und Verfügbarkeit hochwertiger In-vitro-Daten aus dem KeratinoSensTM-Test.
Tabelle 1 Empfohlene Stoffe zum Nachweis der technischen Leistungsfähigkeit beim KeratinoSensTM-Test
| Leistungsstoffe | CAS-Nr. | Physika- lischer Zustand | In-vivo- Vorhersage 1 | KeratinoSensTM Vorhersage 2 | EC1,5 (µM)-Referenz- bereich 3 | IC50 (µM)-Referenz- bereich 3 |
| Isopropanol | 67-63-0 | flüssig | Nicht- sensibilisator | negativ | > 1.000 | > 1.000 |
| Salicylsäure | 69-72-7 | fest | Nicht- sensibilisator | negativ | > 1.000 | > 1.000 |
| Milchsäure | 50-21-5 | flüssig | Nicht- sensibilisator | negativ | > 1.000 | > 1.000 |
| Glyzerin | 56-81-5 | flüssig | Nicht- sensibilisator | negativ | > 1.000 | > 1.000 |
| Zimtalkohol | 104-54-1 | fest | Allergen (schwach) | positiv | 25 - 175 | > 1.000 |
| Ethylenglykoldimethacrylat | 97-90-5 | flüssig | Allergen (schwach) | positiv | 5 - 125 | > 500 |
| 2-Mercaptobenzothiazol | 149-30-4 | fest | Allergen (mäßig) | positiv | 25 - 250 | > 500 |
| Methyldibromglutaronitril | 35691-65-7 | fest | Allergen (stark) | positiv | < 20 | 20 -100 |
| 4-Methylaminophenolsulfat | 55-55-0 | fest | Allergen (stark) | positiv | < 12,5 | 20 - 200 |
| 2,4-Dinitrochlorbenzol | 97-00-7 | fest | Allergen (extrem) | positiv | < 12,5 | 5 - 20 |
| 1) Die Vorhersagen der In-vivo-Gefahr (und Potenz) basieren auf LLNA-Daten (13). Die In-vivo-Potenz wird unter Anwendung der von ECETOC vorgeschlagenen Kriterien abgeleitet (24).
2) Eine KeratinoSensTM-Vorhersage sollte im Rahmen eines IATA sowie gemäß den Nummer 9 und 11 dieser Prüfmethode in Betracht gezogen werden. 3) Basierend auf den historischen beobachteten Werten (12). | ||||||
.
| Qualitätskontrolle bei Lumineszenzmessungen | Anlage 3 |
Grundlagenversuch zur Gewährleistung optimaler Lumineszenzmessungen im KeratinoSensTM-Test
Die drei folgenden Parameter sind von maßgeblicher Bedeutung, um zuverlässige Ergebnisse beim Luminometer zu erhalten:
- ausreichende Empfindlichkeit für einen stabilen Background in den Kontrollmulden;
- kein Gefälle entlang der Platte aufgrund langer Messzeiten;
- keine Lichtkontamination in benachbarten Zellen ausgehend von stark aktiven Mulden.
Vor der Prüfung sollte durch Prüfung eines Kontroll-Plattenaufbaus, wie unten beschrieben, sichergestellt werden, dass geeignete Lumineszenzmessungen durchgeführt werden können (Dreifachanalyse).
Plattenaufbau des ersten Kontrollversuchs
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| A | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| B | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| C | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| D | EGDMA 0,98 | EGDMA 1,95 | EGDMA 3,9 | EGDMA 7,8 | EGDMA 15,6 | EGDMA 31,25 | EGDMA 62,5 | EGDMA 125 | EGDMA 250 | EGDMA 500 | EGDMA 1000 | EGDMA 2000 |
| E | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| F | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| G | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO |
| H | DMSO | DMSO | DMSO | DMSO | DMSO | DMSO | CA 4 | CA 8 | CA 16 | CA 32 | CA 64 | Leer |
EGDMA= Ethylenglykoldimethacrylat (CAS Nr.: 97-90-5), eine stark induzierende Chemikalie
CA= Zimtaldehyd, positive Referenz (CAS Nr.: 104-55-2)
Bei der Qualitätskontrollanalyse sollte Folgendes nachgewiesen werden:
- eindeutige Dosis-Wirkungs-Beziehung in Zeile D, wobei Imax > 20-facher Wert über Background (in den meisten Fällen werden Imax-Werte zwischen 100 und 300 erreicht);
- keine Dosis-Wirkungs-Beziehung in Zeile C und E (kein Induktionswert über 1,5 (idealerweise nicht über 1,3) aufgrund möglicher Lichtkontamination, insbesondere neben stark aktiven Mulden in der EGDMA-Zeile);
- kein statistisch signifikanter Unterschied zwischen den Zeilen A, B, C, E, F und G (d. h. kein Gefälle entlang der Platte);
- die Variabilität in einer der Zeilen A, B, C, E, F und G sowie in den DMSO-Mulden in Zeile H sollte unter 20 % liegen (d. h. stabiler Background).
| weiter . | |
...
X
⍂
↑
↓