
umwelt-online: AMR - Arzneimittel-Richtlinie (2)
 | zurück |  |
Azathioprin
zur Behandlung der Multiplen Sklerose (Imurek®)
Beschluss vom: 03.05.2001
In Kraft getreten am: 24.08.2001
BAnz. 2001, S. 18.422/ 18.424
| Indikation |
Azathioprinhaltige Arzneimittel sind für den Einsatz zur Verhinderung von Abstoßungsreaktionen nach Organtransplantationen und zur Behandlung verschiedener Autoimmunkrankheiten zugelassen, wobei seit September 2000 auch die Multiple Sklerose (MS) zu den zugelassenen Anwendungsgebieten von Imurek® zählt.
Azathioprin ist ein Purinanalogon, welches im Organismus zu 6-Mercaptopurin (6-MP) und Methylnitroimidazol verstoffwechselt wird. Beide Metaboliten sollen immunsuppressiv wirken. Aus 6-MP entstehen Mercaptonukleotide. Durch Kompetition mit DNS-Bausteinen hemmen sie Lymphozyten-Differenzierung und -Aktivierung. Es zeigte sich v.a. ein Effekt auf CD8+ T-Zellen, NK-Zellen und B-Lymphozyten. Blutuntersuchungen erbrachten einen Abfall der TNF-a-Konzentrationen und einen Anstieg der suppressorinducer -Lymphozyten. Die hieraus resultierende immunsuppressive Wirkung beruht höchst wahrscheinlich auf einer Dämpfung von zellvermittelten Hypersensitivitätsreaktionen und Antikörperproduktion; diese Wirkung tritt jedoch erst frühestens nach 2 - 5 Monaten ein.
| Wirksamkeit |
In einer Metaanalyse wurden 7 Einzel- und Doppelblindstudien zur Wirksamkeit des Azathioprin bei Multipler Sklerose erfasst und insgesamt 793 Krankheitsfälle ausgewertet. Die Patienten waren mindestens 1 (n = 719), 2 (n = 563) bzw. 3 Jahre (n = 459) mit Azathioprin oder Placebo behandelt worden. Alle Verlaufsformen der MS waren vertreten. Bewertet wurden 1. die v.a. in der Kurtzke Disability Status Scale (DSS) gemessene Verschlechterung, 2. die Reduktion der Schubzahl verglichen mit der erwarteten Schubrate. Es kam nach 2- bzw. 3-jähriger Behandlung zu einer noch nicht signifikanten Reduktion der Krankheitsprogression (p<0,06 bzw. p<0,09). Die Schubrate wurde auf Signifikanzniveau reduziert, wobei die ermittelten Zahlen einer Reduktion der Schubrate von etwa 30 - 40 % entsprachen (nach 1 Jahr: p<0.01, nach 2 Jahren: p<0,001, nach 3 Jahren: p<0,01).
Diese Ergebnisse wurden in einer weiteren Doppelblindstudie im Wesentlichen bestätigt. Eine retrospektive - und damit weniger aussagekräftige - MRT-Studie zeigte eine 39%ige Reduktion der Zahl zerebraler Entzündungsherde nach Azathioprin verglichen mit einer unbehandelten Kontrollgruppe.
| Risiken ggf. Vorsichtsmaßnahmen |
Unerwünschte Wirkungen von Azathioprin sind allgemeines Krankheitsgefühl, gastrointestinale Funktionsstörungen (Übelkeit, Erbrechen, Durchfall, Leibschmerzen, seltener Anstieg der Leberwerte), Pankreatitis, Überempfindlichkeitsreaktionen wie Fieber, Schüttelfrost, Hautausschlag, Gelenkschmerzen, Blutdruckabfälle. Sie treten meistens schon frühzeitig nach Behandlungsbeginn auf und machen oft einen Therapieabbruch erforderlich. Später werden knochenmarksuppressive Effekte - Leukopenie, seltener Thrombozytopenie oder Anämie - erkennbar. Sie entwickeln sich insbesondere, wenn gleichzeitig Allopurinol gegeben wird. In solchen Fällen ist also eine Dosisreduktion erforderlich. Als weitere unerwünschte Wirkungen sind erhöhtes Infektionsrisiko, seltene interstitielle Pneumonien und Alopezie bekannt.
Folgende Sicherheitsmaßnahmen werden gefordert: 1. Ausschluss von Schwangerschaft und Stillzeit bei Beginn der Azathioprintherapie; während der Therapie soll eine sichere Antikonzeption gewährleistet sein. 2. Regelmäßige Kontrolle von Differential-Blutbild und Leberwerten, die zunächst wöchentlich, später nur noch monatlich erfolgen sollen. 3. Therapieeinstellung auf eine Zielgröße der Leukozytenzahlen (600 - 1200 Lymphozyten pro mm3 bzw. Leukozyten < 3500/mm3). 4. Bei Leukozytenwerten unter 3500/mm3 ist eine 50%ige Dosisreduktion erforderlich, Werte unter 3000/mm3 machen eine Therapiepause bis zum Anstieg über 4000/mm3 erforderlich.
Als Spätfolge der Azathioprintherapie ist ein leicht erhöhtes Malignomrisiko zu beachten. Eine Fall-Kontroll-Studie über das Krebsrisiko bei MS-Patienten, die mit Azathioprin über 5 - 10 Jahre behandelt worden waren, erbrachte ein Risikoerhöhung bei Applikationsdauer unter 5 Jahren auf 1,3, zwischen 5 und 10 Jahren auf 2,0 und über 10 Jahren auf 4,4. Auf der Basis dieser Ergebnisse wird das Krebsrisiko für MS-Patienten während der ersten Behandlungsjahre für unbedeutend eingeschätzt; erst nach über 10jähriger kontinuiertlicher Azathiopringabe in therapieüblichen Dosen könnte sich ein erhöhtes Krebsrisiko ergeben.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
In einer vergleichenden Darstellung wurde gezeigt, dass für das Kriterium Verminderung der Schubzahl durch Azathioprin vergleichbare Ergebnisse erreicht werden können wie durch neuere Therapieprinzipien, zu denen Interferonbeta und Glatirameracetat gehören. Eine signifikante Verlangsamung der Krankheitsprogression ist für Azathioprin nicht belegt. Für i.v. Immunglobulin liegt eine Zulassung zur Behandlung der MS nicht vor.
Die Gabe von Azathioprin bei Multipler Sklerose ist vielfach sinnvoll. Die Zielgruppe sind Patienten mit schubförmiger multipler Sklerose, wenn eine immunprophylaktische Therapie angezeigt ist und eine Therapie mit Beta-Interferon nicht möglich ist oder unter einer bisherigen Therapie mit Azathioprin ein stabiler Verlauf erreicht wurde. Der Vorteil einer oralen Anwendung ist gegeben. Die Startdosis besteht aus 2,5 mg/kg KG, gegeben in 2 oder 3 Einzeldosen. Die weitere Therapiesteuerung erfolgt nach Verträglichkeit und insbesondere den o.a. hämatologischen Kriterien. Der Nutzen einer Kombinationstherapie von Azathioprin mit Interferonbeta-1 ist nicht belegt und Gegenstand der klinischen Forschung.
| Kosten |
Der Kostenvergleich zwischen Azathioprin und Interferonbeta erbringt in etwa um den Faktor 10 günstigere Jahrestherapiekosten für Azathioprin.
Glatirameracetat ist in Deutschland zur Zeit noch nicht zugelassen, hat aber kürzlich im dezentralen EU-Verfahren die Zulassung für Großbritannien erhalten und kann im Rahmen des ® 73 AMG im Einzelfall durch jede öffentliche Apotheke bezogen werden.
| Interferon ß 1-a | Avonex® | 6 Mio I.E. i.m.
1x/Woche | 14.804,97 Euro/Jahr |
| Interferon ß 1-a | Rebif® | 6 Mio I.E.s.c.
3x/Woche | 16.207,44 Euro/Jahr |
| Interferon ß 1-b | Betaferon® | 9,6 Mio I.E.s.c.
jeden 2. Tag | 14.793,21 Euro/Jahr |
| Glatirameracetat | Copaxone® | 20 mg/d s.c. | z. Z. ca. 11.452,94 Euro/Jahr.* |
| Azathioprin | (Imurek®) | 23 mg/kg KG/d p.o. | 2,5 mg, bei 70 kg KG: 1.619,77 Euro/Jahr** |
*) Der Preis für Deutschland nach Markteinführung ist noch offen (evtl. zuzüglich Beschaffungskosten für den Import).
**) Anfallende Laborkosten sind hierbei nicht berücksichtigt.
Becaplermin
(aufgehoben durch BAnz. AT vom 08.06.2016 B1)
Botulinumtoxin A und B
(z.B.: Botox®, Dysport®) / (z.B.: Neurobloc®)
Beschluss vom: 21.09.2004
In Kraft getreten am: 22.01.2005
BAnz. 2005 N r. 14, S. 977
| Indikation |
Von den insgesamt 7 existierenden Typen von Botulinumtoxin sind zur Behandlung verschiedener Formen fokaler Dystonien, spastischer Störungen und der axillären Hyperhidrosis zwei Toxinformen (A und B) zur symptomatischen Therapie zugelassen.
Der Zulassungsstatus der einzelnen Präparate ist unterschiedlich:
Botox® (Clostridium botulinum Toxin Typ A): Behandlung von
- Blepharospasmus, hemifazialem Spasmus und koexistierenden fokalen Dystonien
- idiopathischer rotatorischer zervikaler Dystonie (Torticollis spasmodicus)
- fokaler Spastizität
- in Zusammenhang mit dynamischer Spitzfußstellung infolge von Spastizität bei Patienten mit infantiler Zerebralparese ab dem zweiten Lebensjahr
- des Handgelenkes und der Hand bei erwachsenen Schlaganfallpatienten
- starker, fortbestehender primärer Hyperhidrosis axillaris, die störende Auswirkungen auf die Aktivitäten des täglichen Lebens hat und mit einer topischen Behandlung nicht ausreichend kontrolliert werden kann
Dysport® (Clostridium botulinum Toxin Typ A):
- zur symptomatischen Alternativbehandlung von idiopathischem Blepharospasmus und gleichzeitig bestehenden hemifazialen dystonen Bewegungsabläufen
- zur symptomatischen Behandlung eines einfachen idiopathischen rotierenden Torticollis spasmodicus mit Beginn im Erwachsenenalter
- zur symptomatischen Behandlung einer Armspastik bei Erwachsenen infolge eines Schlaganfalles
Neurobloc® (Clostridium botulinum Toxin Typ B):
- Behandlung von zervikaler Dystonie (Torticollis)
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Entscheidend für eine effektive und wirtschaftliche Therapieplanung ist eine qualifizierte Primärdiagnostik einschließlich umfassender Ausschlussdiagnostik. Die Indikationsstellung und Durchführung der Behandlung soll grundsätzlich durch einen in der Anwendung von Botulinumtoxin erfahrenen Facharzt, im Einzelfall in Absprache mit spezialisierten Zentren, erfolgen. Eine Ausbildung in der Injektionstechnik, sehr gute anatomische und arzneimitteltherapeutische Kenntnisse sollten vorausgesetzt werden. Ergebnisse einer konventionellen physiotherapeutischen, medikamentösen oder operativen Behandlungsstrategie müssen sorgfältig abgewogen werden. Bei Dystonieformen mit besonders störenden Fokalsymptomen ist nach sachgerechter Diagnostik in Abhängigkeit der Vorgeschichte und des klinischen Bildes die selektive periphere Denervierung der betroffenen Muskelgruppen durch lokale Injektion von Botulinumtoxin heute in der Regel Methode der ersten Wahl. Eine ausführliche Patientenaufklärung inklusive realistischer Planung der Therapieziele ist notwendig und sollte gut dokumentiert werden.
Bei der Behandlung der Hyperhidrosen ergeben sich erste Hinweise auf eine mögliche Verbesserung der Lebensqualität unter Botulinumtoxingabe. Studiendesign, -dauer und Datenlage zur Änderung der Lebensqualität lassen einen direkten Vergleich zu den Alternativbehandlungen gegenwärtig nicht zu. Bei der Therapie der axillären Hyperhidrosis ist eine strenge Indikationsstellung erforderlich, da auch Patienten ohne objektivierbare
Störung massive Therapiewünsche äußern. Bei unklarer Situation ist für eine Indikationsstellung ein standardisierter Schweißtest hilfreich. Er kann auch Auswirkungen auf die Dosierung haben. Bei einer Entscheidung für eine Therapie sollte ein Behandlungsversuch mit einer Aluminiumchlorid-Lösung generell vorangestellt werden. Operative Verfahren mit dem Ziel einer dauerhaften Lösung müssen erwogen und mit den Patienten als Alternativen diskutiert werden.
Der Nutzen von Botulinumtoxin bei Insultpatienten mit Spastik der Hand bzw. des Armes hinsichtlich einer echten funktionellen Verbesserung ist zurzeit noch nicht ausreichend gut definiert. Eine sehr konkrete und realistische Planung und Überprüfung der Therapieziele ist in diesem Indikationsbereich unabdingbar. Dies gilt auch für die Behandlung der Spastik mit Spitzfußstellung bei kindlicher Zerebralparese, bei der eine Besserung, nicht jedoch eine Normalisierung der Motorik möglich ist.
| Kosten der ausgewählten Indikationsbereichen |
Aufgrund der sehr differenzierten Vorgehensweise in der Therapie der zervikalen Dystonie ist ein einfacher Kostenvergleich auf der Basis der Herstellerangaben nicht ganz unproblematisch. Für Botox® gibt es zudem keine empfohlene Initialdosierung durch den Hersteller (Fachinformation Juli 2004). Für die Wirtschaftlichkeitsbetrachtung der Therapie der zervikalen Dystonie müssen daher mittlere Dosisangaben als Berechnungsgrundlage als beste Annäherung herangezogen werden. Als Grundlage zur Berechnung der bioäquivalenten Dosis beim Vergleich von Dysport® und Botox® in der Behandlung der zervikalen Dystonie wurde in einer randomisierten doppelblinden Studie eine durchschnittliche Dysport®-Dosis von 240-720 Einheiten (Mittelwert: 477 Einheiten) und für Botox® 70-240 Einheiten (Mittelwert: 152 Einheiten) zugrunde gelegt.
Die Dosierungsbreite liegt nach Herstellerangaben (Fachinformation Juli 2004) für Botox® nach aktuelleren Untersuchungen zwischen 95 und 360 Einheiten (mit einer ungefähren mittleren Dosis von 240 Einheiten), für Dysport® liegt die empfohlene Initialdosis bei 500 Einheiten und die maximale Dosis bei 1.000 Einheiten (Fachinformation November 2003), wobei bei wiederholten Injektionen die Dosis pro Sitzung um 100 bis 250 Einheiten verringert oder erhöht werden kann. Bei Neurobloc® liegt die empfohlene Dosierungsbreite zwischen 5.000 und 10.000 Einheiten (Fachinformation September 2003).
Bei der Wirtschaftlichkeitsbetrachtung muss unter Berücksichtigung der üblichen Praxisbedingungen und der gebotenen Sorgfalt bei der Anwendung weiterhin die beschränkte Lagerungsfähigkeit der gebrauchsfertigen Lösungen nach Anbruch beachtet werden. So beträgt beispielsweise für Botox® die empfohlene Lagerungszeit der gebrauchsfertigen Lösung maximal 4 Stunden.
Vergleich über die indikationsbezogene durchschnittliche Dosierung in Anlehnung an Odegren et al. [1998] und Herstellerangaben.
| Fertigarzneimittel (Wirkstoff) | Hersteller | Mittlere Gesamtdosis | Berechnung inkl. Anbruch | Kosten in Euro |
| Indikation Zervikale Dystonie | ||||
| Botox® (Clostr. bot. Toxin Typ A) | Pharm-Allergan | 152E | 2 x 1 (100 E) (kleinste Packungsgröße) | 665,36 |
| Dysport® (Clostr. bot. Toxin Typ A) | Ipsen Pharma | 477 E | 1 x 1 (500 E) (kleinste Packungsgröße) | 416,82 |
| Neurobloc® (Clostr. bot. Toxin Typ B) | Elan Pharma | 10.000 E | 1 x 2 ml Inj.- Lösg. (10.000 E) | 376,20 |
| Indikation Hyperhidrosis axillaris | ||||
| Botox® (Clostr. bot. Toxin Typ A) | Pharm-Allergan | 100 E | 1 x 1 (100 E) | 332,68 |
(Preis- und Präparatestand: Lauertaxe 15. August 2004)
| Wirkungen |
Botulinumtoxin blockiert die Freisetzung von Acetylcholin an den präsynaptischen Nervenendigungen durch Spaltung eines Proteins (SNAP-25 ), das für die Speicherung und Freisetzung von Acetylcholin wichtig ist. Innerhalb von 2-3 Tagen erfolgt allmählich die (erwünschte) Schwächung der Muskulatur. Die maximale Wirkung wird nach 5-6 Wochen erreicht (bei sehr kleinen Muskeln früher). Durch Reinnervation kommt es innerhalb von ca. 3 Monaten wieder zu einer weitgehenden Reparation der Impulsübertragung.
Schweißbildung wird durch Blockade überaktiver sudomotorischer cholinerger Nervenfasern gehemmt. Auch dieser Effekt ist reversibel.
Die Dosen der verschiedenen Präparate sind nicht äquivalent, so dass bei der Anwendung die Dosis-Empfehlungen des Herstellers unbedingt beachtet werden müssen. Neutralisierende Antikörper, die die Therapie unwirksam machen, sind nach längerer Behandlungsdauer vor allem bei zervikaler Dystonie bei 3-10 % der Patienten beschrieben.
| Wirksamkeit |
Die Wirksamkeit von Botulinumtoxin ist in den verschiedenen Indikationen unterschiedlich evaluiert worden:
- Zur Therapie des Blepharospasmus liegen ausschließlich Anwendungsbeobachtungen mit überwiegend kleinen Patientenzahlen vor. Ein direkter Vergleich mit anderen Therapieoptionen (z.B. Anticholinergika), die als unbefriedigend gelten, fehlt. In allen Arbeiten mit Botulinumtoxin wird von einem guten Ansprechen der Therapie berichtet, die Ansprechrate wird mit 62-100 % angegeben. Eine Studie schildert Langzeiterfahrungen an einem Kollektiv von 239 Patienten, die über 11 Jahre beobachtet wurden. Über % der Patienten wurden auch nach diesem Zeitraum noch regelmäßig und offenbar mit Erfolg behandelt.
- Randomisierte kontrollierte Studien wurden zur Therapie der zervikalen Dystonie durchgeführt. Plazebokontrollierte Doppelblindstudien wurden als Parallgruppenvergleiche, zum Teil auch im Crossover-Design konzipiert. Diese verwendeten standardisierten Torticollis-Scores als objektivierbare Erfolgsparameter. Auch für das nur in diesem Indikationsbereich zugelassene Botulinumtoxin B liegen kontrollierte Arbeiten vor. In allen Studien wurde eine Überlegenheit in Bezug auf Schmerzen und Bewegungsstörungen gegenüber Plazebo gezeigt, allerdings ist der Einfluss auf die Kopfhaltung nicht so eindrucksvoll wie die Wirkung auf die Augenmuskeln bei der Therapie des Blepharospasmus. Der Effekt ist dosisabhängig, mit der Dosis steigt aber auch die Rate an unerwünschten Arzneimittelwirkungen (s. u.). Bei Unwirksamkeit von Botulinumtoxin A durch neutralisierende Antikörper ist eine Wirkung durch Botulinumtoxin B noch erreichbar.
- Botulinumtoxin A bei übermäßigem Schwitzen wurde mit randomisierten, plazebokontrollierten Studien bei der axillären Hyperhidrosis an Patienten, die nicht erfolgreich auf eine Therapie mit Aluminiumchlorid-Lösung ansprachen, geprüft. Gemäß der durchgeführten standardisierten Schweißtests erwies sich die Behandlung als wirksam (Reduktion der Schweißproduktion um mindestens 50 % wurde erreicht bei 94 % der mit Botulinumtoxin A Behandelten, unter Plazebo bei 36 % der Patienten). Der Effekt war in einer offenen Nachbeobachtung nach sieben Monaten noch nachweisbar. Die Lebensqualität wurde in einer Arbeit überprüft und ließ sich nur geringgradig beeinflussen. Langzeitdaten fehlen jedoch, ebenso wie direkte Vergleiche mit anderen etablierten Methoden (z.B. lokale Applikation von Aluminiumchloridhexahydrat). Ein Teil behandlungswilliger Patienten scheint in diesem Indikationsbereich eher ein psychisches als ein körperliches Problem aufzuweisen. Bei der Botulinophilie lässt sich eine übermäßige Schweißbildung durch objektive Tests nicht nachweisen, gleichzeitig besteht jedoch ein überaus starker Behandlungswunsch.
- Der Nutzen von Botulinumtoxin A bei Spastik des Arms und der Finger nach Insult wurde in einer größeren plazebokontrollierten randomisierten Studie bewertet. Untersucht wurde der Effekt einer Behandlung nach einmaliger Injektion an 122 Patienten. Als Erfolgskriterium wurde der Einfluss auf vier Symptomkomplexe erfasst: 1. hygienische Aspekte (assoziiert mit der Handfunktion), 2. Fähigkeit sich anzuziehen, 3. Position des Armes, 4. Schmerzen. Jeder Patient sollte aus diesen das für ihn wichtigste Therapieziel wählen. Bezogen auf den Tonus der Muskulatur sowie ausgewählte funktionelle Parameter war Botulinumtoxin den Plazeboinjektionen überlegen, allerdings wurde bei einer globalen Bewertung der Behandlungserfolg von den Patienten nach 6 Wochen im Mittel als allenfalls moderat bewertet. Ein Effekt war nach 12 Wochen noch (deutlich abgeschwächt) nachweisbar. In einer weiteren randomisierten Doppelblindstudie mit insgesamt 59 Patienten hielt der Spastikreduzierende Effekt der Botulinumtoxin-Typ A-Verabfolgung über mindestens 16 Wochen an. Ausreichende Langzeitdaten liegen zu dieser Indikation noch nicht vor. Der Einfluss auf echte funktionelle Verbesserungen, die zu einer Erleichterung von Alltagsaktivitäten führen, ist nach Studienlage nicht nachgewiesen.
- Kleinere plazebokontrollierte Studien sind auch bei der Therapie von Kindern mit infantiler Zerebralparese publiziert. Verbesserungen der Motorik sind anhand von Videoanalysen des Gangbildes und Messung der motorischen Fähigkeiten belegt. Ein physiotherapeutisches Gehtraining wird z. T. erst durch die Gabe von Botulinumtoxin möglich. Eine längerfristige Normalisierung des Gangbildes, der Motorik oder des Bewegungsumfanges ist nicht ausreichend belegt. Ebenso unklar ist nach gegenwärtiger Studienlage, welche Altersgruppe und welches Störungsbild innerhalb der Krankheitsentität von der Botulinumbehandlung profitiert. Ein differenziertes Vorgehen und sehr gute Kenntnisse der entwicklungs Neurologischen Besonderheiten im Kindesalter sind daher erforderlich.
| Risiken ggf. Vorsichtsmaßnahmen |
Je nach Anwendungsgebiet bestehen u. a. eine Reihe zum Teil häufig bis sehr häufig auftretender unerwünschter Wirkungen:
- Therapie des Blepharospasmus: Ptosis, trockenes Auge, Diplopie, Keratitis superficialis, Photophobie
- Therapie der zervikalen Dystonie: Dysphagie, Schwäche und Parese der regionalen Muskelgruppen einschließlich der Stimm-Muskeln möglich
- Therapie der infantilen Zerebralparese: Virusinfektion, Ohrinfektion, Myalgie, Muskelschwäche, Harninkontinenz, Somnolenz, Unwohlsein
- Therapie der primären Hyperhidrosis axillaris: kompensatorisches Schwitzen außerhalb der Achselhöhle, Schmerzen, Hitzewallungen
- Therapie im Zusammenhang mit Schlaganfall (obere Extremitäten): Ekchymose, Purpura oder Blutung an der Einstichstelle, Schmerzen in den Armen, Muskelschwäche, Muskelhypertonus
Eine Kontraindikation besteht bei nachgewiesener Überempfindlichkeit gegenüber Botulinumtoxin oder einem anderen seiner Bestandteile, bei Schwangerschaft und Stillzeit, Myasthenia gravis, Lambert-Eaton-Syndrom sowie bei anderen Neuromuskulären Erkrankungen.
Aufgrund des Wirkungsmechanismus von Botulinumtoxin ist bei gleichzeitiger Gabe von Wirkstoffen, die die Neuromuskuläre Übertragung beeinträchtigen (z.B. Aminoglycoside), mit Wechselwirkungen zu rechnen, die ein klinisch lebensbedrohliches Ausmaß annehmen können.
Celecoxib
(z.B. Celebrex®)
Beschluss vom: 24.03.2003
In Kraft getreten am: 01.10.2003
BAnz. 2003, Nr. 183 vom 30.09.2003 S. 22.058
| Indikation |
Celecoxib ist in Deutschland in den Wirkstärken 100 mg und 200 mg für die Behandlung von Symptomen bei Reizzuständen degenerativer Gelenkerkrankungen (aktivierte Arthrosen) oder chronischer Polyarthritis (rheumatoide Arthritis) zugelassen. Die Einnahme erfolgt abhängig von der Indikation ein- bis zweimal täglich.
| Wirkungen |
Das NSAR Celecoxib ist ein selektiver COX-2-Hemmer. Diese Substanzgruppe hemmt nur eine Isoform des für die Prostaglandinsynthese wesentlichen Enzyms Cyclooxygenase. COX-2 ist nach gegenwärtigem Kenntnisstand verantwortlich für die Synthese der Prostaglandine, die Entzündung, Schmerz und Fieber vermitteln. Die durch die COX-1-Isoform vermittelten Prostaglandine werden dagegen anders als bei nicht selektiven NSAR - durch Celecoxib nicht gehemmt. Somit wird auch deren protektive Wirkung auf die Magenschleimhaut nicht beeinträchtigt. Ebenso zeigt Celecoxib keine Beeinflussung der COX-1-abhängigen Thrombozytenfunktion.
| Wirksamkeit |
Zur Anwendung bei aktivierter Arthrose liegen Studien mit Tagesdosierungen von 80 - 400 mg Celecoxib versus 1000 mg Naproxen/Tag oder 150 mg Diclofenac/Tag und Placebo bei Patienten mit Knie- oder Hüftgelenksarthrose vor, die eine Verbesserung einzelner Parameter gegenüber Placebo und Gleichwertigkeit gegenüber den konventionellen NSAR ergaben. Als günstigste Dosierung erwies sich 200 mg Celecoxib/Tag.
Weitere publizierte Studien vergleichen die analgetische und antientzündliche Wirksamkeit von Celecoxib bei Patienten mit rheumatoider Arthritis wiederum mit 1000 mg Naproxen/Tag bzw. mit 150 mg Diclofenac/Tag. Während der 12- bzw. 24-wöchigen Beobachtungszeit ergab sich jeweils eine vergleichbare Wirksamkeit für die Substanzen, aber auch unter den eingesetzten Celecoxib-Dosierungen von 200, 400 und 800 mg. Die zugelassene tägl. Höchstdosis bei der rheumatoiden Arthritis beträgt 400 mg.
Die maximale Plasmakonzentration wird nach zwei bis drei Stunden erreicht, ein steadystate-Spiegel stellt sich nach fünf Tagen ein (HWZ 11,2 Std.). Eine Symptomlinderung ist nach den Studien nach 24 bis 48 Stunden zu erwarten. Soweit eine akute Schmerzexazerbation zur Therapieeinleitung führt, sollte dies beachtet werden.
| Risiken ggf. Vorsichtsmaßnahmen |
In der im Jahr 2000 publizierten CLASS-Studie wurden 8.059 Patienten, von denen 4.573 den sechsmonatigen Therapiezeitraum abschlossen, mit der Dosis von 800 mg Celecoxib täglich (doppelte empfohlene Tageshöchstdosis) oder 150 mg Diclofenac oder 2400 mg Ibuprofen über sechs Monate behandelt. Die Rate der Ulkuskomplikationen (definiert als Blutung, Perforation und Obstruktion) lag nach der Publikation unter Celecoxib bei 0,76 %, unter Diclofenac und Ibuprofen bei 1,45 %, standardisiert auf ein Jahr (statistisch nicht signifikanter Unterschied, p = 0,09). Subgruppenanalysen ergaben für Patienten, die ASS als kardiovaskuläre Prophylaxe (< 325 mg/Tag) einnahmen, keine Differenz (2,01 % versus 2,12 %), während für Patienten, die kein ASS einnahmen, eine Rate von Ulkuskomplikationen von 0,44 % unter Celecoxib versus 1,27 % unter Ibuprofen bzw. Diclofenac (statistisch signifikant, p = 0,04) gesehen wurde.
Allerdings wurde die publizierte Auswertung dieser Studie kontrovers diskutiert. Der Vergleich der Publikation mit den der FDA vorliegenden Original-Studienprotokollen (es waren 2 Studien konzipiert) zeige, dass das Protokoll nachträglich geändert wurde. Dies betrifft die Zielsetzungen, die primären Outcomes, die statistische Analyse, die Studiendauer und die Schlussfolgerungen (siehe z.B. Diskussion im British Medical Journal und Cochrane-Library Update November 2002). In der publizierten Auswertung wurden nur die 6 Monatsdaten berücksichtigt. Die Reanalyse der Daten nach dem primär geplanten Protokoll habe für die 12 Monatsdaten dagegen keinen Vorteil mehr für Celecoxib ergeben, da fast alle Ulcuskomplikationen in den zweiten 6 Monaten der Studie in der Celecoxib-Gruppe aufgetreten waren. Die FDA hat bei einer Überarbeitung der US-Gebrauchsinformation im Juni 2002 auch nach der Auswertung der CLASS-Studie die Standardwarnungen bzgl. NSAR-typischer Nebenwirkungen für Celecoxib beibehalten. Die Europäische Arzneimittelbehörde EMEA hat bei der Sitzung des wissenschaftlichen Kommitees am 23.-25.07.2002 ein EU-weites Review zur Arzneimittelsicherheit aller Coxibe Incl. Celecoxib zur Klärung der Frequenz von gastrointestinalen und cardiovaskulären Nebenwirkungen eingeleitet.
Publizierte Langzeitstudien, die eine Aussage über das Auftreten von schweren Komplikationen im oberen Gastrointestinaltrakt möglich machen würden, liegen bisher nicht vor.
Der eventuelle Vorteil von Celecoxib ist auch nicht untersucht für den Vergleich mit niedrigeren Dosierungen von nichtselektiven NSAR (in den Studien: Diclofenac 150 mg/Tag, Ibuprofen 2400 mg/Tag, Naproxen 1000 mg/Tag).
Gastroskopiestudien zeigen eine Reduzierung von Schleimhautläsionen, wobei allerdings abgewartet werden muss, ob die Reduzierung der endoskopischen Ulkusrate unter Celecoxib mit einem klinischen Vorteil verbunden ist, da deren Korrelation mit Symptomen und schwerwiegenden Komplikationen unklar ist.
Eine publizierte Differenzierung nach Hoch- und Niedrigrisikopatienten ergibt, dass 500 Patienten der Niedrigrisiko-Gruppe ein Jahr mit Mehrkosten von 400.000 US-$ auf COX-2-Hemmer umgestellt werden müssen, um 1 schwerwiegende Ulkuskomplikation zu vermeiden. Bei Hochrisikopatienten (älter als 75 Jahre mit Ulkus- oder Blutungsanamnese) beträgt dieses Verhältnis noch 1 : 40.
Diese Beurteilung wird durch ein aktuelles Health Technology Assessment der kanadischen CCOHTA bestätigt. Es kommt zu dem Ergebnis, dass die teuren COX-2-Hemmer nur für bestimmte Patientengruppen (hohes Risiko für gastrointestinale Erkrankungen und bei Celecoxib bei einem Alter über 81 Jahre) kosteneffektiv sind.
Wie andere NSAR ist Celecoxib kontraindiziert bei floridem peptischem Ulkus oder gastrointestinaler Blutung. Weitere Gegenanzeigen sind u. a. gebärfähige Frauen (es sei denn, es wird eine sichere Methode zur Schwangerschaftsverhütung eingesetzt), Schwangerschaft, Stillzeit, entzündliche Darmerkrankungen, höhergradige Nieren- und Leberfunktionsstörungen, Sulfonamidüberempfindlichkeit sowie Asthma, akute Rhinitis, Nasenschleimhautpolypen, angio Neurotisches Ödem, Urtikaria oder sonstige allergische Erkrankungen, die nach Anwendung von ASS oder NSAR beobachtet wurden. Mögliche Wechselwirkungen müssen u. a. bei gleichzeitiger Therapie mit Warfarin, Cyclosporin, Tacrolimus, Antihypertensiva (besonders ACE-Hemmer) und Diuretika beachtet werden.
Unerwünschte Wirkungen sind unter Celecoxib global nicht geringer als unter NSAR. Unter Celecoxib wurden u. a. vermehrt periphere Ödeme bzw. Flüssigkeitsretention, arterielle Hypertonie, abdominelle Beschwerden wie Schmerzen, Diarrhoe, Dyspepsie und Flatulenz, außerdem Schwindelgefühl, Schlaflosigkeit, Hautausschläge und Entzündungen des Nasen-Rachen-Raumes und der oberen Atemwege gesehen (bei mindestens jedem 100. Behandelten).
Bei individuellen Risikokonstellationen (Alter, Multimorbidität, gastrointestinale Blutungen in der Anamnese), zusätzlicher Selbstmedikation mit NSAR, der Einnahme von Thrombozytenaggregationshemmern oder Antikoagulanzien oder einer Komedikation mit Kortison kann auch unter dem ggf. indizierten Einsatz von Celecoxib ein erhöhtes Risiko schwerwiegender gastrointestinaler Nebenwirkungen vorliegen.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
- Nach den Erfahrungen mit anfangs mit großen Erwartungen verbundenen Einführungen anderer neuer NSAR muss abgewartet werden, ob sich die klinische Wirksamkeit und das bezüglich schwerwiegender gastrointestinaler Komplikationen günstigere Nebenwirkungsprofil von Celecoxib in der klinischen Praxis und in der Langzeitbeobachtung bestätigen lassen. Die Arzneimittelkommission der deutschen Ärzteschaft hat am 31.05.02 konstatiert, dass das Sicherheitsprofil der COX-2-Inhibitoren erheblich geringer sei, als angenommen. Bei Patienten mit einer Indikation zur ASS- Langzeitgabe sei ein Vorteil der Behandlung mit COX-2-Inhibitoren gegenüber nicht selektiven NSAR nicht zu erkennen.
- Die Tagestherapiekosten für Celecoxib liegen um ein Mehrfaches über denen nicht selektiver NSAR, wobei bei Anwendung in Risikokonstellationen gegebenenfalls die Zusatzkosten für eine Ulkus-Prophylaxe mit Omeprazol oder Misoprostol bei nicht selektiver NSAR-Therapie beim Vergleich berücksichtigt werden müssen.
- Eine routinemässige Verordnung von COX-II-Hemmern bei Indikation für ein NSAR ist unwirtschaftlich. Die Risikoabwägung für schwerwiegende gastrointestinale Komplikationen ist für den eventuellen Einsatz entscheidend, da ein Vorteil bezüglich der Schmerzlinderung für Celecoxib nicht besteht. Bei Verträglichkeit bisheriger NSAR-Therapie besteht keine Veranlassung zur Umstellung. Bei Neuverordnung ohne Vorliegen einer Risikokonstellation, insbesondere bei bedarfsweiser, niedrig bzw. mittelhoch dosierter oder kurz dauernder Verordnung, und bei kardiovaskulären Erkrankungen sollten nicht selektive NSAR bevorzugt werden.
- Die routinemäßige Verordnung einer Begleittherapie mit Protonenpumpenhemmern, H2-Antagonisten oder Misoprostol ist bei Celecoxib ebenso wenig erforderlich wie bei anderen NSAR. Eine Verbesserung des gastroinstestinalen Nebenwirkungsprofils von Celecoxib durch diese Begleittherapie ist durch Studien nicht belegt.
| Kosten |
Vergleich mit verordnungsstarken Arzneimitteln mit vergleichbarer Indikation:
| Substanz, Wirkstärke, Packungsgröße | Tagestherapiekosten | |
| Diclofenac, 3 x 50 mg, 100 Tabl. | 0,30 Euro bis 0,42 Euro* | 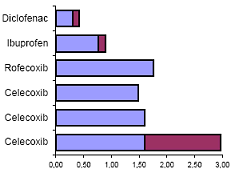 |
| Ibuprofen, 3 x 800mg, 100 Tabl. | 0,76 Euro bis 0,90 Euro | |
| Rofecoxib** 12,5/25mg, 90 Tabl. | 1,76 Euro | |
| Celecoxib, 100 Tabl. | (1 x 200 mg) 1,49 Euro | |
| - Osteoarthrose 1 x täglich | (2 x 100 mg) 1,60 Euro | |
| - Osteoarthrose 2 x täglich | (2 x 100 bis 2 x 200 mg) | |
| - rheumatoide Arthritis | 1,60 Euro bis 2,97 Euro | |
| *(Festbetragsgrenze ) Stand 01.10.2002 | ||
Cilostazol
(z.B. Pletal®, Generika)
Beschluss vom: 21.07.2011
In Kraft getreten am: 22.07.2011
BAnz. 2011, Nr. 154 vom 12.10.2011 S. 3508
| Zugelassene Anwendungsgebiete |
Cilostazol ist seit 2006 in Deutschland zugelassen zur Verlängerung der maximalen und schmerzfreien Gehstrecke bei Patienten mit Claudicatio intermittens, die keinen Ruheschmerz und keine Anzeichen von peripheren Gewebsnekrosen haben (periphere arterielle Verschlusskrankheit Fontaine Stadium II).
Die Überprüfung von Nutzen und Risiken durch die Europäische Arzneimittelagentur (EMA) führte 2013 zur Einschränkung der Zulassung auf den Zweitlinieneinsatz bei Patienten, bei denen Lebensstilumstellungen einschließlich Einstellung des Rauchens und Gehtraining sowie andere angemessene Interventionen die Symptome der Claudicatio intermittens nicht ausreichend verbessern konnten.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Im Stadium II der Peripheren Arteriellen Verschlusskrankheit (PAVK) bzw. Claudicatio intermittens kommt es bei 20 bis 30% der Patienten im Verlauf von 10 Jahren spontan zu einer Symptombesserung. Weitere 20 bis 30 % erleben ein Fortschreiten der Symptomatik. Bei etwa 2 % der Patienten wird eine Amputation notwendig. Die Prognose der Patienten wird entscheidend durch eine deutlich erhöhte kardio- und cerebrovaskuläre Morbidität und Mortalität bestimmt. Sie gelten diesbezüglich als Hochrisikopatienten.
Eine Reduktion des Risikos für kardio- und cerebrovaskuläre Ereignisse ist im Stadium II vorrangiges Behandlungsziel. Weitere Ziele sind eine Hemmung der Progression der AVK, die Vermeidung von Amputationen sowie die Verbesserung der Gehleistung und Verringerung schmerzhafter Symptome.
Therapie der Wahl zur Verbesserung der Gehleistung ist ein Gehtraining. Dabei sind strukturierte, beaufsichtigte Ubungsprogramme am wirksamsten.
Vasoaktive medikamentöse Therapieansätze haben dagegen geringere Effekte. Nur für Naftidrofuryl und Cilostazol konnte eine moderate aber signifikante Wirksamkeit im Hinblick auf die Gehleistung gezeigt werden.
Für keines der zur Behandlung der Claudicatio intermittens zugelassenen Medikamente ist nachgewiesen, dass sie langfristig zu einer verringerten Rate an Amputationen oder vaskulären Interventionen führen. Das deutlich erhöhte kardio- und cerebrovaskuläre Risiko dieser Patienten wird nicht reduziert.
Cilostazol führt gegenüber Placebo zu einer signifikanten Verbesserung der Gehleistung. Der Unterschied zwischen Cilostazol und Placebo entspricht einer Zunahme der maximalen Gehstrecke um ca. 42 m (95 % KI: 21 bis 64 m) auf dem Laufband; dies wird als moderater Behandlungseffekt beurteilt. Dabei ist das individuelle Ansprechen sehr variabel. Nur 11 % der Patienten erfahren nach Abzug des Placeboeffektes eine Steigerung der Gehleistung um mehr als 50 %.
Nur bei ausgewählten Patienten kann ein medikamentöser Therapieversuch wirtschaftlich sein, wenn kumulativ folgende Voraussetzungen vorliegen:
- Die Gehleistung ist nicht durch andere Faktoren limitiert, wie z.B. eine Gonarthrose oder Polyneuropathie.
- Revaskularisierende Interventionen sind nicht angezeigt. Ein Gehtraining ist erfolglos geblieben oder kann nicht durchgeführt werden.
- Eine Das Rauchen wurde eingestellt..
- Aufgrund der Claudicatio bestehen im Alltag relevante Einschränkungen und selbst moderate Verlängerungen der Gehstrecke können ein Zugewinn an Lebensqualität bedeuten. Davon kann bei maximalen Gehstrecken unter 200 m meist ausgegangen werden.
Die Indikation für eine medikamentöse Therapie mit Cilostazol sollte neben anderen Behandlungsmöglichkeiten, wie beispielsweise einer Revaskularisation, sorgfältig geprüft und durch Ärzte mit Erfahrung in der Therapie der PAVK gestellt werden.
Die Indikation für eine zusätzliche Gabe von Cilostazol bei Patienten > 70 Jahre mit bereits bestehender ASS- und Statinmedikation sollte vor dem Hintergrund möglicher Nebenwirkungen, des Interaktionspotenzials und der Tatsache, dass diese Patientengruppe in Studien nicht ausreichend repräsentiert war, besonders streng gestellt werden, um den besonderen Erfordernissen der häufig multimorbiden älteren Patienten und der Wirtschaftlichkeit Rechnung zu tragen.
Eine Überlegenheit von Cilostazol gegenüber Naftidrofuryl ist nicht belegt. Unter dem Aspekt der Wirtschaftlichkeit ist daher die kostengünstigere Therapie zu bevorzugen.
Falls Cilostazol nach drei Monaten keine Wirkung zeigt, soll die Behandlung abgebrochen werden. Bei Fortsetzen der Behandlung soll der Therapieerfolg dokumentiert und jährlich reevaluiert werden. Eine unkritische lebenslange Dauerbehandlung ist nicht indiziert.
Der Behandlungserfolg soll nach 3 Monaten evaluiert werden. Falls Cilostazol keinen klinisch relevanten Effekt zeigt bzw. die Symptome nicht gebessert werden konnten, soll die Behandlung abgebrochen werden. Bei Fortsetzen der Behandlung soll der Therapieerfolg dokumentiert und jährlich reevaluiert werden. Eine lebenslange Dauerbehandlung ohne regelhafte Überprüfung ist nicht indiziert.
| Kosten |
| Wirkstoff | Dosierung | Tagestherapiekosten | Jahrestherapiekosten |
| Naftidrofuryl | 3 x 200 mg | 1,02 Euro | 371 Euro |
| Cilostazol | 2 x 100 mg | 1,64 Euro | 599 Euro |
Kostenberechnung auf Basis der größten verfügbaren Packung bzw. Festbetrag; gesetzliche Pflichtrabatte der Apotheken und pharmazeutischen Unternehmen wurden nicht berücksichtigt.
Stand Lauer Taxe 15. Mai 2014
Andere verschreibungspflichtige durchblutungsfördernde Arzneimittel wie Pentoxifyllin und Buflomedil, welche zur Verlängerung der Gehstrecke bei PAVK zugelassen sind, werden durch die Verordnungseinschränkungen der Anlage III der Arzneimittel-Richtlinie erfasst. Ebenfalls für diese Indikation zugelassene, nicht verschreibungspflichtige ginkgohaltige Arzneimittel gelten nicht als Standardtherapie der Claudicatio und bleiben deshalb von der Verordnung ausgeschlossen.
| Wirkungen |
Cilostazol und mehrere seiner Metaboliten hemmen die Phosphodiesterase-III. Dies führt zur Erhöhung des intrazellulären cAMP-Spiegels in verschiedenen Geweben einschließlich Thrombozyten und Blutgefäßen. Eine reversible Hemmung der Thrombozytenaggregation und vasodilatatorische Effekte wurden nachgewiesen. Die Thrombozytenaggregationshemmung hält bis zu 12 Stunden an und normalisiert sich nach Absetzen innerhalb von 48 bis 96 Stunden ohne Rebound.
Die empfohlene Dosis von Cilostazol sollte jeweils 30 Minuten vor dem Frühstück und Abendessen eingenommen werden. Bei Einnahme zu den Mahlzeiten kommt es zu einem Anstieg der maximalen Plasmakonzentration von Cilostazol, was mit einem vermehrten Auftreten von Nebenwirkungen verbunden sein kann.
Cilostazol wird überwiegend hepatisch durch die Isoenzyme CYP3A4 und in geringerem Ausmaß CYP2C19 metabolisiert. Die Metaboliten werden im Urin ausgeschieden. Eine Dosisreduktion auf zweimal täglich 50 mg wird empfohlen bei Patienten, die gleichzeitig starke Inhibitoren von CYP3A4 (z.B. Azolantimykotika, Makrolide oder Proteasehemmer) oder CYP2C19 (z.B. Omeprazol) einnehmen.
| Wirksamkeit |
Den Zulassungen in Europa ab 2000 und den USA 1999 lagen acht doppelblinde placebokontrollierte Phase-III-Studien über 12 bis 24 Wochen zugrunde. Nach Zulassung wurden zwei weitere große Phase IV-Studien beendet (PACE-, CASTLE-Studie).
Die CASTLE-Studie gehörte zu den Auflagen der FDA bei Zulassung und hatte primär das Ziel, eine überhöhte Mortalität unter Cilostazol auszuschließen. Aufgrund hoher Abbruchraten von 55% und einer geringeren Mortalität als erwartet, erreichte sie dieses Ziel nur knapp. Geringere Unterschiede der Mortalität konnten weder belegt noch sicher ausgeschlossen werden. Daten zur Wirksamkeit trägt diese Studie nicht bei.
Von den weiteren neun Studien sind nur diejenigen sechs Studien publiziert, in denen Cilostazol einem Placebo signifikant überlegen war. Angaben zu den anderen drei Studien lassen sich den Beurteilungsberichten der Zulassungsbehörde entnehmen.
Alle Studien untersuchten die Wirksamkeit von Cilostazol anhand der Veränderung der maximalen Gehstrecke als primären Endpunkt. Eingeschlossen wurden Patienten, deren Gehstrecke aufgrund einer PAVK eingeschränkt war. Patienten mit Ruheschmerzen oder ischämischen Nekrosen wurden ausgeschlossen, ebenso Patienten, deren Mobilität aufgrund anderer Erkrankungen eingeschränkt war. Die Gehstrecken wurden durch Laufbandbelastung bestimmt. Dabei wurden drei unterschiedliche Protokolle für die Laufbandbelastung verwendet. Die Patienten erhielten verblindet über 12 bis 24 Wochen Cilostazol oder ein Placebo.
Zu Studienbeginn lagen die durchschnittlichen maximalen Gehstrecken bei 120 bis 279 m. Unter der zugelassenen Standarddosis von 2 x 100 mg Cilostazol betrugen die durchschnittlichen Steigerungen der maximalen Gehstrecken gegenüber Placebo bei Studienende 2 m bis 106 m. Die Medianwerte lagen bei minus 7 m bis 62 m.
Eine Metaanalyse der neun Studien ergab, dass die maximale Gehstrecke bei Studienende unter Cilostazol im Durchschnitt um 42 m länger war als unter Placebo. In einer IPD-Metaanalyse (IPD = Individual patient data: gepoolte individuelle Patientendaten) zeigte sich eine um 35 % größere Zunahme der maximalen Gehstrecke gegenüber Baseline im Vergleich zu Placebo (Cilostazol 59,4 %, Placebo 24,3 %).
Aus einer IPD-Metaanalyse der Differenzen der logarithmierten Gehstrecken ergab sich ein Behandlungseffekt nach Abzug des Placeboeffektes von 15 % (95 % KI: 11 % bis 19 %) hinsichtlich der maximalen Gehstrecke und von 16 % (95 % KI: 11 % bis 21 %) hinsichtlich der schmerzfreien Gehstrecke.
Zunahme der maximalen Gehstrecke unter Cilostazol 2 x 100 mg in Studien
| Code | Autor/Akronym | Dauer | Zunahme [m] CLZ - PL | P | |||
| [Wo] | n* | Mittel ** | 95% KI | Median*** | |||
| 21-98-213 | PACE**** | 24 | 520 | 1 | -22 bis 24 | - | 0,910 |
| 21-96-202 | Dawson | 24 | 466 | 43 | 15 bis 70 | 24 | 0,003 |
| 21-92-202 | Beebe | 24 | 345 | 102 | 36 bis 168 | 26 | 0,002 |
| 21-94-201 | Strandness | 24 | 262 | 58 | 26 bis 90 | 18 | 0,000 |
| 21-94-301 | **** | 24 | 247 | 34 | -1 bis 68 | 8 | 0,056 |
| 21-94-203 | Money | 16 | 239 | 65 | 34 bis 96 | 62 | 0,000 |
| 21-95-201 | **** | 12 | 142 | -2 | -23 bis 26 | -7 | 0,896 |
| 21-93-201 | Elam | 12 | 189 | 44 | 6 bis 82 | 31 | 0,024 |
| 21-90-201 | Dawson | 12 | 81 | 106 | 30 bis 183 | 27 | 0,007 |
| 42 | 21 bis 64 | <0.0001 | |||||
CLZ: Cilostazol,
PL: Placebo
* randomisierte Patienten, nur Placebo und Cilostazol 2 x100 mg, Assessment Report der EMA
** Angaben nach Pande 2010 bzw. Beurteilungsbericht der FDA
*** nach Beurteilungsbericht der FDA
**** Studien nicht publiziert
Eine Responderanalyse aller 24-Wochen-Studien ergibt, dass der Anteil Patienten, die eine Steigerung der maximalen Gehstrecke um mehr als 50 % erleben, von 26 % unter Placebo auf 37 % unter Cilostazol zunimmt. Neben dem relativ starken Placeboeffekt fällt auf, dass das individuelle Ansprechen sehr variabel ist und Cilostazol bei der überwiegenden Mehrheit der Patienten nicht zu einer klinisch bedeutsamen Zunahme der Gehstrecke führt.
Es finden sich Hinweise, dass in jüngeren Studien mit zunehmender Komedikation von ASS und Statinen die Wirksamkeit von Cilostazol geringer ausfällt. Aufgrund geringer Fallzahlen dieser Subgruppenanalysen zur Komedikation kann bisher jedoch noch keine sichere Aussage gemacht werden.
In einer Subgruppenanalyse über acht Studien war Cilostazol bei Diabetikern weniger stark wirksam als bei Nicht-Diabetikern.
In drei Studien gab es zusätzlich zu Placebo einen aktiv kontrollierten Behandlungsarm mit Pentoxifyllin. In allen drei Studien war die Wirksamkeit von Pentoxifyllin nicht besser als Placebo. Die beiden größeren Studien, welche darauf angelegt waren, statistisch signifikante Unterschiede zwischen Cilostazol und Pentoxifyllin zu erfassen, ergaben inkonsistente Ergebnisse. Während Dawson et al. einen signifikant stärkeren Behandlungseffekt von Cilostazol gegenüber Pentoxifyllin und Placebo berichtet, zeigten sich in der unveröffentlichten PACE-Studie keine Unterschiede.
Direkt vergleichende Studien zu einem Gehtraining fehlen. Eine Metaanalyse von sechs Studien mit insgesamt 391 Patienten zeigt ausgeprägtere Effekte als eine medikamentöse Therapie mit Cilostazol. Gegenüber Patienten, die kein Gehtraining durchführten, kam es zu einer signifikanten Verlängerung der maximalen Gehstrecke um 116 m bei allerdings breitem Konfidenzintervall (95% KI: 27 bis 205 m). Besonders ausgeprägt sind die Effekte eines strukturierten supervidierten Gehtrainings, welches im Vergleich zu einem nicht beaufsichtigten Gehtraining die maximale Gehstrecke um 58 % verlängerte.
| Risiken - ggf. Vorsichtsmaßnahmen |
Andere Phosphodiesterase-III-Hemmer haben bei Patienten mit Herzinsuffizienz arrhythmogene Wirkungen gezeigt und bei oraler Langzeittherapie zu einer Verschlechterung der Herzinsuffizienz mit verkürzter Lebenserwartung geführt.
In der einzigen Langzeitstudie, der sogenannten CASTLE-Studie, wurde eine Übersterblichkeit unter Cilostazol ausgeschlossen. 1439 Patienten mit intermittierender Claudicatio wurden bis zu drei Jahren mit Cilostazol oder Placebo behandelt. In der ITT-Analyse traten 49 Todesfälle unter Cilostazol und 52 unter Placebo auf. Aufgrund hoher Abbruchraten und einer geringeren Mortalität als erwartet, erreichte die Studie nicht die notwendige Anzahl Probanden, um geringe Unterschiede der Mortalität sicher auszuschließen. Nach 36 Monaten nahm die Mehrheit der Teilnehmer die Studienmedikation nicht mehr ein (Cilostazol 68 % vs. Placebo 64 %). Das Studienziel, eine Zunahme des Sterberisikos um 75 % auszuschließen, wurde unter Berücksichtigung der Konfidenzintervalle jedoch erreicht (HR 0,94, 95 % KI 0,64 bis 1,39).
Da Patienten mit klinisch manifester Herzinsuffizienz von der Teilnahme ausgeschlossen wurden, sind die Langzeitwirkungen bei diesen Patienten unbekannt. Cilostazol ist deshalb bei kongestiver Herzinsuffizienz kontraindiziert.
NEUPatienten mit ventrikulärer Tachykardie, Kammerflimmern, multifokalen ventrikulären Ektopien und schwerer Tachyarrythmie in der Vorgeschichte sowie Patienten mit Verlängerung des QTc-Intervalls dürfen ebenfalls kein Cilostazol einnehmen.
Die hohen Abbruchraten in der CASTLE-Studie traten bei 22,5 % der Patienten unter Cilostazol als Folge unerwünschter Ereignisse auf, die auf die Behandlung zurückgeführt wurden (Placebo 14,9 %). Die Inzidenzen von Kopfschmerzen (2,1% vs. 0,3 %), Durchfällen (2,8% vs. 0,7 %), Palpitationen (1,1 % vs. 0 %) und peripheren Ödemen (1,1 % vs. 0 %) waren unter Cilostazol größer als unter Placebo. Hingegen traten Studienabbrüche wegen kardialer Nebenwirkungen in beiden Behandlungsgruppen gleich häufig auf (5,6 % vs. 5,8 %).
Gegenüber Placebo fand sich kein erhöhtes Risiko für schwerwiegende Blutungen. Allerdings war die Inzidenz in der Subgruppe von Patienten, die gleichzeitig ASS und Clopidogrel einnahmen, erhöht (n = 307, 13,3 % Cilostazol vs. 8,7 % Placebo). Kontraindiziert ist nach erneuter Risikobewertung durch die EMA daher die Kombination von Cilostazol mit zwei oder mehr Thrombozytenaggregationshemmern bzw. Antikoagulantien (z.B. Acetylsalicylsäure, Clopidogrel, Heparin, Vitamin-K-Antagonisten wie Phenprocoumon, Dabigatran, Rivaroxaban oder Apixaban). Vorsicht ist bereits geboten bei Kombination von Cilostazol mit einem Wirkstoff dieser Gruppen.
Laut Fachinformation sollen Patienten mit einem Blutungsrisiko (z.B. aktives peptisches Ulkus, hämorrhagischer Schlaganfall innerhalb der letzten sechs Monate, proliferative diabetische Retinopathie, schlecht eingestellte Hypertonie) kein Cilostazol einnehmen.
Aufgrund der thrombozytenaggregationshemmenden Wirkung von Cilostazol ist bei Operationen ein erhöhtes Blutungsrisiko möglich (auch bei kleineren Eingriffen wie z.B. Zahnextraktion). Es wird daher empfohlen, Cilostazol fünf Tage vor elektiven Eingriffen abzusetzen.
Wegen seines Wirkmechanismus kann Cilostazol unter anderem Tachykardie, Palpitationen, Tachyarrhythmie und/oder Hypotonie hervorrufen. Der Anstieg der Herzfrequenz unter Cilostazol kann bei Risikopatienten zu Angina pectoris führen.
Patienten mit einem erhöhten Risiko für schwere unerwünschte kardiale Ereignisse als Folge einer erhöhten Herzfrequenz, z.B. Patienten mit stabiler koronarer Herzkrankheit, sollten während der Behandlung mit Cilostazol engmaschig überwacht werden.
Bei Patienten mit instabiler Angina pectoris oder Myokardinfarkt/Koronarintervention in den letzten 6 Monaten oder einer anamnestisch bekannten schweren Tachyarrhythmie ist Cilostazol nach Risikobewertung durch die EMA kontraindiziert.
Bei Patienten mit atrialer oder ventrikulärer Ektopie sowie bei Patienten mit Vorhofflimmern oder -flattern soll Cilostazol nur mit Vorsicht angewendet werden.
Da Cilostazol überwiegend durch CYP-Enzyme metabolisiert und mit dem Urin ausgeschieden wird, ist es bei Patienten mit mittelschweren bis schweren Leberfunktionsstörungen oder einer Kreatininclearance von < 25 ml/min kontraindiziert. Vielfältige Interaktionsmöglichkeiten mit anderen Arzneimitteln sind zu bedenken.
Starke Inhibitoren der CYP3A4 (z.B. Makrolide wie Erythromycin und Clarithromycin, Azol-Antimykotika wie Ketoconazol oder Protease-Inhibitoren) oder Inhibitoren der CYP2C19 (z.B. Protonenpumpenhemmer) erhöhen die pharmakologische Gesamtaktivität von Cilostazol und können die unerwünschten Wirkungen von Cilostazol verstärken. Aus diesem Grund beträgt die empfohlene Dosis für Patienten, die starke CYP3A4- oder CYP2C19- Inhibitoren einnehmen, 50 mg zweimal täglich.
Es hat selten oder sehr selten Berichte über Blutbildveränderungen einschließlich Thrombozytopenie, Leukopenie, Agranulozytose, Panzytopenie und aplastischer Anämie gegeben. Die meisten Patienten erholten sich nach Absetzen von Cilostazol. Jedoch verliefen einige Fälle von Panzytopenie und aplastischer Anämie tödlich.
Erythropoesestimulierende Wirkstoffe
(zur Behandlung der symptomatischen Anämie bei Tumorpatienten, die eine Chemotherapie erhalten)
Beschluss vom: 17.06.2010
In Kraft getreten am: 18.06.2010
BAnz. 2010, Nr. 158 vom 19.10.2010 S. 3473
| Zugelassene Anwendungsgebiete |
Unter den Erythropoesestimulierenden Wirkstoffen (Erythropoiesis-Stimulating Agents, ESAs) sind in Deutschland Epoetin alfa und Epoetin zeta zur Behandlung der Anämie und zur Reduktion des Transfusionsbedarfs bei Erwachsenen mit soliden Tumoren, malignen Lymphomen und multiplem Myelom, die eine Chemotherapie erhalten und bei denen das Risiko einer Transfusion aufgrund des Allgemeinzustandes besteht, zugelassen. Epoetin betu, Epoetin theta sowie Darbepoetin alfa sind zugelassen zur Behandlung der symptomatischen Anämie bei erwachsenen Tumorpatienten mit nichtmyeloischen Erkrankungen, die eine Chemotherapie erhalten.
Nicht dargestellt an dieser Stelle ist die symptomatische Anämie bei chronischer Niereninsuffizienz, zu deren Behandlung alle ESAs - pegyliertes Epoetin bete und Epoetin delta eingeschlossen - zugelassen sind. Zusätzliche, hier ebenfalls nicht besprochene seltenere Indikationen von Epoetin alfa, Epoetin bete oder Epoetin zeta sind die Steigerung der autologen Blutgewinnung bei Patienten, die an einem autologen Spendeprogramm zur Vermeidung von Fremdblutkonserven teilnehmen, und/oder die Reduktion von Fremdblut bei Patienten mit hohem Risiko von Transfusionskomplikationen, die vor einem elektiven großen orthopädischen Eingriff stehen und nicht an einem autologen Blutspendeprogramm teilnehmen können sowie die Prävention der Frühgeborenenanämie (Epoetin beta).
Tabelle 1: Indikationsübersicht von Präparaten, die zur Behandlung der Anämie onkologischer Patienten zugelassen sind
| Erypo FS 06/2008 | Epoetin alfa Hexal 08/2008 Abseamed 02/2008 Binocrit 03/2008 | Silapo 08/2008 Retacrit 12/2007 | NeoRecormon 500-30.000 I.E. 02/2008 | Biopoin 09/2009 Eporatio 09/2009 | Aranesp 02/2008 | |
| Symptomat. A. bei E u. K mit chron. Nierenversagen | + | + | + | + | + (nur bei E) | + |
| A. und Reduktion des Transfusionsbedarfs bei E (solid. Tu, malig. Lymphome, multiples Myelom) mit Chemotherapie | + | + | + | |||
| Symptomat. A. bei E mit nichtmyeloider malig. Erkrank., mit Chemotherapie | + | + | + | |||
| Steigerung autalog. Blutgewinnung in Eigenblutspendeprogramm | + | + | + | |||
| Redukt. Fremdblut vor gr. elektiv. orthopäd. Eingriffen, hohes Risiko Transfusionskomplikationen, ohne Eigenblutspendeprogramm | + | + | ||||
| Prävention Frühgeb.-Anämie | + | |||||
| Epoetin alfa | Epoetin zeta | Epoetin beta | Epoetin theta | Darbepoetin alfa | ||
A: Anämie; E: Erwachsene:
K: Kinder
Quellen:
Fachinformationen/European Public Aseessment Reports (EPARs)
| Empfehlung zur wirtschaftlichen Verordnungsweise |
ESAs werden intravenös oder subkutan appliziert und stimulieren wie das körpereigen Hormon Erythropoetin (EPO) die Proliferation, Differenzierung und das Überleben von Vorläuferzellen der Erythropoese im Knochenmark. Die biologischen Wirkungen der gentechnologisch hergestellten ESAs werden ebenso wie die des Glykoproteins EPO durch Bindung an den Erythropoetin-Rezeptor (EPO-R) vermittelt, der spezifisch auf Vorläuferzellen der Erythropoese im Knochenmark exprimiert wird.
Zahlreiche randomisierte, kontrollierte Studien (RCT) bei Erwachsenen mit Tumorassoziierter Anämie konnten zeigen, dass unter Chemotherapie ESAs gegenüber Placebo das hämatologische Ansprechen (definiert als transfusionsunabhängiger Anstieg des Hämoglobin (Hb)-Wertes > 2 g/dl) verbessern und den Transfusionsbedarf an Erythrozytenkonzentraten (EK) reduzieren. Die Wirksamkeit von ESAs hinsichtlich des hämatologischen Ansprechens und des Transfusionsbedarfs konnte auch in systematischen Übersichtsarbeiten und Metaanalysen bestätigt werden. Demgegenüber ist die klinische Bedeutung der in einigen Studien berichteten Verbesserung der Lebensqualität durch ESAs aufgrund methodischer Mängel in der Erfassung der Lebensqualität umstritten. Darüber hinaus haben RCTs bei Patienten mit unterschiedlichen Tumorentitäten (maligne Lymphome, Tumore im Kopf-Hals-Bereich, Mamma-, Zervix- und nichtkleinzelliges Bronchialkarzinom) schwerwiegende Risiken (z.B. thromboembolische Komplikationen, verkürztes krankheitsfreies bzw. Gesamtüberleben) unter Gabe von ESAs ergeben. In diese Studien (s. Tabelle 3) wurden euch Tumorpatienten eingeschlossen die weder eine Chemotherapie noch eine Bestrahlung oder nur eine Bestrahlung erhalten hatten, und Hb-Werte angestrebt (z.B. 12-15 (OB, die nicht dem zugelassenen Anwendungsgebiet der ESAs bei Tumorpatienten entsprachen. Sowohl von der US Food and Drug Administration (FDA) als auch von der europäischen Arzneimittelagentur (EMA) wurden daraufhin Warnhinweise und Vorsichtsmaßnahmen für die Anwendung von ESAs bei Tumorpatienten mit Chemotherapieassoziierter Anämie veröffentlicht und zuletzt im Juli 2008 aktualisiert. Bei der Verordnung von ESAs müssen deshalb die folgenden Punkte unbedingt berücksichtigt werden:
- Vor Verordnung der ESAs müssen andere mögliche Ursachen einer Anämie (s. Abschnitt Wirkungen) ausgeschlossen und bei laborchemischen Hinweisen für einen Speichereisen- bzw. funktionellen Eisenmangel eine parenterale Eisensubstitution parallel zur Gabe von ESAs begonnen werden. Auch während der Behandlung mit ESAs sind die Eisenspeicher zu überprüfen und ggf. Eisen zu substituieren.
- Die Verordnung von ESAs bei Tumorpatienten mit symptomatischer Anämie, die keine Chemotherapie erhalten, ist nicht indiziert.
- In einigen klinischen Situationen sollte die symptomatische Anämie bei Tumorpatienten aufgrund der Möglichkeit einer Stimulation des Tumorwachstums oder des gehäuften Auftretens von Thromboembolien bevorzugt mit Bluttransfusionen behandelt werden. Insbesondere Patienten mit symptomatischer Anämie unter Chemotherapie, deren Tumorerkrankung eine gute Prognose aufweist und/oder deren Therapie mit kurativer Zielsetzung erfolgt (z.B. adjuvante Chemotherapie des Mammakarzinoms), sollten bevorzugt EKs verabreicht werden.
- Aussagefähige prädiktive Faktoren, die frühzeitig Hinweise auf ein Ansprechen auf eine Therapie m ESAs geben könnten, wurden in den bisherigen Studien nicht ermittelt.
- Vor Verordnung von ESAs sollte eine Nutzen-Risiko-Abwägung unter Einbeziehung des Patienten erfolgen, die unter anderem folgende Faktoren einschließt: Art, Stadium und Prognose der Erkrankung, Schweregrad der Anämie, klinische Situation (z.B. kardiovaskuläre oder pulmonale Begleiterkrankungen), Behandlungspräferenz der Patienten. Die Patienten müssen über die heute bekannten Risiken bei Gabe von ESAs (thromboembolische Komplikationen, Stimulation des Tumorwachstums, verkürztes krankheitsfreies bzw. Gesamtüberleben) sorgfältig informiert werden.
- Die Verordnung von ESAs zur Behandlung der Anämie sollte nur bei Vorliegen von Anämie-Symptomen und in den zugelassenen Anwendungsgebieten erfolgen. Durch RCTs belegte Therapieziele sind ein Anstieg des Hb-Wertes und eine Verringerung bzw. Vermeidung von Bluttransfusionen.
- Nach den vorliegenden Studienergebnissen müssen in Abhängigkeit vom Basisrisiko, eine Bluttransfusion zu benötigen, z.B. bei einem Risiko von 30% oder 70%, zehn bzw. vier Patienten mit ESAs behandelt werden, um eine Bluttransfusion zu vermeiden. Durchschnittlich wurde der Transfusionsbedarf gegenüber der Kontrollgruppe (durchschnittlicher Verbrauch: 3,39 Konserven) um 1,05 EKs reduziert.
- Für den therapeutischen Einsatz gelten alle heute verfügbaren ESAs entsprechend ihrem zugelassenen Applikationsweg als äquivalent. Für die als sogenannte "Biosimilars" von der Europäischen Kommission zugelassenen ESAs wurde in Zulassungsverfahren nachgewiesen, dass sie in Qualität, Wirksamkeit und Unbedenklichkeit den Zulassungsanforderungen im Vergleich zu dem jeweiligen Referenzarzneimittel entsprechen.
- Die Behandlung der Chemotherapieassoziierten Anämie sollte bei Hb-Werten < 10 g/dl begonnen und ESAs so dosiert werden, dass Hb-Werte zwischen 10 und 12 g/dl bzw. zwischen 6,2 mml/l und 7,45 mmol/l erreicht werden (vgl. Fachinformationen). Bei Hb-Werten >12 g/dl muss eine Dosisanpassung der ESAs entsprechend der Fachinformation erfolgen und bei Hb-Werten > 13 g/dl bzw. 8.07 mmol/l die Gabe von ESAs beendet werden. Nach einem Absinken des Hb-Wertes unter 12 g/dl ist die Therapie mit einer Dosis von 25% unter der vorherigen wieder aufzunehmen. Falls unter Behandlung mit ESAs nach 6-8 Wochen die Therapieziele (hämatologisches Ansprechen, Verringerung des Transfusionsbedarfs) trotz Überprüfung und ggf. Anpassung der Dosis nicht erreicht werden, sollte die Verabreichung von ESAs ebenfalls beendet werden.
- Bei Patienten mit hämatologischen Systemerkrankungen (z.B. multiples Myelom, maligne Lymphome) sollte vor der individuellen Entscheidung über die Gabe von ESAs zunächst eine Behandlung mit Chemotherapie und/oder Kortikosteroiden erfolgen, um die Zahl der Tumorzellen im Knochenmark zu reduzieren. Bei gleichzeitiger Verordnung von Wirkstoffen mit erhöhtem Risiko für thromboembolische Komplikationen (z.B. Thalidomid und Lenalidomid bei multiplem Myelom) ist eine besonders kritische Nutzen-Risiko-Abwägung der Verordnung von ESAs erforderlich.
- Spätestens 4 Wochen nach Beendigung der Chemotherapie sollte die Behandlung mit ESAs abgesetzt werden.
| Kosten |
Die in der nachfolgenden Kostentabelle angegebene Dosierung bezieht sich auf die Anfangsdosis gemäß Fachinformation. Die Kostenberechnung erfolgte anhand des kostengünstigsten Präparates (Apothekenverkaufspreis) einschließlich Import.
Tabelle 2: Kostenübersicht (Stand Lauertaxe:15. September 2010)
| Wirkstoff | Präparat (Injektionslösung in Fertigspritze) | Dosis | Kosten pro Einzeldosis [Euro] 1 | Kosten pro Woche [Euro]1 | Kosten für 8 Wochen [Euro] 1 |
| Epoetin alfa | Erypo® FS 10000 I. E./m1 | 150 I.E./kg KG 3 x/Woche 3 | 133,06 | 399,18 | 3193,40 |
| Eprex ® FS 10000 I. E/ml | 130,55 | 391,65 | 3133,16 | ||
| Abseained® 10000 I.E./1 ml 2 | 101,92 | 305,77 | 2446,12 | ||
| Binocrit® 10000 I.E./1 ml 2 | 101,92 | 305.77 | 2446,12 | ||
| Epoetin alfa Hexal® 10000 I.E./1 ml 1 | 101,92 | 305,77 | 2996,12 | ||
| Epoetin zeta | Silapo® 10000 I.E./1,0 ml 2 | 150 I.E./kg KG 3 x/Woche 3 | 101,92 | 305,77 | 2996,12 |
| Retacrit® 10000 I.E./1,0 ml 2 | 101,92 | 305,77 | 2446,12 | ||
| Epoetin bete | NeuRecormon® 30000 I.E. | 30000 I.E./Woche 4 | 363,83 | 363,83 | 2910,69 |
| Epoetin theta | Biopoin® 20000 I.E./ml 2 | 20000 I.E./Woche 7 | 211,97 | 211,97 | 1695,73 |
| Eporatio® 20000 I.E./ml 2 | 211,97 | 211,97 | 1695,76 | ||
| Darbepoetin alfa | Aranesp® 150 μ g | 2,25 μ g/kg KG/ Woche 5 | 450,32 | 450,32 | 3602,56 |
| Erythrozytenkonzentrat | |||||
| Erythrozytenkonzentrat | 225-375 ml/Beutel 6 | ca. 90,00 Euro/Beutel 6 | |||
1) Kostenberechnung für eine ca. 70 kg schwere Person
2) Biosimilar
3) alternative Dosierung:450 I.E./kg KG einmal pro Woche
4) entsprechend ca. 450 I.E./kg KG pro Woche berechnet auf einen Patienten mit durchschnittlichem Gewicht
5) alternative Dosierung: 500 μ g (6,75 μ g/kg KG) alle 3 Wochen
6) Angaben:
DRK, Blutspendedienst Deutschland, 22. Oktober 2008
7) empfohlene Anfangsdosis, unabhängig vom Körpergewicht einmal pro Woche
Nach Ablauf des Patentschutzes für die erstzugelassenen ESAs stehen als "Biosimilars" derzeit sieben Präparate zur Verfügung. Sie enthalten ebenfalls gentechnologisch hergestelltes Epoetin alfa, Epoetin zeta bzw. Epoetin theta und sind wie die Referenzarzneimittel zur Behandlung der symptomatischen Anämie von Tumorpatienten, die eine Chemotherapie erhalten, zugelassen. Zur Zulassung wurden der Europäischen Kommission entsprechend den für die Nachfolgeprodukte biologischer Arzneimittel entwickelten Richtlinien die Ergebnisse geeigneter Untersuchungen vorgelegt, die nicht nur den enthaltenen Wirkstoff hinreichend charakterisieren, sondern die auch nachweisen, dass die Anforderungen in Bezug auf die Sicherheit und Wirksamkeit des geprüften Arzneimittels erfüllt werden.
| Wirkungen |
In Deutschland sind verschiedene gentechnologisch hergestellte Varianten des körpereigenen Glykoproteins EPO zur Behandlung bei Tumorpatienten der symptomatischen, Chemotherapieassoziierten Anämie zugelassen (s. Tabelle 1). Hierzu zählen: Epoetin alfa, bete, theta, zeta, sowie das Epoetin-Analogon Darbepoetin alfa. Biochemische Unterschiede (z.B. in der Anzahl der Stickstoffgebundenen Kohlenhydratseitenketten, in der Aminosäuresequenz) führen zu Unterschieden im Molekulargewicht, der Affinität zum EPO-R und der Pharmakokinetik. So besitzt Darbepoetin alfa gegenüber Epoetin alfa bzw. bete eine etwa dreifach längere Serumhalbwertszeit und weist gleichzeitig eine um etwa den Faktor vier verminderte Affinität zum EPO-R auf. Diese biochemischen Unterschiede bedingen jedoch keine Unterschiede in den pharmakodynamischen Eigenschaften. Deshalb werden auch alle Wirkstoffe unter der Bezeichnung "Erythropoiesisstimulating agents" (ESAs) zusammengefasst.
Das Hormon EPO wird vorwiegend von der Niere gebildet und ist ein essenzieller Wachstums- und Überlebensfaktor für erythroide Vorläuferzellen. Nach Bindung von EPO an spezifische EPO-R auf Vorläuferzellen der Erythropoese im Knochenmark werden Proliferation und Differenzierung stimuliert sowie das Überleben dieser Zellen verlängert, vor allem durch eine Hemmung des frühzeitigen, programmierten Zelttods (Apoptose). Jede Form von Sauerstoffmangel (Hypoxiel führt zu einer verstärkten Produktion von EPO und somit zu einer Zunahme der Sauerstofftransportkapazität.
Neben seiner wichtigen Funktion als Wachstumsfaktor der Erythropoese haben besonders präklinische Studien in den letzten lehren die Rolle von EPO als pleiotropes Zytokin verdeutlicht. Darüber hinaus wurde an Tumorzelllinien, aber auch an primärem Tumorgewebe von Patienten sowohl eine Expression von EPO als auch von EPO-R gezeigt. Die pathophysiologische Bedeutung dieser Befunde für eine Stimulation des Tumorwachstums, z.B. durch direkte Wirkungen von EPO bzw. ESAs auf Tumorzellen oder indirekt über eine Induktion der Neoangiogenese in Tumoren, ist jedoch noch unklar.
Das Auftreten einer Anämie ist eine häufige Komplikation bei Tumorpatienten, wobei die Angaben zur Prävalenz bzw. Inzidenz in Abhängigkeit von der Definition der Anämie, der Art und dem Stadium der Tumorerkrankung und der durchgeführten Behandlung (z.B. Chemotherapie, Bestrahlung) variieren. Das National Cancer Institute (NCI) hat einheitliche Kriterien für die Klassifikation der Anämie, basierend auf den Hb-Werten, vorgeschlagen:
- Grad 0: Hb-Wert im Normbereich (12,0-16,0 g/dl bei Frauen und 14,0-18,0 g/dl bei Männern)
- Grad 1: milde Anämie (Hb-Wert >10 g/dl bis Normbereich)
- Grad 2: mäßige Anämie (Hb-Wert 8,0-10,0 g/dl)
- Grad 3: schwere Anämie (Hb-Wert 6,5-7,9 g/dl)
- Grad 4: lebensbedrohliche Anämie (Hb-Wert <6,5 g/dl).
In einer großen multinationalen, europäischen, prospektiven Beobachtungsstudie bei Patienten mit soliden Tumoren oder hämatologischen Neoplasien betrug die Prävalenz der Anämie bei Diagnose 39,3 % und lag im Verlauf der Beobachtungsstudie bei 67,0%. Die Patienten wiesen bei Diagnose meistens eine milde Anämie auf, bei etwa 14% der Patienten lagen im weiteren Verlauf die Hb-Werte bei <10,0 g/dl und etwa 50% der mit Chemotherapie behandelten Patienten hatten eine Anämie. In dieser Beobachtungsstudie wurde eine Korrelation zwischen Abnahme der Hb-Werte und Verschlechterung des Allgemeinzustandes beobachtet. Anämien traten besonders häufig bei Patienten mit hämatologischen Systemerkrankungen (maligne Lymphome, multiples Myelom), gynäkologischen Tumorerkrankungen und Lungenkarzinom auf. Die Pathophysiologie der bei Tumorerkrankungen auftretenden Anämie ist multifaktoriell. Auslösende Faktoren können sein: Knochenmarkbeteiligung bei z.B. hämatologischen Systemerkrankungen, Eisen- oder Vitamin B12- bzw. Folsäuremangel, okkulte Blutverluste, Niereninsuffizienz, Hämolyse und Suppression der Erythropoese infolge z.B. autoimmuner Prozesse bzw. Chemo- und/oder Strahlentherapie. Erst nach Ausschluss dieser Ursachen wird von einer Anämie bei chronischen Erkrankungen gesprochen, die nach der Eisenmangelanämie die zweithäufigste Anämieform in Deutschland ist. Als wesentliche pathophysiologische Mechanismen der Anämie bei chronischen Erkrankungen gelten heute eine verminderte Synthese von EPO, eine verminderte Ansprechbarkeit von erythroiden Vorläuferzellen auf EPO, eine verkürzte Überlebenszeit der Erythrozyten sowie ein Einfluss der Tumorerkrankung bzw. chronischen Entzündung auf die Eisenhomöostase. Die hohe Prävalenz der Tumoranämie, deren Auswirkungen auf den Allgemeinzustand bei Tumorpatienten und die an der Entstehung der Anämie bei chronischen Erkrankungen beteiligten pathophysiologischen Faktoren Bind Grund für die Gabe von ESAs bei Tumorpatienten mit Anämie.
| Wirksamkeit |
Die Verordnung von ESAs bei Tumorpatienten mit symptomatischer Anämie hat folgende Therapieziele: Anstieg des Hb- Wertes, Vermeidung von Anämiebedingten Symptomen bzw. Organschäden, Reduktion bzw. Verhinderung der Transfusion von EKs, Verbesserung der Lebensqualität. Zu diesen Zielen wurden in der Vergangenheit zu ESAs zahlreiche randomisierte, kontrollierte Studien an Patienten mit soliden und/oder hämatologischen Neoplasien publiziert. Allerdings zeichneten sich diese klinischen Prüfungen durch eine sehr variable Methodik und Qualität aus. Die untersuchten Patientenkollektive unterschieden sich erheblich hinsichtlich ihrer begleitenden Therapien (Chemotherapie mit/ohne Cisplatin, Strahlenbehandlung, keine spezifische Begleittherapie) sowie Dosierungen bzw. Dosierungsschemata der verabreichten ESAs. Häufig fehlten auch Daten zum Hb-Ausgangswert, der eine Beurteilung des Schweregrades der Anämie ermöglicht hätte, und in keiner Studie wurden unterschiedliche Hb-Schwellenwerte im Hinblick auf die erreichten Endpunkte miteinander verglichen. Auch zu sonstigen, potenziell prädiktiven Faktoren konnten keine validen Schlussfolgerungen aus den Studienergebnissen gezogen werden.
Unter Berücksichtigung dieser methodischen Defizite wurden die vorhandenen Publikationen in den letzten Jahren von mehreren Institutionen im Rahmen von Metaanalysen oder systematischen Reviews ausgewertet Die nachfolgend dargestellten Erkenntnisse der Cochrane-Gruppe aus 57 Studien an 9353 Patienten wurden in aktuellen Leitlinien zum Einsatz von Epoetin und Darbepoetin bei onkologischen Patienten wie auch in aktuellen Fachinformationen und in der Risikoinformation des Bundesinstitutes für Arzneimittel und Medizinprodukte vom 31. Juli 2008 berücksichtigt.
| Hämatologisches Ansprechen |
Hämatologisches Ansprechen, definiert als Anteil der Studienteilnehmer mit einem Hb-Anstieg um mindestens 2 g/dl oder einem Hämatokrit (Hkt)-Anstieg um mindestens sechs Prozentpunkte, wurde für Epoetin und Darbepoetin bei der Mehrzahl von Patienten mit hämatologischen Neoplasien oder soliden Tumoren und einem Hb-Wert <12 g/dl zu Studienbeginn gezeigt [Relatives - Risiko (RR) 3,43; 95%, Konfidenzintervall (IC1) 3,07-3,84; ingesamt = 4307]. In 18 der 22 einbezogenen Studien erhielten die Patienten eine Chemotherapie, davon in neun Studien eine platinhaltige. Ohne begleitende Tumortherapie wurden vier Studien und in keiner Studie wurde eine Strahlentherapie durchgeführt.
| Transfusionsrisiko |
Entsprechend einer Auswertung von 42 Studien mit insgesamt 6510 Patienten konnte das relative Risiko für Bluttransfusionen durch Gabe von ESAs signifikant um 38% gesenkt werden (RR 0,64; 95% K10,60-0,68). Gut doppelt so häufig wurde eine platinhaltige wie eine platinfreie Chemotherapie gegeben (in 22 bzw. 10 Studien). In vier Studien wurde auf eine Chemotherapie verzichtet, in zwei Studien eine Radio- bzw. Radiochemotherapie eingesetzt, vier Studien mussten hinsichtlich der Begleittherapie als unklar eingeordnet werden. Eine differenzierte Berechnung zur Abschätzung der absoluten Risikoreduktion ergab in Abhängigkeit vom Basisrisiko der Patienten, eine Bluttransfusion zu benötigen, unterschiedliche Werte für die Number needed to treat: bei einem Risiko von 30% müssen demnach zehn Patienten mit ESAs behandelt werden, um bei einem Patienten eine Bluttransfusion zu vermeiden, in einer Population mit einem Basisrisiko von 70% sind es nur vier Patienten.
Bezogen auf die Anzahl vermiedener Blutkonserven ergab sich bei einem Gesamtkollektiv von 2353 Studienteilnehmern aus 14 Studien ein durchschnittlich geringerer Verbrauch von 1,05 Erythrozytenkonserven (95 % KI-1,32 bis-0,78) im Vergleich zur Kontrollgruppe (durchschnittlicher Verbrauch: 3,34 Konserven). Insgesamt wird der Einfluss der ESAs auf diesen Zielparameter zwar als statistisch signifikanter, klinisch aber bescheidener Effekt bewertet.
Erythrozytentransfusionen sind daher unter Berücksichtigung des HI> und/oder LW-Wertes sowie des klinischen Gesamtbildes (z.B. kardiovaskuläre oder pulmonale Erkrankungen) - generell als geeignete Behandlungsoption bei schweren Tumorbedingten Anämien anzusehen, besonders auch im Hinblick auf die Risiken einer Therapie mit ESAs. Bei der Notwendigkeit einer sofortigen Korrektur einer Anämie oder im Notfall sind sie das Mittel der Wahl.
| Überlebensdauer |
Als weiteren patientenrelevanten Parameter untersuchte die Cochrane-Gruppe die Gesamtsterblichkeit aus den Ergebnissen von 42 Studien. Unter diesen fanden sich überwiegend Studien an Patienten mit soliden Tumoren (25 Studien), aber auch hämatologischen Neoplasien (8 Studien) sowie Studien mit einem gemischten Kollektiv (8 Studien) und eine Studie an Patienten mit myelodysplastischem Syndrom, zu dessen Behandlung ESAs nicht zugelassen sind. Eine Chemotherapie wurde in 16 Studien mit, in 13 Studien ohne platinhaltige Therapeutika appliziert. Bestrahlt wurden die Patienten in acht Studien, während in drei Studien Patienten ohne begleitende antitumoröse Therapie eingeschlossen waren und in zwei Publikationen die Behandlung unklar blieb. Die durchschnittlichen Hb-Ausgangswerte lagen unter 10 g/dl (20 Studien), zwischen 10 und 12 g/dI (8 Studien), über 12 g/dl (7 Studien). Nicht mitgeteilt wurden sie in sieben weiteren Studien. In 29 Studien lag die mediane Beobachtungsdauer unter einem Jahr, in den restlichen Studien betrug sie mehr als ein Jahr. Bei der Mehrheit der Studien (n = 37) wurden Epoetine (alfa oder bete) untersucht. Insgesamt (n = 8167) fand sich ein Trend zu einem verkürzten Überleben der mit ESAs behandelten Patienten (Hazard Ratio (HR) 1,08; 95% CI 0,99-1,181 wobei keine Hinweise auf besonders gefährdete Subgruppen identifiziert werden konnten.
In einer aktuellen, auf der Auswertung individueller Patientendaten basierenden Metaanalyse bei 13933, in 53 RCTs untersuchten Krebspatienten erhöhte die Gabe von ESAs die Mortalität während der aktiven Studienphase (HR: 1,17; CI: 1,061,30) und verschlechterte das Gesamtüberleben (HR: 1,06; CI: 1,00-1,12). Auch bei den 10.491 Patienten mit Chemotherapie wurde durch die Gabe von ESAs die Mortalität (HR: 1,10; CI: 0,98-1,24) erhöht und das Gesamtüberleben (HR: 1,04; Cl: 0,97-1,11) vermindert.
| Tumoransprechen |
Die von der Cochrane-Gruppe herangezogenen Publikationen (13 Studien, ingesamt = 2.833) lieferten keine validen Daten, die eine zuverlässige Beurteilung des Tumoransprechens erlauben würden.
| Lebensqualität |
Der wichtige Aspekt der Lebensqualitätsverbesserung wurde anhand der Daten von 16 Studien analysiert. Diese umfassten überwiegend Kollektive mit einem Ausgangs-Hb-Wert <10 g/dl und wandten ein breites Instrumentarium von Fragebögen und Messskalen an, um z.B. Parameter wie körperliches, funktionelles, soziales, emotionales Wohlbefinden bzw. Fatigue (FACT: Functional Assessment to Cancer Treatment) oder Kategorien wie Energieniveau, Alltagsaktivitäten, gesamte Lebensqualität (VAS: Visual analogue scale) darzustellen. Zusätzlich kamen eine Vielzahl weiterer Messskalen und deren Subskalen zum Einsatz. Dabei waren die Schwerpunkte und der Detaillierungsgrad der verwendeten Messskalen in den einzelnen Untersuchungen sehr unterschiedlich.
Die Validität der Resultate wird insgesamt kritisch bewertet. Zum Teil fehlte in den Studien eine Randomisierung oder Verblindung und/oder die Erfassung der Lebensqualität stellte keinen primären Endpunkt dar. Die meisten Studien gingen mit hohen Dropout-Raten (>20%) einher. Oft blieb auch unklar in welchem zeitlichen Abstand zu therapeutischen Maßnahmen die Befragung stattgefunden hatte. Lediglich eine numerische Gegenüberstellung signifikanter Ergebnisse der verschiedenen Bewertungsskalen fiel zugunsten von ESAs aus, wobei allerdings ein möglicher Publikationsbias berücksichtigt werden muss und eine quantitative Abschätzung des Effektes nicht erfolgen kann.
| Nebenwirkungen, Risiken und Vorsichtsmaßnahmen |
Die häufigsten unerwünschten Wirkungen von ESAs bei onkologischen Patienten sind - neben dem Auftreten von Kopfschmerzen - ein dosisabhängiger Blutdruckanstieg oder die Verschlechterung einer vorbestehenden Hypertonie. Eine engmaschige Überwachung des Blutdrucks und ggf. seine medikamentöse Einstellung sind erforderlich. Wenn eine ausreichende Blutdruckeinstellung nicht erzielt werden kann, ist die Dosierung der ESAs zu reduzieren bzw. der Wirkstoff abzusetzen.
Selten wurden Überempfindlichkeitsreaktionen der Haut beschrieben, auch über potenziell schwerwiegende anaphylaktische Reaktionen wurde berichtet. Besonders zu Therapiebeginn wurden Influenzaähnliche Symptome mit Gelenk- und Muskelschmerzen, Fieber, Hautausschlag und Reaktionen an der Injektionsstelle beobachtet.
Nach Behandlung von Patienten mit chronischer Niereninsuffizienz mit ESAs trat sehr selten (überwiegend nach subkutaner Gabe) infolge der Bildung neutralisierender Antikörper gegen EPO eine isolierte Erythroblastopenie (pure red cell aplasia, PRCA) auf. Daher sind die Fachinformationen der Arzneimittel zu beachten, nach denen bei nachlassender Wirksamkeit neben den anderen möglichen Ursachen (s. Abschnitt Wirkungen) auch die Retikulozytenzahl sowie ggf. die Anti-EPO-Antikörper zu bestimmen sind und bei Vorliegen einer Erythroblastopenie die Behandlung mit ESAs abzubrechen ist.
Die Expression von EPO und EPO-R wurde nicht nur in normalen, nichthämatologischen Geweben (z.B. Gehirn, Darm, Gefäßendothel), sondern auch auf der Oberfläche von zahlreichen soliden Tumoren wie Prostata-, Lungen- oder gynäkologischen Karzinomen (Brust, Over, Zervix, Endometrium) nachgewiesen. Daraus resultieren Bedenken, dass ESAs des Wachstum von Tumoren anregen können.
Besondere Aufmerksamkeit erzeugte eine Metaanalyse von 51 Phase-III-Studien, die die Gabe von ESAs mit einer Placebo- oder Standardbehandlung bei der Therapie der Anämie von Tumorpatienten verglichen. Die Daten von insgesamt 13.611 Patienten aus diesen 51 klinischen Prüfungen wurden hinsichtlich der Mortalitätsraten evaluiert, die von 8.172 Patienten (38 Studien) im Hinblick auf das Auftreten thromboembolischer Ereignisse. Mit ESA behandelte Tumorpatienten hatten sowohl ein signifikant erhöhtes Risiko für Thromboembolien (339 Ereignisse/9.610 mit ESA behandelte Patienten vs. 173 Ereignisse/3.562 Kontrollpatienten; 7,5% vs. 9,9%; RR: 1,57; 95% KI: 1,31-1,87) als auch ein erhöhtes Sterberisiko (Hazard Ratio 1,10; 95 % KI: 1,01-1,20).
Eine im Juni 2008 veröffentlichte retrospektive Analyse untersuchte Risikofaktoren, die eine leukämische Transformation bei Patienten mit primärer Myelofibrose begünstigen könnten. Unerwartet ergab sich u. a. eine signifikante Assoziation zu einer vorherigen Therapie mit ESAs (p= 0,004), die allerdings außerhalb des zugelassenen Anwendungsbereiches stattgefunden hatte. Dieser Befund bedarf deshalb einer weiteren Abklärung in prospektiven Studien.
Die folgende Tabelle 3 gibt einen Überblick über die Studien, die wegen signifikant erhöhter Tumorprogression und/oder verkürzter Überlebensdauer zu Anwendungseinschränkungen seitens der Behörden geführt haben1:
Tabelle 3 2
| Studie/Tumor (n) | Hämoglobin Zielwert | Erreichter Hb-Wert (Median, Q1, Q3) | Primärer Endpunkt | Negatives Resultat im ESA-Arm |
| Chemotherapie | ||||
| Cancer Study 1 (BEST) Motastatic breast cancer (n = 939) Leyland-Jones B 2005 | 12-19 g/dl | 12,9 g/dl 12,2; 13,3 g/dl | Gesamtüberleben über 12 Monate | Vermindertes Gesamtüberleben nach 12 Monaten |
| Cancer Study 2 (2000-0161) Lymphoid malignancy (n = 344) Hedenus M 2003 | 13-15 g/dl (M) 13-19 g/dl (F) | 11,0 g/dl 9,8; 12,1 g/dl | Rate an Patienten mit hämatologischem Ansprechen | Vermindertes Gesamtüberleben |
| Cancer Study 3 (PREPARE) early breast cancer (n = 733) 3, 4, 5 | 12,5-13 g/dl | 13,1 g/dl 12,5; 13,7g/dl | Rückfallfreies und Gesamtüberleben | Vermindertes rückfallfreies und Gesamtüberleben nach 3 Jahren |
| Cancer Study 4 (GOG 191) cervical cancer (n = 114) Thomas G 2008 | 12-14 g/dl | 12,7 g/dl 12,1; 13,3 g/dl | Progressionsfreies und Gesamtüberleben und lokoregionale Kontrolle | Vermindertes progressionsfreies und Gesamtüberleben und lokoregionale Kontrolle nach 3 Jahren |
| Nur Radiotherapie | ||||
| Cancer Study 5 (ENHANCE) Head and neck cancer (n = 351) Henke M 2003 | > 15 g/dl (M) > 14 g/dl (F) | Nicht verfügbar | Lokoregional progressionsfreies Überleben | Vermindertes lokoregional progressionsfreies Überleben nach 3 Jahren und vermindertes Gesamtüberleben |
| Cancer Study 6 (DAHANCA 10) Head and neck cancer (n = 522) Overgaard J 2006 (Interim A.) | 14-15,5 g/d1 | Nicht verfügbar | Lokoregionale Krankheitskontrolle | Verminderte lokoregionale Krankheitskontrolle |
| Weder Chemo- noch Radiotherapie | ||||
| Cancer Study 7 (EPO-CAN-20) NSCL (n = 70) Wright JR 2007 (Interim A.) | 12-14 g/dl | Nicht verfügbar | Lebensqualität | Vermindertes Gesamtüberleben |
| Cancer Study 8 (2001-0130) Nonmyeloi malignancy (n = 989) Smith RE 2008 | 12-13 g/dl | 10,6 g/dl 9,4; 11,8 g/dl | RBC, Transfusionen | Vermindertes Gesamtüberleben |
g/dl: Gramm pro Deziliter:
M: Männer, F: Frauen
1) http//www.amgen.com/pdfs/misc/healthcare_professionals_letter.pdf
2) http//www.fda.gov/Safety/MedWatch/Safetylnformation/Safety Alertstor HumanMethealgroducts/ ucm200391.htm
3) http://www.fda.gov/ohrms/dockets/ac/07/briefing/2007-4341b2-01-01-Amgen.pdf
4) http://www.medscape.com/viewarticle/571464?src=rss
5) http://www.medicalnewstoday.com /articels/90405.php
Erythropoese - stimulierende Wirkstoffe
(zur Behandlung der symptomatischen renalen Anämie)
Beschluss vom: 23.06.2011
In Kraft getreten am: 24.06.2011
BAnz. 2011, Nr. 143 vom 21.09.2011 S. 3313
| Zugelassene Anwendungsgebiete |
Alle Erythropoese - stimulierenden Wirkstoffe (Erythropoiesis Stimulating Agents, ESAs) sind in Deutschland zur Behandlung der symptomatischen Anämie bei chronischer Niereninsuffizienz zugelassen. Arzneimittel, die Epoetin alfa, Epoetin beta, Epoetin theta, Epoetin zeta, Darbepoetin alfa oder Methoxy - Polyethylenglycol - Epoetin beta enthalten, sind bei Patienten mit renaler Anämie zur intravenösen wie auch zur subkutanen Anwendung zugelassen. Epoetin alfa - haltige Biosimilars sind bei Patienten mit renaler Anämie nur für die intravenöse Verabreichung zugelassen, da die Daten für die Sicherheit der subkutanen Anwendung nicht ausreichten. Mit Ausnahme von Methoxy Polyethylenglycol - Epoetin beta und Epoetin theta bezieht sich die Zulassung auch auf pädiatrische Patienten mit renaler Anämie unter Hämodialysebehandlung.
Weitere Indikationen wie die Behandlung der symptomatischen Anämie und die Reduktion des Transfusionsbedarfs bei onkologischen Patienten, die eine Chemotherapie erhalten, sind an dieser Stelle nicht dargestellt. Diesbezüglich wird auf den Therapiehinweis zu Erythropoese - stimulierenden Wirkstoffen (zur Behandlung der symptomatischen Anämie bei Tumorpatienten, die eine Chemotherapie erhalten [Beschluss vom 17. Juni 2010, BAnz. S. 3473)) verwiesen. Auch zusätzliche, seltenere Indikationen zur Vorbereitung von autologen Bluttransfusionen sind nicht Gegenstand dieses Therapiehinweises.
Tabelle 1: Fertigarzneimittel zur Behandlung der symptomatischen renalen Anämie
| Wirkstoff | Präparat | Art der Verabreichung * | Anwendungsgebiete * |
| Epoetin alfa | Erypo® FS
Eprex® FS | i.v.,
s.c. | Symptomatische Anämie bei chronischer Niereninsuffizienz Kinder + Erwachsene:
Erwachsene:
|
| Abseamed® Binocrit® Epoetin alfa HexalR:\lebensmt\amg\amr2.htm | i.v. | ||
| Epoetin zeta | Silapo®
Retacrit® | i.v.,
s.c. | Symptomatische Anämie bei chronischer Niereninsuffizienz
Kinder + Erwachsene:
Erwachsene:
|
| Epoetin beta | NeoRecormon® | i.v.,
s.c. | Kinder + Erwachsene:
Symptomatische Anämie infolge chronischer |
| Epoetin theta | Biopoin®
Eporatio® | i.v.,
s.c. | Symptomatische Anämie bei chronischer Niereninsuffizienz
Erwachsene:
|
| Darbepoetin alfa | Aranesp® | i.v.,
s.c. | Kinder + Erwachsene:
Symptomatische Anämie bei chronischer Niereninsuffizienz |
| Methoxy - Polyethylenglycol - Epoetin beta | Mircera® | i.v.,
s.c. | Erwachsene:
Symptomatische Anämie bei chronischen |
*) gemäß Fachintormationen
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
ESAs werden intravenös oder subkutan (Ausnahme: Biosimilars Epoetin alfa nur intravenös) appliziert und stimulieren wie das körpereigene Hormon Erythropoetin (EPO) die Proliferation, Differenzierung und das Überleben von Vorläuferzellen der Erythropoese im Knochenmark. Die biologischen Wirkungen der gentechnologisch hergestellten ESAs werden ebenso wie die des Glykoproteins EPO durch Bindung an den Erythropoetin Rezeptor (EPO - R) vermittelt, der spezifisch auf Vorläuferzellen der Erythropoese im Knochenmark exprimiert wird.
Für den therapeutischen Einsatz gelten heute alle verfügbaren ESAs als vergleichbar. Für die als sogenannte "Biosimilars" von der Europäischen Kommission zugelassenen ESAs wurde nachgewiesen, dass ihre Qualität, Wirksamkeit und Sicherheit in den zugelassenen Indikationen ausreichend belegt sind und dem Zulassungsstandard entsprechen.
Durch randomisierte kontrollierte Studien (RCT) belegte Therapieziele sind ein Anstieg des Hämoglobin (Hb) - Wertes und eine Verringerung bzw. Vermeidung von Bluttransfusionen. Eine wesentliche Verbesserung der Lebensqualität konnte anhand der vorliegenden Studien bisher nicht eindeutig gezeigt werden. Dem gegenüber stehen RCTs, die belegen, dass ein zu hoher Hämoglobinzielwert (>12 g/dl) schwerwiegende Risiken (z.B. Erhöhung der Schlaganfallrate, thromboembolische Komplikationen) beinhaltet.
Bei der Verordnung von ESAs zur Behandlung der symptomatischen renalen Anämie müssen folgende Punkte berücksichtigt werden:
- Vor Verordnung der ESAs müssen andere mögliche Ursachen einer Anämie (siehe Abschnitt Wirkungen) ausgeschlossen und bei laborchemischen Hinweisen für einen Eisenmangel bzw. leere Eisenspeicher im Knochenmark eine Eisensubstitution parallel zur Gabe von ESAs begonnen werden. Auch während der Behandlung mit ESAs sind die Eisenspeicher zu überprüfen und ggf. Eisen zu substituieren.
- Vor Verordnung von ESAs sollte unter Einbeziehung der Patienten eine Nutzen - Risiko - Abwägung erfolgen, die unter anderem folgende Faktoren einschließt: Art, Stadium und Prognose der Erkrankung, Schweregrad der Anämie, klinische Situation (z.B. kardiovaskuläre oder pulmonale Begleiterkrankungen), Behandlungspräferenz der Patienten. Die Patienten müssen über die Risiken bei der Gabe von ESAs (erhöhtes Mortalitätsrisiko bei Patienten mit zu hohen Hb - Werten, thromboembolische Komplikationen, erhöhtes Risiko von Schlaganfällen, mögliche Stimulation des Tumorwachstums) sorgfältig und aktuell informiert werden.
- Die Europäische Arzneimittelagentur (European Medicines Agency, EMA) hat nach Abschluss eines Risikobewertungsverfahrens für alle Indikationen einheitliche Therapieziele empfohlen. Danach soll der Zielwert des Hb für Erwachsene zwischen 10 und 12 gdl (entsprechend 6,2 - 7,45 mmo1/1) und für Kinder zwischen 9,5 und 11 g/dl (entsprechend 5,9 - 6,8 mmol/l) liegen und damit den physiologischen Normbereich unterschreiten.
- Die Behandlung der symptomatischen renalen Anämie sollte abhängig von der individuellen klinischen Symptomatik ab Hämoglobin - Werten
< 10,0 g/dl erwogen werden, nachdem andere Ursachen der Anämie ausgeschlossen wurden. - Bei Hämoglobinwerten < 9 g/dl muss das Risiko vermehrt notwendiger Transfusionen gegenüber einem erhöhten Schlaganfallsrisiko abgewogen werden. Insbesondere bei Patienten, die für eine Transplantation infrage kommen, muss die mögliche Bildung von Alloantikörpern gegen Blutgruppenantigene durch Erythrozytenkonzentrate berücksichtigt werden.
- Ein Anheben des Hb - Wertes über 12 g/dl bringt für den Patienten keine messbaren Vorteile, sondern kann mit erhöhten Risiken verbunden sein. Außerdem wäre dafür eine Erhöhung der Epoetin - bzw. Darbepoetin - Dosis erforderlich.
- Die Dosis der ESAs sollte angepasst werden, wenn der Hb - Wert um mehr als 2 g/dl/Monat steigt oder sinkt und/oder wenn der Hb - Wert außerhalb des oben genannten Zielbereichs gerät.
- Für die Biosimilars wurden von der EMA im Vergleich zum Referenzpräparat in den Zulassungsstudien keine klinisch relevanten Dosisunterschiede festgestellt. In den der Zulassung entsprechenden Applikationsformen stellen Biosimilars eine kostengünstige Alternative dar.
- Für Epoetin alfa und beta wurde gezeigt, dass ein Einsparpotential durch Reduktion der Dosis bei subkutaner im Vergleich zur intravenösen Anwendung besteht.
| Kosten |
Die in der nachfolgenden Kostentabelle angegebene Dosierung bezieht sich auf die Anfangsdosis gemäß Fachinformation. Die Kostenberechnung erfolgte anhand des kostengünstigsten Präparates (Apothekenverkaufspreis) einschließlich Import.
Tabelle 2: Kostenübersicht
| Wirkstoff | Präparat | Dosis 1 | Kosten für 12 Wochen 2, 3, 12 |
| Epoetin alfa | Erypo® FS 4.000 I.E./0,4 ml Eprex® FS 4.000 I.E./0,4 ml | 50 I.E./kg KG 3 x/Woche (i.v. / s.c.) 4, 5 | 1362,54 Euro 1321,68 Euro |
| Abseamed® 4.000 I.E./0,4 ml Binocrit® 4.000 I.E./0,4 ml Epoetin alfa Hexal® 4.000 I.E./0,4 ml | 50 I. E./kg KG 3 x/Woche (i.v.) 5, 6 | 1.214,04 Euro 1136,34 Euro 1226,34 Euro | |
| Epoetin zeta | Silapo® 4.000 I.E./0,4 ml Retacrit® 4.000 I.E./0,4 ml | 50 I.E./kg KG 3 x/Woche (i.v. / s.c.) 5 | 1226,34 Euro 1226,34 Euro |
| Epoetin beta | NeoRecormon § 2.000 I.E. | 20 I.E./kg KG 3 x/Woche (s.c.) 7 | 675,36 Euro |
| NeoRecormon § 3.000 I.E. | 40 I.E./kg KG 3 x/Woche (i.v.) 7 | 1008,18 Euro | |
| Epoetin theta | Biopoin § 2.000 I.E./0,5 ml Eporatio § 2.000 I. E./0,5 ml | 20 I. E./kg KG 3 x/Woche (s.c.) 8 | 616,38 Euro 616,38 Euro |
| Biopoin § 3.000 I.E./0,5 ml Eporatio § 3.000 I. E./0,5 m | 40 I. E./kg KG 3 x/Woche (i.v.) 8 | 920,28 Euro 920,28 Euro | |
| Darbepoetin alfa | Aranesp § 40 μ g | 0,45 μ g/kg KG/Woche (i.v. / s.c.) 9 | 1293,15 Euro |
| Methoxyl - Polyethylenglycol - Epoetin beta | Mircera § 50 μ g/0,3 ml | 0,6 μ g/kg KG/alle 2 Wochen (i.v. / s.c.) 10 | 808,10 Euro |
| Erythrozytenkonzentrat (EK) | ||
| Erythrozytenkonzentrat | 225 - 375 ml/Beutel 11 | ca. 88 Euro/Beutel 11 |
Stand Lauertaxe: 1. September 2011
1) Dosierung gemäß Fachinformation (Anfangsdosis bzw. Korrekturphase).
2) Kostenberechnung anhand des kostengünstigsten Präparates einschließlich Import sowie kostengünstiger Stückelungen; gesetzliche Pflichtrabatte der Apotheken und pharmazeutischen Unternehmen wurden nicht berücksichtigt.
3) Kostenberechnung für eine 70 kg schwere Person.
4) Verabreichung vorzugsweise intravenös.
5) Dosisangabe für erwachsene und pädiatrische Hämodialysepatienten und erwachsene Patienten mit Niereninsuffizienz, die noch nicht dialysepflichtig sind.
Dosierung für erwachsene Peritonealdialyse - Patienten: 50 I.E./kg KG, 2 x/Woche.
6) Nur zur intravenösen Applikation zugelassen.
7) Dosisangabe für erwachsene und pädiatrische Patienten.
8) Dosisangabe für erwachsene Patienten.
Es gibt keine Erfahrungen bei Kindern und Jugendlichen.
9) Dosisangabe für erwachsene und pädiatrische Patienten.
Alternative subkutane Gabe bei nicht - dialysepflichtigen erwachsenen und pädiatrischen Patienten: 0,75 pg/kg KG/alle 2 Wochen.
10) Dosisangabe für erwachsene Patienten, die aktuell nicht mit einem Erythropoese - Stimulierenden Wirkstoff (ESA) behandelt werden.
Anwendung wird für Patienten unter 18 Jahren nicht empfohlen.
11) Angaben:
DRK, Blutspendedienst Berlin, 17. August 2009.
12) Die Darstellung des maximalen Zeitraums der Anfangsdosis kann unterschritten werden.
Nach Ablauf des Patentschutzes für die erstzugelassenen ESAs stehen als "Biosimilars" mehrere Präparate zur Verfügung. Sie enthalten ebenfalls gentechnologisch hergestelltes Epoetin und sind wie die Referenzarzneimittel zur Behandlung der renalen Anämie zugelassen. Zur Zulassung wurden der Europäischen Kommission entsprechend den für die Nachfolgeprodukte erythropoetinhaltiger biologischer Arzneimittel entwickelten Richtlinien die Ergebnisse geeigneter Untersuchungen vorgelegt. Diese charakterisieren nicht nur den enthaltenen Wirkstoff hinreichend, sondern weisen auch nach, dass die Anforderungen in Bezug auf die Sicherheit und Wirksamkeit des geprüften Arzneimittels erfüllt werden.
Ein Erythrozytenkonzentrat (EK) kostet ca. 88 Euro. Die Gabe ist nur bei symptomatischer schwerer Anämie gerechtfertigt. Neben dem Infektionsrisiko besteht auch die Gefahr einer Bildung von Alloantikörpern gegen Blutgruppenantigene vor einer geplanten Transplantation.
| Wirkungen |
In Deutschland sind verschiedene gentechnologisch hergestellte Varianten des körpereigenen Glykoproteins EPO zur Behandlung der chronischen Anämie Bei Nierenunsiffizienz zugelassen. (siehe Tabelle 1). Hierzu zählen: Epoetin alfa, beta, theta und zeta, Methoxy - Polyethylenglycol - Epoetin beta sowie das Epoetin - Analogon Darbepoetin alfa. Biochemische Unterschiede (z.B. in der Anzahl der Stickstoff - gebundenen Kohlenhydratseitenketten, in der Aminosäuresequenz) führen zu Unterschieden im Molekulargewicht, der Affinität zum EPO - Rezeptor und der Pharmakokinetik. So besitzt Darbepoetin alfa gegenüber Epoetin alfa bzw. beta eine etwa dreifach längere Serumhalbwertszeit und weist gleichzeitig eine um etwa den Faktor vier verminderte Affinität zum EPO - Rezeptor auf. Diese biochemischen Unterschiede bedingen jedoch keine Unterschiede in den pharmakodynamischen Eigenschaften. Deshalb werden auch alle Wirkstoffe unter der Bezeichnung "Erythropoiesis - stimulating agents" (ESAs) zusammengefasst.
Das Hormon EPO wird vorwiegend von der Niere gebildet und ist ein essentieller Wachstums - und Überlebensfaktor für erythroide Vorläuferzellen. Nach Bindung von EPO an spezifische EPO - Rezeptoren auf Vorläuferzellen der Erythropoese im Knochenmark werden Proliferation und Differenzierung stimuliert sowie das Überleben dieser Zellen verlängert, vor allem durch eine Hemmung des frühzeitigen, programmierten Zelltods (Apoptose). Jede Form von Sauerstoffmangel (Hypoxie) führt zu einer verstärkten Produktion von EPO und somit zu einer Zunahme der Sauerstofftransportkapazität.
| Wirksamkeit |
Die Verordnung von ESAs bei Patienten mit renaler Anämie hat folgende Therapieziele: Anstieg des Hb - Wertes, Vermeidung von Anämie - bedingten Symptomen bzw. Organschäden, Reduktion bzw. Verhinderung der Transfusion von Erythrozytenkonzentraten, Verbesserung der Lebensqualität, Verminderung der Morbidität und Sterblichkeit.
Die Wirksamkeit von ESAs wird bei Patienten mit chronischer Niereninsuffizienz mit und ohne Dialysepflicht im Vergleich zur Nichtgabe oder Placebo und im Vergleich verschiedener Hämoglobinzielwerte dargestellt.
| Hämatologisches Ansprechen |
Die Gabe von ESAs kann die Anämie bei Patienten mit chronischer Niereninsuffizienz verbessern, Hämoglobin und Hämatokrit steigen an. Dies wurde in neun Studien an Prädialysepatienten, in einer Untersuchung bei Peritonealdialysepatienten und in fünf Studien bei Dialysepatienten bestätigt.
| Reduktion der Transfusionsrate |
Entsprechend einer Auswertung von drei Studien mit 300 Probanden konnte das relative Risiko des Bedarfs an Erythrozytenkonzentraten (EK) durch die Gabe von ESAs statistisch signifikant um 85 % reduziert werden (RR: 0,15 [95 % KI: 0,05 - 0,47]).
Hinweise für eine Reduktion der Transfusionsrate ergaben sich ebenfalls bei Prädialysepatienten in der TREAT - Studie, in der die Gabe von EK nur bei 297 Patienten in der Interventionsgruppe (Hämoglobinzielwert > 13 g/dl) im Vergleich zu 496 Patienten in der Placebogruppe notwendig (p <0,001) war.
In einer weiteren Studie waren bei Dialysepatienten mit niedrigem Hämoglobinzielwert (Hb 9,5 - 11,5 g/dl) 0,66 (95% KI: 0,59 - 0,74) Transfusionen pro Jahr und Patient gegenüber 0,26 (95% KI: 0,22 - 0,32) in der Gruppe mit hohem Hämoglobinzielwert (Hb 13,5 - 14,5 g/dl) erforderlich (p = 0,0001).
| Beginn der Dialysepflicht |
In fünf Untersuchungen wurde weder die Zahl der Patienten, die im Studienzeitraum dialysepflichtig wurden, noch die Dauer bis zum Beginn der Nierenersatztherapie (eine Studie) durch den Gebrauch von ESAs signifikant beeinflusst.
In drei Studien war ein Hämoglobinzielwert <12 g/dl nicht mit einem erhöhten Risiko für das Erreichen einer terminalen Niereninsuffizienz, die ein Nierenersatzverfahren notwendig machte, gegenüber einem Hämoglobinzielwert von > 13 g/dl verbunden.
In der CREATE - Studie (Hb - Zielwerte 10,5 - 11,5 vs. 13,0 - 15,0 g/dl) wurden signifikant mehr Patienten in der Gruppe mit hohem Hämoglobinzielwert als in der Gruppe mit niedrigem Hämoglobinzielwert dialysepflichtig (127 vs. 111, p = 0,03). In der CHOIR - Studie (Hb - Zielwerte 11,3 vs. 13,5 g/dl) war ebenso wie in der TREAT - Studie (Zielwerte > 9 g/dl vs. 13,0 g/dl) kein signifikanter Unterschied bezüglich der Einleitung eines Nierenersatzverfahrens nachweisbar.
| Lebensqualität |
Studien zur Lebensqualität, die den Gebrauch von ESAs gegenüber dem Nichtgebrauch oder Placebo bei Dialysepatienten untersuchen, fehlt häufig die Randomisierung oder Verblindung. Außerdem werden sehr unterschiedliche Messverfahren benutzt. Valide Studien könnten nur noch den Einfluss verschiedener Hämoglobinzielwerte auf die Lebensqualität bei Dialysepatienten überprüfen.
In einem Cochrane Review, in dem Studien mit sehr unterschiedlichen Untersuchungsmethoden an Prädialysepatienten zur Lebensqualität ausgewertet wurden, sowie in einer Arbeit bei Dialysepatienten mit dem Kidney Disease Questionnaire und in der CREATE - Studie konnte in einzelnen Parametern (z.B. physische Funktionen, Vitalität) eine Erhöhung der Lebensqualität und der Leistungsfähigkeit gezeigt werden.
Eine randomisierte kontrollierte Studie bei Dialysepatienten (n = 596) ohne symptomatische Herzerkrankung zeigte in einer Gruppe mit einem hohen Hämoglobinzielwert (13,5 - 14,5 g/dl) Verbesserungen lediglich in einem Energie/Ermüdungsscore gegenüber einer Gruppe mit niedrigem Hämoglobinzielwert (9,5 - 11,5 g/dl). Die übrigen 19 untersuchten Variablen zeigten keine Veränderung.
In der CHOIR - Studie (1.432 Probanden, durchschnittliche Behandlungsdauer 16 Monate) kam es zu keiner Verbesserung der Lebensqualität in der Gruppe mit einem hohen Hämoglobin - zielwert gegenüber der Gruppe mit einem niedrigen Hämoglobinzielwert. Eine Meta - Analyse von neun Studien, in der der Einfluss höherer angestrebter Hämoglobinzielwerte (> 12 g/dl) mit denen niedriger und mittlerer Hämoglobinzielwerte (Hb 912 g/dl) auf die gesundheitsbezogene Lebensqualität ausgewertet wurde, bestätigt die Ergebnisse: Die gefundenen Unterschiede hinsichtlich der Lebensqualität werden als gering und klinisch nicht bedeutsam eingestuft.
Die TREAT - Studie (4.038 Prädialysepatienten, durchschnittliche Behandlungsdauer 29,1 Monate), welche als einzige Studie doppelblind und placebokontrolliert durchgeführt wurde und die in oben genannte Meta - Analyse noch nicht eingeflossen ist, zeigt nur eine moderate, klinisch fragliche Verbesserung bezüglich der Verringerung von Fatigue - Symptomen. Die Skalen Vitalität und körperliche Funktionsfähigkeit des SF - 36 weisen keine signifikanten Unterschiede auf. Weitere Ergebnisse des SF - 36 werden in der Publikation nicht berichtet.
| Sterblichkeit |
In zwei Meta - Analysen und in der TREAT - Studie zeigte sich bei der Behandlung mit ESA vs. kein ESA/Placebo bei Patienten mit renaler Anämie keine Verringerung der Gesamtmortalität. Bezüglich der Reduktion kardiovaskulärer Mortalität konnte in einer Meta - Analyse, in die drei ältere Studien minderer Qualität mit 564 Patienten eingingen, eine signifikante Reduktion festgestellt werden. Dem steht die hochwertige TREAT - Studie mit 4.038 Patienten (ausschließlich Prädialysepatienten) gegenüber, die keine Reduktion der kardiovaskulären Mortalität ergab und ein signifikant erhöhtes Schlaganfallrisiko (1,38 - 2,68, p <0,001) zeigte.
Die Behandlung von Patienten mit renaler Anämie zu einem Hämoglobinzielwert > 12 g/dl im Vergleich zu niedrigeren Hämoglobinzielwerten führte zu keinem signifikanten Unterschied sowohl bezüglich der Gesamtmortalität als auch der kardiovaskulären Letalität. In einer weiteren Meta - Analyse, in der hohe (12 - 16 g/dl) mit niedrigen Hämoglobinzielwerten (9,0 - 12 g/dl) bei nierenkranken Patienten mit und ohne Dialyse untersucht wurden, wurde eine signifikant höhere Gesamtmortalität (RR 1,17 [95 % KI: 1,01 - 1,35], p = 0,031) in der Gruppe mit dem hohen Hämoglobinzielwert festgestellt.
| Hämoglobinzielwerte |
Die Frage nach geeigneten Hämoglobinzielwerten wurde in drei Studien bei Patienten, die noch nicht mit einer Nierenersatztherapie behandelt wurden, und in einer Studie bei Dialysepatienten anhand patientenrelevanter Endpunkte untersucht.
Keine dieser Studien konnte ebenso wie zwei Meta - Analysen Vorteile höherer Hämoglobinzielwerte bezüglich Sterblichkeit oder Erreichen einer Dialysepflicht bei chronisch niereninsuffizienten Patienten mit und ohne Dialyse bestätigen. Zwei dieser Studien wurden sogar wegen eines negativen Trends der kardiovaskulären Mortalität in der Gruppe mit höheren Hämoglobinzielwerten abgebrochen.
| Risiken - ggf. Vorsichtsmaßnahmen |
Häufigste unerwünschte Wirkung während der Behandlung mit ESAs ist ein dosisabhängiger Anstieg des Blutdrucks oder die Verschlechterung einer bestehenden Hypertonie. Der Blutdruckanstieg kann medikamentös behandelt werden. Mit den anfangs üblichen sehr hohen Dosen wurden Therapieunterbrechungen wegen schlecht kontrollierter Hypertonie beschrieben. Hypertensive Krisen mit enzephalopathieähnlichen Symptomen (z.B. Kopfschmerzen und Konfusion) und sehr selten generalisierte tonisch - klonische Krampfanfälle traten bei einzelnen Patienten mit normalem oder erniedrigtem Blutdruck auf.
Nach Behandlung von Patienten mit chronischer Niereninsuffizienz mit ESAs trat sehr selten (überwiegend nach subkutaner Gabe) infolge der Bildung neutralisierender Antikörper gegen EPO eine isolierte Erythroblastopenie (pure red cell aplasia, PRCA) auf. Daher sind die Fachinformationen der ESAs zu beachten, nach denen bei nachlassender Wirksamkeit neben den anderen möglichen Ursachen (siehe Abschnitt Wirkungen) auch die Retikulozytenzahl sowie ggf. die Anti - EPO - Antikörper zu bestimmen sind und bei Vorliegen einer Erythroblastopenie die Behandlung mit ESAs abzubrechen ist.
Besonders zu Therapiebeginn wurden Influenza - ähnliche Symptome mit Gelenk - und Muskelschmerzen, Fieber, Hautausschlag und Reaktionen an der Injektionsstelle beobachtet. Weiterführende Informationen zu den unerwünschten Wirkungen sind den Fachinformationen zu entnehmen.
Bei Patienten mit chronischer Niereninsuffizienz sollte die Hämoglobin - Konzentration in der Erhaltungsphase die empfohlene Obergrenze des Hämoglobinzielwertes nicht überschreiten. Ergebnisse der CHOIR - und der CREATE - Studie zeigten, dass bei renaler Anämie eine Anhebung des Hämoglobinzielwertes auf 13 bis 15 g/dl im Vergleich mit niedrigen Hämoglobinzielwerten (10,5 bis 11,5 g/dl) mit einem erhöhten Risiko kardiovaskulärer Ereignisse verbunden sein kann. In die Fachinformationen wurde ein entsprechender Hinweis aufgenommen.
Insbesondere bei Hämodialysepatienten, die eine Tendenz zur Hypotonie aufweisen oder deren arteriovenöse Fisteln Komplikationen aufweisen (z.B. Stenosen, Aneurysmen, etc.), sind Shuntthrombosen aufgetreten. Eine frühzeitige Korrektur des Shunts und eine Thromboseprophylaxe (z.B. Gabe von Acetylsalicylsäure) werden bei dieser Patientengruppe empfohlen.
Hinweise auf ein häufigeres Auftreten von Schlaganfällen (101 versus 53, p <0,001) und venösen Thromboembolien (41 versus 23, p = 0,02) zeigte sich unter der Gabe von Darbepoetin bei noch nicht dialysepflichtigen Diabetikern mit renaler Anämie im Vergleich zu Placebo.
Etanercept
(z.B. Enbrel § )
Beschluss vom: 10.12.1999
In Kraft getreten am: 01.04.2001
BAnz. 2001, S. 2777
| Indikation |
Etanercept wurde am 2. November 1998 unter dem Handelsnamen Enbrel § in den USA zugelassen zur Behandlung von Patienten mit mittelschwerer bis schwerer rheumatoider Arthritis, die zuvor unzureichend auf eine oder mehrere Basistherapien angesprochen haben. Etanercept kann zur Behandlung erwachsener Patienten sowohl als Monotherapie wie auch in Kombination mit Methotrexat eingesetzt werden. Zwischenzeitlich wurde für die USA die Zulassung auf die Behandlung der therapierefraktären juvenilen rheumatoiden Arthritis erweitert. Die Markteinführung in Deutschland wird für März 2000 erwartet.
| Wirkungen |
Tumornekrosefaktor (TNF) alpha und beta sowie Interleukin 1 sind Zytokine, die als wichtige proinflammatorische Faktoren in der Entzündungskaskade der rheumatoiden Arthritis wirken. Das rekombinant hergestellte Fusionsprotein Etanercept ist ein kompetitiver Inhibitor der Bindung von TNF an seine Zelloberflächen-TNF-Rezeptoren und hemmt dadurch die biologische Aktivität von TNF.
| Wirksamkeit |
Etanercept wurde in mehreren klinischen Phase II und Phase III Studien an erwachsenen Patienten mit rheumatoider Arthritis allein oder in Kombination mit Methotrexat erprobt. Gegenüber Plazebo zeigte sich eine signifikante Verbesserung hinsichtlich der Entzündungsaktivität und der Funktionseinschränkungen.
Die Wirksamkeit der Therapie zeigte sich nach ein bis zwei Wochen und war dosisabhängig. Nach Absetzen der Therapie kam es überwiegend innerhalb von 4 Wochen zu einem Wiederaufflammen der Symptome.
Unter der Kombinationsbehandlung mit Etanercept und Methotrexat konnte eine klinische Besserung auch bei Patienten erreicht werden, die zuvor auf Methotrexat allein nicht oder unzureichend angesprochen hatten.
Es liegen bisher keine Erfahrungen zur Langzeitbehandlung über mehr als 36 Monate vor. Weiterhin ist offen, ob es sich ausschließlich um eine kurzfristige symptomatische Therapie handelt oder ob Etanercept den natürlichen Krankheitsverlauf mit Destruktion der Gelenke aufhalten kann.
Die amerikanische Zulassungsbehörde hat dem Hersteller zur Auflage gemacht, langfristig angelegte Phase IV Studien mit Etanercept in verschiedenen Dosierungen und in Kombination mit anderen Basistherapeutika sowie zur Pharmakokinetik zur einmaligen und bei chronischer Verabreichung durchzuführen, um zusätzliche Daten zur Sicherheit und Pharmakokinetik zu generieren.
| Risiken ggf. Vorsichtsmaßnahmen |
Als häufigste Nebenwirkung sind Reaktionen an der Einstichstelle beschrieben, die bei 37% der erwachsenen Patienten vorkommen. Weitere Nebenwirkungen sind u.a. Kopfschmerzen, Müdigkeit, abdominale Schmerzen, Dyspepsie, Obstipation und Hautrötungen.
Infektionen des oberen Respirationstraktes und Sinusitis wurden bei 29 % der erwachsenen Patienten (Placebo = 16 %) beobachtet. Auch andere Infektionen traten auf. Dies ist möglicherweise dadurch begründet, daß Etanercept in das komplizierte Zusammenwirken der Zytokine eingreift, denen eine spezifische biologische Rolle bei der Immunabwehr von Infektionen und Tumoren zukommt. Besondere Vorsicht ist daher bei Personen mit wiederkehrenden Infektionen in der Vorgeschichte oder mit erhöhtem Infektionsrisiko (z.B. Diabetes mellitus) geboten. Patienten mit aktiven Infektionen sollten deshalb Etanercept nicht erhalten. Die Anwendung muß abgebrochen werden, wenn der Patient eine schwere Infektion entwickelt oder die Gefahr einer Sepsis besteht.
Malignome wurden bei 7 von 745 der erwachsenen Patienten während der Behandlung mit Etanercept festgestellt. Dies entspricht der zu erwartenden Anzahl der Neuerkrankungen in der Normalbevölkerung. Eine Antikörperbildung gegen Etanercept wurde bei 1 % der Behandelten beobachtet. Während der Schwangerschaft wird empfohlen, möglichst auf das Präparat zu verzichten. Es ist nicht bekannt, ob Etanercept in die Muttermilch übergeht.
Etanercept kann zusammen mit Methotrexat verabreicht werden. Wechselwirkungen mit anderen immunsuppressiv wirksamen Antirheumatika sind nicht bekannt. Gezielte Studien hierzu sind bisher nicht abgeschlossen. In klinischen Studien wurden keine Wechselwirkungen mit Glucocorticoiden, Salizylaten, nichtsteroidalen Antirheumatika, Analgetika oder Methotrexat beobachtet.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Voraussetzung für den Einsatz von Etanercept als Behandlungsalternative ist das Versagen aller im individuellen therapeutischen Verlauf angemessenen Basismedikationen. Die Erfahrungen mit dem Präparat sind noch begrenzt. Aufgrund der Zytokinhemmung können Langzeitwirkungen bzw. Nebenwirkungen noch nicht abgeschätzt werden. Es ist zu empfehlen, vor Verordnung von Etanercept unter Einbeziehung rheumatologischen Sachverstandes eine strukturierte Zweitmeinung (z.B. Clearingstelle bei der KV) einzuholen.
| Kosten |
Empfohlen wird eine Dosierung von 25 mg subkutan zwei mal pro Woche.
Da Etanercept derzeit in Deutschland keine Zulassung besitzt, kann das Präparat auf der Grundlage des Arzneimittelgesetzes (§ 73) im Einzelfall importiert werden. Die Packung mit 4 Ampullen a 25 mg kostet nach Auskunft verschiedener Importeure zwischen 801,30 Euro und 912,51 Euro. Damit liegen die Jahrestherapiekosten (24 Packungen) zwischen ca. 19.224,57 Euro und 21.985,55 Euro pro Patient (ohne Zuzahlung und Apothekenrabatt).
Exenatide
(z.B. Byetta § )
Beschluss vom: 19.06.2008 / 16.10.2008
In Kraft getreten am: 28.11.2008
BAnz. 2008, Nr. 181 vom 27.11.2008 S.4.261
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Exenatide ist ein Antidiabetikum, welches zur Wirkstoffklasse der Inkretinmimetika gehört. Der Einsatz von Exenatide sollte Typ-2-Diabetikern vorbehalten bleiben, bei denen unter Ausschöpfung einer Therapie mit oralen Antidiabetika eine adäquate Blutzuckerkontrolle nicht erreicht werden konnte und die klinischen Befunde bei massivem Übergewicht (BMI > 30) vorrangig für eine Insulinresistenz sprechen, sodass bei Zugabe von Insulin mit einer weiteren Gewichtszunahme und hohen Insulindosierungen zu rechnen ist. Exenatide ist rund drei- bis viermal teurer als die Therapie mit in vergleichenden Studien eingesetzten Insulin-Analoga und rund fünfmal teurer als eine Therapie mit Humaninsulin in der durchschnittlichen, in diesen Studien verwendeten Insulindosierung. Erst ab einer täglichen Dosis von 80 IE Insulin Glargin, 90 IE biphasischem Insulin Aspart oder 120 IE Humaninsulin schneidet Exenatide im Vergleich der Tagestherapiekosten günstiger ab. Exenatide ist somit in der Regel unwirtschaftlich.
Die Zulassung von Exenatide umfasst nicht die Kombination mit Insulin oder anderen oralen Antidiabetika als Metformin oder Sulfonylharnstoffen. Insbesondere für die Kombination mit Glitazonen besteht in Europa keine Zulassung.
Wird bei adipösen Typ-2-Diabetikern mit Lebensstil-Interventionen, maximal tolerierbaren Dosen von Metformin und Gabe eines weiteren oralen Antidiabetikums eine Senkung des HbA1c-Wertes auf unter 7 % nicht erreicht, wird im Allgemeinen empfohlen, mit einer Insulintherapie zu beginnen.
Für Patienten in dieser Phase ihrer Erkrankung wurde in zwei offenen vergleichenden Studien gezeigt, dass die zusätzliche Gabe von Exenatide der zusätzlichen Gabe von Insulin Glargin oder biphasischem Insulin Aspart hinsichtlich der Senkung des HbA1c-Wertes nicht unterlegen ist. Dies gilt nicht für Patienten mit Versagen der Betazellfunktion oder für Patienten mit ausgeprägter Stoffwechselentgleisung (HbA1c > 11 %).
Die Anwendung von Exenatide ist mit häufigen unerwünschten gastrointestinalen Wirkungen verknüpft, welche unter Studienbedingungen oft zu Therapieabbrüchen führten (6 % gegenüber < 1 % unter Insulin).
Im Gegensatz zu Insulin und anderen insulinotropen Substanzen, wie Sulfonylharnstoffen oder Glitazonen, führt Exenatide bei übergewichtigen Studienteilnehmern (mittlerer BMI 30 - 34) zu einer Gewichtsreduktion (mittlere Gewichtsreduktion 1,6 kg bis 2,6 kg in 30 Wochen bzw. 2,5 kg in 52 Wochen). Inwieweit dieser übereinstimmend in allen Studien beobachtete Effekt in der Langzeitanwendung zu einer Risikoreduktion der kardiovaskulären Morbidität und Mortalität führt, muss in Endpunktstudien untersucht werden. Die Erfahrungen in der Behandlung normalgewichtiger Patienten mit einem BMI < 25 sind sehr begrenzt.
Unter Therapie mit Exenatide ist eine Dosistitration mit häufigen Kontrollen der Blutzuckerwerte nicht erforderlich. In mit Insulin vergleichenden Studien unterscheiden sich die Hypoglykämieraten nicht. Wenn Exenatide nur in Kombination mit Metformin gegeben wird, wurde im Vergleich zu Metformin plus Placebo kein Anstieg der Hypoglykämieinzidenz beobachtet (5,3 % versus 5,3 %). Bei Patienten, die aufgrund eingeschränkter intellektueller Fähigkeiten (z.B. Demenz, geistige Behinderung) nicht in der Lage sind, ihre Lebensführung an eine Hypoglykämiegefährdung anzupassen, kann die Verordnung dieser Kombination im Einzelfall notwendig und wirtschaftlich sein. Voraussetzung ist, dass noch kein absoluter Insulinmangel besteht und mit Kombinationen oraler Antidiabetika - ausgenommen Sulfonylharnstoffen - das Therapieziel nicht erreicht werden konnte.
Kontrollierte Langzeitstudien mit klinischen Endpunkten liegen nicht vor, sodass Nutzen und Sicherheit von Exenatide in der Langzeitanwendung unbekannt sind. Sein Stellenwert in der Behandlung des Typ-2-Diabetes ist noch unklar.
| Kosten |
ZAM: Zylinderampullen, Stand: Lauer: 01.08.2008
*Die Kosten von Metformin und/oder Sulfonylharnstoffen müssen hinzu gerechnet werden. Bei der Verordnung von Analoginsulinen ist Anlage 10 der Arzneimittelrichtlinie zu beachten.
| Indikation |
Exenatide wurde am 20.11.2006 von der Europäischen Zulassungsbehörde (EMEA)
|
|
|
|
| Exenatide* | 2 x tgl. 5 μg | 4,15 Euro | 1515,00 Euro |
| Exenatide* | 2 x tgl. 10 μg | 3,94 Euro | 1438,00 Euro |
| Analoginsuline, langwirksame, 10 x 3 ml, ZAM | 40 IE | 1,96 Euro | 715,00 Euro |
| Analoginsuline, kombiniert intermediär und schnellwirkend, 10 x 3 ml, ZAM | 40 IE | 1,68 Euro | 613,00 Euro |
| humanes Mischinsulin | 40 IE | 1,21 Euro (FB) | 442,00 Euro |
| humanes NPH Insulin | 40 IE | 1,21 Euro (FB) | 442,00 Euro |
zugelassen und wird seit April 2007 in Deutschland vertrieben.
Exenatide ist zugelassen zur Behandlung des Typ-2-Diabetes mellitus in Kombination mit Metformin und/oder Sulfonylharnstoff-Präparaten bei Patienten, bei denen mit der maximal verträglichen Dosis dieser oralen Therapien eine angemessene Blutzuckerkontrolle nicht erreicht werden konnte. Exenatide ist nicht zur Monotherapie zugelassen. Exenatide darf nicht eingesetzt werden bei Typ-2-Diabetikern, bei denen aufgrund eines Betazellversagens eine Insulin-Therapie erforderlich ist. Ebenso wenig darf Exenatide bei Patienten mit Typ-1-Diabetes mellitus oder zur Behandlung der diabetischen Ketoazidose angewendet werden.
Es liegen keine Erfahrungen vor für die Behandlung von Kindern und Jugendlichen unter 18 Jahren. Die Erfahrungen in der Behandlung von älteren Patienten über 75 Jahren sind sehr begrenzt. Die Therapie mit Exenatide wird nicht empfohlen für Patienten mit schweren gastrointestinalen Erkrankungen, wie Gastroparese, oder Patienten mit terminaler Niereninsuffizienz bzw. schwerer Nierenfunktionsstörung.
Exenatide ist in einem als Fertigpen erhältlich mit fixen Einzeldosen von 60 x 5 pg oder 60 x 10 pg. Um die Verträglichkeit zu verbessern, sollte die Behandlung zunächst mit der zweimal täglichen Gabe von 4..ig Exenatide subkutan begonnen und mindestensid einen Monat beibehalten werden. Danach kann auf eine Dosis von zweimal täglich 1Q..ig gesteigert werden. Die Injektionen können zu einem beliebigen Zeitpunkt innerhalb von 60 Minuten vor der Morgen- und Abendmahlzeit, die mindestens 6 Stunden auseinander liegen sollen, appliziert werden. Die Injektion darf nicht nach den Mahlzeiten erfolgen. Bei Patienten über 70 Jahren und Patienten mit mäßig eingeschränkter Nierenfunktion wird zu einer konservativen Dosissteigerung geraten.
Wenn Exenatide zusätzlich zu Metformin gegeben wird, kann die Metformin-Dosis unverändert bleiben. Wird Exenatide zusätzlich zu einem Sulfonylharnstoff gegeben, muss eine Reduktion der Sulfonylharnstoff-Dosis in Betracht gezogen werden, um das Risiko einer Hypoglykämie zu verringern.
| Wirkungen |
Exenatide gehört zu einer Klasse von Wirkstoffen, die unter dem Namen Inkretinmimetika subsumiert werden. Inkretine sind körpereigene Hormone, die im Gastrointestinaltrakt provoziert durch die Nahrungsaufnahme freigesetzt werden und dafür Sorge tragen, dass aus den Betazellen des Pankreas glucoseabhängig Insulin freigesetzt wird. Die Aminosäuresequenz von Exenatide ist teilweise identisch mit der des humanen Glucagon like Peptid (GLP-1). Es aktiviert den humanen GLP-1-Rezeptor und imitiert blutzuckerregulierende Wirkungen von GLP-1. Es hat jedoch im Gegensatz zu GLP-1, welches innerhalb weniger Minuten durch das Enzym Dipeptidyl-Peptidase 4 zu inaktiven Stoffwechselprodukten abgebaut wird, eine deutlich längere Wirkzeit mit einer mittleren Halbwertszeit von 2,4 Stunden.
Exenatide stimuliert glucoseabhängig die Insulinsynthese und -sekretion. Es hemmt die Glucagonsekretion der Alpha-Zellen des Pankreas, wodurch die bei Typ-2-Diabetikern häufig unangemessen erhöhte Glucoseausschüttung aus der Leber gemindert wird. Die gegenregulatorische Glukagonausschüttung bei Hypoglykämie wird dagegen nicht durch Exenatide beeinträchtigt. Exenatide verzögert außerdem die Magenentleerung und reduziert die Nahrungsaufnahme durch ein erhöhtes Sättigungsgefühl und einen verringerten Appetit.
| Wirksamkeit |
Die Wirksamkeit von Exenatide wurde durch drei ähnlich konzipierte randomisierte, placebokontrollierte Wirksamkeitsstudien über 30 Wochen und zwei vergleichende Studien zu Insulin über 26 und 52 Wochen belegt.
In die drei placebokontrollierten Studien über 30 Wochen waren insgesamt 1446 adipöse Typ-2-Diabetiker (durchschnittlicher BMI 34) eingeschlossen, die unter oralen Antidiabetika keine angemessene Blutzuckerkontrolle erreichten (durchschnittlicher HbAlc 8,4 %). Sehr schlecht eingestellte Typ-2-Diabetiker wurden ausgeschlossen (HbAlc > 11 %). In allen drei Studien wurden entweder 5 oder 10 μg Exenatide oder Placeboähnlich zu Metformin und/oder Sulfonylharnstoffen gegeben.
Der primäre Endpunkt war die Veränderung des HbA1c-Wertes. Sekundäre Endpunkte waren der Anteil Patienten mit HbA1c-Werten < 7 % und die Veränderung des Körpergewichtes. Die kombinierte Auswertung aller drei Studien ergab eine signifikante, klinisch relevante dosisabhängige Reduktion des HbA1c-Wertes um 0,6 % in der niedrigeren und um 0,9 % in der höheren Dosierung. Einen HbA1c-Wert von < 7 % erreichten 29,6 % in der niedrigeren und 33,6 % in der höheren Dosierung.
Weiterhin kam es in allen drei Studien zu einer Gewichtsreduktion. Bei kombinierter Auswertung nahm das Körpergewicht um 1,4 kg in der niedrigeren und 1,9 kg in der höheren Dosierung ab.
Bei Patienten, die an den offenen Verlängerungen dieser Studien über zwei Jahre teilnahmen und in dieser Phase 10 pg Exenatide injizierten, hielt der gewichtsreduzierende Effekt an und die Senkung des HbA1c-Wertes konnte erhalten werden. Nach 2 Jahren betrug die durchschnittliche Gewichtsreduktion bei diesen Patienten 4,7 kg. Allerdings waren die Abbruchraten hoch. 46 % der Patienten der 2-Jahres-Kohorte beendeten die Behandlung in der Verlängerungsphase.
In zwei der genannten Studien erfolgte die Kombination von Exenatide mit einer oralen Monotherapie. Dies entspricht nicht der üblichen Vorgehensweise bei Versagen einer Monotherapie mit oralen Antidiabetika. Empfohlen und üblich ist zunächst die Kombinationstherapie mit einem weiteren oralen Antidiabetikum. In Einzelfällen ist bei nur geringer Überschreitung der Interventionsgrenze (HbAlc > 7%) auch eine Kombination aus drei oralen Antidiabetika zielführend. Studien zur Kombination von Exenatide mit drei oralen Antidiabetika liegen bislang nicht vor.
In zwei offenen vergleichenden Nichtunterlegenheitsstudien über 26 und 52 Wochen wurden 1056 adipöse Typ-2-Diabetiker (BMI 30 - 31) mit unzureichender Blutzuckerkontrolle unter maximal effektiver Therapie mit Metformin und Sulfonylharnstoffen (HbAlc 8,2 % - 8,6 %) randomisiert - entweder zusätzlich mit Exenatide oder Insulin Glargin (Lantus § ) bzw. biphasischem Insulin Aspart (Novomix 30 § ) - behandelt.
In beiden Studien führte Exenatide zu einer mit Insulin vergleichbaren Absenkung des HbA1c-Wertes (Exenatide 1,1 % versus Insulin Glargin 1 %; Exenatide 1,04 % versus biphasischem Insulin Aspart 0,89 %).
Angesichts des offenen Designs dieser Studien und der geringen Dosierungen in den mit Insulin behandelten Therapiearmen (gegen Studienende durchschnittlich 25 IE Insulin Glargin und 24 IE biphasisches Insulin Aspart) ist nach Bewertung der EMEA allerdings nicht sicher auszuschließen, dass die Insulintherapie intensiviert und eine stärkere HbAlc-Senkung unter Insulin hätte erreicht werden können.
In beiden Studien nahmen die Patienten unter Exenatide signifikant an Gewicht ab (- 2,3 kg nach 26 Wochen, -2,5 kg nach 52 Wochen), während die Patienten mit Insulin an Gewicht zunahmen (+ 1,8 kg nach 26 Wochen, + 2,9 kg nach 52 Wochen).
Anhand selbst gemessener 7-Punkt-Blutzuckertagesprofile waren unter Exenatide geringere postprandiale Blutzuckerexkursionen festzustellen, während unter Insulin die präprandialen Blutzuckerkonzentrationen niedriger waren. Die mittleren täglichen Blutzuckerkonzentrationen waren unter Insulin und Exenatide vergleichbar.
Untersuchungen an Subgruppen einiger Studien wiesen anhand von Surrogat-Parametern auf eine verbesserte Betazell-Funktion hin. Es ist unklar, welche klinische Relevanz dieser Beobachtung zukommt.
Zusammengefasst führte Exenatide in einer Dosis vonpy3in allen Studien zu einer HbA1c-Senkung um 0,8 % bis 1,1 %. Übereinstimmend zeigte sich in allen Studien ein anhaltender gewichtsreduzierender Effekt, der, wenn auch etwas weniger stark ausgeprägt, auch bei Patienten beobachtet wurde, die nicht unter gastrointestinalen Nebenwirkungen zu leiden hatten. Es handelt sich somit um einen unabhängigen Effekt, der nur zum Teil durch die häufig auftretende Übelkeit erklärt ist.
| Risiken ggf. Vorsichtsmaßnahmen |
Methodisch hochwertige Studien zur Patientenzufriedenheit unter Exenatide sind nicht verfügbar.
Die Sicherheit von Exenatide wurde bisher an ca. 3000 Patienten evaluiert, die im Mittel über 13,7 Wochen behandelt wurden. Dies wird von der EMEA in Anbetracht einer häufig über Jahrzehnte behandlungsbedürftigen Erkrankung als geringe, aber in Verbindung mit einer gezielten Überwachung nach Zulassung letztlich ausreichende Erfahrung angesehen.
Die häufigste Nebenwirkung unter Exenatide waren gastrointestinale Beschwerden. Zeitweise klagen 50 % der Patienten über Übelkeit, 19 % über Erbrechen und 13 % über Durchfall. Diese unerwünschten Wirkungen nahmen unter fortgesetzter Behandlung an Häufigkeit und Schwere ab. Jedoch gaben am Ende der drei placebokontrollierten 30-Wochen-Studien immer noch 10 % der Patienten an, unter Übelkeit zu leiden. Dies führte bei 6 % aller in Studien mit Exenatide behandelten Patienten zum Behandlungsabbruch.
Die Hypoglykämierate unter Exenatide ist überwiegend abhängig von dem gleichzeitigen Gebrauch von Sulfonylharnstoffen. Eine Dosisreduktion der Sulfonylharnstoffe mindert das Risiko. Die Inzidenz schwerer Hypoglykämien ist niedrig.
Exenatide ist als Eiweißkörper potenziell immunogen. 38 % der Patienten, die an den placebokontrollierten 30-Wochen-Studien teilnahmen, entwickelten niedrige Antikörpertiter, die keinen Einfluss auf die Nebenwirkungsraten oder die Blutzuckerkontrolle hatten. Zusätzliche 6 % hatten nach 30 Wochen höhere Antikörpertiter. Bei der Hälfte dieser Patienten zeigte Exenatide keine Wirksamkeit.
Lokalreaktionen an der Injektionsstelle waren unter Exenatide häufiger als unter Insulin oder Placebo (5,1 % versus 3 %). Diese waren meist gering ausgeprägt und führten nicht zum Absetzen der Therapie.
Nach Markteinführung wurden einige Fälle von akuter Pankreatitis berichtet. Patienten sollten über die charakteristischen Symptome einer akuten Pankreatitis (andauernde, schwere abdominale Schmerzen) informiert werden. Ein Abklingen der Pankreatitis nach Absetzen von Exenatide und symptomatischer Behandlung wurde beobachtet. Weiterhin gab es Spontanberichte über Veränderungen der Nierenfunktion, einschließlich akutes Nierenversagen, das in manchen Fällen eine Hämodialyse erforderlich machte, und Verschlechterung eines chronischen Nierenversagens sowie Hautausschläge mit Pruritus und sehr selten anaphylaktische Reaktionen.
Exenatide verzögert die Magenentleerung. Ausmaß und Geschwindigkeit der Absorption anderer Medikamente können hierdurch vermindert werden. Medikamente mit enger therapeutischer Breite oder Medikamente, bei denen bestimmte Mindestkonzentrationen erreicht werden müssen, sollten deshalb mindestens eine Stunde vor der Injektion oder vier Stunden nach der Injektion eingenommen werden. Medikamente, die mit einer Mahlzeit eingenommen werden sollen, können zu einer Mahlzeit gegeben werden, zu der keine Injektion von Exenatide erfolgt (z.B. Mittagessen). Es wird empfohlen
- Antibiotika mindestens 1 Stunde vor der Injektion einzunehmen.
- Das Gleiche gilt für magensaftresistente Arzneimittelzubereitungen (z.B. Protonenpumpeninhibitoren).
- Für Kumarinderivate werden zu Therapiebeginn und während der Dosiserhöhung von Exenatide engmaschige Laborkontrollen empfohlen.
- Unter Therapie mit CSE-Hemmern sollten die Blutfettwerte regelmäßig kontrolliert werden, da eine Dosisanpassung erforderlich sein kann.
Bei Frauen, die schwanger werden wollen oder bereits sind, soll die Behandlung mit Exenatide abgebrochen werden und durch Insulin ersetzt werden.
Ezetimib
(z.B. Ezetrol § , Inegy § )
Beschluss vom: 17.12.2009
In Kraft getreten am: 24.03.2010
BAnz. 2010, Nr. 45 vom vom 23.03.2010 S. 1090
| Zugelassene Anwendungsgebiete |
Die Monotherapie mit Ezetimib ist begleitend zur Diät zugelassen zur Anwendung bei Patienten mit
- primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie, bei denen ein Statin als ungeeignet erachtet oder nicht vertragen wird,
- homozygoter familiärer Sitosterinämie.
Ezetimib ist in Kombination mit einem HMG-CoA-Reduktasehemmer (Statin) zugelassen begleitend zur Diät bei Patienten mit
- primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie oder gemischter Hyperlipidämie, bei denen die Therapie mit einem Statin allein nicht ausreicht,
- homozygoter familiärer Hypercholesterinämie. Die Patienten können weitere begleitende Therapien (wie LDL-Apherese) erhalten.
| Empfehlung zur wirtschaftlichen Verordnungsweise |
Endpunktstudien zu Ezetimib, die eine Reduktion von Morbidität, Mortalität oder eine Verbesserung der Lebensqualität zeigen könnten, liegen nicht vor.
Die Atherosklerose ist bedingt durch zahlreiche nur zum Teil bekannte Risikofaktoren. Strategien zur Verhinderung kardiovaskulärer Ereignisse sollten daher nicht auf einzelne Faktoren fokussieren. Die Hypercholesterinämie ist ein wichtiger Faktor, dennoch wurde in Anbetracht der großen Zahl an entsprechenden klinischen Prüfungen nur in wenigen Studien in der Sekundärprävention eine signifikante Reduktion harter klinischer Endpunkte gezeigt. Ein klinisch relevanter Nutzen besteht nur bei sehr hohem kardiovaskulärem Risiko (zu erwartende Ereignisrate über 20% in 10 Jahren auf der Basis der zur Verfügung stehenden Risikokalkulatoren oder bei bereits bestehender vaskulärer Erkrankung [KHK, cerebrovaskuläre Manifestation, pAVK)). Andererseits wurde gezeigt, dass Simvastatin (20 mg bis 40 mg) allein die Häufigkeit kardiovaskulärer Ereignisse reduziert.
Für Ezetimib allein und in Kombination liegen keine harte Endpunkte erfassenden Studien vor.
Bezüglich des Vorgehens im Rahmen der lipidsenkenden Therapien werden zwei verschiedene Strategien angewandt:
- LDL-Cholesterin-Senkung auf Zielwerte,
- Strategie der festen Dosis.
Zwar ist in epidemiologischen Studien das Risiko für kardiovaskuläre Ereignisse umso geringer, je niedriger der LDL-Cholesterinspiegel ist. Cholesterinwerte sind aber Surrogatparameter mit begrenztem Aussagewert. Entscheidend ist, inwieweit Herzinfarkte, Schlaganfälle und dadurch bedingte Todesfälle reduziert werden.
Diese Einschränkung wird durch die Studienlage bestätigt: In den Zulassungsstudien wurde eine feste Dosis eines oder mehrerer Wirkstoffe gegeben. Selbst unter Studienbedingungen erreichten weniger als die Hälfte der Studienteilnehmer die heute empfohlenen Zielwerte. Eine Titrierungsstrategie zur Erreichung der Zielwerte, wie sie in vielen aktuellen Leitlinien empfohlen wird, wurde in keiner der großen Lipidsenkerstudien direkt vergleichend randomisiert geprüft. Ein Beleg für den Nutzen einer derartigen Therapie fehlt.
Die Verordnung von Ezetimib als Monotherapie bei der Behandlung von Hypercholesterinämien ist nur wirtschaftlich bei den wenigen Patienten, bei denen Statine nicht eingesetzt werden können (Unverträglichkeit, Nebenwirkungen). Die Kombination bleibt Patienten mit schwerwiegenden Fettstoffwechselstörungen vorbehalten, die das genannte hohe Risiko für Ereignisse haben. Zusätzlich ist für eine wirtschaftliche Verordnung zu fordern, dass bei den Patienten
- eine homozygote familiäre Sitosterinämie vorliegt oder
- eine ausgeprägte, nicht anders zu behandelnde familiäre homozygote Hypercholesterinämie vorliegt oder
- das Therapieziel die Verhinderung einer LDL-Apherese ist oder
- eine Unverträglichkeit oder Kontraindikation für Statine nachgewiesen wurde.
| Kosten |
| Wirkstoff | Dosierung/d | Tagestherapiekosten * | Jahrestherapiekosten |
| Ezetimib | 10 mg | 1,94 Euro | 707 Euro |
| Ezetimib/Simvastatin | 10 mg/10 mg | 1,83 Euro | 669 Euro |
| (Kombinationspräparat) | 10 mg/20 mg | 2,05 Euro | 748 Euro |
| 10 mg/40 mg | 2,38 Euro | 868 Euro | |
| 10 mg/80 mg | 2,51 Euro | 915 Euro | |
| Simvastatin | 10 mg | 0,26 Euro | 96 Euro |
| 20 mg | 0,34 Euro | 124 Euro | |
| 40 mg | 0,50Euro | 183Euro | |
| 80 mg | 0,74Euro | 271Euro | |
| Pravastatin | 10-40 mg | 0,26-0,50 Euro | 96-183 Euro |
| Fluvastatin | 20-80 mg | 0,23-0,48 Euro | 85-176 Euro |
| Lovastatin | 10-40 mg | 0,26-0,52 Euro | 96-190 Euro |
| Atorvastatin | 10-80 mg | 0,26-0,76 Euro | 96-276 Euro. |
| Fenofibrat | 200-300 mg | 0,48-0,61 Euro | 175-225 Euro |
| Bezafibrat | 400 mg | 0,36 Euro | 133 Euro |
| Gemfibrozil | 900-1.200 mg | 0,44-0,63 Euro | 162-229 Euro |
| Colestyramin | 4,0-24 gr. | 0,67-4,00 Euro | 244-1459 Euro |
| Nikotinsäure | 1.000-2.000 mg | 1,10-2,19 Euro | 401-801 Euro |
*) Berechnungsgrundlage N3-Packungen auf Basis der Festbeträge soweit vorhanden.
Es gibt Arzneimittel, die unter Festbetrag angeboten werden.
Preisstand (Lauertaxe): 1. Februar 2010
Die Tagestherapiekosten der fixen Kombination von Ezetimib und Simvastatin im Vergleich zur freien Kombination der Einzelstoffe führt unter Berücksichtigung der Festbeträge von Simvastatin und von N3-Packungen zu den tabellarisch dargestellten Differenzen. Allerdings ist zu beachten, dass Simvastatin meistens preiswerter, unter Festbetrag, angeboten wird. Ein aktueller Preisvergleich für jede Wirkstärke von Simvastatin ist anzuraten, sofern die fixe Kombination nicht unter patientenindividuellen Aspekten aus medizinischer Sicht unverzichtbar ist.
In der Regel ist die freie Kombination der Wirkstoffe kostengünstiger als die Fixkombination.
| Wirkungen |
Ezetimib wirkt lipidsenkend durch selektive Hemmung der intestinalen Resorption von Cholesterin und verwandten Phytosterinen.
Das molekulare Ziel von Ezetimib ist der Steroltransporter, das Niemann-Pick-C1-Like1-(NPC1L1-)Protein, der für die intestinale Aufnahme von Cholesterin und Phytosterinen verantwortlich ist.
Ezetimib lagert sich am Bürstensaum des Dünndarms an und hemmt die Cholesterinresorption, was zu einem verminderten Transport von Cholesterin aus dem Darm in die Leber führt.
Statine dagegen reduzieren die Cholesterinsynthese in der Leber. Werden die Wirkstoffe kombiniert, führen die unterschiedlichen Wirkungsmechanismen zu einer komplementären Cholesterinsenkung.
| Wirksamkeit |
Eine zusätzliche LDL-C-Reduktion von zirka 13,9 % (95 %-CI 14,90; 12,98, p < 0,00001) und des Gesamtcholesterins von 10,36 % (95 %-CI 11,9; 9,63, p < 0,00001) wird durch die Kombination von Ezetimib mit einem Statin im Vergleich zur alleinigen Statinbehandlung erreicht. Demgegenüber führt eine Verdoppelung der Statindosis lediglich zu einer weiteren Reduktion des LDL von ungefähr 6 % bis 8 %.
Studien, die Effekte auf die Mortalität oder Morbidität untersuchten, sind zu Ezetimib nicht publiziert. Angesichts des Fehlens von Endpunktstudien finden sich veröffentlichte Studien mit einer Dauer von > 12 Wochen lediglich zu Surrogatparametern. Sie zeigen, dass Ezetimib effektiv LDL-C und Gesamt-LDL senkt.
In der ENHANCE-Studie wurde Ezetimib 10 mg plus Simvastatin 80 mg gegen 80 mg Simvastatin geprüft. Primärer Endpunkt war die Veränderung der Intimadicke der Carotis. Für die Kombinationstherapie betrug der Wert 0,0111 mm und für die Monotherapie mit Simvastatin 0,0058 mm, Differenz 0,053 mm. Der p-Wert war mit 0,29 nach 24 Monaten nicht signifikant, obgleich auch in dieser Studie ein signifikanter Unterschied im Hinblick auf die LDL-Werte gesehen wurde (192,7 § 60,3 mg/dl versus 141,3 § 52,6 mg/dl; Gruppenunterschied 16,5 %, p < 0,01) sowie signifikante Gruppenunterschiede für Triglyceride und CRP von 6,6 % und 25,7%. Auch diese Studie war weder gepowert noch konzipiert, um klinische Endpunkte zu untersuchen.
Die Intimadicke ist gleichfalls nur ein Surrogatparameter.
| Risiken und Vorsichtsmaßnahmen |
Ezetimib ist kontraindiziert bei Überempfindlichkeit gegenüber dem arzneilich wirksamen Bestandteil oder einem der sonstigen Bestandteile der Arzneimittel, ebenso in der Schwangerschaft und Stillzeit. Das Gleiche gilt für Patienten mit aktiver Lebererkrankung oder ungeklärter persistierender Erhöhung der SerumTransaminasen.
Patienten mit der seltenen hereditären Galaktoseintoleranz, Lactasemangel oder Glukose-Galaktose-Malabsorption sollten dieses Arzneimittel nicht einnehmen.
Nach Markteinführung von Ezetimib wurden Fälle von Myopathie und Rhabdomyolyse berichtet. Die meisten Patienten, die eine Rhabdomyolyse entwickelten, nahmen gleichzeitig ein Statin ein. Jedoch wurde eine Rhabdomyolyse sehr selten unter Monotherapie mit Ezetimib sowie sehr selten nach Zugabe von Arzneimitteln berichtet, die bekanntermaßen mit einem erhöhten Rhabdomyolyserisiko in Verbindung stehen. Alle Patienten, die auf Ezetimib eingestellt werden, sollten über das Risiko einer Myopathie aufgeklärt und aufgefordert werden, unklare Muskelschmerzen, -empfindlichkeit oder -schwäche umgehend mitzuteilen.
Eine Therapie mit Ezetimib ist bei Patienten, die mit Ciclosporin behandelt werden, mit Vorsicht einzuleiten. Die Ciclosporinkonzentrationen sollten überwacht werden.
Bei Gabe von Ezetimib zu Warfarin, einem anderen Cumarin Antikoagulans, oder Fluindion ist die "International Normalized Ratio" (INR) entsprechend zu überwachen.
Fibrate können die Cholesterinausscheidung über die Galle erhöhen und so zu Cholelithiasis führen. Bei Patienten unter Fenofibrat und Ezetimib sollte der Arzt über das mögliche Risiko einer Cholelithiasis und einer Gallenblasenerkrankung informiert sein.
Die gleichzeitige Anwendung von Colestyramin verkleinerte die mittlere Fläche unter der Kurve (AUC) von Gesamt-Ezetimib um zirka 55 %. Die gesteigerte Senkung des LDL-Cholesterins durch Hinzufügen von Ezetimib zu Colestyramin könnte durch diese Interaktion vermindert werden.
Die Einnahme von Ezetimib sollte mindestens zwei Stunden vor oder mindestens vier Stunden nach der Einnahme eines Anionenaustauschers erfolgen.
In klinischen Studien über 8 bis 14 Wochen wurden 3.366 Patienten mit 10 mg Ezetimib allein, zusammen mit einem Statin oder mit Fenofibrat (185 Patienten) behandelt. Nebenwirkungen waren normalerweise leicht ausgeprägt und von vorübergehender Natur.
Imiglucerase
(z.B. Cerezyme § ) bei Morbus Gaucher Typ I
Beschluss vom: 01.12.2003
In Kraft getreten am: 28.03.2004
BAnz. 2004, Nr. 61 vom 27.03.2004 S. 6.503
| Indikation |
Imiglucerase ist zugelassen zur langfristigen Enzymsubstitution bei Patienten mit gesicherter Diagnose und klinischer Manifestation der Typ 1 Gaucher-Krankheit, der - nach heutiger Nomenklatur - nichtneuronopathischen (= ohne neurologische Beteiligung einhergehenden) Verlaufsform. Gesichert wird die Diagnose durch Nachweis verminderter Beta-Glucocerebrosidaseaktivität in Leukozyten oder Fibroblasten und ggf. durch Nachweis des Gendefekts.
Das Manifestationsspektrum des Typ 1 reicht von asymptomatischen Formen bis hin zu schwerer Beeinträchtigung und Behinderung, z.B. schwere Skelettveränderungen und seltener vitale Gefährdung, meist bei Lungenbeteiligung. Als typische Manifestationen gelten gemäß der Zulassung: Anämie nach Ausschluss anderer Ursachen, Thrombozytopenie, Knochenerkrankung nach Ausschluss anderer Ursachen, z.B. Eisenmangel, Hepatomegalie und Splenomegalie. Bei Kindern kommt die anhaltende Wachstumsverzögerung hinzu.
Außerdem liegt eine Zulassung für Typ 3 der Gaucher-Krankheit (chronisch neuronopathische Verlaufsform) vor. Die Anwendung von Imiglucerase bei dieser Indikation ist nicht Gegenstand dieses Hinweises.
Die Therapie sollte von einem Arzt überwacht werden, der mit der Behandlung der Gaucher-Krankheit vertraut ist.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
In Anbetracht der außergewöhnlich hohen Kosten des Arzneimittels bezogen auf Jahrestherapiekosten je Fall- und der in der Regel lebenslang notwendigen Enzymsubstitution ist bei der Wahl der Einstiegsdosis und Erhaltungsdosis entsprechend dem Wirtschaftlichkeitsgebot der GKV stets die niedrigste individuell wirksame Dosis einzusetzen. Zur Frage des optimalen Therapiebeginns und zur Dosiswahl sind neben nationalen Empfehlungen auch international publizierte Kriterien und Vorschläge zur Dosierung zu berücksichtigen (z.B. aus den Niederlanden, Israel, Australien, Kanada).
Die nach der Fachinformation und in publizierten Studien untere wirksame Dosis von (10 bis) 15 I.U./KG Körpergewicht alle 2 Wochen ist sowohl in der Initial-, als auch in der Dauertherapie nur zu überschreiten, wenn dies durch die individuelle Manifestation und ggf. den Verlauf anhand des vorliegenden international publizierten Erkenntnismaterials eindeutig begründet ist. Diese Überlegungen sind fallbezogen zu dokumentieren. Eine initiale Hochdosistherapie (60 I.U./KG Körpergewicht alle 2 Wochen) ist nur in besonderen Fällen, z.B. bei gravierenden Knochenveränderungen erforderlich.
Eine evtl. Erhöhung der Dosis setzt eine ausreichend lange Behandlung mit der niedrigeren Dosis voraus. Sie ist ebenfalls anhand klinisch relevanter Zielkriterien zu begründen. Die Möglichkeit einer Dosisreduzierung ist im Rahmen regelmässiger Überprüfungen (i.d.R. Überwachung alle 6 Monate durch mit dem Krankheitsbild vertraute Ärzte) anhand klinisch relevanter Zielkriterien zu überprüfen.
Wenn trotz des Einsatzes der in der Zusammenfassung der Merkmale des Arzneimittels genannten hohen Dosis von 60 I.U./Kg KG alle 2 Wochen über 12 Monate keine relevante Besserung derjenigen klinischen Manifestation, die die Therapie begründet haben, zu verzeichnen ist, muss das Absetzen von Imiglucerase geprüft werden.
Eine probatorische Behandlung mit Imiglucerase bei unklarer Diagnose oder unklarem ätiologischem Zusammenhang der Symptomatik mit der Typ 1-Gaucher-Krankheit ist unwirtschaftlich.
Eine Behandlung asymptomatischer, z.B. in Familienuntersuchungen identifizierter Gendefektträger ist nicht indiziert.
Bei Übergewicht (Erhöhung des BMI) sind ernährungsmedizinische Maßnahmen angezeigt, da eine Gewichtszunahme direkte Auswirkungen auf die zu applizierende Dosis hat.
Die Anwendung von Imiglucerase bei akuten neuronopathischen Verlaufsformen (ehemals M. Gaucher Typ 2) ist ebenfalls nicht indiziert.
| Kosten |
Die jährlichen Therapiekosten (ohne Diagnostik) für Imiglucerase betragen lt. Lauertaxe bei 70 kgKörpergewicht (N3, 400 I.U,):
| Bei ca. 15 I.U./kg Körpergewicht alle 2 Wochen: | ca. 172.000 Euro |
| Bei ca. 18 I.U./kg Körpergewicht alle 2 Wochen: | ca. 230.000 Euro |
| Bei 60 I.U./kg Körpergewicht alle 2 Wochen: | ca. 630.000 Euro |
Bei der Dosiswahl müssen die Packungsgrößen/Stückelungen beachtet werden. Z. B würden bei Verordnung von 15 I.U./kg Körpergewicht bei 70 kg Körpergewicht alle 2 Wochen 150 I.U. Imiglucerase (Kosten: 825,-- Euro je Infusion) verworfen werden. Kleinere Dosisanpassungen können gelegentlich erfolgen, wenn das Verwerfen von Teilmengen vermieden werden soll. Die Dosis kann bis zur nächsten vollen Flasche gerundet werden, solange die monatliche Dosis insgesamt unverändert bleibt.
Kumuliert entstehen z.B. für einen Erwachsenen mit einem Körpergewicht von 70 kg mit einer Lebenserwartung von 40 Jahren und einer mittelhohen Dosis von 30 I.U./kg Körpergewicht alle 2 Wochen prospektiv Lebenszeitkosten - bei heutigem Preisniveau - von 14 Mio. Euro. Eine Erhöhung oder Reduzierung der Dosis um 10 I.U./Kg Körpergewicht bedeutet in der Dauertherapie durchschnittliche Mehr- oder Minderkosten von 4,5 Mio. Euro auf Lebenszeit berechnet.
| Wirkungen |
Beim Morbus Gaucher handelt es sich um eine autosomalrezessiv vererbte Speicherkrankheit mit einem Defekt der Glucocerebrosidase. Die Substitution des defekten Enzyms durch das gentechnisch gewonnene Produkt Imiglucerase kann die Rückbildung der hämatologischen, visceralen und ossären Veränderungen bewirken, soweit diese potentiell reversibel sind und das Allgemeinbefinden verbessern. Erfolgreich behandelbar sind zudem krankheitsbedingte Wachstumsverzögerungen bei Kindern.
| Wirksamkeit |
Die Zulassung des aus menschlicher Plazenta gewonnenen Vorgängerprodukts Alglucerase (Ceredase § ) erfolgte zunächst in den USA und 1994 in Deutschland aufgrund einer kleinen nicht kontrollierten Pilotstudie mit der hohen Dosierung von 2 x 60 I.U./kg Körpergewicht alle 2 Wochen. Eine klinische Wirksamkeit der Dauertherapie mit Alglucerase wurde dokumentiert durch Reduzierung der Spleno- und Hepatomegalie , die Verbesserung hämatologischer Mangelerscheinungen , eine Verbesserung der Knochenmineralisation und eine Verbesserung von Kachexie und Verfall bei Kindern . Das Ansprechen der Knochenveränderungen ist weniger regelhaft und z.T. deutlich verzögert zu beobachten.
Alglucerase wurde ab 1998 von dem rekombinanten Produkt Imiglucerase (Cerezyme § ) abgelöst. Die Wirksamkeit der Enzymsubstitutionstherapie bei manifestem M. Gaucher Typ 1 ist unstrittig. Die hohen Kosten von Alglucerase/Imiglucerase führten bereits früh zu weiteren kleinen, bzgl. der Einschluss- und Zielkriterien usw. heterogenen Studien/Anwendungsbeobachtungen mit z.T. wesentlich niedrigeren Dosierungen (bis zu 1,15 IU./kg Körpergewicht) und verkürzten Applikationsintervallen (3x / Woche). Die Spanne der in Studien / Anwendungsbeobachtungen eingesetzten Dosierungen reicht von 14 I.U./kg Körpergewicht / 4 Wochen bis zu 120 I.U./kg Körpergewicht / 4 Wochen.
Die Beurteilung der Ergebnisse wird international kontrovers diskutiert. Anders als z.B. in den Niederlanden oder in Israel wurde in deutschen Zentren z.T. auch in der Dauertherapie die Hochdosistherapie bevorzugt. In einer Empfehlung der deutschen Therapiezentren aus dem Jahr 2000 werden - abhängig von der Manifestation - mittlerweile Dosierungen in einem Spektrum von 20-60 I.U. pro kg Körpergewicht alle 2 Wochen vorgeschlagen.
Randomisierte kontrollierte Studien zur Dosishöhe und zu den verschiedenen Intervallen liegen nicht vor. Dass derartige Studien grundsätzlich möglich sind, zeigt die einzige publizierte randomisierte Studie, die den Beleg der Gleichwertigkeit von Alglucerase und Imiglucerase als Ziel hatte.
Eine im Jahr 2000 publizierte, nicht randomisierte, vergleichende Studie (10 vers. 60 I.U./kg Körpergewicht alle 2 Wochen) des NIH ergab Hinweise auf einen unteren Schwellenwert von 10-15 I.U./kg Körpergewicht alle 2 Wochen für eine Wirksamkeit der Enzymsubstitutionstherapie bezogen auf die nichtskelettale Symptomatik. Die Hochdosistherapie, die ein rascheres klinisches Ansprechen zeigte, soll danach eher bei aggressiven Verläufen erwogen werden.
Das Vorliegen einer potentiell reversiblen, deutlichen Skelettmanifestation wird von vielen Autoren und in der Fachinformation als Begründung für eine initiale Hochdosistherapie mit ggf. nachfolgender Dosisreduzierung gesehen. Eine nationale und internationale Analyse der pathogenetisch entscheidenden Knochenmarksveränderungen mittels MRT ergab allerdings keinen signifikanten Zusammenhang zwischen Dosis und Ansprechen der Knochenmarksveränderungen.
| Risiken ggf. Vorsichtsmaßnahmen |
Die Verträglichkeit ist insgesamt gut. 15 % der mit Imiglucerase Behandelten entwickeln IgGAntikörper. Bei diesen Patienten sind besondere Vorsichtsmaßnahmen zu beachten. Klinische Zeichen einer Überempfindlichkeit gegen Imiglucerase werden bei 3 % beobachtet, die Behandlung muss dann ggf. aus- bzw. abgesetzt werden.
In der Schwangerschaft soll Imiglucerase nur eingesetzt werden, wenn dies unbedingt nötig ist.
Infliximab bei Rheumatoider Arthritis
(z.B. Remicade § )
Beschluss vom: 6.02.2002
In Kraft getreten am: 22.06.2002
BAnz. 2002, Nr. 112 vom 21.06.2002 S. 13.577
| Indikation |
Infliximab ist ein chimärer, monoklonaler IgG1-Antikörper, der in Deutschland zugelassen ist zur Behandlung:
- der Reduktion der Symptomatik und Verbesserung der körperlichen Funktionsfähigkeit bei Patienten mit aktiver rheumatoider Arthritis, die nur unzureichend auf krankheitsmodifizierende Präparate, einschließlich Methotrexat (MTX), angesprochen haben
- sowie zur Behandlung des Morbus Crohn (siehe gesonderten Therapiehinweis, veröffentlicht im Bundesanzeiger am 21.04.2001).
Die Wirksamkeit und die Verträglichkeit bei rheumatoider Arthritis sind nur für die kombinierte Anwendung mit MTX belegt.
| Wirkungen |
Der monoklonale, humanmurine Antikörper bindet mit hoher Affinität sowohl an lösliche als auch an transmembrane Formen von Tumornekrosefaktor alpha (TNFα), aber nicht an Lymphotoxin-a (TNFβ). Infliximab bildet rasch stabile Komplexe mit menschlichem TNFα, ein Vorgang, der mit der Hemmung von TNFα-Bioaktivität einhergeht. Neben der nachweisbaren Reduktion von TNFα kommt es zu einer Verminderung des normalerweise bei Patienten mit rheumatoider Arthritis erhöhten Entzündungsmarkers im Serum, dem C-reaktiven Protein (CRP) und zu einer Absenkung des Serum-Interleukin 6 (IL-6) Spiegels.
| Wirksamkeit |
Die Wirksamkeit wurde bei 428 Patienten, die trotz Behandlung mit Methotrexat eine rheumatoide Arthritis haben, in einer fünfarmigen randomisierten, placebokontrollierten Doppelblindstudie untersucht. Eingesetzt wurden Placebo oder Infliximab in unterschiedlichen Dosierungen (3 mg/kg oder 10 mg/kg) und Infusionsintervallen (4 bzw. 8 Wochen nach jeweils 3 initialen Infusionen in den Wochen 0-2-6). Die vorbestehende Therapie mit MTX (im Median 15 mg/Tag) wurde beibehalten.
Eine Verbesserung der klinischen Symptomatik (definiert als mindestens 20%ige Reduktion nach den ACR-Kriterien) wurde bei der später zugelassenen Applikationsweise (3 mg / Kg Körpergewicht alle 8 Wochen + MTX) nach 54 Wochen bei 36 von 86 Patienten (42 %) beobachtet,(Placebo + MTX: 15 / 88). Auch ein radiologischer Score (Hemmung der Progression der Gelenkzerstörung) und die Beurteilung der körperlichen Funktionsfähigkeit zeigten einen günstigeren Verlauf in der Therapiegruppe. Das klinische Ansprechen war ab der 2. Woche zu verzeichnen. Es konnte im bisher vorgestellten Zeitraum (bis zur 102. Woche) aufrechterhalten werden. Eine Dosissteigerung oder die Verkürzung der Infusionsintervalle führten zu keinem signifikant besseren Ansprechen. (Fachinfo, Lipsky)
| Risiken ggf. Vorsichtsmaßnahmen |
Infliximab ist kontraindiziert bei Patienten mit Tuberkulose oder anderen opportunistischen Infektionen, schweren Infektionen wie Sepsis, Abszessen, Infektionen sowie mittelschwerer oder schwerer Herzinsuffizienz. Bei Patienten mit leichter Herzinsuffizienz ist besondere Vorsicht geboten.
Gebärfähige Frauen sollten eine adäquate Empfängnisverhütung betreiben und diese über mindestens sechs Monate nach der letzten Infusion fortführen. Bei Kindern unter 17 Jahren ist die Wirksamkeit und Verträglichkeit bisher nicht geprüft.
Die Patienten müssen vor, während und nach der Behandlung mit Remicade § hinsichtlich des Auftretens von Infektionen, einschließlich einer Tuberkulose, genau beobachtet werden. Bei Auftreten einer schweren Infektion oder Sepsis muss die Behandlung mit Remicade § abgesetzt werden.
Bevor die Therapie mit Remicade § begonnen wird, müssen alle Patienten hinsichtlich einer aktiven oder inaktiven ("latenten") Tuberkulose beurteilt werden. Diese Beurteilung muss eine ausführliche klinische Anamnese, einschließlich einer Tuberkulosevorerkrankung oder möglichem Kontakt zu Tuberkulose-Kranken und einer vorherigen und/oder derzeitigen immunsuppressiven Therapie, umfassen. Geeignete Screeningtests, d. h. ein Tuberkulintest und eine Röntgenaufnahme des Thorax, sollten bei allen Patienten durchgeführt werden. Ärzte, die Remicade § verordnen, sollten daran denken, dass es bei schwer kranken oder immungeschwächten Patienten zu falschnegativen Ergebnissen beim Tuberkulintest kommen kann. Bei Diagnose einer aktiven Tuberkulose darf auf keinen Fall eine Behandlung mit Remicade § eingeleitet werden.
Bei Bestehen einer inaktiven Tuberkulose muss eine sorgfältige Nutzen/Risiko-Abwägung für den Patienten vorgenommen werden. Für den Fall einer positiven Therapieentscheidung muß zunächst eine Anti-Tuberkulose-Behandlung erfolgen.
Infliximab wird sowohl mit akuten infusionsbedingten Reaktionen, einschließlich anaphylaktischem Schock als auch einer verzögerten Überempfindlichkeitsreaktion in Zusammenhang gebracht. Akute infusionsbedingte Reaktionen treten während oder innerhalb von zwei Stunden nach der Infusion auf und sind während der ersten und zweiten Infusion am wahrscheinlichsten.
Bei einigen Patienten können gegen Infliximab gebildete Antikörper schwerwiegende allergische Reaktionen verursachen. Diese Antikörper können nicht immer in Serumproben nachgewiesen werden. Bei 25 % der Patienten wurde eine verzögerte Überempfindlichkeitsreaktion beobachtet, wenn Infliximab nach einem Zeitraum von zwei bis vier Jahren erneut eingesetzt wurde. Eine erneute Anwendung nach einer Therapiepause von mehr als 15 Wochen wird nicht empfohlen.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Die Behandlung mit Infliximab stellt eine Alternative zur Reduktion der Symptomatik und Verbesserung der körperlichen Funktionsfähigkeit bei Patienten mit aktiver rheumatoider Arthritis dar, wenn eine Therapie mit allen individuell indizierten DMARD § s (Basistherapeutika) einschließlich MTX bis zu einer Dosis von 25 mg pro Woche (+ ggf. Folinsäure) und deren Kombinationen erfolglos geblieben ist (Smolen 1999, Furst 2000). Diese müssen lang genug (in der Regel mindestens 6 Monate), in adäquater Dosis und unter fachlich kompetenter Überwachung eingesetzt worden sein (Empf.)
Bei der Indikation Rheumatoide Arthritis erfolgt die Infusion nach der Initialphase (0. -2. 6. Woche) alle 8 Wochen. Für höhere Dosierungen als 3mg/kg oder eine Verkürzung des Infusionsintervalls auf z.B. 4 oder 6 Wochen liegt keine Zulassung vor.
Infliximab muss zusammen mit MTX verabreicht werden. Soweit eine Indikation für eine Therapie mit einem TNF alpha-Hemmer besteht und keine gesicherte Kontraindikation für MTX oder Infliximab vorliegt, ist derzeit eine Prioritätensetzung aufgrund der Studienlage nicht möglich (Klippel). Damit stellt die Wirtschaftlichkeit einen wesentlichen Gesichtspunkt
bei der Produktwahl dar. Die Praxisausstattung (Unterbringung für die 2stündige Infusion und die 1-2stündige Nachüberwachung) begründet keine unwirtschaftliche Produktwahl.
Die Therapie ist zu beenden, wenn nach 8 - 12 Wochen keine signifikante Besserung der klinischen und humoralen Entzündungsaktivität zu verzeichnen ist (Furst).
Infliximab ist weder zugelassen noch hinreichend evaluiert für die Anwendung bei weiteren entzündlichrheumatischen Erkrankungen wie der Juvenilen Chronischen Arthritis oder seronegativen Spondarthritiden (z.B. Ankylosierende Spondylitis).
Bei rheumatoider Arthritis werden für einen Erwachsenen pro Infusion 2 bzw. 3 Trockenampullen benötigt (3mg/kg Körpergewicht). Somit ist pro Infusion mit Kosten von ca. 1.660 Euro bzw. (häufiger) 2.390,- Euro zuzüglich der Kosten für MTX zu rechnen.
| Wirkstoff | Dosierung | pro Infusion | Jahrestherapiekosten * im 1. Jahr + MTX 15mg oral/pro Woche |
| Infliximab | bis 66 kg 2 Durchstechflaschen pro Infusion | rd. 1.660,- Euro | rd.13.500,- Euro |
| Infliximab | bis 100 kg 3 Durchstechflaschen pro Infusion | rd. 2.390,- Euro | rd.19.300,- Euro |
(*Die Berechnung erfolgte für das erste Jahr mit 8 Infusionen. 0.-2.-6.-14.-22.-30.-38.-46. KW plus 15 mg MTX/oral pro Woche; Kosten ca. 180,- Euro )
Infliximab bei Morbus Crohn
(z.B. Remicade § )
Beschluss vom: 16.10.2000
In Kraft getreten am: 22.04.2001
BAnz. 2001, S. 7.478
| Indikation |
Infliximab ist ein chimärer, monoklonaler IgG1-Antikörper, der in Deutschland zugelassen ist zur Behandlung:
- einer schwergradigen, aktiven Form des Morbus Crohn bei Patienten, die trotz einer vollständigen und adäquaten Therapie mit einem Kortikosteroid und / oder einem Immunsuppressivum auf diese Behandlung nicht ansprechen und
- von Morbus Crohn mit Fistelbildung bei Patienten, die trotz einer vollständigen und adäquaten Therapie mit einer konventionellen Behandlung auf diese nicht ansprechen.
- zur Therapie der rheumatoiden Arthritis (gesonderter Therapiehinweis hierzu folgt)
Von der europäischen Zulassungsbehörde wurde dem Hersteller die Zulassung mit der Auflage erteilt, zusätzliche Informationen über die Erhaltungstherapie bereitzustellen.
| Wirkungen |
Der monoklonale, humanmurine Antikörper bindet mit hoher Affinität sowohl an lösliche als auch an transmembrane Formen von Tumornekrosefaktor alpha (TNFα), aber nicht an Lymphotoxin-a (TNFβ). Infliximab bildet rasch stabile Komplexe mit menschlichen TNFα, ein Vorgang, der mit dem Verlust von TNFα-Bioaktivität einhergeht. Neben der nachweisbaren Reduktion von TNFα kommt es zu einer Verminderung des normalerweise bei Crohn-Patienten erhöhten Entzündungsmarkers im Serum, dem C-reaktiven Protein (CRP).
| Wirksamkeit |
Aktiver Morbus Crohn
Die Wirksamkeit wurde bei 108 Patienten mit einer mäßig bis schwergradigen, aktiven Form von Morbus Crohn (Morbus Crohn-Aktivindex (CDAI) zwischen 220 und 400) in einer randomisierten, doppelt verblindeten, placebokontrollierten Studie überprüft. Zusätzlich mussten die Patienten zuvor erfolglos über mindestens acht Wochen mit Mesalazin oder zumindest über die letzten acht Wochen mit einer Maximaldosis von 40 mg / Tag Kortikosteroide oder über mindestens sechs Monate 6-Mercaptopurin beziehungsweise Azathioprin vorbehandelt sein. Der Beginn des Ansprechens wurde innerhalb von zwei Wochen beobachtet, das maximale Ansprechen wurde nach vier Wochen erreicht.
Fistelbildung bei Morbus Crohn
Die Wirksamkeit wurde auch in einer randomisierten, placebokontrollierten Doppelblindstudie an 94 Patienten mit Morbus Crohn mit Fistelbildung überprüft, deren Fistel mindestens drei Monate bestand. Die gleichzeitige Verabreichung stabiler Dosen von konventionellen Therapien mit Kortikosteroiden, Aminosalizylaten, Antibiotika, Methotrexat, 6-Mercaptopurin oder Azathioprin war zulässig und 83 % erhielten weiterhin mindestens eines dieser Arzneimittel.
Die mittlere Zeit bis zum Einsetzen eines Ansprechens der mit Infliximab behandelten Gruppe betrug zwei Wochen. Die mittlere Dauer des Ansprechens betrug zwölf Wochen; nach 22 Wochen bestand bezüglich der Anzahl der Patienten mit Ansprechen keine Differenz zwischen Placebo und den beiden Infliximabdosierungen. Es wurden zu wenig
Patienten mit Abdominalfisteln in den Studien behandelt, um die Wirksamkeit zu beurteilen. Neue Fisteln bildeten sich bei ungefähr 15 % der Patienten unabhängig davon, ob sie mit Placebo oder Infliximab behandelt wurden.
| Risiken ggf. Vorsichtsmaßnahmen |
Infliximab ist kontraindiziert bei Patienten mit Sepsis oder mit klinisch manifesten Infektionen und/oder Abszessen. Außerdem darf das Medikament nicht Patienten verabreicht werden, die in der Anamnese eine vorbestehende Überempfindlichkeit gegenüber Infliximab, murinen Proteinen oder irgendeinem Hilfsstoff aufweisen.
Gebärfähige Frauen sollten eine adäquate Empfängnisverhütung betreiben und diese über mindestens sechs Monate nach der letzten Infusion fortführen. Bei Kindern unter 17 Jahren ist die Wirksamkeit und Verträglichkeit bisher nicht geprüft; eine solche Behandlung sollte vermieden werden.
Die häufigsten Nebenwirkungen unter Infliximab waren insgesamt Kopfschmerzen (23 %), gastrointestinale Nebenwirkungen (17 %) und Infektionen des oberen Respirationstraktes (16 %).
Infliximab wurde mit akuten infusionsbedingten Reaktionen und einer verzögerten Überempfindlichkeitsreaktion in Zusammenhang gebracht. Diese unterscheiden sich im Zeitpunkt des Auftretens.
Akute infusionsbedingte Reaktionen treten während oder innerhalb von zwei Stunden nach der Infusion auf und sind während der ersten und zweiten Infusion am wahrscheinlichsten. Einige Reaktionen können mäßig bis schwerwiegend sein und einer symptomatischen Behandlung bedürfen. Deshalb sollten eine Notfallausrüstung und Notfallmedikation zur Behandlung dieser Reaktionen zum Sofortgebrauch zur Verfügung stehen.
Bei einigen Patienten können sich gegen Infliximab gerichtete Antikörper bilden und schwerwiegende allergische Reaktionen verursachen. Diese Antikörper können nicht immer in Serumproben nachgewiesen werden.
Eine verzögerte Überempfindlichkeitsreaktion wurde bei 25 % der Patienten beobachtet, die mit Infliximab nach einem Zeitraum von zwei bis vier Jahren ohne Infliximab-Behandlung wieder behandelt wurden. Anzeichen und Symptome beinhalten Myalgie und/oder Arthralgie mit Fieber, Pruritus, Gesichts-, Hand- oder Lippenödem, Dysphagie, Urtikaria, Halsund/oder Kopfschmerzen.
Über schwere Infektionen, einschließlich Sepsis und tödliche Infektionen, ist bei Patienten mit TNF-blockierenden Substanzen berichtet worden. Die meisten dieser schweren Infektionen bei mit Infliximab behandelten Patienten sind unter gleichzeitiger immunsuppressiver Therapie aufgetreten. Klinische Symptome, die auf ein Lupusähnliches Syndrom hinwiesen, traten in seltenen Fällen auf.
Bei Patienten mit Morbus Crohn und rheumatoider Arthritis, die mit Infliximab behandelt wurden, traten in einzelnen Fällen Lymphome und Myelome auf. Es ist unbekannt, ob die wiederholte Verabreichung von Infliximab die Entstehung dieser Erkrankungen verursachen kann.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Die Behandlung mit Infliximab ist nur bei schwergradigen, aktiven und therapieresistenten Formen des Morbus Crohn sowie beim Morbus Crohn mit Fistelbildung nach Versagen einer vollständigen und adäquaten konventionellen Behandlung indiziert. Zu den anerkannten Behandlungen gehört die Gabe von Kortikosteroiden, Aminosalizylaten, Immunsuppressiva und Antibiotika so wie Ernährungsumstellung. Erst wenn diese Behandlungsstrategien in Art und Dosierung für den individuellen Patienten angepasst erfolglos durchgeführt wurden, ist eine einmalige beziehungsweise bei Fisteln dreimalige Infusion von Infliximab zu erwägen. Häufigere Gaben beziehungsweise höhere Dosierungen als 5 mg/kg Körpergewicht sind unwirtschaftlich. Patienten, die auf eine Infusion nicht angesprochen haben, werden im allgemeine auch nicht auf weitere reagieren.
Falls die Anzeichen und Symptome des Morbus Crohn wieder auftreten, kann Infliximab entsprechend der Fachinformation innerhalb von 14 Wochen nach der letzten Infusion erneut verabreicht werden. Bei der schwergradigen, aktiven Form des Morbus Crohn wurden Wirksamkeit und Sicherheit über eine einmalige Behandlung hinaus bisher jedoch nicht nachgewiesen. Bei Patienten mit Morbus Crohn und Fistelbildung wurden die Sicherheit und Wirksamkeit über die dreimalige Gabe hinaus nicht untersucht. Mit schwerwiegenden Nebenwirkungen muss bei erneuter Behandlung gerechnet werden.
| Kosten |
Remicade kostet je Trocken-Ampulle 100 mg 926,79 Euro.
Bei Fisteln ist mit Kosten für 3 Infusionen in Höhe von ca. 8.180,67 Euro zu rechnen (bezogen auf einen 60 kg schweren Patienten).
Dem stehen folgende Monatskosten für in der Behandlung des Morbus Crohn anerkannte Wirkstoffe qeqenüber:
| Wirkstoff | Dosierung | Kosten 30 Tage |
| Prednisolon | 50 mg/d | 20,45 Euro |
| Budesonid | 9 mg/d | 180,49 Euro |
| Azathioprin | ca. 140 mg/d | 114,02 Euro |
| Mesalazin | 4,0 g/d | 166,08 Euro |
| Sulfasalazin | 4,0 g/d | 90,50 Euro |
Inhalierbares, kurzwirksames Humaninsulin
(aufgehoben durch BAnz. AT vom 08.06.2016 B1)
Leflunomid
(z.B. Arava § )
Beschluss vom: 16.08.2007 / 15.05.2008
In Kraft getreten am: 21.12.2007 / 03.09.2008
BAnz. 2007, Nr. 238 vom 20.12.2007 S. 8.316 / BAnz. 2008, Nr. 132 vom 02.09.2008 S. 3.216
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Rheumatoide Arthritis
Patienten mit rheumatoider Arthritis (RA) sollten möglichst frühzeitig mit Basistherapeutika (Basistherapeutikum = Disease Modifying Antirheumatic Drug = DMARD) behandelt werden, um die Entwicklung von Funktionseinschränkungen zu vermeiden.
Ein engmaschiges Monitoring der Krankheitsaktivität unter Therapie mit DMARDs ist wichtig für die Prognose der Erkrankung. Ein unzureichendes Ansprechen sollte umgehend zu Therapie-Modifikationen führen.
Bei früher RA sind Methotrexat (MTX) und Sulfasalazin (SSZ) sowohl hinsichtlich ihres Effektivitäts- und Toxizitätsprofils als auch unter wirtschaftlichem Aspekt DMARDs der ersten Wahl. Chloroquin, Auranofin und Hydroxychloroquin werden bei früher RA als schwächer wirksam eingeschätzt als MTX, SSZ, parenterales Gold, Ciclosporin und Penicillamin.
Leflunomid ist dagegen bisher nicht untersucht bei Patienten im frühen Stadium der RA.
In fortgeschritteneren Krankheitsstadien hat sich Leflunomid als ähnlich wirksam erwiesen wie MTX oder SSZ (s.u. Abschnitt Wirksamkeit). Unter wirtschaftlichen Gesichtspunkten bietet es sich als Mittel der zweiten oder dritten Wahl an.
Bei therapierefraktären Verläufen kann sein Einsatz erwogen werden bevor auf einen TNF Alpha Blocker umgestellt wird.
Die Überlegenheit einer Kombination von Leflunomid mit einem Tumornekrosefaktor (TNF) Alpha Blocker gegenüber einer TNF Alpha Blocker Monotherapie ist durch randomisierte kontrollierte Studien nicht belegt. Vergleichende Studien zur Kombination von TNF Alpha Blockern mit MTX gibt es nicht. Es ist bisher kein TNF Alpha Blocker explizit für eine Kombinationstherapie mit Leflunomid zugelassen.
Bei ungesichertem Nutzen und erhöhtem Risiko für toxische Nebenwirkungen ist eine Kombinationstherapie von Leflunomid mit TNF Alpha Blockern in der Regel unwirtschaftlich. Für den Fall einer Unverträglichkeit von MTX auch in niedrigeren Dosierungen bzw. Vorliegen von Kontraindikationen, die den Einsatz von MTX ausschließen, sind die TNF Alpha Inhibitoren Adalimumab und Etanercept auch als Monotherapie zugelassen. Bei Versagen einer Therapie mit TNF Alpha Blockern stehen für diese Situation zugelassene Biologicals wie Abatacept oder Rituximab zur Verfügung.
Psoriasis-Arthritis
Die Wirkung aller bisher untersuchten DMARDs bei der Psoriasis-Arthritis wird generell als gering bis mittelmäßig eingeschätzt. Im Gegensatz zur rheumatoiden Arthritis konnte für kein DMARD in dieser Indikation eine Verzögerung der Progression von Gelenkdestruktionen belegt werden. Es existieren bisher keine vergleichenden Studien von Leflunomid mit anderen Basistherapeutika zur Wirksamkeit bei Psoriasis-Arthritis.
Patienten mit Psoriasis-Arthritis, die gleichzeitig systemisch behandlungsbedürftige Hautläsionen aufweisen, sollten primär mit MTX oder Ciclosporin behandelt werden, da bei diesen Substanzen eine gute Wirksamkeit nicht nur bezüglich der dermatologischen Symptome, sondern auch bezüglich der arthritischen Symptome belegt ist.
Bei der kleinen Gruppe von Patienten mit Psoriasis-Arthritis ohne wesentliche dermatologische Symptomatik kommt, sofern eine Therapie mit NSAR nicht ausreichend ist, unter Berücksichtigung des Zulassungsstatus der Einsatz von Leflunomid oder MTX in Betracht.
| Kosten |
Vergleich der Jahrestherapiekosten verschiedener DMARDs und TNF Alpha Blocker:
| Wirkstoff | geniche Dosierungen | Jahrestherapiekosten Anmerkung | |
| Leflunomid | 10 mg / d 20 mg / d | 948 Euro 1.333 Euro | |
| MTX oral* | 10 mg /Woche 15 mg / Woche | 96s 150 Euro | zusätzl. Folsäure 5mg/ Woche (rd.10 Euro ) |
| Sulfasalazin* | 2 g / d | 382 Euro | |
| parenterales Gold | 2 x 50 mg / Monat | 558 Euro | |
| Ciclosporin* | 2 x 100 mg | 4.226 Euro | 2,5 bis max. 5 mg / kg KG |
| Infliximab | 3mg / kg KG | 20.061 Euro | Kombination mit MTX obligat, Kosten müssen dazu gerechnet werden |
| 3mg / kg KG | 35.107 Euro | Dosiseskalation, ab 12. W alle 4 Wochen | |
| 7,5 mg / kg KG | 41.415 Euro |
Dosiseskalation, ab 12. W alle 8 Wochen | |
| Etanercept | 50 mg / Woche | 22.845 Euro | |
| Adalimumab | 40 mg / 2 Wochen | 22.845 Euro | Kombination mit MTX bei dieser Dosis obligat, Kosten müssen dazu gerechnet werden |
| 40 mg / Woche | 45.690 Euro | Monotherapie |
Preisstand 01.07.2007, berücksichtigt wurde die jeweils größte verfügbare Packungsgröße
* Berechnung erfolgte auf Basis von Festbeträgen, Generika unterhalb Festbetrag verfügbar Weitere Informationen finden Sie in den Therapiehinweisen zu TNF Alpha Inhibitoren. Die Preiskalkulation von Infliximab erfolgte auf der Basis eines Patienten mit einem Standardgewicht von 70 kg.
| Indikation |
Leflunomid ist ein antirheumatisches Basistherapeutikum. Es ist zugelassen zur Behandlung Erwachsener mit aktiver rheumatoider Arthritis und aktiver Psoriasis-Arthritis. Eine Zulassung für die Indikation der juvenilen rheumatoiden Arthritis wurde von der EMEA im November 2005 abgelehnt.
Die Behandlung mit Leflunomid wird mit einer Dosierung von 100 mg täglich über drei Tage begonnen. Die empfohlene Erhaltungsdosis bei rheumatoider Arthritis beträgt 10 bis 20 mg Leflunomid täglich. Die Dosis von 20 mg hat Vorteile hinsichtlich der Wirksamkeit, während die 10-mg-Dosierung ein günstigeres Sicherheitsprofil aufweist.
Bei Psoriasis-Arthritis beträgt die Erhaltungsdosis 20 mg täglich.
Die Therapie sollte nur von Fachärzten, die über ausreichende Erfahrung in der Behandlung von rheumatoider Arthritis und Psoriasis-Arthritis verfügen, eingeleitet und überwacht werden (in der Regel von internistischen Rheumatologen).
| Wirkungen |
Leflunomid wirkt antiproliferativ und hat immunsuppressive und antiphlogistische Eigenschaften. Als wesentlicher Wirkmechanismus gilt die Hemmung des Enzyms Dihydroorotat-Dehydrogenase. Hierdurch wird die Synthese von Pyrimidin bzw. Nukleinsäuren beeinträchtigt. Die Proliferation aktivierter Lymphozyten wird gehemmt.
| Wirksamkeit |
Rheumatoide Arthritis
Die Wirksamkeit und Effektivität von Leflunomid bei RA wurde in 3 großen randomisierten kontrollierten doppelblinden multizentrischen Studien über 6 und 12 Monate untersucht (US 301, MN 301, MN 302). Diese Studien wurden anschließend bei erhaltener Verblindung mit verbliebenen Patienten bis zu zwei Jahren fortgeführt (US 301, MN 303 / MN 305, MN 304). Primäre Effektivitätsparameter waren in allen Studien die Verbesserungen nach den Kriterien des American College of Rheumatology (ACR: Reduzierung der Anzahl geschwollener und druckschmerzhafter Gelenke zzgl. 3 von 5 weiteren Kriterien).
Zwei Studien hatten in der Anfangsphase einen Placebo-Arm. Direkte Vergleiche mit anderen Basistherapeutika erfolgten in diesen Studien mit MTX und SSZ.
Gegenüber Placebo erwies sich Leflunomid als effektives Basistherapeutikum mit signifikanter Verbesserung der ACR Response, des Funktionsstatus sowie einer Verminderung der radiologisch erfassbaren Progression (MN 301, US 301).
In der hauptsächlich in Europa durchgeführten Studie MN 302/ MN 304 und der nordamerikanischen Studie US 301 wurden Leflunomid vs. MTX bzw. Leflunomid vs. MTX vs. Placebo untersucht.
In MN 302/ MN 304 (1-Jahreskohorte = 999; 2-Jahreskohorte = 612) waren die ACR 20 Responder-Raten der 1-Jahreskohorte unter MTX signifikant höher. Dieser Unterschied blieb im zweiten Jahr numerisch bestehen, war aber nicht mehr signifikant.
Bezogen auf den Funktionsstatus gemessen am Health Assessment Questionaire (HAQ) war MTX dem Leflunomid in der 1-Jahreskohorte ebenfalls überlegen. Der Unterschied war in der 2-Jahreskohorte nicht mehr signifikant.
Die Auswertung radiologischer Befunde von 64% der Patienten nach dem Larsen-Score ergab in beiden Behandlungsgruppen im ersten Jahr eine geringe Progression der radiologischen Veränderungen. Im zweiten Jahr wurde unter MTX eine Verminderung, unter
Leflunomid keine weitere Progression der radiologisch erkennbaren Gelenkschäden beobachtet. Der Unterschied zugunsten MTX war statistisch signifikant.
Im Gegensatz zur europäischen Studie waren in der nordamerikanischen Studie US 301 (1-Jahreskohorte = 482; 2-Jahreskohorte = 235) die ACR Responder-Raten in der ITT-Analyse unter MTX und Leflunomid statistisch nicht unterschiedlich.
Der Funktionsstatus - gemessen am HAQ - war dagegen sowohl in der 1- als auch in der 2-Jahreskohorte signifikant gebessert bei den mit Leflunomid gegenüber MTX behandelten Patienten.
Die Auswertung radiologischer Befunde von 73% der Patienten nach dem Sharp-Score ergab eine Verlangsamung der Progression unter Leflunomid und MTX gegenüber Placebo. Dieser Effekt war unter Leflunomid und MTX ähnlich stark ausgeprägt (Veränderung des Sharp-Scores in der 1 Jahreskohorte 0,53 Leflunomid vs. 0,88 MTX vs. 2,16 PL, Leflunomid vs. MTX p=0,05, Veränderung des Sharp-Scores in der 2-Jahreskohorte 1,6 Leflunomid vs. 1,2 MTX p=0,65).
Während also in MN 302 / 304 Leflunomid weniger effektiv war als MTX, zeigte sich in US 301 eine vergleichbare Wirksamkeit. Als mögliche Erklärungen für die unterschiedlichen Studienergebnisse wurden sowohl Unterschiede der Häufigkeit einer Folsäure-Supplementation (90% in US 301, 10% in MN 302/ 304), des Studiendesigns (nur in US 301 bestand die Möglichkeit des Therapiewechsels nach 4 Monaten bei ungenügendem Ansprechen) und der Studienpopulation (durchschnittlich längere Krankheitsdauer in US 301) diskutiert.
In der europäischen Studie MN 301 / MN 303 / MN 305 wurde Leflunomid vs. SSZ vs. Placebo geprüft (6-Monatskohorte = 358; 1-Jahreskohorte = 168; 2-Jahreskohorte = 146). Die 6-Monats- und 1-Jahres-Daten zeigten anhand der primären Endpunkte (ACR 20 Response-Kriterien, radiologische Progression anhand des Sharp Scores) eine äquivalente Wirksamkeit von Leflunomid und SSZ. Nach zwei Jahren waren die ACR 20 Response-Raten signifikant größer unter Leflunomid. Der Funktionsstatus war nach sechs Monaten unter beiden DMARDs klinisch bedeutsam gebessert. Die Verbesserung war unter Leflunomid ausgeprägter als unter SSZ (nach sechs Monaten Leflunomid -0,50 vs. SSZ - 0,29 p<0,03). Die Aussagekraft der 2-Jahres-Daten ist beeinträchtigt durch das Studiendesign und die relativ hohen Abbruchraten (nur 116 Patienten beendeten die Studie, zweimalige Verlängerung jeweils nur mit den Patienten, die einer Verlängerung zustimmten).
ACR Response-Raten in den Hauptstudien zu Leflunomid, angegeben ist die Signifikanz des Unterschiedes von Leflunomid (LEF) zum jeweils aktiven Studienarm
| MN 301 | 6. Mo | US 301 | 1 Jahr | MN 302 | 1 Jahr | ||||||
| LEF | SSZ | PL | LEF | MTX | PL | LEF | MTX | ||||
| n | 133 | 133 | 92 | 182 | 182 | 118 | 501 | 498 | |||
| ACR 20% | 55% | 56% | 29% | n.s | 52% | 46% | 26% | n.s | 51% | 65% | s. |
| ACR 50% | 33% | 30% | 14% | n.s | 34% | 23% | 8% | n.s | 22% | 34% | s. |
| ACR 70% | 10% | 8% | 2% | n.s | 20% | 9% | 4% | 15% | 20% | ||
| 2 Jahre | MN 305 | US 301 | MN 304 | ||||||||
| n | 60 | 57 | 98 | 101 | 290 | 317 | |||||
| ACR 20% | 82% | 60% | - | s. | 53% | 48% | - | n.s | 64% | 72% | n.s. |
| ACR 50% | 52% | 25% | - | s. | 34% | 28% | - | n.s | 34% | 38% | n.s. |
| ACR 70% | 25% | 17% | - | 17% | 12% | - | n.s | 15% | 20% | n.s. | |
Eine Metanalyse, in die hauptsächlich die oben genannten Studien eingingen, kommt zu dem Schluss, dass die klinische Effektivität von Leflunomid und MTX vergleichbar ist. Dagegen ergeben sich beim Vergleich von Leflunomid zu SSZ nach zwei Jahren Anhaltspunkte für eine Überlegenheit der klinischen Effektivität von Leflunomid. Alle drei Basistherapeutika zeigen nach radiologisch erfassbaren Kriterien einen gleichermaßen verlangsamenden Effekt.
In randomisierten klinischen Studien wurden bisher die Kombinationen von Leflunomid mit MTX und SSZ untersucht. Eine Überlegenheit dieser Kombinationstherapien gegenüber einer Monotherapie mit Leflunomid wird durch diese Studien nicht belegt. Kombinationsbehandlungen mit Biologika sind nicht ausreichend untersucht. Hierzu existieren bisher nur retrospektive und prospektive Fallserien mit überwiegend kleiner Fallzahl.
Psoriasis-Arthritis
Die Wirksamkeit von Leflunomid bei Psoriasis-Arthritis wurde in einer randomisierten kontrollierten doppelblinden multizentrischen Studie bei 188 Patienten untersucht. Primärer Endpunkt war eine Verbesserung in mindestens 2 Komponenten des PsARC ohne Verschlechterung anderer Komponenten (PsARC = Psoriasis Arthritis Treatment Response Criteria, enthalten als Komponenten die Anzahl druckschmerzhafter und geschwollener Gelenke sowie die globale Einschätzung von Behandler und Patient). Nach sechs Monaten erwies sich Leflunomid gegenüber Placebo überlegen hinsichtlich der Linderung arthritischer Symptome (Verbesserung des PsARC betrug 59 % unter Leflunomid vs. 29,7 % unter Placebo).
Die Wirkung auf Hautläsionen und den Funktionsstatus war zwar statistisch signifikant wurde aber von der EMEA klinisch nur als mäßig bedeutsam gewertet.
Eine 75%ige Besserung des PASI (Psoriasis Area and Severity Index), welche als klinisch relevanter Parameter bei der Beurteilung der Wirksamkeit eines Medikaments in der Psoriasis-Therapie gilt, erreichten nur 17 % der Studienteilnehmer.
Vergleichende Studien zu anderen zugelassenen Arzneimitteln (z.B. MTX) liegen nicht vor.
Pharmakologisch zeichnet sich Leflunomid durch eine lange Halbwertszeit und eine bis zu zwei Jahre anhaltende Verweildauer des aktiven Metaboliten im Organismus aus. Dies muss bei Umstellungen auf andere potentiell toxische DMARDs beachtet werden. Bei der Anwendung der Substanz sind die Empfehlungen zur Therapiesicherheit strikt zu beachten. Gegebenenfalls muss ein Auswaschverfahren mit Cholestyramin erfolgen.
Leflunomid verursacht vermutlich schwerwiegende fetale Schädigungen und ist deshalb in der Schwangerschaft kontraindiziert. Frauen, die schwanger werden wollen, müssen die Therapie beenden und entweder ein Auswaschverfahren anwenden oder eine Wartezeit bis zu 2 Jahren einhalten. Erst wenn nachweislich die Plasmakonzentrationen unter 0,02 mg/I liegen, besteht kein teratogenes Risiko mehr.
Überwiegend traten Nebenwirkungen in der Anfangsphase der Therapie auf. Sie führten in Studien bei 14% - 22% der Patienten zum Therapieabbruch. Nebenwirkungen, die in Studien häufiger zum Therapieabbruch führten, waren Durchfall, Übelkeit, Transaminasenerhöhung, reversible Alopezie und Hypertonie.
Eine Hypertonie oder die Verschlechterung einer vorbestehenden Hypertonie, welche unter Leflunomid in 6% 11% der Fälle und damit 2 3mal häufiger als in den Vergleichsgruppen auftrat, manifestierte sich nicht selten erst im zweiten Behandlungsjahr.
Nach Fachinformation ist Leflunomid u. a. kontraindiziert bei:
- eingeschränkter Leberfunktion,
- schwerem Immundefekt,
- schweren Infektionen,
- deutlich eingeschränkter Knochenmarksfunktion oder ausgeprägter Anämie, Leukopenie oder Thrombozytopenie, die andere Ursachen haben als die rheumatoide Arthritis,
- schwerer Hypoproteinämie,
- mittlerer bis schwerer Niereninsuffizienz sowie
- schwangeren oder stillenden Frauen und
- Frauen, die keinen ausreichenden Empfängnisschutz praktizieren.
Hepatoxizität
Die EMEA hatte 2001 auf ein möglicherweise erhöhtes Risiko schwerwiegender hepatotoxischer Reaktionen unter Leflunomid hingewiesen und zunächst ein monatliches Monitoring der Leberwerte in den ersten 6 Monaten einer Behandlung mit Leflunomid empfohlen. 2003 wurde eine Erhöhung der Frequenz auf 2-wöchentliche Kontrollen empfohlen.
Nachbeobachtungen der Zulassungsstudien bis zu 5 Jahren sowie klinische Studien zu Kombinationen von Leflunomid mit MTX oder SSZ zeigen, dass Leflunomid mit einer relevanten Erhöhung der Leberenzyme (größer 3x Norm) bei 1,5 4,4 % der Patienten verbunden ist. Diese waren jedoch im Verlauf nach Dosisreduzierung oder Abbruch der Behandlung reversibel.
Eine Auswertung der Aetna-US-Datenbank (retrospektive Kohortenanalyse von ca. 40.000 RA-Patienten) ergab für hepatozelluläre Nekrosen Inzidenzen unter Leflunomid von 2,4 pro 10.000 Patientenjahre (1 Fall bei 2.633 Pat.) und unter MTX von 0,6 pro 10.000 Patientenjahre (2 Fälle bei 9.514 Pat.).
Weitere Auswertungen großer Datenbanken amerikanischer Krankenversicherungen und nationaler Register von Rheumapatienten durch die amerikanische Zulassungsbehörde (FDA) 2003 bestätigten die insgesamt geringe Rate ernsthafter, stationär behandlungsbe-
dürftiger hepatotoxischer Reaktionen von ca. 2 pro 10.000 Patientenjahre, die sich nicht signifikant von den Raten anderer DMARDs wie MTX unterscheidet.
Nach Prüfung der in spontanen Meldesystemen berichteten Einzelfällen von akutem Leberversagen kommt die FDA zu dem Schluss, dass nur vereinzelt ein Zusammenhang mit Leflunomid wahrscheinlich war. Angesichts dieses sehr geringen, jedoch lebensbedrohlichen Risikos ist eine sorgfältige Überwachung der Leberwerte entsprechend den Empfehlungen der EMEA auch weiterhin notwendig. Dies gilt besonders bei Kombination mit weiteren hepatotoxischen Substanzen, wie z.B. MTX. Bei Erhöhung der Leberenzyme auf das Zweifache der Norm sollte eine Dosisreduzierung auf 10 mg Leflunomid, bei Persistenz oder Erhöhung über das Dreifache der Norm muss ein Therapieabbruch mit Auswaschverfahren erfolgen.
Montelukast
(Singulair § )
Beschluss vom: 15.11.2007
In Kraft getreten am: 04.04.2008
BAnz. 2008, Nr. 51 vom 03.04.2008 S. 1.200
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Die Therapie der ersten Wahl des Asthmas ist im Erwachsenenalter die Kombination von inhalativen Kortikosteroiden (ICS) mit langwirksamen Betasympathomimetika, wenn ICS in niedriger bis mittlerer Dosis beim mittelgradig persistierenden Asthma nicht ausreichend ist. Es stehen neben der Erhöhung der ICS-Dosis weitere Alternativen zur Verfügung. Die Auswahl richtet sich primär nach dem Nebenwirkungsprofil und sekundär nach dem Preis.
Montelukast verteuert die Therapie erheblich und ist von daher nur angezeigt, eine Monotherapie mit ICS nicht ausreichend ist oder eine Kombinationstherapie von ICS mit langwirksamen Betasympathomimetika nicht in Betracht kommt. Der Einsatz ist nur wirtschaftlich in Kombination mit ICS, wenn eine Monotherapie mit ICS in niedriger bis mittlerer Dosis beim mittelgradig persistierenden Asthma nicht ausreichend ist. Montelukast ist im Erwachsenenalter weder zur Behandlung des Asthmas - auch nicht des Belastungsasthmas - noch der saisonalen allergischen Rhinitis als Komorbidität des Asthmas Therapie der ersten Wahl.
Der Einsatz von Montelukast als Monotherapie des Asthmas ist ab einem Alter von 15 Jahren nicht zugelassen. Das Gleiche gilt für schwergradiges persistierendes Asthma in allen Altersstufen und die chronisch obstruktive Lungenerkrankung (COPD).
Vor dem Hintergrund, dass eine Überlegenheit gegenüber ICS bei Kindern nicht belegt ist und auch das Längenwachstum in der Regel nur unerheblich verzögert wird bei ansonsten vergleichbaren Nebenwirkungen, ist die Monotherapie mit Montelukast im Alter zwischen 2 und 14 Jahren mit leichtem persistierendem Asthma nur indiziert, wenn die Kinder nicht in der Lage sind, Kortikosteroide zu inhalieren oder Nebenwirkungen auftreten, wie zum Beispiel ein erheblich verzögertes Längenwachstum, die gegen den Einsatz von ICS sprechen. Dies entspricht der aktuellen Zulassung des Arzneimittels. Angesichts der heutigen Möglichkeiten zur Inhalation dürfte diese Ausnahme sehr selten sein.
Für alle Altersgruppen gilt, dass beim Belastungsasthma der hohe Preis in der Regel nur gerechtfertigt ist bei Unverträglichkeit gegen inhalative kurzwirksame Betasympathomimetika.
| Kosten (Stand: 01.01.2008) |
Erwachsene
Die Kosten wurden berechnet analog Stufe 3 der Versorgungsleitlinie Asthma (Stand: Januar 2008).
| Zusatzmedikation zur Basistherapie ICS in niedriger bis mittlerer Dosis | DDD | Tagestherapiekosten* | Kosten für 1 Jahr* |
| plus Montelukast | 1 Filmtabl. 10 mg | 1,92 Euro | 700,80 Euro |
| plus Formoterol | 2 Hübe | 0,94 Euro - 1,48 Euro | 343,10 Euro - 540,40 Euro |
| plus Salmeterol | 2 x 50 μg | 1,88 Euro | 686,20 Euro |
| z.B. Salbutamol * | 8 mg | 0,28 Euro | 102,20 Euro |
| ICS Dosissteigerung z.B. Budesonid *, ** | 2 x 400 μg | 0,50 Euro | 182,50 Euro |
| ICS Dosissteigerung z.B. Beclometason *, ** | 5 x 200 μg | 0,85 Euro | 310,25 Euro |
| plus Theophyllin ret. * (75 kg Körpergewicht) | 750 mg | 0,62 Euro | 226,30 Euro |
Zu den hier aufgeführten Kosten kommen jeweils noch die Kosten für die Basistherapie (ICS mittlere Dosis bis z.B. Budesonid 800 oder Beclometason 1000).
*) Berechnung auf Basis der Festbeträge soweit vorhanden; es gibt Arzneimittel, die unter Festbetrag angeboten werden.
**) Bei den ICS wurde die Hälfte einer hohen Gesamtdosis gerechnet, da bei allen anderen Optionen die niedrige bis mittlere ICS-Dosis nicht aufgeführt wurde. Hinsichtlich der Vergleichbarkeit der ICS siehe auch die Hinweise in den entsprechenden Leitlinien.
Kinder
Die Kosten wurden berechnet analog Stufe 3 der Versorgungsleitlinie Asthma (Stand: Januar 2008)
| Zusatzmedikation zur Basistherapie ICS (mittlere Dosis) | DDD | Tagestherapiekosten* | Kosten für 1 Jahr* |
| plus Montelukast | 1 Kautabl. 5 mg | 1,92 | 700,80 |
| 1 Kautabl. 4 mg | 2,14 | 781,10 | |
| Granulat 4 mg | 2,25 | 821,25 | |
| plus Formoterol ** | 2 Hübe | 0,94 - 1,48 | 343,10 - 540,20 |
| plus Salmeterol ** | 2 x 50 pg | 1,88 | 686,20 |
| ICS Dosissteigerung z.B. Budesonid *, *** | 400 pg | 0,28 | 120,45 |
| ICS Dosissteigerung z.B. Beclometason *, *** | 400 pg | 0,34 | 124,10 |
| plus Theophyllin * 12 - 16 mg/kg | 400 mg (Kind 25 kg) | 0,38 | 138,70 |
Zu den hier aufgeführten Kosten kommen jeweils noch die Kosten für die Basistherapie (ICS mittlere Dosis bis z.B. Budesonid 400 oder Beclometason 400).
*) Berechnung auf Basis der Festbeträge soweit vorhanden; es gibt Arzneimittel, die unter Festbetrag angeboten werden.
**) Salmeterol ist für Kinder ab 4 Jahren zugelassen und Formoterol für Kinder ab 6 Jahren. ***Bei den ICS wurde die Hälfte einer hohen Gesamtdosis gerechnet, da bei allen anderen Optionen die mittlere ICS-Dosis nicht aufgeführt wurde. Hinsichtlich der Vergleichbarkeit der ICS siehe auch die Hinweise in den entsprechenden Leitlinien.
Die Kosten wurden berechnet analog Stufe 2 der Versorgungsleitlinie Asthma (Stand: Januar 2008).
| DDD | Tagestherapiekosten* | Kosten für 1 Jahr* | |
| Montelukast | 1 Kautabl. 5 mg | 1,92 Euro | 700,80 Euro |
| 1 Kautabl. 4 mg | 2,14 Euro | 781,10 Euro | |
| Granulat 4 mg | 2,25 Euro | 821,25 Euro | |
| Budesonid 300 * | 300 μg | 0,39 Euro | 142,35 Euro |
| Beclometason 300* | 300 μg | 0,36 Euro | 131,40 Euro |
| DNCG * | 80 mg | 1,60 Euro | 584,00 Euro |
* Berechnung auf Basis der Festbeträge soweit vorhanden; es gibt Arzneimittel, die unter Festbetrag angeboten werden.
(Preisstand Lauertaxe 1. Januar 2008)
| Indikation |
Montelukast ist zugelassen als Zusatzbehandlung bei Patienten, die unter einem leichten bis mittelgradigen persistierenden Asthma leiden, das mit einem ICS nicht ausreichend behandelt und das durch die bedarfsweise Anwendung von kurzwirksamen Betasympathomimetika nicht ausreichend unter Kontrolle gebracht werden kann.
Es kann auch eine Behandlungsalternative zu niedrig dosierten ICS bei Patienten zwischen 2 und 14 Jahren mit leichtem persistierenden Asthma sein, die in letzter Zeit keine schwerwiegenden, mit systemischen Kortikosteroiden zu behandelnden Asthmaanfälle hatten und zeigten, dass sie nicht imstande sind, ICS anzuwenden.
Bei den Patienten ab 15 Jahren, für die Montelukast bei Asthma angezeigt ist, können die 10-mg-Filmtabletten auch die Symptome einer saisonalen allergischen Rhinitis lindern. Außerdem kann Montelukast zur Vorbeugung von Belastungsasthma eingesetzt werden, dessen überwiegende Komponente die durch körperliche Belastung ausgelöste Bronchokonstriktion darstellt.
Die Dosierung für Erwachsene und Jugendliche ab 15 Jahren mit Asthma oder mit allergischer Rhinitis und Asthma beträgt eine 10-mg-Filmtablette täglich am Abend. Bei Kindern zwischen 6 und 14 Jahren liegt die Dosis bei einer 5-mg-Kautablette, für Kinder von 2 bis 5 Jahren bei einer 4-mg-Kautablette und für Kinder zwischen 6 Monaten und 5 Jahren beträgt sie einen Beutelinhalt mit 4 mg Granulat.
| Wirkungen |
Montelukast ist eine oral wirksame Substanz, die sich mit hoher Affinität und Selektivität an vorhandene Cysteinyl-Leukotrien-Rezeptoren (CysLT) bindet.
Cysteinyl-Leukotriene werden von Mastzellen und eosinophilen Granulozyten freigesetzt, die sich an in den Atemwegen des Menschen vorhandenen CysLT binden und dort u. a. eine Verengung der Bronchien, Schleimsekretion, Gefäßpermeabilität und Anreicherung von eosinophilen Granulozyten bewirken.
| Wirksamkeit |
In Studien an Erwachsenen konnte gezeigt werden, dass unter Montelukast die klinische Wirkung von ICS verstärkt werden kann (% Veränderung zum Ausgangswert für inhalatives Beclometason in Kombination mit Montelukast vs. Beclometason für FEV1: 5,43 % vs. 1,04 % bzw. Bedarf an Betaagonisten: 8,70 % vs. +2,64 %). Verglichen mit inhalativem Beclometason (200 pg zweimal täglich mittels Inhalationshilfe) konnte für Montelukast zwar ein initial rascheres Ansprechen auf die Therapie nachgewiesen werden, jedoch war der Therapieeffekt unter Beclometason über die gesamte zwölfwöchige Studiendauer im Durchschnitt größer (% Veränderung zum Ausgangswert für Montelukast vs. Beclometason für FEV1: 7,49 % vs. 13,3 % bzw. Bedarf an Betaagonisten: 28,28 % vs. 43,89 %). Allerdings erreichte ein hoher Prozentsatz der mit Montelukast behandelten Patienten ähnliche klinische Resultate wie die mit Beclometason behandelten Patienten. So erzielten 50 % der mit Beclometason und 42 % der mit Montelukast behandelten Patienten im Vergleich zum Ausgangswert eine Verbesserung des FEV1 von ca. 11 % und mehr.
Untersuchungen, die Montelukast/ICS vs. ICS in einer höheren Dosierung ausschließlich bei Patienten mit leichtem bis mittelgradigem Asthma geprüft haben, liegen nicht vor.
Unter der Kombination von Montelukast und ICS konnte die ICS-Dosis bei Erhalt der Symptomkontrolle stärker reduziert werden als unter ICS-Monotherapie. Aussagen darüber, ob die Absenkung der ICS-Dosis zu einer Verminderung der ICS-spezifischen Nebenwirkungen führt, lassen sich aus den Studien nicht treffen. Es liegen keine Daten vor, die belegen, dass unter der zusätzlichen Therapie mit Montelukast orale Kortikosteroide reduziert werden können.
Bei dem Vergleich Montelukast zu Salmeterol (beide in Kombination mit ICS) zeigen sich bezüglich der Wirksamkeit Vorteile für die Kombinationstherapie mit Salmeterol. Dies betraf die Asthmasymptomatik, gemessen an der Wirkung auf die Symptome bei Tag, beim nächtlichen Erwachen und den symptomfreien Tagen. Auch das Risiko einer Exazerbation war unter Montelukast-Kombinationstherapie gegenüber Salmeterol-Kombinationstherapie erhöht. Die Zahl der Krankenhausaufnahmen, der Arztbesuche und die Lebensqualitätsuntersuchungen ergaben keine signifikanten Unterschiede. Eine endgültige vergleichende Bewertung der potenziell schädlichen Effekte von Montelukast und Salmeterol ist wegen des Fehlens dezidierter Sicherheitsstudien nicht möglich. Die Nutzen-Schaden-Abwägung ergibt keine eindeutige Überlegenheit oder Unterlegenheit von Montelukast im Vergleich zu Salmeterol.
Der Nutzen von Montelukast im Vergleich zu Formoterol als weiterem Beta-2-Rezeptoragonisten oder zu Theophyllin, die ebenfalls als additive Therapie zu ICS empfohlen werden, sowie zu Cromonen kann aus Mangel an direkten Vergleichsstudien nicht bewertet werden.
Beim Belastungsasthma und der allergischen Rhinitis ist eine Überlegenheit gegenüber Mitteln, die dem allgemein anerkannten Stand entsprechen, in direkt vergleichenden Studien nicht belegt. Der therapeutische Stellenwert ist unklar.
Bei Kindern wurden direkt vergleichende Studien zwischen einer Monotherapie mit Montelukast und ICS durchgeführt. Garcia zeigte über 52 Monate eine statistische Nichtunterlegenheit im Hinblick auf die Rate asthmafreier Tage (Montelukast 84 % vs. Fluticason 86,7 %). Allerdings erlitten mehr Kinder Asthmaattacken unter Montelukast und benötigten auch häufiger systemische Kortikosteroide. Für sekundäre Zielparameter wie FEV1, Gebrauch von Betaagonisten und Lebensqualität zeigen sich Vorteile für ICS. In die gleiche Richtung zeigt die Untersuchung von Sorkness, die nach 48 Wochen eine statistisch signifikante Überlegenheit für asthmafreie Tage von Fluticason gegenüber Montelukast belegt, allerdings keine Überlegenheit der Kombination von Fluticason und Salmeterol gegenüber einer Monotherapie mit Montelukast zeigt. Eine weitere Untersuchung, allerdings lediglich mit einer Beobachtungsdauer von 12 Wochen, sowie eine Crossover-Studie bestätigen ebenfalls Vorteile für ICS bei vergleichbaren Nebenwirkungen. Szefler untersuchte Kinder im Alter von 6 bis 17 Jahren und fand, dass 23 % der Kinder ausschließlich auf Fluticason und nur 5 % ausschließlich auf Montelukast ansprachen. Primärer Auswertungspunkt war die FEV1. Die Längenwachstumsrate betrug nach 52 Wochen 0,41 cm mehr unter Montelukast im Vergleich zu inhalierten 100 μg/Tag Fluticason, nach 48 Wochen im Vergleich zu Fluticason 200 μg/Tag 0,40 cm und 0,81 cm mehr nach 56 Wochen als unter inhaliertem Beclometason in einer Dosis von 400 μg/Tag.
| Risiken ggf. Vorsichtsmaßnahmen |
Bei Überempfindlichkeit gegenüber dem Wirkstoff oder einem der Hilfsstoffe dieser Arzneimittel ist Montelukast kontraindiziert.
Die 4-mg- und 5-mg-Kautabletten enthalten Aspartam, aus welchem im Körper Phenylalanin freigesetzt wird. Patienten mit Phenylketonurie sollten berücksichtigen, dass eine 4-mg-Kautablette eine 0,674 mg und eine 5-mg-Kautablette eine 0,842 mg entsprechende Menge Phenylalanin pro Dosis enthält. Patienten mit einer der seltenen Erbkrankheiten Galaktoseintoleranz, Lapp-Laktasemangel oder Glukose-Galaktose-Malabsorption dürfen Montelukast 10-mg-Filmtabletten nicht einnehmen.
Die häufigsten Nebenwirkungen zwischen 1 % und 10 % sind Kopf- und Bauchschmerzen, alle anderen Nebenwirkungen traten in weniger als 1 % der Fälle auf. In seltenen Fällen kann bei Patienten unter der Therapie mit Antiasthmatika, einschließlich Montelukast, eine systemische Eosinophilie, manchmal mit klinischen Zeichen einer Vaskulitis wie bei Churg-Strauss-Syndrom auftreten.
Nach Markteinführung und im Rahmen von klinischen Studien wurden akute Überdosierungen mit Montelukast berichtet. Darunter sind Berichte über Erwachsene und Kinder mit einer Dosis bis zu 1000 mg (ca. 61 mg/kg für ein Kind von 42 Monaten). Die dabei beobachteten klinischen und Labor-Parameter entsprachen dem Nebenwirkungsprofil bei Erwachsenen, Jugendlichen und Kindern. In den meisten Berichten zu Überdosierungen wurden keine Nebenwirkungen beobachtet. Die am häufigsten aufgetretenen Nebenwirkungen entsprachen dem Sicherheitsprofil von Montelukast und umfassten Bauchschmerzen, Schläfrigkeit, Durst, Kopfschmerzen, Erbrechen und psychomotorische Hyperaktivität.
In Tierversuchen war Montelukast weder karzinogen noch mutagen oder teratogen. Montelukast sollte jedoch nicht während der Schwangerschaft und der Stillzeit eingesetzt werden, außer wenn es als absolut erforderlich erachtet wird.
Patienten mit Analgetika-Intoleranz müssen auch unter der Behandlung mit Montelukast die Einnahme von ASS und anderen nichtsteroidalen Antiphlogistika vermeiden.
Die Kautabletten sollten 1 Stunde vor oder 2 Stunden nach der Nahrungsaufnahme eingenommen werden.
Unbedenklichkeit und Wirksamkeit von 4 mg Granulat Montelukast bei Kindern unter 6 Monaten wurden bislang nicht untersucht. Das Gleiche gilt für die 4-mg-Kautabletten bei Kindern unter 2 Jahren.
Natalizumab
(z.B. Tysabri § )
Beschluss vom: 16. Oktober 2009
In Kraft getreten am: 10. April 2009
BAnz. 2009, Nr. 55 vom 9. April 2009, S. 1.304
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Zur Wirksamkeit und Sicherheit einer Therapie mit Natalizumab über einen Behandlungszeitraum von zwei Jahren hinaus sind keine Studien publiziert. Vergleichsstudien zwischen Natalizumab und etablierten Basistherapeutika bei MS (Interferon beta, Glatiameracetat) bzw. Mitoxantron in der Eskalationstherapie liegen nicht vor.
Natalizumab hat wegen des Risikos der Entwicklung einer progressiven multifokalen Leukoenzephalopathie (PML), einer opportunistischen Virusinfektion, welche gewöhnlich zum Tod oder zu einer schweren Behinderung führt, und anderen gravierenden unerwünschten Begleitwirkungen eine streng zu beachtende enge Zulassung erhalten.
Natalizumab ist ausschließlich für die Monotherapie zugelassen und nur zur Behandlung der hochaktiven schubförmig remittierend verlaufenden Multiplen Sklerose (MS).
Es sind in jedem Einzelfall und über die gesamte Therapiedauer mögliche Risiken gegen den Nutzen einer Therapie mit Natalizumab abzuwägen. Bei unklaren Risiken und Wirkungen einer Langzeittherapie und vor dem Hintergrund, dass in Kohortenstudien bis zu 17 % der Patienten mit MS auch ohne immunmodulatorische Therapie einen günstigen Verlauf zeigen, muss die Behandlung mit Natalizumab auf Patienten mit hochaktiver Erkrankung, für die andere angemessene Therapien nicht zur Verfügung stehen, beschränkt bleiben. Gesicherte Prognosekriterien sind nicht vorhanden.
Es sollten deshalb nur solche Patienten mit Natalizumab behandelt werden, bei denen Kontraindikationen oder Unverträglichkeiten für Interferon (IFN) beta oder/und Glatirameracetat bestehen oder die im Verlauf eines Jahres auf Interferon beta oder/und Glatiameracetat nicht ausreichend angesprochen haben und die für eine Eskalationstherapie mit Mitoxantron unter Berücksichtigung seiner Zulassung und Risiken nicht geeignet sind.
Der Einsatz von Natalizumab ist nur wirtschaftlich, wenn die Patienten folgende Krankheits- und Verlaufsmerkmale erfüllen:
- Gesicherte Diagnose einer MS nach den Kriterien von McDonald et al. (immer einschließlich eines Liquorbefundes mit Bestimmung oligoklonaler Banden und/oder des IgG-Indexes)
- Hochaktive schubförmige MS mit mindestens zwei Schüben bei inkompletter Remission und resultierender Behinderungsprogression im vorangegangenen Jahr
- Aktivitätszeichen im kranialen MRT: mindestens eine Gadoliniumanreichernde Läsion oder signifikante Erhöhung von T2-Läsionen im Vergleich zu einer früheren wenige Monate zurückliegenden MRT-Aufnahme
Weitere Voraussetzungen für die Anwendung sind:
- Die Einleitung und Überwachung der Therapie muss durch einen in der Diagnosestellung und Behandlung von neurologischen Erkrankungen erfahrenen Spezialisten (Facharzt) mit raschem Zugang zur MRT-Diagnostik erfolgen.
- Ein kraniales MRT, welches nicht älter als 3 Monate sein sollte, muss vor Behandlungsbeginn mit Natalizumab vorliegen.
- Kein Anhalt für eine geschwächte Immunkompetenz, unauffälliges weißes Blutbild.
- Keine zusätzliche Einnahme von Immunsuppressiva oder anderen immunmodulatorisch wirkenden Arzneimitteln.
- Therapiemöglichkeiten zur Behandlung von anaphylaktischen Reaktionen bei Applikation des Arzneimittels müssen zur Verfügung stehen.
- Der Patient muss über mögliche Risiken aufgeklärt werden und in der Lage sein, eine Entscheidung im Sinne des informed consent treffen zu können. Er muss den von der EMEA vorgeschriebenen Patientenpass ausgehändigt bekommen.
Die Teilnahme an einer der im Pharmakovigilanz-Plan der Europäischen Zulassungsbehörde (EMEA) vorgesehenen Anwendungsbeobachtungen zur Sicherheit von Natalizumab soll angestrebt werden (TYGRIS ROW [Tysabri Global Observational Program in Safety Rest of World] oder TOP [Tysabri Observational Program]).
Bei immungeschwächten Patienten mit erhöhtem Risiko für opportunistische Infektionen ist die Therapie kontraindiziert.
Bei Patienten, die nach 6-monatiger Behandlung noch keinerlei Hinweise auf einen Behandlungserfolg zeigen, ist die Therapie zu beenden.
Natalizumab erbrachte in der akuten Schubbehandlung der Multiplen Sklerose keine schnellere Remission der Symptome als Placebo. Es ist daher für die Schubtherapie nicht geeignet und zugelassen. Für die Therapie chronisch progredienter Verlaufsformen ist Natalizumab ebenfalls nicht zugelassen.
Natalizumab anwendende Ärzte sind verpflichtet, sich regelmäßig über aktuelle Studienergebnisse zu Natalizumab zu informieren.
Die Zulassung von Natalizumab für die Behandlung des Morbus Crohn wurde von der EMEA am 19. Juli 2007 aufgrund einer negativen Nutzen-Risiko-Bewertung abgelehnt.
| Kosten |
Gegenüber einer verlaufsmodifizierenden Therapie der schubförmig remittierenden MS mit Interferon beta oder Glatirameracetat können sich die Kosten bis auf fast das Doppelte erhöhen.
| Wirkstoff | Präparat | Dosierung 1 | Tagestherapiekosten | Jahrestherapiekosten |
| Glatirameracetat | Copaxone | 20 mg/ 1 x tgl. s.c. | 49,96 Euro - 53,27 Euro | 18.235 Euro - 19.444 Euro |
| IFN beta la (CHO-Zellen) | Avonex | 30 pg (= 6 Mio. I.E.)/ 1 x pro Woche i.m. | 48,71 Euro - 62,15 Euro | 17.779 Euro - 22.685 Euro |
| IFN beta la (CHO-Zellen) | Rebif | 22 pg (= 6 Mio. I.E.)/ 3 x pro Woche s.c. | 49,27 Euro - 52,73 Euro | 17.984 Euro - 19.246 Euro |
| IFN beta la (CHO-Zellen) | Rebif | 44 μg (= 12 Mio. I.E.) / 3 x pro Woche s.c. | 60,20 Euro - 64,61 Euro | 21.973 Euro - 23.583 Euro |
| IFN beta 1 b (E.coli) | z.B. Betaferon | 250 μg (= 8 Mio. I.E.) / alle 2 Tage s.c. | 46,67 Euro - 58,71 Euro | 17.035 Euro - 21.429 Euro |
| Azathioprin | z.B. Imurek | 2 mg - 3 mg/ kg KG 3/ tgl. oral | bei 75 kg (= 225 mg)2: bis zu 3,60 Euro | bei 75 kg (= 225 mg) 2: bis zu 1.314 A |
| Mitoxantron | Ralenova | 12 mg/m2 K04 alle 3 Monate i.v. | 1,68 - 2,08 m2 KO: bis zu 6,21 Euro | 1,68 - 2,08 m2 KO: bis zu 2.267 Euro |
| Natalizumab | Tysabri | 300 mg alle 4 Wochen i.v. | 82,41 - 86,67 Euro | 30.080 Euro - 31.635 Euro |
Preisstand 15. Februar 2009
1) Standard-Erhaltungsdosis
2) Kostenberechnung auf Basis des Festbetrages, Therapiekosten der maximalen Dosierung
3) KG = Körpergewicht
4) KO = Körperoberfläche
| Indikation |
Natalizumab ist für die krankheitsmodifizierende Monotherapie von hochaktiver, schubförmig remittierend verlaufender Multipler Sklerose (RRMS) bei folgenden Patientengruppen zur Prävention von Schüben und zur Verlangsamung der Behinderungsprogression zugelassen:
- bei Patienten, die nicht auf einen vollständigen und angemessenen Zyklus einer Interferon beta-Therapie angesprochen haben. Bei den Patienten sollte es während der Therapie im vorangegangenen Jahr zu mindestens einem Schub unter Interferon beta gekommen sein und sie sollten mindestens 9 T2-hyperintense Läsionen in der kraniellen MRT oder mindestens eine Gadoliniumanreichernde Läsion aufweisen, oder
- bei Patienten mit rasch fortschreitender schubförmig remittierend verlaufender Multipler Sklerose, definiert durch zwei oder mehr Schübe mit Behinderungsprogression in einem Jahr, und mit einem oder mehr Gadoliniumanreichernden Läsionen in der MRT des Gehirns oder mit einer signifikanten Erhöhung der T2-Läsionen im Vergleich zu einer früheren, in jüngerer Zeit angefertigten MRT.
Vor Beginn der Behandlung mit Natalizumab sollte eine aktuelle MRT-Aufnahme vorliegen (gewöhnlich nicht älter als drei Monate).
Die Diagnose einer MS muss nach den Kriterien von McDonald et al. gesichert sein.
Die Therapie muss von Fachärzten, die über ausreichende Erfahrung in der Diagnostik und Behandlung von Multipler Sklerose verfügen, eingeleitet und kontinuierlich überwacht werden (Fachärzte für Neurologie oder Nervenärzte). Der rasche Zugang zur kraniellen MRT muss gewährleistet sein. Die Zusammenarbeit mit einem in der MRT-Diagnostik der MS erfahrenen Radiologen oder Neuroradiologen sollte angestrebt werden.
Es ist vorgeschrieben, den Patienten einen speziellen Patientenpass auszuhändigen.
Die vorherige Gabe von Immunsuppressiva (z.B. Mitoxantron, Cyclophosphamid, Azathioprin) kann zu einer anhaltenden Immunsuppression führen (auch wenn ihre Gabe bereits beendet wurde) und somit ein erhöhtes Risiko für die Entwicklung einer PML bedeuten. Der behandelnde Arzt hat sich daher vor Einleitung der Therapie mit Natalizumab zu vergewissern, dass diese Patienten nicht mehr immungeschwächt sind. Es gibt keine Daten dazu, ob eine washout-Phase das Risiko reduziert. Abhängig vom Immunsuppressivum, von Dauer der immunsuppressiven Therapie und Dringlichkeit der Indikation sollte eine washout-Phase von drei bis sechs Monaten in Betracht gezogen werden.
Der Immunstatus soll normalisiert sein, ein Blutbild mit normaler Leukozyten- und Lymphozyten-Zahl ist erforderlich. Akute oder chronisch rezidivierende Infektionen dürfen nicht vorliegen.
Ob eine washout-Phase vor Umstellung von Immunmodulatoren auf Natalizumab das Risiko für eine PML reduziert, ist unklar. Sofern keine Anzeichen relevanter behandlungsbedingter Auffälligkeiten, wie z.B. eine Neutropenie, vorliegen, können die Patienten direkt von Interferon beta oder Glatirameracetat auf Natalizumab umgestellt werden. Anzeichen behandlungsbedingter Auffälligkeiten müssen sich wieder normalisiert haben, bevor die Behandlung mit Natalizumab begonnen werden kann.
In klinischen MS-Studien der Phase III war die begleitende Behandlung von Schüben mit einer kurzzeitigen Gabe von Kortikosteroiden nicht mit einer erhöhten Infektionsrate assoziiert.
Die Gabe von Natalizumab ist kontraindiziert bei:
- Überempfindlichkeit gegen Natalizumab oder einen der sonstigen Bestandteile
- Progressiver multifokaler Leukoenzephalopathie
- erhöhtem Risiko für opportunistische Infektionen
- Kombination mit Interferon beta oder Glatiameracetat
- Kombination mit anderen Immunsuppressiva
- bekannten aktiven Malignomen (Ausnahme: Basaliom)
- Kindern und Jugendlichen
- Schwangeren, es sei denn, dies ist eindeutig erforderlich.
Natalizumab wird alle vier Wochen in einer fixen Dosierung von 300 mg als Infusion gegeben. Für die Infusion werden 15 ml des Konzentrats mit 100 ml Kochsalzlösung verdünnt. Die Infusionsdauer darf eine Stunde nicht unterschreiten. Der Patient muss nach Infusionsende eine Stunde (ggf. mit über diesen Zeitraum belassenem i.v.-Zugang) nachbeobachtet werden. Als biologische Vorprobe sollte zumindest bei der ersten und besonders bei der zweiten Applikation zunächst nur 1 ml der Infusionslösung infundiert werden, um dann den Patienten 15 Minuten auf eine Anaphylaxie hin zu beobachten. Möglichkeiten zur Therapie von Hypersensitivitäts- bzw. Anaphylaxie-Reaktionen müssen zur Verfügung stehen (z.B. i.v.-Antihistaminikum, i.v.-Glukocorticosteroid, i.v.-Adrenalin, Guedeltubus, Beatmungsbeutel).
| Wirkungen |
Bei Natalizumab handelt es sich um einen monoklonalen humanisierten Antikörper, der spezifisch an das α4β1-Integrin (Synonym: Very-Late-Antigen-4) bindet, einem Adhäsionsmolekül, das auf der Oberfläche von T-Lymphozyten exprimiert wird. Die Migration von Lymphozyten durch die Blut-Hirn-Schranke gilt als ein entscheidender früher Schritt bei der Entstehung von Läsionen im Rahmen der MS. Für diese Leukozytenwanderung ist eine Interaktion zwischen Adhäsionsmolekülen auf mononukleären Leukozyten sowie spezifischen Rezeptoren auf den Endothelzellen der Gefäßwand notwendig. Natalizumab unterbindet diese molekularen Interaktionen und verhindert dadurch die transendotheliale Migration von aktivierten T-Lymphozyten in entzündliches Parenchymgewebe.
Ein weiterer Wirkungsmechanismus von Natalizumab liegt möglicherweise in der Unterdrückung von bestehenden entzündlichen Reaktionen in erkranktem Gewebe und der Hemmung einer Rekrutierung von Immunzellen in entzündetem Gewebe.
Die pharmakokinetischen Daten zeigen bei einer 4-wöchentlichen Gabe von 300 mg Natalizumab eine 70% - 80%ige Sättigung der Alpha-4-Integrinmoleküle auf Leukozyten. Nach Absetzen sind noch bis zu etwa 12 Wochen nach der letztmaligen Gabe pharmakodynamische Wirkungen (z.B. eine erhöhte Lymphozytenzahl) nachzuweisen. Die Einleitung anderer Therapien in diesem Zeitraum ist zwangsläufig mit einer begleitenden Exposition gegenüber Natalizumab verbunden.
| Wirksamkeit |
Für die Zulassung wurde die Wirksamkeit und Sicherheit von Natalizumab in zwei großen multizentrischen randomisierten kontrollierten doppelblinden Phase-Ill-Studien geprüft. In beiden Studien wurden Patienten mit schubförmig verlaufender MS aufgenommen, die mindestens einen Schub im Jahr zuvor erlebt hatten. Die Diagnose einer MS war nach den Kriterien von Mc Donald et al. gesichert. Im MRT lagen mit einer MS vereinbare radiologische Veränderungen vor. Weitere Einschlusskriterien waren ein Alter zwischen 18 und 55 Jahren und ein Score von 0 - 5 auf der Expanded Disability Status Scale (EDSS). Ausschlusskriterien waren eine immunsuppressive Therapie innerhalb der letzten sechs Monate, ein entzündlicher Schub oder Gabe von Glucosteroiden in den letzten 50 Tagen sowie eine primär oder sekundär progressive Verlaufsform der MS.
Bei der AFFIRM-Studie (Polman et al.) handelte es sich um eine Natalizumab-Monotherapie-Studie mit Patienten, die innerhalb der letzten sechs Monate nicht mit Interferonen behandelt worden waren und auch insgesamt nicht länger als sechs Monate Interferone erhalten hatten. Die Patienten wurden im Verhältnis 2:1 randomisiert den Behandlungsarmen mit Natalizumab 300 mg (n = 627) bzw. Placebo (n = 315) alle vier Wochen zugeteilt.
Primäre Endpunkte waren die Schubrate nach einem Jahr und die Progression der Behinderung nach zwei Jahren, definiert als eine für mindestens 12 Wochen anhaltende Erhöhung um mindestens 1,0 auf der EDSS bei einem Ausgangs-EDSS >=1,0 oder eine Erhöhung um mindestens 1,5 auf der EDSS bei einem Ausgangs-EDSS = 0. Sekundäre Endpunkte waren der Anteil schubfreier Patienten sowie radiologische Veränderungen in der MRT.
Nach einem Jahr reduzierte sich die Schubrate unter Natalizumab signifikant auf 0,26 gegenüber 0,81 unter Placebo entsprechend einer relativen Risikoreduktion um 68 %. Diese Verminderung der Schubrate setzte sich im zweiten Behandlungsjahr fort.
Nach zwei Jahren sank das Risiko einer Progression der Behinderung signifikant um 12 %. Während es unter Placebo bei 29 % der Patienten zu einer Progression kam, waren es unter Natalizumab nur 17 %. Dies entspricht einer Number Needed to Treat (NNT) von 9 und einer relativen Risikoreduktion von 42 %.
Der Anteil schubfreier Patienten betrug unter Placebo 41 % und unter Natalizumab 67 %. Unter Natalizumab zeigten im MRT 97 % der Patienten keine Gadoliniumanreichernden Läsionen, unter Placebo waren es 72 %. Das Ausbleiben neuer hyperintenser T2-Läsionen wurde bei 57 % der Patienten unter Natalizumab und bei 15 % unter Placebo beobachtet.
Posthoc-Subgruppenanalysen ergaben in der kleinen Gruppe von Patienten mit weniger als neun hyperintensen T2-Läsionen keine Veränderung der Progression der Behinderung.
In der Subgruppe von Patienten mit hochaktiver schubförmig remittierender MS, definiert durch mindestens zwei Schübe im vorangegangen Jahr und mindestens eine Gadolinium-anreichernde Läsion, ergab sich eine jährliche Schubrate von 0,28 (n=148) unter Natalizumab gegenüber 1,5 (n=61) unter Placebo. Die relative Risikoreduktion für eine Behinderungsprogression betrug 64 %.
Die EMEA bewertet das Vorgehen einer nachträglichen Subgruppenanalyse durchaus kritisch. Der Therapieeffekt in der Subgruppe der Patienten mit hochaktiver schubförmig remittierender MS wurde jedoch als so hoch eingeschätzt, dass für diese Patientengruppe eine Zulassung auch ohne Vortherapie mit Immunmodulatoren erging.
Bei der SENTINEL-Studie (Rudick et al.) handelte es sich um eine Kombinationstherapie-Studie, in der Patienten, die trotz einer Behandlung mit Interferon beta mindestens einen Schub im vorangegangenen Jahr erlitten hatten, zusätzlich Natalizumab erhielten. Hierunter traten zwei Fälle einer PML auf, sodass aus Sicherheitsgründen eine Zulassung für diese Kombination nicht erfolgte.
Die Studie hatte die gleichen Endpunkte wie die AFFIRM Studie.
Das Risiko einer Behinderungsprogression wurde nach zwei Jahren ebenfalls signifikant - jedoch weniger stark - um 6 % gesenkt. Während es unter Monotherapie mit Interferon beta bei 29 % der Patienten zu einer Progression kam, waren es unter Natalizumab in Kombination mit Interferon beta nur 23 %. Dies entspricht einer NNT von 17 und einer relativen Risikoreduktion von 24 %.
Nach Einschätzung der EMEA ist der Anteil, den Natalizumab an diesem Ergebnis hat, nicht bestimmbar, da ein Natalizumab-Monotherapiearm in der Studie fehlte. Dennoch war dieses Studienergebnis Grundlage der Zulassung als Monotherapie für Patienten mit nur einem Schub im vorangegangen Jahr unter Interferontherapie.
Natalizumab erhöht das Risiko für eine PML, einer opportunistischen Infektion des Gehirnes, welche gewöhnlich zu schwerer Behinderung oder zum Tod führt. Es handelt sich um eine seltene, rasch progrediente multifokale Demyelinisierungserkrankung des zentralen Nervensystems, die durch das JC-Polyomavirus (JCV) verursacht wird. Die Infektion mit diesem Virus erfolgt zumeist bereits im Kindesalter, verläuft hier aber klinisch inapparent. Bei 20 % - 80 % aller gesunden Erwachsenen sind Antikörper gegen das JCV nachweisbar. Zum Ausbruch einer PML kann es bei immungeschwächten Patienten kommen, z.B. durch längerfristige Behandlung mit Methotrexat, Cyclophosphamid oder Azathioprin, längerfristige Therapie mit Immunsuppressiva nach Transplantation, aber auch bei einer HIV-Infektion oder malignen hämatologischen Erkrankungen.
In der SENTINEL-Studie traten zwei PML-Fälle bei MS-Patienten auf, die begleitend mehr als zwei Jahre mit Interferon beta behandelt worden waren; einer der Patienten verstarb. In einer anderen Studie entwickelte ein Patient mit Morbus Crohn, der immunsuppressiv behandelt worden war und eine damit assoziierte Lymphozytopenie aufwies, nach Behandlung mit Natalizumab ebenfalls eine PML und starb.
Obwohl jeder dieser PML-Fälle entweder bei Patienten mit begleitender Gabe von immunmodulierenden Substanzen oder mit Hinweisen auf eine Immunsuppression auftrat, sind die Erfahrungen zu begrenzt, um ein erhöhtes Risiko unter einer Natalizumab-Monotherapie auszuschließen. Weiterhin ist unbekannt, welches Risiko bei einem Therapiezeitraum von mehr als zwei Jahren besteht.
Eine Nachuntersuchung (Yousry et al.) von 3116 Patienten, darunter ein Großteil der Teilnehmer aller MS-Studien, die im Schnitt 17,9 Monate Natalizumab erhalten hatten, ergab keine weiteren gesicherten Fälle einer PML. Anhand dieser Nachuntersuchung wird das Risiko für eine PML mit 1,0 per 1000 Patienten (95 % Cl 0,2 2,8) bei 17,9 Monaten Natalizumab-Exposition angegeben.
Zwischenzeitlich sind zwei weitere PML-Erkrankungen unter Therapie mit Natalizumab bekannt geworden. Bei den betroffenen Patienten wurde die Diagnose 14 und 17 Monate nach Beginn der Natalizumab-Therapie gestellt. Einer der beiden Patienten hatte zuvor keine anderen immunsuppressiven oder immunmodulierenden Therapien erhalten.
Die Symptome einer PML können denen eines MS-Schubs ähneln. Bei neu auftretenden oder sich verschlechternden klinischen Symptomen ist deshalb eine eingehende neurologische Kontrolluntersuchung durch einen in der MS-Diagnostik und -Therapie erfahrenen Facharzt erforderlich. Dabei ist besonders auf die Entwicklung PML-assoziierter neurologischer Ausfallssymptome zu achten, wie z.B. kognitive Störungen, Verhaltensänderungen, Aphasie, Apraxie und kortikale Blindheit. Ergibt sich der Verdacht auf eine sich entwickelnde PML, muss die Therapie mit Natalizumab unterbrochen werden, bis eine PML ausgeschlossen werden kann. Zur Diagnostik werden ein kranielles MRT und eine Liquorentnahme zum Nachweis von JCV-DNS mittels polymerase chain reaction (PCR) empfohlen.
Ob das Risiko auch für andere opportunistische Infektionen erhöht ist, ist unklar. In MS-Studien wurde über einen einzelnen Fall einer unkompliziert verlaufenden, durch Cryptosporidium ausgelösten Durchfallerkrankung berichtet. In klinischen Studien zum Morbus Crohn wurde über weitere opportunistische Infektionen berichtet, von denen einige tödlich verliefen. Am Anfang der Markteinführung kam es zu einem tödlich verlaufenden Fall von Herpesenzephalitis.
Die biologischen Effekte einer Natalizumab-Behandlung sind bis drei Monate nach Beendigung der Therapie nachzuweisen. In diesem Zeitraum ist noch mit einem Risiko für opportunistische Infektionen zu rechnen.
Unter Natalizumab zeigten sich Hypersensitivitätsreaktionen einschließlich schwerer systemischer Reaktionen bei bis zu 4 % der Patienten. Bei weniger als 1 % der Patienten handelte es sich um anaphylaktische bzw. anaphylaktoide Ereignisse. Die Überempfindlichkeitsreaktionen ereigneten sich in der Regel während der Infusion oder in der ersten Stunde nach Infusionsende, vor allem bei der zweiten Natalizumab-Infusion. Eine ausgeprägte, verzögerte allergische Typ-Ill-Reaktion einige Tage nach Gabe der zweiten Natalizumab-Infusion ist nach Zulassung beschrieben worden.
Bei Hypersensivitäts- oder Anaphylaxiereaktionen muss die Natalizumab-Therapie abgebrochen werden.
Während der zweijährigen klinischen Studien bildeten sich bei ca. 10 % der Patienten Antikörper gegen Natalizumab, bei ca. 6 % persistierend. Patienten mit persistierenden Antikörpern gegen Natalizumab zeigten einen signifikanten Rückgang der Wirksamkeit und ein erhöhtes Risiko für Hypersensitivitätsreaktionen. Besteht der Verdacht auf ein Therapieversagen, sollte eine Bestimmung der neutralisierenden Antikörper erfolgen. Angesichts einer möglicherweise herabgesetzten Wirksamkeit oder erhöhten Inzidenz für Überempfindlichkeitsreaktionen sollte die Behandlung bei Patienten, die persistierende Antikörper entwickeln, beendet werden.
Nebenwirkungen, für die unter Behandlung mit Natalizumab eine um 0,5 % höhere Inzidenz gegenüber Placebo berichtet wurde, waren Harnwegsinfektionen, Nasopharyngitis, Überempfindlichkeit, Urticaria, Kopfschmerzen, Schwindel, Erbrechen, Übelkeit, Arthralgie, Rigor, Fieber und Abgeschlagenheit.
Nach Markteinführung wurden spontane schwere Nebenwirkungen in Form von Leberschädigungen berichtet. Diese können jederzeit während der Behandlung auftreten, sodass regelmäßige Kontrollen der Leberfunktion erfolgen müssen. Bei signifikanter Leberschädigung ist die Behandlung mit Natalizumab abzubrechen.
Weiterhin wurde über zwei Fälle berichtet, bei denen in zeitlichem Zusammenhang mit einer Natalizumab-Behandlung Melanome aufgetreten sind.
| weiter . | |
...
X
⍂
↑
↓