

umwelt-online: AMR - Arzneimittel-Richtlinie (3)
| zurück | |
Oseltamivir
(z.B. Tamiflu®)
Beschluss vom: 24.03.2003
In Kraft getreten am: 10.08.2003
BAnz. 2003, Nr. 147 vom 09.08.2003 S. 17.978
| Indikation |
Oseltamivir wurde am 20.06.2002 durch die europäische Zulassungsbehörde für folgende Anwendungsgebiete zugelassen:
Therapie der Influenza bei Kindern ab einem Jahr und Erwachsenen mit influenzatypischen Symptomen, wenn das Influenzavirus in der Bevölkerung auftritt. Die Wirksamkeit konnte nur nachgewiesen werden, wenn die Behandlung innerhalb von zwei Tagen nach erstmaligem Auftreten der Symptome begonnen wurde.
Prophylaxe der Influenza
Oseltamivir ist kein Ersatz für eine Grippeschutzimpfung.
Die Behandlung erfolgt bei Erwachsenen mit 2 x täglich 75 mg Oseltamivir über fünf Tage. Bei Kindern liegt die empfohlene Dosis in Abhängigkeit vom Körpergewicht zwischen 30 und 75 mg 2 x täglich.
Zur Postexpositionsprophylaxe wird Oseltamivir 75 mg 1 x täglich über mindestens sieben Tage und zur Prophylaxe während einer Influenzaepidemie in der Bevölkerung mit 75 mg Oseltamivir 1 x täglich über einen Zeitraum bis zu sechs Wochen dosiert.
Oseltamivir wird als Hartkapsel ä 75 mg sowie als Pulver ä 30 mg zur Herstellung einer Suspension mit der Konzentration von 12 mg/ml angeboten.
| Wirkungen |
Oseltamivir ist ein prodrug und wird nach oraler Einnahme rasch im Magen-Darm-Trakt resorbiert und überwiegend durch hepatische Esterasen in den aktiven Metaboliten Oseltamivircarboxylat umgewandelt. Dieser hemmt selektiv die Neuraminidasen von Influenzaviren. Neuraminidasen sind Glykoproteine, die auf der Oberfläche des Virions lokalisiert sind. Die enzymatische Aktivität der viralen Neuraminidasen ist entscheidend für die Freisetzung von neu gebildeten Viruspartikeln aus infizierten Zellen und damit für die weitere Verbreitung infektiöser Viren im Körper.
Die Elimination erfolgt zu über 90 % über den Urin.
| Wirksamkeit |
Zum Beleg der Wirksamkeit sind vier wesentliche Studien zur Therapie und drei zur Prophylaxe durchgeführt worden. Die beiden Studien mit negativen Resultaten (bei Patienten mit chronischen kardialen und respiratorischen Erkrankungen sowie bei Kindern mit Asthma) zur Therapie sind zurzeit nicht publiziert.
Therapiestudien
Einschlusskriterien der klinischen Studien bei Erwachsenen waren Fieber über 37,8°C, mindestens ein respiratorisches Symptom wie Husten, Schnupfen oder Halsschmerzen und mindestens ein systemisches Symptom wie Müdigkeit, Schüttelfrost/Schwitzen, Myalgie oder Kopfschmerzen. Bei Kindern war Einschlusskriterium ebenfalls Fiebek 37,8°C und entweder Husten oder Schnupfen.
Primäre Wirksamkeitsvariable war die mittlere Zeit bis zur Symptomfreiheit über mindestens 24 Stunden.
Bei Erwachsenen und Jugendlichen ab 13 Jahren verkürzt die Behandlung mit Oseltamivir 75 mg 2 x täglich über einen Zeitraum von fünf Tagen die mediane Erkrankungsdauer um ungefähr einen Tag von 5,2 Tage auf 4,2 Tage, bei ansonsten gesunden Kindern um 1,5 Tage.
Die mediane Erkrankungsdauer wurde bei älteren Patienten (> 65 Jahre) und bei Patienten mit chronischen kardialen und/oder respiratorischen Erkrankungen nicht signifikant verkürzt. Auch bei asthmatischen Kindern wurde die mediane Dauer der Erkrankung nicht signifikant verkürzt.
Spezielle Studien zur Beurteilung der Verringerung des Risikos von Komplikationen der Influenza wurden nicht durchgeführt.
Die Wirksamkeit wurde im Wesentlichen belegt bei Patienten mit Influenza-A-Virus. Influenza B trat über alle Studien hinweg lediglich in 11 % der nachgewiesenen Grippefällen auf. Bei einer Analyse aller erwachsenen Patienten zeigte sich eine nicht signifikante Verkürzung der Erkrankung um 16 Stunden. Die größte Häufigkeit der Influenza B von 33 % wurde in einer einzigen Studie mit Kindern gefunden. Auch hier war die Zeit bis zu einer Symptomfreiheit nicht statistisch signifikant verkürzt, sodass insgesamt der Effekt von Oseltamivir gegen Influenza B allenfalls als moderat bezeichnet werden kann.
Prophylaxestudien
In den Studien zur Untersuchung der Wirksamkeit von Oseltamivir bei der Prophylaxe wurde die primäre Zielvariable klinische und laborbestätigte Influenza durch den Virusnachweis beziehungsweise mindestens vierfachen Anstieg des Antikörpertiters sowie gleichzeitigem Vorliegen der auch bei der Behandlung auftretenden klinischen Symptomatik definiert, jedoch war die Körpertemperatur mit z 37,2°C niedriger angesetzt.
Zur Postexpositionsprophylaxe müssen sieben Tage lang 16 Patienten mit 75 mg 1 x täglich behandelt werden, um eine Influenzaerkrankung zu verhindern. Zur allgemeinen Prophylaxe während einer Influenzaepidemie in der Bevölkerung müssen sechs Wochen lang 28 Patienten mit 75 mg 1 x täglich behandelt werden, um einen Grippefall zu vermeiden. Um das Gleiche bei älteren Einwohnern in Pflegeheimen zu erreichen, liegt die Zahl der zu behandelnden Patienten bei 25.
Eine Prophylaxe ist ohne verlässliche epidemiologische Daten nicht sinnvoll.
Insgesamt ist die Anzahl der Patienten, die zur Prophylaxe behandelt werden müssen, um einen Therapieerfolg zu erzielen, hoch.
| Risiken ggf. Vorsichtsmaßnahmen |
Als Gegenanzeige gilt die Überempfindlichkeit gegen Oseltamivir oder andere Bestandteile des Arzneimittels. Bezüglich Schwangerschaft und Stillzeit gelten besondere Vorsichtsmaßnahmen.
Unbedenklichkeit und Wirksamkeit von Oseltamivir zur Therapie ist bei Kindern unter einem Jahr noch nicht gesichert, das Gleiche gilt für die Prophylaxe der Influenza bei Kindern unter 12 Jahren.
Eine Dosisanpassung ist sowohl bei der Prophylaxe als auch bei der Therapie bei einer Kreatininclearance zwischen < 10 bis < 30 ml/Minute notwendig. Bei einer Kreatininclearance < 10 ml/Minute beziehungsweise Dialysepflichtigkeit wird die Therapie nicht empfohlen. Obgleich bedeutsame Wechselwirkungen um die renale tubuläre Sekretion mit anderen Arzneimitteln nicht beobachtet wurden, ist bei der gleichzeitigen Anwendung von Substanzen mit geringer therapeutischer Breite, zum Beispiel Chlorpropamid, Methotrexat, Phenylbutazon, Vorsicht geboten.
Die Anwendung bei Patienten mit Leberfunktionsstörungen wurde bisher nicht ausreichend untersucht.
Die häufigsten Nebenwirkungen betreffen Übelkeit, Erbrechen und Bauchschmerzen. Todesfälle wurden bei den bisherigen Untersuchungen nicht beobachtet.
Bisher gibt es keine ausreichenden Daten, um das Risiko von Oseltamivir-Resistenzen bei klinischen Anwendungen zu beurteilen.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Oseltamivir ist nur gegen Erkrankungen, die durch Influenzaviren verursacht werden, wirksam. Es gibt keinen Hinweis darauf, dass Oseltamivir bei durch andere Erreger als Influenzaviren hervorgerufenen Krankheiten wirksam ist. Die Wirksamkeit von Oseltamivir ist bei einer Behandlung 40 Stunden nach Auftreten der ersten Symptome nicht belegt.
Wegen der unspezifischen Symptomatik der Influenza ist es schwierig, sie von anderen respiratorischen Erkrankungen sicher zu unterscheiden. Aufgrund des klinischen Bildes wurde in den Studien die Diagnose in 60 bis 70 % der Fälle korrekt gestellt. Dies ist ein Szenario, welches mit der Anwendung in der Routine nicht vergleichbar ist. So ist davon auszugehen, dass in der Routinepraxis die Diagnose weitaus seltener sichergestellt wird, am schwierigsten ist die Diagnosestellung während der frühen und späten Phase von Influenzaepidemien. Informationen zur Influenza sind zum Beispiel beim Robert-Koch-Institut im Internet unter www.rki.de abrufbar.
In den meisten Fällen ist die Influenza eine selbstlimitierende Erkrankung. Insofern sind unterstützende und symptomatische Therapien ausreichend. Bis heute ist nicht belegt, dass Oseltamivir die Komplikationen der Influenza, beziehungsweise Krankenhausbehandlungsbedürftigkeit, mindern oder den Tod verhindern kann.
Anders als die Impfung ist die Wirksamkeit von Oseltamivir in Hochrisikogruppen noch nicht ausreichend gesichert und auch ein Nutzen bei asthmatischen Kindern bisher nicht überzeugend gelungen. Das Gleiche gilt für immunsupprimierte Patienten.
Der Einsatz von Oseltamivir ersetzt nicht die Impfung und ist in der Regel nicht wirtschaftlich. Er ist lediglich angezeigt, wenn eine Impfung nicht möglich oder nicht effektiv ist, wie zum Beispiel bei einer rasch auftretenden Pandemie beziehungsweise einem Antigendrift. Insofern sind Neuramidasehemmer wie Oseltamivir auf der Basis des bisher verfügbaren Wissens nicht von wesentlicher Bedeutung bei einer typischen Influenzasaison.
Kreuzresistenz wurde mit Zanamivirresistenten Influenzamutanten und Oseltamivirresistenten Influenzamutanten in vitro beobachtet. Zurzeit liegen noch keine hinreichenden Informationen vor, um das Risiko der Entstehung einer Oseltamivir-Resistenz und Kreuzresistenz in der klinischen Praxis zu beurteilen.
Bisher ist die Sicherheit und Wirksamkeit bei wiederholten Behandlungen nicht etabliert.
| Kosten |
Zur Behandlung der Influenza ist neben Oseltamivir als weiterer Neuraminidaseinhibitor Zanamivir zugelassen. Amantadin ist für die gleiche Indikation im Markt und wirkt über die Hemmung des Matrix-(M2-) lonenkanalproteins. Die Wirksamkeit von Amantadin und Zanamivir in der Behandlung der Influenza ist ebenfalls als mäßig einzustufen: Die Dauer der Symptomatik wird um ein bis zwei Tage verkürzt. Auch hier muss die Behandlung innerhalb von 48 Stunden nach Auftreten der Symptome begonnen werden. Direkt vergleichende Studien fehlen.
Tabelle 1: Zugelassene Behandlungsoptionen der Influenza
| Oseltamivir | Zanamivir | Amantadin | |
| Zulassung | Erwachsene und Kinder ab 1 Jahr | Erwachsene und Jugendliche ab 12 Jahren | Erwachsene und Kinder ab 1 Jahr |
| Wirkmechanismus | Neuraminidaseinhibitor | Neuraminidaseinhibitor | Blockiert die virale M2-Proteinaktivität |
| Hemmung von Influenzavirus | A & B | A & B | Nur A |
| Dosierung | 75 mg über 5 Tage 2 x täglich (variiert in Abhängigkeit von Körpergewicht und Nierenfunktion) | 10 mg über 5 Tage 2 x täglich | 100 mg oder 10 ml über 5 Tage 2 x täglich (variiert in Abhängigkeit von Körpergewicht und Nierenfunktion) |
| Applikationsweg | oral (Kapseln oder Suspension) | inhalativ | oral (Kapsel oder Sirup) |
| Wichtige Nebenwirkungen | gastrointestinale Symptome | keine arzneimittelbezogenen - Bronchospasmus bei Patienten mit COPD bei der Inhalation | ZNS & gastrointestinale Symptome |
| Virale Resistenz | möglich | möglich | dokumentierte, sich rasch entwickelnde Resistenz |
| Kosten pro Behandlungsfall in Euro | 34,70 | 29,74 | Tabl.: 7,15 - 13,10 Saft: 13,15 - 26,30 |
Tabelle 2: Zugelassene Prophylaxeoptionen der Influenza
| Arzneimittel | Kosten in Euro pro Patient |
| Impfstoffe | 10,31 - 14,13 |
| Oseltamivir 75 mg/Tag bis zu 6 Wochen | 173,50 bei 6-wöchiger Anwendung |
Omalizumab 16
(z.B. Xolair® bei Asthma bronchiale)
Beschluss vom: 11.11.2010
In Kraft getreten am: 12.11.2010
BAnz. 2011 Nr. 2, S. 723
| Zugelassene Anwendungsgebiete 16 |
Omalizumab ist zugelassen als Zusatztherapie zur verbesserten Asthmakontrolle bei:
1. Erwachsenen und Jugendlichen (ab 12 Jahren)
2. Kindern (6 bis < 12 Jahre)
Omalizumab (150 mg Injektionslösung) ist zugelassen als Zusatztherapie für die Behandlung der chronischen spontanen Urtikaria bei Erwachsenen und Jugendlichen (ab 12 Jahren) mit unzureichendem Ansprechen auf eine Behandlung mit H1-Antihistaminika.1
Es gibt nur begrenzt Erfahrungen mit der Selbstverabreichung von Omalizumab. Daher ist die Verabreichung durch medizinisches Fachpersonal vorgesehen.
Ein im Juni 2000 gestellter Antrag auf Zulassung für die Behandlung der saisonalen allergischen Rhinitis ist aufgrund der negativen Bewertung durch die europäische Zulassungsbehörde vom Hersteller zurückgezogen worden.
In diesem Anwendungsgebiet ist ein Off-Label-Use grundsätzlich durch die Rechtsprechung des Bundessozialgerichts ausgeschlossen.
| Empfehlungen zur wirtschaftlichen Verordnungsweise 16 |
Der Therapiehinweis bezieht sich ausschließlich auf die Indikation Asthma bronchiale.
Die Verordnung von Omalizumab ist als Zusatztherapie bei Jugendlichen ab 12 Jahren und erwachsenen Patienten nur wirtschaftlich, die kumulativ folgende Voraussetzungen erfüllen:
Die Verordnung von Omalizumab ist als Zusatztherapie bei Kindern zwischen 6 und 12 Jahren nur wirtschaftlich, die kumulativ folgende Voraussetzungen erfüllen:
Die Dosierung erfolgt in Abhängigkeit vom Körpergewicht und dem Basis IgE-Spiegel. Die empfohlene Maximaldosis beträgt 600 mg Omalizumab alle zwei Wochen oder 600 mg alle vier Wochen, eine Überschreitung ist unzweckmäßig.
Die Behandlung mit Omalizumab sollte nur durch einen Arzt mit Erfahrung in der Diagnose und der Behandlung von schwerem persistierendem Asthma begonnen werden.
Die Entscheidung zur Weiterbehandlung mit Omalizumab sollte auf einer merklichen Verbesserung der allgemeinen Asthmakontrolle basieren. Als ausreichende Verbesserung ist beispielsweise ein selteneres nächtliches Erwachen oder eine Verbesserung der Symptome über den Tag, die mit Wiederaufnahme von Tätigkeiten im Alltag einhergeht, oder eine Reduktion der Notfallmedikation anzusehen. Dies ist durch das sorgfältige Führen geeigneter Tagebücher durch den Patienten zu dokumentieren.
Die weitere Behandlungsnotwendigkeit sollte spätestens 16 Wochen nach Beginn der Therapie mit Omalizumab durch den Arzt überprüft werden.
Sollte eine Dosisreduktion des inhalativen Kortikosteroids auf eine mittlere bis niedrige Dosis möglich sein, ohne dass Exazerbationen auftreten, ist die Therapiestrategie zu überdenken, spätestens jedoch alle 12 Monate.
Omalizumab ist nicht angezeigt für die Behandlung von akuten Asthmaexazerbationen, akuten Bronchospasmen oder eines Status asthmatints.
Omalizumab wurde nicht untersucht bei Patienten mit Hyperimmunglobulin-E-Syndrom oder allergischer bronchopulmonarer Aspergillose oder zur Vorbeugung von anaphylaktischen Reaktionen, einschließlich durch Nahrungsmittelallergien ausgelöster Anaphylaxien.
Die Verordnung von Omalizumab ist nur unter den zuvor genannten Voraussetzungen wirtschaftlich
Bezüglich der Zweckmäßigkeit ist darüber hinaus zu beachten, dass die doppelblinde randomisierte Zulassungsstudie (Humbert 2005) für Jugendliche und Erwachsene bei Asthma keine statistisch signifikante Überlegenheit für den primären Endpunkt der Asthmaexazerbationsrate ergab. Nicht alle Patienten erhielten einen zusätzlichen Kontroller, wie es nach aktuellen Versorgungsleitlinien gefordert wird. Die Ergebnisse der Studien, die auch Patienten mit mittelschwerem Asthma aufnahmen, sind widersprüchlich in Hinsicht auf die Rate der Asthmaexazerbationen. Bei der Therapieentscheidung ist auch die mangelnde Konsistenz der Ergebnisse zu berücksichtigen (siehe Abschnitt Wirksamkeit, Jugendliche und Erwachsene).
40% der in die Hauptstudie (Lanier 2009) aufgenommenen Kinder hatten eine der Zulassung entsprechende Indikation für die Therapie mit Omalizumab.
Der primäre Endpunkt, Rate der Exazerbationen, wurde erreicht; (siehe Abschnitt Wirksamkeit, Kinder im Alter von 6 bis 12 Jahren) allerdings findet sich für eine Vielzahl von weiteren vom primären Endpunkt klinisch differierend definierten sekundären Zielgrößen, die auch als klinisch relevant einzuschätzen sind, keine statistisch signifikante Überlegenheit gegenüber Placebo, so dass die Ergebnisse hinsichtlich der tatsächlichen klinischen Überlegenheit und Relevanz hinterfragt werden können.
| Kosten 16 |
Die geeignete Dosierung und Behandlungsfrequenz von Omalizumab wird anhand des vor Behandlungsbeginn gemessenen IgE-Basiswertes (I.E./ml) und des Körpergewichts (kg) bestimmt. Zur Dosisfestlegung ist es erforderlich, vor der ersten Anwendung den IgE-Wert des Patienten mit einem handelsüblichen Gesamt-Serum-IgE-Test zu bestimmen. Ausgehend von diesen Messungen können pro Verabreichung 75 bis 600 mg Omalizumab benötigt werden.
Damit entstehen je nach Dosierintervall (alle zwei beziehungsweise vier Wochen) Jahrestherapiekosten zwischen rund 3.400 Euro und 50.300 Euro.
Injektion alle 4 Wochen (13 Injektionen pro Jahr)
| IgE Basiswert | Körpergewicht | Kosten pro Behandlung * | Jahrestherapie-Kosten * |
| > 30 - 100 I.E./ml | > 20 - 0 kg > 40 - 90 kg > 90 - 150 kg | 263,67 Euro 484,00 Euro 968,00 Euro | 3.427,71 Euro 6.292,00 Euro 12.584,00 Euro |
| > 100 - 200 I.E./ml | > 20 - 40 kg > 40 - 90 kg > 90 - 125 kg > 125 - 150 kg | 484,00 Euro 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 6.292,00 Euro 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro |
| > 200 - 300 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 60 kg > 60 - 90 kg > 90 - 125 kg | 484,00 Euro 747,67 Euro 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 6.292,00 Euro 9.719,71 Euro 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro |
| > 300 - 400 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 70 kg > 70 - 90 kg | 747,67 Euro 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 9.719,71 Euro 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro |
| > 400 - 500 I.E./ml | > 20 - 25 kg > 25 - 30 kg > 30 - 50 kg > 50 - 70 kg | 747,67 Euro 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 9.719,71 Euro 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro |
| > 500 - 600 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 60 kg | 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro |
| > 600 - 700 I.E./ml | > 20 - 25 kg > 30 - 40 kg > 40 - 50 kg | 968,00 Euro 1.452,00 Euro 1.936,00 Euro | 12.584,00 Euro 18.876,00 Euro 25.168,00 Euro" |
*) Berechnungsgrundlage Xolair® 150 mg N3 10 Fertigspritzen, Xolair® 75 mg N1 1 Fertigspritze; gesetzliche Pflichtrabatte der Apotheken und pharmazeutischen Unternehmen wurden nicht berücksichtigt.
Stand Lauer-Taxe 1. Oktober 2015#
Injektion alle 2 Wochen (26 Injektionen pro Jahr)
| IgE Basiswert | Körpergewicht | Kosten pro Behandlung* | Jahrestherapie-Kosten* |
| > 200 - 300 I.E./ml |
> 125 - 150 kg |
1.231,67 Euro |
32.023,42 Euro |
| > 300 - 400 I.E./ml | > 90 - 125 kg > 125 - 150 kg | 1.452,00 Euro 1.715,67 Euro | 37.752,00 Euro 44.607,42 Euro |
| > 400 - 500 I.E./ml | > 70 - 90 kg > 90 - 125 kg > 125 - 150 kg | 1.231,67 Euro 1.715,67 Euro 1.936,00 Euro | 32.023,42 Euro 44.607,42 Euro 50.336,00 Euro |
| > 500 - 600 I.E./ml | > 60 - 70 kg > 70 - 90 kg > 90 - 125 kg | 1.231,67 Euro 1.452,00 Euro 1.936,00 Euro | 32.023,42 Euro 37.752,00 Euro 50.336,00 Euro |
| > 600 - 700 I.E./ml | > 25 - 30 kg > 50 - 60 kg > 60 - 80 kg > 80 - 90 kg | 747,67 Euro 1.231,67 Euro 1.452,00 Euro 1.715,67 Euro | 19.439,42 Euro 32.023,42 Euro 37.752,00 Euro 44.607,42 Euro |
| > 700 - 800 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 50 kg > 50 - 70 kg > 70 - 80 kg > 80 - 90 kg | 747,67 Euro 968,00 Euro 1.231,67 Euro 1.452,00 Euro 1.715,67 Euro 1.936,00 Euro | 19.439,42 Euro 25.168,00 Euro 32.023,42 Euro 37.752,00 Euro 44.607,42 Euro 50.336,00 Euro |
| > 800 - 900 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 50 kg > 50 - 60 kg > 60 - 70 kg > 70 - 80 kg | 747,67 Euro 968,00 Euro 1.231,67 Euro 1.452,00 Euro 1.715,67 Euro 1.936,00 Euro | 19.439,42 Euro 25.168,00 Euro 32.023,42 Euro 37.752,00 Euro 44.607,42 Euro 50.336,00 Euro |
| > 900 - 1.000 I.E./ml | > 20 - 25 kg > 25 - 30 kg > 30 - 40 kg > 40 - 50 kg > 50 - 60 kg > 60 - 70 kg | 747,67 Euro 968,00 Euro 1.231,67 Euro 1.452,00 Euro 1.715,67 Euro 1.936,00 Euro | 19.439,42 Euro 25.168,00 Euro 32.023,42 Euro 37.752,00 Euro 44.607,42 Euro 50.336,00 Euro |
| > 1.000 - 1.100 I.E./ml | > 20 - 25 kg > 25 - 30 kg > 30 - 40 kg > 40 - 50 kg > 50 - 60 kg | 747,67 Euro 968,00 Euro 1.231,67 Euro 1.452,00 Euro 1.936,00 Euro | 19.439,42 Euro 25.168,00 Euro 32.023,42 Euro 37.752,00 Euro 50.336,00 Euro |
| > 1.100 - 1.200 I.E./ml | > 20 - 30 kg > 30 - 40 kg > 40 - 50 kg > 50 - 60 kg | 968,00 Euro 1.452,00 Euro 1.715,67 Euro 1.936,00 Euro | 25.168,00 Euro 37.752,00 Euro 44.607,42 Euro 50.336,00 Euro |
| > 1.200 - 1.300 I.E./ml | > 20 - 25 kg > 25 - 30 kg > 30 - 40 kg > 40 - 50 kg | 968,00 Euro 1.231,67 Euro 1.452,00 Euro 1.715,67 Euro | 25.168,00 Euro 32.023,42 Euro 37.752,00 Euro 44.607,42 Euro |
| > 1.300 - 1.500 I.E./ml | > 20 - 25 kg > 25 - 30 kg > 30 - 40 kg > 40 - 50 kg | 968,00 Euro 1.231,67 Euro 1.715,67 Euro 1.936,00 Euro | 25.168,00 Euro 32.023,42 Euro 44.607,42 Euro 50.336,00 Euro" |
*) Berechnungsgrundlage Xolair® 150 mg N3 10 Fertigspritzen, Xolair® 75 mg N1 1 Fertigspritze; gesetzliche Pflichtrabatte der Apotheken und pharmazeutischen Unternehmen wurden nicht berücksichtigt.
Stand Lauer-Taxe 1. Oktober 2015
| Wirkungen |
Omalizumab ist ein rekombinanter, aus DNA abgeleiteter, humanisierter monoklonaler Antikörper, der selektiv an das menschliche Immunglobulin E (IgE) bindet.
Es handelt sich uni einen IgG1kappa-Antikörper mit einem humanen Grundgerüst, dessen komplementaritätsbestimmende Regionen muriner Herkunft sind und an IgE binden.
Omalizumab bindet an IgE und verhindert somit die Bindung von IgE an den hochaffinen Fcε RI-Rezeptor, wodurch die Menge an freiem IgE reduziert wird, das zum Auslösen der allergischen Kaskade verfügbar ist. In klinischen Studien wurde der Serumspiegel an freiem IgE dosisabhängig innerhalb einer Stunde nach der ersten Dosis reduziert.
Ein Jahr nach Absetzen von Omalizumab kehrten die IgE-Spiegel zu den Werten vor der Behandlung zurück, wobei nach dem Auswaschen des Arzneimittels kein Rebound beobachtet wurde.
| Wirksamkeit 16 |
Jugendliche ab 12 Jahren und Erwachsene
1Die Wirksamkeit und Verträglichkeit von Omalizumab wurde in einer doppelblinden placebokontrollierten Studie (Humbert 2005) über 28 Wochen in der Zielpopulation für die Zulassung geprüft. Eingeschlossen wurden 419 Patienten mit allergischem Asthma im Alter von 12 bis 79 Jahren. Die Patienten hatten eine reduzierte Lungenfunktion (FEV1 40 bis 80"/o des Reterenzwertes) und wiesen trotz einer Therapie mit hochdosierten inhalativen Kortikosteroiden und einem lang wirkenden inhalativen Beta-2-Agonisten eine schlechte Kontrolle der Asthma-Symptome auf. Die Patienten hatten im letzten Jahr trotz einer kontinuierlichen Behandlung mit hochdosierten inhalativen Korn kosteroiden (> 1.000 Mikrogramm Beclometasondipropionat oder Äquivalent) und einem lang wirkenden inhalativen Beta-2-Agon isten mehrere Asthmaexazerbationen erfahren, die eine Behandlung mit svstemischen Korn kosteroiden nötig machten oder wurden wegen einer schweren Asthmaexazerbation hospitalisiert oder Waren in einer Notfallambulanz. Zusätzliche Therapien mit oralen Kortikoiden, Theophyllin und Leukotrien-Rezeptor-Antagonisten waren erlaubt (je 22 %, 27% und 35% der Patienten).
2Den primären Endpunkt stellte die Rate der Asthmaexazerbationen dar, bei denen eine Akutbehandlung mit systemischen Kortikosteroiden nötig war. Omalizumab reduzierte die Rate der Asthmaexazerbationen gegenüber Placebo, sie lag bei 0,74 unter Omalizumab und 0,92 unter Placebo. Das Ergebnis war für den primären Endpunkt der Studie statistisch nicht signifikant (p = 0,153). Eine nicht geplante Posthoc-Adjustierung an die Exazerhationsrate vor der Behandlung führt zu signifikanten Ergebnissen. Nicht geplante Posthoc-Adjustierungen werden vom G-BA in methodischer Hinsicht kritisch gesehen.
3Weitere Auswertungen sekundärer Endpunkte zeigten statistische Signifikanz (p < 0,05) zugunsten von Omalizumab für schwere Exazerbationen (bei denen die Lungenfunktion des Patienten auf weniger als 60% des persönlichen Bestwertes reduziert war und systemische Kortikosteroide benötigt wurden) und asthmabedingtes Aufsuchen einer Notfallambulanz (einschließlich Hospitalisierungen, Notfallambulanz und nicht geplante Arztbesuche) sowie für Verbesserungen der ärztlichen Gesamtbewertung der Wirksamkeit der Behandlung, der Asthmasymptome und Parameter der Lungenfunktion. Die Lebensqualität bezüglich Asthma (AQL) zeigte eine statistisch signifikante Verbesserung unter Omalizumab im Vergleich zu vor der Behandlung. Allerdings ist die klinische Relevanz der durchschnittlichen Behandlungsdifferenz von 0,35 Punkten fraglich, da erst eine Differenz von 0,5 Punkten als klinisch bedeutsam betrachtet wird. Die Notfallmedikation war statistisch nicht signifikant reduziert unter Omalizumab.
4In einer Suhgruppenanalyse bei Patienten mit einem IgE-Gesamtwert > 76 I.E./ml vor der Behandlung war ein klinisch relevanter Nutzen von Omalizumab wahrscheinlicher.
5In vier weiteren großen placebokontrollierten unterstützenden Studien wurde die Wirksamkeit von Omalizumab bei Patienten mit mittelschwerem bis schwerem persistierendem Asthma untersucht. Die Rate der Asthmaexazerbationen war in der Hälfte der Studien nicht signifikant verbessert unter Omalizumab und in den anderen beiden Untersuchungen signifikant niedriger als unter Placebo. Das Gleiche gilt für die Rate schwerer Asthmaexazerbationen.
| Studie | Rate der Asthmaexazerbationen | Ratio (95 % Cl) | p-Wert | ||
| Omalizumab | Placebo | Treatment difference | |||
| Vignola 2004 (N=405) | 0.454 | 0.670 | 0.216 | 0.678 (0.432, 1.062) | 0.090 |
| Busse 2001 (N=525) | 0.468 | 0.842 | 0.373 | 0.556 (0.409, 0.756) | < 0.001 |
| Solör 2001 (N=546) | 0.376 | 0.898 | 0.522 | 0.419 (0.309, 0.568) | < 0.001 |
| Holgate 2004 (N=339) | 0.878 | 1.266 | 0.388 | 0.694 (0.432, 1.114) | 0.130 |
(In Anlehnung an den Bericht der europäischen Zulassungsbehörde. Tabelle 20/34 der wissenschaftlichen Bewertung)
Eine offene Studie untersuchte, inwieweit das Ansprechen nach 16-wöchiger Behandlung ein Vorhersagewert für eine anhaltende Response nach einer Behandlung über 32 Wochen ist. Dies war in 83 % der Fälle zutreffend. Sekundäre Endpunkte wie schwere Exazerbationen oder Hospitalisierung und Notfallaufnahme wegen Exazerbationen zeigen eine statistische Überlegenheit von Omalizumab addon zu OAT (optimized asthma therapy) im Vergleich zu OAT alleine (Bousquet 2011).
Es gibt eine weitere placebokontrollierte Studie zu Omalizumab in einem addon Design bei Patienten über 12 Jahren, die der Zulassung entspricht und in der patientenrelevante Endpunkte als primäre Outcome-Parameter gewählt wurden (Rubin 2012). Die Studie wurde vom pharmazeutischen Unternehmer unterstützt.
Rubin untersuchte bei einer kleineren Patientenpopulation die Lebensqualität mit schwerem allergischem Asthma unter Omalizumab (als primären Endpunkt) und fand nach 20 Wochen signifikant bessere Werte unter Omalizumab (n = 72) im Vergleich zu Placebo (n = 36): Asthma Quality of Life (AQLQ)-Score-Veränderung Gruppenunterschied 1,4 (+1,3 vs. -0,1), p < 0,001, keine Angabe des 95 % CI. Diese Studie wurde nicht verblindet durchgeführt, was in Hinblick auf eine mögliche Verzerrung der Bewertung des primären Endpunktes kritisch zu sehen ist. Eine Registrierung der Studie findet sich in der Publikation nicht.
Kinder im Alter von 6 bis 12 Jahren
Die grundlegenden Daten für die Sicherheit und Wirksamkeit von Omalizurnab in der Altersgruppe von 6 bis 12 Jahren stammen aus einer randomisierten, doppelblinden, placebokontrollierten multizentrischen Studie (Lanier 2009). Eine weitere Studie in dieser Altersgruppe über eine siebenmonatige doppelverblindete Phase mit fünfmonatiger offener Verlängerung wird von den Zulassungsbehörden als "unterstützend" betrachtet.
627 Kinder mit mittelschwerem bis schwerem Asthma, die nicht ausreichend auf eine mittlere Dosis von Fluticason > 200 μ g/d einstellbar waren, erhielten nach 2:1 Randomisierung Omalizumab (421) oder Placebo (192). Die Beobachtungsphasen bestanden aus einer Periode von 24 Wochen mit stabiler Dosis des inhalativen Kortikosteroids (ICS) und einer sich daran anschließenden Phase über 28 Wochen, in der die Kortisondosis schrittweise reduziert wurde. Der primäre Studienendpunkt waren Exazerbationen in der Phase der stabilen ICS-Dosis, definiert als Verschlechterung der Asthma-Symptomatik, so dass die ICS-Dosis mindestens drei Tage verdoppelt wurde und/oder systemische Kortikosteroide notwendig waren. Die Rate der Exazerbationen betrug im Mittel 0,45 unter Omalizumab und 0,64 unter Placebo; Differenz 0,19, RR 0,69, 95% CI 0,53-0,90; p = 0,007. Die sekundären Endpunkte - die Rate der nächtlichen Asthmasymptomatik, der Gebrauch von Beta-Agonisten, Lungenfunktionsparameter (FEV1) und die Lebensqualität gemessen mit PAQLQ (Pediatric Asthma Quality of Life Questionaire) - unterschieden sich nicht signifikant von Placebo
Unter klinischen Aspekten ergibt sich aus den Daten der US-amerikanischen Zulassungsbehörde FDA, dass ein Patient 2,34 Jahre behandelt werden muss, um eine Exazerhation zu verhindern, die eine Verdopplung der ICS-Dosis und/oder eine Intervention mit systemischen Kortikosteroiden Tiber mindestens drei Tage verursacht hätte.
Die europäische Zulassungsbehörde European Medicines Agency - EMA stützt die Zulassung unter anderem auf eine prädefinierte Subgruppenanalyse derselben Studie von Kindern, die > 500 µg/d Fluticason oder Äquivalent erhielten sowie LABA. Die Subgruppe bestand aus 235 Patienten von 570 Kindern. In dieser seien die Ergebnisse denen der Erwachsenen mit schwerem Asthma vergleichbar.
Bei Kindern konnte laut EMA bei initialen IgE-Spiegeln < 200 I.E./ml kein "Benefit" nachgewiesen werden.
Untersuchungen i in Vergleich zu anderen Arzneimitteln, die bei diesem Schweregrad des Asthmas empfohlen werden, fehlen für alle zugelassenen Altersgruppen.
| Risiken - ggf. Vorsichtsmaßnahmen 16 |
Omalizmnab ist bei Überempfindlichkeit gegen den arzneilich wirksamen Bestandteil oder einen der sonstigen Bestandteile kontraindiziert.
Während der Schwangerschaft darf Omalizumab nicht verwendet werden, es sei denn, dies ist eindeutig erforderlich. Unter der Therapie soll nicht gestillt werden.
Die am häufigsten berichteten Nebenwirkungen bei Erwachsenen und Jugendlichen ab 12 Jahren sind Reaktionen an der Injektionsstelle einschließlich Schmerzen, Schwellungen, Erythem und Pruritus sowie Kopfschmerzen. In klinischen Studien mit Kindern im Alter von 6 bis < 12 Jahren waren die am häufigsten berichteten unerwünschten Wirkungen, die vermutlich mit dem Arzneimittel in Zusammenhang stehen, Kopfschmerzen, Fieber und Schmerzen im Oberbauch. Die Schwere der meisten Reaktionen war leicht bis mittelschwer
Unter Omalizumab können anaphylaktische Reaktionen vom Typ I bis hin zum anaphylaktischen Schock, auch noch nach längerer Behandlungsdauer und auch in zeitlichem Abstand zur Injektion, auftreten Daher sollten Arzneimittel für die Behandlung einer anaphylaktischen Reaktion zum sofortigen Einsatz nach der Verabreichung von Omalizumab vorhanden sein und die Patienten entsprechend informiert werden.
Bei Patienten, die mit humanisierten monoklonalen Antikörpern wie Omalizumab behandelt wurden Serumkrankheit und serumkrankheitähnliche Reaktionen, die verzögerte allergische Typ-III-Reaktionen sind, festgestellt. Patienten sollen angehalten werden, sämtliche vermutete Symptome zu melden.
Patienten mit schwerem allergischem Asthma können selten ein systemisches hypereosinophiles Syndrom oder eine allergische eosinophile granulomatöse Vaskulitis (Churg-Strauss-Syndrom) aufweisen, die beide üblicherweise mit systemischen Kortikosteroiden behandelt werden.
In seltenen Fällen können Patienten, die mit einem Mittel gegen Asthma einschließlich Omalizumab behandelt werden, eine systemische Eosinophilie oder Vaskulitis aufweisen oder entwickeln. Diese Ereignisse sind häufig mit der Reduktion einer oralen Kortikosteroid-Therapie vergesellschaftet.
Das Absetzen von Omalizumab sollte bei allen schwerwiegenden Fällen der oben erwähnten Erkrankungen des Immunsystems in Erwägung gezogen werden.
Es liegen begrenzte Daten zur Anwendung von Omalizumab bei Patienten über 65 Jahren vor, jedoch gibt es keine Hinweise, dass bei älteren Patienten eine andere Dosierung erforderlich ist als bei jüngeren erwachsenen Patienten.
Die Sicherheit und Wirksamkeit von Omalizumab bei Kindern unter sechs Jahren ist nicht erwiesen. Es liegen keine Daten vor.
Die Therapie mit Omalizumab wurde bei Patienten mit Autoimmunkrankheiten, immunkomplexvermittelten Erkrankungen sowie mit vorgeschädigter Niere oder Leber nicht untersucht. Bei der Verabreichung an diese Patienten ist Vorsicht geboten.
In einer placebokontrollierten Studie an Patienten mit hohem Risiko für eine Wurminfektion zeigte sich ein geringer Anstieg der Infektionsrate unter Omalizumab, obgleich der Verlauf, die Schwere und das Ansprechen auf die Behandlung der Infektion unverändert waren. Bei Patienten mit einem hohen Risiko für eine Wurminfektion kann jedoch Vorsicht geboten sein, insbesondere bei Reisen in Gebiete mit endemischen Wurminfektionen. Wenn Patienten nicht auf die empfohlene Antiwurmbehandlung ansprechen, sollte ein Absetzen der Behandlung mit Omalizumab erwogen werden.
In kontrollierten klinischen Studien und bei Interimsanalysen einer Beobachtungsstudie wurde ein numerisches Ungleichgewicht von arteriellen thromboembolischen Ereignissen (ATE) beobachtet. ATE beinhalteten Schlaganfall, transitorische ischämische Attacke, Herzinfarkt, instabile Angina Pectoris und kardiovaskulären Tod (einschließlich Tod unbekannter Ursache). In einer neuen gepoolten Analyse betrug das Verhältnis der Häufigkeiten von Omalizumab im Vergleich zu Placebo 1,13 mit einem breiten Konfidenzintervall von 0,24 - 5,71
In den klinischen Studien hatten wenige Patienten Blutplättchenzahlen unterhalb des Normalbereiches. Keine dieser Änderungen war mit dem Auftreten von Blutungen oder einem Abfall des Hämoglobins verbunden.
Es ergab sich bei Menschen kein Muster einer anhaltenden Verringerung der Plättchenzahlen, wie dies bei Primaten beobachtet wurde.
_____
1) Dieses Indikationsgebiet ist nicht Gegenstand dieses Therapiehinweises.
Palivizumab
(z.B. Synagis TM)
Beschluss vom: 19.06.2008
In Kraft getreten am: 28.11.2008
BAnz. 2008, Nr. 181 vom 27.11.2008 S. 4.260
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Die Saison für Infektionen mit dem Respiratory-Syncytial-Virus (RSV) beginnt typischerweise im November und hält bis April an. Eine wirksame kausale Behandlung der RSV-Infektion existiert nicht. Die RSV-Infektion ist häufig. Es wird geschätzt, dass 50 % bis 70 % aller Kinder im 1. Lebensjahr die Infektion durchmachen. In Europa sind 60 % bis 90 % der Krankenhausbehandlungen von Kindern wegen einer Bronchiolitis Folge einer RS-Virus Infektion. Die dokumentierte Letalität von RSV-Erkrankungen im Kindesalter in Deutschland ist, bezogen auf die Häufigkeit der RSV-Infektion, die bis zum 2. Lebensjahr praktisch jedes Kind betrifft, als relativ gering anzusehen. Sie liegt unter heutigen intensivmedizinischen Bedingungen bei etwa 1 % der Risikopatienten.
Zu den Risikogruppen für schwere Verlaufsformen zählen Frühgeborene sowie Kinder mit vorgeschädigter Lunge und/oder Herzfehler sowie immunsupprimierte Patienten. Die deutsche Leitlinie nennt darüber hinaus weitere, in Kohortenstudien identifizierte Risikofaktoren, wie schwere neurologische Erkrankung, Vorhandensein von Geschwistern im Kindergarten- oder Schulalter, Entlassung aus der Neonatalogie zwischen Oktober und Dezember sowie männliches Geschlecht.
Bislang ist kein Impfstoff zur aktiven Immunisierung verfügbar. Zur passiven Immunisierung - zur Prophylaxe der RSV-Infektion - und nicht zur Therapie ist Palivizumab angezeigt. Es reduziert nach heutiger Kenntnis lediglich die Hospitalisierungsraten und nicht die Mortalität. Weder die Häufigkeit noch Dauer einer erforderlichen intensivmedizinischen Therapie oder künstlichen Beatmung werden durch Gabe von Palivizumab vermindert. In welchem Maße die überwiegend im Ausland gewonnenen Daten zur Krankenhausaufnahme auf deutsche Verhältnisse übertragbar sind, ist nicht untersucht.
Die Kosten-Nutzen-Bewertungen basieren zurzeit nicht auf validen Untersuchungen in Deutschland, sondern wurden bisher unter Zuhilfenahme der Zulassungsstudien kalkuliert. Bei vielen Kindern, die unter die zugelassenen Indikationen fallen, ist anzunehmen, dass das Risiko für schwerwiegende Erkrankungsverläufe mit Krankenhausaufnahme gering ist und damit vermutlich auch der potenzielle Nutzen der Gabe von Palivizumab.
Entsprechend sind die Empfehlungen in internationalen Leitlinien und ökonomischen Bewertungen heterogen (Dunfield 2007).
Der Einsatz von Palivizumab erscheint, wie unter anderem in der Leitlinie der deutschen Fachgesellschaften beschrieben, nur unter Einschränkung des Einsatzes gegenüber der Zulassung auf Kinder mit höherem Risiko für schwere Infektionsverläufe wirtschaftlich bei:
Kindern mit hohem Risiko im Alter von < 24 Lebensmonaten zum Beginn der RSV-Saison,
Darüber hinaus erscheint die Gabe unter wirtschaftlichen Aspekten noch vertretbar bei:
Kindern im Alter von < 6 Monaten bei Beginn der RSV-Saison,
Zusätzliche nichtmedikamentöse Maßnahmen sind Rauchverbot in der Umgebung von Hochrisikokindern, Stillen, infektionshygienische Allgemeinmaßnahmen zur Vermeidung der RSV-Exposition wie regelmäßiges Händewaschen und das Meiden von Personenansammlungen sowie Kinderkrippen.
Es fehlen valide Erkenntnisse bei Kindern unter Immunsuppression beziehungsweise bei Kindern mit Immundefekten.
Das Medikament ist für Erwachsene nicht zugelassen.
Gemäß Fachinformation ist der Nutzen für mehr als fünf Dosen ebenso wenig belegt wie für die Prophylaxe in einem zweiten Behandlungszyklus während einer darauf folgenden Saison.
| Kosten |
Die übliche Dosierung beträgt 15 mg/kg Körpergewicht intramuskulär, vorzugsweise in die anterolaterale Seite des Oberschenkels. Injektionsvolumen von mehr als 1 ml sollten als geteilte Dosen verabreicht werden.
| Gewicht des Kindes | Dosis 15 mg/kg KG | Kosten pro Gabe | Kosten pro Saison (5-malige Gabe) |
| bis 3,3 kg | 50 mg | 731,87 Euro | 3.659,35 Euro |
| bis 6,6 kg | 100 mg | 1.250,92 Euro | 6.254,60 Euro |
| bis 10 kg | 150 mg | 1.982,79 Euro | 9.913,95 Euro |
| bis 13,3 kg | 200 mg | 2.501,84 Euro | 12.509,20 Euro |
Stand: 15.09.2008
| Indikation |
Palivizumab ist in Deutschland zugelassen zur Prävention der durch das Respiratory-Syncytial-Virus (RSV) hervorgerufenen schweren Erkrankungen der unteren Atemwege, die Krankenhausaufenthalte erforderlich machen, bei Kindern mit hohem Risiko für RSVErkrankungen:
| Wirkungen |
Palivizumab ist ein humanisiertes lgG und monoklonaler Antikörper, der das A-Epitop des Fusionsproteins des RS-Virus bindet. Der humanisierte monoklonale Antikörper setzt sich aus humanen (95 %) und murinen (5 %) Antikörpersequenzen zusammen. Er besitzt eine neutralisierende und fusionsinhibitorische Aktivität gegenüber den beiden RSV-Untertypen A und B.
| Wirksamkeit |
Es sind zwei randomisierte, placebokontrollierte, verblindete Phase-Ill-Studien publiziert.
In der IMpact-Studie, einer placebokontrollierten Studie zur Prophylaxe der RSV-Erkrankung bei 1502 Kindern mit erhöhtem Infektionsrisiko (1002 Palivizumab; 500 Placebo) führten 5 monatliche Dosen von 15 mg Palivizumab/kg KG in 55% der Fälle (p 50,001) zu einer Reduzierung der RSV-bedingten Krankenhausaufnahmen. In der Placebogruppe betrug die RSV-Hospitalisierungsrate 10,6 % und in der Palivizumab-Gruppe 4,8 %. Auf der Basis dieser Daten beträgt die absolute Risikoreduzierung 5,8 %, woraus folgt, dass 17 Kinder behandelt werden müssen, um einer Krankenhausaufnahme vorzubeugen. Unter Berücksichtigung des Konfidenzintervalls liegt die Zahl zwischen 11 und 36 Kindern. Eingeschlossen waren sowohl Frühgeborene als auch Kinder mit bronchopulmonaler Dysplasie (BPD). Die Subqruppenanalyse ergab für die Hospitalisation folqende Ergebnisse:
| Gruppe | Rate % | Absolute Risikoreduktion % | p-Wert | |
| Alle Kinder | Placebo | Palivizumab | ||
| 10,60 | 4,80 | 5,80 | 0,00004 | |
| Frühgeborene ohne BPD | 8,10 | 1,80 | 6,30 | < 0,001 |
| Kinder mit BPD | 12,80 | 7,90 | 4,90 | 0,038 |
Die Schwere der RSV-Erkrankung bei hospitalisierten Kindern, bezogen auf den Aufenthalt (Tage) auf der Intensivstation pro 100 Kinder und Tage unter künstlicher Beatmung pro 100 Kinder, wurde durch die Prophylaxe mit Palivizumab nicht beeinflusst. Auch die Otitis media war in beiden Behandlungsgruppen gleich häufig. Unter der Behandlung mit Palivizumab traten vier Todesfälle (0,4%) und unter Placebo fünf (1 %) auf. Kein Todesfall wurde mit Palivizumab in Verbindung gebracht.
In einer zweiten placebokontrollierten, verblindeten Studie (Feltes) mit 1287 Patienten im Alter von < 24 Monaten mit hämodynamisch signifikanten, angeborenen Herzfehlern (639 Palivizumab; 648 Placebo) reduzierte eine monatliche Dosis von 15 mg/kg Palivizumab über 5 Monate die Inzidenz der RSV-bedingten Krankenhausaufnahme um 45 % (p = 0,003), absolute Risikoreduktion 4,4 %, woraus folgt, dass 23 Kinder behandelt werden müssen, um einer Krankenhausaufnahme vorzubeugen. Die Gruppen waren hinsichtlich der zyanotischen und azyanotischen Patienten ausgeglichen. Die RSV-Hospitalisierungsrate lag bei 9,7 % in der Placebogruppe und 5,3 % in der Palivizumab-Gruppe. Sekundäre Endpunkte der Wirksamkeit zeigten signifikante Reduzierung in der Palivizumab-Gruppe verglichen mit Placebo hinsichtlich der Gesamtzahl der Tage eines RSV-bedingten Krankenhausaufenthaltes (56 % Reduzierung, p = 0,003) und der Gesamtzahl der RSV-Tage mit einem erhöhten zusätzlichen Sauerstoffbedarf (73 % Reduzierung, p = 0,014) pro 100 Kinder. Nicht signifikante Unterschiede wiesen sekundäre Outcome-Parameter wie RSV-bedingte Inzidenz und Tage auf der Intensivstation und Inzidenz und Tage der Beatmung auf. 48 Kinder verstarben in der Studie, 21 in der Palivizumab-Gruppe, 27 in der Placebogruppe.
Eine Reduktion der Mortalität wurde in beiden Studien nicht untersucht und ist von daher nicht belegt.
Der Nutzen einer über 5 Monate hinausgehenden Behandlung ist gemäß Fachinformation des Herstellers nicht gesichert.
Die Wirksamkeit von Palivizumab in einem zweiten Behandlungszyklus während einer darauf folgenden RSV-Saison wurde nicht formell in einer Studie mit dieser Zielsetzung untersucht. Auch ist ein denkbares Risiko bei zuvor behandelten Patienten, in der darauf folgenden Saison verstärkt an einer RSV-Infektion zu erkranken, nicht endgültig durch Studien ausgeschlossen.
| Risiken ggf. Vorsichtsmaßnahmen |
Palivizumab ist kontraindiziert bei bekannter Überempfindlichkeit gegen den Wirkstoff oder weitere Bestandteile des Arzneimittels oder gegen andere humanisierte Antikörper.
Über allergische Reaktionen, einschließlich sehr seltener Fälle von Anaphylaxie, nach der Verabreichung von Palivizumab wurde berichtet. Es sollten Medikamente zur sofortigen Behandlung von schwerwiegenden Überempfindlichkeitsreaktionen, einschließlich Anaphylaxie, nach der Verabreichung von Palivizumab vorhanden sein.
Bei Patienten mit mäßig bis schweren akuten Infektionen oder fieberhaften Erkrankungen kann eine zeitlich verschobene Anwendung von Palivizumab gerechtfertigt sein. In den pädiatrischen Studien zur Prophylaxe traten sowohl in den Placebo- als auch in den Palivizumab-Gruppen vergleichbare Nebenwirkungen auf. Die häufigsten Nebenwirkungen waren Fieber, Reaktionen an der Injektionsstelle und Nervosität. Selten kam es zur Erhöhung der Transaminasen.
Pimecrolimus
(z.B. Elidel ®)
Beschluss vom: 04.09.2003
In Kraft getreten am: 07.01.2004
BAnz. Nr. 2 vom 06.01.2004 S. 68
| Indikation |
Pimecrolimus ist zugelassen bei Patienten ab 2 Jahren mit leichtem bis mittelschwerem atopischen Ekzems zur
Die Behandlung erfolgt zweimal täglich bis zur vollständigen Abheilung und sollte dann abgesetzt werden. Nach Unterbrechung beziehungsweise bei Langzeittherapie sollte die Behandlung beim ersten Wiederauftreten der Symptome erneut begonnen werden, um das Auftreten weiterer Krankheitsschübe zu verhindern.
Neben dem Wirkstoff sind folgende Hilfsstoffe enthalten: mittelkettige Triglyceride, (Z)-Octadec-9-en-1-ol, Propylenglycol, Stearylalkohol, Cetylalkohol, Glycerolmono/dispeisefettsäureester, Natriumcetylstearylsulfat, Benzylalkohol, Citronensäure, Natriumhydroxid und gereinigtes Wasser.
Pimecrolimus sollte nur von Ärzten verschrieben werden, die Erfahrung in der topischen Behandlung des atopischen Ekzems haben.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Der Einsatz als First-Line-Therapie ist unwirtschaftlich.
Angesicht des fehlenden Nachweises einer Überlegenheit gegenüber schwach wirksamen topischen Steroiden und fehlender hinreichend aussagekräftiger placebokontrollierter Studien bei Erwachsenen ist die Anwendung nur wirtschaftlich bei leichtem bis mittelschwerem atopischen Ekzem
Insgesamt dürfte dies nur auf wenige Patienten zutreffen, dies gilt auch für den Einsatz als Second-Line-Behandlung.
Die bisherigen verblindeten, placebovergleichenden Studien gingen nicht über sechs Wochen hinaus, sodass eine abschließende Beurteilung der unterschiedlichen Behandlungsoptionen, insbesondere zu Langzeitnebenwirkungen, zurzeit nicht möglich ist.
Pimecrolimus ist mittelstark bis stark wirksamen Glukokortikoiden unterlegen. Ob es eine vergleichbare Wirksamkeit zu schwach wirksamen Kortikosteroiden hat, ist nicht belegt. Direkt vergleichende Untersuchungen zu schwach wirksamen Steroiden fehlen. Der Stellenwert der Behandlung mit Pimecrolimus, insbesondere im direktem Vergleich zum optimierten Einsatz von schwach wirksamen Glukokortikoiden, auch im Wechsel mit wirkstofffreien Mitteln in der erscheinungsarmen Zeit, ist unklar.
Ein kortisonsparender Effekt zu einem solchen Therapieregime ist nicht belegt.
Es fehlen zurzeit direkt vergleichende Studien zu anderen topischen Makrolidimmunsuppressiva. Aufgrund der jetzigen Datenlage wird angenommen, dass Pimecrolimus eher weniger wirksam als Tacrolimus ist.
Pimecrolimus ist nur zugelassen für Kinder ab 2 Jahren, bei jüngeren traten vermehrt Nebenwirkungen auf. Der Einsatz ist daher nicht vertretbar und somit unwirtschaftlich.
Kombinationsbehandlungen von Pimecrolimus
| Kosten |
Es werden die Preise der Festbeträge von topischen Glukokortikosteroiden angegeben. Innerhalb der einzelnen Festbetragsgruppen variieren die Preise jedoch um bis zu 50 %.
Preisvergleich (Preise in Euro )
| Menge in Gramm | Pimecrolimus Creme 1 % | topische Glukokortikosteroide schwach | Tacrolimus Creme | |
| 0,03 % | 0,1 % | |||
| 15 | 26,66 | 4,91 | ||
| 30 | 49,32 | 8,25 | 49,32 | 53,93 |
| 60 | 13,89 | 91,49 | 102,90 | |
| 100 | 155,02 | 20,39 | ||
| Wirkungen |
Pimecrolimus ist ein lipophiles Macrolacatam-Derivat von Ascomycin mit antiinflammatorischen Eigenschaften. Es ist ein zellselektiver Inhibitor der Produktion und Freisetzung von proinflammatorischen Zytokinen. Pimecrolimus bindet mit hoher Affinität an Macrophilin-12 und inhibiert die kalziumabhängige Phosphatase Calcineurin. Als Folge wird die Synthese von inflammatorischen Zytokinen in T-Zellen blockiert. Es wurde eine Hemmung der Freisetzung von Entzündungsmediatoren und Zytokinen aus Mastzellen nach Stimulation in vitro beobachtet. Pimecrolimus zeigt im Tiermodell an entzündeter Haut hohe antiinflammatorische Aktivität nach topischer und systemischer Anwendung. Es beeinflusst nicht die Langerhans-Zellen in der Haut von Mäusen.
Der Wirkmechanismus von Pimecrolimus ist nicht vollständig geklärt. Die klinische Bedeutung der beschriebenen Mechanismen für die Behandlung des atopischen Ekzems ist nicht bekannt.
| Wirksamkeit |
Es wurden drei placebokontrollierte Hauptstudien zum Beleg der Wirksamkeit durchgeführt, die alle über sechs Wochen eine Doppelblindphase enthielten und eine sich anschließende 20-wöchige Phase, in der offen behandelt wurde. Endpunkt aller drei Studien war die Gesamtbewertung durch den Prüfarzt (IGA = Investigator Global Assessment) nach sechs Wochen. In allen Studien wurde Pimecrolimus 1 % zweimal täglich gegen die Cremegrundlage getestet. Es erfolgte jeweils eine 2:1-Randomisierung.
In zwei Studien wurden Patienten in identischen Designs im Alter von 2 bis 17 Jahren behandelt. Der primäre Endpunkt zeigte eine statistischsignifikante Überlegenheit gegenüber Cremegrundlage an dem prädefinierten Endpunkt in einer Studie, während dies
in der anderen Studie nicht erreicht wurde. Die kombinierte Auswertung, die auch publiziert wurde, zeigt eine signifikante Überlegenheit von Pimecrolimus gegenüber Placebo. Die dritte Studie wurde in fast identischem Design bei Kindern im Alter von 3 bis 21 Monaten durchgeführt. Auch hier zeigte sich eine statistisch signifikante Überlegenheit gegenüber der Cremegrundlage. Allerdings näherte sich der Anteil der Kleinkinder, die in der Verumgruppe in der Doppelblindphase unter Nebenwirkungen litten, dem Niveau statistisch signifikant (p = 0,052). In der sich anschließenden offenen Phase blieb die Rate der Nebenwirkungen unter Pimecrolimus 1 % weitgehend konstant (79,5 %), während die Kinder, die von Cremegrundlage auf Pimecrolimus umgestellt wurden, eine deutliche Zunahme an Nebenwirkungen erlitten. Signifikant häufiger waren Fieber (31,7 % versus 12,7 %), Durchfall (8,1 % versus 0 %) und Otitis media (4,1 % versus 0 %). Gehäuft traten auch Infektionen des oberen Respirationstraktes auf (Differenz 9,3 %), Nasopharynx-Infektionen (6,7 %), Gastroenteritis (4,1 %) etc. Beispielhaft kann hier die Inzidenz der Otitis media aufgeführt werden. Während der Doppelblindphase lag sie bei 0 % in der Gruppe der Kinder, die mit Cremegrundlage behandelt wurden. Nach Umstellung auf Pimecrolimus in der offenen Phase stieg sie auf 7,1 % an. In der Behandlungsgruppe, die durchgängig mit Pimecrolimus behandelt wurde, stieg sie von 4,1 % auf 9,4 %, sodass angenommen werden kann, dass das Risiko mit der Dauer der Behandlung ansteigt.
Die gepoolte Analyse aller drei Studien erreichte statistische Überlegenheit am 43. Tag (p < 0,001), als 160 Patienten (41 %) der mit Pimecrolimus behandelten Patienten erfolgreich behandelt waren im Vergleich zu lediglich 40 (20,1 %) der mit Placebo behandelten. Die drei Hauptstudien wurden einer gemeinsamen Subgruppenanalyse unterzogen. Hierbei zeigte sich eine Überlegenheit von Pimecrolimus 1 % in allen Subgruppen bis auf einen TBSA von > 60 % (total body surface area).
Eine vergleichbare placebokontrollierte Studie wurde bei Erwachsenen nicht durchgeführt.
In einer sechsarmigen Studie an Erwachsenen wurden vier Wirkstärken Pimecrolimus versus Cremegrundlage versus ein stark wirksames Kortikosteroid (0,1 % Betamethasonvalerat) an 260 Patienten über drei Wochen geprüft. Betamethason war in dieser Dosisfindungsstudie wirksamer als Pimecrolimus.
In einer doppelblind randomisierten, 12-monatigen Studie an Erwachsenen wurde bei 658 Patienten Pimecrolimus im Vergleich zu einem mittelstark wirksamen Kortikosteroid (Triamcinolonacetonid 0,1 %) beziehungsweise für Gesicht, Nacken und intertrigeniöse Areale mit einem schwach wirksamen Kortikosteroid (Hydrocortisonacetat 1 %) verglichen. In dieser multizentrischen Studie mit 1:1-Randomisierung war zu allen Beobachtungszeitpunkten das Kortikosteroid statistisch signifikant Pimecrolimus überlegen. Entsprechend unterbrachen Patienten unter Kortikosteroiden deutlich seltener die Therapie als unter Pimecrolimus (8,2 % versus 36,3 %).
Zudem sprachen die Patienten, die mit topischen Kortikosteroiden behandelt wurden, deutlich schneller auf die Therapie an.
In einer randomisierten und multizentrischen, doppelblind placebokontrollierten (Cremegrundlage) Studie mit einer Randomisierung von 2:1 wurde in beiden Therapiearmen beim Schub die Studienmedikation zusammen mit blanden Emollentien verabreicht. Bei einem Schub wurde mit Kortikosteroiden behandelt. Im Anschluss daran wurde wiederum über sieben Tage mit Pimecrolimus beziehungsweise Placebo therapiert. Primärer Endpunkt der Studie war die Schubrate nach sechs Monaten (Schub wurde definiert als IGA von 4 oder 5 sowie einer Second-Line-Kortikoid-Therapie innerhalb von drei Tagen nach klinischer Visite). Die Studie wurde über insgesamt 12 Monate fortgeführt. Eingeschlossen wurden Patienten im Alter von 2 bis 18 Jahren. Die Anzahl der Patienten, die keinerlei Schübe erlitten, war nach sechs Monaten fast doppelt so hoch wie in der placebokontrollierten Gruppe (61 % versus 34,2 %). Dies veränderte sich nicht wesentlich nach 12 Monaten (50,8 % versus 28,3 %). Die Anzahl der Patienten, die Schübe erlitten, unterschied sich in den zwei Armen nach sechs Monaten nicht wesentlich, ein Schub erlitten in beiden Armen 10,1 % der Patienten, zwei Schübe unter Pimecrolimus 2,5 % und unter Placebo 5,1 % und mehr als zwei Schübe 1,9 % beziehungsweise 2,5 %. Die Aussagekraft der Studie wird eingeschränkt dadurch, dass schwach, mittelstark und stark wirksame Glukokortikosteroide eingesetzt wurden und häufig das Protokoll verletzt wurde, insgesamt 53,6 % Protokollverletzungen in der Gruppe, die Pimecrolimus erhielten, und 58,6 % der Patienten, die Placebo erhielten. Ganz wesentlicher Mangel der Studie ist der verpflichtende Gebrauch von Pimecrolimus oder Placebo über sieben Tage nach der Behandlung des Schubs mit topischen Kortikosteroiden, der dazu führt, dass die mit Pimecrolimus behandelte Gruppe insgesamt sieben Tage länger eine aktive Arzneimitteltherapie im Vergleich zu Placebo erhält. Unter diesen Aspekten ist die Studie nicht in der Lage, für sich in Anspruch zu nehmen, nachweisen zu können, dass Pimecrolimus die Anzahl der Schübe bei atopischer Dermatitis im Vergleich zur Standardbehandlung zu vermindern oder insbesondere auch eine Reduktion des Gebrauchs von topischen Kortikosteroiden zu induzieren vermag. Im Studiendesign vergleichbare Studien wurden bei Kindern im Alter von 3 bis 23 Monaten und Erwachsenen durchgeführt. Auf sie trifft die gleiche Kritik zu.
| Risiken ggf. Vorsichtsmaßnahmen |
Die am häufigsten vorkommenden Nebenwirkungen waren Reaktionen am Anwendungsort, die von zirka 19 % der mit Elidel behandelten Patienten und von zirka 16 % der Patienten der Kontrollgruppe berichtet wurden. Diese Reaktionen traten vor allem zu Beginn der Behandlung auf, sie waren schwach bis mäßig stark und von kurzer Dauer.
In klinischen Untersuchungen kam es in 0,9 % zu Lymphadenopathien. In der Mehrzahl waren sie auf Infektionen zurückzuführen, die unter einer angemessenen Antibiotikabehandlung abklangen. Patienten, die eine Lymphadenopathie entwickeln, sollten überwacht werden, um sicherzustellen, dass die Lymphadenopathie abklingt. Die Ätiologie ist zu klären. Kann die Krankheitsursache nicht eindeutig ermittelt werden oder liegt eine akute infektiöse Mononukleose vor, so ist die Unterbrechung der Behandlung mit Pimecrolimus in Erwägung zu ziehen.
Bei Patienten mit ausgedehnter atopischer Dermatitis wird empfohlen, Impfungen während behandlungsfreier Intervalle durchzuführen. Pimecrolimus sollte nicht gleichzeitig mit topischen Kortikosteroiden oder anderen topischen antiinflammatorischen Produkten appliziert werden. Es gibt keine Erfahrungen zur gleichzeitigen Anwendung von immunosuppressiven Therapien bei atopischem Ekzem, wie Azathioprin oder Ciclosporin.
Gemäß US-amerikanischer Fachinformation zeigte sich in Photokanzerogenitätsstudien beim Tier eine Verkürzung der Zeitspanne bis zum Auftreten von Tumorformationen durch die Cremegrundlage. Da die Relevanz dieser Daten für den Menschen nicht bekannt ist, sollten während der Behandlung mit Pimecrolimus-Creme ausgedehnte Bestrahlungen der Haut mit ultravioletten Licht, wie beispielsweise in Solarien, oder die Therapie mit PUVA, UVA oder UVB vermieden werden. Der Arzt sollte die Patienten auf angemessene Sonnenschutzmaßnahmen hinweisen, wie eine Minimierung der Aufenthaltszeit in der Sonne, Benutzung von Sonnenschutzprodukten und Bedeckung der Haut mit entsprechender Kleidung.
Bei Kindern unter zwei Jahren traten, wie dargestellt, vermehrt Nebenwirkungen auf. Die Anwendung von Pimecrolimus bei Kindern unter 2Jahren wird nicht empfohlen.
Eine Behandlung mit Pimecrolimus kann mit einem erhöhten Risiko für eine Herpessimplex-Infektion oder Eczema herpeticum einhergehen (erkennbar an einer schnellen Ausbreitung von bläschenartigen und erosiven Läsionen). Bei Vorhandensein einer Herpessimplex-Infektion sollte an der betroffenen Stelle die Behandlung nicht fortgesetzt werden, bis die virale Infektion abgeklungen ist. Obwohl bei Patienten, die mit Pimecrolimus behandelt wurden, bakterielle Autoinfektionen seltener waren als bei Patienten, die mit Placebo behandelt wurden, kann bei Patienten mit schwerem atopischen Ekzem das Risiko für bakterielle Hautinfektionen (Impetigo) während der Behandlung mit Elidel erhöht sein.
Pimecrolimus darf nicht auf Bereiche aufgetragen werden, die von akuten viralen Hautinfektionen betroffen sind (Herpes simplex, Windpocken).
Die Anwendung in der Schwangerschaft und Stillzeit sowie bei Patienten mit genetisch bedingten Schädigungen der Epidermisschranke (z.B. Netherton-Syndrom) und generalisierter Erythrodermie wird nicht empfohlen. Kontakt mit Augen und Schleimhäuten ist zu vermeiden, das Gleiche gilt für Okklusionsverbände.
Prasugrel
(z.B. Efienü")
Beschluss vom: 17.06.2010
In Kraft getreten am: 11.09.2010
BAnz. Nr. 137 vom 10.09.2010 S. 3108
| Zugelassene Anwendungsgebiete |
Prasugrel ist in Kombination mit Acetylsalicylsäure (ASS) zugelassen zur Prävention atherothrombotischer Ereignisse bei Patienten mit akutem Koronarsyndrom, bei denen eine primäre oder verzögerte perkutane Koronarintervention (PCI) erfolgt.
Die Behandlung wird mit einer 60 mg Aufsättigungsdosis begonnen und dann mit einer Erhaltungsdosis von 10 mg täglich fortgesetzt.
| Empfehlung zur wirtschaftlichen Verordnungsweise |
Gesamtmortalität und kardiovaskuläre Mortalität werden durch eine duale Thrombozytenaggregationshemmung mit Prasugrel und ASS im Vergleich zu Clopidogrel und ASS nicht signifikant verringert. Ein Vorteil zeigt sich in erster Linie durch eine signifikante Reduktion nicht-tödlicher Herzinfarkte. Dein steht ein signifikant erhöhtes Risiko für schwerwiegende Blutungen gegenüber, darunter auch tödlich verlaufende.
Die Behandlung ist laut Zulassung auf 12 Monate zu beschränken, da das Blutungsrisiko darüber hinaus weiter zunimmt, hingegen ein vorteilhafter klinischer Effekt durch die klinischen Daten nicht gestützt wird.
Der Vorteil von Prasugrel ist in der akuten Phase der Behandlung am größten. Insbesondere beim ST-Hebungsinfarkt (STEMI) wird der maximale Effekt in der 3.-4. Woche nach PCI erzielt und nimmt danach gegenüber Clopidogrel nicht mehr weiter zu. Bei Patienten mit instabiler Angina Pectoris oder Nicht-STHebungsinfarkt (UA/NSTEMI) wird der maximale Effekt ebenfalls im ersten Monat der Behandlung gesehen. Im Gegensatz zum STEMI nimmt er darüber hinaus im Verlauf von 12 Monaten gegenüber Clopidogrel noch etwas zu.
In der einzigen Zulassungsstudie der Phase III, der TRITON-38Studie, wurden Patienten mit einem erhöhten Blutungsrisiko ausgeschlossen, zum Beispiel Patienten unter Langzeittherapie mit nicht-steroidalen Antiphlogistika bzw. Coxiben oder Vitamin-K-Antagonisten sowie Patienten, die kürzlich eine gastrointestinale Blutung, ein Magengeschwür oder ein Trauma erlitten hatten. In diesem selektierten Patientengut mussten dennoch gegenüber Clopidogrel je 1.000 Patienten 5 zusätzliche schwer- wiegende Blutungen (darunter 2 tödliche) in Kauf genommen werden, um 22 nicht-tödliche Herzinfarkte zu verhindern.
| Kosten |
Die Kosten einer dualen Thrombozytenaggregationshemmung bei akutem Koronarsyndrom mit primärer oder verzögerter perkutaner Koronarintervention sind bei Einsatz von Prasugrel anstelle von Clopidogrel deutlich höher. Deshalb ist die Therapie mit Prasugrel für Patienten der Altersgruppe 75 Jahre oder mit einem Gewicht unter 60 kg auch als unwirtschaftlich anzusehen.
| Thrombozytenaggregationshemmer | Tagestherapiekosten | Therapiezyklus über 12 Monate |
| Prasugrel plus ASS | 2,98 Euro | 1 087 Euro |
| Clopidogrel plus ASS | 0,54 Euro-2,83 Euro | 197 Euro-1034 Euro |
| Ticlopidin | 0,74 Euro | 271 Euro |
| ASS | 0,03 Euro | 13 Euro |
Berechnung auf Basis der größten verfügbaren Packung bzw. Festbetrag Stand Lauer Taxe 15. August 2010
| Wirkungen |
Prasugrel gehört wie Clopidogrel oder Ticlopidin zu den Thienopyridinen und ist ein Prodrug. Sein aktiver Metabolit bindet irreversibel an den P2Y12 Adenosindiphosphat-Rezeptor auf Thrombozyten. Die Aktivierung und Aggregation von Thrombozyten wird dadurch gehemmt.
Prasugrel wird schnell zu einem Thiolacton hydrolysiert und anschließend in einer Ein-Schritt-Metabolisierung über Cytochrom P450 in seinen aktiven Metaboliten umgewandelt, in erster Linie durch CYP3A4 und CYP2B6 und in einem geringfügigeren Ausmaß durch CYP2C9 und CYP2C19. Die maximale Plasmakonzentration des aktiven Metaboliten wird innerhalb von ca. 30 Minuten erreicht. Sie kann verzögert werden durch Medikamente, welche den pH-Wert des Magens erhöhen (H2-Blocker und Protonenpumpenhemmer), CYP3A Inhibitoren (z.B. AzolAntimykotika) und fetthaltige, kalorienreiche Mahlzeiten. Der schnellste Wirkeintritt kann daher erzielt werden, wenn die Prasugrel-Aufsättigungsdosis nüchtern eingenommen wird. Grundsätzlich kann die Einnahme jedoch unabhängig von der Nahrungsaufnahme erfolgen.
Bei Patienten älter als 75 Jahre oder mit einem Gewicht unter 60 kg war die Exposition gegenüber dem aktiven Metaboliten größer als bei jüngeren Patienten oder solchen mit höherem Gewicht und mit einem erhöhten Risiko für Blutungen verbunden.
Wenn die Behandlung unterbrochen wird, kehrt die Thrombozytenaggregation nach 7 bis 10 Tagen zum Ausgangswert zurück.
Mit Prasugrel wurde in vergleichenden klinischen Studien mit Clopidogrel in der jeweiligen Aufsättigungs- bzw. Erhaltungsdosis eine schnellere und stärkere Hemmung der Thrombozytenaggregation erreicht.
| Wirksamkeit |
13.619 Patienten mit akutem Koronarsyndrom, bei denen primär oder verzögert nach 14 Tagen eine PCI erfolgen sollte, erhielten in der TRITON-38-Studie randomisiert entweder Clopidogrel (Aufsättigungsdosis 300 mg, Erhaltungsdosis 75 mg) oder Prasugrel (Aufsättigungsdosis 60 mg, Erhaltungsdosis 10 mg) jeweils kombiniert mit ASS. Durch stratifizierte Randomisation wurden zwei Subgruppen gebildet von Patienten mit ST-Hebungsinfarkt (STEMI) und Patienten mit instabiler Angina Pectoris oder NichtST-Hebungsinfarkt (UA/NSTEMI). Die Aufsättigungsdosis konnte bis zu einer Stunde nach Verlassen des Katheterlabors gegeben werden. Die größere Gruppe der UA/NSTEMI-Patienten erhielt die Aufsättigungsdosis erst nach koronarangiographischer Klärung der Eignung für eine PCI, sodass die meisten Patienten (74%) diese entgegen gängigen Leitlinienempfehlungen während oder nach PCI erhielten.
Nach einer medianen Behandlungsdauer von 14,5 Monaten trat der kombinierte primäre Endpunkt (kardiovaskulär bedingte Todesfälle oder nicht-tödliche Myokardinfarkte und Schlaganfälle) in der mit Prasugrel plus ASS behandelten Gruppe signifikant seltener auf (9,4 % vs. 11,5 %, p < 0,001). Die absolute Risikoreduktion von 2,1 % entspricht einem NNT-Wert von 49 Patienten und einer relativen Risikoreduktion um 18 %. Diese Risikoreduktion kam in erster Linie durch eine geringere Inzidenz nicht-tödlicher Herzinfarkte zustande (7,3 % vs. 9,5 %). Dabei wurden auch periprozedurale Herzinfarkte berücksichtigt, die nur anhand eines erneuten Anstieges der CK-MB diagnostiziert wurden und deren klinische Bedeutsamkeit nicht unumstritten ist. Die anderen Komponenten des primären Endpunktes unterschieden sich nicht (kardiovaskulär bedingte Todesfälle 2 % vs. 2,2 %, nicht-tödliche Schlaganfälle 0,9 % vs. 0,88 %). Auch die Gesamtmortalität war in beiden Behandlungsgruppen gleich (2,8 % vs. 2,9 %). Bei 1.000 Patienten können demnach durch eine duale Thrombozytenaggregationshemmung mit Prasugrel anstelle von Clopidogrel 22 nicht-tödliche Herzinfarkte verhindert werden.
Dabei ist nicht ausgeschlossen, dass dieses Ergebnis auch durch die späte Gabe der Aufsättigungsdosis beeinflusst wurde, wodurch das schneller wirksame Prasugrel bevorzugt wurde.
Demgegenüber erhöhte sich das Risiko für schwerwiegende, nicht mit einer Bypass-Operation assoziierten Blutung, einschließlich lebensbedrohlicher und letaler Blutungen, unter dualer Thrombozytenaggregationshemmung mit Prasugrel und ASS signifikant (2,2 % vs. 1,7 %, p = 0,03, darunter lebensbedrohliche 1,3 % vs. 0,8 %, p = 0,01 und tödliche 0,3 % vs. 0,1 %, p = 0,002). Wurden auch weniger schwerwiegende Blutungen mit einem Hb-Abfall von 3-5 g/dl betrachtet, so ergab sich ein erhöhtes absolutes Risiko von 1 % entsprechend einer relativen Risikoerhöhung um 31 %. Diese Zahlen bedeuten, dass von 1 000 behandelten Patienten zusätzlich 2 eine letale, 3 eine schwerwiegende, jedoch nicht-tödliche und 6 eine weniger schwerwiegende Blutung erleiden.
Der Vorteil von Prasugrel wurde überwiegend zu Behandlungsbeginn erzielt. In der gesamten Studienpopulation traten fast 80 % der gegenüber Clopidogrel und ASS zusätzlich verhinderten primären Endpunkte in den ersten 30 Tagen nach PCI auf. Bei einer vorab nicht geplanten Analyse unterschied sich der Verlauf hinsichtlich des primären Endpunktes jenseits der ersten 30 Tage nur noch im Trend und war nicht mehr signifikant. Insbesondere in der Subgruppe mit ST-Hebungsinfarkt kam es 3-4 Wochen nach PCI nicht mehr zu einer weiteren Risikoreduktion. Dagegen hielten die Blutungsrisiken weiter an.
In vorab festgelegten Subgruppenanalysen zeigten sich Reduktionen des primären Endpunktes unabhängig von Geschlecht, Alter, Stent-Typ, Körpergewicht und Verwendung von GP-IIb/ IIIa-Inhibitoren. Frauen machten ca. 27 % der Studienteilnehmer aus. Bei ihnen war die Ausprägung des Effektes weniger stark (10,4 % vs. 11,8 %). Bei Patienten 75 Jahre war der beobachtete klinische Effekt besonders gering (16 % vs. 17 %), wohingegen das Risiko für letale Blutungen signifikant erhöht war (1 % vs. 0,1 %).
In der Gesamtstudienpopulation verhinderte Prasugrel keine Schlaganfälle (1,1 % vs. 1 %). Jedoch stieg das Schlaganfallrisiko signifikant (6,5 % vs. 1,2 %, p < 0,002) bei Patienten, die mehr
als 3 Monate vor Studienbeginn einen ischämischen Schlaganfall oder eine TIA erlitten hatten. Der primäre Endpunkt wurde tendenziell ungünstig beeinflusst (17,9% vs. 13,7%, p = 0,15). Für Patienten mit cerebrovaskulären Erkrankungen ist daher ein ungünstiger Effekt von Prasugrel wahrscheinlich, insbesondere unter dem Aspekt, dass Patienten mit hämorrhagischem Schlaganfall in der Vorgeschichte oder ischämischem Schlaganfall in den letzten 3 Monaten an der Studie nicht teilnehmen durften.
| Risiken und Vorsichtsmaßnahmen |
Blutungskomplikationen machen das Hauptrisiko einer Behandlung mit Prasugrel aus. Die Behandlung soll deshalb nicht über 12 Monate hinaus fortgesetzt werden.
Obwohl Patienten mit möglichen Risiken für eine Blutung von der Studienteilnahme ausgeschlossen wurden (z.B. Thrombozytopenie, Anämie, Langzeittherapie mit nichtsteroidalen Antirheumatika bzw. Coxiben, Antikoagulation mit Vitamin-K-Antagonisten, cerebrale Befunde wie Aneurysmen, arteriovenöse Malformationen oder Tumore, kürzlich zurückliegende gastrointestinale Blutungen, akute Magen-Darmgeschwüre), traten hämorrhagische Ereignisse insgesamt sehr häufig und signifikant vermehrt gegenüber Clopidogrel auf (29,7 % vs. 22 %, p < 0,001). An Blutungen verstarben unter Behandlung mit Prasugrel 24 (0,36 %) und unter Clopidogrel 6 (0,09 %) Patienten. Unabhängige Risikofaktoren waren Körpergewicht unter 60 kg, Alter 75 Jahre, Schlaganfall oder TIA in der Vorgeschichte oder die Anwendung von GP-IIb/IIIa-Inhibitoren.
Prasugrel ist für Patienten mit Schlaganfall oder TIA in der Vorgeschichte vor dem Hintergrund des bei diesen Patienten beobachteten erhöhten Schlaganfall- und Blutungsrisikos kontra-indiziert. Bei Patienten 75 Jahre wird die Anwendung im Allgemeinen nicht empfohlen. In Einzelfällen wird für diese Patienten und für Patienten unter 60 kg Körpergewicht eine reduzierte Dosis von 5 mg empfohlen, obwohl für diese Dosis keine klinischen Daten zur Unbedenklichkeit vorliegen.
Auch Patienten, die gleichzeitig Arzneimittel einnehmen, die das Risiko für Blutungen erhöhen können einschließlich oraler Antikoagulantien, Clopidogrel, nicht-steroidaler Antiphlogistika und Fibrinolytika oder mit einer Blutungsneigung (z.B. durch ein kürzlich erlittenes Trauma, kürzlich vorgenommene Operation, kürzlich aufgetretene oder wieder auftretende gastrointestinale Blutung oder ein akutes Magengeschwür), sollen nur nach individueller Nutzen-Risiko-Abwägung Prasugrel erhalten.
Ein besonders hohes Risiko schwerwiegender Blutungen hatten Patienten, bei denen eine Bypass-Operation notwendig wurde (11,3 % vs. 3,6 %). Die Gabe von Prasugrel soll deshalb nach Möglichkeit erst erfolgen, wenn die Koronaranatomie bekannt ist und eine dringliche Bypass-Operation unwahrscheinlich ist.
Die Europäische Zulassungsbehörde (EMA) wertet die erhöhte Inzidenz von Kolonkarzinomen (0,17% vs. 0,03 %) unter Prasugrel als Folge des höheren Risikos gastrointestinaler Blutungen, wodurch es zur diagnostischen Abklärung und früheren Diagnose dieser Karzinome kommen kann. Die Amerikanische Zulassungsbehörde (FDA) hält es dagegen zwar für unwahrscheinlich, jedoch nicht für ausgeschlossen, dass Prasugrel die Ausbreitung vorhandener Tumore stimuliert. Der Hersteller wurde verpflichtet, zusätzliche Daten in laufenden Studien zu erheben.
Prasugrel soll während einer Schwangerschaft nur eingesetzt werden, wenn der mögliche Nutzen für die Mutter das mögliche Risiko für den Fötus rechtfertigt. Die Anwendung während der Stillzeit wird nicht empfohlen.
Vor geplanten operativen Eingriffen sollte Prasugrel spätestens 7 Tage vor der Operation abgesetzt werden.
Raloxifen
(z.B. Evista ®)
Beschluss vom: 16.02.2000
In Kraft getreten am: 31.05.2000
BAnz. 2000, S. 10.094/ 10.095
| Indikation |
Das Benzothiophenderivat Raloxifen wurde 1998 zur Prävention atraumatischer Wirbelbrüche bei postmenopausalen Frauen mit einem erhöhten Osteoporose-Risiko zugelassen. Die empfohlene Dosis in der naturgemäß Langzeitbehandlung - ggf. mit Calciumsubstitution bei nicht ausreichender diätetischer Zufuhr - beträgt 60 mg Raloxifen-Hydrochlorid/d (= 56 mg Raloxifen-Base/d; = 1 Tablette/d), die zu jeder Tageszeit unabhängig von den Mahlzeiten und ohne Anpassung an das Lebensalter per os appliziert wird.
| Wirkungen |
Als ein selektiver Oestrogen Rezeptor Modulator (SERM) zeigt Raloxifen selektive agonistische oder antagonistische Wirkungen; es wirkt als ein Agonist auf den Knochen- und teilweise auf den Cholesterin-Stoffwechsel (Gesamt- und LDL-Cholesterin .L), nicht aber auf Hypothalamus, Uterus- und Brustgewebe. Die biologischen Wirkungen von Raloxifen werden analog zu denen der Oestrogene vermittelt, indem es mit hoher Affinität an Oestrogenrezeptoren bindet und die Genexpression reguliert; diese Bindung führt zu einer differenzierten Expression verschiedenartiger oestrogenregulierter Gene in unterschiedlichen Geweben.
| Wirksamkeit |
Die Wirksamkeit bzgl. der Knochenmineraldichte (bone mineral density = BMD; Surrogatparameter) von einmal täglich verabreichten 60 mg Raloxifen bei postmenopausalen Frauen von bis zu 60 Jahren mit oder ohne Uterus wurde in insgesamt 3 Studien (n = 1.764) in einer zweijährigen Behandlungsperiode nachgewiesen. Raloxifen führte im Vergleich zu Plazebo zu einem signifikanten Anstieg der Knochendichte der Hüfte und der Wirbelsäule sowie zu einem Anstieg des Gesamtkörpermineralgehaltes; der Anstieg der BMD betrug im Mittel 2% im Vergleich zu Plazebo [in einer Studie unter Oestrogenen 5% (Wirbelsäule) bzw. 3% (Hüfte)]; jedoch zeigten 29% (Hüfte) bzw. 37% (Wirbelsäule) der Patientinnen unter Raloxifen eine Abnahme der BMD.
Nach der Zwischenauswertung (3 Jahre) einer Studie mit 7.705 postmenopausalen Frauen (Durchschnittsalter 66 Jahre) reduzierte Raloxifen nach 36-monatiger Therapie die Inzidenz vertebraler Frakturen um ca. 30-50 %. Dabei handelte es sich um 436 Frauen mit klinisch stummen Frakturen, die durch radiologisches Screening ermittelt wurden, und um 65 Frauen mit klinisch symptomatischen Frakturen. Im Gegensatz zu den Östrogenen liegen zur Zeit für Raloxifen keine Daten über die Reduktion nichtvertebraler Frakturen vor. In einer Vergleichsstudie zwischen Raloxifen und Oestrogenen war der Knochen unabhängig vom Medikament histologisch normal. Die klinischen Studien zeigen eine Reduktion von Gesamt-(3-6%) und LDL- (4-10%) Cholesterin und keinen stimulierenden Effekt auf die postmenopausale Gebärmutterschleimhaut. Raloxifen hat keine stimulierende Wirkung auf die Brustdrüse; in klinischen Studien an insgesamt über 12.000 Patientinnen (mind. 30 Monate Behandlung) war das relative Risiko eines neu diagnostizierten Brustkrebses im Vergleich zur Plazebogruppe vermindert (relatives Risiko 0,47). Jedoch ist die Langzeitwirkung von Raloxifen auf das Brustkrebsrisiko nicht bekannt.
Raloxifen wird nach oraler Gabe rasch resorbiert (ca. 60% der applizierten Dosis) und unterliegt einem enterohepatischen Kreislauf und einem ausgeprägten firstpass-Metabolismus zu den entsprechenden Glucuronid-Konjugaten; die Plasmahalbwertszeit beträgt 27,7 Stunden.
| Risiken ggf. Vorsichtsmaßnahmen |
Raloxifen ist kontraindiziert bei noch gebärfähigen Frauen, bestehender oder in der Vorgeschichte aufgetretenen venösen thromboembolischen Ereignissen, eingeschränkter Leberfunktion einschl. Cholestase, schweren Nierenschädigungen und ungeklärten Uterusblutungen.
Die Einnahme von Raloxifen ist mit einem erhöhten Risiko für venöse thromboembolische Ereignisse verbunden; das beobachtete relative Risiko betrug 2,49 im Vergleich zu Plazebo und 1,0 im Vergleich zur Hormonersatztherapie. Bei Immobilisation sollte Raloxifen daher abgesetzt werden.
An weiteren, über Plazebo erhöhten Begleitwirkungen sind insbesondere zu nennen: Hitzewallungen, Wadenkrämpfe, periphere Oedeme und leicht erniedrigte Thrombozytenkonzentrationen.
Gleichzeitige Anwendung von kationischen Antacida (Ca, Mg, Al) beeinflußt die systemische Verfügbarkeit von Raloxifen nicht. Bei gleichzeitiger Anwendung von oralen Antikoagulantien kann die Prothrombinzeit leicht verkürzt sein; die Resorption und Verfügbarkeit von Raloxifen wird durch Anionenaustauscher vermindert.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Raloxifen kann eingesetzt werden zur Prävention atraumatischer Wirbelbrüche bei postmenopausalen Frauen mit manifester Osteoporose, welche durch entsprechende Anamnese und Untersuchungsbefunde zu dokumentieren ist. Die Wirkungen von Raloxifen am Knochen sind qualitativ vergleichbar mit denen einer Oestrogen-Ersatztherapie, sind aber weniger ausgeprägt. Klimakterische Beschwerden werden durch Raloxifen nicht beeinflußt, Hitzewallungen eher vermehrt, andererseits führt Raloxifen nicht zu Schmierblutungen, Blutungen oder Hyperplasie des Endometriums; in klinischen Studien mit Raloxifen zeigten sich eine leichte Senkung von Gesamt- und LDL-Cholesterin und eine Verminderung des relativen Risikos eines neu diagnostizierten Brustkrebses. Wenn in einem individuellen Fall über die Gabe von Raloxifen oder Hormonersatztherapie zu entscheiden ist, sind klimakterische Symptome, Auswirkungen auf das Brustgewebe sowie kardiovaskuläre Risiken und Nutzen zu berücksichtigen. Wegen der Langfristigkeit der Therapie ist insbesondere die Bereitschaft zur Compliance jeweils sorgfältig abzuklären.
Die gleichzeitige Gabe von Raloxifen und systemisch wirkenden Östrogenen wird nicht empfohlen und es wird keine Indikation für prämenopausale Frauen gesehen.
Bei einem Preis von 275,24 DM für die N3 Packung mit 84 Filmtabletten entstehen Tagestherapiekosten von 3,28 DM bzw. Jahrestherapiekosten von 1.195,98 DM.
Repaglinid (aufgehoben) 16
Sitagliptin
(aufgehoben durch BAnz. AT vom 08.06.2016 B1)
Beschluss vom: 12.09.1996
In Kraft getreten am: 14.11.1996
BAnz. 1996, S. 12.015
Somatropin ist ein Protein aus 191 Aminosäuren, das die Struktur des von der Hypophyse des Menschen produzierten Hauptbestandteils des Wachstumshormons besitzt (Monographie des Europ. Arzneibuchs - BAnz. v. 26.01.1994, S. 606).
Seit etwa 10 Jahren wird es gentechnologisch hergestellt (z.B. Genotropin®, Humatrope®, Norditropin®, Saizen® und Zomacton®). Die zunächst aus menschlichen Hypophysen gewonnenen Präparate wurden im Rahmen eines Stufenplanverfahrens wegen des Verdachts der Übertragung der Creutzfeld-Jakob-Erkrankung aus dem Markt genommen.
Somatropin hat ein breites Wirkungsspektrum. Teilweise werden die Wirkungen nicht selbst ausgelöst, sondern über die Bildung von Somatomedinen (z.B. das Knorpel-, Knochen- und Muskelwachstum). Bei Kindern und Jugendlichen kommt es so zu einem verstärkten Längenwachstum des Knochens. Bei Erwachsenen beeinflußt es u.a. die Knochendichte positiv. Somatropin zeigt eine anabole und lipolytische Wirkung, außerdem greift es in den Glucosestoffwechsel ein.
Alle am Markt befindlichen Präparate sind zugelassen für die Behandlung von Kindern mit Minderwuchs infolge nicht ausreichender körpereigener Wachstumshormone durch Hypophysenvorderlappeninsuffizienz.
Indikationserweiterungen einzelner Warenzeichen betreffen:
Insbesondere die in jüngster Zeit verstärkt proklamierte Anwendung zur Verbesserung der Lebensqualität bei Erwachsenen, die aufgrund eines festgestellten Wachstumhormon-Mangels Symptome einer herabgesetzten Vitalität, Energiemangel, Angstzustände und verstärkter sozialer Isolation zeigen, ist durch die o.g. Zulassungs-Kriterien nicht gedeckt und nicht Gegenstand der vertragsärztlichen Versorgung.
Nach wie vor besteht der Verdacht, Somatropin könne die Krebsentwicklung fördern.
Strontiumranelat (z.B. Protelos®, Osseor®) (aufgehoben 040215B1)
Tacrolimus
(zum Beispiel Protopic®)
| Indikation |
Tacrolimus ist zugelassen zur Behandlung des mittelschweren bis schweren atopischen Ekzems bei Erwachsenen, die auf herkömmliche Therapie nicht ausreichend ansprechen oder diese nicht vertragen, sowie bei Kindern ab 2 Jahren, die nicht ausreichend auf die herkömmliche Therapie angesprochen haben.
Es kann zur Kurzzeitbehandlung und intermittierenden Langzeitbehandlung angewendet werden.
Die Behandlung erfolgt zweimal täglich bis zu drei Wochen und wird dann auf einmal täglich reduziert und bis zur Abheilung fortgeführt, danach abgesetzt. Bei Kindern ist nur die Wirkstärke 0,03 % indiziert. Bei Erwachsenen (ab 16 Jahren) sollte mit der 0,1 % Salbe begonnen werden bei zweimal täglicher Anwendung für eine Dauer von bis zu drei Wochen. Danach sollte die Stärke auf 0,03 % bei zweimal täglicher Anwendung reduziert werden. Wenn der klinische Zustand es erlaubt, sollte versucht werden, die Anwendungshäufigkeit zu verringern.
Ist nach zweiwöchiger Behandlung keine Besserung zu erkennen, sind andere Therapiemöglichkeiten in Betracht zu ziehen.
Neben dem Wirkstoff sind folgende Hilfsstoffe enthalten: weißes Vaselin, dickflüssiges Paraffin, Propylencarbonat, gebleichtes Wachs und Hartparaffin.
Tacrolimus darf nur von Dermatologen beziehungsweise Ärzten mit umfangreicher Erfahrung in der Behandlung des atopischen Ekzems mit immunmodulierenden Therapien verschrieben werden.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Tacrolimus ist nur zugelassen zur Behandlung des mittelschweren bis schweren atopischen Ekzems
Die zur Zulassung führenden vergleichenden Studien haben solche Patienten nicht explizit eingeschlossen. Insgesamt dürfte dies nur auf wenige Patienten zutreffen.
Der Einsatz als First-Line-Therapie ist unwirtschaftlich.
In den direkt vergleichenden Untersuchungen traten mehr lokale Nebenwirkungen unter Tacrolimus-Salbe und auch unter der Salbengrundlage allein als unter Kortikosteroidbehandlung auf. Die bisherigen vergleichenden Studien gingen nicht über drei Wochen hinaus, sodass eine abschließende Beurteilung insbesondere zu Langzeitnebenwirkungen der unterschiedlichen Behandlungsoptionen zurzeit nicht möglich ist.
Der Stellenwert der Behandlung mit Tacrolimus, insbesondere im direktem Vergleich zum optimierten Einsatz von topischen Glukokortikoiden, auch im Wechsel mit wirkstofffreien Mitteln in der erscheinungsarmen Zeit, ist unklar. Tacrolimus scheint eine vergleichbare Wirksamkeit wie mittelstark bis stark wirksame Glukokortikoide zu haben.
Es fehlen zurzeit direkt vergleichende Studien zu anderen topischen Makrolidimmunsuppressiva. Aufgrund der jetzigen Datenlage wird angenommen, dass Pimecrolimus eher weniger wirksam als Tacrolimus ist.
Da keine Erfahrungen bei Kindern unter zwei Jahren vorliegen, ist hier eine Behandlung nicht indiziert.
Kombinationsbehandlungen von Tacrolimus
| Kosten |
Es werden die Preise der Festbeträge von topischen Glukokortikosteroiden angegeben. Innerhalb der einzelnen Festbetragsgruppen variieren die Preise jedoch um bis zu 50 %.
Preisvergleich (Preise in Euro )
| Menge in Gramm | Tacrolimus Salbe | topische Glukokortikosteroide | Pimecrolimus Creme 1 % | ||
| 0,03 % | 0,1 % | mittel | stark | ||
| 15 | 6,94 | 7,69 | 26,66 | ||
| 30 | 49,32 | 53,93 | 11,72 | 13,44 | 49,32 |
| 60 | 91,49 | 102,90 | 19,88 | 23,52 | |
| 100 | 29,32 | 35,48 | 155,02 | ||
| Wirkungen |
Tacrolimus wird aus Streptomyces tsukubaensis gewonnen. Es bindet an ein spezifisches Zellplasma-Immunophilin (FKBP12) und hemmt dadurch in den T-Zellen calciumabhängige Wege der Signaltransduktion, wodurch die Synthese von IL-2, IL-3, IL-4, IL-5 und anderen Zytokinen wie GM-CSF, TNF-a und IFN-y verhindert wird. Es wurde eine Hemmung der Freisetzung von Entzündungsmediatoren aus Mastzellen der Haut sowie aus basophilen und eosinophilen Granulozyten nachgewiesen. Bei Patienten mit atopischem Ekzem ging die Besserung der Hautschäden während der Behandlung mit einer Beeinträchtigung der Fc-Rezeptor-Expression auf den Langerhans-Zellen und einer Reduzierung ihrer übermäßig stimulierenden Wirkung auf T-Zellen einher.
Der Wirkmechanismus von Tacrolimus ist nicht vollständig geklärt. Die klinische Bedeutung der beschriebenen Mechanismen für die Behandlung des atopischen Ekzems ist nicht bekannt.
| Wirksamkeit |
Die Wirksamkeit wurde in fünf maßgeblichen Phase-Ill-Studien geprüft, die in Europa und Amerika durchgeführt wurden. Bei den eingeschlossenen Patienten war im Durchschnitt ein Drittel der Körperoberfläche erkrankt und ungefähr die Hälfte der Patienten litten unter einer schweren Erkrankung.
Die Behandlung mit Tacrolimus-Salbe zeigte im Vergleich zur Salbengrundlage in direkt vergleichenden Studien über eine Behandlungsdauer von drei bis zwölf Wochen signifikant bessere Ergebnisse. Ungefähr drei- bis viermal mehr Patienten sprachen auf Tacrolimus versus Salbengrundlage an (Salbengrundlage 7-8 %, 0,03 % Tacrolimus zirka 35 %, 0,1 % Tacrolimus zirka 40 %).
Vergleichende Untersuchungen zu topischen Glukokortikoiden wurden durchgeführt. Bei Kindern war Tacrolimus dem schwach wirksamen 1 % Hydrocortisonacetat in zwei Studien überlegen. Allerdings wird die Wahl des schwach wirksamen Referenzsteroids wegen dessen begrenzter Wirksamkeit als nicht optimal angesehen. Im Vergleich zu einem mittelstarken Kortikosteroid (0,1 % Hydrocortisonbutyrat) ergab sich bei Erwachsenen kein signifikanter Unterschied der Wirksamkeit. In zwei vergleichenden japanischen Studien der Phase III mit insgesamt 329 Patienten war die Wirksamkeit von 0,1 % Tacrolimus dem stark wirksamen topischen Kortikosteroid (0,12 % Betamethasonvalerat) vergleichbar und dem mittelstark wirksamen 0,1 % Alcometasondipropionat überlegen.
Unter Tacrolimus und auch unter der Salbengrundlage allein traten mehr lokale Nebenwirkungen auf als unter Kortikosteroiden.
Das Wiederauftreten der Erkrankung war bisher nicht Ziel von Untersuchungen. In den US-amerikanischen Studien kam es bei ungefähr der Hälfte der Patienten zwei Wochen nach Absetzen der Therapie zu einem erneuten Schub. In den europäischen Untersuchungen hielt eine moderate Verbesserung bei etwa der Hälfte der Patienten zwei Wochen nach Absetzen an.
| Risiken ggf. Vorsichtsmaßnahmen |
Bei 50 % aller Patienten traten Nebenwirkungen in Form von Hautreizungen verschiedener Art im behandelten Bereich auf. Brennen, Jucken und Hautrötung traten sehr häufig auf und verschwanden in der Regel innerhalb einer Woche. Erhöhte Empfindlichkeit in der Haut und Prickeln sowie Hyperästhesie wurden ebenso wie lokale Unverträglichkeit gegenüber Alkohol häufig beobachtet. Unter den häufigen Nebenwirkungen finden sich auch Follikulitis, Akne und Herpes simplex (Herpes, Fieberbläschen, Eczema herpeticatum [Kaposi varicelliforme Eruption]).
In klinischen Untersuchungen kam es in 0,8 % zu Lymphadenopathien. In der Mehrzahl handelte es sich um Infektionen, die unter einer angemessenen Antibiotikabehandlung abklangen. Bei transplantierten, mit Immunsuppressiva behandelten Patienten ist das Risiko der Entstehung eines Lymphoms erhöht; daher sind mit Tacrolimus behandelte Patienten, die eine Lymphadenopathie entwickeln, zu überwachen, um sicherzustellen, dass die Lymphadenopathie abklingt. Die Ätiologie ist zu klären. Kann die Krankheitsursache nicht eindeutig ermittelt werden oder liegt eine infektiöse Mononukleose vor, so ist die Unterbrechung der Behandlung mit Tacrolimus in Erwägung zu ziehen.
Die Auswirkungen der Behandlung auf das sich entwickelnde Immunsystem bei Kindern ist nicht bekannt. Impfungen sollten nicht während der Behandlung mit Tacrolimus verabreicht werden. Bei abgeschwächten Lebendimpfstoffen (z.B. gegen Masern, Mumps, Röteln, oder Kinderlähmung) beträgt die Karenzzeit 28 Tage, bei inaktivierten Impfstoffen (z.B. gegen Tetanus, Diphtherie, Keuchhusten oder Grippe) 14 Tage.
In einer Photokanzerogenitätsstudie wurden haarlose Albinomäuse chronisch mit Tacrolimussalbe und UV-Bestrahlung behandelt. Die mit Tacrolimussalbe behandelten Tiere zeigten eine statistisch signifikante Verkürzung der Zeitspanne bis zum Auftreten von Hauttumoren (Plattenepithelkarzinome) und eine erhöhte Anzahl von Tumoren. Inwieweit diese Befunde auf den Menschen übertragbar sind, ist unbekannt. Nach der Fachinformation des Herstellers sollte während der Behandlung mit Tacrolimus-Salbe die Haut möglichst nicht dem Sonnenlicht ausgesetzt werden. Die Anwendung von ultraviolettem (UV) Licht in Solarien sowie die Therapie mit UVB oder UVA in Kombination mit Psoralenen (PUVA) sollte vermieden werden. Der Arzt muss die Patienten über geeignete Lichtschutzmaßnahmen beraten (z.B. Vermeidung von Aufenthalt in der Sonne, Anwendung von Lichtschutzmitteln und Abdeckung der Haut mit entsprechender Kleidung).
Ob eine Behandlungsdauer von mehr als zwei Jahren mit dem Risiko einer lokalen, eventuell zu Infektionen oder kutanen Malignomen führenden Immunsuppression verbunden ist, ist nicht bekannt.
Hautpflegemittel dürfen innerhalb von zwei Stunden vor beziehungsweise nach Applikation von Tacrolimus nicht im gleichen Hautbereich angewendet werden. Über die gleichzeitige Verwendung anderer topischer Präparate und systemischer Steroide oder Immunsuppressiva liegen keine Erfahrungen vor. Die gleichzeitige systemische Verabreichung von CYP3A4-Hemmern (z.B. Erythromycin, Itraconazol, Ketoconazol und Diltiazem) bei Patienten mit ausgedehnter und/oder erythrodermischer Erkrankung sollte mit Vorsicht erfolgen.
Die Anwendung in der Schwangerschaft und Stillzeit sowie bei Patienten mit genetisch bedingten Schädigungen der Epidermisschranke (z.B. Netherton-Syndrom) und generalisierter Erythrodermie wird nicht empfohlen. Das Gleiche gilt für Okklusiwerbände. Der Kontakt mit Augen und Schleimhaut ist zu vermeiden. Die Salbe darf auf infizierten Hautstellen nicht angewendet werden.
Teriparatid
(zum Beispiel Forsteo®)
Beschluss vom: 21.11.2006
In Kraft getreten am: 24.03.2007
BAnz. 2007, S. 3.121
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Teriparatid ist zur Behandlung der manifesten Osteoporose bei postmenopausalen Frauen nur ein Mittel der zweiten Wahl. Die Verordnung bleibt lediglich definierten Ausnahmefällen vorbehalten. Hinsichtlich der Frakturrate hat Teriparatid gegenüber anderen Osteoporosemitteln, insbesondere Bisphosphonaten keine nachgewiesene Überlegenheit. Die Inzidenz von mit hoher Morbiditätslast verbundenen Hüftfrakturen wird nicht signifikant reduziert (siehe Fachinformation).
Teriparatid ist wegen der im Vergleich zu Bisphosphonaten bis zu 35-fach höheren Tagestherapiekosten in der Regel unwirtschaftlich.
Unter folgenden kumulativen Bedingungen ist eine Verordnung von Teriparatid möglich:
Die maximal zugelassene Behandlungsdauer von 18 Monaten darf nicht überschritten werden (siehe Fachinformation). Im amerikanischen Pflichttext wird der Hinweis gegeben, dass Teriparatid wegen der unsicheren Relevanz der Befunde von Osteosarkomen bei der Ratte für den Menschen nur für solche Patienten verordnet werden sollte, bei denen der mögliche Nutzen die möglichen Risiken überwiegt.
Während der Behandlung mit Teriparatid sollte für eine ausreichende Calcium- und Vitamin-D-Aufnahme gesorgt werden. Die parallele Kombination mit anderen Arzneimitteln zur Osteoporosetherapie ist unwirtschaftlich.
| Kosten (Stand: 15.01.2007) |
Die pharmakodynamisch unterschiedlichen Therapiealternativen zur Behandlung der manifesten Osteoporose stellen sich kostenmäßig wie folgt dar:
| Arzneimittelwirkstoff | Tagestherapiekosten | Kosten für 78 Wochen |
| Teriparatid | 20,28 Euro | 11.072,- Euro |
Forsteo enthält in dem mit 3 ml vorgefüllten Injektor 750 μg Teriparatid. Die empfohlene Tagesdosierung von 20 μg wird einmal täglich in das subkutane Fettgewebe des Oberschenkels oder der Bauchdecke appliziert. Der Injektor ist für eine 28-tägige Behandlung vorgesehen. Somit werden von den 750 μg lediglich 560 μg verbraucht, d. h. 25 % der Substanz verbleiben im Injektor beziehungsweise in den Einmalnadeln.
| Wirkstoffgruppe | Tagestherapiekosten | Kosten für 78 Wochen |
| Bisphosphonate | 0,56 Euro bis 1,70 Euro | 305,- Euro bis 928,- Euro |
| Wirkstoffgruppe | Tagestherapiekosten | Kosten für 78 Wochen |
| SERM | 1,52 Euro | 830,- Euro |
Teriparatid wurde von der europäischen Zulassungsbehörde EMEA im Juli 2003 zur Behandlung der manifesten Osteoporose bei postmenopausalen Frauen für eine Therapiedauer von maximal 18 Monaten zugelassen. Nach Fachinformation ist eine signifikante Reduktion der Inzidenz (zu 80 Prozent asymptomatisch verlaufender) vertebraler Frakturen nachgewiesen. Die Inzidenz von Hüftfrakturen (verbunden mit einer hohen Morbiditätslast wie einer 20 % Mortalität und einer hohen nachfolgenden Pflegebedürftigkeit) wurde nichtsignifikant reduziert.
| Wirkungen |
Teriparatid ist das rekombinant hergestellte, biologisch aktive N-terminale Fragment (rhPTH 1-34) des humanen endogenen Parathormons [PTH (1-84)]. Die einmal tägliche subkutane Gabe von Teriparatid erhöht den Anbau von neuem Knochengewebe auf trabekuläre und kortikale Knochenoberflächen (am Endost und am Periost) durch stärkere Stimulation der Osteoblasten-Aktivität im Vergleich zur Stimulation der Osteoklasten-Aktivität. Im Gegensatz dazu können kontinuierliche supraphysiologische Spiegel von endogenem Parathormon wie beim Hyperparathyreodismus zu einer Schädigung des Skelettsystems führen, da die Knochenresorption stärker stimuliert wird als der Knochenanbau.
Beim Menschen führt der anabole Effekt von Teriparatid zu einem Anstieg der Knochenmasse, einem Anstieg der Marker für Knochenanbau und -resorption und einer Zunahme der Knochenstärke.
| Wirsamkeit |
Vier Hauptstudien der Phase III wurden zur Zulassung durchgeführt, davon allerdings nur zwei mit der zugelassenen Dosis von 20 μg. Von diesen beiden bezieht sich nur eine auf die zugelassene Population, nämlich postmenopausale Frauen.
Alle Studien wurden am 8. Dezember 1998 gestoppt, weil in einer Rattenstudie ein gehäuftes Auftreten von Osteosarkomen gefunden wurde. Den Patienten wurde angeboten, die Studie als Nachbeobachtung fortzuführen.
In der maßgeblichen Zulassungsstudie wurden 9347 Patientinnen gescreent und 1637 postmenopausale Frauen mit manifester Osteoporose, d. h. mindestens einer moderaten osteoporotischen Fraktur (25-40 % Abnahme der Wirbelkörperhöhe) oder zwei milden Frakturen (20 % Abnahme) und zusätzlich einer Knochendichte von -1 SD der Hüfte oder des Lumbalbereichs randomisiert. Somit wurden nach Angaben der FDA 82 % der gescreenten Patientinnen nicht eingeschlossen, ohne dass die Gründe dafür nachvollziehbar sind. Die Wirbelsäule wurde zur Erfassung von Frakturen bei Studienbeginn und nach zwei Jahren bzw. bei Studienende geröntgt. Extravertebrale Frakturen wurden bei klinischer Symptomatik radiologisch verifiziert.
80 % der eingeschlossenen randomisierten Patientinnen konnten ausgewertet werden. Verglichen wurde mit einer Placebobehandlung. Alle Patientinnen erhielten als Supplement 400 bis 1200 IU Vitamin D und ca. 1000 mg Calcium. Unter Teriparatid traten während einer mittleren Behandlungsdauer von 19 Monaten 22/444 (5 %) neue Wirbelkörperfrakturen auf gegenüber 64/448 (14 %) unter Placebo (p < 0,001). Die absolute Risikoreduktion (ARR) liegt bei 9,3 %, das relative Risiko (RR) bei 0,35 (95%iges Konfidenzintervall 0,22; 0,55). Es müssten 11 Patientinnen (95%iges Konfidenzintervall 8-18) 19 Monate behandelt werden, um eine morphometrische Fraktur zu verhindern.
Ein prädefinierter sekundärer Endpunkt war auch die Auswertung nichttraumatischer nichtvertebraler Frakturen. 30/544 Patienten (5,5 %) erlitten solche Frakturen unter Placebo und 14/541 (2,5 %) unter Teriparatid, ARR 3,0 %, RR 0,47 (95%iges Konfidenzintervall 0,25-0,88). Es müssten also 19 Monate lang 33 Patientinnen behandelt werden, um eine nichttraumatische extravertebrale Fraktur zu verhindern. Wegen der geringen Anzahl der Frakturen insgesamt sind Aussagen bezüglich der Lokalisationen der Frakturen nicht möglich.
Nimmt man alle nichtvertebralen Frakturen zusammen, beträgt die absolute Risikoreduktion 3,4 % oder es müssten 29 Patientinnen behandelt werden, um eine Fraktur zu verhindern (95%iges Konfidenzintervall 15; 469).
Ein exakter Zeitbezug zu vertebralen Frakturraten ist nicht herstellbar. Adäquate Berechnungen sind von daher nicht möglich.
Die mittels DXA-gemessene Knochenmineraldichte stieg im Behandlungszeitraum unter Placebo versus Teriparatid signifikant
Insgesamt zeigt die Auswertung der Knochenmineraldichte bis auf den Lumbalbereich keinen größeren Anstieg unter Teriparatid als unter einer Behandlung mit 10 mg Alendronat über 12 bis 24 Monate.
Der im Rahmen der Zulassungsstudie miterfasste Lebensqualitätsparameter Rückenschmerz kann nicht als Verbesserung unter einer Therapie mit Teriparatid herangezogen werden, sondern muss in die Nebenwirkungserfassung einbezogen werden.
Die Ergebnisse der Lebensqualitätsuntersuchungen konnten keine bedeutsamen Verbesserungen, auch nicht unter Osteoporosekorrelierten Aspekten, für Teriparatid aufzeigen.
Direkt vergleichende Studien zur Frakturrate mit der zugelassenen Dosierung zu anderen therapeutischen Optionen fehlen. Es gibt eine Studie, in der die doppelte Dosis von Teriparatid mit Alendronat verglichen wurde.
Relatives Risiko im Vergleich zu Placebo bei Patientinnen mit schwerer Osteoporosis:
| Wirkstoff | Morphometrische Wirbelsäulenfrakturen | Hüftfrakturen | Nichtvertebrale Frakturen |
| Alendronat | 0,53 (0,41 bis 0,68) | 0,49 (0,24 bis 1,01) | 0,81 (0,65 bis 1,01) |
| Etidronat | 0,43 (0,20 bis 0,91) | 0,50 (0,05 bis 5,34) | 1,04 (0,64 bis 1,69) |
| Risedronat | 0,63 (0,51 bis 0,78) | 0,60 (0,42 bis 0,88) | 0,67 (0,50 bis 0,90) |
| Raloxifen | 0,69 (0,56 bis 0,86) | No Data | No Data |
| Teriparatid | 0,35 (0,22 bis 0,55) | 0,50 (0,09 bis 2,73) | 0,65 (0,43 bis 0,98) |
| HRT | 0,58 (0,26 bis 1,30) | No Data | 0,67 (0,12 bis 3,93) |
| Relatives Risiko (95% Cl) | |||
Nach: M. Stevenson, M. Lloyd Jones, E. De Nigris et al.: A systematic review and economic evaluation of alendronate, etidronate, risedronate, raloxifene and teriparatide for the prevention and treatment of postmenopausal osteoporosis. Health Technol. Assess. 9 (2005) Nr. 22.
| Risiken ggf. Vorsichtsmaßnahmen |
Teriparatid ist kontraindiziert bei vorbestehender Hyperkalzämie, ungeklärter Erhöhung der alkalischen Phosphatase, schwerer Niereninsuffizienz, bei Kindern und Jugendlichen mit offenen Epiphysen, vorausgegangener Strahlentherapie des Skeletts sowie bei metabolischen Knochenkrankheiten, wie z.B. Hyperparathyreoidismus oder Morbus Paget.
Bei 3 % der Studienpatientinnen wurde eine Erhöhung der Serum-Calciumspiegel über 11 mg/dl (~ 2,7 mmol/l) beobachtet, die nach 16 bis 24 Stunden wieder auf den Ausgangswert zurückfiel, außerdem ein geringer Anstieg der Calciumausscheidung im Urin. Klinische Symptome einer Hyperkalzämie oder urie traten jedoch nicht auf, sodass eine regelmäßige Überwachung der Calciumspiegel während der Therapie nicht erforderlich ist. Teriparatid wurde bei Patienten mit einer bestehenden Urolithiasis nicht untersucht. Teriparatid muss bei Patienten mit einer derzeit oder vor kurzem bestehenden Urolithiasis mit Vorsicht angewendet werden, da die Möglichkeit bestehen könnte, dass sich dieser Zustand verschlechtert.
Vereinzelt wurden während der ersten Anwendungen Episoden einer orthostatischen Hypotonie innerhalb von 4 Stunden nach der Injektion beschrieben. Gelegentliche Wadenkrämpfe wurden von 3 % der behandelten Frauen berichtet (Placebo: 1 %).
Die in Studien zur Toxizität beobachteten erhöhten Inzidenzen von Osteosarkomen bei Ratten sind im Hinblick auf die klinische Relevanz unklar. Die Firma Lilly hat deshalb auf Anordnung der Zulassungsbehörden zur weiteren Bewertung des kanzerogenen Potentials Langzeitbeobachtungen zugesagt. Bisher wurde ein Fall eines Osteosarkoms unter der Behandlung beobachtet.
Wechselwirkungen mit anderen Arzneimitteln wurden nicht beobachtet. Da Teriparatid vorübergehend den Serumcalciumspiegel erhöht, sollte das Präparat bei digitalisierten Patienten jedoch nur mit Vorsicht angewandt werden.
Thiazolidindione (Glitazone )
(z.B. Pioglitazon (Actos®) und Rosiglitazon (Avandia®))
Beschluss vom: 03.05.2001
In Kraft getreten am: 24.08.2001
BAnz. 2001, S. 18.422/ 18.423
| Indikation |
Rosiglitazon (Avandia®) und Pioglitazon (Actos®) erhielten im Juli bzw. Oktober 2000 die europäische Zulassung und sind seit 19.07.2000 bzw. 01.11.2000 in Deutschland im Handel. Sie sind zugelassen zur oralen Kombinationsbehandlung des Typ 2 Diabetes mellitus bei Patienten, deren Blutzuckerkontrolle trotz einer oralen Monotherapie mit maximal verträglichen Dosen von Metformin oder Sulfonylharnstoffen ungenügend ist:
Glitazone sind zur Monotherapie nicht zugelassen; die Kombinationstherapie mit Insulin ist kontraindiziert. Über Dreifachkombinationen mit anderen oralen Antidiabetika liegen keine Erfahrungen vor.
| Wirkungen |
Thiazolidindione (Glitazone) aktivieren den nukleären PPARy (peroxisomal proliferatoractivated receptor gamma), der überwiegend im Fettgewebe exprimiert wird. Die Insulinwirkung wird verbessert, indem die Transkription von Genen der Adipozyten-Differenzierung sowie des Lipid- und Glukose-Metabolismus gesteigert wird. Die Insulin-Resistenz wird reduziert. Die Blut-Glukose-Konzentrationen gehen zusammen mit der Konzentration zirkulierenden Insulins zurück.
| Wirksamkeit |
In klinischen Studien reduzierte die Kombination von Thiazolidindionen mit Metformin bzw. Sulfonylharnstoff-Derivaten die Glukose-Konzentration. Auch bei unbefriedigender Stoffwechseleinstellung mit Sulfonylharnstoffen, Metformin, Insulin oder einer Kombination dieser Wirkstoffe kann die zusätzliche Gabe von Thiazolidindionen zu einer Reduktion von Serum-Konzentrationen von Glukose, Insulin und HbA,c führen. Etwa ein Viertel der Patienten spricht nicht durch Senkung der Blut-Glukose-Konzentration auf diese neuen Arzneimittel an. Diese Non-Responder haben vorrangig eine verminderte sekretorische Kapazität des Pankreas.
Die Wirkung von Thiazolidindionen auf den Lipidstoffwechsel ist ausgeprägt. Da sie die Lipolyse von Triglyceriden in verylowdensity-Lipoproteinen (VLDL) steigern, reduzieren sie die Triglycerid- und erhöhen die HDL-Cholesterin-Spiegel. Die Kombination der beschriebenen Effekte auf das kardiovaskuläre Risiko kann beim gegenwärtigen Stand der Erkenntnisse nicht vorhergesagt werden. Ergebnisse von Langzeitstudien mit Thiazolidindionen zu Metabolismus und Atherosklerose stehen aus.
| Risiken ggf. Vorsichtsmaßnahmen |
Über die Sicherheit der Thiazolidindion-Medikation wird erst eine Langzeitanwendung Aufschluss geben.
Troglitazon (Rezulin®) wurde in den U.S.A. im März 2000 vom Markt genommen, nachdem Leberfunktionsstörungen bei etwa 2 % der mit Troglitazon behandelten Patienten beobachtet und der amerikanischen Zulassungsbehörde 61 Todesfälle und 7 Lebertransplantationen im Zusammenhang mit Troglitazon berichtet wurden, in Großbritannien war es 1997 nach nur wenigen Wochen zurückgezogen worden. Zu Rosiglitazon und Pioglitazon liegen - auch aus Deutschland - Einzelbeobachtungen über schwere Leberschäden bzw. hepatozelluläre Dysfunktion vor. Ein Monitoring der Leberfunktion ist erforderlich. Derzeit wird dazu geraten, die Therapie bei Patienten mit erhöhten Serumaktivitäten der Leberenzyme (Alanin-Aminotransferase = Glutamat-Pyruvat-Transaminase >2,5-Faches der oberen Normgrenze) oder bei jedem anderen Anzeichen einer Lebererkrankung nicht zu beginnen.
Wegen der häufigen Gewichtszunahme (ca. 4 bis 6% im ersten Jahr) bedarf das Körpergewicht der Kontrolle. Als Ursache ist unter Sicherheitsaspekten die Flüssigkeitsretention wichtiger als die Zunahme des Fettgewebes. Eine Herzinsuffizienz kann durch Thiazolidindione ausgelöst bzw. eine bestehende Herzinsuffizienz verschlechtert werden. Thiazolidindione sind daher bei allen Graden der Herzinsuffizienz kontraindiziert. Eine erhöhte Inzidenz von Herzinsuffizienz wurde im Rahmen klinischer Studien, bei denen Rosiglitazon in Kombination mit Insulin verwendet wurde, beobachtet. Die Kombination von Rosiglitazon mit Insulin ist deshalb kontraindiziert. Mit einem weiterhin erhöhten Risiko der Flüssigkeitsretention mit Ödembildung ist bei gleichzeitiger Gabe nichtsteroidaler Antiphlogistika (NSAR) zu rechnen, deshalb müssen Patienten engmaschig kontrolliert werden.
Die Hämoglobinkonzentration im Blut kann unter der Therapie mit Thiazolidindionen abnehmen, am ehesten als Folge der Flüssigkeitsretention.
Weitere Interaktionen der Thiazolidindione mit anderen Arzneistoffen sind nicht hinreichend untersucht.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Entsprechend der Zulassung und im Hinblick auf die Nebenwirkungen kommt Thiazolidindionen derzeit nur ein limitierter Einsatzbereich zu; es handelt sich um eine Untergruppe von Diabetespatienten, für die das therapeutische Potential von Metformin bzw. Sulfonylharnstoffen ausgeschöpft ist;und die keiner Insulinbehandlung bedürfen.
Die langfristigen Vorteile einer Therapie mit Thiazolidindionen wurden nicht nachgewiesen. Das gilt insbesondere für die bei Diabetes relevanten Endpunkte Mikro-/Makroangiopathie oder kardiovaskuläre bzw. Gesamt-Mortalität.
Voraussetzungen für den wirtschaftlichen Einsatz einer medikamentösen Therapie ist die konsequente Einhaltung einer entsprechenden Diät (vgl. auch Arzneimittel-Richtlinie Ziffer 18 in Verbindung mit 6 und 10 der Richtlinie).
| Kosten |
Eine Therapie mit Rosiglitazon wird üblicherweise mit 4 mg/Tag eingeleitet. In Kombination mit Metformin kann die Dosis nach 8 Wochen auf 8 mg Rosiglitazon/Tag sofern erforderlich - erhöht werden. Bei Kombination mit Sulfonylharnstoffen liegen keine Erfahrungen mit höheren Tagesdosen als 4 mg Rosiglitazon vor. Pioglitazon in Kombination mit Metformin oder Sulfonylharnstoffen kann in einer Dosis von 15 mg oder 30 mg einmal täglich angewendet werden.
| Wirkstoff | Dosierung | Jahrestherapiekosten d. Monotherapie | Jahrestherapiekosten d. Kombinationstherapie mit | |
| Metformin | Sulfonylharnstoffen | |||
| Rosiglitazon | 4 mg/Tag | 582,87 Euro | 752,62 Euro | 681,55 Euro |
| 8 mg/Tag | 891,69 Euro | 1.061,44 Euro | 1.160,12 Euro | |
| Pioglitazon | 15 mg/Tag | 582,87 Euro | 752,62 Euro | 681,55 Euro |
| 30 mg/Tag | 892,20 Euro | 1.061,95 Euro | 1.160,63 Euro | |
Zusätzlich sind Arzneikosten für Metformin bzw. Sulfonylharnstoffe zu berücksichtigen sowie die indirekten Behandlungskosten durch das empfohlene Monitoring der Leberfunktion.
Tibolon
(z.B. Liviella®)
Beschluss vom: 26.02.2002
In Kraft getreten am: 22.06.2002
BAnz. 2002, Nr. 112 vom 21.06.2002 S. 13.577
| Indikation |
Tibolon ist in der Bundesrepublik Deutschland seit 1999 zur Behandlung klimakterischer Beschwerden als Folge des natürlichen und therapeutischen Eintritts der Menopause zugelassen; im Vereinigten Königreich und in den Niederlanden besteht eine Zulassung seit den achtziger Jahren.
Die Behandlung mit Tibolon sollte vorzugsweise nicht früher als zwölf Monate nach der letzten natürlichen Menstruationsblutung beginnen. Bei einer früheren Einnahme kann die Häufigkeit des Auftretens unregelmäßiger Blutungen erhöht sein. Vor Wechsel von einem anderen Präparat zur Hormonsubstitution auf Tibolon ist zu empfehlen, mit einem Gestagen eine Abbruchblutung herbeizuführen.
Die tägliche Dosierung beträgt 2,5 mg.
| Wirkungen |
Das synthetische Steroid Tibolon, ein 19-Testosteron-Derivat, das strukturell dem Norethisteron ähnelt, besitzt, insbesondere über seine aktiven Metaboliten, estrogene, gestogene und schwach androgenanabole Wirkungen.
Da Tibolon und insbesondere seine Metaboliten infolge ihrer zusätzlichen gestagenen Wirkung das Endometrium nicht stimulieren, ist anders als bei einer reinen Estrogen-Monotherapie die zusätzliche Gabe eines Gestagens bei Frauen mit einem intakten Uterus nicht erforderlich. Es besteht daher, wie bei einer Kombinationsbehandlung keine Notwendigkeit des regelmäßigen Auslösens von Abbruchblutungen. Nach oraler Einnahme wird Tibolon rasch metabolisiert, wobei auch Verbindungen entstehen, die zur Wirkung von Tibolon beitragen. Pharmakologisch besitzt die Muttersubstanz estrogene und androgenanabole Eigenschaften, zwei der entstehenden Metaboliten wirken überwiegend estrogen, während der dritte Metabolit in stärkerem Maße als Tibolon und die übrigen Verbindungen gestagene, androgenanabole und estrogene Eigenschaften aufweist.
| Wirksamkeit |
In klinischen Studien reduziert Tibolon in einer Dosierung von 2,5 mg täglich aufgrund seiner estroqenen Wirkungen klimakterische Beschwerden wie Hitzewallungen, Schweißausbrüche und Urogenitalatrophien vergleichbar gut wie die klassische Estrogen-Gestagen-Kombination (z.B. 2 mg 1713-Estradiol und 1 mg Norethisteronacetat).
Die Rate an Durchbruch- und Schmierblutungen ist zunächst um 50 % geringer, unterscheidet sich nach sechs Zyklen jedoch nicht mehr signifikant zwischen den Behandlungsgruppen.
Es treten signifikant weniger Brustschmerzen auf.
Eine weitere größere vergleichende Studie, die allerdings nicht verblindet war, zeigte ebenfalls in den ersten Monaten eine verminderte vaginale Blutung unter Tibolon, während die symptomatische Besserung der Beschwerden in beiden Gruppen vergleichbar war.
Im Vergleich zur Standardtherapie zeigte bezüglich einer Libido-Steigerung der Gesamtscore der McCoy-Skala keine signifikante Überlegenheit der Tibolontherapie nach 24 oder 48 Wochen. Lediglich zwei Einzelitems wiesen eine signifikante Differenz auf. Die Studie weist nicht unerhebliche Mängel auf. So wurde beispielsweise nicht die vollständige McCoy-Skala eingesetzt, die Gruppen unterschieden sich bereits zu Beginn der Untersuchung in einigen Items signifikant und es wurden lediglich 60% aller Patientinnen ausgewertet.
Vergleichende Studien mit Standardtherapien zur Reduktion von Frakturen bei Osteoporose fehlen.
| Risiken ggf. Vorsichtsmaßnahmen |
Es gelten die bei der Gabe von Oestrogen-Gestagen-Kombinationen bekannten Risiken und Vorsichtsmaßnahmen.
Absolute Kontraindikationen sind: Überempfindlichkeit gegen Tibolon bestehende hormonabhängige Tumoren, z.B. Brustkrebs, oder Verdacht darauf kardiovaskuläre oder zerbrovaskuläre Störungen, z.B. Thrombophlebitis, Thromboembolie, auch in der Anamnese ungeklärte Leberfunktionsstörungen Lebertumoren Schwangerschaft Stillzeit.
An unerwünschten Arzneimittelwirkungen (UAW s) sind beschrieben: uterine Blutungen (auch Schmierblutungen), insbesondere in den ersten Behandlungsmonaten Bauchschmerzen, insbesondere in den ersten Behandlungsmonaten Brustspannen, insbesondere in den ersten Behandlungsmonaten Kopfschmerzen Migräne Ödeme Benommenheit Pruritus Gewichtszunahme Übelkeit Ausschlag depressive Verstimmungen
Hirsutismus.
Eine strenge Indikationsstellung ist bei Hypercholesterinämie erforderlich.
Es kommt zu einer Erhöhung der fibrinolytischen Aktivität des Blutes, so dass es die Wirkung von Antikoagulanzien verstärken kann.
Beobachtet wird ein Absinken des Plasma-HDL-Cholesterins, der Triglyceride, des Lipoproteins (a). Die Bedeutung dieser bezüglich des Arterioskleroserisikos heterogenen metabolischen Veränderungen ist unklar.
Epidemiologische Langzeiterhebungen beziehungsweise Interventionsstudien zur Inzidenz und Mortalität kardiovaskulärer Erkrankungen stehen ebenso aus wie zu weiblichen Organneoplasien.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Symptome der Menopause sind in der Regel nicht lebensbedrohend, sie können aber die Lebensqualität beeinträchtigen. Die Mehrzahl der Frauen betrachten die Menopause als natürliches Ereignis. Eine routinemäßige Anwendung von Hormonen in der Menopause wird nicht empfohlen. Vielmehr sollte die Therapieentscheidung unter individueller Abwägung im Hinblick auf den Schweregrad der Symptome, die Art und Schwere von Begleiterkrankungen unter Einbeziehung von Präferenzen der betroffenen Frau getroffen werden.
Tibolon ist pharmakologisch keine Monotherapie, sondern eine Kombination aktiver Metabolite, und auch pharmakodynamisch oder zeitlich keine Innovation. Therapeutisch besteht Vergleichbarkeit zu Oestrogen-Gestagen-Kombinationen. Zur Osteoporose-Prävention bzw. Therapie besteht in Deutschland im Gegensatz zu vielen Kombinationspräparaten keine Zulassung.
Die Therapiedauer einer ausschließlich symptomatisch orientierten Behandlung menopausaler Beschwerden und damit auch von Tibolon sollte die Fünfjahresdauer nicht überschreiten.
Da die Libido durch eine Vielzahl von nichthormonellen Faktoren beeinflusst wird, ist eine detaillierte Diagnostik vor einer Hormontherapie unumgänglich. Abzuwägen ist auch, inwieweit Änderungen der Lebensführung wie diätetische Maßnahmen, körperliche Aktivität, Reduktion von Stress oder auch Einstellen des Rauchens, zur psychischen und physischen Stabilisierung von Frauen in der Menopause führt, bevor unter besonderer Beachtung des Schweregrads menopausaler Symptome eine medikamentöse Therapie in Betracht gezogen wird.
Die klinische Bedeutung der Senkung des HDL-Cholesterin-Spiegels ist derzeit unklar. Es bleibt im Gegenteil abzuklären, ob die HDL-Senkung durch Tibolon eine kardiovaskuläre Schutzfunktion möglicherweise sogar verschlechtert.
Besondere pharmakotherapeutische Vorteile von Tibolon gegenüber der Gabe einer klassischen Oestrogen-Gestagen-Kombination bei der Behandlung klimakterischer Beschwerden als Folge des natürlichen oder therapeutischen Eintritts der Menopause werden nicht gesehen. Tibolon ist beim gegenwärtigen Kenntnisstand auch unter dem Aspekt eines zwei- bis dreifach höheren Monatspreises nur bei nachgewiesener Unverträglichkeit gegenüber der Standardtherapie unter wirtschaftlichen Aspekten vertretbar.
| Kosten |
Liviella® 1 x 28 (NI) kostet 32,10 A 3 x 28 (N3) 86,25 Euro . Die Jahrestherapiekosten von Liviella® sind im Vergleich zur Standardtherapie mit Östrogen-Gestagen-Kombinationspräparaten um das Dreifache erhöht.
Preisvergleich für N1
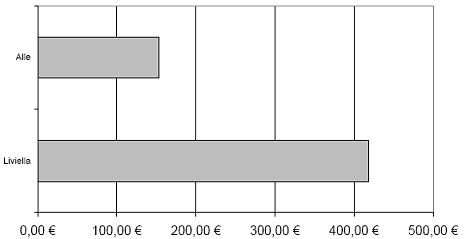
Preisvergleich für N3
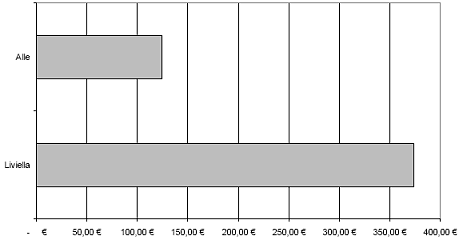
* preiswerte Importarzneimittel vorhanden
Vildagliptin
(aufgehoben durch BAnz. AT vom 08.06.2016 B1)
Zanamivir
(z.B. Relenza®)
Beschluss vom: 16.02.2000
In Kraft getreten am: 31.05.2000
BAnz. 2000, S. 10.094/ 10.095
| Indikation |
In Deutschland ist Zanamivir seit dem 1. Oktober 1999 zur Behandlung der Influenza A und B bei Erwachsenen und Jugendlichen ab 12 Jahren mit typischen Grippe-Symptomen, wenn Influenza in der Region zirkuliert, unter dem Namen Relenza® im Handel. Es ist ausschließlich zur Behandlung und nicht zur Prophylaxe zugelassen. Zanamivir wird zweimal täglich über fünf Tage inhaliert. Die Behandlung sollte so früh wie möglich innerhalb von 48 Stunden nach Einsetzen der ersten Symptome beginnen.
| Wirkungen |
Zanamivir ist ein selektiver, kompetitiver Hemmstoff des Enzyms Neuraminidase, das sich auf der Oberfläche der Influenza-Viren befindet. Die virale Neuraminidase spielt u.a. eine wichtige Rolle bei der Freisetzung von neu gebildeten Viren aus infizierten Zellen. Durch die Blockade kann das Virus sich nicht von der Zelle lösen und somit keine weiteren Zellen infizieren. Zudem scheint der Zugang der Viren durch den Mucus zur Oberfläche von Epithelzellen beeinträchtigt zu werden, so daß eine Virusinfektion anderer Zellen erschwert wird und die Replikation auf das Oberflächenepithel des Respirationstraktes begrenzt bleibt.
| Wirksamkeit |
In drei Placebokontrollierten Studien der Phase III mit über 1.500 Patienten waren ca. 1.160 mit Grippeviren infiziert, davon 89 % mit Influenza A und 11% mit Influenza B. Primärer Endpunkt der Untersuchungen war die Zeit bis zur Symptomerleichterung. Zwei Studien konnten eine signifikante Überlegenheit der Zanamivirbehandlung gegenüber Placebo nachweisen, während die größte Studie ebenso wie eine kleinere Phase II Untersuchung kein Signifikanzniveau erreichte. Eine kombinierte Analyse aller behandelten Patienten zeigte im Median eine Reduktion der subjektiven Grippesymptome um 1,5 Tage. Ein Behandlungserfolg bei Patienten mit afebrilem Krankheitsbild (< 37,8C) ist nicht dokumentiert worden. Risikopatienten, die eine erhöhte Komplikations- und Mortalitätsrate bei Influenza aufweisen, wie Säuglinge, Kleinkinder, ältere Personen, Patienten mit prädisponierenden Grunderkrankungen wie eingeschränkte Lungenfunktion, angeborenen oder erworbenen Herzkrankheiten und Immundefekten, wurden nur in geringer Anzahl in die Untersuchung eingeschlossen, so daß valide Aussagen bezüglich der Wirksamkeit bei diesen besonders gefährdeten Patientengruppen fehlen.
Das Ausmaß der Resistenzentwicklung bei klinischer Anwendung ist noch unklar.
| Risiken ggf. Vorsichtsmaßnahmen |
Die Substanz erwies sich als gut verträglich. Die häufigsten unerwünschten Ereignisse betrafen typische Anzeichen und Symptome der Influenza. Allerdings liegen bisher keine ausreichenden Erfahrungen zur Verträglichkeit bei Patienten über 65 und unter 12 Jahren sowie insbesondere mit chronisch pulmonalen Erkrankungen vor.
Kontraindiziert ist Zanamivir bei bekannter Überempfindlichkeit gegen einen der Bestandteile des Medikamentes. Während der Stillzeit wird die Anwendung nicht empfohlen. Auch während der Schwangerschaft ist Vorsicht geboten.
Für Kinder unter 12 Jahren ist die Wirksamkeit und Verträglichkeit bisher nicht ausreichend geprüft.
| Empfehlungen zur wirtschaftlichen Verordnungsweise |
Klinisch ist z. Zt. zu Beginn eines fieberhaften Infekts die exakte Diagnose einer Influenza A oder B unter Praxisbedingungen nicht zu stellen. Voraussetzung für den Einsatz von Zanamivir ist jedoch die frühzeitige klinische Diagnose einer echten Influenza A oder B.
Bisher ist, anders als für die Impfung, eine Reduktion von Komplikationen und Sterblichkeit in der Gruppe der Risikopatienten nicht belegt. Die Wirksamkeit setzt einen fehlerfreien Gebrauch des Diskhalers voraus, was eine hinreichende Schulung des Patienten bedingt. Die Daten zur Influenza B sind spärlich.
Die Behandlung mit Zanamivir ist, außer in besonderen Fallkonstellationen (z.B. bei Patienten, die aufgrund individueller Unverträglichkeit nicht geimpft werden können oder bei einer Epidemie mit nachgewiesenem Virusshift) nicht zweckmäßig.
Die fünftägige Anwendung von Zanamivir (zweimal täglich zwei Inhalationen) dient der Behandlung der aktuellen Infektion und bietet, anders als die Impfstoffe, keinen Schutz vor einer erneuten Infektion in der gleichen Saison.
Zur Prophylaxe ist Zanamivir in Deutschland nicht zugelassen und kann eine Grippeschutzimpfung nicht ersetzen.
Kosten der einmaligen Behandlung
| Packungsgröße | Kosten pro Behandlung (Euro) | |
| Relenza® (nur Behandlung) | 5 x 4 Einzeldosen | 29,75 |
| weiter . | |