
umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (17)
 | zurück |
|
B.40.bis Invitro-Prüfung auf hautätzende Wirkung: Prüfmethode mit rekonstruierter humaner Epideris (RHE) 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
1. Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie 431 (2016). Als Hautverätzung wird das Auslösen einer irreversiblen Hautschädigung, d. h. einer sichtbaren, bis in das Corium reichenden Nekrose der Epidermis nach Applikation einer Prüfchemikalie [im Sinne der Definition des Global harmonisierten Systems zur Einstufung und Kennzeichnung von Gefahrstoffen der UN (GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) 1] bezeichnet. Diese aktualisierte Prüfmethode B.40bis besteht in einem Invitro-Verfahren zur Identifizierung nicht ätzender und ätzender Stoffe und Gemische nach dem UN GHS und der CLP-Verordnung. Sie ermöglicht auch eine partielle Unterkategorisierung ätzender Chemikalien. 2. Die Bewertung des Hautverätzungspotenzials von Chemikalien erfolgte in der Regel unter Verwendung von Labortieren (PM B.4, entsprechend OECD TG 404, ursprünglich angenommen im Jahr 1981 und geändert in den Jahren 1992, 2002 und 2015) (2). Neben dieser Prüfmethode B.40bis wurden noch zwei weitere Invitro-Prüfmethoden zur Untersuchung des Verätzungspotenzials von Chemikalien als Prüfmethode B.40 (entsprechend OECD TG 430) (3) und PM B.65 (entsprechend OECD TG 435) validiert und angenommen (4). Außerdem wurde die Invitro-Prüfmethode B.46 (entsprechend OECD TG 439) (5) als Methode zur Untersuchung des Hautreizungspotenzials angenommen. In einem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von Hautverätzungen und -reizungen werden mehrere Module beschrieben, in denen Informationsquellen und Analyseinstrumente zu Gruppen zusammengefasst werden. Sie enthalten i) Leitlinien dazu, wie vorhandene Daten aus Untersuchungen und aus anderen Quellen integriert und verwendet werden können, um das Hautreizungs- und das Hautverätzungspotenzial von Chemikalien zu bewerten, und ii) einen Vorschlag zur Durchführung ggf. erforderlicher weiterer Untersuchungen (6). 3. Mit dieser Prüfmethode werden Hautverätzungen mit dem Endpunkt der menschlichen Gesundheit untersucht. Für die Prüfmethode wird (aus menschlichen, nicht transformierten Keratinozyten) rekonstruierte humane Epidermis (RhE) verwendet, die die histologischen, morphologischen, biochemischen und physiologischen Eigenschaften der menschlichen Oberhaut, d. h. der Epidermis weitgehend widerspiegelt. Die entsprechende OECD-Prüfrichtlinie wurde ursprünglich 2004 angenommen und 2013 unter Berücksichtigung weiterer Prüfmethoden unter Verwendung der RhE-Modelle und der Möglichkeit zur Verwendung der Methoden zur Unterstützung der Unterkategorisierung ätzender Chemikalien zunächst im Jahr 2013 und anschließend nochmals im Jahr 2015 unter Verweis auf das IATA-Leitliniendokument und unter Einführung der Verwendung eines alternativen Verfahrens zur Messung der Viabilität aktualisiert. 4. Bei dieser Prüfmethode kommen vier validierte im Handel erhältliche RhE-Modelle zum Einsatz. Nach Vorvalidierungsstudien (7) wurde an zwei dieser im Handel erhältichen Prüfmodelle - dem EpiSkinTM-Standardmodell (SM) und dem EpiDermTM Skin Corrosivity Test (SCT) (EPI-200) (im Folgenden,validierte Referenzmodelle" (VRM) - eine förmliche Validierungsstudie zur Bewertung der Hautverätzung (8)(9)(10) durchgeführt (11) (12). Die Ergebnisse dieser Studien führten zur Empfehlung, dass die beiden genannten VRM für rechtliche Zwecke zur Unterscheidung zwischen ätzenden (C) und nicht ätzenden (NC) Stoffen verwendet werden können und dass mit EpiSkinTM zudem die Unterkategorisierung ätzender Stoffe unterstützt werden kann (13)(14)(15). Zwei weitere im Handel erhältliche Invitro-Verfahren auf der Grundlage von RhE-Modellen zeigten nach Validierungsstudien auf Basis der Leistungsstandards ähnliche Ergebnisse wie das VRM EpiDermTM; (16)(17)(18): die RhE-Modelle SkinEthicTM 2 und epiCS® (früher auch als EST-1000 bezeichnet), die auch für rechtliche Zwecke zur Unterscheidung zwischen ätzenden und nicht ätzenden Stoffen verwendet werden können (19)(20). Post-Validierungsstudien der Hersteller des RhE-Modells in den Jahren 2012 bis 2014 mit einem verbesserten Protokoll, bei dem Interferenzen durch unspezifische MTT-Reduktion durch die Prüfchemikalien korrigiert werden, ermöglichten zum einen eine bessere Unterscheidung zwischen den Wirkungen C und NC und unterstützten zum anderen die Unterkategorisierung ätzender Chemikalien (21)(22). Die in der Post-Validierung mit dem EpiDermTM SCT, der SkinEthicTM RHE und EpiCS® gewonnenen Daten wurden weiteren statistischen Analysen unterzogen, um alternative Vorhersagemodelle mit besserer Vorhersagefähigkeit für Unterkategorisierungen zu entwickeln (23). 5. Bevor eine vorgeschlagene ähnliche oder modifizierte Invitro-RhE-Prüfmethode zur Untersuchung auf Hautverätzungen außer den VRM für rechtliche Zwecke verwendet werden kann, sollten nach den Anforderungen der nach den Grundsätzen des OECD-Leitliniendokuments Nr. 34 (25) entwickelten Leistungsstandards (24) die Verlässlichkeit, die Relevanz (Genauigkeit) und die Grenzen des Verfahrens für den vorgeschlagenen Verwendungszweck bestimmt werden, um sicherzustellen, dass es mit den VRM vergleichbar ist. Die gegenseitige Anerkennung der Daten wird erst dann garantiert, wenn vorgeschlagene neue oder aktualisierte Prüfmethoden auf der Grundlage der Leistungsstandards überprüft und in die entsprechende Prüfrichtlinie aufgenommen wurden. Die in diese Prüfrichtlinie aufgenommenen Prüfmodelle können verwendet werden, um sowohl länderbezogene Anforderungen an die Prüfergebnisse der Invitro-Prüfmethode zur Untersuchung von Hautverätzungen zu erfüllen als auch die gegenseitige Anerkennung von Daten beanspruchen zu können. Begriffsbestimmungen 6. Es gelten die Begriffsbestimmungen in Anlage 1. Ausgangsüberlegungen 7. Diese Prüfmethode ermöglicht die Identifizierung nicht ätzender und ätzender Stoffe und Gemische nach dem UN GHS und der CLP-Verordnung. Außerdem unterstützt diese Prüfmethode die Zuordnung ätzender Stoffe und Gemische zur optionalen Unterkategorie 1A nach Maßgabe des UN GHS (1) sowie eine Kombination der Unterkategorien 1B und 1C (21)(22)(23). Eine Einschränkung bei dieser Prüfmethode besteht darin, dass bei Hautverätzungen wegen der begrenzten Auswahl gut bekannter Chemikalien der Unterkategorie 1C in Invivo-Untersuchungen nicht zwischen den Unterkategorien 1B und 1C nach dem UN GHS und der CLP-Verordnung unterschieden werden kann. Mithilfe der Prüfmodelle EpiSkinTM, EpiDermTM SCT, SkinEthicTM RHE und epiCS® können Unterkategorisierungen (d. h. Unterscheidungen zwischen 1A und 1B- und-1C gegenüber NC) vorgenommen werden. 8. In der Validierung zur Unterstützung der in diese Prüfmethode einbezogenen Prüfmodelle zur Identifizierung nicht ätzender und ätzender Chemikalien wurden zahlreiche Chemikalien (in erster Linie Stoffe) geprüft, und die empirische Datenbank der Validierungsstudie umfasste 60 Chemikalien aus zahlreichen Chemikalienklassen (8)(9)(10). Zum Nachweis der Sensitivität, der Spezifizität, der Genauigkeit und der laborinternen Reproduzierbarkeit des Versuchs zur Unterkategorisierung haben die Entwickler der Prüfungen Untersuchungen durchgeführt; die Ergebnisse dieser Untersuchungen wurden von der OECD geprüft (21)(22)(23). Auf der Grundlage der insgesamt verfügbaren Daten ist festzustellen, dass die Prüfmethode bei zahlreichen Chemikalienklassen und Aggregatzuständen (u. a. Flüssigkeiten, halbfeste Stoffe, Feststoffe und Wachse) anwendbar ist. Flüssigkeiten können wässrig oder nicht wässrig und Feststoffe in Wasser löslich oder unlöslich sein. Sofern möglich, sollten Feststoffe vor der Applikation zu Feinpulver gemahlen werden; eine weitere Vorbehandlung der Probe ist nicht nötig. Wenn nachgewiesen werden kann, dass die in diese Prüfmethode einbezogenen Prüfmodelle bei einer bestimmten Kategorie von Prüfchemikalien nicht anwendbar sind, sollten sie bei dieser Kategorie von Prüfchemikalien nicht verwendet werden. Diese Prüfmethode kann jedoch nicht nur bei Stoffen verwendet werden, sondern wird auch als für Gemische geeignet betrachtet. Da Gemische allerdings vielfältigen Kategorien zuzurechnen sein und unterschiedliche Zusammensetzungen haben können, und da gegenwärtig nur begrenzte Informationen über die Prüfung von Gemischen verfügbar sind, sollten Prüfmethoden, bei denen nachgewiesen werden kann, dass sie für eine bestimmte Kategorie von Gemischen nicht geeignet sind (beispielsweise durch ein Verfahren entsprechend dem in (26) vorgeschlagenen Verfahren), für die betreffende Kategorie von Gemischen nicht verwendet werden. Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist. Gase und Aerosole wurden bislang noch nicht in Validierungsstudien bewertet (8)(9)(10). Obwohl deren Prüfung mit der RhE-Technologie prinzipiell vorstellbar ist, erlaubt die vorliegende Prüfmethode das Untersuchen von Gasen und Aerosolen nicht. 9. Prüfchemikalien, die Licht im selben Spektrum absorbieren können wie MTT-Formazan, und Prüfchemikalien, die in der Lage sind, den lebenswichtigen Farbstoff MTT zu reduzieren (zu MTT-Formazan), können die Messungen der Gewebeviabilität stören und erfordern die Verwendung geeigneter Kontrollen zur Korrektur. Die Art der möglicherweise erforderlichen geeigneten Kontrollen hängt von der Art der durch die Prüfchemikalie ausgelösten Interferenzen und vom Verfahren zur Messung von MTT-Formazan ab (Nummern 25-31). 10. Diese Prüfmethode vermittelt keine geeigneten Informationen über Hautreizungen; mit PM B.46, die auf demselben RhE-Prüfsystem (allerdings mit einem anderen Protokoll) beruht, werden jedoch gezielt die gesundheitlichen Auswirkungen von Hautreizungen in vitro untersucht (5). Eine umfassende Evaluierung lokaler Auswirkungen auf die Haut nach einer einmaligen dermalen Exposition ist dem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA) zu entnehmen (6). Dieser IATA-Ansatz umfasst die Durchführung von Invitro-Prüfungen auf hautätzende Wirkung (wie sie in dieser Methode beschrieben werden) und auf Hautreizungen, bevor Prüfungen an lebenden Tieren in Betracht gezogen werden. Die Verwendung menschlicher Haut unterliegt anerkanntermaßen ethischen Überlegungen und Bedingungen auf nationaler und internationaler Ebene. Prinzip der Prüfmethode 11. Die Prüfchemikalie wird oberflächlich auf ein dreidimensionales RhE-Modell aufgetragen, das aus nicht veränderten humanen epidermalen Keratinozyten besteht, die zu einem mehrschichtigen, ausdifferenzierten Modell humaner Epidermis kultiviert wurden. Das Modell besteht aus geordneten Basal-, Stachel- und Körnerzellschichten und einer mehrlagigen Hornschicht (stratum corneum), die interzelluläre lamellare Fettschichten enthält, deren dominierende Lipidklassen dem in vivo gefundenen Lipidmuster entsprechen. 12. Diese RhE-Prüfmethode basiert auf der Voraussetzung, dass ätzende Chemikalien in der Lage sind, durch Diffusion oder Erosion die Hornhaut zu durchdringen, und darüber hinaus eine zytotoxische Wirkung auf die darunter liegenden Zellschichten ausüben. Die Zellviabilität wird durch Enzymkonversion des Vitalfarbstoffs MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolyl-Blau-Tetrazoliumbromid; CAS-Nummer 298-93-1] zu einem blauen Formazan-Salz gemessen, das nach seiner Extraktion aus Geweben quantifiziert wird (27). Ätzende Chemikalien werden anhand ihrer Fähigkeit erkannt, die Zellviabilität unter vorgegebene Schwellenwerte zu senken (Nummern 35 und 36). Die Prüfmethode auf der Grundlage von RhE-Modellen zur Untersuchung von Hautverätzungen hat sich bei der Prüfung von Kaninchen nach PM B.4 (2) als für die Vorhersage von Invivo-Hautkorrosionswirkungen geeignet erwiesen. Nachweis der Leistungsfähigkeit 13. Vor der routinemäßigen Anwendung eines der vier validierten RhE-Prüfmodelle nach dieser Prüfmethode sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Klassifizierung der zwölf in Tabelle 1 empfohlenen Leistungsstoffe nachweisen. Bei Verwendung der Methode zur Unterklassifizierung konnten zudem die richtigen Unterkategorien nachgewiesen werden. Wenn ein dort genannter Stoff nicht verfügbar ist bzw. wenn dies gerechtfertigt ist, kann ein anderer Stoff verwendet werden, für den geeignete Invivo- und Invitro-Referenzdaten verfügbar sind (z.B. aus der Liste der Referenzchemikalien (24)), sofern die in Tabelle 1 beschriebenen Auswahlkriterien angewendet werden. Tabelle 1 Liste der Leistungsstoffe 1
14. Im Rahmen des Nachweises der Leistungsfähigkeit sollte der Benutzer die vom Hersteller des Hautmodells spezifizierten Barriereeigenschaften der Gewebe nach Erhalt überprüfen. Dies ist besonders dann wichtig, wenn die Gewebe über große Entfernungen/Zeiträume transportiert werden. Sobald eine Methode erfolgreich etabliert und ihre Leistungsfähigkeit demonstriert wurde, ist diese Überprüfung nicht mehr routinemäßig erforderlich. Allerdings empfiehlt es sich auch bei routinemäßig angewandten Prüfmethoden, die Barriereeigenschaften in regelmäßigen Abständen zu kontrollieren. Verfahren 15. Im Folgenden werden die Elemente und die Verfahren der in dieser Prüfmethode verwendeten RhE-Prüfmodelle zur Bewertung von Hautverätzungen allgemein beschrieben. Die als zur Verwendung im Rahmen dieser Prüfmethode wissenschaftlich fundiert bewerteten und unterstützten RhE-Modelle (d. h. die Modelle EpiSkinTM (SM), EpiDermTM (EPI-200), SkinEthicTM RHE und epiCS®) (16)(17)(19)(28)(29)(30)(31)(32)(33) sind im Handel erhältlich. Standardarbeitsanweisungen (SOPs) für diese vier RhE-Modelle sind verfügbar (34)(35)(36)(37), und die jeweiligen wesentlichen Elemente der Prüfmethoden werden in Anlage 2 kurz beschrieben. Bei der Einführung und Verwendung eines dieser Modelle im Labor sollten die relevanten SOPs berücksichtigt werden. Die vier RhE-Prüfmodelle dieser Prüfmethode sollten die folgenden Anforderungen erfüllen: Elemente der RHE-Prüfmethode Allgemeine Bedingungen 16. Die Epithelschicht sollte aus normalen menschlichen Keratinozyten gebildet werden. Unter der funktionsfähigen Hornschicht (stratum corneum) sollten mehrere Lagen lebensfähiger Epithelzellen (Basalzellschicht, stratum spinosum, stratum granulosum) vorhanden sein. Die Hornschicht sollte mehrlagig sein und das zur Erzeugung einer funktionsfähigen Barriere essenzielle Lipidprofil aufweisen und muss robust genug sein, um das schnelle Eindringen zytotoxischer Referenzchemikalien wie Natriumdodecylsulfat (SDS) oder Triton X-100 zu verhindern. Die Barrierefunktion kann entweder durch Bestimmung der Konzentration, bei der eine Referenzchemikalie die Viabilität der Gewebe nach einer vorgegebenen Expositionsdauer um 50 % verringert (IC50), oder durch die Bestimmung der Expositionszeit bewertet werden, die erforderlich ist, um die Zellviabilität bei Anwendung der Referenzchemikalie in einer vorgegebenen festen Konzentration um 50 % zu reduzieren (ET50) (Nummer 18). Die Rückhalteeigenschaften des RhE-Modells müssen ausschließen, dass Material rund um die Hornschicht in lebensfähiges Gewebe eindringt und die Modellierung der Hautexposition beeinträchtigt. Das RhE-Modell sollte nicht mit Bakterien, Viren, Mykoplasma oder Pilzen kontaminiert sein. Funktionale Bedingungen Viabilität 17. Die Größenordnung der Viabilität der Gewebe wird mit der MTT-Prüfung bestimmt (27). Die lebensfähigen Zellen des RhE-Gewebemodells reduzieren den lebenswichtigen Farbstoff MTT zu einem blauen MTT-Formazan-Niederschlag, der dann mit Isopropanol (oder einem ähnlichen Lösungsmittel) aus dem Gewebe extrahiert wird. Die optische Dichte (OD) des Extraktionslösungsmittels allein sollte ausreichend gering sein, d. h. OD < 0,1. Das extrahierte MTT-Formazan kann durch eine Standard-(OD)-Absorptionsmessung oder mit einem HPLC/UPLC-Spektrometrieverfahren (38) quantifiziert werden. Die Anwender des RhE-Modells sollten sicherstellen, dass jede Charge des verwendeten Modells die vorgegebenen Kriterien für die Negativkontrolle erfüllt. Der Entwickler/Hersteller des Hautmodells sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für die OD-Werte der Negativkontrolle festlegen. Die Akzeptanzspanne der OD-Werte der Negativkontrolle für die vier validierten RhE-Prüfmodelle dieser Prüfmethode sind Tabelle 2 zu entnehmen. Bei Durchführung einer HPLC/UPLC-Spektrophotometrie sollten die in Tabelle 2 genannten OD-Spannen der Negativkontrolle als Akzeptanzkriterium für die Negativkontrolle verwendet werden. Es sollte dokumentiert werden, dass die mit der Negativkontrolle behandelten Gewebe über die gesamte Dauer der Exposition stabil bleiben (bzw. vergleichbare OD-Messungen ergeben). Tabelle 2 Akzeptanzspannen für OD-Werte der Negativkontrolle zur Kontrolle der Chargenqualität
Barrierefunktion 18. Das stratum corneum und die Zusammensetzung seiner Fette sollten das schnelle Eindringen bestimmter zytotoxischer Referenzchemikalien wie z.B. SDS oder Triton X-100 verhindern. Dies wird durch die Ermittlung von IC50 oder ET50 bestimmt (Tabelle 3). Die Barrierefunktion der einzelnen Chargen des verwendeten RhE-Modells sollte nach der Lieferung der Gewebe an den Endverwender vom Entwickler/Hersteller des RhE-Modells nachgewiesen werden (Nummer 21). Morphologie 19. Die histologische Untersuchung des RhE-Modells sollte unter Nachweis einer aus mehreren Lagen bestehenden und der menschlichen Hornschicht (epidermis) ähnelnden Struktur mit stratum basale, stratum spinosum, stratum granulosum und stratum corneum erfolgen; das Modell sollte ein Lipidprofil ähnlich dem Lipidprofil der menschlichen Epidermis aufweisen. Die histologische Untersuchung der einzelnen zum Nachweis einer geeigneten Morphologie der Gewebe verwendeten Chargen des RhE-Modells sollte jeweils nach der Lieferung der Gewebe an den Endverwender vom Entwickler/Hersteller des RhE-Modells nachgewiesen werden (Nummer 21). Reproduzierbarkeit. 20. Die Anwender der Prüfmethode sollten die Reproduzierbarkeit der Prüfmethoden mit den Positiv- und den Negativkontrollen über längere Zeit nachweisen. Außerdem sollte die Prüfmethode nur dann verwendet werden, wenn der Entwickler/Hersteller des RhE-Modells Daten vorlegt, die die Reproduzierbarkeit mit ätzenden und mit nicht ätzenden Chemikalien z.B. aus der Liste der Leistungsstoffe (Tabelle 1) über längere Zeit belegen. Bei Verwendung einer Prüfmethode zur Unterkategorisierung sollte zudem die Reproduzierbarkeit in Bezug auf Unterkategorien nachgewiesen werden. Qualitätskontrolle (QK) 21. Das RhE-Modell sollte nur dann verwendet werden, wenn der Entwickler/Hersteller nachweist, dass jede Charge des verwendeten Hautmodells bestimmten Freigabekriterien genügt, von denen die Kriterien der Viabilität (Nummer 17), der Barrierefunktion (Nummer 18) und der Morphologie (Nummer 19) die wichtigsten sind. Diese Daten werden den Anwendern der Prüfmethode zur Verfügung gestellt werden, damit diese sie in den Prüfbericht aufnehmen können. Nur mit freigegebenen Gewebechargen erzielte Ergebnisse können für eine zuverlässige Vorhersage für die Einstufung von Verätzungen akzeptiert werden. Der Entwickler/Hersteller des Hautmodells (oder - bei Verwendung eines hauseigenen Modells - der Prüfer) legt eine Akzeptanzspanne (oberer und unterer Grenzwert) für IC50 bzw. ET50 fest. Die Akzeptanzspannen der vier validierten Prüfmodelle sind in Tabelle 3 aufgeführt. Tabelle 3 Chargenfreigabekriterien im Rahmen der Qualitätskontrolle
Applikation der Prüf- und Kontrollchemikalien 22. Für jede Prüfchemikalie und für die Kontrollen sollten für jeden Expositionszeitraum mindestens zwei Gewebereplikate verwendet werden. Bei flüssigen und festen Chemikalien sollte eine ausreichende Menge der Prüfchemikalie gleichmäßig auf die gesamte Oberfläche der Epidermis aufgetragen werden; überschüssige Dosen sind zu vermeiden, d. h. es sollten mindestens 70 µl/cm2 oder 30 mg/cm2 verwendet werden. Je nach Modell sollte die Epidermis-Oberfläche vor der Applikation fester Chemikalien mit deionisiertem oder destilliertem Wasser angefeuchtet werden, um guten Hautkontakt zu gewährleisten (34)(35)(36)(37). Feststoffe sollten nach Möglichkeit als Feinpulver geprüft werden. Es ist eine für die Prüfchemikalie geeignete Applikationsmethode zu wählen (siehe z.B. Nummern 34-37). Am Ende des Expositionszeitraums sollte die Prüfchemikalie mit einer wässrigen Pufferlösung oder 0,9 % NaCl von der Epidermis abgewaschen werden. Je nachdem, welches der vier validierten RhE-Prüfmodelle verwendet wird, werden pro Prüfchemikalie zwei oder drei Expositionszeiträume verwendet (für alle vier validen RhE-Modelle: 3 Minuten und 1 Stunde; bei EpiSkinTM eine zusätzliche Expositionszeit von nochmals 4 Stunden). Je nach verwendetem RhE-Prüfmodell und je nach bewertetem Expositionszeitraum kann die Inkubationstemperatur während der Exposition zwischen der Raumtemperatur und 37 °C liegen. 23. Bei jeder Studie sollten gleichzeitig Negativkontrollen und Positivkontrollen (PK) verwendet werden, um nachweisen zu können, dass die Viabilität (mit der NK), die Barrierefunktion und die daraus resultierende Empfindlichkeit der Gewebe (mit der PK) innerhalb einer vorgegebenen historischen Akzeptanzspanne liegen. Als PK-Chemikalien werden je nach verwendetem RhE-Modell Eisessigsäure oder 8N KOH vorgeschlagen. 8N KOH ist ein direktes MTT-Reduktionsmittel, für das möglicherweise geeignete Kontrollen benötigt werden wie in den Nummern 25 und 26 beschrieben. Für die Negativkontrollen wird 0,9 %ige (w/v) NaCl-Lösung oder Wasser empfohlen. Zellviabilitätsmessungen 24. Die MTT-Prüfung sollte als quantitative Methode zur Messung der Zellviabilität nach dieser Prüfmethode verwendet werden (27). Die Gewebeprobe wird für 3 Stunden in eine MTT-Lösung mit geeigneter Konzentration (z.B. 0,3 oder 1 mg/ml) gelegt. Der blaue Formazan-Niederschlag wird sodann mit Hilfe eines Lösungsmittels (z.B. Isopropanol, Säure-Isopropanol) aus dem Gewebe extrahiert, und die Formazankonzentration wird durch Bestimmung des OD-Werts bei 570 nm innerhalb einer Filter-Bandbreite von maximal ± 30 nm oder mit einem HPLC/UPLC-Spektrophotometrieverfahren gemessen (Nummern 30 und 31) (38). 25. Prüfchemikalien können die MTT-Untersuchung durch direkte Reduzierung des MTT zu blauem Formazan und/oder Farbinterferenzen stören, wenn die Prüfchemikalie an sich oder infolge von Behandlungsverfahren in derselben OD-Spanne wie Formazan (570 ± 30 nm, hauptsächlich blaue und violette Chemikalien) absorbiert. Zusätzliche Kontrollen (z.B. die nicht spezifische MTT-Reduktionskontrolle (NSMTT) oder die nicht spezifische Farbkontrolle (NSC) sollten verwendet werden, um eine potenzielle Interferenz aufgrund dieser Prüfchemikalien zu erkennen und zu korrigieren (Nummern 26 bis 30). Dies ist besonders dann wichtig, wenn eine bestimmte Prüfchemikalie nicht vollständig aus dem Gewebe ausgewaschen wurde oder die Epidermis durchdringt und daher bei Durchführung der MTT-Viabilitätsprüfung auch in den Geweben enthalten ist. Für eine genaue Beschreibung der richtigen Durchführung der Korrektur der direkten MTT-Reduktion und der Interferenzen durch Färbemittel siehe die Standardarbeitsanweisungen für die Prüfmodelle (34) (35) (36) (37). 26. Um direkte MTT-Reduktionsmittel zu identifizieren, sollten die Prüfchemikalien jeweils zu frisch hergestelltem MTT-Medium hinzugegeben werden (34) (35) (36) (37). Wenn sich das MTT-Gemisch mit der Prüfchemikalie blau/violett verfärbt, ist davon auszugehen, dass die Prüfchemikalie das MTT direkt reduziert; anschließend sollte unabhängig von der Durchführung der Standardabsorptions-(OD-)Messung oder einer HPLC/UPLC-Spektrophotometrie eine weitere Funktionsprüfung vorgenommen werden. Bei dieser zusätzlichen Funktionsprüfung werden abgetötete Gewebe mit nur noch residualer metabolischer Aktivität eingesetzt, die die Prüfchemikalie in ähnlichem Umfang wie lebensfähige Gewebe absorbieren. Jede MTT reduzierende Chemikalie wird auf mindestens zwei abgetötete Gewebereplikate pro Expositionszeitraum aufgetragen, die der vollständigen Prüfung auf hautätzende Wirkungen unterzogen werden. Anschließend wird die tatsächliche Viabilität des Gewebes als Prozentanteil der bei lebenden Geweben nach Exposition gegenüber dem MTT-Reduktionsmittel gemessenen Viabilität abzüglich des Prozentanteils der nicht spezifischen MTT-Reduktion durch dasselbe MTT-Reduktionsmittel bei abgetöteten Geweben bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung durchgeführte Negativkontrolle (%NSMTT) berechnet. 27. Um potenzielle Interferenzen durch Prüffarbstoffe oder durch Prüfchemikalien zu ermitteln, die sich verfärben, wenn sie mit Wasser oder mit Isopropanol in Berührung kommen, und um über die Notwendigkeit weiterer Kontrollen entscheiden zu können, sollte die Prüfchemikalie in Wasser (Umgebung während der Exposition) und/oder Isopropanol (Extraktionslösung) einer Spektralanalyse unterzogen werden. Wenn die Prüfchemikalie in Wasser und/oder Isopropanol Licht im Bereich von 570 ± 30 nm absorbiert, sollten weitere Farbkontrollen durchgeführt oder eine HPLC/UPLC-Spektrophotometrie vorgenommen werden. Im letztgenannten Fall sind diese Kontrollen nicht erforderlich (Nummern 30 und 31). Bei Durchführung der Standardabsorptions-(OD-)Messung wird jeder interferierende Prüffarbstoff auf mindestens zwei lebensfähige Gewebereplikate pro Expositionszeit aufgetragen; diese Replikate werden der vollständigen Prüfung auf Hautverätzungen unterzogen, aber während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert, um eine nicht spezifische Farbkontrolle (NSClebend) herzustellen. Wegen der inhärenten Variabilität lebender Gewebe muss pro Expositionszeitraum und pro Prüffarbstoff (bei jedem Prüflauf) gleichzeitig die NSClebend-Kontrolle durchgeführt werden. Die tatsächliche Viabilität des Gewebes wird dann als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit der MTT-Lösung abzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die bei gleichzeitig mit der zu korrigierenden Prüfung untersuchten lebenden Geweben nach Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist (%NSClebend). 28. Wenn festgestellt wurde, dass Prüfchemikalien sowohl eine direkte MTT-Reduktion (Nummer 26) als auch Farbinterferenzen (Nummer 27) bewirken, wird bei der Durchführung der Standardabsorptions-(OD-)Messung neben den in vorstehenden Nummern beschriebenen Kontrollen (NSMTT und NSClebend) eine dritte Kontrollreihe benötigt. Dies ist gewöhnlich bei dunklen Prüfchemikalien der Fall, die beim MTT-Versuch Interferenzen verursachen (z.B. blau, violett oder schwarz), weil deren Fähigkeit zur direkten MTT-Reduktion durch die inhärente Farbqualität dieser Chemikalien beeinträchtigt wird (Nummer 26). Diese Prüfchemikalien können Bindungen sowohl mit lebenden als auch mit abgetöteten Geweben eingehen. Daher kann bei der NSMTT-Kontrolle eine Korrektur nicht nur der potentiellen direkten MTT-Reduktion durch die Prüfchemikalie, sondern auch der Farbinterferenz infolge der Bindung der Prüfchemikalie an abgetötete Gewebe erfolgen. Dies kann eine doppelte Korrektur der Farbinterferenz zur Folge haben, da mit der NSClebend-Kontrolle bereits die Farbinterferenz aufgrund der Bindung der Prüfchemikalie an lebende Gewebe erfolgt. Um eine mögliche doppelte Korrektur von Farbinterferenzen zu vermeiden, muss eine dritte Kontrolle mit nicht spezifischen Farben mit abgetöteten Geweben (NSCabgetötet) untersucht werden. Bei dieser zusätzlichen Kontrolle wird die Prüfchemikalie auf mindestens zwei abgetötete Gewebereplikate pro Expositionszeitraum aufgetragen, die dem vollständigen Prüfverfahren zu unterziehen sind, wobei die Gewebe jedoch während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert werden. Unabhängig von der Anzahl der durchgeführten unabhängigen Prüfungen/Prüfläufe ist pro Prüfchemikalie eine einzige NSCabgetötet-Kontrolle hinreichend. Diese Kontrolle sollte jedoch gleichzeitig mit der NSMTT-Kontrolle sowie möglichst mit derselben Gewebecharge durchgeführt werden. Die tatsächliche Viabilität des Gewebes wird dann als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der Prüfchemikalie abzüglich %NSMTT und abzüglich %NSClebend zuzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die nach Exposition der abgetöteten Gewebe gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist; dieser Prozentanteil wird bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung untersuchte Kontrolle (%NSCabgetötet) ermittelt. 29. Wichtig ist die Feststellung, dass nicht spezifische MTT-Reduktionen und nicht spezifische Farbinterferenzen bei den Gewebextrakten Ergebnisse oberhalb der Linearitätsspanne des Photospektrometers begünstigen können. Daher sollte jedes Labor vor der Untersuchung von Prüfchemikalien für rechtliche Zwecke die Linearitätsspanne seines Spektrophotometers mit im Handel bezogenem MTT-Formazan (CAS-Nr. 57360-69-7) ermitteln. Insbesondere durch die Standardabsorptions-(OD-)Messung mit einem Spektrophotometer können direkte MTT-Reduktionsmittel und Farbinterferenzen verursachende Prüfchemikalien in den Fällen untersucht werden, in denen die mit einer Prüfchemikalie ermittelten und nicht um die direkte MTT-Reduktion und/oder um Farbinterferenzen korrigierten ODs der Gewebeextrakte innerhalb der Linearitätsspanne des Spektrophotometers liegen oder in denen eine Prüfchemikalie aufgrund des nicht korrigierten Prozentanteils der Viabilität bereits als hautätzend eingestuft wurde (Nummern 35 und 36). Ergebnisse bei Prüfchemikalien mit %NSMTT und/oder %NSClebend > 50 % der Negativkontrolle sind jedoch mit Sorgfalt zu bewerten. 30. Wenn die Ergebnisse von Prüffarbstoffen wegen übermäßiger Interferenzen beim MTT-Versuch mit der Standardabsorptions-(OD-)Messung nicht vereinbar sind, kann alternativ MTT-Formazan durch HPLC/UPLC-Spektrophotometrie gemessen werden (Nummer 31) (37). Bei der HPLC/UPLC-Spektrophotometrie kann das MTT-Formazan vor der Quantifizierung von der Prüfchemikalie getrennt werden (38). Daher werden unabhängig von der zu prüfenden Chemikalie keine NSClebend- oder NSCabgetötet-Kontrollen benötigt, wenn eine HPLC/UPLC-Spektrophotometrie vorgenommen wird. NSMTT-Kontrollen sollten jedoch verwendet werden, wenn vermutet wird, dass eine Prüfchemikalie MTT direkt reduziert oder wenn die Farbe einer Prüfchemikalie die Beurteilung ihrer Fähigkeit zur direkten MTT-Reduktion beeinträchtigt (Nummer 26). Wenn MTT-Formazan durch HPLC/UPLC-Spektrophotometrie gemessen wird, ist der Prozentanteil der Viabilität des Gewebes als Prozentanteil der MTT-Formazan-Peak-Fläche, die mit lebenden Geweben nach Exposition gegenüber der Prüfchemikalie ermittelt wurde, bezogen auf die MTT-Formazan-Peak-Fläche der gleichzeitigen Negativkontrolle zu berechnen. Bei Prüfchemikalien, die MTT direkt reduzieren können, wird die tatsächliche Gewebeviabilität als Prozentanteil der Gewebeviabilität von lebenden Geweben nach der Exposition gegenüber der Prüfchemikalie abzüglich %NSMTT berechnet. Und schließlich ist festzustellen, dass auch direkte MTT-Reduktionsmittel (die nach der Behandlung in den Geweben gebunden sein und MTT so stark reduzieren können, dass es (bei Standard-OD-Messungen) zu ODs oder (bei der Untersuchung der Gewebeextracte durch UPLC-/HPLC-Spektrophotometrie) zu Peakflächen außerhalb der Linearitätsspanne des Spektrophotometers und zu Farbinterferenzen kommen kann) nicht bewertet werden können. Solche Fälle sind allerdings sehr selten. 31. MTT-Formazan-Messungen können bei allen Arten von Prüfchemikalien (gefärbt, nicht gefärbt, MTT-Reduktionsmittel und Mittel, die keine MTT-Reduktion bewirken) auch durch HPLC/UPLC-Spektrophotometrie vorgenommen werden (38). Wegen der Vielfalt der HPLC/UPLC-Spektrophotometriesysteme sollte die Eignung des jeweiligen HPLC/UPLC-Spektrophotometriesystems vor der Verwendung zur Quantifizierung von MTT-Formazan in Gewebextrakten durch Erfüllung der Akzeptanzkriterien für eine Reihe von Standard-Qualifikationsparametern auf der Grundlage der Branchenleitlinien der US-amerikanischen Food and Drug Administration (FDA) für die Validierung bioanalytischer Methoden nachgewiesen werden (38)(39). Diese Schlüsselparameter sowie die jeweiligen Akzeptanzkriterien sind Anlage 4 zu entnehmen. Wenn die in Anlage 4 beschriebenen Akzeptanzkriterien erfüllt sind, wird das betreffende HPLC/UPLC-Spektrophotometriesystem als geeignet für die Messung von MTT-Formazan unter den in dieser Prüfmethode beschriebenen Versuchsbedingungen betrachtet. Akzeptanzkriterien 32. Bei allen Prüfmethoden unter Verwendung valider RhE-Modelle sollten die mit der Negativkontrolle behandelten Gewebe OD-Werte entsprechend der Qualität der Gewebe ergeben (siehe Tabelle 2) und die historisch ermittelten Grenzen nicht unterschreiten. Mit der PK, d. h. mit Eisessigsäure oder 8N KOH, behandelte Gewebe sollten unter den Bedingungen des Prüfmodells auf eine hautätzende Chemikalie reagieren können (siehe Anlage 2). Die Variabilität zwischen Gewebereplikaten für die Prüfchemikalie und/oder Kontrollchemikalien sollten innerhalb der akzeptierten Grenzen der Anforderungen des jeweiligen validen RhE-Modells liegen (siehe Anlage 2), d. h. beispielsweise die Differenz der Viabilität zwischen den beiden Gewebereplikaten sollte nicht mehr als 30 % betragen. Wenn die Negativ- oder die Positivkontrolle bei einem Prüflauf Werte außerhalb der akzeptierten Spannen ergibt, ist der Prüflauf als nicht geeignet zu betrachten und sollte wiederholt werden. Liegt die Variabilität von Prüfchemikalien außerhalb des definierten Bereichs, sollte sie nochmals geprüft werden. Auswertung der Ergebnisse und Prädiktionsmodell 33. Die für jede Prüfchemikalie ermittelten OD-Werte sollten zur Berechnung einer prozentualen Viabilität im Vergleich zur Negativkontrolle herangezogen werden, die gleich 100 % gesetzt wird. Wenn eine HPLC/UPLC-Spektrophotometrie durchgeführt wird, ist der Prozentanteil der Viabilität des Gewebes als Prozentanteil der MTT-Formazan-Peak-Fläche, die mit lebenden Geweben nach Exposition gegenüber der Prüfchemikalie ermittelt wurde, bezogen auf die MTT-Formazan-Peak-Fläche der gleichzeitigen Negativkontrolle zu berechnen. Den folgenden Nummern 35 und 36 sind die prozentualen Schwellenwerte der Zellviabilität für die Prüfmodelle dieser Prüfmethode zu entnehmen, nach denen hautätzende von nicht hautätzenden Prüfchemikalien unterschieden werden (bzw. nach denen zwischen Unterkategorien hautätzender Chemikalien unterschieden wird). Diese Schwellenwerte sollten bei der Auswertung der Ergebnisse zugrunde gelegt werden. 34. Kann eine eindeutige Klassifizierung vorgenommen werden, so ist ein einziger, aus mindestens zwei Geweben bestehender Prüflauf ausreichend. Bei Grenzergebnissen, wie z.B. nicht übereinstimmenden Replikatmessungen, kann ein zweiter Prüflauf in Betracht gezogen werden bzw. ein dritter bei abweichenden Ergebnissen der ersten beiden Prüfläufe. 35. In der folgenden Tabelle 4 wird das Vorhersagemodell für die das Prüfmodell EpiSkinTM zur Feststellung von Hautätzungen (9)(34)(22) in Verbindung mit dem Klassifizierungssystem des UN GHS bzw. der CLP-Verordnung erläutert: Tabelle 4 Vorhersagemodell EpiSkinTM
36. Die Vorhersagemodelle der Prüfmodelle EpiDermTM SCT (10)(23)(35), SkinEthicTM RHE (17)(18)(23)(36) und epiCS® (16)(23)(37) zur Bewertung von Hautverätzungen nach der Klassifizierung des UN GHS bzw. der CLP-Verordnung sind Tabelle 5 zu entnehmen: Tabelle 5 EpiDermTM SCT, SkinEthicTM RHE und epiCS®
Daten und Berichterstattung Daten 37. Für jede Prüfung sollten Daten aus einzelnen Gewebereplikaten (z.B. OD-Werte und Daten über die berechnete prozentuale Zellviabilität für jede Prüfchemikalie, einschließlich Einstufung), gegebenenfalls auch Daten aus Wiederholungsversuchen, tabellarisch zusammengefasst werden. Außerdem sollten die Mittelwerte und die Spannen der Viabilität und die Variationskoeffizienten zwischen Gewebereplikaten der einzelnen Prüfungen angegeben werden. Für jede geprüfte Chemikalie sollten zudem festgestellte Interaktionen mit dem MTT-Reagens durch direkte MTT-Reduktionsmittel oder Prüffarbstoffe berichtet werden. Prüfbericht 38. Der Prüfbericht sollte folgende Angaben enthalten: Prüfchemikalien und Kontrollchemikalien
Verwendetes RhE-Modell und verwendetes Protokoll und Gründe für die Auswahl (wenn relevant) Prüfbedingungen:
Prüfverfahren:
Akzeptanzkriterien für Prüfläufe und Prüfungen
Ergebnisse:
Erörterung der Ergebnisse Schlussfolgerungen Literatur (1) UN (2013). GHS (Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung) der Vereinten Nationen, fünfte überarbeitete Auflage, UN New York und Genf. Abrufbar unter:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html (2) Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung. (3) Kapitel B.40 dieses Anhangs, Invitro-Hautreizung: (4) Kapitel B.65 dieses Anhangs, Invitro-Methode zur Untersuchung der Membranbarriere. (5) Kapitel B.46 dieses Anhangs, Invitro-Hautreizung: Prüfmethode mit rekonstruierter humaner Epidermis. (6) OECD (2014). Guidance Document on Integrated Approaches to Testing and Assessment of Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (7) Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A Prevalidation Study on In Vitro Skin Corrosivity Testing. The report and Recommendations of ECVAM Workshop 6.ATLA 23:219-255. (8) Barratt M.D., Brantom P.G., Fentem J.H., Gerner I., Walker A.P., and Worth A.P. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 1. Selection and distribution of the Test Chemicals. Toxicol.In Vitro 12:471-482. (9) Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G., and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests for SkinCorrosivity. 2. Results and Evaluation by the Management Team. Toxicol.in Vitro 12:483-524. (10) Liebsch, M., Traue, D., Barrabas, C., Spielmann, H., Uphill, P., Wilkins, S., Wiemann, C., Kaufmann, T., Remmele, M. und Holzhütter, H.-G. (2000). The ECVAM Prevalidation Study on the Use of EpiDerm for Skin Corrosivity Testing, ATLA 28: 371-401. (11) Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, CA., Repetto, G., Sladowski, D., Spielmann, H. und Zucco, F. (1995). Practical Aspects of the Validation of Toxicity Test Procedures. The Report and Recommendations of ECVAM Workshops, ATLA 23:129-147. (12) ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods) (1997). Validation and Regulatory Acceptance of Toxicological TestMethods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. (13) ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods) (2002). ICCVAM evaluation of EpiDermTM (EPI-200), EPISKINTM (SM), and the Rat Skin Transcutaneous Electrical Resistance (TER) Assay: In Vitro Test Methods for Assessing Dermal Corrosivity Potential of Chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA. (14) EC-ECVAM (1998). Statement on the Scientific Validity of the EpiSkinTM Test (an In Vitro Test for Skin Corrosivity), herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC10), 3. April 1998. (15) EC-ECVAM (2000). Statement on the Application of the EpiDermTM Human Skin Model for Skin Corrosivity Testing, herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC14), 21. März 2000. (16) Hoffmann, J., Heisler, E., Karpinski, S., Losse, J., Thomas, D., Siefken, W., Ahr, H.J., Vohr, H.W. und Fuchs, H.W. (2005). Epidermal-Skin-Test 1000 (EST-1000)-A New Reconstructed Epidermis for In Vitro Skin Corrosivity Testing. Toxicol.In Vitro 19: 925-929. (17) Kandárová, H., Liebsch, M., Spielmann, H., Genschow, E., Schmidt, E., Traue, D., Guest, R., Whittingham, A., Warren, N, Gamer, A.O., Remmele, M., Kaufmann, T., Wittmer, E., De Wever, B. und Rosdy, M. (2006). Assessment of the Human Epidermis Model SkinEthic RHE for In Vitro Skin Corrosion Testing of Chemicals According to New OECD TG 431. Toxicol.In Vitro 20: 547-559. (18) Tornier, C., Roquet, M. und Fraissinette, A.B. (2010). Adaptation of the Validated SkinEthicTM Reconstructed Human Epidermis (RHE) Skin Corrosion Test Method to 0,5 cm2 Tissue Sample. Toxicol. In Vitro 24: 1379-1385. (19) EC-ECVAM (2006). Statement on the Application of the SkinEthicTM Human Skin Model for Skin Corrosivity Testing, herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC25), 17. November 2006. (20) EC-ECVAM (2009). ESAC Statement on the Scientific Validity of an In-Vitro Test Method for Skin Corrosivity Testing: the EST-1000, herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC30), 12. Juni 2009. (21) OECD (2013). Summary Document on the Statistical Performance of Methods in OECD Test Guideline 431 for Subcategorisation. Environment, Health, and Safety Publications, Series on Testing and Assessment (No 190). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (22) Alépée, N., Grandidier, M.H. und Cotovio, J. (2014). Sub-Categorisation of Skin Corrosive Chemicals by the EpiSkinTM Reconstructed Human Epidermis Skin Corrosion Test Method According to UN GHS: Revision of OECD Test Guideline 431. Toxicol. In Vitro 28:131-145. (23) Desprez, B., Barroso, J., Griesinger, C., Kandárová, H., Alépée, N. und Fuchs, H. (2015). Two Novel Prediction Models Improve Predictions of Skin Corrosive Subcategories by Test Methods of OECD Test Guideline No 431. Toxicol. In Vitro 29:2055-2080. (24) OECD (2015). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Epidermis (RHE) Test Methods For Skin Corrosion in Relation to OECD TG 431. Environmental Health and Safety Publications, Series on Testing and Assessment (No 219). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (25) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (26) Eskes, C., u. a. (2012). Regulatory Assessment of In Vitro Skin Corrosion and Irritation Data Within the European Framework: Workshop Recommendations. Regul.Toxicol.Pharmacol. Pharmacol., 62:393-403. (27) Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 65:55-63. (28) Tinois, E., u. a. (1994). The Episkin Model: Successful Reconstruction of Human Epidermis In Vitro. In: In Vitro Skin Toxicology. Rougier, A., Goldberg, A.M. und Maibach, H.I. (Hrsg.) 133-140. (29) Cannon, C. L., Neal, P.J., Southee, J.A., Kubilus, J. und Klausner, M. (1994), New Epidermal Model for Dermal Irritancy Testing. Toxicol.in Vitro 8:889 - 891. (30) Ponec, M., Boelsma, E, Weerheim, A, Mulder, A., Bouwstra, J. und Mommaas, M. (2000). Lipid and Ultrastructural Characterization of Reconstructed Skin Models. Inter. J. Pharmaceu. 203:211 - 225. (31) Tinois, E., Tillier, J., Gaucherand, M., Dumas, H., Tardy, M. und Thivolet, J. (1991). In Vitro and Post - Transplantation Differentiation of Human Keratinocytes Grown on the Human Type IV Collagen Film of a Bilayered Dermal Substitute. Exp. Cell Res. 193:310-319. (32) Parenteau, N.L., Bilbo, P, Nolte, C.J., Mason, V.S. und Rosenberg, M. (1992). The Organotypic Culture of Human Skin Keratinocytes and Fibroblasts to Achieve Form and Function. Cytotech. 9:163-171. (33) Wilkins, L.M., Watson, S.R., Prosky, S.J., Meunier, S.F. und Parenteau, N.L. (1994). Development of a Bilayered Lebend Skin Construct for Clinical Applications. Biotech. Bioeng. 43/8:747-756. (34) EpiSkinTM SOP (Dezember 2011). INVITTOX Protocol (No 118). EpiSkinTM Skin Corrosivity Test. (35) EpiDermTM SOP (Februar 2012). Version MK-24-007-0024 Protocol for: In Vitro EpiDermTM Skin Corrosion Test (EPI-200-SCT), for Use with MatTek Corporation's Reconstructed Human Epidermal Model EpiDerm. (36) SkinEthicTM RHE SOP (Januar 2012). INVITTOX Protocol SkinEthicTM Skin Corrosivity Test. (37) EpiCS® SOP (Januar 2012). Version 4.1 In Vitro Skin Corrosion: Human Skin Model Test Epidermal Skin Test 1000 (epiCS® ) CellSystems. (38) Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M. und McNamee, P. Use of HPLC/UPLC- spectrophotometry for Detection of MTT Formazan in In Vitro Reconstructed Human Tissue (RhT)- based Test Methods Employing the MTT Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Toxicol. In Vitro 29: 741-761. (39) US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. (Mai 2001). Abrufbar unter:[http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf]. ________________ (2) Die Abkürzung RhE (Reconstructed human Epidermis = rekonstruierte humane Epidermis) wird für alle auf der RhE-Technologie beruhenden Modelle verwendet. In Verbindung mit dem SkinEthic-Modell wird die Abkürzung RHE im gleichen Sinn verwendet, jedoch ausschließlich in Großbuchstaben geschrieben, da sie in dieser Schreibung Bestandteil des Namens ist, unter dem diese spezifische Prüfmethode auf dem Markt angeboten wird.
Die folgende Tabelle bietet einen Überblick über die Leistung der vier Prüfmodelle, die auf der Grundlage einer Reihe von 80 von den vier Entwicklern der Prüfmodelle geprüften Chemikalien berechnet wurde. Die Berechnungen wurden vom OECD-Sekretariat vorgenommen und von einer Experten-Untergruppe geprüft und angenommen (21) (23). Mithilfe der Prüfmodelle EpiSkinTM, EpiDermTM, SkinEthicTM und epiCS® können Unterkategorisierungen (d. h. Unterscheidungen zwischen 1A und 1B- und-1C gegenüber NC) vorgenommen werden. Die Leistungsangaben, die Überklassifizierungsraten, die Unterqualifizierungsraten und die Genauigkeit (Vorhersagefähigkeit) der vier Prüfmodelle beruhen auf einer Reihe von 80 Chemikalien, die für jedes Prüfmodell jeweils in 2 oder 3 Prüfläufen untersucht wurden:
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.41 In-vitro-3T3-NRU-Fototoxizitätstest 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| 1. Methode
Diese Methode entspricht OECD TG 432 (2004). 1.1 Einleitung Unter Fototoxizität versteht man die toxische Reaktion eines auf den Körper aufgebrachten chemischen Stoffs, die bei anschließender Lichtexposition (in einer bei niedrigerer Dosierung wahrnehmbaren Form) entsteht oder verstärkt wird oder die durch Bestrahlung der Haut nach systematischer Verabreichung eines chemischen Stoffs induziert wird. Der In-vitro-3T3-NRU-Fototoxizitätstest dient zur Erfassung des fototoxischen Potenzials einer Testsubstanz, das nach Lichtexposition durch die erkannte Chemikalie induziert wird. Bei diesem Test wird die Fotozytotoxizität anhand der relativen Reduktion der Viabilität der Zellen, die der Chemikalie ausgesetzt sind, unter Einwirkung bzw. Nichteinwirkung von Licht beurteilt. Die durch diesen Test festgestellten Substanzen zeigen nach systemischer Verabreichung und Aufbringung auf der Haut oder nach topischer Anwendung in vivo voraussichtlich fototoxische Wirkung. Fototoxische Wirkung wurde bei zahlreichen Arten von Chemikalien beobachtet (1) (2) (3) (4). Gemeinsames Merkmal dieser Chemikalien ist, dass sie Lichtenergie im Sonnenlichtbereich absorbieren können. Nach dem ersten fotochemischen Gesetz (dem Grotthaus-Draperschen-Gesetz) ist für eine Fotoreaktion eine ausreichende Lichtquantenabsorption erforderlich. Bevor biologische Tests in Frage kommen, muss daher nach OECD Test Guideline 101 ein UV/vis-Absorptionsspektrum der Testchemikalie ermittelt werden. Es wird davon ausgegangen, dass - wenn der molare Extinktions-/Absorptionskoeffizient unter 10 Liter x mol-1 x cm-1 liegt - der chemische Stoff kein fotoreaktives Potenzial besitzt. Diese Chemikalie braucht dann evtl. nicht durch den In-vitro-3T3-NRU-Fototoxizitätstest oder einen anderen biologischen Test auf schädliche fotochemische Wirkungen getestet zu werden (1) (5). Siehe auch Anlage 1. Zuverlässigkeit und Relevanz des In-vitro-3T3-NRU-Fototoxizitätstests wurden in neuerer Zeit untersucht (6) (7) (8) (9). Es wurde nachgewiesen, dass mit dem In-vitro-3T3-NRU-Fototoxizitätstest akute fototoxische Wirkungen bei Tieren und Menschen in vivo vorhergesagt werden können. Der Test ist nicht für die Vorhersage schädlicher Wirkungen ausgelegt, die sich aus einer kombinierten Wirkung eines chemischen Stoffs und von Licht ergeben, d. h., Fotogenotoxizität, Fotoallergie oder Fotokarzinogenität und andere Erscheinungen werden durch diesen Test nicht erfasst, und auch die Beurteilung der fototoxischen Potenz ist damit nicht möglich. Auch indirekte Mechanismen der Fototoxizität, Wirkungen der Stoffwechselprodukte der Testsubstanz oder Wirkungen der Gemische deckt dieser Test nicht ab. Die Verwendung von metabolisierenden Systemen ist bei sämtlichen In-vitro-Tests eine generelle Voraussetzung für die Vorhersage des genotoxischen und karzinogenen Potenzials; allerdings liegen bis jetzt in der Fototoxikologie nur seltene Fälle vor, in denen eine metabolische Transformation erforderlich ist, damit die Chemikalie als In-vivo- oder In-vitro-Fototoxin wirken kann. Daher ist es derzeit weder notwendig noch wissenschaftlich gerechtfertigt, dass der vorliegende Test mit einem Stoffwechselaktivierungssystem durchgeführt wird. 1.2 Definitionen Bestrahlungsstärke: die Intensität des auf eine Oberfläche auftreffenden ultravioletten (UV) oder sichtbaren Lichts, gemessen in W/m2 oder mW/cm2. Lichtdosis: die Menge (= Intensität x Zeit) der auf eine Oberfläche auftreffenden ultravioletten (UV) oder sichtbaren Strahlung, ausgedrückt in Joule (= W x s) je Fläche, z.B. J/m2 oder J/cm2. UV-Licht, Bandbreiten: Die von der Internationalen Beleuchtungskommission (CIE) empfohlenen Bezeichnungen sind: UVA (315-400 nm), UVB (280-315 nm) und UVC (100-280 nm). Andere Bezeichnungen werden ebenfalls verwendet; die Trennung zwischen UVB und UVA erfolgt oft bei 320 nm; UVA kann in UV-A1 und UV-A2 unterteilt werden, wobei die Trennung bei ca. 340 nm liegt. Zellviabilität: Parameter zur Messung der Gesamtaktivität einer Zellpopulation (z.B. Aufnahme des Vitalfarbstoffs "Neutralrot" in Zell-Lysosomen), die je nach dem gemessenen Endpunkt und der angewandten Versuchsauslegung mit der Gesamtzahl und/oder der Vitalität der Zellen korreliert. Relative Zellviabilität: Zellviabilität, ausgedrückt in Relation zu Negativkontrollen (Lösemittel), die das gesamte Testverfahren (entweder +Irr oder -Irr) durchlaufen, jedoch nicht mit einer Testchemikalie behandelt wurden. PIF (Fotoirritationsfaktor): ein Faktor, der durch Vergleich von zwei gleich wirksamen zytotoxischen Konzentrationen (IC50) der chemischen Testsubstanz in Abwesenheit (-Irr) und in Anwesenheit (+Irr) nnich zytotoxischer Strahlung mit UVA/vis-Licht ermittelt wird. IC50: Konzentration der Testchemikalie, bei der die Zellviabilität um 50 % reduziert wird. MPE (Mean Photo Effect): eine Messgröße, die von einer mathematischen Analyse der Konzentrations-Wirkungs-Kurven hergeleitet wurde, die in Abwesenheit (-Irr) und in Anwesenheit (+Irr) nicht zytotoxischer Strahlung mit UVA/vis-Licht erzielt wurden. Fototoxizität: eine akute toxische Reaktion, die nach der ersten Exposition der Haut mit bestimmten chemischen Stoffen bei anschließender Lichtexposition eintritt oder die auf ähnliche Weise durch Bestrahlung der Haut nach systemischer Verabreichung eines chemischen Stoffs induziert wird. 1.3 Prinzip der Testmethode Der In-vitro-3T3-NRU-Fototoxizitätstest basiert auf dem Vergleich der Zytotoxizität eines chemischen Stoffs, der mit Exposition und ohne Exposition einer nicht zytotoxischen Dosis simulierten Sonnenlichts getestet wird. Die Zytotoxizität wird bei diesem Test ausgedrückt als konzentrationsabhängige Reduktion der Aufnahme des Vitalfarbstoffs Neutralrot, wenn diese 24 Stunden nach der Behandlung mit der Testchemikalie und Bestrahlung gemessen wird (10). Neutralrot (NR) ist ein schwach kationischer Farbstoff, der durch Nichtdiffusion rasch in Zellmembranen eindringt und sich intrazellulär in Lysosomen ansammelt. Veränderungen der Zellenoberfläche der sensitiven Lysosomalmembran führen zu lysosomaler Fragilität und weiteren Veränderungen, die nach und nach irreversibel werden. Derartige, durch die Wirkung von Xenobiotika verursachte Veränderungen bewirken eine verringerte Aufnahme und Bindung von Neutralrot (NR). Auf diese Weise ist eine Unterscheidung zwischen lebensfähigen (viablen), geschädigten und toten Zellen möglich; auf dieser Unterscheidung baut auch der hier beschriebene Test auf. Balb/c-3T3-Zellen werden zwecks Bildung eines Monolayers 24 Stunden kultiviert. Zwei "96-well"-Platten je Testchemikalie werden sodann mit acht verschiedenen Konzentrationen der Chemikalie 1 Stunde lang vorinkubiert. Daraufhin wird eine der zwei Platten mit der höchsten nicht zytotoxischen Bestrahlungsdosis bestrahlt, während die andere Platte im Dunkeln aufbewahrt wird. In beiden Platten wird dann das Behandlungsmedium durch ein Kulturmedium ersetzt, und nach weiteren 24 Stunden Inkubation wird die Zellviabilität anhand der Neutralrot-Aufnahme bestimmt. Die Zellviabilität, ausgedrückt als Prozentsatz unbehandelter Lösemittelkontrollen, wird für jede Testkonzentration berechnet. Um das fototoxische Potenzial vorherzusagen, wird die mit und ohne Strahlung erzielte Konzentrations-Wirkungs-Kurve verglichen, in der Regel für den IC50-Wert (d. h. die Konzentration, die die Zellviabilität gegenüber unbehandelten Kontrollen um 50 % vermindert). 1.4 Beschreibung der Testmethode 1.4.1 Zubereitungen 1.4.1.1 Zellen Eine permanente Mäuse-Fibroblastenzelllinie - Balb/c 3T3, Klon 31 - entweder von der American Type Culture Collection (ATCC), Manassas, VA, USA, oder von der European Collection of Cell Cultures (ECACC), Salisbury, Wiltshire, Vereinigtes Königreich - wurde in der Validierungsstudie verwendet und wird daher zur Beschaffung aus einem qualifizierten Zellendepot empfohlen. Andere Zellen oder Zelllinien können erfolgreich mit demselben Testverfahren verwendet werden, wenn die Kulturbedingungen den spezifischen Bedürfnissen der Zellen angepasst werden, jedoch muss die gleichwertige Eignung der Zellen nachgewiesen werden. Zellen sollten regelmäßig auf die Abwesenheit von Mykoplasma-Kontamination hin geprüft und nur verwendet werden, wenn keine derartige Kontamination festgestellt wird (11). Die UV-Empfindlichkeit der Zellen muss regelmäßig nach den in der vorliegenden Verfahrensbeschreibung dargestellten Qualitätssicherungsverfahren kontrolliert werden. Da die UVA-Sensitivität von Zellen mit der erreichbaren Passagenzahl zunehmen kann, sollten Balb/c-3T3-Zellen mit einer möglichst niedrigen Passagenzahl, vorzugsweise weniger als 100, verwendet werden (siehe 1.4.2.2.2 und Anlage 2). 1.4.1.2 Medien und Kulturbedingungen Für Routine-Zellpassagen und während des Testverfahrens sollten geeignete Kulturmedien und Inkubationsbedingungen angewendet werden. Bei Balb/c-3T3-Zellen sind dies DMEM (Dulbecco's Modified Eagle's Medium) mit einem Zusatz von 10 % Serum neugeborener Kälber, 4 mM Glutamin, Penicillin (100 IU) und Streptomycin (100 (µg/ml) sowie Feuchtinkubation bei 37 °C, 5-7,5 % CO2 (je nach Puffer, siehe Abschnitt 1.4.1.4, zweiter Absatz). Es ist dabei besonders wichtig, dass die Zell-Kulturbedingungen eine Zellzykluszeit innerhalb des normalen historischen Bereichs der verwendeten Zellen oder Zelllinien sicherstellen. 1.4.1.3 Ansetzen der Kulturen Zellen aus tiefgefrorenen Mutterkulturen werden in einem Kulturmedium in angemessener Dichte ausgesät und mindestens einmal vor ihrer Verwendung im In-vitro-3T3-NRU-Fototoxizitätstest passagiert. Für den Fototoxizitätstest werden Zellen in einem Kulturmedium in einer geeigneten Dichte ausgesät, die sicherstellt, dass die Kulturen zum Ende des Tests, d. h. wenn die Zellviabilität 48 Stunden nach dem Aussäen der Zellen bestimmt wird, ihre maximale Dichte (Konfluenz) noch nicht erreicht haben. Für Balb/c 3-T3-Zellen, die in "96-well"-Platten kultiviert werden, beträgt die empfohlene Zelldichte 1 x 104 Zellen je "well". Für jede Testchemikalie werden Zellen identisch in zwei separate "96-well"-Platten ausgesät, die ssodan gleichzeitig während des gesamten Testverfahrens unter identischen Kulturbedingungen behandelt werden, abgesehen von dem Zeitraum, in dem eine der Platten (+Irr) bestrahlt und die andere im Dunkeln (-Irr) aufbewahrt wird. 1.4.1.4 Zubereitung der Testsubstanzen Testchemikalien müssen unmittelbar vor ihrer Verwendung frisch zubereitet werden, es sei denn, Haltbarkeitsdaten rechtfertigen eine Lagerung der Präparation. Es wird empfohlen, die Behandlung sämtlicher Chemikalien und die Erstbehandlung der Zellen unter Ausleuchtungsbedingungen durchzuführen, bei denen eine Fotoaktivierung oder Degradation der Testsubstanz vor der Bestrahlung vermieden wird. Die Testchemikalien sollten in einer gepufferten Salzlösung, z.B. EBSS (Earle's Balanced Salt Solution), oder anderen physiologisch ausgewogenen Pufferlösungen, gelöst werden, die, um Interferenzen während der Bestrahlung zu vermeiden, frei sein müssen von Proteinbestandteilen und Licht absorbierenden Bestandteilen (z.B. pH-Indikationsfarben und Vitaminen). Da die Zellen während der Bestrahlung ca. 50 Minuten lang außerhalb des CO2-Inkubators aufbewahrt werden, ist sorgfältig darauf zu achten, dass eine Alkalisierung vermieden wird. Werden schwache Pufferlösungen wie EBSS verwendet, lässt sich dies erreichen, indem die Inkubation der Zellen unter 7,5 %igem CO2 erfolgt. Werden die Zellen bei nur 5 %igem CO2 inkubiert, ist eine stärkere Pufferlösung zu verwenden. Testchemikalien mit eingeschränkter Wasserlöslichkeit sind in einem geeigneten Lösemittel zu lösen. Wird ein Lösemittel verwendet, muss es in allen Kulturen mit konstantem Volumen vorhanden sein, d. h. in den Negativkontrollen (Lösemittelkontrollen) sowie in sämtlichen Konzentrationen der Testchemikalie, und darf bei dieser Konzentration keine zytotoxische Wirkung zeigen. Die Konzentrationen der Testchemikalien sind so zu wählen, dass Ausfällungen oder Trübungen der Lösung vermieden werden. Dimethylsulfoxid (DMSO) und Ethanol (EtOH) werden als Lösemittel empfohlen. Andere Lösemittel mit geringer Zytotoxizität sind unter Umständen ebenfalls geeignet. Vor der Verwendung sind jedoch alle Lösemittel sorgfältig auf spezifische Eigenschaften zu untersuchen, wie Reaktion mit der Testchemikalie, Auslöschen der fototoxischen Wirkung, Einfangen von Radikalen und/oder Stabilität der Chemikalie im Lösemittel. Wirbelmischung (Vortex) und/oder Anwendung von Ultraschall und/oder Erwärmung auf geeignete Temperaturen kommen als Möglichkeiten in Betracht, um die Löslichkeit zu fördern, sofern hierdurch nicht die Stabilität der Testchemikalie beeinträchtigt wird. 1.4.1.5 Bestrahlungsbedingungen 1.4.1.5.1 Lichtquelle Die Wahl der geeigneten Lichtquelle und geeigneter Filter ist ein kritischer Faktor bei den Fototoxizitätstests. Für fototoxische In-vivo-Reaktionen sind in der Regel UVA und sichtbare Bereiche verantwortlich (3) (12), während UVB weniger relevant, aber selbst hochgradig zytotoxisch ist, da seine Zytotoxizität zwischen 313 und 280 nm um ein Tausendfaches zunimmt (13). Kriterien für die Wahl einer geeigneten Lichtquelle müssen die wesentliche Voraussetzung einschließen, dass die Lichtquelle Wellenlängen aussendet, die von der chemischen Testsubstanz absorbiert werden (Absorptionsspektrum), und dass die (in einer akzeptablen Zeit erreichbare) Lichtdosis für den Nachweis bekannter fotozytotoxischer Chemikalien ausreichen muss. Darüber hinaus dürfen die jeweils verwendeten Wellenlängen und Dosen, z.B. der Wärmeemission (Infrarotbereich), für das Testsystem nicht unnötig schädlich sein. Die Simulierung von Sonnenlicht mit Solarsimulatoren wird als optimale Kunstlichtquelle betrachtet. Die Strahlungsleistungsverteilung des gefilterten Solarsimulators muss der in (14) dargestellten Verteilung von Tageslicht im Freien entsprechen. Sowohl Xenon-Lichtbogenlampen als auch (dotierte) Quecksilber-Metallhalogenid-Lichtbogenlampen werden in Solarsimulatoren verwendet (15). Letztere haben den Vorteil, dass sie weniger Wärme emittieren und preiswerter sind, doch ist die Übereinstimmung mit dem Sonnenlicht weniger perfekt als bei Xenon-Lichtbogenlampen. Da alle Solarsimulatoren signifikante Mengen UVB aussenden, müssen sie mit geeigneten Filtern versehen werden, um die hoch zytotoxischen UVB-Wellenlängen zu mindern. Da die Kunststoffmaterialien für Zellkulturen UV-Stabilisatoren enthalten, muss das Spektrum durch den gleichen "96-well"-Plattendeckel gemessen werden, wie er auch in dem Versuch verwendet wird. Unabhängig von Maßnahmen zur Dämpfung eines Teils des Spektrums durch Filterung oder durch unvermeidbare Filtereffekte der Testeinrichtungen darf das unterhalb dieser Filter aufgezeichnete Spektrum nicht vom standardisierten Tageslicht im Freien abweichen (14). Die spektrale Energieverteilung des in der Validierungsstudie zum In-vitro-3T3-NRU-Fototoxizitätstest verwendeten gefilterten Solarsimulators wurde als Beispiel veröffentlicht (8) (16). Siehe dazu auch Anlage 2, Abbildung 1. 1.4.1.5.2 Dosimetrie Die Lichtintensität (Bestrahlungsstärke) sollte regelmäßig vor jedem Fototoxizitätstest unter Verwendung eines geeigneten Breitband-UV-Meters geprüft werden. Die Lichtintensität ist durch die gleiche Ausführung der "96-well"-Plattendeckel zu messen, wie sie auch in dem Versuch verwendet wird. Das UV-Meter muss für die Lichtquelle kalibriert sein. Die Leistungsfähigkeit des UV-Meters ist sicherzustellen. Zu diesem Zweck wird die Verwendung eines zweiten Referenz-UV-Meters des gleichen Typs und der gleichen Kalibrierung empfohlen. Im Idealfall sollte in größeren Abständen mit einem Spektroradiometer die spektrale Energieverteilung der gefilterten Lichtquelle gemessen und die Kalibrierung des Breitband-UV-Meters geprüft werden. Eine Dosis von 5 J/cm2 (im UVA-Bereich gemessen) wurde als nicht zytotoxisch gegenüber Balb/c-3T3-Zellen und als ausreichend stark ermittelt, um fototoxische Chemikalien zu erkennen und damit fototoxische Reaktionen auszulösen (6) (17); so wurde beispielsweise die Bestrahlungsstärke auf 1,7 mW/cm2 eingestellt, um innerhalb von 50 Minuten 5 J/cm2 zu erreichen. Siehe hierzu Anlage 2, Abbildung 2. Wenn eine andere Zelllinie oder eine unterschiedliche Lichtquelle verwendet wird, muss die UVA-Dosis ggf. so angepasst werden, dass Unschädlichkeit gegenüber Zellen und eine ausreichende Stärke zur Erkennung von Standardfototoxinen gegeben ist. Die Zeit der Lichtexposition wird auf folgendem Wege berechnet:
1.4.2 Testbedingungen 1.4.2.1 Konzentrationen der Testsubstanz Die in Anwesenheit (+Irr) und in Abwesenheit (-Irr) von Licht zu testenden Konzentrationsbereiche eines chemischen Stoffs sollten in Dosis-Vorversuchen auf angemessene Weise ermittelt werden. Ggf. empfiehlt es sich, die Löslichkeit eingangs und nach 60 min (bzw. nach der jeweils zugrunde gelegten Behandlungszeit) zu ermitteln, da sich die Löslichkeit zeitabhängig oder im Belichtungsverlauf ändern kann. Um eine durch ungeeignete Bedingungen der Kulturen oder hochgradig saure oder alkalische Chemikalien verursachte Toxizität zu vermeiden, muss der pH-Wert der Zellkulturen mit zugesetzter Testchemikalie im Bereich zwischen 6,5 und 7,8 liegen. Die höchste Konzentration der Testsubstanz muss innerhalb der physiologischen Testbedingungen liegen, d. h., osmotischer Stress und pH-Stress sind zu vermeiden. Je nach Testchemikalie sind ggf. andere physikalischchemische Eigenschaften als Faktoren in Betracht zu ziehen, durch die die höchste Testkonzentration begrenzt wird. Bei relativ unlöslichen Substanzen, die bei Konzentrationen bis zum Sättigungspunkt nicht toxisch sind, ist die höchste erreichbare Konzentration zu verwenden. Grundsätzlich ist eine Ausfällung der Testchemikalie bei einer der Testkonzentrationen zu vermeiden. Die Maximalkonzentration der Testsubstanz darf 1.000 (μg/ ml nicht überschreiten; die Osmolalität darf 10 mmol nicht überschreiten. Es ist eine geometrische Verdünnungsreihe von 8 Testsubstanzenkonzentrationen mit konstantem Verdünnungsfaktor zu verwenden (siehe Abschnitt 2.1, zweiter Absatz). Liegen (aus Vorversuchen) Informationen darüber vor, dass die Testchemikalie bis zur Grenzkonzentration im Dunkelversuch (-Irr) nicht zytotoxisch ist, aber bei Bestrahlung (+Irr) hochgradig zytotoxisch ist, können die Konzentrationsbereiche, die für den (+Irr)-Versuch zugrunde gelegt werden müssen, von den Bereichen für den (-Irr)-Versuch abweichen, damit die Forderung einer ausreichenden Datenqualität erfüllt ist. 1.4.2.2 Kontrollen 1.4.2.2.1 Strahlungsempfindlichkeit der Zellen, Ermittlung historischer Daten Die Zellen sind regelmäßig (etwa jede fünfte Passage) auf Empfindlichkeit gegenüber der Lichtquelle zu kontrollieren; dazu wird ihre Viabilität nach Belichtung mit steigenden Strahlungsdosen beurteilt. Bei dieser Beurteilung sind mehrere verschiedene Strahlungsdosen - einschließlich Dosen, die deutlich über denen des 3T3-NRU-Fototoxizitätstests liegen - zu verwenden. Eine Quantifizierung dieser Dosen ist am einfachsten durch Messung der UV-Anteile der Lichtquelle möglich. Die Zellen werden mit der im In-vitro-3T3-NRU-Fototoxizitätstest verwendeten und am darauf folgenden Tag bestrahlten Dichte ausgesät. Die Zellviabilität wird einen Tag später anhand der Aufnahme von Neutralrot ermittelt. Es muss sich nachweisen lassen, dass die resultierende höchste nicht zytotoxische Dosis (z.B. in der Validierungsstudie: 5 J/cm2 (UVA)) für eine korrekte Klassifizierung der Referenzchemikalien (Tabelle 1) ausreicht. 1.4.2.2.2 Strahlungsempfindlichkeit, Kontrolle des laufenden Tests Der Test erfüllt die Qualitätskriterien, wenn die bestrahlten Negativ-/Lösemittelkontrollen im Vergleich zu nicht bestrahlten Negativ-/Lösemittelkontrollen eine Viabilität von mehr als 80 % aufweisen. 1.4.2.2.3 Viabilität der Lösemittelkontrollen Die absolute optische Dichte (OD540 NRU) des aus den Lösemittelkontrollen extrahierten Neutralrot gibt an, ob die je "well" ausgesäten 1 x 104 Zellen bei normaler Verdopplungszeit während der zwei Tages des Tests gut gewachsen sind. Der Test erfüllt die Akzeptanzkriterien, wenn der mittlere OD540 NRU der unbehandelten Kontrollen > 0,4 ist (d. h. rund das Zwanzigfache des Hintergrund-Lösemittelabsorptionsmaßes). 1.4.2.2.4 Positivkontrolle Ein bekannter fototoxischer chemischer Stoff soll zeitgleich mit jedem In-vitro-3T3-NRU-Fototoxizitätstest getestet werden. Chlorpromazin (CPZ) wird empfohlen. Für mit dem Standardprotokoll im In-vitro-3T3-NRUFototoxizitätstest getestetes CPZ wurden folgende Test-Akzeptanzkriterien festgelegt: bestrahltes CPZ (+Irr): IC50 = 0,1 bis 2,0g/ml, nicht bestrahltes CPZ (-Irr): IC50 = 7,0 bis 90,0g/ml. Der Fotoirritationsfaktor (PIF) sollte > 6 sein. Das historische Verhalten der Positivkontrolle ist zu überwachen. Andere bekannte, für die Chemikalienklasse oder Löslichkeitsmerkmale der bewerteten Chemikalie geeignete fototoxische Chemikalien können an Stelle von Chlorpromazin als gleichzeitige Positivkontrollen verwendet werden. 1.4.3 Testverfahren (6) (7) (8) (16) (17) 1.4.3.1 l. Tag 100l d Kulturmedium werden in die peripheren "wells" einer "96-well"-Gewebekultur-Mikrotiterplatte gegeben (= Blindproben). In die übrigen "wells" werden 100 l der Zellsuspension von 1 x 105 Zellen/ml (= 1 x 104 Zellen/well) pipettiert. Für jede Reihe einzelner Testsubstanzkonzentrationen werden zwei Platten zubereitet; eine weitere Platte wird für die Lösemittel- und Positivkontrollen zubereitet. Die Zellen werden 24 Stunden lang (siehe 1.4.1.2) inkubiert, bis sie einen halbkonfluenten Monolayer bilden. Diese Inkubationszeit ermöglicht die Erholung der Zellen, das Anhaften und ein exponentielles Wachstum. 1.4.3.2 2. Tag Nach der Inkubation wird das Kulturmedium dekantiert. Darauf folgt ein Waschgang mit der für die Inkubation verwendeten Pufferlösung. 100l der Pufferlösung, welche die geeignete Konzentration der Testchemikalie oder des Lösemittels (Lösemittelkontrolle) enthält, werden hinzugefügt. Es sind 8 verschiedene Konzentrationen der Testchemikalie zu verwenden. Die Zellen werden mit der Testchemikalie im Dunkeln 60 Minuten lang inkubiert (siehe Abschnitt 1.4.1.2 und Abschnitt 1.4.1.4, zweiter Absatz). Aus den zwei für jede Reihe der Testsubstanzkonzentrationen und Kontrollen vorbereiteten Platten wird - normalerweise willkürlich - eine Platte für die Bestimmung der Zytotoxizität (Irr) ausgewählt (d. h. die Kontrollplatte) sowie eine Platte (die Behandlungsplatte) für die Bestimmung der Fotozytotoxizität (+Irr). Zur Durchführung der +Irr-Belichtung werden die Zellen bei Raumtemperatur 50 Minuten lang durch den Deckel der "96-well"-Platte mit der höchsten Strahlungsdosis bestrahlt, die nicht zytotoxisch ist (siehe auch Anlage 2). Nicht bestrahlte Platten (-Irr) werden 50 Minuten lang (= Lichtexpositionszeit) bei Raumtemperatur in einem dunklen Kasten gehalten. Die Testflüssigkeit wird dekantiert. Es folgen zwei Waschgänge mit 150 µl der für die Inkubation verwendeten Pufferlösung, jedoch ohne das Testmaterial. Die Pufferlösung wird durch ein Kulturmedium ersetzt und über Nacht (18-22 h) inkubiert (siehe 1.4.1.2). 1.4.3.3 3. Tag 1.4.3.3.1 Mikroskopische Evaluierung Die Zellen werden unter einem Phasen-Kontrast-Mikroskop auf Wachstum, Morphologie und Intaktheit der Monolayer untersucht. Morphologische Veränderungen der Zelle und Wirkungen auf das Zellwachstum sind aufzuzeichnen. 1.4.3.3.2 Neutralrot-Aufnahme-Test Die Zellen werden mit 150 µl vorgewärmter Pufferlösung gewaschen. Die Waschlösung wird durch vorsichtiges Absaugen entfernt. 100 µl einer 50g/ml Neutralrotsubstanz (NR) (3-Amino-7-Dimethylamino-2-Methylphenazin-Hydrochlorid, EINECS-Nummer 209-035-8, CAS-Nummer 553-24-2, C.I. 50040) in einem Medium ohne Serum werden hinzugefügt (16) und 3 Stunden lang gemäß den Angaben in Abschnitt 1.4.1.2 inkubiert. Nach der Inkubation wird das NR-Medium entfernt, und die Zellen werden mit 150 µl Pufferlösung gewaschen. Die überschüssige Pufferlösung wird vollkommen dekantiert und durch Absaugen oder Zentrifugieren entfernt. Es werden genau 150 µl NR-Desorptionslösung (frisch zubereitet aus 49 Teilen Wasser + 50 Teilen Ethanol + 1 Teil Essigsäure) hinzugefügt. Die Mikrotiter-Platte wird 10 Minuten lang auf einem Mikrotiter-Platten-Schüttler vorsichtig geschüttelt, bis das NR aus den Zellen extrahiert ist und eine homogene Lösung bildet. Die optische Dichte der NR-Extrakte wird bei 540 nm in einem Spektralfotometer gemessen, wobei die Blindproben als Referenz verwendet werden. Die Daten sind in angemessenem Dateiformat für spätere Analysen aufzuzeichnen. 2. Daten 2.1 Qualität und Quantität der Daten Die Testdaten sollten eine sinnvolle Analyse der in Anwesenheit und in Abwesenheit von Strahlung ermittelten Konzentrations-Wirkungs-Reaktionen sowie, wenn möglich, die Konzentration der Testchemikalien, bei der die Zellviabilität auf 50 % (IC50) sinkt, ermöglichen. Sofern Zytotoxizität festgestellt wird, sollten sowohl der Konzentrationsbereich als auch der Abstand einzelner Konzentrationen so gewählt sein, dass die experimentellen Daten in einer Kurve dargestellt werden können. Bei klar positiven und klar negativen Ergebnissen (siehe Abschnitt 2.3, erster Absatz) ist gegebenenfalls der Hauptversuch - begleitet von einem oder mehreren Vorversuchen - ausreichend. Mehrdeutige, grenzwertige oder unklare Ergebnisse sind durch weitere Tests abzuklären (siehe hierzu auch Abschnitt 2.4, zweiter Absatz). In derartigen Fällen ist auch eine Änderung der Versuchsbedingungen in Betracht zu ziehen. Zu den Versuchsbedingungen, die geändert werden könnten, zählen der Konzentrationsbereich und -raum, die Vorinkubationszeit und die Strahlungsexpositionszeit. Für Chemikalien, die in Wasser instabil sind, ist evtl. eine kürzere Expositionszeit ausreichend. 2.2 Ergebnisbewertung Zur Evaluierung der Daten kann ein Fotoirritationsfaktor (PIF) oder der Mean Photo Effect (MPE) berechnet werden. Zur Berechnung der Maße für die Fotozytotoxizität (siehe unten) müssen die Dosis-Wirkungs-Werte näherungsweise durch eine geeignete kontinuierliche Dosis-Wirkungs-Kurve (Modell) dargestellt werden. Die Anpassung der Kurve an die Daten erfolgt normalerweise nach dem nichtlinearen Regressionsverfahren (18). Zur Beurteilung des Einflusses der Datenvariabilität auf die angepasste Kurve wird die Verwendung eines Bootstrap-Verfahrens empfohlen. Der Fotoirritationsfaktor (PIF) wird nach der folgenden Formel berechnet: PIF = IC50 (-Irr) / IC50 (+Irr) Ist die Berechnung von IC50 bei Anwesenheit oder Abwesenheit von Licht nicht möglich, kann der PIF des Testmaterials nicht ermittelt werden. Der MPE (Mean Photo Effect) basiert auf dem Vergleich der vollständigen Konzentrations-Wirkungs-Kurven (19). Er wird definiert als der gewichtete Durchschnitt einer repräsentativen Gruppe von Fotoeffektwerten. Der Fotoeffekt PEc bei einer beliebigen Konzentration C ist definiert als das Produkt des Wirkungseffekts REc und des Dosiseffekts DEc, d. h. PEc = REc x DEc. Der Wirkungseffekt REc bezeichnet die Differenz zwischen den bei Abwesenheit und Anwesenheit von Licht ermittelten Wirkungen, d. h. REc = Rc (-Irr) - Rc (+Irr). Der Dosiseffekt wird ausgedrückt durch Dabei stellt C* die Äquivalenzkonzentration dar, d. h. die Konzentration, bei der die +Irr-Wirkung der -Irr-Wirkung bei Konzentration C entspricht. Kann C* nicht ermittelt werden, weil die Wirkungswerte der +Irr-Kurve systematisch über oder unter RC(-Irr) liegen, wird der Dosiseffekt gleich 1 gesetzt. Die Gewichtungsfaktoren wi werden durch den höchsten Wirkungswert ausgedrückt, d. h. wi = MAX {Ri (+Irr), Ri (-Irr)}. Das Konzentrationsgitter Ci wird so gewählt, dass die gleiche Anzahl Punkte in jedes der Konzentrationsintervalle fällt, die durch die im Versuch verwendeten Konzentrationswerte definiert sind. Die Berechnung von MPE wird auf den maximalen Konzentrationswert beschränkt, bei dem mindestens eine der beiden Kurven noch einen Wirkungswert von mindestens 10 % aufweist. Liegt diese Maximalkonzentration über der höchsten Konzentration, die im +Irr-Versuch verwendet wurde, wird der restliche Teil der +Irr-Kurve gleich dem Wirkungswert "0" gesetzt. Je nachdem, ob der MPE-Wert größer als ein auf geeignete Weise gewählter Schwellenwert (MPEc = 0,15) ist oder nicht, wird die Chemikalie als fototoxisch eingestuft. Ein Softwarepaket für die Berechnung von PIF und MPE ist bei (20) erhältlich. 2.3 Interpretation der Ergebnisse Nach der Validierungsstudie (8) bedeutet eine Testsubstanz mit PIF < 2 oder MPE < 0,1 die Prognose "keine Fototoxizität". Ein PIF > 2 und < 5 oder ein MPE > 0,1 und < 0,15 entspricht der Prognose: "Fototoxizität wahrscheinlich"; ein PIF > 5 oder ein MPE > 0,15 bedeutet: "Fototoxizität". Bei jedem Labor, das den einleitenden Aufbau dieses Versuchs vornimmt, sind die in Tabelle 1 aufgeführten Referenzmaterialien zu testen, bevor die Testsubstanzen zur fototoxischen Beurteilung getestet werden. Die PIF- oder MPE-Werte müssen in Nähe der in Tabelle 1 angegebenen Werte liegen.
2.4 Interpretation der Daten Werden fototoxische Wirkungen nur bei der höchsten Testkonzentration festgestellt (insbesondere bei wasserlöslichen Testchemikalien), sind zur Gefahrenbeurteilung evtl. noch weitere Gesichtspunkte in Betracht zu ziehen, so z.B. Daten zur Absorption über die Haut sowie zur Ansammlung der Chemikalie in der Haut und/oder Daten aus anderen Tests, z.B. In-vitro-Tests der Chemikalie auf tierischer bzw. menschlicher Haut oder Hautmodellen. Wird keine Toxizität nachgewiesen (+Irr und -Irr) und sind die Konzentrationen, die getestet werden können, durch mangelhafte Löslichkeit begrenzt, ist die Vergleichbarkeit der Testsubstanz mit dem durchgeführten Test zweifelhaft; in diesem Fall ist die Durchführung konfirmativer Tests, z.B. mit einem anderen Modell, in Betracht zu ziehen. 3. Berichterstattung Testbericht Der Testbericht muss mindestens folgende Informationen umfassen: Testsubstanz:
Lösemittel:
Zellen:
Testbedingungen (1); Inkubation vor und nach der Behandlung:
Testbedingungen (2); Behandlung mit der Chemikalie:
Testbedingungen (3); Bestrahlung:
Testbedingungen (4); Neutralrot-Viabilitätstest:
Ergebnisse:
Diskussion der Ergebnisse; Schlussfolgerungen. 4. Literaturhinweise (1) Lovell W.W. (1993). A scheme for in vitro screening of substances for photoallergenic potential. Toxic. In Vitro 7, 95-102. (2) Santamaria, L. and Prino, G. (1972). List of the photodynamic substances. InResearch Progress in Organic, Biological and Medicinal ChemistryVol. 3 part 1. North Holland Publishing Co. Amsterdam, XI-XXXV. (3) Spielmann, H., Lovell, W.W., Hölzle, E., Johnson, B.E., Maurer, T., Miranda, M.A., Pape, W.J.W., Sapora, O., and Sladowski, D. (1994). In vitro phototoxicity testing: The report and recommendations of ECVAM Workshop 2. ATLA, 22, 314-348. (4) Spikes, J.D. (1989). Photosensitization. In "The science of Photobiology" Edited by K.C. Smith. Plenum Press, New York. 2nd edition, 79-110. (5) OECD (1997) Environmental Health and Safety Publications, Series on Testing and Assessment No.7 ,Guidance Document On Direct Phototransformation Of Chemicals In WaterEnvironment Directorate, OECD, Paris. (6) Spielmann, H., Balls, M., Döring, B., Holzhütter, H.G., Kalweit, S., Klecak, G., L'Eplattenier, H., Liebsch, M., Lovell, W.W., Maurer, T., Moldenhauer. F. Moore. L., Pape, W., Pfannbecker, U., Potthast, J., De Silva, O., Steiling, W., and Willshaw, A. (1994). EEC/COLIPA project on in vitro phototoxicity testing: First results obtained with a Balb/c 3T3 cell phototoxicity assay. Toxic. In Vitro 8, 793-796. (7) Anon (1998). Statement on the scientific validity of the 3T3 NRU PT test (an in vitro test for phototoxicity), European Commission, Joint Research Centre: ECVAM and DGXI/E/2, 3. November 1997, ATLA, 26, 7-8. (8) Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., Pechovitch, G. De Silva, O., Holzhütter, H.G., Clothier, R., Desolle, P., Gerberick, F., Liebsch, M., Lovell, W.W., Maurer, T., Pfannenbecker, U., Potthast, J. M., Csato, M., Sladowski, D., Steiling, W., and Brantom, P. (1998). The international EU/COLIPA In vitro phototoxicity validation study: results of phase II (blind trial), part 1: the 3T3 NRU phototoxicity test. Toxic. In Vitro 12, 305-327. (9) OECD (2002) Extended Expert Consultation Meeting on The In Vitro 3T3 NRU Phototoxicity Test Guideline Proposal, Berlin, 30th-31th Otober 2001, Secretariat's Final Summary Report, 15th March 2002, OECD ENV/EHS; auf Anfrage beim Sekretariat erhältlich. (10) Borenfreund, E., and Puerner, J.A. (1985). Toxicity determination in vitro by morphological alterations and neutral red absorption. Toxicology Lett., 24, 119-124. (11) Hay, RJ. (1988) The seed stock concept and quality control for cell lines. Analytical Biochemistry 171, 225-237. (12) Lambert L.A, Warner W.G., and Kornhauser A. (1996) Animal models for phototoxicity testing. In ,Dermatotoxicology, edited by F.N. Marzulli and H.I. Maibach. Taylor & Francis, Washington DC. 5th Edition, 515-530. (13) Tyrrell R.M., Pidoux M (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet and violet regions of sunlight on epidermal keratinocytes. Cancer Res., 47, 1825-1829. (14) ISO 10977. (1993). Photography - Processed photographic colour films and paper prints - Methods for measuring image stability. (15) Sunscreen Testing (UV.B) TECHNICALREPORT, CIE, International Commission on Illumnation, Publication No. 90, Vienna, 1993, ISBN 3900734275 (16) ZEBET/ECVAM/COLIPA - Standard Operating Procedure: In Vitro 3T3 NRU Phototoxicity Test. Final Version, 7 September, 1998. 18 S. (17) Spielmann, H., Balls, M., Dupuis, J., Pape, W.J.W., De Silva, O., Holzhütter, H.G., Gerberick, F., Liebsch, M., Lovell, W.W., and Pfannenbecker, U. (1998) A study on UV filter chemicals from Annex VII of the European Union Directive 76/768/EEC, in the in vitro 3T3 NRU phototoxicity test. ATLA 26, 679-708. (18) Holzhütter, H.G., and Quedenau, J. (1995) Mathematical modeling of cellular responses to external signals. J. Biol. Systems 3, 127-138. (19) Holzhütter, H.G. (1997). A general measure of in vitro phototoxicity derived from pairs of doseresponse curves and its use for predicting the in vivo phototoxicity of chemicals. ATLA, 25, 445-462. (20) http://www.oecd.org/document/55/0.2340.en_2649_34377_2349687_l_l_l_1.00. html
(Siehe 1.4.1.5, zweiter Absatz) Abbildung 1 zeigt ein Beispiel für eine akzeptable spektrale Leistungsverteilung eines gefilterten Solarsimulators. Sie stammt aus der Quelle mit dotiertem Metallhalid, die im Validierungsversuch des 3T3-NRU-PT verwendet wurde (6) (8) (17). Die Wirkung der beiden verschiedenen Filter und der zusätzliche Filtereffekt des Deckels einer "96-well"-Zellkulturplatte werden hier dargestellt. Der H2-Filter wurde nur mit Testsystemen verwendet, die eine größere UVB-Menge tolerieren können (Hautmodelltest und Fotohämolysetest mit roten Blutzellen). Im 3T3-NRU-PT wurde der H1-Filter verwendet. Die Abbildung zeigt, dass der zusätzliche Filtereffekt des Plattendeckels in erster Linie im UVB-Bereich beobachtet wurde, so dass im Bestrahlungsspektrum noch genug UVB verbleibt, um Chemikalien zu erkennen, die typischerweise im UVB-Bereich absorbiert werden, z.B. Amiodaron (siehe Tabelle 1). Abbildung 2 Bestrahlungsempfindlichkeit von Balb/c-3T3-Zellen (im UVA-Bereich gemessen) Zellviabilität (Aufnahme von Neutralrot in % bei Dunkelkontrollen) (Siehe 1.4.1.5.2, zweiter Absatz, 1.4.2.2.1, 1.4.2.2.2) Empfindlichkeit der Balb/c-3T3-Zellen gegen Bestrahlung durch den Solarsimulator, der im Validierungsversuch des 3T3-NRU-Fototoxizitätstests (nach Messung im UVA-Bereich) verwendet wird. Die Abbildung zeigt die in sieben verschiedenen Labors in der Vorvalidierungsstudie ermittelten Ergebnisse (1). Die beiden Kurven mit offenen Symbolen wurden mit gealterten Zellen ermittelt (hohe Passagenzahl), die durch neue Zellenbestände ersetzt werden mussten. Die Kurven mit fett gedruckten Symbolen zeigen Zellen mit einer ausreichenden Bestrahlungstoleranz. Aus diesen Daten wurde die höchste nicht zytotoxische Bestrahlungsdosis von 5 J/cm2 abgeleitet (senkrechte gestrichelte Linie). Die waagerechte gestrichelte Linie zeigt zusätzlich die in Abschnitt 1.4.2.2 angegebene maximal zulässige Bestrahlungswirkung. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||


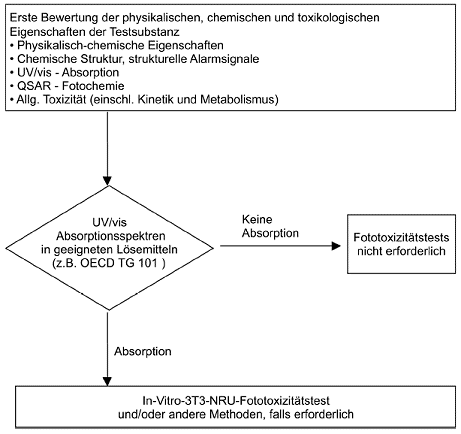


| weiter . |  |
...
X
⍂
↑
↓