
umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (16)
 | zurück |
|
B.37 Verzögerte Neurotoxizität phosphororganischer Substanzen nach akuter Exposition
1. Methode
1.1 Einleitung
Bei der Abschätzung und Bewertung der toxischen Wirkungen von Substanzen muss auch das Potenzial bestimmter Substanzklassen für spezifische neurotoxische Wirkungen, die in anderen Toxizitätsstudien möglicherweise nicht nachweisbar sind, untersucht werden. Bei einigen phosphororganischen Verbindungen wurden verzögerte neurotoxische Wirkungen beobachtet, diese Substanzen kommen für eine Untersuchung mit der vorliegenden Methode in Frage.
In-vitro-Screening-Tests könnten verwendet werden, um Substanzen zu identifizieren, die eine verzögerte Polyneuropathie hervorrufen können; allerdings lassen negative Ergebnisse von In-vitro-Studien nicht darauf schließen, dass die Prüfsubstanz kein Neurotoxikum ist.
Siehe auch allgemeine Einleitung zu Teil B.
1.2 Definitionen
Zu den phosphororganischen Substanzen zählen ungeladene phosphororganische Ester, Thioester oder Anhydride von Phosphinsäuren, Phosphonsäuren bzw. Phosphoramidsäuren oder die engverwandten Thiophosphinsäuren, Thiophosphonsäuren bzw. Thiophosphoramidsäuren oder sonstige Substanzen, welche die bei dieser Substanzklasse bisweilen zu beobachtende verzögerte Neurotoxizität aufweisen können.
Verzögerte Neurotoxizität ist ein Syndrom, das mit langsamem, verzögertem Auftreten von Ataxien, distalen Axonopathien in Rückenmarks- und peripheren Nerven sowie einer Hemmung und Alterung der Neuropathy Target Esterase (NTE) im Nervengewebe verbunden ist.
1.3 Referenzsubstanzen
Eine Referenzsubstanz kann mit einer positiven Kontrollgruppe getestet werden, um nachzuweisen, dass sich die Reaktion der geprüften Spezies unter den Laborversuchsbedingungen nicht wesentlich verändert hat.
Ein Beispiel für ein weit verbreitetes Neurotoxikum ist das Tri-otolylphosphat (CAS 78-30-8, EINECS 201-103-5, CAS-Bezeichnung: Tris(2-methylphenyl)phosphorsäureester), das auch als Tris-ocresylphosphat bezeichnet wird.
1.4 Prinzip der Prüfmethode
Die Prüfsubstanz wird oral als Einmalgabe an Haushühner verabreicht, die ggf. gegen akute cholinergische Wirkungen geschützt wurden. Die Tiere werden für die Dauer von 21 Tagen auf Verhaltensauffälligkeiten, Ataxien und Paralysen hin beobachtet. Biochemische Parameter, insbesondere die Neuropathy Target Esterase (NTE), werden im allgemeinen 24 und 48 Stunden nach Verabreichung bei Hennen bestimmt, die nach Zufallskriterien aus jeder Gruppe ausgewählt werden. 21 Tage nach der Exposition werden die verbliebenen Hennen getötet, und ausgewählte Nervengewebe werden histopathologisch untersucht.
1.5 Beschreibung der Prüfmethode
1.5.1 Vorbereitungen
Gesunde junge erwachsene Hennen, die frei von störenden Viruserkrankungen und Arzneimitteln sind und keine Gangstörungen aufweisen, werden randomisiert, der Behandlungs- oder der Kontrollgruppe zugeteilt und für die Dauer von mindestens 5 Tagen vor Beginn der Studie an die Laborbedingungen akklimatisiert.
Die Käfige oder Gehege sollten so groß sein, dass sich die Tiere frei bewegen können und ihr Gangbild gut zu beobachten ist.
Die Gabe der Prüfsubstanz erfolgt normalerweise auf oralem Wege mittels Magensonde, Gelatinekapseln oder einer vergleichbaren Methode. Flüssigkeiten können unverdünnt oder aufgelöst in einem geeigneten Vehikel wie z.B. Maisöl verabreicht werden. Feste Substanzen sollten nach Möglichkeit aufgelöst werden, da größere Dosen fester Stoffe in Gelatinekapseln möglicherweise nicht wirksam resorbiert werden. Bei nichtwässrigen Vehikeln sollten deren toxische Eigenschaften bekannt sein. Ist dies nicht der Fall, müssen sie vor der Prüfung bestimmt werden.
1.5.2 Prüfbedingungen
1.5.2.1 Versuchstiere
Junge erwachsene Legehennen (Gallus gallus domesticus) im Alter von 8 bis 12 Monaten werden für den Test empfohlen. Normalgroße Rassen sollten verwendet werden, und die Hennen sollten in der Regel unter Bedingungen aufgewachsen sein, unter denen sie sich frei bewegen konnten.
1.5.2.2 Anzahl und Geschlecht
Neben der Behandlungsgruppe sollte eine Vehikelkontrollgruppe und eine positive Kontrollgruppe getestet werden. Die Vehikelkontrollgruppe ist, abgesehen von der Verabreichung der Prüfsubstanz, genauso zu behandeln wie die Behandlungsgruppe.
Die Anzahl der Tiere in jeder Gruppe sollte so bemessen sein, dass mindestens sechs Tiere zur Bestimmung biochemischer Parameter (jeweils drei Tiere zu zwei Beobachtungszeitpunkten) getötet werden können und sechs Tiere für die pathologische Untersuchung bis zum 21. Tag überleben.
Die positive Kontrollgruppe kann gleichzeitig getestet werden oder es kann sich um eine bereits zuvor untersuchte Kontrollgruppe handeln. Die Gruppe sollte aus mindestens sechs Hennen bestehen, die mit einem bekannten verzögerten Neurotoxikum behandelt wurden. Drei der Tiere sind für biochemische, drei für pathologische Untersuchungen nach Ende des Tests vorgesehen. Es empfiehlt sich eine regelmäßige Aktualisierung der Archivdaten. Neue positive Kontrolldaten sollten erarbeitet werden, wenn ein wesentlicher Aspekt des Testablaufs (z.B. Rasse, Futter, Haltungsbedingungen) vom durchführenden Labor geändert wurde.
1.5.2.3 Dosierungen
In einer Vorstudie an einer geeigneten Zahl von Tieren und Dosisgruppen ist die in der Hauptstudie zu verwendende Dosis zu ermitteln. Hierbei sind in der Regel einige Todesfälle erforderlich, um eine angemessene Dosis für die Hauptstudie festlegen zu können. Um aber Todesfälle durch akute cholinerge Wirkungen zu vermeiden, kann Atropin oder ein anderer schützender Wirkstoff, von dem man weiß, dass er keinen Einfluss auf verzögerte neurotoxische Wirkungen hat, verabreicht werden. Zur Abschätzung der maximalen nicht letalen Dosis der Prüfsubstanz stehen eine Reihe verschiedener Methoden zur Auswahl (siehe Methode B.1 bis). Bereits vorliegende frühere Daten für Hennen oder andere toxikologische Daten können ebenfalls bei der Wahl der Dosis von Bedeutung sein.
Die Dosis der Prüfsubstanz in der Hauptstudie sollte unter Berücksichtigung der Ergebnisse der Vorstudie zur Dosisfindung sowie der oberen Grenzdosis von 2.000 mg/kg Körpergewicht möglichst hoch sein. Auch wenn Todesfälle auftreten, sollten bis zum 21. Tag genügend Tiere für die biochemischen (sechs) und die histologischen Untersuchungen (sechs) überleben. Um Todesfälle infolge akuter cholinergischer Wirkungen auszuschließen, sollte Atropin oder ein anderer schützender Wirkstoff, der nachgewiesenermaßen keinen Einfluss auf verzögerte neurotoxische Wirkungen hat, gegeben werden.
1.5.2.4 Limit-Test
Verursacht die Prüfung einer Dosis von mindestens 2.000 mg/kg Körpergewicht pro Tag unter Verwendung der für diese Studie beschriebenen Verfahren keine feststellbaren toxischen Wirkungen und ist aufgrund der Daten strukturverwandter Substanzen keine Toxizität zu erwarten, kann auf eine Studie mit einer höheren Dosis gegebenenfalls verzichtet werden. Der Limit-Test findet allerdings keine Anwendung, wenn die Expositionswirkungen beim Menschen die Prüfung in einer höheren Dosis angezeigt erscheinen lassen.
1.5.2.5 Beobachtungszeitraum
Der Beobachtungszeitraum sollte 21 Tage betragen.
1.5.3 Versuchsdurchführung
Nach Verabreichung eines schützenden Wirkstoffs zur Verhinderung von Todesfällen durch akute cholinergische Wirkungen wird die Prüfsubstanz als Einmalgabe verabreicht.
1.5.3.1 Allgemeine Beobachtungen
Die Beobachtungen sollten unmittelbar nach der Exposition beginnen. Alle Hennen sind in den ersten beiden Tagen mehrmals am Tag und danach bis zum 21. Tag oder bis zur vorgesehenen Tötung mindestens einmal täglich zu beobachten. Alle Toxizitätszeichen, einschließlich Zeitpunkt des Beginns, Art, Schweregrad und Dauer von Verhaltensauffälligkeiten, sind zu dokumentieren. Die Ataxie wird auf einer Ordinalskala aus mindestens vier Stufen bewertet, und Paralysen werden dokumentiert. Mindestens zweimal wöchentlich sollten die für die pathologische Untersuchung vorgesehenen Hennen aus dem Käfig genommen und für einen bestimmten Zeitraum zu verstärkter motorischer Aktivität, z.B. durch Begehen einer Leiter, gezwungen werden, um die Beobachtung bereits geringster toxischer Wirkungen zu erleichtern. Moribunde Tiere und Tiere, bei denen schweres Leiden oder starke Schmerzen festgestellt werden, sollten ausgesondert, unter Verwendung einer tierschutzgerechten Methode getötet und seziert werden.
1.5.3.2 Körpergewicht
Alle Hennen müssen unmittelbar vor der Verabreichung der Prüfsubstanz und danach mindestens einmal wöchentlich gewogen werden.
1.5.3.3 Biochemische Untersuchungen
Je sechs Hennen, die nach Zufallskriterien aus der Behandlungs- und der Vehikelkontrollgruppe ausgewählt werden, sowie drei Hennen aus der positiven Kontrollgruppe (wenn diese Gruppe gleichzeitig getestet wird), sollten einige Tage nach der Gabe der Prüfsubstanz getötet werden, um Gehirn und lumbales Rückenmark zu präparieren und auf hemmende Aktivität gegen Neuropathy Target Esterase zu untersuchen. Auch die Präparation und Untersuchung des Ischiasnervengewebes auf hemmende Aktivität gegen Neuropathy Target Esterase kann aufschlussreich sein. In der Regel werden drei Tiere der Negativkontrolle sowie jeder Behandlungsgruppe nach 24 Stunden und drei nach 48 Stunden getötet, während die drei Hennen der Positivkontrolle nach 24 Stunden getötet werden sollten. Zeigt die Beobachtung der klinischen Zeichen der Intoxikation (z.B. durch Beobachtung des zeitlichen Beginns der cholinergen Zeichen), dass der toxische Wirkstoff möglicherweise ausgesprochen langsam eliminiert wird, empfiehlt es sich gegebenenfalls, von je drei Tieren zu jeweils zwei Zeitpunkten innerhalb von 24 bis spätestens 72 Stunden nach der Verabreichung der Prüfsubstanz Gewebeproben zu entnehmen.
An diesen Proben können auch Bestimmungen der Acetylcholinesterase (AChE) vorgenommen werden, wenn dies angezeigt erscheint. Allerdings kann es in vivo zur spontanen Reaktivierung von AChE kommen, so dass die Wirksamkeit der Prüfsubstanz als AChE-Hemmer unterschätzt wird.
1.5.3.4 Autopsie
Alle Versuchstiere (sowohl die nach Plan getöteten als auch die aufgrund ihres moribunden Zustands getöteten) werden einer Autopsie zur Untersuchung von Gehirn und Rückenmark unterzogen.
1.5.3.5 Histopathologische Untersuchung
Nervengewebe von Tieren, die die Beobachtungsdauer überleben und nicht für biochemische Untersuchungen vorgesehen sind, wird mikroskopisch untersucht. Die Gewebe werden in situ mittels Perfusionsverfahren fixiert. Gewebeschnitte werden unter anderem von Cerebellum (mittlere Längsebene), Medulla oblongata, Rückenmark und peripheren Nerven angefertigt. Die Rückenmarkschnitte sind vom oberen Halswirbelsegment, der mittleren thorakalen und der lumbosakralen Region zu entnehmen. Auch vom distalen Bereich des Nervus tibialis und seiner Verzweigungen zum Musculus gastrocnemius sowie vom Nervus sciaticus sollten Schnitte angefertigt werden. Die Schnitte werden mit geeigneten myelin- und axonspezifischen Färbemitteln angefärbt.
2. Daten
Negative Ergebnisse für die in dieser Methode gewählten Endpunkte (Biochemie, Histopathologie und Verhaltensbeobachtung) erfordern normalerweise keine weiteren Untersuchungen auf eine verzögerte Neurotoxizität. Zweifelhafte oder unklare Ergebnisse für diese Endpunkte erfordern ggf. weitere Untersuchungen.
Die Daten für die einzelnen Tiere sollten aufgeführt werden. Ferner sollten alle Daten in tabellarischer Form zusammengefasst werden. Daraus müssen für jede Prüfgruppe folgende Angaben hervorgehen: die Anzahl der verwendeten Tiere bei Beginn der Prüfung, die Anzahl der Tiere mit Läsionen, verhaltensbezogenen oder biochemischen Wirkungen, die Arten und Schweregrade dieser Läsionen bzw. Wirkungen sowie der Anteil der von den verschiedenen Arten und Schweregraden der Läsionen bzw. Wirkungen betroffenen Tiere.
Die Ergebnisse dieser Studie sind im Hinblick auf Häufigkeit, Schweregrad und Korrelation der verhaltensbezogenen, biochemischen und histopathologischen Wirkungen sowie sonstiger verzeichneter Wirkungen in den Behandlungs- und Kontrollgruppen zu bewerten.
Die numerischen Daten sind nach Möglichkeit durch geeignete und allgemein akzeptierte statistische Verfahren auszuwerten. Diese statistischen Verfahren sollten bei der Festlegung des Designs der Studie ausgewählt werden.
3. Abschlussbericht
Prüfbericht
Der Prüfbericht soll nach Möglichkeit folgende Angaben enthalten:
Versuchstiere:
- verwendete Rasse;
- Anzahl und Alter;
- Herkunft, Haltungsbedingungen usw.;
- Gewicht der einzelnen Tiere bei Prüfungsbeginn.
Prüfbedingungen:
- Angaben zur Zubereitung der Prüfsubstanz, zur Stabilität und ggf. zur Homogenität;
- Begründung für die Wahl des Vehikels;
- Angaben zur Verabreichung der Prüfsubstanz;
- Angaben über Futter- und Wasserqualität;
- Begründung für die Wahl der Dosis;
- Angabe der verabreichten Dosen mit Angaben zu Vehikel, Volumen und physikalischer Form des verabreichten Stoffes;
- Identität und Angaben zur Verabreichung eventueller schützender Wirkstoffe.
3.3 Ergebnisse:
- Angaben zum Körpergewicht;
- Daten der toxischen Reaktionen nach Gruppen, einschließlich Todesfälle;
- Art, Schweregrad und Dauer der klinischen Beobachtungen (mit Angaben zur Reversibilität);
- ausführliche Beschreibung der biochemischen Methoden und Befunde;
- Sektionsbefunde;
- ausführliche Beschreibung aller histopathologischen Befunde;
- ggf. statistische Auswertung der Ergebnisse.
Diskussion der Ergebnisse.
Schlussfolgerungen.
4. Literatur
Diese Methode entspricht der OHCD TG 418.
B.38 Verzögerte Neurotoxizität phosphororganischer Substanzen bei wiederholter Gabe über 28 Tage
1. Methode
1.1 Einleitung
Bei der Abschätzung und Bewertung der toxischen Wirkungen von Substanzen muss auch das Potenzial bestimmter Substanzklassen für spezifische neurotoxische Wirkungen, die in anderen Toxizitätsstudien möglicherweise nicht nachweisbar sind, untersucht werden. Bei einigen phosphororganischen Verbindungen wurden verzögerte neurotoxische Wirkungen beobachtet; diese Substanzen kommen für eine Untersuchung mit der vorliegenden Methode in Frage.
In-vitro-Screening-Tests könnten verwendet werden, um Substanzen zu identifizieren, die eine verzögerte Polyneuropathie hervorrufen können; allerdings lassen negative Ergebnisse von In-vitro-Studien nicht darauf schließen, dass die Prüfsubstanz kein Neurotoxikum ist.
Diese Prüfung auf verzögerte Neurotoxizität nach 28-tägiger Gabe gibt Aufschluss über mögliche Gesundheitsgefahren infolge wiederholter Exposition über einen begrenzten Zeitraum. Die Prüfung erlaubt Rückschlüsse auf die Dosis-Wirkungs-Beziehung und ermöglicht die Abschätzung einer Schwellendosis, unterhalb der keine toxische Wirkung mehr feststellbar ist (NOAEL). Mit Hilfe des NOAEL lassen sich Sicherheitskriterien für die Exposition beim Menschen festlegen.
Siehe auch allgemeine Einleitung zu Teil B.
1.2 Definitionen
Zu den phosphororganischen Substanzen zählen ungeladene phosphororganische Ester, Thioester oder Anhydride von Phosphinsäuren, Phosphonsäuren bzw. Phosphoramidsäuren oder die eng verwandten Thiophosphinsäuren, Thiophosphonsäuren bzw. Thiophosphoramidsäuren oder sonstige Substanzen, welche die bei dieser Substanzklasse bisweilen zu beobachtende verzögerte Neurotoxizität hervorrufen können.
Verzögerte Neurotoxizität ist ein Syndrom, das mit langsamem, verzögertem Auftreten von Ataxien, distalen Axonopathien in Rückenmarks- und peripheren Nerven sowie einer Hemmung und Alterung der Neuropathy Target Esterase (NTE) im Nervengewebe verbunden ist.
1.3 Prinzip der Prüfmethode
Die Prüfsubstanz wird 28 Tage lang einmal täglich oral an Haushühner verabreicht. Die Tiere werden bis 14 Tage nach der letzten Gabe mindestens einmal täglich auf auffälliges Verhalten, Ataxie und Paralysen hin beobachtet. Biochemische Parameter, insbesondere die Neuropathy Target Esterase (NTE), werden im allgemeinen 24 und 48 Stunden nach Verabreichung der letzten Dosis bei Hennen bestimmt, die nach Zufallskriterien aus jeder Gruppe ausgewählt werden. Zwei Wochen nach der letzten Gabe werden die verbliebenen Hennen getötet, und ausgewählte Nervengewebe werden histopathologisch untersucht.
1.4 Beschreibung der Prüfmethode
1.4.1 Vorbereitungen
Gesunde junge erwachsene Hennen, die frei von störenden Viruserkrankungen und Arzneimitteln sind und keine Gangstörungen aufweisen, werden randomisiert, der Behandlungs- oder der Kontrollgruppe zugeteilt und für die Dauer von mindestens 5 Tagen vor Beginn der Studie an die Laborbedingungen akklimatisiert.
Die Käfige oder Gehege sollten so groß sein, dass sich die Tiere frei bewegen können und ihr Gangbild gut zu beobachten ist.
Die orale Gabe der Prüfsubstanz an 7 Tagen in der Woche sollte vorzugsweise mittels Magensonde oder Verabreichung von Gelatinekapseln erfolgen. Flüssigkeiten können unverdünnt oder aufgelöst in einem geeigneten Vehikel wie z.B. Maisöl verabreicht werden. Feste Substanzen sollten nach Möglichkeit aufgelöst werden, da größere Dosen fester Stoffe in Gelatinekapseln möglicherweise nicht wirksam resorbiert werden. Bei nichtwässrigen Vehikeln sollten deren toxische Eigenschaften bekannt sein. Ist dies nicht der Fall, müssen sie vor der Prüfung bestimmt werden.
1.4.2 Prüfbedingungen
1.4.2.1 Versuchstiere
Junge erwachsene Legehennen (Gallus gallus domesticus) im Alter von 8 bis 12 Monaten werden für den Test empfohlen. Normalgroße Rassen sollten verwendet werden, und die Hennen sollten normalerweise unter Bedingungen aufgewachsen sein, unter denen sie sich frei bewegen konnten.
1.4.2.2 Anzahl und Geschlecht
In der Regel sollten mindestens drei Behandlungsgruppen sowie eine Vehikelkontrollgruppe getestet werden. Die Vehikelkontrollgruppe ist, abgesehen von der Verabreichung der Prüfsubstanz, genauso zu behandeln wie die Behandlungsgruppen.
Die Anzahl der Tiere in jeder Gruppe sollte so bemessen sein, dass mindestens sechs zur Bestimmung biochemischer Parameter (jeweils drei Tiere zu zwei Beobachtungszeitpunkten) getötet werden können und mindestens sechs Tiere für die pathologische Untersuchung bis 14 Tage nach der letzten Gabe überleben.
1.4.2.3 Dosierungen
Bei der Wahl der Dosisstufen sind die Ergebnisse einer Akutprüfung auf verzögerte Neurotoxizität sowie weitere vorliegende Daten zur Toxizität oder Kinetik der Prüfsubstanz berücksichtigt worden. Die höchste Dosis sollte so gewählt werden, dass zwar toxische Wirkungen, vorzugsweise eine verzögerte Neurotoxizität, aber keine Todesfälle oder schweres Leiden hervorgerufen werden. Anschließend sollte eine absteigende Folge von Dosisstufen gewählt werden, um dosisabhängige Wirkungen und die niedrigste Dosis ohne zu beobachtende unerwünschte Wirkungen nachzuweisen.
1.4.2.4 Limit-Test
Verursacht die Prüfung einer Dosis von mindestens 1.000 mg/kg Körpergewicht pro Tag unter Verwendung der für diese Studie beschriebenen Verfahren keine feststellbaren toxischen Wirkungen und ist aufgrund der Daten strukturverwandter Substanzen keine Toxizität zu erwarten, kann auf eine Studie mit einer höheren Dosis gegebenenfalls verzichtet werden. Der Limit-Test findet allerdings keine Anwendung, wenn die Expositionswirkungen beim Menschen die Prüfung in einer höheren Dosis angezeigt erscheinen lassen.
1.4.2.5 Beobachtungszeitraum
Alle Tiere werden während der Expositionsdauer und bis 14 Tage danach mindestens täglich beobachtet, sofern sie nicht für die Sektion vorgesehen sind.
1.4.3 Versuchsdurchführung
Die Tiere erhalten die Prüfsubstanz für die Dauer von 28 Tagen an 7 Tagen in der Woche.
1.4.3.1 Allgemeine Beobachtungen
Die Beobachtungen sollten unmittelbar nach Behandlungsbeginn beginnen. Alle Hennen sind an jedem der 28 Behandlungstage und an den 14 Tagen nach der letzten Gabe oder bis zur vorgesehenen Tötung mindestens einmal täglich zu beobachten. Alle Toxizitätszeichen, einschließlich Zeitpunkt ihres Beginns sowie Art, Schweregrad und Dauer, sind zu dokumentieren. Die Beobachtungen sollten auch, jedoch nicht nur, Verhaltensauffälligkeiten einschließen. Die Ataxie wird auf einer Ordinalskala aus mindestens vier Stufen bewertet, und Paralysen werden ebenfalls dokumentiert. Mindestens zweimal wöchentlich sollten die Hennen aus dem Käfig genommen und für einen bestimmten Zeitraum zu verstärkter motorischer Aktivität, z.B. durch Begehen einer Leiter, gezwungen werden, um die Beobachtung bereits geringster toxischer Wirkungen zu erleichtern. Moribunde Tiere und Tiere, bei denen schweres Leiden oder starke Schmerzen festgestellt werden, sollten ausgesondert, unter Verwendung einer tierschutzgerechten Methode getötet und seziert werden.
1.4.3.2 Körpergewicht
Alle Hennen müssen unmittelbar vor der ersten Gabe der Prüfsubstanz und danach mindestens einmal wöchentlich gewogen werden.
1.4.3.3 Biochemische Untersuchungen
Je sechs Hennen, die nach Zufallskriterien aus der Behandlungs- und der Vehikelkontrollgruppe ausgewählt werden, sollten einige Tage nach der letzten Gabe der Prüfsubstanz getötet werden, um Gehirn und lumbales Rückenmark zu präparieren und auf hemmende Aktivität gegen Neuropathy Target Esterase (NTE) zu untersuchen. Auch die Präparation und Untersuchung des Ischiasnervengewebes auf hemmende Aktivität gegen Neuropathy Target Esterase kann aufschlussreich sein. In der Regel werden drei Tiere der Kontrollgruppe sowie jeder Behandlungsgruppe nach 24 Stunden und drei 48 Stunden nach der letzten Dosis getötet. Sind aufgrund der Daten aus der Akutstudie oder aus anderen Untersuchungen (z.B. zur Toxikokinetik) andere Tötungszeiten nach der letzten Gabe der Prüfsubstanz angezeigt, sollte entsprechend vorgegangen und das geänderte Vorgehen begründet werden.
An diesen Proben können auch Bestimmungen der Acetylcholinesterase (AChE) vorgenommen werden, wenn dies angezeigt erscheint. Allerdings kann es in vivo zur spontanen Reaktivierung von AChE kommen, so dass die Wirksamkeit der Prüfsubstanz als AChE-Hemmer unterschätzt wird.
1.4.3.4 Autopsie
Alle Versuchstiere (sowohl die nach Plan getöteten als auch die aufgrund ihres moribunden Zustands getöteten) werden einer Autopsie zur Untersuchung von Gehirn und Rückenmark unterzogen.
1.4.3.5 Histopathologische Untersuchung
Nervengewebe von Tieren, die die Beobachtungsdauer überleben und nicht für biochemische Untersuchungen vorgesehen sind, wird mikroskopisch untersucht. Die Gewebe werden in situ mittels Perfusionsverfahren fixiert. Gewebeschnitte werden unter anderem von Cerebellum (mittlere Längsebene), Medulla oblongata, Rückenmark und peripheren Nerven angefertigt. Die Rückenmarkschnitte sind vom oberen Halswirbelsegment, der mittleren thorakalen und der lumbosakralen Region zu entnehmen. Auch vom distalen Bereich des Nervus tibialis und seiner Verzweigungen zum Musculus gastrocnemius sowie vom Nervus sciaticus sollten Schnitte angefertigt werden. Die Schnitte werden mit geeigneten myelin- und axonspezifischen Färbemitteln angefärbt. Die mikroskopischen Untersuchungen sind zunächst an den konservierten Geweben aller Tiere in der Kontrollgruppe sowie der höchsten Dosisgruppe vorzunehmen. Ergeben sich Anhaltspunkte für Wirkungen in der höchsten Dosisgruppe, sollte die mikroskopische Untersuchung auch bei den Hennen aus der mittleren und niedrigen Dosisgruppe erfolgen.
2. Daten
Negative Ergebnisse für die in dieser Methode gewählten Endpunkte (Biochemie, Histopathologie und Verhaltensbeobachtung) erfordern normalerweise keine weiteren Untersuchungen auf eine verzögerte Neurotoxizität. Zweifelhafte oder unklare Ergebnisse für diese Endpunkte erfordern ggf. weitere Untersuchungen.
Die Daten für die einzelnen Tiere sollten aufgeführt werden. Ferner sollten alle Daten in tabellarischer Form zusammengefasst werden. Daraus müssen für jede Prüfgruppe folgende Angaben hervorgehen: die Anzahl der verwendeten Tiere bei Beginn der Prüfung, die Anzahl der Tiere mit Läsionen, verhaltensbezogenen oder biochemischen Wirkungen, die Arten und Schweregrade dieser Läsionen bzw. Wirkungen sowie der Anteil der von den verschiedenen Arten und Schweregraden der Läsionen bzw. Wirkungen betroffenen Tiere.
Die Ergebnisse dieser Studie sind im Hinblick auf Häufigkeit, Schweregrad und Korrelation der verhaltensbezogenen, biochemischen und histopathologischen Wirkungen sowie sonstiger verzeichneter Wirkungen in den Behandlungs- und Kontrollgruppen zu bewerten.
Die numerischen Daten sind nach Möglichkeit durch geeignete und allgemein akzeptierte statistische Verfahren auszuwerten. Diese statistischen Verfahren sollten bei der Festlegung des Designs der Studie ausgewählt werden.
3. Abschlussbericht
Prüfbericht
Der Prüfbericht soll nach Möglichkeit folgende Angaben enthalten: Versuchstiere:
- verwendete Rasse;
- Anzahl und Alter der Tiere;
- Herkunft, Haltungsbedingungen usw.;
- Gewicht der einzelnen Tiere bei Prüfungsbeginn.
Prüfbedingungen:
- Angaben zur Zubereitung der Prüfsubstanz, zur Stabilität und zur Homogenität;
- Begründung für die Wahl des Vehikels;
- Angaben zur Verabreichung der Prüfsubstanz;
- Angaben über Futter- und Wasserqualität;
- Begründung für die Wahl der Dosis;
- Angabe der verabreichten Dosen mit Angaben zu Vehikel, Volumen und physikalischer Form des verabreichten Stoffes;
- Begründung für die Wahl anderer Zeitpunkte zur Bestimmung der biochemischen Parameter (falls nicht nach 24 und 48 Stunden).
Ergebnisse:
- Angaben zum Körpergewicht;
- Daten der toxischen Reaktionen nach Dosisstufen, einschließlich Todesfälle;
- NOAEL;
- Art, Schweregrad und Dauer der klinischen Beobachtungen (mit Angaben zur Reversibilität);
- ausführliche Beschreibung der biochemischen Methoden und Befunde;
- Sektionsbefunde;
- ausführliche Beschreibung aller histopathologischen Befunde;
- ggf. statistische Auswertung der Ergebnisse.
Diskussion der Ergebnisse.
Schlussfolgerungen.
4. Literatur
Diese Methode entspricht der OECD TG 419.
B.39 In-vivo-Test zur unplanmäßigen DNA-Synthese (UDS) in Säugetierleberzellen 23
Diese Prüfmethode wurde gestrichen, da sie nicht mehr für die Gewinnung von Informationen über die toxikologischen Eigenschaften von Chemikalien für die Zwecke der Verordnung (EG) Nr. 1907/2006 als geeignet anerkannt ist. Die anzuwendenden Prüfmethoden für den betreffenden Endpunkt sind in Teil 0 Tabelle 2 aufgeführt.
| 1. Methode
Diese Methode entspricht der OECD TG 486, Unscheduled DNA Synthesis (UDS) Test with Mammalian Liver Cells In Vivo (1997). 1.1 Einleitung Der In-vivo-Test zur unplanmäßigen DNA-Synthese in Säugetierleberzellen dient zum Nachweis von Agenzien, die in den Leberzellen der behandelten Tiere eine DNA-Reparatur auslösen (1) (2) (3) (4). Dieser In-vivo-Test ermöglicht die Untersuchung der gentoxischen Wirkung von Chemikalien in der Leber. Der ermittelte Endpunkt deutet auf eine DNA-Schädigung und anschließende Reparatur in Leberzellen hin. Die Leber ist in der Regel Hauptort des Stoffwechsels der resorbierten Verbindungen. Sie eignet sich also gut zur In-vivo-Bestimmung einer DNA-Schädigung. Wenn Anzeichen dafür bestehen, dass die Prüfsubstanz das Zielgewebe nicht erreicht, ist dieser Test nicht geeignet. Der Endpunkt der unplanmäßigen DNA-Synthese (UDS) wird durch die Bestimmung der Aufnahme markierter Nukleoside in Zellen, die keine planmäßige (S-Phasen-)DNA-Synthese durchlaufen, ermittelt. Das gängigste Verfahren ist die Bestimmung der Aufnahme von tritiummarkiertem Thymidin 3 H-TdR) durch Autoradiografie. Für In-vivo-UDS-Tests wird vorzugsweise die Leber von Ratten verwendet. Neben Lebergewebe kann auch anderes Gewebe Verwendung finden, doch ist dieses nicht Gegenstand dieser Methode. Der Nachweis einer UDS-Reaktion hängt von der Zahl der ausgeschnittenen und ersetzten DNA-Basen am Ort der Schädigung ab. Der UDS-Test eignet sich daher insbesondere zum Nachweis der von einer Substanz induzierten "Longpatch"-Reparatur (20-30 Basen). Die "Shortpatch"-Reparatur (1-3 Basen) lässt sich hingegen nur mit einem wesentlich geringeren Empfindlichkeitsgrad feststellen. Zudem können mutagene Ereignisse aus der Nichtreparatur, fehlerhaften Reparatur oder fehlerhaften Replikation der DNA-Läsionen herrühren. Das Ausmaß der UDS-Reaktion gibt keinen Aufschluss über die Genauigkeit des Reparaturprozesses. Darüber hinaus ist es möglich, dass ein Mutagen mit der DNA reagiert, die DNA-Schädigung aber nicht über einen Exzisionsreparaturprozess behoben wird. Die Tatsache, dass der UDS-Test keine spezifischen Informationen zur mutagenen Aktivität liefert, wird durch die potenzielle Empfindlichkeit dieses Endpunkts kompensiert, denn er wird im gesamten Genom ermittelt. Siehe auch allgemeine Einleitung zu Teil B. 1.2 Definitionen In Reparatur befindliche Zellen: Nettokörnerzahl über dem Zellkern (NNG) oberhalb eines vorher festgelegten Schwellenwerts, der von dem jeweiligen Labor zu begründen ist. Nettokörnerzahl über dem Zellkern (NNG): quantitative Maßzahl der UDS-Aktivität von Zellen bei autoradiografischen UDS-Tests, errechnet durch Subtraktion der durchschnittlichen Körnerzahl über kernäquivalenten Bereichen des Zytoplasmas (CG) von der Körnerzahl über dem Zellkern (NG): NNG = NG - CG. Die NNG wird für einzelne Zellen bestimmt, dann für alle Zellen in einer Kultur, in Parallelkulturen usw. Unplanmäßige DNA-Synthese (UDS): DNA-Reparatursynthese nach Ausschneiden und Entfernen des Abschnitts eines DNA-Strangs, der eine Region mit einer durch chemische Substanzen oder physikalische Agenzien induzierten Schädigung enthält. 1.3 Prinzip der Prüfmethode Der In-vivo-UDS-Test an Säugetierleberzellen erbringt den Nachweis einer DNA-Reparatursynthese nach Ausschneiden und Entfernen eines DNA-Abschnitts, der eine Region mit einer durch chemische Substanzen oder physikalische Agenzien induzierten Schädigung enthält. Der Test beruht gewöhnlich auf dem Einbau von 3H-TdR in die DNA von Leberzellen, wo sich nur wenige Zellen in der S-Phase des Zellzyklus befinden. Die Aufnahme von 3H-TdR wird in der Regel durch Autoradiografie bestimmt, da dieses Verfahren nicht so anfällig für eine Einwirkung durch S-Phasen-Zellen ist wie beispielsweise die Flüssigkeitsszintillationszählung (LSC). 1.4 Beschreibung der Prüfmethode 1.4.1 Vorbereitungen 1.4.1.1 Versuchstiere Gewöhnlich werden Ratten verwendet, doch kommen auch andere geeignete Säugetierarten in Frage. Es sollten junge gesunde und geschlechtsreife Tiere üblicher Labortierstämme zum Einsatz kommen. Zu Beginn des Versuchs sollte die Abweichung des Körpergewichts der Tiere vom Mittelwert so gering wie möglich sein und bei beiden Geschlechtern nicht mehr als ± 20 % betragen. 1.4.1.2 Haltungs- und Fütterungsbedingungen Es gelten die allgemeinen Bedingungen in der allgemeinen Einleitung zu Teil B, doch ist bei der Luftfeuchtigkeit ein Wert von 50-60 % anzustreben. 1.4.1.3 Vorbereitung der Tiere Gesunde und geschlechtsreife junge Tiere werden randomisiert und den einzelnen Kontroll- bzw. Behandlungsgruppen zugeteilt. Die Käfige sind so anzuordnen, dass sich ihre Position möglichst wenig auswirkt. Es erfolgt eine Einzelidentifizierung der Tiere. Vor Beginn der Studie werden die Tiere in ihren Käfigen über einen Zeitraum von mindestens 5 Tagen unter Laborbedingungen eingewöhnt. 1.4.1.4 Prüfsubstanz/Vorbereitung Feste Prüfsubstanzen sollten vor der Verabreichung an die Tiere in geeigneten Lösungsmitteln oder Vehikeln gelöst oder suspendiert und ggf. verdünnt werden. Flüssige Prüfsubstanzen können direkt verabreicht oder zuvor verdünnt werden. Es sind frische Zubereitungen der Prüfsubstanz zu verwenden, wenn die Stabilitätsdaten gegen eine Lagerung sprechen. 1.4.2 Prüfbedingungen 1.4.2.1 Lösungsmittel/Vehikel Das Lösungsmittel/Vehikel sollte bei den gewählten Dosierungen keine toxischen Wirkungen hervorrufen und nicht im Verdacht stehen, mit der Prüfsubstanz eine chemische Reaktion einzugehen. Werden keine allgemein bekannten Lösungsmittel/Vehikel verwendet, so sind Referenzdaten zur Kompatibilität beizubringen. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. 1.4.2.2 Kontrollen Für jeden gesondert vorgenommenen Teil des Versuchs sind gleichzeitig Positiv- und Negativ-(Lösungsmittel-/ Vehikel-)Kontrollen anzulegen. Bis auf die Verabreichung der Prüfsubstanz sind die Tiere der Kontrollgruppe ebenso zu behandeln wie die Tiere der Behandlungsgruppen. Die als Positivkontrollen verwendeten Substanzen sollten erwiesenermaßen eine UDS bei Expositionskonzentrationen hervorrufen, die voraussichtlich eine erkennbare Zunahme gegenüber dem Hintergrund ergeben.
Positivkontrollen, die eine Stoffwechselaktivierung benötigen, sollten in Dosen verwendet werden, die eine mäßige Reaktion hervorrufen (4). Die Dosen sollten so gewählt werden, dass die Wirkungen eindeutig sind, aber beim Auswerten nicht sofort die Identität der kodierten Objektträger erkennen lassen.
Es kommen beispielsweise folgende Positivkontrollsubstanzen in Frage:
Auch andere geeignete Positivkontrollsubstanzen kommen in Betracht. Es ist vertretbar, dass die Positivkontrolle auf anderem Weg als die Prüfsubstanz verabreicht wird. 1.5 Verfahren 1.5.1 Anzahl und Geschlecht der Tiere Es ist eine ausreichende Anzahl von Tieren zu verwenden, um die natürliche biologische Schwankungsbreite der Testreaktion zu berücksichtigen. Jede Gruppe sollte mindestens 3 analysierbare Tiere umfassen. Liegt ein größerer Bestand an historischen Daten vor, so sind für die gleichzeitigen Negativ- und Positivkontrollgruppen nur 1 oder 2 Tiere erforderlich. Wenn zum Zeitpunkt der Studie Daten aus Untersuchungen zur gleichen Spezies und Expositionsform vorliegen, die belegen, dass zwischen den Geschlechtern kein nennenswerter Unterschied der Toxizität feststellbar ist, reicht die Prüfung von Tieren nur eines - vorzugsweise des männlichen - Geschlechts aus. Sollte es sich beim Menschen um eine geschlechtsspezifische Exposition handeln, z.B. bei bestimmten pharmazeutischen Wirkstoffen, ist der Versuch an Tieren des betreffenden Geschlechts durchzuführen. 1.5.2 Behandlungsplan Die Prüfsubstanzen werden in der Regel als Einmalgabe verabreicht. 1.5.3 Dosierungen Im Normalfall sind mindestens zwei Dosisstufen zu verwenden. Unter der Höchstdosis ist jene Dosis zu verstehen, die so deutliche Toxizitätszeichen hervorruft, dass höhere Dosisstufen bei gleichem Verabreichungsschema voraussichtlich zum Tode führen. Die geringere Dosis sollte in der Regel 50 % bis 25 % der hohen Dosis entsprechen. Substanzen mit spezifischen biologischen Aktivitäten bei geringen nichttoxischen Dosen (wie Hormone und Mitogene) entsprechen möglicherweise nicht den Dosierungskriterien und sollten anhand einer Einzelfallprüfung bewertet werden. Wird eine Studie zur Dosisfindung durchgeführt, weil keine geeigneten Daten verfügbar sind, so sollte sie im gleichen Labor unter Verwendung der gleichen Spezies, des gleichen Stammes und Geschlechts und der gleichen Behandlungsform wie im Hauptversuch erfolgen. Die Höchstdosis kann auch als jene Dosis definiert werden, die bestimmte Anzeichen von Toxizität in der Leber hervorruft (z.B. pyknotische Kerne). 1.5.4 Limit-Test Verursacht die Prüfung einer Dosis von mindestens 2.000 mg/kg Körpergewicht bei Einmalgabe oder Gabe von zwei Teilmengen am gleichen Tag keine feststellbaren toxischen Wirkungen und ist aufgrund der Daten strukturverwandter Substanzen keine Gentoxizität zu erwarten, kann auf eine vollständige Studie verzichtet werden. Die voraussichtlichen Expositionswirkungen beim Menschen können aber beim Limit-Test eine höhere Dosis angezeigt erscheinen lassen. 1.5.5 Verabreichung Die Prüfsubstanz wird in der Regel mittels Magen- oder Schlundsonde verabreicht. Auch andere Expositionsformen können bei entsprechender Begründung vertretbar sein. Eine intraperitoneale Injektion ist allerdings nicht zu empfehlen, da die Leber damit möglicherweise der Prüfsubstanz direkt und nicht über das Kreislaufsystem ausgesetzt wird. Das maximale Flüssigkeitsvolumen, das jeweils durch Sonde oder Injektion verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 2 ml/100 g Körpergewicht nicht übersteigen. Die Verwendung eines höheren Volumens ist zu begründen. Abgesehen von Reizstoffen oder ätzenden Stoffen, die in der Regel bei höheren Konzentrationen eine verstärkte Wirkung hervorrufen, ist die Variabilität des Prüfvolumens dadurch auf ein Mindestmaß zu reduzieren, dass eine Konzentration gewählt wird. die auf allen Dosisstufen ein konstantes Volumen gewährleistet. 1.5.6 Präparation der Leberzellen Die Präparation der Leberzellen behandelter Tiere erfolgt in der Regel 12-16 Stunden nach der Verabreichung. Im Allgemeinen ist zusätzlich eine frühere Aufarbeitung (gewöhnlich 2-4 Stunden nach der Behandlung) erforderlich, wenn nicht nach 12-16 Stunden eine eindeutige positive Reaktion vorliegt. Auf der Grundlage der toxikokinetischen Daten sind aber bei entsprechender Begründung auch Aufarbeitungen zu anderen Zeitpunkten möglich. Kurzzeitkulturen von Säugetier-Leberzellen werden in der Regel dadurch angelegt, dass eine Perfusion der Leber in situ mit Collagenase erfolgt und es frisch gewonnenen Leberzellen ermöglicht wird, sich an eine geeignete Wachstumsfläche festzuheften. Die Leberzellen von Tieren der Negativkontrollgruppe sollten eine Lebensfähigkeit (5) von mindestens 50 % aufweisen. 1.5.7 Bestimmung der UDS Frisch isolierte Säugetier-Leberzellen werden über einen angemessenen Zeitraum, z.B. 3-8 Stunden, in einem 3 H-TdR enthaltenden Medium inkubiert. Nach Ablauf der Inkubationszeit sollte das Medium aus den Zellen entfernt werden, die dann mit einem überschüssiges unmarkiertes Thymidin enthaltenden Medium inkubiert werden können, um die nicht inkorporierte Radioaktivität zu verringern (cold chase). Anschließ werden die Zellen gewaschen, fixiert und getrocknet. Bei längeren Inkubationszeiten ist eine "cold chas" möglicherweise nicht erforderlich. Die Objektträger werden in Autoradiografieemulsion getaucht, im Dunkeln "belichtet" (z.B. gekühlt für 7-14 Tage), entwickelt und gefärbt, und es werden die belichteten Silberkörner gezählt. Von jedem Tier werden zwei bis drei Objektträger hergestellt. 1.5.8 Analyse Die Objektträgerpräparate sollten eine ausreichende Anzahl von morphologisch normalen Zellen enthalten, um eine aussagefähige Bewertung der UDS zu ermöglichen. Die Präparate werden mikroskopisch auf Zeichen offener Zytotoxizität (z.B. Pyknose, verringerte Werte der radioaktiven Markierung) untersucht. Vor der Bestimmung der Körnerzahl sind die Objektträger zu kodieren. In der Regel werden je Tier 100 Zellen von mindestens zwei Objektträgern ausgewertet. Die Auswertung von weniger als 100 Zellen/Tier ist zu begründen. Bei der Körnerzahlbestimmung erfolgt keine Zählung der S-Phasen-Kerne, doch kann der Anteil der S-Phasen-Zellen erfasst werden. Das Ausmaß des 3H-TdR-Einbaus im Zellkern und Zytoplasma morphologisch normaler Zellen, das an der Ablagerung von Silberkörnern ablesbar ist, sollte durch geeignete Verfahren ermittelt werden. Die Körnerzahl wird über dem Zellkern (NG) und über kernäquivalenten Bereichen des Zytoplasmas (CG) ermittelt. Als CG-Wert kommt entweder die für den am stärksten markierten Bereich des Zytoplasmas ermittelte Körnerzahl oder der Durchschnittswert von zwei bis drei nach dem Zufallsprinzip ausgewählten Bereichen in Nähe des Zellkerns in Frage. Auch andere Zählverfahren (z.B. Ganzzellenzählung) können bei entsprechender Begründung angewendet werden (6). 2. Daten 2.1 Aufbereitung der Ergebnisse Es sind die Daten für die einzelnen Objektträger und Tiere zu dokumentieren. Zusätzlich sollten alle Daten in tabellarischer Form zusammengefasst werden. Die Nettokörnerzahl über dem Zellkern (NNG) ist für jede Zelle, für jedes Tier und für jede Dosis und jeden Zeitpunkt zu bestimmen, indem der CG-Wert vom NG-Wert subtrahiert wird. Werden die "in Reparatur befindlichen Zellen" gezählt, so sollten die Kriterien für die Definition der "in Reparatur befindlichen Zellen" begründet werden und auf historischen oder gleichzeitigen Negativkontrolldaten beruhen. Numerische Ergebnisse können mit Hilfe statistischer Verfahren bewertet werden. Kommen statistische Tests zur Anwendung, so sind sie vor Durchführung der Studie auszuwählen und zu begründen. 2.2 Bewertung und Interpretation der Ergebnisse Als Beispiele für Kriterien zur Bestimmung positiver/negativer Reaktionen seien genannt:
Es ist die biologische Relevanz der Daten zu untersuchen, d. h., es sind Parameter wie Variabilität der Tiere, Dosis-Wirkungs-Verhältnis und Zytotoxizität zu berücksichtigen. Als Hilfsmittel bei der Bewertung der Versuchsergebnisse können statistische Methoden dienen. Die statistische Signifikanz sollte aber nicht der einzige bestimmende Faktor für eine positive Reaktion sein. Auch wenn die meisten Versuche eindeutig positive oder negative Ergebnisse liefern, erlaubt der Datensatz in seltenen Fällen keine definitive Aussage über die Aktivität der Prüfsubstanz. Es kommt vor, dass sich die Ergebnisse unabhängig davon, wie oft der Versuch wiederholt wird, weiterhin als nicht eindeutig oder als fragwürdig erweisen. Ein positiver Befund des In-vivo-UDS-Tests an Säugetierleberzellen deutet darauf hin, dass die Prüfsubstanz in vivo bei Säugetierleberzellen eine DNA-Schädigung hervorruft, die in vitro durch unplanmäßige DNA-Synthese repariert werden kann. Ein negativer Befund ist ein Anzeichen dafür, dass die Prüfsubstanz unter diesen Versuchsbedingungen keine mit diesem Test nachweisbare DNA-Schädigung induziert. Zu erörtern ist die Wahrscheinlichkeit, dass die Prüfsubstanz in den allgemeinen Blutkreislauf bzw. in das spezifische Zielgewebe gelangt (z.B. systemische Toxizität). 3. Abschlussbericht Prüfbericht Der Prüfbericht muss die folgenden Angaben enthalten: Lösungsmittel/Vehikel:
Versuchstiere:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse. Schlussfolgerungen. 4. Literaturhinweise (1) Ashby, J., Lefevre, P. A., Burlinson, B. and Penman, M. G. (1985), An Assessment of the In Vivo Rat Hepatocyte DNA Repair Assay, Mutation Res., 156, 1-18. (2) Butterworth, B. E., Ashby, J., Bermudez, E., Casciano, D., Mirsalis, J., Probst G. and Williams, G. (1987), A Protocol and Guide for the In Vivo Rat Hepatocyte DNA-Repair Assay, Mutation Res., 189, 123-133. (3) Kennelly. J. C, Waters, R., Ashby, J., Lefevre, P. A., Burlinson B., Benford, D. J., Dean, S. W. and Mitchell I. de G. (1993), In Vivo Rat Liver UDS Assay, in: Kirkland D. J. and Fox M., (eds.), Supplementary Mutagenicity Tests: UKEM Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing Report. Part II revised, Cambridge University Press, Cambridge, New York, Port Chester, Melbourne, Sydney, 52-77. (4) Madle, S., Dean, S. W., Andrae, U., Brambilla, G., Burlinson, B., Doolittle, D. J., Furihata, C., Hertner, T., McQueen, C. A. and Mori, H. (1993), Recommendations for the Performance of UDS Tests In Vitro and In Vivo. Mutations Res., 312, 263-285. (5) Fautz, R., Hussain, B., Efstathiou, E. and Hechenberger-Freudl, C. (1993), Assessment of the Relation Between the Initital Viability and the Attachment of Freshly Isolated Rat Hepatocytes Used for the In Vivo/ ln Vitro DNA Repair Assay (UDS), Mutation Res., 291, 21-27. (6) Mirsalis, J. C, Tyson, C. K. and Butterworth, B. E. (1982), Detection of Genotoxic Carcinogens in the In Vivo/In Vitro Hepatocyte DNA Repair Assay, Environ Mutagen, 4, 553-562. |
B.40 In vitro-Hautverätzung: TER-Prüfmethode
Einleitung
1. Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie 430 (2015). Als Hautverätzung wird das Auslösen einer irreversiblen Hautschädigung, d. h. einer sichtbaren, bis in das Corium reichenden Nekrose der Epidermis nach Applikation einer Prüfchemikalie [im Sinne der Definition des Global harmonisierten Systems zur Einstufung und Kennzeichnung von Gefahrstoffen der UN (GHS) 1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) 1] bezeichnet. Diese aktualisierte Prüfmethode B.40 besteht in einem Invitro-Verfahren zur Identifizierung nicht hautätzender und hautätzender Stoffe und Gemische nach dem UN GHS (1) und nach der CLP-Verordnung.
2. Die Bewertung von Hautverätzungen erfolgte in der Regel unter Verwendung von Labortieren (PM B.4, entsprechend OECD TG 404, ursprünglich angenommen im Jahr 1981 und geändert in den Jahren 1992, 2002 und 2015) (2). Neben dieser PM B.40 wurden noch weitere Invitro-Prüfmethoden zur Untersuchung des Hautverätzungspotenzials von Chemikalien als PM B.40bis (entsprechend OECD TG 431) (3) und PM B.65 (entsprechend OECD TG 435) validiert und angenommen (4), mit denen ggf. ebenfalls Unterkategorien ätzender Chemikalien identifiziert werden können. Mehrere validierte Invitro-Prüfmethoden wurden als PM B.46 (entsprechend OECD TG 439 (5) angenommen, die zur Untersuchung von Hautreizungen verwendet werden können. In einem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von Hautverätzungen und -reizungen werden mehrere Module beschrieben, in denen mehrere Informationsquellen und Analyseinstrumente zu Gruppen zusammengefasst werden. Sie enthalten i) Leitlinien dazu, wie existierende Daten aufgrund von Versuchen und aus sonstigen Quellen integriert und verwendet werden können, um das Hautreizungs- und das Hautverätzungspotenzial von Chemikalien zu bewerten, und ii) einen Vorschlag zur Durchführung ggf. erforderlicher weiterer Versuche (6).
3. Mit dieser Prüfmethode werden Hautverätzungen mit dem Endpunkt der menschlichen Gesundheit untersucht. Sie beruht auf der Methode zur Messung des elektrischen Widerstands der Haut (TER-Prüfung), bei der ätzende Chemikalien anhand ihrer Fähigkeit ermittelt werden, einen Verlust der normalen Beschaffenheit und Barrierefunktion der Hornhaut (stratum corneum) herbeizuführen. Die entsprechende OECD-Prüfrichtlinie wurde ursprünglich im Jahr 2004 angenommen und 2015 unter Berücksichtigung des IATA-Leitliniendokuments aktualisiert.
4. Um Invitro-Prüfungen auf Hautverätzungen für rechtliche Zwecke zu evaluieren, wurden Vorvalidierungsstudien (7) mit anschließender formeller Validierungsstudie der TER-Prüfung mit Rattenhaut zur Bewertung von Hautverätzungen durchgeführt (8) (9) (10) (11). Die Ergebnisse dieser Studien führten zu der Empfehlung, dass die TER-Prüfmethode (auch als validierte Referenzmethode (VRM) bezeichnet) für rechtliche Zwecke zur Bewertung von Invivo-Hautverätzungen verwendet werden kann (12) (13) (14).
5. Bevor eine vorgeschlagene ähnliche oder modifizierte Invitro-TER-Prüfmethode zur Untersuchung auf Hautverätzungen außer der VRM für rechtliche Zwecke verwendet werden kann, sollten nach den Anforderungen der Leistungsstandards (15) Verlässlichkeit, Relevanz (Genauigkeit) und die Grenzen des Verfahrens für den vorgeschlagenen Verwendungszweck bestimmt werden, um sicherzustellen, dass es mit der VRM vergleichbar ist. Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird erst dann garantiert, wenn vorgeschlagene neue oder aktualisierte Prüfmethoden auf der Grundlage der Leistungsstandards überprüft und in die entsprechende OECD-Prüfrichtlinie aufgenommen wurden.
Begriffsbestimmungen
6. Es gelten die Begriffsbestimmungen der Anlage.
Ausgangsüberlegungen
7. In einer Validierungsstudie (10) und in anderen veröffentlichten Studien (16) (17) wurde festgestellt, dass die TER-Prüfmethode zur Untersuchung von Rattenhaut geeignet ist, mit einer Gesamtempfindlichkeit von 94 % (51/54) und einer Spezifizität von 71 % (48/68) zwischen bekannten hautätzenden Chemikalien und nicht hautätzenden Chemikalien aus einer Datenbank mit 122 Stoffen zu unterscheiden.
8. Gegenstand dieser Prüfmethode sind Invitro-Hautverätzungen. Mit der Prüfmethode können nicht ätzende und ätzende Prüfchemikalien nach dem UN GHS bzw. der CLP-Verordnung festgestellt werden. Eine Einschränkung bei dieser Prüfmethode besteht den Validierungsstudien (8) (9) (10) (11) zufolge darin, dass eine Unterkategorisierung ätzender Stoffe und Gemische nach Maßgabe des UN GHS bzw. der CLP-Verordnung nicht möglich ist. Mit dem geltenden Rechtsrahmen wird festgelegt, wie diese Prüfmethode zu verwenden ist. Diese Prüfmethode vermittelt keine geeigneten Informationen über Hautreizungen; mit PM B.46 werden jedoch gezielt die gesundheitlichen Auswirkungen von Hautreizungen in vitro untersucht (5). Eine umfassende Evaluierung lokaler Auswirkungen auf die Haut nach einer einmaligen dermalen Exposition ist dem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA) zu entnehmen (6).
9. In der dieser Prüfmethode zugrunde liegenden Validierung wurden zahlreiche Chemikalien (in erster Linie Stoffe) geprüft, und die empirische Datenbank der Validierungsstudie umfasste 60 Stoffe aus zahlreichen Chemikalienklassen (8) (9). Auf der Grundlage der insgesamt verfügbaren Daten ist festzustellen, dass die Prüfmethode bei zahlreichen Chemikalienklassen und Aggregatzuständen (u. a. Flüssigkeiten, halbfeste Stoffe, Feststoffe und Wachse) anwendbar ist. Da für bestimmte Aggregatzustände jedoch nicht ohne Weiteres Prüfstoffe mit geeigneten Referenzdaten verfügbar sind, ist darauf hinzuweisen, dass bei der Validierung eine vergleichsweise geringe Anzahl an Wachsen und ätzenden Feststoffen bewertet wurde. Flüssigkeiten können wässrig oder nicht wässrig und Feststoffe in Wasser löslich oder unlöslich sein. Wenn nachgewiesen werden kann, dass die Prüfmethode bei einer bestimmten Kategorie von Stoffen nicht anwendbar ist, sollte sie bei diesen nicht verwendet werden. Diese Prüfmethode kann jedoch nicht nur bei Stoffen verwendet werden, sondern wird auch als für Gemische geeignet betrachtet. Da Gemische jedoch vielfältigen Kategorien zuzurechnen sein und unterschiedliche Zusammensetzungen haben können, und da gegenwärtig nur begrenzte Informationen über die Prüfung von Gemischen verfügbar sind, sollten Prüfmethoden, bei denen nachgewiesen werden kann, dass sie für eine bestimmte Kategorie von Gemischen nicht geeignet sind (beispielsweise durch ein Verfahren entsprechend dem von Eskes u. a. (2012) vorgeschlagenen Verfahren) (18), für die betreffende Kategorie von Gemischen nicht verwendet werden. Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist. Gase und Aerosole wurden bislang noch nicht in Validierungsstudien bewertet (8)(9). Obwohl deren Prüfung mit der TER-Prüfmethode prinzipiell vorstellbar ist, erlaubt die vorliegende Prüfmethode das Untersuchen von Gasen und Aerosolen nicht.
Prinzip der Prüfmethode
10. Die Prüfchemikalie wird in einem Zweikammersystem bis zu 24 Stunden lang auf den Epidermisflächen von Hautstücken aufgebracht, wobei die Hautstücke als Trennwand zwischen den beiden Kammern fungieren. Die Hautstücke werden 28-30 Tage alten Ratten entnommen, die zuvor tierschutzgerecht getötet wurden. Hautätzende Chemikalien werden anhand ihrer Fähigkeit ermittelt, einen Verlust der normalen Beschaffenheit und Barrierefunktion der Hornhaut (stratum corneum) herbeizuführen, welcher als Senkung des TER unter einen bestimmten Schwellenwert (16) gemessen wird (Nummer 32). Beim TER der Haut von Ratten wurde ein Schwellenwert von 5 kΩ auf der Grundlage umfangreicher Datenbestände für ein breites Spektrum unterschiedlicher Chemikalien gewählt, wobei die überwiegende Mehrzahl der Werte entweder eindeutig deutlich oberhalb dieses Schwellenwerts (oft > 10 k Ω) oder aber deutlich unterhalb dieses Werts (oft < 3 kΩ) lag (16). Im Allgemeinen sinkt der TER bei Prüfchemikalien, die bei Tieren nicht hautätzend wirken, aber hautreizende bzw. nicht hautreizende Eigenschaften aufweisen, nicht bis unter diesen Schwellenwert. Außerdem kann sich durch die Verwendung anderer Hautherstellungen oder anderer Prüfgeräte der Schwellenwert verändern, wodurch eine zusätzliche Validierung notwendig wird.
11. Im Prüfverfahren ist zur Bestätigung der Prüfungen auf positive Ergebnisse im TER (einschließlich Werten um ca. 5 kΩ) ein Farbbindungsschritt vorgesehen. Durch den Farbbindungsschritt wird festgestellt, ob die steigende Ionenpermeabilität auf die materielle Zerstörung der Hornhaut (stratum corneum) zurückzuführen ist. Durch die TER-Methode mit Rattenhaut konnte eine gute Vorhersagegenauigkeit der hautätzenden Invivo-Wirkung bei dem nach Prüfmethode B.4 (2) untersuchten Kaninchen nachgewiesen werden.
Nachweis der Leistungfähigkeit
12. Vor der routinemäßigen Anwendung der TER-Prüfmethode mit Rattenhaut nach dieser Prüfmethode sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Klassifizierung der zwölf in Tabelle 1 empfohlenen Leistungsstoffe nachweisen. Wenn ein dort genannter Stoff nicht verfügbar ist bzw. wenn dies gerechtfertigt ist, kann ein anderer Stoff verwendet werden, für den geeignete Invivo- und Invitro-Referenzdaten verfügbar sind (z.B. aus der Liste der Referenzchemikalien (16)), sofern die in Tabelle 1 beschriebenen Auswahlkriterien angewendet werden.
Tabelle 1 Liste der Leistungsstoffe 1
| Stoff | CAS-Nr. | Chemikalienklasse 2 | UN GHS/CLP Kat. nach Invivo-Ergebnissen 3 | VRM Kat. nach Invitro-Ergebnissen | Aggregatzustand | pH 4 |
| In vivo ätzende Chemikalien | ||||||
| N,N'-Dimethyl dipropylenetriamin | 10563-29-8 | organische Base | 1A | 6 × C | L | 8,3 |
| 1,2-Diaminopropan | 78-90-0 | organische Base | 1A | 6 × C | L | 8,3 |
| Chlorwasserstoffsäure (10 %) | 7664-93-9 | anorganische Säure | (1A/)1B/1C | 5 × C 1 × NC | L | 1,2 |
| Kaliumhydroxid (10 % aq.) | 1310-58-3 | anorganische Base | (1A/)1B/1C | 6 × C | L | 13,2 |
| Octansäure (Caprylsäure) | 124-07-2 | organische Säure | 1B/1C | 4 × C 2 × NC | L | 3,6 |
| 2-tert-Butylphenol | 88-18-6 | Phenol | 1B/1C | 4 × C 2 × NC | L | 3,9 |
| In Vivo nicht ätzende Chemikalien | ||||||
| Isostearinsäure | 2724-58-5 | organische Säure | NC | 6 × NC | L | 3,6 |
| 4-Amino-1,2,4-Triazol | 584-13-4 | organische Base | NC | 6 × NC | S | 5,5 |
| Phenethylbromid | 103-63-9 | Elektrophil | NC | 6 × NC | L | 3,6 |
| 4-(Methylthio)-Benzaldehyd | 3446-89-7 | Elektrophil | NC | 6 × NC | L | 6,8 |
| 1,9-Decadien | 1647-16-1 | neutral organisch | NC | 6 × NC | L | 3,9 |
| Tetrachlorethylen | 127-18-4 | neutral organisch | NC | 6 × NC | L | 4,5 |
| Abkürzungen: aq = wässrig; CAS-Nr. (CAS Registry Number = CAS-Registrierungsnummer); VRM = validierte Referenzmethode; C = ätzend; NC = nicht ätzend.
1) Die Leistungsstoffe, sortiert zunächst nach ätzenden und nicht ätzenden Stoffen, anschließend nach Unterkategorien ätzender Stoffe und schließlich nach Chemikalienklassen, wurden unter den in der Validierungsstudie des Europäischen Zentrum zur Validierung alternativer Methoden (EZVAM) zur TER-Prüfmethode mit Rattenhäuten ausgewählt (8) (9). Wenn nicht anderweitig angegeben, wurden die Stoffe in der Reinheit geprüft, in der sie im Handel gekauft wurden (8). Die Auswahl beinhaltete nach Möglichkeit Stoffe, die (i) repräsentativ für die Spanne der Ätzwirkungen sind (z.B. nicht ätzend, schwach ätzend usw. bis hin zu stark ätzend), die mit der VRM gemessen oder vorhergesagt werden können; (ii) repräsentativ für die in der Validierungsstudie verwendeten Chemikalienklassen sind; (iii) Aufschluss über die Leistungsmerkmale der VRM geben; (iv) durch gut definierte chemische Strukturen gekennzeichnet sind; (v) bei der Invivo -Referenzprüfmethode klare Ergebnisse induzieren; (vi) im Handel erhältlich sind; (vii) nicht mit nicht tragbaren Entsorgungskosten verbunden sind. 2) Von Barratt u. a. zugewiesene Chemikalienklasse (8). 3) Den UN-GHS-/CLP-Kategorien 1A, 1B und 1C entsprechen die UN-Verpackungsgruppen I, II und III. 4) Die pH-Werte wurden von Fentem u. a. (9) und Barratt u. a. (8) ermittelt. | ||||||
Verfahren
13. Für die TER-Prüfmethode zur Feststellung der Ätzwirkung bei Rattenhaut sind Standardarbeitsanweisungen (SOPs) verfügbar (19). Die in dieser Prüfmethode verwendeten TER-Prüfmethoden mit Rattenhaut sollten die folgenden Anforderungen erfüllen:
Versuchstiere
14. Für die Prüfung sollten Ratten verwendet werden, weil die Empfindlichkeit von Rattenhaut gegenüber den bei dieser Prüfmethode verwendeten Stoffen bereits nachgewiesen wurde (12) und weil nur Rattenhaut bislang förmlich validiert wurde (8) (9). Alter (wenn sich die Haut vollständig gebildet hat) und Abstammung der Ratten sind von besonderer Bedeutung, damit gewährleistet ist, dass sich die Fellfollikel in der Ruhephase befinden, bevor das Fellwachstum der erwachsenen Tiere beginnt.
15. An den jungen, ca. 22 Tage alten männlichen oder weiblichen Ratten (Wistar-Ratten oder vergleichbare Abstammung) wird das Fell am Rücken und an den Flanken mit einem geeigneten Instrument sorgfältig geschoren. Anschließend werden die Tiere mit vorsichtigen Wischbewegungen gewaschen, wobei die geschorenen Flächen in eine Antibiotikalösung getaucht werden (die Lösung enthält beispielsweise Streptomycin, Penicillin, Chloramphenicol und Amphotericin in Konzentrationen, die ein bakterielles Wachstum verhindern). Die Tiere werden dann am dritten oder vierten Tag nach dem ersten Waschvorgang erneut gewaschen und innerhalb von 3 Tagen nach dem zweiten Waschvorgang verwendet, wenn sich die Hornhaut vom Schervorgang erholt hat.
Gewinnung der Hautstücke
16. Die Versuchstiere werden im Alter von 28-30 Tagen tierschutzgerecht getötet; dieses Alter ist von entscheidender Bedeutung. Die dorsallaterale Haut der Tiere wird entfernt und von überschüssigem subkutanem Fett befreit, das von der Haut sorgfältig abgeschält wird. Es werden Hautstücke mit einem Durchmesser von je ca. 20 mm entnommen. Die Haut kann vor der Verwendung der Hautstücke eingelagert werden, wenn sich zeigt, dass die positiven und negativen Kontrolldaten mit den an frischer Haut ermittelten Daten übereinstimmen.
17. Jedes Hautstück wird so über das Ende eines PTFE-(Polytetrafluorethylen)-Rohres gelegt, dass die Epidermisoberfläche auf dem Ende des Rohres aufliegt. Zur Fixierung des Hautstücks wird ein O-Ring aus Gummi straff über das Rohrende gezogen, so dass die Haut fixiert wird. Überflüssiges Hautgewebe wird abgeschnitten. Der O-Ring wird sodann mit gereinigter Naturvaseline zum Ende des PTFE-Rohrs hin gründlich abgedichtet. Das Rohr wird durch eine Federklemme in einer Rezeptorkammer gehalten, die MgSO4-Lösung (154 mM) enthält (Abbildung 1). Das Hautstück ist vollständig in die MgSO4-Lösung einzutauchen. Aus einer einzigen Rattenhaut können bis zu 10-15 Hautstücke entnommen werden. Die Abmessungen des Rohrs und des O-Rings sind in Abbildung 2 angegeben.
18. Vor Beginn der Prüfung wird zu Qualitätssicherungszwecken bei jeder Tierhaut der TER an zwei Hautstücken gemessen. Beide Hautstücke müssen einen Widerstand von mehr als 10 kΩ aufweisen, damit sie für die Prüfmethode verwendet werden können. Liegt der Widerstand unter 10 kΩ, sind die übrigen Hautstücke der betreffenden Tierhaut zu vernichten.
Applikation der Prüfchemikalie und Kontrollstoffe
19. Zu jedem Prüflauf (Versuch) sind gleichzeitige Positiv- und Negativkontrollen durchzuführen, damit eine angemessene Eignung des Experimentalmodells gewährleistet ist. Bei jedem Prüflauf (Versuch) sind Hautstücke eines einzigen Tieres zu verwenden. Als Prüfchemikalien für die Positiv- und Negativkontrolle sind 10 M Chlorwasserstoffsäure bzw. destilliertes Wasser zu verwenden.
20. Die flüssigen Prüfchemikalien (150 µl) werden im Rohrinneren gleichmäßig auf die epidermale Oberfläche aufgebracht. Bei Prüfungen an Feststoffen wird eine ausreichende Menge des Feststoffs gleichmäßig auf das Hautstück aufgebracht, so dass gewährleistet ist, dass die gesamte Epidermisoberfläche bedeckt ist. Auf den Feststoff wird entionisiertes Wasser (150 µl) aufgebracht und das Rohr vorsichtig bewegt. Um optimalen Kontakt der Prüfchemikalien mit der Haut zu erreichen, müssen einige Feststoffe gegebenenfalls auf 30 °C erwärmt werden, so dass die Prüfchemikalie schmilzt oder weich wird, oder die Prüfchemikalie muss zu einem Granulat oder Pulver zermahlen werden.
21. Für jede Prüf- und Kontrollchemikalie werden bei jedem Prüflauf (Versuch) je drei Hautstücke verwendet. Die Prüfchemikalien werden für 24 h bei 20-23 °C aufgebracht. Anschließend wird die Prüfchemikalie mit fließendem Leitungswasser höchstens mit Raumtemperatur gewaschen, bis kein weiteres Material mehr entfernt werden kann.
TER-Messungen
22. Der Hautwiderstand wird als TER mit einer Wheatstone'schen Wechselstrom-Messbrücke mit niedriger Spannung gemessen (18). Die Kenndaten der Messbrücke lauten: Betriebsspannung 1-3 V, sinus- oder rechteckförmiger Wechselstrom mit 50-1 000 Hz, Messbereich mindestens 0,1-30 kΩ. Die in der Validierungsstudie verwendete Datenbrücke misst Induktivität, Kapazität und Widerstand bis 2 000 H, 2 000 ¼F bzw. 2 M© bei Frequenzen von 100 Hz oder 1 kHz unter Verwendung serieller oder paralleler Werte. Für die TER-Messungen werden Messungen der hautätzenden Wirkung in Widerständen bei einer Frequenz von 100 Hz und unter Verwendung serieller Werte verwendet. Vor der Messung des elektrischen Widerstands wird die Oberflächenspannung der Haut verringert, indem so viel 70 %iges Ethanol hinzugegeben wird, dass die Epidermis bedeckt ist. Nach einigen Sekunden wird das Ethanol aus dem Rohr entfernt und das Hautgewebe durch Hinzufügen von 3 ml MgSO4-Lösung (154 mM) befeuchtet. Zur Messung des Widerstands in k Ω/Hautstück (Abbildung 1) werden die Elektroden der Datenbrücke auf beiden Seiten des Hautstücks angebracht. Die Gesamtabmessungen der Elektroden sowie die Länge der Elektrode unterhalb der Abgreifklemmen sind in Abbildung 2 dargestellt. Die an der Innenelektrode angebrachte Abgreifklemme liegt während der Widerstandsmessung oben auf dem PTFE-Rohr auf, so dass stets eine gleich bleibende Elektrodenlänge in die MgSO4-Lösung eingetaucht bleibt. Die äußere Elektrode ist in der Rezeptorkammer so angeordnet, dass sie am Kammerboden ansteht. Der Abstand zwischen der Federklemme und dem unteren Ende des PTFE-Rohrs bleibt konstant (Abbildung 2), da dieser Abstand den erzielten Widerstandswert beeinflusst. Folglich muss auch der Abstand zwischen der Innenelektrode und dem Hautstück konstant und möglichst gering sein (1-2 mm).
23. Ist der gemessene Widerstand größer als 20 kΩ, so kann dies daran liegen, dass ein Rest der Prüfchemikalie die Epidermisoberfläche des Hautstücks bedeckt. Zur weiteren Entfernung dieser Schicht kann beispielsweise versucht werden, das PTFE-Rohr mit dem Daumen (geschützt durch einen Gummihandschuh) zu verschließen und das Rohr ca. 10 Sekunden lang zu schütteln; anschließend wird die MgSO4-Lösung weggeschüttet und die Widerstandsmessung mit frischer MgSO4-Lösung wiederholt.
24. Eigenschaften und Abmessungen der Prüfapparatur und das verwendete Versuchsverfahren können die ermittelten TER-Werte beeinflussen. Der Schwellenwert von 5 kΩ für hautätzende Wirkung wurde aus Daten abgeleitet, die mit der/dem in dieser Methode beschriebenen speziellen Versuchsanordnung und Versuchsverfahren ermittelt wurden. Abweichende Schwellen- und Kontrollwerte gelten möglicherweise, wenn sich die Prüfbedingungen ändern oder eine andere Versuchsanordnung verwendet wird. Daher müssen die Schwellenwerte für die Methodik und den Widerstand durch Prüfung einer Reihe von Leistungsstoffen kalibriert werden, die aus den in der Validierungsstudie (8) (9) verwendeten Stoffen oder aus Chemikalienklassen, die den untersuchten Stoffen ähneln, ausgewählt wurden. Tabelle 1 gibt eine Übersicht über geeignete Leistungsstoffe.
Farbbindungsmethoden
25. Bei bestimmten nicht hautätzenden Materialien, denen die Haut ausgesetzt ist, kann der Widerstand unter den Schwellenwert von 5 kΩ sinken, wodurch es zum Ionendurchtritt durch die Hornhaut kommt und der elektrische Widerstand absinkt (9). So können beispielsweise neutrale organische Stoffe und Chemikalien mit oberflächenaktiven Eigenschaften (u. a. Detergenzien, Emulgatoren und andere Tenside) die Hautlipide entziehen, wodurch die Trennschicht ionendurchlässiger wird. Liegen die TER-Werte dieser Prüfchemikalien also unter oder um 5 kΩ und sind keine optischen Anzeichen einer Schädigung der Hautstücke zu erkennen, ist eine Beurteilung des Farbeindringvermögens an den behandelten Hautgeweben und den Kontrollgeweben durchzuführen, um festzustellen, ob die ermittelten TER-Werte durch gestiegene Hautpermeabilität oder durch hautätzende Wirkung zustande kamen (7) (9). In letzterem Fall dringt, wenn eine Unterbrechung der Hornhaut vorliegt, der auf die Hautoberfläche aufgetragene Farbstoff Sulforhodamin B rasch ein und verursacht eine Verfärbung des darunter liegenden Hautgewebes. Dieser Farbstoff ist gegenüber einer breiten Palette von Stoffen stabil und wird durch das nachstehend beschriebene Extraktionsverfahren nicht beeinflusst.
Anwendung und Entfernung von Sulforhodamin-B-Farbstoff
26. Nach der Beurteilung der TER-Prüfungen wird das Magnesiumsulfat aus dem Rohr abgeschüttet und die Haut sorgfältig auf erkennbare Schäden untersucht. Wenn keine offensichtliche größere Schädigung (z.B. eine Perforation) zu erkennen ist, werden 150 µl einer 10 %igen (w/v) Verdünnung des Farbstoffs Sulforhodamin B (Acid Red 52, CI-Nr. 45100; CAS-Nr. 3520-42-1) 2 h in destilliertem Wasser auf die Epidermis der einzelnen Hautstücke aufgetragen. Diese Hautstücke werden anschließend höchstens bei Raumtemperatur unter fließendem Wasser ca. 10 Sekunden lang abgewaschen, um überschüssige/ungebundene Farbe zu entfernen. Jedes Hautstück wird sorgfältig vom PTFE-Rohr entfernt und in ein Gefäß (z.B. ein 20-ml-Szintillationsfläschchen) eingelegt, das entionisiertes Wasser (8 ml) enthält. Die Fläschchen werden 5 Minuten lang vorsichtig geschüttelt, um restliche ungebundene Farbe zu entfernen. Nach Wiederholung des Spülvorgangs werden die Hautstücke entnommen und in Fläschchen eingelegt, die 5 ml 30 %iges (w/v) Natriumdodecylsulfat (SDS) in destilliertem Wasser enthalten, und über Nacht bei 60 °C aufbewahrt.
27. Nach dieser Inkubation werden die einzelnen Hautstücke entnommen und entsorgt und die verbleibende Lösung 8 Minuten lang bei 21 °C zentrifugiert (relative Zentrifugalkraft ~175 × g). Eine 1-ml-Probe des oberflächenaktiven Stoffs wird im Verhältnis 1:5 (v/v) (d. h. 1 ml + 4 ml) mit 30 %igem (w/v) SDS in destilliertem Wasser verdünnt. Die optische Dichte (OD) der Lösung wird bei 565 nm gemessen.
Berechnung des Farbgehalts
28. Der Gehalt an Sulforhodamin-B-Farbstoff je Hautstück wird mittels der OD-Werte (9) berechnet (molarer Extinktionskoeffizient von Sulforhodamin B bei 565 nm = 8,7 × l04, Molekulargewicht = 580). Der Farbstoffgehalt wird für jedes der drei Hautstücke durch eine entsprechende Kalibrierungskurve bestimmt und dann der mittlere Farbstoffgehalt für die Wiederholungs-Gleichtests berechnet.
Akzeptanzkriterien
29. Die mittleren TER-Werte werden akzeptiert, wenn die Ergebnisse der gleichzeitigen Positiv- und Negativkontrollen innerhalb der akzeptablen Bereiche für die Methode im Prüflabor liegen. Die akzeptablen Widerstandsbereiche für die oben beschriebene Methode und Versuchsanordnung lauten wie folgt:
| Kontrolle | Stoff | Widerstandsbereich (kΩ) |
| Positivkontrolle | 10 M Chlorwasserstoffsäure | 0,5-1,0 |
| Negativkontrolle | Destilliertes Wasser | 10-25 |
30. Die ermittelten Mittelwerte der Farbbindung werden nur akzeptiert, wenn die Ergebnisse der gleichzeitig geprüften Kontrollen innerhalb definierter Akzeptanzgrenzen der betreffenden Methode liegen. Die für die Kontrollstoffe der oben beschriebenen Methodik und Versuchsanordnung vorgeschlagenen Akzeptanzspannen sind der folgenden Tabelle zu entnehmen:
| Kontrolle | Stoff | Farbgehaltsbereich (µg/Hautstück) |
| Positivkontrolle | 10 M Chlorwasserstoffsäure | 40-100 |
| Negativkontrolle | Destilliertes Wasser | 15-35 |
Auswertung der Ergebnisse
31. Der TER-Schwellenwert zur Unterscheidung zwischen ätzenden und nicht ätzenden Prüfchemikalien wurde bei der Optimierung der Prüfmethode ermittelt, in einer Vorvalidierungsphase geprüft und in einer förmlichen Validierungsstudie bestätigt.
32. Im Folgenden wird das Vorhersagemodell für die TER-Prüfmethode zur Feststellung von Hautätzungen bei Rattenhaut (9)(19) in Verbindung mit dem Klassifizierungssystem des UN GHS bzw. der CLP-Verordnung erläutert:
Die Prüfchemikalie gilt als "nicht hautätzend":
- wenn der Mittelwert des für die Prüfchemikalie ermittelten TER-Werts größer als 5 kΩ ist oder
- wenn der Mittelwert des für die Prüfchemikalie ermittelten TER-Werts gleich 5 kΩ oder kleiner ist und
- die Hautstücke keine wahrnehmbaren Veränderungen (z.B. Perforationen) erkennen lassen, und
- der mittlere Farbstoffgehalt des Hautstücks unter (<) dem mittleren Farbstoffgehalt des Hautstücks der gleichzeitig mit 10M HCl durchgeführten Positivkontrolle liegt (siehe Nummer 30 zu Werten der Positivkontrolle).
Die Prüfchemikalie gilt als "hautätzend":
- wenn der für die Prüfchemikalie ermittelte Mittelwert des TER-Werts gleich 5 kΩ oder kleiner ist (≤) und das Hautstück offensichtliche Schädigungen aufweist (z.B. eine Perforation) oder
- wenn der Mittelwert des für die Prüfchemikalie ermittelten TER-Werts gleich 5 kΩ oder kleiner ist, und
- die Hautstücke keine wahrnehmbaren Veränderungen (z.B. Perforationen) erkennen lassen, aber
- der mittlere Farbstoffgehalt des Hautstücks größer oder gleich (≥) dem mittleren Farbstoffgehalt des Hautstücks der gleichzeitig mit 10M HCl durchgeführten Positivkontrolle ist (siehe Nummer 30 zu Werten der Positivkontrolle).
33. Vorausgesetzt die Prüfung liefert ein eindeutiges Ergebnis, genügt ein einziger Prüflauf (Versuch) mit drei parallel geprüften Replikat-Hautstücken. Bei Grenzergebnissen, wie z.B. nicht übereinstimmenden Replikatmessungen und/oder einer mittleren TER-Werten von 5 ± 0,5 kΩ, sollte ein zweiter unabhängiger Prüflauf (Versuch) in Betracht gezogen werden bzw. ein dritter bei abweichenden Ergebnissen der ersten beiden Prüfläufe (Versuche).
Daten und Berichterstattung
Daten
34. Die Widerstandswerte (k) und ggf. die Farbstoffgehalte (µg/Hautstück) für die Prüfchemikalie sowie für Positiv- und Negativkontrollen sind in Tabellenform einschließlich der Daten für die einzelnen Replikat-Hautstücke pro Prüflauf (Versuch) und der Mittelwerte ± Standardabweichung in Berichten zusammenzufassen. Alle Wiederholungsversuche sollten vermerkt werden. Für jede Prüfchemikalie sind festgestellte Schädigungen der Hautstücke zu vermerken.
Prüfbericht
35. Der Prüfbericht sollte folgende Angaben enthalten:
Prüfchemikalien und Kontrollchemikalien:
- Einkomponentiger Stoff: chemische Bezeichnung, wie z.B. IUPAC- oder CAS-Bezeichnung, CAS-Nummer, SMILES- oder InChI-Code, Strukturformel, Reinheit, chemische Zusammensetzung von Verunreinigungen, soweit zutreffend und praktisch durchführbar, usw.;
- mehrkomponentiger Stoff, UVCB und Gemische: So weit wie möglich charakterisiert durch die chemische Zusammensetzung (siehe oben), das quantitative Vorkommen und die relevanten physikalisch-chemischen Eigenschaften der einzelnen Komponenten;
- physikalisches Erscheinungsbild, Wasserlöslichkeit und weitere relevante physikalisch-chemische Eigenschaften;
- Herkunft, Chargennummer, sofern vorhanden;
- Behandlung der Prüfchemikalien/Kontrollstoffe vor der Prüfung, sofern relevant (z.B. Erwärmen, Mahlen);
- Stabilität der Prüfchemikalie, letztes Verwendungsdatum oder Datum für erneute Analyse, soweit bekannt;
- Lagerungsbedingungen.
Versuchstiere:
- Abstammung und Geschlecht;
- Alter der Tiere zum Zeitpunkt der Verwendung als Spendertiere;
- Herkunft der Tiere, Haltungsbedingungen, Futter usw.;
- Details zur Herstellung der Hautstücke.
Prüfbedingungen:
- Eichkurven für die Versuchsapparaturen;
- Eichkurven für die Durchführung der Farbbindungsprüfungen, verwendete Bandbreite für die Messung von OD-Werten und die OD-Linearitätsspanne des Messgeräts (z.B. eines Spektrophotometers) (sofern relevant);
- Details des Prüfverfahrens für die TER-Messungen;
- Details des Prüfverfahrens für die Beurteilung der Farbbindefähigkeit (sofern relevant);
- verwendete Prüfdosierungen, Expositionszeitraum (-zeiträume) und -temperatur(en);
- Details zum verwendeten Waschverfahren nach dem Expositionszeitraum;
- Anzahl der je Prüfchemikalie und Kontrolle (Positiv- und Negativkontrolle) verwendeten Replikat-Hautstücke;
- Beschreibung etwaiger Änderungen am Prüfverfahren;
- Verweis auf historische Modelldaten.
Dabei sollten unter anderem die folgenden Aspekte berücksichtigt werden:
- Akzeptanz der TER-Werte der Positiv- und Negativkontrolle (in k Ω) mit Verweis auf die Spannen der Widerstandswerte der Positiv- und Negativkontrollen,
- Akzeptanz der Farbstoffgehaltwerte der Positiv- und Negativkontrolle (in µg/Hautstück) mit Verweis auf die Spannen der Farbstoffgehaltwerte der Positiv- und Negativkontrollen,
- Akzeptanz der Prüfergebnisse mit Verweis auf die historischen Schwankungen zwischen Replikat-Hautstücken;
- Beschreibung der berücksichtigten Entscheidungskriterien und des verwendeten Vorhersagemodells.
Ergebnisse:
- Tabellarische Darstellung der Daten der Versuche zur Beurteilung der TER-Werte und der Farbbindefähigkeit (wenn relevant) für die einzelnen Prüfchemikalien und Kontrollen, für jeden einzelnen Prüflauf (Versuch) und für jedes Replikat-Hautstück (einzelne Tiere und einzelne Hautproben), Mittel, Standardabweichungen und Variationskoeffizienten;
- Beschreibung etwaiger beobachteter Wirkungen;
- abgeleitete Einstufung mit Bezug auf das Vorhersagemodell und/oder angewandte Entscheidungskriterien.
Erörterung der Ergebnisse
Schlussfolgerungen
Literatur
(1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), fünfte überarbeitete Ausgabe, UN New York und Genf, 2013. Abrufbar unter:[http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html].
(2) Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung.
(3) Kapitel B.40bis dieses Anhangs, Invitro-Hautmodell.
(4) Kapitel B.65 dieses Anhangs, Invitro-Methode zur Untersuchung der Membranbarriere.
(5) Kapitel B.46 dieses Anhangs, Invitro-Hautreizung: Prüfmethode mit rekonstruierter humaner Epidermis.
(6) OECD (2014). Guidance document on Integrated Approaches to Testing and Assessment for Skin Irritation/Corrosion, Environment, Health and Safety Publications, Series on Testing and Assessment, (No 203), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
(7) Botham, P.A., Chamberlain, M., Barratt, M.D., Curren, R.D., Esdaile, D.J., Gardner, J.R., Gordon, V.C., Hildebrand, B., Lewis, R.W., Liebsch, M., Logemann, P., Osborne, R., Ponec, M., Regnier, J.F., Steiling, W., Walker, A.P., and Balls, M. (1995). A Prevalidation Study on In Vitro Skin Corrosivity Testing. The Report and Recommendations of ECVAM Workshop 6.ATLA 23, 219-255.
(8) Barratt, M.D., Brantom, P.G., Fentem, J.H., Gerner, I., Walker, A.P. und Worth, A.P. (1998). The ECVAM International Validation Study on In Vitro Tests for Skin Corrosivity. 1. Selection and Distribution of the Test Chemicals. Toxic.In Vitro 12, 471-482.
(9) Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Esdaile, D.J., Holzhütter, H.-G., and Liebsch, M. (1998). The ECVAM International Validation Study on In Vitro Tests For Skin Corrosivity. 2. Results and Evaluation by the Management Team. Toxic.In Vitro12, 483- 524.
(10) Balls, M., Blaauboer, B.J., Fentem, J.H., Bruner, L., Combes, R.D., Ekwall, B., Fielder, R.J., Guillouzo, A., Lewis, R.W., Lovell, D.P., Reinhardt, CA., Repetto, G., Sladowski, D., Spielmann, H. und Zucco, F. (1995). Practical Aspects of the Validation of Toxicity Test Procedures. The Report and Recommendations of ECVAM Workshops.ATLA23, 129-147.
(11) ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods). (1997). Validation and Regulatory Acceptance of Toxicological Test Methods. NIH Publication No. 97-3981. National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA.
(12) EC-ECVAM (1998). Statement on the Scientific Validity of the Rat Skin Transcutaneos Electrical Resistance (TER) Test (an In Vitro Test for Skin Corrosivity), herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC10), 3. April 1998.
(13) ECVAM (1998). ECVAM News & Views. ATLA 26, 275-280.
(14) ICCVAM (Interagency Coordinating Committee on the Validation of Alternative Methods) (2002). ICCVAM Evaluation of EpiDermTM (EPI-200), EPISKINTM (SM), and the Rat Skin Transcutaneous Electrical Resistance (TER) Assay: In Vitro Test Methods for Assessing Dermal Corrosivity Potential of Chemicals. NIH Publication No. 02-4502. National Toxicology Program Interagency center for the Evaluation of Alternative Toxicological Methods, National Institute of Environmental Health Sciences, Research Triangle Park, NC, USA.
(15) OECD (2015). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Transcutaneous Electrical Resistance (TER) Test Method for Skin Corrosion in Relation to TG 430. Environmental Health and Safety Publications, Series on Testing and Assessment No 218. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
(16) Oliver, G.J.A., Pemberton, M.A. und Rhodes, C. (1986). An In Vitro Skin Corrosivity Test -Modifications and Validation. Fd. Chem. Toxicol. 24, 507-512.
(17) Botham, P.A., Hall, T.J., Dennett, R., McCall, J.C., Basketter, D.A., Whittle, E., Cheeseman, M., Esdaile, D.J. und Gardner, J. (1992). The Skin Corrosivity Test In Vitro: Results of an Interlaboratory Trial. Toxicol. In Vitro 6,191-194.
(18) Eskes, C., Detappe, V., Koëter, H., Kreysa, J., Liebsch, M., Zuang, V., Amcoff, P., Barroso, J., Cotovio, J., Guest, R., Hermann, M., Hoffmann, S., Masson, P., Alépée, N., Arce, L.A., Brüschweiler, B., Catone, T., Cihak, R., Clouzeau, J., D'Abrosca, F., Delveaux, C., Derouette, J.P., Engelking, O., Facchini, D., Fröhlicher, M., Hofmann, M., Hopf, N., Molinari, J., Oberli, A., Ott, M., Peter, R., Sá-Rocha, V.M., Schenk, D., Tomicic, C., Vanparys, P., Verdon, B., Wallenhorst, T., Winkler, G.C. und Depallens, O. (2012). Regulatory Assessment of In Vitro Skin Corrosion and Irritation Data Within the European Framework: Workshop Recommendations. Regul.Toxicol.Pharmacol. 62, 393-403.
(19) TER SOP (December 2008). INVITTOX Protocol (No 115) Rat Skin Transcutaneous Electrical Resistance (TER) Test.
(20) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 34), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris.
____________________
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. L 353 vom 31.12.2008 S. 1).
Abbildung 1 Apparatur Für Die Ter-Prüfung Mit Rattenhaut
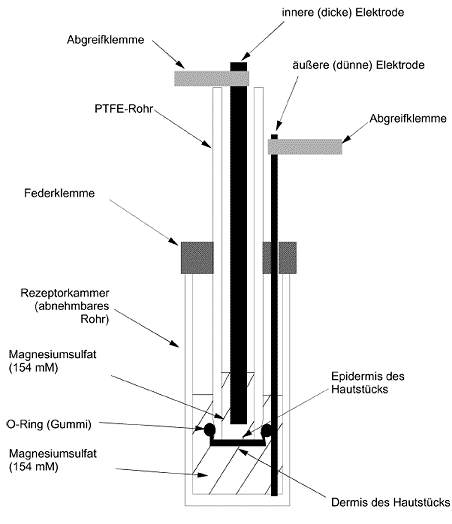
Abbildung 2 Abmessungen Der Polytetrafluorethylen- (PTFE-) und Rezeptorrohre und Der Verwendeten Elektroden
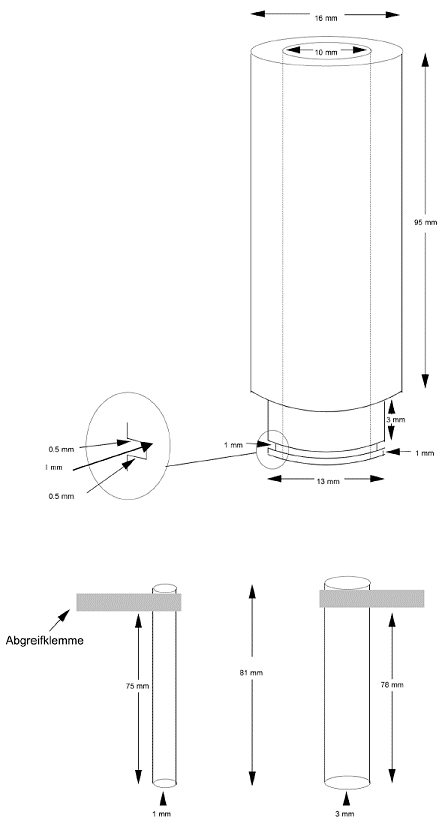
Kritische Faktoren der obigen Apparaturen:
- Innendurchmesser des PTFE-Rohrs,
- Länge der Elektroden in Relation zum PTFE-Rohr und zum Rezeptorrohr, so dass die Hautscheibe nicht mit den Elektroden in Kontakt kommen sollte und die Elektrode auf einer Standardlänge mit der MgSO4-Lösung in Kontakt steht,
- die Menge an MgSO4-Lösung im Rezeptorrohr muss in Relation zum Füllstand im PTFE-Rohr den in Abbildung 1 dargestellten Flüssigkeitsstand ergeben,
- das Hautstück muss so am PTFE-Rohr befestigt sein, dass der elektrische Widerstand ein richtiges Maß für die Hauteigenschaften darstellt.
| Begriffsbestimmungen | Anlage |
| C : | Hautätzend |
| Chemikalie : | Ein Stoff oder ein Gemisch. |
| Einkomponentiger Stoff : | Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorliegt. |
| Gemisch : | Ein Gemisch oder eine Lösung, die aus zwei oder mehr Stoffen besteht. |
| Genauigkeit : | Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von,Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (20). |
| GHS: (Globally Harmonized System of Classification and Labelling of Chemicals = Global harmonisiertes System zur Einstufung und Kennzeichnung) (UN) | Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenbögen, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1). |
| Hautverätzung, in vivo : | Das Auslösen einer irreversiblen Hautschädigung, d. h. einer sichtbaren, bis in das Corium reichenden Nekrose der Epidermis nach Applikation einer Prüfchemikalie für die Dauer von bis zu 4 Stunden. Verätzungsreaktionen sind gekennzeichnet durch Geschwüre, Blutungen, blutige Verschorfungen und am Ende des Beobachtungszeitraums von 14 Tagen durch eine auf ein Ausbleichen der Haut zurückzuführende Verfärbung, komplett haarlose Bereiche und Narben. Bei der Beurteilung fragwürdiger Schädigungen ist die Histopathologie mit zu berücksichtigen. |
| IATA : | Integrated Approach to Testing and Assessment = integrierter Prüfungs- und Bewertungsansatz. |
| Leistungsstandards (PS) : | Auf einer validierten Prüfmethode beruhende Normen, auf deren Grundlage die Vergleichbarkeit einer vorgeschlagenen, mechanistisch und funktionell ähnlichen Prüfmethode bewertet werden kann. Sie umfassen (i) wesentliche Elemente der Prüfmethode; (ii) ein Mindestverzeichnis von Referenzchemikalien, ausgewählt aus den Chemikalien, die zum Nachweis der akzeptablen Leistung der validierten Referenzmethode verwendet werden; und (iii) je nach den für die validierte Referenzmethode erzielten Ergebnissen die vergleichbaren Zuverlässigkeits- und Genauigkeitswerte, die die vorgeschlagene Prüfmethode bei der Bewertung anhand des Mindestverzeichnisses von Referenzchemikalien demonstrieren sollte. |
| Mehrkomponentiger Stoff : | Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens ≥ 10 % w/w und < 80 % w/w vorliegt. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet. |
| NC : | Nicht hautätzend |
| OD : | Optische Dichte. |
| PK : | Positivkontrolle, ein Replikat, das alle Komponenten eines Prüfsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. |
| Prüfchemikalie : | Stoff oder Gemisch, der/das mit dieser Prüfmethode untersucht wird. |
| Prüflauf : | Eine einzelne Prüfchemikalie, die gleichzeitig an mindestens drei Replikat-Hautstücken geprüft wird. |
| Relevanz : | Dieser Begriff bezieht sich auf das Verhältnis zwischen der Prüfmethode und der betreffenden Wirkung und auf die Frage, ob er aussagekräftig und nützlich für einen bestimmten Zweck ist. Er beschreibt das Ausmaß, in dem die Prüfmethode die untersuchte biologische Wirkung korrekt misst oder vorhersagt. Die Relevanz schließt eine Beurteilung der Genauigkeit (Übereinstimmung) einer Prüfmethode ein (20). |
| Sensitivität : | Der Anteil aller positiven/wirkenden Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Sensitivität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (20). |
| Spezifizität : | Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (20). |
| Stoff : | Ein chemisches Element und seine Verbindungen in natürlicher Form oder gewonnen durch ein Herstellungsverfahren, einschließlich der zur Wahrung seiner Stabilität notwendigen Zusatzstoffe und der durch das angewandte Verfahren bedingten Verunreinigungen, aber mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können. |
| TER (Transcutaneous Electrical Resistance) : | Ein Maß für den elektrischen Widerstand der Haut, angegeben als Widerstand in Ohm. Ein einfaches und stabiles Verfahren zur Beurteilung der Barriere funktion, indem der Ionendurchtritt durch die Haut mithilfe einer Wheatstone-Messbrücke aufgezeichnet wird. |
| Übereinstimmung : | Die Übereinstimmung ist ein Maß der Leistung einer Prüfmethode für Prüfverfahren mit kategorialen Ergebnissen und ein Aspekt der Relevanz. Der Begriff wird gelegentlich gleichbedeutend mit Genauigkeit verwendet und als Anteil aller geprüften Chemikalien definiert, die korrekt als positiv oder negativ eingestuft werden. Die Übereinstimmung hängt in hohem Maße von der Prävalenz positiver Ergebnisse bei den Typen der untersuchten Prüfchemikalien ab (20). |
| UVCB : | Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien. |
| Zuverlässigkeit : | Maß der Verlässlichkeit der Reproduzierbarkeit der Prüfmethode innerhalb von und zwischen Laboratorien in einem bestimmten Zeitintervall bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet (20). |
| weiter . |  |
...
X
⍂
↑
↓