 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2014, Chemikalien - EU Bund
| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2014, Chemikalien - EU Bund |
Verordnung (EU) Nr. 260/2014 der Kommission vom 24. Januar 2014 zur Änderung der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zwecks Anpassung an den technischen Fortschritt
(Text von Bedeutung für den EWR)
(ABl. Nr. L 81 vom 19.03.2014 S. 1)
Die Europäische Kommission -
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Chemikalienagentur, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission 1, insbesondere auf Artikel 13 Absatz 3,
in Erwägung nachstehender Gründe:
(1) In der Verordnung (EG) Nr. 440/2008 der Kommission 2 sind die in der Verordnung (EG) Nr. 1907/2006 vorgesehenen Prüfmethoden zur Bestimmung der physikalisch-chemischen Eigenschaften, der Toxizität und der Ökotoxizität von Stoffen festgelegt.
(2) Es ist angezeigt, die Verordnung (EG) Nr. 440/2008 zu aktualisieren und vorrangig um neue und aktualisierte alternative Prüfmethoden zu ergänzen, die von der OECD kürzlich angenommen wurden mit dem Ziel, die Zahl der für Versuchszwecke verwendeten Tiere gemäß der Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere 3 und der Richtlinie 86/609/EWG des Rates vom 24. November 1986 zur Annäherung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten zum Schutz der für Versuche und andere wissenschaftliche Zwecke verwendeten Tiere 4 zu verringern.
(3) Die Anpassung betrifft zwei Methoden zur Bestimmung physikalisch-chemischer Eigenschaften (einschließlich einer Aktualisierung der Prüfmethode zur Bestimmung der Wasserlöslichkeit und einer neuen Prüfmethode zur Bestimmung des Verteilungskoeffizienten, die für die Bewertung persistenter, bioakkumulierbarer und toxischer (persistent, bioaccumulative and toxic, PBT) Stoffe relevant sind), vier neue und eine aktualisierte Methode zur Bestimmung der Ökotoxizität sowie des Verbleibs und Verhaltens von Stoffen in der Umwelt, neun Methoden zur Bestimmung der Toxizität und anderer gesundheitlicher Auswirkungen von Stoffen einschließlich vier Inhalationstoxizitätsmethoden (darunter eine Aktualisierung von drei bestehenden Methoden und eine neue Methode zur Verringerung der Zahl von Versuchstieren und zur Verbesserung der Wirkungsabschätzung, eine Aktualisierung der 28-Tage-Prüfung auf orale Toxizität bei wiederholter Verabreichung zwecks Einbeziehung von Parametern für die Bewertung der endokrinen Aktivität, einer Aktualisierung der Toxikokinetik-Prüfmethode, die für die Ausrichtung und das Verständnis toxikologischer Studien relevant ist, sowie eine Aktualisierung der Prüfung auf chronische Toxizität und der Prüfung auf Karzinogenität und der kombinierten Prüfung auf chronische Toxizität und Karzinogenität).
(4) Die Verordnung (EG) Nr. 440/2008 sollte daher entsprechend geändert werden.
(5) Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des mit Artikel 133 der Verordnung (EG) Nr. 1907/2006 eingesetzten Ausschusses
- hat folgende Verordnung erlassen:
Der Anhang der Verordnung (EG) Nr. 440/2008 wird nach Maßgabe des Anhangs der vorliegenden Verordnung geändert.
Diese Verordnung tritt am dritten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
____
1) ABl. Nr. L 396 vom 30.12.2006 S. 1.
2) ABl. Nr. L 142 vom 31.05.2008 S. 1.
3) ABl. Nr. L 276 vom 20.10.2010 S. 33.
4) ABl. Nr. L 358 vom 18.12.1986 S. 1.
| Anhang |
Der Anhang der Verordnung (EG) Nr. 440/2008 wird wie folgt geändert:
1. Das Kapitel A.6 erhält folgende Fassung:
"A.6 Wasserlöslichkeit
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 105 (1995). Sie ist eine überarbeitete Fassung der ursprünglichen TG 105, die 1981 angenommen wurde. Es gibt keine inhaltlichen Unterschiede zwischen der derzeitigen Fassung und der von 1981. Geändert wurde hauptsächlich das Format. Die Überarbeitung stützte sich auf die EU-Prüfmethode 'Wasserlöslichkeit' 1.
Ausgangsüberlegungen
2. Die Wasserlöslichkeit eines Stoffs kann durch Verunreinigungen erheblich beeinflusst werden. Diese Prüfmethode behandelt die Bestimmung der Wasserlöslichkeit von im Wesentlichen reinen Substanzen, die in Wasser stabil und nicht flüchtig sind. Vor Bestimmung der Wasserlöslichkeit sollten Vorinformationen über die Strukturformel, den Dampfdruck, die Dissoziationskonstante und das Hydrolyseverhalten (als Funktion des pH-Wertes) des Stoffes vorliegen.
3. Die beiden nachstehend beschriebenen Methoden, d. h. die Säulen-Elutions-Methode und die Kolbenmethode, decken Löslichkeiten von unter bzw. über 10-2 g/l ab. Ein einfacher Vorversuch wird ebenfalls beschrieben. Er ermöglicht die Bestimmung der bei der eigentlichen Prüfung zu verwendenden ungefähren Probenmenge und die zum Erreichen der Sättigungskonzentration notwendige Zeit.
Definitionen und Einheiten
4. Die Wasserlöslichkeit einer Substanz wird durch ihre Massen-Sättigungskonzentration in Wasser bei einer bestimmten Temperatur angegeben.
5. Die Wasserlöslichkeit wird als Masse des gelösten Stoffs je Lösungsvolumen ausgedrückt. Die SI-Einheit ist kg/m3, aber g/l kann auch verwendet werden.
Referenzsubstanzen
6. Bei der Untersuchung der Prüfsubstanz brauchen keine Referenzsubstanzen verwendet zu werden.
Beschreibung der Methoden
Prüfbedingungen
7. Die Prüfung wird vorzugsweise bei 20 °C ± 0,5 °C durchgeführt. Die gewählte Temperatur ist in allen wichtigen Teilen der Apparatur konstant zu halten.
Vorversuch
8. Etwa 0,1 g der Probe (feste Prüfsubstanzen müssen pulverisiert sein) werden in einen mit Glasstopfen verschließbaren 10-ml-Messzylinder gegeben. Dann werden portionsweise zunehmende Volumen Wasser von Raumtemperatur zugesetzt. Nach jedem Zusatz einer Wassermenge wird die Mischung 10 Minuten geschüttelt und mit bloßem Auge auf ungelöste Teilchen der Probe untersucht. Wenn nach Zusatz von 10 ml Wasser die Probe oder Teile von ihr ungelöst bleiben, ist der Versuch in einem 100-ml-Messzylinder zu wiederholen. Die ungefähre Löslichkeit ist in Tabelle 1 unter demjenigen Volumen Wasser angegeben, bei dem die Probe vollständig gelöst wird. Bei geringer Löslichkeit kann es lange (bis zu 24 Stunden) dauern, bis die Prüfsubstanz gelöst ist. Ist die Prüfsubstanz nach 24 Stunden noch nicht gelöst, sollte der Versuch verlängert werden (maximal 96 Stunden) oder es sollte weiter verdünnt werden, um festzustellen, ob die Säulen-Elutions- oder die Kolben-Methode zu benutzen ist.
Tabelle 1
| ml Wasser auf 0,1 g lösliche Substanz | 0,1 | 0,5 | 1 | 2 | 10 | 100 | > 100 |
| Ungefähre Löslichkeit in g/l | > 1.000 | 1.000-200 | 200-100 | 100-50 | 50-10 | 10-1 | < 1 |
Säulen-Elutions-Methode
Prinzip
9. Diese Methode basiert auf der Elution einer Prüfsubstanz mit Wasser aus einer Mikrosäule, die mit einem inerten Trägermaterial gefüllt ist, welches mit einem Überschuss an Prüfsubstanz beschichtet ist 2. Die Wasserlöslichkeit wird durch die Massenkonzentration des Eluats ausgedrückt, wenn dieses ein Plateau in Abhängigkeit von der Zeit erreicht hat.
Apparatur
10. Die Apparatur besteht aus einer Mikrosäule (Abbildung 1), die auf konstanter Temperatur gehalten wird. Sie ist entweder mit einer Umwälzpumpe (Abbildung 2) oder mit einem Niveaugefäß (Abbildung 3) verbunden. Die Mikrosäule enthält ein inertes Trägermaterial, das durch einen kleinen Pfropfen aus Glaswolle in der Säule gehalten wird, der gleichzeitig zum Herausfiltern von Partikeln dient. Als Trägermaterial können Glaskugeln, Diatomeenerde oder andere inerte Stoffe verwendet werden.
11. Die Mikrosäule in Abbildung 1 eignet sich für die Apparatur mit Umwälzpumpe. Sie hat einen Kopfraum für fünf Säulenbett-Volumina (werden zu Beginn des Versuchs verworfen) und das Volumen von fünf Proben (werden während des Versuchs entnommen). Der Kopfraum kann kleiner gehalten werden, wenn während des Versuchs Wasser zugegeben werden kann, um die anfänglich mit Verunreinigungen entnommenen fünf Säulenbett-Volumina zu ersetzen. Die Säule ist durch einen Schlauch aus inertem Material mit der Umwälzpumpe verbunden, die mit einem Fluss von etwa 25 ml/h fördern kann. Die Umwälzpumpe kann z.B. eine Schlauch- oder eine Membranpumpe sein. Dabei ist darauf zu achten, dass es nicht zu einer Verunreinigung und/oder Absorption durch das Schlauchmaterial kommt.
12. Abbildung 3 zeigt die schematische Darstellung mit Verwendung eines Niveaugefäßes. In dieser Darstellung ist die Mikrosäule mit einem Einweghahn versehen. Sie wird mit einem Glasschliff-Verbindungsstück und einem Schlauch aus inertem Material an das Niveaugefäß angeschlossen. Die Durchflussrate vom Niveaugefäß sollte etwa 25 ml/h betragen.
Abbildung 1
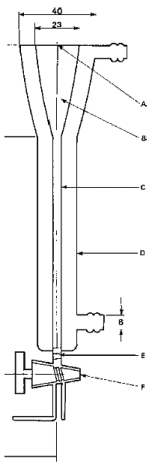
Abmessungen in mm
A. Anschluss für Glasschliff-Stopfen
B. Kopfraum
C. Innenmaß 5
D. Außenmaß 19
E. Glaswollpfropfen
F. Absperrhahn
Abbildung 2
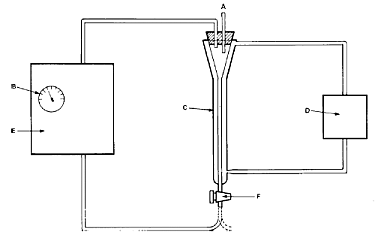
A. atmosphärischer Druckausgleich
B. Durchflussmesser
C. Mikrosäule
D. thermostatgeregelte Pumpe
E. Umwälzpumpe
F. 2-Wege-Hahn zur Probenentnahme
Abbildung 3
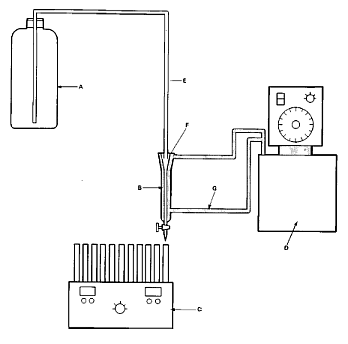
A. Niveaugefäß (z.B. 2,5-Liter-Kolben)
B. Säule
C. Fraktionssammler
D. Thermostat
E. Teflonschlauch
F. Glasschliff-Stopfen
G. Wasserschlauch (zwischen Thermostat und Säule, Innendurchmesser ungefähr 8 mm)
13. Etwa 600 mg Trägermaterial werden in einen 50-ml-Rundkolben eingefüllt. Eine geeignete Menge Prüfsubstanz wird in einem flüchtigen Lösungsmittel von Analysenqualität gelöst, und eine ausreichende Menge dieser Lösung wird zum Trägermaterial hinzugefügt. Das Lösungsmittel muss vollständig abgezogen werden, z.B. in einem Rotationsverdampfer, da sonst während der Elutionsphase wegen Verteilungseffekten auf der Oberfläche keine vollständige Sättigung dieses Materials mit Wasser erzielt wird. Das beladene Trägermaterial lässt man etwa 2 Stunden lang in etwa 5 ml Wasser quellen. Dann wird die Suspension in die Mikrosäule gefüllt. Es ist auch möglich, das trockene, beladene Trägermaterial in die mit Wasser gefüllte Mikrosäule zu geben. Auch hier wird der Quellvorgang von etwa 2 Stunden abgewartet.
14. Das Aufbringen der Prüfsubstanz auf das Trägermaterial kann problematisch werden und zu fehlerhaften Ergebnissen führen, z.B. wenn sich die Prüfsubstanz ölartig auf dem Träger niederschlägt. Diese Probleme sollten untersucht und Einzelheiten dazu dokumentiert werden.
Verfahren mit Umwälzpumpe
15. Der Säulenfluss wird in Gang gesetzt. Eine Durchflussleistung von etwa 25 ml/h wird empfohlen (etwa zehn Säulenbett-Volumina/h bei der beschriebenen Säule). Mindestens die ersten fünf Säulenbett-Volumina werden verworfen, um wasserlösliche Verunreinigungen zu entfernen. Danach lässt man die Umwälzpumpe bis zur Einstellung des Gleichgewichts laufen. Das Gleichgewicht ist erreicht, wenn bei fünf aufeinander folgenden Proben die Konzentrationen um nicht mehr als ± 30 % streuen. Diese Proben sollten in solchen zeitlichen Abständen genommen werden, in denen mindestens zehn Säulenbett-Volumina die Säule durchlaufen haben. Je nach verwendeter Analysemethode kann es ratsam sein, eine Konzentrations-Zeit-Kurve zu erstellen, um zu zeigen, dass das Gleichgewicht erreicht ist.
Verfahren mit Niveaugefäß
16. Aufeinander folgende Eluatfraktionen werden gesammelt und ihre Konzentrationen mit der gewählten Analysemethode bestimmt. Fraktionen des mittleren Eluatbereichs, bei denen die Konzentrationen in mindestens fünf aufeinander folgenden Proben konstant bleiben (± 30 %), werden zur Bestimmung der Wasserlöslichkeit benutzt.
17. Das bevorzugte Elutionsmittel ist bidestilliertes Wasser. Es kann auch deionisiertes Wasser mit einem spezifischen Widerstand von mehr als 10 Megaohm/cm und einem Gesamtgehalt an organischem Kohlenstoff unter 0,01 % verwendet werden.
18. Bei beiden Verfahren wird ein zweiter Durchlauf mit halber Durchflussrate durchgeführt. Stimmen die Ergebnisse der beiden Versuche überein, wird das Prüfergebnis als zufriedenstellend betrachtet. Ist die gemessene Löslichkeit bei dem niedrigeren Durchfluss höher, muss die Durchflussleistung so lange weiter halbiert werden, bis zwei aufeinander folgende Versuchsdurchläufe die gleiche Löslichkeit ergeben.
19. Bei beiden Verfahren sollten die Fraktionen durch Prüfung des Tyndall-Effekts auf kolloidale Substanzpartikel untersucht werden. Wenn solche Substanzpartikel vorkommen, ist das Prüfergebnis unbrauchbar. Die Prüfung sollte dann wiederholt werden, nachdem die Filterfunktion der Säule verbessert wurde.
20. Der pH-Wert jeder Probe sollte vorzugsweise mit speziellen Indikatorstäbchen bestimmt werden.
Kolben-Methode
Prinzip
21. Die Prüfsubstanz (Feststoffe müssen pulverisiert werden) wird bei einer Temperatur in Wasser aufgelöst, die leicht über der Prüftemperatur liegt. Wenn die Sättigung erreicht ist, wird die Lösung abgekühlt und auf der Prüftemperatur gehalten. Alternativ kann die Messung direkt bei der Prüftemperatur durchgeführt werden, wenn durch entsprechende Probenahme gesichert ist, dass das Sättigungsgleichgewicht erreicht ist. Dann wird die Massenkonzentration der Prüfsubstanz in der wässrigen Lösung, die keine ungelösten Substanzpartikel enthalten darf, mit einer geeigneten Analysemethode 3 bestimmt.
Geräte
22. Folgende Geräte werden benötigt:
Verfahren
23. Die zur Sättigung des vorgegebenen Wasservolumens erforderliche Prüfsubstanzmenge wird anhand der Ergebnisse des Vorversuches abgeschätzt. Etwa das Fünffache dieser Menge wird jeweils in drei mit Glasstopfen versehene Glasgefäße eingewogen (z.B. Zentrifugenröhrchen oder Kolben). Jedem Gefäß wird ein je nach Analysemethode und Löslichkeitsbereich gewähltes Wasservolumen zugesetzt. Die Gefäße werden fest verschlossen und dann bei 30 °C geschüttelt. Hierzu sollte ein Schüttel- oder Rührgerät verwendet werden, das bei einer konstanten Temperatur arbeitet, z.B. Magnetrührstäbe in einem thermostatisierten Wasserbad. Nach einem Tag wird eines der Gefäße 24 Stunden unter gelegentlichem Schütteln bei Prüftemperatur stehen gelassen, bis sich das Gleichgewicht eingestellt hat. Dann wird der Inhalt des Gefäßes bei Prüftemperatur zentrifugiert und die Konzentration der Prüfsubstanz in der klaren wässrigen Phase mit einem geeigneten Analyseverfahren bestimmt. Mit den beiden anderen Kolben wird nach zwei bzw. drei Tagen genauso verfahren, nachdem zuvor das Sättigungsgleichgewicht bei 30 °C eingestellt wurde. Weichen die gemessenen Konzentrationen bei mindestens den letzten beiden Gefäßen um nicht mehr als 15 % voneinander ab, ist die Prüfung als zufriedenstellend anzusehen. Wenn die Prüfergebnisse der Gefäße 1, 2 und 3 eine steigende Tendenz aufweisen, sollte die gesamte Prüfung unter Verlängerung der Zeiten für die Gleichgewichtseinstellung wiederholt werden.
24. Die Prüfung kann auch ohne Präinkubation bei 30 °C durchgeführt werden. Um den Grad des erreichten Sättigungsgleichgewichts zu bestimmen, werden so lange Proben entnommen, bis die gemessenen Konzentrationen nicht länger von der Rührzeit beeinflusst werden.
25. Der pH-Wert jeder Probe sollte vorzugsweise mit speziellen Indikatorstäbchen bestimmt werden.
Analytische Bestimmungen
26. Eine substanzspezifische Methode ist vorzuziehen, da bereits kleine Mengen von löslichen Verunreinigungen große Fehler bei der Bestimmung der Löslichkeit verursachen können. Beispiele für solche Analysemethoden sind: Gas- oder Flüssigchromatographie, Titrierverfahren, fotometrische Methoden, voltametrische Verfahren.
Daten und Berichterstattung
Daten
Säulen-Elutions-Methode
27. Für jeden Durchlauf werden der Mittelwert und die Standardabweichung von mindestens fünf aufeinander folgenden Proben aus dem Bereich des Sättigungsplateaus berechnet. Die für zwei Prüfungen mit unterschiedlichen Durchflussleistungen berechneten Mittelwerte sollten nicht um mehr als 30 % voneinander abweichen.
Kolben-Methode
28. Die einzelnen Ergebnisse für jeden der drei Kolben, die nicht um mehr als 15 % voneinander abweichen sollten, werden gemittelt.
Prüfbericht
Säulen-Elutions-Methode
29. Der Prüfbericht muss folgende Informationen enthalten:
Kolben-Methode
30. Der Prüfbericht muss folgende Informationen enthalten:
Literatur:
1. Richtlinie 92/69/EWG der Kommission vom 31. Juli 1992 zur siebzehnten Anpassung der Richtlinie 67/548/EWG des Rates zur Angleichung der Rechts- und Verwaltungsvorschriften für die Einstufung, Verpackung und Kennzeichnung gefährlicher Stoffe an den technischen Fortschritt, ABl.L 383 vom 29.12.1992 S. 113.
2. NF T 20-045 (AFNOR) (September 1985). Chemical products for industrial use - Determination of water solubility of solids and liquids with low solubility - Column elution method.
3. NF T 20-046 (AFNOR) (September 1985). Chemical products for industrial use - Determination of water solubility of solids and liquids with high solubility - Flask method."
2. Das Kapitel A.23 wird angefügt:
"A.23 1-Octanol/Wasser-Verteilungskoeffizient: Methode zur Prüfung unter langsamem Rühren
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 123 (2006). Die POW-Werte (POW = 1-Octanol/ Wasser-Verteilungskoeffizient) konnten mit der Methode zur Prüfung unter langsamem Rühren bis zu log POW 8,2 genau bestimmt werden (1). Entsprechend kommt diese Methode für die direkte Bestimmung der POW-Werte stark hydrophober Substanzen in Betracht.
2. Weitere Methoden zur Bestimmung des 1-Octanol/Wasser-Verteilungskoeffizienten (POW) sind die 'Schüttelmethode' (2) und die Bestimmung des POW aufgrund des Retentionsverhaltens bei der HPLC mit Phasenumkehr (3). Die 'Schüttelmethode' ist wegen der Übertragung von Octanol-Mikrotröpfchen in die wässrige Phase jedoch fehleranfällig. Mit steigenden POW-Werten führt das Vorhandensein dieser Tröpfchen in der wässrigen Phase zu einer zunehmenden Überschätzung der Konzentration der Prüfsubstanz im Wasser. Daher ist diese Methode nur bei Substanzen mit log POW < 4 geeignet. Die zweite Methode beruht auf direkt bestimmten POW-Werten, die zur Kalibrierung der Beziehung zwischen dem Retentionsverhalten in der HPLC und den gemessenen POW- Werten verwendet werden. Es gab einen Entwurf einer OECD-Richtlinie zur Bestimmung des 1-Octanol/Wasser-Verteilungskoeffizienten ionisierbarer Substanzen (4), der aber nicht mehr verwendet werden soll.
3. Diese Prüfmethode wurde in den Niederlanden entwickelt. Die Genauigkeit der in dieser Prüfmethode beschriebenen Verfahren wurde in einer Ringtest-Validierungsstudie unter Beteiligung von 15 Labors validiert und optimiert (5).
Ausgangsüberlegungen
Bedeutung und Anwendung
4. Bei inerten organischen Substanzen wurden hoch signifikante Beziehungen zwischen den 1-Octanol/Wasser- Verteilungskoeffizienten (POW) und der jeweiligen Bioakkumulation in Fischen festgestellt. Außerdem wurde eine Korrelation zwischen den POW-Werten und der Toxizität für Fische sowie zwischen den POW-Werten und der Sorption chemischer Stoffe in Feststoffen wie z.B. in Böden und in Sedimenten nachgewiesen. Das Literaturverzeichnis enthält eine umfassende Übersicht über die verschiedenen Zusammenhänge (6).
5. Zwischen dem 1-Octanol/Wasser-Verteilungskoeffizienten und sonstigen für die Umwelttoxizität und für das chemische Verhalten erheblichen Merkmalen von Substanzen wurden vielfältige Beziehungen festgestellt. Entsprechend hat sich der 1-Octanol/Wasser-Verteilungskoeffizient zum Schlüsselparameter für die Bewertung des mit chemischen Stoffen verbundenen Umweltrisikos sowie zur Prognose der Persistenz chemischer Stoffe in der Umwelt entwickelt.
Anwendungsbereich
6. Bei der Methode zur Prüfung unter langsamem Rühren soll die Bildung von 1-Octanol-Mikrotröpfchen in der wässrigen Phase verringert werden. Entsprechend ist ausgeschlossen, dass die Konzentration in der wässrigen Phase wegen der ansonsten mit diesen Tröpfchen verbundenen Moleküle der Prüfsubstanz überschätzt wird. Daher eignet sich die Methode zur Prüfung unter langsamem Rühren insbesondere zur Bestimmung der POW-Werte von Substanzen, bei denen log POW-Werte im Bereich von mindestens 5 zu erwarten sind und bei denen die Schüttelmethode (2) eher fehleranfällig wäre.
Definitionen und Einheiten
7. Der Verteilungskoeffizient einer Substanz für Wasser und ein lipophiles Lösungsmittel (1-Octanol) beschreibt die Gleichgewichtsverteilung des jeweiligen chemischen Stoffs zwischen den beiden Phasen. Der Verteilungskoeffizient für Wasser und 1-Octanol (POW) wird definiert als Verhältnis der Gleichgewichtskonzentrationen der Prüfsubstanz in mit Wasser gesättigtem 1-Octanol (CO) und in mit 1-Octanol gesättigtem Wasser (CW).
POW = CO/CW
Für das Konzentrationsverhältnis wird keine Einheit angegeben. Meist wird das Verhältnis als Zehnerlogarithmus (log POW) ausgedrückt. POW ist temperaturabhängig; entsprechend sollte die Messtemperatur berücksichtigt werden.
Prinzip der Methode
8. Um den Verteilungskoeffizienten zu bestimmen, werden Wasser, 1-Octanol und die Prüfsubstanz bei einer konstanten Temperatur in ein Gleichgewicht gebracht. Anschließend werden die Konzentrationen der Prüfsubstanz in den beiden Phasen bestimmt.
9. Die bei der Prüfung mit der Schüttelmethode auftretenden Schwierigkeiten infolge der Entstehung von Mikrotröpfchen können mit der hier vorgeschlagenen Methode unter langsamem Rühren verringert werden. Bei der Prüfung unter langsamem Rühren werden Wasser, 1-Octanol und die Prüfsubstanz in einem thermostatgeregelten Rührbehälter in ein Gleichgewicht gebracht. Durch das Rühren wird der Austausch zwischen den Phasen beschleunigt. Beim Rühren entstehen begrenzte Turbulenzen, welche den Austausch zwischen 1-Octanol und Wasser begünstigen, ohne dass Mikrotröpfchen entstehen können (1).
Anwendbarkeit der Prüfmethode
10. Da sich das Vorhandensein anderer Substanzen auf den Aktivitätskoeffizienten der Prüfsubstanz auswirken kann, sollte die Prüfsubstanz als reine Substanz untersucht werden. Daher sollte die jeweilige Substanz für die Messung der 1-Octanol/Wasser-Verteilung in der höchsten auf dem Markt verfügbaren Reinheit verwendet werden.
11. Diese Methode ist anzuwenden bei reinen Substanzen, bei denen weder eine Dissoziation noch eine Assoziation erfolgt und die keine erhebliche Grenzflächenaktivität aufweisen. Die Methode kann zur Bestimmung der 1- Octanol/Wasser-Verteilung dieser Substanzen und von Gemischen eingesetzt werden. Wenn die Methode für Gemische eingesetzt wird, hängt die 1-Octanol/Wasser-Verteilung von der chemischen Zusammensetzung des zu prüfenden Gemischs sowie von der Zusammensetzung des als wässrige Phase genutzten Elektrolyten ab. Wenn die Methode um verschiedene Schritte ergänzt wird, kann sie auch für dissoziierende und assoziierende Verbindungen eingesetzt werden (siehe Nummer 12).
12. Aufgrund der unterschiedlichen Gleichgewichte in Wasser und 1-Octanol, die sich bei der 1-Octanol-/Wasser- Verteilung dissoziierender Substanzen wie z.B. organischer Säuren und Phenole, organischer Basen und organometallischer Substanzen ergeben, ist das Verteilungsverhältnis für 1-Octanol/Wasser ebenfalls in hohem Maße von der Zusammensetzung des Elektrolyten abhängig (7)(8). Die Bestimmung des Verteilungsverhältnisses für 1- Octanol/Wasser setzt voraus, dass pH-Wert und Elektrolytzusammensetzung während der Messung überwacht und protokolliert werden. Die Bewertung der Verteilungsverhältnisse muss durch Fachleute erfolgen. Unter Berücksichtigung der Dissoziationskonstante(n) müssen geeignete pH-Werte so ausgewählt werden, dass für jedes Ionisierungsstadium ein Verteilungsverhältnis bestimmt werden kann. Zur Prüfung organometallischer Verbindungen müssen Pufferlösungen eingesetzt werden, die nicht als Komplexbildner wirken (8). Unter Berücksichtigung des aktuellen Kenntnisstandes bezüglich der Chemie der wässrigen Phase (Komplexbildungskonstanten, Dissoziationskonstanten) sollten die Versuchsbedingungen so gewählt werden, dass die Speziation der Prüfsubstanz in der wässrigen Phase bestimmt werden kann. Die Ionenkonzentration sollte in allen Prüfungen gleich sein; um dies zu gewährleisten, sollte ein Hintergrundelektrolyt eingesetzt werden.
13. Schwierigkeiten können sich bei der Prüfung in Verbindung mit der Untersuchung von Substanzen mit geringer Wasserlöslichkeit oder mit hohem POW ergeben, weil die Konzentrationen im Wasser sehr gering werden und eine genaue Bestimmung entsprechend problematisch ist. Diese Prüfmethode erläutert, wie diesem Problem begegnet werden kann.
Informationen zur Prüfsubstanz
14. Die chemischen Reagenzien sollten mindestens Analysequalität besitzen. Es wird empfohlen, nicht markierte Prüfsubstanzen mit bekannter chemischer Zusammensetzung und einer Reinheit von mindestens 99 % oder radioaktiv markierte Prüfsubstanzen mit bekannter chemischer Zusammensetzung und radiochemischer Reinheit einzusetzen. Beim Einsatz von Indikatorsubstanzen mit kurzer Halbwertszeit sollten Korrekturen unter Berücksichtigung des Zerfallsverhaltens vorgenommen werden. Wenn die Prüfsubstanzen radioaktiv markiert wurden, sollte eine speziell für den jeweiligen chemischen Stoff vorgesehene Analysemethode eingesetzt werden, um sicherzustellen, dass die gemessene Radioaktivität unmittelbar auf die Prüfsubstanz zurückzuführen ist.
15. Log POW kann mit einer im Handel erhältlichen und für diesen Zweck vorgesehenen Software geschätzt werden; alternativ kann die Schätzung auch aufgrund des Verhältnisses der Löslichkeiten in beiden Lösungsmitteln erfolgen.
16. Bevor POW durch eine Prüfung unter langsamem Rühren bestimmt wird, sollten die folgenden Informationen zur Prüfsubstanz bekannt sein:
Beschreibung der Methode
Geräte und Apparatur
17. Für die Prüfungen werden Standardlaborgeräte benötigt; insbesondere sind folgende Geräte erforderlich:
Log POW = 0,88 log SR + 0,41
Dabei ist
SR = SOCt/SW (Molarität)
sowie auf der von Lyman (15) definierten Beziehung für die Prognose der Löslichkeit in Wasser. Die mit der in Anlage 1 genannten Formel bestimmten Wasserlöslichkeiten sind als erste Schätzung zu betrachten. Der Prüfende kann die Wasserlöslichkeit auch aufgrund einer sonstigen Beziehung schätzen, die er für besser geeignet hält, Auskunft über die Beziehung zwischen Hydrophobizität und Löslichkeit zu geben. Bei festen Verbindungen wird z.B. die Berücksichtigung des Schmelzpunktes bei der Prognose der Löslichkeit empfohlen. Wenn eine modifizierte Formel verwendet wird, sollte sichergestellt werden, dass die Gleichung zur Berechnung der Löslichkeit in Octanol noch gültig ist. In Anlage 2 ist ein Rührgefäß mit Glasmantel und einem Inhalt von etwa einem Liter dargestellt. Die Proportionen des in Anlage 2 dargestellten Gefäßes haben sich als günstig erwiesen und sollten auch dann beibehalten werden, wenn eine anders dimensionierte Apparatur verwendet wird,
18. Die Gefäße sollten aus einem inerten Material bestehen, damit die Adsorption durch die Oberfläche der Gefäße vernachlässigt werden kann.
Herstellung der Prüflösungen
19. Der Wert für POW sollte mit 1-Octanol der höchsten im Handel erhältlichen Reinheit (mindestens + 99 %) bestimmt werden. Die Reinigung von 1-Octanol durch die Extraktion mit einer Säure, einer Base und Wasser und eine anschließende Trocknung werden empfohlen. Außerdem kann 1-Octanol auch durch Destillation gereinigt werden. Zur Herstellung der Standardlösungen der Prüfsubstanzen ist gereinigtes 1-Octanol zu verwenden. Das für die Bestimmung von POW zu verwendende Wasser sollte durch Glas oder Quarz destilliert oder mit einem Reinigungssystem hergestellt worden sein; alternativ kann auch HPLC-Wasser verwendet werden. Destilliertes Wasser sollte durch ein 0,22-µm-Filter gefiltert werden; mit Blindproben sollte sichergestellt werden, dass die konzentrierten Extrakte keine Verunreinigungen enthalten, welche die Prüfsubstanz verändern könnten. Wenn ein Glasfaserfilter verwendet wird, sollte das Filter durch mindestens dreistündige Erhitzung auf 400 °C gereinigt werden.
20. Beide Lösungsmittel werden vor Durchführung der Prüfung wechselseitig gesättigt, indem sie in einem hinreichend großen Gefäß in ein Gleichgewicht gebracht werden. Dazu wird das aus zwei Phasen bestehende System zwei Tage lang langsam gerührt.
21. Eine geeignete Konzentration der Prüfsubstanz wird ausgewählt und in 1-Octanol (mit Wasser gesättigt) gelöst. Der 1-Octanol/Wasser-Verteilungskoeffizient ist in verdünnten Lösungen mit 1-Octanol und Wasser zu bestimmen. Daher sollte die Konzentration der Prüfsubstanz höchstens 70 % ihrer Löslichkeit bei einer Höchstkonzentration von 0,1 M in beiden Phasen betragen (1). Die für die Prüfungen verwendeten 1-Octanol-Lösungen dürfen keine suspendierten Feststoffe aus der Prüfsubstanz enthalten.
22. Eine geeignete Menge der Prüfsubstanz wird in 1-Octanol (mit Wasser gesättigt) gelöst. Wenn die Schätzung für log POW einen Wert über fünf ergibt, ist besonders darauf zu achten, dass die für die Prüfung verwendeten 1- Octanol-Lösungen keine suspendierten Feststoffe aus der Prüfsubstanz enthalten. Dazu ist bei chemischen Stoffen mit einem geschätzten log POW > 5 wie folgt zu verfahren:
Extraktion und Analyse der Proben
23. Für die Untersuchung der Prüfsubstanz sollte eine validierte Analysemethode verwendet werden. Der Prüfende muss nachweisen, dass die Konzentrationen in dem mit Wasser gesättigten 1-Octanol sowie in der mit 1-Octanol gesättigten wässrigen Phase während der Prüfung über der bei dem eingesetzten Analyseverfahren für die Methode festgesetzten Quantifizierungsgrenze liegen. In Fällen, in denen Extraktionsmethoden erforderlich sind, muss die analytische Wiederfindung der Prüfsubstanzen aus der wässrigen Phase und aus der 1-Octanol-Phase vor der Prüfung erfolgen. Die Anforderungen an die Analysesignale sind aufgrund von Blindwerten zu korrigieren; dabei ist darauf zu achten, dass keine Analytübertragungen zwischen den Proben vorkommen können.
24. Bei hydrophoben Prüfsubstanzen sind wegen der eher niedrigen Konzentrationen der Prüfsubstanzen in der wässrigen Phase vor der Analyse wahrscheinlich eine Extraktion der wässrigen Phase mit einem organischen Lösungsmittel und eine Vorkonzentration des Extrakts vorzunehmen. Aus demselben Grund müssen die eventuell verwendeten Blindprobenkonzentrationen reduziert werden. Dazu sind hochreine und vorzugsweise für Rückstandsanalysen vorgesehene Lösungsmittel zu verwenden. Die Verwendung sorgfältig vorgereinigter Glasgeräte (z.B. durch Spülen mit einem Lösungsmittel oder unter starker Erwärmung) kann zusätzlich helfen, Kreuzkontamination zu vermeiden.
25. Log POW kann mit einem geeigneten Programm ermittelt oder von Fachleuten mit entsprechender Erfahrung geschätzt werden. Bei Werten über 6 müssen Blindwert-Korrekturen und Analyteintragungen sorgfältig überwacht werden. Wenn der geschätzte Wert für log POW über 6 liegt, muss ein Surrogatstandard für die Wiederfindungskorrektur verwendet werden, damit die erforderlichen hohen Vorkonzentrationsfaktoren erreicht werden können. Auf dem Markt werden verschiedene Computer-Programme zur Schätzung von log POW angeboten 1 (z.B. Clog P(16), KOWWIN(17), ProLogP(18) und ACD log P(19)). Das Literaturverzeichnis enthält Beschreibungen der verschiedenen Schätzverfahren (20-22).
26. Die Quantifizierungsgrenzen (LOQ) für die Bestimmung der Prüfsubstanz in 1-Octanol und Wasser werden mit anerkannten Methoden bestimmt. Als Faustregel kann die Konzentration in Wasser oder 1-Octanol, bei der sich ein Signal-/Rauschverhältnis von 10 ergibt, als Quantifizierungsgrenze für die betreffende Methode angenommen werden. Entsprechend sollten eine geeignete Methode zur Extraktion und zur Vorkonzentration ausgewählt und Verfahren für die analytische Wiederfindung spezifiziert werden. Der Vorkonzentrationsfaktor ist so auszuwählen, dass sich bei der Analyse ein Signal der geforderten Stärke ergibt.
27. Ausgehend von den Parametern der Analysemethode und von den erwarteten Konzentrationen wird die Probengröße bestimmt, die für eine genaue Bestimmung der Konzentration der jeweiligen Verbindung ungefähr erforderlich ist. Wasserproben, die so klein sind, dass kein hinreichendes Analysesignal festgestellt werden kann, sollten nicht verwendet werden. Ebenso sollte die Verwendung zu großer Wasserproben vermieden werden, weil ansonsten möglicherweise zu wenig Wasser für die erforderlichen Analysen (n = 5) verbleibt. In Anlage 1 wird das Proben-Mindestvolumen abhängig vom Gefäßvolumen, von der Nachweisgrenze der Prüfsubstanz und von der Löslichkeit der Prüfsubstanz angegeben.
28. Die Quantifizierung der Prüfsubstanzen erfolgt durch den Vergleich mit Kalibrierungskurven der entsprechenden Verbindung. Die Konzentrationen in den analysierten Proben müssen innerhalb der Bandbreite der Standardkonzentrationen liegen.
29. Bei Prüfsubstanzen mit log POW-Werten über 6 muss der Wasserprobe vor der Extraktion ein Surrogatstandard zugesetzt werden, um die bei der Extraktion und bei der Vorkonzentration der Wasserprobe auftretenden Verluste zu erfassen. Für exakte Wiederfindungskorrekturen müssen die Surrogate Merkmale aufweisen, die denen der Prüfsubstanz möglichst ähnlich sind oder vollständig mit deren Merkmalen übereinstimmen. Dazu werden vorzugsweise mit einem (stabilen) Isotop markierte, den Prüfsubstanzen analoge Verbindungen verwendet (z.B. deuterierte oder mit 13C markierte Verbindungen). Wenn mit einem stabilen Isotop (d. h. mit 13C oder 2H) markierte Verbindungen nicht verwendet werden können, sollte anhand zuverlässiger Daten in der Fachliteratur nachgewiesen werden, dass die physikalisch-chemischen Merkmale der Surrogate den Merkmalen der Prüfsubstanzen sehr nahekommen. Bei der Flüssigflüssig-Extraktion der wässrigen Phase können Emulsionen entstehen. Diese Emulsionen können durch die Zugabe von Salz und Ausfällen der Emulsion über Nacht verringert werden. Die Methoden zur Extraktion und zur Vorkonzentration der Proben sind zu protokollieren.
30. Aus der 1-Octanol-Phase gezogene Proben können vor der Analyse erforderlichenfalls mit einem geeigneten Lösungsmittel verdünnt werden. Die Verwendung von Surrogatstandards zur Wiederfindungskorrektur wird für Substanzen empfohlen, bei denen in Wiederfindungsprüfungen starke Schwankungen zu verzeichnen waren (relative Standardabweichung > 10 %).
31. Die Analysemethode ist detailliert zu protokollieren. Zu erfassen sind unter anderem die Extraktionsmethode, Vorkonzentrations- und Verdünnungsfaktoren, Geräteparameter, die Kalibrierungsroutine, der Kalibrierungsbereich, Angaben zur analytischen Wiederfindung der Prüfsubstanzen aus dem Wasser, Zugaben von Surrogatstandards zur Wiederfindungskorrektur, Blindwerte, Nachweisgrenzen und Quantifizierungsgrenzen.
Durchführung der Prüfung
Optimale 1-Octanol-/Wasser-Verhältnisse
32. Bei der Auswahl der Volumina von Wasser und 1-Octanol sollten die folgenden Punkte berücksichtigt werden: die Quantifizierungsgrenze (LOQ) in 1-Octanol und Wasser, die Vorkonzentrationsfaktoren der Wasserproben, das Volumen der aus 1-Octanol und aus Wasser genommenen Proben und die zu erwartenden Konzentrationen. Aus durchführungspraktischen Gründen sollte das 1-Octanol-Volumen bei dem System zur Prüfung unter langsamem Rühren so gewählt werden, dass die 1-Octanol-Schicht hinreichend stark (> 0,5 cm) ist, damit die Proben ohne Beeinträchtigung aus der 1-Octanol-Phase entnommen werden können.
33. Typische Phasenverhältnisse für die Bestimmung von Verbindungen mit log POW-Werten von mindestens 4,5 sind 20 bis 50 ml 1-Octanol und 950 bis 980 ml Wasser in einem 1-l-Gefäß.
Prüfbedingungen
34. Während der Prüfung wird das Reaktionsgefäß mit einem Thermostaten so geregelt, dass die Temperaturschwankungen unter 1 °C liegen. Die Untersuchung sollte bei einer Temperatur von 25 °C durchgeführt werden.
35. Das Prüfsystem sollte vor Tageslichteinfall geschützt werden, indem die Prüfungen entweder im Dunkeln durchgeführt werden oder das Reaktionsgefäß mit einer Aluminiumfolie bedeckt wird.
36. Außerdem sollte die Prüfung in (möglichst) staubfreier Umgebung erfolgen.
37. Das 1-Octanol-Wasser-System wird gerührt, bis das erforderliche Gleichgewicht hergestellt ist. In einem Pilottest unter langsamem Rühren bei regelmäßiger Entnahme von Proben aus der wässrigen Phase und aus der 1- Octanol-Phase wird die Zeitspanne bis zur Herstellung des Gleichgewichts ermittelt. Zwischen den verschiedenen Probenahmen sollten jeweils mindestens 5 Stunden liegen.
38. Die POW-Werte sind in mindestens drei unabhängig voneinander durchzuführenden Prüfungen unter langsamem Rühren zu bestimmen.
Bestimmung der zur Herstellung des Gleichgewichts erforderlichen Zeitspanne
39. Es wird angenommen, dass das Gleichgewicht dann erreicht ist, wenn das Konzentrationsverhältnis für 1-Octanol und Wasser in einem Zeitraum mit vier Messzeitpunkten bei einem p-Wert von 0,05 so weit zurückgegangen ist, dass der Endwert nicht mehr signifikant von 0 abweicht. Die Zeitspanne zur Herstellung des Gleichgewichts beträgt mindestens einen Tag. Erst dann kann mit der Probenahme begonnen werden. Als Faustregel kann davon ausgegangen werden, dass bei Substanzen, bei denen log POW auf unter 5 geschätzt wird, die Probenahme am zweiten und am dritten Tag erfolgen kann. Bei stärker hydrophoben Verbindungen dauert die Herstellung des Gleichgewichts unter Umständen länger. Bei einer Verbindung mit log POW 8,23 (Decachlorbiphenyl) wurde das Gleichgewicht nach 144 Stunden erreicht. Ob das Gleichgewicht hergestellt ist, wird anhand mehrfacher Probenahmen aus einem einzigen Gefäß beurteilt.
Beginn der Prüfung
40. Zu Beginn der Prüfung wird das Reaktionsgefäß Wasser befüllt, das mit 1-Octanol gesättigt wurde. Dabei sollte genügend Zeit zum Erreichen der thermostatgeregelten Temperatur gelassen werden.
41. Die gewünschte Menge der Prüfsubstanz (im erforderlichen Volumen des mit Wasser gesättigten 1-Octanol) wird vorsichtig in das Reaktionsgefäß gegeben. Dies ist ein entscheidender Schritt bei der Prüfung, da bei der Mischung der beiden Phasen Verwirbelungen vermieden werden müssen. Dazu kann die 1-Octanol-Phase langsam mit einer Pipette dicht über der Wasseroberfläche gegen die Wand des Prüfgefäßes getropft werden. Die 1- Octanol-Phase fließt dann an der Glaswand hinab und bildet einen Film auf der wässrigen Phase. Eine direkte Dekantierung von 1-Octanol in den Kolben sollte in jedem Fall vermieden werden; unter keinen Umständen sollten 1-Octanol-Tropfen direkt auf die Wasseroberfläche fallen.
42. Nach dem Beginn des Rührvorgangs sollte die Rührgeschwindigkeit langsam gesteigert werden. Wenn die Rührmotoren nicht in geeigneter Weise eingestellt werden können, sollte der Einsatz eines Transformators in Erwägung gezogen werden. Die Rührgeschwindigkeit sollte so eingestellt werden, dass am Übergang zwischen Wasser und 1-Octanol ein Wirbel mit einer Tiefe von 0,5 cm bis höchstens 2,5 cm entsteht. Die Rührgeschwindigkeit ist zu verringern, wenn der Wirbel tiefer als 2,5 cm wird; andernfalls können 1-Octanol-Mikrotröpfchen in der wässrigen Phase entstehen und dazu führen, dass die Konzentration der Prüfsubstanz im Wasser überschätzt wird. Die Rührgeschwindigkeit, bei der ein Wirbel mit einer Tiefe von höchstens 2,5 cm entsteht, wird ausgehend von den Ergebnissen der Ringtest-Validierungsstudie empfohlen (5). Diese Rührgeschwindigkeit gewährleistet einen Kompromiss zwischen der möglichst raschen Herstellung des erforderlichen Gleichgewichts und der Vermeidung von 1-Octanol-Mikrotröpfchen.
Probenahme und Behandlung der Proben
43. Vor der Probenahme sollte der Rührmotor ausgeschaltet und gewartet werden, bis die Flüssigkeiten zur Ruhe gekommen sind. Nach der Probenahme wird der Rührmotor bei geringer Rührgeschwindigkeit wieder eingeschaltet und die Rührgeschwindigkeit wie oben beschrieben langsam gesteigert.
44. Die Probenahme aus der wässrigen Phase erfolgt aus einem Absperrhahn unten am Reaktionsgefäß. Dabei ist das Totvolumen des im Hahn enthaltenen Wassers (in dem in Anlage 2 abgebildeten Gefäß etwa 5 ml) zu verwerfen. Das im Hahn enthaltene Wasser wurde nicht umgerührt und befindet sich daher nicht im Gleichgewicht mit der übrigen Flüssigkeit. Das Volumen der Wasserproben wird protokolliert; außerdem ist die Menge der im verworfenen Wasservolumen enthaltenen Prüfsubstanz bei der Berechnung der Massenbilanz zu berücksichtigen. Verdampfungsverluste sollten vermieden werden; dazu sollte dem Wasser Gelegenheit gegeben werden, ruhig in den Abscheidetrichter abzufließen, damit die Wasser-1-Octanol-Schicht nicht gestört wird.
45. Die Probenahme aus der 1-Octanol-Phase erfolgt, indem eine kleine Aliquote (ca. 100 µl) mit einer 100- Mikroliter-Glas-Metall-Spritze aus der 1-Octanol-Schicht gezogen wird. Dabei ist darauf zu achten, dass der Übergangsbereich nicht berührt wird. Anschließend wird das Volumen der als Probe entnommenen Flüssigkeit protokolliert. Eine kleine Aliquote ist ausreichend, da die 1-Octanol-Probe verdünnt wird.
46. Bei der Handhabung der Proben sollten unnötige Übertragungsschritte vermieden werden. Daher sollte das Probenvolumen gravimetrisch bestimmt werden. Bei Wasserproben kann dies geschehen, indem die Wasserproben in einem Abscheidetrichter gesammelt werden, der bereits die erforderliche Lösungsmittelmenge erhält.
Daten und Berichterstattung
47. Bei der hier beschriebenen Prüfmethode wird POW aufgrund von drei Prüfungen der zu untersuchenden Verbindung (drei Prüfeinheiten) unter langsamem Rühren bestimmt; dabei müssen jeweils identische Bedingungen gegeben sein. Die zum Nachweis des hergestellten Gleichgewichts vorgenommene Regression sollte auf den Ergebnissen von mindestens vier CO/CW-Werten zu aufeinanderfolgenden Zeitpunkten beruhen. Anhand dieser Ergebnisse kann eine Varianz als Maß für die Unsicherheit des pro Prüfeinheit ermittelten Durchschnittswerts berechnet werden.
48. POW kann über die Varianz der in den einzelnen Prüfeinheiten ermittelten Daten beschrieben werden. Aufgrund dieser Information wird POW nämlich als gewichteter Durchschnitt der Ergebnisse der einzelnen Prüfeinheiten berechnet. Dazu wird der Kehrwert der Varianz der Ergebnisse der Prüfeinheiten als Gewichtung angenommen. Dies hat zur Folge, dass sich Daten mit großen Schwankungen (ausgedrückt in einer hohen Varianz) und entsprechend geringerer Zuverlässigkeit weniger auf das Ergebnis auswirken als Daten mit niedriger Varianz.
49. In entsprechender Weise wird die gewichtete Standardabweichung berechnet. Die Standardabweichung beschreibt die Wiederholbarkeit der POW-Messung. Eine niedrige gewichtete Standardabweichung ist Ausdruck einer sehr hohen Wiederholbarkeit der POW-Bestimmung in ein und demselben Labor. Im Folgenden wird die formale statistische Behandlung der Daten beschrieben.
Auswertung der Ergebnisse
Nachweis der Herstellung des Gleichgewichts
50. Für jeden Probenahmezeitpunkt wird der Logarithmus des Verhältnisses der Konzentration der Prüfsubstanz in 1-Octanol und Wasser (log (CO/CW)) berechnet. Die Herstellung des chemischen Gleichgewichts wird nachgewiesen, indem dieses Verhältnis bezogen auf die betreffende Zeit dargestellt wird. Ein Plateau in der aus Messungen zu mindestens vier aufeinanderfolgenden Zeitpunkten erstellten Kurve zeigt, dass ein Gleichgewicht erreicht und die Verbindung tatsächlich in 1-Octanol gelöst ist. Andernfalls sind die Prüfungen fortzusetzen, bis sich aus den Werten von vier aufeinanderfolgenden Zeitpunkten eine Kurve ergibt, deren Steigung sich bei einem p-Wert von 0,05 nicht mehr signifikant von null unterscheidet, und die entsprechend zeigt, dass log CO/CW nicht mehr zeitabhängig ist.
Berechnung von Log POW
51. Log POW der Versuchseinheit wird als gewichteter Durchschnitt von log Co/Cw für den Teil der Kurve zur Darstellung des Verhältnisses von log Co/Cw zur Zeitspanne berechnet, in dem das Gleichgewicht nachgewiesen wurde. Zur Berechnung des gewichteten Durchschnitts werden die Daten mit dem Kehrwert der Varianz so berechnet, dass der Einfluss der Daten auf das Endergebnis umgekehrt proportional zur Unsicherheit der Daten ist.
Durchschnittlicher Wert für log POW
52. Der durchschnittliche Wert für log POW bei verschiedenen Versuchseinheiten wird als Durchschnitt der Ergebnisse der einzelnen mit der jeweiligen Varianz gewichteten Versuchseinheiten berechnet.
Die Berechnung erfolgt nach der nachstehenden Formel:
Log POW,Av = (Σwi x log POW,i) x (Σwi)-1
Dabei ist
Log POW,i = log POW der jeweiligen Versuchseinheit i,
log POW, Av = gewichteter Durchschnitt der jeweils ermittelten Werte für log POW,
wi = statistische Gewichtung von log POW der Versuchseinheit i.
Der Kehrwert der Varianz von log POW,i wird als wi eingesetzt (wi = var(log POW,i)-1).
53. Der Fehler des Durchschnitts von log POW wird geschätzt als die in den einzelnen Versuchseinheiten während der Gleichgewichtsphase ermittelte Wiederholbarkeit von log CO/CW und als gewichtete Standardabweichung von log POW, Av (σlog POW, Av) ausgedrückt, die wiederum ein Maß für den mit log POW, Av verbundenen Fehler ist. Die gewichtete Standardabweichung kann wie folgt aus der gewichteten Varianz (varlog POW, Av) berechnet werden:
varlog Pow, Av = (Σwi x (log POW,i - log POW, Av)2) x (Σwi x (n - 1))-1
σlog Pow, Av = (varlog Pow, Av)0,5
Dabei steht n für die Anzahl der Versuchseinheiten.
Prüfbericht
54. Der Prüfbericht sollte folgende Informationen enthalten:
Prüfsubstanz:
Prüfbedingungen:
Ergebnisse:
Literatur:
1. De Bruijn JHM, Busser F, Seinen W, Hermens J. (1989). Determination of octanol/water partition coefficients with the 'slow-stirring' method. Environ. Toxicol. Chem. 8: 499-512.
2. Kapitel A.8 dieses Anhangs, Verteilungskoeffizient.
3. Kapitel A.8 dieses Anhangs, Verteilungskoeffizient.
4. OECD (2000). OECD Draft Guideline for the Testing of Chemicals: 122 Partition Coefficient (n-Octanol/Water): pH-Metric Method for Ionisable Substances. Paris.
5. Tolls J (2002). Partition Coefficient 1-Octanol/Water (Pow) Slow-Stirring Method for Highly Hydrophobic Chemicals, Validation Report. RIVM contract-Nrs 602730 M/602700/01.
6. Boethling RS, Mackay D (eds.) (2000). Handbook of property estimation methods for chemicals. Lewis Publishers Boca Raton, FL, USA.
7. Schwarzenbach RP, Gschwend PM, Imboden DM (1993). Environmental Organic Chemistry. Wiley, New York, NY.
8. Arnold CG, Widenhaupt A, David MM, Müller SR, Haderlein SB, Schwarzenbach RP (1997). Aqueous speciation and 1-octanol-water partitioning of tributyl- and triphenyltin: effect of pH and ion composition. Environ. Sci. Technol. 31: 2596-2602.
9. OECD (1981) OECD Guidelines for the Testing of Chemicals: 112 Dissociation Constants in Water. Paris.
10. Kapitel A.6 dieses Anhangs, Wasserlöslichkeit.
11. Kapitel C.7 dieses Anhangs, Abbaubarkeit - abiotischer Abbau: Hydrolyse in Abhängigkeit vom pH-Wert.
12. Kapitel C.4 - Teile II - VII (Methoden A bis F) dieses Anhangs -Bestimmung der 'leichten' biologischen Abbaubarkeit.
13. Kapitel A.4 dieses Anhangs, Dampfdruck.
14. Pinsuwan S, Li A and Yalkowsky S.H. (1995). Correlation of octanol/water solubility ratios and partition coefficients, J. Chem. Eng. Data. 40: 623-626.
15. Lyman WJ (1990). Solubility in water. In: Handbook of Chemical Property Estimation Methods: Environmental Behavior of Organic Compounds, Lyman WJ, Reehl WF, Rosenblatt DH, Eds. American Chemical Society, Washington, DC, 2-1 to 2-52.
16. Leo A, Weininger D (1989). Medchem Software Manual. Daylight Chemical Information Systems, Irvine, CA.
17. Meylan W (1993). SRC-LOGKOW for Windows. SRC, Syracuse, N.Y.
18. Compudrug L (1992). ProLogP. Compudrug, Ltd, Budapest.
19. ACD. ACD logP; Advanced Chemistry Development: Toronto, Ontario M5H 3V9, Canada, 2001.
20. Lyman WJ (1990). Octanol/water partition coefficient. In Lyman WJ, Reehl WF, Rosenblatt DH, eds, Handbook of chemical property estimation, American Chemical Society, Washington, D.C.
21. Rekker RF, de Kort HM (1979). The hydrophobic fragmental constant: An extension to a 1.000 data point set. Eur. J. Med. Chem. Chim. Ther. 14: 479-488.
22. Jübermann O (1958). Houben-Weyl, ed, Methoden der Organischen Chemie: 386-390.
_____
1) Die Angaben dienen nur zur Information. Wenn mit anderen Programmen nachweislich dieselben Ergebnisse ermittelt werden, können auch diese Programme verwendet werden.
Anlage 1 Tabelle zur Berechnung der für den Nachweis von prüßubstanzen mit unterschiedlichen logPOW-Werten in der wässrigen Phase mindestens erforderlichen Volumina
Voraussetzungen:
Geschätzter Wert für Sw
| log POW | Formel | log Sw | Sw (mg/l) |
| 4 | (-)0,922 x log POW + 4,184 | 0,496 | 3,133E+00 |
| 4,5 | (-)0,922 x log POW + 4,184 | 0,035 | 1,084E+00 |
| 5 | (-)0,922 x log POW + 4,184 | - 0,426 | 3,750E-01 |
| 5,5 | (-)0,922 x log POW + 4,184 | - 0,887 | 1,297E-01 |
| 6 | (-)0,922 x log POW + 4,184 | - 1,348 | 4,487E-02 |
| 6,5 | (-)0,922 x log POW + 4,184 | - 1,809 | 1,552E-02 |
| 7 | (-)0,922 x log POW + 4,184 | - 2,270 | 5,370E-03 |
| 7,5 | (-)0,922 x log POW + 4,184 | - 2,731 | 1,858E-03 |
| 8 | (-)0,922 x log POW + 4,184 | - 3,192 | 6,427E-04 |
Geschätzter Wert für Soct
| log POW | Formel | Soct (mg/l) |
| 4 | log POW = 0,88log SR + 0,41 | 3,763E+04 |
| 4,5 | log POW = 0,88log SR + 0,42 | 4,816E+04 |
| 5 | log POW = 0,88log SR + 0,43 | 6,165E+04 |
| 5,5 | log POW = 0,88log SR + 0,44 | 7,890E+04 |
| 6 | log POW = 0,88log SR + 0,45 | 1,010E+05 |
| 6,5 | log POW = 0,88log SR + 0,46 | 1,293E+05 |
| 7 | log POW = 0,88log SR + 0,47 | 1,654E+05 |
| 7,5 | log POW = 0,88log SR + 0,48 | 2,117E+05 |
| 8 | log POW = 0,88log SR + 0,49 | 2,710E+05 |
| Gesamtmasse der Prüfsubstanz (mg) | MasseOct/MasseWasser | MasseH2O (mg) | KonzH2O (mg/l) | Masseoct (mg) | Konzoct (mg/l) |
| 1.319 | 526 | 2,5017 | 2,6333 | 1.317 | 26.333 |
| 1.686 | 1.664 | 1,0127 | 1,0660 | 1.685 | 33.709 |
| 2.158 | 5.263 | 0,4099 | 0,4315 | 2.157 | 43.149 |
| 2.762 | 16.644 | 0,1659 | 0,1747 | 2.762 | 55.230 |
| 3.535 | 52.632 | 0,0672 | 0,0707 | 3.535 | 70.691 |
| 4.524 | 166.436 | 0,0272 | 0,0286 | 4.524 | 90.480 |
| 5.790 | 526.316 | 0,0110 | 0,0116 | 5.790 | 115.807 |
| 7.411 | 1.664.357 | 0,0045 | 0,0047 | 7.411 | 148.223 |
| 9.486 | 5.263.158 | 0,0018 | 0,0019 | 9.486 | 189.713 |
Berechnung der Volumina
Erforderliches Mindestvolumen der H2O-Phase bei den verschiedenen LOD-Konzentrationen
| log Kow | LOD (¼g/l)' | 0,001 | 0,01 | 0,10 | 1,00 | 10 |
| 4 | 0,04 | 0,38 | 3,80 | 38 | 380 | |
| 4,5 | 0,09 | 0,94 | 9,38 | 94 | 938 | |
| 5 | 0,23 | 2,32 | 23,18 | 232 | 2.318 | |
| 5,5 | 0,57 | 5,73 | 57,26 | 573 | 5.726 | |
| 6 | 1,41 | 14,15 | 141 | 1.415 | 14.146 | |
| 6,5 | 3,50 | 34,95 | 350 | 3.495 | 34.950 | |
| 7 | 8,64 | 86,35 | 864 | 8.635 | 86.351 | |
| 7,5 | 21,33 | 213 | 2.133 | 21.335 | 213.346 | |
| 8 | 52,71 | 527 | 5.271 | 52.711 | 527.111 | |
| Volumen für Nachweisgrenze LOD (l) | 0,1 |
Erläuterungen
< 10 % des Gesamtvolumens der wässrigen Phase, 1-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 2-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 5-l-Ausgleichsgefäß.
< 10 % des Gesamtvolumens der wässrigen Phase, 10-l-Ausgleichsgefäß.
Mehr als 10 % des Volumens eines 10-l-Ausgleichsgefäßes.
Übersicht über die benötigten Volumina abhängig von der Wasserlöslichkeit und von log POW
Erforderliches Mindestvolumen der H2O-Phase bei den verschiedenen LOD-Konzentrationen (ml)
| log POW | Sw (mg/l) | LOD (μg/l) -> | 0,001 | 0,01 | 0,10 | 1,00 | 10 |
| 4 | 10 | 0,01 | 0,12 | 1,19 | 11,90 | 118,99 | |
| 5 | 0,02 | 0,24 | 2,38 | 23,80 | 237,97 | ||
| 3 | 0,04 | 0,40 | 3,97 | 39,66 | 396,62 | ||
| 1 | 0,12 | 1,19 | 11,90 | 118,99 | 1.189,86 | ||
| 4,5 | 5 | 0,02 | 0,20 | 2,03 | 20,34 | 203,37 | |
| 2 | 0,05 | 0,51 | 5,08 | 50,84 | 508,42 | ||
| 1 | 0,10 | 1,02 | 10,17 | 101,68 | 1.016,83 | ||
| 0,5 | 0,20 | 2,03 | 20,34 | 203,37 | 2.033,67 | ||
| 5 | 1 | 0,09 | 0,87 | 8,69 | 86,90 | 869,01 | |
| 0,5 | 0,17 | 1,74 | 17,38 | 173,80 | 1.738,02 | ||
| 0,375 | 0,23 | 2,32 | 23,18 | 231,75 | 2.317,53 | ||
| 0,2 | 0,43 | 4,35 | 43,45 | 434,51 | 4.345,05 | ||
| 5,5 | 0,4 | 0,19 | 1,86 | 18,57 | 185,68 | 1.856,79 | |
| 0,2 | 0,37 | 3,71 | 37,14 | 371,36 | 3.713,59 | ||
| 0,1 | 0,74 | 7,43 | 74,27 | 742,72 | 7.427,17 | ||
| 0,05 | 1,49 | 14,85 | 148,54 | 1.485,43 | 14.854,35 | ||
| 6 | 0,1 | 0,63 | 6,35 | 63,48 | 634,80 | 6.347,95 | |
| 0,05 | 1,27 | 12,70 | 126,96 | 1.269,59 | 12.695,91 | ||
| 0,025 | 2,54 | 25,39 | 253,92 | 2.539,18 | 25.391,82 | ||
| 0,0125 | 5,08 | 50,78 | 507,84 | 5.078,36 | 50.783,64 | ||
| 6,5 | 0,025 | 2,17 | 21,70 | 217,02 | 2.170,25 | 21.702,46 | |
| 0,0125 | 4,34 | 43,40 | 434,05 | 4.340,49 | 43.404,93 | ||
| 0,006 | 9,04 | 90,43 | 904,27 | 9.042,69 | 90.426,93 | ||
| 0,003 | 18,09 | 180,85 | 1.808,54 | 18.085,39 | 180.853,86 | ||
| 7 | 0,006 | 7,73 | 77,29 | 772,89 | 7.728,85 | 77.288,50 | |
| 0,003 | 15,46 | 154,58 | 1.545,77 | 15.457,70 | 154.577,01 | ||
| 0,0015 | 23,19 | 231,87 | 2.318,66 | 23.186,55 | 231.865,51 | ||
| 0,001 | 46,37 | 463,73 | 4.637,31 | 46.373,10 | 463.731,03 | ||
| 7,5 | 0,002 | 19,82 | 198,18 | 1.981,77 | 19.817,73 | 198.177,33 | |
| 0,001 | 39,64 | 396,35 | 3.963,55 | 39.635,47 | 396.354,66 | ||
| 0,0005 | 79,27 | 792,71 | 7.927,09 | 79.270,93 | 792.709,32 | ||
| 0,00025 | 158,54 | 1.585,42 | 15.854,19 | 158.541,86 | 1.585.418,63 | ||
| 8 | 0,001 | 33,88 | 338,77 | 3.387,68 | 33.876,77 | 338.767,72 | |
| 0,0005 | 67,75 | 677,54 | 6.775,35 | 67.753,54 | 677.535,44 | ||
| 0,00025 | 135,51 | 1.355,07 | 13.550,71 | 135.507,09 | 1.355.070,89 | ||
| 0,000125 | 271,01 | 2.710,14 | 27.101,42 | 271.014,18 | 2.710.141,77 | ||
| Volumen für Nachweisgrenze LOD (l) | 0,1 | ||||||
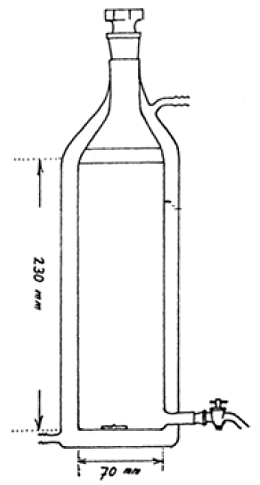
Anlage 2 Beispiel eines Prüfgefäßes mit Glasmantel zur Bestimmung von POW unter langsamem Rühren
3. Das Kapitel B.2 erhält folgende Fassung:
"B.2 Akute Inhalationstoxizität
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 403 (2009) (1). Die ursprüngliche Prüfrichtlinie 403 (TG 403) zur akuten Inhalation wurde 1981 angenommen. Diese überarbeitete Prüfmethode B.2 (die der überarbeiteten TG 403 entspricht) ist dazu ausgelegt, mehr Flexibilität zu bieten, die Verwendung von Versuchstieren zu reduzieren und die Regulierungsanforderungen zu erfüllen. Die überarbeitete Prüfmethode umfasst zwei Arten von Versuchen: ein traditionelles LC50-Protokoll und ein Konzentration-x-Zeit-Protokoll (c x t). Die Hauptmerkmale dieser Prüfmethode sind die Möglichkeit, eine Konzentrations-Wirkungs-Beziehung aufzustellen, die von nichtletalen zu letalen Ergebnissen reicht, um eine mittlere letale Konzentration (LC50), eine nichtletale Schwellenkonzentration (z.B. LC01) und die Steigung der Kurve zu bestimmen und eine eventuelle geschlechtsspezifische Empfindlichkeit festzustellen. Das c-x-t-Protokoll sollte angewendet werden, wenn wegen einer spezifischen Regelung oder aus wissenschaftlicher Notwendigkeit eine Prüfung von Tieren über unterschiedliche Zeiträume erforderlich ist, z.B. für Zwecke der Notfallplanung [z.B. zur Ableitung von Störfallbeurteilungswerten (Acute Exposure Guideline Levels - AEGL, Emergency Response Planning Guidelines - ERPG oder Acute Exposure Threshold Levels - AETL)] oder für die Flächennutzungsplanung.
2. Eine Anleitung für die Durchführung und Auswertung dieser Prüfmethoden ist im Guidance Document on Acute Inhalation Toxicity Testing (GD 39 (2)) enthalten.
3. Die im Zusammenhang mit dieser Prüfmethode verwendeten Begriffe werden am Ende dieses Kapitels und im GD 39 (2) definiert.
4. Diese Prüfmethode ermöglicht die Charakterisierung von Prüfsubstanzen und eine quantitative Risikobewertung und sie erlaubt die Einstufung von Prüfsubstanzen nach der Verordnung (EG) Nr. 1272/2008 (3). Das GD 39 (2) enthält Hinweise zur Auswahl der geeigneten Prüfmethode für akute Prüfungen. Wenn nur Informationen über die Einstufung und Kennzeichnung benötigt werden, wird im Allgemeinen Kapitel B.52 dieses Anhangs (4) empfohlen [siehe GD 39 (2)]. Diese Prüfmethode B.2 ist nicht speziell für die Prüfung von Spezialmaterialien wie schwer löslichen isometrischen oder Fasermaterialien oder hergestellten Nanomaterialien bestimmt.
Ausgangsüberlegungen
5. Bevor Versuche nach dieser Prüfmethode in Betracht gezogen werden, sollte das Prüflabor alle verfügbaren Informationen über die Prüfsubstanz, einschließlich bereits vorliegender Studien (z.B. Kapitel B.52 dieses Anhangs (4)), deren Ergebnisse darauf hindeuten, dass auf weitere Versuche verzichtet werden kann, auswerten, damit möglichst wenig Tiere verwendet werden. Für die Auswahl der in Bezug auf Art, Stamm und Geschlecht am besten geeigneten Tiere sowie der geeigneten Expositionsart und Prüfkonzentrationen sollten u. a. Informationen wie die Identität, die chemische Struktur und die physikalisch-chemischen Eigenschaften der Prüfsubstanz, Ergebnisse jeglicher In-vitro- oder In-vivo-Toxizitätsprüfungen, vorgesehene Verwendungen und die Möglichkeit der Exposition des Menschen, (Q)SAR-Daten und toxikologische Daten über strukturverwandte Substanzen herangezogen werden [siehe GD 39 (2)].
6. Die Prüfung hautätzender und/oder reizender Prüfsubstanzen in Konzentrationen, die voraussichtlich starke Schmerzen und/oder Qualen verursachen, sollte soweit wie möglich vermieden werden. Die hautätzenden/reizenden Eigenschaften sollten von Fachleuten auf der Grundlage von Erfahrungswerten bei Mensch und Tier (z.B. Studien mit wiederholter Verabreichung in nicht hautätzenden/reizenden Konzentrationen), vorliegenden In- vitro-Daten (z.B. aus den Kapiteln B.40 (5), B.40bis (6) dieses Anhangs oder OECD TG 435 (7)), den pH- Werten sowie Informationen über ähnliche Substanzen oder anderen sachdienlichen Daten beurteilt werden, damit untersucht werden kann, ob auf weitere Versuche verzichtet werden kann. Bei spezifischen Regulierungsanforderungen (z.B. für Zwecke der Notfallplanung) kann diese Prüfmethode verwendet werden, um Tiere diesen Stoffen auszusetzen, weil die Methode dem Studienleiter oder Hauptprüfer die Kontrolle über die Auswahl der Zielkonzentrationen gibt. Die Zielkonzentrationen sollten jedoch keine schwerwiegenden reizenden/hautätzenden Wirkungen hervorrufen; sie sollten aber ausreichen, um die Konzentrations-Wirkungs-Kurve so zu erweitern, dass das regulatorische und wissenschaftliche Ziel der Prüfung erreicht wird. Diese Konzentrationen sollten von Fall zu Fall ausgewählt werden; die Wahl der Konzentration ist zu begründen [siehe GD 39 (2)].
Prinzip der Prüfmethode
7. Diese überarbeitete Prüfmethode B.2 wurde entwickelt, um ausreichende Informationen über die akute Toxizität einer Prüfsubstanz zu gewinnen, damit sie eingestuft werden kann, und um Letalitätsdaten (z.B. LC50, LC01 und Steigung) für eines oder beide Geschlechter zu erhalten, die für quantitative Risikobewertungen benötigt werden. Die Prüfmethode umfasst zwei Methoden. Bei der ersten Methode handelt es sich um ein traditionelles Protokoll, bei dem Gruppen von Tieren für eine im Voraus festgelegte Dauer von üblicherweise 4 Stunden einer Grenzkonzentration (Limit-Test) oder schrittweise einer Reihe von Konzentrationen ausgesetzt werden. Für spezifische Regulierungszwecke sind andere Expositionszeiten möglich. Die zweite Methode ist ein Protokoll (c x t), bei dem Gruppen von Tieren über mehrere unterschiedliche Zeiträume einer Konzentration (Grenzkonzentration) oder einer Reihe unterschiedlicher Konzentrationen ausgesetzt werden.
8. Moribunde Tiere oder Tiere, die offensichtlich unter Schmerzen leiden oder Anzeichen von schwerem und anhaltendem Leiden zeigen, sollten auf humane Weise getötet werden und sind bei der Auswertung der Testergebnisse auf die gleiche Weise zu werten wie während des Tests gestorbene Tiere. Kriterien für die Entscheidung, moribunde oder schwer leidende Tiere zu töten, sowie Hinweise zur Erkennung des absehbaren oder bevorstehenden Todes sind Gegenstand des OECD Guidance Document No. 19 on Humane Endpoints (8).
Beschreibung der Methode
Auswahl von Versuchstierarten
9. Es sind junge, gesunde, adulte Tiere aus üblicherweise eingesetzten Laborstämmen zu verwenden. Bevorzugtes Versuchstier ist die Ratte. Die Verwendung anderer Tierarten ist zu begründen.
Vorbereitung der Tiere
10. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Am Tag der Exposition sollten die jungen, adulten Tiere 8 bis 12 Wochen alt sein; ihr Körpergewicht sollte innerhalb von ± 20 % des mittleren Gewichts für jedes Geschlecht aller zuvor exponierten Tiere desselben Alters liegen. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert und vor Beginn der Prüfung für einen Zeitraum von mindestens fünf Tagen in ihren Käfigen an die Laborbedingungen gewöhnt. Die Tiere sollten auch für einen kurzen Zeitraum vor der Prüfung an die Versuchsapparatur gewöhnt werden, da dies den Stress durch die Umsetzung in eine neue Umgebung verringert.
Tierhaltung
11. Die Temperatur in dem Raum, in dem die Versuchstiere gehalten werden, sollte 22 ± 3 °C betragen. Die relative Luftfeuchtigkeit sollte im Idealfall zwischen 30 und 70 % liegen; bei Verwendung von Wasser als Vehikel könnte dies jedoch unmöglich sein. Die Tiere sollten vor und nach den Expositionen im Allgemeinen nach Geschlecht und Konzentration in Käfigen gruppiert werden, wobei aber die Anzahl der Tiere pro Käfig noch eine genaue Beobachtung der einzelnen Tiere ermöglichen muss und Verluste aufgrund von Kannibalismus oder Kämpfen minimiert werden sollten. Wenn die Tiere der Prüfsubstanz nur mit der Nase ausgesetzt werden sollen, müssen sie möglicherweise an die Restrainer gewöhnt werden. Die Restrainer sollten die Tiere weder körperlich noch in Bezug auf Wärme oder Fixierung übermäßig beeinträchtigen. Die Fixierung kann physiologische Endpunkte wie Körpertemperatur (Hyperthermie) und/oder das Atemminutenvolumen beeinflussen. Wenn generische Daten zeigen, dass keine derartigen Veränderungen in nennenswertem Ausmaß vorkommen, ist eine Eingewöhnung an die Restrainer nicht erforderlich. Bei der Ganzkörperexposition gegen ein Aerosol sollten die Tiere während der Exposition einzeln untergebracht sein, damit sie die Prüfsubstanz nicht durch das Fell ihrer Käfiggenossen filtriert einatmen. Außer während der Exposition kann herkömmliches und zertifiziertes Labortierfutter verwendet werden bei uneingeschränkter Versorgung mit Trinkwasser. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
Inhalationskammern
12. Bei der Auswahl einer Inhalationskammer sind die Art der Prüfsubstanz und das Ziel der Prüfung zu berücksichtigen. Das bevorzugte Verfahren ist die 'Nose-only'-Exposition (dieser Begriff umfasst 'nur Kopf', 'nur Nase' oder 'nur Schnauze'). Für die Untersuchung von Flüssigkeits- oder Feststoffaerosolen und für Dämpfe, die zu Aerosolen kondensieren können, wird im Allgemeinen die 'Nose-only'-Exposition bevorzugt. Besondere Ziele der Untersuchung können möglicherweise mit einer Ganzkörperexposition besser erreicht werden, doch dies sollte im Prüfbericht begründet werden. Um bei Verwendung einer Ganzkörperkammer die Stabilität der Atmosphäre sicherzustellen, sollte das Gesamtvolumen der Versuchstiere 5 % des Volumens der Kammer nicht übersteigen. Die Prinzipien der 'Nose-only'- und der Ganzkörperexposition sowie ihre jeweiligen Vor- und Nachteile sind in GD 39 (2) beschrieben.
Expositionsbedingungen
Verabreichung der Konzentrationen
13. 'Nose-only'-Expositionen können bei Ratten bis zu 6 Stunden dauern. Bei Mäusen sollten 'Nose-only'-Expositionen im Allgemeinen 4 Stunden nicht überschreiten. Wenn Studien mit längerer Expositionsdauer erforderlich sind, ist dies zu begründen [siehe GD 39 (2)]. Bei Ganzkörperexpositionen gegen Aerosole sollten die Tiere einzeln untergebracht sein, um eine Aufnahme der Prüfsubstanz durch das Putzen von Käfiggenossen zu verhindern. Während der Exposition sollte kein Futter verabreicht werden. Wasser kann während einer Ganzkörperexposition angeboten werden.
14. Die Tiere werden der Prüfsubstanz in Form von Gas, Dampf, Aerosol oder einer Kombination dieser Formen ausgesetzt. Der zu prüfende Aggregatzustand hängt von den physikalisch-chemischen Eigenschaften der Prüfsubstanz, der gewählten Konzentration und/oder der physikalischen Form ab, in der die Prüfsubstanz bei der Handhabung und Verwendung am wahrscheinlichsten vorliegt. Hygroskopische und chemisch reaktive Prüfsubstanzen sollten bei geringer Luftfeuchtigkeit geprüft werden. Dabei ist darauf zu achten, dass keine explosionsfähigen Konzentrationen erzeugt werden.
Partikelgrößenverteilung
15. Bei allen Aerosolen und bei Dämpfen, die zu Aerosolen kondensieren können, sollte die Partikelgröße bestimmt werden. Damit alle relevanten Regionen der Atemwege der Prüfsubstanz ausgesetzt werden, werden mittlere aerodynamische Massendurchmesser (Mass Median Aerodynamic Diameter - MMAD) von 1 bis 4 pm mit einer geometrischen Standardabweichung (σg) von 1,5 bis 3,0 empfohlen (2) (9) (10). Wenngleich nach Möglichkeit versucht werden sollte, diese Werte zu erreichen, ist Fachwissen erforderlich, falls sie nicht erzielt werden können. Metalldämpfe können z.B. unter diesen Werten liegen, und geladene Partikel, Fasern und hygroskopische Stoffe (die sich in der feuchten Umgebung der Atemwege ausdehnen) können diese Werte überschreiten.
Vorbereitung der Prüfsubstanz in einem Vehikel
16. Um die gewünschte Konzentration und Partikelgröße der Prüfsubstanz in der Atmosphäre herzustellen, kann ein Vehikel verwendet werden. Hierbei ist in der Regel Wasser zu bevorzugen. Partikel können durch mechanische Prozesse auf die erforderliche Partikelgrößenverteilung gebracht werden. Dabei ist darauf zu achten, dass die Prüfsubstanz nicht zersetzt oder verändert wird. Wenn angenommen wird, dass die Zusammensetzung der Prüfsubstanz durch mechanische Prozesse verändert wurde (z.B. hohe Temperaturen aufgrund von Reibung durch übermäßiges Mahlen), sollte die Zusammensetzung der Prüfsubstanz analytisch überprüft werden. Es ist darauf zu achten, dass die Prüfsubstanz nicht kontaminiert wird. Nicht brüchige Granulate, die speziell so formuliert sind, dass sie nicht eingeatmet werden können, brauchen nicht geprüft zu werden. Mit einem Abriebtest sollte nachgewiesen werden, dass beim Umgang mit dem Granulat keine lungengängigen Partikel entstehen. Entstehen bei einem Abriebtest lungengängige Partikel, so sollte eine Inhalationstoxizitätsprüfung durchgeführt werden.
Kontrolltiere
17. Eine gleichzeitige negative (Luft-)Kontrollgruppe ist nicht erforderlich. Wenn zur Erzeugung der Prüfatmosphäre ein anderes Vehikel als Wasser verwendet wird, sollte nur dann eine Vehikelkontrollgruppe verwendet werden, wenn keine historischen Daten über Inhalationstoxizität vorliegen. Ergibt eine Toxizitätsstudie einer in einem Vehikel formulierten Prüfsubstanz, dass keine Toxizität vorliegt, ist das Vehikel folglich in der geprüften Konzentration nicht toxisch. Daher ist keine Vehikelkontrolle erforderlich.
Überwachung der Expositionsbedingungen
Luftstrom in der Inhalationskammer
18. Der Luftstrom durch die Kammer sollte während jeder Exposition sorgfältig geregelt, kontinuierlich überwacht und mindestens stündlich protokolliert werden. Die Überwachung der Konzentration (oder Stabilität) der Prüfatmosphäre ist eine integrale Messung aller dynamischen Parameter und gibt indirekt die Möglichkeit, alle relevanten dynamischen Parameter der Erzeugung der Prüfatmosphäre zu messen. Es sollte besonders darauf geachtet werden, das erneute Einatmen in 'Nose-only'-Expositionskammern zu vermeiden, wenn die Luftströmung durch das Expositionssystem nicht ausreicht, um eine dynamische Strömung der Prüfsubstanzatmosphäre zu erreichen. Es gibt festgelegte Methoden, mit denen nachgewiesen werden kann, dass es unter den gewählten Bedingungen nicht zu erneutem Einatmen kommt (2) (11). Die Sauerstoffkonzentration sollte mindestens 19 % betragen, und die Kohlendioxidkonzentration sollte 1 % nicht überschreiten. Gibt es Grund zu der Annahme, dass diese Werte nicht eingehalten werden können, sind die Sauerstoff- und die Kohlendioxidkonzentrationen zu messen.
Temperatur und relative Luftfeuchtigkeit in der Inhalationskammer
19. Die Temperatur in der Inhalationskammer sollte 22 ± 3 °C betragen. Sowohl bei der 'Nose-only'- als auch bei der Ganzkörperexposition sollte die relative Luftfeuchtigkeit im Atembereich der Tiere für Zeiträume von bis zu 4 Stunden mindestens dreimal und für kürzere Zeiträume stündlich überwacht und dokumentiert werden. Die relative Luftfeuchtigkeit sollte im Idealfall zwischen 30 und 70 % liegen, was jedoch möglicherweise nicht erreichbar ist (z.B. bei der Prüfung von wasserbasierten Mischungen) oder wegen chemischer Interferenz mit der Prüfmethode nicht gemessen werden kann.
Prüfsubstanz: nominale Konzentration
20. Die nominale Konzentration in der Expositionskammer sollte möglichst berechnet und protokolliert werden. Die nominale Konzentration ist die Masse der erzeugten Prüfsubstanz dividiert durch das Gesamtvolumen der durch das Kammersystem geleiteten Luft. Sie wird nicht zur Beschreibung der Exposition der Tiere verwendet; vielmehr gibt ein Vergleich der nominalen Konzentration und der tatsächlichen Konzentration Aufschluss über die Effizienz des Prüfsystems bei der Erzeugung der Prüfkonzentration und kann daher für die Aufdeckung von Problemen bei dieser Erzeugung verwendet werden.
Prüfsubstanz: tatsächliche Konzentration
21. Die tatsächliche Konzentration ist die Konzentration der Prüfsubstanz im Atembereich der Tiere in einer Inhalationskammer. Die tatsächlichen Konzentrationen können durch spezifische Methoden (z.B. direkte Probenahme, adsorptive Methoden oder chemische Reaktionsverfahren mit anschließender analytischer Charakterisierung) oder durch unspezifische Methoden wie Gravimetrie bestimmt werden. Die gravimetrische Methode ist lediglich für Aerosole mit nur einem Bestandteil in Pulverform oder Aerosole von Flüssigkeiten mit geringer Flüchtigkeit akzeptabel und sollte sich auf geeignete, vor der Studie zu erstellende und für die Prüfsubstanz spezifische Beschreibungen stützen. Die Konzentration von Aerosolen mit mehreren Bestandteilen in Pulverform kann ebenfalls gravimetrisch bestimmt werden. Hierzu muss jedoch mit Analysedaten belegt werden, dass die Schwebstoffe eine ähnliche Zusammensetzung haben wie das Ausgangsmaterial. Liegen diese Angaben nicht vor, muss die Prüfsubstanz (im Idealfall im Schwebezustand) möglicherweise im Verlauf der Studie in regelmäßigen Abständen neu analysiert werden. Bei aerosolisierten Agenzien, die verdunsten oder sublimieren können, sollte gezeigt werden, dass alle Phasen von der gewählten Methode erfasst wurden. Im Prüfbericht sollten die Zielkonzentration sowie die nominale und die tatsächliche Konzentration angegeben werden, aber nur die tatsächlichen Konzentrationen werden für statistische Analysen zur Berechnung letaler Konzentrationswerte verwendet.
22. Es sollte möglichst eine Partie der Prüfsubstanz verwendet werden; die Probe sollte unter Bedingungen aufbewahrt werden, die ihre Reinheit, Homogenität und Stabilität gewährleisten. Die Prüfsubstanz sollte vor Beginn der Studie mit Angaben zur Reinheit und, falls technisch machbar, zur Identität sowie zu den Mengen identifizierter Schadstoffe und Verunreinigungen beschrieben werden. Hierzu können unter anderem die folgenden Daten verwendet werden: Retentionszeit und relative Peakfläche, durch Massenspektrometrie oder Gaschromatographie bestimmtes Molekulargewicht oder andere Werte. Das Prüflabor ist zwar nicht für die Identität der Probe verantwortlich, doch es kann ratsam sein, dass es die Beschreibung des Auftraggebers zumindest in gewissen Grenzen (z.B. Farbe, physikalische Beschaffenheit usw.) überprüft.
23. Die Expositionsatmosphäre ist so konstant wie möglich zu halten und je nach Analysemethode kontinuierlich und/oder intermittierend zu überwachen. Bei der intermittierenden Probenahme sollten in einer vierstündigen Studie mindestens zweimal Proben der Atmosphäre in der Kammer genommen werden. Ist dies wegen begrenzter Luftdurchflussraten oder niedriger Konzentrationen nicht möglich, kann während der gesamten Expositionszeit eine einzige Probe genommen werden. Weichen die einzelnen Proben stark voneinander ab, sollten bei den nächsten geprüften Konzentrationen vier Proben je Exposition gezogen werden. Die einzelnen Proben der Konzentration in der Kammer sollten bei Gasen und Dämpfen nicht mehr als ± 10 % und bei Flüssig- oder Feststoffaerosolen nicht mehr als ± 20 % von der mittleren Kammerkonzentration abweichen. Die Zeit bis zum Erreichen eines Gleichgewichts in der Kammer (t95) ist zu berechnen und zu dokumentieren. Die Expositionsdauer erstreckt sich über den Zeitraum, in dem die Prüfsubstanz erzeugt wird; dazu gehört die zur Erreichung von t95 erforderliche Zeit. GD 39 (2) enthält Hinweise zur Einschätzung von t95.
24. Bei sehr komplexen Mischungen aus Gasen/Dämpfen und Aerosolen (z.B. Verbrennungsatmosphären und Prüfsubstanzen, die aus hierzu bestimmten Endverbraucherprodukten/-geräten gesprüht werden), kann sich jede Phase in einer Inhalationskammer anders verhalten, so dass mindestens eine Indikatorsubstanz (Analyt), normalerweise der wichtigste Wirkstoff in der Mischung, von jeder Phase (Gas/Dampf und Aerosol) ausgewählt werden sollte. Wenn die Prüfsubstanz eine Mischung ist, sollte die Analysekonzentration für die Mischung und nicht nur für den Wirkstoff oder den Bestandteil (Analyt) dokumentiert werden. Weitere Informationen zu tatsächlichen Konzentrationen sind in GD 39 (2) zu finden.
Prüfsubstanz: Partikelgrößenverteilung
25. Die Partikelgrößenverteilung von Aerosolen sollte während jeder 4-stündigen Exposition mindestens zweimal mit einem Kaskaden-Impaktor oder einem anderen Messgerät wie einem APS bestimmt werden. Kann nachgewiesen werden, dass die mit einem Kaskaden-Impaktor und einem alternativen Messgerät erzielten Ergebnisse gleichwertig sind, so kann das alternative Instrument während der gesamten Studie verwendet werden. Parallel zum Hauptinstrument ist ein zweites Gerät wie ein Gravimetriefilter oder eine Gaswaschflasche zu verwenden, um den Abscheidegrad des Hauptinstruments zu bestätigen. Die durch die Partikelgrößenanalyse bestimmte Massenkonzentration sollte innerhalb vertretbarer Grenzen um die durch die Filteranalyse bestimmte Massenkonzentration liegen [siehe DG 39 (2)]. Wenn die Gleichwertigkeit zu Beginn der Studie nachgewiesen werden kann, kann auf weitere bestätigende Messungen verzichtet werden. Aus Tierschutzgründen sollten Vorkehrungen getroffen werden, um unklare Daten zu minimieren, die dazu führen könnten, dass eine Exposition wiederholt werden muss. Wenn die Möglichkeit besteht, dass Dampfkondensation zur Bildung eines Aerosols führen kann, oder wenn in einer Dampfatmosphäre mit dem Potenzial für gemischte Phasen Partikel nachgewiesen werden, sollte eine Partikelgrößenbestimmung für Dämpfe vorgenommen werden (siehe Nummer 15).
Verfahren
26. Nachstehend werden zwei Prüfungsarten beschrieben: das traditionelle Protokoll und das C-x-t-Protokoll. Beide Protokolle können eine Vorstudie, eine Hauptstudie und/oder einen Limit-Test (traditionelles Protokoll) bzw. eine Prüfung bei einer Grenzkonzentration (c x t) umfassen. Wenn bekannt ist, dass ein Geschlecht empfindlicher reagiert, kann der Studienleiter entscheiden, diese Prüfungen nur unter Verwendung dieses Geschlechts durchzuführen. Wenn für eine 'Nose-only'-Exposition andere Nagetierarten als Ratten verwendet werden, kann die maximale Expositionsdauer angepasst werden, um artenspezifisches Leiden zu minimieren. Damit möglichst wenig Tiere verwendet werden, sollten vor Beginn der Prüfung alle verfügbaren Daten ausgewertet werden. Mit Ergebnissen der Methode gemäß Kapitel B.52 dieses Anhangs (4) kann möglicherweise die Notwendigkeit einer Vorstudie ausgeräumt und gezeigt werden, ob ein Geschlecht empfindlicher reagiert als das andere [siehe GD 39 (2)].
Traditionelles Protokoll
Allgemeine Überlegungen: traditionelles Protokoll
27. In einer traditionellen Studie werden Gruppen von Tieren einer Prüfsubstanz für einen festgelegten Zeitraum (im Allgemeinen 4 Stunden) entweder in einer 'Nose-only'- oder einer Ganzkörperexpositionskammer ausgesetzt. Die Tiere werden entweder einer Grenzkonzentration (Limit-Test) oder schrittweise mindestens drei Konzentrationen (Hauptstudie) ausgesetzt. Wenn nicht bereits Informationen über die Prüfsubstanz vorliegen, wie z.B. aus einer zuvor durchgeführten Prüfung nach Kapitel B.52, kann vor der Hauptstudie eine Vorstudie durchgeführt werden [siehe GD 39 (2)].
Vorstudie: traditionelles Protokoll
28. Eine Vorstudie dient dazu, die Wirkstärke der Prüfsubstanz einzuschätzen, geschlechtsspezifische Unterschiede bei der Empfindlichkeit festzustellen und die Festlegung der Expositionskonzentrationen für die Hauptstudie oder den Limit-Test zu erleichtern. Die Auswahl der Konzentrationen für die Vorstudie sollte sich auf alle verfügbaren Informationen, einschließlich verfügbarer (Q)SAR-Daten und Daten für ähnliche Chemikalien stützen. Jeder Konzentration der Prüfsubstanz sollten höchstens drei männliche und drei weibliche Tiere ausgesetzt werden (drei Tiere je Geschlecht können erforderlich sein, um einen geschlechtsspezifischen Unterschied festzustellen). Es ist möglich, dass in einer Vorstudie nur eine einzige Konzentration geprüft wird, erforderlichenfalls können aber auch mehr Konzentrationen geprüft werden. In einer Vorstudie sollten nicht so viele Tiere und Konzentrationen untersucht werden, dass sie quasi einer Hauptstudie gleichkommt. Statt einer Vorstudie kann auf die Ergebnisse einer zuvor durchgeführten Prüfung gemäß Kapitel B.52 (4) zurückgegriffen werden [siehe GD 39 (2)].
Limit-Test: traditionelles Protokoll
29. Ein Limit-Test wird verwendet, wenn bekannt oder zu erwarten ist, dass die Prüfsubstanz praktisch nicht toxisch ist, d. h. nur über der regulatorisch festgelegten Grenzkonzentration eine toxische Wirkung hervorruft. Beim Limit-Test wird eine einzige Gruppe von drei männlichen und drei weiblichen Tieren der Prüfsubstanz bei einer Grenzkonzentration ausgesetzt. Informationen über die Toxizität der Prüfsubstanz können aus Kenntnissen über ähnliche geprüfte Stoffe gewonnen werden, wobei Art und prozentualer Anteil der Komponenten zu berücksichtigen sind, deren toxikologische Relevanz bekannt ist. In den Fällen, in denen nur wenige oder keine Informationen über die Toxizität der Prüfsubstanz vorliegen oder in denen von einer Toxizität der Prüfsubstanz ausgegangen wird, sollte der Haupttest durchgeführt werden.
30. Die Wahl der Grenzkonzentrationen hängt im Allgemeinen von den Regulierungsanforderungen ab. Wird die Verordnung (EG) Nr. 1272/2008 zugrunde gelegt, so betragen die Grenzkonzentrationen für Gase, Dämpfe und Aerosole 20.000 ppm, 20 mg/l bzw. 5 mg/l (oder die höchste erreichbare Konzentration) (3). Bei bestimmten Prüfsubstanzen, insbesondere bei Dämpfen und Aerosolen, kann es technisch schwierig sein, Grenzkonzentrationen zu erzeugen. Bei der Prüfung von Aerosolen sollte das Hauptziel darin bestehen, eine lungengängige Partikelgröße (d. h. ein MMAD von 1-4 µm) zu erreichen. Dies ist bei den meisten Prüfsubstanzen bei einer Konzentration von 2 mg/l der Fall. Aerosole sollten nur dann bei mehr als 2 mg/l geprüft werden, wenn eine lungengängige Partikelgröße erreicht werden kann [siehe DG 39 (2)]. Die Verordnung (EG) Nr. 1272/2008 rät aus Tierschutzgründen von der Prüfung über einer Grenzkonzentration ab (3). Die Grenzkonzentration sollte nur in Betracht gezogen werden, wenn eine hohe Wahrscheinlichkeit besteht, dass die Ergebnisse einer solchen Prüfung von unmittelbarer Relevanz für den Schutz der menschlichen Gesundheit sind (3); dies ist im Prüfbericht zu begründen. Bei potenziell explosiven Prüfsubstanzen ist darauf zu achten, dass keine explosionsfördernden Bedingungen geschaffen werden. Um eine unnötige Verwendung von Versuchstieren zu vermeiden und sicherzustellen, dass die Kammerbedingungen für einen Limit-Test erreicht werden können, sollte vor dem Limit-Test ein Probedurchlauf ohne Tiere vorgenommen werden.
31. Werden bei der Grenzkonzentration Mortalität oder Siechtum beobachtet, können die Ergebnisse der Prüfung bei dieser Konzentration als Vorstudie für weitere Prüfungen bei anderen Konzentrationen dienen (siehe Hauptstudie). Wenn eine Grenzkonzentration wegen der physikalischen oder chemischen Eigenschaften einer Prüfsubstanz nicht erreicht werden kann, sollte die höchste erreichbare Konzentration geprüft werden. Wenn bei der höchsten erreichbaren Konzentration eine Letalität von weniger als 50 % auftritt, sind keine weiteren Prüfungen erforderlich. Konnte die Grenzkonzentration nicht erreicht werden, sollte der Prüfbericht eine Erklärung und entsprechende Daten enthalten. Wenn die höchste erreichbare Konzentration eines Dampfs keine Toxizität hervorruft, muss die Prüfsubstanz möglicherweise als Flüssigkeitsaerosol angewendet werden.
Hauptstudie: traditionelles Protokoll
32. Eine Hauptstudie wird normalerweise mit fünf männlichen und fünf weiblichen Tieren (oder, falls bekannt, fünf Tieren des empfindlicheren Geschlechts) je Konzentrationsstufe mit mindestens drei Konzentrationsstufen durchgeführt. Für eine robuste statistische Analyse sollten ausreichend hohe Konzentrationsstufen verwendet werden. Der Zeitabstand zwischen den einzelnen Expositionsgruppen richtet sich nach Einsetzen, Dauer und Schweregrad der toxischen Zeichen. Die Tiere sollten erst dann der nächsten Konzentrationsstufe ausgesetzt werden, wenn mit angemessener Gewissheit vom Überleben der zuvor getesteten Tiere ausgegangen werden kann. Dies gibt dem Studienleiter die Möglichkeit, die Zielkonzentration für die nächste Expositionsgruppe anzupassen. Wegen der Abhängigkeit von hochentwickelter Technologie ist dies in Inhalationsstudien nicht immer praktikabel. Deshalb sollte die Exposition von Tieren gegen die nächste Konzentrationsstufe auf Erfahrungswerten und wissenschaftlichem Sachverstand beruhen. Bei der Prüfung von Mischungen ist das GD 39 (2) zu konsultieren.
Konzentration-x-Zeit-Protokoll (C x T)
Allgemeine Überlegungen: C-x-t-Prokokoll
33. Bei der Bewertung der Inhalationstoxizität kann als Alternative zu einem traditionellen Protokoll eine schrittweise C-x-t-Studie in Betracht gezogen werden (12) (13) (14). Bei dieser Methode werden die Tiere der Prüfsubstanz in unterschiedlichen Konzentrationsstufen und für unterschiedliche Zeiträume ausgesetzt. Für alle Prüfungen werden 'Nose-only'-Kammern verwendet (Ganzkörperkammern sind bei diesem Protokoll nicht praktikabel). Ein Fließdiagramm in Anlage 1 veranschaulicht dieses Protokoll. Eine Simulationsanalyse hat gezeigt, dass sowohl das traditionelle Protokoll und als auch das C-x-t-Protokoll robuste LC50-Werte ergeben können, aber das C-x-t-Protokoll bei robusten LC01- und LC10-Werten im Allgemeinen überlegen ist (15).
34. Einer Simulationsanalyse zufolge ist die Verwendung von zwei Tieren je C-x-t-Intervall (ein Tier je Geschlecht bei Verwendung beider Geschlechter oder zwei Tiere des empfindlicheren Geschlechts) im Allgemeinen ausreichend, wenn in einer Hauptstudie 4 Konzentrationen und 5 Expositionszeiträume geprüft werden. Unter bestimmten Umständen kann der Studienleiter beschließen, je zwei Ratten beider Geschlechter je C-x-t-Intervall zu verwenden (15). Die Verwendung von zwei Tieren je Geschlecht je Konzentration und Zeitpunkt kann Verzerrungen und Schwankungen der Schätzungen verringern, die Quote der zutreffenden Schätzungen erhöhen und die Abdeckung des Konfidenzintervalls verbessern. Wenn die Daten jedoch keine ausreichende Annäherung für eine Schätzung erlauben (bei Verwendung von je einem Tier beider Geschlechter oder von zwei Tieren des empfindlicheren Geschlechts), kann auch eine fünfte Expositionskonzentration ausreichen. Das GD 39 (2) enthält weitere Hinweise zur Zahl der Tiere und zu den Konzentrationen, die in einer C-x-t-Studie zu verwenden sind.
Vorstudie: C-x-t-Protokoll
35. Eine Vorstudie dient dazu, die Wirkstärke der Prüfsubstanz einzuschätzen und die Festlegung der Expositionskonzentrationen für die Hauptstudie zu erleichtern. Um eine geeignete Ausgangskonzentration für die Hauptstudie festzulegen und möglichst wenig Tiere zu verwenden, kann eine Vorstudie mit bis zu drei Tieren/Geschlecht/Konzentration [Einzelheiten siehe Anlage III von GD 39 (2)] erforderlich sein. Zur Feststellung eines geschlechtsspezifischen Unterschieds müssen gegebenenfalls drei Tiere je Geschlecht verwendet werden. Diese Tiere sind für einen einzigen Zeitraum, im Allgemeinen 240 Minuten, zu exponieren. Die Erzeugung geeigneter Prüfatmosphären ist in technischen Vorversuchen ohne Tiere zu erproben. Wenn Mortalitätsdaten aus einer Studie nach B.52 (4) vorliegen, braucht normalerweise keine Vorstudie durchgeführt zu werden. Bei der Festlegung der anfänglichen Zielkonzentration einer Studie nach B.2 sollte der Studienleiter die in allen verfügbaren B.52-Studien (4) beobachteten Mortalitätsmuster für beide Geschlechter und für alle geprüften Konzentrationen berücksichtigen [siehe GD 39 (2)].
Ausgangskonzentration: C-x-t-Protokoll
36. Die Ausgangskonzentration (Exposition I) (Anlage 1) ist entweder eine Grenzkonzentration oder eine vom Studienleiter auf Basis der Vorstudie gewählte Konzentration. Gruppen von je einem Tier beider Geschlechter werden dieser Konzentration für unterschiedliche Zeiträume ausgesetzt (z.B. 15, 30, 60, 120 oder 240 Minuten), so dass insgesamt 10 Tiere verwendet werden (Exposition I) (Anlage 1).
37. Die Wahl der Grenzkonzentrationen hängt im Allgemeinen von den Regulierungsanforderungen ab. Wird die Verordnung (EG) Nr. 1272/2008 zugrunde gelegt, so betragen die Grenzkonzentrationen für Gase, Dämpfe und Aerosole 20.000 ppm, 20 mg/l bzw. 5 mg/l (oder die höchste erreichbare Konzentration) (3). Bei bestimmten Prüfsubstanzen, insbesondere bei Dämpfen und Aerosolen, kann es technisch schwierig sein, Grenzkonzentrationen zu erzeugen. Bei der Prüfung von Aerosolen sollte eine lungengängige Partikelgröße (d. h. ein MMAD von 1-4 µm) bei einer Grenzkonzentration von 2 mg/l erreicht werden. Dies ist bei den meisten Prüfsubstanzen möglich. Aerosole sollten nur dann bei mehr als 2 mg/l geprüft werden, wenn eine lungengängige Partikelgröße erreicht werden kann [siehe DG 39 (2)]. Die Verordnung (EG) Nr. 1272/2008 rät aus Tierschutzgründen von der Prüfung über einer Grenzkonzentration ab (3). Prüfungen über der Grenzkonzentration sollten nur in Betracht gezogen werden, wenn eine hohe Wahrscheinlichkeit besteht, dass die Ergebnisse einer solchen Prüfung von unmittelbarer Relevanz für den Schutz der menschlichen Gesundheit sind (3); dies ist im Prüfbericht zu begründen. Bei potenziell explosiven Prüfsubstanzen ist darauf zu achten, dass keine explosionsfördernden Bedingungen geschaffen werden. Vor der Prüfung bei der Ausgangskonzentration sollte ein Probedurchlauf ohne Tiere vorgenommen werden, um eine unnötige Verwendung von Versuchstieren zu vermeiden und sicherzustellen, dass die Kammerbedingungen für diese Konzentration erreicht werden können.
38. Werden bei der Ausgangskonzentration Mortalität oder Siechtum beobachtet, können die Ergebnisse dieser Konzentration als Ausgangspunkt für weitere Prüfungen bei anderen Konzentrationen dienen (siehe Hauptstudie). Wenn eine Grenzkonzentration wegen der physikalischen oder chemischen Eigenschaften einer Prüfsubstanz nicht erreicht werden kann, sollte die höchste erreichbare Konzentration geprüft werden. Wenn bei der höchsten erreichbaren Konzentration eine Letalität von weniger als 50 % auftritt, sind keine weiteren Prüfungen erforderlich. Konnte die Grenzkonzentration nicht erreicht werden, sollte der Prüfbericht eine Erklärung und entsprechende Daten enthalten. Wenn die höchste erreichbare Konzentration eines Dampfs keine Toxizität hervorruft, muss die Prüfsubstanz möglicherweise als Flüssigkeitsaerosol angewendet werden.
Hauptstudie: C-x-t-Protokoll
39. Die in der Hauptstudie geprüfte Ausgangskonzentration (Exposition I) (Anlage 1) ist entweder eine Grenzkonzentration oder eine vom Studienleiter auf Basis der Vorstudie gewählte Konzentration. Wenn während oder nach der Exposition I Mortalität beobachtet wurde, gilt die Mindestexposition (c x t), die zu Mortalität führt, als Anhaltspunkt für die Festlegung der Expositionskonzentration und -dauer für die Exposition II. Jede folgende Exposition hängt von der vorhergehenden Exposition ab (siehe Anlage 1).
40. Bei vielen Prüfsubstanzen sind die bei der Ausgangskonzentration erzielten Ergebnisse zusammen mit drei zusätzlichen Expositionen mit einem kleineren Zeitraster (das ist der geometrische Abstand von Expositionsperioden, der angegeben wird durch den Faktor zwischen aufeinanderfolgenden Perioden, im Allgemeinen √2) ausreichend, um die C-x-t-Mortalitätsbeziehung festzulegen (15), aber es kann hilfreich sein, eine fünfte Expositionskonzentration zu verwenden [siehe Anlage 1 und GD 39 (2)]. Anlage 1 enthält Hinweise zur mathematischen Aufbereitung der Ergebnisse für das C-x-t-Protokoll.
Beobachtungen
41. Die Tiere sollten während der Exposition häufig auf klinische Zeichen beobachtet werden. Nach der Exposition sollten klinische Beobachtungen mindestens zweimal am Tag der Exposition oder, falls es aufgrund der Reaktion der Tiere auf die Behandlung angezeigt erscheint, häufiger und danach für einen Zeitraum von insgesamt 14 Tagen mindestens einmal täglich vorgenommen werden. Die Länge des Beobachtungszeitraums ist nicht festgelegt; sie sollte nach Art und Zeitpunkt des Einsetzens klinischer Zeichen und der Länge der Erholungsphase bestimmt werden. Der Zeitpunkt, zu dem die Toxizitätszeichen auftreten und wieder abklingen, ist von Bedeutung, insbesondere dann, wenn Anzeichen für ein verzögertes Auftreten von Toxizitätszeichen erkennbar sind. Sämtliche Beobachtungen werden systematisch in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Tiere, bei denen ein moribunder Zustand festgestellt wird, sowie Tiere, die starke Schmerzen haben oder anhaltende Anzeichen von schwerem Leiden zeigen, sollten aus Tierschutzgründen auf humane Weise getötet werden. Bei den Untersuchungen auf klinische Toxizitätszeichen ist darauf zu achten, dass ein anfänglich schlechtes Aussehen und vorübergehende Atemveränderungen, die auf das Expositionsverfahren zurückzuführen sind, nicht mit einer durch die Prüfsubstanz bedingten Toxizität verwechselt werden, die eine vorzeitige Tötung der Tiere erfordern würde. Die im Guidance Document on Humane Endpoints (GD 19) zusammengefassten Prinzipien und Kriterien sind zu berücksichtigen (7). Wenn Tiere aus humanen Gründen getötet werden oder ihr Tod festgestellt wird, sollte der Todeszeitpunkt so genau wie möglich registriert werden.
42. Bei den Beobachtungen ist auf Veränderungen von Haut, Fell, Augen und Schleimhäuten sowie Atmung, Kreislauf, autonomes und zentrales Nervensystem, Somatomotorik und Verhaltensmuster zu achten. Soweit möglich, ist auf Differenzierungen zwischen lokalen und systemischen Wirkungen zu achten. Besonderes Augenmerk ist auf Tremor, Konvulsionen, Salivation, Diarrhö, Lethargie, Schlaf und Koma zu richten. Die Messung der Rektaltemperatur kann zusätzliche Belege für mit der Behandlung oder Unterbringung zusammenhängende Reflex- Bradypnoe oder Hypo-/Hyperthermie liefern.
Körpergewicht
43. Das Körpergewicht der einzelnen Tiere sollte einmal während der Eingewöhnungszeit, am Tag der Exposition vor der Exposition (Tag 0) und mindestens an den Tagen 1, 3 und 7 (und danach wöchentlich) sowie zum Zeitpunkt des Todes oder der Tötung, falls später als Tag 1, dokumentiert werden. Das Körpergewicht gilt als kritischer Indikator für Toxizität; Tiere, die gegenüber ihrem Gewicht vor der Prüfung eine dauerhafte Abnahme um > 20 % aufweisen, sollten sorgfältig überwacht werden. Die überlebenden Tiere werden gewogen und am Ende der Post-Expositionsphase auf humane Weise getötet.
Pathologie
44. Alle Versuchstiere (einschließlich der Tiere, die während des Tests sterben oder aus Tierschutzgründen getötet und aus der Studie genommen werden) sind auf makroskopische Veränderungen zu untersuchen. Kann die Nekropsie nicht unmittelbar nach Auffinden eines toten Tieres erfolgen, sollte der Körper auf eine Temperatur gekühlt (nicht eingefroren) werden, die tief genug ist, um die Autolyse zu minimieren. Die Nekropsie ist baldmöglichst, in der Regel innerhalb von einem oder zwei Tagen durchzuführen. Alle makroskopischen Veränderungen sollten für jedes Tier protokolliert werden, wobei besonders auf Veränderungen der Atemwege zu achten ist.
45. Es kann in Betracht gezogen werden, von vorneherein zusätzliche Untersuchungen in die Studienauslegung aufzunehmen, die die Aussagekraft der Studie erhöhen, z.B. die Messung des Lungengewichts überlebender Ratten und/oder den Nachweis einer Reizwirkung durch mikroskopische Untersuchung der Atemwege. Untersucht werden können auch diejenigen Organe, die bei 24 Stunden oder länger überlebenden Tieren makroskopische Befunde aufweisen, sowie Organe, die bekanntermaßen oder vermutlich betroffen sind. Eine mikroskopische Untersuchung des gesamten Atemtrakts kann nützliche Informationen über Prüfsubstanzen liefern, die mit Wasser reagieren, z.B. Säuren und hygroskopische Prüfsubstanzen.
Daten und Berichterstattung
Daten
46. Das Körpergewicht der einzelnen Tiere und Sektionsbefunde sollten angegeben werden. Die Daten der klinischen Beobachtung sollten in tabellarischer Form zusammengefasst werden. Daraus müssen für jede Prüfgruppe die Anzahl der verwendeten Tiere, die Anzahl der Tiere mit spezifischen Toxizitätszeichen, die Anzahl der Tiere, die während der Prüfung tot aufgefunden oder vorzeitig getötet wurden, der Todeszeitpunkt der einzelnen Tiere, eine Beschreibung und der zeitliche Verlauf der toxischen Wirkungen und deren Reversibilität sowie die Sektionsbefunde ersichtlich sein.
Prüfbericht
47. Der Prüfbericht sollte, soweit zutreffend, die folgenden Informationen enthalten:
Versuchstiere und Tierhaltung
Prüfsubstanz
Vehikel
Inhalationskammer
Expositionsdaten
Prüfbedingungen
Ergebnisse
Diskussion und Auswertung der Ergebnisse
Literatur:
1. OECD (2009). Acute Inhalation Toxicity Testing. OECD Guideline for Testing of Chemicals No. 403, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
2. OECD (2009). Guidance Document on Acute Inhalation Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment No. 39, OECD, Paris. Abrufbar unter: [http://www.oecd.org/ env/testguidelines]
3. Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
4. Kapitel B.52 dieses Anhangs, Akute Inhalationstoxizität - akut toxische Klassenmethode.
5. Kapitel B.40 dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: TER-Test (transcutaneous electrical resistance test)
6. Kapitel B.40bis dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: Test mit menschlichem Hautmodell)
7. OECD (2005), In Vitro Membrane Barrier Test Method For Skin Corrosion. OECD Guideline for Testing of Chemicals No. 435, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
8. OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
9. SOT (1992). Technical Committee of the Inhalation Specialty Section, Society of Toxicology (SOT). Recommendations for the Conduct of Acute Inhalation Limit Tests. Fund. Appl. Toxicol. 18: 321-327.
10. Phalen RF (2009). Inhalation Studies: Foundations and Techniques. (2nd Edition) Informa Healthcare, New York.
11. Pauluhn J and Thiel A (2007). A Simple Approach to Validation of Directed-Flow Nose-Only Inhalation Chambers. J. Appl. Toxicol. 27: 160-167.
12. Zwart JHE, Arts JM, ten Berge WF, Appelman LM (1992). Alternative Acute Inhalation Toxicity Testing by Determination of the Concentration-Time-Mortality Relationship: Experimental Comparison with Standard LC50 Testing. Reg. Toxicol. Pharmacol. 15: 278-290.
13. Zwart JHE, Arts JM, Klokman-Houweling ED, Schoen ED (1990). Determination of Concentration-Time-Mortality Relationships to Replace LC50 Values. Inhal. Toxicol. 2: 105-117.
14. Ten Berge WF and Zwart A (1989). More Efficient Use of Animals in Acute Inhalation Toxicity Testing. J. Haz. Mat. 21: 65-71.
15. OECD (2009). Performance Assessment: Comparison of 403 and C x t Protocols via Simulation and for Selected Real Data Sets. Environmental Health and Safety Monograph Series on Testing and Assessment No. 104, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
16. Finney DJ (1977). Probit Analysis, 3rd ed. Cambridge University Press, London/New York.
Definition
Prüfsubstanz: jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird.
Anlage 1 C-x-t-Protokoll
1. Bei der Bewertung der Inhalationstoxizität kann als Alternative zu einem traditionellen Protokoll eine schrittweise Konzentrations-x-Zeit-Studie (c x t) in Betracht gezogen werden (12) (13) (14). Sie sollte vorzugsweise dann angewendet werden, wenn wegen einer spezifischen Regelung oder aus wissenschaftlicher Notwendigkeit eine Prüfung von Tieren über unterschiedliche Zeiträume erforderlich ist, z.B. für Zwecke der Notfallplanung oder für die Flächennutzungsplanung. Diese Methode beginnt normalerweise mit der Prüfung der Prüfsubstanz in einer Grenzkonzentration (Exposition I), bei der die Tiere der Prüfsubstanz für fünf Zeiträume ausgesetzt werden (z.B. 15, 30, 60, 120 und 240 Minuten), so dass innerhalb einer Exposition Ergebnisse für mehrere Zeiträume erzielt werden (siehe Abbildung 1). Wird die Verordnung (EG) Nr. 1272/2008 zugrunde gelegt, so betragen die Grenzkonzentrationen für Gase, Dämpfe und Aerosole 20.000 ppm, 20 mg/l bzw. 5 mg/l. Diese Werte dürfen nur überschritten werden, wenn eine regulatorische oder wissenschaftliche Notwendigkeit für die Prüfung mit diesen höheren Werten besteht (siehe Nummer 37 im Haupttext von Kapitel B.2).
2. Liegen nur wenig oder keine Informationen über die Toxizität einer Prüfsubstanz vor, sollte eine Vorstudie durchgeführt werden, bei der Gruppen von höchstens drei Tieren beider Geschlechter den vom Studienleiter festgelegten Zielkonzentrationen im Allgemeinen für 240 Minuten ausgesetzt werden.
3. Wenn bei der Exposition I eine Grenzkonzentration geprüft wird und die Mortalität bei unter 50 % liegt, sind keine weiteren Prüfungen erforderlich. Besteht eine regulatorische oder wissenschaftliche Notwendigkeit zur Festlegung der Konzentrations-/Zeit-/Wirkungs-Beziehung bei höheren Werten als der angegebenen Grenzkonzentration, sollte die nächste Exposition auf einem höheren Niveau, z.B. dem Doppelten der Grenzkonzentration, durchgeführt werden (d. h. 2L in Abbildung 1).
4. Wenn bei der Grenzkonzentration Toxizität beobachtet wird, sind zusätzliche Prüfungen (Hauptstudie) erforderlich. Diese zusätzlichen Expositionen werden entweder bei niedrigeren Konzentrationen (in Abbildung 1: Expositionen II, III oder IV') oder bei höheren Konzentrationen von kürzerer Dauer (in Abbildung 1: Exposition IV) durchgeführt, wobei angepasste Zeiträume mit geringerem Abstand verwendet werden.
5. Die Prüfung (Ausgangskonzentration und zusätzliche Konzentrationen) wird mit je einem Tier beider Geschlechter je Konzentration/Zeitpunkt oder zwei Tieren des empfindlicheren Geschlechts je Konzentration/Zeitpunkt durchgeführt. Unter bestimmten Umständen kann der Studienleiter entscheiden, je zwei Ratten beider Geschlechter je Konzentration/Zeitpunkt (oder vier Tiere des empfindlicheren Geschlechts je Konzentration/Zeitpunkt) zu verwenden (15). Die Verwendung von zwei Tieren je Geschlecht je Konzentration/Zeitpunkt verringert im Allgemeinen Verzerrungen und Schwankungen der Schätzungen, erhöht die Quote der zutreffenden Schätzungen und verbessert die Abdeckung des Konfidenzintervalls gegenüber dem hier beschriebenen Protokoll. GD 39 (2) enthält nähere Hinweise.
6. Im Idealfall wird jede Exposition innerhalb eines Tages durchgeführt. Bei dieser Vorgehensweise kann die nächste Exposition auf einen Zeitpunkt gelegt werden, an dem mit angemessener Gewissheit vom Überleben der Tiere ausgegangen werden kann. Außerdem kann der Studienleiter die Zielkonzentration und Dauer der nächsten Exposition anpassen. Es empfiehlt sich, jede Exposition mit der Gruppe zu beginnen, die am längsten exponiert sein wird, z.B. die 240-Minuten-Gruppe, gefolgt von der 120-Minuten-Gruppe usw. Wenn beispielsweise Tiere in der 240- Minuten-Gruppe nach 90 Minuten sterben oder deutliche Toxizitätszeichen aufweisen (wie extreme Veränderungen der Atmung, z.B. Atemnot), wäre es nicht sinnvoll, eine Gruppe für 120 Minuten der Prüfsubstanz auszusetzen, da die Mortalität wahrscheinlich 100 % betragen würde. Der Studienleiter sollte dann für diese Konzentration eine kürzere Expositionsdauer festlegen (z.B. 90, 65, 45, 33 und 25 Minuten).
7. Die Konzentration in der Kammer sollte häufig gemessen werden, um für jede Expositionsdauer die zeitgewichtete Durchschnittskonzentration zu bestimmen. Soweit möglich, sollte bei jedem Tier der Todeszeitpunkt (nicht die Expositionsdauer) für die statistische Analyse verwendet werden.
8. Die Ergebnisse der ersten vier Expositionen sollten auf Datenlücken in der Konzentrations-Zeit-Kurve geprüft werden (siehe Abbildung 1). Wenn eine unzureichende Annäherung gegeben ist, kann eine zusätzliche Exposition (fünfte Konzentration) durchgeführt werden. Konzentration und Expositionsdauer der fünften Exposition sollten so gewählt werden, dass diese Lücke geschlossen wird.
9. Die Konzentrations-Zeit-Wirkungsbeziehung wird durch statistische Analyse unter Einbeziehung aller Expositionen (einschließlich der Exposition I) berechnet (16). Soweit möglich, sollten für jedes c-x-t-Intervall die zeitgewichtete Durchschnittskonzentration und die Expositionsdauer bis zum Tod (falls der Tod während der Exposition eintritt) verwendet werden.
Abbildung 1 Hypothetische Darstellung einer Konzentrations-Zeit-Mortalitäts-Beziehung bei Ratten
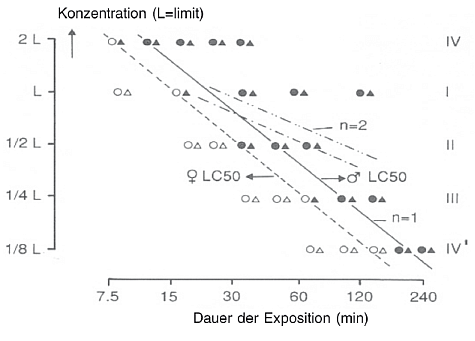
Offene Symbole = überlebende Tiere; geschlossene Symbole = tote Tiere
Dreiecke = Weibchen; Kreise = Männchen
Durchgezogene Linie = LC50 -Werte (Bereich 7,5-240 min) für männliche Tiere mit n = 1
Gestrichelte Linie = LC50-Werte (Bereich 7,5-240 min) für weibliche Tiere mit n = 1
Gestrichelt-gepunktete Linien = hypothetische LC 50 -Werte für männliche und weibliche Tiere, falls n = 2 war (12).
Glossar
Konzentration:
Dauer der Exposition:
10. Beispiel für das schrittweise Verfahren:
Exposition I - Prüfung bei der Grenzkonzentration (siehe Abbildung 1)
↓
Exposition II c - Hauptstudie
↓
Exposition III - Hauptstudie
↓
Exposition IV' - Hauptstudie
↓ oder
Exposition IV - Hauptstudie
Mathematische Aufbereitung der Ergebnisse für das c-x-t-Protokoll
11. Ein c-x-t-Verfahren mit 4 oder 5 Expositionskonzentrationen und fünf Expositionszeiträumen ergibt 20 bzw. 25 Datenpunkte. Anhand dieser Datenpunkte kann die c-x-t-Beziehung durch statistische Analyse berechnet werden (16):
Gleichung 1
Probit(P) = b0 + b1ln C + b2ln t
dabei ist: C = Konzentration; t = Expositionsdauer oder
Gleichung 2
Response = ƒ(Cnt)
dabei ist: n = b1/b2:
Mit der Gleichung 1 kann der LC50-Wert für einen bestimmten Zeitraum (z.B. 4 Stunden, 1 Stunde, 30 Minuten oder jeden Zeitraum innerhalb des geprüften Zeitbereichs) unter Verwendung von P = 5 (50 % Response) berechnet werden. Die Habersche Regel gilt nur, wenn n = 1. Der LC01-Wert kann unter Verwendung von P = 2,67 berechnet werden.
____
a) Liegen keine Informationen über geschlechtsspezifische Empfindlichkeit vor, sind Ratten beider Geschlechter zu verwenden, d. h. 1 Tier/Geschlecht je Konzentration. Auf der Grundlage der vorhandenen Informationen, oder falls sich im Laufe der Exposition zeigt, dass ein Geschlecht empfindlicher ist, werden während der weiteren Prüfung auf jeder Konzentrationsstufe 10 Tiere des empfindlichen Geschlechts (2 Tiere je Konzentration/Zeitpunkt) verwendet.
b) Wird die Verordnung (EG) Nr. 1272/2008 zugrunde gelegt, so betragen die Grenzkonzentrationen für Gase, Dämpfe und Aerosole 20.000 ppm, 20 mg/l bzw. 5 mg/l. Wenn mit Toxizität gerechnet wird oder die Ergebnisse der Vorstudie darauf hindeuten, sollten niedrigere Ausgangskonzentrationen gewählt werden. Bei regulatorischer oder wissenschaftlicher Notwendigkeit können höhere Konzentrationen verwendet werden.
c) Im Idealfall sollten die Tiere erst dann der nächsten Konzentrationsstufe ausgesetzt werden, wenn mit angemessener Gewissheit vom Überleben der zuvor getesteten Tiere ausgegangen werden kann. Dies gibt dem Studienleiter die Möglichkeit, Zielkonzentration und Dauer für die nächste Exposition anzupassen.
d) Die kleinste Dosis (Konzentration x Zeit), die während der Prüfung bei der Ausgangskonzentration (Exposition I) zu einem Todesfall geführt hat, dient als Anhaltspunkt für die Festlegung der nächsten Kombination von Konzentration und Expositionsdauer. Normalerweise wird die Konzentration halbiert (1/2L), und die Tiere werden für eine neue, feiner unterteilte Zeitspanne exponiert, wobei die Expositionszeiten eine geometrische Folge mit dem Faktor 1,4 (√2, siehe Literaturangabe 11) bilden, in deren Mitte diejenige Zeitdauer liegt, die der kleinsten letalen Dosisstufe (Zeit x Konzentration) entspricht, die bei der ersten Exposition beobachtet wurde. In dieser Abbildung 1 wurde bei der Exposition I nach 15 Minuten der erste Todesfall beobachtet. Die Zeiträume der Exposition II liegen daher um 30 Minuten und betragen 15, 21, 30, 42 und 60 Minuten. Es wird dringend empfohlen, die Daten nach den ersten beiden Expositionen wie in der obigen Abbildung grafisch darzustellen und zu prüfen, ob die Beziehung zwischen Konzentration und Zeit einen Winkel von 45° (n = 1) aufweist oder ob die Konzentrations-Zeit-Wirkungsbeziehung weniger steil (z.B. n = 2) oder steiler (z.B. n = 0,8) ist. In den letztgenannten Fällen wird dringend empfohlen, die nächsten Konzentrationen und Expositionszeiträume entsprechend anzupassen.
e) In bestimmten Fällen muss möglicherweise die Konzentration erhöht (2L) und eine neue, noch feiner unterteilte Zeitspanne festgelegt werden, wobei die Expositionszeiten eine geometrische Folge mit dem Faktor 1,4 (√2) bilden, in deren Mitte diejenige Zeitdauer liegt, die der kleinsten letalen Dosis entspricht, die bei der ersten Exposition beobachtet wurde. Die Mindestexpositionsdauer sollte möglichst mehr als 5 Minuten betragen; die Höchstexpositionsdauer sollte 8 Stunden nicht überschreiten."
4. Die Kapitel B.7 und B.8 erhalten folgende Fassung:
"B.7 28-Tage-Toxizitätsstudie mit wiederholter oraler Verabreichung an Nagetieren
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 407 (2008). Die ursprüngliche Prüfrichtlinie 407 wurde 1981 angenommen. 1995 wurde eine überarbeitete Fassung angenommen mit dem Ziel, insbesondere in Bezug auf Neurotoxizität und Immunotoxizität zusätzliche Informationen von den in der Studie verwendeten Tieren zu gewinnen.
2. Die OECD leitete 1998 mit hoher Priorität eine Überarbeitung der bestehenden Prüfrichtlinien und die Ausarbeitung neuer Prüfrichtlinien für Screening und Prüfung potenzieller endokriner Disruptoren ein (8). Ein Aspekt dieser Maßnahme war die Aktualisierung der bestehenden OECD-Prüfrichtlinie für eine '28-Tage-Toxizitätsstudie mit wiederholter oraler Verabreichung an Nagetieren' (TG 407) durch Parameter, mit denen eine endokrine Wirkung von Prüfsubstanzen festgestellt werden kann. Dieses Verfahren wurde in einem umfangreichen internationalen Programm auf Relevanz und Praktikabilität der zusätzlichen Parameter, ihre Leistungsfähigkeit in Bezug auf chemische Stoffe mit (anti)östrogener, (anti)androgener und (anti)thyroider Wirkung, die Intra- und Interlabor-Reproduzierbarkeit und die Interferenz der neuen Parameter mit den zuvor nach der TG 407 vorgesehenen Parametern geprüft. Die dabei gewonnene umfangreiche Datenmenge wurde in einem umfassenden OECD-Bericht zusammengestellt und ausführlich bewertet (9). Diese aktualisierte Prüfmethode B.7 (die der TG 407 entspricht) ist das Ergebnis der im internationalen Prüfprogramm gesammelten Erfahrungen und Erkenntnisse. Mithilfe dieser Prüfmethode können bestimmte endokrin vermittelte Wirkungen in einen Gesamtzusammenhang mit anderen toxikologischen Wirkungen gestellt werden.
Ausgangsüberlegungen und Begrenzungen
3. Bei der Beurteilung und Bewertung der toxischen Merkmale eines chemischen Stoffs kann die orale Toxizität nach wiederholter Verabreichung des Stoffs bestimmt werden, nachdem zunächst durch Prüfungen auf akute Toxizität erste Toxizitätsdaten gewonnen wurden. Diese Prüfmethode ist dazu bestimmt, die Wirkungen auf ein sehr breites Spektrum potenzieller Toxizitätsziele zu untersuchen. Sie gibt Aufschluss über die möglichen Gesundheitsgefahren, einschließlich der Wirkungen auf das Nervensystem, das Immunsystem und das endokrine System, die durch wiederholte Exposition über einen relativ begrenzten Zeitraum auftreten können. In Bezug auf diese speziellen Endpunkte sollte die Methode es ermöglichen, chemische Stoffe mit neurotoxischem Potenzial, die in dieser Hinsicht eingehender untersucht werden sollten, und chemische Stoffe, die die Physiologie der Schilddrüse beeinflussen, zu identifizieren. Sie kann auch Daten über Stoffe liefern, die die Fortpflanzungsorgane männlicher und/oder weiblicher junger, adulter Tiere beeinträchtigen, und Hinweise auf immunologische Wirkungen geben.
4. Die Ergebnisse dieser Prüfmethode B.7 sollten für die Identifizierung von Gefahren und zur Risikobewertung verwendet werden. Die Ergebnisse in Bezug auf die endokrinen Parameter sind im Kontext des 'OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals' (11) zu interpretieren. Die Methode umfasst die Basisstudie zur Prüfung auf Toxizität bei wiederholter Verabreichung, die für chemische Stoffe, bei denen eine 90-Tage-Studie nicht gerechtfertigt ist (z.B. wenn das Produktionsvolumen bestimmte Grenzen nicht überschreitet), oder als Vorstudie zu einer Langzeitstudie verwendet werden kann. Die Expositionsdauer sollte 28 Tage betragen.
5. Das internationale Programm für die Validierung der Parameter, die geeignet sind, möglicherweise eine endokrine Wirkung einer Prüfsubstanz nachzuweisen, hat gezeigt, dass die Qualität der mit dieser Prüfmethode B.7 gewonnenen Daten weitgehend von der Erfahrung des Prüflabors abhängt. Dies gilt insbesondere für die histopathologische Bestimmung von zyklischen Veränderungen der weiblichen Fortpflanzungsorgane und für die Gewichtsbestimmung bei den kleinen hormonabhängigen Organen, die schwierig zu sezieren sind. Es wurde ein Leitfaden zur Histopathologie entwickelt (19), der auf der öffentlichen OECD-Website für Prüfrichtlinien abrufbar ist. Er soll Pathologen bei Untersuchungen helfen und die Empfindlichkeit der Prüfungen verbessern. Es wurde festgestellt, dass verschiedene Parameter auf endokrine Toxizität hindeuten; diese wurden in die Prüfrichtlinie aufgenommen. Andere Parameter, deren Nutzen wegen unzureichender Daten nicht erwiesen ist oder deren Fähigkeit zur Feststellung endokriner Disruptoren im Validierungsprogramm nur unzureichend belegt wurde, werden als fakultative Endpunkte vorgeschlagen (siehe Anlage 2).
6. Die Ergebnisse des Validierungsprozesses deuten darauf hin, dass diese Prüfung nicht empfindlich genug ist, um alle Stoffe mit (anti)androgener oder (anti)östrogener Wirkungsweise zu identifizieren (9). Diese Prüfmethode wird nicht in einer für endokrine Störungen sehr empfindlichen Lebensphase durchgeführt. Während des Validierungsprozesses konnten zwar Substanzen mit schwacher oder starker Wirkung auf die Schilddrüsenfunktion und solche, die das endokrine System über östrogene oder androgene Rezeptoren mehr oder weniger stark beeinflussen, identifiziert werden; in den meisten Fällen konnten Stoffe mit endokriner Wirkung, die diese Rezeptoren nur geringfügig beeinträchtigen, mit der Methode aber nicht erkannt werden. Aus diesem Grund kann sie nicht als Screening-Test für endokrine Aktivität bezeichnet werden.
7. Die Tatsache, dass keine mit diesen Wirkungsweisen verbundenen Effekte festgestellt werden, ist daher kein Nachweis dafür, dass es keine Wirkungen auf das endokrine System gibt. Die Charakterisierung der Substanz in Bezug auf endokrin vermittelte Wirkungen darf sich deshalb nicht allein auf die Ergebnisse dieser Prüfmethode stützen; es ist vielmehr ein WoE-Ansatz anzuwenden, der alle vorhandenen Daten über einen chemischen Stoff zur Bestimmung seiner potenziellen endokrinen Aktivität einbezieht. Aus diesem Grund dürfen sich Regulierungsentscheidungen über endokrine Aktivität (Charakterisierung von Substanzen) nicht allein auf die Ergebnisse aus der Anwendung dieser Prüfmethode stützen, sondern müssen auf einer breiten Grundlage basieren.
8. Bei allen Verfahren, bei denen Tiere verwendet werden, sind die örtlichen Standards der Versuchstierpflege einzuhalten. Die nachstehenden Beschreibungen der Tierpflege und -behandlung sind Mindeststandards; wenn örtliche Bestimmungen strenger sind, gehen diese vor. Die OECD hat einen Leitfaden über die humane Behandlung von Versuchstieren herausgegeben (14).
9. Die verwendeten Begriffe sind in Anlage 1 definiert.
Prinzip der Prüfmethode
10. Die Prüfsubstanz wird mehreren Gruppen von Versuchstieren über einen Zeitraum von 28 Tagen täglich in abgestuften Dosen oral verabreicht, und zwar eine Dosisstufe je Gruppe. Während des Verabreichungszeitraums werden die Tiere täglich sorgfältig auf Toxizitätszeichen beobachtet. Tiere, die im Verlauf der Prüfung sterben, und vorzeitig getötete Tiere werden seziert; die nach Abschluss des Tests überlebenden Tiere werden getötet und ebenfalls seziert. Eine 28-Tage-Studie gibt Aufschluss über die Wirkungen einer wiederholten oralen Exposition und kann Aufschluss darüber geben, ob weitere Studien über längere Zeiträume erforderlich sind. Sie kann auch Informationen über die Wahl der Konzentrationen für Langzeitstudien liefern. Die mit dieser Prüfmethode gewonnenen Daten sollten es ermöglichen, die Toxizität der Prüfsubstanz zu beschreiben, Aufschluss über die Dosis-Wirkungs-Beziehung zu erhalten und den NOAEL (No Observed Adverse Effect Level) zu bestimmen.
Beschreibung der Methode
Auswahl von Versuchstierarten
11. Die bevorzugte Nagetierart ist die Ratte, aber auch andere Nagetierarten sind geeignet. Wenn die in dieser Prüfmethode B.7 genannten Parameter an einer anderen Nagetierart untersucht werden, ist dies ausführlich zu begründen. Es ist zwar biologisch plausibel, dass andere Arten ähnlich auf toxische Stoffe reagieren dürften wie die Ratte, aber die Verwendung kleinerer Arten kann wegen der technisch schwierigeren Sektion kleinerer Organe zu größeren Schwankungen bei den Ergebnissen führen. Im internationalen Validierungsprogramm für den Nachweis von endokrinen Disruptoren wurde nur die Ratte als Versuchstier verwendet. Es sind junge, gesunde, adulte Tiere aus üblicherweise eingesetzten Laborstämmen zu verwenden. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Mit der Dosierung sollte möglichst bald nach dem Absetzen begonnen werden, auf jeden Fall jedoch, bevor die Tiere 9 Wochen alt sind. Bei Beginn der Studie sollten die Gewichtsunterschiede der Tiere möglichst gering sein und ± 20 % des geschlechtsspezifischen Durchschnittsgewichts nicht überschreiten. Wenn eine Studie mit wiederholter oraler Gabe als Vorstudie zu einer Langzeitstudie durchgeführt wird, sollten in beiden Studien vorzugsweise Tiere desselben Stamms und derselben Herkunft verwendet werden.
Haltung und Fütterung
12. Bei allen Verfahren sind die örtlichen Standards der Versuchstierpflege einzuhalten. Die Temperatur im Tierversuchsraum sollte 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und - außer beim Reinigen des Raums - 70 % nicht überschreiten. Angestrebt werden sollte eine Luftfeuchtigkeit von 50-60 %. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung der Prüfsubstanz sichergestellt werden muss, wenn die Prüfsubstanz auf diese Art verabreicht werden soll. Die Tiere sollten in kleinen gleichgeschlechtlichen Gruppen untergebracht werden; sie können auch einzeln gehalten werden, wenn dies wissenschaftlich gerechtfertigt ist. Bei Gruppenhaltung sollten maximal fünf Tiere in einem Käfig untergebracht sein.
13. Das Futter ist regelmäßig auf Schadstoffe zu analysieren. Eine Probe des Futters ist bis zur Fertigstellung des Abschlussberichts aufzubewahren.
Vorbereitung der Tiere
14. Gesunde, junge, adulte Tiere werden randomisiert und den einzelnen Kontroll- bzw. Behandlungsgruppen zugeteilt. Die Käfige sollten so angeordnet werden, dass etwaige Einflüsse der Käfigplatzierung minimiert werden. Die Tiere werden eindeutig gekennzeichnet und vor Beginn der Behandlungsstudie in ihren Käfigen über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt.
Herstellung der Dosen
15. Die Prüfsubstanz wird über eine Schlundsonde, mit der Nahrung oder dem Trinkwasser verabreicht. Die Methode der oralen Verabreichung hängt vom Zweck der Studie und von den physikalischen / chemischen / toxikokinetischen Eigenschaften der Prüfsubstanz ab.
16. Bei Bedarf wird die Prüfsubstanz in einem geeigneten Vehikel gelöst oder suspendiert. Es empfiehlt sich, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspension, dann eine Lösung/Suspension in Öl (z.B. Maisöl) und erst dann eine Lösung in einem anderen Vehikel in Betracht zu ziehen. Bei anderen Vehikeln als Wasser müssen seine toxischen Merkmale bekannt sein. Die Stabilität der Prüfsubstanz im Vehikel sollte bestimmt werden.
Verfahren
Zahl und Geschlecht der Versuchstiere
17. Für jede Dosisstufe sind mindestens zehn Tiere (fünf weibliche und fünf männliche) zu verwenden. Sollen im Verlauf der Prüfung Tiere getötet werden, ist die Zahl der Tiere um die Zahl zu erhöhen, die vor Abschluss der Studie getötet werden sollen. Zur Beobachtung der Reversibilität, der Persistenz oder des verzögerten Auftretens toxischer Wirkungen für mindestens 14 Tage nach der Behandlung ist die Einbeziehung einer zusätzlichen Satellitengruppe von zehn Tieren (fünf je Geschlecht) in der Kontrollgruppe und in der Gruppe mit der höchsten Dosis in Betracht zu ziehen.
Dosierung
18. Im Allgemeinen sind mindestens drei Prüfgruppen und eine Kontrollgruppe zu verwenden; wenn aber angesichts der Beurteilung anderer Daten bei einer Dosis von 1.000 mg pro kg Körpergewicht und Tag keine Wirkungen zu erwarten sind, kann ein Limit-Test durchgeführt werden. Liegen keine entsprechenden Daten vor, kann eine Dosisfindungsstudie (Tiere desselben Stamms und derselben Herkunft) durchgeführt werden, um die zu verwendenden Dosen zu bestimmen. Abgesehen von der Behandlung mit der Prüfsubstanz sollen die Tiere in der Kontrollgruppe unter identischen Bedingungen behandelt werden wie die Versuchstiere in der Prüfgruppe. Wird die Prüfsubstanz mit einem Vehikel verabreicht, muss die Kontrollgruppe das Vehikel im höchsten verwendeten Volumen erhalten.
19. Bei der Wahl der Dosisstufen sind sämtliche für die Prüfsubstanz oder verwandte Stoffe vorliegenden Daten zur Toxizität und (Toxiko-)Kinetik zu berücksichtigen. Die höchste Dosisstufe ist so zu wählen, dass zwar toxische Wirkungen, aber keine Todesfälle oder schweres Leiden hervorgerufen werden. Anschließend ist eine absteigende Folge von Dosisstufen zu wählen, um dosisabhängige Wirkungen und die niedrigste Dosisstufe ohne zu beobachtende unerwünschte Wirkungen (NOAEL) nachzuweisen. Zwei- bis vierfache Abstände erweisen sich häufig als optimale Dosisabstufungen, und meist ist eine zusätzliche vierte Prüfgruppe der Verwendung von sehr großen Dosisabständen (z.B. um mehr als den Faktor 10) vorzuziehen.
20. Bei allgemeiner Toxizität (z.B. vermindertes Körpergewicht, Wirkungen auf Leber, Herz, Lungen oder Nieren usw.) oder anderen Veränderungen, die möglicherweise keine toxischen Reaktionen sind (z.B. verminderte Futteraufnahme, Lebervergrößerung) sind die beobachteten Wirkungen auf immunologische, neurologische oder endokrine Endpunkte mit Vorsicht zu interpretieren.
Limit-Test
21. Verursacht die Prüfung bei einer Dosisstufe von mindestens 1.000 mg/kg Körpergewicht pro Tag bzw. eine entsprechende Konzentration im Futter oder Trinkwasser (in Abhängigkeit vom jeweiligen Körpergewicht) unter Verwendung der für diese Studie beschriebenen Verfahren keine feststellbaren toxischen Wirkungen und ist aufgrund der Daten strukturverwandter Substanzen keine Toxizität zu erwarten, kann auf eine vollständige Studie mit drei Dosisstufen gegebenenfalls verzichtet werden. Der Limit-Test findet allerdings keine Anwendung, wenn die Exposition des Menschen die Prüfung in einer höheren Dosisstufe angezeigt erscheinen lassen.
Verabreichung der Dosen
22. Die Tiere erhalten die Prüfsubstanz an sieben Tagen in der Woche über einen Zeitraum von 28 Tagen. Wird die Prüfsubstanz über eine Sonde verabreicht, so sollte dies in einer einmaligen Dosis unter Verwendung einer Schlundsonde oder einer geeigneten Intubationskanüle erfolgen. Das maximale Flüssigkeitsvolumen, das einem Versuchstier jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 1 ml/100 g Körpergewicht nicht überschreiten, außer bei wässrigen Lösungen, von denen 2 ml/100 g Körpergewicht gegeben werden können. Abgesehen von reizenden oder ätzenden Stoffen, die in der Regel bei höheren Konzentrationen eine verstärkte Wirkung hervorrufen, ist die Variabilität des Prüfvolumens dadurch auf ein Mindestmaß zu reduzieren, dass eine Konzentration gewählt wird, die auf allen Dosisstufen ein konstantes Volumen gewährleistet.
23. Bei mit dem Futter oder dem Trinkwasser verabreichten Stoffen ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfsubstanz die normale Nahrungsaufnahme oder den Wasserhaushalt nicht beeinträchtigen. Wenn die Prüfsubstanz im Futter verabreicht wird, kann entweder eine konstante Konzentration im Futter (ppm) oder eine konstante Dosis, bezogen auf das Körpergewicht des Tieres, verwendet werden; die jeweils gewählte Verfahrensweise muss angegeben werden. Eine mit einer Schlundsonde verabreichte Dosis sollte jeweils zu denselben Tageszeiten gegeben und so angepasst werden, dass eine konstante Dosis in Relation zum Körpergewicht aufrechterhalten wird. Wenn eine Studie mit wiederholter Gabe als Vorstudie zu einer Langzeitstudie durchgeführt wird, sollten die beiden Studien bezüglich des Futters der Tiere vergleichbar sein.
Beobachtungen
24. Die Tiere sollten 28 Tage beobachtet werden. Tiere in einer Satellitengruppe, bei denen Nachfolgebeobachtungen vorgesehen sind, sollten für mindestens weitere 14 Tage ohne Behandlung gehalten werden, um ein verzögertes Auftreten, die Persistenz oder die Reversibilität von toxischen Wirkungen festzustellen.
25. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich erfolgen, vorzugsweise jeweils zur gleichen Zeit und unter Berücksichtigung des Zeitraums nach der Verabreichung, in dem voraussichtlich der Wirkungsgipfel zu erwarten ist. Der Gesundheitszustand der Tiere ist zu dokumentieren. Mindestens zweimal täglich werden alle Tiere auf Erkrankungen oder Todesfälle überprüft.
26. Alle Tiere sollten einmal vor der ersten Exposition (um intraindividuelle Vergleiche zu ermöglichen) und danach mindestens einmal wöchentlich eingehend klinisch untersucht werden. Die Beobachtungen sind außerhalb des Käfigs, in dem die Tiere gehalten werden, in stets gleicher Umgebung und vorzugsweise stets zur gleichen Tageszeit vorzunehmen. Sie sind sorgfältig zu dokumentieren, am besten nach einer speziell vom Prüflabor entwickelten Bewertungsskala. Durch geeignete Maßnahmen ist sicherzustellen, dass die Prüfbedingungen möglichst konstant bleiben und dass die Beobachtungen vorzugsweise von Personen vorgenommen werden, denen die Behandlung der Tiere nicht bekannt ist. Zu achten ist insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, auf Sekrete und Exkrete sowie autonome Aktivitäten (z.B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster). Gang- und Haltungsstörungen, ferner Reaktionen auf den Umgang mit den Tieren sowie etwaige klonische oder tonische Bewegungen, Stereotypien (z.B. übermäßiges Putzen, wiederholte Kreisbewegungen) oder abnormes Verhalten (z.B. Selbstverstümmelung, Rückwärtsgehen) sollten auch dokumentiert werden (2).
27. In der vierten Expositionswoche sollten die sensorische Reaktivität auf Reize verschiedener Art (2) (z.B. akustische, visuelle und propriozeptive Reize) (3)(4)(5), die Greifkraft (6) und die motorische Aktivität (7) bewertet werden. Weitere Einzelheiten zu den möglichen Untersuchungen finden sich in der Literatur. Allerdings können auch andere als dort genannte Verfahren angewendet werden.
28. Die funktionellen Beobachtungen in der vierten Expositionswoche können entfallen, wenn es sich um eine Vorstudie für eine nachfolgende Prüfung auf subchronische Toxizität (90 Tage) handelt. In diesem Fall sollten die funktionellen Beobachtungen im Rahmen dieser Folgestudie vorgenommen werden. Andererseits könnten die Daten über funktionelle Beobachtungen aus der Studie mit wiederholter Verabreichung aber die Wahl der Dosisstufen für eine nachfolgende Prüfung auf subchronische Toxizität erleichtern.
29. In Ausnahmefällen können funktionelle Beobachtungen auch bei Gruppen entfallen, die so starke sonstige Toxizitätsanzeichen aufweisen, dass die Leistungen in Funktionstests dadurch signifikant beeinträchtigt würden.
30. Bei der Nekropsie kann der Östruszyklus aller weiblichen Tiere (fakultativ) durch Vaginalabstriche bestimmt werden. Diese Beobachtungen geben Aufschluss über die Phase des Östruszyklus zum Zeitpunkt der Tötung und erleichtern die histologische Beurteilung östrogenempfindlicher Gewebe [siehe Leitfaden zur Histopathologie (19)].
Körpergewicht und Futter-/Trinkwasseraufnahme
31. Alle Tiere sind mindestens einmal wöchentlich zu wiegen. Die Futteraufnahme wird mindestens wöchentlich gemessen. Wenn die Prüfsubstanz über das Trinkwasser verabreicht wird, wird auch die Wasseraufnahme mindestens einmal wöchentlich gemessen.
Hämatologische Untersuchung
32. Die folgenden hämatologischen Parameter sind am Ende der Prüfung zu bestimmen: Hämatokrit, Hämoglobinkonzentration, Erythrozytenzahl, Retikulozyten, Gesamt- und Differential-Leukozytenzahl, Thrombozytenzahl und Blutgerinnungszeit/-fähigkeit. Wenn die Prüfsubstanz oder ihre möglichen Metaboliten oxidierende Eigenschaften haben oder diese vermutet werden, sollten zusätzlich die Methämoglobinkonzentration und die Heinz-Körper bestimmt werden.
33. Die Blutproben müssen an einer benannten Stelle unmittelbar vor oder bei der Tötung der Tiere entnommen und fachgerecht gelagert werden. In der Nacht vor der Tötung sollten die Tiere kein Futter erhalten 1.
Klinisch-biochemische Untersuchungen
34. Zur Untersuchung der wesentlichen toxischen Wirkungen in Geweben und insbesondere der Wirkungen auf Leber und Nieren sollten klinisch-biochemische Parameter in Blutproben aller Tiere bestimmt werden, die unmittelbar vor oder bei der Tötung der Tiere (mit Ausnahme von Tieren, die moribund aufgefunden und/oder vor Beendigung der Studie getötet werden) entnommen werden. Die Plasma- oder Serumuntersuchungen umfassen die Parameter Natrium, Kalium, Glucose, Gesamtcholesterin, Harnstoff, Kreatinin, Gesamtprotein und Albumin, mindestens zwei Enzyme, die auf hepatozelluläre Wirkungen schließen lassen (wie Alanin-Aminotransferase, Aspartat-Aminotransferase, alkalische Phosphatase, γ-Glutamyltranspeptidase und Glutamatdehydrogenase) sowie Gallensäuren. Die Bestimmung weiterer Enzyme (aus Leber oder anderen Organen) sowie von Bilirubin kann unter bestimmten Umständen ebenfalls wertvolle Hinweise liefern.
35. Optional können in der letzten Woche der Studie am Urin, der zu festgelegten Zeiten gesammelt wird, folgende Analysebestimmungen durchgeführt werden: Aussehen, Volumen, Osmolalität oder spezifisches Gewicht, pH- Wert, Protein, Glucose und Blut/Blutzellen.
36. Darüber hinaus sollten Untersuchungen zur Bestimmung von Plasma- oder Serummarkern für eine allgemeine Gewebsschädigung erwogen werden. Des Weiteren sollten, wenn die bekannten Eigenschaften der Prüfsubstanz im Verdacht stehen, die entsprechenden Stoffwechselprofile zu beeinflussen, die Parameter Calcium, Phosphat, Triglyzeride, spezifische Hormone und Cholinesterase bestimmt werden. Die jeweiligen Parameter sind je nach Prüfsubstanzklasse bzw. von Fall zu Fall zu bestimmen.
37. Wenngleich in der internationalen Bewertung der endokrinen Endpunkte kein klarer Vorteil der Bestimmung von Schilddrüsenhormonen (T3, T4) und TSH nachgewiesen werden konnte, kann es hilfreich sein, Plasma- oder Serumproben zur Messung von T3, T4 und TSH (fakultativ) aufzubewahren, wenn es einen Hinweis auf eine Wirkung auf die Hypophysen-Schilddrüsen-Achse gibt. Diese Proben können zur Lagerung bei - 20 °C eingefroren werden. Die folgenden Faktoren können die Variabilität und die absoluten Konzentrationen der Hormonbestimmungen beeinflussen:
Schilddrüsenaktive Substanzen können durch histopathologische Untersuchungen zuverlässiger identifiziert werden als über die Hormonspiegel.
38. Plasmaproben, die speziell zur Hormonbestimmung vorgesehen sind, sollten immer zur gleichen Tageszeit gewonnen werden. Es wird empfohlen, die durch histopathologische Veränderungen der Schilddrüse verursachten T3-, T4- und TSH-Spiegel zu bestimmen. Die verschiedenen im Handel erhältlichen Assay-Kits können bei der Analyse der Hormonkonzentration unterschiedliche numerische Werte ergeben. Aus diesem Grund können möglicherweise keine Leistungskriterien auf der Grundlage einheitlicher historischer Daten angegeben werden. Die Labors sollten stattdessen anstreben, die Variationskoeffizienten für T3 und T4 unter 25 und für TSH unter 35 zu halten. Alle Konzentrationen sind in ng/ml zu protokollieren.
39. Erweisen sich die Daten aus vorangegangenen Versuchen als ungeeignet, sollte eine Bestimmung hämatologischer und klinisch-biochemischer Parameter vor Versuchsbeginn oder vorzugsweise an einer nicht in die Versuchsgruppen einbezogenen Gruppe von Tieren in Betracht gezogen werden.
Pathologie
Makroskopische Untersuchung
40. Alle an der Studie beteiligten Tiere müssen einer vollständigen, eingehenden makroskopischen Untersuchung unterzogen werden, die die sorgfältige Untersuchung der äußeren Körperoberfläche, aller Körperöffnungen sowie der Schädel-, Brust- und Bauchhöhlen und ihres Inhalts umfasst. Leber, Nieren, Nebennieren, Hoden, Nebenhoden, Prostata und Samenbläschen mit Koagulationsdrüsen als Ganzes, Thymus, Milz, Gehirn und Herz aller Tiere (mit Ausnahme von Tieren, die moribund aufgefunden und/oder vor Beendigung der Studie getötet werden) sind in angemessener Form von anhaftendem Gewebe zu befreien, und ihr Nassgewicht ist so rasch wie möglich nach der Sektion festzustellen, um ein Austrocknen zu verhindern. Bei der Entfernung der anhaftenden Gewebe von der Prostata ist sorgfältig darauf zu achten, die mit Flüssigkeit gefüllten Samenbläschen nicht zu perforieren. Alternativ können Samenbläschen und Prostata auch nach der Fixierung von anhaftendem Gewebe befreit und gewogen werden.
41. Darüber hinaus können fakultativ auch zwei weitere Organe baldmöglichst nach der Sektion gewogen werden, um ein Austrocknen zu verhindern: die Ovarien (Nassgewicht) und der Uterus mit Zervix (Anleitung zur Entfernung und Aufbereitung des Uterusgewebes für die Gewichtsbestimmung in OECD TG 440 (18)).
42. Das Gewicht der Schilddrüse (fakultativ) kann nach der Fixierung bestimmt werden. Anhaftendes Gewebe ist sehr vorsichtig und erst nach der Fixierung zu entfernen, um Gewebeschäden zu vermeiden. Eine Gewebeschädigung könnte die histopathologische Analyse beeinträchtigen.
43. Die folgenden Gewebe sind in dem für die betreffende Gewebeart und die vorgesehene anschließende histopathologische Untersuchung am besten geeigneten Fixierungsmittel aufzubewahren (siehe Nummer 47): alle Gewebe mit makroskopischen Veränderungen, Hirn (typische Regionen einschließlich Cerebrum, Cerebellum und Pons), Rückenmark, Auge, Magen, Dünn- und Dickdarm (mit Peyer'schen-Platten), Leber, Nieren, Nebennieren, Milz, Herz, Thymus, Schilddrüse, Trachea und Lungen (konserviert durch Inflation mit Fixierungsmittel und Immersion), Gonaden (Hoden und Ovarien), akzessorische Geschlechtsorgane (Uterus und Zervix, Nebenhoden, Prostata und Samenbläschen mit Koagulationsdrüsen), Vagina, Harnblase, Lymphknoten [je nach Erfahrung des Labors sollte neben dem proximalsten drainierenden Lymphknoten ein weiterer Lymphknoten entnommen werden (15)], periphere Nerven (N. ischiadicus oder N. tibialis) vorzugsweise in der Nähe des Muskels, Skelettmuskel und Knochen mit Knochenmark (Schnitt oder wahlweise ein frisch fixiertes Knochenmarkaspirat). Es wird empfohlen, die Hoden durch Immersion in Bouin'scher Lösung oder modifizierter Davidson-Lösung zu fixieren (16) (17). Damit das Fixierungsmittel rasch eindringen kann, muss die Tunica albuginea an beiden Enden des Organs vorsichtig und flach mit einer Nadel punktiert werden. Die klinischen und sonstigen Befunde können weitere Gewebeuntersuchungen erforderlich machen. Auch Organe, die aufgrund der bekannten Eigenschaften der Prüfsubstanz als mögliche Zielorgane in Frage kommen, sollten aufbewahrt werden.
44. Die folgenden Gewebe können wichtige Hinweise auf endokrine Wirkungen geben: Gonaden (Ovarien und Hoden), akzessorische Geschlechtsorgane (Uterus mit Zervix, Nebenhoden, Samenbläschen mit Koagulationsdrüsen, Prostata dorsolateral und ventral), Vagina, Hypophose, männliche Brustdrüse, Schilddrüse und Nebennieren. Veränderungen der männlichen Brustdrüsen sind nicht ausreichend dokumentiert, aber dieser Parameter kann sehr empfindlich auf Stoffe mit östrogener Wirkung reagieren. Die Beobachtung von nicht unter Nummer 43 genannten Organen/Geweben ist fakultativ (siehe Anlage 2).
45. Der Leitfaden zur Histopathologie (19) enthält zusätzliche Informationen zur Sektion, Fixierung, Schnittherstellung und Histopathologie endokriner Gewebe.
46. Das internationale Prüfprogramm hat Hinweise darauf ergeben, dass subtile endokrine Wirkungen chemischer Stoffe, die die Homöostase der Geschlechtshormone geringfügig beeinträchtigen können, eher durch die Störung der Synchronisation des Östruszyklus als durch deutliche histopathologische Veränderungen in weiblichen Geschlechtsorganen identifiziert werden können. Wenngleich diese Wirkungen nicht eindeutig nachgewiesen werden konnten, wird empfohlen, Hinweise auf eine mögliche Asynchronie des Östruszyklus bei der Auswertung der histopathologischen Untersuchung von Ovarien (Follikelzellen, Theca-Zellen und Granulosazellen), Uterus, Zervix und Vagina zu berücksichtigen. Falls die Phase des Östruszyklus durch Vaginalabstriche bestimmt wird, kann sie auch in diesen Vergleich einbezogen werden.
Histopathologie
47. Bei allen Tieren der Kontrollgruppe und der Hochdosisgruppe sind die konservierten Organe und Gewebe umfassend histopathologisch zu untersuchen. Diese Untersuchungen sind auch auf die Tiere aller anderen Dosisgruppen auszudehnen, wenn behandlungsbedingte Veränderungen in der Hochdosisgruppe festgestellt werden.
48. Alle makroskopischen Läsionen sind zu untersuchen.
49. Umfasst eine Prüfung auch eine Satellitengruppe, sind bei den Tieren dieser Gruppe die Gewebe und Organe histopathologisch zu untersuchen, bei denen in den Behandlungsgruppen Wirkungen aufgetreten sind.
Daten und Berichterstattung
Daten
50. Es sollten Einzeldaten zur Verfügung gestellt werden. Darüber hinaus sollten alle Daten in Form einer Tabelle zusammengefasst werden, aus der für jede Prüfgruppe folgende Angaben hervorgehen: die Zahl der Tiere bei Beginn der Prüfung und die Zahl der während der Prüfung tot aufgefundenen oder aus Tierschutzgründen getöteten Tiere, ferner der Zeitpunkt des Todes oder der Tötung, die Zahl der Tiere, die Toxizitätszeichen aufweisen, eine Beschreibung der beobachteten Toxizitätszeichen, einschließlich des Zeitpunkts, zu dem die toxischen Wirkungen erstmalig aufgetreten sind, ihrer Dauer und ihres Schweregrads, die Zahl der Tiere mit Läsionen, die Art der Läsionen und ihr Schweregrad sowie der Prozentsatz der von jeder Läsion betroffenen Tiere.
51. Wenn möglich, sollen die numerischen Ergebnisse mittels einer geeigneten und allgemein anerkannten statistischen Methode ausgewertet werden. Durch Vergleiche der Wirkungen in einem Dosisbereich sollte die Anwendung multipler t-Tests vermieden werden. Die Statistikmethoden sind bei der Planung der Studie festzulegen.
52. Für die Qualitätskontrolle wird vorgeschlagen, historische Kontrolldaten zu sammeln und Variationskoeffizienten für numerische Daten zu berechnen, insbesondere für die Parameter, die mit dem Nachweis der Störung des endokrinen Systems zusammenhängen. Diese Daten können für Vergleichszwecke verwendet werden, wenn tatsächliche Studien bewertet werden.
Prüfbericht
53. Der Prüfbericht muss folgende Informationen enthalten:
Prüfsubstanz:
Vehikel (wenn verwendet):
Versuchstiere:
Prüfbedingungen:
Untersuchte fakultative Endpunkte
Ergebnisse:
Erörterung der Ergebnisse.
Schlussfolgerungen.
_____
1) Für eine Reihe von Serum- und Plasmabestimmungen, insbesondere der Glucose, ist eine Futterkarenz der Tiere über Nacht zu empfehlen. Der Hauptgrund dafür ist, dass die bei fehlender Futterkarenz unweigerlich zunehmende Variabilität zu einer Maskierung subtilerer Wirkungen führen und die Interpretation erschweren könnte. Andererseits jedoch kann die nächtliche Futterkarenz den allgemeinen Stoffwechsel der Tiere beeinflussen und, insbesondere in Futterstudien, die tägliche Exposition gegen die Prüfsubstanz beeinträchtigen. Wenn man sich für die nächtliche Futterkarenz entscheidet, sollten die klinisch-biochemischen Parameter nach Durchführung der funktionellen Beobachtungen in Woche 4 der Studie bestimmt werden.
Anlage 1 Definitionen
Androgene Wirkung: die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches androgenes Hormon zu wirken (z.B. Testosteron).
Antiandrogene Wirkung: die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen androgenen Hormons (z.B. Testosteron) in einem Säugetierorganismus zu unterdrücken.
Antiöstrogene Wirkung: die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen östrogenen Hormons (z.B. Östradiol 17ß) in einem Säugetierorganismus zu unterdrücken.
Antithyroide Wirkung: die Fähigkeit einer Chemikalie, die Wirkung eines natürlichen Schilddrüsenhormons (z.B. T 3) in einem Säugetierorganismus zu unterdrücken.
Dosierung: ein allgemeiner Begriff, der die Dosis, ihre Häufigkeit und die Dauer der Verabreichung umfasst.
Dosis: die Menge der verabreichten Prüfsubstanz. Die Dosis wird ausgedrückt als Masse der Prüfsubstanz je Einheit Körpergewicht des Versuchstiers pro Tag (z.B. mg/kg Körpergewicht/Tag) oder als konstante Konzentration im Futter.
Offensichtliche Toxizität: ein allgemeiner Begriff zur Beschreibung deutlicher Toxizitätszeichen nach Verabreichung einer Prüfsubstanz. Diese Zeichen sollten für eine Bewertung der Gefährdung ausreichen und so schwerwiegend sein, dass bei einer Steigerung der verabreichten Dosis die Entwicklung schwerer Toxizitätszeichen und der wahrscheinliche Tod zu erwarten wären.
NOAEL: die Abkürzung für no observed adverse effect level. Dies ist die höchste Dosis, bei der keine schädigenden behandlungsbedingten Wirkungen festgestellt werden.
Östrogene Wirkung: die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches östrogenes Hormon zu wirken (z.B. Östradiol 17ß).
Prüfsubstanz: jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird.
Thyroide Wirkung: die Fähigkeit einer Chemikalie, in einem Säugetierorganismus wie ein natürliches Schilddrüsenhormon (z.B. T3) zu wirken.
Validierung: ein wissenschaftlicher Prozess zur Beschreibung der operationellen Anforderungen und Grenzen einer Prüfmethode und zum Nachweis ihrer Zuverlässigkeit und Eignung für einen bestimmten Zweck.
Anlage 2 Empfohlene Endpunkte für den Nachweis endokriner Disruptoren in dieser Prüfmethode b.7
| Obligatorische Endpunkte | Fakultative Endpunkte |
| Gewicht | |
|
|
| Histopathologie | |
|
|
| Hormonbestimmung | |
| |
Literatur:
1. OECD (Paris, 1992). Chairman's Report of the Meeting of the ad hoc Working Group of Experts on Systemic Short- term and (Delayed) Neurotoxicity.
2. IPCS (1986). Principles and Methods for the Assessment of Neurotoxicity Associated with Exposure to Chemicals. Environmental Health Criteria Document No. 60
3. Tupper DE, Wallace RB (1980). Utility of the Neurologic Examination in Rates. Acta Neurobiol. Exp. 40: 999-1003.
4. Gad SC (1982). A Neuromuscular Screen for Use in Industrial Toxicology. J. Toxicol Environ. Health 9: 691-704.
5. Moser VC, McDaniel KM, Phillips PM (1991). Rat Strain and Stock Comparisons Using a Functional Observational Battery: Baseline Values and Effects of Amitraz. Toxicol. Appl. Pharmacol. 108: 267-283.
6. Meyer OA, Tilson HA, Byrd WC, Riley MT (1979). A Method for the Routine Assessment of Fore- and Hindlimb Grip Strength of Rates and Mice. Neurobehav. Toxicol. 1: 233-236.
7. Crofton KM, Howard JL, Moser VC, Gill MW, Reiter LW, Tilson HA, MacPhail RC (1991). Interlaboratory Comparison of Motor Activity Experiments: Implication for Neurotoxicological Assessments. Neurotoxicol. Teratol. 13: 599- 609.
8. OECD (1998). Report of the First Meeting of the OECD Endocrine Disrupter Testing and Assessment (EDTA) Task Force, 10th-11 March 1998, ENV/MC/CHEM/RA(98)5.
9. OECD. (2006). Report of the Validation of the Updated Test Guideline 407: Repeat Dose 28-day Oral Toxicity Study in Laboratory Rates. Series on Testing and Assessment No 59, ENV/JM/MONO(2006)26.
10. OECD (2002). Detailed Review Paper on the Appraisal of Test Methods for Sex Hormone Disrupting Chemicals. Series on Testing and Assessment No 21, ENV/JM/MONO(2002)8.
11. OECD (2012).Conceptual Framework for Testing and Assessment of Endocrine Disrupting Chemicals. http://www. oecd.org/document/58/0,3343,fr_2649_37407_2348794_1_1_1_37407,00.html
12. OECD (2006). Final Summary report of the meeting of the Validation Management Group for mammalian testing. ENV/JM/TG/EDTA/M(2006)2.
13. OECD. Draft Summary record of the meeting of the Task Force on Endocrine Disrupters Testing and Assessment. ENV/JM/TG/EDTA/M(2006)3.
14. OECD (2000). Guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. Series on Testing and Assessment No 19. ENV/JM/MONO(2000)7.
15. Haley P, Perry R, Ennulat D, Frame S, Johnson C, Lapointe J-M, Nyska A, Snyder PW, Walker D, Walter G (2005). STP Position Paper: Best Practice Guideline for the Routine Pathology Evaluation of the Immune System. Toxicol Pathol 33: 404-407.
16. Hess RA, Moore BJ (1993). Histological Methods for the Evaluation of the Testis. In: Methods in Reproductive Toxicology, Chapin RE and Heindel JJ (eds). Academic Press: San Diego, CA, pp. 52-85.
17. Latendresse JR, Warbrittion AR, Jonassen H, Creasy DM (2002). Fixation of testes and eyes using a modified Davidson's fluid: comparison with Bouin's fluid and conventional Davidson's fluid. Toxicol. Pathol. 30, 524-533.
18. OECD (2007). OECD Guideline for Testing of Chemicals No 440: Uterotrophic Bioassay in Rodents: A short-term screening test for oestrogenic properties.
19. OECD (2009). Guidance Document 106 on Histologic evaluation of Endocrine and Reproductive Tests in Rodents ENV/JM/Mono(2009)11.
B.8 Prüfung auf subakute Toxizität nach Inhalation - 28-Tage-Test
Zusammenfassung
Diese überarbeitete Prüfmethode B.8 wurde entwickelt, um die Toxizität der Prüfsubstanz nach wiederholter Exposition durch Inhalation über einen begrenzten Zeitraum (28 Tage) umfassend zu beschreiben und um Daten für die Bewertung des quantitativen Inhalationsrisikos zu gewinnen. Gruppen von mindestens fünf männlichen und fünf weiblichen Nagern werden über einen Zeitraum von 28 Tagen 6 Stunden am Tag a) der Prüfsubstanz in drei oder mehr Konzentrationsstufen, b) gefilterter Luft (negative Kontrolle) und/oder c) dem Vehikel (Vehikelkontrolle) ausgesetzt. Im Allgemeinen werden die Tiere der Prüfsubstanz an fünf Tagen pro Woche ausgesetzt, aber auch sieben Tage pro Woche sind zulässig. Es werden immer männliche und weibliche Tiere geprüft, aber sie können unterschiedlichen Konzentrationsstufen ausgesetzt werden, wenn bekannt ist, dass ein Geschlecht empfindlicher auf eine bestimmte Prüfsubstanz reagiert als das andere. Bei dieser Methode hat der Studienleiter die Möglichkeit, Satellitengruppen (Reversibilitätsprüfung) aufzunehmen sowie eine bronchoalveoläre Lavage (BAL), neurologische Tests, zusätzliche klinische Pathologieuntersuchungen und histopathologische Untersuchungen durchzuführen, um die Toxizität einer Prüfsubstanz besser beschreiben zu können.
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 412 (2009). Die ursprüngliche Prüfrichtlinie 412 (TG 412) zur subakuten Inhalation wurde 1981 angenommen (1). Diese Prüfmethode B.8 (die der überarbeiteten TG 412 entspricht) wurde aktualisiert, um dem neuesten Stand der Wissenschaft Rechnung zu tragen und derzeitige und künftige Regulierungsanforderungen zu erfüllen.
2. Die Methode ermöglicht die Beschreibung der schädlichen Wirkungen nach wiederholter täglicher inhalativer Exposition gegen eine Prüfsubstanz für einen Zeitraum von 28 Tagen. Die in Prüfungen auf subakute Toxizität nach Inhalation über 28 Tage gewonnenen Daten können für quantitative Risikobewertungen verwendet werden [wenn im Anschluss keine Prüfung auf subchronische Toxizität nach Inhalation über 90 Tage erfolgt (Kapitel B.29 dieses Anhangs)]. Die Daten können auch Informationen für die Wahl der Konzentrationen für längerfristige Studien wie die Prüfung auf subchronische Toxizität nach Inhalation über 90 Tage liefern. Diese Prüfmethode ist nicht speziell für die Prüfung von Nanomaterialien bestimmt. Die im Zusammenhang mit dieser Prüfmethode verwendeten Begriffe werden am Ende dieses Kapitels und im Guidance Document 39 (2) definiert.
Ausgangsüberlegungen
3. Um die Qualität der Studie zu verbessern und möglichst wenig Versuchstiere zu verwenden, sollte das Prüflabor vor Durchführung der Studie alle verfügbaren Informationen über die Prüfsubstanz berücksichtigen. Für die Auswahl der am besten geeigneten Prüfkonzentrationen könnten u. a. Informationen wie die Identität, die chemische Struktur und die physikalisch-chemischen Eigenschaften der Prüfsubstanz, Ergebnisse jeglicher In- vitro- oder In-vivo-Toxizitätsprüfungen, vorgesehene Verwendungen und die Möglichkeit der Exposition des Menschen, (Q)SAR-Daten und toxikologische Daten über strukturverwandte Substanzen sowie Daten aus Versuchen zur Prüfung der akuten Toxizität nach Inhalation herangezogen werden. Falls Neurotoxizität erwartet oder im Verlauf der Studie beobachtet wird, kann der Studienleiter beschließen, geeignete Untersuchungen wie eine FOB (functional observational battery) und die Messung der motorischen Aktivität aufzunehmen. Obwohl die Expositionszeit bei bestimmten Untersuchungen ein kritischer Aspekt sein kann, darf die Durchführung dieser zusätzlichen Versuche die Auslegung der Hauptstudie nicht beeinträchtigen.
4. Verdünnungen ätzender oder reizender Prüfsubstanzen können in Konzentrationen geprüft werden, die den gewünschten Toxizitätsgrad erzielen [siehe GD 39 (2)]. Wenn Versuchstiere diesen Stoffen ausgesetzt werden, sollten die Zielkonzentrationen so niedrig sein, dass sie keine starken Schmerzen oder Leiden verursachen; sie sollten aber ausreichen, um die Konzentrations-Wirkungs-Kurve so zu erweitern, dass das regulatorische und wissenschaftliche Ziel der Prüfung erreicht wird. Diese Konzentrationen sollten von Fall zu Fall festgelegt werden, möglichst auf Basis einer entsprechend ausgelegten Dosisfindungsstudie, die Informationen über den kritischen Endpunkt, eine etwaige Reizschwelle und den Zeitpunkt des Einsetzens der Wirkung liefert (siehe Nummern 11, 12 und 13). Die Wahl der Konzentration ist zu begründen.
5. Moribunde Tiere oder Tiere, die Anzeichen starker und andauernder Qualen zeigen, sollten tierschutzgerecht getötet werden. Moribunde Tiere werden auf die gleiche Weise gewertet wie während des Tests gestorbene Tiere. Kriterien für die Entscheidung, moribunde oder schwer leidende Tiere zu töten, sowie Hinweise zur Erkennung des absehbaren oder bevorstehenden Todes sind Gegenstand des OECD Guidance Document on Humane Endpoints (3).
Beschreibung der Methode
Auswahl von Versuchstierarten
6. Es sind junge, gesunde, adulte Nagetiere aus üblicherweise eingesetzten Laborstämmen zu verwenden. Bevorzugtes Versuchstier ist die Ratte. Wird eine andere Tierart eingesetzt, ist dies zu begründen.
Vorbereitung der Tiere
7. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Am Tag der Randomisierung sollten die jungen, adulten Tiere 7 bis 9 Wochen alt sein; ihr Körpergewicht sollte innerhalb von ± 20 % des mittleren Gewichts für jedes Geschlecht liegen. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert und vor Beginn der Prüfung für einen Zeitraum von mindestens fünf Tagen in ihren Käfigen an die Laborbedingungen gewöhnt.
Tierhaltung
8. Um die Beobachtungen zu erleichtern und Verwechslungen auszuschließen, sollten die Tiere möglichst mit einem subkutanen Transponder einzeln gekennzeichnet werden. Die Temperatur in dem Raum, in dem die Versuchstiere gehalten werden, sollte 22 ± 3 °C betragen. Die relative Luftfeuchtigkeit sollte im Idealfall im Bereich zwischen 30 und 70 % liegen; bei Verwendung von Wasser als Vehikel könnte dies jedoch unmöglich sein. Die Tiere sollten vor und nach den Expositionen im Allgemeinen nach Geschlecht und Konzentration in Käfigen gruppiert werden, wobei aber die Anzahl der Tiere pro Käfig noch eine genaue Beobachtung der einzelnen Tiere ermöglichen muss und Verluste aufgrund von Kannibalismus oder Kämpfen minimiert werden sollten. Wenn die Tiere der Prüfsubstanz nur mit der Nase ausgesetzt werden sollen, müssen sie möglicherweise an die Restrainer gewöhnt werden. Die Restrainer sollten die Tiere weder körperlich noch in Bezug auf Wärme oder Fixierung übermäßig beeinträchtigen. Die Fixierung kann physiologische Endpunkte wie Körpertemperatur (Hyperthermie) und/oder das Atemminutenvolumen beeinflussen. Wenn generische Daten zeigen, dass keine derartigen Veränderungen in nennenswertem Ausmaß vorkommen, ist eine Eingewöhnung an die Restrainer nicht erforderlich. Bei der Ganzkörperexposition gegen ein Aerosol sollten die Tiere während der Exposition einzeln untergebracht sein, damit sie die Prüfsubstanz nicht durch das Fell ihrer Käfiggenossen filtriert einatmen. Außer während der Exposition kann herkömmliches und zertifiziertes Labortierfutter bei uneingeschränkter Versorgung mit Trinkwasser verwendet werden. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
Inhalationskammern
9. Bei der Auswahl einer Inhalationskammer sind die Art der Prüfsubstanz und der Gegenstand der Prüfung zu berücksichtigen. Das bevorzugte Verfahren ist die 'Nose-only'-Exposition (dieser Begriff umfasst 'nur Kopf', 'nur Nase' oder 'nur Schnauze'). Für die Untersuchung von Flüssigkeits- oder Feststoffaerosolen und für Dämpfe, die zu Aerosolen kondensieren können, wird im Allgemeinen die 'Nose-only'-Exposition bevorzugt. Besondere Ziele der Untersuchung können möglicherweise mit einer Ganzkörperexposition besser erreicht werden, doch dies sollte im Prüfbericht begründet werden. Um bei Verwendung einer Ganzkörperkammer die Stabilität der Atmosphäre sicherzustellen, sollte das 'Gesamtvolumen' der Versuchstiere 5 % des Volumens der Kammer nicht übersteigen. Die Grundzüge der 'Nose-only'- und der Ganzkörperexposition sowie ihre jeweiligen Vor- und Nachteile sind in GD 39 (2) beschrieben.
Toxizitätsstudien
Grenzkonzentrationen
10. Anders als bei Studien zur akuten Toxizität gibt es bei Prüfungen auf subakute Toxizität nach Inhalation über 28 Tage keine festgelegten Grenzkonzentrationen. Bei der Festlegung der maximalen Prüfkonzentration ist Folgendes zu beachten: 1) die höchste erreichbare Konzentration, 2) das maximale Expositionsniveau von Menschen ('worst case), 3) die Notwendigkeit, eine ausreichende Sauerstoffversorgung aufrechtzuerhalten, und/oder 4) Tierschutzerwägungen. Gibt es keine auf Daten basierenden Grenzwerte, können die akuten Grenzwerte der Verordnung (EG) Nr. 1272/2008 (13) zugrunde gelegt werden (d. h. bis zu einer Höchstkonzentration von 5 mg/l bei Aerosolen, 20 mg/l bei Dämpfen und 20.000 ppm bei Gasen); siehe GD 39 (2). Wenn diese Grenzen bei der Prüfung von Gasen oder hochflüchtigen Prüfsubstanzen (z.B. Kältemitteln) überschritten werden müssen, ist dies zu begründen. Die Grenzkonzentration muss eine eindeutige Toxizität hervorrufen, ohne den Tieren übermäßigen Stress zu bereiten oder ihre Lebensdauer zu beeinträchtigen (3).
Dosisfindungsstudie
11. Vor Beginn der Hauptstudie muss möglicherweise eine Dosisfindungsstudie durchgeführt werden. Diese ist umfassender als eine Vorstudie, weil sie nicht auf die Wahl der Konzentration begrenzt ist. Die in einer Dosisfindungsstudie gewonnenen Erkenntnisse können zu einer erfolgreichen Hauptstudie führen. Sie kann z.B. technische Informationen zu den Analysemethoden, zur Partikelgröße, zur Erkennung toxischer Mechanismen, klinische Pathologiedaten und histopathologische Daten sowie Schätzungen möglicher NOAEL- und MTC-Konzentrationen in einer Hauptstudie liefern. Der Studienleiter kann entscheiden, mithilfe der Dosisfindungsstudie die Schwelle für die Reizung der Atemwege (z.B. durch histopathologische Untersuchung der Atemwege, Lungenfunktionsprüfung oder bronchoalveoläre Lavage), die höchste Konzentration, die von den Tieren ohne übermäßigen Stress toleriert wird, und die Parameter, die die Toxizität der Prüfsubstanz am besten beschreiben, zu identifizieren.
12. Eine Dosisfindungsstudie kann eine oder mehrere Konzentrationsstufen umfassen. Auf jeder Konzentrationsstufe sollten höchstens drei männliche und drei weibliche Tiere der Prüfsubstanz ausgesetzt werden. Eine Dosisfindungsstudie sollte mindestens fünf Tage und im Allgemeinen nicht mehr als 14 Tage dauern. Die Wahl der Konzentrationen für die Hauptstudie ist im Prüfbericht zu begründen. Ziel der Hauptstudie ist es, nachzuweisen, dass eine Beziehung zwischen der Konzentration und der Wirkung besteht, die am voraussichtlich empfindlichsten Endpunkt auftritt. Die niedrigste Konzentration sollte im Idealfall eine Konzentration sein, bei der keine zu beobachtenden schädlichen Wirkungen auftreten, und die höchste Konzentration sollte eine eindeutige Toxizität hervorrufen, ohne den Tieren übermäßigen Stress zu bereiten oder ihre Lebensdauer zu beeinträchtigen (3).
13. Bei der Auswahl der Konzentrationsstufen für die Dosisfindungsstudie sollten alle verfügbaren Informationen berücksichtigt werden, einschließlich der Struktur-Wirkungs-Beziehungen und der Daten über ähnliche Stoffe (siehe Nummer 3). Eine Dosisfindungsstudie kann bestätigen/widerlegen, welche Endpunkte nach mechanistischen Kriterien als die empfindlichsten Endpunkte angesehen werden, z.B. die Cholinesterasehemmung durch Organophosphate, die Methämoglobinbildung durch für Erythrozyten toxische Stoffe, Schilddrüsenhormone (T3, T4) im Fall von thyrotoxischen Stoffen, Proteine, LDH oder Neutrophile in bronchoalveolärer Lavage im Fall schwach löslicher unschädlicher Partikel oder lungenreizender Aerosole.
Hauptstudie
14. Die Hauptstudie zur Prüfung auf subakute Toxizität umfasst im Allgemeinen drei Konzentrationsstufen sowie, falls erforderlich, eine gleichzeitige negative (Luft-)Kontrolle und/oder eine Vehikelkontrolle (siehe Nummer 17). Die Festlegung der geeigneten Expositionsstufen sollte sich auf alle verfügbaren Daten stützen, einschließlich der Ergebnisse systemischer Toxizitätsprüfungen, des Metabolismus und der Kinetik (hohe Konzentrationsstufen, die kinetische Prozesse sättigen, sind zu vermeiden). Jede Prüfgruppe umfasst mindestens zehn Nagetiere (fünf Männchen und fünf Weibchen), die der Prüfsubstanz für einen Zeitraum von 4 Wochen an fünf Tagen in der Woche jeweils 6 Stunden pro Tag ausgesetzt werden (Gesamtdauer der Prüfung 28 Tage). Die Tiere können auch an sieben Tagen in der Woche exponiert werden (z.B. wenn inhalierte Arzneimittel geprüft werden). Ist bekannt, dass ein Geschlecht empfindlicher auf eine bestimmte Prüfsubstanz reagiert, können die Geschlechter unterschiedlichen Konzentrationsstufen ausgesetzt werden, um die Konzentrations-Wirkungs-Beziehung genauer zu bestimmen (siehe Nummer 15). Wenn für eine 'Nose-only'-Exposition andere Nagetierarten als Ratten verwendet werden, kann die maximale Expositionsdauer angepasst werden, um artenspezifisches Leiden zu minimieren. Eine Expositionsdauer von weniger als 6 Stunden/Tag oder die Notwendigkeit einer Ganzkörperexpositionsstudie mit Langzeitexposition (z.B. 22 Stunden/Tag) ist zu begründen [siehe GD 39 (2)]. Während der Exposition sollte kein Futter verabreicht werden, es sei denn die Exposition dauert länger als 6 Stunden. Wasser kann während einer Ganzkörperexposition jederzeit angeboten werden.
15. Die gewählten Zielkonzentrationen sollen es ermöglichen, die Zielorgane zu identifizieren und eine deutliche Konzentrations-Wirkungs-Beziehung zu belegen:
Satellitenstudie (Reversibilität)
16. Es kann eine Satellitenstudie durchgeführt werden, um die Tiere für einen angemessenen Zeitraum nach der Behandlung (mindestens 14 Tage) auf Reversibilität, Persistenz oder ein verzögertes Auftreten von Toxizität zu beobachten. Satellitengruppen bestehen aus fünf männlichen und fünf weiblichen Tieren, die der Prüfsubstanz gleichzeitig mit den Versuchstieren der Hauptstudie ausgesetzt werden. Dabei sollten sie der Prüfsubstanz auf der höchsten Konzentrationsstufe ausgesetzt werden; erforderlichenfalls sollte es auch gleichzeitige Luft- und/oder Vehikelkontrollen geben (siehe Nummer 17).
Kontrolltiere
17. Eine gleichzeitige negative (Luft-)Kontrollgruppe ist genauso zu behandeln wie die Prüfgruppe, außer dass die Tiere nicht der Prüfsubstanz, sondern gefilterter Luft ausgesetzt werden. Wenn die Prüfatmosphäre mithilfe von Wasser oder einem anderen Stoff erzeugt wird, ist statt der negativen (Luft-)Kontrollgruppe eine Vehikelkontrollgruppe zu verwenden. Wenn möglich sollte Wasser als Vehikel benutzt werden. In diesem Fall sind die Tiere Luft mit derselben relativen Luftfeuchtigkeit auszusetzen wie die exponierten Gruppen. Das geeignete Vehikel ist auf der Grundlage einer geeigneten Vorstudie oder von historischen Daten auszuwählen. Ist die Toxizität eines Vehikels unklar, kann der Studienleiter beschließen, sowohl eine negative (Luft-)Kontrolle als auch eine Vehikelkontrolle zu verwenden; hiervon wird jedoch dringend abgeraten. Wenn historische Daten belegen, dass ein Vehikel nicht toxisch ist, besteht keine Notwendigkeit für eine negative (Luft-)Kontrollgruppe und es sollte nur eine Vehikelgruppe verwendet werden. Ergibt eine Vorstudie einer in einem Vehikel formulierten Prüfsubstanz, dass keine Toxizität vorliegt, ist das Vehikel folglich in der geprüften Konzentration nicht toxisch, und diese Vehikelkontrolle sollte verwendet werden.
Expositionsbedingungen
Verabreichung der Konzentrationen
18. Die Tiere werden der Prüfsubstanz in Form von Gas, Dampf, Aerosol oder einer Kombination dieser Formen ausgesetzt. Der zu prüfende Aggregatzustand hängt von den physikalisch-chemischen Eigenschaften der Prüfsubstanz, der gewählten Konzentration und/oder der physikalischen Form ab, in der die Prüfsubstanz bei der Handhabung und Verwendung am wahrscheinlichsten vorliegt. Hygroskopische und chemisch reaktive Prüfsubstanzen sollten bei geringer Luftfeuchtigkeit geprüft werden. Dabei ist darauf zu achten, dass keine explosionsfähigen Konzentrationen erzeugt werden. Bei Partikeln kann die Partikelgröße durch mechanische Prozesse verringert werden. GD 39 (2) enthält nähere Hinweise.
Partikelgrößenverteilung
19. Bei allen Aerosolen und bei Dämpfen, die zu Aerosolen kondensieren können, sollte die Partikelgröße bestimmt werden. Damit alle relevanten Regionen der Atemwege der Prüfsubstanz ausgesetzt werden, werden mittlere aerodynamische Massendurchmesser (Mass Median Aerodynamic Diameter - MMAD) von 1 bis 3 μm mit einer geometrischen Standardabweichung (σg) von 1,5 bis 3,0 empfohlen (4). Wenngleich nach Möglichkeit versucht werden sollte, diese Werte zu erreichen, ist Fachwissen erforderlich, falls sie nicht erzielt werden können. Metalldampfpartikel können z.B. unter diesen Werten liegen, geladene Partikel und Fasern dagegen können diese Werte überschreiten.
Vorbereitung der Prüfsubstanz in einem Vehikel
20. Im Idealfall sollte die Prüfsubstanz ohne ein Vehikel geprüft werden. Wenn für die Erzeugung einer geeigneten Prüfsubstanzkonzentration oder Partikelgröße ein Vehikel verwendet werden muss, ist Wasser zu bevorzugen. Wird eine Prüfsubstanz in einem Vehikel gelöst, so ist seine Stabilität nachzuweisen.
Überwachung der Expositionsbedingungen
Luftstrom in der Inhalationskammer
21. Der Luftstrom durch die Kammer sollte während jeder Exposition sorgfältig geregelt, kontinuierlich überwacht und mindestens stündlich protokolliert werden. Die Echtzeit-Überwachung der Konzentration (oder zeitliche Stabilität) der Prüfatmosphäre ist eine integrale Messung aller dynamischen Parameter und gibt indirekt die Möglichkeit, alle relevanten dynamischen Inhalationsparameter zu messen. Wenn die Konzentration in Echtzeit überwacht wird, kann die Frequenz der Messung der Luftströme auf eine einzige Messung je Exposition und Tag reduziert werden. Es sollte besonders darauf geachtet werden, das erneute Einatmen in 'Nose-only'-Expositionskammern zu vermeiden. Die Sauerstoffkonzentration sollte mindestens 19 % betragen, und die Kohlendioxidkonzentration sollte 1 % nicht überschreiten. Gibt es Grund zu der Annahme, dass diese Werte nicht eingehalten werden können, sind die Sauerstoff- und Kohlendioxidkonzentrationen zu messen. Wenn die Messungen am ersten Expositionstag die richtigen Werte dieser Gase bestätigen, sollten keine weiteren Messungen erforderlich sein.
Temperatur und relative Luftfeuchtigkeit in der Inhalationskammer
22. Die Temperatur in der Inhalationskammer sollte 22 ± 3 °C betragen. Sowohl bei der 'Nose-only'- als auch bei der Ganzkörperexposition sollte die relative Luftfeuchtigkeit im Atembereich der Tiere kontinuierlich überwacht und während jeder Exposition möglichst stündlich dokumentiert werden. Die relative Luftfeuchtigkeit sollte möglichst zwischen 30 und 70 % liegen, was jedoch möglicherweise nicht erreichbar ist (z.B. bei der Prüfung von wasserbasierten Mischungen) oder wegen chemischer Interferenz mit der Prüfmethode nicht gemessen werden kann.
Prüfsubstanz: nominale Konzentration
23. Die nominale Konzentration in der Expositionskammer sollte möglichst berechnet und protokolliert werden. Die nominale Konzentration ist die Masse der erzeugten Prüfsubstanz dividiert durch das Gesamtvolumen der durch die Inhalationskammer geleiteten Luft. Sie wird nicht zur Beschreibung der Exposition der Tiere verwendet; vielmehr gibt ein Vergleich der nominalen Konzentration und der tatsächlichen Konzentration Aufschluss über die Effizienz des Prüfsystems bei der Erzeugung der Prüfkonzentration und kann daher für die Aufdeckung von Problemen bei dieser Erzeugung verwendet werden.
Prüfsubstanz: tatsächliche Konzentration
24. Die tatsächliche Konzentration ist die Konzentration der Prüfsubstanz im Atembereich der Tiere in einer Inhalationskammer. Die tatsächlichen Konzentrationen können entweder durch spezifische Methoden (z.B. direkte Probenahme, adsorptive Methoden oder chemische Reaktionsverfahren mit anschließender analytischer Charakterisierung) oder durch unspezifische Methoden wie Gravimetrie bestimmt werden. Die gravimetrische Methode ist lediglich für Aerosole mit nur einem Bestandteil in Pulverform oder Aerosole von Flüssigkeiten mit geringer Flüchtigkeit akzeptabel und sollte sich auf geeignete, vor der Studie zu erstellende und für die Prüfsubstanz spezifische Beschreibungen stützen. Die Konzentration von Aerosolen mit mehreren Bestandteilen in Pulverform kann ebenfalls gravimetrisch bestimmt werden. Hierzu muss jedoch mit Analysedaten belegt werden, dass die Schwebstoffe eine ähnliche Zusammensetzung haben wie das Ausgangsmaterial. Liegen diese Angaben nicht vor, muss die Prüfsubstanz (im Idealfall im Schwebezustand) möglicherweise im Verlauf der Studie in regelmäßigen Abständen neu analysiert werden. Bei aerosolisierten Agenzien, die verdunsten oder sublimieren können, sollte gezeigt werden, dass alle Phasen von der gewählten Methode erfasst wurden.
25. Für die gesamte Dauer der Studie sollte möglichst eine Partie der Prüfsubstanz verwendet werden; die Probe sollte unter Bedingungen aufbewahrt werden, die ihre Reinheit, Homogenität und Stabilität gewährleisten. Die Prüfsubstanz sollte vor Beginn der Studie mit Angaben zur Reinheit und, falls technisch machbar, zur Identität sowie zu den Mengen identifizierter Schadstoffe und Verunreinigungen beschrieben werden. Hierzu können unter anderem die folgenden Daten verwendet werden: Retentionszeit und relative Peakfläche, durch Massenspektrometrie oder Gaschromatographie bestimmtes Molekulargewicht oder andere Werte. Das Prüflabor ist zwar nicht für die Identität der Probe verantwortlich, doch es kann ratsam sein, dass es die Beschreibung des Auftraggebers zumindest in gewissen Grenzen (z.B. Farbe, physikalische Beschaffenheit usw.) überprüft.
26. Die Expositionsatmosphäre ist so konstant wie möglich zu halten. Um die Stabilität der Expositionsbedingungen nachzuweisen, kann ein Echtzeitüberwachungsgerät verwendet werden, z.B. ein Aerosol-Photometer für Aerosole oder ein Gesamtkohlenwasserstoff-Analysator (THC) für Dämpfe. Die tatsächliche Konzentration in der Kammer sollte an jedem Expositionstag für jede Expositionsstufe mindestens dreimal gemessen werden. Falls dies wegen geringer Luftdurchflussraten oder niedriger Konzentrationen nicht möglich ist, reicht eine Probe je Expositionsperiode. Im Idealfall sollte diese Probe dann über die gesamte Expositionszeit gewonnen werden. Die einzelnen Proben der Konzentration in der Kammer sollten bei Gasen und Dämpfen nicht mehr als ± 10 % und bei Flüssig- oder Feststoffaerosolen nicht mehr als ± 20 % von der mittleren Kammerkonzentration abweichen. Die Zeit bis zum Erreichen eines Gleichgewichts in der Kammer (t95) ist zu berechnen und zu dokumentieren. Die Expositionsdauer erstreckt sich über den Zeitraum, in dem die Prüfsubstanz erzeugt wird. Dies schließt die Zeiten zur Erreichung des Gleichgewichts in der Kammer (t95) und zum Abbau der Konzentrationen ein. GD 39 (2) enthält Hinweise zur Einschätzung von t95.
27. Bei sehr komplexen Mischungen aus Gasen/Dämpfen und Aerosolen (z.B. Verbrennungsatmosphären und Prüfsubstanzen, die aus hierzu bestimmten Endverbraucherprodukten/-geräten gesprüht werden) kann sich jede Phase in einer Inhalationskammer anders verhalten. Daher sollte mindestens eine Indikatorsubstanz (Analyt), normalerweise der wichtigste Wirkstoff in der Mischung, von jeder Phase (Gas/Dampf und Aerosol) ausgewählt werden. Wenn die Prüfsubstanz eine Mischung ist, sollte die Analysekonzentration für die gesamte Mischung und nicht nur für den Wirkstoff oder die Indikatorsubstanz (Analyt) dokumentiert werden. Weitere Informationen zu tatsächlichen Konzentrationen sind in GD 39 (2) zu finden.
Prüfsubstanz: Partikelgrößenverteilung
28. Die Partikelgrößenverteilung von Aerosolen sollte auf jeder Konzentrationsstufe mindestens wöchentlich mit einem Kaskaden-Impaktor oder einem anderen Messgerät wie einem APS bestimmt werden. Kann nachgewiesen werden, dass die mit einem Kaskaden-Impaktor und dem alternativen Messgerät erzielten Ergebnisse gleichwertig sind, so kann das alternative Instrument während der gesamten Studie verwendet werden.
29. Parallel zum Hauptinstrument ist ein zweites Gerät wie ein Gravimetriefilter oder eine Gaswaschflasche zu verwenden, um den Abscheidegrad des Hauptinstruments zu bestätigen. Die durch die Partikelgrößenanalyse bestimmte Massenkonzentration sollte innerhalb vertretbarer Grenzen um die durch die Filteranalyse bestimmte Massenkonzentration liegen [siehe GD 39 (2)]. Wenn die Gleichwertigkeit bei allen geprüften Konzentrationen zu Beginn der Studie nachgewiesen werden kann, kann auf weitere bestätigende Messungen verzichtet werden. Aus Tierschutzgründen sollten Vorkehrungen getroffen werden, um unklare Daten zu minimieren, die dazu führen könnten, dass eine Studie wiederholt werden muss.
30. Wenn die Möglichkeit besteht, dass Dampfkondensation zur Bildung eines Aerosols führen kann, oder wenn in einer Dampfatmosphäre mit dem Potenzial für gemischte Phasen Partikel nachgewiesen werden, sollte eine Partikelgrößenbestimmung für Dämpfe vorgenommen werden.
Beobachtungen
31. Die Tiere sollten vor, während und nach der Exposition auf klinische Zeichen beobachtet werden. Je nach Reaktion der Tiere während der Exposition können häufigere Beobachtungen angezeigt sein. Wenn die Beobachtung der Tiere durch die Verwendung von Restrainern, wegen schlecht beleuchteter Ganzkörperkammern oder getrübter Atmosphäre erschwert ist, sind die Tiere nach der Exposition sorgfältig zu beobachten. Durch Beobachtungen vor der Exposition am folgenden Tag kann beurteilt werden, ob toxische Wirkungen sich zurückgebildet oder verschlimmert haben.
32. Sämtliche Beobachtungen werden in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Wenn Tiere aus humanen Gründen getötet werden oder ihr Tod festgestellt wird, sollte der Todeszeitpunkt so genau wie möglich registriert werden.
33. Die Beobachtungen der Tiere sollten sich insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, des Atmungs- und Kreislaufsystems, des Nervensystems sowie auf Somatomotorik und Verhaltensmuster erstrecken. Besonderes Augenmerk ist auf Tremor, Konvulsionen, Salivation, Diarrhö, Lethargie, Schlaf und Koma zu richten. Die Messung der Rektaltemperatur kann zusätzliche Belege für mit der Behandlung oder Unterbringung zusammenhängende Reflex-Bradypnoe oder Hypo-/Hyperthermie liefern. Darüber hinaus können zusätzliche Aspekte wie Kinetik, Biomonitoring, Lungenfunktion, Retention schlecht löslicher Stoffe, die im Lungengewebe akkumulieren, und Verhaltensstörungen in das Studienprotokoll aufgenommen werden.
Körpergewicht
34. Das Körpergewicht der einzelnen Tiere sollte kurz vor der ersten Exposition (Tag 0), danach zweimal wöchentlich (z.B. freitags und montags, um die Erholung während eines expositionsfreien Wochenendes nachzuweisen, oder in einem Zeitintervall, das die Beurteilung der systemischen Toxizität ermöglicht) und zum Zeitpunkt des Todes oder der Tötung dokumentiert werden. Treten in den ersten zwei Wochen keine Wirkungen auf, kann das Körpergewicht während der restlichen Studiendauer wöchentlich gemessen werden. Satellitentiere (Reversibilitätsprüfung) (falls verwendet) sollten während der gesamten Erholungsphase weiterhin wöchentlich gewogen werden. Am Ende der Studie sollten alle Tiere kurz vor der Tötung gewogen werden, um eine objektive Berechnung der Organ-Körpergewicht-Verhältnisse zu ermöglichen.
Futter- und Wasseraufnahme
35. Die Futteraufnahme sollte wöchentlich gemessen werden. Auch die Wasseraufnahme kann gemessen werden.
Klinische Pathologie
36. An allen Tieren, auch an Kontroll- und Satellitentieren (Reversibilitätprüfung), sollten klinische Pathologieuntersuchungen durchgeführt werden, wenn sie getötet werden. Der Zeitraum zwischen dem Ende der Exposition und der Blutentnahme ist zu protokollieren, insbesondere wenn der betreffende Endpunkt rasch zu seinem ursprünglichen Wert zurückkehrt. Für Parameter mit einer kurzen Plasmahalbwertszeit (z.B. COHb, CHE und MetHb) ist die Probenahme nach Ende der Exposition angezeigt.
37. In Tabelle 1 sind die im Allgemeinen für alle Toxikologiestudien erforderlichen klinischen Pathologieparameter aufgeführt. In der Regel ist eine Urinanalyse nicht notwendig, kann aber durchgeführt werden, wenn sie wegen erwarteter oder festgestellter Toxizität für nützlich gehalten wird. Der Studienleiter kann beschließen, zusätzliche Parameter zu bestimmen, um die Toxizität einer Prüfsubstanz genauer zu beschreiben (z.B. Cholinesterase, Lipide, Hormone, Säure-Basen-Gleichgewicht, Methämoglobin oder Heinz-Körper, Creatin-Kinase, Verhältnis von myeloiden zu erythroiden Zellen, Troponin, arterielle Blutgase, Lactatdehydrogenase, Sorbitdehydrogenase, Glutamatdehydrogenase und γ-Glutamyltranspeptidase).
Tabelle 1: Klinische Standardpathologieparameter
| Hämatologische Untersuchung | |
| Erythrozytenzahl
Hämatokrit Hämoglobinkonzentration Mittleres korpuskuläres Hämoglobin Mittleres Erythrozyteneinzelvolumen Mittlere korpuskuläre Hämoglobinkonzentration Retikulozyten | Gesamtleukozytenzahl
Differentialleukozytenzahl Thrombozytenzahl Gerinnungsfähigkeit (einen Wert wählen):
|
| Klinische Chemie | |
| Glucose *)
Gesamtcholesterin Triglyceride Harnstoff-N Gesamtbilirubin Kreatinin Gesamtprotein Albumin Globulin | Alanin-Aminotransferase
Aspartat-Aminotransferase Alkalische Phosphatase Kalium Natrium Calcium Phosphor Chlorid |
| Urinuntersuchung (fakultativ) | |
| Aussehen (Farbe und Trübung)
Menge Spezifisches Gewicht oder Osmolarität pH-Wert | Gesamtprotein
Glucose Blut/Blutzellen |
| *) Da ein längerer Futterentzug die Glucosemessungen bei den behandelten gegenüber den Kontrolltieren verzerren kann, sollte der Studienleiter entscheiden, ob eine Futterkarenz angezeigt ist. Die Dauer des Futterentzugs muss auf die verwendete Art abgestimmt sein; bei der Ratte kann sie 16 Stunden betragen (nächtliche Futterkarenz). Der Nüchternglucosewert kann nach nächtlicher Futterkarenz in der letzten Expositionswoche oder nach nächtlicher Futterkarenz vor der Nekropsie (in letzterem Fall zusammen mit allen anderen klinischen Pathologieparametern) bestimmt werden. | |
38. Gibt es Anhaltspunkte dafür, dass die unteren Atemwege (d. h. die Alveolen) die Hauptablagerungs- und Retentionsorte sind, kann die bronchoalveoläre Lavage (BAL) die Methode der Wahl sein, um hypothesenbasierte Dosis-Wirkungs-Parameter quantitativ zu analysieren, wobei Alveolitis, Lungenentzündung und Phospholipidose im Vordergrund stehen. Auf diese Weise können Veränderungen der Dosis-Wirkungs-Beziehung und des zeitlichen Verlaufs alveolärer Läsionen angemessen untersucht werden. Die BAL-Flüssigkeit kann auf Gesamt- und Differenzialleukozytenzahl, Gesamtprotein und Laktatdehydrogenase analysiert werden. In Betracht gezogen werden können auch Parameter, die auf lysosomale Schäden, Phospholipidose, Fibrose und reizende oder allergische Entzündung hindeuten; dazu kann auch die Bestimmung entzündungsfördernder Zytokine/Chemokine gehören. BAL-Messungen dienen im Allgemeinen zur Ergänzung der Ergebnisse histopathologischer Untersuchungen, können sie aber nicht ersetzen. Eine Anleitung zur Durchführung der Lungenlavage ist in GD 39 (2) enthalten.
Makroskopische Pathologie und Organgewichte
39. Alle Versuchstiere, einschließlich der Tiere, die während der Prüfung sterben oder aus Tierschutzgründen getötet und aus der Studie genommen werden, sind (falls möglich) vollständig zu entbluten und auf makroskopische Veränderungen zu untersuchen. Der Zeitabstand zwischen dem Ende der letzten Exposition eines Tiers und seiner Tötung ist zu dokumentieren. Kann die Nekropsie nicht unmittelbar nach Auffinden eines toten Tieres erfolgen, sollte der Körper auf eine Temperatur gekühlt (nicht eingefroren) werden, die tief genug ist, um die Autolyse zu minimieren. Die Nekropsie ist baldmöglichst, in der Regel innerhalb von einem oder zwei Tagen durchzuführen. Alle makroskopischen Veränderungen sollten für jedes Tier protokolliert werden, wobei besonders auf Veränderungen der Atemwege zu achten ist.
40. In Tabelle 2 sind die Organe und Gewebe aufgeführt, die bei der Nekropsie zur histopathologischen Untersuchung in einem geeigneten Medium aufbewahrt werden sollten. Die Aufbewahrung der in [Klammern] gesetzten Organe und Gewebe sowie aller sonstigen Organe und Gewebe liegt im Ermessen den Studienleiters. Die durch Fettdruck hervorgehobenen Organe sind so bald wie möglich nach der Sektion von anhaftendem Gewebe zu befreien und feucht zu wiegen, um ein Austrocknen zu verhindern. Die Schilddrüse und die Nebenhoden sind nur zu wiegen, wenn dies notwendig ist, da ihre Befreiung von anhaftendem Gewebe die histopathologische Bewertung erschweren kann. Gewebe und Organe sind unmittelbar nach der Nekropsie und je nach verwendetem Fixierungsmittel mindestens 24-48 Stunden vor der Befreiung von anhaftendem Gewebe in 10 %ig gepuffertem Formalin oder einem anderen geeigneten Fixierungsmittel zu fixieren.
Tabelle 2: Bei der Nekropsie aufbewahrte Organe und Gewebe
| Nebennieren
Knochenmark (und/oder frisches Aspirat) Gehirn (mit Schnitten von Cerebrum, Cerebellum und Medulla/Pons) [Augen (Netzhaut, Sehnerv) und Lider] Herz Nieren Larynx (3 Ebenen, 1 Ebene, die die Basis der Epiglottis enthält) Leber Lunge (alle Lungenlappen auf einer Ebene, einschließlich der Hauptbronchien) Lymphknoten aus der Hilusregion der Lunge, insbesondere bei schlecht löslichen Prüfsubstanzen, die in Partikelform vorliegen. Für gründlichere Untersuchungen und/oder Studien mit immunologischem Schwerpunkt können zusätzliche Lymphknoten in Betracht gezogen werden, z.B. aus der mediastinalen, der cervicalen/submandibulären und/oder der aurikularen Region. Nasopharyngeale Gewebe (mindestens 4 Ebenen; 1 Ebene muss den Nasen-Rachen-Gang und das Lymphgewebe des Nasen-Rachen-Raums (NALT) umfassen. Speiseröhre [Riechkolben] Ovarien | Samenbläschen
Rückenmark (zervical, mittlerer Thoraxbereich und lumbar) Milz Magen Hoden Thymus Schilddrüse Trachea (mindestens 2 Ebenen mit einem Längsschnitt durch die Carina und 1 Querschnitt) [Harnblase] Uterus Alle makroskopischen Veränderungen |
41. Die Lungen sind in intaktem Zustand zu entfernen, zu wiegen und mit einem geeigneten Fixierungsmittel bei einem Druck von 20-30 cm Wasser zu behandeln, damit die Lungenstruktur erhalten bleibt (5). Die Schnitte werden bei allen Lungenlappen auf einer Ebene einschließlich der Hauptbronchien hergestellt; wenn eine Lungenlavage durchgeführt wird, ist der nicht gewaschene Lappen jedoch auf drei Ebenen zu schneiden (keine seriellen Schnitte).
42. Mindestens vier Ebenen der nasopharyngealen Gewebe sind zu untersuchen; eine der Ebenen sollte den Nasen- Rachen-Gang umfassen (5, 6, 7, 8, 9), damit das Plattenepithel, das (nicht Zilientragende respiratorische) Übergangsepithel, das (Zilientragende respiratorische) Flimmerepithel und das Riechepithel sowie das Lymphgewebe (NALT; 10,11) gründlich untersucht werden können. Drei Ebenen des Larynx sind zu untersuchen; eine dieser Ebenen sollte die Basis der Epiglottis enthalten (12). Mindestens zwei Ebenen der Trachea sind zu untersuchen, darunter ein Längsschnitt durch die Carina der Bifurkation der extrapulmonalen Bronchien und ein Querschnitt.
Histopathologie
43. Die in Tabelle 2 aufgeführten Organe und Gewebe der Tiere in der Kontrollgruppe und der Gruppe mit der höchsten Konzentration und aller während der Studie gestorbenen oder getöteten Tiere sollten histopathologisch untersucht werden. Besonderes Augenmerk ist auf Atemwege, Zielorgane und makroskopische Veränderungen zu richten. Die Organe und Gewebe, die in der höchsten Konzentrationsgruppe makroskopische Veränderungen aufweisen, sollten in allen Gruppen untersucht werden. Der Studienleiter kann beschließen, histopathologische Untersuchungen bei zusätzlichen Gruppen durchzuführen, um eine eindeutige Konzentrationswirkung nachzuweisen. Umfasst eine Prüfung auch eine Satellitengruppe (Reversibilitätsprüfung), sind alle Gewebe und Organe histopathologisch zu untersuchen, bei denen in den Behandlungsgruppen Wirkungen aufgetreten sind. Treten in der Gruppe mit der höchsten Konzentration übermäßig viele frühzeitige Todesfälle oder andere Probleme auf, die die Signifikanz der Daten beeinträchtigen, so ist die Gruppe mit der nächstniedrigeren Konzentration histopathologisch zu untersuchen. Man sollte versuchen, die makroskopischen Befunde mit den Ergebnissen der mikroskopischen Untersuchung zu korrelieren.
Daten und Berichterstattung
Daten
44. Körpergewichte, Futteraufnahme, Ergebnisse der klinischen Pathologie, makroskopische Befunde, Organgewichte und Ergebnisse der Histopathologie sind für die einzelnen Tiere anzugeben. Die Daten der klinischen Beobachtung sind in tabellarischer Form zusammenzufassen. Daraus müssen für jede Prüfgruppe die Anzahl der verwendeten Tiere, die Anzahl der Tiere mit spezifischen Toxizitätszeichen, die Anzahl der Tiere, die während der Prüfung tot aufgefunden oder vorzeitig getötet wurden, der Todeszeitpunkt der einzelnen Tiere, eine Beschreibung und der zeitliche Verlauf der toxischen Wirkungen und deren Reversibilität sowie die Sektionsbefunde ersichtlich sein. Sowohl die quantitativen als auch die gelegentlich erzielten Ergebnisse sind anhand eines geeigneten statistischen Verfahrens zu bewerten. Hierzu ist eine allgemein anerkannte Statistikmethode heranzuziehen; die Statistikmethoden sind bei der Auslegung der Studie festzulegen.
Prüfbericht
45. Der Prüfbericht sollte, soweit zutreffend, die folgenden Informationen enthalten:
Versuchstiere und Tierhaltung
Prüfsubstanz
Vehikel
Inhalationskammer
Expositionsdaten
Prüfbedingungen
Ergebnisse
Diskussion und Auswertung der Ergebnisse
Literatur:
1. OECD (1981). Subchronic Inhalation Toxicity Testing, Original Test Guideline No 412, Environment Directorate, OECD, Paris.
2. OECD (2009). Guidance Document on Acute Inhalation Toxicity Testing, Environmental Health and Safety Monograph Series on Testing and Assessment No. 39, ENV/JM/MONO(2009)28, OECD, Paris.
3. OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris.
4. Whalan JE and Redden JC (1994). Interim Policy for Particle Size and Limit Concentration Issues in Inhalation Toxicity Studies. Office of Pesticide Programs, United States Environmental Protection Agency.
5. Dungworth DL, Tyler WS, Plopper CE (1985). Morphological Methods for Gross and Microscopic Pathology (Chapter 9) in Toxicology of Inhaled Material, Witschi, H.P. and Brain, J.D. (eds), Springer Verlag Heidelberg, pp. 229-258.
6. Young JT (1981). Histopathological examination of the rat nasal cavity. Fundam. Appl. Toxicol. 1: 309-312.
7. Harkema JR (1990). Comparative pathology of the nasal mucosa in laboratory animals exposed to inhaled irritants. Environ. Health Perspect. 85: 231-238.
8. Woutersen RA, Garderen-Hoetmer A, van Slootweg PJ, Feron VJ (1994). Upper respiratory tract carcinogenesis in experimental animals and in humans. In: Waalkes MP and Ward JM (eds) Carcinogenesis. Target Organ Toxicology Series, Raven Press, New York, 215-263.
9. Mery S, Gross EA, Joyner DR, Godo M, Morgan KT (1994). Nasal diagrams: A tool for recording the distribution of nasal lesions in rats and mice. Toxicol. Pathol. 22: 353-372.
10. Kuper CF, Koornstra PJ, Hameleers DMH, Biewenga J, Spit BJ, Duijvestijn AM, Breda Vriesman van PJC, Sminia T (1992). The role of nasopharyngeal lymphoid tissue. Immunol. Today 13: 219-224.
11. Kuper CF, Arts JHE, Feron VJ (2003). Toxicity to nasal-associated lymphoid tissue. Toxicol. Lett. 140-141: 281- 285.
12. Lewis DJ (1981). Mitotic Indices of Rat Laryngeal Epithelia. Journal of Anatomy 132(3): 419-428.
13. Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
Anlage 1 Definition
Prüfsubstanz: jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird."
5. Die Kapitel B.29 und B.30 erhalten folgende Fassung:
"B.29 Prüfung auf sub-chronische Toxizität nach Inhalation - 90-Tage-Test
Zusammenfassung
Diese überarbeitete Prüfmethode B.29 wurde entwickelt, um die Toxizität der Prüfsubstanz nach Inhalation über einen subchronischen Zeitraum (90 Tage) umfassend zu beschreiben und um robuste Daten für die quantitative Bewertung des Inhalationsrisikos zu gewinnen. Gruppen von zehn männlichen und zehn weiblichen Nagern werden über einen Zeitraum von 90 Tagen (13 Wochen) 6 Stunden am Tag a) der Prüfsubstanz in drei oder mehr Konzentrationsstufen, b) gefilterter Luft (negative Kontrolle) und/oder c) dem Vehikel (Vehikelkontrolle) ausgesetzt. Im Allgemeinen werden die Tiere der Prüfsubstanz an fünf Tagen pro Woche ausgesetzt, aber auch sieben Tage pro Woche sind zulässig. Es werden immer männliche und weibliche Tiere geprüft, aber sie können unterschiedlichen Konzentrationsstufen ausgesetzt werden, wenn bekannt ist, dass ein Geschlecht empfindlicher auf eine bestimmte Prüfsubstanz reagiert als das andere. Bei dieser Methode hat der Studienleiter die Möglichkeit, Satellitengruppen (Reversibilitätsprüfung) aufzunehmen, Tiere im Verlauf der Studie zu töten, sowie eine bronchoalveoläre Lavage (BAL), neurologische Tests, zusätzliche klinische Pathologieuntersuchungen und histopathologische Untersuchungen durchzuführen, um die Toxizität einer Prüfsubstanz besser beschreiben zu können.
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 413 (2009). Die ursprüngliche Prüfrichtlinie 413 (TG 413) zur subchronischen Inhalation wurde 1981 angenommen (1). Diese Prüfmethode B.29 (die der überarbeiteten TG 413 (2009) entspricht), wurde aktualisiert, um dem neuesten Stand der Wissenschaft Rechnung zu tragen und derzeitige und künftige Regulierungsanforderungen zu erfüllen.
2. Prüfungen auf subchronische Toxizität nach Inhalation haben hauptsächlich den Zweck, die in Vorschriften festzusetzenden Konzentration zu bestimmen, nach denen das Risiko für Arbeitnehmer im beruflichen Umfeld beurteilt wird. Sie werden auch verwendet, um das mit einer Exposition verbundene Risiko für den Menschen am Wohnort, im Verkehr und in der Umwelt zu beurteilen. Diese Methode ermöglicht die Beschreibung der schädlichen Wirkungen nach wiederholter täglicher inhalativer Exposition gegen eine Prüfsubstanz für einen Zeitraum von 90 Tagen (etwa 10 % der Lebenszeit einer Ratte). Die in Prüfungen auf subchronische Toxizität nach Inhalation gewonnenen Daten können für quantitative Risikobewertungen und für die Auswahl von Konzentrationen für chronische Prüfungen verwendet werden. Diese Prüfmethode ist nicht speziell für die Prüfung von Nanomaterialien bestimmt. Die im Zusammenhang mit dieser Prüfmethode verwendeten Begriffe werden am Ende dieses Kapitels und im Guidance Document (GD 39 (2)) definiert.
Ausgangsüberlegungen
3. Um die Qualität der Studie zu verbessern und möglichst wenig Versuchstiere zu verwenden, sollte das Prüflabor vor Durchführung der Studie alle verfügbaren Informationen über die Prüfsubstanz berücksichtigen. Für die Auswahl der am besten geeigneten Prüfkonzentrationen könnten u. a. Informationen wie die Identität, die chemische Struktur und die physikalisch-chemischen Eigenschaften der Prüfsubstanz, Ergebnisse jeglicher In- vitro- oder In-vivo-Toxizitätsprüfungen, vorgesehene Verwendungen und die Möglichkeit der Exposition des Menschen, (Q)SAR-Daten und toxikologische Daten über strukturverwandte Substanzen sowie Daten aus anderen Studien mit wiederholter Exposition herangezogen werden. Falls Neurotoxizität erwartet oder im Verlauf der Studie beobachtet wird, kann der Studienleiter beschließen, geeignete Untersuchungen wie eine FOB (functional observational battery) und die Messung der motorischen Aktivität aufzunehmen. Obwohl die Expositionszeit bei bestimmten Untersuchungen ein kritischer Aspekt sein kann, darf die Durchführung dieser zusätzlichen Versuche die Auslegung der Hauptstudie nicht beeinträchtigen.
4. Verdünnungen ätzender oder reizender Prüfsubstanzen können in Konzentrationen geprüft werden, die den gewünschten Toxizitätsgrad erzielen. Weitere Informationen sind GD 39 (2) zu entnehmen. Wenn Versuchstiere diesen Stoffen ausgesetzt werden, sollten die Zielkonzentrationen so niedrig sein, dass sie keine starken Schmerzen oder Leiden verursachen; sie sollten aber ausreichen, um die Konzentrations-Wirkungs-Kurve so zu erweitern, dass das regulatorische und wissenschaftliche Ziel der Prüfung erreicht wird. Diese Konzentrationen sollten von Fall zu Fall festgelegt werden, möglichst auf Basis einer entsprechend ausgelegten Dosisfindungsstudie, die Informationen über den kritischen Endpunkt, eine etwaige Reizschwelle und den Zeitpunkt des Einsetzens der Wirkung liefert (siehe Nummern 11, 12 und 13). Die Wahl der Konzentration ist zu begründen.
5. Moribunde Tiere oder Tiere, die Anzeichen starker und andauernder Qualen zeigen, sollten tierschutzgerecht getötet werden. Moribunde Tiere werden auf die gleiche Weise gewertet wie während des Tests gestorbene Tiere. Kriterien für die Entscheidung, moribunde oder schwer leidende Tiere zu töten, sowie Hinweise zur Erkennung des absehbaren oder bevorstehenden Todes sind Gegenstand des OECD Guidance Document on Humane Endpoints (3).
Beschreibung der Methode
Auswahl von Versuchstierarten
6. Es sind junge, gesunde, adulte Nagetiere aus üblicherweise eingesetzten Laborstämmen zu verwenden. Bevorzugtes Versuchstier ist die Ratte. Wird eine andere Tierart eingesetzt, ist dies zu begründen.
Vorbereitung der Tiere
7. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Am Tag der Randomisierung sollten die jungen, adulten Tiere 7 bis 9 Wochen alt sein; ihr Körpergewicht sollte innerhalb von ± 20 % des mittleren Gewichts für jedes Geschlecht liegen. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert und vor Beginn der Prüfung für einen Zeitraum von mindestens fünf Tagen in ihren Käfigen an die Laborbedingungen gewöhnt.
Tierhaltung
8. Um die Beobachtungen zu erleichtern und Verwechslungen auszuschließen, sollten die Tiere möglichst mit einem subkutanen Transponder einzeln gekennzeichnet werden. Die Temperatur in dem Raum, in dem die Versuchstiere gehalten werden, sollte 22 ± 3 °C betragen. Die relative Luftfeuchtigkeit sollte im Idealfall im Bereich zwischen 30 und 70 % liegen; bei Verwendung von Wasser als Vehikel könnte dies jedoch unmöglich sein. Die Tiere sollten vor und nach den Expositionen im Allgemeinen nach Geschlecht und Konzentration in Käfigen gruppiert werden, wobei aber die Anzahl der Tiere pro Käfig noch eine genaue Beobachtung der einzelnen Tiere ermöglichen muss und Verluste aufgrund von Kannibalismus oder Kämpfen minimiert werden sollten. Wenn die Tiere der Prüfsubstanz nur mit der Nase ausgesetzt werden sollen, müssen sie möglicherweise an die Restrainer gewöhnt werden. Die Restrainer sollten die Tiere weder körperlich noch in Bezug auf Wärme oder Fixierung übermäßig beeinträchtigen. Die Fixierung kann physiologische Endpunkte wie Körpertemperatur (Hyperthermie) und/oder das Atemminutenvolumen beeinflussen. Wenn generische Daten zeigen, dass keine derartigen Veränderungen in nennenswertem Ausmaß vorkommen, ist eine Eingewöhnung an die Restrainer nicht erforderlich. Bei der Ganzkörperexposition gegen ein Aerosol sollten die Tiere während der Exposition einzeln untergebracht sein, damit sie die Prüfsubstanz nicht durch das Fell ihrer Käfiggenossen filtriert einatmen. Außer während der Exposition kann herkömmliches und zertifiziertes Labortierfutter verwendet werden bei uneingeschränkter Versorgung mit Trinkwasser. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
Inhalationskammern
9. Bei der Auswahl einer Inhalationskammer sind die Art der Prüfsubstanz und der Gegenstand der Prüfung zu berücksichtigen. Das bevorzugte Verfahren ist die 'Nose-only'-Exposition (dieser Begriff umfasst 'nur Kopf', 'nur Nase' oder 'nur Schnauze'). Für die Untersuchung von Flüssigkeits- oder Feststoffaerosolen und für Dämpfe, die zu Aerosolen kondensieren können, wird im Allgemeinen die 'Nose-only'-Exposition bevorzugt. Besondere Ziele der Untersuchung können möglicherweise mit einer Ganzkörperexposition besser erreicht werden, doch dies sollte im Prüfbericht begründet werden. Um bei Verwendung einer Ganzkörperkammer die Stabilität der Atmosphäre sicherzustellen, sollte das Gesamtvolumen der Versuchstiere 5 % des Volumens der Kammer nicht übersteigen. Die Grundzüge der 'Nose-only'- und der Ganzkörperexposition sowie ihre jeweiligen Vor- und Nachteile sind in GD 39 (2) beschrieben.
Toxizitätsstudien
Grenzkonzentrationen
10. Anders als bei Studien zur akuten Toxizität gibt es bei Prüfungen auf subchronische Toxizität nach Inhalation keine festgelegten Grenzkonzentrationen. Bei der Festlegung der maximalen Prüfkonzentration ist Folgendes zu beachten: 1) die höchste erreichbare Konzentration, 2) das maximale Expositionsniveau von Menschen ('worst case), 3) die Notwendigkeit, eine ausreichende Sauerstoffversorgung aufrechtzuerhalten, und 4) Tierschutzerwägungen. Gibt es keine auf Daten basierenden Grenzwerte, können die akuten Grenzwerte der Verordnung (EG) Nr. 1272/2008 (13) zugrunde gelegt werden (d. h. bis zu einer Höchstkonzentration von 5 mg/l bei Aerosolen, 20 mg/l bei Dämpfen und 20.000 ppm bei Gasen); siehe GD 39 (2). Wenn diese Grenzen bei der Prüfung von Gasen oder hochflüchtigen Prüfsubstanzen (z.B. Kältemitteln) überschritten werden müssen, ist dies zu begründen. Die Grenzkonzentration muss eine eindeutige Toxizität hervorrufen, ohne den Tieren übermäßigen Stress zu bereiten oder ihre Lebensdauer zu beeinträchtigen (3).
Dosisfindungsstudie
11. Vor Beginn der Hauptstudie muss in der Regel eine Dosisfindungsstudie durchgeführt werden. Diese ist umfassender als eine Vorstudie, weil sie nicht auf die Wahl der Konzentration begrenzt ist. Die in einer Dosisfindungsstudie gewonnenen Erkenntnisse können zu einer erfolgreichen Hauptstudie führen. Sie kann z.B. technische Informationen zu den Analysemethoden, zur Partikelgröße, zur Erkennung toxischer Mechanismen, klinische Pathologiedaten und histopathologische Daten sowie Schätzungen möglicher NOAEL- und MTC-Konzentrationen in einer Hauptstudie liefern. Der Studienleiter kann entscheiden, mithilfe der Dosisfindungsstudie die Schwelle für die Reizung der Atemwege (z.B. durch histopathologische Untersuchung der Atemwege, Lungenfunktionsprüfung oder bronchoalveoläre Lavage), die höchste Konzentration, die von den Tieren ohne übermäßigen Stress toleriert wird, und die Parameter, die die Toxizität der Prüfsubstanz am besten beschreiben, zu identifizieren.
12. Eine Dosisfindungsstudie kann aus einer oder mehreren Konzentrationsstufen bestehen. Auf jeder Konzentrationsstufe sollten je nach den gewählten Endpunkten drei bis sechs männliche und drei bis sechs weibliche Tiere der Prüfsubstanz ausgesetzt werden. Eine Dosisfindungsstudie sollte mindestens fünf Tage und im Allgemeinen nicht mehr als 28 Tage dauern. Die Wahl der Konzentrationen für die Hauptstudie ist im Prüfbericht zu begründen. Ziel der Hauptstudie ist es, nachzuweisen, dass eine Beziehung zwischen der Konzentration und der Wirkung besteht, die am voraussichtlich empfindlichsten Endpunkt auftritt. Die niedrigste Konzentration sollte im Idealfall eine Konzentration sein, bei der keine zu beobachtenden schädlichen Wirkungen auftreten, und die höchste Konzentration sollte eine eindeutige Toxizität hervorrufen, ohne den Tieren übermäßigen Stress zu bereiten oder ihre Lebensdauer zu beeinträchtigen (3).
13. Bei der Auswahl der Konzentrationsstufen für die Dosisfindungsstudie sollten alle verfügbaren Informationen berücksichtigt werden, einschließlich der Struktur-Wirkungs-Beziehungen und der Daten über ähnliche Stoffe (siehe Nummer 3). Eine Dosisfindungsstudie kann bestätigen/widerlegen, welche Endpunkte nach mechanistischen Kriterien als die empfindlichsten Endpunkte angesehen werden, z.B. die Cholinesterasehemmung durch Organophosphate, die Methämoglobinbildung durch für Erythrozyten toxische Stoffe, Schilddrüsenhormone (T3, T4) im Fall von thyrotoxischen Stoffen, Proteine, LDH oder Neutrophile in bronchoalveolärer Lavage im Fall schwach löslicher unschädlicher Partikel oder lungenreizender Aerosole.
Hauptstudie
14. Die Hauptstudie zur Prüfung auf subchronische Toxizität umfasst im Allgemeinen drei Konzentrationsstufen sowie, falls erforderlich, eine gleichzeitige negative (Luft-)Kontrolle und/oder eine Vehikelkontrolle (siehe Nummer 18). Die Festlegung der geeigneten Expositionsstufen sollte sich auf alle verfügbaren Daten stützen, einschließlich der Ergebnisse systemischer Toxizitätsprüfungen, des Metabolismus und der Kinetik (hohe Konzentrationsstufen, die kinetische Prozesse sättigen, sind zu vermeiden). Jede Prüfgruppe umfasst zehn männliche und zehn weibliche Nager, die der Prüfsubstanz für einen Zeitraum von 13 Wochen an fünf Tagen in der Woche jeweils 6 Stunden pro Tag ausgesetzt werden (Gesamtdauer der Prüfung 90 Tage). Die Tiere können auch an sieben Tagen in der Woche exponiert werden (z.B. wenn inhalierte Arzneimittel geprüft werden). Ist bekannt, dass ein Geschlecht empfindlicher auf eine bestimmte Prüfsubstanz reagiert, können die Geschlechter unterschiedlichen Konzentrationsstufen ausgesetzt werden, um die Konzentrations-Wirkungs-Beziehung genauer zu bestimmen (siehe Nummer 15). Wenn für eine 'Nose-only'-Exposition andere Nagetierarten als Ratten verwendet werden, kann die maximale Expositionsdauer angepasst werden, um artenspezifisches Leiden zu minimieren. Eine Expositionsdauer von weniger als 6 Stunden/Tag oder die Notwendigkeit einer Ganzkörperexpositionsstudie mit Langzeitexposition (z.B. 22 Stunden/Tag) ist zu begründen (siehe GD 39 (2). Während der Exposition sollte kein Futter verabreicht werden, es sei denn die Exposition dauert länger als 6 Stunden. Wasser kann während einer Ganzkörperexposition jederzeit angeboten werden.
15. Die gewählten Zielkonzentrationen sollen es ermöglichen, die Zielorgane zu identifizieren und eine deutliche Konzentrations-Wirkungs-Beziehung zu belegen:
Tötungen im Verlauf der Studie
16. Sollen im Verlauf des Versuchs Tiere getötet werden, so muss die Zahl der Tiere jeder Expositionsstufe um die Zahl der Tiere erhöht werden, die schon vor Versuchsende getötet werden sollen. Tötungen im Verlauf der Studie sind zu begründen und in den statistischen Analysen entsprechend zu berücksichtigen.
Satellitenstudie (Reversibilität)
17. Es kann eine Satellitenstudie durchgeführt werden, um die Tiere für einen angemessenen Zeitraum nach der Behandlung (mindestens 14 Tage) auf Reversibilität, Persistenz oder ein verzögertes Auftreten von Toxizität zu beobachten. Satellitengruppen bestehen aus zehn männlichen und zehn weiblichen Tieren, die der Prüfsubstanz gleichzeitig mit den Versuchstieren der Hauptstudie ausgesetzt werden. Dabei sollten sie der Prüfsubstanz auf der höchsten Konzentrationsstufe ausgesetzt werden, und erforderlichenfalls sollte es gleichzeitige Luft- und/oder Vehikelkontrollen geben (siehe Nummer 18).
Kontrolltiere
18. Eine gleichzeitige negative (Luft-)Kontrollgruppe ist genauso zu behandeln wie die Prüfgruppe, außer dass die Tiere nicht der Prüfsubstanz, sondern gefilterter Luft ausgesetzt werden. Wenn die Prüfatmosphäre mithilfe von Wasser oder einem anderen Stoff erzeugt wird, ist statt der negativen (Luft-)Kontrollgruppe eine Vehikelkontrollgruppe zu verwenden. Wenn möglich sollte Wasser als Vehikel benutzt werden. In diesem Fall sind die Tiere Luft mit derselben relativen Luftfeuchtigkeit auszusetzen wie die exponierten Gruppen. Das geeignete Vehikel ist auf der Grundlage einer geeigneten Vorstudie oder von historischen Daten auszuwählen. Ist die Toxizität eines Vehikels unklar, kann der Studienleiter beschließen, sowohl eine negative (Luft-)Kontrolle als auch eine Vehikelkontrolle zu verwenden; hiervon wird jedoch dringend abgeraten. Wenn historische Daten belegen, dass ein Vehikel nicht toxisch ist, besteht keine Notwendigkeit für eine negative (Luft-)Kontrollgruppe und es sollte nur eine Vehikelgruppe verwendet werden. Ergibt eine Vorstudie einer in einem Vehikel formulierten Prüfsubstanz, dass keine Toxizität vorliegt, ist das Vehikel folglich in der geprüften Konzentration nicht toxisch, und diese Vehikelkontrolle sollte verwendet werden.
Expositionsbedingungen
Verabreichung der Konzentrationen
19. Die Tiere werden der Prüfsubstanz in Form von Gas, Dampf, Aerosol oder einer Kombination dieser Formen ausgesetzt. Der zu prüfende Aggregatzustand hängt von den physikalisch-chemischen Eigenschaften der Prüfsubstanz, den gewählten Konzentrationen und/oder der physikalischen Form ab, in der die Prüfsubstanz bei der Handhabung und Verwendung am wahrscheinlichsten vorliegt. Hygroskopische und chemisch reaktive Prüfsubstanzen sollten bei geringer Luftfeuchtigkeit geprüft werden. Dabei ist darauf zu achten, dass keine explosionsfähigen Konzentrationen erzeugt werden. Bei Partikeln kann die Partikelgröße durch mechanische Prozesse verringert werden. GD 39 (2) enthält nähere Hinweise.
Partikelgrößenverteilung
20. Bei allen Aerosolen und bei Dämpfen, die zu Aerosolen kondensieren können, sollte die Partikelgröße bestimmt werden. Damit alle relevanten Regionen der Atemwege der Prüfsubstanz ausgesetzt werden, werden mittlere aerodynamische Massendurchmesser (Mass Median Aerodynamic Diameter - MMAD) von 1 bis 3 µm mit einer geometrischen Standardabweichung (σg) von 1,5 bis 3,0 empfohlen (4). Wenngleich nach Möglichkeit versucht werden sollte, diese Werte zu erreichen, ist Fachwissen erforderlich, falls sie nicht erzielt werden können. Metalldampfpartikel liegen z.B. unter diesen Werten, geladene Partikel und Fasern dagegen können diese Werte überschreiten.
Vorbereitung der Prüfsubstanz in einem Vehikel
21. Im Idealfall sollte die Prüfsubstanz ohne ein Vehikel geprüft werden. Wenn für die Erzeugung einer geeigneten Prüfsubstanzkonzentration oder Partikelgröße ein Vehikel verwendet werden muss, ist Wasser zu bevorzugen. Wird eine Prüfsubstanz in einem Vehikel gelöst, so ist seine Stabilität nachzuweisen.
Überwachung der Expositionsbedingungen
Luftstrom in der Inhalationskammer
22. Der Luftstrom durch die Kammer sollte während jeder Exposition sorgfältig geregelt, kontinuierlich überwacht und mindestens stündlich protokolliert werden. Die Echtzeit-Überwachung der Konzentration (oder zeitlichen Stabilität) der Prüfatmosphäre ist eine integrale Messung aller dynamischen Parameter und gibt indirekt die Möglichkeit, alle relevanten dynamischen Inhalationsparameter zu messen. Wenn die Konzentration in Echtzeit überwacht wird, kann die Frequenz der Messung der Luftströme auf eine einzige Messung je Exposition und Tag reduziert werden. Es sollte besonders darauf geachtet werden, das erneute Einatmen in 'Nose-only'-Expositionskammern zu vermeiden. Die Sauerstoffkonzentration sollte mindestens 19 % betragen, und die Kohlendioxidkonzentration sollte 1 % nicht überschreiten. Gibt es Grund zu der Annahme, dass diese Werte nicht eingehalten werden können, sind die Sauerstoff- und Kohlendioxidkonzentrationen zu messen. Wenn die Messungen am ersten Expositionstag die richtigen Werte dieser Gase bestätigen, sollten keine weiteren Messungen erforderlich sein.
Temperatur und relative Luftfeuchtigkeit in der Inhalationskammer
23. Die Temperatur in der Inhalationskammer sollte 22 ± 3 °C betragen. Sowohl bei der 'Nose-only'- als auch bei der Ganzkörperexposition sollte die relative Luftfeuchtigkeit im Atembereich der Tiere kontinuierlich überwacht und während jeder Exposition möglichst stündlich dokumentiert werden. Die relative Luftfeuchtigkeit sollte möglichst zwischen 30 und 70 % liegen, was jedoch möglicherweise nicht erreichbar ist (z.B. bei der Prüfung von wasserbasierten Mischungen) oder wegen chemischer Interferenz mit der Prüfmethode nicht gemessen werden kann.
Prüfsubstanz: nominale Konzentration
24. Die nominale Konzentration in der Expositionskammer sollte möglichst berechnet und protokolliert werden. Die nominale Konzentration ist die Masse der erzeugten Prüfsubstanz dividiert durch das Gesamtvolumen der durch die Inhalationskammer geleiteten Luft. Sie wird nicht zur Beschreibung der Exposition der Tiere verwendet; vielmehr gibt ein Vergleich der nominalen Konzentration und der tatsächlichen Konzentration Aufschluss über die Effizienz des Prüfsystems bei der Erzeugung der Prüfkonzentration und kann daher für die Aufdeckung von Problemen bei dieser Erzeugung verwendet werden.
Prüfsubstanz: tatsächliche Konzentration
25. Die tatsächliche Konzentration ist die Konzentration der Prüfsubstanz im Atembereich der Tiere in einer Inhalationskammer. Die tatsächlichen Konzentrationen können entweder durch spezifische Methoden (z.B. direkte Probenahme, adsorptive Methoden oder chemische Reaktionsverfahren mit anschließender analytischer Charakterisierung) oder durch unspezifische Methoden wie Gravimetrie bestimmt werden. Die gravimetrische Methode ist lediglich für Aerosole mit nur einem Bestandteil in Pulverform oder Aerosole von Flüssigkeiten mit geringer Flüchtigkeit akzeptabel und sollte sich auf geeignete, vor der Studie zu erstellende und für die Prüfsubstanz spezifische Beschreibungen stützen. Die Konzentration von Aerosolen mit mehreren Bestandteilen in Pulverform kann ebenfalls gravimetrisch bestimmt werden. Hierzu muss jedoch mit Analysedaten belegt werden, dass die Schwebstoffe eine ähnliche Zusammensetzung haben wie das Ausgangsmaterial. Liegen diese Angaben nicht vor, muss die Prüfsubstanz (im Idealfall im Schwebezustand) möglicherweise im Verlauf der Studie in regelmäßigen Abständen neu analysiert werden. Bei aerosolisierten Agenzien, die verdunsten oder sublimieren können, sollte gezeigt werden, dass alle Phasen von der gewählten Methode erfasst wurden.
26. Für die gesamte Dauer der Studie sollte möglichst eine Partie der Prüfsubstanz verwendet werden; die Probe sollte unter Bedingungen aufbewahrt werden, die ihre Reinheit, Homogenität und Stabilität gewährleisten. Die Prüfsubstanz sollte vor Beginn der Studie mit Angaben zur Reinheit und, falls technisch machbar, zur Identität sowie zu Mengen identifizierter Schadstoffe und Verunreinigungen beschrieben werden. Hierzu können unter anderem die folgenden Daten verwendet werden: Retentionszeit und relative Peakfläche, durch Massenspektrometrie oder Gaschromatographie bestimmtes Molekulargewicht oder andere Werte. Das Prüflabor ist zwar nicht für die Identität der Probe verantwortlich, doch es kann ratsam sein, dass es die Beschreibung des Auftraggebers zumindest in gewissen Grenzen (z.B. Farbe, physikalische Beschaffenheit usw.) überprüft.
27. Die Expositionsatmosphäre ist so konstant wie möglich zu halten. Um die Stabilität der Expositionsbedingungen nachzuweisen, kann ein Echtzeitüberwachungsgerät verwendet werden, z.B. ein Aerosol-Photometer für Aerosole oder ein Gesamtkohlenwasserstoff-Analysator (THC) für Dämpfe. Die tatsächliche Konzentration in der Kammer sollte an jedem Expositionstag für jede Expositionsstufe mindestens dreimal gemessen werden. Falls dies wegen geringer Luftdurchflussraten oder niedriger Konzentrationen nicht möglich ist, reicht eine Probe je Expositionsperiode. Im Idealfall sollte diese Probe dann über die gesamte Expositionszeit gewonnen werden. Die einzelnen Proben der Konzentration in der Kammer sollten bei Gasen und Dämpfen nicht mehr als ± 10 % und bei Flüssig- oder Feststoffaerosolen nicht mehr als ± 20 % von der mittleren Kammerkonzentration abweichen. Die Zeit bis zum Erreichen eines Gleichgewichts in der Kammer (t95) ist zu berechnen und zu dokumentieren. Die Expositionsdauer erstreckt sich über den Zeitraum, in dem die Prüfsubstanz erzeugt wird. Dies schließt die Zeiten zur Erreichung des Gleichgewichts in der Kammer (t95) und zum Abbau der Konzentrationen ein. GD 39 (2) enthält Hinweise zur Einschätzung von t95.
28. Bei sehr komplexen Mischungen aus Gasen/Dämpfen und Aerosolen (z.B. Verbrennungsatmosphären und Prüfsubstanzen, die aus hierzu bestimmten Endverbraucherprodukten/-geräten gesprüht werden), kann sich jede Phase in einer Inhalationskammer anders verhalten. Daher sollte mindestens eine Indikatorsubstanz (Analyt), normalerweise der wichtigste Wirkstoff in der Mischung, von jeder Phase (Gas/Dampf und Aerosol) ausgewählt werden. Wenn die Prüfsubstanz eine Mischung ist, sollte die Analysekonzentration für die gesamte Mischung und nicht nur für den Wirkstoff oder die Indikatorsubstanz (Analyt) dokumentiert werden. Weitere Informationen zu tatsächlichen Konzentrationen sind in GD 39 (2) zu finden.
Prüfsubstanz: Partikelgrößenverteilung
29. Die Partikelgrößenverteilung von Aerosolen sollte auf jeder Konzentrationsstufe mindestens wöchentlich mit einem Kaskaden-Impaktor oder einem anderen Messgerät wie einem APS bestimmt werden. Kann nachgewiesen werden, dass die mit einem Kaskaden-Impaktor und dem alternativen Messgerät erzielten Ergebnisse gleichwertig sind, so kann das alternative Instrument während der gesamten Studie verwendet werden.
30. Parallel zum Hauptinstrument ist ein zweites Gerät wie ein Gravimetriefilter oder eine Gaswaschflasche zu verwenden, um den Abscheidegrad des Hauptinstruments zu bestätigen. Die durch die Partikelgrößenanalyse bestimmte Massenkonzentration sollte innerhalb vertretbarer Grenzen um die durch die Filteranalyse bestimmte Massenkonzentration liegen [siehe GD 39 (2)]. Wenn die Gleichwertigkeit bei allen geprüften Konzentrationen zu Beginn der Studie nachgewiesen werden kann, kann auf weitere bestätigende Messungen verzichtet werden. Aus Tierschutzgründen sollten Vorkehrungen getroffen werden, um unklare Daten zu minimieren, die dazu führen könnten, dass eine Exposition wiederholt werden muss.
31. Wenn die Möglichkeit besteht, dass Dampfkondensation zur Bildung eines Aerosols führen kann, oder wenn in einer Dampfatmosphäre mit dem Potenzial für gemischte Phasen Partikel nachgewiesen werden, sollte eine Partikelgrößenbestimmung für Dämpfe vorgenommen werden.
Beobachtungen
32. Die Tiere sollten vor, während und nach der Exposition auf klinische Zeichen beobachtet werden. Je nach Reaktion der Tiere während der Exposition können häufigere Beobachtungen angezeigt sein. Wenn die Beobachtung der Tiere durch die Verwendung von Restrainern, wegen schlecht beleuchteter Ganzkörperkammern oder getrübter Atmosphäre erschwert ist, sind die Tiere nach der Exposition sorgfältig zu beobachten. Durch Beobachtungen vor der Exposition am folgenden Tag kann beurteilt werden, ob toxische Wirkungen sich zurückgebildet oder verschlimmert haben.
33. Sämtliche Beobachtungen werden in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Wenn Tiere aus humanen Gründen getötet werden oder ihr Tod festgestellt wird, sollte der Todeszeitpunkt so genau wie möglich registriert werden.
34. Die Beobachtungen der Tiere sollten sich insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, des Atmungs- und Kreislaufsystems, des Nervensystems sowie auf Somatomotorik und Verhaltensmuster erstrecken. Besonderes Augenmerk ist auf Tremor, Konvulsionen, Salivation, Diarrhö, Lethargie, Schlaf und Koma zu richten. Die Messung der Rektaltemperatur kann zusätzliche Belege für mit der Behandlung oder Unterbringung zusammenhängende Reflex-Bradypnoe oder Hypo-/Hyperthermie liefern. Darüber hinaus können zusätzliche Aspekte wie Kinetik, Biomonitoring, Lungenfunktion, Retention schlecht löslicher Stoffe, die im Lungengewebe akkumulieren, und Verhaltensstörungen in das Studienprotokoll aufgenommen werden.
Körpergewicht
35. Das Körpergewicht der einzelnen Tiere sollte kurz vor der ersten Exposition (Tag 0), danach zweimal wöchentlich (z.B. freitags und montags, um die Erholung während eines expositionsfreien Wochenendes nachzuweisen, oder in einem Zeitintervall, das die Beurteilung der systemischen Toxizität ermöglicht) und zum Zeitpunkt des Todes oder der Tötung dokumentiert werden. Treten in den ersten vier Wochen keine Wirkungen auf, kann das Körpergewicht während der restlichen Studiendauer wöchentlich gemessen werden. Satellitentiere (Reversibilitätsprüfung) (falls verwendet) sollten während der gesamten Erholungsphase weiterhin wöchentlich gewogen werden. Am Ende der Studie sollten alle Tiere kurz vor der Tötung gewogen werden, um eine objektive Berechnung der Organ-Körpergewicht-Verhältnisse zu ermöglichen.
Futter- und Wasseraufnahme
36. Die Futteraufnahme sollte wöchentlich gemessen werden. Auch die Wasseraufnahme kann gemessen werden.
Klinische Pathologie
37. An allen Tieren, auch an Kontroll- und Satellitentieren (Reversibilitätsprüfung) sollten klinische Pathologieuntersuchungen durchgeführt werden, wenn sie getötet werden. Der Zeitraum zwischen dem Ende der Exposition und der Blutentnahme ist zu protokollieren, insbesondere wenn der betreffende Endpunkt rasch zu seinem ursprünglichen Wert zurückkehrt. Für Parameter mit einer kurzen Plasmahalbwertszeit (z.B. COHb, CHE und MetHb) ist die Probenahme nach Ende der Exposition angezeigt.
38. In Tabelle 1 sind die im Allgemeinen für alle Toxikologiestudien erforderlichen klinischen Pathologieparameter aufgeführt. In der Regel ist eine Urinanalyse nicht notwendig, kann aber durchgeführt werden, wenn sie wegen erwarteter oder festgestellter Toxizität für nützlich gehalten wird. Der Studienleiter kann beschließen, zusätzliche Parameter zu bestimmen, um die Toxizität einer Prüfsubstanz genauer zu beschreiben (z.B. Cholinesterase, Lipide, Hormone, Säure-Basen-Gleichgewicht, Methämoglobin oder Heinz-Körper, Creatin-Kinase, Verhältnis von myeloiden zu erythroiden Zellen, Troponin, arterielle Blutgase, Lactatdehydrogenase, Sorbitdehydrogenase, Glutamatdehydrogenase und γ-Glutamyltranspeptidase).
Tabelle 1: Klinische Standardpathologieparameter
| Hämatologische Untersuchung | |
| Erythrozytenzahl
Hämatokrit Hämoglobinkonzentration Mittleres korpuskuläres Hämoglobin Mittleres Erythrozyteneinzelvolumen Mittlere korpuskuläre Hämoglobinkonzentration Retikulozyten | Gesamtleukozytenzahl
Differentialleukozytenzahl Thrombozytenzahl Gerinnungsfähigkeit (einen Wert wählen):
|
| Klinische Chemie | |
| Glucose *)
Gesamtcholesterin Triglyceride Harnstoff-N Gesamtbilirubin Kreatinin Gesamteiweiß Albumin Globulin | Alanin-Aminotransferase
Aspartat-Aminotransferase Alkalische Phosphatase Kalium Natrium Calcium Phosphor Chlorid |
| Urinuntersuchung (fakultativ) | |
| Aussehen (Farbe und Trübung)
Menge Spezifisches Gewicht oder Osmolarität pH-Wert | Gesamtprotein
Glucose Blut/Blutzellen |
| *) Da ein längerer Futterentzug die Glucosemessungen bei den behandelten gegenüber den Kontrolltieren verzerren kann, sollte der Studienleiter entscheiden, ob eine Futterkarenz angezeigt ist. Die Dauer des Futterentzugs muss auf die verwendete Art abgestimmt sein; bei der Ratte kann sie 16 Stunden betragen (nächtliche Futterkarenz). Der Nüchternglucosewert kann nach nächtlicher Futterkarenz in der letzten Expositionswoche oder nach nächtlicher Futterkarenz vor der Nekropsie (in letzterem Fall zusammen mit allen anderen klinischen Pathologieparametern) bestimmt werden. | |
39. Gibt es Anhaltspunkte dafür, dass die unteren Atemwege (d. h. die Alveolen) die Hauptablagerungs- und Retentionsorte sind, kann die bronchoalveoläre Lavage (BAL) die Methode der Wahl sein, um hypothesenbasierte Dosis-Wirkungs-Parameter quantitativ zu analysieren, wobei Alveolitis, Lungenentzündung und Phospholipidose im Vordergrund stehen. Auf diese Weise können Veränderungen der Dosis-Wirkungs-Beziehung und des zeitlichen Verlaufs alveolärer Läsionen angemessen untersucht werden. Die BAL-Flüssigkeit kann auf Gesamt- und Differenzialleukozytenzahl, Gesamtprotein und Laktatdehydrogenase analysiert werden. In Betracht gezogen werden können auch Parameter, die auf lysosomale Schäden, Phospholipidose, Fibrose und reizende oder allergische Entzündung hindeuten; dazu kann auch die Bestimmung entzündungsfördernder Zytokine/Chemokine gehören. BAL-Messungen dienen im Allgemeinen zur Ergänzung der Ergebnisse histopathologischer Untersuchungen, können sie aber nicht ersetzen. GD 39 (2) enthält eine Anleitung zur Durchführung der Lungenlavage.
Ophthalmologische Untersuchung
40. Mit einem Ophthalmoskop oder einem gleichwertigen Gerät werden vor der Verabreichung der Prüfsubstanz bei allen Tieren und nach Abschluss der Prüfung bei allen Hochkonzentrations- und Kontrollgruppen der Augenhintergrund, die brechenden Medien, die Iris und die Bindehaut untersucht. Werden Veränderungen der Augen festgestellt, sind alle Tiere in den anderen Gruppen, einschließlich der Satellitengruppe (Reversibilität) ebenfalls zu untersuchen.
Makroskopische Pathologie und Organgewichte
41. Alle Versuchstiere, einschließlich der Tiere, die während der Prüfung sterben oder aus Tierschutzgründen getötet und aus der Studie genommen werden, sind (falls möglich) vollständig zu entbluten und auf makroskopische Veränderungen zu untersuchen. Der Zeitabstand zwischen dem Ende der letzten Exposition des Tiers und seiner Tötung ist zu dokumentieren. Kann die Nekropsie nicht unmittelbar nach Auffinden eines toten Tieres erfolgen, sollte der Körper auf eine Temperatur gekühlt (nicht eingefroren) werden, die tief genug ist, um die Autolyse zu minimieren. Die Nekropsie ist baldmöglichst, in der Regel innerhalb von einem oder zwei Tagen durchzuführen. Alle makroskopischen Veränderungen sollten für jedes Tier protokolliert werden, wobei besonders auf Veränderungen der Atemwege zu achten ist.
42. In Tabelle 2 sind die Organe und Gewebe aufgeführt, die bei der Sektion zur histopathologischen Untersuchung in einem geeigneten Medium aufbewahrt werden sollten. Die Aufbewahrung der in [Klammern] gesetzten Organe und Gewebe sowie aller sonstigen Organe und Gewebe liegt im Ermessen des Studienleiters. Die durch Fettdruck hervorgehobenen Organe sind so bald wie möglich nach der Sektion von anhaftendem Gewebe zu befreien und feucht zu wiegen, um ein Austrocknen zu verhindern. Die Schilddrüse und die Nebenhoden sind nur zu wiegen, wenn dies notwendig ist, da ihre Befreiung von anhaftendem Gewebe die histopathologische Bewertung erschweren kann. Gewebe und Organe sind unmittelbar nach der Nekropsie und je nach verwendetem Fixierungsmittel mindestens 24-48 Stunden vor der Befreiung von anhaftendem Gewebe in 10 %ig gepuffertem Formalin oder einem anderen geeigneten Fixierungsmittel zu fixieren.
Tabelle 2: Bei der Nekropsie aufbewahrte Organe und Gewebe
| Nebennieren
Aorta Knochenmark (und/oder frisches Aspirat) Gehirn (mit Schnitten von Cerebrum, Cerebellum und Medulla/Pons) Caecum Kolon Duodenum [Nebenhoden] [Augen (Netzhaut, Sehnerv) und Lider] Femur und Kniegelenk Gallenblase (falls vorhanden) [Hardersche Drüsen] Herz Ileum Jejunum Nieren [Tränendrüsen (extraorbital)] Larynx (3 Ebenen einschließlich der Basis der Epiglottis) Leber Lunge (alle Lungenlappen auf einer Ebene, einschließlich der Hauptbronchien) Lymphknoten aus der Hilusregion der Lunge, insbesondere bei schlecht löslichen Prüfsubstanzen, die in Partikelform vorliegen. Für gründlichere Untersuchungen und/oder Studien mit immunologischem Schwerpunkt können zusätzliche Lymphknoten in Betracht gezogen werden, z.B. aus der mediastinalen, der cervicalen/submandibulären und/oder der aurikularen Region. Lymphknoten (distal vom Eingangsort) Brustdrüsen (weibliche) Muskel (Oberschenkel) Nasopharyngeale Gewebe (mindestens 4 Ebenen; 1 Ebene muss den Nasen-Rachen-Gang und das Lymphgewebe des Nasen-Rachen-Raums (NALT) umfassen. | Speiseröhre
[Riechkolben] Ovarien Pankreas Nebenschilddrüsen Periphere Nerven (N. ischiadicus oder N. tibialis, vorzugsweise in der Nähe des Muskels) Hypophyse Prostata Rectum Speicheldrüsen Samenbläschen Haut Rückenmark (zervical, mittlerer Thoraxbereich und lumbar) Milz Brustbein Magen Zähne Hoden Thymus Schilddrüse [Zunge] Trachea (mindestens 2 Ebenen mit einem Längsschnitt durch die Carina und 1 Querschnitt) [Harnleiter] [Harnröhre] Harnblase Uterus Zielorgane Alle makroskopischen Läsionen und Massen |
43. Die Lungen sind in intaktem Zustand zu entfernen, zu wiegen und mit einem geeigneten Füierungsmittel bei einem Druck von 20-30 cm Wasser zu behandeln, damit die Lungenstruktur erhalten bleibt (5). Die Schnitte werden bei allen Lungenlappen auf einer Ebene einschließlich der Hauptbronchien hergestellt; wenn eine Lungenlavage durchgeführt wird, ist der nicht gewaschene Lappen jedoch auf drei Ebenen zu schneiden (keine seriellen Schnitte).
44. Mindestens vier Ebenen der nasopharyngealen Gewebe sind zu untersuchen; eine der Ebenen sollte den Nasen- Rachen-Gang umfassen (5, 6, 7, 8, 9), damit das Plattenepithel, das (nicht Zilientragende respiratorische) Übergangsepithel, das (Zilientragende respiratorische) Flimmerepithel und das Riechepithel sowie das Lymphgewebe (NALT; 10, 11) gründlich untersucht werden können. Drei Ebenen des Larynx sind zu untersuchen; eine dieser Ebenen sollte die Basis der Epiglottis enthalten (12). Mindestens zwei Ebenen der Trachea sind zu untersuchen, darunter ein Längsschnitt durch die Carina der Bifurkation der extrapulmonalen Bronchien und ein Querschnitt.
Histopathologie
45. Die in Tabelle 2 aufgeführten Organe und Gewebe der Tiere in der Kontrollgruppe und der Gruppe mit der höchsten Konzentration und aller während der Studie gestorbenen oder getöteten Tiere sollten histopathologisch untersucht werden. Besonderes Augenmerk ist auf Atemwege, Zielorgane und makroskopische Veränderungen zu richten. Die Organe und Gewebe, die in der Gruppe mit der höchsten Konzentration makroskopische Veränderungen aufweisen, sollten in allen Gruppen untersucht werden. Der Studienleiter kann beschließen, histopathologische Untersuchungen bei zusätzlichen Gruppen durchzuführen, um eine eindeutige Konzentrationswirkung nachzuweisen. Umfasst eine Prüfung auch eine Satellitengruppe (Reversibilitätsprüfung), sind alle Gewebe und Organe histopathologisch zu untersuchen, bei denen in den Behandlungsgruppen Wirkungen aufgetreten sind. Treten in der Gruppe mit der höchsten Konzentration übermäßig viele frühzeitige Todesfälle oder andere Probleme auf, die die Signifikanz der Daten beeinträchtigen, so ist die nächstniedrigere Konzentration histopathologisch zu untersuchen. Man sollte versuchen, die makroskopischen Befunde mit den Ergebnissen der mikroskopischen Untersuchung zu korrelieren.
Daten und Berichterstattung
Daten
46. Körpergewichte, Futteraufnahme, Ergebnisse der klinischen Pathologie, makroskopische Befunde, Organgewichte und Ergebnisse der Histopathologie sind für die einzelnen Tiere anzugeben. Die Daten der klinischen Beobachtung sollten in tabellarischer Form zusammengefasst werden. Daraus müssen für jede Prüfgruppe die Anzahl der verwendeten Tiere, die Anzahl der Tiere mit spezifischen Toxizitätszeichen, die Anzahl der Tiere, die während der Prüfung tot aufgefunden oder vorzeitig getötet wurden, der Todeszeitpunkt der einzelnen Tiere, eine Beschreibung und der zeitliche Verlauf der toxischen Wirkungen und deren Reversibilität sowie die Sektionsbefunde ersichtlich sein. Sowohl die quantitativen als auch die gelegentlich erzielten Ergebnisse sind anhand eines geeigneten statistischen Verfahrens zu bewerten. Hierzu ist eine allgemein anerkannte Statistikmethode heranzuziehen; die Statistikmethoden sind bei der Auslegung der Studie festzulegen.
Prüfbericht
47. Der Prüfbericht sollte, soweit zutreffend, die folgenden Informationen enthalten:
Versuchstiere und Tierhaltung
Prüfsubstanz
Vehikel
Inhalationskammer
Expositionsdaten
Prüfbedingungen
Ergebnisse
Diskussion und Auswertung der Ergebnisse
Literatur:
1. OECD (1981). Subchronic Inhalation Toxicity Testing, Original Test Guideline No 413, Environment Directorate, OECD, Paris.
2. OECD (2009). Guidance Document on Acute Inhalation Toxicity Testing, Environmental Health and Safety Monograph Series on Testing and Assessment No. 39, ENV/JM/MONO(2009)28, OECD, Paris.
3. OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris.
4. Whalan E and Redden JC (1994). Interim Policy for Particle Size and Limit Concentration Issues in Inhalation Toxicity Studies. Office of Pesticide Programs, United States Environmental Protection Agency.
5. Dungworth DL, Tyler WS, Plopper CE (1985). Morphological Methods for Gross and Microscopic Pathology (Chapter 9) in Toxicology of Inhaled Material, Witschi, H.P. and Brain, J.D. (eds), Springer Verlag Heidelberg, pp. 229-258.
6. Young JT (1981). Histopathological examination of the rat nasal cavity. Fundam. Appl. Toxicol. 1: 309-312.
7. Harkema JR (1990). Comparative pathology of the nasal mucosa in laboratory animals exposed to inhaled irritants. Environ. Health Perspect. 85: 231-238.
8. Woutersen RA, Garderen-Hoetmer A, van Slootweg PJ, Feron VJ (1994). Upper respiratory tract carcinogenesis in experimental animals and in humans. In: Waalkes MP and Ward JM (eds) Carcinogenesis. Target Organ Toxi-cology Series, Raven Press, New York, 215-263.
9. Mery S, Gross EA, Joyner DR, Godo M, Morgan KT (1994). Nasal diagrams: A tool for recording the distribution of nasal lesions in rats and mice. Toxicol. Pathol. 22: 353-372.
10. Kuper CF, Koornstra PJ, Hameleers DMH, Biewenga J, Spit BJ, Duijvestijn AM, Breda Vriesman van PJC, Sminia T (1992). The role of nasopharyngeal lymphoid tissue. Immunol. Today 13: 219-224.
11. Kuper CF, Arts JHE, Feron VJ (2003). Toxicity to nasal-associated lymphoid tissue. Toxicol. Lett. 140-141: 281- 285.
12. Lewis DJ (1981). Mitotic Indices of Rat Laryngeal Epithelia. Journal of Anatomy 132(3): 419-428.
13. Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
Anlage 1 Definition
Prüfsubstanz: jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird.
B.30 Prüfungen auf chronische Toxizität
Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 452 (2009). Die ursprüngliche TG 452 wurde 1981 angenommen. Die Entwicklung dieser überarbeiteten Prüfmethode B.30 wurde für notwendig gehalten, um neueren Entwicklungen auf dem Gebiet des Tierschutzes sowie Regulierungsanforderungen Rechnung zu tragen (1) (2) (3) (4). Diese Prüfmethode B.30 wurde parallel zur Überarbeitung von Kapitel B.32 dieses Anhang 'Prüfungen auf Kanzerogenität' und Kapitel B.33 dieses Anhangs 'Kombinierte Studien zur Prüfung auf chronische Toxizität und Kanzerogenität' aktualisiert mit dem Ziel, zusätzliche Informationen von den in der Untersuchung verwendeten Tieren und detailliertere Angaben zur Wahl der Dosis zu gewinnen. Diese Prüfmethode ist dazu ausgelegt, für die Prüfung eines breiten Spektrums von chemischen Stoffen, einschließlich Pestiziden und Industriechemikalien, verwendet zu werden.
2. Bei den meisten Prüfungen auf chronische Toxizität werden Nagetierarten verwendet; diese Prüfmethode gilt daher hauptsächlich für Prüfungen an diesen Arten. Wenn solche Prüfungen an Nichtnagern erforderlich sind, können die in dieser Prüfmethode beschriebenen Grundsätze und Verfahren in Kombination mit denen des Kapitels B.27 dieses Anhangs 'Prüfung auf subchronische orale Toxizität - 90-Tage-Toxizitätsstudie mit wiederholter Verabreichung an Nicht-Nagetieren' (5), mit entsprechenden Änderungen, wie im OECD Guidance Document No. 116 on the Design and Conduct of Chronic Toxicity and Carcinogenicity Studies (6) beschrieben, ebenfalls angewendet werden.
3. Bei Prüfungen auf chronische Toxizität finden im Wesentlichen die orale, die dermale und die inhalative Verabreichung Anwendung. Die Wahl des Verabreichungswegs hängt von den physikalischen und chemischen Eigenschaften der Prüfsubstanz und der vorherrschenden Art der Exposition beim Menschen ab. Zusätzliche Informationen zur Wahl des Expositionswegs sind im OECD Guidance Document No. 116 (6) enthalten.
4. Bei dieser Prüfmethode steht die Exposition auf oralem Weg im Vordergrund. Dies ist der bei Prüfungen auf chronische Toxizität am häufigsten verwendete Expositionsweg. Zwar sind Prüfungen auf Langzeittoxizität mit Exposition über die Haut oder durch Inhalation möglicherweise auch notwendig, um das Risiko für die menschliche Gesundheit zu beurteilen, und/oder im Rahmen bestimmter Regelungen vorgeschrieben, aber diese beiden Expositionswege sind aus technischer Sicht außerordentlich komplex. Derartige Prüfungen sind von Fall zu Fall zu konzipieren, wobei die hier beschriebene Prüfmethode für die Bewertung und Evaluierung chronischer Toxizität bei oraler Verabreichung allerdings mit Bezug auf Empfehlungen für Behandlungszeiten, klinische und Pathologieparameter usw. als Grundlage für ein Protokoll für Inhalations- und/oder dermale Prüfungen dienen könnte. Es gibt OECD-Leitlinien für die Verabreichung von Prüfsubstanzen durch Inhalation (6) (7) und über die Haut (6). Bei der Konzeption länger dauernder Prüfungen mit Exposition durch Inhalation sind insbesondere Kapitel B.8 dieses Anhangs (8) und Kapitel B.29 dieses Anhangs (9) sowie das OECD Guidance Document über Prüfungen auf akute Toxizität nach Inhalation (7) zu berücksichtigen. Bei Prüfungen mit dermaler Applikation ist Kapitel B.9 dieses Anhangs (10) zu beachten.
5. Die Prüfung auf chronische Toxizität liefert Informationen über mögliche gesundheitliche Schädigungen, die durch wiederholte Exposition über einen beträchtlichen Teil der Lebensdauer der verwendeten Art entstehen können. Sie liefert Informationen über die toxischen Wirkungen der Prüfsubstanz und gibt Hinweise auf Zielorgane und die Möglichkeit der Akkumulation. Außerdem kann sie zur Ableitung eines NOAEL-Werts (Dosis ohne beobachtete schädigende Wirkung) beitragen, der zur Festlegung von Sicherheitskriterien für die Humanexposition herangezogen werden kann. Ferner wird die Notwendigkeit sorgfältiger klinischer Beobachtung der Tiere hervorgehoben, damit so viele Informationen wie möglich gewonnen werden können.
6. Zu den Zielen von Prüfungen nach dieser Prüfmethode gehören
Ausgangsüberlegungen
7. Bei der Beurteilung und Bewertung der toxikologischen Eigenschaften einer Prüfsubstanz sollte das Prüflabor vor Durchführung der Studie alle verfügbaren Informationen über die Prüfsubstanz berücksichtigen, damit die Studienauslegung auf eine effizientere Prüfung des Potenzials für chronische Toxizität ausgerichtet und die Verwendung von Versuchstieren minimiert werden kann. Für die Auslegung der Studie könnten u. a. Informationen wie die Identität, die chemische Struktur und die physikalisch-chemischen Eigenschaften der Prüfsubstanz, sämtliche Informationen über die Wirkungsweise, Ergebnisse jeglicher In-vitro- oder In-vivo-Toxizitätsprüfungen, vorgesehene Verwendungen und die Möglichkeit der Exposition des Menschen, (Q)SAR-Daten und toxikologische Daten über strukturverwandte Substanzen, toxikokinetische Daten (Kinetik bei Einzel- und Mehrfachdosierung) sowie Daten aus anderen Studien mit wiederholter Exposition herangezogen werden. Die chronische Toxizität sollte erst bestimmt werden, wenn erste Informationen über die Toxizität aus Toxizitätsprüfungen mit wiederholter Gabe über 28 Tage und/oder 90 Tage vorliegen. Im Rahmen der Gesamtbewertung der potenziellen schädlichen Wirkungen einer bestimmten Prüfsubstanz auf die Gesundheit sollte bei der Prüfung auf chronische Toxizität etappenweise vorgegangen werden (11) (12) (13) (14).
8. Vor Beginn der Prüfung ist festzulegen, welche Statistikmethoden angesichts der Versuchsauslegung und der Ziele am besten für die Analyse der Ergebnisse geeignet sind. Dabei ist unter anderem festzulegen, ob bei der statistischen Auswertung Anpassungen in Bezug auf die Überlebensrate zu berücksichtigen sind und welche Art der Analyse bei vorzeitigem Tod der Tiere einer oder mehrerer Gruppen durchzuführen ist. Hinweise zu den geeigneten statistischen Analysen und wichtige Literaturverweise zu international anerkannten Statistikmethoden sind im Guidance Document No. 116 (6) sowie im Guidance Document No. 35 on the analysis and evaluation of chronic toxicity and carcinogenicity studies (15) zu finden.
9. Bei der Durchführung einer Prüfung auf chronische Toxizität sind stets die im OECD Guidance Document No. 19 on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation (16), insbesondere in Absatz 62, genannten Grundsätze und Erwägungen zu befolgen. In diesem Absatz heißt es: Wenn ein Tier in Studien mit wiederholter Verabreichung progressive klinische Anzeichen für eine fortschreitende Verschlechterung seines Zustands aufweist, ist eine informierte Entscheidung zu treffen, ob das Tier tierschutzgerecht getötet werden sollte oder nicht. Bei dieser Entscheidung sind Faktoren wie der Wert der Informationen, die bei Verbleiben des Tiers in der Studie gewonnen werden können, und der allgemeine Gesundheitszustand des Tiers gegeneinander abzuwägen. Wird beschlossen, das Tier in der Studie zu belassen, ist es entsprechend den Erfordernissen häufiger zu beobachten. Zur Linderung von Schmerzen oder Qualen kann auch die Verabreichung der Prüfsubstanz unterbrochen oder die Dosis verringert werden, sofern dabei der Zweck der Prüfung nicht beeinträchtigt wird.
10. Ausführliche Informationen und eine Diskussion der Prinzipien der Dosiswahl bei Prüfung auf chronische Toxizität und Kanzerogenität sind im Guidance Document No. 116 (6) sowie in zwei Veröffentlichungen des International Life Sciences Institute (17) (18) zu finden. Die grundlegende Strategie der Dosiswahl hängt von den Hauptzielen der Studie ab (Nummer 6). Bei der Auswahl der geeigneten Dosisstufen ist ein Gleichgewicht zwischen Gefahrenidentifizierung einerseits und der Beschreibung von Wirkungen bei geringer Dosierung und ihrer Relevanz andererseits herzustellen. Dieses Gleichgewicht ist bei einer kombinierten Studie zur Prüfung auf chronische Toxizität und auf Kanzerogenität (Kapitel B.3 3 dieses Anhangs) besonders wichtig (Nummer 11).
11. Es sollte in Betracht gezogen werden, statt der getrennten Durchführung einer Prüfung auf chronische Toxizität (diese Prüfmethode B.30) und einer Prüfung auf Kanzerogenität (Kapitel B.32 dieses Anhangs) eher eine kombinierte Prüfung auf chronische Toxizität und Kanzerogenität (Kapitel B.33 dieses Anhangs) durchzuführen. Die kombinierte Methode ist zeit- und kosteneffizienter als zwei getrennte Studien, ohne dass dabei die Qualität der Daten der chronischen Komponente oder der Kanzerogenitätskomponente beeinträchtigt würde. Bei Durchführung einer kombinierten Prüfung auf chronische Toxizität und Kanzerogenität (Kapitel B.33 dieses Anhangs) sind die Grundsätze der Dosiswahl (Nummer 9 und Nummern 20-25) jedoch genau zu beachten. Es wird allerdings anerkannt, dass im Rahmen bestimmter Regelungen möglicherweise getrennte Prüfungen vorgeschrieben sind.
12. Die im Zusammenhang mit dieser Prüfmethode verwendeten Begriffe werden am Ende dieses Kapitels und im Guidance Document 116 (6) definiert.
Prinzip der Prüfmethode
13. Die Prüfsubstanz wird mehreren Gruppen von Versuchstieren täglich in abgestuften Dosen verabreicht, und zwar normalerweise über einen Zeitraum von 12 Monaten, wobei je nach Regulierungsanforderungen (siehe Nummer 33) auch eine längere oder kürzere Dauer gewählt werden kann. Die Dauer sollte ausreichend lang sein, damit sich etwaige Wirkungen kumulativer Toxizität manifestieren können, ohne dass sich die verzerrenden Wirkungen geriatrischer Veränderungen bemerkbar machen. Abweichungen von der 12-monatigen Expositionsdauer sind zu begründen, insbesondere wenn eine kürzere Dauer gewählt wird. Die Prüfsubstanz wird in der Regel oral verabreicht, aber auch die Verabreichung durch Inhalation oder auf dermalem Weg kann angebracht sein. Die Studienauslegung kann auch eine oder mehrere zwischenzeitliche Tötungen vorsehen, z.B. nach 3 und 6 Monaten; außerdem können zu diesem Zweck auch zusätzliche Gruppen von Versuchstieren aufgenommen werden (siehe Nummer 19). Während des Verabreichungszeitraums werden die Tiere sorgfältig auf Toxizitätszeichen beobachtet. Tiere, die im Verlauf der Prüfung sterben, und vorzeitig getötete Tiere werden seziert; die nach Abschluss der Prüfung überlebenden Tiere werden getötet und ebenfalls seziert.
Beschreibung der Methode
Auswahl von Versuchstierarten
14. Diese Prüfmethode betrifft hauptsächlich die Bewertung und Evaluierung chronischer Toxizität bei Nagetieren (Nummer 2). Bestimmte Regelungen können jedoch ähnliche Studien an Nichtnagern vorschreiben. Die Wahl der Tierart ist zu begründen. Wenn Prüfungen auf chronische Toxizität bei Nichtnagern erforderlich sind, sollten Auslegung und Durchführung der Versuche auf den Grundsätzen der vorliegenden Prüfmethode sowie denen von Kapitel B.27 dieses Anhangs - Prüfung auf subchronische orale Toxizität - 90-Tage-Toxizitätsstudie mit wiederholter Verabreichung an Nicht-Nagetieren (5) basieren. Zusätzliche Informationen zur Wahl der Tierart und des Stamms sind im Guidance Document No. 116 (6) enthalten.
15. Bei dieser Prüfmethode ist die Ratte die bevorzugte Nagetierart, doch können auch andere Nagetierarten, z.B. die Maus, verwendet werden. Ratten und Mäuse sind wegen ihrer relativ kurzen Lebenszeit, der weit verbreiteten Verwendung in pharmakologischen und toxikologischen Studien, ihrer Anfälligkeit für die Entstehung von Tumoren und der Verfügbarkeit ausreichend beschriebener Stämme die bevorzugten Versuchsmodelle. Daher liegen über ihre Physiologie und Pathologie umfangreiche Informationen vor. Es sind junge, gesunde, adulte Tiere aus üblicherweise eingesetzten Laborstämmen zu verwenden. Für die Prüfung auf chronische Toxizität sollten Tiere desselben Stamms und derselben Herkunft verwendet werden wie für die kürzere(n) Toxizitätsvorstudie(n). Die weiblichen Tiere dürfen weder bereits geworfen haben noch trächtig sein.
Haltungs- und Fütterungsbedingungen
16. Die Tiere können entweder einzeln oder in kleinen gleichgeschlechtlichen Gruppen in Käfigen untergebracht werden. Eine Einzelunterbringung ist nur in Betracht zu ziehen, wenn sie wissenschaftlich gerechtfertigt ist (19) (20) (21). Die Käfige sollten so angeordnet werden, dass etwaige Einflüsse der Käfigplatzierung minimiert werden. Die Temperatur im Versuchstierraum sollte 22 °C (± 3 °C) betragen. Die relative Luftfeuchtigkeit sollte mindestens 30 % betragen und - außer beim Reinigen des Raums - 70 % nicht überschreiten. Angestrebt werden sollte eine Luftfeuchtigkeit von 50-60 %. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Das Futter sollte den Nährstoffbedarf der eingesetzten Tierart decken und möglichst wenig Schadstoffe wie z.B. Pestizidrückstände, persistente organische Schadstoffe, Phytoöstrogene, Schwermetalle und Mykotoxine enthalten, die das Ergebnis der Prüfung beeinflussen könnten. Das Futter ist regelmäßig, und zwar zumindest zu Beginn der Prüfung und bei Verwendung einer anderen Charge, auf den Nährstoff- und Schadstoffgehalt hin zu analysieren; die Ergebnisse sind im Abschlussbericht anzugeben. Ergebnisse von Analysen des in der Prüfung verwendeten Trinkwassers sind ebenfalls anzugeben. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung einer Prüfsubstanz und die Deckung des Nährstoffbedarfs der Tiere sichergestellt werden muss, wenn die Prüfsubstanz mit dem Futter verabreicht wird.
Vorbereitung der Tiere
17. Es sind gesunde Tiere zu verwenden, die mindestens sieben Tage an die Laborbedingungen gewöhnt und zuvor nicht für andere Experimente verwendet wurden. Bei Nagetieren sollte die Verabreichung so bald wie möglich nach dem Absetzen und der Eingewöhnung beginnen, vorzugsweise bevor die Tiere 8 Wochen alt sind. Art, Stamm, Herkunft, Geschlecht, Gewicht und Alter der Versuchstiere sind anzugeben. Bei Beginn der Studie sollten die Gewichtsunterschiede bei den Tieren beider Geschlechter möglichst gering sein und ± 20 % des geschlechtsspezifischen Durchschnittsgewichts aller Tiere in der Studie nicht überschreiten. Die Tiere werden nach dem Zufallsprinzip in Kontroll- und Behandlungsgruppen eingeteilt. Nach der Randomisierung sollte sich das Durchschnittsgewicht der Tiere desselben Geschlechts von Gruppe zu Gruppe nicht signifikant unterscheiden. Gibt es statistisch signifikante Unterschiede, sollte die Randomisierung möglichst wiederholt werden. Jedes Versuchstier erhält zur sicheren Identifizierung eine eigene Nummer und wird durch Tätowierung, Mikrochipimplantat oder auf eine andere geeignete Weise mit dieser Nummer gekennzeichnet.
Verfahren
Zahl und Geschlecht der Versuchstiere
18. Es sind Tiere beider Geschlechter zu verwenden. Es sind so viele Tiere zu verwenden, dass am Ende der Studie in jeder Gruppe genug Tiere für eine gründliche biologische und statistische Auswertung zur Verfügung stehen. Bei Nagern sind in der Regel auf jeder Dosisstufe je Gruppe mindestens je 20 Tiere beider Geschlechter zu verwenden, bei Nichtnagern werden pro Gruppe mindestens je 4 Tiere beider Geschlechter empfohlen. Bei Studien an Mäusen werden in jeder Dosisgruppe möglicherweise zusätzliche Tiere benötigt, um alle erforderlichen hämatologischen Bestimmungen vornehmen zu können.
Tötungen im Verlauf der Studie, Satellitengruppen und Sentineltiere
19. Es kann vorgesehen werden, dass im Verlauf der Studie, z.B. nach sechs Monaten, Tiere getötet werden (mindestens 10 Tiere/Geschlecht/Gruppe), um Erkenntnisse über den Fortgang toxikologischer Veränderungen und mechanistische Informationen zu gewinnen, falls dies wissenschaftlich gerechtfertigt ist. Wenn diese Informationen bereits aus vorherigen Studien mit wiederholter Gabe der Prüfsubstanz vorliegen, sind zwischenzeitliche Tötungen möglicherweise wissenschaftlich nicht gerechtfertigt. Es können auch Satellitengruppen aufgenommen werden, um die Reversibilität etwaiger toxikologischer Veränderungen zu beobachten, die durch die Prüfsubstanz hervorgerufen werden. Satellitengruppen sind normalerweise auf die höchste Dosisgruppe der Studie zuzüglich einer Kontrolle beschränkt. Der Krankheitsstatus während der Studie kann erforderlichenfalls auch mit einer zusätzlichen Gruppe von Sentineltieren (normalerweise je fünf Tiere beider Geschlechter) überwacht werden (22). Sollen im Verlauf der Prüfung Tiere getötet werden oder Satelliten- oder Sentinelgruppen aufgenommen werden, ist die Zahl der Tiere in der Studienauslegung um die Zahl zu erhöhen, die vor Abschluss der Studie getötet werden sollen. Diese Tiere sind normalerweise in Bezug auf die Bestimmung des Körpergewichts und der Futter-/Wasseraufnahme, hämatologische und klinisch-biochemische Bestimmungen sowie pathologische Untersuchungen genauso zu behandeln wie die Tiere in der chronischen Toxizitätsphase der Hauptstudie; es kann allerdings vorgesehen werden, die Messungen (in den Gruppen mit im Verlauf der Studie zu tötenden Tieren) auf bestimmte Schlüsselaspekte wie Neurotoxizität oder Immunotoxizität zu beschränken.
Dosisgruppen und Dosierung
20. Das Guidance Document No. 116 (6) enthält Hinweise zu allen Aspekten der Dosiswahl und zu den Abständen der Dosisstufen. Es sollten mindestens drei Dosisstufen und eine gleichzeitige Kontrolle verwendet werden, es sei denn, ein Limit-Test wird durchgeführt (siehe Nummer 27). Die Dosisstufen werden im Allgemeinen auf der Grundlage der Ergebnisse von Studien mit kurzzeitiger wiederholter Verabreichung oder Dosisfindungsstudien festgelegt und sollten alle vorliegenden toxikologischen und toxikokinetischen Daten für die Prüfsubstanz oder verwandte Stoffe berücksichtigen.
21. Soweit keine Beschränkungen aufgrund der physikalisch-chemischen Beschaffenheit oder der biologischen Wirkungen der Prüfsubstanz bestehen, ist die höchste Dosisstufe normalerweise so zu wählen, dass zwar die Hauptzielorgane und die toxischen Wirkungen identifiziert werden können, aber Leiden, schwere Toxizität, Morbidität oder Tod der Tiere vermieden werden. Unter Berücksichtigung der unter Nummer 22 beschriebenen Faktoren ist die höchste Dosisstufe so zu wählen, dass Anzeichen von Toxizität hervorgerufen werden, die sich z.B. in einer verzögerten Körpergewichtsentwicklung äußern (etwa 10 %).
22. Je nach den Zielen der Studie (siehe Nummer 6) kann jedoch eine Höchstdosis festgelegt werden, die unter der zu Toxizitätszeichen führenden Dosis liegt, z.B. wenn eine Dosis eine besorgniserregende Wirkung auslöst, die sich aber nur geringfügig auf Lebensdauer oder Körpergewicht auswirkt. Die Höchstdosis darf 1.000 mg/kg Körpergewicht/Tag nicht übersteigen (Grenzdosis siehe Nummer 27).
23. Die Dosisstufen und die Abstände der Dosisstufen können so gewählt werden, dass auf der niedrigsten Dosisstufe eine Dosis-Wirkungs-Beziehung und ein NOAEL-Wert oder andere angestrebte Studienergebnisse, z.B. ein BMD (siehe Nummer 25), festgestellt werden können. Zu den Faktoren, die bei der Festlegung niedrigerer Dosen zu berücksichtigen sind, gehören die voraussichtliche Steigung der Dosis-Wirkungs-Kurve, die Dosen, bei denen wichtige Änderungen des Metabolismus oder der toxischen Wirkungsweise eintreten können, und das Niveau, bei dem eine Schwelle oder ein Ausgangspunkt für eine Extrapolation niedriger Dosen erwartet wird.
24. Welche Dosisstufenabstände gewählt werden, hängt von den Merkmalen der Prüfsubstanz ab und kann in dieser Prüfmethode nicht vorgeschrieben werden. Abstände mit einem Faktor von 2 bis 4 erweisen sich jedoch oft als gut geeignet für die Festsetzung abnehmender Dosisstufen. Außerdem ist es oft besser, statt der Verwendung sehr großer Intervalle (z.B. mit einem Faktor zwischen 6 und 10) zwischen den Dosierungen eine vierte Prüfgruppe einzurichten. Die Verwendung von Faktoren über 10 sollte im Allgemeinen vermieden bzw. gegebenenfalls begründet werden.
25. Entsprechend dem Guidance Document No. 116 (6) sind bei der Dosisfestlegung u. a. folgende Faktoren zu berücksichtigen:
26. Die Kontrollgruppe ist eine unbehandelte Gruppe oder eine Vehikelkontrollgruppe, sofern ein Vehikel zur Verabreichung der Prüfsubstanz verwendet wird. Abgesehen von der Behandlung mit der Prüfsubstanz sollten die Tiere der Kontrollgruppe genauso behandelt werden wie die Tiere in den Prüfgruppen. Wird ein Vehikel verwendet, erhält die Kontrollgruppe das Vehikel im höchsten bei den Dosisgruppen verwendeten Volumen. Wird eine Prüfsubstanz mit dem Futter verabreicht, und führt dies aus geschmacklichen Gründen zu einer verminderten Futteraufnahme, kann eine paarweise gefütterte Kontrollgruppe nützlich und eine bessere Kontrolle sein.
27. Wenn aufgrund von Informationen aus Vorstudien davon ausgegangen werden kann, dass eine Prüfung bei einer einzigen Dosisstufe von mindestens 1.000 mg/kg Körpergewicht/Tag unter Anwendung der für diese Studie beschriebenen Verfahren wahrscheinlich keine schädlichen Wirkungen hervorrufen wird und wenn aufgrund der Daten strukturverwandter Stoffe keine Toxizität zu erwarten ist, kann auf eine vollständige Studie mit drei Dosisstufen gegebenenfalls verzichtet werden. Sofern die Exposition des Menschen nicht die Prüfung bei einer höheren Dosis erforderlich erscheinen lässt, kann eine Grenze von 1.000 mg/kg Körpergewicht/Tag angewandt werden.
Zubereitung der Dosen und Verabreichung der Prüfsubstanz
28. Die Prüfsubstanz wird in der Regel oral, d. h. mit der Nahrung oder dem Trinkwasser, oder über eine Schlundsonde verabreicht. Zusätzliche Informationen zu Verabreichungswegen und -methoden sind im Guidance Document No. 116 (6) enthalten. Verabreichungsweg und -methode richten sich nach dem Zweck der Studie, den physikalisch-chemischen Eigenschaften der Prüfsubstanz, ihrer Bioverfügbarkeit und der vorherrschenden Art der Exposition beim Menschen. Der gewählte Verabreichungsweg und die Methode sollten begründet werden. Aus Tierschutzgründen sollte die Schlundsonde nur für Agenzien gewählt werden, bei denen dieser Weg und diese Methode der Verabreichung die potenzielle Humanexposition annähernd repräsentieren (z.B. Arzneimittel). Nahrungs- oder Umweltchemikalien einschließlich Pestizide werden normalerweise mit dem Futter oder dem Trinkwasser verabreicht. Für bestimmte Szenarios, z.B. berufliche Exposition, können andere Applikationswege besser geeignet sein.
29. Bei Bedarf wird die Prüfsubstanz in einem geeigneten Vehikel gelöst oder suspendiert. Zu berücksichtigen sind gegebenenfalls folgende Merkmale des Vehikels und anderer Additive: Auswirkungen auf die Resorption, die Verteilung, den Stoffwechsel oder die Retention der Prüfsubstanz, Auswirkungen auf die chemischen Eigenschaften der Prüfsubstanz, die deren toxische Eigenschaften verändern können, und ferner Auswirkungen auf die Futter- oder Wasseraufnahme oder den Ernährungszustand der Versuchstiere. Es empfiehlt sich, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspenion, sodann eine Lösung/Emulsion in Öl (z. 8. Maisöl) und erst dann eine Lösung in anderen Vehikeln in Betracht zu ziehen. Bei anderen Vehikeln als Wasser müssen deren toxische Merkmale bekannt sein. Es sollten Informationen über die Stabilität der Prüfsubstanz und die Homogenität der Lösungen oder Futterrationen, die (je nach Fall) die Dosierung enthalten, unter den Verabreichungsbedingungen (z.B. mit dem Futter) vorliegen.
30. Für mit dem Futter oder dem Trinkwasser verabreichte Stoffe ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfsubstanz die normale Nahrungsaufnahme oder den Wasserhaushalt nicht beeinträchtigen. Um eine unausgewogene Ernährung zu vermeiden, sollte die Konzentration der Prüfsubstanz in Langzeittoxizitätsstudien mit Verabreichung über die Nahrung normalerweise eine Obergrenze von 5 % der Gesamtnahrung nicht übersteigen. Wenn die Prüfsubstanz mit dem Futter verabreicht wird, kann entweder eine konstante Futterkonzentration (mg/kg Futter oder ppm) oder eine konstante Dosierung in Relation zum Körpergewicht des Tiers (mg/kg Körpergewicht), berechnet auf Wochenbasis, verwendet werden. Die jeweils gewählte Verfahrensweise ist anzugeben.
31. Bei oraler Verabreichung erhalten die Tiere die Prüfsubstanz täglich (sieben Tage in der Woche), in der Regel für einen Zeitraum von 12 Monaten (siehe auch Nummer 33); je nach Regulierungsanforderungen kann auch eine längere Dauer erforderlich sein. Jede Abweichung von diesem Dosierungsplan, z.B. fünf Tage pro Woche, ist zu begründen. Bei dermaler Applikation werden die Tiere, wie in Kapitel B.9 dieses Anhangs (10) beschrieben, für einen Zeitraum von 12 Monaten an sieben Tagen in der Woche mindestens 6 Stunden pro Tag mit der Prüfsubstanz behandelt. Die Inhalationsexposition wird an sieben Tagen in der Woche für 6 Stunden pro Tag durchgeführt, aber auch fünf Tage in der Woche sind möglich, wenn dies gerechtfertigt ist. Die Expositionsdauer beträgt normalerweise 12 Monate. Wenn für eine 'Nose-only'-Exposition andere Nagetierarten als Ratten verwendet werden, kann die maximale Expositionsdauer angepasst werden, um artenspezifisches Leiden zu minimieren. Eine Expositionsdauer von weniger als 6 Stunden pro Tag ist zu begründen. Siehe auch Kapitel B.8 dieses Anhangs (8).
32. Wird die Prüfsubstanz über eine Sonde verabreicht, so sollte dies unter Verwendung einer Schlundsonde oder einer geeigneten Intubationskanüle jeweils zur gleichen Tageszeit erfolgen. Normalerweise wird einmal am Tag eine einzige Dosis verabreicht; wenn es sich bei der Prüfsubstanz z.B. um einen lokal reizenden Stoff handelt, kann die Tagesdosis auf zwei Teilmengen aufgeteilt werden. Das maximale Flüssigkeitsvolumen, das einem Versuchstier jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte so gering wie möglich sein und bei Nagetieren normalerweise 1 ml/100 g Körpergewicht nicht überschreiten (22). Die Variabilität der Prüfvolumina sollte durch Anpassung der Konzentration auf ein konstantes Volumen bei allen Dosen möglichst gering gehalten werden. Davon ausgenommen sind potenziell ätzende oder reizende Stoffe, die zur Vermeidung schwerwiegender lokaler Wirkungen verdünnt werden müssen. Die Prüfung in Konzentrationen, die wahrscheinlich eine ätzende oder reizende Wirkung für den Magen-Darm-Trakt haben, ist zu vermeiden.
Versuchsdauer
33. Diese Prüfmethode ist zwar hauptsächlich zur Prüfung auf chronische Toxizität über 12 Monate ausgelegt, aber je nach den Anforderungen bestimmter Regulierungsregelungen oder für spezifische mechanistische Zwecke sind auch kürzere (z.B. 6 oder 9 Monate) oder längere (z.B. 18 oder 24 Monate) Zeiträume möglich. Abweichungen von der 12-monatigen Expositionsdauer sind zu begründen, insbesondere wenn eine kürzere Dauer gewählt wird. Satellitengruppen zur Beobachtung der Reversibilität von toxikologischen Veränderungen, die durch die Prüfsubstanz hervorgerufen werden, sollten nach Beendigung der Exposition noch für einen Zeitraum von mindestens 4 Wochen, aber nicht länger als ein Drittel der Gesamtstudiendauer ohne Behandlung beibehalten werden. Weitere Hinweise, insbesondere in Bezug auf das Überleben der Versuchstiere, sind im Guidance Document No. 116 (6) zu finden.
Beobachtungen
34. Alle Tiere sind in der Regel jeden Tag morgens und abends, auch am Wochenende und an Feiertagen, auf Anzeichen von Morbidität und Mortalität hin zu untersuchen. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich, vorzugsweise zur selben Tageszeit, unter Berücksichtigung des Zeitraums, in dem der Wirkungsgipfel nach Verabreichung der Dosis über eine Schlundsonde zu erwarten ist, vorgenommen werden.
35. Bei allen Tieren sind mindestens einmal vor der ersten Exposition (für intraindividuelle Vergleiche), am Ende der ersten Studienwoche und danach monatlich gründliche klinische Beobachtungen vorzunehmen. Das Beobachtungsprotokoll ist so zu gestalten, dass Abweichungen zwischen den Beobachtern minimiert werden und von der Prüfgruppe unabhängig stattfinden. Diese Beobachtungen sollten außerhalb des Käfigs erfolgen, in dem die Tiere gehalten werden, und zwar vorzugsweise in einem Standardgehege jeweils zu denselben Zeiten. Sie sind sorgfältig zu dokumentieren, am besten nach einer speziell vom Prüflabor entwickelten Bewertungsskala. Die Beobachtungsbedingungen sollten möglichst konstant sein. Zu achten ist insbesondere auf Veränderungen an Haut, Fell, Augen, Schleimhäuten, auf Sekrete und Exkrete sowie autonome Aktivitäten (z.B. Tränensekretion, Piloerektion, Pupillengröße, ungewöhnliche Atemmuster). Gang- und Haltungsstörungen, ferner Reaktionen auf den Umgang mit den Tieren sowie etwaige klonische oder tonische Bewegungen, Stereotypien (z.B. übermäßiges Putzen, wiederholte Kreisbewegungen) oder abnormes Verhalten (z.B. Selbstverstümmelung, Rückwärtsgehen) sollten auch dokumentiert werden (24).
 | weiter. |