umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (21)
| zurück |
|
B.56 Erweiterte Ein-Generationen-Prüfung auf Reproduktionstoxizität 14 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
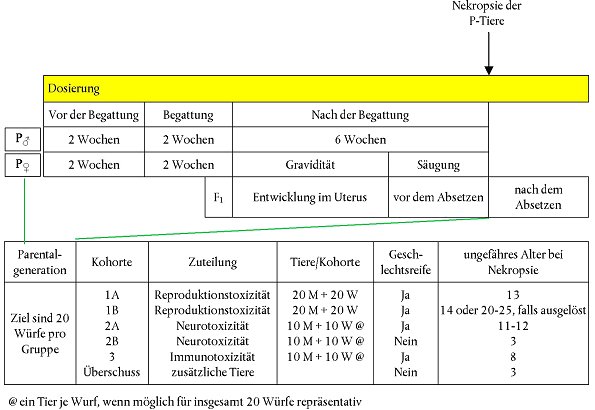
| Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 443 (2012). Sie basiert auf dem Vorschlag des Fachausschusses Agricultural Chemical Safety Assessment (ACSA) des ILSI Health and Environmental Sciences Institute (HESI) für eine in Bezug die Lebensphase F1 erweiterte Ein-Generationen-Prüfung auf Reproduktionstoxizität (EOGRTS), wie in Cooper et al., 2006 (1) veröffentlicht. Der Prüfplan wurde in einigen Punkten verbessert und präzisiert, um Flexibilität zu gewährleisten und deutlich zu machen, dass auf vorhandenem Wissen aufgebaut wird, gleichzeitig jedoch Beobachtungen am lebenden Tier genutzt werden, um den Prüfungsablauf zu steuern und zu maßschneidern. Die nachstehende Prüfmethode beschreibt die Verfahrensschritte einer EOGRTS-Prüfung im Einzelnen. Sie basiert auf drei Kohorten von F1-Tieren: Kohorte 1: Bewertung reproduktionstoxischer/entwicklungstoxischer Endpunkte; diese Kohorte kann auf eine F2-Generation erweitert werden. Kohorte 2: Bewertung der potenziellen Auswirkung der Exposition gegenüber der Prüfsubstanz auf das sich entwickelnde Nervensystem. Kohorte 3: Bewertung der potenziellen Auswirkung der Exposition gegenüber der Prüfsubstanz auf das sich entwickelnde Immunsystem. 2. Bei Entscheidungen darüber, ob auch die zweite Generation bewertet und die Kohorten für Entwicklungsneurotoxizität und/oder Entwicklungsimmuntoxizität ausgelassen werden sollten, sollte vorhandenes Wissen über die Prüfsubstanz ebenso berücksichtigt werden wie die Bedürfnisse der verschiedenen Regulierungsbehörden. Zweck der Prüfmethode ist es, die möglichen Verfahrenschritte für die Bewertung der einzelnen Kohorten im Detail zu beschreiben. 3. Für Regulierungsbehörden, die zur Produktion einer zweiten Generation interne Auslöser (trigger) verwenden, enthält das OECD Guidance Document Nr. 117 (39) entsprechende Verfahrensvorschriften. Ausgangsüberlegungen und Ziele 4. Hauptziel der EOGRTS-Prüfung ist es, bestimmte Lebensphasen zu evaluieren, die von anderen Arten von Toxizitätsstudien nicht abgedeckt werden, und nach etwaigen Wirkungen einer prä- und postnatalen Chemikalienexposition zu suchen. Zur Bestimmung der reproduktionstoxischen Endpunkte ist vorgesehen, in einem ersten Schritt - und soweit sie vorliegen - Informationen aus Prüfungen auf Toxizität bei wiederholter Verabreichung einer Prüfsubstanz (einschließlich Screeningstests auf Reproduktionstoxizität, z.B. OECD TG 422 (32)), oder kurzfristige Screeningtests auf endokrine Disruptoren, (z.B. Uterotropher Bioassay - Prüfmethode B.54 (36); und Hershberger-Test - Prüfmethode B.55 (37)) heranzuziehen, um Auswirkungen auf die Fortpflanzungsorgane von männlichen und weiblichen Tieren festzustellen. Diese könnten bei männlichen Tieren Daten über die Spermatogenese (Testikular-Histopathologie) und bei weiblichen Tieren Daten über Östruszyklen, Follikelzahlen/Eizellenreifung und die Integrität der Eierstöcke (Histopathologie) umfassen. Anschließend werden durch die EOGRTS-Prüfung die reproduktionstoxischen Endpunkte untersucht, die die Interaktion von männlichen mit weiblichen Tieren, von weiblichen Tieren mit befruchteten Eizellen (Conceptus) und von weiblichen Tieren mit ihren Nachkommen und der F1-Generation bis nach der Geschlechtsreife voraussetzen (siehe OECD Guidance Document Nr. 151, das diese Prüfmethode (40) unterstützt). 5. Der Prüfplan ermöglicht die Bewertung der prä- und postnatalen Auswirkungen von Chemikalien auf die Entwicklung sowie eine detaillierte Bewertung der systemischen Toxizität bei trächtigen und laktierenden weiblichen Tieren sowie jungen und adulten Nachkommen. Durch ausführliche Untersuchung der wichtigsten entwicklungstoxischen Endpunkte (wie Lebensfähigkeit der Nachkommen, Gesundheit der Neugeborenen, Entwicklungsstand bei der Geburt und körperliche sowie funktionale Entwicklung bis zum Erwachsenenalter) sollen spezifische Zielorgane bei den Nachkommen ermittelt werden. Die Prüfung liefert und/oder bestätigt außerdem Informationen über die Auswirkungen einer Prüfsubstanz auf die Integrität und Leistungsfähigkeit der Fortpflanzungsorgane adulter männlicher und weiblicher Tiere. Es werden speziell - aber nicht ausschließlich - die folgenden Parameter geprüft: Gonadenfunktion, Östruszyklus, epididymale Spermienreifung, Paarungsverhalten, Empfängnis, Trächtigkeit, Geburt und Laktation. Außerdem werden die Erkenntnisse aus den Untersuchungen der Entwicklungsneurotoxizität und Immuntoxizität potenzielle Wirkungen in diesen Systemen charakterisieren. Die aus diesen Prüfungen gewonnenen Daten dürften die Bestimmung der NOAEL-Werte (No Observed Adverse Effect Level), der LOAEL-Werte (Lowest Observed Adverse Effect Level) und/oder Benchmark-Dosen für die verschiedenen Endpunkte ermöglichen und/oder sollten zur Charakterisierung der in früheren Untersuchungen der Toxizität bei wiederholter Verabreichung (repeatdose studies) festgestellten Wirkungen verwendet werden und/oder als Richtwerte für spätere Tests dienen. 6. Ein Schema des Protokolls ist in Abbildung 1 gegeben. Die Prüfsubstanz wird in abgestuften Dosen kontinuierlich an mehrere Gruppen geschlechtsreifer männlicher und weiblicher Versuchstiere verabreicht. Diese Parentalgeneration (P) erhält die Dosen vor der Paarung während eines vorgegebenen Expositionszeitraums (ausgewählt auf Basis der für die Prüfsubstanz vorliegenden Informationen; mindestens aber zwei Wochen) und während eines zweiwöchigen Paarungszeitraums. P-Männchen werden mindestens bis zum Entwöhnen der F1-Nachkommen weiterbehandelt. Die Behandlung sollte mindestens zehn Wochen dauern. Sie können auch länger behandelt werden, falls Auswirkungen auf die Fortpflanzung unschlüssig sind. Die Behandlung der P-Weibchen wird während Trächtigkeit und Laktation bis nach der Entwöhnung ihrer Würfe fortgesetzt (d. h. acht- bis zehnwöchige Behandlung). Die F1-Nachkommen werden vom Zeitpunkt ihrer Entwöhnung bis zum Erwachsenenalter mit der Prüfsubstanz weiterbehandelt. Wird eine zweite Generation untersucht (siehe OECD Guidance Document Nr. 117 (39)), so werden die F1-Nachkommen so lange behandelt, bis die F2-Generation entwöhnt ist bzw. bis die Prüfung abgeschlossen ist. 7. Alle Tiere werden klinisch und pathologisch auf Anzeichen von Toxizität untersucht, wobei insbesondere auf die Integrität und Leistungsfähigkeit der Fortpflanzungsorgane der männlichen und der weiblichen adulten Tiere sowie auf Gesundheit, Wachstum, Entwicklung und Fortpflanzungsfähigkeit der Nachkommen geachtet wird. Am Tag des Absetzens werden ausgewählte Nachkommen für weitere Untersuchungen, einschließlich Untersuchungen der Geschlechtsreife, der Integrität und Funktionsfähigkeit der Fortpflanzungsorgane, der neurologischen Endpunkte und der Verhaltensendpunkte sowie der Immunfunktionen in bestimmte Untergruppen (Kohorten 1-3, siehe die Nummern 33 und 34 sowie Abbildung 1) eingeteilt. 0 Bei der Prüfung sollten die im OECD Guidance Document Nr. 19 on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluation (34) genannten Grundsätze und Erwägungen beachtet werden. 9. Wurden genügend Prüfungen durchgeführt, um über die Eignung dieses neuen Prüfplans zu befinden, wird die Prüfmethode überprüft und auf der anhand der gewonnenen Erfahrungen gegebenenfalls überarbeitet. Schema der erweiterten Ein-Generationen-Prüfung auf Reproduktionstoxizität Beschreibung der Prüfmethode/Vorbereitungen für den Test Versuchstiere Auswahl von Versuchstierart und -stamm 10. Die Art der Versuchstiere für die Prüfung auf Reproduktionstoxizität sollte unter Berücksichtigung aller verfügbaren Informationen sorgfältig ausgewählt werden. Wegen der Fülle der vorliegenden Hintergrundinformationen und der Vergleichbarkeit mit allgemeinen Toxizitätsprüfungen ist die Ratte in der Regel das Versuchstier der Wahl, und die in dieser Prüfmethode angegebenen Kriterien und Empfehlungen beziehen sich auf diese Tierart. Falls eine andere Tierart verwendet wird, ist dies zu begründen, und das Protokoll ist entsprechend abzuändern. Stämme mit geringer Fruchtbarkeit oder allgemein anerkanntem spontanem Vorkommen von Entwicklungsstörungen sollten nicht verwendet werden. Alter, Körpergewicht und Aufnahmekriterien 11. Es sind gesunde Elterntiere zu verwenden, die zuvor nicht in anderen Versuchen verwendet wurden. Es sind sowohl männliche als auch weibliche Tiere zu untersuchen, wobei die weiblichen Tiere nicht geworfen haben noch trächtig sein dürfen. Die P-Tiere sollten geschlechtsreif sein, bei der ersten Dosisverabreichung über ein ähnliches Gewicht (innerhalb des Geschlechts) verfügen, bei der Paarung etwa gleich alt (ungefähr 90 Tage) und für die Prüfart und den Prüfstamm repräsentativ sein. Die Tiere sind nach ihrem Eintreffen im Labor mindestens fünf Tage lang einzugewöhnen. Sie werden nach dem Zufallsprinzip so auf die Kontroll- und Behandlungsgruppen verteilt, dass die verschiedenen Gruppen vergleichbare mittlere Körpergewichtswerte aufweisen (d. h. ± 20 % des Mittelwerts). Unterbringungs- und Fütterungsbedingungen 12. Die Temperatur im Versuchstierraum sollte 22 °C (± 3°) betragen. Die relative Luftfeuchtigkeit sollte bei 30-70 % liegen, wobei eine Luftfeuchtigkeit von 50-60 % ideal ist. Die Hell- und Dunkelphasen der künstlichen Beleuchtung sollten jeweils 12 Stunden dauern. Es kann herkömmliches Laborfutter verfüttert werden, die Tiere sollten jedoch unbegrenzten Zugang zu Trinkwasser haben. Es ist besonders auf den Phytoöstrogengehalt des Futters zu achten, da einige reproduktionstoxische Endpunkte durch einen zu hohen Phytoöstrogengehalt im Futter beeinträchtigt werden könnten. Es wird standardisiertes Futter mit offener Rezeptur empfohlen, dessen Gehalt an östrogen wirkenden chemischen Substanzen reduziert wurde (2) (30). Die Auswahl des Futters kann eventuell dadurch beeinflusst werden, dass eine geeignete Menge Prüfsubstanz beigemischt werden muss, wenn die Prüfsubstanz über das Futter verabreicht werden soll. Der Gehalt, die Homogenität und die Stabilität der Prüfsubstanz in den Futterrationen sind zu überprüfen. Futter und Trinkwasser sind regelmäßig auf Schadstoffe zu analysieren. Proben jeder im Laufe des Versuchs verwendeten Futtercharge sind bis zur Fertigstellung des Abschlussberichts unter geeigneten Bedingungen (z.B. durch Einfrieren bei - 20 °C) aufzubewahren, falls die Ergebnisse eine weitere Analyse der Futterbestandteile erfordern. 13. Die Tiere sollten in kleinen Gruppen gleichen Geschlechts und gleicher Behandlung untergebracht werden. Sie können auch einzeln untergebracht werden, um potenzielle Verletzungen zu vermeiden (z.B. männliche Tiere nach der Paarungszeit). Die Paarung sollte in zweckbestimmten Käfigen erfolgen. Nach dem nachweislich erfolgten Deckakt sind weibliche Tiere, von denen angenommen wird, dass sie trächtig sind, in separaten Käfigen speziell für trächtige Tiere bzw. in Wurfkäfigen zu halten, in denen ihnen vorgegebene und geeignete Nistmaterialien zur Verfügung stehen. Würfe werden bis zur Entwöhnung bei ihren Müttern gehalten. F1-Tiere sollten ab dem Tag ihres Absetzens bis zum Versuchsende in kleinen Gruppen gleichen Geschlechts und gleicher Behandlung gehalten werden. Sie können auch einzeln gehalten werden, soweit dies wissenschaftlich gerechtfertigt ist. Der Phytoöstrogengehalt der gewählten Einstreu sollte so gering wie möglich sein. Zahl und Kennzeichnung der Versuchstiere 14. Normalerweise sollte sich in jeder Prüf- und Kontrollgruppe eine ausreichende Menge Begattungspaare befinden, um pro Dosisgruppe mindestens 20 trächtige Weibchen zu ergeben. Das Ziel besteht darin, so viele trächtige Weibchen zu erhalten, dass eine aussagekräftige Bewertung des Potenzials der Prüfsubstanz, die Fruchtbarkeit, die Trächtigkeit und das Verhalten der Muttertiere der P-Generation sowie Wachstum und Entwicklung der F1-Nachkommen von der Befruchtung bis hin zur Geschlechtsreife zu beeinträchtigen, gewährleistet ist. Wird die gewünschte Zahl trächtiger Tiere nicht erzielt, so wird die Studie dadurch nicht zwangsläufig unbrauchbar. Hier ist der jeweilige Einzelfall zu bewerten, wobei zu prüfen ist, ob eine mögliche kausale Beziehung zur Prüfsubstanz hergestellt werden kann. 15. Jedes P-Tier erhält eine individuelle Kennnummer, bevor mit der Dosisverabreichung begonnen wird. Wenn historische Labordaten darauf hindeuten, dass ein erheblicher Teil der weiblichen Tiere möglicherweise keinen regelmäßigen (4- oder 5-tägigen) Östruszyklus aufweist, wird empfohlen, die Östruszyklen vor Beginn der Behandlung zu untersuchen. Alternativ kann die Gruppe vergrößert werden, um sicherzustellen, dass zu Beginn der Behandlung mindestens 20 weibliche Tiere in jeder Gruppe einen regelmäßigen (4- oder 5-tägigen) Zyklus aufweisen. Alle F1-Nachkommen werden bei der ersten Untersuchung der Neugeborenen am Tag der Geburt (Tag 0) oder am Tag 1 nach der Geburt (postnatal day, PND) einzeln gekennzeichnet. Aufzeichnungen, aus denen der Wurf, dem sie entstammen, hervorgeht, sind für alle F1-Tiere und gegebenenfalls auch für alle F2-Tiere während der gesamten Versuchsdauer aufzubewahren. Prüfsubstanz Verfügbare Informationen über die Prüfsubstanz 16. Die Überprüfung vorliegender Informationen ist für die Wahl des Verabreichungswegs, des Vehikels der Tierart und der Dosisabstufungen sowie für potenzielle Änderungen des Verabreichungszeitplans wichtig. Daher sollten bei der Planung der EOGRTS-Prüfung alle über die Prüfsubstanz vorliegenden sachdienlichen Informationen wie physikalisch-chemische Eigenschaften, toxikokinetische Eigenschaften (einschließlich des artenspezifischen Stoffwechsels), toxikodynamische Eigenschaften, Struktur-Wirkungs-Beziehungen (SAR), Invitro-Stoffwechselprozesse, Ergebnisse früherer Toxizitätsstudien und sachdienliche Informationen über strukturelle Analogien berücksichtigt werden. Vorläufige Informationen über Resorption, Verteilung, Metabolismus und Elimination (ADME) sowie Bioakkumulation können aus der chemischen Struktur, physikalisch-chemischen Daten, dem Umfang der Plasmaproteinbindung oder toxikokinetischen Studien abgeleitet werden, während Ergebnisse aus Toxizitätsstudien zusätzliche Informationen, z.B. über den NOAEL-Wert, den Stoffwechsel oder eine Stoffwechselinduktion liefern. Berücksichtigung toxikokinetischer Daten 17. Obwohl nicht erforderlich, können toxikokinetische Daten (TK-Daten) aus früheren Dosisfindungs- oder anderen Studien bei der Erstellung des Prüfplans, der Dosisabstufung und der Auswertung der Ergebnisse äußerst nützlich sein. Ganz besonders zweckdienlich sind dabei Daten, die 1) die Exposition von Föten und Jungtieren gegenüber der Prüfsubstanz (oder relevanten Metaboliten) bestätigen, 2) zur Ableitung einer internen Dosimetrie beitragen und 3) eine potenzielle dosisabhängige Sättigung kinetischer Prozesse bewerten. Falls weitere TK-Daten vorliegen, beispielsweise Metabolitprofile, Konzentrations-Zeit-Kurven usw. sind diese ebenfalls zu berücksichtigen. Ergänzende TK-Daten können auch während des Hauptversuchs erhoben werden, vorausgesetzt die Erhebung und Interpretation der wichtigsten Versuchsendpunkte wird dadurch nicht beeinträchtigt. Grundsätzlich wären folgende TK-Datensätze bei der Planung der EOGRTS-Prüfung nützlich:
Die Bestimmung der spezifischen Analyten (wie Ausgangssubstanz und/oder Metaboliten) und des Probenahmeplans sollte flexibel gehandhabt werden. Beispielsweise hängen Zahl und Zeitpunkt der Probenahmen an einem beliebigen Entnahmetag vom Expositionsweg und der Vorabkenntnis der toxikokinetischen Merkmale nicht trächtiger Tiere ab. Bei Versuchen mit Verabreichung über das Futter sind Probenahmen stets zum selben Zeitpunkt an jedem dieser Tage ausreichend, wohingegen bei Verabreichungen über eine Schlundsonde unter Umständen zusätzliche Probenahmen erforderlich sind, um einen besseren Schätzwert für den Bereich der internen Dosen zu erhalten. Eine vollständige Konzentrations-Zeit-Kurve muss jedoch nicht für jeden Probenahmetag erstellt werden. Erforderlichenfalls können Blutproben nach Geschlechtern innerhalb der Würfe für fetale und neonatale Analysen gepoolt werden. Verabreichungsweg 18. Bei der Wahl des Verabreichungswegs sollten die Routen berücksichtigt werden, die für die menschliche Exposition am relevantesten sind. Obwohl das Protokoll für die Verabreichung der Prüfsubstanz über das Futter ausgelegt ist, kann es für eine Verabreichung über andere Wege (Trinkwasser, Schlundsonde, Inhalation, dermal) je nach Eigenschaften der Prüfsubstanz und den erforderlichen Informationen abgeändert werden. Wahl des Vehikels 19. Bei Bedarf wird die Prüfsubstanz in einem geeigneten Vehikel gelöst oder suspendiert. Es wird empfohlen, nach Möglichkeit zunächst die Verwendung einer wässrigen Lösung/Suspension und erst dann eine Lösung/Emulsion in Öl (z.B. Maisöl) in Betracht zu ziehen. Bei anderen Vehikeln als Wasser sollten deren toxische Merkmale bekannt sein. Die Verwendung von Vehikeln mit einer potenziellen intrinsischen Toxizität (z.B. Aceton, DMSO) ist zu vermeiden. Die Stabilität der Prüfsubstanz im Vehikel sollte bestimmt werden. Wird ein Vehikel oder ein anderes Additiv zur Erleichterung der Dosierung verwendet, so sollten folgende Aspekte berücksichtigt werden: die Auswirkungen auf die Resorption, die Verteilung, die Verstoffwechselung oder die Retention der Prüfsubstanz, Auswirkungen auf die chemischen Eigenschaften der Prüfsubstanz, die deren toxische Eigenschaften verändern können, und ferner Auswirkungen auf die Futter- oder Wasseraufnahme oder den Ernährungszustand der Versuchstiere. Wahl der Dosis 20. Normalerweise sollten im Versuch mindestens drei Dosisstufen und eine gleichzeitige Kontrolle verwendet werden. Bei der Wahl der geeigneten Dosisstufen sollte der Prüfer alle vorliegenden Informationen berücksichtigen, einschließlich der Dosierungsinformationen aus früheren Versuchen, TK-Daten zu trächtigen und nicht trächtigen Tieren, das Ausmaß der Übertragung über die Muttermilch und die Schätzwerte für die menschliche Exposition. Falls toxikokinetische Daten vorliegen, die auf eine dosisbedingte Sättigung der toxikokinetischen Prozesse hindeuten, ist darauf zu achten, dass hohe Dosisstufen vermieden werden, die eindeutig eine hohe Sättigung aufweisen, natürlich nur unter der Voraussetzung, dass menschliche Expositionen möglichst deutlich unter dem Sättigungspunkt liegen. In solchen Fällen sollte die höchste Dosisstufe auf oder knapp über dem Wendepunkt zum Übergang zu nichtlinearem toxikokinetischem Verhalten liegen. 21. Sind keine relevanten toxikokinetischen Daten verfügbar, sollten die Dosisstufen auf der Grundlage der toxischen Wirkungen gewählt werden, es sei denn, es bestehen Beschränkungen aufgrund der physikalisch-chemischen Beschaffenheit der Prüfsubstanz. Werden die Dosisstufen auf der Grundlage der Toxizität gewählt, ist die höchste Dosisstufe so anzusetzen, dass bei den Tieren zwar toxische Wirkungen, aber keine Todesfälle oder schwere Leiden hervorgerufen werden. 22. Die Dosisstufen sind in absteigender Reihenfolge zu wählen, um etwaige dosisabhängige Wirkungen nachzuweisen und NOAEL-Werte oder Dosen an der Nachweisgrenze zu ermitteln, über die eine Benchmark-Dosis (BMD) für die empfindlichsten Endpunkte abgeleitet werden kann. Zwei- bis vierfache Abstände erweisen sich häufig als optimale Dosisabstufungen, um große Dosisstufenabstände zwischen NOAEL- und LOAEL-Werten zu vermeiden. Außerdem ist es oft besser, eine vierte Prüfgruppe einzurichten, statt zwischen den Dosen sehr große Abstände (z.B. mehr als × 10) zu verwenden. 23. Bis auf die Behandlung mit der Prüfsubstanz sind die Tiere in der Kontrollgruppe genauso zu behandeln wie die Tiere in den Prüfgruppen. Diese Gruppe sollte eine unbehandelte oder scheinbehandelte Gruppe oder eine Vehikelkontrollgruppe sein, sofern ein Vehikel zur Verabreichung der Prüfsubstanz verwendet wird. Wird ein Vehikel verwendet, erhält die Kontrollgruppe das Vehikel im höchsten verwendeten Volumen. Limit-Test 24. Wenn in Versuchen mit wiederholter Verabreichung bei einer Dosis von mindestens 1.000 mg/kg Körpergewicht/ Tag keine Anzeichen von Toxizität zu erkennen sind oder wenn aufgrund von Daten über struktur- und/oder stoffwechselverwandte Stoffe, die auf ähnliche Invivo/Invitro-Stoffwechseleigenschaften hindeuten, keine Toxizität zu erwarten ist, kann möglicherweise auf einen Versuch mit mehreren Dosisstufen verzichtet werden. In solchen Fällen könnte die EOGRTS-Prüfung mit einer Kontrollgruppe und mit einer einzelnen Dosis von mindestens 1.000 mg/kg Körpergewicht/Tag durchgeführt werden. Sollten jedoch bei dieser Grenzdosis Nachweise einer Reproduktions- oder Entwicklungstoxizität festgestellt werden, sind zur Ermittlung eines NOAEL-Wertes weitere Versuche bei niedrigeren Dosisstufen erforderlich. Das Kriterium des Limit-Tests findet jedoch nur Anwendung, wenn das Niveau der menschlichen Exposition keine höhere Dosisstufe erfordert. Exposition der Nachkommen 25. Die bevorzugte Verabreichungsmethode ist die Aufnahme über die Nahrung. Falls die Untersuchungen mit Schlundsonden durchgeführt werden, wird darauf hingewiesen, dass die Jungtiere die Prüfsubstanz in der Regel nur indirekt über die Milch erhalten, bis nach der Entwöhnung auch für sie die Direktverabreichung beginnt. Bei Futter- oder Trinkwasserversuchen erhalten die Jungtiere die Prüfsubstanz zusätzlich direkt, sobald sie während der letzten Woche der Laktationszeit beginnen, selbst zu fressen. Wenn zu wenig Prüfsubstanz in die Muttermilch übergeht und nicht nachwiesen werden kann, dass die Nachkommen kontinuierlich exponiert sind, sollte eine Änderung des Prüfplans in Betracht gezogen werden. In diesem Fall sollte auf der Grundlage vorliegender toxikokinetischer Erkenntnisse, der Toxizität bei den Nachkommen oder veränderter Biomarker bei Jungtieren während der Laktationszeit eine Direktverabreichung in Betracht gezogen werden (3) (4). Vor der Durchführung von Direktverabreichungsversuchen mit säugenden Jungtieren sind die Vor- und Nachteile sorgfältig gegeneinander abzuwägen (5). Verabreichungszeitplan und Verabfolgung der Dosen 26. Unter Umständen liegen aus früheren ausreichend langen Prüfungen mit wiederholter Verabreichung Informationen zum Östruszyklus, zur Histopathologie des männlichen und weiblichen Fortpflanzungsapparats und zur Analyse testikulärer/epididymaler Spermien vor. Daher soll bei der EOGRTS-Prüfung die Dauer der Behandlung vor der Paarung die Feststellung von Auswirkungen auf funktionale Veränderungen ermöglichen, die das Paarungsverhalten und die Fruchtbarkeit beeinträchtigen können. Die Behandlung vor der Paarung sollte so lange dauern, bis bei P-Männchen und P-Weibchen ein Expositionsgleichgewicht (Steady-State) erreicht ist. Eine zweiwöchige Behandlung vor der Paarung wird in den meisten Fällen für beide Geschlechter als angemessen angesehen. Bei Weibchen werden damit drei bis vier vollständige Östruszyklen abgedeckt, was genügen sollte, um etwaige Auswirkungen auf den Zyklus festzustellen. Bei männlichen Tieren entspricht dies der Zeit, die für den epididymalen Übergang der reifenden Spermien erforderlich ist; dieser Zeitraum sollte für die Feststellung posttestikulärer Auswirkungen auf Spermien (während der Endphasen der Spermiation und der epididymalen Spermienreifung) bei der Paarung ausreichen. Am Ende des Versuchs, d. h. der Zeitpunkt, für den die testikuläre und epididymale Histopathologie und die Analyse der Spermienparameter anberaumt sind, waren die P- und F1-Männchen zumindest für die Dauer eines vollständigen Spermatogeneseprozesses exponiert ((6) (7) (8) (9) und OECD Guidance Document Nr. 151 (40)). 27. Die Expositionsszenarien für Männchen vor der Paarung könnten angepasst werden, wenn in früheren Untersuchungen nachweislich testikuläre Toxizität (Beeinträchtigung der Spermatogenese) oder Beeinträchtigungen der Integrität und Funktionsfähigkeit der Spermien festgestellt wurden. Gleichermaßen können auch bei Weibchen vor der Paarung andere Expositionsszenarien gerechtfertigt sein, wenn Auswirkungen der Prüfsubstanz auf den Östruszyklus und damit auf die Empfänglichkeit bekannt sind. In besonderen Fällen kann es unter Umständen akzeptabel sein, dass mit der Behandlung der P-Weibchen erst nach einem spermienpositiven Abstrich begonnen wird (siehe OECD Guidance Document Nr. 151 (40)). 28. Sobald die Phase der Verabreichung vor der Paarung feststeht, sollten die Tiere bis zur Nekropsie nach einem 7 Tage/Woche-Schema mit der Prüfsubstanz behandelt werden. Allen Tieren sollte die Prüfsubstanz nach derselben Methode verabreicht werden. Die Verabreichung sollte während der zweiwöchigen Paarungszeit und - bei P-Weibchen - während der gesamten Trächtigkeitsdauer und Laktationsperiode bis zu ihrer Tötung am Tag nach dem Absetzen fortgeführt werden. Männchen sollten bis zu ihrer Tötung am Tag der Absetzung der F1-Tiere in derselben Weise zu behandeln. Bei der Nekropsie ist weiblichen Tieren Priorität einzuräumen; sie sollten am selben/einem ähnlichen Laktationstag seziert werden. Die Nekropsie der männlichen Tiere kann sich je nach Verfügbarkeit der Laboreinrichtungen über mehrere Tage erstrecken. Sofern mit der direkten Verabreichung der Prüfsubstanz nicht bereits während der Laktationsperiode begonnen wurde, sollte die Direktverabfolgung an ausgewählte F1-Männchen und F1-Weibchen am Tag des Absetzens beginnen und bis zur geplanten Nekropsie (je nach in Kohorteneinteilung) fortgesetzt werden. 29. Bei über das Futter oder das Trinkwasser verabreichten Prüfsubstanzen ist unbedingt sicherzustellen, dass die Mengen der jeweiligen Prüfsubstanz den normalen Nährstoff- oder Wasserhaushalt nicht beeinträchtigen. Wird die Prüfsubstanz über die Nahrung verabreicht, kann entweder eine konstante Konzentration im Futter/Wasser (ppm) oder eine konstante Dosis, bezogen auf das Körpergewicht des Tieres, verwendet werden. Dabei ist anzugeben, welche Option gewählt wurde. 30. Wird die Prüfsubstanz über eine Schlundsonde verabreicht, sollte die Menge der jeweils verabreichten Flüssigkeit grundsätzlich 1 ml/100 g Körpergewicht nicht übersteigen (0,4 ml/100 g Körpergewicht ist der Höchstwert für Öl, z.B. Maisöl). Außer bei reizenden oder ätzenden Stoffen, die in der Regel in höheren Konzentrationen eine verstärkte Wirkung hervorrufen, sollten Abweichungen bei den verabreichten Prüfsubstanzmengen minimiert werden, indem die Konzentration so angepasst wird, dass auf allen Dosisstufen ein konstantes Volumen gewährleistet ist. Die Behandlung sollte jeden Tag etwa zur gleichen Uhrzeit erfolgen. Die Dosis für jedes Tier sollte sich auf das aktuelle Körpergewichtstützen und ist bei adulten Männchen und nichtträchtigen adulten Weibchen mindestens wöchentlich und bei trächtigen Weibchen und F1-Tieren alle zwei Tage anzupassen, wenn die Verabreichung vor dem Absetzen und während der zwei Wochen nach der Entwöhnung erfolgt. Falls toxikokinetische Daten auf eine schwache plazentare Übertragung der Prüfsubstanz hindeuten, muss die in der der letzten Trächtigkeitswoche über die Schlundsonde verabreichte Dosis unter Umständen angepasst werden, um die Verabreichung einer zu giftigen Dosis an das Muttertier zu verhindern. Weibliche Tiere sollten am Wurftag nicht mit einer Schlundsonde oder auf andere Weise behandelt werden, die eine Hantierung der Tiere erfordert; es ist besser, am Wurftag keine Prüfsubstanz zu verabreichen, als den Geburtsvorgang zu beeinträchtigen. Paarung 31. Jedes P-Weibchen sollte so lange einem einzigen nach dem Zufallsprinzip ausgewählten nicht verwandten Männchen aus derselben Dosisgruppe zugeführt werden (1:1-Paarung), bis die Kopulation nachweislich stattgefunden hat oder zwei Wochen abgelaufen sind. Falls nicht genügend Männchen vorhanden sind, weil männliche Tiere beispielsweise noch vor der Paarung gestorben sind, können Männchen, die bereits ein Weibchen gedeckt haben (1:1) mit einem zweiten Weibchen angepaart werden, damit alle Weibchen gedeckt werden. Tag 0 der Trächtigkeit wird als der Tag festgelegt, an dem die Deckung (durch Vaginalpfropf oder Spermaspuren) nachgewiesen werden kann. Die Tiere sind nach der nachweislichen Kopulation so bald wie möglich zu trennen. Falls nach zwei Wochen keine Paarung stattgefunden hat, sollten die Tiere ohne weitere Paarungsmöglichkeit getrennt werden. Begattungspaare sind in den Aufzeichnungen als solche zu vermerken. Wurfgröße 32. An PND 4 kann die Größe eines jeden Wurfs angepasst werden, indem überschüssige Jungtiere nach dem Zufallsprinzip aussortiert werden, um pro Wurf nach Möglichkeit fünf männliche und fünf weibliche Tiere zu erhalten. Die selektive Eliminierung von Jungtieren, z.B. auf der Grundlage des Körpergewichts, wird nicht empfohlen. Wenn es wegen der Anzahl männlicher bzw. weiblicher Jungtiere nicht möglich ist, pro Wurf jeweils fünf Jungtiere eines jeden Geschlechts zu erhalten, ist auch eine grobe Anpassung (beispielsweise sechs Männchen und vier Weibchen) akzeptabel. Auswahl von Jungtieren für Untersuchungen nach der Entwöhnung (siehe Abbildung 1) 33. Zum Zeitpunkt der Entwöhnung (um PND 21) werden aus allen verfügbaren Würfen bis zu 20 Jungtiere pro Dosis- und Kontrollgruppe für weitere Untersuchungen ausgewählt und bis zur Geschlechtsreife gehalten (sofern keine früheren Tests erforderlich sind). Die Jungtiere werden nach dem Zufallsprinzip ausgewählt, wobei offensichtliche Kümmerlinge (Tiere mit mehr als zwei Standardabweichungen unter dem mittleren Jungtiergewicht des betreffenden Wurfes) grundsätzlich nicht einbezogen werden, da sie für die Behandlungsgruppe kaum repräsentativ sind. An PND 21 werden die ausgewählten F1-Jungtiere nach dem Zufallsprinzip in eine der drei folgenden Tierkohorten eingeteilt: Kohorte 1 (1A und 1B) = Testung auf Reproduktions-/Entwicklungstoxizität, Kohorte 2 (2A und 2B) = Testung auf Entwicklungsneurotoxizität, Kohorte 3 = Testung auf Entwicklungsimmuntoxizität. Kohorte 1A: Ein Männchen und ein Weibchen pro Wurf/Gruppe (20 pro Geschlecht/Gruppe): prioritäre Auswahl für die primäre Untersuchung der Auswirkungen auf die Fortpflanzungsorgane und der allgemeinen Toxizität. Kohorte 1B: Ein Männchen und ein Weibchen pro Wurf/Gruppe (20 pro Geschlecht/Gruppe): prioritäre Auswahl für Folgeuntersuchungen der Reproduktionsleistung durch Paarung von F1-Tieren (siehe OECD Guidance Document Nr. 117 (39)) und zur Generierung zusätzlicher histopathologischer Daten in Fällen mutmaßlich reproduktionstoxischer oder endokrin wirksamer Stoffe oder wenn Ergebnisse für Kohorte 1A unschlüssig sind. Kohorte 2A: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf), eingeteilt für Untersuchungen neurologisch bedingten Verhaltens mit anschließender neurohistopathologischer Untersuchung im adulten Alter. Kohorte 2B: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf), eingeteilt für neurohistopathologische Untersuchungen am Tag des Absetzens (PND 21 oder 22). Wenn nicht genügend Tiere vorhanden sind, sollten Tiere vorrangig der Kohorte 2A zugewiesen werden. Kohorte 3: Insgesamt 20 Jungtiere pro Gruppe (10 Männchen und 10 Weibchen pro Gruppe; ein Männchen oder ein Weibchen pro Wurf, soweit möglich). Unter Umständen werden zusätzliche Jungtiere aus der Kontrollgruppe benötigt, die im TDAR-Assay (T-Zellabhängige Antikörperantwort) an PND 56 ± 3 Tage als positive Kontrolltiere dienen. 34. Sollte ein Wurf nicht genügend Tieren umfassen, um alle Kohorten zu bedienen, so wird Kohorte 1 Vorrang eingeräumt, da sie zur Produktion einer F2-Generation erweitert werden kann. Zusätzliche Jungtiere können bei speziellem Bedarf, z.B. wenn es sich bei einer Chemikalie möglicherweise um ein Neurotoxin, ein Immuntoxin oder ein Reproduktionstoxin handelt, jeder beliebigen Kohorte zugewiesen werden. Diese Jungtiere können für Untersuchungen zu anderen Zeitpunkten oder ergänzender Endpunkte verwendet werden. Jungtiere, die keinen Kohorten zugeteilt werden, werden klinisch und biochemisch (Nummer 55) untersucht und seziert (Nummer 68) untersucht. Zweite Paarung von P-Tieren 35. Eine zweite Paarung wird für P-Tiere in der Regel nicht empfohlen, denn sie geht mit dem Verlust wichtiger Informationen über die Zahl der Implantationsstellen (und somit Angaben über Abgänge nach der Implantation und über perinatale Abgängen - Indikatoren eines teratogenen Potenzials) für den ersten Wurf einher. Wirkungen in exponierten weiblichen Tieren ließen sich durch eine Erweiterung der Prüfung durch Paarung der F1-Generation leichter bestätigen oder klären. Eine zweite Paarung der P-Männchen mit unbehandelten weiblichen Tieren ist jedoch stets eine Möglichkeit, um unschlüssige Ergebnisse zu klären oder um bei der ersten Paarung festgestellte Auswirkungen auf die Fruchtbarkeit näher zu charakterisieren. Beobachtungen am lebenden Tier Klinische Beobachtungen 36. Bei den P-Tieren und den ausgewählten F1-Tieren wird einmal täglich eine allgemeine klinische Untersuchung vorgenommen. Bei Verabreichung der Prüfsubstanz über eine Schlundsonde sind die klinischen Beobachtungen (auf mögliche Toxizitätsanzeichen, die Plasmakonzentrationsspitzen zugeordnet werden) vor und nach der Verabreichung vorzunehmen. Pertinente Verhaltensänderungen, Anzeichen für eine schwierige oder langwierige Geburt und alle Toxizitätsanzeichen werden festgehalten. Zweimal täglich - am Wochenende einmal täglich - werden alle Tiere auf Anzeichen von schwerer Toxizität, Morbidität und Mortalität untersucht. 37. Darüber hinaus werden alle P- und F1 -Tiere (nach dem Entwöhnen) einmal wöchentlich eingehender untersucht; diese Untersuchung könnte praktischerweise durchgeführt werden, wenn das Tier gewogen wird, um den umgangsbedingten Stress auf ein Mindestmaß zu reduzieren. Die Untersuchungen sind sorgfältig durchzuführen und nach einer speziell vom Prüflabor entwickelten Bewertungsskala zu dokumentieren. Die Testbedingungen sollten möglichst konstant sein. Die festzuhaltenden Anzeichen umfassen, ohne darauf beschränkt zu sein, u. a. Veränderungen an Haut, Fell, Augen, Schleimhäuten sowie Sekrete und Exkretionen und autonome Körperfunktionen (wie Tränensekretion, Piloerektion, Pupillenveränderung, ungewöhnliche Atmungsmuster). Gang- und Haltungsstörungen, Reaktionen auf Berührungen sowie etwaige klonischtonische Anfälle, stereotype Verhaltensweisen (z.B. übermäßiges Putzen, wiederholte Kreisbewegungen) oder abnormes Verhalten (z.B. Selbstverstümmelung, Rückwärtsgehen) sollten ebenfalls dokumentiert werden. Körpergewicht und Futter-/Trinkwasseraufnahme 38. P-Tiere werden am ersten Tag der Verabreichung der Prüfsubstanz und danach mindestens einmal wöchentlich gewogen. Darüber hinaus werden P-Weibchen während der Laktation an denselben Tagen gewogen wie die Jungtiere in ihren Würfen (siehe Nummer 44). Alle F1-Tiere werden am Tag des Absetzens (PND 21) und danach mindestens einmal wöchentlich einzeln gewogen. Das Körpergewicht wird auch an dem Tag aufgezeichnet, an dem die Pubertät eintritt (Abschluss der Präputialseparation oder Öffnung der Vagina). Alle Tiere werden bei der Tötung gewogen. 39. Im Laufe des Versuchs werden Futter- und Wasseraufnahme (bei Verabreichung der Prüfsubstanz im Trinkwasser) mindestens einmal wöchentlich stets an denselben Tagen gemessen und erfasst, an denen auch das Körpergewicht gemessen und dokumentiert wird (außer während der Kohabitation). Die Futteraufnahme der F1-Tiere jedes Käfigs wird ab der Einteilung in eine Kohorte einmal wöchentlich dokumentiert. Östruszyklen 40. Es existieren möglicherweise bereits vorläufige Informationen über die Auswirkungen der Prüfsubstanz auf den Östruszyklus aus früheren Prüfungen auf Toxizität bei wiederholter Verabreichung, die zur Festlegung eines prüfsubstanzspezifischen Protokolls für die EOGRTS-Prüfung herangezogen werden können. In der Regel beginnt die Bewertung der weiblichen Zyklizität (durch Vaginalzytologie) zu Beginn des Behandlungszeitraums und wird bis zur Paarungsbestätigung bzw. bis zum Ende der zweiwöchigen Paarungszeit fortgesetzt. Falls weibliche Tiere vor der Behandlung auf einen normalen Östruszyklus hin untersucht wurden, ist es zweckdienlich, die Abstriche auch nach Beginn der Behandlung fortzusetzen; wenn jedoch zu Beginn der Behandlung Bedenken in Bezug auf unspezifische Auswirkungen bestehen (beispielsweise eine beginnende ausgeprägte Futterverweigerung), kann den Tieren bis zu zwei Wochen Zeit gelassen werden, um sich der Behandlung anzupasen, bevor die zweiwöchige Abstrichperiode anläuft, die der Paarung vorausgeht. Wenn die Behandlungszeit für die weiblichen Tiere auf diese Art und Weise (d. h. auf eine vierwöchige Behandlung vor der Paarung) verlängert wird, sollte der Erwerb jüngerer Tiere und die Verlängerung des Behandlungszeitraums für männliche Tiere vor der Paarung in Erwägung gezogen werden. Bei der Entnahme vaginaler/zervikaler Zellen ist sorgfältig darauf zu achten, dass die Schleimhaut nicht gereizt und infolgedessen eine Pseudogravidität eingeleitet wird (10) (11). 41. Vaginalabstriche sind bei allen F1-Weibchen in der Kohorte 1A nach beginnender Öffnung der Vagina bis zur Erfassung des ersten verhornten Abstrichs täglich zu untersuchen, um den zeitlichen Abstand zwischen diesen beiden Ereignissen zu bestimmen. Außerdem sollten die Östruszyklen bei allen F1-Weibchen in Kohorte 1A über einen Zeitraum von zwei Wochen, etwa ab PND 75, überwacht werden. Sollte sich die Paarung der F1-Generation als nötig erweisen, wird die Vaginalzytologie in Kohorte 1B vom Zeitpunkt der Paarung an bis zur nachweislichen Deckung überwacht. Deckung und Gravidität 42. Zusätzlich zu den Standardendpunkten (z.B. Körpergewicht, Futteraufnahme, klinische Beobachtungen, einschließlich Mortalitäts-/Morbiditätskontrollen) werden die Zeitpunkte der Paarung, der Befruchtung und der Geburt aufgezeichnet und das präkoitale Intervall (Paarung bis Befruchtung) sowie die Dauer der Gravidität (Befruchtung bis Geburt) berechnet. Die P-Weibchen sind zum Zeitpunkt des voraussichtlichen Partus sorgfältig auf Anzeichen von Dystokie zu untersuchen. Alle Abnormitäten beim Nistverhalten oder bei der Säugeleistung sind zu dokumentieren. 43. Der Wurftag ist für das Muttertier Laktationstag (LD) 0 und für die Nachkommen der postnatale Tag (PND) 0. Alternativ dazu können auch die Tage nach der Kopulation herangezogen werden, um Fehler bei den Daten über die postnatale Entwicklung zu vermeiden, die auf Unterschiede bei der Trächtigkeitsdauer zurückzuführen sind; die Zeit nach der Geburt ist jedoch ebenfalls zu dokumentieren. Dies ist vor allem wichtig, wenn sich die Prüfsubstanz auf die Trächtigkeitsdauer auswirkt. Parameter für die Nachkommen 44. Jeder Wurf ist so bald wie möglich nach der Geburt (PND 0 oder 1) zu untersuchen, um die Anzahl und Geschlecht der Jungtiere, Totgeburten, Lebendgeburten und etwaige grobe Anomalien (extern sichtbare Abnormalitäten, einschließlich Gaumenspalten; subkutane Hämorrhagien; anormale Hautfarbe oder Hautbeschaffenheit; Vorhandensein der Nabelschnur; Abwesenheit von Milch im Magen; Präsenz von trockener Absonderungen) festzustellen. Darüber hinaus sollte die erste klinische Untersuchung der Neugeborenen auch eine qualitative Beurteilung der Körpertemperatur, des Bewegungsmusters und der Reaktion auf Berührungen beinhalten. Jungtiere, die am PND 0 oder zu einem späteren Zeitpunkt tot aufgefunden werden, sind auf mögliche Defekte und auf die Todesursache hin zu untersuchen. Lebende Jungtiere werden am PND 0 oder 1 gezählt und danach regelmäßig, mindestens jedoch an PND 4, 7, 14 und 21, einzeln gewogen. Klinische Untersuchungen, die je nach dem Alter der Tiere durchzuführen sind, sollten wiederholt werden, wenn die Nachkommen gewogen werden, oder auch öfter, wenn bei ihrer Geburt fallspezifische Befunde festgestellt wurden. Zu achten ist insbesondere auf Veränderungen an Haut, Fell, Augen und Schleimhäuten, auf Absonderungen und Ausscheidungen sowie autonome Körperfunktionen. Gang- und Haltungsstörungen sowie Reaktionen auf Berührungen und etwaige klonischtonische Anfälle, stereotype oder bizarre Verhaltensweisen sind ebenfalls zu dokumentieren. 45. Der anogenitale Abstand (AGD) sollte bei jedem Jungtier mindestens einmal (zwischen PND 0 und PND 4) gemessen werden. Das Körpergewicht des Jungtiers wird am Tag der Messung des AGD erfasst, der auf Jungtiergröße - vorzugsweise die Quadratwurzel des Körpergewichts - genormt sein sollte (12). Das Vorhandensein von Brustwarzen/Warzenhöfen bei männlichen Jungtieren ist am PND 12 oder 13 zu kontrollieren. 46. Alle ausgewählten F1-Tiere werden täglich auf Vorhaut-Eichel-Trennung bei männlichen Tieren bzw. Öffnung der Vagina bei weiblichen Tieren untersucht, und zwar vor dem Tag, an dem das Erreichen dieser Endpunkte erwartet wird, um festzustellen, ob die Geschlechtsreife eventuell früh eintritt. Alle Abnormalitäten der Geschlechtsorgane (wie persistente Vaginalfilamente, Hypospadie oder Spaltpenis) sind festzuhalten. Die Geschlechtsreife der F1-Tiere wird mit der körperlichen Entwicklung verglichen, indem Alter und Körpergewicht zum Zeitpunkt der Vorhaut- Eichel-Trennung bei männlichen Tieren bzw. der Öffnung der Vagina bei weiblichen Tieren bestimmt werden (13). Bewertung der potenziellen Entwicklungsneurotoxizität (Kohorten 2A und 2B) 47. Für Bewertungen der Neurotoxizität sind aus jeder Behandlungsgruppe zehn männliche und zehn weibliche Tiere der Kohorte 2A sowie zehn männliche und zehn weibliche Tiere der Kohorte 2B zu verwenden (für jede Kohorte: ein männliches oder ein weibliches Tier pro Wurf; alle Würfe müssen durch mindestens ein nach dem Zufallsprinzip ausgewähltes Jungtier vertreten sein). Tiere der Kohorte 2A sind FOB-Tests (Functional Observational Battery) sowie Untersuchungen auf akustische Schreckreaktion und motorische Aktivität (siehe Nummern 48-50) sowie neuropathologischen Tests (siehe Nummern 74-75) zu unterziehen. Dabei ist nach Möglichkeit sicherzustellen, dass die Prüfbedingungen möglichst wenig variieren und dass diese Variationen nicht systematisch mit der Behandlung zusammenhängen. Zu den Variablen, die das Verhalten beeinflussen können, gehören Geräuschpegel (beispielsweise intermittierender Lärm), Temperatur, Feuchtigkeit, Beleuchtung, Gerüche, Tageszeit und Ablenkungen aus der Umgebung. Die Ergebnisse der Neurotoxizitätstests sind bezogen auf entsprechende historische Kontrollreferenzbereiche auszulegen. Tiere der Kohorte 2B sollten an PND 21 oder 22 neuropathologisch untersucht werden (siehe Nummern 74-75). 48. Ein Test auf akustische Schreckreaktion sollte am PND 24 (± 1 Tag) mit Tieren der Kohorte 2A durchgeführt werden. Die Untersuchung der Behandlungsgruppe und der Kontrollgruppe ist ausgewogen über den Tag zu verteilen. Jede Testreihe umfasst 50 Prüfungen. Bei diesen Tests wird die mittlere Reaktionsamplitude für jeden Block von 10 Prüfungen (5 Blöcke mit je 10 Prüfungen) berechnet, dessen Prüfbedingungen optimiert wurden, um während der Testreihe Habituation zu erzeugen. Diese Verfahren sollten mit der Prüfmethode B.53 (35) übereinstimmen. 49. Zu einem geeigneten Zeitpunkt zwischen PND 63 und PND 75 werden die Tiere der Kohorte 2A einem FOB-Test und einem automatisierten Motoriktest unterzogen. Diese Verfahren sollten mit den Prüfmethoden B.43 (33) und B.53 (35) übereinstimmen. Der FOB-Test beinhaltet eine ausführliche Beschreibung des äußeren Erscheinungsbildes, des Verhaltens und der funktionalen Integrität des Versuchstiers. Diese Bewertungen beruhen auf Beobachtungen im Haltungskäfig, in einem Standardgehege (offenes Feld), in dem sich das Tier frei bewegen kann, sowie durch Manipulationstests. Die Untersuchungen sollten in aufsteigender Reihenfolge der Interaktivität durchgeführt werden. Anhang 1 enthält eine Liste von Maßnahmen. Alle Tiere sollten von geschulten Beobachtern untersucht werden, denen die Behandlungsphase des jeweiligen Tieres nicht bekannt ist. Dabei sind standardisierte Verfahren anzuwenden, um beobachterbedingte Abweichungen auf ein Mindestmaß zu begrenzen. Nach Möglichkeit sollten die Tiere in einem bestimmten Test stets durch denselben Beobachter untersucht werden. Falls dies nicht möglich ist, ist die Zuverlässigkeit bei verschiedenen Beobachtern auf andere Weise nachzuweisen. Für jeden Parameter der Verhaltensprüfbatterie sind Skalen und Bewertungskriterien zu verwenden, deren Anwendung genau festgelegt ist. Nach Möglichkeit sind für Beobachtungsendpunkte, die subjektive Einstufungen beinhalten, objektive quantitative Messungen durchzuführen. In Bezug auf die Motorik wird jedes Tier einzeln getestet. Die Testreihe sollte so lange dauern, bis bei Kontrollen eine Habituation innerhalb der Testreihe nachgewiesen werden kann. Die Motorik sollte mithilfe eines automatischen Bewegungsmessgeräts untersucht werden, das in der Lage ist, sowohl eine Zunahme als auch ein Nachlassen der Bewegung zu erfassen (d. h., die vom Gerät gemessene Basisbewegung sollte weder so schwach sein, dass dadurch die Erfassung eines Nachlassen der Bewegung unmöglich wird, noch so stark, dass Bewegungszunahmen nicht erfasst werden können). Jedes einzelne Gerät sollte im Rahmen standardisierter Verfahren geprüft worden sein, um bei Einsatz mehrerer Geräte über mehrere Tage eine möglichst hohe Betriebssicherheit zu gewährleisten. Die Behandlungsgruppen sollten so weit wie möglich gleichmäßig auf die Geräte verteilt werden. Die Untersuchung Beobachtung der Behandlungsgruppen sollte über den Tag verteilt werden, um zirkadianen Aktivitätsrhythmus zu berücksichtigen. 50. Falls vorhandene Informationen auf die Notwendigkeit hindeuten, weitere funktionelle Tests durchzuführen (z.B. sensorische, soziale oder kognitive Tests), sind diese zu berücksichtigen, ohne die Integrität der anderen durchgeführten Untersuchungen zu beeinträchtigen. Falls diese Tests an denselben Tieren durchgeführt werden, die auch für den Standardtest auf akustische Schreckreaktion, den FOB-Test und den Motoriktest verwendet wurden, sind andere Tests einzuplanen, um das Risiko, dass die Integrität dieser Tests beeinträchtigt wird, auf ein Mindestmaß zu begrenzen. Zusätzliche Prüfverfahren können insbesondere dann sinnvoll sein, wenn die empirische Beobachtung, die erwarteten Wirkungen oder der Wirkmechanismus/die Wirkungsweise auf eine bestimmte Art von Neurotoxizität hindeuten. Bewertung einer potenziellen Entwicklungsimmuntoxizität (Kohorte 3) 51. Am postnatalen PND 56 (± 3 Tage) sollten aus jeder Behandlungsgruppe zehn männliche und zehn weibliche Tiere der Kohorte 3 (ein männliches oder ein weibliches Tier pro Wurf; alle Würfe müssen durch mindestens ein nach dem Zufallsprinzip ausgewähltes Jungtier vertreten sein) im Einklang mit den aktuellen Immuntoxizitätstestverfahren (14) (15) einem Test auf T-Zellabhängige Antikörperantwort (d. h. auf primäre IgM-Antikörperantwort auf ein T-Zellabhängiges Antigen, wie zum Beispiel rote Blutkörperchen von Schafen oder Schlitzschnecken- Hämocyanin (KLH)) unterzogen werden. Die Reaktion kann bestimmt werden durch Zählung spezifischer plaquebildender Zellen (PFC) in der Milz oder durch Bestimmung des Titerwertes für SRBC- oder KLH-spezifische IgM-Antikörper im Serum mittels ELISA-Test auf dem Höhepunkt der Reaktion. Die Maximalreaktionen lassen sich in der Regel vier (PFC-Reaktion) oder fünf (ELISA-Test) Tage nach der intravenösen Beimpfung festzustellen. Wird die primäre Antikörperreaktion durch Auszählen der plaquebildenden Zellen bestimmt, so ist die Bewertung von Untergruppen von Tieren an getrennten Tagen zulässig, sofern die Immunisierung der Untergruppe und die Tötung der Tiere zeitlich so geplant sind, dass die plaquebildenden Zellen zum Zeitpunkt der Höchstreaktion gezählt werden, die Untergruppen aus ebenso vielen männlichen wie weiblichen Nachkommen aller Dosisgruppen, einschließlich Kontrolltieren, bestehen und die Tiere der Untergruppen ungefähr im selben postnatalen Alter untersucht werden.Die Exposition gegenüber der Prüfsubstanz wird bis zum Tag vor der Entnahme der Milz zur Bestimmung der PFC-Reaktion oder des Serums für den ELISA-Test fortgesetzt. Folgeuntersuchung auf potenzielle Reproduktionstoxizität (Kohorte 1B) 52. Tiere der Kohorte 1B können gegebenenfalls auch nach PND 90 weiterbehandelt und gezüchtet werden, um erforderlichenfalls eine F2-Generation zu produzieren. Männliche und weibliche Tiere derselben Dosisgruppen sind ab oder nach PND 90 bis zu zwei Wochen lang, allerdings nicht über PND 120 hinaus, zusammenzuführen (wobei die Paarung von Geschwistern zu vermeiden ist). Es sollten genau so vorgegangen werden wie bei den P-Tieren. Sofern dies nachgewiesen weren kann, reicht es jedoch unter Umständen jedoch aus, die Würfe an PND 4 zu töten, statt sie bis zur Entwöhnung oder darüber hinaus weiter zu untersuchen. Klinischbiochemische/Hämatologische Untersuchungen 53. Systemische Wirkungen in P-Tieren sollten überwacht werden. An einer vorgegebenen Stelle werden von zehn nach dem Zufallsprinzip ausgewählten P-Männchen und -Weibchen pro Dosisgruppe am Versuchsende Nüchternblutproben entenommen, unter angemessenen Bedingungen gelagert und teilweise oder vollständig hämatologischen, klinischen, biochemischen Untersuchungen, einer T4- und TSH-Analyse oder anderen Tests unterzogen, die aufgrund des bekannten Wirkungsprofils der Prüfsubstanz naheliegen (siehe OECD Guidance Document Nr. 151 (40)). Die folgenden hämatologischen Parameter sollten dabei untersucht werden: Hämatokrit, Hämoglobinkonzentration, Erythrozytenzahl, Gesamt- und Differential-Leukozytenzahl, Thrombozytenzahl und Blutgerinnungszeit/-fähigkeit. Die Plasma- oder Serumuntersuchungen sollten Folgendes umfassen: Glucose, Gesamtcholesterin, Harnstoff, Kreatinin, Gesamtprotein und Albumin sowie mindestens zwei Enzyme, die auf hepatozelluläre Wirkungen schließen lassen (wie Alanin-Aminotransferase, Aspartat-Aminotransferase, alkalische Phosphatase, γ-Glutamyltranspeptidase und Glutamatdehydrogenase). Die Bestimmung weiterer Enzyme und Gallens´uren kann unter bestimmten Umständen ebenfalls wertvolle Hinweise liefern. Darüber hinaus können Blutproben von allen Tieren entnommen und für eine spätere Analyse aufbewahrt werden, um unschlüssige Wirkungsergebnisse zu klären oder interne Expositionsdaten zu generieren. Ist keine zweite Paarung der P-Tiere beabsichtigt, werden die Blutproben unmittelbar vor oder bei der geplanten Tötung der Tiere gezogen. Falls Tiere behalten werden, sind die Blutproben einige Tage vor der zweiten Paarung der Tiere zu ziehen. Sofern aus vorliegenden Daten aus Untersuchungen mit wiederholter Verabreichung nicht hervorgeht, dass der Parameter nicht durch die Prüfsubstanz beeinträchtigt wird, sollten vor Abschluss der Studie eine Urinuntersuchung durchgeführt und die folgenden Parameter bewertet werden: Aussehen, Volumen, Osmolalität oder Dichte, pH-Wert, Protein, Glucose, Blut und Blutzellen, Zelltrümmer. Urin kann auch gesammelt werden um die Ausscheidung der Prüfsubstanz und/oder Metaboliten zu überwachen. 54. Systemische Wirkungen sind auch in F1-Tieren zu überwachen. Von zehn nach dem Zufallsprinzip ausgewählten Männchen und Weibchen der Kohorte 1A pro Dosisgruppe werden am Versuchsende an einer vorgegebenen Stelle Nüchternblutproben entnommen, unter angemessenen Bedingungen gelagert und klinischen biochemischen Standarduntersuchungen, einschließlich einer Bewertung der Serumspiegel auf Schilddrüsenhormone (T4- und TSH), hämatologischen Untersuchungen (Gesamt- und Differenzialleukozytenzahl) sowie Urinuntersuchungen unterzogen. 55. Die an PND 4 überzähligen Jungtiere werden makroskopisch untersucht, wobei erwogen werden kann, die Konzentration der Schilddrüsenhormone (T4) im Serum zu bestimmen. Erforderlichenfalls können nach Würfen Blutproben Neugeborener (PND 4) für biochemische Analysen und zur Bestimmung der Konzentration der Schilddrüsenhormone gepoolt werden. Blutproben für die T4- und TSH-Analyse wird außerdem von gerade entwöhnten Tieren gezogen, die am PND 22 makroskopisch untersucht werden (F1-Jungtiere, die nicht für Kohorten ausgewählt werden). Spermienparameter 56. Spermienparameter sollten in allen männlichen Tieren der P-Generation gemessen werden, sofern keine Daten vorliegen, die nachweisen, dass Spermienparameter in einem 90-Tage-Versuch nicht beeinträchtigt werden. Die Untersuchung der Spermienparameter sollte bei allen männlichen Tieren der Kohorte 1A durchgeführt werden. 57. Bei Versuchsabschluss wird für alle männlichen P- und F1-Tieren (Kohorte 1A) das Gewicht der Hoden und Nebenhoden aufgezeichnet. Mindestens ein Hoden und ein Nebenhoden werden für die histopathologische Untersuchung konserviert. Der verbleibende Nebenhoden wird zur Auszählung von Spermienreserven im Nebenhodenschwanz (Cauda epididymis) (16) (17) verwendet. Darüber hinaus werden Spermien aus dem Nebenhodenschwanz bzw. aus dem Samenleiter (Vas deferens) mithilfe von Methoden gewonnen, die Schäden für die Bewertung der Motilität und Morphologie der Spermien auf ein Mindestmaß begrenzen (18). 58. Die Spermienmotilität kann entweder unverzüglich nach der Tötung bewertet oder für eine spätere Analyse aufgezeichnet werden. Der Anteil der progressiv beweglichen Spermien könnte entweder subjektiv oder mithilfe einer computergestützten Bewegungsanalyse objektiv bestimmt werden (19) (20) (21) (22) (23) (24). Für die Bewertung der Morphologie der Spermien sollte eine Spermienprobe aus dem Nebenhoden (oder aus dem Samenleiter) als Fest- oder Feuchtpräparat (25) untersucht werden, wobei mindestens 200 Spermien pro Probe entweder als normal (sowohl Kopf als auch Mittelstück/Schwanz erscheinen normal) oder abnormal einzustufen sind. Morphologische Abnormalitäten der Spermien wären beispielsweise die Verschmelzung von Köpfen, isolierte Köpfe und Kopf- und/oder Schwanzmissbildungen (26). Missgebildete oder große Spermienköpfe können auf Störungen bei der Spermiation hindeuten. 59. Werden zum Zeitpunkt der Sektion Spermienproben eingefroren, Abstriche fixiert und Bilder zur Analyse der Spermienmotilität dokumentiert (27), kann die anschließende Analyse auf männliche Tiere der Kontrollgruppe und der Hochdosisgruppe beschränkt werden. Werden jedoch behandlungsbezogene Wirkungen beobachtet, sind auch die niedrigeren Dosisgruppen zu bewerten. Makroskopische Untersuchung 60. Bei Versuchsende oder bei vorzeitigem Tod werden alle P- und F1-Tiere seziert und makroskopisch auf etwaige strukturelle Abnormalitäten oder pathologische Veränderungen hin untersucht. Dabei ist besonders auf die Organe des Fortpflanzungssystems zu achten. Jungtiere, die in moribundem Zustand auf humane Weise getötet werden, und tote Jungtiere sind zu dokumentieren und sollten - wenn sie nicht mazeriert werden - auf mögliche Defekte und/oder die Ursache des Todes untersucht und konserviert werden. 61. Bei adulten P- und F1-Weibchen ist am Tag der Sektion ein Vaginalabstrich zu untersuchen, um das Stadium des Östruszyklus zu bestimmen und eine Korrelation zur histopathologischen Untersuchung der Fortpflanzungsorgane zu ermöglichen. Die Uteri aller P-Weibchen (und gegebenenfalls auch aller F1-Weibchen) sind so auf Vorhandensein und Anzahl von Implantationsstellen zu untersuchen, dass die histopathologische Bewertung nicht beeinträchtigt wird. Wiegen der Organe und Konservation von Gewebe -P-Tiere und adulte F1-Tiere 62. Bei Versuchsende werden von allen P-Tieren und von allen adulten F1-Tieren der relevanten (nachstehend angeführten) Kohorten möglichst bald nach der Sektion Körpergewicht und Nassgewicht der nachstehend angeführten Organe bestimmt, um Austrocknen zu vermeiden. Die genannten Organe sind anschließend unter geeigneten Bedingungen zu konservieren. Sofern keine anderslautenden Vorgaben vorliegen, können paarige Organe einzeln oder zusammen entsprechend der üblichen Praxis des ausführenden Labors gewogen werden.
63. Zusätzlich zu den oben angeführten Organen sollten auch Proben des peripheren Nervengewebes, der Muskeln, des Rückenmarks, des Auges mit Sehnerv, des Magen-Darm-Trakts, der Harnblase, der Lunge, der Trachea (mit anhaftender Schilddrüse und Nebenschilddrüse), des Knochenmarks, des Samenleiters (bei männlichen Tieren), der Brustdrüse (bei männlichen und weiblichen Tieren) und der Vagina unter geeigneten Bedingungen zu konservieren. 64. Bei Tieren der Kohorte 1A sind alle Organe zu wiegen und für die histopathologische Untersuchung zu konservieren. 65. Für die Ermittlung prä- und postnatal induzierter immuntoxischer Wirkungen sind zehn männliche und zehn weibliche Tiere der Kohorte 1A einer jeden Behandlungsgruppe (ein Männchen oder ein Weibchen pro Wurf; alle Würfe werden durch mindestens ein nach dem Zufallsprinzip ausgewählten Jungtier repräsentiert) bei Versuchsende den folgenden Untersuchungen zu unterziehen:
Die Analyse der Lymphozytensubpopulationen in der Milz nicht immunisierter Tiere (Kohorte 1A) zeigt, ob die Exposition mit einer Veränderung der immunologischen Steady-State-Verteilung von, Helferzellen" (CD4+) oder von zytotoxischen T-Lymphozyten (CD8+) oder von natürlichen Killerzellen (NK) (schnelle Reaktionen auf neoplastische Zellen und Pathogene) zusammenhängt. 66. Bei Tieren der Kohorte 1B sollten die folgenden Organe gewogen und die entsprechenden Gewebe zum Blockstadium umgewandelt werden:
Histopathologische Untersuchungen von Kohorte 1B sollten dann durchgeführt werden, wenn die Ergebnisse der Kohorte 1A unschlüssig sind oder ein Verdacht auf Reproduktionstoxine oder endokrine Disruptoren besteht. 67. Kohorten 2A und 2B: Tests auf Entwicklungsneurotoxizität (PND 21 oder PND 22 sowie adulte Nachkommen). Tiere der Kohorte 2A werden nach den Verhaltenstests getötet, das Gewicht ihres Gehirns wird aufgezeichnet und sie werden zur Bewertung der Neurotoxizität vollständig neurohistopathologisch untersucht. Tiere der Kohorte 2B werden am PND 21 oder PND 22 getötet; das Gewicht ihres Gehirns wird aufgezeichnet und das Gehirn wird zur Bewertung der Neurotoxizität mikroskopisch untersucht. Für Tiere der Kohorte 2A und optional für Tiere der Kohorte 2B ist eine Perfusionsfixierung im Sinne der Prüfmethode B.53 (35) erforderlich. Wiegen der Organe und Konservierung von Gewebe - entwöhnte F1-Tiere 68. Nicht für die Kohorten ausgewählte Jungtiere, einschließlich Kümmerlinge, werden nach dem Absetzen an PND 22 getötet, sofern die Ergebnisse nicht darauf hindeuten, dass weitere Beobachtungen an lebenden Tieren erforderlich sind. Getötete Jungtiere werden seziert und ihre Fortpflanzungsorgane werden, wie unter den Nummern 62 und 63 beschrieben, untersucht. Bei bis zu zehn Jungtieren pro Geschlecht und Gruppe aus möglichst vielen Würfen sollten Gehirn, Milz und Thymusdrüse gewogen und unter geeigneten Bedingungen konserviert werden. Darüber hinaus kann von diesen männlichen und weiblichen Jungtieren Brustdrüsengewebe für eine weitere mikroskopische Analyse 1 (siehe OECD Guidance Document Nr. 151 (40)) konserviert werden. Massive Abnormalitäten und Zielgewebe sollten für eine etwaige spätere histologische Untersuchungen sichergestellt werden. Histopathologische Untersuchung - P-Tiere 69. Eine vollständige histopathologische Untersuchung der unter den Nummern 62 und 63 genannten Organe wird bei allen P-Tieren der Hochdosisgruppen und der Kontrollgruppen durchgeführt. Organe, die behandlungsbedingte Veränderungen aufweisen, sollten auch bei allen Tieren in Niedrigdosisgruppen untersucht werden, um die Bestimmung eines NOAEL-Werts zu unterstützen. Darüber hinaus sollten die Fortpflanzungsorgane aller Tiere mit mutmaßlich verringerter Fruchtbarkeit histopathologisch zu untersuchen, wie Tiere, die sich nicht gepaart haben, die nicht empfangen haben, nicht gedeckt wurden, keine gesunden Nachkommen geboren haben oder bei denen der Östruszyklus oder die Zahl, die Motilität oder die Morphologie der Spermien beeinträchtigt waren, sowie alle makroskopischen Läsionen histopathologisch untersucht werden. Histopathologische Untersuchung - F1-Tiere Tiere der Kohorte 1 70. Eine vollständige histopathologische Untersuchung der unter den Nummern 62 und 63 genannten Organe wird bei allen adulten Tieren der Hochdosisgruppen und der Kontrollgruppen der Kohorte 1A durchgeführt. Alle Würfe sollten durch mindestens ein Jungtier pro Geschlecht repräsentiert sein. Organe und Gewebe, die behandlungsbedingte Veränderungen aufweisen, sind auch von allen Tieren in Niedrigdosisgruppen zu untersuchen, um die Bestimmung eines NOAEL-Werts zu unterstützen. Für die Bewertung der prä- und postnatal induzierter Wirkungen auf Lymphorgane sollten neben einer bereits in allen 1A-Tieren durchgeführten histopathologischen Bewertung der Thymusdrüse, der Milz und der Nebennieren auch die gesammelten Lymphknoten und das Knochenmark von zehn männlichen und zehn weiblichen Tieren der Kohorte 1A histopathologisch untersucht werden. 71. Bei mutmaßlichen Reproduktionstoxinen oder endokrinen Disruptoren sollten nach der Beschreibung unter Nummer 66 zu Blockstadien umgewandeltes Gewebe der Fortpflanzungsorgane und des endokrinen Gewebes von allen Tieren der Kohorte 1B histopathologisch untersucht werden. Falls die Ergebnisse der Kohorte 1A unschlüssig sind, sollte auch die Kohorte 1B histologisch untersucht werden. 72. Ovarien adulter Weibchen sollten Primordialfollikel und heranreifende Follikel sowie Gelbkörper enthalten; daher sollte eine histopathologische Untersuchung darauf abzielen, Primordialfollikel und kleine heranreifende Follikel sowie Gelbkörper in F1-Weibchen quantitativ zu bestimmen; die Zahl der Tiere, die Wahl der Ovarsektion und der Umfang der Stichprobe für die Sektion sollten für das angewandte Bewertungsverfahren statistisch aussagekräftig sein. Zunächst können die Follikel bei Tieren der Kontrollgruppe und der Hochdosisgruppe gezählt werden; wenn bei letzteren eine Schadwirkung festgestellt wird, sind auch Tiere der Niedrigdosisgruppen zu untersuchen. Die Untersuchung sollte auch die Zählung der Primordialfollikel umfassen, die mit der Zählung der kleinen heranwachsenden Follikel kombiniert werden kann, um Eierstöcke behandelter Tiere mit Eierstöcken unbehandelter Tiere vergleichen zu können (siehe OECD Guidance Document Nr. 151 (40)). Die Gelbkörperuntersuchung sollte parallel zur Untersuchung der Östruszyklizität stattfinden, damit das Zyklusstadium bei der Bewertung berücksichtigt werden kann. Ovidukt, Uterus und Vagina sind auf eine angemessene organtypische Entwicklung hin zu untersuchen. 73. Eine eingehende histopathologische Untersuchung der Hoden wird bei männlichen F1-Tieren durchgeführt, um behandlungsbedingte Wirkungen auf die Entwicklung der Hoden (Differenzen) sowie auf die Spermatogenese festzustellen (38). Nach Möglichkeit sind Sektionen des Rete testis zu untersuchen. Caput, Corpus und Cauda des Nebenhodens und der Samenleiter werden auf angemessene organtypische Entwicklung sowie auf die für die P-Männchen erforderlichen Parameter hin untersucht. Tiere der Kohorte 2 74. Nach dem Abschluss der Untersuchungen neurologisch bedingter Verhaltensweisen (nach PND 75 aber vor PND 90) werden alle Tiere der Hochdosisgruppen und der Kontrollgruppen der Kohorte 2A nach Geschlecht neurohistopathologisch untersucht. Das Gehirn aller Tiere der Hochdosisgruppen und der Kontrollgruppen der Kohorte 2B wird an PND 21 oder 22 nach Geschlecht histopathologisch untersucht. Organe oder Gewebe, die behandlungsbedingte Veränderungen aufweisen, sollten auch bei Tieren in den Niedrigdosisgruppen untersucht werden, um die Bestimmung eines NOAEL-Wertes zu unterstützen. Bei Tieren der Kohorten 2A und 2B werden multiple Sektionen des Gehirns geprüft, um eine Untersuchung von Riechkolben, Großhirnrinde (Cortex cerebri), Hippocampus, Basalganglien, Thalamus, Hypothalamus, Mittelhirn (Tectum, Tegmentum und Pedunculus cerebri), Pons, Medulla oblongata, Kleinhirn) zu gewährleisten. Nur bei Tieren der Kohorte 2A werden die Augen (Netzhaut und Sehnerv) sowie Proben des peripheren Nervengewebes, der Muskeln und des Rückenmarks untersucht. Alle neurohistologischen Verfahren sollten mit der Prüfmethode B.53 (35) übereinstimmen. 75. Morphometrische (quantitative) Bewertungen sollten an repräsentativen Regionen des Gehirns (homologe und sorgfältig auf der Grundlage zuverlässiger mikroskopischer Messpunkte ausgewählte Sektionen) durchgeführt werden und können auch lineare und/oder areale Messungen der spezifischen Gehirnregionen umfassen. An jedem Orientierungspunkt (Ebene) sind mindestens drei konsekutive Schnitte vorzunehmen, damit der einheitlichste und repräsentativste Schnitt für die spezifische Hirnregion bewertet werden kann. Der Neuropathologe sollte mit angemessenem Urteilsvermögen bewerten, ob die für die Messung präparierten Schnitte mit den anderen Schnitten in der Probenreihe homolog sind und sich daher für die Einbeziehung eignen, da sich insbesondere lineare Messungen über einen relativ kurzen Abstand ändern können (28). Nicht homologe Schnitte sollten nicht verwendet werden. Das Ziel besteht zwar darin, Proben von allen diesem Zweck vorbehaltenen Tieren (10 je Geschlecht und Dosisstufe) zu entnehmen, es kann aber auch eine kleinere Anzahl an Proben angemessen sein. Proben von weniger als 6 Tieren je Geschlecht und Dosisstufe gelten für die Zwecke der vorliegenden Prüfmethode in der Regel jedoch nicht als ausreichend. Mithilfe der Stereologie können behandlungsbedingte Wirkungen auf bestimmte Parameter wie Volumen oder Zellzahl für bestimmte neuroanatomische Regionen festgestellt werden. Bei allen die Gewebepräparation betreffenden Aspekten sollte auf Ausgewogenheit geachtet werden, d. h. von der Gewebefixierung über das Schneiden der Gewebeproben und die Probenvorbereitung bis hin zur Färbung der Objektträger sollte jeder Satz repräsentative Proben einer jeden Dosisgruppe enthalten. Bei morphometrischen oder stereologischen Analysen sollte Hirngewebe bei allen Dosisstufen zur gleichen Zeit in ein geeignetes Medium eingebettet werden, um ein Schrumpfen der Prüfgegenstände zu vermeiden, was bei zu langer Aufbewahrung im Fixativ auftreten kann. Daten 76. Die Daten sind sowohl einzeln zu protokollieren als auch in tabellarischer Form zusammenzufassen. Gegebenenfalls sind für jede Prüfgruppe und für jede Generation die folgenden Angaben aufzuzeichnen: die Zahl der Tiere zu Beginn der Prüfung und die Zahl der während der Prüfung tot aufgefundenen oder aus Tierschutzgründen getöteten Tiere, ferner der Zeitpunkt des Todes oder der Tötung, die Zahl der fruchtbaren Tiere, die Zahl der trächtigen Weibchen, die Zahl der Weibchen, die Jungtiere werfen, und die Zahl der Tiere, die Toxizitätszeichen aufweisen, sowie eine Beschreibung der beobachteten Toxizität, einschließlich des Zeitpunkts, zu dem die toxischen Wirkungen erstmalig aufgetreten sind, ihrer Dauer und ihres Schweregrads. 77. Die numerischen Daten sollten nach einem geeigneten statistischen Verfahren ausgewertet werden. Die Statistikmethoden sollten Teil des Prüfplans sowie geeignet sein, um Nichtnormaldaten (z.B. Zähldaten), zensierte Daten (z.B. eingeschränkte Beobachtungszeit), Unabhängigkeit (z.B. Wirkungen der Würfe und wiederholte Messungen) sowie ungleiche Varianzen zu bewältigen. Allgemeingültige lineare gemischte Modelle und Dosis-Wirkungs- Modelle decken ein breites Spektrum an Analysetools ab, die für die im Rahmen dieser Prüfmethode erzeugten Daten geeignet sein können. Der Bericht sollte ausreichende Informationen über das angewandte Analyseverfahren und Computerprogramm enthalten, damit ein unabhängiger Überprüfer/Statistiker die Analyse bewerten und nachvollziehen kann. Auswertung der Ergebnisse 78. Die Befunde sind im Hinblick auf die beobachteten Wirkungen, einschließlich der Befunde der makroskopischen und mikroskopischen Untersuchungen, zu bewerten. Ausgewertet werden u. a. die Beziehung oder die fehlende Beziehung zwischen der Dosis und dem Vorliegen, dem Auftreten und dem Schweregrad von Abnormalitäten, einschließlich makroskopischer Veränderungen. Zielorgane, Fruchtbarkeit, klinische Abnormalitäten, Reproduktionsleistung und Wurfleistung, Veränderungen des Körpergewichts, Mortalität und alle sonstigen toxischen Auswirkungen und Auswirkungen auf die Entwicklung sind ebenfalls zu bewerten. Besonderes Augenmerk gilt dabei geschlechtsspezifischen Veränderungen. Bei der Auswertung der Testergebnisse sind die physikalisch-chemischen Eigenschaften der Prüfsubstanz und - falls verfügbar - toxikokinetische Daten, einschließlich plazentarer Übertragung und Milchausscheidung, zu berücksichtigen. Prüfbericht 79. Der Prüfbericht sollte die folgenden, zu den in dieser Prüfung untersuchten P-, F1-Tieren und gegebenenfalls F2-Tieren generierten Daten enthalten: Prüfsubstanz:
Vehikel (falls verwendet):
Versuchstiere:
Prüfungsbedingungen:
Ergebnisse (Sammel- und Einzeldaten, aufgeschlüsseltnach Geschlecht und Dosis):
Parameter für Kohorte 2:
Parameter für die Kohorte 3:
Diskussion der Ergebnisse Schlussfolgerungen, einschließlich der NOAEL-Werte für Wirkungen bei Elterntieren und deren Nachkommen Alle Informationen, die nicht während des Versuchs generiert wurden, für die Auswertung der Ergebnisse jedoch zweckdienlich sind (z.B. Ähnlichkeiten der Wirkungen mit bekannten Neurotoxinen) sind ebenfalls anzuführen. Auswertung der Ergebnisse 80. Eine erweiterte Ein-Generationen-Prüfung auf Reproduktionstoxizität (EOGRTS) generiert Daten über die Wirkungen wiederholter Verabreichungen einer Chemikalie während aller Phasen des Fortpflanzungszyklus. Sie gibt insbesondere Auskunft über das Fortpflanzungssystem sowie über Entwicklung, Wachstum, Überleben und funktionale Endpunkte der Nachkommen bis zum postnatalen Tag (PND) 90. 81. Bei der Auswertung der Prüfungsergebnisse sollten alle verfügbaren Daten über die Prüfsubstanz, einschließlich physikalisch-chemischer, toxikokinetischer und toxikodynamischer Eigenschaften, sowie verfügbare relevante Informationen über strukturelle Analogien und Ergebnisse vorausgegangener Toxizitätsstudien mit der Prüfsubstanz (z.B. akute Toxizität, Toxizität bei wiederholter Verabreichung, mechanistische Studien und Studien, in denen bewertet wird, ob bei den Invivo-/Invitro-Stoffwechseleigenschaften erhebliche qualitative und quantitative artspezifische Unterschiede vorliegen) berücksichtigt werden. Die Ergebnisse der makroskopischen Untersuchung (Nekropsie) und die Organgewichte sollten nach Möglichkeit im Kontext der Beobachtungen bewertet werden, die bereits in anderen Versuchen mit wiederholter Verabreichung gemacht wurden. Wachstumsverlangsamung bei Nachkommen könnte auf den Einfluss der Prüfsubstanz auf die Milchzusammensetzung zurückgeführt werden (29). Kohorte 2 (Entwicklungsneurotoxizität) 82. Neurologisch bedingtes Verhalten und neuropathologische Ergebnisse sollten unter Berücksichtigung aller Befunde von Fachleuten nach einem Weightof-evidence-Ansatz ausgewertet werden. Die Muster verhaltensbedingter oder morphologischer Befunde sollten, sofern vorhanden, ebenso erörtert werden wie eine nachweisliche Dosis-Wirkungs-Beziehung. In diese Charakterisierung sollte die Beurteilung der Entwicklungsneurotoxizität, einschließlich epidemiologischer Untersuchungen beim Menschen oder Fallberichte sowie tierexperimentelle Studien (z.B. toxikokinetische Daten, Daten zur Struktur-Wirkungs-Beziehung, Daten aus anderen Toxizitätsstudien) einfließen. Die Datenauswertung sollte ferner eine Diskussion sowohl der biologischen als auch der statistischen Signifikanz beinhalten. Sofern ein Zusammenhang zwischen neuropathologischen Veränderungen und Verhaltensänderungen beobachtet wurde, sollte dieser in die Beurteilung einbezogen werden. Leitlinien zur Auswertung der entwicklungsneurotoxischen Ergebnisse finden sich in der Prüfmethode B.53 (35) und in Tyl et al., 2008 (31). Kohorte 3 (Entwicklungsimmuntoxizität) 83. Die mithilfe des TDAR-Assay (T-Zellabhängigen Antikörperantwort) ermittelte Unterdrückung oder Verstärkung der Immunfunktion sollte im Kontext aller festgestellten Beobachtungen bewertet werden. Die Signifikanz des TDAR-Ergebnisses kann durch andere Wirkungen auf immunologisch verwandte Indikatoren (z.B. Knochenmarkzellularität, Gewicht und histopathologische Untersuchung des Lymphdrüsengewebes, Lymphozyten-Teilmengenverteilung) untermauert werden. Die im TDAR ermittelten Wirkungen sind möglicherweise weniger aussagekräftig bei anderen Toxizitäten, die bei niedrigeren Expositionskonzentrationen festgestellt werden. 84. Als Hilfe bei der Auswertung der reproduktionstoxischen und neurotoxischen Ergebnisse ist das OECD Guidance Document Nr. 43 zu Rate zu ziehen (26). (1) Cooper, R.L., J.C. Lamb, S.M. Barlow, K. Bentley, A.M. Brady, N. Doerr, D.L. Eisenbrandt, P.A. Fenner-Crisp, R.N. Hines, L.F.H. Irvine, C.A. Kimmel, H. Koeter, A.A. Li, S.L. Makris, L.P. Sheets, G.J.A. Speijers and K.E. Whitby (2006).,A Tiered Approach to Life Stages Testing for Agricultural Chemical Safety Assessment", Critical Reviews in Toxicology, 36, S. 69-98. (2) Thigpen, J.E., K.D.R. Setchell, K.B. Ahlmark, J. Locklear, T. Spahr, G.F. Leviness, M.F. Goelz, J.K. Haseman, R.R. Newbold, and D.B. Forsythe (1999)., Phytoestrogen Content of Purified Open and Closed Formula Laboratory Animal Diets", Lab. Anim. Sci., 49, S. 530-536. (3) Zoetis, T. and I. Walls (2003). Principles and Practices for Direct Dosing of Pre-Weaning Mammals in Toxicity Testing and Research, ILSI Press, Washington, DC. (4) Moser, V.C., I. Walls and T. Zoetis (2005)., Direct Dosing of Preweaning Rodents in Toxicity Testing and Research: Deliberations of an ILSI RSI Expert Working Group", International Journal of Toxicology, 24, S. 87-94. (5) Conolly, R.B., B.D. Beck, and J.I. Goodman (1999)., Stimulating Research to Improve the Scientific Basis of Risk Assessment", Toxicological Sciences, 49, S. 1-4. (6) Ulbrich, B. and A.K. Palmer (1995)., Detection of Effects on Male Reproduction - a Literature Survey", Journal of the American College of Toxicologists, 14, S. 293-327. (7) Mangelsdorf, I., J. Buschmann and B. Orthen (2003)., Some Aspects Relating to the Evaluation of the Effects of Chemicals on Male Fertility", Regulatory Toxicology and Pharmacology, 37, S. 356-369. (8) Sakai, T., M. Takahashi, K. Mitsumori, K. Yasuhara, K. Kawashima, H. Mayahara und Y. Ohno (2000)., Collaborative work to evaluate toxicity on male reproductive organs by repeated dose studies in rats - overview of the studies", Journal of Toxicological Sciences, 25, S. 1-21. (9) Creasy, D.M. (2003)., Evaluation of Testicular Toxicology: A Synopsis and Discussion of the Recommendations Proposed by the Society of Toxicologic Pathology", Birth Defects Research, Part B, 68, S. 408-415. (10) Goldman, J.M., A.S. Murr, A.R. Buckalew, J.M. Ferrell and R.L. Cooper (2007)., The Rodent Estrous Cycle: Characterization of Vaginal Cytology and its Utility in Toxicological Studies", Birth Defects Research, Part B, 80 (2), S. 84-97. (11) Sadleir, R.M.F.S. (1979)., Cycles and Seasons", in C.R. Auston and R.V. Short (eds.), Reproduction in Mammals: I. Germ Cells and Fertilization, Cambridge, New York. (12) Gallavan, R.H. Jr, J.F. Holson, D.G. Stump, J.F. Knapp and V.L. Reynolds (1999)., Interpreting the Toxicologic Significance of Alterations in Anogenital Distance: Potential for Confounding Effects of Progeny Body Weights", Reproductive Toxicology, 13: S. 383-390. (13) Korenbrot, C.C., I.T. Huhtaniemi and R.I. Weiner (1977)., Preputial Separation as an External Sign of Pubertal Development in the Male Rat", Biological Reproduction, 17, S. 298-303. (14) Ladics, G.S. (2007)., Use of SRBC Antibody Responses for Immunotoxicity Testing", Methods, 41, S. 9-19. (15) Gore, E.R., J. Gower, E. Kurali, J.L. Sui, J. Bynum, D. Ennulat and D.J. Herzyk (2004)., Primary Antibody Response to Keyhole Limpet Hemocyanin in Rat as a Model for Immunotoxicity Evaluation", Toxicology, 197, S. 23-35. (16) Gray, L.E., J. Ostby, J. Ferrell, G. Rehnberg, R. Linder, R. Cooper, J. Goldman, V. Slott and J. Laskey (1989).,A Dose-Response Analysis of Methoxychlor-Induced Alterations of Reproductive Development and Function in the Rat", Fundamental and Applied Toxicology, 12, S. 92-108. (17) Robb, G.W., R.P. Amann and G.J. Killian (1978)., Daily Sperm Production and Epididymal Sperm Reserves of Pubertal and Adult Rates", Journal of Reproduction and Fertility,54, S. 103-107. (18) Klinefelter, G.R., L.E. Jr Gray and J.D. Suarez (1991)., The Method of Sperm Collection Significantly Influences Sperm Motion Parameters Following Ethane Dimethanesulfonate Administration in the Rat". Reproductive Toxicology,5, S. 39-44. (19) Seed, J., R.E. Chapin, E.D. Clegg., L.A. Dostal, R.H. Foote, M.E. Hurtt, G.R. Klinefelter, S.L. Makris, S.D. Perreault, S. Schrader, D. Seyler, R. Sprando, K.A. Treinen, D.N. Veeramachaneni and L.D. Wise (1996)., Methods for Assessing Sperm Motility, Morphology, and Counts in the Rat, Rabbit, and Dog: a Consensus Report", Reproductive Toxicology, 10, S. 237-244. (20) Chapin, R.E., R.S. Filler, D. Gulati, J.J. Heindel, D.F. Katz, C.A. Mebus, F. Obasaju, S.D. Perreault, S.R. Russell and S. Schrader (1992)., Methods for Assessing Rat Sperm Motility", Reproductive Toxicology, 6, S. 267-273. (21) Klinefelter, G.R., N.L. Roberts and J.D. Suarez (1992)., Direct Effects of Ethane Dimethanesulphonate on Epididymal Function in Adult Rates: an In Vitro Demonstration", Journal of Andrology, 13, S. 409-421. (22) Slott, V.L., J.D. Suarez and S.D. Perreault (1991)., Rat Sperm Motility Analysis: Methodologic Considerations", Reproductive Toxicology, 5, S. 449-458. (23) Slott, V.L., and S.D. Perreault (1993)., Computer-Assisted Sperm Analysis of Rodent Epididymal Sperm Motility Using the Hamilton-Thorn Motility Analyzer", Methods in Toxicology, Part A, Academic, Orlando, Florida, S. 319-333. (24) Toth, G.P., J.A. Stober, E.J. Read, H. Zenick and M.K. Smith (1989)., The Automated Analysis of Rat Sperm Motility Following Subchronic Epichlorhydrin Administration: Methodologic and Statistical Considerations", Journal of Andrology, 10, S. 401-415. (25) Linder, R.E., L.F. Strader, V.L. Slott and J.D. Suarez (1992)., Endpoints of Spermatoxicity in the Rat After Short Duration Exposures to Fourteen Reproductive Toxicants", Reproductive Toxicology, 6, S. 491-505. (26) OECD (2008). Guidance Document on Mammalian Reproductive Toxicity Testing and Assessment, Series on Testing and Assessment, Nr. 43, ENV/JM/MONO(2008)16, OECD, Paris. (27) Working, P.K., M. Hurtt (1987)., Computerized Videomicrographic Analysis of Rat Sperm Motility", Journal of Andrology, 8, S. 330-337. (28) Bolin, B., R. Garman, K. Jensen, G. Krinke, B. Stuart, and an ad Hoc Working Group of the STP Scientific and Regulatory Policy Committee (2006).,A, Best Practices" Approach to Neuropathologic Assessment in Developmental Neurotoxicity Testing - for Today", Toxicological Pathology, 34, S. 296-313. (29) Stütz, N., B. Bongiovanni, M. Rassetto, A. Ferri, A.M. Evangelista de Duffard, and R. Duffard (2006)., Detection of 2,4-dichlorophenoxyacetic Acid in Rat Milk of Dams Exposed During Lactation and Milk Analysis of their Major Components", Food Chemicals Toxicology, 44, S. 8-16. (30) Thigpen, JE, K.D.R. Setchell, J.K. Haseman, H.E. Saunders, G.F. Caviness, G.E. Kissling, M.G. Grant and D.B. Forsythe (2007)., Variations in Phytoestrogen Content between Different Mill Dates of the Same Diet Produces Significant Differences in the Time of Vaginal Opening in CD-1 Mice and F344 Rates but not in CD Sprague Dawley Rates", Environmental health perspectives, 115(12), S. 1717-1726. (31) Tyl, R.W., K. Crofton, A. Moretto, V. Moser, L.P. Sheets and T.J. Sobotka (2008)., Identification and Interpretation of Developmental Neurotoxicity Effects: a Report from the ILSI Research Foundation/Risk Science Institute Expert Working Group on Neurodevelopmental Endpoints", Neurotoxicology and Teratology, 30: S. 349-381. (32) OECD (1996). Combined Repeated Dose Toxicity Study with the Reproduction/Developmental Toxicity Screening Test, OECD Guideline for Testing of Chemicals, Nr. 422, OECD, Paris. (33) Kapitel B.43 dieses Anhangs, Prüfung auf Neurotoxizität bei Nagetieren. (34) OECD (2000). Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations, Series on Testing and Assessment, Nr. 19, ENV/JM/MONO(2000)7, OECD, Paris. (35) Kapitel B.53 dieses Anhangs, Prüfung auf Entwicklungsneurotoxizität. (36) Kapitel B.54 dieses Anhangs: Uterotropher Bioassay an Nagetieren: Ein Kurzzeit-Screening-Test auf östrogene Eigenschaften. (37) Kapitel B.55 dieses Anhangs: Hershberger-Bioassay an Ratten: Ein Kurzzeit-Screening-Test auf (anti-)androgene Eigenschaften. (38) OECD (2009). Guidance Document for Histologic Evalution of Endocrine and Reproductive Test in Rodents, Series on Testing and Assessment, Nr. 106, OECD, Paris. (39) OECD (2011). Guidance Document on the Current Implementation of Internal Triggers in the Extended One Generation Reproductive Toxicity Study in the United States and Canada, Series on Testing and Assessment, Nr. 117, ENV/JM/MONO(2011)21, OECD, Paris. (40) OECD (2013). Guidance Document supporting TG 443: Extended One Generation Reproductive Toxicity Study, Series on Testing and Assessment, Nr. 151, OECD, Paris. ____________ 1) Die Forschung hat gezeigt, dass die Brustdrüse, insbesondere in ihrer frühen Entwicklungsphase, einen empfindlichen Endpunkt für die Östrogenwirkung darstellt. Endpunkte, die Brustdrüsen von Jungtieren beiderlei Geschlechts betreffen, sollten deshalb, sobald sie validiert wurden, in diese Prüfmethode einbezogen werden.
Maßnahmen und Beobachtungen im Rahmen der FOB (Functional Observational Battery) (Kohorte 2A)
Chemikalie: ein Stoff oder eine Mischung. Prüfsubstanz: jede(r) mittels dieser Prüfmethode getestete Stoff bzw. Mischung. |
B.57. H295R-Steroidgenese-Assay 14 24
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 456 (2011). Die OECD setzte sich 1998 zur Priorität, bestehende Prüfrichtlinien für Screening und Testung potenziell endokriner Disruptoren zu überarbeiten und neue Richtlinien zu entwickeln. Das Rahmenkonzept der OECD für die Testung und Bewertung endokriner Disruptoren von 2002 umfasst fünf Stufen, wobei jede Stufe einem anderen Grad der biologischen Komplexität entspricht (1). Bei dem in der vorliegenden Prüfmethode beschriebenen Invitro Testsystem - dem H295R-Steroidgenese-Assay - wird eine menschliche Adenokarzinom-Zelllinie (NCI-H295R-Zellen) verwendet. Der Assay stellt einen, Invitro- Assay der Stufe 2 dar, und liefert mechanistische Daten", die zum Screening und für Priorisierungszwecke zu verwenden sind. Die Ausarbeitung und Standardisierung des Assays als Screeningmethode für chemische Auswirkungen auf die Steroidgenese, insbesondere auf die Produktion von 17β -Östradiol (E2) und Testosteron (T), erfolgte in mehreren Schritten. Der H295R-Assay ist optimiert und validiert worden (2) (3) (4) (5). 2. Das Ziel des H295R-Steroidgenese-Assays besteht darin, Chemikalien nachzuweisen, durch die die Produktion von E2 und T beeinflusst wird. Mit dem H295R-Assay sollen Xenobiotika ermittelt werden, die diejenigen endogenen Bestandteile als Zielstelle(n) haben, die den intrazellularen biochemischen Pfad bilden, der mit Cholesterol beginnt und bis zur Produktion von E2 und/oder T führt. Mit dem H295R-Assay sollen keine Chemikalien bestimmt werden, die die Steroidgenese aufgrund von Auswirkungen auf die Hypothalamus-Hypophysen-Gonaden-Achse (HPG- Achse) beeinträchtigen. Der Assay zielt vielmehr darauf ab, im Hinblick auf das Potenzial einer Chemikalie, die Produktion von T und E2 zu induzieren oder zu hemmen, eine JA/NEIN-Antwort zu liefern; in einigen Fällen können jedoch auch quantitative Ergebnisse erzielt werden (siehe Nummern 53 und 54). Die Ergebnisse des Assays werden als relative Veränderungen in der Hormonproduktion im Vergleich zu den Lösungsmittelkontrollen (LK) angegeben. Der Assay zielt nicht darauf ab, spezifische mechanistische Informationen über die Interaktion der Prüfsubstanz mit dem endokrinen System zu liefern. Anhand der Zelllinie wurden Forschungen durchgeführt, um Auswirkungen auf bestimmte Enzyme und Zwischenhormone, wie zum Beispiel Progesteron, zu bestimmen (2). 3. Die in der vorliegenden Prüfmethode verwendeten Begriffe und Abkürzungen sind in der Anlage beschrieben. Ein detailliertes Protokoll mit Anweisungen zur Herstellung von Lösungen, Kultivierung von Zellen und Durchführung verschiedener Aspekte der Prüfung ist als Anhang I-III des OECD-Dokuments, Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production" verfügbar (4). Ausgangsüberlegungen und Grenzen 4. An der Biosynthese sexueller Steroidhormone sind fünf verschiedene Enzyme, die sechs unterschiedliche Reaktionen katalysieren, beteiligt. Die enzymatische Umwandlung von Cholesterin in Pregnenolon durch die Cytochrom P450-abhängige Cholesterin-Monooxygenase (CYP11A) ist der erste Schritt in einer Reihe biochemischer Reaktionen, die in der Synthese steroider Endprodukte gipfeln. Je nach Reihenfolge der beiden nächsten Reaktionen teilt sich der Pfad der Steroidgenese in zwei Pfade auf: den Δ5-Hydroxysteroid-Pfad und den Δ4-Ketosteroid-Pfad, die in der Produktion von Androstenedion (Abbildung 1) zusammenlaufen. 5. Androstenedion wird durch 17β-Hydroxysteroid Dehydrogenase (17β-HSD) in Testosteron (T) umgewandelt. Testosteron ist sowohl ein Zwischen- als auch ein Endhormonprodukt. Im männlichen Organismus kann T durch 5α-Reduktase, die in Zellmembranen, Kernhülle und im Retikulum von Zielgewebe mit androgener Wirkung, wie zum Beispiel Prostata und Samenbläschen, gefunden wird, in Dihydrotestosteron (DHT) umgewandelt werden. DHT ist als Androgen erheblich wirksamer als T und gilt ebenfalls als Endprodukthormon. Der H295R-Assay misst kein DHT (siehe Nummer 10). 6. Das Enzym im Pfad der Steroidgenese, das androgene Chemikalien in östrogene Chemikalien umwandelt, ist Aromatase (CYP19). CYP19 wandelt T in 17β -Östradiol (E2) und Androstenedion in Östron um. E2 und T gelten als Endprodukthormone des Steroidgenesepfads. 7. Die Spezifizität der Lyaseaktivität von CYP17 ist bei den verschiedenen Intermediaten von Tierart zu Tierart unterschiedlich. Im Menschen bevorzugt das Enzym Substrate des Δ5-Hydroxysteroid-Pfads (Pregnenolon); dagegen werden in der Ratte Substrate im Δ4-Ketosteroid-Pfad (Progesteron) begünstigt (19). Solche Unterschiede in der CYP17-Lyaseaktivität können einige artspezifische Unterschiede in der Reaktion auf Chemikalien erklären, die die Steroidgenese in vivo verändern (6). Es hat sich erwiesen, dass die H295-Zellen die Expression des humanen adulten Nebennierenenzyms und das Muster der Steroidproduktion am genauesten widerspiegeln (20), aber auch dafür bekannt sind, Enzyme sowohl für den Δ5-Hydroxysteroid als auch für den Δ4-Ketosteroid-Pfad für Androgensynthese zu exprimieren (7) (11) (13) (15). Abbildung 1 Pfad der Steroidgenese in H295R-Zellen Anmerkung: 8. Die menschliche H295R-Adenokarzinom-Zelllinie ist ein nützliches Invitro-Modell für die Ermittlung der Auswirkungen auf die Synthese von Steroidhormonen (2) (7) (8) (9) (10). Die H295R-Zelllinie exprimiert Gene, die alle wichtigen Enzyme für die oben genannte Steroidgenese verschlüsseln (11) (15) (Abbildung 1). Das ist eine einzigartige Eigenschaft, weil die Invivo-Expression dieser Gene gewebe- und entwicklungsstadiumspezifisch ist, d. h., kein Gewebe- oder Entwicklungsstadium exprimiert alle an der Steroidgenese beteiligten Gene (2). H295R-Zellen weisen physiologische Eigenschaften zonal undifferenzierter Nebennierenzellen menschlicher Föten auf (11). Die Zellen stellen ein einzigartiges Invitro-System dar, da sie die Fähigkeit besitzen, alle in der adulten Nebennierenrinde und den Gonaden gefundenen Steroidhormone zu produzieren. Mit ihnen können Auswirkungen sowohl auf die Corticosteroidsynthese als auch auf die Produktion von sexuellen Steroidhormonen, wie zum Beispiel Androgene und Östrogene, geprüft werden, auch wenn der Assay nur für den Nachweis von T und E2 validiert wurde. Die durch das Prüfsystem erfassten Änderungen in Form einer Veränderung der Produktion von T und E2 können das Ergebnis einer Vielzahl unterschiedlicher Interaktionen der Prüfsubstanzen mit Steroidgenesefunktionen sein, die durch die H295R-Zellen exprimiert werden. Dazu gehört die Modulation der Expression, die Synthese oder die Funktion von bei der Produktion, Umwandlung oder Eliminierung von Steroidhormonen beteiligten Enzymen (12) (13) (14). Die Hemmung der Hormonproduktion kann auf eine direkte kompetitive Bindung an ein Enzym, einen Einfluss auf Ko-Faktoren, wie zum Beispiel NADPH (Nicotinamidadenindinukleotidphosphat) und cAMP (zyklisches Adenosinmonophosphat), und/oder eine Steroidstoffwechselerhöhung oder die Reprimierung der Genexpression bestimmter Enzyme im Steroidgenesepfad zurückgeführt werden. Während die Hemmung sowohl von direkten als auch indirekten an der Hormonproduktion beteiligten Prozessen abhängig sein kann, erfolgt die Induktion in der Regel indirekt, beispielsweise durch beeinflussende Ko-Faktoren wie NADPH und cAMP (wie etwa bei Forskolin), einen sinkenden Steroidstoffwechsel (13) und/oder eine Hochregulierung der Genexpression der Steroidgenese. 9. Der H295R-Assay weist mehrere Vorteile auf:
10. Die wichtigsten Einschränkungen des Assays sind folgende:
Prinzip der Prüfmethode 11. Zweck des Assays ist der Nachweis von Chemikalien, die die Produktion von T und E2 beeinträchtigen. T ist außerdem ein Zwischenprodukt im Produktionspfad von E2. Mit dem Assay können Chemikalien nachgewiesen werden, die die Enzyme des Steroidgenesepfades in der Regel hemmen oder auslösen. 12. Der Assay wird in der Regel unter Standardzellkulturbedingungen in 24-Mulden-Kulturplatten durchgeführt. Alternativ dazu können andere Plattengrößen für die Durchführung des Assays verwendet werden; die Saat- und Versuchsbedingungen sind dabei jedoch so anzupassen, dass die Leistungskriterien erfüllt sind. 13. Nach einer Akklimatisierungsphase von 24 Stunden in Multiwellplatten werden die Zellen 48 Stunden lang mindestens dreifach sieben Konzentrationen der Prüfsubstanz ausgesetzt. Als Negativ- und Positivkontrollen dienen Testreihen mit Lösungsmittel und jeweils einem bekannten Stoff, der die Hormonproduktion hemmt bzw. induziert, in einer festgelegten Konzentration. Am Ende der Expositionszeit wird das Medium aus jeder Mulde entfernt. Unmittelbar nach Entfernen des Mediums wird in jeder Mulde die Zellviabilität untersucht. Die Hormonkonzentrationen im Medium können mit einer Vielzahl von Methoden gemessen werden, wozu auch im Handel erhältliche Kits zur Hormonmessung und/oder Gerätetechniken wie die Flüssigchromatographie-Massenspektrometrie gehören. Die Daten werden als,fold change" (xfache Änderung) bezüglich der Lösungsmittelkontrolle und der niedrigsten Konzentration mit messbarer Wirkung (Lowest-Observed-Effect-Concentration, LOEC) angegeben. Falls der Assay negativ ist, wird die höchste geprüfte Konzentration als geprüfte Konzentration ohne messbare schädliche Wirkung (No-Observed-Effect-Concentration, NOEC) dokumentiert. Schlussfolgerungen bezüglich der Fähigkeit einer Chemikalie, die Steroidgenese zu beeinträchtigen, sollten sich auf mindestens zwei unabhängige Testreihen stützen. Die erste Testreihe kann zur Dosisfindung, gegebenenfalls mit anschließender Anpassung der Konzentrationen für die Testreihen 2 und 3, dienen, wenn Probleme mit der Löslichkeit oder der Zytotoxizität auftreten oder die Aktivität der Chemikalie am Ende des geprüften Konzentrationsbereichs zu liegen scheint. Zelllinie 14. Die NCI-H295R-Zellen sind nach Unterzeichnung einer Materialübertragungsvereinbarung (Material Transfer Agreement, MTA) 1 im Handel von der American Type Culture Collections (ATCC) erhältlich. Einleitung 15. Wegen Änderungen in der E2-Produktionskapazität der Zellen mit zunehmendem Alter/steigender Passagenzahl (2) sollten die Zellen vor ihrer Verwendung unter Befolgung eines speziellen Protokolls gezüchtet werden, und die Zahl der Passagen nach dem Auftauen der Zellen sollte ebenso dokumentiert werden wie die Zahl der Passagen, bei denen die Zellen eingefroren und in flüssigem Stickstoff gelagert wurden. Die erste Zahl gibt die tatsächliche Anzahl Zellpassagen an und die zweite Zahl beschreibt die Anzahl Passagen, bei denen die Zellen eingefroren und eingelagert wurden. So würden beispielsweise Zellen, die nach Passage fünf eingefroren und aufgetaut und anschließend dreimal geteilt wurden (vier Passagen, wenn die frisch aufgetauten Zellen als Passage 1 gezählt werden), nachdem sie wieder gezüchtet wurden, als Passage 4.5 gekennzeichnet. Ein Beispiel für ein Nummerierungsschema ist in Anhang 1 des Validierungsberichts angeführt (4). 16. Als Basis für angereichertes Medium und Einfriermedium wird Stammmedium verwendet. Angereichertes Medium ist zur Züchtung von Zellen ein notwendiger Bestandteil. Einfriermedium wurde speziell für ein auswirkungsfreies Einfrieren von Zellen für die langfristige Lagerung konzipiert. Vor der Verwendung sollte Nu-Serum (oder ein vergleichbares Serum mit gleichen Eigenschaften, das nachgewiesenermaßen Daten erzeugt, die die Anforderungen an die Prüfleistung und die Qualitätskontrolle erfüllen), das ein Bestandteil des angereicherten Mediums ist, auf Hintergrundkonzentrationen von T und E2 untersucht werden. Die Zubereitung dieser Lösungen wird im Anhang II des Validierungsberichts beschrieben (4). 17. Nach Ansetzen einer H295R-Zellkultur aus einer ursprünglichen ATCC-Charge sind die Zellen über fünf Passagen zu vermehren (d. h.,die Zellen werden vier Mal geteilt). Passage-Fünf-Zellen werden dann in flüssigem Stickstoff zur Lagerung eingefroren. Vor dem Einfrieren der Zellen wird eine Probe der vorangegangenen Passage-Vier-Zellen in einer Qualitätskontrollplatte getestet (siehe die Nummern 36 und 37), um zu überprüfen, ob die Basalhormonproduktion und die Reaktion auf positive Kontrollchemikalien die in Tabelle 5 festgelegten Qualitätskontrollkriterien für den Assay erfüllen. 18. H295R-Zellen müssen gezüchtet, eingefroren und in flüssigem Stickstoff gelagert werden, um sicherzustellen, dass für die Kultivierung und die Verwendung stets Zellen der richtigen Passage/des richtigen Alters verfügbar sind. Die maximale Anzahl Passagen nach Übernahme einer neuen 2 oder gefrorenen 3 Zellcharge in die Kultur, die zur Verwendung im H295R-Assay akzeptabel ist, sollte 10 nicht übersteigen. Akzeptable Passagen für Kulturen von Zellen aus einer bei Passage 5 eingefrorenen Charge wären beispielsweise 4.5 bis 10.5. Bei Zellen, mit denen von diesen eingefrorenen Chargen begonnen wurde, ist das in Nummer 19 beschriebene Verfahren zu befolgen. Diese Zellen sind in mindestens vier (4) zusätzlichen Passagen (Passage 4.5) zu züchten, bevor sie in Tests eingesetzt werden können. Begin einer neuer Zellkultur mit Zellen aus der Gefrierlagerung 19. Das Verfahren mit zum Start einer neuen Zellkultur mit Zellen aus der Gefrierlagerung ist anzuwenden, wenn eine neue Charge Zellen aus der Lagerung in flüssigem Stickstoff zu Zucht- und Testzwecken entnommen wird. Dieses Verfahren ist in Anhang III des Validierungsberichts ausführlich beschrieben (4). Die Zellen werden aus der Kryokonservierung entnommen, rasch aufgetaut, in angereichertem Medium in ein Zentrifugenröhrchen überführt, bei Zimmertemperatur abzentrifugiert, in angereichertem Medium resuspendiert und in eine Kulturflasche übertragen. Das Medium sollte am nächsten Tag gewechselt werden. Die H295R-Zellen werden in einem Inkubator bei 37 °C in einer mit 5 % CO2-angereicherten Luftatmosphäre kultiviert und das Medium wird 2-3 Mal pro Woche gewechselt. Wenn die Zellen zu ungefähr 85-90 % konfluent sind, sollten sie aufgeteilt werden. Die Zellen müssen aufgeteilt werden, um die Lebensfähigkeit und das Wachstum der Zellen sicherzustellen und um ausreichend Zellen für die Durchführung von Bioassays zur Verfügung zu haben. Die Zellen werden drei Mal mit Phosphatgepufferter Salzlösung (PBS, ohne Ca2+ Mg2+.) ausgewaschen und durch Zugabe eines geeigneten Enzyms, z.B. Trypsin, in PBS (ohne Ca2+ Mg2+) aus der Kulturflasche herausgelöst. Die Reaktion sollte nach Ablösen der Zellen von der Kulturflasche durch Zugabe eines dreifachen Volumens an angereichertem Medium, im Verhältnis zu dem für die Enzymbehandlung verwendeten Volumen, gestoppt werden. Die Zellen werden in ein Zentrifugenröhrchen überführt, bei Zimmertemperatur zentrifugiert; der Überstand wird entfernt und das Pellet in angereichertem Medium resuspendiert. Eine entsprechende Menge Zellsuspension wird in die neue Kulturflasche gegeben. Die Menge an Zellsuspension sollte sollte so gewählt werden, dass die Zellen innerhalb von fünf bis sieben Tagen Konfluenz erreichen. Das empfohlene Subkultivierungsverhältnis beträgt 1:3 bis 1:4. Die Platte ist sorgfältig zu beschriften. Die Zellen sind jetzt für die Verwendung im Assay bereit. Überschüssige Zellen sollten in flüssigem Stickstoff eingelagert werden so wie in Paragraph 20 beschrieben. Einfrieren von H295R-Zellen (Vorbereitung von Zellen für die Kryokonservierung) 20. Um H295R-Zellen zum Einfrieren vorzubereiten, ist das oben beschriebene Verfahren für die Aufteilung von Zellen bis zu dem Schritt der Resuspendierung des Zellenpellets am Boden des Zentrifugenröhrchens zu befolgen. Hier wird das Zellenpellet in Einfriermedium resuspendiert. Die Lösung wird in ein entsprechend etikettiertes Kryogenfläschchen übertragen und bei - 80 °C 24 Stunden lang eingefroren. Danach wird das Kryogenfläschchen zur Lagerung in flüssigen Stickstoff gegeben. Dieses Verfahren ist in Anhang III des Validierungsberichts ausführlich beschrieben (4). Plattierung und Vorinkubation von Zellen für die Durchführung der Tests 21. Die benötigte Anzahl an nach den Angaben in Absatz 19 vorbereiteten 24-Muldenplatten hängt von der Zahl der zu prüfenden Chemikalien und der Konfluenz der Zellen in den Kulturschalen ab. In der Regel bietet eine Kulturflasche (75 cm2) mit 80-90 % konfluenten Zellen genügend Zellen für eine bis eineinhalb Platten (24-Mulden) mit einer angestrebten Dichte von 200.000 bis 300.000 Zellen pro ml Medium, was innerhalb von 24 Stunden zu ungefähr 50-60 % Konfluenz in den Mulden führt (Abbildung 2). Dies ist typischerweise die optimale Zelldichte für die Hormonproduktion im Assay. Bei höheren Dichten verändern sich sowohl die T- als auch die Muster der T- als auch der E2-Produktion. Bevor der Assay das erste Mal durchgeführt wird, empfiehlt es sich, unterschiedliche Einsaatdichten zwischen 200.000 und 300.000 Zellen pro ml zu prüfen und die Dichte, die sich bei 50-60 % Konfluenz in der Mulde nach 24 Stunden ergibt, für weitere Versuche auszuwählen. Mikrofotografie von H295R-Zellen bei einer Einsaatdichte von 50 % in einer 24-Mulden-Kulturplatte nach 24 Stunden, aufgenommen am Rand (A) und in der Mitte (B) einer Mulde. 22. Das Medium wird aus der Kulturflasche abpipettiert und die Zellen werden drei Mal mit steriler PBS (ohne Ca2+Mg2+) gespült. Es wird eine Enzymlösung (in PBS) zugegeben, um die Zellen von der Kulturflasche abzulösen. Nachdem ein angemessener Zeitraum zur Ablösung der Zellen verstrichen ist, sollte die Enzymwirkung durch Zugabe eines angereicherten Mediums im dreifachen Verhältnis zu dem für die Enzymbehandlung verwendeten Volumen gestoppt werden. Die Zellen werden in ein Zentrifugenröhrchen überführt und bei Zimmertemperatur zentrifugiert; der Überstand wird entfernt und das Zellpellet in angereichertem Medium resuspendiert. Anschließend wird die Zelldichte bestimmt, z.B. mittels einer Zählkammer oder eines Zellzählers. Die Zelllösung sollte entsprechend der gewünschten Dichte für das Ausplattieren verdünnt und gründlich gemischt werden, um eine homogene Zelldichte sicherzustellen. Die Zellen sollten mit 1 ml Zelllösung/Mulde plattiert und die Platten und Mulden beschriftet werden. Die ausgesäten Platten werden 24 Stunden lang bei 37 °C und 5 % CO2 in Luft inkubiert, damit die Zellen an den Mulden anwachsen können. Anforderungen an die Qualitätskontrolle 23. Es ist wichtig, bei der Dosierung exakte Volumina der Lösungen und Proben in die Mulden zu geben, weil diese Volumina die Konzentrationen bestimmen, die in den Berechnungen der Assay-Ergebnisse verwendet werden. 24. Vor dem Ansetzen einer Zellkultur und der späteren Durchführung von Tests, hat jedes Labor die Empfindlichkeit seines Hormonmesssystems nachzuweisen (Nummern 29-31). 25. Werden antikörperbasierte Hormonmessungsassays verwendet, sind die Prüfsubstanzen vor Beginn der Tests, wie unter Nummer 32 beschrieben, darauf zu prüfen, ob sie das für die quantitative Bestimmung von T und E2 verwendete Bestimmungssystem unter Umständen beeinträchtigen können. 26. Für den Assay wird DMSO als Lösungsmittel empfohlen. Falls ein alternatives Lösungsmittel verwendet wird, ist Folgendes zu bestimmen:
Es wird empfohlen, dass die maximal zulässige Lösungsmittelkonzentration eine 10-fache Verdünnung der am wenigsten zytotoxischen Konzentration des Lösungsmittels nicht übersteigen sollte. 27. Bevor die Tests zum ersten Mal durchgeführt werden, hat das Labor einen Eignungsversuch durchzuführen und nachzuweisen, dass es die entsprechende Zellkultur und die Versuchsbedingungen, die für die chemischen Prüfungen laut den Beschreibungen in den Absätzen 33-35 erforderlich sind, erzielen und aufrechterhalten kann. 28. Wenn Testreihen begonnen werden, bei denen eine neue Charge verwendet wird, ist vor der Verwendung einer neuen Zellcharge eine Testreihe mit einer Kontrollplatte durchzuführen, um die Leistungsfähigkeit der Zellen, wie unter den Nummern 36 und 37 beschrieben, zu bewerten. Leistung des Hormonmesssystems Methodenempfindlichkeit, -genauigkeit, -präzision und Kreuzreaktivität mit der Probenmatrix 29. Jedes Labor kann für die Analyse der Produktion von T und E2 durch H295R-Zellen ein Hormonmesssystem seiner Wahl verwenden, so lange dieses die Leistungskriterien, einschließlich der Quantifizierungsgrenze (Limit of Quantification, LOQ) erfüllt. Nominal liegt die Quantifizierungsgrenze bei 100 pg/ml für T bzw. bei 10 pg/ml für E2. Diese Grenzen basieren auf den in den Validierungsstudien beobachteten Basalhormonspiegeln. In Abhängigkeit zu den im durchführenden Labor erzielten Basalhormonspiegeln können jedoch auch höhere oder niedrigere Werte angemessen sein. Vor dem Ansetzen einer Qualitätskontrollplatte und der Einleitung von Testreihen hat das Labor nachzuweisen, dass der Hormonassay, der verwendet werden soll, Hormonkonzentrationen in angereichertem Medium so genau und präzise bestimmen kann, dass die in den Tabellen 1 und 5 angegebenen Qualitätskontrollkriterien erfüllt werden. Dieser Nachweis erfolgt durch die Analyse eines mit einer internen Hormonkontrolle versetzten angereicherten Mediums. Das angereicherte Medium ist mit mindestens drei Konzentrationen eines jeden Hormons zu versetzen (z.B. 100, 500 und 2.500 pg/ml von T; 10, 50 und 250 pg/ml von E2; oder für die niedrigsten Spikekonzentrationen für T und E2 können die auf den Nachweisgrenzen des gewählten Hormonmesssystems gestützten niedrigstmöglichen Konzentrationen verwendet werden) und zu analysieren. Die gemessenen Hormonkonzentrationen der nicht extrahierten Proben sollten innerhalb von 30 % der Nominalkonzentrationen liegen und die Variation zwischen wiederholten Messungen derselben Probe sollte 25 % nicht übersteigen (weitere Kriterien für die Qualitätskontrolle finden sich auch in Tabelle 8). Werden diese Qualitätskontrollkriterien erfüllt, wird davon ausgegangen, dass der ausgewählte Hormonassay ausreichend genau und präzise ist und nicht zu Kreuzreaktionen mit Mediumsbestandteilen (Probenmatrix) führt, zumindest nicht in einem derartigen Ausmaß, dass von einer deutlichen Beeinflussung des Assayergebnisses auszugehen ist. In einem solchen Fall ist keine Extraktion von Proben vor der Messung der Hormone erforderlich. 30. Für den Fall, dass die in den Tabellen 1 und 8 angeführten Qualitätskontrollkriterien nicht erfüllt werden, kann es zu einer erheblichen Matrixwirkung kommen und es ist ein Versuch mit extrahiertem und mit Spike (T oder E2) versetztem Medium durchzuführen. Ein Beispiel für ein Extraktionsverfahren ist in Anhang 1 des Validierungsberichts angeführt (4). Die Messungen der Hormonkonzentrationen in den extrahierten Proben sind dreifach durchzuführen 4. Wenn gezeigt werden kann, dass die Bestandteile des Mediums nach der Extraktion die Hormonbestimmungsmethode nach den Festlegungen durch die Qualitätskontrollkriterien nicht beeinträchtigen, sind alle weiteren Versuche mithilfe extrahierter Proben durchzuführen. Falls die Qualitätskontrollkriterien nach der Extraktion nicht erfüllt werden können, ist das verwendete Hormonmesssystem für den Zweck des H295R Steroidgenese- Assay nicht geeignet und es ist eine alternative Hormonbestimmungsmethode zu verwenden. Standardkurve 31. Die Hormonkonzentrationen der Lösungsmittelkontrollen (LK) sollten innerhalb des linearen Teils der Standardkurve liegen. Die Werte der Lösungsmittelkontrollen sollten vorzugsweise nahe an den Mittelpunkt des linearen Anteils fallen, um sicherzustellen, dass die Stimulation und die Hemmung der Hormonsynthese gemessen werden kann. Die zu messenden Verdünnungen des Mediums (oder Extrakte) sind entsprechend auszuwählen. Die lineare Beziehung ist durch einen geeigneten statistischen Ansatz zu bestimmen. Test auf chemische Interferenz 32. Sollen zur Messung der Hormone antikörperbasierte Assays, wie zum Beispiel Enzyme-Linked Immunosorbent-Assays (ELISA) oder Radio-Immuno-Assays (RIA) verwendet werden, ist jede Chemikalie vor Beginn der tatsächlichen Prüfung der Chemikalien darauf zu prüfen, ob sie das einzusetzende Hormonmesssystem möglicherweise beeinträchtigt (Anhang III des Validierungsberichts (4)), da diese Tests durch einige Chemikalien beeinträchtigt werden können (17). Wenn aus der Bestimmung der Hormonanalyse hervorgeht, dass eine Beeinflussung ≥ 20 % der Basalhormonproduktion für T und/oder E2 erfolgt, ist der Test auf chemische Beeinflussung des Hormonassay (wie im Anhang III des Validierungsberichts (4) Abschnitt 5.0 beschrieben) für alle Stammlösungsverdünnungen der Prüfsubstanz durchzuführen, um die Schwellendosis zu ermitteln, bei der eine signifikante Beeinflussung (≥ 20 %) erfolgt. Wenn die Beeinflussung geringer ist als 30 % können die Ergebnisse um die Beeinflussung korrigiert werden. Wenn die Beeinflussung größer ist als 30 %, sind die Daten ungültig und die Daten bei diesen Konzentrationen sind zu verwerfen. Wenn es bei mehr als einer nicht zytotoxischen Konzentration zu einer signifikanten Beeinflussung des Hormonmesssystems durch die Prüfsubstanz kommt, muss ein anderes Hormonmesssystem verwendet werden. Um Beeinflussungen von kontaminierenden Chemikalien zu vermeiden, wird empfohlen, dass Hormone aus dem Medium mithilfe eines geeigneten Lösungsmittels extrahiert werden. Geeignete Methoden finden sich im Validierungsbericht (4). Tabelle 1: Leistungskriterien für Hormonmesssysteme
Eignungsprüfung des Prüflabors 33. Bevor unbekannte Chemikalien geprüft werden, hat ein Labor nachzuweisen, dass es die Kompetenz besitzt, die entsprechenden Zellkultur- und die Versuchsbedingungen, die für die erfolgreiche Durchführung des Assays erforderlich sind, herzustellen und aufrechtzuerhalten. Dieser Nachweis erfolgt im Rahmen einer Eignungsprüfung. Da die Leistung eines Assays unmittelbar mit den durchführenden Labortechnikern zusammenhängt, sollten diese Verfahren teilweise wiederholt werden, wenn es zu einem Wechsel des Laborpersonals kommt. 34. Diese Eignungsprüfung wird unter denselben Bedingungen durchgeführt, wie sie unter den Nummern 38 bis 40 beschrieben sind, d. h. Zellen werden sieben zunehmenden Konzentrationen starker, mittlerstarker und schwacher Induktoren und Inhibitoren sowie einer negativen Chemikalie ausgesetzt werden (siehe Tabelle 2). Zu den zu prüfenden Chemikalien gehören im Einzelnen der starke Induktor Forskolin (CAS-Nr. 66575-29-9), der starke Inhibitor Prochloraz (CAS-Nr. 67747-09-5), der mittelstarke Induktor Atrazin (CAS-Nr. 1912-24-9), der mittelstarke Inhibitor Aminoglutethimid (CAS-Nr. 125-84-8), der schwache Induktor (E2-Produktion) und der schwache Inhibitor (T-Produktion) Bisphenol A (CAS-Nr. 80-05-7) sowie die negative Chemikalie Human-Choriongonadotropin (hCG) (CAS-Nr. 9002-61-3), wie in Tabelle 2 dargestellt. Testreihen mit separaten Platten werden für alle Chemikalien durchgeführt; dabei ist das in Tabelle 6 angegebene Format zu verwenden. Eine Qualitätskontrollplatte (Tabelle 4, Nummern 36-37) ist bei den täglichen Testreihen für die Eignungsprüfungschemikalien einzubeziehen. Tabelle 2: Eignungsprüfungschemikalien und Expositionskonzentrationen
Die Exposition von H295R-Zellen gegenüber Eignungsprüfungschemikalien sollte bei der Eignungsprüfung des Labors in 24-Mulden-Platten durchgeführt werden. Die Dosierung erfolgt für alle Prüfsubstanzdosen in µM. Die Dosen sind in DMSO bei 0,1 % v/v pro Mulde zu verabreichen. Alle Prüfkonzentrationen sind in Triplikaten zu testen (Tabelle 6). Für jede Chemikalie werden separate Platten verwendet. Eine Qualitätskontrollplatte wird in alle täglichen Testreihen einbezogen. 35. Zellviabilitäts- und Hormonanalysen sind nach den Vorgaben der Nummern 42 bis 46 durchzuführen. Der Schwellenwert [niedrigste Konzentration mit messbarer Wirkung (Lowest-Observed-Effect-Concentration, LOEC)] und das Einstufungsergebnis sind zu dokumentieren und mit den Werten in Tabelle 3 zu vergleichen. Die Daten gelten als akzeptabel, wenn sie die Bedingungen für den LOEC-Wert und das Einstufungsergebnis in Tabelle 3 erfüllen. Tabelle 3: Schwellenwerte (LOEC-Werte) und Einstufungergebnisse für Eignungsprüfungschemikalien
Qualitätskontrollplatte 36. Die Qualitätskontrollplatte (QK-Platte) wird zur Überprüfung der Leistung der H295R-Zellen unter Standardkulturbedingungen und zur Errichtung einer historischen Datenbasis für frühere Daten über Hormonkonzentrationen in Lösungsmittelkontrollen und Positiv- und Negativkontrollen sowie zur Festlegung anderer im Laufe der Zeit durchzuführenden Qualitätskontrollmaßnahmen verwendet.
37. Der Qualitätskontrolltest wird in einer 24-Muldenplatte durchgeführt und folgt für die Inkubation, die Dosierung, die Zellviabilität/Zytotoxizität, die Hormonextraktion und die Hormonanalyse denselben Verfahren, die sie bereits unter den Nummern 38 bis 46 für Chemikalientestungen beschrieben wurden. Die Qualitätskontrollplatte enthält Blindproben, Lösungsmittelkontrollen und zwei Konzentrationen eines bekannten Induktors (Forskolin, 1, 10 µM) und Inhibitors (Prochloraz, 0,1, 1 µM) für die E2- und T-Synthese. Darüber hinaus wird in ausgewählten Mulden als Positivkontrolle für den Viabilitäts-/Zytotoxizitäts-Assay MeOH verwendet. Eine ausführliche Beschreibung der Plattenanordnung findet sich in Tabelle 4. Die Kriterien, die bei der QK-Platte erfüllt sein müssen, sind in Tabelle 5 aufgeführt. Die Mindestwerte für die Basalhormonproduktion für T und E2 sind sowohl bei den Mulden mit der Lösungsmittelkontrolle als auch bei den Blindproben einzuhalten. Anordnung der Qualitätskontrollplatte für die Prüfung der Leistung nicht exponierter H295R-Zellen und Zellen, die gegenüber einem bekannten Inhibitor (PRO = Prochloraz) und Induktor (FOR = Forskolin) der E2- und T-Produktion exponiert wurden. Nach abgeschlossenem Expositionsversuch und der Entfernung des Mediums wird eine 70 %ige Methanollösung in alle MeOH-Mulden gegeben, die als Positivkontrolle für die Zytotoxizität dienen (siehe Zytotoxizitäts-Assay in Anhang III des Validierungsberichts (4)).
Leistungskriterien für die Qualitätskontrollplatte
Verfahren der Chemikalienexposition 38. Die vorinkubierten Zellen werden aus dem Inkubator entnommen (Nummer 21) und vor der Zudosierung unter einem Mikroskop kontrolliert, um sicherzustellen, dass sie sich in gutem Zustand befinden (Anhaftung, Morphologie). 39. Die Zellen werden in eine Biosicherheitswerkbank platziert und das angereicherte Medium wird entfernt und durch ein neues angereichertes Medium ersetzt (1 ml/Mulde). Für diese Prüfmethode wird DMSO als Lösungsmittel empfohlen. Sollten jedoch Gründe für die Verwendung anderer Lösungsmittel vorliegen, ist dies wissenschaftlich zu begründen. Die Zellen werden durch Zugabe von 1 µl der geeigneten Stammlösung in DMSO (siehe Anhang II des Validierungsberichts (4)) pro 1 ml angereichertes Medium (Muldenvolumen) gegenüber der Prüfsubstanz exponiert. Dadurch ergibt sich eine Endkonzentration von 0,1 % DMSO in den Mulden. Um eine angemessene Vermischung sicherzustellen, wird im Allgemeinen empfohlen, die geeignete Stammlösung der Prüfsubstanz in DMSO mit angereichertem Medium zu mischen, um so für jede Dosis die gewünschte Endkonzentration zu erhalten, und die Mischung unverzüglich nach Entfernen des alten Mediums in die Mulden zu geben. Bei einer solchen Vorgehensweise sollte die Konzentration von DMSO (0,1 %) in allen Mulden gleich bleiben. Die Mulden mit den beiden höchsten Konzentrationen werden mithilfe eines Stereomikroskops visuell auf Bildung von Niederschlag oder Trübung (Hinweis auf die unvollständige Löslichkeit der Prüfsubstanz) untersucht. Werden solche Bedingungen (Trübung, Niederschlagsbildung) beobachtet, sind die Mulden mit den nächstniedrigeren Konzentrationen ebenfalls zu untersuchen (und so weiter), und Konzentrationen, die sich nicht vollständig gelöst haben, sind aus der weiteren Bewertung und Analyse auszuschließen. Die Platte wird bei 37 °C und 5 % CO2 in der Luftatmosphäre 48 Stunden lang in den Inkubator zurückgestellt. Die Anordnung der Prüfsubstanzplatte ist in Tabelle 6 dargelegt. Die Stämme 1-7 zeigen Bestückung mit zunehmenden Dosen Prüfsubstanz. Dosierungsschema für die Exposition von H295R-Zellen gegenüber Prüfsubstanzen in einer 24-Muldenplatte
40. Nach 48 Stunden werden die Expositionsplatten aus dem Inkubator genommen und jede Mulde wird unter dem Mikroskop auf den Zustand der Zellen (Anhaftung, Morphologie, Grad der Konfluenz) und Anzeichen von Zytotoxizität untersucht. Das Medium aus jeder Mulde wird in zwei gleiche Mengen (von jeweils ungefähr 490 µl) unterteilt und in zwei separate, entsprechend gekennzeichnete Fläschchen gegeben (d. h. ein Aliquot als Ersatzprobe für jede Mulde). Um zu verhindern, dass die Zellen austrocknen, wird jeweils immer nur aus einer Reihe oder einer Kolonne Medium entfernt und durch das Medium für die Zellviabilitäts-/Zytotoxizitätsprüfung ersetzt. Wenn die Zellviabilität/Zytotoxizität nicht unverzüglich bestimmt wird, werden in jede Mulde 200 µl PBS mit Ca2+ und Mg2+ zugegeben. Die Medien werden bis zur weiteren Analyse der Hormonkonzentrationen (siehe Nummern 44-46) bei - 80 °C eingefroren. Obwohl in Medium bei - 80 °C aufbewahrtes T und E2 in der Regel zwar mindestens drei Monate lang stabil bleiben, sollte die Hormonstabilität während der Lagerung in jedem Labor dennoch dokumentiert werden. 41. Unmittelbar nachdem das Medium entfernt wurde, wird für jede Expositionsplatte die Zellviabilität/Zytotoxizität bestimmt. Bestimmung der Zellviabilität 42. Zur Bestimmung der potenziellen Auswirkung der Prüfsubstanz auf die Zellviabilität/Zytotoxizität kann ein beliebiger Zellviabilitäts-/Zytotoxizitäts-Assay durchgeführt werden. Der Assay sollte eine reale Messung des Anteils der in einer Mulde präsenten lebensfähigen Zellen gewährleisten oder es sollte nachgewiesen werden, dass er direkt mit dem (einer linearen Funktion des) Live/Dead®-Assay vergleichbar ist (siehe Anhang III des Validierungsberichts (4)). Ein alternativer Assay, der sich ebenfalls bewährt hat, ist der MTT-Test [3-(4,5-Dimethylthiazol-2-yl)- 2,5-Diphenyltetrazoliumbromid] (18). Die Bewertung der Zellviabilität nach den vorgenannten Methoden entspricht einer relativen Messung, die nicht unbedingt lineare Beziehungen zur absoluten Zahl der Zellen in einer Mulde aufweist. Daher sollte der Prüfer parallel dazu jede Mulde einer subjektiven visuellen Beurteilung unterziehen und Digitalfotos der Lösungsmittelkontrollen und der beiden höchsten nicht zytotoxischen Konzentrationen aufnehmen und archivieren, um so eine spätere Beurteilung der genauen Zelldichte zu ermöglichen, sollte eine solche erforderlich werden. Sollten Sichtkontrolle oder Viabilitäts-/Zytotoxizitäts-Assay auf eine Erhöhung der Zellenanzahl hindeuten, so muss diese überprüft werden. Wird die Erhöhung der Zellenanzahl bestätigt, ist dies im Prüfbericht anzugeben. Die Zellviabilität wird bezogen auf die durchschnittliche Reaktion in den Lösungsmittelkontrollen ausgedrückt (bei der davon ausgegangen wird, dass sie zu 100 % lebensfähigen Zellen entspricht) und in einer für den jeweils verwendeten Zellviabilitäts-/Zytotoxizitäts-Assay geeigneten Weise berechnet. Für den MTT-Assay kann die folgende Formel verwendet werden: % lebensfähige Zellen = (Reaktion in der Mulde - durchschnittliche Reaktion in mit MeOH [= 100 % tote Zellen] behandelten Mulden) _ (durchschnittliche Reaktion in Mulden mit Lösungsmittelkontrolle - durchschnittliche Reaktion in mit MeOH [= 100 % tote Zellen] behandelten Mulden) 43. Mulden mit einer niedrigeren Viabilität als 80 % im Verhältnis zu der durchschnittlichen Viabilität in den Lösungsmittelkontrollen (= 100 % Viabilität) sollten in die endgültige Datenanalyse nicht einbezogen werden. Kommt es bei einer Zytotoxizität von beinahe 20 % zur Hemmung der Steroidgenese, muss sich der Prüfer vergewissern, dass die Ursache für die Hemmung nicht die Zytotoxizität ist. Hormonanalyse 44. Jedes Labor kann für die Analyse von T und E2 ein Hormonmesssystem seiner Wahl verwenden. Überschüssige Aliquote von Medium aus den einzelnen Behandlungsgruppen können für die Zubereitung von Verdünnungen verwendet werden, um die Konzentration in den linearen Teil der Standardkurve zu bringen. Gemäß Nummer 29 sollte jedes Labor nachweisen, dass sein Hormonmesssystem (z.B. ELISA, RIA, LC-MS, LC-MS/MS) den Qualitätskontrollkriterien entspricht. Dieser Nachweis erfolgt durch die Analyse eines mit einer internen Hormonkontrolle versetzten angereicherten Mediums, bevor Qualitätskontrolltestreihen durchgeführt oder Chemikalien geprüft werden. Um sicherzustellen, dass die Bestandteile des Prüfsystems die Hormonmessung nicht beeinträchtigen, müssen die Hormone vor ihrer Messung möglicherweise aus dem Medium extrahiert werden (für die Bedingungen, unter denen eine Extraktion erforderlich oder nicht erforderlich ist, siehe Nummer 30). Es wird empfohlen, bei der Extraktion die in Anhang III des Validierungsberichts angegebenen Verfahrensvorschriften zu befolgen (4). 45. Wird ein im Handel erhältliches Testkit für die Messung der Hormonproduktion verwendet, sollte die Hormonanalyse nach den Vorgaben in den Bedienungsanleitungen des jeweiligen Herstellers durchgeführt werden. Die meisten Hersteller verfügen über ein spezielles Verfahren für die Durchführung von Hormonanalysen. Verdünnungen von Proben müssen so angepasst werden, dass die erwarteten Hormonkonzentrationen für die Lösungsmittelkontrollen in die Mitte des linearen Bereichs der Standardkurve des einzelnen Assays fallen (Anhang III des Validierungsberichts (4)). Werte außerhalb des linearen Bereichs der Standardkurve sind zu verwerfen. 46. Die endgültigen Hormonkonzentrationen werden folgendermaßen berechnet: Beispiel:
Wahl der Prüfkonzentrationen 47. Es sind mindestens zwei unabhängige Assay-Testreihen durchzuführen. Sofern für die Wahl der Prüfkonzentrationen nicht bereits Informationen (wie Angaben zu Löslichkeitsgrenzen oder zur Zytotoxizität) vorliegen, wird empfohlen, die Prüfkonzentrationen für die erste Testreihe in log10-Abständen festzulegen, wobei 10-3 M die Höchstkonzentration darstellt. Ist die Chemikalie löslich und bei keiner der geprüften Konzentrationen zytotoxisch und war die erste Testreihe bei allen Konzentrationen negativ, so ist dies in einer weiteren Testreihe, die unter denselben Bedingungen wie die erste Testreihe durchgeführt wird, zu bestätigen (Tabelle 7). Sind die Ergebnisse der ersten Testreihe unschlüssig (d. h., der fold change [x-fache Änderung] ist im Vergleich zur Lösungsmittelkontrolle nur für eine einzige Konzentration statistisch signifikant) oder positiv (d. h., der fold change ist für zwei oder mehr nebeneinanderliegende Konzentrationen statistisch signifikant), so sollte der Test, wie in Tabelle 7 angegeben, mit verfeinerten Prüfkonzentrationen wiederholt werden. Die Prüfkonzentrationen in den Testreihen zwei und drei (falls zutreffend) sind auf der Grundlage der Ergebnisse aus der ersten Testreihe anzupassen, indem die Bracketing- Konzentrationen, die eine Wirkung hervorgerufen haben, mit in 1/2-log-Abständen verfeinert werden (wenn z.B. die ursprüngliche Testreihe mit 0,001, 0,01, 0,1, 1, 10, 100, 1000 µM zu Induktionen bei 1 und 10 µM geführt hat, sollten für die zweite Testreihe die Konzentrationen 0,1, 0,3, 1, 3, 10, 30, 100 µM verwendet werden), sofern keine niedrigeren Konzentrationen eingesetzt werden müssen, um einen LOEC-Wert zu erhalten. Im letztgenannten Fall sollten in der zweiten Testreihe mindestens fünf Konzentrationen unter der in der Testreihe geprüften niedrigsten Konzentration mit 1/2-log-Abständen verwendet werden. Wird die erste Testreihe durch die zweite Testreihe nicht bestätigt, (d. h., die zuvor positiv getestete Konzentration ± 1 Konzentrationssteigerung ergibt keine statistische Signifikanz), muss ein dritter Versuch unter den anfänglichen Testbedingungen durchgeführt werden. Unschlüssige Ergebnisse in der ersten Testreihe werden als negative Ergebnisse angesehen, wenn die gemessene Wirkung in keiner der beiden folgenden Testreihen bestätigt werden konnte. Unschlüssige Ergebnisse aus der ersten Testreihe werden als positive Reaktionen (Wirkungen) gewertet, wenn die Reaktion in mindestens einer weiteren Testreihe innerhalb einer Konzentrationssteigerung von ± 1 bestätigt werden kann (das Verfahren zur Datenauswertung findet sich in Abschnitt 55). Entscheidungsmatrix für mögliche Ergebnisszenarien
Qualitätskontrolle der Testplatte 48. Neben den Qualitätskriterien für die Qualitätskontrollplatte sind weitere Qualitätskriterien einzuhalten, die in Tabelle 8 dargelegt sind und zulässige Abweichungen zwischen Replikatmulden, Wiederholungsversuche, die Linearität und Empfindlichkeit der Hormonmesssysteme, die Variabilität zwischen Wiederholungs- Hormonmessungen derselben Probe und die prozentuale Wiederfindung gespikter Hormone nach der Mediumextraktion (falls zutreffend; die Anforderungen an die Extraktion finden sich in Nummer 30) betreffen. Die Daten sollten innerhalb der Bereiche liegen, die für jeden Parameter, der für die weitere Auswertung zu berücksichtigen ist, festgelegt wurden. Werden diese Kriterien nicht erfüllt, ist auf dem Arbeitsblatt anzugeben, dass die Qualitätskontrollkriterien bei der fraglichen Probe nicht eingehalten wurden, und die Probe ist entweder erneut zu analysieren oder aus dem Datensatz zu streichen. Tabelle 8: Akzeptanzbereiche und/oder Variation (in %) für die H295R-Assay-Testplattenparameter (LOQ: Quantifizierungsgrenze des Hormonmesssystems. VK: Variationskoeffizient; LK: Lösungsmittelkontrolle; DPM: Disintegrationen pro Minute)
Datenanalyse und Berichterstattung Datenanalyse 49. Zur Bewertung der relativen Zunahme/Abnahme der chemisch veränderten Hormonproduktion, sind die Ergebnisse auf den mittleren Lösungsmittelkontrollwert jeder Testplatte zu standardisieren und als Änderungen bezogen auf die Lösungsmittelkontrolle in jeder Testplatte anzugeben. Alle Daten sind als Mittelwert ± 1 Standardabweichung anzugeben. 50. Nur Hormondaten für Mulden, bei denen die Zytotoxizität niedriger war als 20 %, sind in die Datenanalyse aufzunehmen. Relative Änderungen sind folgendermaßen zu berechnen: Relative Veränderung = (Hormonkonzentration in jeder Mulde) ÷ (Mittlere Hormonkonzentration in allen Mulden mit Lösungsmittelkontrolle). 51. Sollte die Sichtkontrolle der Mulde oder der unter Nummer 42 beschriebene Viabilitäts-/Zytotoxizitäts-Assay auf eine Erhöhung der Zellanzahl hindeuten, muss diese offensichtliche Erhöhung überprüft werden. Wird die Erhöhung der Zellanzahl bestätigt, ist dies im Prüfbericht anzugeben. 52. Bevor statistische Analysen durchgeführt werden, sind die Normalitäts- und Varianzhomogenitätshypothesen zu bewerten. Die Normalität ist mittels Normalwahrscheinlichkeitsplot (Standard-Probability-Plot)oder nach anderen geeigneten statistischen Methoden (z.B. Shapiro-Wilk"s Test) zu bewerten. Sind die Daten (Fold Changes) nicht normal verteilt, sollte versucht werden, die Daten umzuwandeln, um eine approximative Normalverteilung zu erhalten. Wenn die Daten normalverteilt oder approximativ normalverteilt sind, sollten die Unterschiede zwischen den verschiedenen Gruppen chemischer Konzentrationen und den Lösungsmittelkontrollen anhand eines parametrischen Tests (z.B. Dunnett"s Test) analysiert werden, wobei die Konzentration die unabhängige und die Reaktion (Fold Change) die abhängige Variable darstellt. Sind die Daten nicht normalverteilt, ist ein geeigneter nichtparametrischer Test zu verwenden (z.B. Kruskal-Wallis-Test, Steel"s Many-One Rank Test). Unterschiede gelten als signifikant bei p ≤ 0,05. Die statistischen Bewertungen erfolgen auf Grundlage der durchschnittlichen Werte der Mulden, die unabhängige Wiederholungsdatenpunkte darstellen. Es wird davon ausgegangen, dass es aufgrund der großen Dosisstufenabstände in der ersten Testreihe (log10-Staffelung) nicht möglich sein wird, eindeutige Konzentration- Wirkungs-Beziehungen zu beschreiben, bei denen die beiden höchsten Dosen im linearen Bereich der Sigmoidkurve liegen. Daher finden bei der ersten Testreihe oder etwaigen anderen Datensätzen, bei denen dieser Fall eintritt (z.B. wenn keine maximale Wirksamkeit geschätzt werden kann), statistische Daten mit fester Variable des Typs I Anwendung. 53. Wenn mehr als zwei Datenpunkte im linearen Bereich der Kurve liegen und soweit die maximalen Wirksamkeiten berechnet werden können (wie dies für bestimmte zweite Testreihen mit 1/2-log-Abstand zwischen den Expositionskonzentrationen erwartet wird), ist ein Probit-, Logit- oder ein anderes geeignetes Regressionsmodell für die Berechnung der wirksamen Konzentrationen zu verwenden (z.B. EC50 und EC20). 54. Die Ergebnisse sind sowohl in grafischer Form (Balkendiagramme, die Mittelwert ± 1 Standardabweichung darstellen) als auch in tabellarischer Form (LOEC-/NOEC-Wert, Wirkungsrichtung und Stärke der maximalen Reaktion, die Teil des Dosis-Wirkung-Teils der Daten ist) vorzulegen (siehe Beispiel in Abbildung 3). Die Datenauswertung wird nur dann als gültig angesehen, wenn sie auf mindestens zwei unabhängig durchgeführten Testreihen basiert. Ein Versuch oder eine Testreihe gilt als unabhängig, wenn er/sie an einem anderen Tag mit neuen Lösungen und Kontrollen durchgeführt wurde. Der für die Testreihen 2 und 3 (falls erforderlich) verwendete Konzentrationsbereich kann auf der Grundlage der Ergebnisse aus der ersten Testreihe angepasst werden, um den Dosis-Wirkungs- Bereich mit der niedrigsten Konzentration mit messbarer Wirkung (LOEC) (siehe Nummer 47) besser bestimmen zu können. Beispiel für die Präsentation und Bewertung der bei der Durchführung des H295R-Assays generierten Daten in grafischer und tabellarischer Form (Sternchen zeigen statistisch signifikante Unterschiede zur Lösungsmittelkontrolle (p< 0,05) an. LOEC: niedrigste Konzentration mit gemessener Wirkung; Max. Änderung: Maximale Stärke der bei den einzelnen Konzentration gemessenen Reaktion, bezogen auf die durchschnittlichen Reaktion der Lösunsgsmittelkontrolle (= 1))
Verfahren für die Datenauswertung 55. Eine Prüfsubstanz gilt als positiv, wenn die xfache Induktion bei zwei nebeneinander liegenden Konzentrationen in mindestens zwei unabhängigen Testreihen (Tabelle 7) statistisch gesehen von der Lösungsmittelkontrolle abweicht (p ≤ 0,05). Eine Prüfsubstanz gilt nach zwei unabhängigen negativen Testreihen oder nach drei Testreihen, von denen zwei negative Ergebnisse und einer entweder unschlüssige oder positive Ergebnisse aufweisen, als negativ. Wenn die in drei unabhängigen Versuchen generierten Daten die in Tabelle 7 angeführten Entscheidungskriterien nicht erfüllen, können die Versuchsergebnisse nicht ausgewertet werden. Ergebnisse bei Konzentrationen, die über den Löslichkeitsgrenzen liegen, oder bei zytotoxischen Konzentrationen sollten nicht in die Datenauswertung einfließen. Prüfbericht 56. Der Prüfbericht sollte folgende Angaben enthalten: Prüfanstalt
Prüfsubstanz, Reagenzien und Kontrollen,
Zellen
Vorbedingungen für den Test (falls zutreffend),
Prüfbedingungen
Prüfergebnisse
Datenauswertung
Diskussion
Schlussfolgerungen (1) OECD (2002). Rahmenkonzept der OECD für die Testung und Bewertung endokrin wirksamer Substanzen (endokrine Disruptoren) (,OECD Conceptual Framework for the Testing and Assessment of Endocrine Disrupting Chemicals"), in Kapitel B.54 Anlage 2 dieses Anhangs. (2) Hecker, M., Newsted, J.L., Murphy, M.B., Higley, E.B., Jones, P.D., Wu, R. and Giesy, J.P. (2006). Human adrenocarcinoma (H295R) cells for rapid in vitro determination of effects on steroidogenesis: Hormone production. Toxicol. Appl. Pharmacol., 217, 114-124. (3) Hecker, M., Hollert, H., Cooper, R., Vinggaard, A.-M., Akahori, Y., Murphy, M., Nellemann, C., Higley, E., Newsted, J., Wu, R., Lam, P., Laskey, J., Buckalew, A., Grund, S., Nakai, M., Timm, G., and Giesy, J. P. (2007). The OECD validation program of the H295R steroidgenesis assay for the identification of in vitro inhibitors or inducers of testosterone and estradiol production, Phase 2: inter laboratory prevalidation studies. Env. Sci. Pollut. Res., 14, 23-30. (4) OECD (2010). Multi-Laboratory Validation of the H295R Steroidogenesis Assay to Identify Modulators of Testosterone and Estradiol Production. OECD Series of Testing and Assessment No. 132, ENV/JM/MONO(2010)31, Paris. Abrufbar unter [http://www.oecd.org/document/30/0,3746,en_2649_34377_1916638_1_1_1_1,00.html]. (5) OECD (2010). Peer Review Report of the H295R Cell-Based Assay for Steroidogenesis. OECD Series of Testing and Assessment No. 133, ENV/JM/MONO(2010)32, Paris. Abrufbar unter [http://www.oecd.org/document/30/0,3746, en_2649_34377_1916638_1_1_1_1,00.html]. (6) Battelle (2005). Detailed Review Paper on Steroidogenesis. Abrufbar unter [http://www.epa.gov/endo/pubs/edmvs/ steroidogenesis_drp_final_3_29_05.pdf]. (7) Hilscherova, K., Jones, P. D., Gracia, T., Newsted, J. L., Zhang, X., Sanderson, J. T., Yu, R. M. K., Wu, R. S. and Giesy, J. P. (2004). Assessment of the Effects of Chemicals on the Expression of Ten Steroidogenic Genes in the H295R Cell Line Using Real-Time PCR, Toxicol. Sci., 81, 78-89. (8) Sanderson, J. T., Boerma, J., Lansbergen, G. and Van den Berg, M. (2002). Induction and inhibition of aromatase (CYP19) activity by various classes of pesticides in H295R human adrenocortical carcinoma cells, Toxicol. Appl. Pharmacol., 182, 44-54. (9) Breen, M.S., Breen, M., Terasaki, N., Yamazaki, M. and Conolly, R.B. (2010). Computational model of steroidogenesis in human H295R cells to predict biochemical response to endocrineactive chemicals: Model development for metyrapone. Environ. Health Perspect., 118: 265-272. (10) Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010). Assessment of chemical effects on aromatase activity using the H295R cell line, Environ. Sci. Poll. Res., 17:1137-1148. (11) Gazdar, A. F., Oie, H. K., Shackleton, C. H., Chen, T. R., Triche, T. J., Myers, C. E., Chrousos, G. P., Brennan, M. F., Stein, C. A. and La Rocca, R. V. (1990). Establishment and characterization of a human adrenocortical carcinoma cell line that expresses Multiple pathways of steroid biosynthesis. Cancer Res., 50, 5488-5496. (12) He, Y.H., Wiseman, S.B., Zhang, X.W., Hecker, M., Jones, P.D., El-Din, M.G., Martin, J.W. and Giesy, J.P. (2010). Ozonation attenuates the steroidogenic disruptive effects of sediment free oil sands process water in the H295R cell line. Chemosphere, 80:578-584. (13) Zhang, X.W., Yu, R.M.K., Jones, P.D., Lam, G.K.W., Newsted, J.L., Gracia, T., Hecker, M., Hilscherova, K., Sanderson, J.T., Wu, R.S.S. and Giesy, J.P. (2005). Quantitative RT-PCR methods for evaluating toxicantinduced effects on steroidogenesis using the H295R cell line. Environ. Sci. Technol., 39:2777-2785. (14) Higley, E.B., Newsted, J.L., Zhang, X., Giesy, J.P. and Hecker, M. (2010). Differential assessment of chemical effects on aromatase activity, and E2 and T production using the H295R cell line. Environ. Sci. Pol. Res., 17:1137-1148. (15) Rainey, W. E., Bird, I. M., Sawetawan, C., Hanley, N. A., Mccarthy, J. L., Mcgee, E. A., Wester, R. and Mason, J. I. (1993). Regulation of human adrenal carcinoma cell (NCI-H295) production of C19 steroids. J. Clin. Endocrinol. Metab., 77, 731-737. (16) Kapitel B.55 dieses Anhangs: Hershberger Bioassay mit Ratten: Ein Kurzzeit Screening-Test auf (anti-)androgene Eigenschaften. (17) Shapiro, R., and Page, L.B. (1976). Interference by 2,3-dimercapto-1-propanol (BAL) in angiotensin I radioimmunoassay, J. Lab. Clin. Med., 2, 222-231. (18) Mosmann, T. (1983). Rapid colorimetric assay for growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods., 65, 55-63. (19) Brock, B.J., Waterman, M.R. (1999). Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species. Biochemistry. 38:1598-1606. (20) Oskarsson, A., Ulleras, E., Plant, K., Hinson, J. Goldfarb, P.S., (2006). Steroidogenic gene expression in H295R cells and the human adrenal gland: adrenotoxic effects of lindane in vitro. J. Appl. Toxicol., 26:484-492. ___________ 1) ATCC CRL-2128; ATCC, Manassas, VA, USA, [http://www.1gcstandardsatcc.org/] 2), Neue Charge" bezieht sich auf eine frische Charge Zellen, die von ATCC bezogen wurde. 3), Eingefrorene Charge" bezieht sich auf Zellen, die zuvor gezüchtet und anschließend in einem anderen Labor als ATCC eingefroren wurden. 4) Anmerkung:Wenn die Extraktion erforderlich ist, werden für jeden Extrakt drei wiederholte Messungen durchgeführt. Jede Probe wird nur einmal extrahiert.
Angereichertes Medium: Stammmedium plus BD Nu-Serum und ITS+ Premium Mix (siehe Anhang II des Validierungsberichts) (4). Chemikalie: ein Stoff oder eine Mischung. CYP: Cytochrom-P450-Monooxygenase, eine Familie von Genen und den daraus hervorgegangenen Enzymen, die bei der Katalyse vielfältiger biochemischer Reaktionen, einschließlich Synthese und Verstoffwechselung von Steroidhormonen, beteiligt sind. DPM: Disintegration (Zerfall) pro Minute, Anzahl Atome in einer bestimmten Menge radioaktiven Materials, deren Zerfall minütlich nachgewiesen wird. E2: 17β-Estradiol; wichtigstes Östrogen in Säugetiersystemen. Einfriermedium: Mediumzum Einfrieren von Zellen und zur Lagerung gefrorener Zellen, bestehend aus Kulturmedium plus BD NuSerum und Dimethylsulfoxid. H295R-Zellen: humane Adenokarzinom-Zellen, die die physiologischen Merkmale zonal undifferenzierter Nebennierenzellen menschlicher Föten aufweisen und die alle Enzyme des steroidogenen Pfades exprimieren. Sie sind bei der ATCC erhältlich. Konfluenz: Bedeckung der Oberfläche eines Kulturmediums mit Zellen oder Proliferation von Zellen im Kulturmedium. Linearer Bereich: Bereich innerhalb der Standardkurve eines Hormonmesssystems mit Ergebnissen proportional zur Konzentration des in der Probe präsenten Analyts. LOEC (Lowest Observed Effect Concentration): die niedrigste Konzentrationstufe, bei der die Assay-Reaktion statistisch gesehen von der Reaktion der Lösungsmittelkontrolle abweicht. LOQ (Limit of Quantification): Quantifizierungsgrenze; kleinste Menge einer Chemikalie, die innerhalb eines bestimmten Konfidenzintervalls im Gegensatz zur Leermessung (Blindwert) gerade noch nachgewiesen werden kann. Für die Zwecke der vorliegenden Methode und falls nicht anders angegeben, wird der LOQ-Wert in der Regel vom Hersteller der Testsysteme vorgegeben. NOEC (No Observed Effect Concentration): die höchste getestete Konzentration, bei der im Test keine signifikante Wirkung festgestellt wird. Passage: die Anzahl Zellteilungen nach dem Ansetzen einer Kultur aus gefrorenen Zellen, wobei der Ausgangspassage der gefrorenen Zellen die Nummer eins (1) zugeordnet wird. Zellen, die einmal gesplittet wurden, werden als Passage 2 bezeichnet, usw. PBS (Phosphate Buffered Saline): Dulbeccos phosphatgepufferte Salzlösung. Prüfsubstanz: ein(e) nach dieser Prüfmethode geprüfter Stoff/geprüfte Mischung. Qualitätskontrolle (QK): die Maßnahmen, die nötig sind, um aussagekräftige Daten zu gewährleisten. Qualitätskontrollplatte: eine 24-Muldenplatte mit zwei Konzentrationen der positiven und negativen Kontrollen zur Überwachung der Leistung einer neuen Charge Zellen oder zur Bereitstellung der positiven Kontrollen für den Assay, wenn Chemikalien geprüft werden. Stammmedium: die Basis für die Zubereitung anderer Reagenzien, bestehend aus einer 1:1-Mischung von Dulbecco"s Modified Eagle"s Medium (DMEM) und Ham"s F-12 Nutrient Mixture (DMEM/F12) in 15 mM-HEPES-Puffer ohne Phenolrot oder Natriumhydrogenkarbonat. Natriumhydrogenkarbonat wird als Puffer zugegeben (siehe Anhang II des Validierungsberichts) (4). Steroidgenese: Synthesepfad vom Cholesterol zu verschiedenen Steroidhormonen. Bestimmte Zwischenprodukte auf dem steroiden Synthesepfad, wie Progesteron und Testosteron, sind eigenständige wichtige Hormone, dienen aber auch als Vorläufer für nachgeschaltete Hormone. T: Testosteron, eines der beiden wichtigsten Androgene bei Säugern. Testplatte: Platte, auf der H295R-Zellen der Prüfsubstanz ausgesetzt werden. Testplatten enthalten die Lösungsmittelkontrolle und die Prüfsubstanz in sieben Konzentrationsstufen in Dreifachmulden. Testreihe: ein unabhängiger Versuch, gekennzeichnet durch einen neuen Satz Lösungen und Kontrollen. Trypsin 1X: eine verdünnte Lösung des Enzyms Trypsin, einer pankreatischen Serin-Protease, die zur Ablösung von Zellen von einer Zellkulturplatte verwendet wird (siehe Anhang III des Validierungsberichts) (4). VK: Variationskoeffizient, definiert als Verhältnis der Standardabweichung einer Verteilung zu ihrem arithmetischen Mittelwert. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.58 Genmutations-Assays an somatischen Zellen und Keimzellen transgener Nagetiere 14 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 488 (2013). EU-Prüfmethoden stehen für eine Vielzahl von Invitro-Assays zum Nachweis von Chromosomen- und/oder Genmutationen zur Verfügung. Es gibt Prüfmethoden für Invivo-Endpunkte (d. h. Chromosomenaberrationen und nicht geplante DNA-Synthesen), die jedoch keine Genmutationen messen. Mutationsassays an transgenen Nagetieren (transgenic rodents, TGR) decken den Bedarf an praktischen und zugänglichen Invivo-Genmutationstests. 2. Die TGR-Mutationsassays sind gründlich überprüft worden (24) (33). Für diese Tests werden transgene Ratten und Mäuse verwendet, die mehrere Kopien chromosomal integrierten Plasmids oder Phagen-Shuttle-Vektoren enthalten. Die Transgene enthalten Reporter-Gene zum Nachweis verschiedener Arten von Mutationen, die in vivo durch Prüfsubstanzen induziert wurden. 3. Mutationen bei Nagetieren werden bewertet, indem das Transgen wiedergefunden und der Phänotyp des Reporter- Gens in einer bakteriellen Wirtszelle ohne Reporter-Gen analysiert wird. Mit TGR-Mutationsassays werden Mutationen gemessen, die in genetisch neutralen Genen induziert wurden, die sich in praktisch jedem Gewebe des Nagetiers wiederfinden. Mit diesen Assays können somit viele der bestehenden Grenzen, die die Invivo-Untersuchung von Genmutationen in endogenen Genen derzeit noch behindern (z.B. begrenzte Menge geeigneten Gewebes für die Analyse, Negativ-/Positivselektion im Vergleich zu den Mutationen). 4. Die vorliegenden Daten deuten darauf hin, dass Transgene in ähnlicher Weise wie endogene Gene auf Mutagene reagieren, insbesondere was den Nachweis des Austauschs einer Base gegen eine andere, Rasterschubmutationen und kleine Deletionen und Insertionen betrifft(24). 5. Die Internationalen Workshops für Genotoxizitätsprüfungen (International Workshop on Genotoxicity Testing, IWGT) haben die Berücksichtigung von TGR-Mutationsassays für den Invivo-Nachweis von Genmutationen befürwortet und ein Durchführungsprotokoll empfohlen (15) (29). Die vorliegende Prüfmethode stützt sich auf diese Empfehlungen. Eine weitergehende Analyse, die die Verwendung dieses Protokolls unterstützt, findet sich unter (16). 6. Es wird davon ausgegangen, dass es in Zukunft möglich sein wird, TGR-Mutationsassays mit einer Prüfung auf Toxizität bei wiederholten Dosisverabreichungen (Kapitel B.7 dieses Anhangs) zu kombinieren. Es sind jedoch weitere Daten erforderlich, um sicherzustellen, dass die Empfindlichkeit der TGR-Mutationsassays durch den kürzeren - eintägigen - Zeitabstand zwischen dem Ende des Verabreichungszeitraums und dem Zeitpunkt der Probenahme (wie sie bei Prüfungen auf Toxizität bei wiederholter Verabreichung üblich ist) im Vergleich zu dem bei TGR-Mutationsassays üblichen Drei-Tage-Abstand nicht beeinträchtigt wird. Es sind auch Daten erforderlich, die belegen, dass die Leistung der Assays mit wiederholter Verabreichung durch die Verwendung eines transgenen Nagerstamms anstelle der traditionellen Nagerstämme nicht beeinträchtigt wird. Sobald diese Daten vorliegen, wird die vorliegende Prüfmethode aktualisiert. 7. Die wichtigsten Begriffe sind im Anhang bestimmt. 8. Für die nachstehend angeführten TGR-Mutationsassays liegen ausreichende Daten vor, die ihre Verwendung im Rahmen der vorliegenden Prüfmethode, soweit sie unter Standardbedingungen durchgeführt werden, unterstützen: lacZ Bakteriophage Maus (MutaTMMouse); lacZ Plasmid Maus; gpt Delta (gpt und Spi-) Maus und Ratte; lacI Maus und Ratte (Big Blue®). Außerdem kann der cII-Positivselektionsassay zur Bewertung von Mutationen in den Modellen Big Blue® und MutaTMMouse verwendet werden. Die Mutagenese in den TGR-Modellen wird in der Regel als Mutantenhäufigkeit bewertet; erforderlichenfalls kann eine Molekularanalyse der Mutationen aber zusätzliche Informationen liefern (siehe Nummer 24). 9. Diese Invivo-Genmutationstests an Nagetieren sind insbesondere für die Bewertung des mutagenen Risikos von Relevanz, da die Assay-Reaktionen vom Invivo-Stoffwechsel, von der Pharmakokinetik, den DNA-Reparaturprozessen und der Transläsionssynthese abhängen, auch wenn diese bei den einzelnen Tierarten, Geweben und Arten der DNA-Schädigungen unterschiedlich sein können. Ein Invivo-Genmutationsassay ist auch für die weitere Untersuchung einer bei einer Invitro-Prüfung festgestellten mutagenen Wirkung und zur weiteren Berücksichtigung von Testergebnissen von Nutzen, bei denen andere Invivo-Endpunkte verwendet werden (24). Abgesehen davon, dass zwischen Genmutationen und der Auslösung von Krebs ein kausaler Zusammenhang besteht, ist die Genmutation ein relevanter Endpunkt für die Prognose anderer mutationsbedingter Erkrankungen somatischer Gewebe als Krebs (12) (13) und von über die Keimbahn übertragenen Krankheiten. 10. Wenn Anzeichen dafür bestehen, dass die Prüfsubstanz oder ein reaktiver Metabolit keines der Zielgewebe erreicht, ist die Durchführung eines TGR-Mutationsassays nicht sinnvoll. 11. In den unter Nummer 8 beschriebenen Assays ist das Zielgen bakterieller oder bakteriophager Herkunft, und seine Wiederfindung anhand der genomischen Nagetier-DNA erfolgt durch Einführung des Transgens in einen λ-Bakteriophagen oder Plasmid-Shuttle-Vektor. Bei dem Verfahren wird aus dem zu untersuchenden Nagetiergewebe genomische DNA extrahiert; diese genomische DNA wird in vitro verarbeitet (d. h. die λ-Vektoren werden verpackt, bzw. die Plasmiden werden ligiert und elektroporiert, um den Shuttle-Vektor wiederzufinden), und anschließend werden unter geeigneten Bedingungen Mutationen in bakteriellen Wirtszellen nachgewiesen. Bei den Assays werden neutrale Transgene eingesetzt, die sich ohne weiteres in den meisten Geweben wiederfinden lassen. 12. Bei dem grundlegenden TGR-Mutationsversuch wird das Nagetier über einen bestimmten Zeitraum mit einer Chemikalie behandelt. Die Chemikalien können in jeder geeigneten Form, auch durch Implantation, verabreicht werden (z.B. Prüfung von Medizinprodukten). Der Gesamtdauer, während der die Chemikalie einem Tier verabreicht wird, wird Verabreichungszeitraum genannt. Auf die Verabreichung folgt - vor der Tötung des Tieres - in der Regel ein Zeitraum, in dem die Chemikalie nicht verabreicht wird und in dem nicht reparierte DNA-Läsionen in stabile Mutationen fixiert werden. In der Literatur wird dieser Zeitraum unterschiedlich entweder als Manifestationszeit, Fixierungszeit oder Expressionszeit bezeichnet; das Ende dieses Zeitraums markiert den Probenahmezeitpunkt (15) (29). Nach dem Töten des Tieres wird die genomische DNA aus dem zu untersuchenden Gewebe isoliert und gereinigt. 13. Daten zu jeweils ein und demselben Gewebe/Tier aus mehreren Verpackungen/Ligationen werden normalerweise aggregiert und die Mutantenhäufigkeit wird in der Regel mithilfe von insgesamt 105 bis 107 plaquebildenden oder kolonienbildenden Einheiten bewertet. Bei Anwendung von Positivselektionsmethoden wird die Gesamtzahl der plaquebildenden Einheiten mit einem separaten Satz nichtselektiver Platten bestimmt. 14. Es wurden Positivselektionsmethoden entwickelt, um den Nachweis von Mutationen sowohl im gpt-Gen[gpt Delta Maus und Ratte, gpt- Phänotyp (20) (22) (28)] als auch im lacZ-Gen [MutaTMMaus oder lacZ-Plasmid Maus (3) (10) (11) (30)] zu erleichtern; hingegen werden Mutationen des lacI-Gens in Big Blue®-Tieren über eine nicht- selektive Methode nachgewiesen, bei der Mutanten durch die Erzeugung (blau) gefärbter Plaques gekennzeichnet werden. Eine Positivselektionsmethode liegt auch für den Nachweis von Punktmutationen vor, zu denen es im cII- Gen des λ-Bakteriophagen-Shuttle-Vektors kommt [Big Blue® Maus oder Ratte und MutaTMMaus (17)] sowie von Deletionen in den λ rot und gam [Spi- Auswahl in gpt Delta Maus und Ratte (21) (22) (28)]. Die Mutantenhäufigkeitwird berechnet, indem die Zahl der Plaques/Plasmiden mit Mutationen im Transgen durch die Gesamtzahl der aus derselben DNA-Probe gewonnenen Plaques/Plasmiden geteilt wird. In TGR-Genmutationsstudien ist die Mutantenhäufigkeit der gemessene Parameter. Darüber hinaus kann eine Mutationshäufigkeit als Anteil der Zellen mit unabhängigen Mutationen bestimmt werden; bei dieser Berechnung muss die klonale Expansiondurch Sequenzierung der wiedergefundenen Mutanten korrigiert werden (24). 15. Die in den lacI-, lacZ-, cII- und gpt- Punktmutationsassays gezählten Mutationen bestehen hauptsächlich aus Substitutionsmutationen (Basenpaare), Rasterschubmutationen und kleinen Insertionen/Deletionen. Der relative Anteil dieser Mutationstypen unter spontanen Mutationen ist vergleichbar mit dem Anteil, der im endogenen Hprt-Gen gemessen wird. Große Deletionen werden nur mit der Spi--Auswahl und den lacZ-Plasmidassays nachgewiesen (24). Zu untersuchende Mutationen sind Invivo-Mutationen, zu denen es in der Maus oder in der Ratte kommt. Invitro und Exvivo-Mutationen, zu denen es bei der Phagen-/Plasmidwiederfindung, Replikation oder Reparatur kommen kann, sind relativ selten und können in einigen Systemen durch die bakterielle Wirtszelle/das System der positiven Selektion eindeutig bestimmt oder ausgeschlossen werden. Vorbereitungen Auswahl der Versuchstierarten 16. Derzeit ist eine Vielzahl von Modellen für den Nachweis von Genmutationen bei transgenen Mäusen erhältlich und diese Systeme sind gängiger als Modelle für transgene Ratten. Wenn die Ratte eindeutig ein geeigneteres Modell ist als die Maus (beispielsweise wenn der Mechanismus der Karzinogenese bei einem nur in Ratten vorkommenden Tumor untersucht wird, um einen Bezug zu einer Rattentoxizitätsstudie herzustellen, oder wenn bekannt ist, dass der Stoffwechsel der Ratte für den menschlichen Stoffwechsel repräsentativer ist) ist die Verwendung von Modellen für den Nachweis von Genmutationen bei transgenen Ratten in Erwägung zu ziehen. Haltungs- und Fütterungsbedingungen 17. Die Temperatur im Versuchstierraum sollte idealerweise 22 °C (± 3 °C) betragen. Auch wenn die relative Luftfeuchtigkeit mindestens 30 % betragen und - außer beim Reinigen des Raums - 70 % nicht überschreiten sollte, ist eine Luftfeuchtigkeit von 50-60 % anzustreben. Die Beleuchtung sollte künstlich sein, und die täglichen Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung der Prüfsubstanz sichergestellt werden muss, wenn die Prüfsubstanz auf diese Art verabreicht werden soll. Die Tiere sollten in kleinen gleichgeschlechtlichen Gruppen (nicht mehr als fünf Tiere) untergebracht werden, wenn kein aggressives Verhalten erwartet wird. Sie können auch einzeln gehalten werden, wenn dies wissenschaftlich gerechtfertigt ist. Vorbereitung der Tiere 18. Gesunde, jungadulte geschlechtsreife Tiere (acht bis zwölf Wochen alt zu Beginn der Behandlung) werden nach dem Zufallsprinzip in die Kontroll- bzw. Behandlungsgruppen eingeteilt. Die Tiere werden individuell gekennzeichnet. Sie werden über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt. Die Käfige sollten so angeordnet werden, dass etwaige Wirkungen aufgrund des Käfigstandorts minimiert werden. Bei Versuchsbeginn sollten die Gewichtsunterschiede der Tiere möglichst gering sein und ± 20 % des geschlechtsspezifischen Durchschnittsgewichts nicht überschreiten. Herstellung der Dosen 19. Feste Prüfsubstanzen sollten vor der Verabreichung an die Tiere in geeigneten Lösungsmitteln oder Vehikeln gelöst oder suspendiert oder in das Futter bzw. das Trinkwasser gegeben werden. Flüssige Prüfsubstanzen können direkt verabreicht oder zuvor verdünnt werden. Für inhalative Expositionen können die Prüfsubstanzen in Abhängigkeit zu ihren physikalisch-chemischen Eigenschaften als Gas, Dampf oder festes/flüssiges Aerosol verabreicht werden. Es sind frische Zubereitungen der Prüfsubstanz zu verwenden, es sei denn, die Stabilität der Substanz bei Lagerung ist erwiesen. Prüfbedingungen Lösungsmittel/Vehikel 20. Das Lösungsmittel/Vehikel sollte in der verwendeten Dosierung keine toxischen Wirkungen hervorrufen und nicht im Verdacht stehen, mit der Prüfsubstanz eine chemische Reaktion einzugehen. Werden keine allgemein anerkannten Lösungsmittel/Vehikel verwendet, so sollte ihre Einbeziehung durch Kompatibilitätsdaten untermauert werden. Es empfiehlt sich, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. Positivkontrollen 21. In der Regel sind parallel Positivkontrolltiere zu verwenden. Bei Laboren, die ihre Kompetenz unter Beweis gestellt haben (siehe Nummer 23) und diese Assays routinemäßig verwenden, kann DNA von zuvor behandelten Positivkontrolltieren in jeden Versuch einbezogen werden, um den Erfolg der Methode zu bestätigen. Derartige DNA aus vorausgegangenen Versuchen ist von derselben Tierart und denselben Zielgeweben zu gewinnen und ordnungsgemäß zu lagern (siehe Nummer 36). Werden parallel Positivkontrollen verwendet, müssen diese nicht auf dem gleichen Verabreichungsweg gegeben werden wie die Prüfsubstanz; von den Positivkontrollen sollte jedoch bekannt sein, dass sie Mutationen in einem oder mehreren für die Prüfsubstanz relevanten Geweben hervorrufen. Die Dosen der Chemikalien für die Positivkontrolle sind so auszuwählen, dass sie schwache oder moderate Wirkungen hervorrufen, mit denen die Leistung und Empfindlichkeit des Assays kritisch bewertet werden können. Beispiele für derartige Chemikalien und bestimmte Zielgewebe finden sich in Tabelle 1. Beispiele für Positivkontrollchemikalien und bestimmte Zielgewebe
Negativkontrollen 22. In jede Probenahme sind Negativkontrolltiere einzubeziehen, die nur ein Lösungsmittel oder ein Vehikel erhalten und ansonsten in derselben Weise behandelt werden wie die Behandlungsgruppen. Um die Eignung der Vehikelkontrolle festzustellen, sollten darüber hinaus in jede Probenahme auch unbehandelte Kontrolltiere einbezogen werden, soweit keine historischen oder veröffentlichten Kontrolldaten vorliegen, aus denen hervorgeht, dass das gewählte Lösungsmittel/Vehikel keine schädlichen oder mutagenen Wirkungen hervorruft. Eignungsprüfung des Prüflaboratoriums 23. Das Labor sollte seine Kompetenz unter Beweis stellen, erwartete Ergebnisse aus veröffentlichten Daten (24) in folgenden Bereichen zu reproduzieren: 1) Mutantenhäufigkeiten bei Positivkontrollchemikalien (einschließlich schwacher Reaktionen), wie den in Tabelle 1 angeführten Chemikalien, Nicht-Mutagenen und Vehikelkontrollen; und 2) die Wiederfindung von Transgenen aus genomischer DNA (z.B. Verpackungseffizienz). Sequenzierung von Mutanten 24. Für Zulassungsanträge ist keine DNA-Sequenzierung von Mutanten erforderlich, insbesondere, wenn ein eindeutiges positives oder negatives Ergebnis erzielt wird. Sequenzierungsdaten können sich jedoch als nützlich erweisen, wenn eine hohe interindividuelle Streuung festgestellt wird. In solchen Fällen können durch Sequenzierung, Jackpot"-Mutationen oder klonale Ereignisse ausgeschlossen werden, indem der Anteil eines bestimmten Gewebes an eindeutigen Mutanten ermittelt wird. Die Sequenzierung von ungefähr zehn Mutanten pro Gewebe und Tier sollte für die einfache Bestimmung, ob klonale Mutanten zur Mutantenhäufigkeit beitragen, ausreichen; um die Mutantenhäufigkeit mathematisch um die Klonalität zu berichtigen, kann es sich als notwendig erweisen, bis zu 25 Mutanten zu sequenzieren. Die Sequenzierung von Mutanten kann auch in Betracht gezogen werden, wenn kleinere Zunahmen der Mutantenhäufigkeit (d. h. Zunahmen knapp über den Werten der unbehandelten Kontrolltiere) festgestellt werden. Unterschiede im Mutantenspektrum zwischen den Mutantenkolonien aus behandelten und unbehandelten Tieren können eine mutagene Wirkung unterstützen (29). Mutationsspektra können außerdem für die Aufstellung mechanistischer Hypothesen nützlich sein. Wenn Sequenzierung als fester Bestandteil in das Versuchsprotokoll aufgenommen werden soll, ist der Versuch besonders sorgfältig zu planen, insbesondere im Hinblick auf die Anzahl der pro Probe sequenzierten Mutanten, um entsprechend dem verwendeten statistischen Modell eine angemessene Aussagekraft zu erzielen (siehe Nummer 43). Zahl und Geschlecht der Versuchstiere 25. Es sollten genügend Tiere pro Gruppe vorgesehen sein, damit die statistische Aussagekraft gewährleistet ist, die nötig ist, um mindestens eine Verdopplung der Mutantenhäufigkeit nachzuweisen. Die Gruppen bestehen aus mindestens fünf Tieren; falls die statistische Aussagekraft jedoch zu gering ist, ist die Zahl der Tiere gegebenenfalls zu erhöhen. In der Regel sind männliche Tiere zu verwenden. In einigen Fällen kann die Durchführung von Tests ausschließlich an weiblichen Tieren gerechtfertigt sein; beispielsweise wenn frauenspezifische Humanmedikamente getestet werden oder wenn spezielle Untersuchungen zum weiblichen Stoffwechsel durchgeführt werden. Liegen im Hinblick auf Toxizität oder Stoffwechsel signifikante Unterschiede zwischen den Geschlechtern vor, sind für die Tests sowohl männliche als auch weibliche Tiere erforderlich. Verabreichungszeitraum 26. Gestützt auf die Beobachtung, dass sich Mutationen mit jeder Behandlung steigern, ist ein Schema für wiederholte Verabreichungen erforderlich, wobei über einen Zeitraum von 28 Tagen täglich behandelt wird. Dies wird allgemein sowohl für das Hervorrufen einer ausreichenden Akkumulation von Mutationen durch schwache Mutagene als auch für die Bereitstellung einer angemessenen Expositionsdauer für den Nachweis von Mutationen in langsam proliferierenden Organen als akzeptabel angesehen. Bei einigen Bewertungen mögen alternative Behandlungsschemata angebracht sein. Diese alternativen Dosierungspläne sind im Protokoll wissenschaftlich zu begründen. Die Behandlungen sollten nicht kürzer sein als die Zeit, die für die vollständige Induktion aller relevanten metabolisierender Enzyme erforderlich ist, und kürzere Behandlungen erfordern möglicherweise mehrere Probenahmen, die sich für Organe mit unterschiedlicher Proliferation eignen. In jedem Fall sind zur Begründung eines Protokolls alle vorliegenden Informationen (z.B. zur allgemeinen Toxizität bzw. zum Stoffwechsel und zur Pharmakokinetik) zu verwenden, vor allem, wenn von den vorgenannten Standardempfehlungen abgewichen wird. Länger als acht Wochen andauernde Behandlungszeiten können zwar die Empfindlichkeit erhöhen, müssen aber klar erläutert und begründet werden, weil lange Behandlungszeiten durch klonale Expansion zu einem scheinbaren Anstieg der Mutantenhäufigkeit führen können (29). Dosisstufen 27. Dosisstufen sollten sich auf die Ergebnisse einer Dosisfindungsstudie stützen, bei der die allgemeine Toxizität gemessen und die über den gleichen Expositionsweg durchgeführt wurde oder aber auf bereits vorliegende Ergebnisse aus Untersuchungen der subakuten Toxizität. Zur Bestimmung der Dosisbereiche können nicht transgene Tiere desselben Nagetierstamms verwendet werden. Der Haupttest sollte eine vollständige Untersuchung einer Negativkontrollgruppe beinhalten, um Informationen über Dosisreaktionen zu gewinnen (siehe Nummer 22) und mindestens drei angemessen abgestufte Dosierungen umfassen, außer bei Verwendung der Grenzdosis (siehe Nummer 28). Die höchste Dosis sollte die maximal verträgliche Dosis (maximum tolerated dose, MTD) sein. Die MTD wird als die Dosis festgelegt, die Toxizitätsanzeichen in einem Ausmaß hervorruft, das darauf schließen lässt, dass nach demselben Dosierungsplan verabreichte höhere Dosen voraussichtlich zum Tod des Tieres führen würden. Chemikalien mit spezifischen biologischen Aktivitäten bei niedrigen, nicht toxischen Dosen (wie Hormone und Mitogene) und Chemikalien mit gesättigten toxikokinetischen Eigenschaften können als Ausnahmen von den Kriterien zur Dosisfestsetzung angesehen werden und sind auf Fallbasis zu bewerten. Die verwendeten Dosisstufen sollten von der Dosis mit der höchsten Toxizität bis zur Dosis mit der geringsten oder überhaupt keiner Toxizität reichen. Limit-Test 28. Falls Dosisfindungsversuche oder vorliegende Daten aus verwandten Nagetierstämmen darauf hinweisen, dass ein Behandlungsplan, der zumindest die Grenzdosis vorsieht (siehe unten), keine messbaren toxischen Wirkungen hervorruft,und wenn aufgrund von Daten über strukturverwandte Chemikalien keine Genotoxizität erwartet wird, kann unter Umständen auf eine vollständige Untersuchung mit drei Dosisstufen verzichtet werden. Für einen Verabreichungszeitraum von 28 Tagen (d. h. 28 tägliche Behandlungen) beträgt die Grenzdosis 1.000 mg/kg Körpergewicht/Tag. Für Verabreichungszeiträume von 14 Tagen oder weniger beträgt die Grenzdosis 2.000 mg/kg/Körpergewicht/Tag (von 28 täglichen Behandlungen abweichende Dosierungspläne sind im Protokoll wissenschaftlich zu begründen; siehe Nummer 26). Verabreichung der Dosen 29. In der Regel wird die Prüfsubstanz über eine Schlundsonde oder eine geeignete Intubationskanüle verabreicht. Bei der Planung eines Assays ist grundsätzlich die Art der Humanexposition zu berücksichtigen. Folglich sind unter Umständen auch andere Expositionswege (subkutan, intravenös, topisch, inhalativ oder intratracheal oder über das Trinkwasser/Futter oder Implantation) akzeptabel, soweit sie begründet werden können. Eine Intraperitonealinjektion wird nicht empfohlen, weil sie beim Menschen keinen physiologisch relevanten Expositionsweg darstellt. Das maximale Flüssigkeitsvolumen, das einem Versuchstier über eine Schlundsonde oder über eine Injektion jeweils verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Dieses Volumen sollte 2 ml/100 g Körpergewicht nicht überschreiten. Die Verwendung höherer Volumina muss gerechtfertigt werden. Abgesehen von reizenden oder ätzenden Stoffen, die in der Regel bei höheren Konzentrationen eine verstärkte Wirkung hervorrufen, sollte die Variabilität des Prüfvolumens auf ein Mindestmaß begrenzt werden, indem die Konzentration so angepasst wird, dass auf allen Dosisstufen ein konstantes Volumen gewährleistet ist. Probenahmezeitpunkt Somatische Zellen 30. Der Probenahmezeitpunkt stellt einen kritischen Parameter dar, weil er von dem für die Fixierung der Mutationen erforderlichen Zeitraum abhängt. Dieser Zeitraum ist gewebespezifisch und hängt offensichtlich mit der Turnover- Zeit der Zellpopulation zusammen, wobei Knochenmark und Darm schnell und die Leber erheblich langsamer reagieren. Ein angemessener Kompromiss für die Messung der Mutantenhäufigkeiten sowohl bei schnell als auch bei langsam proliferierenden Geweben wären tägliche Behandlungen an 28 aufeinanderfolgenden Tagen (wie unter Nummer 26 angegeben) mit Probenahme drei Tage nach der letzten Behandlung; dabei kann es vorkommen, dass die maximale Mutantenhäufigkeit in langsam proliferierendem Gewebe unter diesen Bedingungen nicht sichtbar wird. Wenn langsam proliferierende Gewebe von ausschlaggebender Bedeutung sind, ist eine spätere Probenahme 28 Tage nach dem 28-tägigen Verabreichungszeitraum möglicherweise angemessener (16) (29). In solchen Fällen würde die Probenahme drei Tage nach der letzten Behandlung durch die spätere Probenahme ersetzt und bedürfte einer wissenschaftlichen Begründung. Keimzellen 31. TGR-Assays sind für die Untersuchung von Genmutationen in männlichen Keimzellen (7) (8) (27), für die der Zeitpunkt und die Kinetik der Spermatogenese genau bestimmt wurden (27), gut geeignet. Die geringe Zahl der - selbst nach einer Superovulation - für die Analyse verfügbaren Eizellen und mangelnde DNA-Synthese in der Eizelle schließen die Bestimmung von Mutationen in weiblichen Keimzellen in Studien mit transgenen Tieren von vorne herein aus (31). 32. Der Probenahmezeitpunkt ist bei männlichen Keimzellen so auszuwählen, dass Proben aus dem während der gesamten Keimzellenentwicklung exponierten Zelltypbereich entnommen werden und die Phase, auf die die Probenahme abzielt, ausreichend exponiert wurde. Die Entwicklungszeit der Keimzellen von spermatogonialen Stammzellen zu reifen Spermien, die den Samenleiter/Nebenhodenschwanz erreichen,beträgt ~ 49 Tage bei der Maus (36) und ~ 70 Tage bei der Ratte (34) (35). Nach einer 28-tägigen Exposition und einer anschließenden dreitägigen Probenahme repräsentieren die aus dem Samenleiter/Nebenhodenschwanz (7) (8) entnommenen Spermien eine Population von Zellen, die ungefähr in der zweiten Hälfte der Spermatogenese exponiert wurden; in diesen Zeitraum fallen auch die meiotische und die postmeiotische Phase, nicht aber die spermatogoniale oder Stammzellenphase. Um adäquate Proben von Zellen aus dem Samenleiter/Nebenhodenschwanz entnehmen zu können, die während des Expositionszeitraums spermatogoniale Stammzellen waren, ist frühestens sieben Wochen (Mäuse) oder zehn Wochen (Ratten) nach Ende der Behandlung eine zusätzliche Probenahme erforderlich. 33. Zellen, die nach dem Schema,28 + 3"-Tage aus den Hodenkanälchen herausgepresst wurden, bilden eine für alle Entwicklungsphasen der Keimzellen angereicherte Mischpopulation (7) (8). Die Phasen, in denen Keimzellmutationen induziert werden, lassen sich durch Beprobung derartiger Zellen zum Nachweis von Genmutationen weniger präzise bewerten als durch die Beprobung von Spermatozoen aus dem Samenleiter/Nebenhodenschwanz (weil die Proben aus den Kanälchen aus einer ganzen Reihe von Keimzelltypen bestehen und diese Zellpopulation in einem gewissen Maß mit somatischen Zellen kontaminiert ist). Bei der Beprobung von Zellen aus Hodenkanälchen UND von Spermatozoen aus dem Samenleiter/Nebenhodenschwanz nach nur,28 + 3"-Probenahmetagen würden in gewissem Umfang jedoch auch Zellen erfasst, die in fast allen Keimzellentwicklungsphasen exponiert wurden, was für den Nachweis bestimmter Keimzellmutagene von Nutzen sein könnte. Beobachtungen 34. Allgemeine klinische Beobachtungen sollten mindestens einmal täglich erfolgen, vorzugsweise jeweils zur gleichen Zeit und unter Berücksichtigung des Zeitraums nach der Verabreichung, in dem die Wirkungsspitzen erwartet werden. Der Gesundheitszustand der Tiere ist zu dokumentieren. Mindestens zweimal täglich sind alle Tiere auf Morbiditäts- und Mortalitätsanzeichen zu überprüfen. Alle Tiere sind mindestens einmal wöchentlich sowie zum Zeitpunkt ihrer Tötung zu wiegen. Die Futteraufnahme wird mindestens wöchentlich gemessen. Wenn die Prüfsubstanz über das Trinkwasser verabreicht wird, wird die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen. Tiere, die nichtletale Anzeichen einer übermäßigen Toxizität aufweisen, sind vor Testende zu euthanasieren (23). Gewebeentnahme 35. Die Argumente für eine Gewebeentnahme sollten fundiert sein. Da Mutationsinduktionen in praktisch jedem beliebigen Gewebe untersucht werden können, sollten die zu entnehmenden Gewebe nach dem Versuchsmotiv und unter Berücksichtigung aller vorliegenden Daten zur Mutagenität, Karzinogenität oder Toxizität der Prüfsubstanz ausgewählt werden. Zu den wichtigen zu berücksichtigenden Faktoren sollten auch der Verabreichungsweg (gestützt auf den (die) wahrscheinlichen Expositionsweg(e) beim Menschen), die voraussichtliche Gewebeverteilung und der mögliche Wirkungsmechanismus gehören. Liegen keinerlei Hintergrundinformationen vor, sind einige somatische Gewebe zu entnehmen, die für die Untersuchung von Interesse sind. Bei diesem Gewebe sollte es sich um schnell und langsam proliferierendes Gewebe und um Kontaktstellengewebe handeln. Darüber hinaus sind (nach der Beschreibung unter den Nummern 32 und 33) Spermatozoen aus dem Samenleiter/Nebenhodenschwanz und in der Entwicklung befindliche Keimzellen aus den Hodenkanälchen zu entnehmen und für den Fall, dass eine künftige Analyse der Keimzellenmutagenität erforderlich wird, aufzubewahren. Die Organe sind zu wiegen, und bei größeren Organen sollten von allen Tieren Gewebe aus demselben Organbereich entnommen werden. Lagerung von Gewebe und DNA 36. Gewebe (oder Gewebehomogenate) sind bei oder unter einer Temperatur von - 70 °C zu lagern und für die DNA-Isolierung innerhalb von fünf Jahren zu verwenden. Bei einer Temperatur von 4 °C in einem geeigneten Puffer kühl gelagerte isolierte DNA sollte idealerweise innerhalb eines Jahres für Mutationsanalysen verwendet werden. Gewebeauswahl für Mutantenanalysen 37. Die Gewebe sind unter anderem nach folgenden Krierien auszuwählen: 1) Verabreichungsweg oder Stelle des Erstkontakts (z.B. Drüsenmagen, wenn die Verabreichung oral erfolgt; Lunge, wenn die Verabreichung inhalativ erfolgt, oder Haut bei einer topischen Anwendung), und 2) in allgemeinen Toxizitätsstudien gemessene pharmakokinetische Parameter, die die Gewebedisposition, -retention oder -akkumulation oder Zielorgane für die Toxizität anzeigen. Wenn Versuche aufgrund von Ergebnissen aus Karzinogenitätsuntersuchungen durchgeführt werden, sind Zielgewebe für die Karzinogenität in Erwägung zu ziehen. Die für die Analyse ausgewählten Gewebe sollten den Nachweis von Chemikalien maximieren, die in vitro direkt mutagen wirken, schnell verstoffwechselt werden, hochreaktiv sind oder schlecht absorbiert werden, oder von Chemikalien, bei denen das Zielgewebe über den Verabreichungsweg bestimmt wird (6). 38. In Ermangelung von Hintergrundinformationen und unter Berücksichtigung der durch den Verabreichungsweg bedingten Kontaktstelle sind die Leber und mindestens ein sich schnell teilendes Gewebe (z.B. Drüsenmagen, Knochenmark) auf Mutagenität zu untersuchen. In den meisten Fällen kann den oben angeführten Anforderungen durch die Analyse zweier sorgfältig ausgewählter Gewebe genüge getan werden. In einigen Fällen sind möglicherweise drei oder mehr Gewebe nötig. Wenn es spezifische Besorgnis hinsichtlich Keimzellmutationen gibt, auch aufgrund positiver Befunde in Somazellen, sollten Keimzellgewebe auf Mutationen untersucht werden. Messmethoden 39. Für die empfohlenen transgenen Modelle zum Nachweis von Mutanten stehen folgende Standardlabormethoden bzw. veröffentlichte Methoden zur Verfügung: lacZ-Bakteriophage lambda und Plasmid (30); lacI Maus (2) (18); gpt Delta Maus (22); gpt Delta Ratte (28); cII (17). Änderungen sind zu begründen und ordnungsgemäß zu dokumentieren. Daten aus multiplen Verpackungen können zusammengefasst und zum Erzielen einer angemessenen Anzahl Plaques oder Kolonien verwendet werden. Wird eine große Anzahl Verpackungsreaktionen benötigt, um die geeignete Anzahl Plaques zu erzielen, kann dies jedoch ein Hinweis auf schlechte DNA-Qualität sein. In solchen Fällen sind die Daten mit Vorsicht zu behandeln, weil sie unzuverlässig sein könnten. Die optimale Gesamtzahl an Plaques oder Kolonien pro DNA-Probe richtet sich nach der statistischen Wahrscheinlichkeit des Nachweises einer ausreichenden Anzahl Mutanten bei einer gegebenen Häufigkeit spontaner Mutanten. In der Regel sind mindestens 125.000 bis 300.000 Plaques erforderlich, wenn die Häufigkeit spontaner Mutanten bei 3 × 10-5 (15) liegt. Beim Big Blue® lacI Assay muss unbedingt gewährleistet sein, dass durch Einbeziehung geeigneter Farbkontrollen parallel zu allen Ausstrichen der gesamte Bereich der Mutanten-Farbphänotypen nachgewiesen werden kann. Gewebe und die resultierenden Proben (Items) sind nach einem Blockschema zu bearbeiten und zu analysieren, bei dem Items aus der Vehikel-/Lösungsmittel-Kontrollgruppe, der Positivkontrollgruppe (falls verwendet) oder der DNA-Positivkontrolle (falls zutreffend) und jede Behandlungsgruppe zusammen verarbeitet werden. Auswertung der Ergebnisse 40. Die Daten zu den einzelnen Tieren sind in tabellarischer Form vorzulegen. Die Versuchseinheit ist das Tier. Der Bericht sollte folgende Angaben enthalten: Gesamtzahl der plaquebildenden Einheiten (Plaque-Forming Units, PFU) bzw. der koloniebildenden Einheiten (Colony-Forming Units, CFU), Anzahl Mutanten und Mutantenhäufigkeit für jedes Gewebe und Tier. Bei multiplen Verpackungs-/Rettungsreaktionen ist die Zahl der Reaktionen pro DNA-Probe anzugeben. Auch wenn es sich empfiehlt, die Daten über jede einzelne Reaktion aufzubewahren, müssen lediglich die Gesamt-PFU bzw. -CFU schriftlich festgehalten werden. Daten zur Toxizität und klinische Anzeichen gemäß Nummer 34 sind zu dokumentieren. Sequenzierungsergebnisse sind für jeden einzelnen analysierten Mutanten vorzulegen, wobei die entsprechend durchgeführten Mutationshäufigkeitsberechnungen für jedes Tier und jedes Gewebe anzugeben sind. Statistische Auswertung und Interpretation der Ergebnisse 41. Es gibt mehrere Kriterien für die Entscheidung über ein positives Ergebnis, wie eine dosisabhängigee Zunahme der Mutantenhäufigkeit oder eine deutliche Zunahme der Mutantenhäufigkeit in einer einzigen Dosisgruppe im Vergleich zur Lösungsmittel-/Vehikelkontrollgruppe. Es sind mindestens drei behandelte Dosisgruppen zu analysieren, um ausreichende Daten für Dosis-Wirkungs-Analysen zu liefern. Das Hauptaugenmerk sollte auf der biologischen Relevanz der Ergebnisse liegen, wobei geeignete statistische Methoden als Hilfsmittel für die Auswertung der Testergebnisse verwendet werden können (4) (14) (15) (25) (26). Die verwendeten statistischen Tests müssen das Tier als Versuchseinheit berücksichtigen. 42. Eine Prüfsubstanz, bei der die Ergebnisse die oben angeführten Kriterien in keinem Gewebe erfüllen, gilt im vorliegenden Test als nicht mutagen. Für die biologische Relevanz eines negativen Ergebnisses ist die Gewebeexposition zu bestätigen. 43. Für DNA-Sequenzierungsanalysen stehen eine Reihe statistischer Konzepte zur Erleichterung der Auswertung der Ergebnisse zur Verfügung (1) (5) (9) (19). 44. Indem ermittelt wird, ob die gemessenen Werte innerhalb oder außerhalb des historischen Kontrollbereichs liegen, lassen sich Leitlinien für die Bewertung der biologischen Signifikanz der Reaktion herleiten (32). Prüfbericht 45. Der Prüfbericht sollte folgende Angaben enthalten: Prüfsubstanz:
Lösungsmittel/Vehikel:
Versuchstiere:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse Schlussfolgerung (1) Adams, W.T. and T.R. Skopek (1987)., Statistical Test for the Comparison of Samples from Mutational Spectra". J. Mol. Biol., 194: 391-396. (2) Bielas, J.H. (2002).,A more Efficient Big Blue® Protocol Improves Transgene Rescue and Accuracy in an Adduct and Mutation Measurement". Mutation Res., 518: 107-112. (3) Boerrigter, M.E., M.E. Dollé, H.-J. Martus, J.A. Gossen and J. Vijg (1995)., Plasmidbased Transgenic Mouse Model for Studying in vivo Mutations" Nature, 377(6550): 657-659 (4) Carr, G.J. and N.J. Gorelick (1995)., Statistical Design and Analysis of Mutation Studies in Transgenic Mice", Environ. Mol. Mutagen, 25(3): 246-255. (5) Carr, G.J. and N.J. Gorelick (1996)., Mutational Spectra in Transgenic Animal Research: Data Analysis and Study Design Based upon the Mutant or Mutation Frequency", Environ. Mol. Mutagen, 28: 405-413. (6) Dean, S.W., T.M. Brooks, B. Burlinson, J. Mirsalis, B. Myhr, L. Recio and V. Thybaud (1999)., Transgenic Mouse Mutation Assay Systems can Play an important Role in Regulatory Mutagenicity Testing in vivo for the Detection of Siteof-contact Mutagens", Mutagenesis, 14(1): 141-151. (7) Douglas, G.R., J. Jiao, J.D. Gingerich, J.A. Gossen and L.M. Soper(1995)., Temporal and Molecular Characteristics of Mutations Induced by Ethylnitrosourea in Germ Cells Isolated from Seminiferous Tubules and in Spermatozoa of lacZ Transgenic Mice", Proc. Natl. Acad. Sci. USA, 92: 7485-7489. (8) Douglas, G.R., J.D. Gingerich, L.M. Soper and J. Jiao (1997)., Toward an Understanding of the Use of Transgenic Mice for the Detection of Gene Mutations in Germ Cells", Mutation Res., 388(2-3): 197-212. (9) Dunson, D.B. and K.R. Tindall (2000)., Bayesian Analysis of Mutational Spectra", Genetics, 156: 1411-1418. (10) Gossen, J.A., W.J. de Leeuw, C.H. Tan, E.C. Zwarthoff, F. Berends, P.H. Lohman, D.L. Knook and J. Vijg(1989)., Efficient Rescue of Integrated Shuttle Vectors from Transgenic Mice: a Model for Studying Mutations in vivo", Proc. Natl. Acad. Sci. USA, 86(20): 7971-7975. (11) Gossen, J.A. and J. Vijg (1993),,A Selective System for lacZ-Phage using a Galactosesensitive E. coli Host", Biotechniques, 14(3): 326, 330. (12) Erikson, R.P. (2003)., Somatic Gene Mutation and Human Disease other than Cancer", Mutation Res., 543: 125-136. (13) Erikson, R.P. (2010)., Somatic Gene Mutation and Human Disease other than Cancer: an Update", Mutation Res., 705: 96-106. (14) Fung, K.Y., G.R. Douglas and D. Krewski (1998)., Statistical Analysis of lacZ Mutant Frequency Data from MutaTMMouse Mutagenicity Assays", Mutagenesis, 13(3): 249-255. (15) Heddle, J.A., S. Dean, T. Nohmi, M. Boerrigter, D. Casciano, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.C. Mirsalis, H.-J Martus, T.R. Skopek, V. Thybaud, K.R.Tindall and N. Yajima (2000)., In vivo Transgenic Mutation Assays", Environ. Mol. Mutagen., 35: 253-259. (16) Heddle, J.A., H.-J. Martus and G.R. Douglas (2003)., Treatment and Sampling Protocols for Transgenic Mutation Assays", Environ. Mol. Mutagen., 41: 1-6. (17) Jakubczak, J.L., G. Merlino, J.E. French, W.J. Muller, B. Paul, S. Adhya and S. Garges (1996)., Analysis of Genetic Instability during Mammary Tumor Progression using a novel Selectionbased Assay for in vivo Mutations in a Bacteriophage ≫ Transgene Target", Proc. Natl. Acad. Sci. USA, 93(17): 9073-9078. (18) Kohler, S.W., G.S. Provost, P.L. Kretz, A. Fieck, J.A. Sorge and J.M. Short (1990)., The Use of Transgenic Mice for Shortterm, in vivo Mutagenicity Testing", Genet. Anal. Tech. Appl., 7(8): 212-218. (19) Lewis P.D., B. Manshian, M.N. Routledge, G.B. Scott and P.A. Burns (2008)., Comparison of Induced and Cancer- associated Mutational Spectra using Multivariate Data Analysis", Carcinogenesis, 29(4): 772-778. (20) Nohmi, T., M. Katoh, H. Suzuki, M. Matsui, M. Yamada, M. Watanabe, M. Suzuki, N. Horiya, O. Ueda, T. Shibuya, H. Ikeda and T. Sofuni (1996).,A new Transgenic Mouse Mutagenesis Test System using Spi - and 6-thioguanine Selections", Environ. Mol. Mutagen., 28(4): 465-470. (21) Nohmi, T., M. Suzuki, K. Masumura, M. Yamada, K. Matsui, O. Ueda, H. Suzuki, M. Katoh, H. Ikeda and T. Sofuni (1999)., Spi- Selection: an Efficient Method to Detect ³-rayinduced Deletions in Transgenic Mice", Environ. Mol. Mutagen., 34(1): 9-15. (22) Nohmi, T., T. Suzuki and K.I. Masumura (2000)., Recent Advances in the Protocols of Transgenic Mouse Mutation Assays", Mutation Res., 455(1-2): 191-215. (23) OECD (2000). Guidance Document on the recognition, assessment, and use of clinical signs as humane endpoints for experimental animals used in safety evaluations, Series on Testing and Assessment, Nr. 19, ENV/JM/MONO(2000)7, OECD, Paris. (24) OECD (2009). Detailed Review Paper on Transgenic Rodent Mutation Assays, Series on Testing and Assessment, No 103, ENV/JM/MONO(2009)7, OECD, Paris. (25) Piegorsch, W.W., B.H. Margolin, M.D. Shelby, A. Johnson, J.E. French, R.W. Tennant and K.R. Tindall (1995)., Study Design and Sample Sizes for a lacI Transgenic Mouse Mutation Assay", Environ. Mol. Mutagen., 25(3): 231-245. (26) Piegorsch, W.W., A.C. Lockhart, G.J. Carr, B.H. Margolin, T. Brooks, ... G.R. Douglas, U.M. Liegibel, T. Suzuki, V. Thybaud, J.H. van Delft and N.J. Gorelick (1997)., Sources of Variability in Data from a Positive Selection lacZ Transgenic Mouse Mutation Assay: an Interlaboratory Study", Mutation. Res., 388(2-3): 249-289. (27) Singer, T.M., I.B. Lambert, A. Williams, G.R. Douglas and C.L. Yauk(2006)., Detection of Induced Male Germline Mutation: Correlations and Comparisons between Traditional Germline Mutation Assays, Transgenic Rodent Assays and Expanded Simple Tandem Repeat Instability Assays", Mutation. Res., 598: 164-193. (28) Toyoda-Hokaiwado, N., T. Inoue, K. Masumura, H. Hayashi, Y. Kawamura, Y. Kurata, M. Takamune, M. Yamada, H. Sanada, T. Umemura, A. Nishikawa and T. Nohmi (2010)., Integration of in vivo Genotoxicity and Shortterm Carcinogenicity Assays using F344 gpt delta Transgenic Rates: in vivo Mutagenicity of 2,4-diaminotoluene and 2,6-diaminotoluene Structural Isomers", Toxicol. Sci., 114(1): 71-78. (29) Thybaud, V., S. Dean, T. Nohmi, J. de Boer, G.R. Douglas, B.W. Glickman, N.J. Gorelick, J.A. Heddle, R.H. Heflich, I. Lambert, H.-J. Martus, J.C. Mirsalis, T. Suzuki and N. Yajima (2003)., In vivo Transgenic Mutation Assays", Mutation Res., 540: 141-151. (30) Vijg, J. and G.R. Douglas (1996),, Bacteriophage ≫ and Plasmid lacZ Transgenic Mice for studying Mutations in vivo" in: G. Pfeifer (ed.), Technologies for Detection of DNA Damage and Mutations, Part II, Plenum Press, New York, NY, USA, pp. 391-410. (31) Yauk, C.L., J.D. Gingerich, L. Soper, A. MacMahon, W.G. Foster and G.R. Douglas (2005).,A lacZ Transgenic Mouse Assay for the Detection of Mutations in Follicular Granulosa Cells", Mutation Res., 578(1-2): 117-123. (32) Hayashi, M., K. Dearfield, P. Kasper, D. Lovell, H.-J. Martus, V. Thybaud (2011)., Compilation and Use of Genetic Toxicity Historical Control Data", Mutation Res., doi:10.1016/j.mrgentox.2010.09.007. (33) OECD (2011). Retrospective Performance Assessment of OECD Test Guideline on Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays, Series on Testing and Assessment, No 145, ENV/JM/MONO(2011)20, OECD, Paris. (34) Clermont, Y. (1972)., Kinetics of spermatogenesis in mammals seminiferous epithelium cycle and spermatogonial renewal". Physiol. Rev. 52: 198-236. (35) Robaire, B., Hinton, B.T., and Oregbin-Crist, M.-C. (2006)., The Epididymis", in Neil, J.D., Pfaff, D.W., Chalis, J.R.G., de Kretser, D.M., Richards, J.S., and P. M, Wassarman (eds.), Physiology of Reproduction, Elsevier, the Netherlands, pp. 1071-1148. (36) Russell, L.B. (2004)., Effects of male germcell stage on the frequency, nature, and spectrum of induced specific- locus mutations in the mouse", Genetica, 122: 25-36.
Basenpaarsubstitution: eine Art Mutation, die dazu führt, dass eine einzelne DNA-Nukleotidbase gegen eine andere DNA-Nukleotidbase ausgetauscht wird. Chemikalie: ein Stoff oder eine Mischung. Cos-Stelle: ein 12-Nukleotidsegment einer Einzelstrang-DNA, das an beiden Enden des Doppelstrang-Genoms des Bakteriophagen vorkommt. Deletion: eine Mutation, bei der ein oder mehrere (sequenzielle) Nukleotiden vom Genom verloren werden. Elektroporation: Anwendung elektrischer Impulse zur Erhöhung der Durchlässigkeit von Zellmembranen. Endogenes Gen: ein Gen, das aus dem Genom selbst stammt. Extrabinomiale Variation: größere Variabilität bei Wiederholungsschätzungen eines Populationsanteils als bei einer Binomialverteilung der Population erwartet würde. Große Deletionen: Deletionen mehrerer Kilobasen in der DNA (die durch Spi - Auswahl und die lacZ-Plasmidassays gezielt nachgewiesen werden). Insertion: Hinzufügen eines oder mehrerer Nukleotidenbasenpaare in eine DNA-Sequenz. Jackpot: große Anzahl Mutanten, durch klonale Expansion aus einer einzigen Mutation entstanden. Kapsid: die ein Viruspartikel umschließende Proteinhülle. Klonale Expansion: die Erzeugung vieler Zellen aus einer einzigen (Mutanten-)Zelle. Koloniebildende Einheit (Colony-Forming Unit, CFU): ein Größe zur Quantifizierung lebensfähiger Bakterien. Konkatamer: eine Wiederholungssequenz einer DNA-Kette (Biomolekül). Ligation: die kovalente Anbindung von zwei DNA-Molekülenden durch DNA-Ligase. Mitogen: eine Chemikalie, die eine Zelle zur Zellteilung anregt und Mitose auslöst (d. h. Zellteilung). Neutrales Gen: ein Gen, das weder von positivem, noch von negativem Selektionsdruck beeinflusst wird. Plaquebildende Einheit (Plaque Forming Unit, PFU): eine Größe zur Quantifizierung lebensfähiger Bakteriophagen. Positivselektion: eine Methode, bei der nur Mutanten überleben. Probenahmezeitpunkt: das Ende des Zeitraums vor der Tötung des Tieres, in dem die Chemikalie nicht verabreicht wird und in dem nicht reparierte DNA-Läsionen sich in Form stabiler Mutationen fixieren. Prüfsubstanz: jede(r) nach dieser Prüfmethode getesteter Stoff bzw. Mischung. Punktmutation: allgemeiner Begriff für eine Mutation, die nur eine kleine Sequenz der DNA betrifft, einschließlich kleiner Insertionen, Deletionen und Basenpaarsubstitutionen. Rasterschubmutation: eine genetische Mutation innerhalb einer ein Protein/Peptid codierenden DNA-Sequenz, verursacht durch Insertionen oder Deletionen einer Reihe von Nukleotiden, die nicht gleichmäßig durch drei geteilt werden können. Reporter-Gen: ein Gen, dessen Produkt (mutantes Gen) einfach nachzuweisen ist. Shuttle-Vektor: ein Vektor, der so strukturiert ist, dass er sich in zwei verschiedenen Wirtsarten vermehren kann; entsprechend kann in einen Shuttle-Vektor eingebrachte DNA in zwei unterschiedlichen Zelltypen oder zwei unterschiedlichen Organismen geprüft oder manipuliert werden. Transgen: bezogen auf einen Organismus oder der Organismus selbst, dessen Genom durch die Übertragung eines oder mehrerer Gene einer anderen Spezies verändert worden ist. Verabreichungszeitraum: die Gesamtdauer, während der einem Tier Prüfsubstanz verabreicht wird. Verpackung: die Synthese infektiöser Phagenpartikel aus der Zubereitung von Phagenkapsid und Schwanzproteinen und einem Konkatamer von Phagen-DNA-Molekülen. Wird gemeinhin zur Verpackung von auf einen Lambda-Vektor geklonter DNA (die durch Cos-Stellen getrennt ist) in infektiöse Lambdapartikel verwendet. Verpackungseffizienz: die Effizienz, mit der verpackte Bakteriophagen in Wirtsbakterien wiedergefunden werden. | |||||||||||||||||||||||||||||
| weiter . | |