umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (22)
| zurück |
|
B.59 In-chemico-Hautsensibilisierung: Direkt-Peptidreaktivitätstest (DPRA) 17 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 442C (2015). Ein Hautallergen ist gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP) 1 ein Stoff, der bei Hautkontakt eine allergische Reaktion auslöst. Diese Prüfmethode ist ein In-chemico-Verfahren (Direkt-Peptidreaktivitätstest - DPRA) zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren gemäß UN-GHS und CLP. Es besteht allgemeines Einvernehmen über die der Hautsensibilisierung zugrunde liegenden biologischen Vorgänge. Das vorhandene Wissen über die chemischen und biologischen Mechanismen im Zusammenhang mit der Hautsensibilisierung wurde in Form eines Adverse Outcome Pathway (AOP) (2), ausgehend von dem auslösenden molekularen Ereignis über die Zwischenvorgänge bis hin zur schädlichen Auswirkung auf die Gesundheit, nämlich die allergische Kontaktdermatitis beim Menschen oder die Kontakt-Überempfindlichkeit bei Nagetieren, zusammengefasst. Innerhalb des Hautsensibilisierungs-AOP ist das auslösende molekulare Ereignis die kovalente Bindung von elektrophilen Stoffen an nukleophile Zentren in Hautproteinen. Die Bewertung der Hautsensibilisierung erfolgte normalerweise an Labortieren. Bei den klassischen Methoden an Meerschweinchen, dem Maximierungstest an Meerschweinchen (Guinea Pig Maximisation Test, GMPT) nach Magnusson/Kligman und dem Bühler-Test (Kapitel B.6 (3)), werden die Induktions- und die Auslösephase der Hautsensibilisierung untersucht. Ein Test an der Maus, der Lokale Lymphknotentest (LLNA, Kapitel B.42 (4)) und die beiden nicht radioaktiven Abwandlungen dieses Tests, LLNA: DA (Kapitel B.50 (5)) und LLNA: BrdU-ELISA (Kapitel B.51 (6)), bei denen jeweils nur die Induktionsreaktion bewertet wird, haben ebenfalls an Akzeptanz gewonnen, da sie sowohl in Bezug auf den Tierschutz als auch die objektive Messung der Induktionsphase der Hautsensibilisierung Vorteile gegenüber Tests am Meerschweinchen bieten. Vor kurzem wurden mechanistisch basierte In-chemico- und In-vitro-Prüfmethoden für die Bewertung der Gefahr einer Hautsensibilisierung durch Chemikalien als wissenschaftlich fundiert befunden. Allerdings sind Methoden ohne Tierversuche (in silico, in chemico, in vitro) im Rahmen von integrierten Test- und Bewertungsansätzen (Integrated Approaches to Testing and Assessment, IATA) erforderlich, um die gegenwärtig angewandten Tierversuche angesichts der eingeschränkten mechanistischen AOP-Abdeckung der gegenwärtig verfügbaren Prüfmethoden ohne Tierversuche zu ersetzen (2) (7). Der DPRA wird für das auslösende molekulare Ereignis im AOP der Hautsensibilisierung, nämlich die Proteinreaktivität, vorgeschlagen, indem die Reaktivität von Prüfchemikalien gegenüber synthetischen lysin- oder cysteinhaltigen Modellpeptiden quantifiziert wird (8). Anhand der prozentualen Peptid-Depletionswerte für Cystein und Lysin wird ein Stoff dann in eine von vier Reaktivitätsklassen eingestuft, um die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren zu unterstützen (9). Der DPRA wurde in einer Validierungsstudie unter der Leitung eines Europäischen Referenzlabors für Alternativen zu Tierversuchen (European Union Reference Laboratory for Alternatives to Animal Testing, EURL ECVAM) und in einem anschließenden unabhängigen Peer-Review des Wissenschaftlich Beratenden Ausschusses (ESAC) des EURL ECVAM bewertet und aus wissenschaftlicher Sicht für zulässig befunden (10), um im Rahmen eines IATA die Unterscheidung zwischen Hautallergenen und Nichtsensibilatoren im Hinblick auf die Gefahreneinstufung und -kennzeichnung zu unterstützen. Beispiele für die Verwendung von DPRA-Daten in Kombination mit anderen Informationen werden in der Literatur beschrieben (11) (12) (13) (14). Definitionen sind Anlage I zu entnehmen. Vorbemerkungen, Anwendbarkeit und Einsatzgrenzen Die Korrelation zwischen Proteinreaktivität und Hautsensibilisierungspotenzial ist gut nachgewiesen (15) (16) (17). Da die Proteinbindung jedoch nur einen einzigen Schlüsselvorgang, wenn auch das auslösende molekulare Ereignis für den AOP der Hautsensibilisierung, darstellt, reichen die mit Prüfmethoden und Nicht-Prüfmethoden ermittelten Informationen alleine möglicherweise nicht aus, um zu dem Schluss zu gelangen, dass Chemikalien kein Hautsensibilisierungspotenzial besitzen. Daher sollten die mit dieser Prüfmethode ermittelten Daten im Rahmen integrierter Ansätze, wie z.B. IATA, betrachtet und mit anderen ergänzenden Informationen, die beispielsweise aus In-vitro-Tests in Bezug auf andere Schlüsselvorgänge des Hautsensibilisierungs-AOP abgeleitet werden, sowie mit anderen Nicht-Prüfmethoden, einschließlich der Übertragung von Informationen (read across) zu chemischen Analoga, kombiniert werden. Diese Prüfmethode kann zusammen mit anderen ergänzenden Informationen zur Unterstützung der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kategorie 1) und Nichtsensibilisatoren im Rahmen eines IATA eingesetzt werden. Diese Prüfmethode kann alleine weder zur Einstufung von Hautallergenen in die Unterkategorien 1A und 1B gemäß Definition in UN-GHS/CLP noch zur Vorhersage des Potenzials im Rahmen von Sicherheitsbewertungsentscheidungen verwendet werden. Jedoch kann ein positives Ergebnis mit dem DPRA je nach Rechtsrahmen alleine zur Einstufung einer Chemikalie in die UN-GHS/CLP-Kategorie 1 herangezogen werden. Es hat sich gezeigt, dass die DPRA-Prüfmethode Labors mit Erfahrung auf dem Gebiet der Hochleistungsflüssigchromatographie (HPLC) übertragen werden kann. Der Grad der Reproduzierbarkeit bei Vorhersagen, der bei der Prüfmethode erwartet werden kann, liegt in der Größenordnung von 85 % innerhalb von Labors und von 80 % zwischen Labors (10). Die Ergebnisse der Validierungsstudie (18) und veröffentlichter Studien (19) weisen insgesamt darauf hin, dass die Genauigkeit des DPRA bei der Unterscheidung von Sensibilatoren (d. h. UN-GHS-/CLP-Kategorie 1) gegenüber Nichtsensibilisatoren 80 % (N = 157) bei einer Empfindlichkeit von 80 % (88/109) und einer Spezifität von 77 % (37/48) im Vergleich zu den Ergebnissen des LLNA beträgt. Beim DPRA ist die Wahrscheinlichkeit einer Unterschätzung bei Chemikalien mit geringem bis mittlerem Hautsensibilisierungspotenzial (d. h. UN-GHS/CLP-Unterkategorie 1B) größer als bei Chemikalien mit hohem Hautsensibilisierungspotenzial (d. h. UN-GHS/CLP-Unterkategorie 1A) (18) (19). Jedoch sind die Genauigkeitswerte, die hier für den DPRA als eigenständige Testmethode angegeben werden, lediglich als Anhaltspunkte zu betrachten, da die Prüfmethode in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 9 betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA-Test sowie andere Tierversuche die Situation bei den relevanten Arten, d. h. Menschen, nicht vollständig widerspiegeln. Auf der Grundlage der verfügbaren Gesamtdaten über den DPRA wurde nachgewiesen, dass der Test bei Prüfchemikalien, die eine Vielzahl an organischen funktionellen Gruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzialen (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (8) (9) (10) (19). Insgesamt deuten diese Informationen auf die Zweckmäßigkeit des DPRA für die Erkennung der Gefahr einer Hautsensibilisierung hin. Der Begriff "Prüfchemikalie" bezeichnet bei dieser Prüfmethode das, was getestet wird, und bezieht sich nicht auf die Anwendbarkeit des DPRA für die Prüfung von Stoffen und/oder Gemischen. Diese Prüfmethode ist nicht für die Untersuchung von Metallverbindungen geeignet, da diese bekanntermaßen mit Proteinen reagieren, die über andere Mechanismen als die kovalente Bindung wirken. Eine Prüfchemikalie sollte in einem geeigneten Lösungsmittel in einer Endkonzentration von 100 mM löslich sein (siehe Nummer 18). Prüfchemikalien, die in dieser Konzentration nicht löslich sind, können unter Umständen jedoch trotzdem in geringeren löslichen Konzentrationen getestet werden. In einem solchen Fall könnte ein positives Ergebnis dennoch zur Identifizierung der Prüfchemikalie als Hautallergen unterstützend herangezogen werden, während bei einem negativen Ergebnis keine verbindlichen Rückschlüsse auf die fehlende Reaktivität gezogen werden dürfen. Gegenwärtig stehen nur begrenzte Informationen über die Anwendbarkeit des DPRA auf Gemische mit bekannter Zusammensetzung zur Verfügung (18) (19). Dennoch gilt der DPRA für die Prüfung von mehrkomponentigen Substanzen und Gemischen mit bekannter Zusammensetzung als technisch geeignet (siehe Nummer 18). Bevor diese Prüfmethode auf ein Gemisch angewendet wird, um zu Regulierungszwecken Daten zu gewinnen, sollte geprüft werden, ob, und falls ja, warum, sie für diesen Zweck geeignete Ergebnisse liefert. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs gesetzlich vorgeschrieben ist. Das gegenwärtige Vorhersagemodell kann aufgrund des festgelegten Molverhältnisses von Prüfchemikalie und Peptid nicht bei komplexen Gemischen mit unbekannter Zusammensetzung oder bei Stoffen mit unbekannter oder variabler Zusammensetzung, komplexen Reaktionsprodukten oder biologischen Materialien (z.B. UVCB-Stoffen) eingesetzt werden. Für diesen Zweck wird es erforderlich sein, ein neues Vorhersagemodell auf der Grundlage eines gravimetrischen Ansatzes zu entwickeln. Wenn nachgewiesen werden kann, dass die Prüfmethode bei anderen spezifischen Chemikalienkategorien nicht anwendbar ist, sollte sie bei diesen nicht verwendet werden. Diese Prüfmethode ist eine In-chemico-Methode, die kein Stoffwechselsystem beinhaltet. Chemikalien, die ihr Hautsensibilisierungspotenzial erst durch eine enzymatische Bioaktivierung erlangen (d. h. Prohaptene), können mit dieser Prüfmethode nicht erkannt werden. Chemikalien, die erst nach einer abiotischen Umwandlung zu Sensibilisatoren werden (d. h. Prähaptene), werden Berichten zufolge in einigen Fällen mit der Prüfmethode richtig erkannt (18). Angesichts der vorstehenden Ausführungen sollten mit der Prüfmethode erhaltene negative Ergebnisse im Kontext der angegebenen Grenzen und in Verbindung mit anderen Informationsquellen im Rahmen eines IATA interpretiert werden. Prüfchemikalien, die nicht kovalent an das Peptid binden, sondern seine Oxidation (d. h. Cystein-Dimerisierung) fördern, könnten zu einer potenziellen Überschätzung der Peptid-Depletion führen, mit der Folge möglicher falscher Positiv-Vorhersagen und/oder einer Einstufung in eine höhere Reaktivitätsklasse (siehe Nummern 29 und 30). Der DPRA unterstützt, wie bereits erwähnt, die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren. Er kann jedoch bei Verwendung im Rahmen von integrierten Ansätzen wie IATA möglicherweise auch zur Bewertung der Sensibilisierungspotenz beitragen (11). Es sind allerdings weitere Untersuchungen notwendig, vorzugsweise auf der Grundlage von Humandaten, um herauszufinden, inwieweit die Ergebnisse des DPRA möglicherweise zur Potenzbewertung herangezogen werden können. Der DPRA ist eine In-chemico-Methode, bei der die Restkonzentration eines cystein- oder lysinhaltigen Peptids nach 24-stündiger Inkubation mit der Prüfchemikalie bei 25 ± 2,5 °C quantifiziert wird. Die synthetischen Peptide enthalten Phenylalanin als Hilfsmittel zum Nachweis. Die relative Peptidkonzentration wird mittels Hochleistungsflüssigchromatographie (HPLC) mit Gradientenelution und UV-Detektion bei 220 nm gemessen. Anschließend werden die prozentualen Peptid-Depletionswerte für Cystein und Lysin berechnet, die als Eingangsparameter in ein Vorhersagemodell (siehe Nummer 29) zur Einstufung der Prüfchemikalie in eine von vier Reaktivitätsklassen einfließen, die zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilatoren verwendet werden. Vor der routinemäßigen Verwendung des unter dieser Prüfmethode beschriebenen Verfahrens sollten Laboratorien ihre technische Kompetenz anhand der zehn in Anlage 2 aufgeführten Leistungsstoffe nachweisen. Diese Prüfmethode basiert auf dem DPRA DB-ALM-Protokoll Nr. 154 (20), das für die vom EURL ECVAM koordinierte Validierungsstudie verwendet wurde. Es wird empfohlen, dieses Protokoll bei der Durchführung und Anwendung der Methode im Labor zugrunde zu legen. Nachfolgend werden die wichtigsten Komponenten und Verfahren des DPRA beschrieben. Bei der Verwendung eines anderen HPLC-Aufbaus ist dessen Gleichwertigkeit mit dem im DB-ALM-Protokoll beschriebenen validierten Aufbau nachzuweisen (z.B. durch Prüfung der Leistungsstoffe in Anlage 2). Vorbereitung der cystein- oder lysinhaltigen Peptide Die Stammlösungen von cysteinhaltigen (Ac-RFAACAA-COOH) und lysinhaltigen (Ac-RFAAKAA-COOH) synthetischen Peptiden mit einer Reinheit von mehr als 85 % (vorzugsweise im Bereich von 90-95 %) sollten kurz vor ihrer Inkubation mit der Prüfchemikalie frisch zubereitet werden. Die Endkonzentration des Cysteinpeptids sollte 0,667 mM in einem Phosphatpuffer mit pH 7,5 und die Endkonzentration des Lysinpeptids 0,667 mM in einem Ammoniumacetat-Puffer mit pH 10,2 betragen. Die HPLC-Sequenz ist so einzustellen, dass die HPLC-Analyse in weniger als 30 Stunden abgeschlossen ist. Bei dem in der Validierungsstudie verwendeten und in dieser Prüfmethode beschriebenen HPLC-Aufbau können in einem einzigen HPLC-Durchlauf bis zu 26 Analyseproben (bestehend aus der Prüfchemikalie, der Positivkontrolle und einer entsprechenden Anzahl an Lösungsmittelkontrollen, abhängig von der Anzahl der einzelnen im Test verwendeten Lösungsmittel, jeweils dreifach getestet) abgearbeitet werden. Alle im gleichen Durchlauf analysierten Replikate sollten die gleichen Cystein- und Lysinpeptid-Stammlösungen verwenden. Es wird empfohlen, vor der Verwendung der einzelnen Peptidchargen ihre Löslichkeit entsprechend nachzuweisen. Vorbereitung der Prüfchemikalie Vor der Durchführung des Tests sollte die Löslichkeit der Prüfchemikalie in einem geeigneten Lösungsmittel gemäß dem im DPRA DB-ALM-Protokoll (20) beschriebenen Solubilisierungsverfahren bewertet werden. Ein geeignetes Lösungsmittel löst die Prüfchemikalie vollständig auf. Da die Prüfchemikalie beim DPRA mit hohem Überschuss mit den Cystein- oder den Lysinpeptiden inkubiert wird, gilt die visuelle Prüfung auf Bildung einer klaren Lösung als ausreichend, um festzustellen, dass die Prüfchemikalie (und - bei Testung einer mehrkomponentigen Substanz oder eines Gemischs - alle Bestandteile) aufgelöst wurde(n). Geeignete Lösungsmittel sind Acetonitril, Wasser, ein 1:1-Gemisch aus Wasser und Acetonitril, Isopropanol, Aceton oder ein 1:1-Gemisch aus Aceton und Acetonitril. Andere Lösungsmittel können verwendet werden, solange sie die Stabilität des Peptids nicht beeinträchtigen. Dies wird mithilfe von Referenzkontrollen C (d. h. Proben, bei denen das Peptid alleine im jeweiligen Lösungsmittel aufgelöst ist; siehe Anlage 3) überwacht. Ist die Prüfchemikalie in keinem dieser Lösungsmittel löslich, sollte als letzte Möglichkeit versucht werden, sie in 300 µL DMSO zu lösen und die erhaltene Lösung mit 2.700 µL Acetonitril zu verdünnen. Ist die Prüfchemikalie auch in diesem Gemisch unlöslich, sollte versucht werden, sie in gleicher Menge in 1.500 µL DMSO zu lösen und die erhaltene Lösung mit 1.500 µL Acetonitril zu verdünnen. Zum Ansetzen einer 100-mM-Lösung wird die Prüfchemikalie vorgewogen in Glasgefäße gegeben und unmittelbar vor dem Test in einem geeigneten Lösungsmittel aufgelöst. Bei Gemischen und mehrkomponentigen Substanzen mit bekannter Zusammensetzung wird aus der Summe der Anteile der einzelnen Bestandteile (ausgenommen Wasser) ein einziger Wert für die Reinheit und anhand der Molekulargewichte der einzelnen Bestandteile im Gemisch (ausgenommen Wasser) und ihres jeweiligen Anteils ein einziges scheinbares Molekulargewicht bestimmt. Aus den erhaltenen Werten für die Reinheit und das scheinbare Molekulargewicht wird dann das erforderliche Gewicht der Prüfchemikalie für die Zubereitung einer 100-mM-Lösung berechnet. Bei Polymeren, für die sich kein vorherrschendes Molekulargewicht bestimmen lässt, kann das Molekulargewicht des Monomers (oder das scheinbare Molekulargewicht der verschiedenen Monomere, aus denen sich das Polymer zusammensetzt) für die Zubereitung einer 100-mM-Lösung herangezogen werden. Bei der Prüfung von Gemischen, mehrkomponentigen Substanzen oder Polymeren mit bekannter Zusammensetzung sollte jedoch auch in Erwägung gezogen werden, die unverdünnte Chemikalie zu testen. Bei Flüssigkeiten wird die unverdünnte Chemikalie so, wie sie ist, ohne vorherige Verdünnung getestet und in einem molaren Verhältnis von 1:10 und 1:50 mit den Cystein- bzw. Lysinpeptiden inkubiert. Bei festen Stoffen wird die Prüfchemikalie auf ihre höchste lösliche Konzentration in dem gleichen Lösungsmittel aufgelöst, das zum Ansetzen der scheinbaren 100-mM-Lösung verwendet wurde. Anschließend wird sie so, wie sie ist, ohne weitere Verdünnung getestet und in einem molaren Verhältnis von 1:10 und 1:50 mit den Cystein- bzw. Lysinpeptiden inkubiert. Übereinstimmende Ergebnisse (reaktiv oder nicht reaktiv) zwischen der scheinbaren 100-mM-Lösung und der unverdünnten Chemikalie sollten eine verbindliche Schlussfolgerung bezüglich des Ergebnisses zulassen. Vorbereitung der Positivkontrolle, Referenzkontrollen und Koelutionskontrollen Als Posivivkontrolle (PC) sollte Zimtaldehyd (CAS 104-55-2; > 95 % von lebensmitteltauglicher Reinheit) mit einer Konzentration von 100 mM in Acetonitril verwendet werden. Andere geeignete Positivkontrollen, die vorzugsweise Depletionswerte im mittleren Bereich ergeben, können verwendet werden, sofern historische Daten für die Ableitung vergleichbarer Akzeptanzkriterien für einen Testdurchlauf zur Verfügung stehen. Außerdem sollte die HPLC-Sequenz auch Referenzkontrollen (d. h. Proben, die nur das im entsprechenden Lösungsmittel gelöste Peptid enthalten) umfassen. Diese dienen dazu, die Eignung des HPLC-Systems vor der Analyse (Referenzkontrollen A) und die Stabilität der Referenzkontrollen im Zeitverlauf (Referenzkontrollen B) zu verifizieren und zu bestätigen, dass das zur Lösung der Prüfchemikalie verwendete Lösungsmittel die prozentuale Peptid-Depletion nicht beeinflusst (Referenzkontrollen C) (siehe Anlage 3). Mithilfe der geeigneten Referenzkontrolle für jede Chemikalie wird die prozentuale Peptid-Depletion für diese Chemikalie berechnet (siehe Nummer 26). Darüber hinaus sollte für jede analysierte Prüfchemikalie eine Koelutionskontrolle, bestehend aus der Prüfchemikalie alleine, in die Ablaufsequenz aufgenommen werden, um eine mögliche Koelution der Prüfchemikalie mit dem Lysinpeptid bzw. dem Cysteinpeptid zu erkennen. Inkubation der Prüfchemikalie mit den Cystein- und Lysinpeptid-Lösungen Die Cystein- und Lysinpeptid-Lösungen sollten in Autosampler-Glasgefäßen in einem Verhältnis von 1:10 bzw. 1:50 mit der Prüfchemikalie inkubiert werden. Wenn es unmittelbar nach der Zugabe der Prüfchemikalie zur Peptidlösung aufgrund der geringen Wasserlöslichkeit der Prüfchemikalie zu einer Ausfällung kommt, kann nicht mit Sicherheit festgestellt werden, welche Menge der Prüfchemikalie in der Lösung verblieben ist, um mit dem Peptid zu reagieren. In diesem Fall könnte ein positives Ergebnis trotzdem verwendet werden. Ein negatives Ergebnis ist hingegen unsicher und sollte entsprechend vorsichtig interpretiert werden (siehe auch die Vorschriften unter Nummer 11 für die Prüfung von Chemikalien, die bis zu einer Konzentration von 100 mM unlöslich sind). Die Reaktionslösung wird bei 25 ± 2,5 °C 24 ± 2 Stunden vor der Durchführung der HPLC-Analyse im Dunkeln gelassen. Jede Prüfchemikalie wird für beide Peptide in dreifacher Ausfertigung analysiert. Die Proben sind vor der HPLC einer Sichtprüfung zu unterziehen. Bei festgestellter Ausfällung oder Phasentrennung können die Proben als Vorsichtsmaßnahme bei geringer Geschwindigkeit (100-400 x g) zentrifugiert werden, damit die Ausfällung auf den Boden des Gefäßes sinkt, da große Ausfällungsmengen die Leitungen oder Säulen der HPLC-Apparatur verstopfen können. Wird nach der Inkubationszeit eine Ausfällung oder Phasentrennung festgestellt, kann die Peptid-Depletion unterschätzt werden. In diesem Fall kann bei einem negativen Ergebnis nicht mit hinreichender Sicherheit auf das Fehlen von Reaktivität geschlossen werden. Erstellung der HPLC-Standardkalibrierungskurve Sowohl für die Cystein- als auch die Lysinpeptide sollte eine Standardkalibrierungskurve erstellt werden. Die Peptidstandards werden in einer Lösung mit 20 % oder 25 % Acetonitril/Puffer unter Verwendung eines Phosphatpuffers (pH 7,5) für das Cysteinpeptid und eines Ammoniumacetat-Puffers (pH 10,2) für das Lysinpeptid zubereitet. Aus Verdünnungsreihen-Standards der Peptid-Stammlösung (0,667 mM) werden sechs Kalibrierungslösungen hergestellt, die den Bereich von 0,534 bis 0,0167 mM abdecken. Eine Blindkontrolle des Verdünnungspuffers sollte ebenfalls in die Standardkalibrierungskurve aufgenommen werden. Geeignete Kalibrierungskurven sollten einen Wert von r2 > 0,99 aufweisen. Vorbereitung des HPLC-Systems und Analyse Vor der Durchführung der Analyse ist die Eignung des HPLC-Systems zu verifizieren. Die Peptid-Depletion wird durch ein mit einem UV-Detektor (Fotodiodenarray-Detektor oder Festwellenlängen-Absorptionsdektektor mit 220-nm-Signal) verbundenes HPLC-Gerät überwacht. Die entsprechende Säule wird in das HPLC-System eingebaut. Bei dem im validierten Protokoll beschriebenen HPLC-Aufbau wird das Modell Zorbax SB-C-18 2,1 mm x 100 mm x 3,5 µm als bevorzugte S´ule verwendet. Bei dieser Säule für die Umkehrphasen-HPLC sollte das gesamte System mindestens 2 Stunden vor dem Betrieb bei 301 °C mit 50 % Phase A (0,1 % v/v Trichloressigsäure in Wasser) und 50 % Phase B (0,085 % v/v Trichloressigsäure in Acetonitril) äquilibriert werden. Die HPLC-Analyse erfolgt mit einer Flussrate von 0,35 ml/min und einem linearen Gradienten von 10 % bis 25 % Acetonitril über einen Zeitraum von 10 Minuten, gefolgt von einem schnellen Anstieg auf 90 % Acetonitril, um andere Materialien zu entfernen. Standard, Probe und Kontrolle werden jeweils in gleicher Menge injiziert. Zwischen den Injektionen wird die Säule unter den Ausgangsbedingungen 7 Minuten lang neu äquilibriert. Bei Verwendung einer anderen Säule für die Umkehrphasen-HPLC müssen die oben genannten Einstellparameter möglicherweise angepasst werden, um eine angemessene Elution und Integration der Cystein- und Lysinpeptide sicherzustellen. Dies gilt auch für die Injektionsmenge, die je nach verwendetem System variieren kann (normalerweise im Bereich von 3-10 µl). Wichtig ist auch, dass bei der Verwendung eines anderen Aufbaus der HPLC-Apparatur dessen Gleichwertigkeit mit dem vorstehend beschriebenen validierten Aufbau nachgewiesen wird (z.B. durch Prüfung der Leistungsstoffe in Anlage 2). Die Extinktion wird bei 220 nm überwacht. Bei Verwendung eines Fotodiodenarray-Detektors sollte die Extinktion bei 258 nm ebenfalls aufgezeichnet werden. Es wird darauf hingewiesen, dass einige Acetonitril-Chargen die Peptidstabilität beeinträchtigen können. Dies muss bei der Verwendung einer neuen Acetonitril-Charge bewertet werden. Als Indikator für eine Koelution kann das Verhältnis der Peakfläche bei 220 nm und der Peakfläche bei 258 nm verwendet werden. Beispielsweise wäre bei jeder Probe ein Verhältnis im Bereich von 90 % < mittleres 2 Flächenverhältnis der Kontrollproben < 100 % ein guter Anhaltspunkt dafür, dass keine Koelution eingetreten ist. Es gibt unter Umständen Prüfchemikalien, die die Oxidation des Cysteinpeptids fördern könnten. Der Peak des dimerisierten Cysteinpeptids kann visuell überwacht werden. Scheint eine Dimerisierung erfolgt zu sein, sollte dies vermerkt werden, da die prozentuale Peptid-Depletion überschätzt werden kann, was zu falschen Positiv-Vorhersagen und/oder einer Einstufung in eine höhere Reaktivitätsklasse führen kann (siehe Nummer 29 und 30). Die HPLC-Analyse der Cystein- und Lysinpeptide kann gleichzeitig (wenn zwei HPLC-Systeme zur Verfügung stehen) oder an verschiedenen Tagen durchgeführt werden. Erfolgt die Analyse an verschiedenen Tagen, sind alle Prüfchemikalienlösungen für beide Tests am jeweiligen Tag frisch herzustellen. Die Analyse wird zeitlich so angesetzt, dass die Injektion der ersten Probe 22 bis 26 Stunden nach Mischung der Prüfchemikalie mit der Peptidlösung stattfindet. Die HPLC-Sequenz ist so einzustellen, dass die HPLC-Analyse in weniger als 30 Stunden abgeschlossen ist. Bei dem in der Validierungsstudie verwendeten und bei dieser Prüfmethode beschriebenen HPLC-Aufbau können in einem einzigen HPLC-Durchlauf bis zu 26 Analyseproben abgearbeitet werden (siehe auch Nummer 17). Ein Beispiel für eine HPLC-Analysesequenz ist Anlage 3 zu entnehmen. Datenauswertung Die Konzentration des Cystein- oder Lysinpeptids der einzelnen Proben wird bei 220 nm fotometrisch bestimmt. Hierzu wird die Peakfläche (Fläche unter der Kurve (Area Under the Curve, AUC) der entsprechenden Peaks gemessen, und anhand der aus den Standards abgeleiteten linearen Kalibrierungskurve wird die Peptidkonzentration berechnet. Die prozentuale Peptid-Depletion der einzelnen Proben erhält man nach folgender Formel durch Messung der Peakfläche und deren Division durch die mittlere Peakfläche der jeweiligen Referenzkontrollen C (siehe Anlage 3) Akzeptanzkriterien Ein Testdurchlauf gilt als gültig, wenn die folgenden Kriterien erfüllt sind:
Wenn eines oder mehrere dieser Kriterien nicht erfüllt sind, ist der Testdurchlauf zu wiederholen. Die Ergebnisse für eine Prüfchemikalie gelten als gültig, wenn die folgenden Kriterien erfüllt sind:
Vorhersagemodell Für jede Prüfchemikalie wird der Mittelwert der prozentualen Cystein- und Lysin-Depletion berechnet. Eine negative Depletion wird bei der Berechnung des Mittelwerts mit "0" angenommen. Unter Verwendung des Vorhersagemodells Cystein 1:10/Lysin 1:50 in Tabelle 1 wird eine durchschnittliche Peptid-Depletion von 6,38 % als Schwelle zur Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren im Rahmen eines IATA herangezogen. Die Anwendung des Vorhersagemodells für die Einstufung einer Prüfchemikalie in eine Reaktivitätsklasse (d. h. geringe, mittlere und hohe Reaktivität) kann ggf. als nützliche Information für die Potenzbewertung im Rahmen eines IATA dienen. Tabelle 1 Vorhersagemodell Cystein 1:10/Lysin 1:50 1
Es könnte Fälle geben, in denen die Prüfchemikalie (der Stoff oder einer oder mehrere Bestandteile einer mehrkomponentigen Substanz oder eines Gemischs) eine starke Extinktion bei 220 nm aufweist und die gleiche Retentionszeit wie das Peptid besitzt (Koelution). Eine Koelution kann vermieden werden, indem der HPLC-Aufbau so verändert wird, dass die Elutionszeit der Prüfchemikalie und des Peptids weiter auseinander liegen. Wird ein anderer HPLC-Aufbau verwendet, um eine Koelution aufzulösen, so ist seine Gleichwertigkeit mit dem validierten Aufbau nachzuweisen (z.B. durch Prüfung der Leistungsstoffe in Anlage 2). Bei Vorliegen einer Koelution kann der Peak des Peptids nicht integriert und die prozentuale Peptid-Depletion nicht berechnet werden. Koeluieren solche Prüfchemikalien sowohl mit den Cystein- als auch den Lysinpeptiden, ist die Analyse als "nicht aussagekräftig" anzugeben. Liegt eine Koelution nur mit dem Lysinpeptid vor, kann das Vorhersagemodell Cystein 1:10 in Tabelle 2 verwendet werden. Tabelle 2 Vorhersagemodell Cystein 1:10 1
Es könnte andere Fälle geben, in denen sich die Retentionszeiten der Prüfchemikalie und eines der beiden Peptide nicht vollständig überlappen. In solchen Fällen können die Peptid-Depletionswerte geschätzt und im Vorhersagemodell Cystein 1:10/Lysin 1:50 verwendet werden. Eine genaue Einstufung der Prüfchemikalie in eine Reaktivitätsklasse ist jedoch nicht möglich. Bei einem eindeutigen Ergebnis ist eine einzige HPLC-Analyse sowohl für das Cystein- als auch das Lysinpeptid pro Prüfchemikalie ausreichend. Liegen die Ergebnisse jedoch nahe an der Schwelle zur Unterscheidung zwischen positivem und negativem Ergebnis (d. h. handelt es sich um grenzwertige Ergebnisse), sind möglicherweise weitere Tests erforderlich. Fällt der Mittelwert der prozentualen Depletion in den Bereich von 3 % bis 10 % (Vorhersagemodell Cystin 1:10/Lysin 1:50) bzw. die prozentuale Cystein-Depletion in den Bereich von 9 % bis 17 % (Vorhersagemodell Cystin 1:10), sind ein zweiter Testdurchlauf sowie ein dritter Durchlauf (bei abweichenden Ergebnissen zwischen den ersten beiden Durchläufen) in Erwägung zu ziehen. Prüfbericht Der Prüfbericht sollte folgende Angaben enthalten: Prüfchemikalie
Kontrollen
Vorbereitung von Peptiden, Positivkontrolle und Prüfchemikalie
Einstellung des HPLC-Geräts und Analyse
Eignung des Systems
Ablauf der Analyse
Leistungstests
Erörterung der Ergebnisse
Schlussfolgerung (1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS). 5. überarbeitete Auflage, UN New York und Genf, 2013. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html (2) OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. Series on Testing and Assessment, Nr. 168, OECD, Paris. (3) Kapitel B.6: Sensibilisierung der Haut (4) Kapitel B.42: Lokaler Lymphknotentest (5) Kapitel B.50: Hautsensibilisierung: Lokaler Lymphknotentest: DA. (6) Kapitel B.51: Hautsensibilisierung: Lokaler Lymphknotentest BrdU-ELISA (7) Adler et al. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Archives of Toxicology, 85:367-485. (8) Gerberick et al. (2004). Development of a peptide reactivity assay for screening contact allergens. Toxicological Sciences, 81:332-343. (9) Gerberick et al. (2007). Quantification of chemical peptide reactivity for screening contact allergens: A classification tree model approach. Toxicological Sciences, 97:417-427. (10) EC EURL-ECVAM (2013). Recommendation on the Direct Peptide Reactivity Assay (DPRA) for skin sensitisation testing. Verfügbar unter: https://eurl-ecvam.jrc.ec.europa.eu/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-direct-peptide-reactivity-assay-dpra (11) Jaworska et al. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: from theory to practice. Journal of Applied Toxicology, Online-Veröffentlichung, 14. Mai 2013, DOI: 10.1002/jat.2869. (12) Bauch et al. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potential. Regulatory Toxicology and Pharmacology, 63: 489-504. (13) Nukada et al. (2013). Data integration of non-animal tests for the development of a test battery to predict the skin sensitizing potential and potency of chemicals. Toxicology in Vitro, 27:609 618. (14) Ball et al (2011). Evaluating the sensitization potential of surfactants: integrating data from the local lymph node assay, guinea pig maximization test, and in vitro methods in a weight-of-evidence approach. Regulatory Toxicology and Pharmacology, 60:389-400. (15) Landsteiner und Jacobs (1936). Studies on the sensitization of animals with simple chemical compounds. Journal of Experimental Medicine, 64:625-639. (16) Dupuis and Benezra (1982). Allergic contact dermatitis to simple chemicals: a molecular approach. New York und Basel: Marcel Dekker Inc. (17) Lepoittevin et al. (1998). Allergic contact dermatitis: the molecular basis. Springer, Berlin. (18) EC EURL ECVAM (2012). Direct Peptide Reactivity Assay (DPRA) Validation Study Report, 74pp. Verfügbar unter: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations/eurl-ecvam-recommendation-on-the-direct-peptide-reactivity-assay-dpra (19) Natsch et al. (2013). A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. Journal of Applied Toxicology, Online-Veröffentlichung, 9. April 2013, DOI:10.1002/jat.2868. (20) DB-ALM (INVITTOX) Protokoll 154. Direct Peptide Reactivity assay (DPRA) for skin sensitisation testing, 17pp. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/ (21) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Series on Testing and Assessment, Nr. 34. Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris, Frankreich. (22) FDA (Food and Drug Administration) (2001). Guidance for Industry: Bioanalytical Method Validation, 22pp. Verfügbar unter: www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidance/ucm070107.pdf -138 (23) ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology and Toxicology of Chemicals (Technical Report Nr. 87). _____ 2) Unter dem Begriff "Mittelwert" oder "mittlere/r/s" ist im gesamten Dokument das arithmetische Mittel zu verstehen.
Definitionen AOP (Adverse Outcome Pathway): Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den auslösenden molekularen Vorgang bis in zu einem In-vivo-Ergebnis von Interesse (2). Auslösender molekularer Vorgang: Chemisch induzierte Störung eines biologischen Systems auf molekularer Ebene, die als Ausgangspunkt des Adverse Outcome Pathway identifiziert wird. Chemikalie: Stoff oder Gemisch. Eignung des Systems: Feststellung der Leistung (z.B. Empfindlichkeit) eines Instruments durch Analyse eines Referenzstandards vor dem Durchlauf der zu analysierenden Charge (22). Einkomponentige Substanz: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist. Empfindlichkeit: Der Anteil aller positiven/wirkenden Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Empfindlichkeit ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (21). Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen. Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (1). Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (21). Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeitnehmer, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1). Gültige Prüfmethode: Eine Prüfmethode, die eine ausreichende Relevanz und Zuverlässigkeit für einen bestimmten Zweck aufweist und auf wissenschaftlich fundierten Grundsätzen beruht. Eine Prüfmethode ist nie im absoluten Sinn, sondern nur in Bezug auf einen definierten Zweck (21) gültig. IATA (Integrated Approach to Testing and Assessment - Integrierter Test- und Bewertungsansatz): Strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbewertung (Potenzial/Potenz und Exposition) einer Chemikalie oder Chemikaliengruppe, bei dem alle maßgeblichen Daten strategisch integriert und gewichtet werden, um als Grundlage für fundierte regulatorische Entscheidungen über potenzielle Gefahren und/oder Risiken und/oder die Notwendigkeit weiterer gezielter und somit minimaler Testungen herangezogen zu werden. Kalibrierungskurve: Beziehung zwischen dem im Versuch ermittelten Reaktionswert und der Analysekonzentration (auch als Standardkurve bezeichnet) eines bekannten Stoffs. Mehrkomponentige Substanz: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens > 10 % w/w und < 80 % w/w vorhanden sind. Eine mehrkomponentige Substanz ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einer mehrkomponentigen Substanz besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Eine mehrkomponentige Substanz wird durch eine chemische Reaktion gebildet. Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. Prüfchemikalie: Der Begriff "Prüfchemikalie" bezeichnet das, was getestet wird. Referenzkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert. Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Sie berücksichtigt auch die Genauigkeit (Übereinstimmung) einer Prüfmethode (21). Reproduzierbarkeit: Übereinstimmung der Ergebnisse von Tests, die an der gleichen Chemikalie bei einheitlichem Prüfprotokoll durchgeführt werden (siehe Zuverlässigkeit) (21). Spezifität: Der Anteil aller negativen/wirkungslosen Chemikalien, die durch die Prüfmethode korrekt eingestuft werden. Die Spezifität ist ein Maß der Genauigkeit einer Prüfmethode mit kategorialen Ergebnissen und ein wichtiger Aspekt bei der Bewertung ihrer Relevanz (21). Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (1). UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien. Variationskoeffizient: Kennwert der Varianz. Er wird für eine Gruppe von Replikatdaten mittels Division der Standardabweichung durch den Mittelwert berechnet. Multipliziert mit 100 ergibt sich ein Prozentwert. Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Labors über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (21).
Leistungsstoffe In-chemico-Hautsensibilisierung: Direkt-Peptidreaktivitätstest Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Labors ihre technische Kompetenz nachweisen, indem sie die erwartete DPRA-Vorhersage für die zehn in Tabelle 1 empfohlenen Leistungsstoffe richtig treffen und bei acht von zehn Leistungsstoffen für jedes Peptid Werte für die Cystein- und Lysin-Depletion erhalten, die im jeweiligen Referenzbereich liegen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Stoffe im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger In-vitro-Daten aus dem DPRA und dass diese in der vom EURL ECVAM koordinierten Validierungsstudie verwendet wurden, um die erfolgreiche Durchführung der Prüfmethode in den an der Studie beteiligten Labors nachzuweisen. Tabelle 1 Empfohlene Leistungsstoffe für den Nachweis der technischen Kompetenz zur Durchführung des Direkt-Peptidreaktivitätstests
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.60 In-vitro-Hautsensibilisierung: ARE-Nrf2 Luciferase-Prüfmethode 17 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 442D (2015). Ein Hautallergen ist gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (1) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 (CLP) ein Stoff, der bei Hautkontakt eine allergische Reaktion auslöst. Diese Prüfmethode ist ein In-vitro-Verfahren (ARE-Nrf2 Luciferase-Test) zur Unterstützung der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren gemäß UN-GHS (1) und CLP. Es besteht allgemeines Einvernehmen über die der Hautsensibilisierung zugrunde liegenden biologischen Schlüsselvorgänge. Das vorhandene Wissen über die chemischen und biologischen Mechanismen im Zusammenhang mit der Hautsensibilisierung wurde in Form eines Adverse Outcome Pathway (AOP) (2) - vom molekularen auslösenden Vorgang über die dazwischen liegenden Vorgänge bis hin zur schädlichen Auswirkung auf die Gesundheit, z.B. allergische Kontaktdermatitis beim Menschen oder Kontakt-Überempfindlichkeit bei Nagetieren, - zusammengefasst (2) (3). Der molekulare auslösende Vorgang ist die kovalente Bindung von elektrophilen Stoffen an nukleophile Zentren in Hautproteinen. Der zweite Schlüsselvorgang in diesem AOP findet in den Keratinozyten statt und umfasst entzündliche Reaktionen sowie Genexpression in Verbindung mit spezifischen Zellsignalketten wie beispielsweise ARE (Antioxidant/Electrophile Response Element)-abhängigen Ketten. Der dritte Schlüsselvorgang ist die Aktivierung von dendritischen Zellen, die normalerweise durch Expression von spezifischen Zelloberflächenmarkern, Chemokinen und Cytokinen bewertet werden. Der vierte Schlüsselvorgang ist die T-Zellproliferation, die indirekt im Lokalen Lymphknotentest (LLNA) an der Maus (4) bewertet wird. Die Bewertung der Hautsensibilisierung erfolgte normalerweise an Labortieren. Bei den klassischen Methoden an Meerschweinchen, dem Maximierungstest an Meerschweinchen (GMPT) nach Magnusson/Kligman und dem Bühler-Test (TM B.6 (5)), werden die Induktions- und die Auslösephase der Hautsensibilisierung untersucht. Ein Test an der Maus, der Lokale Lymphknotentest (LLNA) (TM B.42 (4)) sowie die beiden nicht radioaktiven Abwandlungen dieses Tests, LLNA: DA (TM B.50 (6)) und LLNA: BrdU-ELISA (TM B.51 (7)), bei denen jeweils nur die Induktionsreaktion bewertet wird, haben ebenfalls an Akzeptanz gewonnen, da sie sowohl in Bezug auf den Tierschutz als auch in Bezug auf die objektive Messung der Induktionsphase der Hautsensibilisierung Vorteile gegenüber Tests am Meerschweinchen bieten. Vor Kurzem wurden mechanistisch basierte In-chemico- und In-vitro-Prüfmethoden für die Bewertung der Gefahr einer Hautsensibilisierung durch Chemikalien als wissenschaftlich fundiert befunden. Allerdings sind Methodenkombinationen ohne Tierversuche (in silico, in chemico, in vitro) im Rahmen von Integrierten Test- und Bewertungsansätzen (Integrated Approaches to Testing and Assessment, IATA) erforderlich, um die gegenwärtig verwendeten Tierversuche angesichts der eingeschränkten mechanistischen AOP-Abdeckung der gegenwärtig verfügbaren Prüfmethoden ohne Tierversuche vollständig zu ersetzen (2) (3). Diese Prüfmethode (ARE-Nrf2 Luciferase-Test) wird für den unter Nummer 2 erläuterten zweiten Schlüsselvorgang vorgeschlagen. Es wurde berichtet, dass Hautallergene Gene induzieren können, die durch das Antioxidant Response Element (ARE) reguliert werden (8) (9). Kleinmolekulare elektrophile Stoffe wie beispielsweise Hautallergene können auf das Sensorprotein Keap1 (Kelch-like ECH-associated protein 1) einwirken, beispielsweise durch kovalente Modifizierung seines Cysteinrests, was zu einer Dissoziation vom Transkriptionsfaktor Nrf2 (nuclear factor-erythroid 2-related factor2) führt. Das dissoziierte Nrf2 kann dann ARE-abhängige Gene wie beispielsweise jene, die Phase-II-Entgiftungsenzyme codieren, aktivieren (8) (10) (11). Gegenwärtig ist der einzige In-vitro-ARE-Nrf2-Luciferase-Test, der durch diese Prüfmethode abgedeckt wird, der KeratinoSensTM-Test, für den Validierungsstudien abgeschlossen wurden (9) (12) (13), denen ein unabhängiger Peer-Review des Europäischen Referenzlabors für Alternativen zu Tierversuchen (European Union Reference Laboratory for Alternatives to Animal Testing, EURL ECVAM) folgte (14). Der KeratinoSensTM-Test wurde als aus wissenschaftlicher Sicht für die Anwendung als Teil eines IATA zulässig befunden, um die Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren im Hinblick auf die Gefahreneinstufung und -kennzeichnung zu unterstützen (14). Labors, die die Prüfmethode durchführen möchten, können die im KeratinoSensTM-Test verwendete rekombinante Zelllinie durch Abschluss einer Lizenzvereinbarung mit dem Entwickler der Prüfmethode erhalten (15). Definitionen sind Anlage 1 zu entnehmen. Vorbemerkungen, Anwendbarkeit und Einsatzgrenzen Da sich die Aktivierung der Keap1-Nrf2-ARE-Kette nur auf den zweiten Schlüsselvorgang des Hautsensibilisierungs-AOP bezieht, reichen Informationen aus Prüfmethoden auf der Grundlage der Aktivierung dieser Kette wahrscheinlich alleine nicht aus, um Schlussfolgerungen über das Hautsensibilisierungspotenzial von Chemikalien zu ziehen. Daher sollten die mit der gegenwärtigen Prüfmethode ermittelten Daten im Rahmen integrierter Ansätze, wie z.B. IATA, betrachtet und mit anderen ergänzenden Informationen, die beispielsweise aus In-vitro-Tests in Bezug auf andere Schlüsselvorgänge des Hautsensibilisierungs-AOP abgeleitet werden, sowie anderen Nicht-Prüfmethoden, einschließlich chemischer Analogien, kombiniert werden. Beispiele für die Verwendung der ARE-Nrf2-Luciferase-Prüfmethode in Kombination mit anderen Informationen werden in der Literatur beschrieben (13) (16) (17) (18) (19). Diese Prüfmethode kann zur Unterstützung der Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kategorie 1) und Nichtsensibilisatoren im Rahmen eines IATA eingesetzt werden. Diese Prüfmethode kann alleine weder zur Einstufung von Hautallergenen in die Unterkategorien 1A und 1B gemäß Definition in UN-GHS/CLP noch zur Vorhersage der Potenz im Rahmen von Sicherheitsbewertungsentscheidungen verwendet werden. Jedoch kann ein positives Ergebnis je nach Rechtsrahmen alleine zur Einstufung einer Chemikalie in die UN-GHS/CLP-Kategorie 1 herangezogen werden. Die Daten aus der Validierungsstudie und den internen Prüfungen im Rahmen des unabhängigen Peer-Review der Prüfmethode haben ergeben, dass der KeratinoSensTM-Test an Labors mit Erfahrung auf dem Gebiet der Zellkultur übertragen werden kann. Der Grad der Reproduzierbarkeit, der bei der Prüfmethode erwartet werden kann, liegt in der Größenordnung von 85 % innerhalb und zwischen Labors (14). Die Genauigkeit (77 % -155/201), Empfindlichkeit (78 % - 71/91) und Spezifität (76 % - 84/110) des KeratinoSensTM-Tests für die Unterscheidung zwischen Hautallergenen (d. h. UN-GHS/CLP-Kat. 1) und Nichtsensibilisatoren wurden unter Berücksichtigung aller Daten, die vom EURL ECVAM für die Bewertung und das Peer-Review der Prüfmethode vorgelegt wurden, im Vergleich zu den LLNA-Ergebnissen ermittelt (14). Diese Zahlen sind vergleichbar mit den Zahlen, die kürzlich basierend auf internen Prüfungen von ungefähr 145 Stoffen veröffentlicht wurden (Genauigkeit 77 %, Empfindlichkeit 79 %, Spezifität 72 %) (13). Beim KeratinoSensTM-Test ist die Wahrscheinlichkeit einer Unterschätzung bei Chemikalien mit geringer bis mittlerer Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1B) größer als bei Chemikalien mit hoher Hautsensibilisierungspotenz (d. h. UN-GHS/CLP-Unterkategorie 1A) (13) (14). Insgesamt deuten diese Informationen auf die Zweckmäßigkeit des KeratinoSensTM-Tests für die Erkennung der Gefahr einer Hautsensibilisierung hin. Jedoch sind die Genauigkeitswerte, die hier für den KeratinoSensTM-Test als eigenständiger Test angegeben werden, lediglich als Anhaltspunkte zu betrachten, da die Prüfmethode in Kombination mit anderen Informationsquellen im Rahmen eines IATA sowie gemäß den Bestimmungen unter Nummer 9 oben betrachtet werden sollte. Darüber hinaus sollte bei der Bewertung von Prüfmethoden zur Hautsensibilisierung ohne Tierversuche beachtet werden, dass der LLNA sowie andere Tierversuche die Situation bei der untersuchten Spezies, d. h. Menschen, nicht vollständig widerspiegeln. Der Begriff "Prüfchemikalie" bezeichnet bei dieser Prüfmethode das, was getestet wird, und bezieht sich nicht auf die Anwendbarkeit der ARE-Nrf2-Luciferase-Prüfmethode hinsichtlich der Prüfung von Stoffen und/oder Gemischen. Auf der Grundlage der gegenwärtig verfügbaren Daten über den KeratinoSensTM-Test wurde nachgewiesen, dass der Test bei Prüfchemikalien, die eine Vielzahl an organischen Funktionsgruppen, Reaktionsmechanismen, Hautsensibilisierungspotenzen (wie in In-vivo-Studien festgestellt) und physikalisch-chemischen Eigenschaften abdecken, anwendbar ist (9) (12) (13) (14). Es wurden zwar hauptsächlich einkomponentige Stoffe getestet, jedoch liegen auch begrenzte Daten über die Prüfung von Gemischen vor (20). Die Prüfmethode ist dennoch für die Prüfung von mehrkomponentigen Stoffen und Gemischen technisch geeignet. Bevor diese Prüfmethode jedoch für die Generierung von Daten für einen bestimmten Regelungszweck verwendet wird, sollte geprüft werden, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefert, und wenn dem so ist, warum. Diese Überlegungen erübrigen sich, sofern die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist. Zudem sollte bei mehrkomponentigen Stoffen oder Gemischen die mögliche Interferenz der zytotoxischen Komponenten mit den beobachteten Reaktionen beachtet werden. Die Prüfmethode ist bei löslichen Prüfchemikalien oder solchen Prüfchemikalien anwendbar, die eine stabile Dispersion (d. h. ein Kolloid oder eine Suspension, worin sich die Prüfchemikalie nicht absetzen oder in anderen Phasen vom Lösungsmittel trennen kann) in Wasser oder DMSO (einschließlich aller technischen Komponenten im Falle der Prüfung eines mehrkomponentigen Stoffs oder Gemischs) bilden. Prüfchemikalien, die diese Bedingungen bei der höchsten erforderlichen Konzentration von 2.000 µM (siehe Nummer 22) nicht erfüllen, können trotzdem bei niedrigeren Konzentrationen geprüft werden. In einem solchen Fall könnten Ergebnisse, die die unter Nummer 39 beschriebenen Bedingungen für die Positivität erfüllen, dennoch unterstützend zur Identifizierung der Prüfchemikalie als Hautallergen herangezogen werden, während ein negatives Ergebnis, das bei Konzentrationen < 1.000 µM erzielt wird, als nicht aussagekräftig zu betrachten ist (siehe Vorhersagemodell unter Nummer 39). Im Allgemeinen wurden Stoffe mit einem LogP-Wert bis 5 erfolgreich geprüft, während extrem hydrophobe Stoffe mit LogP > 7 außerhalb der bekannten Anwendbarkeit der Prüfmethode liegen (14). Für Stoffe mit einem LogP-Wert zwischen 5 und 7 liegen nur beschränkte Informationen vor. Negative Ergebnisse sind mit Vorsicht auszulegen, da Stoffe mit ausschließlicher Reaktivität gegenüber Lysinresten durch die Prüfmethode als negativ erkannt werden können. Außerdem können Prohaptene (d. h. Chemikalien, die beispielsweise über P450-Enzyme aktiviert werden müssen) und Prähaptene (d. h. Chemikalien, die durch Selbstoxidation aktiviert werden) aufgrund der begrenzten Stoffwechselfähigkeit der verwendeten Zelllinie (21) sowie der Versuchsbedingungen, insbesondere bei geringer Oxidationsgeschwindigkeit, zu negativen Ergebnissen führen. Andererseits können Prüfchemikalien, die zwar nicht als Allergene, aber dennoch als chemische Stressoren wirken, zu falschen positiven Ergebnissen führen (14). Zudem lassen sich hochgradig zytotoxische Prüfchemikalien nicht immer zuverlässig bewerten. Schließlich können Prüfchemikalien, die mit dem Luciferase-Enzym interferieren, die Aktivität der Luciferase in zellbasierten Tests stören, was entweder zu scheinbarer Hemmung oder verstärkter Lumineszenz führt (22). Beispielweise wurde berichtet, dass Phytoöstrogenkonzentrationen > 1 µM die Lumineszenzsignale in anderen auf Luciferase basierenden Reporter-Gen-Tests aufgrund der Überaktivierung des Luciferase-Reporter-Gens stören (23). Infolgedessen muss die Luciferase-Expression, die bei hohen Konzentrationen von Phytoöstrogenen oder ähnlichen Chemikalien, die vermutlich eine mit Phytoöstrogen vergleichbare Überaktivierung des Luciferase-Reporter-Gens bewirken, sorgfältig untersucht werden (23). In Fällen, in denen die Nichtanwendbarkeit der Prüfmethode bei anderen spezifischen Kategorien von Prüfchemikalien nachgewiesen werden kann, sollte die Prüfmethode bei diesen spezifischen Kategorien nicht verwendet werden. Abgesehen von der Unterstützung bei der Unterscheidung zwischen Hautallergenen und Nichtsensibilisatoren liefert der KeratinoSensTM-Test auch Konzentrations-/Wirkungs-Informationen, die potenziell zur Bewertung der Sensibilisierungspotenz bei Verwendung in integrierten Ansätzen wie beispielsweise IATA beitragen können (19). Darüber hinaus sind weitere Untersuchungen, vorzugsweise auf der Grundlage verlässlicher Humandaten, notwendig, um herauszufinden, inwieweit die Ergebnisse des KeratinoSensTM-Tests zur Potenzbewertung (24) und Einstufung von Allergenen in Unterkategorien gemäß UN-GHS/CLP beitragen können. Die ARE-Nrf2-Luciferase-Prüfmethode basiert auf einer immortalisierten, adhärenten Zelllinie, die aus humanen und mit einem wählbaren Plasmid stabil transfizierten HaCaT-Keratinozyten abgeleitet wurde. Die Zelllinie enthält das Luciferase-Gen unter der transkriptionalen Kontrolle eines konstitutiven Promoters, verschmolzen mit einem ARE-Element aus einem Gen, das bekanntermaßen durch Kontaktsensibilisatoren hochreguliert wird (25) (26). Das Luciferase-Signal spiegelt die Aktivierung durch Sensibilisatoren der endogenen Nrf2-abhängigen Gene wider, wobei die Abhängigkeit des Luciferase-Signals in der rekombinanten Zelllinie von Nrf2 nachgewiesen wurde (27). Dies ermöglicht die quantitative Messung (durch Lumineszenzerkennung) der Luciferase-Geninduktion unter Verwendung von bekannten lichterzeugenden Luciferase-Substraten als Indikator für die Aktivität des Nrf2-Transkriptionsfaktors in Zellen nach Exposition gegenüber elektrophilen Stoffen. Prüfchemikalien werden im KeratinoSensTM-Test als positiv angesehen, wenn sie eine statistisch signifikante Induktion der Luciferase-Aktivität oberhalb einer gegebenen Schwelle (d. h. > 1,5-facher Wert oder Anstieg um 50 %) bewirken, und zwar unterhalb einer festgelegten Konzentration, die sich nicht signifikant auf die Zellviabilität auswirkt (d. h. unter 1.000 µM und bei einer Konzentration, bei der die Zellviabilität mehr als 70 % beträgt (9) (12)). Zu diesem Zweck wird die maximal-fache Induktion der Luciferase-Aktivität über eine (Negativ-)Lösungsmittel-Kontrolle (Imax) bestimmt. Da die Zellen ferner verschiedenen Konzentrationen der Prüfchemikalien ausgesetzt werden, sollte die erforderliche Konzentration für eine statistisch signifikante Induktion der Luciferase-Aktivität oberhalb der Schwelle (d. h. EC1.5-Wert) aus der Dosis-Wirkungs-Kurve interpoliert werden (für Berechnungen siehe Nummer 32). Schließlich sollten parallele zytotoxische Messungen durchgeführt werden, um zu bewerten, ob die Induktionswerte der Luciferase-Aktivität bei subzytotoxischen Konzentrationen auftreten. Vor der routinemäßigen Anwendung des ARE-Nrf2-Luciferase-Tests, der den Anforderungen der vorliegenden Prüfmethode genügt, sollte die technische Leistungsfähigkeit der Labors anhand der in Anlage 2 aufgeführten zehn Leistungsstoffe nachgewiesen werden. Es stehen Leistungsnormen (28) zur Verfügung, die die Validierung neuer oder geänderter In-vitro-ARE-Nrf2-Luciferase-Prüfmethoden ähnlich dem KeratinoSensTM-Test sowie die rechtzeitige Anpassung dieser Prüfmethoden für deren Einbeziehung ermöglichen. Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird nur für Prüfmethoden garantiert, die gemäß diesen Leistungsnormen validiert wurden, sofern diese Prüfmethoden von der OECD überprüft und in die entsprechende Prüfrichtlinie aufgenommen wurden. Gegenwärtig ist die einzige unter diese Prüfmethode fallende Methode der wissenschaftlich validierte KeratinoSensTM-Test (9) (12) (13) (14). Es liegen Standardarbeitsanweisungen für den KeratinoSensTM-Test vor, die bei der Umsetzung und Verwendung dieser Prüfmethode im Labor angewendet werden sollten (15). Labors, die die Prüfmethode durchführen möchten, können die im KeratinoSensTM-Test verwendete rekombinante Zelllinie durch Abschluss einer Lizenzvereinbarung mit dem Entwickler der Prüfmethode erhalten. Nachfolgend werden die Hauptkomponenten und Verfahren der ARE-Nrf2-Luciferase-Prüfmethode beschrieben. Vorbereitung der Keratinozytenkulturen Es sollte eine transgene Zelllinie mit einer stabilen Insertion des Luciferase-Reporter-Gens unter der Kontrolle des ARE-Elements verwendet werden (z.B. KeratinoSensTM-Zelllinie). Bei Erhalt werden die Zellen propagiert (z.B. 2 bis 4 Passagen) und als homogener Stamm eingefroren aufbewahrt. Zellen aus diesem ursprünglichen Stamm können bis zu einer Höchstzahl von Passagen (z.B. 25 im Fall von KeratinoSensTM) propagiert und für routinemäßige Tests unter Verwendung des geeigneten Trägermediums eingesetzt werden (im Fall von KeratinoSensTM ist dies serum- und Geneticin-haltiges DMEM). Für die Prüfung sollten die Zellen zu 80 bis 90 % konfluent sein, und es sollte darauf geachtet werden, dass die Zellen nicht zu vollständiger Konfluenz zusammengewachsen sind. Einen Tag vor der Prüfung werden die Zellen gewonnen und in 96-Mulden-Platten (10.000 Zellen/Mulde im Fall von KeratinoSensTM) verteilt. Eine Sedimentation der Zellen bei der Beimpfung ist zu vermeiden, um eine homogene quantitative Verteilung der Zellen in den Mulden zu gewährleisten. Ist dies nicht der Fall, kann dieser Schritt zu einer hohen Variabilität zwischen den Mulden führen. Bei jeder Wiederholung werden drei Replikate für die Messungen der Luciferase-Aktivität und ein paralleles Replikat für den Zellviabilitätstest verwendet. Vorbereitung der Prüfchemikalie und Kontrollstoffe Die Prüfchemikalie sowie die Kontrollstoffe werden am Tag der Prüfung vorbereitet. Für den KeratinoSensTM-Test werden die Prüfchemikalien in Dimethylsulfoxid (DMSO) bis zur gewünschten Endkonzentration gelöst (z.B. 200 mM). Die DMSO-Lösungen können als selbststerilisierend angesehen werden, sodass keine sterile Filtration erforderlich ist. Prüfchemikalien, die nicht in DMSO löslich sind, werden in sterilem Wasser oder Kulturmedium gelöst und die Lösungen beispielsweise durch Filtration sterilisiert. Bei einer Prüfchemikalie ohne festgelegtes Molekulargewicht wird im KeratinoSensTM-Test eine Stammlösung mit einer Standardkonzentration hergestellt (40 mg/ml oder 4 % (w/v)). Falls andere Lösungsmittel als DMSO, Wasser oder Kulturmedium zum Einsatz kommen, sollten ausreichende wissenschaftliche Gründe vorgelegt werden. Basierend auf den DMSO-Stammlösungen der Prüfchemikalie werden Verdünnungsreihen unter Verwendung von DMSO hergestellt, um 12 Hauptkonzentrationen der zu prüfenden Chemikalie zu erhalten (von 0,098 bis 200 mM im KeratinoSensTM-Test). Bei einer nicht in DMSO löslichen Prüfchemikalie werden die Verdünnungen zum Erhalt der Hauptkonzentrationen unter Verwendung von sterilem Wasser oder Kulturmedium hergestellt. Unabhängig vom verwendeten Lösungsmittel werden die Hauptkonzentrationen dann weiter 25-fach im serumhaltigen Kulturmedium verdünnt und schließlich für die Behandlung mit einem weiteren 4-fachen Verdünnungsfaktor verwendet, sodass die Endkonzentrationen der geprüften Chemikalie im KeratinoSensTM-Test zwischen 0,98 und 2.000 µM liegen. Andere Konzentrationen kÆnnen verwendet werden, sofern dies gerechtfertigt ist (z.B. bei Zytotoxizität oder schlechter Löslichkeit). Die Negativ-(Lösungsmittel-)Kontrolle, die im KeratinoSensTM-Test verwendet wird, ist DMSO (CAS Nr. 67-68-5, > 99 % Reinheit), wobei sechs Mulden pro Platte vorbereitet werden. Dabei werden die gleichen Verdünnungen wie für die Hauptkonzentrationen unter Nummer 22 hergestellt, sodass die Endkonzentration der Negativ-(Lösungsmittel-)Kontrolle 1 % beträgt. Diese Konzentration wirkt sich bekanntermaßen nicht auf die Zellviabilität aus und entspricht der DMSO-Konzentration in der geprüften Chemikalie und der Positivkontrolle. Bei einer nicht in DSMO löslichen Chemikalie, bei der Verdünnungen in Wasser hergestellt wurden, muss der DMSO-Wert in allen Mulden der endgültigen Prüflösung wie bei den anderen Prüfchemikalien und Kontrollstoffen an 1 % angepasst werden. Die Positivkontrolle, die im Fall des KeratinoSensTM-Tests verwendet ist, ist Zimtaldehyd (CAS Nr. 14371-10-9, > 98 % Reinheit), wobei eine Reihe von 5 Hauptkonzentrationen zwischen 0,4 und 6,4 mM in DMSO (ausgehend von einer Stammlösung mit 6,4 mM) hergestellt und wie für die Hauptkonzentrationen unter Nummer 22 beschrieben verdünnt wird, sodass die Endkonzentrationen der Positivkontrolle zwischen 4 und 64 µM liegen. Andere geeignete Positivkontrollen, vorzugsweise mitEC1,5-Werten im mittleren Bereich, kÆnnen verwendet werden, sofern historische Daten vorliegen, aus denen vergleichbare Versuchsakzeptanzkriterien abgeleitet werden können. Applikation der Prüfchemikalie und Kontrollstoffe Für jede Prüfchemikalie und jeden Positivkontrollstoff ist ein Versuch erforderlich, um eine Vorhersage (positiv oder negativ), bestehend aus mindestens zwei unabhängigen Wiederholungen mit je drei Replikaten (d. h. n = 6), abzuleiten. Weichen die Ergebnisse zwischen den beiden unabhängigen Wiederholungen ab, sollte eine dritte Wiederholung mit drei Replikaten (d. h. n = 9) durchgeführt werden. Jede unabhängige Wiederholung wird an einem anderen Tag mit einer frischen Stammlösung der Prüfchemikalie und unabhängig voneinander gewonnenen Zellen vorgenommen. Die Zellen können jedoch aus derselben Passage stammen. Nach der Beimpfung wie unter Nummer 20 beschrieben lässt man die Zellen 24 Stunden in den 96-Mulden-Mikrotiterplatten wachsen. Das Medium wird dann entfernt und durch frisches Kulturmedium (150 µl serumhaltiges Kulturmedium, jedoch ohne Geneticin im Fall von KeratinoSensTM) ersetzt, dem 50 µl der25-fach verdünnten Prüfchemikalie und Kontrollstoffe zugegeben werden. Mindestens eine Mulde pro Platte sollte zur Bewertung der Background-Werte leer gelassen werden (ohne Zellen und Behandlung). Die behandelten Platten werden im KeratinoSensTM-Test ungefähr 48 Stunden bei 37±1 1 °C in der Gegenwart von 5 % CO2 inkubiert. Es ist darauf zu achten, dass eine Verdunstung der flüchtigen Prüfchemikalien sowie eine Kreuzkontamination zwischen Mulden durch die Prüfchemikalien vermieden wird, indem die Platten beispielsweise vor der Inkubation mit den Prüfchemikalien mit einer Folie abgedeckt werden. Messungen der Luciferase-Aktivität Zur Gewährleistung angemessener Lumineszenz-Messwerte sind drei Faktoren entscheidend:
Vor der Prüfung sollte ein Kontrollversuch wie in Anlage 3 beschrieben durchgeführt werden, um sicherzustellen, dass die drei genannten Bedingungen erfüllt sind. Nach einer Expositionsdauer von 48 Stunden mit der Prüfchemikalie und den Kontrollstoffen im KeratinoSensTM-Test werden die Zellen mit einer phosphatgepufferten Kochsalzlösung gewaschen. Ferner wird jeder Mulde der relevante Lysepuffer für Lumineszenzmessungen 20 Minuten bei Zimmertemperatur hinzugefügt. Die Platten mit dem Zelllysat werden dann zur Messung in das Luminometer gestellt, das im KeratinoSensTM-Test wie folgt programmiert wird: i) Hinzufügen des Luciferase-Substrats zu jeder Mulde (d. h. 50 µl), ii) Abwarten von 1 Sekunde und iii) Integration der Luciferase-Aktivit´t für2 Sekunden. Bei Verwendung anderer Einstellungen, z.B. je nach verwendetem Luminometermodell, sollten diese begründet werden. Darüber hinaus kann auch ein Glimmsubstrat verwendet werden, sofern der in Anlage 3 beschriebene Qualitätssicherungsversuch erfolgreich durchgeführt wird. Bewertung der Zytotoxizität Für den KeratinoSensTM-Zellviabilitätstest wird das Medium nach der Expositionsdauer von 48 Stunden durch frisches Medium ersetzt. Dieses Medium enthält MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolylblau Tetrazoliumbromid; CAS Nr. 298-93-1) und Zellen, die 4 Stunden bei 37 1 °C in der Gegenwart von 5 % CO2 inkubiert wurden. Das MTT-Medium wird dann entfernt und die Zellen werden über Nacht lysiert (z.B. durch Hinzufügen von 10 %iger SDS-Lösung zu jeder Mulde). Nach dem Schütteln wird die Absorption bei 600 nm mit einem Photometer gemessen. Datenauswertung Die folgenden Parameter werden im KeratinoSensTM-Test berechnet:
Dabei sind: LProbe: Lumineszenz-Messwert in der Prüfchemikalien-Mulde LLeer: Lumineszenz-Messwert in der leeren Mulde (ohne Zellen und Behandlung) LLösungsmittel: durchschnittlicher Lumineszenz-Messwert in den Mulden, die Zellen und (Negativ-)Lösungsmittel-Kontrolle enthalten EC1,5 wird durch lineare Interpolation anhand von Gleichung 2 und der EC1,5-Gesamtwert als geometrisches Mittel der einzelnen Wiederholungen berechnet. Dabei sind: Ca: niedrigste Konzentration in µM bei > 1,5-facher Induktion Cb: höchste Konzentration in µM bei < 1,5-facher Induktion Ia: n-fache Induktion, gemessen bei der niedrigsten Konzentration bei > 1,5-facher Induktion (Mittel von drei Replikat-Mulden) Ib: n-fache Induktion, gemessen bei der höchsten Konzentration bei < 1,5-facher Induktion (Mittel von drei Replikat-Mulden) Die Viabilität wird durch Gleichung 3 berechnet:
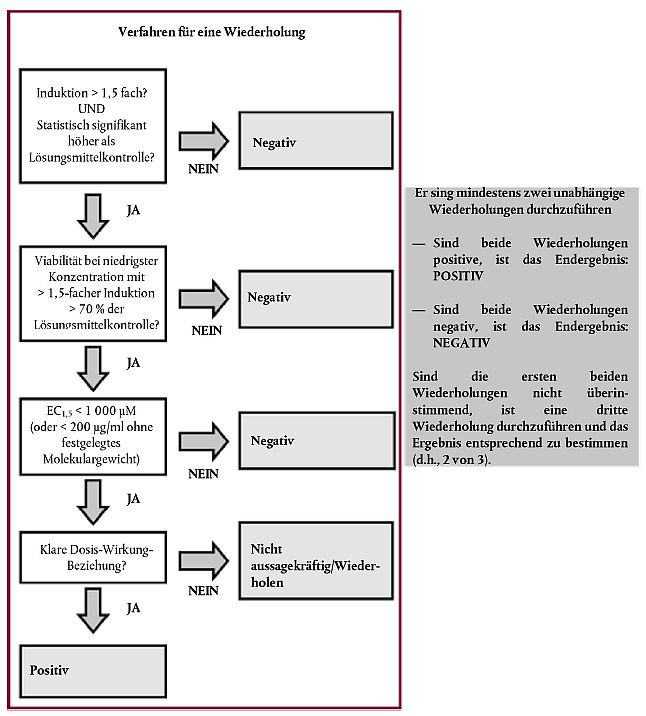
Dabei sind: VProbe: MTT-Absorptions-Messwert in der Prüfchemikalien-Mulde Vleer: MTT-Absorptions-Messwert in der leeren Mulde (ohne Zellen und Behandlung) VLösungsmittel: durchschnittlicher MTT-Absorptions-Messwert in den Mulden, die Zellen und (Negativ-)Lösungsmittel-Kontrolle enthalten IC50 und IC30 werden durch lineare Interpolation anhand von Gleichung 4 und die IC50- und IC30-Gesamtwerte als geometrisches Mittel der einzelnen Wiederholungen berechnet. Dabei sind: X: prozentuale Verringerung bei der zu berechnenden Konzentration (50 und 30 bei IC50 und IC30) Ca: niedrigste Konzentration in µM bei > x % Verringerung der Viabilität Cb: höchste Konzentration in µM bei < x % Verringerung der Viabilität Va: prozentuale Viabilität bei der niedrigsten Konzentration bei > x % Verringerung der Viabilität Vb: prozentuale Viabilität bei der höchsten Konzentration bei < x % Verringerung der Viabilität Für jede Konzentration, bei der die Induktion der Luciferase-Aktivität mehr als das 1,5-fache beträgt, wird die statistische Signifikanz berechnet (z.B. durch einen zweiseitigen Student-t-Test), wobei die Lumineszenzwerte für die drei Replikate mit den Lumineszenzwerten in den Mulden der (Negativ-)Lösungsmittel-Kontrolle verglichen werden, um zu ermitteln, ob die Induktion der Luciferase-Aktivität statistisch signifikant ist (p < 0,05). Die niedrigste Konzentration, bei der die Induktion der Luciferase-Aktivität mehr als das 1,5-fache beträgt, ist der Wert zur Bestimmung des EC1,5-Werts. Es wird in jedem Fall geprüft, ob dieser Wert unter dem IC30-Wert liegt, was darauf hindeutet, dass die Verringerung der Zellviabilität bei der bestimmenden Konzentration für den EC1,5-Wert weniger als 30 % beträgt. Es wird empfohlen, die Daten anhand von Diagrammen visuell zu überprüfen. Wenn keine eindeutige Dosis-Wirkungs-Kurve beobachtet wird oder wenn die ermittelte Dosis-Wirkungs-Kurve pfadspezifisch ist (d. h. die Schwelle von 1,5 zweimal überschritten wird), sollte der Versuch wiederholt werden, um zu prüfen, ob dies für die Prüfchemikalie spezifisch oder auf einen Versuchsartefakt zurückzuführen ist. Falls die biphasige Wirkung in einem unabhängigen Versuch reproduziert werden kann, ist der niedrigere EC1,5-Wert (Konzentration, bei der die Schwelle von 1,5 erstmalig überschritten wird) anzugeben. In den seltenen Fällen, in denen eine statistisch nicht signifikante Induktion über dem 1,5-fachen gefolgt von einer höheren Konzentration mit einer statistisch signifikanten Induktion beobachtet wird, werden die Ergebnisse aus dieser Wiederholung nur dann als gültig und positiv angesehen, wenn die statistisch signifikante Induktion über der Schwelle von 1,5 bei einer nicht zytotoxischen Konzentration ermittelt wurde. Bei Prüfchemikalien, die bereits bei der niedrigsten Testkonzentration von 0,98 µM eine 1,5-fache oder hÆhere Induktion erzeugen, wird der EC1,5-Wert < 0,98 basierend auf der visuellen Überprüfung der Dosis-Wirkungs-Kurve festgelegt. Akzeptanzkriterien Bei der Verwendung des KeratinoSensTM-Tests sollten die folgenden Akzeptanzkriterien erfüllt werden. Erstens sollte die Induktion der Luciferase-Aktivität, die bei der Positivkontrolle (Zimtaldehyd) erzeugt wird, bei mindestens einer der geprüften Konzentrationen (4 bis 64 µM) statistisch signifikant über der Schwelle von 1,5 (z.B. bei Verwendung eines T-Tests) liegen. Zweitens sollte der EC1,5-Wert innerhalb von zwei Standardabweichungen vom historischen Mittelwert der Prüfanstalt liegen (z.B. zwischen 7 µM und 30 µM basierend auf dem Validierungsdatensatz), der regelmäßig aktualisiert werden sollte. Darüber hinaus sollte die durchschnittliche Induktion in den drei Replikaten für Zimtaldehyd bei 64 µM zwischen 2 und 8 liegen. Ist letzteres Kriterium nicht erfüllt, sollte die Dosis-Wirkung von Zimtaldehyd sorgfältig überprüft werden. Tests sind nur dann akzeptabel, wenn die Dosis-Wirkungs-Beziehung eindeutig ist, wobei die Induktion der Luciferase-Aktivität bei zunehmenden Konzentrationen in der Positivkontrolle ansteigt. Schließlich sollte der durchschnittliche Variationskoeffizient des Lumineszenz-Messwerts für die Negativ-(Lösungsmittel-)Kontrolle (DMSO) bei jeder Wiederholung, bestehend aus 6 dreifach geprüften Mulden, unter 20 % liegen. Liegt die Variabilität über diesem Wert, sollten die Ergebnisse ignoriert werden. Interpretation der Ergebnisse und Vorhersagemodell Eine KeratinoSensTM-Vorhersage wird als positiv betrachtet, wenn die folgenden vier Bedingungen alle bei 2 von 2 oder bei denselben 2 von 3 Wiederholungen erfüllt sind. Andernfalls wird die KeratinoSensTM-Vorhersage als negativ erachtet (Abbildung 1):
Wenn bei einer gegebenen Wiederholung die ersten drei Bedingungen erfüllt sind, jedoch keine eindeutige Dosis-Wirkungs-Beziehung für die Induktion der Luciferase-Aktivität beobachtet werden kann, ist das Ergebnis dieser Wiederholung als nicht aussagekräftig anzusehen, sodass eventuell weitere Prüfungen erforderlich sind (Abbildung 1). Zudem ist ein negatives Ergebnis bei Konzentrationen < 1.000 µM (oder < 200 µg/ml bei Prüfchemikalien ohne festgelegtes Molekulargewicht) ebenfalls als nicht aussagekräftig zu betrachten (siehe Nummer 11). Abbildung 1 Vorhersagemodell des KeratinoSensTM-Tests. Eine KeratinoSensTM-Vorhersage sollte im Rahmen eines IATA sowie gemäß den Nummern 9 und 11 in Betracht gezogen werden In seltenen Fällen können Prüfchemikalien, bei denen die Induktion der Luciferase-Aktivität sehr nahe bei zytotoxischen Werten erfolgt, in einigen Wiederholungen bei nicht zytotoxischen Werten (d. h. bestimmende Konzentration für den EC1,5-Wert unter (<) IC30) und in anderen Wiederholungen nur bei zytotoxischen Werten (d. h. bestimmende Konzentration für den EC1,5-Wert größer als (>) IC30) positiv sein. Solche Prüfchemikalien sind erneut im Rahmen einer sehr genauen Dosis-Wirkungs-Analyse unter Verwendung eines geringeren Verdünnungsfaktors (z.B. 1,33 oder Ö2 (= 1,41)-fache Verdünnung zwischen Mulden) zu prüfen, um festzustellen, ob die Induktion bei zytotoxischen Werten aufgetreten ist oder nicht (9). Prüfbericht Der Prüfbericht muss folgende Angaben enthalten: Prüfchemikalie
Kontrollen
Prüfbedingungen
Prüfverfahren
Ergebnisse
Erörterung der Ergebnisse
Schlussfolgerung (1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), fünfte überarbeitete Ausgabe, UN New York und Genf, 2013. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html. (2) OECD (2012). The Adverse Outcome Pathway for Skin Sensitisation Initiated by Covalent Binding to Proteins. Part 1: Scientific Evidence. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 168. OECD, Paris. (3) Adler S., Basketter D., Creton S., Pelkonen O., van Benthem J., Zuang V., Andersen K.E., Angers-Loustau A., Aptula A., Bal-Price A., Benfenati E., Bernauer U., Bessems J., Bois F.Y., Boobis A., Brandon E., Bremer S., Broschard T., Casati S., Coecke S., Corvi R., Cronin M., Daston G., Dekant W., Felter S., Grignard E., Gundert-Remy U., Heinonen T., Kimber I., Kleinjans J., Komulainen H., Kreiling R., Kreysa J., Leite S.B., Loizou G., Maxwell G., Mazzatorta P., Munn S., Pfuhler S., Phrakonkham P., Piersma A., Poth A., Prieto P., Repetto G., Rogiers V., Schoeters G., Schwarz M., Serafimova R., Tähti H., Testai E., van Delft J., van Loveren H., Vinken M., Worth A., Zaldivar J.M. (2011). Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Archives of Toxicology 85, 367-485. (4) Kapitel B.42 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest. (5) Kapitel B.6 dieses Anhangs: Sensibilisierung der Haut. (6) Kapitel B.50 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest: DA. (7) Kapitel B.51 dieses Anhangs: Hautsensibilisierung: Lokaler Lymphknotentest: BrdU-ELISA. (8) Natsch A. (2010). The Nrf2-Keap1-ARE Toxicity Pathway as a Cellular Sensor for Skin Sensitizers-Functional Relevance and Hypothesis on Innate Reactions to Skin Sensitizers. Toxicological Sciences 113, 284-292. (9) Emter R., Ellis G., Natsch A. (2010). Performance of a novel keratinocyte-based reporter cell line to screen skin sensitizers in vitro. Toxicology and Applied Pharmacology 245, 281-290. (10) Dinkova-Kostova A.T., Holtzclaw W.D., Kensler T.W. (2005). The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 18, 1779-1791. (11) Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. (2013). The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 1(1), 45-49. (12) Natsch A., Bauch C., Foertsch L., Gerberick F., Normann K., Hilberer A., Inglis H., Landsiedel R., Onken S., Reuter H., Schepky A., Emter R. (2011). The intra- and inter-laboratory reproducibility and predictivity of the KeratinoSens assay to predict skin sensitizers in vitro: results of a ring-study in five laboratories. Toxicol. In Vitro 25, 733-744. (13) Natsch A., Ryan C.A., Foertsch L., Emter R., Jaworska J., Gerberick G.F., Kern P. (2013). A dataset on 145 chemicals tested in alternative assays for skin sensitization undergoing prevalidation. Journal of Applied Toxicology, 33, 1337-1352. (14) EURL-ECVAM (2014). Recommendation on the KeratinoSensTM assay for skin sensitisation testing, 42 ff. Verfügbar unter: http://ihcp.jrc.ec.europa.eu/our_labs/eurl-ecvam/eurl-ecvam-recommendations/recommendation-keratinosens-skin-sensitisation. (15) DB-ALM (INVITTOX) (2013) Protocol 155: KeratinoSensTM., 17 ff. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/beta/index.cfm/methods AndProtocols/index (16) Natsch A., Emter R., Ellis G. (2009). Filling the concept with data: integrating data from different in vitro and in silico assays on skin sensitizers to explore the battery approach for animal-free skin sensitization testing. Toxicol. Sci. 107, 106-121. (17) Ball N., Cagen S., Carrillo J.C., Certa H., Eigler D., Emter R., Faulhammer F., Garcia C., Graham C., Haux C., Kolle S.N., Kreiling R., Natsch A., Mehling A. (2011). Evaluating the sensitization potential of surfactants: integrating data from the local lymph node assay, guinea pig maximization test, and in vitro methods in a weight-of-evidence approach. Regul. Toxicol. Pharmacol., 60, 389-400. (18) Bauch C., Kolle S.N., Ramirez T., Eltze T., Fabian E., Mehling A., Teubner W., van Ravenzwaay B., Landsiedel R. (2012). Putting the parts together: combining in vitro methods to test for skin sensitizing potentials. Regul. Toxicol. Pharmacol., 63, 489-504. (19) Jaworska J., Dancik Y., Kern P., Gerberick F., Natsch A. (2013). Bayesian integrated testing strategy to assess skin sensitization potency: from theory to practice. J Appl Toxicol. 33, 1353-1364. (20) Andres E., Sa-Rocha V.M., Barrichello C., Haupt T., Ellis G., Natsch A. (2013). The sensitivity of the KeratinoSensTM assay to evaluate plant extracts: A pilot study. in Toxicology in Vitro, 27, 1220-1225. (21) Fabian E., Vogel D., Blatz V., Ramirez T., Kolle S., Eltze T., van Ravenzwaay B., Oesch F., Landsiedel R. (2013). Xenobiotic metabolizin enzyme activities in cells used for testing skin sensitization in vitro. Arch. Toxicol. 87, 1683-1969. (22) Thorne N., Inglese J., Auld D.S. (2010). Illuminating Insights into Firefly Luciferase and Other Bioluminescent Reporters Used in Chemical Biology. Chemistry and Biology 17, 646-657. (23) OECD (2012). BG1Luc Estrogen Receptor Transactivation Test Method for Identifying Estrogen Receptor Agonists and Antagonists. OECD Guidelines for Chemical Testing No. 457. OECD, Paris. (24) ECETOC (2003). Contact sensitization: Classification according to potency. European Centre for Ecotoxicology and Toxicology of Chemicals (Technical Report Nr. 87). (25) Gildea L.A., Ryan C.A., Foertsch L.M., Kennedy J.M., Dearman R.J., Kimber I., Gerberick G.F. (2006). Identification of gene expression changes induced by chemical allergens in dendritic cells: opportunities for skin sensitization testing. J. Invest. Dermatol., 126, 1813-1822. (26) Ryan C.A., Gildea L.A., Hulette B.C., Dearman R.J., Kimber I., Gerberick G.F. (2004). Gene expressing changes in peripheral blood-derived dendritic cells following exposure to a contact allergen. Toxicol. Lett. 150, 301-316. (27) Emter R., van der Veen J.W., Adamson G., Ezendam J., van Loveren H., Natsch A. (2013). Gene expression changes induced by skin sensitizers in the KeratinoSensTM cell line: Discriminating Nrf2-dependent and Nrf2-independent events. Toxicol. in vitro 27, 2225-2232. (28) OECD (2015). Performance Standards for assessment of proposed similar or modified in vitro skin sensitisation ARE-Nrf2 luciferase test methods. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 213, OECD, Paris. (29) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. OECD Environment, Health and Safety publications, Series on Testing and Assessment No. 34. OECD, Paris, Frankreich. (30) NAFTA (North American Free Trade Agreement) (2012). Technical Working Group on Pesticides - (Quantitative) Structure Activity Relationship ((Q)SAR) Guidance Document. 186 S. http://www.epa.gov/oppfead1/international/naftatwg/guidance/qsar-guidance.pdf _____
Definitionen AOP (Adverse Outcome Pathway): Abfolge von Vorgängen, ausgehend von der chemischen Struktur einer Zielchemikalie oder Zielgruppe ähnlicher Chemikalien, über den molekularen auslösenden Vorgang bis hin zu einem In-vivo-Ergebnis von Interesse (2). ARE: Antioxidant Response Element (auch als EpRE (elektrophile Response Element) bezeichnet), ist ein Response-Element, das in der vorgelagerten Promoterregion vieler zytoprotektiven und Phase-II-Gene anzutreffen ist. Bei Aktivierung durch Nfr2 steuert es die transkriptionelle Induktion dieser Gene. Chemikalie: Stoff oder Gemisch. EC1,5: Interpolierte Konzentration bei einer 1,5-fachen Induktion der Luciferase-Aktivität. Einkomponentiger Stoff: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem ein Hauptbestandteil in einer Konzentration von mindestens 80 % w/w vorhanden ist. Empfindlichkeit: Anteil aller positiven/aktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (29). Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen. Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (1). Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (29). Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (1). Gültige Prüfmethode: Eine Prüfmethode, die eine ausreichende Relevanz und Zuverlässigkeit für einen bestimmten Zweck aufweist und auf wissenschaftlich fundierten Grundsätzen beruht. Eine Prüfmethode ist niemals gültig im absoluten Sinn, sondern nur in Bezug auf einen festgelegten Zweck (29). IATA (Integrated Approach to Testing and Assessment - Integrierter Test- und Bewertungsansatz): Strukturierter Ansatz zur Gefahrenidentifizierung (Potenzial), Gefahrencharakterisierung (Potenz) und/oder Sicherheitsbewertung (Potenzial/Potenz und Exposition) einer Chemikalie oder Chemikaliengruppe, bei dem alle maßgeblichen Daten strategisch integriert und gewichtet werden, um als Grundlage für fundierte regulatorische Entscheidungen über potenzielle Gefahren und/oder Risiken und/oder die Notwendigkeit weiterer gezielter und somit minimaler Testungen herangezogen zu werden. IC30: Konzentration, die eine Verringerung der Zellviabilität um 30 % bewirkt. IC50: Konzentration, die eine Verringerung der Zellviabilität um 50 % bewirkt. Imax: Maximaler Induktionsfaktor der Luciferase-Aktivität im Vergleich zur (Negativ-)Lösungsmittel-Kontrolle bei jeder Prüfchemikalienkonzentration. Keap1: Kelch-like ECH-associated protein 1 ist ein Sensorprotein, durch das die Nrf2-Aktivität reguliert werden kann. Unter nicht induzierten Bedingungen wirkt das Keap1-Sensorprotein auf den Transkriptionsfaktor Nrf2 für eine Ubiquitinylierung und den proteolytischen Abbau im Proteasom ein. Die kovalente Modifizierung der reaktiven Cysteinreste von Keap1 durch kleine Moleküle kann zur Nrf2-Dissoziation von Keap1 führen (8) (10) (11)). Lösungsmittel-/Vehikelkontrolle: Replikat, das alle Komponenten eines Testsystems, einschließlich des verwendeten Lösungsmittels, jedoch außer der Prüfchemikalie, enthält. Dies wird verwendet, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel aufgelöst wurden, zu bestimmen. Mehrkomponentiger Stoff: Ein nach seiner quantitativen Zusammensetzung definierter Stoff, bei dem mehr als ein Hauptbestandteil in einer Konzentration von mindestens > 10 % w/w und < 80 % w/w vorhanden sind. Ein mehrkomponentiger Stoff ist das Ergebnis eines Herstellungsprozesses. Der Unterschied zwischen einem Gemisch und einem mehrkomponentigen Stoff besteht darin, dass ein Gemisch durch die Mischung von zwei oder mehr Stoffen ohne chemische Reaktion entsteht. Ein mehrkomponentiger Stoff wird durch eine chemische Reaktion gebildet. Nrf2: Nuclear factor (erythroid-derived 2)-like 2 ist ein Transkriptionsfaktor, der am Antioxidant Response Pathway beteiligt ist. Wenn Nrf2 nicht ubiquitinyliert ist, baut es sich im Zytoplasma auf und transloziert in den Kern, wo es sich mit dem ARE in der vorgelagerten Promoterregion vieler zytoprotektiven Gene vereint, wobei deren Transkription initiiert wird (8) (10) (11). Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einem Stoff behandelt wird, der bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. Prüfchemikalie: Der Begriff "Prüfchemikalie" bezeichnet das, was getestet wird. Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Sie beinhaltet die Prüfung der Genauigkeit (Übereinstimmung) einer Prüfmethode (29). Reproduzierbarkeit: Übereinstimmung zwischen den Ergebnissen, die bei Prüfungen der gleichen Chemikalie bei einheitlichem Protokoll erzielt werden (siehe Zuverlässigkeit) (29). Spezifität: Anteil aller negativen/inaktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (29). Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (1). UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien. Variationskoeffizient: Kennwert der Varianz. Er wird für eine Gruppe replizierter Daten mittels Division der Standardabweichung durch den Mittelwert berechnet. Multipliziert mit 100 ergibt sich ein Prozentwert. Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet (29).
Leistungsstoffe In-vitro-Hautsensibilisierung: ARE-Nrf2 Luciferase-Prüfmethode Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Labors ihre technische Leistungsfähigkeit demonstrieren, indem sie die erwartete KeratinoSensTM-Vorhersage für die in Tabelle 1 empfohlenen 10 Leistungsstoffe korrekt ermitteln und die EC1,5- und IC50-Werte, die in den betreffenden Referenzbereich fallen, bei mindestens 8 der 10 Leistungsstoffe bestimmen. Diese Leistungsstoffe wurden so ausgewählt, dass sie die Bandbreite von Reaktionen im Hinblick auf die Gefahr einer Hautsensibilisierung repräsentieren. Weitere Auswahlkriterien waren kommerzielle Verfügbarkeit, Verfügbarkeit hochwertiger In-vivo-Referenzdaten und Verfügbarkeit hochwertiger In-vitro-Daten aus dem KeratinoSensTM-Test. Tabelle 1 Empfohlene Stoffe zum Nachweis der technischen Leistungsfähigkeit beim KeratinoSensTM-Test
Grundlagenversuch zur Gewährleistung optimaler Lumineszenzmessungen im KeratinoSensTM-Test Die drei folgenden Parameter sind von maßgeblicher Bedeutung, um zuverlässige Ergebnisse beim Luminometer zu erhalten:
Vor der Prüfung sollte durch Prüfung eines Kontroll-Plattenaufbaus, wie unten beschrieben, sichergestellt werden, dass geeignete Lumineszenzmessungen durchgeführt werden können (Dreifachanalyse). Plattenaufbau des ersten Kontrollversuchs
EGDMA= Ethylenglykoldimethacrylat (CAS Nr.: 97-90-5), eine stark induzierende Chemikalie CA= Zimtaldehyd, positive Referenz (CAS Nr.: 104-55-2) Bei der Qualitätskontrollanalyse sollte Folgendes nachgewiesen werden:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.61 Fluorescein-Leckage-Prüfmethode zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung 17 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 460 (2012). Bei der Fluorescein-Leckage-Prüfmethode handelt es sich um eine In-vitro-Prüfmethode, die unter bestimmten Umständen und mit bestimmten Einschränkungen zur Einstufung von Chemikalien (Stoffen und Gemischen) als Stoffe mit augenverätzender und stark augenreizender Wirkung gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN-GHS) (Kategorie 1), in der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 (CLP) (Kategorie 1) und durch die US-Umweltschutzbehörde (EPA) (Kategorie I) eingesetzt werden kann (1) (2). Im Sinne dieser Prüfmethode sind Stoffe mit stark augenreizender Wirkung als Chemikalien definiert, die nach Applikation eine Gewebeschädigung im Auge verursachen, die nicht innerhalb von 21 Tagen ausheilt, oder eine massive Verschlechterung des Sehvermögens auslösen, während Stoffe mit augenverätzender Wirkung als Chemikalien definiert sind, die eine irreversible Gewebeschädigung im Auge verursachen. Diese Chemikalien werden in UN-GHS-Kategorie 1, EU-CLP-Kategorie 1 oder US-EPA-Kategorie I eingestuft. Obwohl die FL-Prüfmethode den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird sie als Teil einer gestuften Prüfstrategie zu gesetzgeberischen Einstufungs- und Kennzeichnungszwecken empfohlen. Somit wird die FL-Prüfmethode als erster Schritt innerhalb eines Top-Down-Ansatzes zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung, insbesondere bei bestimmten Arten von Chemikalien (z.B. wasserlösliche Stoffe und Gemische), empfohlen (3) (4). Gegenwärtig herrscht allgemeines Einvernehmen darüber, dass der In-vivo-Augentest (TM B.5 (5)) in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Reizungen für verschiedene Chemikalienklassen vorherzusagen. Allerdings kann der In-vivo-Augentest eventuell durch strategische Kombination verschiedener alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie ersetzt werden (4). Der Top-Down-Ansatz (4) sollte zum Einsatz kommen, wenn die vorliegenden Informationen darauf schließen lassen, dass eine Chemikalie ein hohes Reizungspotenzial besitzt. Basierend auf dem Vorhersagemodell unter Nummer 35 können durch die FL-Prüfmethode Chemikalien innerhalb eines begrenzten Anwendungsbereichs ohne weitere Testung als Stoffe mit augenverätzender oder stark augenreizender Wirkung (UN-GHS Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) identifiziert werden. Gleiches wird für Gemische angenommen, obwohl Gemische bei der Validierung nicht verwendet wurden. Daher kann mithilfe der FL-Prüfmethode die augenverätzende/-reizende Wirkung von Chemikalien entsprechend der sequenziellen Prüfstrategie TM B.5 bestimmt werden (5). Jedoch müsste eine Chemikalie, die nach der FL-Prüfmethode nicht als Stoff mit augenverätzender oder stark augenreizender Wirkung vorhergesagt wurde, mit einer oder mehreren weiteren Prüfmethoden (in vitro und/oder in vivo) geprüft werden, mit denen folgende Chemikalien identifiziert werden können: i) Chemikalien, die nach der FL-Prüfmethode in vitro falsch-negative Stoffe mit augenverätzender/-reizender Wirkung sind (UN-GHS Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I); ii) Chemikalien, die nicht als augenverätzend/-reizend eingestuft sind (UN-GHS "Keine Einstufung", EU-CLP "Keine Einstufung", US-EPA-Kategorie IV); und/oder iii) Chemikalien, die als Stoffe mit leichter/mäßiger augenreizender Wirkung eingestuft sind (UN-GHS-Kategorien 2A und 2B, EU-CLP-Kategorie 2, US-EPA-Kategorien II und III). Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung der potenziellen augenverätzenden oder -reizenden Wirkung einer Prüfchemikalie, gemessen als ihre Fähigkeit, an einem impermeablen, konfluenten epithelialen Monolayer eine Schädigung hervorzurufen. Die Integrität der transepithelialen Permeabilität ist eine wichtige Funktion eines Epithels, das beispielsweise in der Bindehaut und Hornhaut zu finden ist. Die transepitheliale Permeabilität wird durch verschiedene undurchlässige Verbindungen (Tight Junctions) kontrolliert. Es wurde nachgewiesen, dass eine Zunahme der Permeabilität des Hornhautepithels in vivo mit der Entzündungsaktivität und Oberflächenschädigung, die mit fortschreitender Augenreizung zu beobachten sind, im Zusammenhang steht. In der FL-Prüfmethode werden die toxischen Wirkungen nach kurzer Exposition gegenüber der Prüfchemikalie anhand einer Zunahme der Permeabilität für Fluorescein-Natrium durch das epitheliale Monolayer von Madin-Darby Canine Kidney (MDCK)-Zellen, die auf permeablen Inserts kultiviert werden, bestimmt. Die auftretende Fluorescein-Leckage ist proportional zu der von der Chemikalie induzierten Schädigung an den Tight Junctions, desmosomalen Verbindungen und Zellmembranen. Anhand der Leckagemenge kann das augentoxische Potenzial einer Prüfchemikalie geschätzt werden. Anlage 1 zeigt ein Diagramm von auf einer Insertmembran kultivierten Zellen für die FL-Prüfmethode. Definitionen sind Anlage 2 zu entnehmen. Vorbemerkungen und Einsatzgrenzen Diese Prüfmethode basiert auf dem INVITTOX-Protokoll Nr. 71 (6), das in einer internationalen Validierungsstudie des Europäischen Zentrums zur Validierung alternativer Methoden (ECVAM) in Zusammenarbeit mit dem US-amerikanischen Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) und dem Japanese center for the Validation of Alternative Methods (JaCVAM) bewertet wurde. Die FL-Prüfmethode wird weder zur Identifizierung von Chemikalien empfohlen, die als Chemikalien mit leichter/mäßiger Reizwirkung eingestuft werden sollten, noch von Chemikalien, die nicht als augenreizend eingestuft werden sollten (Stoffe und Gemische) (d. h. GHS-Kat. 2A/2B, "keine Einstufung"; EU-CLP-Kat. 2, "keine Einstufung"; US-EPA-Kat. II/III/IV), wie durch die Validierungsstudie nachgewiesen (3) (7). Die Prüfmethode ist nur an wasserlöslichen Chemikalien (Stoffen und Gemischen) anwendbar. Durch die FL-Prüfmethode wird das stark augenreizende Potenzial von Chemikalien, die wasserlöslich sind und/oder bei denen die toxische Wirkung nicht durch Verdünnung verändert wird, in der Regel exakt vorhergesagt (7). Um eine Chemikalie als wasserlöslich unter Versuchsbedingungen einzustufen, sollte sie in einer sterilen calciumhaltigen (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien Hanks-Salzlösung (Hanks' Balanced Salt Solution, HBSS) mit einer Konzentration von > 250 mg/ml löslich sein (eine Dosis über der Schwelle von 100 mg/ml). Ist die Prüfchemikalie jedoch unter der Konzentration 100 mg/ml löslich, bewirkt aber bereits eine FL-Induktion von 20 % bei dieser Konzentration (was bedeutet: FL20 < 100 mg/ml), kann sie dennoch in GHS-Kat. 1 oder EPA-Kat. I eingestuft werden. Aufgrund der anerkannten Einsatzgrenzen dieser Prüfmethode sind starke Säuren und Basen, Zellfixierlösungen und stark flüchtige Chemikalien aus dem Anwendungsbereich ausgeschlossen. Diese Chemikalien weisen Mechanismen auf, die von der FL-Prüfmethode nicht gemessen werden, z.B. übermäßige Koagulation, Verseifung oder spezifische chemische Reaktionen. Andere anerkannte Einsatzgrenzen dieser Methode beruhen auf den Ergebnissen der Vorhersagefähigkeit bei gefärbten und viskosen Prüfchemikalien (7). Es ist zu beachten, dass beide Arten von Chemikalien nach kurzer Expositionsdauer schwer aus dem Monolayer zu entfernen sind und dass die Vorhersagefähigkeit der Prüfmethode verbessert werden könnte, wenn eine größere Anzahl von Spülschritten verwendet wird. Feste, in Flüssigkeit suspendierte Chemikalien neigen zu Ausfällung und die Endkonzentration, der die Zellen ausgesetzt sind, kann schwer zu bestimmen sein. Durch Ausschluss von Chemikalien dieser chemischen und physikalischen Klassen aus der Datenbank wird die Genauigkeit der FL-Prüfmethode bei den EU-, EPA- und GHS-Klassifizierungssystemen erheblich verbessert (7). Basierend auf dem Zweck dieser Prüfmethode (Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung) sind Falsch-Negativ-Raten (siehe Nummer 13) unkritisch, da solche Chemikalien anschließend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequenziellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz (weight-of-evidence approach) (5) im Rahmen anderer angemessen validierter In-vitro-Tests oder an Kaninchen geprüft werden (siehe auch Nummern 3 und 4). Andere anerkannte Einsatzgrenzen der FL-Prüfmethode beruhen auf Falsch-Negativ- und Falsch-Positiv-Raten. Bei Einsatz als erster Schritt innerhalb eines Top-Down-Ansatzes zur Identifizierung von wasserlöslichen Stoffen und Gemischen mit augenverätzender oder stark augenreizender Wirkung (UN-GHS-Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) lag die Falsch-Positiv-Rate der FL-Prüfmethode im Bereich von 7 % (7/103; UN-GHS und EU-CLP) bis 9 % (9/99; US-EPA) und die Falsch-Negativ-Rate im Bereich von 54 % (15/28; US-EPA) bis 56 % (27/48; UN-GHS und EU-CLP) im Vergleich zu den In-vivo-Ergebnissen. Chemikaliengruppen, die bei der FL-Prüfmethode zu Falsch-Positiv- und/oder Falsch-Negativ-Ergebnissen führen, werden hier nicht definiert. Einige technische Einsatzgrenzen sind für die MDCK-Zellkultur spezifisch. Die Tight Junctions, die die Passage des Fluorescein-Natrium-Farbstoffs durch den Monolayer hemmen, werden mit steigender Zellpassagenzahl zunehmend beschädigt. Die unvollständige Ausbildung der Tight Junctions führt zu einer verstärkten Fluorescein-Leckage in der unbehandelten Kontrolle. Daher muss eine zulässige maximale Leckage in den unbehandelten Kontrollen festgelegt werden (siehe Nummer 38: 0 % Leckage). Wie bei allen In-vitro-Tests besteht die potenzielle Möglichkeit, dass die Zellen im Laufe der Zeit eine Transformation durchlaufen, sodass die Passagenzahlbereiche für die Tests unbedingt angegeben werden müssen. Der gegenwärtige Anwendungsbereich könnte in einigen Fällen ausgedehnt werden, aber erst nach Analyse eines erweiterten Datenbestands von untersuchten Prüfchemikalien, der vorzugsweise durch Testung zusammengetragen wurde (3). Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Laboratorien, die diese Prüfmethode erstmals anwenden, sollen die in Anlage 3 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung der FL-Prüfmethode nachzuweisen, bevor sie FL-Testdaten zum Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen. Die FL-Prüfmethode ist ein zellbasierter In-vitro-Zytotoxizitätstest an einem konfluenten Monolayer von tubulären MDCK CB997-Epithelzellen, die auf semipermeablen Inserts kultiviert wurden und den nicht proliferierenden Zustand des In-vivo-Hornhautepithels abbilden. Die MDCK-Zelllinie ist gründlich erprobt und bildet Tight Junctions und desmosomale Verbindungen, die mit denjenigen auf der apikalen Seite des Bindehaut- und Hornhautepithels vergleichbar sind. Die Tight Junctions und desmosomalen Verbindungen verhindern in vivo, dass gelöste Stoffe und Fremdstoffe das Hornhautepithel durchdringen. Der Verlust der transepithelialen Impermeabilität aufgrund beschädigter Tight Junctions und desmosomaler Verbindungen ist eines der ersten Anzeichen einer chemikalieninduzierten Augenreizung. Die Prüfchemikalie wird auf den konfluenten Zellenlayer auf der apikalen Seite des Inserts appliziert. Eine kurze Expositionsdauer von einer Minute wird routinemäßig angewandt, um die normale Ausscheidungsrate bei Exposition am Menschen widerzuspiegeln. Ein Vorteil der kurzen Expositionsdauer ist, dass wasserbasierte Stoffe und Gemische unverdünnt getestet werden können, falls sie nach der Expositionsdauer einfach zu entfernen sind. Dies ermöglicht einen direkteren Vergleich der Ergebnisse mit den Wirkungen der Chemikalie beim Menschen. Die Prüfchemikalie wird dann entfernt und der nicht toxische, stark fluoreszierende Fluorescein-Natrium-Farbstoff wird 30 Minuten lang auf die apikale Seite des Monolayers appliziert. Die von der Prüfchemikalie hervorgerufene Schädigung an den Tight Junctions wird anhand der Fluorescein-Menge bestimmt, die während einer festgelegten Zeitdauer durch die Zellschicht dringt. Die Menge an Fluorescein-Natrium-Farbstoff, die durch den Monolayer und die Insertmembran in eine festgelegte Lösungsmenge in der Mulde (in die der Fluorescein-Natrium-Farbstoff durchtritt) gelangt, wird durch spektrofluorometrische Messung der Fluorescein-Konzentration in der Mulde bestimmt. Die Menge der Fluorescein-Leckage (FL) wird bezogen auf die gemessene Fluoreszenzintensitität (FI) aus zwei Kontrollen berechnet: einer Blindkontrolle und einer Maximal-Leckage-Kontrolle. Der Leckageprozentsatz und somit das Ausmaß der Schädigung an den Tight Junctions wird bezogen auf diese Kontrollen für jede der festgelegten Konzentrationen der Prüfchemikalie bestimmt. Dann wird der FL20-Wert (d. h. die Konzentration, bei der es zu 20 % FL relativ zu dem erfassten Wert für den unbehandelten konfluenten Monolayer und Inserts ohne Zellen kommt) berechnet. Der FL20-Wert (mg/ml) wird im Vorhersagemodell zur Identifizierung von Stoffen mit augenverätzender und schwer augenreizender Wirkung verwendet (siehe Nummer 35). Der Erholungsfaktor ist ein wichtiger Teil des Toxizitätsprofils einer Prüfchemikalie, der auch im In-vivo-Augenreizungstest bewertet wird. Vorläufige Analysen haben ergeben, dass Daten zur Erholung (bis zu 72 h nach Exposition gegenüber der Chemikalie) die Vorhersagekapazität des INVITTOX-Protokolls 71 potenziell verbessern könnten, jedoch ist eine weitergehende Bewertung erforderlich, bei der zusätzliche, vorzugsweise aus weiteren Prüfungen ermittelte Daten von Vorteil wären (6). Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Vorbereitung des Zellmonolayers Das MDCK CB997-Zellmonolayer wird unter Verwendung von subkonfluenten Zellen hergestellt, die in Zellkulturflaschen in DMEM/F12-Nährstoffmischung (1x-Konzentrat mit L-Glutamin, 15 mM HEPES, Calcium (mit einer Konzentration von 1,0-1,8 mM) und 10 % hitzeinaktiviertem FCS/FBS) kultiviert wurden. Wichtig ist, dass alle Medien/Lösungen, die im FL-Test verwendet werden, Calcium mit einer Konzentration zwischen 1,8 mM (200 mg/l) und 1,0 mM (111 mg/l) enthalten, um die Bildung und Integrität der Tight Junctions sicherzustellen. Die Anzahl der Zellpassagen sollte Gegenstand einer Kontrolle sein, damit eine gleichmäßige und reproduzierbare Bildung der Tight Junctions gewährleistet ist. Vorzugsweise sollten Zellen verwendet werden, die nach 3-30 Passagen ab dem Auftauen gewonnen wurden, da Zellen in diesem Bereich eine ähnliche Funktionalität aufweisen, was zur Reproduzierbarkeit der Testergebnisse beiträgt. Vor Durchführung der FL-Prüfmethode werden die Zellen durch Trypsinisation aus der Flasche entnommen und zentrifugiert. Eine geeignete Menge an Zellen wird in die Inserts von 24-Mulden-Platten ausgesät (siehe Anlage 1). Zum Aussäen der Zellen sollten Inserts (12 mm Durchmesser) mit einer Zellulosemischester-Membran, einer Dicke von 80-150 µm und einer Porengröße von 0,45 µm verwendet werden. In der Validierungsstudie wurden Millicell-HA-Inserts (12 mm) verwendet. Die Eigenschaften des Insert- und Membrantyps sind wichtig, da sich diese auf das Zellwachstum und die Bindung der Chemikalie auswirken. Bestimmte Arten von Chemikalien können eine Bindung mit der Millicell-HA-Insertmembran eingehen, was die Interpretation der Ergebnisse beeinflussen könnte. Es sollten Leistungschemikalien (siehe Anlage 3) verwendet werden, um die Gleichwertigkeit bei Verwendung anderer Membrane nachzuweisen. Die Bindung der Chemikalie an die Insertmembran ist bei kationischen Chemikalien, wie beispielsweise Benzalkoniumchlorid, die von der geladenen Membran angezogen werden, häufiger anzutreffen (7). Durch die Bindung der Chemikalie an die Insertmembran kann sich die Dauer der Exposition gegenüber der Chemikalie verlängern, was zu einer Überschätzung des toxischen Potenzials der Chemikalie führt; andererseits kann sich aber auch die Leckage von Fluorescein durch das Insert physikalisch bedingt verringern, da das Färbemittel an die kationische, an die Insertmembran gebundene Chemikalie gebunden wird, was wiederum zu einer Unterschätzung des toxischen Potenzials der Chemikalie führt. Dies lässt sich überwachen, indem nur die Membran der höchsten Konzentration der geprüften Chemikalie ausgesetzt und dann Fluorescein-Natrium-Farbstoff bei normaler Konzentration während der Standarddauer beigegeben wird (ohne Zellkontrolle). Wenn der Fluorescein-Natrium-Farbstoff gebunden wird, hat die Insertmembran nach dem Abspülen des Prüfmaterials die Farbe Gelb. Daher müssen die Bindungseigenschaften der Prüfchemikalie unbedingt bekannt sein, damit die Wirkung der Chemikalie auf die Zellen ausgewertet werden kann. Durch die Beimpfung der Inserts mit den Zellen sollte zum Zeitpunkt der Exposition gegenüber der Chemikalie ein konfluenter Monolayer gewonnen worden sein. 1,6 x 105 Zellen sollten pro Insert beigegeben werden (400 µl einer Zellsuspension mit einer Dichte von 4 x 105 Zellen/ml). Unter diesen Umständen wird ein konfluenter Monolayer in der Regel nach 96 Stunden in Kultur gewonnen. Die Inserts sollten vor der Beimpfung sichtgeprüft werden, um sicherzustellen, dass etwaige Schäden, die bei der unter Nummer 30 beschriebenen visuellen Kontrolle erfasst werden, der Handhabung zuzuschreiben sind. Die MDCK-Zellkulturen sollten in Inkubatoren in einer befeuchteten Atmosphäre bei 5 % ± 1 % CO2 und 37 ± 1 °C kultiviert werden. Die Zellen sollten nicht durch Bakterien, Viren, Mycoplasmen oder Pilze kontaminiert sein. Applikation der Prüf- und Kontrollchemikalien Für jede Versuchsdurchführung sollte eine frische Stammlösung der Prüfchemikalie hergestellt und nach der Zubereitung innerhalb von 30 Minuten verwendet werden. Die Prüfchemikalien sollten in einer calciumhaltigem (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien Hanks-Salzlösung (HBBS) vorbereitet werden, um die Bindung von Serumproteinen zu vermeiden. Die Löslichkeit der Chemikalie bei 250 mg/ml in HBSS sollte vor der Prüfung bewertet werden. Wenn die Chemikalie bei dieser Konzentration 30 Minuten lang eine stabile Suspension oder Emulsion bildet (d. h. gleichförmig bleibt und sich nicht absetzt oder in mehrere Phasen trennt), kann HBSS als Lösungsmittel verwendet werden. Stellt sich jedoch heraus, dass die Chemikalie bei dieser Konzentration nicht in HBSS löslich ist, sollte die Verwendung anderer Prüfmethoden anstelle des FL-Tests in Erwägung gezogen werden. Der Einsatz von leichtem Mineralöl als Lösungsmittel in Fällen, in denen die Chemikalie nachweislich nicht in HBSS löslich ist, sollte mit Bedacht geprüft werden, da keine ausreichenden Daten vorliegen, die eine Schlussfolgerung hinsichtlich der Leistungsfähigkeit des FL-Tests unter solchen Bedingungen zulassen. Alle zu prüfenden Chemikalien werden in einer sterilen, calciumhaltigen (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreien HBSS aus der Stammlösung bei fünf festgelegten Konzentrationen, die auf Gewichts-/Volumenbasis verdünnt werden, hergestellt: 1, 25, 100, 250 mg/ml sowie eine unverdünnte oder gesättigte Lösung. Beim Prüfen einer festen Chemikalie sollte eine sehr hohe Konzentration von 750 mg/ml eingeschlossen werden. Bei dieser Konzentration muss für die Applikation auf die Zellen möglicherweise eine Direktverdrängerpipette verwendet werden. Wenn die ermittelte Toxizität zwischen 25 und 100 mg/ml liegt, sollten die folgenden zusätzlichen Konzentrationen zweimal geprüft werden: 1, 25, 50, 75, 100 mg/ml. Der FL20-Wert sollte aus diesen Konzentrationen abgeleitet werden, sofern die Akzeptanzkriterien erfüllt waren. Die Prüfchemikalien werden nach Entfernung des Zellkulturmediums und zweimaliger Spülung mit steriler, warmer (37 °C), calciumhaltiger (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreier HBSS auf den konfluenten Zellmonolayer appliziert. Zuvor wurden die Filter visuell auf bereits vorhandene Schäden kontrolliert, die fälschlicherweise potenziellen Unverträglichkeiten gegenüber Prüfchemikalien zugeschrieben werden könnten. Es sollten mindestens drei Replikate für jede Konzentration der Prüfchemikalie sowie für die Kontrollen bei der Durchführung verwendet werden. Nach einer Minute Exposition bei Zimmertemperatur sollte die Prüfchemikalie vorsichtig abgesaugt werden, der Monolayer zweimal mit steriler, warmer (37 °C), calciumhaltiger (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreier HBSS abgespült und die Fluorescein-Leckage sofort bestimmt werden. Bei jeder Durchführung sollten gleichzeitige Negativ- und Positivkontrollen verwendet werden, um nachzuweisen, dass die Monolayer-Integrität (Negativkontrolle) und die Empfindlichkeit der Zellen (Positivkontrolle) innerhalb eines festgelegten historischen Akzeptanzbereichs liegen. Als Positivkontrolle wird Brij 35 (CAS Nr. 9002-92-0) mit einer Konzentration von 100 mg/ml vorgeschlagen. Diese Konzentration sollte eine Fluorescein-Leckage von ungefähr 30 % bewirken (akzeptabler Bereich 20-40 % Fluorescein-Leckage, d. h. Beschädigung der Zellschicht). Die vorgeschlagene Negativkontrolle ist calciumhaltige (mit einer Konzentration von 1,0-1,8 mM), phenolrotfreie HBSS (unbehandelt, Blindkontrolle). Ferner sollte eine Maximal-Leckage-Kontrolle bei jeder Durchführung eingeschlossen sein, um die Berechnung von FL20-Werten zu ermöglichen. Die Maximal-Leckage wird über ein Kontroll-Insert ohne Zellen bestimmt. Bestimmung der Fluorescein-Permeabilität Unmittelbar nach Entfernung der Prüf- und Kontrollchemikalien werden 400 µl einer 0,1-mg/ml-Fluorescein-Natrium-Lösung (0,01 % (w/v) in calciumaltiger [mit einer Konzentration von 1,0-1,8 mM], phenolrotfreier HBSS) den Inserts beigegeben (z.B. Millicell-HA). Die Kulturen werden 30 Minuten bei Zimmertemperatur gehalten. Am Ende der Inkubation mit Fluorescein werden die Inserts vorsichtig aus den Mulden entnommen. Jeder Filter wird visuell kontrolliert und etwaige Schäden, die eventuell während der Handhabung aufgetreten sind, werden protokolliert. Die Menge an Fluorescein, die durch Monolayer und Insert gedrungen ist, wird in der Lösung bestimmt, die nach Entfernen der Inserts in den Mulden verblieben ist. Die Messungen werden in einem Spektrofluorometer bei einer Anregungs- bzw. Emissionswellenlänge von 485 nm bzw. 530 nm durchgeführt. Die Empfindlichkeit des Spektrofluorometers sollte so eingestellt werden, dass die numerische Differenz zwischen der maximalen FL (Insert ohne Zellen) und der minimalen FL (Insert mit konfluentem Monolayer, behandelt mit Negativkontrolle) am größten ist. Aufgrund der Unterschiede bei dem verwendeten Spektrofluorometer wird die Verwendung einer Empfindlichkeit vorgeschlagen, die eine Fluoreszenzintensitität > 4.000 bei der maximalen Fluorescein-Leckage-Kontrolle ergibt. Der maximale FL-Wert sollte nicht größer als 9.999 sein. Die Fluoreszenzintensitität bei maximaler Leckage sollte im linearen Bereich des verwendeten Spektrofluorometers liegen. Interpretation der Ergebnisse und Vorhersagemodell Die FL-Menge ist proportional zu der durch die Chemikalie hervorgerufenen Schädigung an den Tight Junctions. Der FL-Prozentsatz für jede geprüfte Konzentration der Chemikalie wird aus den FL-Werten berechnet, die für die Prüfchemikalie mit Bezug auf die FL-Werte aus der Negativkontrolle (Messwert aus dem konfluenten Monolayer der mit der Negativkontrolle behandelten Zellen) und einer maximalen Leckage-Kontrolle (Messwert für die FL durch ein Insert ohne Zellen) ermittelt wurden. Mittlere Fluoreszenzintensität bei maximaler Leckage = x Mittlere Fluoreszenzintensität bei 0 % Leckage (Negativkontrolle) = y Die mittlere 100 % Leckage wird durch Subtraktion der mittleren 0 % Leckage von der mittleren maximalen Leckage ermittelt, d. h. x - y = z Die prozentuale Leckage für jede festgelegte Dosis wird durch Subtraktion der 0 % Leckage von der mittleren Fluoreszenzintensität der drei Replikatmesswerte (m) und Division dieses Werts durch die 100 % Leckage bestimmt, d. h. % FL = [(m-y)/z] x 100 %. Dabei sind: m = mittlere Fluoreszenzintensität der drei Replikatmessungen für die verwendete Konzentration % FL = Prozentsatz des Fluorescein, das durch die Zellschicht dringt Für die Berechnung der Chemikalienkonzentration, die 20 % FL bewirkt, sollte folgende Gleichung herangezogen werden: FLD = [(A-B)/(C-B)] x (MC - MB) + MB Dabei sind: D = prozentuale Hemmung A = prozentuale Schädigung (20 % Fluorescein-Leckage) B = prozentuale Fluorescein-Leckage < A C = prozentuale Fluorescein-Leckage > A MC = Konzentration (mg/ml) von C MB = Konzentration (mg/ml) von B Der FL20-Grenzwert für die Vorhersage von Chemikalien als Stoffe mit augenverätzender/schwer augenreizender Wirkung ist nachfolgend angegeben:
Die FL-Prüfmethode wird nur für die Identifizierung von wasserlöslichen Stoffen mit augenverätzender oder schwer augenreizender Wirkung empfohlen (UN-GHS-Kategorie 1, EU-CLP-Kategorie 1, US-EPA-Kategorie I) (siehe Nummern 1 und 10). Zur Identifizierung von wasserlöslichen Chemikalien (Stoffen und Gemischen) (3) (6) (7) als Stoff mit schwer augenschädigender Wirkung (UN-GHS/EU-CLP-Kategorie 1) oder als Stoff mit augenverätzender oder stark augenreizender Wirkung (US-EPA-Kategorie I) sollte die Prüfchemikalie einen FL20-Wert von < 100 mg/ml induzieren. Akzeptanz der Ergebnisse Der mittlere maximale Fluorescein-Leckage-Wert (x) sollte größer 4.000 (siehe Nummer 31) sein, der mittlere 0 % Leckage-Wert (y) kleiner-gleich 300 sein und der mittlere 100 %-Leckage-Wert (z) zwischen 3.700 und 6.000 liegen. Eine Prüfung gilt als akzeptabel, wenn die Positivkontrolle 20 % bis 40 % Schädigung an der Zellschicht verursacht hat (gemessen als prozentuale Fluorescein-Leckage). Daten Je Versuchsdurchführung sollten die Daten von einzelnen Replikat-Mulden (z.B. Fluoreszenzintensitätswerte und berechnete prozentuale FL-Werte für jede Prüfchemikalie, einschließlich Einstufung) in tabellarischer Form angegeben werden. Darüber hinaus sollten die Mittelwerte ± Standardabweichung von einzelnen Replikatmessungen angegeben werden. Prüfbericht Der Prüfbericht muss folgende Angaben enthalten: Prüfchemikalien und Kontrollchemikalien
Rechtfertigung der Prüfmethode und des verwendeten Protokolls
Prüfbedingungen
Ergebnisse
Erörterung der Ergebnisse
Schlussfolgerungen (1) UN (2009), Globally Harmonized System of Classification and Labelling of Chemicals (GHS), dritte überarbeitete Auflage, New York und Genf: United Nations Publications. ISBN: 978-92-1-117006-1. Verfügbar unter: [http://www.unece.org/trans/danger/publi/ghs/ghs_rev03/03files_e.html] (2) U.S. EPA (1996), Label Review Manual: 2nd Edition, EPA737-B-96-001, Washington, DC: U.S., Environmental Protection Agency. (3) EC-ECVAM (2009), Statement on the scientific validity of cytotoxicity/cell-function based in vitro assays for eye irritation testing. (4) Scott, L. et al. (2010), A proposed eye irritation testing strategy to reduce and replace in vivo studies using Bottom-Up and Top-Down approaches, Toxicol. In Vitro 24, 1-9 (5) Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung. (6) EC-ECVAM (1999), INVITOX Protocol 71: Fluorescein Leakage Test, Ispra, Italien: European Centre for the Validation of Alternative Methods (ECVAM). Verfügbar unter: [http://ecvam-dbalm.jrc.ec.europa.eu (7) EC-ECVAM (2008), Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing. (8) OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, OECD Series on Testing and Assessment No. 34. OECD, Paris. _____
Eine konfluente Schicht von MDCK-Zellen wird auf der semipermeablen Membran eines Inserts kultiviert. Die Inserts werden in die Mulden von 24-Mulden-Platten eingesetzt. Abbildung entnommen aus: Wilkinson, P.J. (2006), Development of an in vitro model to investigate repeat ocular exposure, Ph.D. Thesis, University of Nottingham, UK.
Definitionen Chemikalie: Stoff oder Gemisch. Empfindlichkeit: Anteil aller positiven/aktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode (8). EPA-Kategorie I: Chemikalien mit verätzender Wirkung (irreversible Schädigung des Augengewebes) oder Hornhautkomplikation oder -reizwirkung, die länger als 21 Tage anhält (2). Ersatzprüfung: Eine Prüfung, die eine routinemäßig angewandte Prüfung zur Identifikation von Gefahren und/oder zur Risikobewertung ersetzen soll und die im Vergleich zur akzeptierten Prüfung nachweislich in allen denkbaren Prüfsituationen und mit allen Chemikalien einen gleichwertigen oder besseren Schutz der Gesundheit von Mensch oder Tier oder der Umwelt gewährleistet, je nachdem, was zutrifft. EU CLP (Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen): Umsetzung des UN-GHS-Systems zur Einstufung von Chemikalien (Stoffen und Gemischen) in der Europäischen Union (EU). Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Chemikalie entscheiden zu können und diese Entscheidung zu untermauern. Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. FL20: Kann durch Bestimmung der Konzentration geschätzt werden, bei der die geprüfte Chemikalie 20 % Fluorescein-Leckage durch die Zellschicht bewirkt. Fluorescein-Leckage: Fluorescein-Menge, die durch die Zellschicht dringt, spektrofluorometrisch gemessen. Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen. Gemisch: Wird im UN-GHS verwendet und bezeichnet ein Gemisch oder eine Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren. Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode. Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann. GHS (Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN)): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten. GHS-Kategorie 1: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Leistungschemikalien: Teilmenge der Referenzchemikalien, die von einem Labor verwendet werden können, um seine Kompetenz hinsichtlich der validierten Referenzprüfmethode nachzuweisen. Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert. Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert. Nicht eingestuft: Chemikalien, die nicht als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorien 1, 2A oder2B, der EU-CLP-Kategorien 1 oder2 oder der US-EPA-Kategorien I, II oder III eingestuft sind. Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird. Relevanz: Beschreibung der Beziehung zwischen dem Test und der untersuchten Wirkung und ob der Test aussagekräftig und nützlich für einen bestimmten Zweck ist. Die Relevanz gibt an, inwieweit der Test die untersuchte biologische Wirkung richtig misst oder vorhersagt. Die Relevanz beinhaltet die Prüfung der Genauigkeit (Übereinstimmung) einer Prüfmethode (8). Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Spezifität: Anteil aller negativen/inaktiven Chemikalien, die durch die Prüfmethode korrekt klassifiziert werden. Dies ist ein Maß der Genauigkeit für eine Prüfmethode, die zu kategorischen Ergebnissen führt, und ein wichtiger Aspekt bei der Beurteilung der Relevanz einer Prüfmethode. Stoff: Wird im UN-GHS verwendet und bezeichnet chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können. Stoff mit augenreizender Wirkung: a) Chemikalie, die nach Applikation auf die Vorderfläche des Auges eine reversible Augenschädigung verursacht; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorien 2A oder2B, der EU-CLP-Kategorie 2 oder der US-EPA-Kategorien II oder III eingestuft sind. Stoff mit augenverätzender Wirkung: a) Chemikalie, die das Augengewebe irreversibel schädigt; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorie 1, der EU-CLP-Kategorie 1 oder der US-EPA-Kategorie I eingestuft sind. Stoff mit stark augenreizender Wirkung: a) Chemikalie, die nach Applikation auf die Vorderfläche des Auges das Augengewebe derart schädigt, dass die Schädigung nicht innerhalb von 21 Tagen nach der Applikation des Stoffs zurückgeht oder dass das Sehvermögen schwer beeinträchtigt ist; b) Chemikalien, die als Stoffe mit augenreizender Wirkung der UN-GHS-Kategorie 1, der EU-CLP-Kategorie 1 oder der US-EPA-Kategorie I eingestuft sind. Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden (8). Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Laboratorien ihre technische Kompetenz nachweisen, indem sie die in Tabelle 1 empfohlenen acht Chemikalien in die richtige Augenschädigungsklasse einstufen. Diese Chemikalien wurden so ausgewählt, dass sie die Bandbreite der lokalen Augenreizungen/-verätzungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405, TM B.5 (5)) (d. h. den Kategorien 1, 2A, 2B oder "Keine Einstufung" gemäß UN-GHS) basieren. Angesicht der validierten Zweckmäßigkeit des FL-Tests (d. h. zur Identifizierung von Stoffen mit augenverätzender und stark augenreizender Wirkung) lässt sich die Leistungsfähigkeit lediglich anhand von zwei Testergebnissen zu Einstufungszwecken (verätzend/stark reizend oder nicht verätzend/nicht stark reizend) nachweisen. Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger Daten aus der FL-Prüfmethode. Aus diesem Grund wurden die Leistungschemikalien aus der Publikation Fluorescein Leakage Assay Background Review Document as an Alternative Method for Eye Irritation Testing (8) ausgewählt, die zur retrospektiven Validierung der FL-Prüfmethode herangezogen wurde. Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des FL-Tests
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.62 Alkalischer In-vivo-Comet-Assay an Säugetierzellen 17
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 489 (2016). Der alkalische In-vivo-Comet-Assay (Einzelzell-Gelelektrophorese (Single Cell Gel Electrophoresis, SCGE)) (im Folgenden "Comet-Assay") wird für die Erkennung von DNA-Strangbrüchen in Zellen oder Zellkernen eingesetzt, die aus Geweben von Tieren, gewöhnlich Nagern, isoliert wurden, die potenziell toxischen Stoffen ausgesetzt wurden. Der Comet-Assay wurde überprüft, und verschiedene Expertengruppen haben Empfehlungen abgegeben (1) (2) (3) (4) (5) (6) (7) (8) (9) (10). Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (11).
Der Comet-Assay dient zum Nachweis von Chemikalien, die DNA-Schäden auslösen. Durch den Comet-Assay können unter alkalischen Bedingungen (> pH 13) Einzel- und Doppelstrangbrüche nachgewiesen werden, die beispielsweise auf direkte Interaktionen mit der DNA, alkali-labile Stellen oder transiente DNA-Strangbrüche aufgrund einer DNA-Exzisionsreparatur zurückzuführen sind. Diese Strangbrüche können repariert werden, ohne dass eine anhaltende Wirkung eintritt, können tödlich für die Zelle sein oder können zu einer Mutation führen, die eine dauerhafte lebensfähige Veränderung bewirkt. Zudem können sie Chromosomenschäden hervorrufen, die mit vielen Erkrankungen des Menschen, darunter auch Krebs, im Zusammenhang stehen.
Eine formale Validierungsstudie des In-vivo-Comet-Assays an Nagetieren wurde 2006-2012 unter Koordinierung des Japanischen Zentrums zur Validierung alternativer Methoden (JaCVAM) in Zusammenarbeit mit dem Europäischen Zentrum zur Validierung alternativer Methoden (ECVAM) und den US-amerikanischen Validierungszentren Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) und NTP Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM) durchgeführt (12). Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des Comet-Assays und basiert auf dem bei der Validierungsstudie angewandten Schlussprotokoll (12) sowie auf weiteren einschlägigen veröffentlichten und unveröffentlichten (laboreigenen) Daten.
Die Definitionen der Schlüsselbegriffe sind Anlage 1 zu entnehmen. Es ist zu beachten, dass für diesen Assay viele verschiedene Plattformen eingesetzt werden können (Objektträger, Gelproben, 96-Mulden-Platten usw.). Zur Vereinfachung wird im Folgenden der Begriff "Objektträger" verwendet, wobei aber alle anderen Plattformen darin eingeschlossen sind.
Vorbemerkungen und Einsatzgrenzen
Der Comet-Assay ist eine Methode zur Bestimmung von DNA-Strangbrüchen in eukaryotischen Zellen. In Agarose eingebettete Einzelzellen/Zellkerne auf einem Objektträger werden mit Detergenzien und hoher Salzkonzentration lysiert. Durch diesen Lyseschritt werden die Zell- und Zellkernmembrane zerstört, sodass die spiralisierten DNA-Schleifen (allgemein als Nukleoide bezeichnet) und die DNA-Fragmente freigelegt werden können. Die Elektrophorese bei hohem pH-Wert führt zu kometartigen Strukturen, die bei Verwendung geeigneter Farbstoffe unter dem Fluoreszenzmikroskop beobachtet werden können; die DNA-Fragmente wandern je nach Größe vom "Kopf" in den "Schweif" und die Intensität des Kometenschweifs relativ zur Gesamtintensität (Kopf plus Schweif) entspricht dem Grad des DNA-Bruchs (13) (14) (15).
Der alkalische In-vivo-Comet-Assay ist insbesondere für die Bewertung der genotoxischen Wirkung relevant, da die Reaktionen im Rahmen des Assays von der In-vivo-ADME (Absorption, distribution, metabolism and excretion, Resorption, Verteilung, Metabolisierung und Ausscheidung) sowie von DNA-Reparaturprozessen abhängig sind. Diese unterscheiden sich je nach Tierart, Gewebetyp und Art der DNA-Schädigung.
Zur Einhaltung der Tierschutzanforderungen, insbesondere zur Verringerung der Verwendung von Versuchstieren (3R-Prinzip: Replace, Reduce, Refine - Vermeiden, Verringern, Verbessern) kann dieser Test auch mit anderen toxikologischen Studien, z.B. Toxizitätsstudien mit wiederholter Verabreichung (10) (16) (17), integriert werden oder der Endpunkt kann mit anderen genotoxischen Endpunkten, wie beispielsweise dem In-vivo-Erythrozyten-Mikrokerntest bei Säugern, kombiniert werden (18) (19) (20). Der Comet-Assay wird überwiegend an Nagetieren durchgeführt, wurde aber auch bei anderen Säugetier- und Nichtsäugetierarten angewandt. Die Verwendung von Nichtnagetierarten sollte im Einzelfall wissenschaftlich und ethisch begründet werden. Es wird dringend empfohlen, den Comet-Assay an anderen Arten als Nagetieren nur im Rahmen einer anderen Toxizitätsstudie und nicht als eigenständigen Test durchzuführen.
Expositionspfad und zu untersuchende Gewebe sollten basierend auf allen verfügbaren/vorliegenden Informationen über die Prüfchemikalien, z.B. beabsichtigter/erwarteter Expositionspfad beim Menschen, Metabolisierung und Verteilung, Potenzial für Wirkungen an Kontaktstellen, strukturelle Warnungen, andere Daten zur Genotoxizität oder Toxizität und Zweck der Studie, gewählt werden. Das genotoxische Potenzial der Prüfchemikalien kann daher gegebenenfalls im Zielgewebe bzw. in den Zielgeweben karzinogener und/oder anderer toxischer Wirkungen untersucht werden. Der Test gilt ferner als hilfreich für weitere Untersuchungen zur Genotoxizität, die in einem In-vitro-System nachgewiesen wurde. Es ist zweckmäßig, einen In-vivo-Comet-Assay an einem bestimmten Gewebe durchzuführen, wenn begründeterweise von einer entsprechenden Exposition des Gewebes ausgegangen werden kann.
Der Test wurde am ausführlichsten im Rahmen von Kooperationsstudien wie beispielsweise der JaCVAM-Studie (12) und in Rothfuss et al., 2010 (10) am somatischen Gewebe männlicher Ratten validiert. In der internationalen Validierungsstudie von JaCVAM wurden Leber und Magen verwendet. Die Leber, weil sie das aktivste Organ in der Metabolisierung von Chemikalien und häufig ein Zielorgan für Karzinogenität ist. Der Magen, weil er gewöhnlich die erste Kontaktstelle für Chemikalien nach oraler Aufnahme ist, obwohl andere Bereiche des Magen-Darm-Trakts wie Zwölffingerdarm und Dünndarm ebenfalls als Kontaktstellengewebe untersucht werden sollten und beim Menschen unter Umständen relevanter sind als der Drüsenmagen von Nagetieren. Es muss unbedingt darauf geachtet werden, dass solches Gewebe nicht übermäßig hohen Prüfchemikalienkonzentrationen ausgesetzt wird (21). Das Verfahren ist grundsätzlich bei allen Geweben anwendbar, von denen analysierbare Einzelzell-/Zellkernsuspensionen gewonnen werden können. Proprietäre Daten verschiedener Labors belegen die erfolgreiche Anwendung bei verschiedenen Geweben, und es liegen zahlreiche Publikationen vor, in denen die Anwendbarkeit des Verfahrens bei anderen Organen oder Geweben als Leber und Magen, z.B. Dünndarm (22), Niere (23) (24), Haut (25) (26) oder Harnblase (27) (28), Lunge und bronchoalveoläre Lavage-Zellen (relevant bei Untersuchungen zu eingeatmeten Chemikalien) (29) (30), aufgezeigt wurde. Ferner wurden Tests an mehreren Organen gleichzeitig durchgeführt (31) (32).
Obwohl die genotoxischen Wirkungen in Keimzellen durchaus von Interesse sein mögen, ist zu beachten, dass der standardmäßige alkalische Comet-Assay, der in der Prüfmethode beschrieben wird, für die Messung von DNA-Strangbrüchen in reifen Keimzellen als ungeeignet angesehen wird. Da im Rahmen einer Auswertung der Fachliteratur zum Einsatz des Comet-Assays bei der Untersuchung der Genotoxizität in Keimzellen von hohen und variablen Background-Werten der DNA-Schädigung berichtet wurde (33), werden Änderungen des Protokolls sowie verbesserte Standardisierungs- und Validierungsstudien für notwendig erachtet, bevor der Comet-Assay an reifen Keimzellen (z.B. Spermien) in die Prüfmethode aufgenommen werden kann. Darüber hinaus ist das Expositionsschema, das in dieser Prüfmethode beschrieben wird, nicht optimal. Für eine aussagekräftige Analyse der DNA-Strangbrüche in reifen Spermien wären längere Expositions- oder Probenahmezeiten erforderlich. Genotoxische Wirkungen, die durch den Comet-Assay in Hodenzellen in verschiedenen Differenzierungsstadien gemessen wurden, wurden in der Fachliteratur beschrieben (34) (35). Allerdings sollte beachtet werden, dass Gonaden eine Mischung aus somatischen und Keimzellen enthalten. Aus diesem Grund spiegeln positive Ergebnisse an einer Gonade (Hoden) nicht unbedingt eine Keimzellenschädigung wider, deuten aber darauf hin, dass die geprüfte(n) Chemikalie(n) und/oder ihre Metaboliten die Gonade erreicht haben.
Vernetzungen können mit den Standard-Versuchsbedingungen des Comet-Assays nicht zuverlässig identifiziert werden. Unter bestimmten geänderten Versuchsbedingungen könnten DNA-DNA- und DNA-Protein-Vernetzungen sowie sonstige Basenveränderungen wie beispielsweise oxidierte Basen erkannt werden (23) (36) (37) (38) (39). Jedoch wären weitere Arbeiten notwendig, um die notwendigen Protokolländerungen angemessen zu charakterisieren. Daher ist der Nachweis von Vernetzungsmitteln nicht der primäre Zweck des hierin beschriebenen Tests. Der Test eignet sich, auch mit Änderungen, nicht zum Nachweis von Aneugenen.
Nach aktuellem Kenntnisstand hat der In-vivo-Comet-Assay verschiedene zusätzliche Einsatzgrenzen (siehe Anlage 3). Es wird davon ausgegangen, dass die Prüfmethode in Zukunft überprüft und gegebenenfalls angesichts der gewonnenen Erkenntnisse überarbeitet werden wird.
Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Diese Überlegungen erübrigen sich, wenn die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist.
Die Prüfchemikalie wird den Tieren über einen geeigneten Applikationsweg verabreicht. Dosierung und Probenahme sind unter den Nummern 36-40 ausführlich beschrieben. Zu den ausgewählten Probenahmezeitpunkten werden die untersuchten Gewebe seziert und Einzelzell-/Zellkernsuspensionen hergestellt (In-situ-Perfusion kann vorgenommen werden, sofern sie als sinnvoll angesehen wird, z.B. Leber) und in Softagar eingebettet, um sie auf Objektträgern zu immobilisieren. Die Zellen/Zellkerne werden mit Lysepuffer behandelt, um die Zell- und/oder Zellkernmembran zu entfernen, und einer starken Lauge, z.B. pH > 13, ausgesetzt, um das "Entwinden" der DNA und die Freisetzung der relaxierten DNA-Schleifen und -Fragmente zu ermöglichen. Die nukleare DNA im Agar wird dann der Elektrophorese unterzogen. Normale nichtfragmentierte DNA-Moleküle bleiben an der Stelle, an der sich die nukleare DNA im Agar befand, während fragmentierte DNA und relaxierte DNA-Schleifen zur Anode wandern. Nach der Elektrophorese wird die DNA durch einen geeigneten fluoreszierenden Farbstoff sichtbar gemacht. Die Präparate sollten mikroskopisch und mittels voll- oder halbautomatischer Bildanalysesysteme untersucht werden. Der Umfang der DNA-Wanderung bei der Elektrophorese und der Migrationsabstand entsprechen der Menge und Größe der DNA-Fragmente. Beim Comet-Assay gibt es verschiedene Endpunkte. Es wurde empfohlen, den DNA-Inhalt im Schweif (% DNA im Schweif oder % Schweif-Intensität) zur Bewertung der DNA-Schädigung heranzuziehen (12) (40) (41) (42). Nach der Analyse einer ausreichenden Anzahl von Zellkernen werden die Testergebnisse mit geeigneten Analysemethoden analysiert.
Es ist zu beachten, dass Änderungen verschiedener Aspekte der Methodik, einschließlich Vorbereitung der Proben, Elektrophoresebedingungen, visuelle Analyseparameter (z.B. Farbstoffintensität, Lichtintensität der Mikroskoplampe und Einsatz von Mikroskopfiltern und Kameradynamik), und der Umgebungsbedingungen (z.B. Hintergrundbeleuchtung), untersucht wurden und sich auf die DNA-Wanderung auswirken können (43) (44) (45) (46).
Überprüfung der Eignung des Labors
Jedes Labor sollte seine experimentelle Kompetenz in Bezug auf den Comet-Assay belegen, indem es seine Fähigkeit zur Herstellung von Einzelzell- oder Zellkernsuspensionen von ausreichender Qualität für jedes Zielgewebe und jede verwendete Tierart nachweist. Die Qualität der Zubereitungen wird in erster Linie anhand des prozentualen DNA-Anteils im Schweif bei mit Vehikel behandelten Tieren bewertet, deren Werte in einen reproduzierbaren niedrigen Bereich fallen. Die aktuellen Daten deuten darauf hin, dass der prozentuale DNA-Anteil im Schweif im Gruppenmittel (basierend auf dem Mittelwert der Mediane - Erläuterungen zu diesen Begriffen unter Nummer 57) in der Rattenleber vorzugsweise 6 % nicht überschreiten sollte, was mit den Werten aus der JaCVAM-Validierungsstudie (12) sowie aus anderen veröffentlichten und proprietären Daten im Einklang stehen würde. Zum gegenwärtigen Zeitpunkt liegen nicht genügend Daten vor, um Empfehlungen für optimale oder akzeptable Bereiche bei anderen Geweben abzugeben. Dies schließt die Verwendung anderer Gewebe, sofern gerechtfertigt, nicht aus. Der Prüfbericht sollte eine Beurteilung der Leistung des Comet-Assays bei diesen Geweben unter Bezugnahme auf die veröffentliche Literatur oder proprietäre Daten beinhalten. Erstens ist ein niedriger prozentualer DNA-Anteil im Schweif in den Kontrollen wünschenswert, um eine hinreichend dynamische Bandbreite für den Nachweis einer positiven Wirkung zu gewährleisten. Zweitens sollte jedes Labor die erwarteten Reaktionen für direkte Mutagene und Promutagene für verschiedene Wirkungsweisen gemäß den Beispielen in Tabelle 1 (Nummer 29) reproduzieren können.
Beispielsweise können positive Stoffe aus der JaCVAM-Validierungsstudie (12) oder anderen veröffentlichten Daten (siehe Nummer 9), gegebenenfalls mit Begründung, ausgewählt werden, wobei die eindeutigen positiven Wirkungen in den untersuchten Geweben nachgewiesen werden. Zudem sollte die Fähigkeit zum Nachweis von schwachen Wirkungen bekannter Mutagene, wie z.B. niedrig dosiertem EMS, nachgewiesen werden, indem beispielsweise Dosis-Wirkungs-Beziehungen an einer hinreichenden Anzahl von Dosierungen mit entsprechendem Abstand aufgestellt werden. Anfänglich sollte sich das Labor auf den Nachweis der Kompetenz bei den am häufigsten verwendeten Geweben (z.B. Rattenleber) konzentrieren, wobei ein Vergleich zwischen vorliegenden Daten und erwarteten Ergebnissen angestellt werden könnte (12). Die Daten von anderen Geweben wie z.B. Magen, Zwölffingerdarm, Dünndarm, Blut usw. könnten gleichzeitig erfasst werden. Das Labor muss seine Kompetenz für sämtliche Gewebe aller Tierarten, die untersucht werden sollen, nachweisen und aufzeigen, dass eine akzeptable positive Reaktion bei einem bekannten Mutagen (z.B. EMS) im jeweiligen Gewebe erreicht werden kann.
Vehikel-/Negativkontrolldaten sollten erfasst werden, um die Reproduzierbarkeit negativer Reaktionen nachzuweisen und sicherzustellen, dass die technischen Aspekte des Assays vorschriftsmäßig kontrolliert wurden, oder darauf hinzuweisen, dass historische Kontrollbereiche erneut festgelegt werden sollten (siehe Nummer 22).
Es ist zu beachten, dass das Labor, da mehrere Gewebe bei der Sektion gewonnen und für die Comet-Analyse aufgearbeitet werden können, in der Lage sein muss, mehrere Gewebe von einem einzelnen Tier zu gewinnen und somit sicherzustellen, dass keine potenzielle DNA-Läsion verloren geht und die Comet-Analyse nicht beeinträchtigt wird. Die Zeitdauer zwischen der Tötung des Tiers und der Gewinnung der Gewebe zur Aufbereitung kann kritisch sein (siehe Nummer 44).
Da bei der Entwicklung der Kompetenz für diesen Test Tierschutzbelange berücksichtigt werden müssen, können Gewebe von in anderen Tests verwendeten Tieren zur Entwicklung der Kompetenz bei den verschiedenen Aspekten des Tests eingesetzt werden. Darüber hinaus muss während der verschiedenen Phasen der Etablierung einer neuen Prüfmethode in einem Labor nicht unbedingt eine vollständige Studie durchgeführt werden. Ferner können weniger Tiere oder Prüfkonzentrationen bei der Entwicklung der notwendigen Fähigkeiten verwendet werden.
Historische Kontrolldaten
Im Verlauf der Untersuchungen zum Nachweis der Kompetenz sollte das Labor eine Datenbank mit historischen Daten aufbauen, um Positiv- und Negativkontrollbereiche und -verteilungen für die betreffenden Gewebe und Tierarten zu erfassen. Empfehlungen zur Erstellung und Verwendung historischer Daten (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus der Datenbank und die Akzeptanzkriterien für einen bestimmten Versuch) sind der Literatur zu entnehmen (47). Verschiedene Gewebe und Tierarten sowie verschiedene Vehikel und Verabreichungswege können unterschiedliche prozentuale Schweif-DNA-Werte in den Negativkontrollen ergeben. Daher müssen unbedingt Negativkontrollbereiche für jedes Gewebe und jede Tierart erfasst werden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder Xbar-Karten (48)), um zu ermitteln, wie variabel ihre Daten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist. Die Auswahl der für Positivkontrollen geeigneten Stoffe, der Dosisbereiche und der Versuchsbedingungen (z.B. Elektrophoresebedingungen) muss zum Nachweis schwacher Wirkungen vielleicht ebenfalls optimiert werden (siehe Nummer 17).
Sämtliche Änderungen am Versuchsprotokoll sind auf ihre Übereinstimmung mit den bereits vorhandenen Datenbanken historischer Kontrolldaten des Labors zu prüfen. Bei größeren Inkonsistenzen sollte eine neue Datenbank historischer Kontrolldaten erstellt werden.
Vorbereitungen
Auswahl der Tierart
In der Regel werden junge gesunde und geschlechtsreife Nagetiere üblicher Laborstämme verwendet (die bei Behandlungsbeginn 6 bis 10 Wochen alt sind, wenngleich etwas ältere Tiere auch zulässig sind). Die Auswahl der Nagetierarten sollte basieren auf i) Arten, die in anderen Toxizitätsstudien verwendet werden (um eine Korrelation zwischen den Daten herstellen zu können und integrierte Studien zu ermöglichen), ii) Arten, die in einer Kanzerogenitätsstudie Tumore entwickelt haben (bei der Untersuchung des Mechanismus der Karzinogenese) oder iii) Arten, deren Metabolismus dem des Menschen am ähnlichsten ist, soweit bekannt. In diesem Test werden routinemäßig Ratten verwendet, aber es können auch andere Arten zum Einsatz kommen, sofern dies ethisch und wissenschaftlich begründet ist.
Haltungs- und Fütterungsbedingungen
Bei Nagern soll die Temperatur im Versuchstierraum 22 1 °C (± 3 1 °C) betragen. Die relative Luftfeuchte sollte bei 50 bis 60 % liegen, wenigstens 30 % betragen und außer bei Reinigung des Raumes 70 % vorzugsweise nicht übersteigen. Der Raum sollte künstlich beleuchtet sein, wobei die Beleuchtung im 12-Stunden-Rhythmus ein- und ausgeschaltet werden sollte. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Auswahl des Futters wird eventuell dadurch beeinflusst, dass eine geeignete Beimischung einer Prüfchemikalie gewährleistet werden muss, wenn diese über das Futter verabreicht wird. Nagetiere sollten in kleinen gleichgeschlechtlichen Gruppen (maximal fünf pro Käfig) untergebracht werden, sofern kein aggressives Verhalten zu erwarten ist. Die Tiere sind nur dann einzeln zu halten, wenn dies wissenschaftlich begründet ist. Es sind möglichst feste Böden zu verwenden, da Maschendrahtböden Verletzungen verursachen können (49). Es muss für eine entsprechende Ausgestaltung des Lebensumfelds gesorgt werden.
Vorbereitung der Versuchstiere
Die Tiere werden den Kontroll- und Behandlungsgruppen nach dem Zufallsprinzip zugeordnet. Sie werden individuell gekennzeichnet und über einen Zeitraum von mindestens fünf Tagen vor Behandlungsbeginn unter Laborbedingungen eingewöhnt. Zur individuellen Kennzeichnung der Tiere muss eine minimalinvasive Methode gewählt werden. Geeignete Methoden sind Anbringen von Ringen, Marken oder Mikrochips oder eine biometrische Identifizierung. Kupieren der Ohren oder Zehen ist bei diesen Tests wissenschaftlich nicht gerechtfertigt. Die Käfige sind so anzuordnen, dass sich ihre Position möglichst wenig auswirkt. Zu Beginn des Versuchs sollte die Abweichung des Körpergewichts der Tiere möglichst gering sein und nicht mehr als ± 20 % betragen.
Vorbereitung der Dosierung
Feste Prüfchemikalien sollten vor der Verabreichung an die Tiere in geeigneten Vehikeln gelöst oder suspendiert oder dem Futter oder Trinkwasser beigemischt werden. Flüssige Prüfchemikalien können direkt verabreicht oder zuvor verdünnt werden. Bei Exposition durch Inhalation können die Prüfchemikalien je nach ihren physikalisch-chemischen Eigenschaften als Gas, Dampf oder festes/flüssiges Aerosol verabreicht werden (50) (51).
Es sind frische Zubereitungen der Prüfchemikalie zu verwenden, es sei denn, die Stabilität der Chemikalie bei Lagerung wird nachgewiesen und die entsprechenden Lagerbedingungen werden definiert.
Prüfbedingungen
Vehikel
Das Vehikel sollte bei den gewählten Dosierungen keine toxischen Wirkungen hervorrufen und nicht im Verdacht stehen, mit den Prüfchemikalien eine chemische Reaktion einzugehen. Werden keine allgemein bekannten Vehikel verwendet, so sind Daten zu deren Kompatibilität in Bezug auf Versuchstiere, Verabreichungsweg und Endpunkt beizubringen. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. Es ist zu beachten, dass einige Vehikel (insbesondere zähflüssige Vehikel) Entzündungen hervorrufen und die Background-Werte der DNA-Strangbrüche an der Kontaktstelle, insbesondere bei mehrfachen Verabreichungen, erhöhen können.
Kontrollen
Positivkontrollen
Zum gegenwärtigen Zeitpunkt sollte jeder Versuch eine Gruppe von mindestens drei analysierbaren Tieren eines Geschlechts oder je Geschlecht, sofern beide Geschlechter verwendet werden (siehe Nummer 32), die mit einem Positivkontrollstoff behandelt wurden, umfassen. Möglicherweise kann die Eignung künftig adäquat nachgewiesen werden, sodass weniger Positivkontrollen erforderlich sind. Bei Verwendung mehrerer Probenahmezeitpunkte (z.B. bei Protokoll mit Einzelverabreichung) müssen nur bei einer Probenahme Positivkontrollen einbezogen werden. In einem solchen Fall sollte für einen ausgewogenen Versuchsplan gesorgt werden (siehe Nummer 48). Die gleichzeitig als Positivikontrollen verwendeten Stoffe müssen nicht auf demselben Weg verabreicht werden wie die Prüfchemikalie. Allerdings ist es wichtig, dass bei der Messung der Wirkungen an Kontaktstellen derselbe Verabreichungsweg angewandt wird. Die Positivkontrollstoffe sollten nachweislich DNA-Strangbrüche in allen für die Prüfchemikalie untersuchten Geweben induzieren. EMS ist wahrscheinlich der am besten geeignete Positivkontrollstoff, da er in allen untersuchten Geweben DNA-Strangbrüche hervorgerufen hat. Die Dosierungen der Positivkontrollstoffe sollten jeweils so gewählt werden, dass mäßige Wirkungen für eine kritische Bewertung der Leistungsfähigkeit und Empfindlichkeit des Tests erreicht werden. Sie könnten auf Dosis-Wirkungs-Kurven beruhen, die das Labor während des Nachweises seiner Kompetenz aufgestellt hat. Der prozentuale DNA-Anteil im Schweif bei Tieren, die gleichzeitig mit einer Positivkontrolle behandelt wurden, sollte mit dem vorab ermittelten Laborbereich für jedes Gewebe und jede Probenahmezeit für diese Tierart übereinstimmen (siehe Nummer 16). Beispiele für Positivkontrollstoffe und einige ihrer Zielgewebe (bei Nagern) sind Tabelle 1 zu entnehmen. In wissenschaftlich begründeten Fällen können auch andere als die in Tabelle 1 genannten Stoffe ausgewählt werden.
Tabelle 1 Beispiele für Positivkontrollstoffe und einige der jeweils zutreffenden Zielgewebe
| Stoffe und CAS-Nr. |
| Ethylmethansulfonat (CAS-Nr. 62-50-0) für sämtliche Gewebe |
| Ethylnitrosoharnstoff (CAS-Nr. 759-73-9) für Leber und Magen, Zwölffingerdarm oder Dünndarm |
| Methylmethansulfonat (CAS-Nr. 66-27-3) für Leber, Magen, Zwölffingerdarm oder Dünndarm, Lunge oder bronchoalveoläre Lavage-Zellen, Niere, Harnblase, Lunge, Hoden und Knochenmark/Blut |
| N-Methyl-N2-nitro-N-nitrosoguanidin (CAS-Nr. 70-25-7) für Magen, Zwölffingerdarm oder Dünndarm |
| 1,2-Dimethylhydrazin-2HCl (CAS-Nr. 306-37-6) für Leber und Darm |
| N-methyl-N-nitrosoharnstoff (CAS-Nr. 684-93-5) für Leber, Knochenmark, Blut, Niere, Magen, Dünndarm und Hirn. |
Negativkontrollen
Jeder Versuch sollte für jede Probenahmezeit und jedes Gewebe eine Gruppe von Negativkontrolltieren umfassen, die nur mit dem Vehikel und ansonsten auf dieselbe Weise wie die Behandlungsgruppen behandelt wurden. Der prozentuale DNA-Anteil im Schweif in den Negativkontrolltieren sollte innerhalb des vorab ermittelten Background-Bereichs des Labors für jedes Gewebe und jede Probenahmezeit für diese Tierart liegen (siehe Nummer 16). Liegen keine historischen oder veröffentlichten Kontrolldaten vor, aus denen hervorgeht, dass weder das gewählte Vehikel noch die Zahl der Verabreichungen oder der Verabreichungsweg schädliche oder genotoxische Wirkungen hervorruft, sollten Vorversuche vor der Komplettstudie durchgeführt werden, um die Akzeptanz der Vehikelkontrolle nachzuweisen.
Anzahl und Geschlecht der Tiere
Obwohl nur wenige Daten über weibliche Tiere vorliegen, die bei einem Vergleich zwischen den Geschlechtern in Bezug auf den Comet-Assay herangezogen werden können, reagieren männliche und weibliche Tiere bei anderen In-vivo-Genotoxizitätstest in der Regel ähnlich, sodass die meisten Studien an beiden Geschlechtern durchgeführt werden könnten. Daten, aus denen nennenswerte Unterschiede zwischen männlichen und weiblichen Tieren hervorgehen (z.B. Unterschiede in der systemischen Toxizität, im Stoffwechsel, in der Bioverfügbarkeit usw., einschließlich unter anderem eine Dosisfindungsstudie) legen die Verwendung von Tieren beider Geschlechter nahe. In diesem Fall kann es angebracht sein, eine Studie an männlichen und weiblichen Tieren durchzuführen, z.B. als Teil einer Toxizitätsstudie mit wiederholter Verabreichung. Gegebenenfalls ist die Verwendung eines faktoriellen Versuchsplans geeignet, wenn beide Geschlechter einbezogen werden. Einzelheiten zur Analyse der Daten bei Verwendung eines solchen Versuchsplans sind in Anlage 2 enthalten.
Zu Beginn der Studie (und während des Kompetenznachweises) sollten die Gruppengrößen so festgelegt werden, dass man pro Gruppe mindestens fünf analysierbare Tiere eines Geschlechts - oder je Geschlecht, sofern beide Geschlechter einbezogen werden - erhält (weniger in der gleichzeitigen Positivkontrollgruppe - siehe Nummer 29). Sollte es sich beim Menschen um eine geschlechtsspezifische Exposition handeln, z.B. bei bestimmten Pharmazeutika, ist der Versuch an Tieren des betreffenden Geschlechts durchzuführen. Bei einer gemäß den unter Nummer 33 festgelegten Parametern durchgeführten Studie mit drei Dosisgruppen und gleichzeitigen Negativ- und Positivkontrollgruppen (wobei jede Gruppe aus fünf Tieren desselben Geschlechts besteht) kann als Richtwert für die üblicherweise erforderliche Höchstmenge an Tieren eine Anzahl von 25 bis 35 Versuchstieren angegeben werden.
Die Tiere sollten täglich über einen Zeitraum von mindestens zwei Tagen behandelt werden (d. h. zwei oder mehr Behandlungen in Abständen von ungefähr 24 Stunden), und die Probenahme sollte einmal 2 bis 6 Stunden (oder zum Zeitpunkt Tmax) nach der letzten Behandlung erfolgen (12). Proben aus verlängerten Verabreichungsschemata (z.B. tägliche Dosierung über 28 Tage) sind zulässig. Es wurde nachgewiesen, dass der Comet-Assay erfolgreich mit dem Erythrozyten-Mikrokerntest kombiniert werden kann (10) (19). Jedoch sollte die Logistik im Zusammenhang mit Gewebeproben für die Comet-Analyse unter Berücksichtigung der Anforderungen an Gewebeproben für andere Arten von toxikologischen Bewertungen sorgfältig geplant werden. Die Probenahme 24 Stunden nach der letzten Verabreichung, was bei einer allgemeinen Toxizitätsstudie üblicherweise der Fall ist, ist in den meisten Fällen ungeeignet (siehe Nummer 40 zum Probenahmezeitpunkt). Die Anwendung anderer Behandlungs- und Probenahmepläne sollte begründet werden (siehe Anlage 3). Beispielsweise könnte eine Einzelbehandlung mit mehreren Probenahmen erfolgen, jedoch sollte in einem solchen Fall beachtet werden, dass bei einer Studie mit Einzelverabreichung mehrere Tiere benötigt werden, da mehrere Probenahmezeitpunkte erforderlich sind. Allerdings ist dies gelegentlich vorzuziehen, wenn beispielsweise die Prüfchemikalie eine übermäßige Toxizität nach wiederholter Verabreichung induziert.
Jede Art der Testdurchführung ist akzeptabel, solange die Prüfchemikalie eine positive Wirkung hervorruft oder im Falle einer Negativstudie die Exposition der Zielgewebe oder die Toxizität für diese Zielgewebe durch direkte oder indirekte Hinweise nachgewiesen wurde oder die Limit-Dosis erreicht wird (siehe Nummer 36).
Die Prüfchemikalien können auch in Form von zwei Teilmengen am selben Tag im Abstand von nicht mehr als 2 bis 3 Stunden verabreicht werden, wenn es sich um ein großes Volumen handelt. In diesen Fällen sollte der Zeitpunkt der Probenahme ausgehend vom Zeitpunkt der letzten Verabreichung geplant werden (siehe Nummer 40).
Dosierungen
Wenn zunächst eine Dosisfindungsstudie durchgeführt wird, da keine geeigneten Daten aus anderen einschlägigen Studien zur Dosierungswahl vorliegen, sollte diese im selben Labor unter Verwendung von Tieren derselben Art und Rasse und desselben Geschlechts sowie nach demselben Behandlungsverfahren stattfinden, die nach den gegenwärtigen Ansätzen für die Durchführung von Dosisfindungsstudien in der Hauptstudie zu verwenden sind. Ziel der Studie sollte sein, die maximal verträgliche Dosis (MTD) zu ermitteln, die als die Dosis definiert ist, die für die Dauer der Studie zu leichten toxischen Wirkungen (z.B. eindeutige klinische Anzeichen wie Abweichungen in Verhalten oder Reaktionen, geringer Rückgang des Körpergewichts oder zytotoxische Wirkung auf ein Zielgewebe) führt, jedoch weder zum Tod führt noch Anzeichen von Schmerzen, Leiden oder Ängsten hervorruft, die eine Tötung erforderlich machen würden. Bei einer nicht toxischen Prüfchemikalie beträgt die Höchstdosis (Limit-Dosis) bei einem Behandlungszeitraum von mindestens 14 Tagen 1.000 mg/kg Körpergewicht/Tag. Bei Behandlungszeiträumen von weniger als 14 Tagen beträgt die Höchstdosis (Limit-Dosis) 2.000 mg/kg Körpergewicht/Tag. Bei bestimmten Arten von Prüfchemikalien (z.B. Humanarzneimittel), die spezifischen Vorschriften unterliegen, können andere Grenzwerte gelten.
Chemikalien, die eine Sättigung der toxikokinetischen Eigenschaften aufweisen oder Entgiftungsprozesse einleiten, die nach einer Langzeitgabe möglicherweise zu einem Rückgang der Exposition führen, entsprechen möglicherweise nicht den Dosierungskriterien und sollten anhand einer Einzelfallprüfung bewertet werden.
Bei der akuten und subakuten Version des Comet-Assays sollten neben der Höchstdosis (MTD, maximal mögliche Dosis, maximale Expositionsdosis oder Limit-Dosis) eine absteigende Folge von mindestens zwei zusätzlichen Dosierungen mit geeignetem Abstand (vorzugsweise weniger als 10) für jeden Probenahmezeitpunkt gewählt werden, um dosisbezogene Wirkungen nachzuweisen. Jedoch sollten die verwendeten Dosierungen vorzugsweise einen Bereich vom Höchstwert bis zu einer Dosierung, die wenig oder keine Toxizität erzeugt, abdecken. Werden bei allen untersuchten Dosierungen toxische Wirkungen im Zielgewebe beobachtet, sind weitere Untersuchungen bei nichttoxischen Dosierungen anzuraten (siehe Nummern 54 und 55). Studien, in denen der Verlauf der Dosis-Wirkungs-Kurve umfassender untersucht werden soll, erfordern gegebenenfalls weitere Dosisgruppen.
Verabreichung
Bei der Versuchsplanung ist der zu erwartende Expositionsweg beim Menschen zu berücksichtigen. Daher können mit entsprechender Begründung Verabreichungswege wie etwa eine Aufnahme über die Nahrung, über das Trinkwasser, eine topische, subkutane, intravenöse, orale (über eine Magensonde), intratracheale Verabreichung, durch Inhalation oder Implantation, gewählt werden. In jedem Fall sollte der Verabreichungsweg so gewählt werden, dass eine angemessene Exposition des/der Zielgewebe(s) sichergestellt ist. Eine intraperitoneale Injektion wird in der Regel nicht empfohlen, da sie keinen typischen Expositionsweg beim Menschen darstellt; sie sollte nur mit spezieller Begründung angewandt werden (z.B. bestimmte Stoffe, die als Positivkontrollen verwendet werden, zu Untersuchungszwecken oder für einige intraperitoneal verabreichte Arzneimittel). Die Höchstmenge an Flüssigkeit, die je Gabe über eine Magensonde oder eine Injektion verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen können aber auch 2 ml/100 g Körpergewicht in Betracht gezogen werden. Werden größere Volumina verwendet (sofern durch Tierschutzvorschriften erlaubt), ist dies zu begründen. Soweit möglich sollten verschiedene Dosierungen durch Anpassung der Konzentration der Dosisformulierung erreicht werden, um bei allen Dosen ein konstantes Volumen im Verhältnis zum Körpergewicht sicherzustellen.
Probenahmezeitpunkt
Der Probenahmezeitpunkt ist eine kritische Variable, da er von dem Zeitraum abhängt, der erforderlich ist, bis die Prüfchemikalien ihre maximale Konzentration im Zielgewebe erreichen und DNA-Strangbrüche induziert werden; er muss jedoch vor dem Zeitpunkt liegen, zu dem diese Brüche entfernt oder repariert werden oder zum Zelltod führen. Die Persistenz einiger Läsionen, die zu den im Comet-Assay nachgewiesenen DNA-Strangbrüchen führen, kann sehr kurz sein, zumindest bei einigen in vitro geprüften Chemikalien (52) (53). Wenn solche transienten DNA-Läsionen vermutet werden, sollten Maßnahmen ergriffen werden, um deren Verlust zu verringern, indem sichergestellt wird, dass die Proben der Gewebe ausreichend früh, möglichst vor den nachfolgend angegebenen Standardzeiten, genommen werden. Die optimalen Probenahmezeitpunkte können chemikalien- oder pfadspezifisch sein, was z.B. zu einer raschen Gewebeexposition bei intravenöser Verabreichung oder bei Exposition durch Inhalation führt. Die Probenahmezeitpunkte sollten, sofern vorhanden, anhand von kinetischen Daten (z.B. Zeitpunkt (Tmax), zu dem die höchste Plasma- oder Gewebekonzentration (Cmax) erreicht wird oder im stabilen Zustand bei mehrfacher Verabreichung) bestimmt werden. Liegen keine kinetischen Daten vor, besteht ein akzeptabler Kompromiss für die Messung der Genotoxizität in der Probenahme 2 bis 6 Stunden nach der letzten Behandlung im Falle von zwei oder mehr Behandlungen oder 2 bis 6 und 16 bis 26 Stunden nach einer Einzelverabreichung. In einem solchen Fall ist jedoch darauf zu achten, dass alle Tiere gleichzeitig nach der letzten (oder einzigen) Dosis seziert werden. Ferner können Informationen über das Auftreten toxischer Wirkungen in Zielorganen (falls verfügbar) bei der Wahl geeigneter Probenahmezeitpunkte als Grundlage dienen.
Beobachtungen
Allgemeine klinische Beobachtungen in Bezug auf die Gesundheit der Tiere sollten mindestens einmal täglich, vorzugsweise zum gleichen Zeitpunkt und unter Berücksichtigung des Zeitraums, in dem der Wirkungsgipfel nach Verabreichung der Dosis zu erwarten ist, vorgenommen und protokolliert werden (54). Mindestens zweimal täglich sind alle Tiere auf Morbidität und Mortalität zu überprüfen. Bei länger andauernden Studien sollten alle Tiere mindestens einmal wöchentlich und am Ende des Prüfzeitraums gewogen werden. Die Futteraufnahme sollte bei jedem Futterwechsel und mindestens einmal wöchentlich gemessen werden. Wenn die Prüfchemikalie über das Trinkwasser verabreicht wird, sollte auch die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen werden. Tiere, die nicht letale Anzeichen übermäßiger Toxizität aufweisen, sollten vor Ende des Prüfzeitraums getötet werden und werden in der Regel nicht für die Comet-Analyse verwendet.
Gewebegewinnung
Da die Induktion von DNA-Strangbrüchen (Comets) praktisch in jedem Gewebe untersucht werden kann, sollte die Begründung der Auswahl der zu gewinnenden Gewebe eindeutig definiert werden und auf dem Grund für die Durchführung der Studie sowie vorliegenden Daten zu ADME, Genotoxizität, Karzinogenität oder sonstiger Toxizität für die untersuchte Prüfchemikalie beruhen. Wichtige zu berücksichtigende Faktoren sind der Verabreichungsweg (basierend auf dem bzw. den wahrscheinlichen Expositionspfaden beim Menschen), die vorhergesagte Resorption und Verteilung im Gewebe, die Rolle der Metabolisierung und der mögliche Wirkmechanismus der Prüfchemikalien. Die Leber ist das am häufigsten untersuchte Gewebe, für das auch die meisten Daten vorliegen. Wenn keine Hintergrundinformationen vorliegen und keine spezifischen zu untersuchenden Gewebe identifiziert werden, wäre daher eine Probenahme an der Leber gerechtfertigt, da sie ein primärer Ort des Metabolismus xenobiotischer Stoffe und häufig sowohl der/den ursprünglichen Chemikalie(n) als auch Metaboliten ausgesetzt ist. In einigen Fällen kann jedoch die Untersuchung einer direkten Kontaktstelle (z.B. bei oral verabreichten Chemikalien der Drüsenmagen oder Zwölffingerdarm/Dünndarm oder bei inhalierten Chemikalien die Lunge) am zweckmäßigsten sein. Zusätzliche oder alternative Gewebe sollten basierend auf den jeweiligen Gründen für die Durchführung des Versuchs gewählt werden. Allerdings kann es sinnvoll sein, mehrere Gewebe derselben Tiere zu untersuchen, vorausgesetzt, das Labor hat seine Kompetenz bei solchen Geweben sowie im Umgang mit mehreren Geweben gleichzeitig nachgewiesen.
Vorbereitung der Proben
Für die nachstehend (Nummern 44-49) beschriebenen Prozesse ist es wichtig, dass alle Lösungen oder stabilen Suspensionen vor ihrem Verfallsdatum verwendet oder bei Bedarf frisch zubereitet werden. In der nachfolgenden Beschreibung gelten die Zeiträume, um i) die einzelnen Gewebe nach der Sektion zu entnehmen, ii) die Gewebe in Zell-/Zellkernsuspensionen aufzuarbeiten und iii) die Suspension aufzuarbeiten und die Objektträger vorzubereiten, alle als kritische Variablen (siehe Definitionen in Anlage 1). Für jeden dieser Schritte sollte bei der Festlegung der Methode und beim Kompetenznachweis eine akzeptable Zeitdauer festgelegt worden sein.
Die Tiere werden im Einklang mit den einschlägigen Tierschutzvorschriften und nach dem 3R-Prinzip zu den entsprechenden Zeitpunkten nach der letzten Behandlung mit einer Prüfchemikalie getötet. Die ausgewählten Gewebe werden entfernt und seziert und ein Teil wird für den Comet-Assay gewonnen. Gleichzeitig sollte eine Sektion aus demselben Gewebeteil herausgeschnitten und für eine mögliche histopathologische Analyse (siehe Nummer 55) nach Standardmethoden (12) in eine Formaldehydlösung oder eine geeignete Fixierlösung gelegt werden. Das Gewebe für den Comet-Assay wird in einen Zerkleinerungspuffer gelegt, ausreichend mit kaltem Zerkleinerungspuffer gespült, um Restblut zu beseitigen, und bis zur Verarbeitung in eiskaltem Zerkleinerungspuffer aufbewahrt. Eine In-situ-Perfusion kann ebenfalls durchgeführt werden, z.B. bei Leber, Niere.
Es wurden zahlreiche Methoden für die Zell-/Zellkernisolierung veröffentlicht. Dazu gehören das Zerkleinern von Geweben wie beispielsweise Leber und Niere, das Abschaben der Schleimhautoberflächen des Magen-Darm-Trakts, die Homogenisierung und die enzymatische Verdauung. In der JaCVAM-Validierungsstudie wurden lediglich isolierte Zellen untersucht. Um die Methode festzulegen und um zwecks Nachweis der Kompetenz auf die JaCVAM-Versuchsdaten Bezug nehmen zu können, werden isolierte Zellen bevorzugt. Jedoch wurde gezeigt, dass sich das Testergebnis bei Verwendung von isolierten Zellen oder von Zellkernen nicht wesentlich unterschied (8). Ferner führten unterschiedliche Methoden für die Isolierung der Zellen (z.B. Homogenisierung, Zerkleinerung, enzymatische Verdauung und Siebfiltration) zu vergleichbaren Ergebnissen (55). Folglich können entweder isolierte Zellen oder Zellkerne verwendet werden. Das Labor sollte die gewebespezifischen Methoden zur Einzelzell-/Zellkernisolierung sorgfältig prüfen und validieren. Wie bereits unter Nummer 40 erwähnt, kann die Persistenz einiger Läsionen, die zu den vom Comet-Assay nachgewiesenen DNA-Strangbrüchen führen, sehr kurz sein (52) (53). Daher ist es ungeachtet der Methode für die Zubereitung der Einzelzell-/Zellkernsuspensionen wichtig, dass die Gewebe möglichst bald nach Tötung der Tiere aufbereitet und unter Bedingungen aufbewahrt werden, die den Verlust der Läsionen verringern (z.B. durch Lagerung bei niedriger Temperatur). Die Zellsuspensionen sollten bis zur Verwendung eiskalt aufbewahrt werden, sodass die Abweichungen zwischen Proben minimal sind und angemessene Reaktionen bei den Positiv- und Negativkontrollen nachgewiesen werden können.
Die Objektträger sollten schnellstmöglich (im Idealfall innerhalb einer Stunde) nach Zubereitung der Einzelzell-/Zellkernsuspensionen vorbereitet werden. Die Temperatur und die Zeit zwischen der Tötung der Tiere und der Vorbereitung der Objektträger sollten eng kontrolliert und unter Laborbedingungen validiert werden. Durch das Volumen der Zellsuspension, das der Agarose mit niedrigem Schmelzpunkt (gewöhnlich 0,5-1,0 %) zur Vorbereitung der Objektträger beigegeben wird, sollte der Anteil der Agarose mit niedrigem Schmelzpunkt nicht auf weniger als 0,45 % reduziert werden. Die optimale Zelldichte wird durch das Bildanalysesystem bestimmt, das zur Auswertung der Comets verwendet wird.
Lyse
Die Lysebedingungen stellen ebenfalls eine kritische Variable dar und können sich auf die Strangbrüche, die aus spezifischen Arten von DNA-Veränderungen resultieren (bestimmte Alkylierungen und Bildung von Addukten zu Basen der DNA), auswirken. Daher empfiehlt es sich, die Lysebedingungen bei allen Objektträgern innerhalb eines Versuchs so konstant wie möglich zu halten. Nach der Vorbereitung sollten die Objektträger mindestens eine Stunde (oder über Nacht) bei ungefähr 2-8 1 °C bei gedämpften Lichtverhältnissen (z.B. gelbes Licht (oder vor Licht geschützt)) zur Vermeidung einer Exposition gegenüber weißem Licht, das UV-Komponenten enthalten kann, in gekühlte Lyselösung eingelegt werden. Nach dieser Inkubationszeit sollten die Objektträger abgespült werden, um vor der alkalischen Entwindung Detergenz- und Salzreste zu beseitigen. Dies kann unter Verwendung von gereinigtem Wasser, neutralisierender Pufferlösung oder Phosphat-Pufferlösung erfolgen. Elektrophorese-Pufferlösung kann ebenfalls verwendet werden. Dies würde die alkalischen Bedingungen in der Elektrophoresekammer erhalten.
Entwindung und Elektrophorese
Die Objektträger sollten nach dem Zufallsprinzip auf die Plattform einer Unterwasser-Elektrophoreseapparatur gesetzt werden, die so viel Elektrophoreselösung enthält, dass die Oberflächen der Objektträger vollständig bedeckt sind (die Höhe der Bedeckung sollte bei allen Versuchsdurchführungen gleich sein). Bei anderen Arten von Comet-Assay-Elektrophoreseapparaturen, z.B. mit aktiver Kühlung, Umwälzung und Hochleistungsnetzteil, führt eine höhere Bedeckung durch das Lösungsmittel zu einer höheren Stromstärke bei konstanter Spannung. Die Objektträger sollten nach einem ausgewogenen Versuchsplan in den Elektrophoresetank eingelegt werden, damit die Wirkungen von Trends oder Randeffekte im Tank abgeschwächt und Schwankungen zwischen den Chargen möglichst gering gehalten werden. Dies bedeutet, dass jede Elektrophorese-Versuchsdurchführung die gleiche Anzahl von Objektträgern von jedem in der Studie eingeschlossenen Tier und Proben aus den verschiedenen Dosisgruppen, Negativ- und Positivkontrollen umfassen sollte. Die Objektträger sollten mindestens 20 Minuten im Elektrophoresetank belassen werden, damit die DNA sich entwinden kann, und dann unter kontrollierten Bedingungen elektrophoretisch untersucht werden. Diese Bedingungen sollten eine optimale Empfindlichkeit und dynamische Bandbreite des Tests garantieren (d. h. zu akzeptablen Werten des prozentualen DNA-Anteil im Schweif bei Negativ- und Positivkontrollen zur Optimierung der Empfindlichkeit führen). Das Ausmaß der DNA-Wanderung steht im linearen Verhältnis zur Dauer der Elektrophorese sowie zum Potenzial (V/cm). Auf der Grundlage der JaCVAM-Studie könnte dies 0,7 V/cm bei mindestens 20 Minuten sein. Die Dauer der Elektrophorese wird als kritische Variable betrachtet, und die Elektrophoresezeit sollte so eingestellt werden, dass die dynamische Bandbreite optimiert wird. Längere Elektrophoresezeiten (z.B. 30 oder 40 Minuten zur Maximierung der Empfindlichkeit) bewirken bei bekannten Mutagenen in der Regel stärkere Positivreaktionen. Allerdings können längere Elektrophoresezeiten auch zu einer übermäßigen Wanderung in den Kontrollproben führen. Die Spannung sollte in jedem Versuch konstant und die Variabilität der anderen Parameter in einem schmalen, festgelegten Bereich gehalten werden. In der JaCVAM-Studie ergab beispielsweise ein Wert von 0,7 V/cm eine anfängliche Stromstärke von 300 mA. Die Tiefe der Pufferlösung sollte so eingestellt werden, dass die erforderlichen Bedingungen erreicht und während des gesamten Versuchs eingehalten werden. Die Stromstärke am Anfang und Ende der Elektrophoresezeit sollte protokolliert werden. Daher sollten die optimalen Bedingungen während des anfänglichen Nachweises der Kompetenz im jeweiligen Labor bei jedem untersuchten Gewebe ermittelt werden. Die Temperatur der Elektrophoreselösung sollte während der Entwindung und Elektrophorese niedrig gehalten werden (in der Regel 2-10 1 °C) (10). Die Temperatur der Elektrophoreselösung am Anfang der Entwindung sowie am Anfang und Ende der Elektrophorese ist jeweils zu protokollieren.
Nach der Elektrophorese sollten die Objektträger mindestens 5 Minuten lang in die neutralisierende Pufferlösung eingelegt bzw. darin abgespült werden. Gele können angefärbt und "frisch" (z.B. innerhalb von 1-2 Tagen) ausgewertet oder zur späteren Auswertung (z.B. innerhalb von 1-2 Wochen nach Anfärbung) dehydriert werden (56). Allerdings sollten die Bedingungen während des Nachweises der Kompetenz validiert und historische Daten für jede dieser Bedingungen gesondert erfasst und protokolliert werden. Im letzteren Fall sollten die Objektträger zur Dehydration mindestens 5 Minuten lang in reines Ethanol eingelegt, an der Luft getrocknet und bis zur Auswertung entweder bei Zimmertemperatur oder in einem Behälter in einem Kühlschrank aufbewahrt werden.
Messmethoden
Comets sollten mithilfe eines automatischen oder halbautomatischen Bildanalysesystems quantitativ ausgewertet werden. Die Objektträger werden mit einem geeigneten fluoreszierenden Farbstoff, z.B. SYBR Gold, Green I, Propidiumjodid oder Ethidiumbromid, angefärbt und mit geeigneter Vergrößerung (z.B. 200x) unter einem Mikroskop mit Epi-Fluoreszenz- und entsprechenden Detektoren oder mit einer Digitalkamera (z.B. CCD-Kamera) gemessen.
Die Zellen lassen sich, wie im Atlas of Comet Assay Images (57) beschrieben, in drei Kategorien einteilen, nämlich in auswertbar, nicht auswertbar und Hedgehog (weitere Ausführungen unter Nummer 56). Nur auswertbare Zellen (eindeutig definierter Kopf und Schweif, keine Interferenzen mit Nachbarzellen) sollten für den prozentualen DNA-Anteil im Schweif ausgewertet werden, um Artefakte zu vermeiden. Die Häufigkeit von nicht auswertbaren Zellen muss nicht angegeben werden. Die Häufigkeit von Hedgehogs sollte durch visuelle Auswertung (da das Fehlen eines klar definierten Kopfes bedeutet, dass sie mittels Bildanalyse nicht leicht erkannt werden) von mindestens 150 Zellen pro Probe (näheren Erläuterungen unter Nummer 56) bestimmt und gesondert dokumentiert werden.
Alle zu analysierenden Objektträger, einschließlich der Positiv- und Negativkontrollen, sollten vor der Analyse unabhängig kodiert und "blind" ausgewertet werden, damit die Auswertung ohne Kenntnis der Behandlungsbedingungen erfolgt. Bei jeder Probe (pro Gewebe pro Tier) sollten mindestens 150 Zellen (ohne Hedgehogs - siehe Nummer 56) analysiert werden. Die Auswertung von 150 Zellen pro Tier bei mindestens fünf Tieren pro Dosis (weniger in der gleichzeitigen Positivkontrolle - siehe Nummer 29) gewährleistet entsprechend der Analyse von Smith et al., 2008, (5) eine hinreichende statistische Aussagekraft. Bei Verwendung von Objektträgern könnten dies zwei oder drei ausgewertete Objektträger pro Probe sein, wenn fünf Tiere pro Gruppe zum Einsatz kommen. Es sollten mehrere Bereiche des Objektträgers in einer solchen Dichte untersucht werden, dass eine Überschneidung von Schweifen ausgeschlossen ist. Die Auswertung am Rand der Objektträger ist zu vermeiden.
DNA-Strangbrüche im Comet-Assay können anhand von unabhängigen Endpunkten wie z.B. dem prozentualen DNA-Anteil im Schweif, der Schweiflänge und dem Schweifmoment gemessen werden. Sofern ein geeignetes Bildanalysesystem verwendet wird, können alle drei Messungen durchgeführt werden. Jedoch wird der prozentuale DNA-Anteil im Schweif (auch als prozentuale Schweif-Intensität bezeichnet) für die Bewertung und Interpretation der Ergebnisse empfohlen (12) (40) (41) (42). Dieser Anteil wird anhand der DNA-Fragmentintensität im Schweif, ausgedrückt als Prozentsatz der Gesamtintensität der Zelle, bestimmt (13).
Gewebeschädigung und Zytotoxizität
Positive Ergebnisse im Comet-Assay sind möglicherweise nicht allein auf Genotoxizität zurückzuführen sein. Auch toxische Wirkungen im Zielgewebe können eine Zunahme der DNA-Wanderung hervorrufen (12) (41). Hingegen ist bei bekannten Genotoxinen häufig eine geringe oder mäßige Zytotoxizität zu beobachten (12), was zeigt, dass der Comet-Assay alleine keine Unterscheidung zwischen DNA-Wanderung, die durch Genotoxizität ausgelöst wird, und durch Zytotoxizität hervorgerufener DNA-Wanderung erlaubt. Wenn jedoch eine Zunahme der DNA-Wanderung beobachtet wird, wird empfohlen, eine Untersuchung auf einen oder mehrere Indikatoren für Zytotoxizität durchzuführen, da dies bei der Interpretation der Ergebnisse hilfreich sein kann. Eine Zunahme der DNA-Wanderung ist bei Vorliegen eindeutiger Anzeichen von Zytotoxizität mit Vorsicht zu interpretieren.
Es wurden zahlreiche Messgrößen für die Zytotoxität vorgeschlagen, wobei histopathologische Veränderungen als relevante Messgröße für Gewebetoxizität gelten. Beobachtungen wie Entzündung, Zellinfiltration, apoptotische oder nekrotische Veränderungen wurden mit einer Zunahme der DNA-Wanderung in Verbindung gebracht. Wie die JaCVAM-Validierungsstudie (12) gezeigt hat, gibt es jedoch keine endgültige Liste der mit einer Zunahme der DNA-Wanderung stets einhergehenden, histopathologischen Veränderungen. Änderungen bestimmter biologischer Parameter (z.B. AST, ALT) können ebenfalls nützliche Informationen über die Gewebeschädigung liefern, und ferner können zusätzliche Indikatoren wie z.B. Caspase-Aktivierung, TUNEL-Färbung, Annexin-V-Färbung usw. in Betracht gezogen werden. Allerdings wurden über die Verwendung dieser Indikatoren in In-vivo-Studien nur wenig Daten veröffentlicht, und nicht alle sind gleich zuverlässig.
Hedgehogs (oder Clouds, Geisterzellen) sind Zellen, deren mikroskopisches Bild einen kleinen oder nicht existenten Kopf und lange, diffuse Schweife zeigt und die als schwer beschädigte Zellen gelten, obwohl die Genese der Hedgehogs unsicher ist (siehe Anlage 3). Aufgrund ihres Aussehens sind Messungen des prozentualen Anteils der Schweif-DNA mittels Bildanalyse unzuverlässig, weshalb Hedgehogs gesondert zu bewerten sind. Das Auftreten von Hedgehogs sollte vermerkt und angegeben werden, und jede relevante Zunahme, bei der davon ausgegangen wird, dass sie auf die Prüfchemikalie zurückzuführen ist, ist zu untersuchen und mit Vorsicht zu interpretieren. Bei solchen Überlegungen kann die Kenntnis der potenziellen Wirkungsweise der Prüfchemikalien von Nutzen sein.
Behandlung der Ergebnisse
Die Versuchseinheit ist das Tier. Daher sollten sowohl die Daten für die einzelnen Tiere als auch die zusammengefassten Ergebnisse in tabellarischer Form dargestellt werden. Aufgrund der hierarchischen Art der Daten wird empfohlen, für jeden Objektträger den Median des prozentualen Anteils der Schweif-DNA zu bestimmen und für jedes Tier den Mittelwert der Mediane zu berechnen (12). Anschließend wird der Mittelwert aus den Mittelwerten für die einzelnen Tiere bestimmt, um den Gruppenmittelwert zu erhalten. Alle diese Werte sind in den Bericht aufzunehmen. Alternative Ansätze (siehe Nummer 53) können verwendet werden, sofern dies wissenschaftlich und statistisch begründet ist. Statistische Analysen können nach verschiedenen Ansätzen durchgeführt werden (58) (59) (60) (61). Bei der Auswahl der anzuwendenden statistischen Methoden sollte die Notwendigkeit einer Transformation (z.B. Logarithmus oder Quadratwurzel) der Daten und/oder der Addition einer kleinen Zahl (z.B. 0,001) zu allen Werten (auch Werten ungleich Null), wie in den obigen Referenzdokumenten erörtert, berücksichtigt werden, um die Wirkungen von Null-Zellwerten abzuschwächen. Nähere Informationen zur Analyse von Wechselwirkungen zwischen Behandlung und Geschlecht bei Verwendung beider Geschlechter sowie zur nachfolgenden Analyse der Daten, wenn Unterschiede bestehen oder keine Unterschiede festgestellt werden, sind Anlage 2 zu entnehmen. Daten zur Toxizität bei Tieren und klinische Anzeichen sind ebenfalls anzugeben.
Gültigkeitskriterien
Die Akzeptanz eines Versuchs beruht auf folgenden Kriterien:
Bewertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn
Sind alle diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie DNA-Strangbrüche in den in diesem Versuchssystem untersuchten Geweben auslösen kann. Sind nur ein oder zwei dieser Kriterien erfüllt, siehe Nummer 62.
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn
Es wird dann davon ausgegangen, dass die Prüfchemikalie keine DNA-Strangbrüche in den in diesem Versuchssystem untersuchten Geweben auslösen kann.
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
In den Fällen, in denen die Reaktion, weder eindeutig negativ noch eindeutig positiv ist (d. h. nicht alle unter Nummer 59 oder 60 genannten Kriterien sind erfüllt), und um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung bewertet und/oder weitere Untersuchungen durchgeführt werden, wenn dies wissenschaftlich begründet ist. Die Auswertung weiterer Zellen (soweit dies angebracht ist) oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter optimierten Versuchsbedingungen (z.B. Abstände der Dosierungen, andere Verabreichungswege, andere Probenahmezeitpunkte oder andere Gewebe), könnte hilfreich sein.
In seltenen Fällen erlaubt der Datensatz selbst nach weiteren Untersuchungen keine definitive Aussage zu positiven oder negativen Ergebnissen, so dass die Reaktion als nicht eindeutig eingestuft wird.
Um die biologische Relevanz eines positiven oder mehrdeutigen Ergebnisses zu bewerten, sind Informationen zur Zytotoxizität für das Zielgewebe erforderlich (siehe Nummern 54 und 55). Wenn lediglich im Falle von eindeutigen Anzeichen für zytotoxische Wirkungen positive oder mehrdeutige Ergebnisse beobachtet werden, würde die Studie als nicht eindeutig im Hinblick auf die Genotoxizität abgeschlossen werden, außer wenn genügend Informationen vorliegen, die eine endgültige Schlussfolgerung zulassen. Bei einem negativen Studienergebnis mit Anzeichen für Toxizität bei allen untersuchten Dosierungen sind weitere Untersuchungen bei nichttoxischen Dosierungen anzuraten.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalie:
Einkomponentige Substanz:
Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
Lösungsmittel/Vehikel:
Versuchstiere:
Prüfbedingungen:
Ergebnisse:
Erörterung der Ergebnisse
Schlussfolgerung
Referenzdokumente
(1) Kirkland, D., G. Speit (2008), Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens III. Appropriate follow-up testing in vivo, Mutation Research, Vol. 654/2, 114-32.
(2) Brendler-Schwaab, S. et al. (2005), The in vivo Comet assay: use and status in genotoxicity testing, Mutagenesis, Vol. 20/4, 245-54.
(3) Burlinson, B. et al. (2007), Fourth International Workgroup on Genotoxicity Testing: result of the in vivo Comet assay workgroup, Mutation Research, Vol. 627/1, 31-5.
(4) Burlinson, B. (2012), The in vitro and in vivo Comet assays, Methods in Molecular Biology, Vol. 817, 143-63.
(5) Smith, C.C. et al. (2008), Recommendations for design of the rat Comet assay, Mutagenesis, Vol. 23/3, 233-40.
(6) Hartmann, A. et al. (2003), Recommendations for conducting the in vivo alkaline Comet assay, Mutagenesis, Vol. 18/1, 45-51.
(7) McKelvey-Martin, V.J. et al. (1993), The single cell gel electrophoresis assay (Comet assay): a European review, Mutation Research, Vol. 288/1, 47-63.
(8) Tice, R.R. et al. (2000), Single cell gel/Comet assay: guidelines for in vitro and in vivo genetic toxicology testing, Environmental and Molecular Mutagenesis, Vol. 35/3, 206-21.
(9) Singh, N.P. et al. (1988), A simple technique for quantitation of low levels of DNA damage in individual cells, Experimental Cell Research, Vol. 175/1, 184-91.
(10) Rothfuss, A. et al. (2010), Collaborative study on fifteen compounds in the rat-liver Comet assay integrated into 2- and 4-week repeat-dose studies, Mutation Research, Vol. 702/1, 40-69.
(11) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(12) OECD (2014), Reports of the JaCVAM initiative international pre-validation and validation studies of the in vivo rodent alkaline comet assay for the detection of genotoxic carcinogens, Series on Testing and Assessment, Nos. 195 and 196, OECD Publishing, Paris.
(13) Olive, P.L., J.P. Banath, R.E. Durand (1990), Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells using the "Comet" assay, Radiation Research, Vol. 122/1, 86-94.
(14) Tice, R.R., D.L. Strauss (1995), The single cell gel electrophoresis/Comet assay: a potential tool for detecting radiation-induced DNA damage in humans, Stem Cells, Vol. 13/1, 207-14.
(15) Collins, A.R (2004), The Comet assay for DNA damage and repair: principles, applications, and limitations, Molecular Biotechnology, Vol. 26/3, 249-61.
(16) Rothfuss, A. et al. (2011), Improvement of in vivo genotoxicity assessment: combination of acute tests and integration into standard toxicity testing, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 723/2, 108-20.
(17) Kushwaha, S. et al. (2010), Evaluation of multi-organ DNA damage by Comet assay from 28 days repeated dose oral toxicity test in mice: A practical approach for test integration in regulatory toxicity testing, Regulatory Toxicology and Pharmacology, Vol. 58/1, 145-54.
(18) Vasquez, M.Z. (2010), Combining the in vivo Comet and micronucleus assays: a practical approach to genotoxicity testing and data interpretation, Mutagenesis, Vol. 25/2, 187-99.
(19) Bowen, D.E. (2011), Evaluation of a multi-endpoint assay in rats, combining the bone-marrow micronucleus test, the Comet assay and the flow-cytometric peripheral blood micronucleus test, Mutation Research, Vol. 722/1, 7-19.
(20) Recio, L. et al. (2010), Dose-response assessment of four genotoxic chemicals in a combined mouse and rat micronucleus (MN) and Comet assay protocol, The Journal of Toxicological Science, Vol. 35/2, 149-62.
(21) O'Donovan, M., B. Burlinson (2013), Maximum dose levels for the rodent comet assay to examine damage at the site of contact or to the gastrointestinal tract, Mutagenesis, Vol. 28/6, 621-3.
(22) Hartmann, A. (2004), Use of the alkaline in vivo Comet assay for mechanistic genotoxicity investigations, Mutagenesis, Vol. 19/1, 51-9.
(23) Nesslany, F. (2007), In vivo Comet assay on isolated kidney cells to distinguish genotoxic carcinogens from epigenetic carcinogens or cytotoxic compounds, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 630/1, 28-41.
(24) Brendler-Schwaab, S.Y., B.A. Herbold (1997), A new method for the enrichment of single renal proximal tubular cells and their first use in the Comet assay, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 393/1-2, 175-8.
(25) Toyoizumi, T. et al. (2011), Use of the in vivo skin Comet assay to evaluate the DNA-damaging potential of chemicals applied to the skin, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 726/2, 175-80.
(26) Struwe, M. et al. (2008), Detection of photogenotoxicity in skin and eye in rat with the photo Comet assay, Photochemical and Photobiological Sciences, Vol. 7/2, 240-9.
(27) Wada, K. et al. (2012), A comparison of cell-collecting methods for the Comet assay in urinary bladders of rats, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 742/1-2, 26-30.
(28) Wang, A. et al. (2007), Measurement of DNA damage in rat urinary bladder transitional cells: improved selective harvest of transitional cells and detailed Comet assay protocols, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 634/ 1-2, 51-9.
(29) Burlinson, B. et al. (2007), In Vivo Comet Assay Workgroup, part of the Fourth International Workgroup on Genotoxicity Testing. Fourth International Workgroup on Genotoxicity testing: results of the in vivo Comet assay workgroup, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 627/1, 31-5.
(30) Jackson, P. et al. (2012), Pulmonary exposure to carbon black by inhalation or instillation in pregnant mice: effects on liver DNA strand breaks in dams and offspring, Nanotoxicology, Vol. 6/5, 486-500.
(31) Sasaki, Y.F. et al. (2000), The comet assay with multiple mouse organs: comparison of Comet assay results and carcinogenicity with 208 chemicals selected from the IARC monographs and U.S. NTP Carcinogenicity Database, Critical Reviews in Toxicology, Vol. 30/6, 629-799.
(32) Sekihashi, K. et al. (2002), Comparative investigations of multiple organs of mice and rats in the Comet assay, Mutation Research, Vol. 517/1-2, 53-74.
(33) Speit, G, M. Vasquez, A. Hartmann (2009), The comet assay as an indicator test for germ cell genotoxicity, Mutation Research, Vol. 681/1, 3-12.
(34) Zheng, H., P.L. Olive (1997), Influence of oxygen on radiation-induced DNA damage in testicular cells of C3H mice, International Journal of Radiation Biology, Vol. 71/3, 275-282.
(35) Cordelli, E. et al. (2003), Evaluation of DNA damage in different stages of mouse spermatogenesis after testicular X irradiation, Journal of Radiation Research, Vol. 160/4, 443-451.
(36) Merk, O., G. Speit (1999), Detection of crosslinks with the Comet assay in relationship to genotoxicity and cytotoxicity, Environmental and Molecular Mutagenesis, Vol. 33/2, 167-72.
(37) Pfuhler, S., H.U. Wolf (1996), Detection of DNA-crosslinking agents with the alkaline Comet assay, Environmental and Molecular Mutagenesis, Vol. 27/3, 196-201.
(38) Wu, J.H., N.J. Jones (2012), Assessment of DNA interstrand crosslinks using the modified alkaline Comet assay, Methods in Molecular Biology, Vol. 817, 165-81.
(39) Spanswick, V.J., J.M. Hartley, J.A. Hartley (2010), Measurement of DNA interstrand crosslinking in individual cells using the Single Cell Gel Electrophoresis (Comet) assay, Methods in Molecular Biology, Vol. 613, 267-282.
(40) Kumaravel, T.S., A.N. Jha (2006), Reliable Comet assay measurements for detecting DNA damage induced by ionizing radiation and chemicals, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 605(1-2), 7-16.
(41) Burlinson, B. et al. (2007), Fourth International Workgroup on Genotoxicity Testing: result of the in vivo Comet assay workgroup, Mutation Research, Vol. 627/1, 31-5.
(42) Kumaravel, T.S. et al. (2009), Comet Assay measurements: a perspective, Cell Biology and Toxicology, Vol. 25/1, 53-64.
(43) Ersson, C., L. Möller (2011), The effects on DNA migration of altering parameters in the Comet assay protocol such as agarose density, electrophoresis conditions and durations of the enzyme or the alkaline treatments, Mutagenesis, Vol. 26/6, 689-95.
(44) Møller, P. et al. (2010), Assessment and reduction of Comet assay variation in relation to DNA damage: studies from the European Comet Assay Validation Group, Mutagenesis, Vol. 25/2, 109-11.
(45) Forchhammer, L. et al. (2010), Variation in the measurement of DNA damage by Comet assay measured by the ECVAG inter-laboratory validation trial, Mutagenesis, Vol. 25/2, 113-23.
(46) Azqueta, A. et al. (2011), Towards a more reliable comet assay: Optimising agarose concentration, unwinding time and electrophoresis conditions, Mutation Research, Vol. 724/1-2, 41-45.
(47) Hayashi, M. et al. (2011), Compilation and use of genetic toxicity historical control data, Mutation Research, Vol. 723/2, 87-90.
(48) Ryan, T. P. (2000), Statistical Methods for Quality Improvement, John Wiley and Sons, New York 2nd ed.
(49) Appendix A of the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes (ETS No. 123)
(50) Kapitel B.8 dieses Anhangs: Prüfung auf subakute Toxizität nach Inhalation: 28-Tage-Test.
(51) Kapitel B.29 dieses Anhangs: Prüfung auf subchronische Toxizität nach Inhalation: 90-Tage-Test.
(52) Blakey, D.H., G.R. Douglas (1984), Transient DNA lesions induced by benzo[a]pyrene in Chinese hamster ovary cells, Mutation Research, Vol. 140/2-3, 141-45.
(53) Blakey, D.H., G.R. Douglas (1990), The role of excision repair in the removal of transient benzo[a]pyrene-induced DNA lesions in Chinese hamster ovary cells, Mutation Research, Vol. 236/1, 35-41.
(54) OECD (2002), "Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation", OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, No. 19, OECD Publishing, Paris.
(55) Nakajima, M. (2012), Tissue sample preparation for in vivo rodent alkaline Comet assay, Genes and Environment, Vol. 34/1, 50-4.
(56) Hartmann, A. et al. (2003), Recommendations for conducting the in vivo alkaline Comet assay, Mutagenesis, Vol.18/1, 45-51.
(57) Atlas of Comet Assay Images, Scientist Press Co., Ltd., Tokio, Japan.
(58) Lovell, D.P., G. Thomas, R. Dubow (1999), Issues related to the experimental design and subsequent statistical analysis of in vivo and in vitro Comet studies, Teratogenesis Carcinogenesis Mutagenesis, Vol. 19/2, 109-19.
(59) Wiklund, S.J., E. Agurell (2003), Aspects of design and statistical analysis in the Comet assay, Mutagenesis, Vol. 18/2, 167-75.
(60) Bright, J. et al. (2011), Recommendations on the statistical analysis of the Comet assay, Pharmaceutical Statistics, Vol. 10/6, 485-93.
(61) Lovell, D.P., T. Omori (2008), Statistical issues in the use of the Comet assay, Mutagenesis, Vol. 23/3, 171-82.
| Definitionen | Anlage 1 |
Definitionen
Alkalische Einzelzell-Gelelektrophorese: Empfindliches Verfahren zum Nachweis primärer DNA-Schädigungen auf Einzelzell-/Zellkernebene.
Chemikalie: Stoff oder Gemisch.
Comet: Kometähnliche Form, die Nucleoide nach Einwirkung eines elektrophoretischen Felds annehmen: Der Kopf ist der Zellkern, und der Schweif wird durch die DNA gebildet, die aufgrund des elektrischen Felds aus dem Zellkern gewandert ist.
Kritische Variable/kritischer Parameter: Dies ist eine Protokollvariable, bei der sich eine kleine Änderung erheblich auf die Schlussfolgerung des Tests auswirken kann. Kritische Variablen können gewebespezifisch sein. Kritische Variablen sollten (insbesondere innerhalb eines Tests) nicht geändert werden, ohne zu prüfen, wie sich die Änderung auf die Assay-Reaktion auswirkt, wie beispielsweise durch die Größenordnung und Variabilität bei den Positiv- und Negativkontrollen angedeutet. Im Prüfbericht sollten Änderungen an kritischen Variablen, die während des Tests oder gegenüber dem Standardprotokoll des Labors vorgenommen wurden, angegeben und einzeln begründet werden.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Schweif-Intensität oder % DNA im Schweif: Dies entspricht der Intensität des Comet-Schweifs bezogen auf die Gesamtintensität (Kopf plus Schweif) und spiegelt den als Prozentsatz ausgedrückten Anteil der DNA-Schädigung wider.
UVCB: Stoffe mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien.
| Faktorieller Versuchsplan zur Ermittlung geschlechtsspezifischer Unterschiede beim In-vivo-Comet-Assay | Anlage 2 |
Faktorieller Versuchsplan und Analyse
Bei diesem Versuchsplan werden mindestens fünf männliche und fünf weibliche Tiere je Konzentration getestet. Dies ergibt einen Versuch, bei dem mindestens 40 Tiere verwendet werden (20 männliche und 20 weibliche zzgl. entsprechender Positivkontrollen).
Der Versuchsaufbau, der zu den einfacheren faktoriellen Versuchsplänen zählt, entspricht einer zweifaktoriellen Varianzanalyse, wobei Geschlecht und Konzentration die Hauptfaktoren sind. Die Daten können anhand vieler Standard-Statistiksoftwareanwendungen wie SPSS, SAS, STATA oder Genstat oder auch mit R analysiert werden.
Bei der Analyse wird die Varianz im Datensatz in die zwischen den Geschlechtern, die zwischen den Konzentrationen und die in Zusammenhang mit den Interaktionen zwischen den Geschlechtern und Konzentrationen partinioniert. Jeder der Terme wird anhand eines Schätzwerts der Varianz zwischen den Replikatversuchstieren innerhalb der gleichgeschlechtlichen Tiergruppen überprüft, die die gleiche Konzentration erhalten haben. Die zugrundeliegende Methodik ist in vielen Standardwerken zur Statistik (siehe Referenzdokumente) und in den in Statistiksoftware-Pakten mitgelieferten Hilfe-Funktionen im Einzelnen beschrieben.
Die Analyse erfolgt durch Prüfung des Terms der Interaktion "Geschlecht x Konzentration" in der ANOVA-Tabelle 1. Liegt kein signifikanter Term für die Interaktion vor, ergeben die kombinierten Werte für beide Geschlechter oder mehrere Konzentrationen eine zulässige Grundlage für statistische Tests zwischen den Konzentrationen aufgrund des Terms der ANOVA für die Varianz der innerhalb eines solchen Gruppe zusammengefassten Werte.
Die Analyse wird fortgesetzt mit der Partitionierung der geschätzten Varianz zwischen den Konzentrationen in kontrastierende Klassen, die die Grundlage bilden für einen Test auf lineare und quadratische Abhängigkeit der Reaktionen bei den verschiedenen Konzentrationen. Liegt dagegen eine signifikante Interaktion "Geschlecht x Konzentration" vor, so kann dieser Term auch in Gegenüberstellung der Interaktion "linear x Geschlecht" und "quadratisch x Geschlecht" partitioniert werden. Anhand dieser Terme kann geprüft werden, ob die Reaktionen auf die Konzentrationen bei beiden Geschlechtern parallel verlaufen oder ob die Geschlechter unterschiedlich reagieren.
Der Schätzwert für die Varianz der innerhalb der Gruppen zusammengefassten Werte kann als Grundlage für paarweise Tests zu Abweichungen zwischen den Mittelwerten dienen. Diese Vergleiche könnten zwischen den Mittelwerten für die beiden Geschlechter und zwischen den Mittelwerten für die verschiedenen Konzentrationen durchgeführt werden, beispielsweise um einen Vergleich mit den Negativkontrollen vorzunehmen. Bei signifikanter Interaktion können die Mittelwerte verschiedener Konzentrationen innerhalb eines Geschlechts oder die Mittelwerte beider Geschlechter bei derselben Konzentration verglichen werden.
Referenzdokumente
Es sind viele Werke zur Statistik erhältlich, in denen die Theorie, der Aufbau, die Methodik, die Analyse und die Interpretation faktorieller Versuchspläne erörtert werden, von einfachen Zweifaktorenanalysen bis hin zu komplexeren Formen, wie sie in der "Design of Experiment"-Methode verwendet werden. Die folgende Auflistung ist nicht erschöpfend. Einige Bücher enthalten Beispiele für die Anwendung vergleichbarer Versuchspläne, in einigen Fällen auch mit einem Code zur Durchführung der Analysen unter Verwendung verschiedener Softwarepakete.
(1) Box, G.E.P, Hunter, W.G. and Hunter, J.S. (1978). Statistics for Experimenters. An Introduction to Design, Data Analysis, and Model Building. New York: John Wiley & Sons.
(2) Box G.E.P. & Draper, N.R. (1987) Empirical model-building and response surfaces. John Wiley & Sons Inc.
(3) Doncaster, C.P. & Davey, A.J.H. (2007) Analysis of Variance and Covariance: How to choose and Construct Models for the Life Sciences. Cambridge University Press.
(4) Mead, R. (1990) The Design of Experiments. Statistical principles for practical application. Cambridge University Press.
(5) Montgomery D.C. (1997) Design and Analysis of Experiments. John Wiley & Sons Inc.
(6) Winer, B.J. (1971) Statistical Principles in Experimental Design. McGraw Hill.
(7) Wu, C.F.J & Hamada, M.S. (2009) Experiments: Planning, Analysis and Optimization. John Wiley & Sons Inc.
_____
1) Statistiker, die mit einem Modellierungsansatz wie dem Ansatz der allgemeinen linearen Modelle (GLM) arbeiten, folgen bei der Analyse möglicherweise einem anderen, wenn auch vergleichbarem Ansatz, werden jedoch nicht notwendigerweise eine Herleitung der herkömmlichen Anova-Tabelle vornehmen, die auf algorithmische Lösungswege für statistische Berechnungen aus dem Vor-Computerzeitalter zurückgeht.
| Gegenwärtige Einsatzgrenzen des Tests | Anlage 3 |
Nach aktuellem Kenntnisstand sind mit dem In-vivo-Comet-Assay verschiedene Einsatzgrenzen verbunden. Es wird davon ausgegangen, dass diese Einsatzgrenzen in dem Maße verringert oder enger definiert werden, wie zunehmende Erfahrungen mit der Anwendung des Tests zur Klärung von Sicherheitsfragen in einem regulatorischen Umfeld beitragen.
1. Einige Arten von DNA-Schädigungen können kurzlebig sein, d. h. zu rasch repariert werden, um 24 Stunden oder noch später nach der letzten Dosis beobachtet werden zu können. Es liegt weder eine Liste der Arten von kurzlebigen Schädigungen noch der Chemikalien vor, die diese Art von Schädigung wahrscheinlich auslösen, und es ist zudem nicht bekannt, über welchen Zeitraum diese Art von Schädigung nachweisbar ist. Die optimalen Probenahmezeitpunkte können außerdem spezifisch für die jeweilige Chemikalie oder den Verabreichungsweg sein und sollten auf kinetischen Daten (z.B. Zeitpunkt Tmax, zu dem die höchste Plasma- oder Gewebekonzentration erreicht wird) basieren, falls solche Daten vorliegen. In den meisten Validierungsstudien, auf die sich diese Prüfmethode stützt, wurde eine Sektion 2 oder 3 Stunden nach Verabreichung der letzten Dosis angegeben. In der veröffentlichten Literatur wird beschrieben, dass bei den meisten Studien die letzte Dosis 2 bis 6 Stunden vor der Tötung verabreicht wurde. Daher wurden diese Versuche als Grundlage für die Empfehlung in der Prüfmethode verwendet, wonach in Ermangelung von Daten, die eine andere Vorgehensweise nahelegen, die letzte Dosis zu einem festgelegten Zeitpunkt zwischen 2 und 6 Stunden vor der Sektion verabreicht werden sollte.
2. Es gibt keine validierten Daten zur Empfindlichkeit des Versuchs in Bezug auf den Nachweis kurzlebiger DNA-Schädigungen nach Verabreichung im Futter oder Trinkwasser im Vergleich zur Verabreichung über eine Magensonde. DNA-Schädigungen nach Verabreichung im Futter oder Trinkwasser wurden zwar nachgewiesen, aber es gibt nur wenige solcher Berichte verglichen mit der weitaus größeren Erfahrung mit Magensonden und intraperitonealer Verabreichung. Somit kann die Empfindlichkeit des Tests bei Chemikalien, die bei Verabreichung im Futter oder Trinkwasser kurzlebige Schädigungen auslösen, reduziert sein.
3. Da keine Interlaborstudien an anderen Geweben als Leber und Magen durchgeführt wurden, gibt es keine Empfehlung dazu, wie eine empfindliche und reproduzierbare Reaktion in anderen Geweben als Leber, wie z.B. zu erwartende Bereiche bei Positiv- und Negativkontrollen, zu erreichen ist. Für die Leber konnte ferner keine Einigung über die Festlegung einer niedrigeren Grenze für den Negativkontrollwert erzielt werden.
4. Obwohl in verschiedenen Veröffentlichungen die unklare zytotoxische Wirkung in vitro aufgezeigt wurde, wurden nur sehr wenige In-vivo-Daten veröffentlicht, sodass keine einzelne Messgröße für die Zytotoxizität empfohlen werden konnte. Histopathologische Veränderungen wie Entzündung, Zellinfiltration, apoptotische oder nekrotische Veränderungen wurden mit einer Zunahme der DNA-Wanderung in Verbindung gebracht. Wie die JaCVAM-Validierungsstudie (OECD, 2014) gezeigt hat, führen diese Veränderungen nicht immer zu positiven Comet-Ergebnissen, sodass keine endgültige Liste der stets mit einer Zunahme der DNA-Wanderung einhergehenden, histopathologischen Veränderungen vorliegt. Es wurde vorgeschlagen, Hedgehogs (oder Clouds, Geisterzellen) als Indikator für Zytotoxizität zu verwenden, doch die Genese der Hedgehogs ist unklar. Es liegen Daten vor, die darauf hindeuten, dass sie durch Zytotoxizität der Chemikalien, mechanisch/enzyminduzierte Schäden, die während der Aufbereitung der Proben hervorgerufen werden (Guerard et al., 2014), und/oder eine extremere Wirkung der Genotoxizität der Prüfchemikalie verursacht werden können. Andere Daten scheinen darauf hinzudeuten, dass sie auf umfangreiche, aber vielleicht reparierbare DNA-Schädigungen zurückzuführen sind (Lorenzo et al., 2013).
5. Gewebe oder Zellkerne wurden erfolgreich für eine spätere Analyse eingefroren. Dies führt gewöhnlich zu einem messbaren Effekt bei der Reaktion auf die Vehikel- und Positivkontrolle (Recio et al., 2010; Recio et al., 2012; Jackson et al., 2013). Wenn das Labor dieses Verfahren anwendet, sollte es seine Kompetenz bei Einfriermethoden nachweisen und belegen, dass die Bereiche der prozentualen DNA-Anteile im Schweif in den Zielgeweben bei den mit dem Vehikel behandelten Tieren ausreichend niedrig sind und positive Reaktionen noch nachweisbar sind. In der Literatur wurden verschiedene Methoden für das Einfrieren von Gewebe beschrieben. Allerdings besteht kein Einvernehmen darüber, wie Gewebe am besten einzufrieren und aufzutauen sind und wie zu bewerten ist, ob eine potenziell geänderte Wirkung die Empfindlichkeit des Tests beeinträchtigen kann.
6. Neuere Arbeiten zeigen, dass davon auszugehen ist, dass die Liste der kritischen Variablen weiter verkürzt wird und die Parameter für kritische Variablen näher definiert werden (Guerard et al., 2014).
Referenzdokumente
(1) Guerard, M., C. Marchand, U. Plappert-Helbig (2014), Influence of Experimental Conditions on Data Variability in the Liver Comet Assay, Environmental and Molecular Mutagenesis, Vol. 55/2, pp. 114-21.
(2) Jackson, P. et al. (2013), Validation of use of frozen tissues in high-throughput comet assay with fully-automatic scoring, Mutagenesis, Vol. 28/6, pp. 699-707.
(3) Lorenzo, Y. et al. (2013), The comet assay, DNA damage, DNA repair and cytotoxicity: hedgehogs are not always dead, Mutagenesis, Vol. 28/4, pp. 427-32.
(4) OECD (2014), Reports of the JaCVAM initiative international pre-validation and validation studies of the in vivo rodent alkaline comet assay for the detection of genotoxic carcinogens, Series on Testing and Assessment, Nos. 195 and 196, OECD Publishing, Paris.
(5) Recio L, Hobbs C, Caspary W, Witt KL, (2010), Dose-response assessment of four genotoxic chemicals in a combined mouse and rat micronucleus (MN) and Comet assay protocol, J. Toxicol. Sci. 35:149-62.
(6) Recio, L. et al. (2012), Comparison of Comet assay dose-response for ethyl methanesulfonate using freshly prepared versus cryopreserved tissues, Environmental and Molecular Mutagenesis, Vol. 53/2, pp. 101-13.
| weiter . | |