 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2017, Chemikalien - EU Bund
| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk, EU 2017, Chemikalien - EU Bund |
Verordnung (EU) 2017/735 der Kommission vom 14. Februar 2017 zur Änderung - zwecks Anpassung an den technischen Fortschritt - des Anhangs der Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH)
(Text von Bedeutung für den EWR)
(ABl. Nr. L 112 vom 28.04.2017 S. 1)
Die Europäische Kommission -
gestützt auf den Vertrag über die Arbeitsweise der Europäischen Union,
gestützt auf die Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Chemikalienagentur, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission 1, insbesondere auf Artikel 13 Absatz 2,
in Erwägung nachstehender Gründe:
(1) In der Verordnung (EG) Nr. 440/2008 der Kommission 2 sind die zum Zwecke der Verordnung (EG) Nr. 1907/2006 anzuwendenden Prüfmethoden für die Bestimmung der physikalisch-chemischen Eigenschaften, der Toxizität und der Ökotoxizität von Chemikalien festgelegt.
(2) Die Verordnung (EG) Nr. 440/2008 muss aktualisiert und um die von der Organisation für wirtschaftliche Zusammenarbeit und Entwicklung (OECD) kürzlich angenommenen neuen und aktualisierten Prüfmethoden ergänzt werden, um dem technischen Fortschritt Rechnung zu tragen und sicherzustellen, dass die Zahl der für wissenschaftliche Zwecke verwendeten Tiere im Sinne der Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates 3 verringert wird. Interessenträger wurden zu diesem Entwurf konsultiert.
(3) Diese Anpassung an den technischen Fortschritt betrifft 20 Prüfmethoden: eine neue Methode zur Bestimmung einer physikalisch-chemischen Eigenschaft, fünf neue und eine aktualisierte Prüfmethode zur Bewertung der Ökotoxizität, zwei aktualisierte Prüfmethoden zur Bewertung des Verbleibs und Verhaltens von Stoffen in der Umwelt sowie vier neue und sieben aktualisierte Prüfmethoden zur Bestimmung von Wirkungen auf die menschliche Gesundheit.
(4) Die OECD überprüft ihre Prüfrichtlinien regelmäßig, um jene auszusondern, die wissenschaftlich überholt sind. Mit dieser Anpassung an den technischen Fortschritt werden auch sechs Prüfmethoden gestrichen, deren zugrunde liegende OECD-Prüfrichtlinien ebenfalls gestrichen wurden.
(5) Die Verordnung (EG) Nr. 440/2008 sollte daher entsprechend geändert werden.
(6) Die in dieser Verordnung vorgesehenen Maßnahmen entsprechen der Stellungnahme des mit Artikel 133 der Verordnung (EG) Nr. 1907/2006 eingesetzten Ausschusses
- hat folgende Verordnung erlassen:
Der Anhang der Verordnung (EU) Nr. 440/2008 wird gemäß dem Anhang der vorliegenden Verordnung geändert.
Diese Verordnung tritt am zwanzigsten Tag nach ihrer Veröffentlichung im Amtsblatt der Europäischen Union in Kraft.
Diese Verordnung ist in allen ihren Teilen verbindlich und gilt unmittelbar in jedem Mitgliedstaat.
2) Verordnung (EG) Nr. 440/2008 der Kommission vom 30. Mai 2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (ABl. Nr. L 142 vom 31.05.2008 S. 1).
3) Richtlinie 2010/63/EU des Europäischen Parlaments und des Rates vom 22. September 2010 zum Schutz der für wissenschaftliche Zwecke verwendeten Tiere (ABl. Nr. L 276 vom 20.10.2010 S. 33).
| Anhang |
Der Anhang zur Verordnung (EG) Nr. 440/2008 wird wie folgt geändert:
(1) In Teil A wird das folgende Kapitel hinzugefügt:
"A.25 Dissoziationskonstanten in Wasser (Titrationsverfahren - Spektrofotometrisches Verfahren - Konduktometrisches Verfahren)
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 112 (1981).
Voraussetzungen
Leitlinien
Einschränkende Aussagen
Standarddokumente
Diese Prüfmethode beruht auf den Verfahren, auf die im Abschnitt "Literaturhinweise" verweisen wird, sowie auf dem Leitfaden der EPA "Preliminary Draft Guidance for Premanufacture Notification" vom 18. August 1978.
Methode - Einleitung, Zweck, Anwendungsbereich, Relevanz, Anwendung und Versuchsgrenzen
Die Dissoziation eines Stoffes in Wasser ist wichtig für die Beurteilung seiner Auswirkungen auf die Umwelt. Sie bestimmt die Form des Stoffes, die wiederum für das Verhalten und den Transport des Stoffes maßgeblich ist. Sie beeinflusst möglicherweise die Anlagerung der Chemikalie an Böden und Sedimente und ihr Eindringen in biologische Zellen (Absorption).
Definitionen und Einheiten
Unter Dissoziation versteht man den reversiblen Vorgang der Spaltung einer chemischen Spezies in zwei oder mehrere chemische Stoffe, die auch in ionischer Form vorliegen können. Dieser Vorgang wird im Allgemeinen wie folgt angegeben:
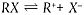
Die der Reaktion zugrunde liegende Gleichgewichtskonstante der Konzentration ist
K = [R+][X-] / [RX]
Handelt es sich bei R beispielsweise um Wasserstoff (eine Säure), ist die Konstante
Kα = [H+] ⋅ ([X-] / [HX])
oder
pKα = pH - log ([X-] / [HX])
Referenzsubstanzen
Die folgenden Referenzstoffe müssen nicht in allen Fällen verwendet werden, in denen eine neue Substanz untersucht wird.
Sie werden hier vor allem angegeben, damit die Methode gelegentlich kalibriert werden kann und um die Ergebnisse mit denen anderer Methoden vergleichen zu können.
| pKa 1 | Temp. in °C | |
| p-Nitrophenol | 7,15 | 25 1 |
| Benzoesäure | 4,12 | 20 |
| p-Chloroanilin | 3,93 | 20 |
| 1) Für 20 °C ist kein Wert verfügbar, aber es kann davon ausgegangen werden, dass die Variabilität der Messergebnisse höher ist als die zu erwartende Temperaturabhängigkeit. | ||
Sinnvoll wäre ein Stoff mit mehreren pKs (siehe "Prinzip der Prüfmethode"), z.B.
| Zitronensäure | pKa (8) | Temp. in °C |
| 1) 3,14 | 20 | |
| 2) 4,77 | 20 | |
| 3) 6,39 | 20 |
Prinzip der Prüfmethode
Das beschriebene chemische Verfahren ist im umweltrelevanten Temperaturbereich in der Regel nur wenig temperaturabhängig. Zur Bestimmung der Dissoziationskonstante ist eine Messung der Konzentrationen des chemischen Stoffes in seiner dissoziierten und undissoziierten Form erforderlich. Aus dem stöchiometrischen Verhältnis der Dissoziationsreaktion (siehe "Definitionen und Einheiten") lässt sich die jeweilige Konstante bestimmen. In dem bei dieser Prüfmethode beschriebenen besonderen Fall verhält sich der Stoff wie eine Säure oder Base, und die Bestimmung erfolgt am besten mittels Bestimmung der relativen Konzentrationen der ionisierten und nichtionisierten Formen des Stoffes und des pH-Werts der Lösung. Welche Beziehung zwischen diesen Begriffen besteht, ist der Gleichung für pKa in "Definitionen und Einheiten" zu entnehmen. Für einige Stoffe ergeben sich mehrere Dissoziationskonstanten, und es lassen sich ähnliche Gleichungen aufstellen. Einige der hier beschriebenen Methoden eignen sich auch für nicht-saure/basische Dissoziationen.
Qualitätskriterien
Wiederholbarkeit
Die Dissoziationskonstante sollte mit einer Toleranz von ± 0,1 log-Einheiten repliziert werden (mindestens dreifache Bestimmung).
Beschreibung der Prüfverfahren
Zur Bestimmung des pKa-Wertes stehen zwei grundlegende Ansätze zur Verfügung - Titration einer bekannten Menge des Stoffes mit einer Standardsäure bzw. Standardbase und Bestimmung der relativen Konzentration der ionisierten und nichtionisierten Formen und ihrer pH-Abhängigkeit.
Vorbereitungen
Die auf diesen Grundsätzen basierenden Methoden können als Titrationsverfahren, spektrofotometrische Verfahren und konduktometrische Verfahren klassifiziert werden.
Testlösungen
Beim Titrier- und konduktometrischen Verfahren sollte die chemische Substanz in destilliertem Wasser gelöst werden. Beim spektrofotometrischen und bei anderen Verfahren werden Pufferlösungen verwendet. Die Konzentration des Prüfstoffs sollte den niedrigeren der beiden Werte - 0,01 M bzw. die Hälfte der Sättigungskonzentration - nicht überschreiten, und bei der Herstellung der Lösungen sollte die reinste verfügbare Form der Substanz verwendet werden. Sofern der Stoff nur schwer löslich ist, kann er in einer kleinen Menge wassermischbaren Lösungsmittels gelöst werden, bevor er den oben genannten Konzentrationen zugegeben wird.
Die Lösungen sollten mithilfe eines Tyndall-Strahls auf Emulsionen überprüft werden, insbesondere wenn ein Zusatzlösungsmittel zur Verbesserung der Löslichkeit verwendet wurde. Sofern Pufferlösungen verwendet werden, sollte die Konzentration 0,05 M nicht überschreiten.
Prüfbedingungen
Temperatur
Temperaturkonstanz sollte mit einer Toleranz von ± 1 °C gewährleistet sein. Die Bestimmung sollte am besten bei 201 °C erfolgen.
Wenn eine erhebliche Temperaturabhängigkeit vermutet wird, sollte die Bestimmung bei mindestens zwei weiteren Temperaturwerten erfolgen. Die Temperaturintervalle sollten im vorliegenden Fall 101 °C betragen, und Temperaturkonstanz sollte mit einer Toleranz von ± 0,11 °C gewährleistet sein.
Analysen
Die Methode richtet sich nach der Art des Prüfstoffs. Er muss ausreichend empfindlich sein, damit eine Bestimmung der unterschiedlichen Spezies bei jeder Konzentration der Testlösung möglich ist.
Durchführung des Tests
Titrationsverfahren
Die Bestimmung der Prüflösung erfolgt durch Titration mit der Standardsäure- bzw. Standardbasenlösung, wobei nach jeder Zugabe des Titranten der pH-Wert gemessen wird. Vor Erreichen des Äquivalenzpunktes sollten mindestens 10 inkrementelle Zugaben erfolgen. Wenn ein Gleichgewicht relativ schnell erreicht wird, kann ein Kompensationschreiber (Potenziometer) verwendet werden. Für dieses Verfahren müssen sowohl die Gesamtmenge des Stoffes als auch seine Konzentration genau bekannt sein. Es müssen Vorkehrungen zum Ausschluss von Kohlendioxid getroffen werden. In den Standardtests sind Verfahren, Vorsichtsmaßnahmen und Berechnungsmethoden im Einzelnen beschrieben (vgl. Literaturhinweise (1), (2), (3), (4)).
Spektrofotometrisches Verfahren
Es wird eine Wellenlänge ermittelt, bei der sich die Extinktionskoeffizienten der ionisierten und nichtionisierten Formen des Stoffes deutlich unterscheiden. Das UV/VIS-Absorptionsspektrum wird aus Lösungen mit konstanter Konzentration bei einem pH-Wert, bei dem der Stoff im Wesentlichen nichtionisiert und bei dem sie vollständig ionisiert vorliegt, sowie bei mehreren pH-Zwischenwerten ermittelt. Dies geschieht entweder durch Zugabe von Inkrementen konzentrierter Säure (Base) zu einem relativ großen Volumen einer Lösung der Substanz in einem Mehrkomponentenpuffer bei einem anfangs hohen (niedrigen) pH-Wert (Literaturhinweis 5) oder durch Zugabe gleicher Mengen einer Stammlösung des Stoffes z.B. in Wasser oder Methanol zu konstanten Volumina verschiedener Pufferlösungen, die den gewünschten pH-Bereich abdecken. Aus den pH- und Absorptionswerten bei der gewählten Wellenlänge wird eine ausreichende Anzahl von Werten für den pKa-Wert berechnet; dabei werden Daten aus mindestens 5 pH-Bereichen verwendet, in denen die Substanz zu mindestens 10 Prozent und zu weniger als 90 Prozent ionisiert ist. Für weitere Einzelheiten zum Versuch und zur Berechnungsmethode siehe Literaturhinweis (1).
Konduktometrisches Verfahren
Anhand einer Zelle mit niedriger, bekannter Zellkonstante wird die Leitfähigkeit einer ca. 0,1 M-Lösung der Substanz in Leitfähigkeitswasser gemessen. Ferner werden die Leitfähigkeiten einiger akkurat hergestellter Verdünnungen dieser Lösung gemessen. Die Konzentration wird jedes Mal halbiert, und die Reihen sollten verschiedene Konzentrationen in abgestufter Reihenfolge umfassen. Die Grenzleitfähigkeit bei unendlicher Verdünnung lässt sich ermitteln, indem ein ähnlicher Versuch mit Na-Salz durchgeführt und extrapoliert wird. Der Dissoziationsgrad lässt sich sodann aus der Leitfähigkeit der jeweiligen Lösung nach der Onsagerschen Gleichung ermitteln, und die Dissoziationskonstante kann anschließend nach dem Ostwaldschen Verdünnungsgesetz als K = α2C/(1 - α) berechnet werden, wobei C der Konzentration in Mol pro Liter undα dem dissoziierten Teil entspricht. Es mÌssen Vorkehrungen zum Ausschluss von CO2 getroffen werden. Für weitere Einzelheiten zum Versuch und zur Berechnungsmethode siehe Standardtexte und Literaturhinweise (1), (6) und (7).
Daten und Berichterstattung
Ergebnisauswertung
Titrationsverfahren
Der pKa-Wert wird für 10 Messpunkte auf der Titrationskurve gemessen. Es werden die Mittelwerte und Standardabweichungen dieser pKa-Werte errechnet. Es sollte eine Korrelationskurve pH-Wert/Volumen der Standardbase oder -säure in tabellarischer Form erstellt werden.
Spektrofotometrisches Verfahren
Das Absorptionsmaß und der pH-Wert werden für jedes Spektrum tabellarisch erfasst. Aus den Datenpunkten der Zwischenspektren werden mindestens fünf Werte für den pKa berechnet, ebenso wie die Mittelwerte und Standardabweichungen dieser Ergebnisse.
Konduktometrisches Verfahren
Die Äquivalentleitfähigkeit Λ wird für jede Säurekonzentration und für jede Konzentration eines Gemisches aus einem Säureäquivalent plus einem 0,98 Äquivalent karbonatfreier Natronlauge ermittelt. Die Säure liegt im Überschuss vor, um einen hydrolysebedingten Überschuss an OH-zu vermeiden. 1/Λ wird gegen Ö_C aufgetragen, und Λo des Salzes lässt sich durch Extrapolation zur Nullkonzentration ermitteln.
Λo der Säure lässt sich anhand von Literaturwerten für H+ und Na+ berechnen. Der pKa lässt sich für jede Konzentration aus α =Λi /Λo und Ka = α2C/(1 - α) ermitteln. Man erhält bessere Werte für Ka, indem Korrekturen für Mobilität und Aktivität vorgenommen werden. Es sollten die Mittelwerte und Standardabweichungen dieser pKa-Werte berechnet werden.
Prüfbericht
Anzugeben sind sämtliche Rohdaten, die berechneten pKa-Werte und die Berechnungsmethode (am besten in tabellarischer Form, wie in Literaturhinweis (1) empfohlen), ebenso wie die oben beschriebenen statistischen Parameter. Bei Titrationsverfahren sind nähere Angaben zur Standardisierung der Titranten zu machen.
Beim spektrofotometrischen Verfahren sind alle Spektren anzugeben. Beim konduktometrischen Verfahren sollten nähere Angaben zur Bestimmung der Zellkonstante gemacht werden. Zudem sind Angaben zum angewandten Verfahren, zur Analysemethode und zur Art des verwendeten Puffers zu machen.
Die Prüftemperatur(en) sollte(n) festgehalten werden.
Literaturhinweise
(1) Albert, A. & Sergeant, E.P., Ionization Constants of Acids and Bases, Wiley, Inc., New York, 1962.
(2) Nelson, N.H. & Faust, S.D., Acidic dissociation constants of selected aquatic herbicides, Env. Sci. Tech. 3, II, S. 1186-1188 (1969).
(3) ASTM D 1293 - Annual ASTM Standards, Philadelphia, 1974.
(4) Standard Method 242. APHA/AWWA/WPCF, Standard Methods for the Examination of Water and Waste Water, 14. Ausgabe, American Public Health Association, Washington, D.C., 1976.
(5) Clark, J. & Cunliffe, A.E., Rapid spectrophotometric measurement of ionisation constants in aqueous solution. Chem. Ind. (London) 281, (März 1973).
(6) ASTM D 1125 - Annual ASTM Standards, Philadelphia, 1974.
(7) Standard Method 205 - APHA/AWWA/NPCF (siehe (4)).
(8) Handbook of Chemistry and Physics, 60th ed. CRC-Press, Boca Raton, Florida, 33431 (1980)."
(2) In Teil B erhält Kapitel B.5 folgende Fassung:
"B.5 Akute Augenreizung/-verätzung
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 405 (2012). Die OECD-Prüfrichtlinien für die Prüfung von Chemikalien werden regelmäßig überprüft, um sicherzustellen, dass sie den besten verfügbaren wissenschaftlichen Erkenntnissen entsprechen. Bei vorangegangenen Überprüfungen dieser Prüfrichtlinien wurde ein besonderes Augenmerk auf mögliche Verbesserungen durch die Auswertung aller vorhandenen Informationen über die Prüfchemikalie gelegt, um unnötige Versuche an Labortieren zu vermeiden und somit auch Belange des Tierschutzes zu berücksichtigen. Die (1981 verabschiedete und 1987, 2002 und 2012 aktualisierte) Prüfrichtlinie 405 beinhaltet die Empfehlung, vor der Durchführung des beschriebenen In-vivo-Tests zur Ermittlung der akuten Reiz-/Ätzwirkung des Stoffs auf die Augen eine evidenzbasierte kritische Analyse (Weight-of-Evidence analysis, WoE-Analyse) (1) der bereits vorhandenen einschlägigen Daten vorzunehmen. Sofern nicht genügend Daten zur Verfügung stehen, können diese mit Hilfe sequenzieller Tests generiert werden (2) (3). Die Prüfstrategie, die ergänzend zu dieser Prüfmethode empfohlen wird, sieht die Durchführung validierter und anerkannter In-vitro-Tests vor. Für die Zwecke der Verordnung EG (Nr.) 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) 1 wird außerdem eine integrierte Prüfstrategie in die entsprechende ECHA-Leitlinie (21) aufgenommen. Tierversuche sollten nur dann durchgeführt werden, wenn deren Notwendigkeit nach Abwägen verfügbarer alternativer Methoden festgestellt und die Anwendung der so ermittelten Methoden für angemessen befunden wurde. Zum Zeitpunkt der Ausarbeitung dieser aktualisierten Prüfmethode gibt es nach wie vor Fälle, in denen die Anwendung dieser Prüfmethode noch immer notwendig oder gesetzlich vorgeschrieben ist.
Bei der letzten Aktualisierung lag Der Schwerpunkt in erster Linie auf der Verwendung von Analgetika und Anästhetika; das grundlegende Konzept und der Aufbau der Prüfrichtlinien blieben unberührt. ICCVAM 2 und eine unabhängige internationale wissenschaftliche Peer-Review-Gruppe überprüften den Nutzen und die Grenzen einer routinemäßigen Verwendung topischer Anästhetika, systemischer Analgetika und humaner Endpunkte bei In-vivo-Sicherheitstests zur Ermittlung von Augenreizungen (12). Die Überprüfung ergab, dass durch die Verwendung topischer Anästhetika und systemischer Analgetika Schmerzen und Leiden ganz oder zu einem Großteil vermieden werden könnten, ohne die Testergebnisse zu beeinträchtigen, und es wurde ein genereller Einsatz dieser Stoffe empfohlen. Bei der vorliegenden Prüfmethode wurde diese Überprüfung berücksichtigt. Topische Anästhetika, systemische Analgetika und humane Endpunkte sollten bei In-vivo-Tests zur Feststellung akuter Augenreizung/-verätzungen routinemäßig eingesetzt werden. Ausnahmen sind zu begründen. Die in dieser Methode beschriebenen Verfeinerungen werden bei den meisten Versuchen, bei denen Sicherheitstests zur Feststellung von Augenreizungen am lebenden Tier nach wie vor erforderlich sind, erheblich zur Verringerung oder Vermeidung von Schmerzen und Leiden bei den Versuchstieren beitragen.
Eine ausgewogene präventive Schmerzbehandlung sollte Folgendes umfassen: i) eine routinemäßige Vorbehandlung mit einem topischen Anästhetikum (z.B. Proparacain oder Tetracain) und einem systemischen Analgetikum (z.B. Buprenorphin), ii) eine routinemäßige Nachbehandlung mit systemischen Analgetika (z.B. Buprenorphin und Meloxicam), iii) eine planmäßige Beobachtung und Überwachung von Tieren mit Aufzeichnung klinischer Anzeichen von Schmerzen und/oder Leiden und iv) eine planmäßige Beobachtung, Überwachung und Erfassung der Art, des Schweregrads und des Verlaufs sämtlicher Augenverletzungen. Für weitere Details siehe die nachfolgend beschriebenen aktualisierten Verfahren. Nach Verabreichung der Prüfchemikalie sollten keine zusätzlichen topischen Anästhetika oder Analgetika gegeben werden, um eine Beeinträchtigung der Studie zu vermeiden. Analgetika mit entzündungshemmender Wirkung (z.B. Meloxicam) sollten nicht topisch aufgetragen werden und systemisch verabreichte Dosen sollten nicht mit den Wirkungen auf die Augen interferieren.
Definitionen finden sich in der Anlage zur Prüfmethode.
Ausgangsüberlegungen
Im Interesse wissenschaftlicher Verlässlichkeit und des Tierschutzes sollen In-vivo-Tests erst dann in Erwägung gezogen werden, wenn alle für das Hautverätzungs-/-reizungspotenzial der Chemikalie zur Verfügung stehenden einschlägigen Daten auf Basis ihrer Beweiskraft (weight-of-evidence, WoE) ausgewertet worden sind. Zu diesen Daten gehören unter anderem Erkenntnisse aus bereits durchgeführten Untersuchungen am Menschen und/oder an Labortieren, Hinweise auf Verätzungen/Reizungen durch eine oder mehrere strukturell verwandte Substanzen oder Gemische aus diesen Substanzen, Daten, die eine starke Azidität oder Alkalinität der Substanz belegen (4) (5) und Ergebnisse validierter und anerkannter In-vitro- oder Ex-vivo-Tests auf Hautverätzungen und -reizungen (6) (13) (14) (15) (16) (17). Diese Studien können sowohl vor als auch nach einer WoE-Analyse durchgeführt worden sein.
Für bestimmte Chemikalien ergibt eine solche Analyse möglicherweise, dass deren Augenverätzungs-/-reizungspotenzial im Rahmen von In-vivo-Studien untersucht werden muss. In all diesen Fällen sollten zunächst die augenverätzenden Wirkungen der Chemikalie in vitro und/oder in vivo untersucht und nach der sequenziellen Prüfstrategie in Prüfmethode B.4 (7) oder nach der in der ECHA-Leitlinie (21) beschriebenen integrierten Prüfstrategie evaluiert werden, bevor ein In-vivo-Augentest in Erwägung gezogen wird.
Ergänzend zu dieser Prüfmethode wird eine sequenzielle Prüfstrategie, die auch validierte In-vitro- oder Ex-vivo-Tests auf Augenverätzungs-/-reizungswirkungen vorsieht, in diese Prüfmethode und, für die Zwecke der REACH-Verordnung, auch in die ECHA-Leitlinie (21) aufgenommen. Es wird empfohlen, dass eine solche sequenzielle Prüfstrategie vor einem In-vivo-Test durchgeführt wird. Für neue Chemikalien wird ein stufenweiser Prüfansatz empfohlen, um wissenschaftliche fundierte Daten über die durch die Chemikalie bedingte Verätzung/Reizung erheben zu können. Bei bereits bekannten Chemikalien, für die nicht genügend Daten zum Hautverätzungs-/-reizungspotenzial bzw. Augenverätzungs-/-reizungspotenzial vorliegen, kann die Strategie genutzt werden, um Datenlücken zu schließen. Die Anwendung einer anderen Prüfstrategie bzw. eines anderen Prüfverfahrens oder die Entscheidung gegen einen stufenweisen Prüfansatz sollten begründet werden.
Prinzip des In-vivo-Tests
Nach einer Vorbehandlung mit einem systemischen Analgetikum und nach Einleitung einer geeigneten topischen Anästhesie wird die zu prüfende Chemikalie als Einzeldosis in ein Auge des Versuchstiers geträufelt; das unbehandelte Auge dient als Kontrolle. Der Grad der Reizung/Verätzung wird bestimmt, indem in zuvor festgelegten Zeitabständen und anhand einer Punkteskala Schädigungen der Bindehaut, der Hornhaut und der Iris bewertet werden. Es werden auch andere Reaktionen des Auges und systemische Schäden erfasst, um die Wirkungen umfassend beurteilen zu können. Die Beobachtungsdauer sollte lang genug sein, um die Reversibilität bzw. Irreversibilität der Wirkungen evaluieren zu können.
Tiere, die zu irgendeinem Zeitpunkt während des Versuchs Anzeichen schweren Leidens und/oder starker Schmerzen oder Läsionen aufweisen, die den in dieser Prüfmethode beschriebenen humanen Endpunkten (siehe Nummer 26) entsprechen, sollten human getötet werden, und die Chemikalie ist entsprechend einzustufen. Kriterien für die Entscheidung über die humane Tötung moribunder Tiere mit starken Leidensanzeichen sind Gegenstand eines OECD-Leitfadens (8).
Vorbereitung des In-vivo-Tests
Auswahl der Tierart
Bevorzugtes Labortier für den Test sind gesunde, junge, geschlechtsreife Albino-Kaninchen. Die Verwendung anderer Stämme oder Arten sollte begründet werden.
Vorbereitung der Versuchstiere
Innerhalb von 24 Stunden vor dem Versuch werden bei jedem der ausgewählten Versuchstiere beide Augen untersucht. Tiere, bei denen bereits eine Augenreizung, okulare Defekte oder eine Hornhautschädigung vorliegen, sollen nicht verwendet werden.
Haltungs- und Fütterungsbedingungen
Die Tiere sollen einzeln gehalten werden. Die Temperatur im Versuchstierraum sollte für Kaninchen 201 °C (± 31 °C) betragen. Obwohl die relative Luftfeuchtigkeit mindestens 30 % und außer während der Raumreinigung maximal 70 % betragen sollte, ist ein Wert von 50-60 % anzustreben. Der Raum soll künstlich beleuchtet sein, mit Hell-/Dunkelphasen im 12-Stunden-Rhythmus. Überhöhte Lichtintensität sollte vermieden werden. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist.
Prüfverfahren
Verwendung topischer Anästhetika und systemischer Analgetika
Es wird empfohlen, zur Verringerung oder Vermeidung von Schmerzen und Leiden bei Augensicherheitsprüfungen wie nachstehend beschrieben vorzugehen. Ersatzweise können auch alternative Verfahren angewendet werden, mit denen sich Schmerzen und Leiden nachweislich ebenso gut oder sogar besser vermeiden oder lindern lassen.
Applikation der Prüfchemikalie
Die Prüfchemikalie sollte bei jedem Tier in den Bindehautsack eines Auges appliziert werden, indem das untere Lid leicht vom Augapfel weggezogen wird. Die Lider dann etwa eine Sekunde lang leicht zusammendrücken, damit kein Prüfmaterial verloren geht. Das andere, unbehandelte Auge dient als Kontrolle.
Ausspülen
Die Augen der Versuchstiere sollten frühestens 24 Stunden nach Applikation der Prüfchemikalie ausgewaschen werden; Ausnahmen gelten für Feststoffe (siehe Nummer 18) und bei sofortigem Eintritt einer Ätz- oder Reizwirkung. Nach 24 Stunden kann eine Augenspülung erfolgen, sofern dies für angemessen gehalten wird.
Untersuchungen an einer zusätzlichen Versuchstiergruppe (Satellitengruppe) zur Erforschung des Einflusses des Ausspülens wird nicht empfohlen, außer in wissenschaftlich begründeten Fällen. Sollte eine Satellitengruppe tatsächlich benötigt werden, sind zwei Kaninchen vorzusehen. Die Bedingungen, unter denen die Augenspülung erfolgte (Zeitpunkt, Zusammensetzung und Temperatur der Spüllösung, Dauer, Volumen und Geschwindigkeit der Applikation) sollten genau dokumentiert werden.
Dosierung
(1) Prüfung von Flüssigkeiten
Bei der Prüfung von Flüssigkeiten eine Dosis von 0,1 ml verwenden. Liegt die Prüfchemikalie als Pumpspray vor, sollte sie nicht direkt ins Auge gesprüht, sondern mittels Sprühstoß entnommen und in einem Behälter aufgefangen werden. Anschließend 0,1 ml in das Auge einträufeln.
(2) Prüfung von Feststoffen
Bei Feststoffen, Pasten und partikelförmigen Chemikalien sollte die Prüfmenge ein Volumen von 0,1 ml oder ein Gewicht von höchstens 100 mg haben. Die Prüfchemikalie sollte zu einem feinen Pulver zermahlen werden. Das Volumen von Feststoffen sollte erst nach vorsichtigem Kompaktieren, z.B. durch Klopfen des Messbehälters, bestimmt werden. Ist die in Form eines Feststoffs vorliegende Prüfchemikalie bis zum ersten Beobachtungszeitpunkt, d. h. 1 Stunde nach der Applikation, nicht aufgrund physiologischer Vorgänge aus dem Auge entfernt worden, kann das Auge mit Kochsalzlösung oder destilliertem Wasser ausgespült werden.
(3) Prüfung von Aerosolen
Es wird empfohlen, alle als Pumpspray oder Aerosol vorliegenden Prüfchemikalien mittels Sprühstoß zu entnehmen, in einem Behälter aufzufangen und anschließend zu applizieren. Einzige Ausnahme sind Chemikalien in Aerosol-Druckbehältern, die aufgrund der Vaporisierung nicht aufgefangen werden können. In diesen Fällen sollte das Auge offen gehalten und die Prüfchemikalie mit einem einzigen Sprühstoß etwa eine Sekunde lang aus 10 cm Entfernung ins Auge gesprüht werden. In Abhängigkeit vom Druck und vom Behälterinhalt kann dieser Abstand variieren. Es ist darauf zu achten, dass das Auge durch den Sprühdruck nicht verletzt wird. Unter Umständen muss das Risiko "mechanischer" Augenschäden, die auf den Sprühdruck zurückzuführen sind, beurteilt werden.
Bei Aerosolen kann die Dosis geschätzt werden, indem der Test folgendermaßen simuliert wird: Die Chemikalie durch eine Öffnung in der Größe eines Kaninchenauges auf Wägepapier sprühen, wobei sich die Öffnung unmittelbar vor dem Papier befindet. Anhand der Gewichtszunahme des Papiers wird ein Näherungswert für die ins Auge gesprühte Menge ermittelt. Bei flüchtigen Chemikalien kann ein Schätzwert für die Dosis ermittelt werden, indem man einen Auffangbehälter vor und nach Entnahme der Prüfchemikalie wiegt.
Vorversuch (In-vivo-Test auf Augenreizung/-verätzung an einem einzigen Tier)
Es empfiehlt sich dringend, den In-vivo-Test zunächst nur an einem Tier durchzuführen (siehe Ergänzung zu dieser Prüfmethode: Eine sequenzielle Strategie für die Prüfung auf Augenreizung/-verätzung). Die Beobachtungen sollen ausreichen, um den Schweregrad und die Reversibilität der Wirkungen zu bestimmen, bevor ein Bestätigungstest an einem zweiten Tier durchgeführt wird.
Sofern das beschriebene Verfahren ergibt, dass der Stoff augenverätzend wirkt oder schwere Augenreizungen auslöst, sollen keine weiteren Prüfungen auf Augenreizung durchgeführt werden.
Bestätigungstest (In-vivo-Test auf augenreizende Wirkungen an zusätzlichen Tieren)
Wird im Vorversuch keine ätzende oder schwer augenreizende Wirkung beobachtet, sollte die Reizungsreaktion bzw. die negative Reaktion an bis zu zwei weiteren Tieren bestätigt werden. Ergibt der Vorversuch eine Reizwirkung, sollte der Bestätigungstest nach Möglichkeit als sequenzieller Versuch an einem Tier einem bestimmten Zeitpunkt und nicht durch gleichzeitige Exposition der zwei weiteren Tiere erfolgen. Der Test ist abzubrechen, wenn beim zweiten Tier Anzeichen einer Verätzung oder einer schweren Reizung festgestellt werden. Wenn die Ergebnisse beim zweiten Tier eine Bestimmung der Gefahrenklasse zulassen, sollten keine weiteren Tests durchgeführt werden.
Beobachtungszeitraum
Die Beobachtungszeit sollte so bemessen sein, dass das Ausmaß und die Reversibilität der festgestellten Wirkungen umfassend bewertet werden können. Allerdings sollte der Versuch abgebrochen werden, sobald das Tier starke Anzeichen von Leiden und Schmerzen zeigt (8). Um feststellen zu können, ob die Wirkungen reversibel sind, sollten die Tiere in der Regel während 21 Tagen nach Applikation der Prüfchemikalie beobachtet werden. Bilden sich die Schäden vor Ablauf dieser 21 Tage zurück, sollte der Versuch zu diesem Zeitpunkt beendet werden.
Klinische Beobachtungen und Einstufung von Augenreaktionen
Die Augen sollten eine Stunde nach Applikation der Prüfchemikalie umfassend auf vorhandene bzw. nicht vorhandene Augenläsionen untersucht werden; danach mindestens eine Untersuchung täglich durchführen. In den ersten 3 Tagen sollten die Tiere mehrmals täglich untersucht werden, damit Entscheidungen, den Versuch zu beenden, zeitnah getroffen werden können. Versuchstiere sollten während der gesamten Studiendauer routinemäßig mindestens zweimal täglich, falls nötig auch häufiger, auf klinische Anzeichen von Schmerzen und/oder Leiden untersucht werden (z.B. wiederholtes Kratzen und Reiben am Auge, übermäßiges Blinzeln, übermäßig tränende Augen) (9) (10) (11), wobei zwischen den Beobachtungen mindestens 6 Stunden liegen sollten. Dies ist nötig, um i) Tiere angemessen auf Anzeichen von Schmerzen und Leiden zu untersuchen und fundierte Entscheidungen im Hinblick auf eine Erhöhung der Dosis von Analgetika treffen zu können, und ii) Tiere auf Anzeichen vorbestimmter humaner Endpunkte zu untersuchen, damit fundierte Entscheidungen darüber getroffen werden können, ob Tiere human getötet werden sollten, und damit solche Entscheidungen zeitnah getroffen werden können. Eine Anfärbung mit Fluorescein sollte routinemäßig erfolgen und gegebenenfalls sind als Hilfsmittel zum Nachweis und zur Messung von Augenschäden und zur Beurteilung, ob vorbestimmte Endpunktkriterien für ein humanes Töten erfüllt sind, eine Spaltlampe und ein Biomikroskop zu verwenden (z.B. bei der Beurteilung der Tiefe einer Schädigung im Falle einer Hornhautulzeration). Digitale Fotografien von beobachteten Schädigungen können zu Referenzzwecken und zur dauerhaften Dokumentation des Ausmaßes der Augenschädigung erfasst werden. Sobald aussagekräftige Informationen vorliegen, sollte der Versuch nicht länger als nötig fortgesetzt werden. Tiere mit starken Anzeichen von Schmerzen oder Leiden sollen unverzüglich human getötet werden, wobei die Chemikalie entsprechend einzustufen ist.
Bei Tieren mit folgenden Augenschädigungen nach Applikation der Prüfchemikalie ist eine humane Tötung angezeigt (siehe Tabelle 1 zur Beschreibung der Schädigungsgrade): Hornhautperforation oder signifikante Hornhautulzeration mit Staphylom; Blut in der vorderen Augenkammer; Hornhauttrübung Grad 4; fehlender Pupillenreflex (Irisreaktion Grad 2) während 72 Stunden; Ulzeration der Bindehaut; Nekrose der Bindehaut oder der Nickhaut oder Gewebsdemarkierung. Diese Schäden sind im Allgemeinen irreversibel. Darüber hinaus wird empfohlen, die folgenden Augenschädigungen als humane Endpunkte zu definieren, bei denen Studien vor Ablauf des planmäßigen 21-tägigen Beobachtungszeitraums beendet werden sollten. Diese Schädigungen gelten als prädiktiv für schwere Reizungen oder Verätzungen und Schädigungen, bei denen davon ausgegangen werden muss, dass sie bis zum Ablauf des 21-tägigen Beobachtungszeitraums nicht vollständig abklingen: sehr tiefe Schädigung (z.B. eine Hornhautulzeration bis in Schichten unterhalb der Stromaoberfläche), Zerstörung des Limbus zu mehr als 50 % (nachgewiesen durch ein Ausbleichen des Bindehautgewebes) und schwere Augeninfektion (eitriger Ausfluss). Eine Kombination von Vaskularisierung der Hornhautoberfläche (d. h. Pannus), sich nicht rückbildender Fluoresceinfärbung (basierend auf einer täglichen Untersuchung) und/oder einer ausbleibenden Reepithelisierung am 5. Tag nach Applikation der Prüfchemikalie könnte ebenfalls als potenziell zweckdienliches Kriterium für die klinische Entscheidung über eine vorzeitige Beendigung der Studie angesehen werden. Einzeln betrachtet sind diese Ergebnisse jedoch nicht ausreichend, um eine vorzeitige Beendigung der Studie zu rechtfertigen. Sobald schwerwiegende Auswirkungen auf die Augen festgestellt werden, sollten ein behandelnder Tierarzt oder ein qualifizierter Labortierarzt oder im Nachweis klinischer Schädigungen geschultes Laborpersonal zur Notwendigkeit einer klinischen Untersuchung zur Feststellung, ob die Kombination dieser Auswirkungen eine vorzeitige Beendigung der Studie rechtfertigt, konsultiert werden. Der jeweilige Grad der Augenreaktion (Bindehaut, Hornhaut und Iris) ist 1, 24, 48 und 72 Stunden nach Applikation der Prüfchemikalie zu ermitteln und zu dokumentieren (Tabelle 1). Tiere, bei denen keine Augenschädigungen auftreten, dürfen frühestens 3 Tage nach der Behandlung getötet werden. Bei leichten bis mäßigen Augenschädigungen sollten die Tiere bis zum Abklingen der Symptome bzw. für die Dauer von 21 Tagen beobachtet werden; erst danach wird die Studie abgeschlossen. Untersuchungen sollen mindestens nach 1 Stunde, 24 Stunden, 48 Stunden, 72 Stunden sowie am 7., 14. und 21. Tag durchgeführt und dokumentiert werden, um den Status der Schädigungen zu ermitteln und zu klären, ob sie reversibel oder irreversibel sind. Erforderlichenfalls sollten häufigere Untersuchungen durchgeführt werden, um zu entscheiden, ob das Versuchstier aus Tierschutzgründen human getötet oder aufgrund von negativen Ergebnissen aus dem Versuch genommen werden sollte.
Der jeweilige Grad der Augenschädigung (vgl. Tabelle 1) sollte für jede Untersuchung erfasst werden. Etwaige weitere Augenschädigungen (z.B. Pannus, Verfärbungen, Veränderungen in der vorderen Augenkammer) oder negative systemische Wirkungen sing ebenfalls zu dokumentieren.
Als Hilfsmittel können bei den Untersuchungen Binokularlupen, Handspaltlampen, Biomikroskope und andere geeignete Geräte benutzt werden. Nach Aufzeichnung der Beobachtungen nach 24 Stunden können die Augen außerdem mit Fluorescein weiter untersucht werden.
Die Bewertung von Augenreaktionen ist zwangsläufig subjektiv. Um die Einstufung von Augenreaktionen stärker zu vereinheitlichen und den Prüflabors und allen an den Versuchen und an der Interpretation der Versuchsergebnisse Beteiligten die Arbeit zu erleichtern, müssen die Prüfer im Umgang mit der Bewertungsskala geschult werden.
Daten und Berichterstattung
Ergebnisauswertung
Die Bewertung der Augenreizung sollte anhand der Art und des Schweregrads der Schädigung und deren Reversibilität bzw. Irreversibilität vorgenommen werden. Die jeweils ermittelten Schweregrade stellen keinen alleingültigen Maßstab für die reizenden Eigenschaften einer Chemikalie dar, denn es werden auch andere Wirkungen der Prüfchemikalie beurteilt. Vielmehr sollten die einzelnen Graduierungswerte als Referenzwerte betrachtet werden, die nur dann sinnvoll sind, wenn sämtliche Beobachtungen genau erfasst und evaluiert werden.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Begründung für den In-vivo-Test: WoE-Analyse von Daten aus früheren Versuchen unter Einbeziehung von Ergebnissen aus der sequenziellen Prüfstrategie:
Prüfchemikalie:
Vehikel:
Versuchstiere:
Anästhetika und Analgetika
Ergebnisse:
Diskussion der Ergebnisse.
Interpretation der Ergebnisse
Eine Extrapolation der Ergebnisse von Untersuchungen auf Augenreizungen an Labortieren auf den Menschen ist nur bedingt möglich. Oftmals reagiert das Albino-Kaninchen empfindlicher als der Mensch auf Stoffe mit augenreizenden oder -verätzenden Eigenschaften.
Bei der Interpretation von Daten ist darauf zu achten, dass Reizungen aufgrund einer sekundären Infektion nicht berücksichtigt werden.
Literaturhinweise
(1) Barratt, M.D., et al. (1995), The Integrated Use of Alternative Approaches for Predicting Toxic Hazard, ECVAM Workshop Report 8, ATLA 23, 410-429.
(2) de Silva, O., et al. (1997), Evaluation of Eye Irritation Potential: Statistical Analysis and Tier Testing Strategies, Food Chem. Toxicol 35, 159-164.
(3) Worth A.P. and Fentem J.H. (1999), A general approach for evaluating stepwise testing strategies ATLA 27, 161-177.
(4) Young, J.R., et al. (1988), Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals, Toxicol. In Vitro, 2, 19-26.
(5) Neun, D.J. (1993), Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH, J. Toxicol. Cut. Ocular Toxicol. 12, 227 - 231.
(6) Fentem, J.H., et al. (1998), The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team, Toxicology in vitro 12, 483-524.
(7) Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung.
(8) OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. OECD Environmental Health and Safety Publications, Series on Testing and Assessment, Nr. 19. (http://www.oecd.org/ehs/test/monos.htm).
(9) Wright EM, Marcella KL, Woodson JF. (1985), Animal pain: evaluation and control, Lab Animal, Mai/Juni, 20-36.
(10) National Research Council (NRC) (2008), Recognition and Alleviation of Distress in Laboratory Animals, Washington, DC: The National Academies Press.
(11) National Research Council (NRC) (2009), Recognition and Alleviation of Distress in Laboratory Animals, Washington, DC: The National Academies Press.
(12) ICCVAM (2010), ICCVAM Test Method Evaluation Report: Recommendations for Routine Use of Topical Anesthetics, Systemic Analgesics, and Humane Endpoints to Avoid or Minimize Pain and Distress in Ocular Safety Testing, NIH Publication No. 10-7514, Research Triangle Park, NC, USA: National Institute of Environmental Health Sciences.
http://iccvam.niehs.nih.gov/methods/ocutox/OcuAnest- TMER.htm
(13) Kapitel B.40 dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: TER-Test (Transcutaneous Electrical Resistance Test).
(14) Kapitel B.40 bis dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: Test mit einem menschlichen Hautmodell.
(15) OECD (2006), Test No. 435: In vitro Membrane Barrier Test Method for Skin corrosion, OECD Guidelines for the Testing of Chemicals, Abschnitt 4, OECD Paris.
(16) Kapitel B.47 dieses Anhangs, Trübungs- und Durchlässigkeitstest an der Rinderhornhaut zwecks Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
(17) Kapitel B.48 dieses Anhangs, Test am isolierten Hühnerauge zur Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
(18) U.S. EPA (2003), Label Review Manual: 3rd Edition, EPA737-B-96-001, Washington, DC: U.S., Environmental Protection Agency.
(19) UN (2011), Globally Harmonized System of Classification and Labelling of Chemicals (GHS), Vierte überarbeitete Ausgabe, New York und Genf: United Nations Publications.
(20) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006. Amtsblatt der Europäischen Union L 353, S. 1-1355.
(21) ECHA Guidance on information requirements and chemical safety assessment, Kapitel R.7a: Endpoint specific guidance.
http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf
Tabelle 1 Einstufung von Augenschädigungen
| Cornea | Grad |
| Trübung: Trübungsgrad (für die Auswertung wird der am stärksten betroffene Bereich genommen) * | |
| Keine Ulzeration oder Trübung | 0 |
| Punktförmige oder diffuse Trübungsbereiche (ohne leichte Trübung des normalen Glanzes); Einzelheiten der Iris deutlich erkennbar | 1 |
| Leicht erkennbarer durchlässiger Bereich, Einzelheiten der Iris etwas verschattet | 2 |
| Perlmuttartige Bereiche, keine Einzelheiten der Iris sichtbar, Größe der Pupille kaum erkennbar | 3 |
| Trübe Hornhaut; Iris aufgrund der Trübung nicht erkennbar | 4 |
| Höchstmögliche Punktzahl: 4 | |
| Iris | |
| Normal | 0 |
| Deutlich vertiefte Rugae, Kongestion, Schwellung, mäßige circumcorneale Hyperämie oder Injektion; Iris reagiert auf Licht (träge Reaktion ist positiv) | 1 |
| Blutungen, großflächige Zerstörung, keine Reaktion auf Licht | 2 |
| Höchstmögliche Punktzahl: 2 | |
| Conjunctivae | |
| Rötung (der Augenlidbindehaut und der Augapfelbindehaut; ohne Hornhaut und Iris) | |
| Normal | 0 |
| Einige Blutgefäße zeigen Hyperämie (Injektion) | 1 |
| Diffuse karmesinrote Farbe; einzelne Gefäße nur schwer erkennbar | 2 |
| Diffuse dunkelrote Verfärbung | 3 |
| Höchstmögliche Punktzahl: 3 | |
| Chemosis | |
| Schwellung (der Augenlider und/oder Nickhäute) | |
| Normal | 0 |
| Über dem Normalen liegende Schwellung | 1 |
| Deutliche Schwellung mit partieller Auswärtskehrung der Lider | 2 |
| Schwellung mit etwa halbgeschlossenen Lidern | 3 |
| Schwellung mit mehr als halbgeschlossenen Lidern | 4 |
| Höchstmögliche Punktzahl: 4 | |
| *1) Die Größe des von der Hornhauttrübung betroffenen Areals sollte dokumentiert werden. | |
_____
1) Verordnung EG (Nr.) 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH), zur Schaffung einer Europäischen Agentur für chemische Stoffe, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission.
ABl. Nr. L 304 vom 22.11.2007 S. 1.
2) Das US-amerikanische Validierungszentrum ("Interagency Coordinating Committee on the Validation of Alternative Methods").
.
| Definitionen | Anlage |
Definitionen
Chemikalie: ein Stoff oder ein Gemisch.
Evidenzbasierte Analyse (Weight-of-Evidence, WoE): der Prozess der Prüfung der Stärken und Schwächen einer Datensammlung als Grundlage für eine Schlussfolgerung, zu der es auf Basis von Einzeldaten möglicherweise nicht gekommen wäre.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Saure/alkalische Reserve: Bei sauren Zubereitungen: zum Erreichen eines bestimmten pH-Werts erforderliche Menge (in g) Natronlauge/100 g Zubereitung. Bei alkalischen Zubereitungen: zum Erreichen eines bestimmten pH-Werts erforderliche Menge (in g) Natronlauge, die der Menge (in g) Schwefelsäure/100 g Zubereitung entspricht, (Young et al. 1988).
Stoffe ohne Reizwirkung: Stoffe, die nicht als augenreizende Stoffe der EPA-Kategorien I, II oder III eingestuft sind, oder augenreizende Stoffe der GHS-Kategorien 1, 2, 2A oder2B oder der EU-Kategorien 1 oder2 (17) (18) (19).
Stoff mit augenätzender Wirkung: a) eine Chemikalie, die eine irreversible Gewebeschädigung am Auge verursacht; b) Chemikalien, die als augenreizende Stoffe der GHS-Kategorie 1 oder als augenreizende Stoffe der EPA-Kategorie I oder der EU-Kategorie 1 eingestuft sind (17) (18) (19).
Stoff mit augenreizender Wirkung: a) eine Chemikalie, die eine reversible Veränderung im Auge hervorruft; b) Chemikalien, die als augenreizende Stoffe der EPA-Kategorien II oder III oder als augenreizende Stoffe der GHS-Kategorien 2, 2A oder2B oder der EU-Kategorie 2 eingestuft sind (17) (18) (19).
Stoff mit schwer augenreizender Wirkung: a) eine Chemikalie, die eine Gewebeschädigung im Auge verursacht, die nicht innerhalb von 21 Tagen nach Applikation ausheilt, oder eine massive Verschlechterung des Sehvermögens auslöst; b) Chemikalien, die als augenreizende Stoffe der GHS-Kategorie 1 oder als augenreizende Stoffe der EPA-Kategorie I oder der EU-Kategorie 1 eingestuft sind (17) (18) (19).
Stufenweiser Ansatz: eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Analyseansatz (Weight-of-Evidence, WoE) vorgegangen wird, um festzustellen, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung möglich ist.
Ergänzung zur Prüfmethode B.5 1
Sequenzielle Strategie für die Prüfung auf Augenreizungen und Augenverätzungen
Allgemeine Überlegungen
Im Interesse wissenschaftlicher Verlässlichkeit und des Tierschutzes muss die unnötige Verwendung von Versuchstieren verhindert und die Durchführung von Versuchen, die bei den Tieren wahrscheinlich schwere Reaktionen hervorrufen, auf ein Mindestmaß beschränkt werden. Bevor ein In-vivo-Test ins Auge gefasst werden kann, sollten zunächst alle einschlägigen Informationen über die potenziell augenreizenden/-verätzenden Wirkungen einer Chemikalie bewertet werden. Möglicherweise liegen bereits genügend Kriterien (Evidence) für die Einstufung einer Prüfchemikalie aufgrund ihres Augenreizungs-/-verätzungspotenzials vor, sodass sich Versuche an Labortieren erübrigen. Folglich schränken WoE-Analysen bereits vorliegender Daten und die Anwendung einer sequenziellen Prüfstrategie die Notwendigkeit von In-vivo-Tests deutlich ein. Dies gilt umso mehr, wenn davon auszugehen ist, dass die Chemikalie schwere Reaktionen hervorruft.
Zur Beurteilung bereits vorhandener Informationen über die augenreizenden/-verätzenden Wirkungen von Chemikalien sollte das Instrument der evidenzbasierten Analyse herangezogen werden. Ausgehend davon ist zu entscheiden, ob als Beitrag zur Charakterisierung dieses Potenzials zusätzliche Studien, bei denen es sich nicht um In-vivo-Augenuntersuchungen handelt, durchgeführt werden sollten. Sofern weitere Studien durchgeführt werden müssen, empfiehlt es sich, zur Generierung sachdienlicher Versuchsdaten die sequenzielle Prüfstrategie anzuwenden. Bei noch nicht geprüften Stoffen soll die sequenzielle Prüfstrategie genutzt werden, um die Datensätze zu generieren, die für die Beurteilung des augenätzenden/-reizenden Potenzials benötigt werden. Die ursprüngliche in dieser Ergänzung beschriebene Prüfstrategie wurde in einem OECD-Workshops (1) entwickelt. Sie wurde im Rahmen des "Harmonised Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances" bestätigt und weiter ausgebaut, das im November 1998 von den Teilnehmern der 28. Gemeinsamen Tagung des Chemikalien-Ausschusses und der Arbeitsgruppe Chemikalien gebilligt (2) und 2011 von einer OECD-Expertengruppe überarbeitet wurde.
Diese Prüfstrategie ist zwar nicht integraler Bestandteil der Prüfmethode B.5, wird aber zur Ermittlung augenreizender/-verätzender Merkmale empfohlen. Dieser Ansatz entspricht sowohl der besten Praxis als auch dem ethischen Richtwert für Augenreizungs/-verätzungsprüfungen am lebenden Tier. Die Prüfmethode ist nicht nur eine Anleitung für die Durchführung des In-vivo-Tests, sondern beschreibt auch die Faktoren, die vor der Durchführung eines solchen Versuchs überprüft werden sollten. Die sequenzielle Prüfstrategie liefert einen Ansatz für die Bewertung bereits vorhandener Daten über die augenreizenden/-verätzenden Eigenschaften von Chemikalien und einen stufenweisen Ansatz für die Generierung sachdienlicher Daten über Chemikalien, die im Rahmen zusätzlicher Studien untersucht werden müssen bzw. noch gar nicht untersucht wurden. Die Strategie sieht die Durchführung validierter und anerkannter In-vitro- und Ex-vivo-Tests vor, in bestimmten Fällen gefolgt von Untersuchungen nach Prüfmethode B.4 (3) (4).
Beschreibung der stufenweisen Prüfstrategie
Alle vorhandenen Informationen sollten vor der Durchführung von Versuchen im Rahmen der sequenziellen Prüfstrategie (Fließbild) bewertet werden, um die Notwendigkeit von In-vivo-Augentests zu klären. Auch wenn wichtige Informationen aus der Beurteilung einzelner Parameter (z.B. extreme pH-Werte) gewonnen werden können, sollten die bereits vorliegenden Angaben in ihrer Gesamtheit betrachtet werden. Alle relevanten Daten über die Wirkungen der betreffenden Chemikalie und Struktur verwandter Verbindungen sollten mittels WoE-Analyse bewertet werden, und die Entscheidung sollte begründet werden. Dabei sollten bereits vorliegende Angaben zur Wirkung der Chemikalie auf Mensch und Tier im Vordergrund stehen, gefolgt von den Ergebnissen von In-vitro- und Ex-vivo-Tests. In-vivo-Untersuchungen mit Chemikalien mit Ätzwirkung sollten nach Möglichkeit immer vermieden werden. Folgende Faktoren werden bei der Prüfstrategie berücksichtigt:
Beurteilung bereits vorliegender Daten zur Wirkung der Chemikalie auf Mensch und/oder Tier und/oder mit validierten und international anerkannten Methoden gewonnener In-vitro-Daten (Stufe 1)
Zunächst sollten vorhandene Humandaten, z.B. aus klinischen Studien oder Studien zur Exposition am Arbeitsplatz, Fallberichte und/oder Ergebnisse von Tierversuchen (Untersuchungen am Auge) und/oder mit validierten und international anerkannten Methoden gewonnene In-vitro-Daten zu augenreizenden/-verätzenden Wirkungen bewertet werden, denn sie liefern Hinweise, die unmittelbar die Wirkungen auf das Auge betreffen. Danach sollten vorhandene Daten aus Untersuchungen an Menschen und/oder Tieren zu Hautverätzungen/-reizungen und/oder mit validierten und international anerkannten Methoden gewonnene In-vitro-Daten zu hautätzenden Wirkungen bewertet werden. Chemikalien mit bekannter augenverätzender oder schwer augenreizender Wirkung sollen nicht in die Augen von Tieren geträufelt werden. Dies gilt ebenso für Chemikalien, die Hautreizungen oder -verätzungen hervorrufen. Alle diese Chemikalien sollten ebenfalls als augenverätzende und/oder -reizende Stoffe betrachtet werden. Auch mit Chemikalien, bei denen in früheren Augenuntersuchungen hinreichend nachgewiesen wurde, dass sie weder Verätzungen noch Reizungen hervorrufen, sollten keine In-vivo-Studien am Auge durchgeführt werden.
Analyse der Struktur-Wirkungs-Beziehungen (Structure-Activity Relationships, SAR) (Stufe 2)
Die gegebenenfalls vorhandenen Ergebnisse von Untersuchungen an strukturell verwandten Chemikalien sollten berücksichtigt werden. Liegen genügend Daten über das augenverätzende/-reizende Potenzial von strukturell verwandten Stoffen oder Gemischen aus diesen Stoffen aus Untersuchungen an Menschen und/oder Tieren vor, kann davon ausgegangen werden, dass die zu beurteilende Prüfchemikalie die gleichen Reaktionen hervorrufen wird. In diesen Fällen braucht die Chemikalie nicht getestet zu werden. Für die Zwecke der sequenziellen Prüfstrategie reichen negative Daten aus Untersuchungen an strukturell verwandten Stoffen oder Gemischen als Nachweis für ein Nichtvorhandensein augenverätzender/-reizender Wirkungen nicht aus. Zur Ermittlung des Verätzungs- und Reizungspotenzials in Bezug auf die Haut und die Augen sollten validierte und anerkannte SAR-Verfahren herangezogen werden.
Physikalisch-chemische Eigenschaften und chemische Reaktivität (Stufe 3)
Stoffe mit extremen pH-Werten (≤ 2,0 bzw. ≥ 11,5) können starke lokale Reaktionen hervorrufen. Gilt ein extremer pH-Wert als Anhaltspunkt für die augenverätzende oder -reizende Wirkung einer Chemikalie, so kann deren saure/alkalische Reserve (Pufferkapazität) ebenfalls berücksichtigt werden (5) (6) (7). Lässt die Pufferkapazität darauf schließen, dass eine Chemikalie möglicherweise keine augenverätzende Wirkung hat (d. h. Chemikalien mit extremem pH-Wert und geringer saurer/alkalischer Reserve), sollten weitere Prüfungen zur Bestätigung dieser Vermutung durchgeführt werden. Dafür eignen sich validierte und anerkannte In-vitro- oder Ex-vivo-Tests (siehe Punkt 10).
Einbeziehung anderer vorhandener Informationen (Stufe 4)
In dieser Phase sollen alle verfügbaren Informationen über die systemische Toxizität bei Applikation auf die Haut bewertet werden. Die akute dermale Toxizität der Prüfchemikalie sollte ebenfalls beurteilt werden. Hat sich die Prüfchemikalie bei Hautkontakt als sehr giftig erwiesen, braucht sie nicht am Auge getestet zu werden. Auch wenn nicht unbedingt ein Zusammenhang zwischen akuter dermaler Toxizität und Augenreizung/-verätzung besteht, kann davon ausgegangen werden, dass ein Stoff, der bei Hautapplikation sehr giftig ist, auch beim Einträufeln in das Auge eine starke Toxizität aufweist. Diese Daten können auch zwischen den Stufen 2 und 3 bewertet werden.
Bewertung des Hautverätzungspotenzials der Chemikalie, sofern auch unter gesetzgeberischen Aspekten erforderlich (Stufe 5)
Zunächst sollte das Potenzial zur Hautverätzung und starken Hautreizung nach Prüfmethode B.4 (4) und der zugehörigen Ergänzung (8) bewertet werden, auch anhand der validierten und international anerkannten In-vitro-Tests auf hautverätzende Wirkung (9) (10) (11). Wird der Nachweis erbracht, dass die Chemikalie Verätzungen oder schwere Hautreizungen verursacht, so kann sie auch als Chemikalie mit augenverätzenden oder stark augenreizenden Eigenschaften betrachtet werden. In diesem Falle sind keine weiteren Tests erforderlich. Verursacht die Chemikalie keine Verätzung oder starke Reizung der Haut, sollte ein In-vitro- oder Ex-vivo-Augentest durchgeführt werden.
Ergebnisse von In-vitro- oder Ex-vivo-Tests (Stufe 6)
Chemikalien, deren verätzende oder stark reizende Eigenschaften in validierten und international anerkannten, auf die Ermittlung der Augenverätzung/-reizung ausgerichteten In-vitro- oder Ex-vivo-Tests (12) (13) nachgewiesen wurden, müssen nicht an Tieren getestet zu werden. Es kann davon ausgegangen werden, dass diese Substanzen in vivo vergleichbar schwere Wirkungen hervorrufen werden. Stehen validierte und anerkannte In-vitro-/Ex-vivo-Tests nicht zur Verfügung, sollte die Stufe 6 übersprungen und es sollte direkt zu Stufe 7 übergegangen werden.
In-vivo-Test an Kaninchen (Stufen 7 und 8)
Vor dem eigentlichen In-vivo-Augentest sollte zunächst ein Vorversuch an nur einem Tier durchgeführt werden. Es sollten keine weiteren Tests erfolgen, wenn der Vorversuch ergibt, dass die Chemikalie schwere Augenreizungen oder -verätzungen hervorruft. Liefert der Vorversuch keine Anhaltspunkte für eine ätzende Wirkung oder schwere Reizungen, wird ein Bestätigungstest an zwei weiteren Tieren durchgeführt. Abhängig von den Ergebnissen des Bestätigungstests müssen gegebenenfalls weitere Tests durchgeführt werden. [siehe Prüfmethode B.5]
Prüf- und Bewertungsstrategie:
Augenreizung/-verätzung
| Stufe | Ergebnis | Schlussfolgerung | |
| 1 | Bereits vorliegende Daten aus Untersuchungen am Menschen und/oder an Tieren und/oder In-vitro-Daten aus validierten und international anerkannten Methoden belegen Wirkungen auf das Auge | Schwere Augenschäden | Apikaler Endpunkt; gilt als augenverätzend. Keine Prüfung erforderlich. |
| Augenreizend | Apikaler Endpunkt; gilt als augenreizend. Keine Prüfung erforderlich. | ||
| Nicht augenverätzend/nicht augenreizend | Apikaler Endpunkt; gilt als nicht augenverätzend oder -reizend. Keine Prüfung erforderlich. | ||
| Bereits vorliegende Daten aus Untersuchungen am Menschen und/oder an Tieren und/oder In-vitro-Daten aus validierten und international anerkannten Methoden belegen ätzende Wirkungen auf die Haut | Hautverätzend | Vermutlich augenverätzend. Keine Prüfung erforderlich. | |
| Bereits vorliegende Daten aus Untersuchungen am Menschen und/oder an Tieren und/oder In-vitro-Daten aus validierten und international anerkannten Methoden belegen schwere hautreizende Wirkungen | Stark hautreizend | Vermutlich augenreizend. Keine Prüfung erforderlich. | |
| ↓ | |||
| Keine Informationen vorhanden bzw. vorhandene Informationen nicht schlüssig | |||
| ↓ | |||
| 2 | Durchführung einer SAR-Analyse auf augenverätzende/-reizende Wirkung | schwere Augenschädigung vorhersehbar | Vermutlich augenverätzend. Keine Prüfung erforderlich. |
| Augenreizung vorherabsehbar | Vermutlich augenreizend. Keine Prüfung erforderlich. | ||
| Prüfung einer SAR-Analyse auf hautverätzende/-reizende Wirkung | Hautverätzung vorhersehbar | Vermutlich augenverätzend. Keine Prüfung erforderlich. | |
| ↓ | |||
| Vorhersagen nicht möglich bzw. Vorhersagen nicht schlüssig oder negativ | |||
| ↓ | |||
| 3 | Messung pH-Wert (ggf. unter Berücksichtigung der Puffer-kapazität) | pH ≤ 2 oder ≥ 11,5 (ggf. mit hoher Pufferkapazität) | Vermutlich augenverätzend. Keine Prüfung erforderlich. |
| ↓ | |||
| 2 < pH < 11,5 oder ggf. pH ≤ 2,0 oder ≥ 11,5 mit geringem/ohne Pufferungsvermögen | |||
| ↓ | |||
| 4 | Prüfung bereits vorliegender Daten zur systemischen Toxizität nach Applikation auf die Haut | Sehr toxisch bei Konzentrationen, die für Augentests genutzt werden. | Kein Test, weil Chemikalie zu giftig. Keine Prüfung erforderlich. |
| ↓ | |||
| Keine entsprechenden Angaben vorhanden bzw. Chemikalie ist nicht sehr giftig. | |||
| ↓ | |||
| 5 | Versuch zur Bewertung des Hautverätzungspotenzials nach Prüfstrategie in Kapitel B.4 dieses Anhangs, sofern auch unter gesetzgeberischen Aspekten erforderlich | Verätzung bzw. schwere Reizung | Vermutlich augenverätzend. Keine weitere Prüfung erforderlich. |
| ↓ | |||
| Chemikalie ruft keine Hautverätzung oder schwere Hautreizung hervor | |||
| ↓ | |||
| 6 | Durchführung validierter und anerkannter In-vitro- oder Ex-vivo- Augentest(s) | Verätzung bzw. schwere Reizung | Vermutlich augenverätzend oder schwer augenreizend, vorausgesetzt, der durchgeführte Test ist zur Ermittlung ätzender/schwer reizender Stoffe geeignet und die Chemikalie fällt in den Anwendungsbereich des Tests. Keine weitere Prüfung erforderlich. |
| Reizung | Vermutlich augenreizend, vorausgesetzt, der/die durchgeführte(n) Test(s) ist (sind) zur Ermittlung ätzender, schwer reizender und reizender Stoffe geeignet und Chemikalie fällt in den Anwendungsbereich des/der Tests. Keine weitere Prüfung erforderlich. | ||
| Keine Reizung | Vermutlich nicht augenreizend, vorausgesetzt, der/die durchgeführte(n) Test(s) ist (sind) zur Ermittlung nichtreizender Stoffe sowie zur Unterscheidung von reizenden, schwer reizenden oder augenverätzenden Stoffen geeignet und die Chemikalie fällt in den Anwendungsbereich des Tests. Keine weitere Prüfung erforderlich. | ||
| ↓ | |||
| Validierte und anerkannte In-vitro- oder Ex-vivo-Augentest(s) eignen sich nicht für Schlussfolgerungen | |||
| ↓ | |||
| 7 | Durchführung des In-vivo-Vorversuchs an einem Kaninchen | Schwere Augenschäden | Vermutlich augenverätzend. Keine weitere Prüfung erforderlich. |
| ↓ | |||
| Keine schwere Schädigung bzw. keine Reaktion | |||
| ↓ | |||
| 8 | Durchführung des Bestätigungstests an ein oder zwei weiteren Tieren | Ätzend oder reizend | Vermutlich augenverätzend oder augenreizend. Keine weitere Prüfung erforderlich. |
| Weder ätzend noch reizend | Vermutlich weder augenreizend noch augenverätzend. Keine weitere Prüfung erforderlich. |
Literaturhinweise
(1) OECD (1996) OECD Test Guidelines Programme: Final Report of the OECD Workshop on Harmonization of Validation and Acceptance Criteria for Alternative Toxicological Test Methods. Solna, Schweden, 22.-24. Januar 1996 (http://www.oecd.org/ehs/test/background.htm).
(2) OECD (1998) Harmonized Integrated Hazard Classification System for Human Health and Environmental Effects of Chemical Substances, bestätigt auf der28. Gemeinsamen Tagung des Chemikalien-Ausschusses und der Arbeitsgruppe Chemikalien, November 1998 (http://www.oecd.org/ehs/Class/HCL6.htm).
(3) Worth, A.P. and Fentem J.H. 1999. A General Approach for Evaluating Stepwise Testing Strategies. ATLA 27, 161-177.
(4) Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung.
(5) Young, J.R., How, M.J., Walker, A.P., Worth W.M.H. (1988) Classification as Corrosive or Irritant to Skin of Preparations Containing Acidic or Alkaline Substance Without Testing on Animals. Toxicol. In Vitro, 2, 19-26.
(6) Fentem, J.H., Archer, G.E.B., Balls, M., Botham, P.A., Curren, R.D., Earl, L.K., Edsail, D.J., Holzhutter, H.G. and Liebsch, M. (1998) The ECVAM international validation study on in vitro tests for skin corrosivity. 2. Results and evaluation by the Management Team. Toxicology in vitro 12, 483-524.
(7) Neun, D.J. (1993) Effects of Alkalinity on the Eye Irritation Potential of Solutions Prepared at a Single pH. J. Toxicol. Cut. Ocular Toxicol. 12, 227-231.
(8) Ergänzung zu Kapitel B.4 dieses Anhangs, Sequenzielle Prüfstrategie für Augenreizungen und -verätzungen.
(9) Kapitel B.40 dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: TER-Test (Transcutaneous Electrical Resistance Test).
(10) Kapitel B.40 dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: Test mit einem menschlichen Hautmodell.
(11) OECD (2006), Test No. 435: In vitro Membrane Barrier Test Method for Skin corrosion, OECD-Prüfrichtlinie für Chemikalienprüfungen, Abschnitt 4, OECD, Paris.
(12) Kapitel B.47 dieses Anhangs, Trübungs- und Durchlässigkeitstest an der Rinderhornhaut zwecks Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
(13) Kapitel B.48 dieses Anhangs, Test am isolierten Hühnerauge zur Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern.
_____
1) Zur Anwendung einer integrierten Strategie für Augenreizungsprüfungen im Rahmen der REACH-Verordnung vgl. auch die ECHA Guidance on information requirements and chemical safety assessment, Kapitel R.7a: Endpoint specific guidance http://echa.europa.eu/documents/10162/13632/information_requirements_r7a_en.pdf"
(3) In Teil B erhält Kapitel B.10 folgende Fassung:
"B.10 In-vitro-Test auf Chromosomenaberrationen in Säugetierzellen
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 473 (2016). Sie ist Teil einer Reihe von Prüfmethoden zur genetischen Toxikologie. Ein neu erstelltes OECD-Dokument enthält kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien (1).
Der In-vitro-Test auf Chromosomenaberrationen dient dem Nachweis von Chemikalien, die in Säugerzellkulturen strukturelle Chromosomenaberrationen auslösen (2) (3) (4). Dabei ist zwischen strukturellen Chromosomentyp- und Chromatidentypaberrationen zu unterscheiden. Bei In-vitro-Tests auf Chromosomenaberrationen könnte es zu Polyploidie (einschließlich Endoreduplikation) kommen. Aneugene können zwar eine Polyploidie hervorrufen, die an sich jedoch kein Hinweis auf ein aneugenisches Potenzial ist und möglicherweise nur auf Störungen des Zellzyklus oder Zytotoxizität hinweist (5). Dieser Test dient nicht der Messung der Aneuploidie; dazu wird ein In-vitro-Mikrokerntest (6) empfohlen.
Für den In-vitro-Chromosomenaberrationstest eignen sich Kulturen von etablierten Zelllinien oder primäre Zellkulturen vom Menschen oder von Nagetieren. Die verwendeten Zellen werden unter dem Gesichtspunkt ihrer Wachstumsfähigkeit in Kultur, der Karyotypstabilität (einschließlich Chromosomenzahl) und der spontanen Häufigkeit von Chromosomenaberrationen ausgewählt (7). Die bisher vorhandenen Daten lassen zwar keine verbindlichen Empfehlungen zu, legen jedoch nahe, dass bei der Bewertung des Gefahrenpotenzials chemischer Stoffe der p53-Status, die genetische (Karyotyp-) Stabilität, die DNA-Reparaturfähigkeit und die Herkunft (Nagetier/Mensch) der für die Tests ausgewählten Zellen berücksichtigt werden müssen. Anwendern dieser Prüfmethode wird daher empfohlen, beim Nachweis der Entwicklung von Chromosomenaberrationen den Einfluss dieser und anderer Zellcharakteristika auf die Leistungsfähigkeit von Zelllinien zu berücksichtigen, da sich die wissenschaftlichen Kenntnisse auf diesem Gebiet ständig weiterentwickeln.
Für Definitionen siehe Anlage 1.
Ausgangsüberlegungen und Grenzen
In vitro durchgeführte Versuche setzen in der Regel eine exogene Metabolisierung voraus, es sei denn, die Zellen sind in Bezug auf die Prüfchemikalie metabolisch kompetent. Mit exogener Metabolisierung lassen sich die In-vivo-Bedingungen jedoch nicht gänzlich nachvollziehen. Es sind unbedingt Bedingungen zu vermeiden, die zu künstlich herbeigeführten Positivergebnissen führen könnten, d. h. zu Chromosomenschäden, die nicht von einer direkten Interaktion zwischen den Prüfchemikalien und den Chromosomen herrühren; zu solchen Bedingungen gehören Veränderungen des pH-Wertes bzw. der Osmolalität (8) (9) (10), eine Interaktion mit einzelnen Komponenten des Mediums (11) (12) oder eine hochgradige Zytotoxizität (13) (14) (15) (16).
Dieser Test dient der Feststellung von Chromosomenaberrationen infolge klastogener Vorgänge. Zur Analyse von Chromosomenaberrationen sollten Metaphasenzellen verwendet werden. Deshalb ist es wichtig, dass Zellen sowohl in behandelten als auch in unbehandelten Kulturen die Mitose erreichen. Für hergestellte Nanomaterialien sind möglicherweise spezielle Anpassungen dieser Prüfmethode erforderlich, die an dieser Stelle jedoch nicht beschrieben werden.
Bevor die Prüfmethode auf ein Gemisch angewendet wird, um Daten für regulatorische Zwecke zu generieren, sollte geprüft werden, ob, und, falls ja, warum sie diesbezüglich zweckdienliche Ergebnisse liefert. Diese Überlegungen erübrigen sich, wenn die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist.
Testprinzip
Die Behandlung der Zellkulturen (humane Zellen oder andere Säugetierzellen) mit der Prüfchemikalie erfolgt mit und ohne exogene Metabolisierung, es sei denn, es werden Zellen verwendet, die über eine entsprechende Stoffwechselkompetenz verfügen (siehe Nummer 13). In bestimmten vorab festgelegten Zeitabständen werden die Zellkulturen mit einem Spindelgift (z.B. Colcemid oder Colchicin) behandelt, geerntet und angefärbt und die Metaphasezellen anschließend mikroskopisch auf Chromatidentyp- und Chromosomentypaberrationen untersucht.
Beschreibung der Methode
Vorbereitungen
Zellen
Es können verschiedene Zelllinien (z.B. Ovarialzellen des chinesischen Hamsters (CHO), V79-Lungenzellen des chinesischen Hamsters (CHL), TK6-Lungenzellen des chinesischen Hamsters (CHL)/IU) oder primäre Zellkulturen, auch menschliche Zellen oder Lymphozyten aus dem peripheren Blut von Menschen oder anderen Säugern, verwendet werden (7). Die Wahl der verwendeten Zelllinien sollte wissenschaftlich begründet sein. Wenn Primärzellen verwendet werden, sollten im Interesse des Tierschutzes Primärzellen menschlichen Ursprungs in Betracht gezogen werden, soweit dies möglich ist und die Entnahme nach humanethischen Grundsätzen und Regeln erfolgt. Lymphozyten aus peripherem Humanblut sollte jungen (etwa 18-35 Jahre alten) Personen entnommen werden, die Nichtraucher sind, bei denen keine Krankheit festgestellt wird und die kürzlich nicht in einem Umfang mit gentoxischen Substanzen (z.B. Chemikalien, ionisierende Strahlungen) in Berührung kamen, der die Hintergrundinzidenz von Chromosomenaberrationen erhöhen würde. Auf diese Weise wird sichergestellt, dass die Hintergrundinzidenz von Chromosomenaberrationen niedrig und konstant ist. Die Baseline-Inzidenz von Chromosomenaberrationen steigt mit dem Alter, wobei dieser Trend bei Frauen ausgeprägter ist als bei Männern (17) (18). Wenn Zellen mehrerer Spender zwecks Verwendung gepoolt werden, ist die Zahl der Spender anzugeben. Es muss nachgewiesen werden, dass sich die Zellen zwischen Behandlungsbeginn und Zellentnahme geteilt haben. Die Zellkulturen werden in einer Phase exponentiellen Wachstums gehalten (Zelllinien) oder zur Teilung angeregt (primäre Lymphozytenkulturen), um Zellen in unterschiedlichen Zyklusstadien zu exponieren, da die Empfindlichkeit der Zellstadien gegenüber den Prüfchemikalien möglicherweise nicht bekannt ist. Die Primärzellen, die mit mitogenen Wirkstoffen zur Teilung angeregt werden müssen, werden in der Regel während der Behandlung mit der Prüfchemikalie nicht weiter synchronisiert (z.B. humane Lymphozyten nach einer 48-stündigen mitogenen Stimulation). Die Verwendung synchronisierter Zellen während der Behandlung wird nicht empfohlen, kann jedoch zulässig sein, sofern gerechtfertigt.
Kulturmedien und Inkubationsbedingungen
Die Kultivierung erfordert geeignete Kulturmedien und Inkubationsbedingungen (Kulturgefäße, ggf. befeuchtete Atmosphäre mit einer CO2-Konzentration von 5 %, Inkubationstemperatur von 371 °C). Zelllinien sind routinemäßig auf Stabilität der modalen Chromosomenzahl und Mycoplasma-Verunreinigung zu überprüfen (7) (19); bei Verunreinigung oder bei veränderter modaler Chromosomenzahl sollten Zellen nicht verwendet werden. Die normale Dauer des Zellzyklus sollte bei den gewählten Zelllinien oder den im Prüflabor verwendeten primären Kulturen bekannt sein und mit veröffentlichten Zellcharakteristiken übereinstimmen (20).
Vorbereitung der Kulturen
Zelllinien: Die Zellen werden aus Stammkulturen gewonnen und im Kulturmedium in einer solchen Dichte überimpft, dass die Zellen in Suspensionen oder Monolayern bis zum Zeitpunkt ihrer Ernte weiterhin exponentiell wachsen (z.B. sollte eine Konfluenz bei in Monolayern gezüchteten Zellen vermieden werden).
Lymphozyten: Mit einem Antikoagulans (z.B. Heparin) behandeltes Vollblut oder separierte Lymphozyten werden einem Kulturmedium beigegeben (z.B. im Fall von humanen Lymphozyten für die Dauer von 48 Stunden), das ein Mitogen (z.B. Phytohämagglutinin (PHA) bei humanen Lymphozyten) enthält, um eine Zellteilung vor der Behandlung mit der Prüfchemikalie herbeizuführen.
Stoffwechselaktivierung
Bei Zellen mit unzulänglicher endogener Stoffwechselkapazität sollten exogene metabolisierende Systeme verwendet werden. Das gängigste und, sofern nicht anders begründet, standardmäßig empfohlene System, ist eine durch Ko-Faktoren ergänzte post-mitochondriale Fraktion (S9) aus der Leber von Nagetieren (in der Regel Ratten), die mit enzyminduzierenden Agenzien wie Aroclor 1254 (21) (22) (23) oder einer Kombination aus Phenobarbiton und β-Naphtoflavon (24) (25) (26) (27) (28) (29) vorbehandelt wurde. Letztere Kombination verstößt nicht gegen das Stockholmer Übereinkommen über persistente organische Schadstoffe (30) und hat sich für die Induktion von Multifunktionsoxidasen als ebenso wirksam wie Aroclor 1254 erwiesen (24) (25) (26) (28). Die S9-Fraktion wird im Endmedium in der Regel in Konzentrationen von 1 bis 2 % v/v verwendet, kann jedoch auf 10 % v/v erhöht werden. Die Verwendung von Produkten, die den Mitoseindex senken, insbesondere Komplexbildner für Calcium (31), sollten während der Behandlung vermieden werden. Die Wahl der Art und Konzentration des exogenen Metabolisierungssystems oder metabolischen Agens ist möglicherweise von der Klasse der geprüften Chemikalien abhängig.
Vorbereitung der Prüfchemikalie
Feste Prüfchemikalien sollten vor der Zellbehandlung in geeigneten Lösungsmitteln gelöst und ggf. verdünnt werden (siehe Nummer 23). Flüssige Prüfchemikalien können dem Versuchssystem vor der Behandlung direkt zugegeben und/oder verdünnt werden. Gasförmige oder flüchtige Prüfchemikalien sind durch entsprechende Modifikationen der Standardprotokolle zu prüfen, z.B. durch Behandlung in hermetisch verschlossenen Kulturgefäßen (32) (33) (34). Zubereitungen der Prüfchemikalie sollten kurz vor der Behandlung hergestellt werden, es sei denn, die Chemikalie ist bei Lagerung nachweislich stabil.
Prüfbedingungen
Lösungsmittel
Das Lösungsmittel sollte so gewählt werden, dass eine optimale Löslichkeit der Prüfchemikalie gewährleistet ist, ohne dass die Durchführung des Versuchs beeinträchtigt wird, z.B. durch Veränderung des Zellwachstums, Beeinträchtigung der Integrität der Prüfchemikalie, Reaktion mit Kulturgefäßen, Behinderung des Metabolisierungssystems. Es ist empfiehlt sich, als erste Wahl die Verwendung eines wässrigen Lösungsmittels (oder Kulturmediums) in Erwägung zu ziehen. Gründlich erprobte Lösungsmittel sind z.B. Wasser oder Dimethylsulfoxid. Organische Lösungsmittel sollten 1 % v/v und wässrige Lösungsmittel (Kochsalzlösung oder Wasser) sollten 10 % v/v im Endmedium möglichst nicht überschreiten. Werden weniger gründlich erprobte Lösungsmittel verwendet (z. B Ethanol oder Aceton), so ist dies durch Daten zu untermauern, die ihre Verträglichkeit mit der Prüfchemikalie und mit dem Versuchssystem sowie ihre mangelnde Gentoxizität in der verwendeten Konzentration belegen. Liegen keine Daten vor, die dies belegen, sollten unbedingt unbehandelte Kontrollen (siehe Anlage 1) einbezogen werden, um nachzuweisen, dass durch die gewählten Lösungsmittel keine schädlichen oder klastogenen Wirkungen ausgelöst werden.
Messung von Zellproliferation und Zytotoxizität und Wahl der Behandlungskonzentrationen
Bei der Bestimmung der höchsten Konzentration der Prüfchemikalie sind Konzentrationen zu vermeiden, die zu künstlich positiven Reaktionen führen können, z.B. zu übermäßiger Zytotoxizität (siehe Nummer 22), Ausfällungen im Kulturmedium (siehe Nummer 23) oder ausgeprägten Veränderungen des pH-Werts oder der Osmolalität (siehe Nummer 5). Sofern die Prüfchemikalie zum Zeitpunkt der Zugabe den pH-Wert des Mediums erheblich verändert, lässt sich dieser auch durch Zugabe eines Puffers ins Endmedium einstellen, damit künstlich positive Reaktionen vermieden und geeignete Kulturbedingungen aufrechterhalten werden.
Es sind Messungen der Zellproliferation vorzunehmen, um sicherzustellen, dass während des Tests eine ausreichende Zahl behandelter Zellen eine Mitose durchlaufen hat und dass die Behandlungen auf geeigneten Zytotoxizitätsniveaus durchgeführt werden (siehe Nummern 18 und 22). Die Zytotoxizität sollte im Hauptversuch mit und ohne Stoffwechselaktivierung unter Verwendung eines geeigneten Indikators für Zelltod und -wachstum bestimmt werden. Wenngleich die Bewertung der Zytotoxizität im Rahmen eines Vorversuchs nützlich sein kann, um eine bessere Bestimmung der im Hauptversuch verwendeten Konzentrationen vornehmen zu können, ist ein Vorversuch nicht zwingend erforderlich. Wird er durchgeführt, ersetzt er nicht die Messung der Zytotoxizität im Hauptversuch.
Die relative Populationsverdopplung (RPD) oder die relative Erhöhung der Zellzahl (RICC) sind geeignete Verfahren zur Bewertung der Zytotoxizität in zytogenetischen Versuchen (13) (15) (35) (36) (55) (Formeln siehe Anlage 2). Bei Langzeitbehandlungen und Probenahmezeitpunkten nach Beginn der Behandlung, die über 1,5 normale Zellzykluslängen (d. h. mehr als 3 Zellzykluslängen insgesamt) andauern, könnte es bei der RPD zu einer Unterschätzung der Toxizität kommen (37). Unter diesen Umständen ist die RICC möglicherweise das bessere Verfahren; anderenfalls erhält man bei Bewertung der Zytotoxizität nach 1,5 normalen Zellzykluslängen beim RPD-Verfahren einen hilfreichen Schätzwert.
Bei Lymphozyten in Primärkulturen ist der Mitoseindex (MI), auch wenn er ein geeigneter Wert zur Messung zytotoxischer/zytostatischer Wirkungen ist, abhängig vom Zeitpunkt der Messung nach der Behandlung, vom verwendeten Mitogen sowie möglichen Störungen des Zellzyklus. Der MI ist jedoch zulässig, da andere Verfahren zur Messung der Zytotoxizität möglicherweise zu komplex und wenig praktikabel und nicht auf die Zielpopulation der Lymphozyten anwendbar sind, die infolge der PHA-Stimulation wachsen.
Zwar sind das RICC- bzw. das RPD-Verfahren für Zelllinien und der MI für Primärkulturen von Lymphozyten die empfohlenen Zytotoxizitätsparameter, weitere Indikatoren (z.B. Zellintegrität, Apoptose, Nekrose, Zellzyklus) könnten jedoch nützliche Zusatzinformationen liefern.
Es sollten mindestens drei Versuchskonzentrationen (ausgenommen Lösungsmittel und Positivkontrollen), die die Akzeptanzkriterien erfüllen (geeignete Zytotoxizität, Anzahl der Zellen usw.), ausgewertet werden. Unabhängig von der Art der Zellen (Zelllinien oder Primärkulturen von Lymphozyten) können für jede überprüfte Konzentration Replikat- oder Einfachkulturen verwendet werden. Wenngleich die Verwendung von Zweifachkulturen ratsam ist, sind Einfachkulturen auch zulässig, vorausgesetzt, es wird für Einfach- oder Zweifachkulturen jeweils die gleiche Gesamt-Zellpopulation ausgewertet. Die Verwendung von Einzelkulturen ist insbesondere dann relevant, wenn mehr als drei Konzentrationen bewertet werden (siehe Nummer 31). Die Ergebnisse aus den unabhängigen Replikatkulturen bei einer gegebenen Konzentration können zu Datenanalysezwecken gepoolt werden (38). Bei Prüfchemikalien mit geringer oder ohne Zytotoxizität sind in der Regel Konzentrationsintervalle mit zwei- bis dreifacher Konzentration geeignet. Wenn Zytotoxizität auftritt, sollten die Versuchskonzentrationen einen Bereich ausgehend von dem Wert, bei dem Zytotoxizität auftritt (siehe Beschreibung unter Nummer 22), bis zu Konzentrationen mit mäßiger und geringer oder nicht vorhandener Toxizität umfassen. Viele Prüfchemikalien zeigen steile Konzentrations-Wirkungs-Kurven, und um Daten bei mäßiger oder geringer Toxizität zu erhalten oder die Dosis-Wirkungs-Beziehung im Einzelnen auszuwerten, wird es erforderlich sein, Konzentrationen mit kleineren Abständen und/oder mehr als drei Konzentrationen zu verwenden (Einfach- oder Replikatkulturen), insbesondere in Fällen, in denen ein Wiederholungsversuch erforderlich ist (siehe Nummer 47).
Beruht die höchste Konzentration auf Zytotoxizität, so sollte versucht werden, unter Verwendung der empfohlenen Zytotoxizitätsparameter (d. h. Verringerung der RICC und RPD bei Zelllinien und Verringerung des MI bei Primärkulturen von Lymphozyten auf 45 ± 5 % der gleichzeitigen Negativkontrolle) mit der höchsten Konzentration eine Zytotoxizität von 55 ± 5 % zu erreichen. Vorsicht ist geboten, positive Ergebnisse dahingehend zu interpretieren, dass sie ausschließlich am oberen Ende dieses zytotoxischen Bereichs von 55 ± 5 % anzutreffen sind (13).
Im Falle schwer löslicher Chemikalien, die bei Konzentrationen unterhalb der niedrigsten unlöslichen Konzentration nicht zytotoxisch sind, sollte die höchste analysierte Konzentration am Ende der Behandlung mit der Prüfchemikalie eine Trübung oder eine mit bloßem Auge oder mithilfe eines inversen Mikroskops erkennbare Ausfällung bewirken. Auch wenn Zytotoxizität oberhalb der niedrigsten unlöslichen Konzentration auftritt, ist es ratsam, nur eine Konzentration zu testen, bei der es zu einer Trübung oder sichtbaren Ausfällung kommt, da künstliche Wirkungen eine Folge dieser Ausfällung sein könnten. Bei der Konzentration, bei der es zu einer Ausfällung kommt, ist unbedingt sicherzustellen, dass die Ausfällung die Durchführung des Versuchs nicht beeinträchtigt (z.B. Färbung oder Auswertung). Es ist möglicherweise sinnvoll, die Löslichkeit im Kulturmedium vor dem Versuch zu bestimmen.
Wird keine Ausfällung bzw. keine grenzwertige Zytotoxizität beobachtet, sollte die höchste Versuchskonzentration 10 mM, 2 mg/ml oder 2 µl/ml entsprechen, je nachdem, welcher Wert der niedrigere ist (39) (40) (41). Sofern die Zusammensetzung der Prüfchemikalie nicht vorgegeben ist, es sich z.B. um einen Stoff mit unbekannter oder schwankender Zusammensetzung, um komplexe Reaktionsprodukte oder biologische Materialien (UVCB) (42), einen Umweltextrakt usw. handelt, muss die höchste Konzentration möglicherweise höher angesetzt werden (z.B. bei 5 mg/ml), sofern keine ausreichende Zytotoxizität vorhanden ist, um die Konzentration der einzelnen Komponenten zu erhöhen. Es sei jedoch darauf hingewiesen, dass diese Anforderungen sich von denen für Humanpharmazeutika unterscheiden können (43).
Kontrollen
Bei jedem Zellerntezeitpunkt sind gleichzeitige Negativkontrollen (siehe Nummer 15) zu berücksichtigen, bei denen das Behandlungsmedium lediglich Lösungsmittel enthält und die auf die gleiche Weise wie die Behandlungskulturen behandelt werden.
Gleichzeitige Positivkontrollen müssen angelegt werden, um die Eignung des Labors zum Nachweis von Klastogenen unter den Bedingungen des verwendeten Prüfprotokolls sowie ggf. die Wirksamkeit des exogenen Metabolisierungssystems nachzuweisen. Beispiele für Positivkontrollen sind Tabelle 1 zu entnehmen. Es können andere geeignete Positivkontrollchemikalien verwendet werden, sofern gerechtfertigt. Da In-vitro-Tests auf Gentoxizität in Säugetierzellen ausreichend standardisiert sind, kann sich die Hinzuziehung von Positivkontrollen auf ein Klastogen beschränken, das eine Stoffwechselaktivierung erfordert. Unter der Voraussetzung, dass diese einzeln durchgeführte Positivkontrolle zeitgleich zu dem nicht aktivierten Versuch mit derselben Behandlungsdauer erfolgt, wird durch ihre Wirkung sowohl die Aktivität des Metabolisierungssystems als auch die Reaktionsfähigkeit des Versuchssystems nachgewiesen. Im Falle einer Langzeitbehandlung (ohne S9) sollte jedoch eine gesonderte Positivkontrolle erfolgen, da die Behandlungsdauer beim Versuch mit Stoffwechselaktivierung eine andere ist. Jede Positivkontrolle sollte bei einer oder mehreren Konzentrationen durchgeführt werden, die voraussichtlich eine reproduzierbare und erkennbare Zunahme gegenüber dem Hintergrund ergeben, womit sich die Empfindlichkeit des Versuchssystems nachweisen lässt (d. h. die Wirkungen sind eindeutig, lassen aber beim Ablesen nicht sofort die Identität der kodierten Objektträger erkennen), und die Wirkung sollte nicht durch einen Zytotoxizitätswert beeinträchtigt werden, der die in der Prüfmethode vorgegebenen Grenzen überschreitet.
Tabelle 1 Zur Beurteilung der Eignung des Labors und zur Wahl der Positivkontrollen empfohlene Referenzchemikalien
| Kategorie | Chemikalie | CAS-Nr. |
| 1. Klastogene, die ohne Stoffwechselaktivierung wirken | ||
| Methylmethansulfonat | 66-27-3 | |
| Mitomycin C | 50-07-7 | |
| 4-Nitroquinolin-N-oxid | 56-57-5 | |
| Cytosinarabinosid | 147-94-4 | |
| 2. Klastogene, die eine Stoffwechselaktivierung erfordern | ||
| Benzo[a]pyren | 50-32-8 | |
| Cyclophosphamid | 50-18-0 | |
Verfahren
Behandlung mit der Prüfchemikalie
Proliferierende Zellen werden mit und ohne Stoffwechselaktivierungssystem mit der Prüfchemikalie behandelt.
Zeitpunkt der Zellernte
Um eine genaue Bewertung zu ermöglichen, die erforderlich wäre, um auf ein negatives Ergebnis schließen zu können, sollte jede der drei nachgenannten Versuchsbedingungen getestet werden - eine Kurzzeitbehandlung mit und ohne Stoffwechselaktivierung und eine Langzeitbehandlung ohne Stoffwechselaktivierung (siehe Nummern 43, 44 und 45):
In Fällen, in denen eine der oben genannten Versuchsbedingungen zu einem positiven Befund führt, kann möglicherweise auf Untersuchungen nach den anderen Behandlungsverfahren verzichtet werden.
Chromosomenpräparation
Die Zellkulturen werden vor der Gewinnung in der Regel ein bis drei Stunden lang mit Colcemid oder Colchicin behandelt. Für die Chromosomenpräparation wird jede Zellkultur gesondert geerntet und aufgearbeitet. Zur Chromosomenpräparation gehören die Behandlung der Zellen mit hypotoner Lösung, die Fixierung und das Anfärben. In Monolayern können am Ende der 3- bis 6-stündigen Behandlung mitotische Zellen vorhanden sein (diese sind daran zu erkennen, dass sie rund sind und sich von der Oberfläche lösen). Da diese mitotischen Zellen sich leicht lösen, können sie bei Entfernung des Mediums mit der Prüfchemikalie verloren gehen. Kann nachgewiesen werden, dass verglichen mit den Kontrollen die Zahl der mitotischen Zellen erheblich zugenommen hat, was mit hoher Wahrscheinlichkeit auf einen mitotischen Arrest hinweist, sollten die Zellen durch Zentrifugieren gesammelt und anschließend den Kulturen wieder zugeführt werden, um zu vermeiden, dass Zellen verlorengehen, die sich in der Mitose befinden und zum Zeitpunkt der Gewinnung dem Risiko einer Chromosomenaberration ausgesetzt sind.
Analyse
Alle Objektträger, auch die für die Positiv- und Negativkontrollen, sollten vor der mikroskopischen Untersuchung von unabhängiger Seite kodiert werden. Da es bei der Fixierung bei einem Teil der Metaphasezellen häufig zum Verlust von Chromosomen kommt, sollten die ausgewerteten Zellen daher eine Zentromerzahl enthalten, die bei allen Zelltypen dem Modalwert ± 2 entspricht.
Es sollten mindestens 300 gut gespreitete Metaphasen je Konzentration und Kontrolle analysiert werden, um schlussfolgern zu können, dass eine Prüfchemikalie eindeutig negativ ist (siehe Nummer 45). Die 300 Zellen sind gleichmäßig auf die Replikate zu verteilen, sofern solche verwendet werden. Bei der Verwendung von Einzelkulturen je Konzentration (siehe Nummer 21) sollten mindestens 300 gut gespreitete Metaphasen in dieser Einzelkultur analysiert werden. Die Analyse von 300 Zellen hat den Vorteil, dass die statistische Aussagekraft des Versuchs erhöht wird; zudem sind dann kaum Nullwerte zu erwarten (erwartungsgemäß nur 5 %) (44). Die Anzahl der zu analysierenden Metaphasen kann verringert werden, wenn eine hohe Zahl von Zellen mit Chromosomenaberrationen beobachtet wird und die Prüfchemikalie als eindeutig positiv gilt.
Zellen mit einer oder mehreren strukturellen Chromosomenaberration(en) mit und ohne Gaps sollten analysiert werden. Brüche und Gaps sind gemäß (45) (46) in Anlage 1 definiert. Chromatidentyp- und Chromosomentypaberration sollten getrennt erfasst und Subtypen zugeordnet werden (Brüche, Austausche). Die im Labor angewandten Verfahren sollten gewährleisten, dass die Analyse von Chromosomenaberrationen von qualifizierten Technikern ausgeführt und ggfs. einer Peer-Review unterzogen wird.
Obwohl es bei dem Test um den Nachweis struktureller Chromosomenaberrationen geht, ist das Auftreten von Polyploidie und Endoreduplikation unbedingt festzuhalten (siehe Nummer 2).
Kompetenz des Labors
Um ausreichende Erfahrung mit der Durchführung des Versuchs nachzuweisen, bevor er für routinemäßige Testungen angewendet wird, sollte das Labor eine Reihe von Versuchen mit positiven Referenzchemikalien durchgeführt haben, die sich unterschiedlicher Mechanismen und verschiedener Negativkontrollen bedienen (unter Verwendung verschiedener Lösungsmittel/Vehikel). Die Reaktionen dieser Positiv- und Negativkontrollen sollten der Literatur entsprechen. Dies gilt nicht für erfahrene Laboratorien, d. h. für Laboratorien, die über eine historische Datenbank im Sinne von Nummer 37 verfügen.
Eine Auswahl von Positivkontrollchemikalien (siehe Tabelle 1 unter Nummer 26) sollte anhand von Kurz- und Langzeitbehandlungen ohne Stoffwechselaktivierung und darüber hinaus anhand einer Kurzzeitbehandlung mit Stoffwechselaktivierung untersucht werden, um die Eignung des Labors zum Nachweis klastogener Chemikalien und zur Bestimmung der Wirksamkeit des Metabolisierungssystems zu belegen. Zum Nachweis der Empfindlichkeit und dynamischen Bandbreite des Versuchssystems sollten mehrere Konzentrationen der ausgewählten Chemikalien ausgewählt werden, um reproduzierbare und konzentrationsbezogene Zunahmen gegenüber dem Hintergrund zu erhalten.
Historische Kontrolldaten
Das Labor sollte Folgendes nachweisen:
Beim erstmaligen Erwerb von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen veröffentlichten Kontrolldaten entsprechen, soweit solche vorhanden sind. Kommen weitere Versuchsdaten zur Verteilung der Kontrollen hinzu, sollten gleichzeitige Negativkontrollen idealerweise innerhalb von 95 % der Kontrollgrenzen der gewählten Verteilung liegen (44) (47). Die Datenbank des Labors für historische Negativkontrollen sollte zunächst mit mindestens 10 Versuchen angelegt werden. Vorzugsweise sollte sie jedoch aus mindestens 20 Versuchen bestehen, die unter vergleichbaren Versuchsbedingungen durchgeführt wurden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder X-Bar-Karten (48)), um zu ermitteln, wie variabel ihre Positiv- und Negativkontrolldaten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist (44). Weitere Empfehlungen zu Aufbau und historischer Datensammlungen (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus historischen Datensammlungen und die Akzeptanzkriterien für einen bestimmten Versuch) sind den Literaturhinweisen zu entnehmen (47).
Etwaige Änderungen am Versuchsprotokoll sollten auf Übereinstimmung mit den bereits vorhandenen historischen Kontrolldatenbanken geprüft werden. Bei größeren Unstimmigkeiten sollte eine neue historische Kontrolldatenbank erstellt werden.
Daten über Negativkontrollen sollten das Auftreten von Zellen mit Chromosomenaberrationen aus einer Einzelkultur oder aus einer Summe von Replikatkulturen umfassen (vgl. Beschreibung unter Punkt 21). Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der Kontrollgrenzen von 95 % der gewählten Verteilung in der Datenbank des Labors zu historischen Negativkontrollen liegen (44) (47). Sofern gleichzeitige Negativkontrolldaten außerhalb der Kontrollgrenzen von 95 % liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um "extreme Ausreißer" handelt und nachgewiesen werden kann, dass das Versuchssystem "unter Kontrolle" ist (siehe Nummer 37) und nachweislich kein technisches oder menschliches Versagen vorliegt.
Daten und Berichterstattung
Präsentation der Ergebnisse
Bewertet werden sollte der Anteil der Zellen mit struktureller Chromosomenaberration bzw. strukturellen Chromosomenaberrationen. Chromatidentyp- und Chromosomentypaberrationen, die Subtypen zugeordnet sind (Brüche, Austausche), sollten dabei unter Angabe ihrer Anzahl und Häufigkeit für Versuchs- und Kontrollkulturen getrennt erfasst werden. Gaps werden getrennt erfasst und angegeben, aber in der Regel nicht bei der Gesamthäufigkeit der Aberrationen berücksichtigt. Der Anteil der Zellen mit Polyploidie und/oder Endoreduplikation wird angegeben, sofern beobachtet.
Erfasst werden sollten auch Maßnahmen, die in den Hauptprüfungen auf Aberrationen gleichzeitig zur Bestimmung der Zytotoxizität aller behandelten und Negativ- sowie Positivkontrollkulturen durchgeführt werden.
Die Daten für die einzelnen Kulturen sollten erfasst dokumentiert werden. Zusätzlich sollten alle Daten in tabellarischer Form zusammengefasst werden.
Gültigkeitskriterien
Die Akzeptanz eines Versuchs beruht auf folgenden Kriterien:
Auswertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn bei einer der getesteten Versuchsbedingungen (siehe Nummer 28):
Sind all diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie Chromosomenaberrationen in Säugerzellkulturen in diesem Versuchssystem auslösen kann. Für Empfehlungen zu den am besten geeigneten statistischen Methoden siehe Literaturhinweise (49) (50) (51).
Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn in allen getesteten Versuchsbedingungen (siehe Punkt 28):
Es wird dann davon ausgegangen, dass die Prüfchemikalie keine Chromosomenaberrationen in Säugerzellkulturen in diesem Versuchssystem auslösen kann.
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
In den Fällen, in denen die Reaktion, wie oben beschrieben, weder eindeutig negativ noch eindeutig positiv ist, oder um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen bewertet werden. Die Auswertung (ggf.) weiterer Zellen oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter veränderten Versuchsbedingungen (z.B. Abstände der Konzentrationen, andere Metabolisierungsbedingungen (d. h. Konzentration (S9) oder Herkunft (S9)), könnten hilfreich sein.
In seltenen Fällen lässt der Datensatz selbst nach weiteren Untersuchungen keine definitive Schlussfolgerung zu positiven oder negativen Ergebnissen zu; in diesem Fall wird die Reaktion der Prüfchemikalie als unschlüssig eingestuft.
Eine zahlenmäßige Zunahme der polyploiden Zellen deutet möglicherweise darauf hin, dass die Prüfchemikalie mitotische Prozesse zu hemmen und numerische Chromosomenaberrationen hervorzurufen vermag (52). Eine zahlenmäßige Zunahme der Zellen mit endoreduplizierten Chromosomen ist möglicherweise ein Anzeichen dafür, dass die Prüfchemikalie die Zellzyklusprogression zu hemmen vermag (53) (54) (siehe Nummer 2). Die Inzidenz von polyploiden Zellen und Zellen mit endoreduplizierten Chromosomen sollte daher getrennt erfasst werden.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Prüfchemikalie:
Einkomponentiger Stoff:
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
Lösungsmittel:
Zellen:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse.
Schlussfolgerungen
Literaturhinweise
(1) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(2) Evans, H.J. (1976), Cytological Methods for Detecting Chemical Mutagens, in Chemical Mutagens, Principles and Methods for their Detection, Band 4, Hollaender, A. (ed.), Plenum Press, New York und London, 1-29
(3) Ishidate, M. Jr., T. Sofuni (1985), The in vitro Chromosomal Aberration Test Using Chinese Hamster Lung (CHL) Fibroblast Cells in Culture in Progress in Mutation Research, Bd. 5, Ashby, J. et al. (eds.), Elsevier Science Publishers, Amsterdam-New York-Oxford, 427-432.
(4) Galloway, S.M. et al. (1987), Chromosomal aberration and sister chromatid exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals, Environmental and Molecular Mutagenesis, Bd. 10/Ergänz. 10, 1-175.
(5) Muehlbauer, P.A. et al. (2008), Improving dose selection and identification of aneugens in the in vitro chromosome aberration test by integration of flow cytometry-based methods, Environmental and Molecular Mutagenesis, Band 49/4, 318-327.
(6) Kapitel B.49 dieses Anhangs: In-vitro-Mikronukleustest an Säugetierzellen.
(7) ILSI paper (draft), Lorge, E., M. Moore, J. Clements, M. O Donovan, F. Darroudi, M. Honma, A. Czich, J van Benthem, S. Galloway, V. Thybaud, B. Gollapudi, M. Aardema, J. Kim, D.J. Kirkland, Recommendations for good cell culture practices in genotoxicity testing.
(8) Scott, D. et al. (1991), Genotoxicity under Extreme Culture Conditions. A report from ICPEMC Task Group 9, Mutation Research/Reviews in Genetic Toxicology, Bd. 257/2, 147-204.
(9) Morita, T. et al. (1992), Clastogenicity of Low pH to Various Cultured Mammalian Cells, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd. 268/2, 297-305.
(10) Brusick, D. (1986), Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations, Environmental and Molecular Mutagenesis, Bd. 8/6, 789-886.
(11) Long, L.H. et al. (2007), Different cytotoxic and clastogenic effects of epigallocatechin gallate in various cell-culture media due to variable rates of its oxidation in the culture medium, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd. 634/1-2, 177-183.
(12) Nesslany, F. et al. (2008), Characterization of the Genotoxicity of Nitrilotriacetic Acid, Environmental and Molecular Mutagenesis, Bd. 49/6, 439-452.
(13) Galloway, S. (2000), Cytotoxicity and chromosome aberrations in vitro: Experience in industry and the case for an upper limit on toxicity in the aberration assay, Environmental and Molecular Mutagenesis, Bd. 35/3, 191-201.
(14) Kirkland, D. et al. (2005), Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens. I: Sensitivity, specificity and relative predictivity, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd.584/1-2, 1-256.
(15) Greenwood, S. et al. (2004), Population doubling: a simple and more accurate estimation of cell growth suppression in the in vitro assay for chromosomal aberrations that reduces irrelevant positive results, Environmental and Molecular Mutagenesis, Bd. 43/1, 36-44.
(16) Hilliard, C.A. et al. (1998), Chromosome aberrations in vitro related to cytotoxicity of nonmutagenic chemicals and metabolic poisons, Environmental and Molecular Mutagenesis, Bd.31/4, 316-326.
(17) Hedner K. et al. (1982), Sister chromatid exchanges and structural chromosomal aberrations in relation to age and sex, Human Genetics, Bd. 62, 305-309.
(18) Ramsey M.J. et al. (1995), The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting, Mutation Research, Bd. 338, 95-106.
(19) Coecke S. et al. (2005), Guidance on Good Cell Culture Practice. A Report of the Second ECVAM Task Force on Good Cell Culture Practice, ATLA, Bd. 33/3, 261-287.
(20) Henderson, L. et al. (1997), Industrial Genotoxicology Group collaborative trial to investigate cell cycle parameters in human lymphocyte cytogenetics studies, Mutagenesis, Bd. 12/3, 163-167.
(21) Ames, B.N., J. McCann, E. Yamasaki (1975), Methods for Detecting Carcinogens and Mutagens with the Salmonella/Mammalian Microsome Mutagenicity Test, Mutation Research/Environmental Mutagenesis and Related Subjects, Bd. 31/6, 347-363.
(22) Maron, D.M., B.N. Ames (1983), Revised Methods for the Salmonella Mutagenicity Test, Mutation Research/Environmental Mutagenesis and Related Subjects, Bd. 113/3-4, 173-215.
(23) Natarajan, A.T. et al. (1976), Cytogenetic Effects of Mutagens/Carcinogens after Activation in a Microsomal System In Vitro, I. Induction of Chromosomal Aberrations and Sister Chromatid Exchanges by Diethylnitrosamine (DEN) and Dimethylnitrosamine (DMN) in CHO Cells in the Presence of Rat-Liver Microsomes, Mutation Research, Bd. 37/1, 83-90.
(24) Matsuoka, A., M. Hayashi, M. Jr. Ishidate (1979), Chromosomal Aberration Tests on 29 Chemicals Combined with S9 Mix in vitro, Mutation Research/Genetic Toxicology, Bd. 66/3, 277-290.
(25) Ong, T.-m. et al. (1980), Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver, Journal of Environmental Pathology and Toxicology, Bd. 4/1, 55-65.
(26) Elliot, B.M. et al. (1992), Report of UK Environmental Mutagen Society Working Party. Alternatives to Aroclor 1254-induced S9 in in vitro Genotoxicity Assays, Mutagenesis, Bd. 7/3, 175-177.
(27) Matsushima, T. et al. (1976), A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems, in In Vitro Metabolic Activation in Mutagenesis Testing, de Serres, F.J. et al. (Herausgeber.), Elsevier, North-Holland, 85-88.
(28) Galloway, S.M. et al. (1994). Report from Working Group on in vitro Tests for Chromosomal Aberrations, Mutation Research/Environmental Mutagenesis and Related Subjects, Bd. 312/3, 241-261.
(29) Johnson, T.E., D.R. Umbenhauer, S.M. Galloway (1996), Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9, Environmental and Molecular Mutagenesis, Bd. 28/1, 51-59.
(30) UNEP (2001), Stockholm Convention on Persistent Organic Pollutants, United Nations Environment Programme (UNEP). Abrufbar unter: http://www.pops.int/.
(31) Tucker, J.D., M.L. Christensen (1987), Effects of anticoagulants upon sister-chromatid exchanges, cell-cycle kinetics, and mitotic index in human peripheral lymphocytes, Mutation Research, Bd. 190/3, 225-8.
(32) Krahn, D.F., F.C. Barsky, K.T. McCooey (1982), CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids, in Genotoxic Effects of Airborne Agents, Tice, R.R., D.L. Costa, K.M. Schaich (eds.), Plenum, New York, 91-103.
(33) Zamora, P.O. et al. (1983), Evaluation of an Exposure System Using Cells Grown on Collagen Gels for Detecting Highly Volatile Mutagens in the CHO/HGPRT Mutation Assay, Environmental and Molecular Mutagenesis, Bd. 5/6, 795-801.
(34) Asakura, M. et al. (2008), An improved system for exposure of cultured mammalian cells to gaseous compounds in the chromosomal aberration assay, Mutation Research, Bd. 652/2, 122-130.
(35) Lorge, E. et al. (2008), Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd. 655/1-2, 1-3.
(36) Galloway, S. et al. (2011), Workshop summary: Top concentration for in vitro mammalian cell genotoxicity assays; and Report from working group on toxicity measures and top concentration for in vitro cytogenetics assays (chromosome aberrations and micronucleus), Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd. 723/2, 77-83.
(37) Honma, M. (2011), Cytotoxicity measurement in in vitro chromosome aberration test and micronucleus test, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Bd. 724/1-2, 86-87.
(38) Richardson, C. et al. (1989), Analysis of Data from In Vitro Cytogenetic Assays. In: Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (ed.) Cambridge University Press, Cambridge, 141-154.
(39) OECD (2014), Document supporting the WNT decision to implement revised criteria for the selection of the top concentration in the in vitro mammalian cell assays on genotoxicity (Test Guidelines 473, 476 and 487) ENV/JM/TG(2014)17. Auf Anfrage erhältlich.
(40) Morita, T., M. Honma, K. Morikawa (2012), Effect of reducing the top concentration used in the in vitro chromosomal aberration test in CHL cells on the evaluation of industrial chemical genotoxicity, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Bd. 741/1-2, 32-56.
(41) Brookmire, L., J.J. Chen, D.D. Levy (2013), Evaluation of the Highest Concentrations Used in the In Vitro Chromosome Aberrations Assay, Environmental and Molecular Muagenesis, Bd. 54/1, 36-43.
(42) EPA, Office of Chemical Safety and Pollution Prevention (2011), Chemical Substances of Unknown or Variable Composition, Complex Reaction Products and Biological Materials: UVCB Substances, http://www.epa.gov/opptintr/newchems/pubs/uvcb.txt.
(43) USFDA (2012), International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use. Abrufbar unter: https://federalregister.gov/a/2012-13774.
(44) OECD (2014), Statistical analysis supporting the revision of the genotoxicity Test Guidelines, OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, Nr. 198, OECD Publishing, Paris.
(45) ISCN (2013), An International System for Human Cytogenetic Nomenclature, Schaffer, L.G., J. MacGowan-Gordon, M. Schmid (Herausgeber), Karger Publishers Inc., Connecticut.
(46) Scott, D. et al. (1990), Metaphase chromosome aberration assays in vitro, in Basic Mutagenicity Tests: UKEMS Recommended Procedures, Kirkland, D.J. (Herausgeber), Cambridge University Press, Cambridge, 62-86.
(47) Hayashi, M. et al. (2011), Compilation and use of genetic toxicity historical control Data, Mutation Research, Bd. 723/2, 87-90.
(48) Ryan, T. P. (2000), Statistical Methods for Quality Improvement, Zweite Ausgabe, John Wiley and Sons, New York.
(49) Fleiss, J. L., B. Levin, M.C. Paik (2003), Statistical Methods for Rates and Proportions, Dritte Ausgabe, John Wiley & Sons, New York.
(50) Galloway, S.M. et al. (1987), Chromosome aberration and sister chromatid exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals, Environmental and Molecular Mutagenesis, Bd. 10/Ergänz. 10, 1-175.
(51) Richardson, C. et al. (1989), Analysis of Data from In Vitro Cytogenetic Assays, in Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Herausgeber), Cambridge University Press, Cambridge, 141-154.
(52) Warr, T.J., E.M. Parry, J.M. Parry (1993), A comparison of two in vitro mammalian cell cytogenetic assays for the detection of mitotic aneuploidy using 10 known or suspected aneugens, Mutation Research, Bd. 287/1, 29-46.
(53) Locke-Huhle, C. (1983), Endoreduplication in Chinese hamster cells during alpha-radiation induced G2 arrest, Mutation Research, Bd. 119/3, 403-413.
(54) Huang, Y., C. Change, J.E. Trosko (1983), Aphidicolin - induced endoreduplication in Chinese hamster cells, Cancer Research, Bd. 43/3, 1362-1364.
(55) Soper, K.A., S.M. Galloway (1994), Cytotoxicity measurement in in vitro chromosome aberration test and micronucleus test, Mutation Research, Bd. 312, 139-149.
.
| Definitionen | Anlage 1 |
Aneuploidie: Abweichung von der normalen diploiden (oder haploiden) Chromosomenzahl durch ein einziges Chromosom oder mehr, nicht aber durch einen ganzen (oder mehrere) Chromosomensatz/-sätze (Polyploidie).
Apoptose: programmierter Zelltod, der durch eine Reihe von Schritten charakterisiert ist, an deren Ende ein Schrumpfen von Zellen zu membrangebundenen Partikeln steht, die schließlich durch Phagozytose oder Shedding abgebaut werden.
Chemikalie: ein Stoff oder ein Gemisch.
Chromatidbruch: Diskontinuität in einem einzelnen Chromatid mit eindeutiger Dislokation eines der Bruchstücke.
Chromatid-Gap: nicht gefärbter Bereich (achromatische Läsion) eines einzelnen Chromatids mit minimaler Dislokation eines der Bruchstücke.
Chromatidentypaberration: strukturelle Chromsomenanomalie, gekennzeichnet durch Bruch einzelner Chromatiden oder Bruch und Reunion zwischen Chromatiden.
Chromosomentypaberration: strukturelle Chromosomenanomalie, gekennzeichnet durch Bruch oder Bruch und Reunion beider Chromatiden an gleicher Position.
Endoreduplikation: Prozess, bei dem der Kern nach einer S-Phase der DNA-Replikation keine Mitose durchläuft, sondern in eine weitere S-Phase eintritt. Das Ergebnis sind Chromosomen mit 4, 8, 16, ... Chromatiden.
Genotoxisch: allgemeiner Begriff, der alle Typen von DNA- oder Chromosomenschädigungen umfasst, einschließlich Brüchen, Deletionen, Addukten, Nukleotidmodifikationen und -verknüpfungen, Rearrangements, Genmutationen, Chromosomenaberrationen sowie Aneuploidie. Nicht alle genotoxischen Effekte führen zu Mutationen oder stabilen Chromosomenschäden.
Klastogen: Chemikalie, die strukturelle Chromosomenaberrationen in Zellpopulationen oder eukaryontischen Organismen auslöst.
Konzentrationen: beziehen sich auf Endkonzentrationen der Prüfchemikalie im Kulturmedium.
Lösungsmittelkontrolle: allgemeiner Begriff zur Bezeichnung der Kontrollkulturen, die nur mit dem Lösungsmittel behandelt werden, das verwendet wird, um die Prüfchemikalie zu lösen.
Mitose: Teilung des Zellkerns, die in der Regel in Prophase, Prometaphase, Metaphase, Anaphase und Telophase gegliedert ist.
Mitoseindex (MI): Anteil der Zellen einer Zellpopulation, die sich zum Beobachtungszeitpunkt in Metaphase befinden; zugleich Hinweis auf den Grad der Zellproliferation in dieser Population.
Mutagen: Auslöser einer Erbgutveränderung der DNA-Basenpaarsequenz(en) in Genen oder in der Chromosomenstruktur (Chromosomenaberrationen).
Numerische Aberration: Abweichung der Chromosomenzahl vom Normalwert, der für die verwendeten Zellen charakteristisch ist.
p53-Status: Das p53-Protein ist an der Regulierung des Zellzyklus, Apoptose und DNA-Reparatur beteiligt. Zellen, denen ein funktionales p53-Protein fehlt und die nicht in der Lage sind, den Zellzyklus aufzuhalten oder beschädigte Zellen über Apoptose oder andere Mechanismen (z.B. Einleitung einer DNA-Reparatur) im Zusammenhang mit Aufgaben des p53-Proteins als Reaktion auf DNA-Schäden zu beseitigen, sollten theoretisch eher zu Genmutationen oder Chromosomenaberrationen neigen.
Polyploidie: zahlenmäßige Chromosomenaberrationen in Zellen oder Organismen, von denen ein oder mehrere ganze Chromosomensätze betroffen sind, im Gegensatz zur Aneuploidie, bei der nur ein oder mehrere einzelne Chromosomen betroffen sind.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Relative Erhöhung der Zellzahl (RICC): zahlenmäßige Zunahme von Zellen in Kulturen, die mit der Chemikalie behandelt sind, verglichen mit der Zunahme in nicht behandelten Kulturen, ausgedrückt als Prozentanteil.
Relative Populationsverdopplung (RPD): Zunahme der Anzahl an Populationsverdopplungen in Kulturen, die mit der Chemikalie behandelt sind, verglichen mit der Zunahme in nicht behandelten Kulturen, ausgedrückt als Prozentanteil.
S9-Leberfraktion: Überstand des Leberhomogenats nach Zentrifugieren bei 9.000g, d. h. Rohleberextrakt.
S9-Gemisch: Gemisch aus der S9-Leberfraktion und für die metabolische Enzymaktivität notwendigen Ko-Faktoren.
Strukturelle Aberration: Veränderung der Chromosomenstruktur, nachweisbar durch mikroskopische Untersuchung des Metaphase-Stadiums der Zellteilung, äußert sich in Form von Deletionen und Fragmenten, intrachromosomalen oder reziproken Translokationen.
Unbehandelte Kontrollen: Kulturen, die nicht behandelt werden (d. h. weder mit der Prüfchemikalie noch mit Lösungsmittel), jedoch gleichzeitig in gleicher Weise aufgearbeitet werden wie die Kulturen, die mit der Prüfchemikalie behandelt werden.
Zellproliferation: Zunahme der Anzahl von Zellen als Ergebnis der mitotischen Zellteilung.
Zytotoxizität: Für die Zwecke der unter diese Prüfmethode fallenden Versuche, bei denen Zelllinien zum Einsatz kommen, bezeichnet Zytotoxizität eine Verringerung der relativen Populationsverdopplung (RPD) bzw. eine relative Erhöhung der Zellzahl (RICC) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 17 und Anlage 2). Für die Zwecke der unter dieser Prüfmethode fallenden Versuche, bei denen Primärkulturen von Lymphozyten zum Einsatz kommen, bezeichnet Zytotoxizität eine Verringerung des Mitoseindex (MI) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 18 und Anlage 2).
.
| Formeln zur Bewertung der Zytotoxizität | Anlage 2 |
Mitoseindex (MI):
| Zahl der mitotischen Zellen | ||
| MI(%) = |
| x 100 |
| Gesamtzahl ausgewerteter Zellen |
Die relative Erhöhung der Zellzahl (RICC) oder die relative Populationsverdopplung (RPD) wird empfohlen, da bei beiden der Anteil der Zellpopulation berücksichtigt wird, der eine Teilung durchlaufen hat.
| (zahlenmäßige Zunahme von Zellen in behandelten Kulturen(Ende-Beginn)) | ||
| RICC(%) = |
| x 100 |
| (zahlenmäßige Zunahme von Zellen in Kontrollkulturen(Ende)Beginn)) | ||
| (Anzahlt von Populationsverdoppelungen in behandelten Kulturen) | ||
| RPD(%) |
| x 100 |
| (Anzahl von Populationsverdoppelungen in Kontrollkulturen) |
Dabei gilt:
Populationsverdopplung = [log ((Zellzahl nach Behandlung ÷ Anfängliche Zellzahl)] ÷ log 2
Beispielsweise deutet eine RICC oder eine RPD von 53 % auf eine Zytotoxizität/Zytostase von 47 % hin, und eine anhand des MI gemessene Zytotoxizität/Zytostase von 55 % bedeutet, dass der tatsächliche MI zu 45 % außer Kontrolle ist.
In jedem Fall sollte die Größe der Zellpopulation vor der Behandlung ermittelt werden; Gleiches gilt für behandelte Kulturen und Negativkontrollkulturen.
Wenngleich der RCC-Wert (d. h. die in behandelten Kulturen/Zellzahl in Kontrollkulturen) in der Vergangenheit als Bestimmungsgröße für die Zytotoxizität herangezogen wurde, wird er nicht mehr empfohlen, da er zu einer Unterschätzung der Toxizität führen kann.
Bei den Negativkontrollkulturen sollte die Populationsverdopplung der Anforderung gerecht werden, dass Zellprobenahmen nach der Behandlung zu einem Zeitpunkt erfolgen müssen, der etwa der 1,5-fachen Dauer des normalen Zellzyklus entspricht, und der Mitoseindex sollte so hoch liegen, dass man eine ausreichend hohe Zellzahl erhält, die die Mitose erreicht, und zuverlässig mit einer 50 %igen Verringerung kalkulieren kann."
(4) In Teil B erhält Kapitel B.11 folgende Fassung:
"B.11 Test auf Chromosomenaberrationen in Knochenmarkzellen von Säugetieren
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 475 (2016). Sie ist Teil einer Serie von Prüfmethoden zur genetischen Toxikologie. Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (1).
Der In-vivo-Test auf Chromosomenaberrationen in Knochenmarkzellen von Säugetieren ist vor allem für die Bewertung der Genotoxizität relevant, da trotz artenspezifischer Unterschiede bestimmte Faktoren den In-vivo-Stoffwechsel, die Pharmakokinetik und die DNA-Reparaturprozesse beeinflussen und zu den Reaktionen beitragen. Ein In-vivo-Versuch ist ferner hilfreich für weitere Untersuchungen zu gentoxischen Wirkungen, die in einem In-vitro-System nachgewiesen wurden.
Der In-vivo-Test auf Chromosomenaberrationen in Säugetierzellen dient dem Nachweis von strukturellen Chromosomenaberrationen, die von Prüfchemikalien in Knochenmarkzellen von Säugetieren, in der Regel Nagetieren, ausgelöst werden (2) (3) (4) (5). Dabei ist zwischen strukturellen Chromosomentyp- und strukturellen Chromatidentypaberrationen zu unterscheiden. Bei der Mehrzahl der auf gentoxischen Chemikalien beruhenden Mutagene sind die Aberrationen dem Chromatidentyp zuzuordnen, doch kommen auch Chromosomentypaberrationen vor. Chromosomenschäden und damit zusammenhängende Prozesse sind die Ursache für zahlreiche humangenetische Erkrankungen, und es gibt wesentliche Anhaltspunkte dafür, dass diese Schäden und Prozesse, wenn sie Veränderungen an Onkogenen und Tumorsuppressorgenen auslösen, an der Entstehung von Krebs beim Menschen und in Versuchssystemen beteiligt sind. Polyploidie (einschließlich Endoreduplikation) könnte bei In-vivo-Versuchen zum Nachweis von Chromosomenaberrationen entstehen. Eine Polyploidie an sich ist jedoch kein Hinweis auf ein aneugenisches Potenzial und weist möglicherweise nur auf Störungen des Zellzyklus oder Zytotoxizität hin. Dieser Test dient nicht der Messung der Aneuploidie, zu deren Nachweis ein In-vivo-Erythrozyten-Mikrokerntest bei Säugern (vgl. Kapitel B.12 dieses Anhangs) oder der In-vitro-Mikronukleustest an Säugetierzellen (vgl. Kapitel B.49 dieses Anhangs) empfohlen würden.
Für Definitionen der verwendeten Begriffe siehe Anlage 1.
Ausgangsüberlegungen
Bei dieser Prüfung werden routinemäßig Nagetiere eingesetzt, aber auch andere Arten können in einigen Fällen geeignet sein, sofern dies wissenschaftlich gerechtfertigt wird. Zielgewebe bei dieser Prüfung ist das Knochenmark, da es sich um ein gefäßreiches Gewebe mit einer Population rasch proliferierender Zellen handelt, die sich leicht isolieren und aufarbeiten lassen. Die Verwendung anderer Versuchstiere als Ratten und Mäuse sollte im Bericht wissenschaftlich begründet werden. Sofern andere Versuchstiere als Nagetiere verwendet werden, wird empfohlen, Chromosomenaberrationen in Knochenmarkzellen im Rahmen anderer geeigneter Toxizitätstests zu messen.
Soweit es Anhaltspunkte dafür gibt, dass die Prüfchemikalie(n) oder (ein) reaktive® Metabolit(en) das Zielgewebe nicht erreicht bzw. erreichen, ist dieser Test nicht geeignet.
Bevor die Prüfmethode auf ein Gemisch angewendet wird, um Daten für Regulierungszwecke zu generieren, sollte geprüft werden, ob, und falls ja, warum sie für diese Zwecke geeignete Ergebnisse liefert. Diese Überlegungen erübrigen sich, wenn die Durchführung von Tests für das betreffende Gemisch gesetzlich vorgeschrieben ist.
Prinzip der Prüfmethode
Die Prüfchemikalie wird den Tieren über einen geeigneten Expositionsweg verabreicht; letztere werden nach der Behandlung zu einem angemessenen Zeitpunkt human getötet. Vor der Tötung werden die Tiere mit einem Spindelgift (z.B. Colchicin oder Colcemid) behandelt. Anschließend werden aus den Knochenmarkzellen Chromosomen präpariert und angefärbt, und die Metaphasezellen werden auf Chromosomenaberrationen untersucht.
Überprüfung der Eignung des Labors
Eignungsprüfungen
Um ausreichende Erfahrung mit der Durchführung des Versuchs nachzuweisen, bevor er für routinemäßige Testungen angewendet wird, sollte das Labor seine Fähigkeit zur Reproduktion erwarteter Ergebnisse aus veröffentlichten Daten (z.B. (6)) über Chromosomenaberrationshäufigkeiten mit mindestens zwei Positivkontrollchemikalien (einschließlich durch geringe Dosen positiver Kontrollen ausgelöste schwache Reaktionen), wie in Tabelle 1 aufgelistet, und mit kompatiblen Vehikel-/Lösungsmittelkontrollen demonstriert haben (siehe Nummer 22). In diesen Versuchen sollten Dosierungen verwendet werden, mit denen reproduzierbare und dosisabhängige Zunahmen erzielt werden und die die Empfindlichkeit und dynamische Bandbreite des Versuchssystems im untersuchten Gewebe (Knochenmark) demonstrieren, wobei nach der die im Labor normalerweise angewandten Auswertungsmethode vorgegangen werden sollte. Diese Anforderung gilt nicht für erfahrene Laboratorien, d. h. für Laboratorien, die über eine historische Datenbank im Sinne der Nummern 10-14 verfügen.
Historische Kontrolldaten
Im Rahmen der Eignungsprüfungen sollte das Labor Folgendes nachweisen:
Beim erstmaligen Erwerb von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen mit veröffentlichten Kontrolldaten übereinstimmen, soweit solche vorhanden sind. Kommen weitere Versuchsdaten zur Verteilung der historischen Kontrollen hinzu, sollten gleichzeitige Negativkontrollen idealerweise innerhalb der 95 %-Kontrollgrenze dieser Verteilung liegen. Die Datenbank des Labors für historische Negativkontrollen sollte statistisch robuste Werte enthalten, die gewährleisten, dass das Labor in der Lage ist, die Verteilung seiner Negativkontrolldaten zu bewerten. Nach der Literatur reicht möglicherweise ein Minimum von 10 Versuchen aus, doch wird empfohlen, unter vergleichbaren Versuchsbedingungen mindestens 20 Versuche durchzuführen. Laboratorien sollten Qualitätskontrollverfahren wie Qualitätsregelkarten (z.B. C-Karten oder X-Bar-Karten (7)) anwenden, um zu ermitteln, wie variabel ihre Daten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist. Für weitere Empfehlungen zu Aufbau und Verwendung historischer Datensammlungen (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus historischen Datensammlungen und die Akzeptanzkriterien für einen bestimmten Versuch) siehe Literaturhinweise (8).
Soweit das Labor während der Eignungsprüfungen (siehe Nummer 9) nicht genügend Versuche abschließt, um eine statistisch robuste Verteilung der Negativkontrollen zu demonstrieren (siehe Punkt 11), kann die Verteilung auch während der ersten routinemäßigen Tests erstellt werden. Diese Vorgehensweise sollte sich an den Literaturempfehlungen orientieren (8), und die bei diesen Versuchen erzielten Negativkontrollergebnisse sollten mit veröffentlichten Negativkontrolldaten übereinstimmen.
Etwaige Änderungen am Versuchsprotokoll sollten hinsichtlich ihrer Auswirkungen auf die resultierenden Daten geprüft werden, die mit den bereits vorhandenen Labordaten über historische Kontrollen übereinstimmen müssen. Nur bei größeren Unstimmigkeiten sollte eine neue Datenbank für historische Kontrollen erstellt werden, soweit eine fachkundige Beurteilung ergibt, dass eine Abweichung von der vorherigen Verteilung besteht (siehe Nummer 11). Während der Neuerstellung muss für die Durchführung eines aktuellen Versuchs ggfs. keine vollständige Datenbank mit Negativkontrollen vorhanden sein, vorausgesetzt, dass Labor kann nachweisen, dass seine Werte aus gleichzeitigen Negativkontrollen entweder mit seiner vorangegangenen Datenbank oder mit den entsprechenden veröffentlichten Daten übereinstimmen.
Daten über Negativkontrollen sollten das Vorkommen struktureller Chromosomenaberrationen (ohne Gaps) bei jedem Tier umfassen. Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der 95 %-Kontrollgrenzen der gewählten Verteilung in der Datenbank des Labors für historische Negativkontrollen liegen. Sofern Daten zu gleichzeitigen Negativkontrollen außerhalb der 95 %-Kontrollgrenzen liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um "extreme Ausreißer"handelt und nachgewiesen werden kann, dass das Versuchssystem "unter Kontrolle" ist (siehe Nummer 11) und kein Hinweis auf technisches oder menschliches Versagen vorliegt.
Beschreibung der Prüfmethode
Vorbereitungen
Auswahl der Tierart
Es sollten junge, gesunde und geschlechtsreife Tiere der üblichen Labortierstämme zum Einsatz kommen. Gewöhnlich werden Ratten verwendet, doch kommen auch Mäuse in Frage. Es können auch andere geeignete Säugetierarten verwendet werden, sofern dies im Bericht wissenschaftlich begründet wird.
Haltungs- und Fütterungsbedingungen
Bei Nagern sollte die Temperatur im Versuchstierraum 221 °C (± 31 °C) betragen. Die relative Luftfeuchte sollte vorzugsweise bei 50 bis 60 % liegen, mindestens aber 40 % betragen und außer bei Reinigung des Raumes 70 % nicht übersteigen. Der Raum sollte künstlich beleuchtet sein, mit Hell-/Dunkelphasen im 12-Stunden-Rhythmus. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Wahl des Futters kann dadurch beeinflusst werden, dass es sich für die Beimischung einer Prüfchemikalie eignen muss, wenn diese über das Futter verabreicht wird. Nagetiere können in kleinen Gruppen von Tieren gleichen Geschlechts und der gleichen Behandlungsgruppen untergebracht werden (maximal fünf pro Käfig), sofern kein aggressives Verhalten zu erwarten ist, vorzugsweise in Käfigen mit festem Boden und entsprechender Ausgestaltung des Lebensumfelds. Die Tiere dürfen nur dann einzeln untergebracht werden, wenn dies wissenschaftlich gerechtfertigt ist.
Vorbereitung der Tiere
In der Regel werden junge, gesunde und geschlechtsreife Tiere verwendet (bei Nagetieren vorzugsweise Tiere, die zu Behandlungsbeginn 6 bis 10 Wochen alt sind, wobei etwas ältere Tiere auch zulässig sind), die den Kontroll- und Behandlungsgruppen nach dem Zufallsprinzip zuzuordnen sind. Die Tiere werden nach einer humanen, minimalinvasiven Methode (z.B. durch Anbringen von Ringen, Kennmarken, Mikrochips oder biometrisch, nicht jedoch durch Kupieren der Ohren oder Zehen) einzeln gekennzeichnet. Die Tiere werden über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt. Die Käfige sind so anzuordnen, dass etwaige standortbedingte Auswirkungen minimal sind. Kreuzkontaminationen zwischen Positivkontrolle und Prüfchemikalie sind zu vermeiden. Zu Beginn der Studie sollten die Körpergewichtsunterschiede zwischen den behandelten Tiere möglichst gering sein und um nicht mehr als ± 20 % vom Durchschnittsgewicht des jeweiligen Geschlechts abweichen.
Vorbereitung der Dosierung
Feste Prüfchemikalien sollten vor ihrer Verabreichung an die Tiere in geeigneten Lösungsmitteln oder Vehikeln gelöst oder suspendiert oder dem Futter oder Trinkwasser beigemischt werden. Flüssige Prüfchemikalien können direkt verabreicht oder zuvor verdünnt werden. Bei Exposition durch Inhalation können die Prüfchemikalien je nach physikalisch-chemischen Eigenschaften als Gase, Dämpfe oder festes/flüssiges Aerosol verabreicht werden. Es sind frische Zubereitungen der Prüfchemikalie zu verwenden, es sei denn, deren die Stabilität bei Lagerung ist erwiesen und die geeigneten Lagerbedingungen sind vorgegeben.
Lösungsmittel/Vehikel
Das Lösungsmittel/Vehikel sollte bei den gewählten Dosisstufen keine toxischen Wirkungen hervorrufen und nicht in Verdacht stehen, mit den Prüfchemikalien eine chemische Reaktion einzugehen. Werden keine gängigen Lösungsmittel/Vehikel verwendet, so sind Referenzdaten über ihre Kompatibilität beizubringen. Es empfiehlt sich, wann immer möglich zunächst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. Beispiele für gängige, kompatible Lösungsmittel/Vehikel sind Wasser, physiologische Kochsalzlösung, Methylcelluloselösung, Carboxymethylcellulose-Natriumsalzlösung, Olivenöl und Maisöl. Liegen keine historischen oder veröffentlichten Kontrolldaten vor, aus denen hervorgeht, dass keine strukturellen Aberrationen oder anderen schädlichen Wirkungen von einem gewählten, nicht gängigen Lösungsmittel/Vehikel ausgehen, sollte ein Vorversuch durchgeführt werden, der die Eignung des Lösungsmittels/Vehikels belegt.
Kontrollen
Positivkontrollen
Jeder Versuch sollte eine Gruppe von Tieren umfassen, die mit einer Positivkontrollchemikalie behandelt wurden. Darauf kann möglicherweise verzichtet werden, wenn das Prüflabor seine Eignung zur Durchführung des Tests demonstriert und eine Serie historischer Positivkontrollen nachgewiesen hat. Umfasst der Versuch keine gleichzeitige Positivkontrolle, sollten Auswertungskontrollen (fixierte und nicht angefärbte Objektträger) einbezogen werden. Dazu eignen sich Referenzproben, die im Rahmen eines separaten Positivkontrollversuchs, der in dem Labor, das den Versuch durchführt, in regelmäßigen Zeitabständen (z.B. alle 6 bis 18 Monate) durchgeführt wird (z.B. bei Eignungsprüfungen und danach ggf. auf regelmäßiger Basis), entnommen und gelagert wurden.
Positivkontrollchemikalien sollten zuverlässig bewirken, dass die Häufigkeit der Zellen mit Chromosomenaberrationen gemessen an der spontan entstehenden Zellmenge nachweislich zunimmt. Positivkontrolldosen sollten so gewählt werden, dass die Wirkungen eindeutig sind, aber beim Ablesen nicht sofort die Identität der kodierten Proben erkennen lassen. Es ist vertretbar, dass die Positivkontrolle im Rahmen eines anderen Behandlungsplans auf anderem Wege als die Prüfchemikalie verabreicht wird und nur eine einzige Probenahme erfolgt. Ggf. könnte eine zusätzliche Positivkontrolle verwendet werden, die derselben chemischen Klasse angehört wie die Prüfchemikalie. Für Beispiele für Positivkontrollchemikalien siehe Tabelle 1.
Tabelle 1 Beispiele für Positivkontrollchemikalien
| Chemikalie | CAS-Nr. |
| Ethylmethansulfonat | 62-50-0 |
| Methylmethansulfonat | 66-27-3 |
| Ethylnitrosoharnstoff | 759-73-9 |
| Mitomycin C | 50-07-7 |
| Cyclophosphamid(monohydrat) | 50-18-0 (6055-19-2) |
| Triethylenmelamin | 51-18-3 |
Negativkontrollen
Tiere der Negativkontrolle sollten bei jeder Probenahme berücksichtigt und genau wie die Behandlungsgruppen behandelt werden, außer dass ihnen keine Prüfchemikalie verabreicht wird. Soweit zur Verabreichung der Prüfchemikalie ein Lösungsmittel/Vehikel verwendet wird, sollte die auch Kontrollgruppe dieses Lösungsmittel/Vehikel erhalten. Demonstrieren jedoch historische Negativkontrolldaten bei jeder Probenahme für das betreffende Prüflabor übereinstimmende Werte zur Variabilität der Tiere und Häufigkeit der Zellen mit Chromosomenaberrationen, so ist für die Negativkontrollen möglicherweise nur eine einzige Probenahme erforderlich. Wird bei den Negativkontrollen nur eine einzige Probe entnommen, so sollte dies zum ersten Probenahmezeitpunkt erfolgen.
Verfahren
Anzahl und Geschlecht der Tiere
Die Mikrokernreaktion verläuft bei männlichen und weiblichen Tieren in der Regel ähnlich (9), und es ist davon auszugehen, dass dies auch für strukturelle Chromosomenaberrationen gilt, sodass die meisten Studien unabhängig vom Geschlecht durchgeführt werden können. Daten, die nennenswerte Unterschiede zwischen männlichen und weiblichen Tieren demonstrieren (z.B. Unterschiede bei der systemischen Toxizität, beim Stoffwechsel, bei der Bioverfügbarkeit, bei der Knochenmarktoxizität usw., einschließlich unter anderem einer Dosisfindungsstudie), würden die Verwendung von Tieren beider Geschlechter nahe legen. In diesem Fall kann es angebracht sein, eine Studie an Tieren beider Geschlechter durchzuführen, z.B. im Rahmen einer Toxizitätsstudie mit wiederholter Verabreichung. Bei Verwendung beider Geschlechter könnte die Anwendung des faktoriellen Modells zweckdienlich sein. Für Einzelheiten zur Analyse der Daten nach diesem Modell siehe Anlage 2.
Zu Beginn der Studie sollten die Gruppengrößen so festgelegt werden, dass jede Gruppe mindestens 5 analysierbare Tiere eines Geschlechts oder beider Geschlechter, sofern beide Geschlechter einbezogen werden, umfasst. Sollte es sich beim Menschen um eine geschlechtsspezifische Exposition handeln, z.B. wie dies bei bestimmten Pharmazeutika der Fall ist, ist der Versuch am Tier ebenfalls geschlechtsspezifisch durchzuführen. Richtwert für eine typische Versuchstiermenge: Für eine Knochenmarkstudie mit zwei Probenahmezeitpunkten, drei Dosisgruppen und einer gleichzeitigen Negativkontrollgruppe zuzüglich einer Positivkontrollgruppe (wobei jede Gruppe aus fünf Tieren jedes Geschlechts besteht) wären maximal 45 Versuchstiere erforderlich.
Dosisstufen
Wird vorab eine Dosisfindungsstudie durchgeführt, da keine geeigneten Daten zur Dosisauswahl verfügbar sind, sollte diese im selben Labor unter Verwendung derselben Spezies und desselben Stamms, Geschlechts und Behandlungsverfahrens wie beim Hauptversuch stattfinden (10). Ziel der Studie sollte sein, die maximal verträgliche Dosis (MTD) zu ermitteln, die definiert ist als die Höchstdosis, die, bezogen auf den Versuchszeitraum, keine Anzeichen von Toxizität hervorruft, die die Studie begrenzen würden (z.B. Rückgang des Körpergewichts oder Zytotoxizität des hämatopoetischen Systems), ausgenommen Tod oder Anzeichen von Schmerzen und Leiden, die eine humane Tötung erforderlich machen würden (11).
Die Höchstdosis kann ferner als die Dosis definiert werden, die bestimmte Anzeichen toxischer Wirkungen auf das Knochenmark hervorruft.
Chemikalien, die zu einer Sättigung der toxikokinetischen Eigenschaften führen oder Entgiftungsprozesse einleiten, die nach einer Langzeitbehandlung möglicherweise zu einem Rückgang der Exposition führen, können von den Dosierungskriterien ausgenommen werden und sollten auf Einzelfallbasis evaluiert werden.
Um Dosis-Wirkungs-Informationen zu erhalten, sollte eine vollständige Studie eine Negativkontrollgruppe und mindestens drei Dosisstufen (Dosis x 2, jedoch maximal x 4) vorsehen. Ruft die Prüfchemikalie in einer Dosisfindungsstudie oder nach bereits vorhandenen Daten keine Toxizität hervor, so sollte die Höchstdosis für eine Einzelgabe 2.000 mg/kg Körpergewicht betragen. Ruft die Prüfchemikalie jedoch Toxizität hervor, sollte die MTD die höchste verabreichte Dosis sein, und die Dosisstufen sollten vorzugsweise einen Bereich zwischen Höchstdosis und der Dosis abdecken, die wenig oder keine Toxizität erzeugt. Wenn für alle untersuchten Dosisstufen Toxizität im Zielgewebe (Knochenmark) beobachtet wird, sind weitere Untersuchungen bei nichttoxischen Dosisstufen ratsam. Studien zur genaueren Charakterisierung der quantitativen Dosis-Wirkungs-Informationen erfordern möglicherweise weitere Dosisgruppen. Bei bestimmten Arten von Prüfchemikalien (z.B. Humanpharmazeutika), für die spezielle Anforderungen gelten, können diese Grenzen variieren.
Limit-Test
Weisen Dosisfindungsstudien oder bereits vorhandene Daten für verwandten Tierstämme darauf hin, dass eine Behandlung zumindest mit der Limit-Dosis (siehe Beschreibung unten) keine feststellbaren toxischen Wirkungen (und auch keinen Rückgang der Proliferation des Knochenmarks oder andere Anzeichen für toxische Wirkungen im Zielgewebe) verursacht, und ist auf Basis von In-vitro-Studien zur Untersuchung der Genotoxizität oder von Daten über strukturell verwandte Chemikalien keine Genotoxizität zu erwarten, so ist möglicherweise keine umfassende Studie mit drei Dosisstufen erforderlich, sofern nachgewiesen wurde, dass die Prüfchemikalie(n) das Zielgewebe (Knochenmark) erreicht bzw. erreichen. In solchen Fällen könnte eine der Limit-Dosis entsprechende Einzeldosis ausreichen. Bei einem Behandlungszeitraum von > 14 Tagen beträgt die Limit-Dosis 1.000 mg/kg Körpergewicht/Tag. Bei einem Behandlungszeitraum von 14 oder weniger Tagen beträgt die Limit-Dosis 2.000 mg/kg Körpergewicht/Tag.
Verabreichung
Bei der Versuchsplanung ist der antizipierte Verabreichungsweg beim Menschen zu berücksichtigen. Routen wie die Aufnahme über die Nahrung oder das Trinkwasser, die topische, subkutane, intravenöse, orale (Magensonde), intratracheale Verabreichung, die Inhalation oder die Implantation sind daher als wissenschaftlich gerechtfertigt zulässig. In jedem Fall sollte der Verabreichungsweg so gewählt werden, dass das (die) Zielgewebe angemessen exponiert werden. Eine intraperitoneale Injektion wird in der Regel nicht empfohlen, da diese nicht als Verabreichungsweg beim Menschen vorgesehen ist, und sollte nur mit entsprechender wissenschaftlicher Begründung angewandt werden. Sofern die Prüfchemikalie der Nahrung oder dem Trinkwasser beigemischt wird, insbesondere im Fall von Einzeldosierungen, ist darauf zu achten, dass ein ausreichender Zeitabstand zwischen der Nahrungsmittel-/Trinkwasseraufnahme und der Probenahme eingehalten wird, damit ein Nachweis der Wirkungen möglich ist (siehe Nummern 33-34). Die Höchstmenge an Flüssigkeit, die jeweils über eine Magensonde verabreicht oder injiziert werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte im Normalfall 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen können aber auch 2 ml/100 g in Betracht gezogen werden. Werden größere Volumina verwendet, ist dies zu begründen. Mit Ausnahme von reizenden oder ätzenden Prüfchemikalien, die normalerweise bei höheren Konzentrationen gravierende Wirkungen zeigen, sollte die Variabilität der Prüfvolumina minimiert werden, indem die Konzentration angepasst wird, dass im Verhältnis zum Körpergewicht bei allen Dosisstufen ein konstantes Volumen verabreicht wird.
Behandlungsplan
Prüfchemikalien werden in der Regel auf einmal verabreicht. Die Gabe kann aber auch in Form von zwei oder mehreren Teilmengen erfolgen (am gleichen Tag im Abstand von nicht mehr als 2 bis 3 Stunden), wenn es sich um eine große Menge handelt. In diesen Fällen, oder wenn die Prüfchemikalie durch Inhalation verabreicht wird, sollte der Zeitpunkt der Probenahme auf Basis der letzten Dosisgabe oder des Zeitpunkts, an dem die Exposition beendet wurde, angesetzt werden.
Es liegen nur wenige Informationen darüber vor, ob sich ein Protokoll für wiederholte Verabreichung für diesen Versuch eignet. In Fällen, in denen es zweckmäßig erscheint, diesen Versuch in einen Toxizitätstest mit wiederholter Verabreichung einzubinden, ist ein Verlust von mitotischen Zellen mit beschädigten Chromosomen zu vermeiden, wozu es bei toxischen Dosierungen kommen kann. Eine solche Einbindung ist zulässig, wenn die höchste Dosis der Limit-Dosis entspricht oder größer ist (siehe Nummer 29) und eine Dosisgruppe für die Dauer des Behandlungszeitraums die Limit-Dosis erhält. Soll eine Einbindung in andere Studien erfolgen, sollte vorrangig der Mikrokerntest (Prüfmethode B.12) als In-vivo-Test für Chromosomenaberrationen gewählt werden.
Knochenmarkproben sollten an zwei verschiedenen Zeitpunkten im Anschluss an Einzelbehandlungen genommen werden. Bei Nagern sollte das erste Probenahmeintervall der Dauer von 1,5 normalen Zellzyklen (die in der Regel 12 bis 18 Stunden nach der Behandlung abgeschlossen sind) entsprechen. Da die für die Aufnahme und Metabolisierung der Prüfchemikalie(n) sowie für die Wirkung auf die Zellzykluskinetik benötigte Zeit den optimalen Zeitpunkt für die Feststellung von Chromosomenaberrationen beeinflussen kann, wird empfohlen, 24 Stunden nach der ersten Probenahme eine weitere Probenahme vorzunehmen. Bei der ersten Probenahme sollten alle Dosisgruppen behandelt, und es sollten Proben für die Analyse aufgearbeitet werden. Bei (einer) weiteren Probenahme(n) muss nur die Höchstdosis verabreicht werden. Werden Verabreichungsschemata gewählt, die über einen Tag hinausgehen, und ist dies wissenschaftlich begründet, sollte die Probenahme im Anschluss an die letzte Behandlung nach Ablauf eines Zeitraums erfolgen, der der l,5-fachen Dauer des normalen Zellzyklus entspricht.
Nach der Behandlung und vor der Aufarbeitung der Proben erhalten die Versuchstiere eine intraperitoneale Injektion mit einer geeigneten Dosis eines Spindelgifts (z.B. Colcemid oder Colchicin), und nach einem angemessenen Zeitraum im Anschluss daran erfolgt die Aufarbeitung. Bei Mäusen beträgt dieser Zeitraum etwa 3 bis 5 Stunden vor der Aufarbeitung und bei Ratten 2 bis 5 Stunden. Aus dem Knochenmark werden Zellen gewonnen, aufgequollen, fixiert und angefärbt und anschließend auf Chromosomenaberrationen untersucht (12).
Beobachtungen
Mindestens einmal täglich sollten allgemeine klinische Beobachtungen der Versuchstiere vorgenommen und vorzugsweise zum gleichen Zeitpunkt und unter Berücksichtigung des Zeitraums, in dem der Wirkungsgipfel nach Verabreichung der Dosis zu erwarten ist, protokolliert werden. Mindestens zweimal täglich während der Verabreichungszeit sind alle Tiere auf Morbidität und Mortalität zu untersuchen. Alle Tiere sollten zu Studienbeginn, mindestens einmal pro Woche bei Studien mit wiederholter Verabreichung sowie bei humaner Tötung gewogen werden. In Studien von mindestens einwöchiger Dauer sollten mindestens wöchentlich Messungen der Futteraufnahme vorgenommen werden. Wenn die Prüfchemikalie über das Trinkwasser verabreicht wird, sollte auch die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen werden. Tiere mit Anzeichen von übermäßiger, jedoch nicht tödlich wirkender Toxizität sollten vor Ende des Prüfzeitraums human getötet werden (11).
Exposition des Zielgewebes
Zu (einem) geeigneten Zeitpunkt(en) sollte eine Blutprobe gezogen werden, um den Plasmaspiegel der Prüfchemikalien untersuchen zu können. Dadurch soll nachgewiesen werden, dass eine Exposition des Knochenmarks stattgefunden hat, wo dies gerechtfertigt erscheint und keine anderen Expositionsdaten vorhanden sind (siehe Nummer 44).
Knochenmark- und Chromosomenpräparate
Die Knochenmarkzellen werden in der Regel unmittelbar nach der humanen Tötung aus den Oberschenkel- oder Schienbeinknochen der Tiere gewonnen, mit hypotoner Lösung behandelt und fixiert. Die Zellen werden anschließend nach anerkannten Verfahren auf Objektträger aufgetropft und angefärbt (siehe (3) (12)).
Analyse
Alle Objektträger, einschließlich der Positiv- und Negativkontrollen, sollten vor der Analyse unabhängig kodiert und randomisiert werden, damit die Auswertung ohne Kenntnis der Behandlungsbedingungen erfolgt.
Bei allen behandelten Tieren (einschließlich der Positivkontrollen), unbehandelten oder mit einem Lösungsmittel/Vehikel behandelten Tieren der Negativkontrollgruppe ist der Mitoseindex als Gradmesser der Zytotoxizität in mindestens 1.000 Zellen pro Tier zu bestimmen.
Pro Tier sollten mindestens 200 Metaphasen auf strukturelle Chromsomenaberrationen, mit und ohne Gaps, analysiert werden (6). Wenn jedoch aus den historischen Negativkontrolldaten hervorgeht, dass die durchschnittliche Hintergrundhäufigkeit für strukturelle Chromsomenaberrationen im Prüflabor < 1 % beträgt, sollte eine Auswertung weiterer Zellen in Erwägung gezogen werden. Chromatidentyp- und Chromosomentypaberrationen sollten gesondert erfasst und Subtypen zugeordnet werden (Brüche, Austausche). Die Laborpraxis sollte gewährleisten, dass die Analyse von Chromosomenaberrationen von gut ausgebildeten Labortechnikern vorgenommen und ggf. einer Peer-Review unterzogen wird. Da es bei der Präparation der Objektträger häufig zum Bruch eines Teils der Metaphasezellen und zum Verlust von Chromosomen kommt, sollten die ausgewerteten Zellen eine Zentromerzahl enthalten, die der Zahl 2n ± 2 entspricht, wobei n die haploide Chromosomenzahl für diese Spezie ist.
Daten und Berichterstattung
Behandlung der Ergebnisse
Die Daten für die einzelnen Tiere sollten tabellarisch erfasst werden. Für jedes Tier sollten der Mitoseindex, die Anzahl der bewerteten Metaphasezellen, die Zahl der Aberrationen pro Metaphasezelle und der Anteil der Zellen mit strukturellen Chromosomenaberrationen angegeben werden. Für die Versuchs- und Kontrollgruppen sollten die unterschiedlichen Typen struktureller Chromsomenaberrationen unter Angabe ihrer Anzahl und Häufigkeit aufgeführt werden. Gaps sowie polyploide Zellen und Zellen mit endoreduplizierten Chromosomen werden getrennt erfasst. Die Häufigkeit von Gaps ist anzugeben, wird in der Regel bei der Analyse der Gesamthäufigkeit der Aberrationen jedoch nicht berücksichtigt. Gibt es keine Anhaltspunkte für unterschiedliche Reaktionen der Geschlechter, so können die Daten für beide Geschlechter für die statistische Analyse zusammengefasst werden. Daten zur Toxizität und klinische Anzeichen bei Tieren sind ebenfalls anzugeben.
Gültigkeitskriterien
Die folgenden Kriterien entscheiden über die Gültigkeit des Versuchs:
Auswertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn
Wird zu einem bestimmten Probenahmezeitpunkt nur die Höchstdosis untersucht, so gilt eine Prüfchemikalie als eindeutig positiv, wenn, verglichen mit der gleichzeitigen Negativkontrolle, eine statistisch signifikante Zunahme vorliegt und die Ergebnisse außerhalb der Verteilung der historischen Negativkontrolldaten liegen (z.B. auf einer Poisson-Verteilung beruhende Kontrollgrenzen von 95 %). Empfehlungen zu geeigneten statistischen Methoden sind der Literatur zu entnehmen (13). Bei der Durchführung einer Dosis-Wirkungs-Analyse sollten mindestens drei behandelte Dosisgruppen analysiert werden. Bei statistischen Versuchen sollte das Tier Versuchseinheit sein. Positive Ergebnisse beim Chromosomenaberrationstest deuten darauf hin, dass eine Prüfchemikalie Chromosomenaberrationen im Knochenmark der getesteten Spezies auslöst.
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn unter allen getesteten Versuchsbedingungen
Für Empfehlungen zu den am besten geeigneten statistischen Methoden siehe Literaturhinweis (13). Als Nachweis für eine Exposition des Knochenmarks gegenüber einer Prüfchemikalie kann auch ein Rückgang des Mitoseindex oder eine Untersuchung der Plasma- oder Blutspiegel der Prüfchemikalie(n) dienen. Bei intravenöser Verabreichung ist kein Expositionsnachweis erforderlich. Alternativ können ADME-Daten herangezogen werden, die in einer unabhängigen Studie unter Verwendung der gleichen Verabreichungswege und der gleichen Spezies gewonnen wurden, um nachzuweisen, dass eine Knochenmarkexposition stattgefunden hat. Negative Ergebnisse deuten darauf hin, dass die Prüfchemikalie unter den Versuchsbedingungen keine strukturellen Chromosomenaberrationen im Knochenmark der getesteten Spezies hervorruft.
Bei einer eindeutig positiven oder negativen Reaktion ist keine Verifizierung erforderlich.
In Fällen, in denen die Reaktion, wie oben beschrieben, weder eindeutig negativ noch eindeutig positiv ist, oder um die biologische Relevanz eines Ergebnisses zu untermauern (z.B. eine geringe oder grenzwertige Zunahme), sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen bewertet werden. In einigen Fällen kann die Auswertung weiterer Zellen oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter veränderten Versuchsbedingungen, hilfreich sein.
In seltenen Fällen erlaubt der Datensatz auch nach weiteren Untersuchungen keine definitive Aussage darüber, ob die Prüfchemikalie positive oder negative Ergebnisse zur Folge hat; in diesen Fällen wird die Studie als unschlüssig abgeschlossen.
Die Häufigkeit des Auftretens polyploider und endoreduplizierter Metaphasen gemessen an der Gesamtzahl der Metaphasen sollte getrennt erfasst werden. Eine zahlenmäßige Zunahme der polyploiden/endoreduplizierten Zellen deutet möglicherweise darauf hin, dass die Prüfchemikalie mitotische Prozesse zu hemmen und numerische Chromosomenaberrationen hervorzurufen vermag (siehe Abschnitt 3).
Prüfbericht
Der Prüfbericht sollte folgende Angaben enthalten:
Zusammenfassung
Prüfchemikalie:
Einkomponentige Substanz:
Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
Zubereitung der Prüfchemikalie:
Versuchstiere:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse.
Schlussfolgerung.
Referenzdokumente.
Literaturhinweise
(1) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(2) Adler, I.D. (1984), Cytogenetic Tests in Mammals, in Mutagenicity Testing: A Practical Approach, Venittand, S., J.M. Parry (eds.), IRL Press, Washington, DC, 275-306.
(3) Preston, R.J. et al. (1987), Mammalian in vivo cytogenetic assays. Analysis of chromosome aberrations in bone marrow cells, Mutation Research, Band 189/2, 157-165.
(4) Richold, M. et al. (1990), "In Vivo Cytogenetics Assays", in Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Bericht. Überarbeiteter Teil I, Kirkland, D.J. (ed.), Cambridge University Press, Cambridge, 115-141.
(5) Tice, R.R. et al. (1994), Report from the working group on the in vivo mammalian bone marrow chromosomal aberration test, Mutation Research, Band 312/3, 305-312.
(6) Adler, I.D. et al. (1998), Recommendations for statistical designs of in vivo mutagenicity tests with regard to subsequent statistical analysis, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 417/1, 19-30.
(7) Ryan, T.P. (2000), Statistical Methods for Quality Improvement, 2nd ed., John Wiley and Sons, New York.
(8) Hayashi, M. et al. (2011), Compilation and use of genetic toxicity historical control data, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 723/2, 87-90.
(9) Hayashi, M. et al. (1994), in vivo rodent erythrocyte micronucleus assay, Mutation Research/Environmental Mutagenesis and Related Subjects, Band 312/3, 293-304.
(10) Fielder, R.J. et al. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group. Dose setting in in vivo mutagenicity assays, Mutagenesis, Band 7/5, 313-319.
(11) OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, No19, OECD Publishing, Paris.
(12) Pacchierotti, F., V. Stocchi (2013), Analysis of chromosome aberrations in somatic and germ cells of the mouse, Methods in Molecular Biology, Band 1044, 147-163.
(13) Lovell, D.P. et al. (1989), Statistical Analysis of in vivo Cytogenetic Assays, in Statistical Evaluation of Mutagenicity Test Data. UKEMS SubCommittee on Guidelines for Mutagenicity Testing, Bericht, Teil III, Kirkland, D.J. (ed.), Cambridge University Press, Cambridge, 184-232.
.
| Definitionen | Anlage 1 |
Aneuploidie: jede Abweichung von der normalen diploiden (oder haploiden) Chromosomenzahl um ein oder mehrere Chromosomen, jedoch nicht um ganze Chromosomensätze (vgl. Polyploidie).
Chemikalie: ein Stoff oder ein Gemisch.
Chromatidentypaberration: strukturelle Chromsomenanomalie, gekennzeichnet durch Bruch einzelner Chromatiden oder Bruch und Reunion zwischen Chromatiden.
Chromosomentypaberration: strukturelle Chromosomenanomalie, gekennzeichnet durch Bruch oder Bruch und Reunion beider Chromatiden an gleicher Position.
Endoreduplikation: Prozess, bei dem der Kern nach einer S-Phase der DNA-Replikation keine Mitose durchläuft, sondern in eine weitere S-Phase eintritt. Das Ergebnis sind Chromosomen mit 4,8,16, ... Chromatiden.
Gap: achromatische Läsion von geringerer Breite als eine Chromatide mit minimaler Verlagerung der Chromatiden.
Mitoseindex: Anteil der Zellen einer Zellpopulation, die sich zum Beobachtungszeitpunkt in der Mitose befinden: Gradmesser für den Vermehrungsgrad dieser Population.
Numerische Aberration: Abweichung der Chromosomenzahl vom Normalwert, der für die verwendeten Tiere charakteristisch ist (Aneuploidie).
Polyploidie: numerische Chromosomenaberration, von der ein ganzer Chromosomensatz betroffen ist, im Gegensatz zu einer numerischen Abweichung in einem Teil des Chromosomensatzes (vgl. Aneuploidie).
Prüfchemikalie: ein Stoff oder ein Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Strukturelle Chromosomenaberration: Veränderung der Chromosomenstruktur, nachweisbar durch mikroskopische Untersuchung des Metaphase-Stadiums der Zellteilung; äußert sich in Form von Deletionen und Fragmenten, intra-chromosomalen oder reziproken Translokationen.
Zentromere: Region(en) eines Chromosoms, an die die Spindelfasern während der Zellteilung anhaften, wodurch die ordnungsgemäße Beförderung der Tochterchromosomen zu den Polen der Tochterzellen ermöglicht wird.
.
| Faktorielles Modell zur Ermittlung geschlechtsspezifischer Differenzen beim In-vivo-Test auf Chromosomenaberrationen | Anlage 2 |
Faktorielles Modell und zugehörige Analysen
Bei diesem Modell werden mindestens 5 männliche und 5 weibliche Tiere je Konzentration getestet, d. h. insgesamt mindestens 40 Versuchstiere (20 männliche und 20 weibliche zuzüglich Positivkontrollen).
Das Modell, das zu den einfacheren Faktormodellen zählt, entspricht einer Zweiwege-Varianzanalyse, bei der Geschlecht und Konzentration im Wesentlichen die Wirkung bestimmen. Die Daten können im Rahmen zahlreicher Standard-Statistiksoftwareanwendungen wie SPSS, SAS, STATA oder Genstat oder auch mit "R" analysiert werden.
Mit der Analyse lässt sich die Varianz im Datensatz als Varianz zwischen den Geschlechtern, Varianz zwischen den Konzentrationen und Varianz bezogen auf die Interaktion zwischen den Geschlechtern und Konzentrationen abbilden. Jede Bedingung wird an einem Schätzwert der Varianz zwischen den Replikattieren in den gleichgeschlechtlichen Tiergruppen, die die gleiche Konzentration erhalten, gemessen. Die zugrundeliegende Methodik ist in vielen Standardwerken zur Statistik (siehe Referenzdokumente) und in den in Statistiksoftware-Paketen mitgelieferten Hilfe-Funktionen näher beschrieben.
Die Analyse beginnt mit der Untersuchung der Interaktionsbedingung "Konzentration x Geschlecht" nach der ANOVA-Tabelle 1. Liegt keine signifikante Interaktion vor, liefern die kombinierten Werte für die Geschlechter oder Konzentrationen gültige statistische Tests für die jeweiligen Konzentrationen, und zwar basierend auf der ANOVA-Bedingung der innerhalb der Gruppe gepoolten Varianzen.
Es folgt eine Partitionierung des Schätzwerts für die Varianzen zwischen den Konzentrationen in Kontraste, die einen Test für lineare und quadratische Kontraste aus den Reaktionen der verschiedenen Konzentrationen liefern. Ergibt sich hingegen eine signifikante Interaktion für Term "Konzentration x Geschlecht", so kann dieser Term auch in Interaktionskontraste "linearer Wert x Geschlecht" und "quadratischer Wert x Geschlecht" partitioniert werden. Aufgrund dieser Terme lässt sich prüfen, ob die Reaktionen der jeweiligen Konzentrationen für beide Geschlechter parallel verlaufen oder ob es zwischen den Geschlechtern zu einer differenzierten Reaktion kommt.
Anhand des Schätzwerts für die innerhalb der Gruppe gepoolten Varianzen lassen sich paarweise Tests auf Abweichungen zwischen Mittelwerten durchführen. Diese Vergleiche könnten zwischen den Mittelwerten für die beiden Geschlechter und zwischen den Mittelwerten für die verschiedenen Konzentrationen durchgeführt werden, beispielsweise um einen Vergleich mit den Negativkontrollen vorzunehmen. In Fällen mit signifikanter Interaktion können Vergleiche zwischen den Mittelwerten verschiedener Konzentrationen innerhalb eines Geschlechts oder zwischen den Mittelwerten beider Geschlechter bei gleicher Konzentration vorgenommen werden.
Referenzdokumente
Es sind viele Werke zur Statistik erhältlich, in denen die Theorie, der Aufbau, die Methodik, die Analyse und die Interpretation faktorieller Analysemodelle erörtert werden, von einfachen Zweifaktorenanalysen bis hin zu komplexeren Formen, wie sie in der "Design of Experiment"-Methode verwendet werden. Die folgende Auflistung ist nicht erschöpfend. Einige Bücher enthalten Beispielrechnungen zu vergleichbaren Versuchsplänen, in einigen Fällen auch mit einem Code zur Durchführung der Analysen unter Verwendung verschiedener Softwarepakete.
Box, G.E.P, Hunter, W.G. and Hunter, J.S. (1978). Statistics for Experimenters. An Introduction to Design, Data Analysis, and Model Building. New York: John Wiley & Sons.
Box G.E.P. & Draper, N.R. (1987). Empirical model-building and response surfaces. John Wiley & Sons Inc.
Doncaster, C.P. & Davey, A.J.H. (2007). Analysis of Variance and Covariance: How to Choose and Construct Models for the Life Sciences. Cambridge University Press.
Mead, R. (1990). The Design of Experiments. Statistical principles for practical application. Cambridge University Press.
Montgomery D.C. (1997). Design and Analysis of Experiments. John Wiley &Sons Inc.
Winer, B.J. (1971). Statistical Principles in Experimental Design. McGraw Hill.
Wu, C.F.J & Hamada, M.S. (2009). Experiments: Planning, Analysis and Optimization. John Wiley & Sons Inc.
______
1) Statistiker, die mit einem Modellierungsansatz wie dem Ansatz der allgemeinen linearen Modelle (GLM) arbeiten, folgen bei der Analyse möglicherweise einem anderen, wenn auch vergleichbarem Ansatz, werden jedoch nicht notwendigerweise eine Herleitung der herkömmlichen Anova-Tabelle vornehmen, die auf algorithmische Lösungswege für statistische Berechnungen aus dem Vor-Computerzeitalter zurückgeht."
(5) In Teil B erhält Kapitel B.12 folgende Fassung:
"B.12 Erythrozyten-Mikrokerntest bei Säugern
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 474 (2016). Sie ist Teil einer Reihe von Prüfmethoden zur genetischen Toxikologie. Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (1).
Der In-vivo-Mikrokerntest bei Säugern ist insbesondere für die Bewertung der Genotoxizität relevant, da trotz artenspezifischer Unterschiede Faktoren des In-vivo-Stoffwechsels, der Pharmakokinetik und von DNA-Reparaturprozessen aktiv sind und zu den Reaktionen beitragen. Ein In-vivo-Versuch ist ferner hilfreich für weitere Untersuchungen zu gentoxischen Wirkungen, die in einem In-vitro-System nachgewiesen wurden.
Der In-vivo-Mikrokerntest bei Säugern dient zum Nachweis einer von der Prüfchemikalie in den Chromosomen oder im mitotischen Apparat von Erythroblasten hervorgerufenen Schädigung. Der Test untersucht die Bildung von Mikrokernen in Erythrozyten, von denen Stichproben entweder aus dem Knochenmark oder dem peripheren Blut von Tieren, in der Regel Nagern, entnommen wurden.
Zweck des Mikrokerntests ist der Nachweis von Chemikalien, die zytogenetische Schäden hervorrufen, durch die es zur Bildung von Mikrokernen mit zurückgebliebenen Chromosomenfragmenten oder ganzen Chromosomen kommt.
Wenn sich ein Knochenmarkerythroblast zu einem unreifen Erythrozyten (manchmal auch als polychromatischer Erythrozyt oder Retikulozyt bezeichnet) entwickelt, wird der Hauptkern ausgestoßen. Dabei kann aber ein möglicherweise entstandener Mikrokern im Zytoplasma verbleiben. Die Sichtbarmachung bzw. das Aufspüren der Mikrokerne wird in diesen Zellen dadurch erleichtert, dass sie keinen Hauptkern aufweisen. Eine Zunahme der Häufigkeit von mikrokernhaltigen unreifen Erythrozyten in behandelten Tieren deutet auf die Verursachung struktureller oder numerischer Chromosomenaberrationen hin.
Neu gebildete mikrokernhaltige Erythrozyten werden durch Färben identifiziert und quantifiziert und im Anschluss daran visuell mithilfe eines Mikroskops oder anhand eines automatisierten Verfahrens analysiert. Das Zählen einer ausreichenden Zahl unreifer Erythrozyten im peripheren Blut oder Knochenmark geschlechtsreifer Tiere wird erheblich erleichtert durch die Verwendung eines automatisierten Auswertungssystems. Solche Systeme stellen vertretbare Alternativen zur manuellen Auswertung dar (2). Vergleichende Studien haben ergeben, dass solche Verfahren unter Verwendung geeigneter Kalibrierstandards eine bessere Reproduzierbarkeit und Empfindlichkeit (innerhalb und zwischen Labors) bieten als eine mikroskopische Auswertung (3) (4). Automatisierte Systeme zur Messung der Häufigkeit von mikrokernhaltigen Erythrozyten umfassen unter anderem Durchflusszytometer (5), Bildanalyseplattformen (6) (7) und Laser-Scanning-Zytometer (8).
Auch wenn dies im Rahmen des Tests normalerweise nicht erfolgt, können Chromosomenfragmente von ganzen Chromosomen anhand einiger Kriterien unterschieden werden. Dazu zählt die Feststellung des Vorhandenseins oder Fehlens einer Kinetochor- bzw. Zentromer-DNA in den Mikrokernen; beides ist charakteristisch für intakte Chromosomen. Das Fehlen einer Kinetochor- bzw. Zentromer-DNA deutet darauf hin, dass der Mikrokern nur Fragmente von Chromosomen enthält, wohingegen das Vorhandensein auf einen Chromosomenverlust hinweist.
Die Definitionen zur verwendeten Terminologie sind Anlage 1 zu entnehmen.
Ausgangsüberlegungen
Zielgewebe für genetische Schäden ist in diesem Test das Knochenmark junger geschlechtsreifer Nagetiere, da Erythrozyten in diesem Gewebe gebildet werden. Die Mikrokernuntersuchung in unreifen Erythrozyten in peripherem Blut ist auch bei anderen Säugetierarten vertretbar, für die eine entsprechende Empfindlichkeit zum Nachweis von Chemikalien, die strukturelle oder numerische Chromosomenaberrationen in diesen Zellen auslösen, nachgewiesen (durch Induktion von Mikrokernen in unreifen Erythrozyten) und eine wissenschaftliche Begründung geliefert wurde. Hauptendpunkt ist die Häufigkeit der nicht ausgereiften mikrokernhaltigen Erythrozyten. Werden die Tiere kontinuierlich über einen Zeitraum behandelt, der die Lebensdauer des Erythrozyten in der verwendeten Spezies überschreitet (z.B. 4 Wochen oder länger bei Mäusen), kommt aber auch der Anteil der Mikrokerne enthaltenden ausgereiften Erythrozyten im peripheren Blut von Spezies, bei denen in der Milz kein übermäßiger Abbau von Mikrokernzellen erfolgt, als Endpunkt des Tests in Betracht.
Wenn Anzeichen dafür bestehen, dass die Prüfchemikalie(n) oder ein reaktiver Metabolit bzw. reaktive Metaboliten das Zielgewebe nicht erreicht bzw. erreichen, ist dieser Test nicht geeignet.
Bevor die Prüfmethode auf ein Gemisch angewendet wird, um unter bestimmten gesetzgeberischen Aspekten Daten zu gewinnen, sollte geprüft werden, ob, und falls ja, warum, sie für diesen Zweck geeignete Ergebnisse liefert. Diese Überlegungen erübrigen sich, sofern die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist.
Prinzip der Prüfmethode
Die Prüfchemikalie wird den Tieren über einen geeigneten Applikationsweg verabreicht. Bei Verwendung von Knochenmark werden sie zu einem geeigneten Zeitpunkt nach der Behandlung human getötet. Das Knochenmark wird entnommen, präpariert und gefärbt (9) (10) (11) (12) (13) (14) (15). Findet peripheres Blut Verwendung, so wird es zu geeigneten Zeitpunkten nach der Behandlung entnommen, präpariert und gefärbt (12) (16) (17) (18). Bei akuter Verabreichung ist es wichtig, den Zeitpunkt der Gewinnung des Knochenmarks oder Blutes so zu wählen, dass die behandlungsabhängige Induktion von nicht ausgereiften mikrokernhaltigen Erythrozyten nachweisbar ist. Werden Blutproben aus peripherem Blut entnommen, sollte ausreichend Zeit vergangen sein, damit diese Vorgänge im zirkulierenden Blut nachweisbar sind. Die Präparate werden auf das Vorhandensein von Mikrokernen untersucht, entweder durch mikroskopische Darstellung, Bildanalyse, Durchflusszytometrie oder Laser-Scanning-Zytometrie.
Überprüfung der Eignung des Labors
Untersuchungen zur Eignung
Zum Nachweis ausreichender Erfahrungen mit der Durchführung des Versuchs, bevor er für routinemäßige Testungen verwendet wird, sollte das Labor die Eignung zur Reproduktion erwartbarer Ergebnisse aus den veröffentlichten Daten (17) (19) (20) (21) (22) für Mikrokernhäufigkeiten mit mindestens zwei Positivkontrollchemikalien (einschl. durch geringe Dosen von Positivkontrollen ausgelöste schwache Reaktionen), wie in Tabelle 1 aufgelistet, und mit kompatiblen Vehikel-/Lösungsmittelkontrollen nachgewiesen haben (siehe Nummer 26). In diesen Versuchen sollten Dosierungen verwendet werden, mit denen man reproduzierbare und dosisabhängige Zunahmen erhält und die Empfindlichkeit und dynamische Bandbreite des Versuchssystems im untersuchten Gewebe (Knochenmark oder peripheres Blut) nachweisen kann; außerdem sollte die Auswertungsmethode verwendet werden, die im Labor eingesetzt werden soll. Diese Anforderung gilt nicht für erfahrene Labors, d. h. für Labors, die über historische Daten gemäß Definition unter den Nummern 14-18 verfügen.
Historische Kontrolldaten
Im Rahmen der Untersuchungen zur Eignung des Labors sollte das Labor Folgendes nachweisen:
Bei der erstmaligen Gewinnung von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen mit veröffentlichten Kontrolldaten übereinstimmen, sofern solche vorhanden sind. Werden weitere Versuchsdaten zur Verteilung der historischen Kontrollen hinzugefügt, sollten gleichzeitige Negativkontrollen idealerweise innerhalb der 95 %-Kontrollgrenze der gewählten Verteilung liegen. Die Datenbank des Labors zu historischen Negativkontrollen sollte statistisch solide Werte enthalten, um sicherzustellen, dass das Labor in der Lage ist, die Verteilung seiner Negativkontrolldaten zu bewerten. In der Literatur wird eine Mindestanzahl von 10 Versuchen vorgeschlagen; vorzugsweise sollte der Datensatz jedoch mindestens 20 Versuche umfassen, die unter vergleichbaren Versuchsbedingungen durchgeführt wurden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder X-Bar-Karten (23)), um zu ermitteln, wie variabel ihre Daten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist. Weitere Empfehlungen zum Aufbau und zur Verwendung historischer Datensammlungen (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus historischen Datensammlungen und die Gültigkeitskriterien für einen bestimmten Versuch) sind den Literaturhinweisen zu entnehmen (24).
Wenn das Labor während der Untersuchungen zur Eignung des Labors (siehe Nummer 13) keine ausreichende Zahl von Versuchen zum Nachweis einer statistisch soliden Verteilung der Negativkontrollen abschließt (siehe Nummer 15), kann die Verteilung auch während der ersten routinemäßigen Tests festgelegt werden. Diese Vorgehensweise sollte sich an den in der Literatur vorgegebenen Empfehlungen orientieren (24) und die bei diesen Versuchen erhaltenen Negativkontrollergebnisse sollten mit veröffentlichten Negativkontrolldaten übereinstimmen.
Sämtliche Änderungen am Versuchsprotokoll sind hinsichtlich ihrer Auswirkungen auf die zu gewinnenden Daten zu prüfen, die mit den bereits vorhandenen Daten zu historischen Kontrollen übereinstimmen müssen. Nur bei größeren Inkonsistenzen sollte eine neue Datenbank zu historischen Kontrollen erstellt werden, wenn eine fachkundige Beurteilung eine Abweichung von der vorherigen Verteilung ergibt (siehe Nummer 15). Während der Neuerstellung muss für die Durchführung eines aktuellen Versuchs gegebenenfalls keine vollständige Datenbank mit Negativkontrollen vorhanden sein, vorausgesetzt, das Labor kann nachweisen, dass seine Werte aus gleichzeitigen Negativkontrollen entweder mit seiner vorhergehenden Datenbank oder mit den entsprechenden veröffentlichten Daten übereinstimmen.
Daten zu Negativkontrollen sollten vorhandene nicht ausgereifte mikrokernhaltige Erythrozyten bei jedem Tier erfassen. Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der 95 %-Kontrollgrenzen der Verteilung in der Datenbank des Labors zu historischen Negativkontrollen liegen. Sofern Daten zu den gleichzeitigen Negativkontrollen außerhalb der 95 %-Kontrollgrenzen liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um "extreme Ausreißer" handelt und nachgewiesen werden kann, dass das Versuchssystem "unter Kontrolle" ist (siehe Nummer 15) und kein Hinweis auf ein technisches oder menschliches Versagen vorliegt.
Beschreibung der Methode
Vorbereitungen
Auswahl der Tierspezies
Es sollten junge gesunde und geschlechtsreife Tiere üblicher Labortierstämme zum Einsatz kommen. Es können Mäuse, Ratten oder andere geeignete Säugetierarten verwendet werden. Wird peripheres Blut verwendet, ist nachzuweisen, dass der in der Milz erfolgende Abbau Mikrokerne enthaltender Zellen aus dem Blutkreislauf nicht den Nachweis induzierter Mikrokerne in der gewählten Spezies beeinträchtigt. Bei Mäusen und Ratten konnte dies eindeutig nachgewiesen werden (2). Die wissenschaftliche Begründung für die Verwendung anderer Versuchstiere als Ratten und Mäuse sollte in den Bericht aufgenommen werden. Sofern andere Versuchstiere als Nagetiere verwendet werden, wird empfohlen, die Messung induzierter Mikrokerne in einen anderen geeigneten Toxizitätstest einzubinden.
Haltungs- und Fütterungsbedingungen
Bei Nagern sollte die Temperatur im Versuchstierraum 221 °C (± 31 °C) betragen. Die relative Luftfeuchte sollte vorzugsweise bei 50 bis 60 % liegen, mindestens aber 40 % betragen und außer bei Reinigung des Raumes 70 % nicht übersteigen. Der Raum sollte künstlich beleuchtet sein, wobei die Beleuchtung im 12-Stunden-Rhythmus ein- und ausgeschaltet werden sollte. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, wobei eine unbegrenzte Trinkwasserversorgung zu gewährleisten ist. Die Auswahl des Futters kann dadurch beeinflusst werden, dass eine geeignete Beimischung einer Prüfchemikalie gewährleistet werden muss, wenn diese über das Futter verabreicht wird. Nagetiere sollten in kleinen Gruppen (maximal fünf pro Käfig) von Tieren gleichen Geschlechts und der gleichen Behandlungsgruppen untergebracht werden, sofern kein aggressives Verhalten zu erwarten ist, vorzugsweise in Käfigen mit festem Boden und angemessener Ausgestaltung des Lebensumfelds. Die Tiere können nur dann einzeln untergebracht werden, wenn dies wissenschaftlich begründet ist.
Vorbereitung der Versuchstiere
In der Regel werden junge gesunde Tiere verwendet (bei Nagetieren vorzugsweise Tiere, die bei Behandlungsbeginn 6 bis 10 Wochen alt sind, wobei etwas ältere Tiere auch zulässig sind), die den Kontroll- und Behandlungsgruppen nach dem Zufallsprinzip zuzuordnen sind. Es erfolgt eine Einzelidentifizierung der Tiere unter Verwendung einer humanen, minimalinvasiven Methode (z.B. durch Anbringen von Ringen, Marken, Mikrochips oder durch biometrische Identifizierung, nicht jedoch durch Kupieren der Ohren oder Zehen). Die Tiere werden über einen Zeitraum von mindestens fünf Tagen unter Laborbedingungen eingewöhnt. Die Käfige sind so aufzustellen, dass etwaige durch den Standort bedingte Auswirkungen möglichst gering sind. Eine gegenseitige Kontamination durch die Positivkontrolle und die Prüfchemikalie ist zu vermeiden. Bei Beginn der Studie sollte die Schwankung des Körpergewichts der behandelten Tiere gering sein und um nicht mehr als maximal ± 20 % vom mittleren Gewicht für jedes Geschlecht abweichen.
Vorbereitung der Dosierung
Feste Prüfchemikalien sollten vor der Verabreichung an die Tiere in geeigneten Lösungsmitteln oder Vehikeln gelöst oder suspendiert oder dem Futter oder Trinkwasser beigemischt werden. Flüssige Prüfchemikalien können direkt verabreicht oder zuvor verdünnt werden. Bei Exposition durch Inhalation können die Prüfchemikalien je nach ihren physikalisch-chemischen Eigenschaften als Gase, Dämpfe oder festes/flüssiges Aerosol verabreicht werden. Es sind frische Zubereitungen der Prüfchemikalie zu verwenden, es sei denn, die Stabilität der Chemikalie bei Lagerung wird nachgewiesen und die entsprechenden Lagerbedingungen werden definiert.
Prüfbedingungen
Lösungsmittel/Vehikel
Das Lösungsmittel/Vehikel sollte bei den gewählten Dosierungen keine toxischen Wirkungen hervorrufen und mit den Prüfchemikalien keine chemische Reaktion eingehen. Werden keine allgemein bekannten Lösungsmittel/Vehikel verwendet, so sind Daten zur Kompatibilität beizubringen. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels/Vehikels in Erwägung zu ziehen. Beispiele für üblicherweise verwendete, kompatible Lösungsmittel/Vehikel sind Wasser, physiologische Kochsalzlösung, Methylcelluloselösung, Carboxymethylcellulose-Natriumsalzlösung, Olivenöl und Maisöl. Liegen keine historischen oder veröffentlichten Kontrolldaten vor, aus denen hervorgeht, dass von einem gewählten, üblicherweise nicht verwendeten Lösungsmittel/Vehikel keine Mikrokerne oder andere schädliche Wirkungen induziert werden, sollte ein Vorversuch durchgeführt werden, um die Akzeptanz des Lösungsmittels/Vehikels nachzuweisen.
Kontrollen
Positivkontrollen
Jeder Versuch sollte in der Regel eine Gruppe von Tieren umfassen, die mit einer Positivkontrollchemikalie behandelt wurden. Darauf kann möglicherweise verzichtet werden, wenn das Prüflabor seine Eignung für die Durchführung des Tests nachgewiesen und einen bestimmten Bereich historischer Positivkontrollen etabliert hat. Enthält der Versuch keine gleichzeitige Positivkontrolle, sind Auswertungskontrollen (fixierte und nicht gefärbte Objektträger oder Zellsuspensionen, je nach Auswertungsmethode) in jeden Versuch zu integrieren. Diese erhält man beispielsweise im Rahmen von Untersuchungen zur Eignung des Labors und danach gegebenenfalls auf regelmäßiger Basis, indem bei der Auswertung der Studie geeignete Referenzproben hinzugezogen werden, die bei einem gesonderten, regelmäßig (z.B. alle 6 bis 18 Monate) durchgeführten Positivkontrollversuch gewonnen und aufbewahrt wurden.
Positivkontrollchemikalien sollten zuverlässig eine nachweisliche Zunahme der Häufigkeit von Mikrokernen gegenüber dem spontanen Niveau erzeugen. Erfolgt eine manuelle mikroskopische Auswertung, sind Positivkontrolldosen so zu wählen, dass die Wirkungen eindeutig sind, aber beim Ablesen nicht sofort die Identität der kodierten Proben erkennen lassen. Es ist vertretbar, dass die Positivkontrolle unter Verwendung eines anderen Behandlungsplans auf anderem Wege als die Prüfchemikalie verabreicht wird und nur eine einzige Probenahme erfolgt. Gegebenenfalls könnte auch eine zusätzliche Positivkontrolle herangezogen werden, die der gleichen chemischen Klasse angehört wie die zu prüfende Chemikalie. Beispiele für Positivkontrollchemikalien sind Tabelle 1 zu entnehmen.
Tabelle 1 Beispiele für Positivkontrollchemikalien.
| Chemikalien und CAS-Nr. |
| Ethylmethansulfonat [CAS-Nr. 62-50-0] |
| Methylmethansulfonat [CAS-Nr. 66-27-3] |
| Ethylnitrosoharnstoff [CAS-Nr. 759-73-9] |
| Mitomycin C [CAS-Nr. 50-07-7] |
| Cyclophosphamid(monohydrat) [CAS-Nr. 50-18-0 (CAS-Nr. 6055-19-2)] |
| Triethylenmelamin [CAS-Nr. 51-18-3] |
| Colchicin [CAS-Nr. 64-86-8] oder Vinblastin [CAS-Nr. 865-21-4] - als Aneugen |
Negativkontrollen
Zu jedem Probenahmezeitpunkt sind Tiere der Negativkontrollgruppe einzubeziehen, die - abgesehen davon, dass ihnen die Prüfchemikalie nicht verabreicht wird - ebenso behandelt werden wie die Behandlungsgruppen. Sofern ein Lösungsmittel/Vehikel bei der Verabreichung der Prüfchemikalie verwendet wird, sollte die Kontrollgruppe dieses Lösungsmittel/Vehikel erhalten. Wenn jedoch aus historischen Negativkontrolldaten für das Prüflabor zu jedem Stichprobenzeitpunkt übereinstimmende Werte zur Variabilität der Tiere und zur Häufigkeit der Zellen mit Mikrokernen hervorgehen, ist bei den Negativkontrollen gegebenenfalls nur eine einzige Probenahme erforderlich. Erfolgt bei den Negativkontrollen nur eine einzige Probenahme, so ist diese zum ersten in der Studie vorgesehenen Probenahmezeitpunkt vorzunehmen.
Bei Verwendung von peripherem Blut ist anstelle einer gleichzeitigen Negativkontrolle möglicherweise auch eine vor der Behandlung entnommene Probe vertretbar, jedoch nur bei Kurzzeitstudien unter der Voraussetzung, dass die gewonnenen Daten mit der historischen Kontrolldatenbank für das Prüflabor übereinstimmen. Es wurde belegt, dass bei Ratten die Entnahme von Proben kleiner Volumina (z.B. unter 100 µl/Tag) vor der Behandlung nur minimale Auswirkungen auf die Hintergrundhäufigkeit von Mikrokernen hat (25).
Verfahren
Anzahl und Geschlecht der Tiere
Die Mikrokernreaktion verläuft im Normalfall bei männlichen und weiblichen Tieren ähnlich. Daher können die meisten Studien unabhängig vom Geschlecht durchgeführt werden (26). Daten, aus denen nennenswerte Unterschiede zwischen männlichen und weiblichen Tieren hervorgehen (z.B. Unterschiede bei der systemischen Toxizität, beim Stoffwechsel, bei der Bioverfügbarkeit, bei der Knochenmarktoxizität usw., auch beispielsweise im Rahmen einer Dosisfindungsstudie), würden die Verwendung von Tieren aus jedem Geschlecht nahe legen. In diesem Fall kann es angebracht sein, eine Studie an Tieren beider Geschlechter durchzuführen, z.B. als Teil einer Toxizitätsstudie bei wiederholter Verabreichung. Gegebenenfalls ist die Verwendung eines faktoriellen Versuchsplans geeignet, wenn beide Geschlechter einbezogen werden. Einzelheiten zur Analyse der Daten bei Verwendung eines solchen Versuchsplans sind in Anlage 2 enthalten.
Zu Beginn der Studie sollten die Gruppengrößen so bestimmt werden, dass man pro Gruppe mindestens 5 analysierbare Tiere pro Geschlecht oder aus beiden Geschlechtern, sofern beide Geschlechter einbezogen werden, erhält. Sollte es sich beim Menschen um eine geschlechtsspezifische Exposition handeln, z.B. bei bestimmten Pharmazeutika, ist der Versuch an Tieren des betreffenden Geschlechts durchzuführen. Bei einer gemäß den unter Nummer 37 festgelegten Parametern durchgeführten Knochenmarkstudie mit drei Dosisgruppen und gleichzeitigen Negativ- und Positivkontrollen (wobei jede Gruppe aus fünf Tieren eines einzigen Geschlechts besteht) kann als Richtwert für die üblicherweise erforderliche Höchstmenge an Tieren eine Anzahl von 25 bis 35 Versuchstieren angegeben werden.
Dosierungen
Wenn zunächst eine Dosisfindungsstudie durchgeführt wird, da keine geeigneten Daten zur Dosierungswahl verfügbar sind, sollte diese im gleichen Labor unter Verwendung des/der gleichen Spezies, Rasse, Geschlechts und Behandlungsverfahrens wie im Hauptversuch stattfinden (27). Ziel der Studie sollte sein, die maximal verträgliche Dosis (MTD) zu ermitteln, die definiert ist als die höchste Dosierung, die vertragen wird, ohne dass Anzeichen von Toxizität auftreten, die die Studie, bezogen auf den Untersuchungszeitraum, begrenzen würden (z.B. durch Rückgang des Körpergewichts oder Zytotoxizität des blutbildenden Systems, ausgenommen jedoch Tod oder Anzeichen von Schmerzen und Leiden, die eine humane Tötung erforderlich machen würden (28)).
Die Höchstdosis kann auch als Dosis definiert werden, die Toxizität im Knochenmark hervorruft (z.B. eine Reduzierung des Anteils unreifer Erythrozyten an der Gesamtzahl der Erythrozyten im Knochenmark oder peripheren Blut von mehr als 50 %, nicht jedoch auf weniger als 20 % des Kontrollwerts). Bei der Analyse von CD71-positiven Zellen im peripheren Blutkreislauf (d. h. anhand einer Durchflusszytometrie) reagiert diese sehr junge Fraktion unreifer Erythrozyten jedoch schneller als die größere RNA-positive Kohorte unreifer Erythrozyten auf toxische Einwirkungen. Daher kann sich gegebenenfalls eine höhere scheinbare Toxizität bei Versuchsplänen mit akuter Exposition ergeben, bei denen die Fraktion der CD71-positiven unreifen Erythrozyten untersucht wird, vergleicht man sie mit der von unreifen Erythrozyten mit entsprechendem RNA-Gehalt. Aus diesem Grund kann die Höchstdosis für Toxizität hervorrufende Prüfchemikalien bei Versuchen mit fünf oder weniger Behandlungstagen auch als die Dosis verstanden werden, die eine statistisch signifikante Verringerung des Anteils CD71-positiver unreifer Erythrozyten an der Gesamtzahl der Erythrozyten hervorruft, nicht jedoch auf weniger als 5 % des Kontrollwerts (29).
Chemikalien, die eine Sättigung der toxikokinetischen Eigenschaften aufweisen oder Entgiftungsprozesse einleiten, die nach einer Langzeitgabe möglicherweise zu einem Rückgang der Exposition führen, entsprechen möglicherweise nicht den Dosierungskriterien und sollten anhand einer Einzelfallprüfung bewertet werden.
Um Dosis-Wirkungs-Informationen zu erhalten, sollte eine vollständige Studie eine Negativkontrollgruppe und mindestens drei Dosierungen enthalten, die sich in der Regel um einen Faktor von 2 (maximal von 4) unterscheiden. Ruft die Prüfchemikalie in einer Dosisfindungsstudie oder auf der Basis bereits vorhandener Daten keine Toxizität hervor, sollte die höchste Dosierung für einen Behandlungszeitraum von 14 Tagen oder mehr 1.000 mg/kg Körpergewicht/Tag bzw. für Behandlungszeiträume von weniger als 14 Tagen 2.000 mg/kg Körpergewicht/Tag betragen. Ruft die Prüfchemikalie jedoch Toxizität hervor, sollte die MTD die höchste verabreichte Dosierung sein und die Dosierung sollte vorzugsweise einen Bereich vom Höchstwert bis zu einer Dosierung, die wenig oder keine Toxizität erzeugt, abdecken. Wenn bei allen untersuchten Dosierungen toxische Wirkungen im Zielgewebe (Knochenmark) beobachtet werden, sind weitere Untersuchungen bei nichttoxischen Dosierungen anzuraten. Studien zur ausführlicheren Charakterisierung der quantitativen Dosis-Wirkungs-Informationen erfordern gegebenenfalls weitere Dosisgruppen. Bei bestimmten Arten von Prüfchemikalien (z.B. Humanpharmazeutika), für die spezielle Anforderungen gelten, können die Grenzen variieren.
Limit-Test
Wenn Dosisfindungsstudien oder bereits vorhandene Daten von verwandten Tierstämmen darauf hindeuten, dass eine Behandlung mit mindestens der Limit-Dosis (siehe Beschreibung unten) keine feststellbaren toxischen Wirkungen (und auch keinen Rückgang der Proliferation des Knochenmarks oder andere Anzeichen für toxische Wirkungen im Zielgewebe) verursacht, und wenn auf der Basis von In-vitro-Studien zur Untersuchung der Genotoxizität oder von Daten strukturell verwandter Chemikalien keine Genotoxizität zu erwarten ist, ist eine vollständige Studie mit drei Dosierungen gegebenenfalls nicht erforderlich, vorausgesetzt, es wurde nachgewiesen, dass die Prüfchemikalie(n) das Zielgewebe (Knochenmark) erreicht bzw. erreichen. In solchen Fällen ist eine Einzeldosierung mit der Limit-Dosis gegebenenfalls ausreichend. Bei einem Behandlungszeitraum von 14 Tagen oder mehr beträgt die Limit-Dosis 1.000 mg/kg Körpergewicht/Tag. Bei Behandlungszeiträumen von weniger als 14 Tagen beträgt die Limit-Dosis 2.000 mg/kg Körpergewicht/Tag.
Verabreichung
Bei der Versuchsplanung sind die zu erwartenden Verabreichungswege beim Menschen zu berücksichtigen. Verabreichungswege, wie etwa eine Aufnahme über die Nahrung, über das Trinkwasser, eine topische, subkutane, intravenöse, orale (über eine Magensonde), intratracheale Verabreichung, durch Inhalation oder Implantation, sind mit entsprechender Begründung zu wählen. In jedem Fall sollte der Verabreichungsweg so gewählt werden, dass das bzw. die Zielgewebe eine angemessene Exposition erfährt bzw. erfahren. Eine intraperitoneale Injektion wird in der Regel nicht empfohlen, da diese nicht als Verabreichungsweg beim Menschen vorgesehen ist, und sollte nur mit spezieller wissenschaftlicher Begründung angewandt werden. Sofern die Prüfchemikalie der Nahrung oder dem Trinkwasser beigemischt wird, insbesondere im Fall von Einzeldosierungen, ist darauf zu achten, einen ausreichenden Abstand zwischen der Nahrungsmittel- und Trinkwasseraufnahme und der Probenahme einzuhalten, damit ein Nachweis der Wirkungen möglich ist (siehe Nummer 37). Die Höchstmenge an Flüssigkeit, die jeweils über eine Magensonde oder eine Injektion verabreicht werden kann, hängt von der Größe des Versuchstiers ab. Das Volumen sollte im Normalfall 1 ml/100 g Körpergewicht nicht überschreiten, bei wässrigen Lösungen können aber auch 2 ml/100 g in Betracht gezogen werden. Werden größere Volumina verwendet, ist dies zu begründen. Mit Ausnahme von reizenden oder ätzenden Prüfchemikalien, die normalerweise bei höheren Konzentrationen gravierende Wirkungen zeigen, sollte die Variabilität der Prüfvolumina durch Anpassung der Konzentration auf ein konstantes Volumen im Verhältnis zum Körpergewicht bei allen Dosen möglichst gering gehalten werden.
Behandlungsplan
Vorzugsweise werden 2 oder mehr Behandlungen durchgeführt mit Verabreichung der Prüfchemikalie in Abständen von 24 Stunden, insbesondere wenn dieser Test in andere Toxizitätsstudien eingebunden wird. Alternativ können Einzelbehandlungen erfolgen, wenn dies wissenschaftlich begründet ist (wenn z.B. bekannt ist, dass die Prüfchemikalie den Zellzyklus blockiert). Die Prüfchemikalien können auch in Form von zwei oder mehreren Teilmengen am gleichen Tag im Abstand von nicht mehr als 2 bis 3 Stunden verabreicht werden, wenn es sich um ein großes Materialvolumen handelt. In diesen Fällen, oder wenn die Prüfchemikalie durch Inhalation verabreicht wird, sollte der Zeitpunkt der Probenahme ausgehend von der letzten Dosis oder dem Zeitpunkt, an dem die Exposition beendet wurde, geplant werden.
Die Prüfung kann an Mäusen oder Ratten auf dreierlei Weise durchgeführt werden:
Sofern relevant oder wissenschaftlich begründet und zur leichteren Einbindung in andere Toxizitätstests sind auch andere Dosierungs- oder Probenahmeverfahren vertretbar.
Beobachtungen
Mindestens einmal täglich - vorzugsweise zum gleichen Zeitpunkt und unter Berücksichtigung des Zeitraums, in dem der Wirkungsgipfel nach Verabreichung der Dosis zu erwarten ist - sollten allgemeine klinische Beobachtungen der Versuchstiere vorgenommen und protokolliert werden. Mindestens zweimal täglich während der Verabreichungszeit sind alle Tiere auf Morbidität und Mortalität zu überprüfen. Alle Tiere sollten zu Studienbeginn, mindestens einmal pro Woche während Studien mit Wiederholungsdosen und bei humaner Tötung gewogen werden. In Studien von mindestens einwöchiger Dauer sollten mindestens wöchentlich Messungen der Futteraufnahme vorgenommen werden. Wenn die Prüfchemikalie über das Trinkwasser verabreicht wird, sollte auch die Wasseraufnahme bei jedem Wasserwechsel und mindestens einmal wöchentlich gemessen werden. Tiere mit Anzeichen von übermäßiger, jedoch nicht tödlich wirkender Toxizität sollten vor Ende des Prüfzeitraums human getötet werden (28). Unter bestimmten Umständen kann die Körpertemperatur der Versuchstiere überwacht werden, da behandlungsabhängige Hyper- und Hypothermien eine Rolle bei falschen Ergebnissen gespielt haben mögen (32) (33) (34).
Exposition des Zielgewebes
Zu einem oder mehreren geeigneten Zeitpunkten sollte eine Blutprobe genommen werden, um eine Untersuchung der Plasmaspiegel der Prüfchemikalie zu ermöglichen. Damit soll nachgewiesen werden, dass eine Exposition des Knochenmarks stattgefunden hat, wo dies gerechtfertigt erscheint und keine anderen Expositionsdaten vorhanden sind (siehe Nummer 48).
Knochenmark-/Blutpräparation
Die Knochenmarkzellen werden in der Regel unmittelbar nach der humanen Tötung aus den Oberschenkel- oder Schienbeinknochen gewonnen. Dabei werden die Zellen gewöhnlich entnommen und unter Verwendung erprobter Methoden präpariert und gefärbt. Kleine Mengen peripheren Bluts können, unter Einhaltung entsprechender Tierschutznormen, entweder anhand einer Methode gewonnen werden, die ein Überleben des Versuchstiers ermöglicht, wie z.B. eine Blutentnahme aus der Schwanzvene oder einem anderen geeigneten Blutgefäß, oder durch Kardialpunktion oder Blutentnahme aus einem großen Blutgefäß bei humaner Tötung des Versuchstiers. Bei Erythrozyten sowohl aus dem Knochenmark als auch aus peripherem Blut werden die Zellen, abhängig von der Analysemethode, sofort supravital gefärbt (16) (17) (18), es werden Ausstrichpräparate hergestellt und sie werden dann für mikroskopische Zwecke gefärbt, oder die Zellen werden fixiert und für eine Durchflusszytometrie-Analyse entsprechend gefärbt. Durch Verwendung eines DNA-spezifischen Farbstoffs [z.B. Acridin Orange (35) oder Hoechst 33258 plus pyronin-Y (36)] lassen sich bestimmte Artefakte vermeiden, die bei Verwendung eines nicht DNA-spezifischen Farbstoffs auftreten. Trotz dieses Vorteils ist aber die Verwendung herkömmlicher Farbstoffe (z.B. Giemsa für mikroskopische Analyse) nicht ausgeschlossen. Zusätzliche Systeme [z.B. Cellulosesäulen zur Entfernung kernhaltiger Zellen (37) (38)] können ebenfalls verwendet werden, falls diese Systeme nachweislich für die Aufbereitung von Proben im Labor kompatibel sind.
Sofern diese Verfahren anwendbar sind, können Anti-Kinetochor-Antikörper (39), FISH mit panzentromerischen DNA-Sonden (40) oder eine In-situ-Markierung mit Panzentromer-spezifischen Primern zusammen mit einer geeigneten DNA-Gegenfärbung (41) zur Untersuchung der Art der Mikrokerne (Chromosom/Chromosomenfragment) herangezogen werden, um zu bestimmen, ob der der Mikrokerninduktion zugrunde liegende Mechanismus auf clastogene und/oder aneugene Wirkungen zurückzuführen ist. Es können auch andere Verfahren zur Unterscheidung zwischen Clastogenen und Aneugenen verwendet werden, sofern ihre Wirksamkeit nachgewiesen wurde.
Analyse (manuell und automatisiert)
Alle Objektträger oder Analysenproben, einschließlich der Positiv- und Negativkontrollen, sollten vor der Analyse, gleich welcher Art, unabhängig kodiert und randomisiert werden, damit die Auswertung ohne Kenntnis der Behandlungsbedingungen erfolgt. Eine solche Codierung erübrigt sich, wenn automatisierte Auswertungssysteme verwendet werden, die nicht auf einer Sichtprüfung beruhen und keinen Bedienungseinflüssen unterliegen. Der Anteil unreifer Erythrozyten an der Gesamtzahl (unreife + reife Erythrozyten) wird für jedes Tier bestimmt, indem bei Verwendung von Knochenmark mindestens 500 Erythrozyten und bei Verwendung von peripherem Blut mindestens 2.000 Erythrozyten gezählt werden (42). Je Tier werden mindestens 4.000 unreife Erythrozyten auf das Vorhandensein von unreifen mikrokernhaltigen Erythrozyten untersucht (43). Wenn aus der Datenbank der historischen Negativkontrollen hervorgeht, dass die mittlere Hintergrundhäufigkeit für unreife mikrokernhaltige Erythrozyten im Prüflabor < 0,1 % beträgt, sollte eine Auswertung weiterer Zellen in Erwägung gezogen werden. Bei der Analyse der Proben sollte der Anteil der unreifen Erythrozyten an der Gesamtzahl der Erythrozyten in behandelten Tieren nicht weniger als 20 % des Anteils in der (Vehikel-/Lösungsmittel)kontrolle bei einer mikroskopischen Auswertung und nicht weniger als etwa 5 % des Anteils in der (Vehikel-/Lösungsmittel)kontrolle bei Auswertung der unreifen CD71+-Erythrozyten anhand von zytometrischen Verfahren betragen (siehe Nummer 31) (29). Bei einem mikroskopisch ausgewerteten Knochenmarktest würde die Höchstgrenze der Toxizität beispielsweise bei einem Anteil unreifer Erythrozyten von 10 % liegen, wenn der Anteil der unreifen Erythrozyten im Knochenmark bei den Kontrollen 50 % beträgt.
Da die Milz von Ratten mikrokernhaltige Erythrozyten sequestriert und vernichtet, ist es zur Beibehaltung einer hohen Empfindlichkeit des Tests bei der Analyse von peripherem Blut von Ratten ratsam, die Analyse mikrokernhaltiger unreifer Erythrozyten auf die jüngste Fraktion zu beschränken. Bei der Verwendung automatisierter Analysemethoden können diese am wenigsten ausgereiften Erythrozyten aufgrund ihres hohen RNA-Gehalts oder ihres hohen Anteils an Transferrin-Rezeptoren (CD71+) auf ihrer Oberfläche nachgewiesen werden (31). Der direkte Vergleich verschiedener Färbemethoden hat jedoch ergeben, dass mit unterschiedlichen Methoden zufriedenstellende Ergebnisse erzielt werden können, darunter auch die herkömmliche Färbung mit Acridin Orange (3) (4).
Daten und Berichterstattung
Behandlung der Ergebnisse
Die Daten für die einzelnen Tiere sind in tabellarischer Form darzustellen. Die Anzahl der ausgewerteten unreifen Erythrozyten, die Anzahl mikrokernhaltiger unreifer Erythrozyten und der Anteil unreifer Erythrozyten an der Gesamtzahl sind getrennt für jedes Tier aufzulisten. Werden Mäuse kontinuierlich über einen Zeitraum von 4 Wochen oder länger behandelt, sind auch die Daten zur Anzahl und zum Anteil mikrokernhaltiger reifer Erythrozyten anzugeben, sofern sie erfasst wurden. Daten zur Toxizität bei Tieren und klinische Anzeichen sind ebenfalls anzugeben.
Gültigkeitskriterien
Die folgenden Kriterien entscheiden über die Gültigkeit des Versuchs:
Bewertung und Interpretation der Ergebnisse
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn
Wird zu einem bestimmten Probenahmezeitpunkt nur die höchste Dosierung untersucht, gilt eine Prüfchemikalie als eindeutig positiv, wenn, verglichen mit der gleichzeitigen Negativkontrolle, eine statistisch signifikante Zunahme vorliegt und die Ergebnisse außerhalb der Verteilung der historischen Negativkontrolldaten liegen (z.B. auf einer Poisson-Verteilung beruhende Kontrollgrenzen von 95 %). Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (44) (45) (46) (47). Bei der Durchführung einer Dosis-Wirkungs-Analyse sollten mindestens drei behandelte Dosisgruppen analysiert werden. Bei statistischen Versuchen sollte das Tier als Versuchseinheit zugrunde gelegt werden. Positive Ergebnisse im Mikrokerntest deuten darauf hin, dass eine Prüfchemikalie Mikrokerne induziert, die das Ergebnis von Chromosomenschäden oder einer Schädigung im mitotischen Apparat von Erythroblasten der getesteten Spezies sind. In den Fällen, in denen der Test durchgeführt wurde, um Zentromere in Mikrokernen nachzuweisen, dient eine Prüfchemikalie, die zentromerhaltige Mikrokerne erzeugt (zentromerische DNA oder Kinetochore, die auf den Verlust ganzer Chromosomen hinweisen) als Nachweis dafür, dass es sich bei der Prüfchemikalie um ein Aneugen handelt.
Unter der Voraussetzung, dass alle Gültigkeitskriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn unter allen untersuchten Versuchsbedingungen
Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (44) (45) (46) (47). Als Nachweis für eine Exposition des Knochenmarks gegenüber einer Prüfchemikalie kann ein Rückgang des Verhältnisses von unreifen zu reifen Erythrozyten oder der gemessenen Plasma- oder Blutspiegel der Prüfchemikalie dienen. Im Falle einer intravenösen Verabreichung ist kein Nachweis für eine Exposition zu erbringen. Alternativ dazu können ADME-Daten herangezogen werden, die in einer unabhängigen Studie unter Verwendung der gleichen Verabreichungswege und der gleichen Spezies gewonnen wurden, um nachzuweisen, dass eine Knochenmarkexposition stattgefunden hat. Negative Ergebnisse deuten darauf hin, dass die Prüfchemikalie unter den Versuchsbedingungen keine Mikrokerne in den unreifen Erythrozyten der getesteten Spezies erzeugt.
Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich.
In den Fällen, in denen die Reaktion weder eindeutig negativ noch eindeutig positiv ist, oder um die biologische Relevanz eines Ergebnisses zu untermauern (z.B. eine geringe oder grenzwertige Zunahme), sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen der vorliegenden abgeschlossenen Versuche bewertet werden. In einigen Fällen kann die Auswertung weiterer Zellen oder die Durchführung eines Wiederholungsversuchs unter veränderten Versuchsbedingungen hilfreich sein.
In seltenen Fällen erlaubt der Datensatz auch nach weiteren Untersuchungen keine definitive Aussage darüber, ob die Prüfchemikalie positive oder negative Ergebnisse zur Folge hat, und die Studie wird daher als nicht eindeutig abgeschlossen.
Prüfbericht
Der Prüfbericht muss folgende Angaben enthalten:
Zusammenfassung
Prüfchemikalie:
Einkomponentiger Stoff:
Mehrkomponentiger Stoff, UVCB-Stoffe und Gemische:
Zubereitung der Prüfchemikalie:
Versuchstiere:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse.
Schlussfolgerung.
Referenzdokumente.
Literaturhinweise
(1) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris.
(2) Hayashi, M. et al. (2007), in vivo erythrocyte micronucleus assay III. Validation and regulatory acceptance of automated scoring and the use of rat peripheral blood reticulocytes, with discussion of non-hematopoietic target cells and a single dose-level limit test, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 627/1, 10-30.
(3) MacGregor, J.T. et al. (2006), Flow cytometric analysis of micronuclei in peripheral blood reticulocytes: II. An efficient method of monitoring chromosomal damage in the rat, Toxicology Sciences, Band 94/1, 92-107.
(4) Dertinger, S.D. et al. (2006), Flow cytometric analysis of micronuclei in peripheral blood reticulocytes: I. Intra- and interlaboratory comparison with microscopic scoring, Toxicological Sciences, Band 94/1, 83-91.
(5) Dertinger, S.D. et al. (2011), Flow cytometric scoring of micronucleated erythrocytes: an efficient platform for assessing in vivo cytogenetic damage, Mutagenesis, Band 26/1, 139-145.
(6) Parton, J.W., W.P. Hoffman, M.L. Garriott (1996), Validation of an automated image analysis micronucleus scoring system, Mutation Research, Band 370/1, 65-73.
(7) Asano, N. et al. (1998), An automated new technique for scoring the rodent micronucleus assay: computerized image analysis of acridine orange supravitally stained peripheral blood cells, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 404/1-2, 149-154.
(8) Styles, J.A. et al. (2001), Automation of mouse micronucleus genotoxicity assay by laser scanning cytometry, Cytometry, Band 44/2, 153-155.
(9) Heddle, J.A. (1973), A rapid in vivo test for chromosomal damage, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 18/2, 187-190.
(10) Schmid, W. (1975), The micronucleus test, Mutation Research, Band 31/1, 9-15.
(11) Heddle, J.A. et al. (1983), The induction of micronuclei as a measure of genotoxicity. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Research/Reviews in Genetic Toxicology, Band 123/1, 61-118.
(12) Mavournin, K.H. et al. (1990), The in vivo micronucleus assay in mammalian bone marrow and peripheral blood. A report of the U.S. Environmental Protection Agency Gene-Tox Program, Mutation Research/Reviews in Genetic Toxicology, Band 239/1, 29-80.
(13) MacGregor, J.T. et al. (1983), Micronuclei in circulating erythrocytes: a rapid screen for chromosomal damage during routine toxicity testing in mice, Developments in Toxicology Environmental Science, Band 11, 555-558.
(14) MacGregor, J.T. et al. (1987), Guidelines for the conduct of micronucleus assays in mammalian bone marrow erythrocytes, Mutation Research/Genetic Toxicology, Band 189/2, 103-112.
(15) MacGregor, J.T. et al. (1990), The in vivo erythrocyte micronucleus test: measurement at steady state increases assay efficiency and permits integration with toxicity studies, Fundamental and Applied Toxicology, Band 14/3, 513-522.
(16) Hayashi, M. et al. (1990), The micronucleus assay with mouse peripheral blood reticulocytes using acridine orange-coated slides, Mutation Research/Genetic Toxicology, Band 245/4, 245-249.
(17) CSGMT/JEMS.MMS - The Collaborative Study Group for the Micronucleus Test (1992), Micronucleus test with mouse peripheral blood erythrocytes by acridine orange supravital staining: the summary report of the 5th collaborative study, Mutation Research/Genetic Toxicology, Band 278/2-3, 83-98.
(18) CSGMT/JEMS.MMS - The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan (1995), Protocol recommended by the CSGMT/JEMS.MMS for the short-term mouse peripheral blood micronucleus test. The Collaborative Study Group for the Micronucleus Test (CSGMT) (CSGMT/JEMS.MMS, The Mammalian Mutagenesis Study Group of the Environmental Mutagen Society of Japan), Mutagenesis, Band 10/3, 153-159.
(19) Salamone, M.F., K.H. Mavournin (1994), Bone marrow micronucleus assay: a review of the mouse stocks used and their published mean spontaneous micronucleus frequencies, Environmental and Molecular Mutagenesis, Band 23/4, 239-273.
(20) Krishna, G., G. Urda, J. Paulissen (2000), Historical vehicle and positive control micronucleus data in mice and rats, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 453/1, 45-50.
(21) Hayes, J. et al. (2009), The rat bone marrow micronucleus test--study design and statistical power, Mutagenesis, Band 24/5, 419-424.
(22) Wakata, A. et al. (1998), Evaluation of the rat micronucleus test with bone marrow and peripheral blood: summary of the 9th collaborative study by CSGMT/JEMS. MMS. Collaborative Study Group for the Micronucleus Test. Environmental Mutagen Society of Japan. Mammalian Mutagenicity Study Group, Environmental and Molecular Mutagenesis, Band 32/1, 84-100.
(23) Ryan, T.P. (2000), Statistical Methods for Quality Improvement, 2nd ed., John Wiley and Sons, New York.
(24) Hayashi, M. et al. (2011), Compilation and use of genetic toxicity historical control data, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 723/2, 87-90.
(25) Rothfuss, A. et al. (2011), Improvement of in vivo genotoxicity assessment: combination of acute tests and integration into standard toxicity testing, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 723/2, 108-120.
(26) Hayashi, M. et al. (1994), in vivo rodent erythrocyte micronucleus assay, Mutation Research/Environmental Mutagenesis and Related Subjects, Band 312/3, 293-304.
(27) Fielder, R.J. et al. (1992), Report of British Toxicology Society/UK Environmental Mutagen Society Working Group. Dose setting in in vivo mutagenicity assays, Mutagenesis, Band 7/5, 313-319.
(28) OECD (2000), "Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation", OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, No. 19, OECD Publishing, Paris.
(29) LeBaron, M.J. et al. (2013), Influence of counting methodology on erythrocyte ratios in the mouse micronucleus test, Environmental and Molecular Mutagenesis, Band 54/3, 222-228.
(30) Higashikuni, N., S. Sutou (1995), An optimal, generalized sampling time of 30 +/- 6 h after double dosing in the mouse peripheral blood micronucleus test, Mutagenesis, Band 10/4, 313-319.
(31) Hayashi, M. et al. (2000), in vivo rodent erythrocyte micronucleus assay. II. Some aspects of protocol design including repeated treatments, integration with toxicity testing, and automated scoring, Environmental and Molecular Mutagenesis, Band 35/3, 234-252.
(32) Asanami, S., K. Shimono (1997), High body temperature induces micronuclei in mouse bone marrow, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 390/1-2, 79-83.
(33) Asanami, S., K. Shimono, S. Kaneda (1998), Transient hypothermia induces micronuclei in mice, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 413/1, 7-14.
(34) Spencer, P.J. et al. (2007), Induction of micronuclei by phenol in the mouse bone marrow: I. Association with chemically induced hypothermia, Toxicological Sciences, Band 97/1, 120-127.
(35) Hayashi, M., T. Sofuni, M. Jr. Ishidate (1983), An application of Acridine Orange fluorescent staining to the micronucleus test, Mutation Research Letters, Band 120/4, 241-247.
(36) MacGregor, J.T. Wehr, R.G. Langlois (1983), A simple fluorescent staining procedure for micronuclei and RNA in erythrocytes using Hoechst 33258 and pyronin Y, Mutation Research, Band 120/4, 269-275.
(37) Romagna, F., C.D. Staniforth (1989), The automated bone marrow micronucleus test, Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Band 213/1, 91-104.
(38) Sun, J.T., M.J. Armstrong, S.M. Galloway (1999), Rapid method for improving slide quality in the bone marrow micronucleus assay; an adapted cellulose column procedure, Mutation Research, Band 439/1, 121-126.
(39) Miller, B.M., I.D. Adler (1990), Application of antikinetochore antibody staining (CREST staining) to micronuclei in erythrocytes induced in vivo, Mutagenesis, Band 5/4, 411-415.
(40) Miller, B.M. et al. (1991), Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA, Mutagenesis, Band 6/4, 297-302.
(41) Russo, A. (2002), PRINS tandem labeling of satellite DNA in the study of chromosome damage, American Journal of Medical Genetics, Band 107/2, 99-104.
(42) Gollapudi, B.B., L.G. McFadden (1995), Sample size for the estimation of polychromatic to normochromatic erythrocyte ratio in the bone marrow micronucleus test, Mutation Research, Band 347/2, 97-99.
(43) OECD (2014), Statistical analysis supporting the revision of the genotoxicity Test Guidelines, OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment, No. 198, OECD Publishing, Paris.
(44) Richold, M. et al. (1990), "In Vivo Cytogenetics Assays", in Basic Mutagenicity Tests, UKEMS Recommended Procedures. UKEMS Subcommittee on Guidelines for Mutagenicity Testing. Report. Part I revised, Kirkland, D.J. (ed.), Cambridge University Press, Cambridge, 115-141.
(45) Lovell, D.P. et al. (1989), Statistical Analysis of in vivo"Cytogenetic Assays", in Statistical Evaluation of Mutagenicity Test Data. UKEMS SubCommittee on Guidelines for Mutagenicity Testing, Report, Part III, Kirkland, D.J. (ed.), Cambridge University Press, Cambridge, 184-232.
(46) Hayashi, M. et al. (1994), Statistical analysis of data in mutagenicity assays: rodent micronucleus assay, Environmental Health Perspectives, Band 102/Suppl 1, 49-52.
(47) Kim, B.S., M. Cho, H.J. Kim (2000), Statistical analysis of in vivo rodent micronucleus assay, Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Band 469/2, 233-241.
.
| Definitionen | Anlage 1 |
Chemikalie: Stoff oder Gemisch.
Erythroblast: Frühe Entwicklungsstufe von Erythrozyten, unmittelbar vor der Bildung unreifer Erythrozyten, bei der die Zelle noch einen Zellkern enthält.
Kinetochore: Proteinstruktur, die dem Zentromer eukaryotischer Zellen aufsitzt und die Chromosomen während der Mitose und Meiose mit Mikrotubuli-Polymeren aus dem mitotischen Spindelapparat verbindet, wodurch während der Zellteilung die Schwesterchromatiden auseinander gezogen werden.
Mikrokerne: kleine Kerne zusätzlich zu den Hauptkernen der Zellen und von diesen getrennt, die während der Telophase der Mitose (Meiose) durch zurückgebliebene Chromosomenteile oder ganze Chromosomen gebildet werden.
Normochromatischer oder reifer Erythrozyt: vollständig ausgereifter Erythrozyt, der nicht mehr die nach der Enukleation verbleibende residuale RNA und/oder andere kurzlebige Zellmarker nach der Enukleation im Anschluss an die letzte Teilung in den Erythroblasten enthält.
Polychromatischer oder unreifer Erythrozyt: neu gebildeter Erythrozyt in einer Zwischenstufe der Entwicklung, der sich aufgrund des Vorhandenseins residualer RNA in den neu gebildeten Zellen sowohl mit den blauen als auch den roten Komponenten klassischer Blutfarbstoffe, wie z.B. Giemsa von Wright, färben lässt. Solche neu gebildeten Zellen entsprechen in etwa den Retikulozyten, die mithilfe eines Vitalfarbstoffs sichtbar gemacht werden, durch den die residuale RNA in einem Retikulum ausfällt. Zum Nachweis der neu gebildeten roten Blutkörperchen werden derzeit oft andere Verfahren verwendet, unter anderem die monochromatische Färbung von RNA mit Fluoreszenzfarbstoffen oder die Kennzeichnung kurzlebiger Oberflächenmarker wie CD71 mit fluoreszierenden Antikörpern. Polychromatische Erythrozyten, Retikulozyten und CD71-positive Erythrozyten sind allesamt den unreifen Erythrozyten zuzuordnen, auch wenn sie vom Alter geringfügig verschieden verteilt sind.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Retikulozyt: neu gebildeter Erythrozyt, mit einem Vitalfarbstoff gefärbt, durch den residuale zelluläre RNA in einem charakteristischen Retikulum ausfällt. Bei Retikulozyten und polychromatischen Erythrozyten ist das Zellalter ähnlich verteilt.
Zentromere: Region(en) eines Chromosoms, an die während der Zellteilung die Spindelfasern anhaften, wodurch die ordnungsgemäße Beförderung der Tochterchromosomen zu den Polen der Tochterzellen ermöglicht wird.
.
| Faktorieller Versuchsplan zur Ermittlung geschlechtsspezifischer Unterschiede beim In-vivo-Mikrokerntest | Anlage 2 |
Faktorieller Versuchsplan und zugehörige Analysen
Anhand dieses Versuchsplans werden mindestens 5 männliche und 5 weibliche Tiere je Konzentration getestet. So ergibt sich ein Versuch, bei dem mindestens 40 Tiere verwendet werden (20 männliche und 20 weibliche zzgl. entsprechende Positivkontrollen).
Der Versuchsaufbau, der zu den einfacheren faktoriellen Versuchsplänen zählt, entspricht einer Zweiwege-Varianzanalyse, wobei das Geschlecht und die Konzentration im Wesentlichen die Wirkung bestimmen. Die Daten können anhand vieler Standard-Statistiksoftwareanwendungen wie SPSS, SAS, STATA oder Genstat oder auch mit "R" analysiert werden.
Durch die Analyse lässt sich die Varianz im Datensatz zwischen den Geschlechtern, zwischen den Konzentrationen und in Bezug auf die Wechselwirkungen zwischen den Geschlechtern und Konzentrationen partitionieren. Jede der Bedingungen wird anhand eines Schätzwerts der Varianz zwischen den Wiederholungsversuchstieren innerhalb der gleichgeschlechtlichen Tiergruppen überprüft, die die gleiche Konzentration erhalten haben. Die zugrundeliegende Methodik ist in vielen Standardwerken zur Statistik (siehe Referenzdokumente) und in den in Statistiksoftware-Paketen mitgelieferten Hilfe-Funktionen im Einzelnen beschrieben.
Die Analyse beginnt mit der Untersuchung der Interaktionsbedingung "Konzentration x Geschlecht" anhand der ANOVA-Tabelle 1. Liegt keine signifikante Interaktion vor, liefern die kombinierten Werte für die Geschlechter oder Konzentrationen gültige statistische Tests für die jeweiligen Konzentrationen, und zwar basierend auf der ANOVA-Bedingung der innerhalb der Gruppe gepoolten Varianz.
Es folgt eine Partitionierung des Schätzwerts für die Varianzen zwischen den Konzentrationen in Kontraste, die einen Test für lineare und quadratische Kontraste aus den Reaktionen der verschiedenen Konzentrationen liefern. Ergibt sich hingegen eine signifikante Interaktion für die Bedingung "Konzentration x Geschlecht", kann diese Bedingung auch in Interaktionskontraste "linearer Wert x Geschlecht" und "quadratischer Wert x Geschlecht" partitioniert werden. Diese Bedingungen liefern Tests dazu, ob die Reaktionen der jeweiligen Konzentrationen für die zwei Geschlechter parallel verlaufen oder ob es zu einer zwischen den Geschlechtern differenzierten Reaktion kommt.
Anhand des Schätzwerts für die innerhalb der Gruppen gepoolten Varianzen lassen sich paarweise Tests zu Abweichungen zwischen den Mittelwerten durchführen. Diese Vergleiche könnten zwischen den Mittelwerten für die beiden Geschlechter und zwischen den Mittelwerten für die verschiedenen Konzentrationen durchgeführt werden, beispielsweise um einen Vergleich mit den Negativkontrollen vorzunehmen. In Fällen mit signifikanter Interaktion können Vergleiche zwischen den Mittelwerten verschiedener Konzentrationen innerhalb eines Geschlechts oder zwischen den Mittelwerten beider Geschlechter bei gleicher Konzentration vorgenommen werden.
Referenzdokumente
Es sind viele Werke zur Statistik erhältlich, in denen die Theorie, der Aufbau, die Methodik, die Analyse und die Interpretation faktorieller Versuchspläne erörtert werden, von einfachen Zweifaktorenanalysen bis hin zu komplexeren Formen, wie sie in der "Design of Experiment"-Methode verwendet werden. Die folgende Auflistung ist nicht erschöpfend. Einige Bücher enthalten Beispielrechnungen zu vergleichbaren Versuchsplänen, in einigen Fällen auch mit einem Code zur Durchführung der Analysen unter Verwendung verschiedener Softwarepakete.
Box, G.E.P, Hunter, W.G. and Hunter, J.S. (1978). Statistics for Experimenters. An Introduction to Design, Data Analysis, and Model Building. New York: John Wiley & Sons.
Box G.E.P. & Draper, N.R. (1987). Empirical model-building and response surfaces. John Wiley & Sons Inc.
Doncaster, C.P. & Davey, A.J.H. (2007). Analysis of Variance and Covariance: How to Choose and Construct Models for the Life Sciences. Cambridge University Press.
Mead, R. (1990). The Design of Experiments. Statistical principles for practical application. Cambridge University Press.
Montgomery D.C. (1997). Design and Analysis of Experiments. John Wiley &Sons Inc.
Winer, B.J. (1971). Statistical Principles in Experimental Design. McGraw Hill.
Wu, C.F.J & Hamada, M.S. (2009). Experiments: Planning, Analysis and Optimization. John Wiley & Sons Inc.
_____
1) Statistiker, die mit einem Modellierungsansatz wie dem Ansatz der allgemeinen linearen Modelle (GLM) arbeiten, folgen bei der Analyse möglicherweise einem anderen, wenn auch vergleichbarem Ansatz, werden jedoch nicht notwendigerweise eine Herleitung der herkömmlichen Anova-Tabelle vornehmen, die auf algorithmische Lösungswege für statistische Berechnungen aus dem Vor-Computerzeitalter zurückgeht."
(6) In Teil B wird Kapitel B.15 gestrichen.
(7) In Teil B wird Kapitel B.16 gestrichen.
(8) In Teil B wird Kapitel B.18 gestrichen.
(9) In Teil B wird Kapitel B.19 gestrichen.
(10) In Teil B wird Kapitel B.20 gestrichen.
(11) In Teil B wird Kapitel B.24 gestrichen.
(12) In Teil B erhält Kapitel B.47 folgende Fassung:
"B.47 Trübungs- und Durchlässigkeitstest an der Rinderhornhaut zwecks Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern
Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 437 (2013). Der Trübungs- und Durchlässigkeitstest an der Rinderhornhaut (Bovine Corneal Opacity and Permeability, BCOP) wurde vom Organisationsübergreifenden Koordinationsausschuss zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods, ECVAM) und des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese Centre for the Validation of Alternative Methods, JaCVAM) in den Jahren 2006 und 2010 evaluiert (1) (2). Im Rahmen der ersten Evaluierung wurde der BCOP-Test im Hinblick auf seine Eignung zur Identifizierung von Chemikalien (Stoffen und Gemischen) überprüft, die schwere Augenschäden verursachen (1). Im Rahmen der zweiten Evaluierung wurde der BCOP-Test im Hinblick auf seine Eignung zum Nachweis von Chemikalien (Stoffen und Gemischen) überprüft, die nicht als augenreizende oder schwer augenschädigende Stoffe eingestuft wurden (2). Die BCOP-Validierungsdatenbank enthielt insgesamt 113 Stoffe und 100 Gemische (2)(3). Auf der Grundlage dieser Evaluierungen und zugehöriger Peer-Reviews wurde die Schlussfolgerung gezogen, dass sich die Prüfmethode sowohl zum Nachweis von Chemikalien (Stoffen und Gemischen) eignet, die schwere Augenschäden verursachen (Kategorie 1), als auch von Chemikalien, bei denen keine Einstufung aufgrund von Augenreizung oder schweren Augenschäden gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (4) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 erforderlich ist, weshalb sie für beide Zwecke als wissenschaftlich gültig anerkannt wurde. Unter schweren Augenschäden sind Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges zu verstehen, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Prüfchemikalien, die schwere Augenschäden hervorrufen, werden als Stoffe der UN-GHS-Kategorie 1 eingestuft. Chemikalien, die nicht als augenreizende oder schwer augenschädigende Stoffe eingestuft werden, sind als Stoffe definiert, die nicht den Anforderungen zur Einstufung in die UN-GHS-Kategorie 1 oder 2 (2A oder2B) entsprechen, d. h. sie werden als Stoffe der UN-GHS-Kategorie "Keine Einstufung" bezeichnet. Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des BCOP-Tests auf Grundlage der zugehörigen Evaluierungen. Zu den wichtigsten Unterschieden zwischen der ursprünglichen Fassung der OECD-Prüfrichtlinie aus dem Jahr2009 und der aktualisierten Fassung von 2013 gehören u. a. die Verwendung der BCOP-Prüfmethode zur Identifizierung von Chemikalien, die keine Einstufung nach dem UN-GHS-Klassifizierungssystem erfordern (Nummern 2 und 7), Klarstellungen zur Anwendbarkeit der BCOP-Prüfmethode für die Prüfung von Alkoholen, Ketonen und Feststoffen (Nummern 6 und 7) sowie von Stoffen und Gemischen (Nummer 8), Klarstellungen zur Art der Prüfung von oberflächenaktiven Stoffen und tensidhaltigen Gemischen (Nummer 28), Aktualisierungen und Klarstellungen zu Positivkontrollen (Nummern 39 und 40), eine Aktualisierung der Entscheidungskriterien der BCOP-Prüfmethode (Nummer 47), eine Aktualisierung der Gültigkeitskriterien der Studie (Nummer 48), eine Aktualisierung der Bestandteile des Prüfberichts (Nummer 49), eine Aktualisierung der Anlage 1 zu Definitionen, die Ergänzung von Anlage 2 zur Vorhersagefähigkeit der BCOP-Prüfmethode unter verschiedenen Klassifizierungssystemen, eine Aktualisierung der Anlage 3 zur Liste der Leistungschemikalien und eine Aktualisierung der Anlage 4 zum BCOP-Hornhauthalter (Nummer 1) und zum Opazimeter (Nummern 2 und 3).
Gegenwärtig herrscht allgemeiner Konsens darüber, dass der In-vivo-Draize-Augentest in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Augenreizungen für verschiedene Chemikalienklassen vorherzusagen. Unter Umständen ist es jedoch möglich, den Draize-Augentest durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie zu ersetzen (5). Der Top-Down-Ansatz (5) wird verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie ein hohes Reizpotenzial aufweist. Umgekehrt wird der Bottom-Up-Ansatz (5) verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie keine ausreichende Augenreizung erzeugt und somit keine Einstufung erfordert. Die BCOP-Prüfmethode ist eine In-vitro-Prüfmethode, die unter bestimmten Bedingungen und mit bestimmten Grenzen zur Einstufung und Kennzeichnung von Chemikalien mit augengefährdender Wirkung angewendet werden kann. Auch wenn der Test den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird die BCOP-Prüfmethode als erster Schritt im Rahmen einer Prüfstrategie wie des von Scott et al. vorgeschlagenen Top-Down-Ansatzes (5) empfohlen, um ohne weitere Tests (4) schwer augenschädigende Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, zu identifizieren. Die BCOP-Prüfmethode wird auch für die Identifizierung von Chemikalien empfohlen, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS ("Keine Einstufung") (4) erfordern, und kann daher im Rahmen einer Prüfstrategie z.B. mit Bottom-up-Ansatz verwendet werden (5). Im Falle einer Chemikalie, bei der anhand der BCOP-Prüfmethode nicht vorhergesagt werden kann, dass sie schwere Augenschäden verursacht oder keine Einstufung als augenreizend/schwer augenschädigend erfordert, müssten jedoch weitere Tests (in vitro und/oder in vivo) durchgeführt werden, um eine eindeutige Einstufung vornehmen zu können.
Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie, gemessen als ihre Fähigkeit, bei isolierten Rinderhornhäuten Trübungs- und verstärkte Durchlässigkeitseffekte hervorzurufen. Toxische Effekte für die Hornhaut sind messbar durch: i) verminderte Lichtübertragung (Trübung) und ii) den verstärkten Durchtritt von Fluorescein-Natrium-Farbstoff (Durchlässigkeit). Die Trübungs- und Durchlässigkeitsbewertungen der Hornhaut nach Applikation einer Prüfchemikalie ergeben in Kombination einen In-vitro-Reizwert (In Vitro Irritancy Score, IVIS), der für die Einstufung des Reizpotenzials der Prüfchemikalie maßgeblich ist.
Definitionen sind Anlage 1 zu entnehmen.
Vorbemerkungen und Einsatzgrenzen
Diese Prüfmethode basiert auf dem BCOP-Testprotokoll des Organisationsübergreifenden Koordinationsausschusses zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (6) (7), das ursprünglich auf Informationen des Instituts für In-vitro- Forschung (Institute for In Vitro Sciences, IIVS) und auf dem INVITTOX-Protokoll 124 (8) beruht. Letzteres Protokoll wurde für die von der Europäischen Gemeinschaft finanzierte Prävalidierungsstudie aus den Jahren 1997-1998 herangezogen. Beide Protokolle beruhen auf der BCOP-Prüfmethodik, wie sie von Gautheron et al. 9 erstmals beschrieben wurde.
Die BCOP-Prüfmethode kann zur Identifizierung von schwer augenschädigenden Chemikalien gemäß UN-GHS-Definition, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, angewendet werden (4). Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist die BCOP-Prüfmethode für diesen Zweck eine allgemeine Genauigkeit von 79 % (150/191), eine Falsch-Positiv-Rate von 25 % (32/126) und eine Falsch-Negativ-Rate von 14 % (9/65) auf (3) (siehe Anlage 2, Tabelle 1). Werden Prüfchemikalien einer bestimmten Chemikalienklasse (Alkohole, Ketone) oder mit bestimmten physikalischen Eigenschaften (Feststoffe) aus der Datenbank ausgeschlossen, so liegt die allgemeine Genauigkeit der BCOP-Prüfmethode gemessen am UN-GHS-Klassifizierungssystem bei 85 % (111/131), die Falsch-Positiv-Rate bei 20 % (16/81) und die Falsch-Negativ-Rate bei 8 % (4/50) (3). Potenzielle Mängel der BCOP-Prüfmethode bei der Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) gründen auf hohen Falsch-Positiv-Raten für Alkohole und Ketone und hohen Falsch-Negativ-Raten für Feststoffe, die in den Validierungsdaten beobachtet wurden (1)(2)(3). Da jedoch nicht für alle Alkohole und Ketone durch die BCOP-Prüfmethode zu hohe Werte vorhergesagt werden und einige korrekt als Stoffe der UN-GHS-Kategorie 1 eingestuft werden, werden diese beiden organischen funktionellen Gruppen nicht als Stoffe außerhalb des Anwendungsbereichs dieser Prüfmethode betrachtet. Es muss also bei der Anwendung dieser Prüfmethode entschieden werden, ob eine gegebenenfalls zu hohe Vorhersage für einen Alkohol oder ein Keton vertretbar ist oder ob weitere Tests mit einem evidenzbasierten Bewertungsansatz durchgeführt werden sollten. Bezüglich der Falsch-Negativ-Raten für Feststoffe ist zu beachten, dass Feststoffe beim In-vivo-Draize-Augenreizungstest zu variablen und extremen Expositionsbedingungen führen können, aus denen sich möglicherweise irrelevante Vorhersagen über das tatsächliche Reizungspotenzial ableiten (10). Es ist ferner zu beachten, dass keine der Falsch-Negativ-Raten, die in den ICCVAM-Validierungsdaten (2)(3) im Zusammenhang mit der Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) festgestellt wurden, im Ergebnis zu einem IVIS ≤ 3 führten, dem Kriterium zur Identifizierung einer Prüfchemikalie als Stoff der UN-GHS-Klasse "Keine Einstufung" . Des Weiteren gelten Falsch-Negativ-Raten der BCOP-Prüfmethode in diesem Zusammenhang nicht als kritisch, da alle Prüfchemikalien mit einem Wert von 3 < IVIS ≤ 55 nachfolgend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequentiellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz, anhand weiterer, angemessen validierter In-vitro-Tests oder als letzte Option an Kaninchen getestet werden würden. Angesichts der Tatsache, dass einige feste Chemikalien anhand der BCOP-Prüfmethode korrekterweise als Stoffe der UN-GHS-Kategorie 1 vorhergesagt werden, wird auch nicht davon ausgegangen, dass diese physikalischen Eigenschaften außerhalb des Anwendungsbereichs dieser Prüfmethode liegen. Prüfer können diese Prüfmethode für alle Arten von Chemikalien einsetzen und ein IVIS-Wert > 55 sollte als Indikator für eine schwer augenschädigende Wirkung gelten, nach der eine Einstufung in UN-GHS-Kategorie 1 ohne weitere Tests vorzunehmen ist. Wie bereits erwähnt, sollten Positivergebnisse bei Verwendung von Alkoholen oder Ketonen angesichts des Risikos der Vorhersage zu hoher Werte jedoch mit einer gewissen Zurückhaltung interpretiert werden.
Die BCOP-Prüfmethode kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN-GHS-Klassifizierungssystem erfordern (4). Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist die BCOP-Prüfmethode für diesen Zweck eine allgemeine Genauigkeit von 69 % (135/196), eine Falsch-Positiv-Rate von 69 % (61/89) und eine Falsch-Negativ-Rate von 0 % (0/107) auf (3) (siehe Anlage 2, Tabelle 2). Die erhaltene Falsch-Positiv-Rate (In-vivo-Chemikalien der UN-GHS-Klasse "Keine Einstufung" mit einem IVIS-Wert > 3, siehe Nummer 47) ist zwar recht hoch, gilt in diesem Zusammenhang jedoch nicht als kritisch, da alle Prüfchemikalien mit einem Wert von 3 < IVIS ≤ 55 nachfolgend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequentiellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz, anhand weiterer, angemessen validierter In-vitro-Tests oder als letzte Option an Kaninchen getestet werden würden. Die BCOP-Prüfmethode weist keine nennenswerten Mängel für die Untersuchung von Alkoholen, Ketonen und Feststoffen auf, sofern das Ziel die Identifizierung von Chemikalien ist, die keine Einstufung als augenreizend oder schwer augenschädigend (UN-GHS-Klasse "Keine Einstufung") erfordern (3). Prüfer können diese Prüfmethode jedoch für alle Arten von Chemikalien einsetzen, und ein Negativergebnis (IVIS ≤ 3) sollte als Indikator dafür gelten, dass keine Einstufung vorzunehmen ist (UN-GHS-Klasse "Keine Einstufung"). Da mit der BCOP-Prüfmethode nur 31 % der Chemikalien richtig identifiziert werden können, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern, sollte diese Prüfmethode nicht die erste Wahl sein, wenn als erster Schritt ein Bottom-up-Ansatz verwendet werden soll (5), sofern andere validierte und akzeptierte In-vitro-Verfahren mit ähnlich hoher Empfindlichkeit, jedoch einer höheren Spezifität zur Verfügung stehen.
Die BCOP-Validierungsdatenbank enthielt insgesamt 113 Stoffe und 100 Gemische (2)(3). Die BCOP-Prüfmethode gilt demnach als für die Prüfung von Stoffen und Gemischen gleichermaßen anwendbar.
Aufgrund der beträchtlichen Anzahl von Chemikalien der UN-GHS-Kategorie 1, die zu niedrig in die UN-GHS-Kategorien 2, 2A oder 2B eingestuft sind, und von Chemikalien, die keine Einstufung erfordern, jedoch zu hoch in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, wird die BCOP-Prüfmethode nicht für die Identifizierung von Prüfchemikalien empfohlen, die als augenreizend (d. h. UN-GHS-Kategorie 2 oder Kategorie 2A) oder leicht augenreizend (UN-GHS-Kategorie 2B) eingestuft werden sollten (2)(3). Diesbezüglich können weitere Tests mit einer anderen geeigneten Methode erforderlich sein.
Bei allen Verfahren, die Rinderaugen und Rinderhornhäute involvieren, sollten die geltenden Regeln und Verfahrensvorschriften der Prüfanstalt für den Umgang mit Tiermaterial (u. a. Gewebe und Gewebeflüssigkeiten) eingehalten werden. Universelle Laborregeln sollten beachtet werden (11).
Bindehaut- und Irisverletzungen werden bei der BCOP-Prüfmethode zwar außer Acht gelassen, doch werden Auswirkungen auf die Hornhaut berücksichtigt, die wesentliche Einflussfaktoren für die In-vivo-Klassifizierung nach dem UN-GHS-Klassifizierungssystem bilden. Die Reversibilität von Hornhautläsionen lässt sich per se mit der BCOP-Prüfmethode nicht beurteilen. Doch wurde ausgehend von Kaninchenaugenstudien vorgeschlagen, eine Bewertung der anfänglichen Tiefe der Hornhautverletzung heranzuziehen, um einige Arten irreversibler Wirkungen zu identifizieren (12). Allerdings sind weitere wissenschaftliche Erkenntnisse erforderlich, um zu verstehen, wie irreversible Wirkungen auftreten, die nicht mit einer anfänglichen starken Verletzung im Zusammenhang stehen. Schließlich sei auch erwähnt, dass das Potenzial für eine mit der Augenexposition verbundene systemische Toxizität nach der BCOP-Methode nicht bewertet werden kann.
Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Beispielsweise können histopathologische Befunde potenziell nützlich sein, wenn eine umfassendere Charakterisierung der Hornhautschädigung erforderlich ist. Wie im OECD-Leitfaden Nr. 160 (13) umrissen, sollten Anwender Hornhäute aufbewahren und histopathologische Proben zubereiten, die zum Aufbau einer Datenbank und zur Entwicklung von Entscheidungskriterien herangezogen werden können, mit denen sich die Genauigkeit dieser Prüfmethode weiter verbessern lässt.
Laboratorien, die diese Prüfmethode zum ersten Mal anwenden, sollten die in Anlage 3 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung der BCOP-Prüfmethode nachzuweisen, bevor sie BCOP-Testdaten für Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen.
Testprinzip
Die BCOP-Prüfmethode ist ein organtypisches Modell, das die normalen physiologischen und biochemikalischen Funktionen von Rinderhornhäuten in vitro kurzfristig aufrechterhält. Bei dieser Methode werden durch die Prüfchemikalie hervorgerufene Schäden bewertet, indem Veränderungen von Trübung und Durchlässigkeit der Hornhaut mithilfe eines Opazimeters bzw. eines VIS-Spektrofotometers quantitativ gemessen werden. Die beiden Messwerte dienen der Berechnung eines In-vitro-Reizwertes (IVIS), der seinerseits herangezogen wird, um zwecks Prädiktion des Augenreizpotenzials einer Prüfchemikalie am lebenden Objekt (in vivo) eine Zuordnung zu einer Gefahrenkategorie aufgrund des In-vitro-Reizungspotenzials des Stoffes vorzunehmen (siehe Entscheidungskriterien unter Nummer 48).
Für die BCOP-Prüfmethode werden isolierte Hornhäute der Augen frisch geschlachteter Rinder verwendet. Die Hornhauttrübung wird quantitativ bestimmt als Menge der die Hornhaut durchdringenden Lichtstrahlung. Die Durchlässigkeit der Hornhaut wird quantitativ bestimmt als die Menge an Natrium-Fluorescein, die die gesamte Dicke der Hornhaut durchdringt und im Medium in der Hinterkammer nachgewiesen wird. Die Applikation der Prüfchemikalien auf die Epitheloberfläche der Hornhaut erfolgt durch Zugabe der Stoffe in die Vorderkammer des Hornhauthalters. Anlage 4 enthält eine Beschreibung sowie ein Schaubild einer Halterung, wie sie für die BCOP-Prüfmethode verwendet wird. Hornhauthalter sind im Handel erhältlich oder können als Bausatz bezogen werden.
Bezugsquelle und Alter der Rinderaugen und Auswahl der Tiere
Schlachtrinder werden in der Regel für den menschlichen Verzehr oder für andere gewerbliche Zwecke getötet. Nur gesunde und für die menschliche Nahrungskette geeignet befundene Tiere kommen als Bezugsquelle für Augenhornhäute für den BCOP-Test in Frage. Da Rinder je nach Rasse, Alter und Geschlecht eine große Gewichtsspanne aufweisen, gibt es keine Gewichtsempfehlung für die Tiere zum Zeitpunkt der Schlachtung.
Augenhornhäute von Tieren verschiedener Altersklassen sind mitunter unterschiedlich groß. Hornhäute mit einem horizontalen Durchmesser > 30,5 mm und einer zentralen Hornhautdicke (CCT) mit Werten ≥ 1 100 µm stammen in der Regel von über acht Jahre alten Rindern, während Hornhäute mit einem horizontalen Durchmesser < 28,5 mm und CCT-Werten < 900 µm im Allgemeinen von weniger als fünf Jahre alten Tieren stammen (14). Deshalb werden Augen von über 60 Monate alten Tieren in der Regel nicht verwendet. Traditionsgemäß werden keine Augen von weniger als 12 Monate alten Rindern verwendet, da die Augen dieser Tiere noch nicht ausgewachsen sind und Hornhautdicke sowie Hornhautdurchmesser wesentlich kleiner sind als bei ausgewachsenen Rindern. Hornhäute von Jungtieren (d. h. Tiere im Alter von sechs bis 12 Monaten) sind aufgrund ihrer Vorteile jedoch zulässig: Sie sind beispielsweise leichter erhältlich, die Altersspanne ist geringer und das Laborpersonal ist in geringerem Maße BSE-gefährdet (15). Da eine weitere Evaluierung des Einflusses von Hornhautgröße oder Hornhautdicke auf die Wirkung verätzender oder reizender Stoffe nützlich wäre, sollten Anwender das geschätzte Alter und/oder das Gewicht der Tiere, von denen die für eine Studie verwendeten Hornhäute stammen, mitteilen.
Gewinnung und Beförderung der Augen zum Labor
Die Augen werden im Schlachthof gewonnen. Um jede mechanische und sonstige Schädigung der Augen auf ein Minimum zu beschränken, sollten die Augen nach der Tötung des Tieres so bald wie möglich ausgeschält und unmittelbar danach sowie während der Beförderung gekühlt werden. Damit die Augen nicht mit potenziellen Reizstoffen in Berührung kommen, sollte das Schlachthofpersonal zum Abspülen des Schädels keine Detergenzien verwenden.
Die Augen sollten in einem angemessen großen Behältnis vollständig in HBSS eingelegt und so zum Labor befördert werden, dass sich ihr Zustand möglichst nicht verschlechtert und/oder es möglichst nicht zu einer bakteriellen Kontamination kommt. Da die Augen während des Schlachtprozesses gewonnen werden, könnten sie mit Blut und anderen biologischen Stoffen wie Bakterien und sonstigen Mikroorganismen in Berührung kommen. Deshalb muss unbedingt sichergestellt werden, dass das Kontaminationsrisiko auf ein Mindestmaß begrenzt wird (z.B. indem das für die Beförderung der Augen verwendete Behältnis auf Nasseis gelagert und die zur Lagerung der Augen während der Beförderung verwendete HBSS-Lösung mit Antibiotika (z.B. 100 IU/ml Penizillin und 100 µg/ml Streptomycin) angereichert wird).
Der Zeitabstand zwischen der Gewinnung der Augen und der Verwendung der Hornhäute im BCOP-Test sollte möglichst gering sein (im Idealfall sollten die Hornhäute am selben Tag gewonnen und verwendet werden) und die Testergebnisse nachweislich nicht kompromittieren. Letztere basieren auf den Auswahlkriterien für die Augen sowie auf den Reaktionen der Positiv- und Negativkontrollen. Alle für die Prüfung verwendeten Augen sollten aus einer an ein und demselben Tag gewonnenen Partie Augen stammen.
Auswahlkriterien für Augen, die im BCOP-Test eingesetzt werden
Unmittelbar nach ihrer Ankunft im Labor werden die Augen sorgfältig auf Mängel wie unter anderem verstärkte Trübung, Kratzer und Neovaskularisation untersucht. Nur Hornhäute von Augen, die keine derartigen Mängel aufweisen, dürfen verwendet werden.
Die Qualität jeder Hornhaut wird auch in späteren Testphasen geprüft. Hornhäute, die nach einer ersten einstündigen Äquilibrierung mehr als sieben Trübungseinheiten oder einen vergleichbaren Wert für das verwendete Opazimeter und die verwendeten Hornhauthalterungen aufweisen (ANMERKUNG: der Opazimeter sollte nach den Trübungsnormen kalibriert werden, die auch zur Festlegung der Trübungseinheiten verwendet werden; siehe Anlage 4), sind zu verwerfen.
Jede Behandlungsgruppe (Prüfchemikalie sowie gleichzeitige Negativ- und Positivkontrollen) umfasst mindestens drei Augen. Für die Negativkontrollen des BCOP-Tests sollten drei Hornhäute verwendet werden. Da die Hornhäute operativ vom Augapfel entfernt und in die Hornhautkammern eingespannt werden, kann es infolge dieser Handgriffe bei einzelnen Hornhäuten zu veränderten Trübungs- und Durchlässigkeitswerten kommen (auch bei der Negativkontrolle). Außerdem werden die Trübungs- und Durchlässigkeitswerte von Hornhäuten der Negativkontrolle dazu verwendet, die Trübungs- und Durchlässigkeitswerte der mit der Prüfchemikalie behandelten Hornhäute und der Hornhäute der Positivkontrolle bei den IVIS-Berechnungen zu korrigieren.
Verfahren
Vorbereitung der Augen
Unbeschädigte Hornhäute werden seziert, wobei ein 2 bis 3 mm breiter Sklera-Rand belassen wird, um das anschließende Manipulieren zu erleichtern; dabei ist darauf zu achten, dass die Epithel- und Endothelzellschicht der Hornhaut nicht beschädigt wird. Die isolierten Hornhäute werden in speziell entwickelte Hornhauthalter eingespannt, die aus Vorder- und Hinterkammern bestehen, welche die Schnittstellen zur Epithel- bzw. Endothelseite der Hornhäute bilden. Beide Kammern (Hinterkammer zuerst) werden bis zum Überlaufen mit vorgewärmtem phenolrotfreiem EMEM-Medium gefüllt, wobei sicherzustellen ist, dass sich keine Luftblasen bilden. Das Gerät wird sodann bei 32 ± 11°C für mindestens eine Stunde äquilibriert, um die Hornhäute mit dem Medium zu äquilibrieren und so weit wie möglich eine normale Stoffwechseltätigkeit zu gewährleisten (die ungefähre Temperatur der Hornhautoberfläche beträgt in vivo 321 °C).
Nach der Äquilibrierung wird frisches vorgewärmtes, phenolrotfreies EMEM-Medium in beide Kammern gegeben, und für jede Hornhaut werden Referenztrübungswerte abgelesen. Hornhäute, die makroskopische Gewebeschäden (z.B. Kratzer, Pigmentierung, Neovaskularisation) oder über sieben Trübungseinheiten bzw. einen für das verwendete Opazimeter und die verwendeten Hornhauthalterungen vergleichbaren Wert aufweisen, werden verworfen. Mindestens drei Hornhäute werden für die Negativ-(oder Lösungsmittel-)Kontrolle ausgewählt. Die restlichen Hornhäute werden in Behandlungs- und Positivkontrollgruppen aufgeteilt.
Da die Wärmekapazität von Wasser höher ist als die Wärmekapazität von Luft, bietet Wasser stabilere Temperaturbedingungen für die Inkubation. Deshalb wird ein Wasserbad empfohlen, um Hornhauthalter samt Inhalt auf einer Temperatur von 32 ± 11 °C zu halten. Es können jedoch auch Luftinkubatoren verwendet werden, sofern alle erforderlichen Vorkehrungen getroffen wurden, um die Temperatur stabil zu halten (z.B. durch Vorwärmen der Halterungen und Medien).
Applikation der Prüfchemikalie
Es werden zwei unterschiedliche Behandlungsprotokolle verwendet - eines für Flüssigkeiten und Tenside (Feststoffe oder Flüssigkeiten) und eines für nichttensidische Feststoffe.
Flüssigkeiten werden unverdünnt, halbfeste Stoffe, Cremes und Wachse werden in der Regel als Flüssigkeiten getestet. Unverdünnte Tenside werden in einer Konzentration von 10 % w/v in 0,9 % Natriumchloridlösung, destilliertem Wasser oder einem anderen Lösungsmittel, welches das Testsystem nachweislich nicht beeinträchtigt, getestet. Sofern alternative Lösungskonzentrationen verwendet werden, ist dies angemessen zu begründen. Tensidhaltige Gemische können unverdünnt getestet oder zunächst auf eine geeignete Konzentration entsprechend der jeweiligen In-vivo-Exposition verdünnt werden. Die getestete Konzentration ist angemessen zu begründen. Die Hornhäute werden den Flüssigkeiten und Tensiden für die Dauer von zehn Minuten ausgesetzt. Bei anderen Expositionszeiten sollten diese wissenschaftlich begründet werden. Vgl. Anlage 1 bezüglich einer Definition für "Tensid" und "tensidhaltiges Gemisch" .
Nichttensidische Feststoffe werden in der Regel als Lösungen oder Suspensionen in einer Konzentration von 20 % w/v in 0,9 % Natriumchloridlösung, destilliertem Wasser oder einem anderem Lösungsmittel, welches das Testsystem nachweislich nicht beeinträchtigt, getestet. Unter bestimmten Bedingungen und sofern wissenschaftlich begründet, können Feststoffe auch unverdünnt durch direkte Applikation auf die Hornhautoberfläche getestet werden, wobei die offene Methode (open chamber method, siehe Nummer 32) angewandt wird. Die Hornhäute werden den Feststoffen für die Dauer von vier Stunden ausgesetzt; soweit wissenschaftlich fundiert können jedoch auch, wie bei Flüssigkeiten und Tensiden, alternative Expositionszeiten verwendet werden.
Je nach den physikalischen und chemischen Eigenschaften der Prüfchemikalie (z.B. fest, flüssig, zähflüssig oder nicht zähflüssig) können unterschiedliche Behandlungsmethoden angewandt werden. Der kritische Faktor besteht darin sicherzustellen, dass die Prüfchemikalie die Epitheloberfläche angemessen bedeckt und während der Spülungen sachgemäß entfernt wird. Die geschlossene Methode (closed chamber method) wird in der Regel für nicht zähflüssige bis leicht zähflüssige Prüfchemikalien eingesetzt, während die offene Methode eher für halbzähflüssige und zähflüssige Prüfchemikalien sowie für unverdünnte Feststoffe verwendet wird.
Bei der geschlossenen Methode wird über die Dosieröffnungen auf der Oberseite der Kammer so viel Prüfchemikalie (750 µl) in die Vorderkammer gegeben, dass die Epithelseite der Hornhaut bedeckt ist; die ¦ffnungen werden anschließend für die gesamte Expositionsdauer mit den Kammerverschlüssen abgedichtet. Es muss unbedingt sichergestellt werden, dass alle Hornhäute der Prüfchemikalie für eine angemessene Dauer ausgesetzt werden.
Bei der offenen Methode werden vor der Behandlung Scheibenverschlussring und Glasscheibe von der Vorderkammer entfernt. Die Kontroll- oder die Prüfchemikalie (750 µl bzw. genÌgend Prüfchemikalie, um die Hornhaut komplett zu bedecken) wird mit einer Mikropipette direkt auf die Epitheloberfläche der Hornhaut appliziert. Erweist sich das Pipettieren einer Prüfchemikalie als schwierig, so kann diese zur leichteren Dosierung mittels Druckausgleich in eine Direktverdrängungspipette geladen werden. Die Spitze der Direktverdrängungspipette wird in die Dosierspitze der Spritze eingeführt, damit das Material unter Druck in die Spitze der Verdrängungspipette geladen werden kann. Lässt man den Bedienknopf langsam zurückgleiten, fährt der Kolben der Pipette nach oben. Treten in der Pipettenspitze Luftbläschen auf, wird die Prüfchemikalie verworfen (ausgestoßen) und der Prozess wird so lange wiederholt, bis die Spitze ohne Luftblasen gefüllt ist. Erforderlichenfalls kann eine normale Spritze (ohne Nadel) verwendet werden, denn sie gestattet das Abmessen einer akkuraten Menge Prüfchemikalie und erleichtert die Applikation auf die Epitheloberfläche der Hornhaut. Nach der Dosierung wird die Glasscheibe wieder auf die Vorderkammer gesetzt, um das System wieder zu schließen.
Inkubation nach der Exposition
Nach Ablauf der Expositionszeit werden Prüfchemikalie, Negativkontrolle oder Positivkontrolle aus der Vorderkammer entfernt, und das Epithel wird mindestens drei Mal (oder bis keine Prüfchemikalie mehr sichtbar ist) mit (phenolrothaltigem) EMEM-Medium gewaschen. Phenolrot wird zum Abspülen verwendet, da sich aufgrund der Farbveränderung bei Phenolrot die Wirksamkeit des Abspülens saurer oder alkalischer Prüfchemikalien bestimmen lässt. Die Hornhäute werden mehr als drei Mal gewaschen, wenn die Farbveränderung (ins Gelbe oder Lila) anhält oder wenn die Prüfchemikalie nach wie vor sichtbar ist. Sobald das Medium frei von der Prüfchemikalie ist, werden die Hornhäute ein letztes Mal mit EMEM-Medium (ohne Phenolrot) abgespült. Das EMEM-Medium (ohne Zusatz von Phenolrot) wird als letzte Spülung verwendet, um sicherzustellen, dass der Phenolrot-Farbstoff vor der Trübungsmessung aus der Vorderkammer entfernt wurde. Die Vorderkammer wird sodann wieder mit nicht phenolrothaltigem frischem EMEM-Medium aufgefüllt.
Bei Flüssigkeiten oder Tensiden werden die Hornhäute nach dem Abspülen für weitere zwei Stunden bei 32 ± 11 °C inkubiert. Unter bestimmten Umständen könnte sich nach der Exposition eine längere Inkubationszeit als zweckdienlich erweisen; dies sollte auf Fallbasis geprüft werden. Mit Feststoffen behandelte Hornhäute werden nach Ablauf der vierstündigen Exposition gründlich abgespült; eine längere Inkubation ist nicht erforderlich.
Nach Ablauf der auf die Exposition folgenden Inkubation (Flüssigkeiten und Tenside) bzw. nach Ablauf der vierstündigen Exposition (nichttensidische Feststoffe) werden Trübung und Durchlässigkeit der einzelnen Hornhäute aufgezeichnet. Außerdem wird jede Hornhaut visuell geprüft, und relevante Befunde (z.B. Gewebeabschälung, Reste der Prüfchemikalie, uneinheitliche Trübungsmuster) werden aufgezeichnet. Diese Daten könnten insofern wichtig sein, als sie Abweichungen der Opazimeter-Messwerte bestätigen könnten.
Kontrollchemikalien
Jeder Versuch beinhaltet gleichzeitige Negativ- oder Lösungsmittel-/Vehikelkontrollen und Positivkontrollen.
Für Tests von Flüssigstoffen zu 100 % sieht der BCOP-Test eine gleichzeitige Negativkontrolle (z.B. 0,9 % Natriumchloridlösung oder destilliertes Wasser) vor, damit unspezifische Veränderungen im Testsystem festgestellt werden können und ein Referenzwert für die Test-Endpunkte ermittelt werden kann. Auf diese Weise wird auch sichergestellt, dass die Testbedingungen nicht zu einer unerwünschten Reizwirkung führen.
Für Tests von verdünnten Flüssigkeiten, Tensiden oder Feststoffen sieht der BCOP-Test eine Gruppe gleichzeitiger Lösungsmittel-/Vehikelkontrollen vor, damit unspezifische Veränderungen im Testsystem festgestellt werden können und ein Referenzwert für die Test-Endpunkte ermittelt werden kann. Es sind nur Lösungsmittel/Vehikel zulässig, die das Testsystem nachweislich nicht beeinträchtigen.
Jeder Versuch beinhaltet als gleichzeitige Positivkontrolle eine bekannte augenreizende Chemikalie, damit die Integrität des Testsystems und seine korrekte Durchführung überprüft werden können. Um sicherzustellen, dass im Zeitverlauf auftretende Wirkungsschwankungen bei der Positivkontrolle bewertet werden können, sollte die Reaktion jedoch nicht zu heftig sein.
Beispiele für Positivkontrollen für flüssige Prüfchemikalien sind 100 %iges Ethanol oder 100 %iges Dimethylformamid. Für feste Prüfchemikalien käme 20 %iges (Gewichtsprozent) Imidazol in 0,9 % Natriumchloridlösung als Positivkontrolle in Frage.
Referenzchemikalien sind nützlich für die Evaluierung des Augenreizpotenzials unbekannter Chemikalien einer bestimmten Chemikalien- oder Produktklasse oder zur Evaluierung des relativen Reizpotenzials eines Augenreizstoffes innerhalb einer spezifischen Spanne von Reizwirkungen.
Gemessene Endpunkte
Die Trübung wird durch Messung der durch die Hornhaut durchgehenden Lichtstrahlung bestimmt. Die quantitative Messung der Hornhauttrübung erfolgt mithilfe eines Opazimeters, eines Messgeräts, bei dem die Trübungswerte kontinuierlich gemessen werden.
Die Durchlässigkeit wird durch Messung der Menge Fluorescein-Natrium bestimmt, die alle Zellschichten der Hornhaut (d. h. von der Epithelzellschicht auf der äußeren Hornhautoberfläche bis zur Endothelzellschicht auf der inneren Hornhautfläche) passiert. Die Vorderkammer des Hornhauthalters, die die Verbindung zur Epithelseite der Hornhaut bildet, wird mit 1 ml Fluorescein-Natrium-Lösung (4 oder 5 mg/ml bei Flüssigkeiten und Tensiden bzw. nichttensidischen Feststoffen) aufgefüllt, während die Hinterkammer, die die Verbindung zur Endothelseite der Hornhaut bildet, mit frischem EMEM-Medium gefüllt wird. Die Halterung wird sodann in horizontaler Position für 90 ± 5 Minuten bei 32 ± 11 °C inkubiert. Die Menge an Fluorescein-Natrium, die in die Hinterkammer eindringt, wird mithilfe eines UV/VIS-Spektrofotometers gemessen. Bei 490 nm ausgewertete spektrofotometrische Messungen werden als Werte für optische Dichte (OD490) oder Absorbanzwerte aufgezeichnet, die kontinuierlich gemessen werden. Zur Bestimmung der Fluorescein-Durchlässigkeit werden OD490-Werte herangezogen, die mithilfe eines VIS-Spektrofotometers bei einer Standardpfadlänge von 1 cm ermittelt wurden.
Alternativ kann ein Plattenleser für 96-Mulden-Mikrotiterplatten verwendet werden, sofern i) der lineare Messbereich des Plattenlesers für die Bestimmung der Fluorescein- OD490-Werte festgelegt werden kann und ii) in der 96-Mulden-Mikrotiterplatte Fluorescein-Proben in der richtigen Menge verwendet werden, um OD490-Werte zu ergeben, die der Standardpfadlänge von 1 cm entsprechen (dies könnte vollständig gefüllte Mulden [in der Regel 360 µl] erfordern).
Daten und Berichterstattung
Datenauswertung
Sobald die Trübungs- und die mittleren Durchlässigkeits-(OD490-)Werte um die Hintergrundtrübungswerte und die OD490-Durchlässigkeitswerte der Negativkontrolle korrigiert wurden, werden die mittleren Trübungs- und OD490-Durchlässigkeitswerte für jede Behandlungsgruppe in einer empirisch abgeleiteten Formel kombiniert, um für jede Behandlungsgruppe einen In-vitro-Reizwert (IVIS) zu berechnen:
IVIS = mittlerer Trübungswert + (15 x mittlerer OD490-Durchlässigkeitswert)
Nach Sina et al. (16) wurde diese Formel aus Labor- und Interlaborstudien abgeleitet. Die für eine Serie von 36 Verbindungen in einer Multilaborstudie generierten Daten wurden einer multivariaten Analyse unterzogen, um die Best-fit-Gleichung zwischen In-vivo- und In-vitro-Daten zu ermitteln. Diese Analyse wurde von Wissenschaftlern zweier separater Unternehmen durchgeführt, die zu quasi identischen Gleichungsergebnissen gelangten.
Die Trübungs- und Durchlässigkeitswerte sollten auch unabhängig voneinander bestimmt werden, um feststellen zu können, ob eine Prüfchemikalie für nur einen der beiden Endpunkte (siehe Entscheidungskriterien) eine augenverätzende oder stark augenreizende Wirkung verursachte.
Entscheidungskriterien
Die IVIS-Schwellenwerte zur Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) und Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern (UN-GHS-Klasse "Keine Einstufung") sind nachfolgend aufgeführt:
| IVIS | UN-GHS |
| ≤ 3 | Keine Einstufung |
| > 3; ≤ 55 | Keine Vorhersage möglich |
| > 55 | Kategorie 1 |
Studienakzeptanzkriterien
Ein Test gilt als akzeptabel, wenn die Positivkontrolle einen IVIS-Wert innerhalb von zwei Standardabweichungen des geltenden historischen Mittelwertes ergibt, der mindestens alle drei Monate bzw. immer dann aktualisiert werden muss, wenn Laboratorien, die nur selten Testungen vornehmen (d. h. weniger als einmal im Monat), einen akzeptablen Test durchführen. Die Negativ- oder Lösungsmittel-/Vehikelkontrollen sollten Trübungs- und Durchlässigkeitswerte ergeben, die geringer sind als die Obergrenzen, die für Hintergrundtrübungs- und Durchlässigkeitswerte für Rinderhornhäute vorgegeben sind, welche mit der jeweiligen Negativ- bzw. Lösungsmittel-/Vehikelkontrolle behandelt wurden. Kann eine eindeutige Klassifizierung vorgenommen werden, so ist ein einziger Testlauf, der aus mindestens drei Hornhäuten besteht, ausreichend. Kommt es jedoch im ersten Testlauf zu grenzwertigen Ergebnissen, sollte ein zweiter Testlauf in Erwägung gezogen werden (ist jedoch nicht unbedingt erforderlich) und darüber hinaus ein dritter, sollte es beim ersten und zweiten Testlauf zu abweichenden mittleren IVIS-Ergebnissen kommen. In diesem Zusammenhang gilt ein Ergebnis im ersten Testlauf als grenzwertig, wenn die Vorhersagen aus den 3 Hornhäuten nicht übereinstimmend waren, und zwar wie folgt:
Prüfbericht
Der Prüfbericht sollte die folgenden Informationen umfassen, soweit sie für die Studie relevant sind:
Prüfchemikalien und Kontrollchemikalien
Informationen zu Auftraggeber und Prüfanstalt
Testbedingungen
Testakzeptanzkriterien
Gewinnung und Vorbereitung der Augen
Testverfahren
Ergebnisse
Erörterung der Ergebnisse
Schlussfolgerung
Literaturhinweise
(1) ICCVAM (2006). Test Method Evaluation Report - In Vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm.
(2) ICCVAM (2010). ICCVAM Test Method Evaluation Report: Current Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products. NIH-Veröffentlichung Nr. 10-7553: Research Triangle Park, NC: National Institute of Environmental Health Sciences. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm.
(3) OECD (2013). Streamlined Summary Document supporting the Test Guideline 437 for eye irritation/corrosion. Series on Testing and Assessment, No. 189, OECD, Paris.
(4) UN (2011). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), ST/SG/AC.10/30 Rev 4, New York und Genf: United Nations. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev04/04files_e.html.
(5) Scott, L., Eskes, C., Hoffman, S., Adriaens, E., Alepee, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Hamernik, K., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., Mcnamee, P., Osborn, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielmann, H., Stokes, W., Trouba, K., Vassallo, M., Van den Berghe, C., Van Goethem, F., Vinardell, P., Zuang, V (2010), A proposed eye irritation testing strategy to reduce and replace in vivo studies using Bottom-Up and Top-Down approaches. Toxicol. in Vitro 24:1-9.
(6) ICCVAM (2006). ICCVAM Recommended BCOP Test Method Protocol. In: ICCVAM Test Method Evaluation Report - in vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm.
(7) ICCVAM (2010). ICCVAM Recommended BCOP Test Method Protocol. In: ICCVAM Test Method Evaluation Report - Current Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 10-7553A. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm.
(8) INVITTOX (1999). Protokoll Nr. 124: Bovine Corneal Opacity and Permeability Assay - SOP of Microbiological Associates Ltd. Ispra, Italien: European Centre for the Validation of Alternative Methods (ECVAM).
(9) Gautheron, P., Dukic, M., Alix, D. and Sina, J.F. (1992). Bovine corneal opacity and permeability test: An in vitro assay of ocular irritancy. Fundam. Appl. Toxicol. 18:442-449.
(10) Prinsen, M.K. (2006). The Draize Eye Test and in vitro alternatives; a left-handed marriage? Toxicol. in Vitro 20:78-81.
(11) Siegel, J.D., Rhinehart, E., Jackson, M., Chiarello, L. und das Healthcare Infection Control Practices Advisory Committee (2007), Guideline for Isolation Precautions: Preventing Transmission of Infectious Agents in Healthcare Settings. Verfügbar unter: [http://www.cdc.gov/ncidod/dhqp/pdf].
(12) Maurer, J.K., Parker, R.D. und Jester, J.V. (2002). Extent of corneal injury as the mechanistic basis for ocular irritation: key findings and recommendations for the development of alternative assays. Reg. Tox. Pharmacol., 36:106-117.
(13) OECD (2011). Guidance Document on The Bovine Corneal Opacity and Permeability (BCOP) and Isolated Chicken Eye (ICE) Test Methods: Collection of Tissues for Histological Evaluation and Collection of Data on Non-severe Irritants. Series on Testing and Assessment, No. 160. Angenommen am 25. Oktober2011. Paris: Organisation for Economic Co-operation and Development.
(14) Doughty, M.J., Petrou, S. and Macmillan, H. (1995). Anatomy and morphology of the cornea of bovine eyes from a slaughterhouse. Can. J. Zool. 73:2159-2165.
(15) Collee, J. and Bradley, R. (1997). BSE: A decade on - Part I. The Lancet 349: 636-641.
(16) Sina, J.F., Galer, D.M., Sussman, R.S., Gautheron, P.D., Sargent, E.V., Leong, B., Shah, P.V., Curren, R.D., and Miller, K. (1995). A collaborative evaluation of seven alternatives to the Draize eye irritation test using pharmaceutical intermediates. Fundam. Appl. Toxicol. 26:20-31.
(17) Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung.
(18) ICCVAM (2006). Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Bovine Corneal Opacity and Permeability Test Method. NIH-Veröffentlichung Nr. 06-4512. Research Triangle Park: National Toxicology Program. Verfügbar unter: [http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_brd_bcop.htm].
(19) OECD (1998). Series on Good Laboratory Practice and Compliance Monitoring. No. 1: OECD Principles on Good Laboratory Practice (revised in 1997).
Verfügbar unter: http://www.oecd.org/document/63/0,3343,en_2649_34381_2346175_1_1_1_1,00.html
_____
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
.
| Definitionen | Anlage 1 |
Augenreizung: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Austauschbar mit "Reversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 2" (4).
Bottom-Up-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie keine Einstufung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zuerst Chemikalien, die keine Einstufung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden.
Chemikalie: Stoff oder Gemisch.
Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Prüfchemikalie entscheiden zu können und diese Entscheidung zu untermauern.
Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode.
Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen.
Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (4).
Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode.
Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann.
Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (4).
Hornhaut: Der Iris und Pupille überdeckende transparente vordere Teil des Augapfels, über den Licht ins Augeninnere übertragen wird.
Hornhautdurchlässigkeit: Quantitativer Messwert für die Schädigung der Epithelzellschicht der Hornhaut, ermittelt durch Bestimmung der Menge an Fluorescein-Natrium, die alle Zellschichten der Hornhaut durchdringt.
Hornhauttrübung: Messwert für die Undurchsichtigkeit der Hornhaut nach Applikation einer Prüfchemikalie. Eine verstärkte Hornhauttrübung ist ein Indikator für die Schädigung der Hornhaut. Die Trübung kann subjektiv (Draize-Kaninchenaugentest) oder objektiv (mithilfe eines Messgeräts, z.B. eines Opazimeters) bestimmt werden.
In-vitro-Reizwert (In Vitro Irritancy Score, IVIS): Eine bei der BCOP-Prüfmethode verwendete empirisch abgeleitete Formel, bei der der mittlere Trübungs- und der mittlere Durchlässigkeitswert für jede Behandlungsgruppe in einem einzigen In-vitro-Wert zusammengefasst werden. IVIS = mittlerer Trübungswert + (15 x mittlerer Durchlässigkeitswert).
Irreversible Wirkungen am Auge: Siehe "Schwere Augenschädigung"
Keine Einstufung nach UN-GHS: Chemikalien, die die Voraussetzungen für eine Einstufung in die UN-GHS-Kategorien 1 oder 2 (2A oder2B) nicht erfüllen. Austauschbar mit "Nicht eingestuft".
Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert.
Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert.
Nicht eingestuft: Chemikalien, die nicht als augenreizend (UN-GHS-Kategorie 2, 2A oder 2B) oder schwer augenschädigend (UN-GHS-Kategorie 1) eingestuft sind. Austauschbar mit der UN-GHS-Klasse "Keine Einstufung".
Opazimeter: Ein Instrument zur Messung der "Hornhauttrübung" durch quantitative Evaluierung der Lichtübertragung durch die Hornhaut. Das Gerät besteht in der Regel aus zwei Kammern, beide mit eigener Lichtquelle und Fotozelle. Eine Kammer wird für die behandelte Hornhaut verwendet, die zweite zur Kalibrierung und Nulleinstellung des Instruments. Licht aus einer Halogenleuchte wird durch eine Kontrollkammer (leere Kammer ohne Fenster oder Flüssigkeit) zu einer Fotozelle geleitet und mit dem Lichtstrahl verglichen, der durch die Versuchskammer, welche die Kammer mit der Hornhaut enthält, zu einer Fotozelle geleitet wird. Der Unterschied bei der Lichtübertragung aus den Fotozellen wird errechnet und als numerischer Trübungswert digital angezeigt.
Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein.
Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird.
Referenzchemikalie: Eine zum Vergleich mit einer Prüfchemikalie verwendete Bezugsgröße. Eine Referenzchemikalie sollte die folgenden Eigenschaften aufweisen: i) beständige und zuverlässige Quelle(n); ii) strukturelle und funktionelle Ähnlichkeit zur Klasse der geprüften Chemikalien; iii) bekannte physikalische/chemische Eigenschaften; iv) unterstützende Daten zu bekannten Wirkungen und v) bekannte Potenz innerhalb der gewünschten Wirkungsspanne.
Reversible Wirkungen am Auge: Siehe "Augenreizung".
Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Austauschbar mit "Irreversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 1" (4).
Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (4).
Tensid: Auch als oberflächenaktiver Stoff bezeichnet. Hierbei handelt es sich um Stoffe, wie waschaktive Substanzen (Detergenzien), die die Oberflächenspannung einer Flüssigkeit herabsetzen und so die Bildung von Schaum oder das Eindringen in feste Stoffe ermöglichen; auch bekannt als Netzmittel.
Tensidhaltiges Gemisch: Im Kontext dieser Prüfmethode ein Gemisch mit einem oder mehreren Tensiden in einer Endkonzentration von > 5 %.
Top-Down-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zuerst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden.
UN-GHS-Kategorie 1: Siehe "Schwere Augenschädigung"
UN-GHS-Kategorie 2: Siehe "Augenreizung".
Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden.
Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll.
Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
 | weiter . |  |