
umwelt-online: Verordnung (EG) Nr. 440/2008 zur Festlegung von Prüfmethoden gemäß der VO (EG) Nr. 1907/2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) (19)
 | zurück |
|
B.44 Hautresorption: In-vivo-Methode
1. Methode
Diese Testmethode entspricht OECD TG 427 (2004).
1.1 Einleitung
Die meisten Chemikalien wirken über die Haut auf den Körper ein, während bei den meisten an Labortieren durchgeführten toxikologischen Studien die Chemikalie oral verabreicht wird. Die in dieser Richtlinie beschriebene Untersuchung der perkutanen In-vivo-Resorption stellt die Verbindung her, die bei der Sicherheitsbewertung im Anschluss an dermale Exposition für die Extrapolation der Ergebnisse oraler Studien notwendig ist.
Eine Substanz muss eine große Zahl von Hautschichten durchdringen, bevor sie in den Kreislauf gelangen kann. Bei den meisten Substanzen wird die Penetrationsgeschwindigkeit durch die aus toten Zellen bestehende Hornhaut (Stratum Corneum) bestimmt. Die Permeabilität der Haut wird sowohl von der Lipophilie der Chemikalie als auch von der Dicke der äußeren Epidermisschicht sowie durch Faktoren wie dem Molgewicht und der Stoffkonzentration bestimmt. Im Allgemeinen ist die Haut von Ratten und Hasen permeabler als die menschliche Haut, während die Permeabilität der Haut von Meerschweinchen und Affen der Permeabilität der menschlichen Haut ähnelt.
Die Methoden zur Messung der perkutanen Resorption lassen sich in zwei Kategorien einteilen: in vivo und in vitro. Die In-vivo-Methode liefert bei unterschiedlichen Labortierarten gute Informationen zur Hautresorption. In jüngerer Zeit wurden ergänzend In-vitro-Methoden entwickelt. Bei diesen wird der Transport durch die menschliche oder tierische Haut in ihrer vollständigen oder teilweisen Dicke in einem Flüssigkeitsbehälter untersucht. Die In-vitro-Methode wird in einer separaten Testverfahrensbeschreibung (1) beschrieben. Es wird empfohlen, das OECD Guidance Document for the Conduct of Skin Absorption Studies (2) als Hilfe bei der Wahl der für den jeweiligen Fall geeignetsten Methode zu Rate zu ziehen; dieses Dokument enthält nähere Angaben zur Eignung der In-vivo- und In-vitro-Methoden.
Die hier beschriebene In-vivo-Methode ermöglicht die Bestimmung der Penetration der Testsubstanz durch die Haut in den Kreislauf. Dieses Verfahren findet seit Jahren weithin Anwendung (3) (4) (5) (6) (7). Zwar sind In-vitro-Studien zur perkutanen Resorption in zahlreichen Fällen geeignet, doch können die benötigten Daten in bestimmten Fällen nur durch In-vivo-Studien gewonnen werden.
Die Vorteile der In-vivo-Methode bestehen darin, dass dabei ein physiologisch und metabolisch intaktes System und eine zahlreichen Toxizitätsstudien gemeinsame Tierart verwendet wird und dass diese Methode für die Verwendung mit anderen Arten modifiziert werden kann. Nachteile sind die Verwendung lebender Tiere und die Notwendigkeit, radioaktiv markiertes Material einzusetzen, damit zuverlässige Ergebnisse gewährleistet sind, ferner Schwierigkeiten bei der Bestimmung der Frühresorptionsphase und die unterschiedliche Permeabilität der Haut der bevorzugt verwendeten Art (Ratten) und der menschlichen Haut. Tierische Haut ist im Allgemeinen durchlässiger, d. h., die perkutane Resorption der menschlichen Haut könnte demzufolge überschätzt werden (6) (8) (9). Ätzende Materialien sollten an lebenden Tieren nicht getestet werden.
1.2 Definitionen
Unresorbierte Dosis: Dies entspricht der nach der Exposition von der Hautoberfläche abgewaschenen Dosis sowie der ggf. in der nicht okkludierten Abdeckung enthaltenen Dosis, einschließlich etwaiger Dosen, bei denen nachgewiesen wird, dass sie sich während der Exposition auf der Haut verflüchtigen.
Resorbierte Dosis (in vivo): Diese umfasst die im Urin, in Käfigreinigungsrückständen, Fäzes, ausgeatmeter Luft (soweit diese gemessen wird), im Blut, Gewebe (sofern erfasst) und dem übrigen Körper verbliebene Dosis nach Entfernung der Haut an der Stelle, an der die Substanz aufgetragen wurde.
Resorbierbare Dosis: Diese stellt die nach dem Waschen auf oder in der Haut vorhandene Dosis dar.
1.3 Prinzip der Prüfmethode
Die Testsubstanz wird - möglichst in radioaktiv markierter Form - in einer oder mehreren geeigneten Dosierungen als repräsentative praktische Präparation auf der geschorenen Haut der Tiere aufgebracht. Die Testpräparation bleibt während einer bestimmten Zeitdauer unter einer geeigneten (nicht okklusiven, semiokklusiven oder okklusiven) Abdeckung mit der Haut in Kontakt. Am Ende des Expositionszeit wird die Abdeckung entfernt und die Haut mit einem geeigneten Reinigungsmittel gereinigt; Abdeckung und Reinigungsmittel werden zur Analyse aufbewahrt, und es wird eine frische Abdeckung angebracht. Die Tiere werden vor, während und nach der Expositionszeit in Stoffwechsel-Einzelkäfigen untergebracht, und die während dieses Zeitraums anfallenden Exkremente und die ausgeatmete Luft werden zu Analysezwecken gesammelt. Die Sammlung der ausgeatmeten Luft kann entfallen, wenn ausreichende Angaben darüber vorliegen, dass nur wenig oder gar keine flüchtigen radioaktiven Stoffwechselprodukte gebildet werden. Im Rahmen der Studien werden normalerweise mehrere Tiergruppen der Testpräparation ausgesetzt. Eine Gruppe wird am Ende der Expositionszeit getötet. Andere Gruppen werden zu festgelegten späteren Zeitpunkten getötet (2). Am Ende der Probenahmephase werden die verbleibenden Tiere getötet, Blut zu Analysezwecken entnommen, die Auftragsstelle zur Analyse entnommen und der Körper auf etwaige nicht ausgeschiedene Stoffe untersucht. Die Stichproben werden durch entsprechende Mittel untersucht und der Grad der perkutanen Resorption näherungsweise ermittelt (6) (8) (9).
1.4 Beschreibung der Testmethode
1.4.1 Auswahl von Versuchstierarten
Üblicherweise werden Ratten verwendet, allerdings können auch haarlose Stämme und Arten, deren Hautresorptionsrate denen der menschlichen Haut näherkommt, verwendet werden (3) (6) (7) (8) (9). Es sind junge, ausgewachsene und gesunde Tiere gleichen Geschlechts (standardmäßig männliche Tiere) üblicherweise verwendeter Laborstämme zu verwenden. Zu Beginn der Studie dürfen die Gewichtsunterschiede der verwendeten Tiere eine Grenze von ± 20 % des mittleren Gewichts nicht überschreiten. So sind beispielsweise männliche Ratten mit einem Gewicht von 200-250 g geeignet, insbesondere Tiere in der oberen Hälfte dieses Gewichtsbereichs.
1.4.2 Zahl und Geschlecht der Versuchstiere
Zur Vorbereitung des Tests und für jeden Beendigungszeitpunkt ist je eine Gruppe von mindestens 4 Tieren gleichen Geschlechts zu verwenden. Jede Gruppe von Tieren wird nach unterschiedlichen Zeitintervallen getötet, beispielsweise nach Ende der Expositionszeit (typischerweise 6 oder 24 Stunden) bzw. zu späteren Zeitpunkten (z.B. nach 48 und 72 Stunden). Liegen Daten vor, anhand deren erhebliche Unterschiede der dermalen Toxizität bei männlichen und weiblichen Tieren nachgewiesen werden, ist das empfindlichere Geschlecht zu wählen. Liegen keine diesbezüglichen Daten vor, ist die Wahl des Geschlechts freigestellt.
1.4.3 Unterbringungs- und Fütterungsbedingungen
Die Temperatur im Tierversuchsraum muss 22 °C (± 3 °C) betragen. Die relative Luftfeuchte muss mindestens 30 % betragen und sollte 70 % - außer beim Reinigen des Raums - nicht überschreiten. Angestrebt werden sollte eine Luftfeuchte von 50-60 %. Für die Beleuchtung ist Kunstlicht zu verwenden und so zu schalten, dass sich 12 Stunden Licht mit 12 Stunden Dunkelheit abwechseln. Zur Ernährung sind herkömmliche Labornahrungsmittel zu verwenden, die unrationiert mit einer unbegrenzten Menge Trinkwasser zur Verfügung stehen sollten. Während der Studie sowie vorzugsweise auch während der Eingewöhnungsphase werden die Tiere einzeln in Stoffwechselkäfigen untergebracht. Da die Ergebnisse durch verschüttetes Nahrungsmittel und Wasser beeinträchtigt würden, ist die Wahrscheinlichkeit derartiger Störungen zu minimieren.
1.4.4 Präparation der Tiere
Die Tiere werden markiert, damit sie einzeln identifiziert werden können. Vor Beginn der Studie werden sie mindestens 5 Tage lang in ihren Käfigen gehalten, so dass eine Gewöhnung an die Laborbedingungen erfolgen kann.
Nach der Akklimatisierungsphase und rund 24 Stunden vor der Verabreichung der Dosis wird an jedem Tier ein Hautbereich im Schulter- und Rückenbereich geschoren. Da geschädigte Haut andere Permeationseigenschaften als intakte Haut aufweist, ist Abrieb der Hautoberfläche zu vermeiden. Nach der Schur und rund 24 Stunden vor Auftragen der Testsubstanz auf die Haut (siehe Abschnitt 1.4.7) ist die Hautoberfläche mit Aceton abzuwischen, um das Sebum zu entfernen. Zusätzliches Waschen mit Seife und Wasser ist nicht zu empfehlen, da Seifenrückstände die Resorption der Testsubstanz fördern könnten. Der Bereich muss so groß sein, dass eine zuverlässige Berechnung der resorbierten Menge der Testchemikalie je cm2 Haut gewährleistet ist, vorzugsweise also mindestens 10 cm2. Ein solcher Bereich kann an Ratten mit einem Körpergewicht von 200-250 g hergestellt werden. Nach der Vorbereitung werden die Tiere wieder in die Stoffwechselkäfige platziert.
1.4.5 Testsubstanz
Als Testsubstanz wird der Stoff verwendet, dessen Penetrationseigenschaften untersucht werden sollen. Idealerweise ist die Testsubstanz radioaktiv zu markieren.
1.4.6 Testpräparation
Die Präparation der Testsubstanz (z.B. reines, verdünntes oder formuliertes Material, das die auf der Haut aufgetragene Testchemikalie enthält) muss der Substanz entsprechen (oder ein realistisches Surrogat hiervon bilden), der Menschen oder andere betroffene Gruppen ausgesetzt sein können. Etwaige Abweichungen von der in der Praxis verwendeten Präparation sind zu begründen. Bei Bedarf wird die Prüfsubstanz in einem geeigneten Medium gelöst oder suspendiert. Bei anderen Trägermedien als Wasser müssen die Resorptionseigenschaften und mögliche Wechselwirkungen mit der Testsubstanz bekannt sein
1.4.7 Aufbringen auf die Haut
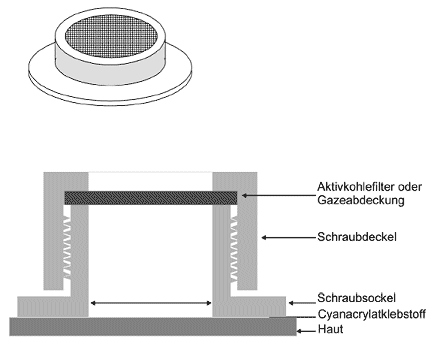
Auf der Hautoberfläche wird eine Applikationsstelle mit einer bestimmten Fläche festgelegt. Eine bekannte Menge der Testpräparation wird gleichmäßig auf dieser Stelle aufgetragen. Diese Menge muss im Regelfall die potenzielle Exposition wiedergeben, der der Mensch ausgesetzt ist, typischerweise bei Feststoffen 1-5 mg/cm2 oder bei Flüssigkeiten bis zu 10 µl/cm2. Abweichende Mengen müssen aufgrund der zu erwartenden Verwendungsbedingungen, der Untersuchungsziele oder der physikalischen Eigenschaften der Testpräparation gerechtfertigt sein. Nach dem Auftrag ist die behandelte Stelle gegen Überstreichen zu schützen. Eine typische Vorrichtung hierfür ist in Abbildung 1 dargestellt; normalerweise wird die Hautstelle, an der der Auftrag erfolgte, durch eine nicht okklusive Abdeckung geschützt (z.B. eine permeable Nylongazeabdeckung). Bei infiniter Applikation ist die Applikationsstelle jedoch zu okkludieren. Falls durch die Verdunstung semiflüchtiger Testsubstanzen die Rückgewinnungsrate der Testsubstanz in einem inakzeptablen Maß verringert wird (siehe auch Abschnitt 1.4.10, erster Absatz), muss die verdunstete Substanz in einem Aktivkohlefilter über der Applikationsvorrichtung aufgefangen werden (siehe Abbildung 1). Derartige Vorrichtungen dürfen die Haut nicht schädigen und die Testpräparation weder absorbieren noch mit ihr reagieren. Die Tiere werden anschließend wieder in ihre Stoffwechsel-Einzelkäfige gesetzt, damit die Exkremente aufgefangen werden können.
1.4.8 Dauer der Exposition und Probenahme
Die Expositionsdauer entspricht der Zeitdauer zwischen Auftrag und Entfernung der Testpräparation durch Waschen der Haut. Dabei ist eine aussagefähige Expositionszeit (normalerweise 6 oder 24 Stunden) entsprechend der beim Menschen zu erwartenden Expositionsdauer einzuhalten. Nach der Expositionsdauer verbleiben die Tiere bis zum planmäßigen Ende der Studie in den Stoffwechselkäfigen. Während der gesamten Dauer der Studie sind die Tiere in regelmäßigen Intervallen auf Anzeichen toxischer Wirkungen/abnormaler Reaktionen zu beobachten. Am Ende der Expositionszeit ist die behandelte Hautfläche auf sichtbare Anzeichen einer Reizung zu untersuchen.
Die Stoffwechselkäfige müssen so eingerichtet sein, dass Urin und Fäzes während der gesamten Untersuchungsdauer separat gesammelt werden können. Die Sammlung von 14C-Kohlendioxid und flüchtigen 14C-Kohlenstoffverbindungen muss möglich sein; diese Verbindungen sind, wenn sie in entsprechender Menge anfallen (> 5 %), zu analysieren. Urin, Exkremente und in der Auffangvorrichtung zurückgehaltene Flüssigkeiten (z.B. 14C-Kohlendioxid und flüchtige 14C-Verbindungen) sind zu den einzelnen Probenahmezeitpunkten aus jeder Gruppe einzeln zu sammeln. Liegen ausreichende Angaben darüber vor, dass kaum oder keine flüchtigen radioaktiven Stoffwechselprodukte gebildet werden, können auch offene Käfige verwendet werden.
Die Exkremente werden während der Dauer der Exposition, bis zu 24 Stunden nach dem erstmaligen Hautkontakt und anschließend täglich bis zum Ende des Versuchs gesammelt. Normalerweise sind drei Intervalle für die Sammlung von Exkrementen ausreichend, je nach beabsichtigtem Zweck der Testpräparation oder vorliegenden kinetischen Daten können jedoch auch geeignetere oder zusätzliche Zeitpunkte für eine Untersuchung in Betracht kommen.
Am Ende des Expositionszeitraums wird die Schutzvorrichtung von den Tieren entfernt und separat zur weiteren Analyse verwahrt. An allen Tieren ist die behandelte Haut mindestens dreimal mit geeigneten Tupfern zu waschen. Dabei ist darauf zu achten, dass keine anderen Körperteile kontaminiert werden. Das Reinigungsmittel muss typisch für die bei normaler Körperhygiene verwendeten Produkte sein, z.B. eine wässrige Seifenlösung. Abschließend ist die Haut zu trocknen. Sämtliche Tupfer und Waschrückstände sind zur Analyse aufzubewahren. Bei Tieren jener Gruppen, die bis zu einem späteren Zeitpunkt untersucht werden, ist zum Schutz der behandelten Hautstelle eine neue Abdeckung anzubringen, bevor diese wieder in ihre Einzelkäfige verbracht werden.
1.4.9 Abschließende Schritte
Die Tiere der einzelnen Gruppen sind zum festgelegten Zeitpunkt zu töten, und das Blut ist zur Analyse aufzufangen. Die Schutzvorrichtung bzw. -abdeckung ist zur Analyse zu entfernen. Die Haut an der Applikationsstelle und ein ähnlicher Bereich unbehandelter, rasierter Haut sind zur separaten Analyse bei jedem Tier zu entfernen. Die Applikationsstelle kann zerlegt werden, wobei das Stratum corneum von der darunter liegenden Epidermis getrennt wird und daraus weitere Informationen zur Verteilung der Testchemikalie gewonnen werden. Durch die Ermittlung der Ablagerung über einen bestimmten Zeitraum nach der Expositionszeit lassen sich Angaben zum Verbleib bzw. Verhalten der einzelnen Testchemikalien im Stratum corneum gewinnen. Um die Fraktionierung der Haut zu erleichtern (nachdem die Haut letztmalig gewaschen und das Tier getötet wurde), werden die einzelnen Schutzabdeckungen entfernt. Die Applikationsstelle auf der Haut sowie ein angrenzender ringförmiger Hautbereich werden aus dem Rattenkörper herausgeschnitten und auf einer Unterlage befestigt. Ein Klebestreifen wird mit leichtem Druck auf der Hautoberfläche angedrückt und das Klebeband zusammen mit einem Teil des Stratum corneum abgezogen. Anschließend werden weitere Klebestreifen aufgebracht, bis das Klebeband nicht mehr an der Hautoberfläche haftet; dann ist das gesamte Stratum corneum entfernt. Sämtliche Klebestreifen eines jeden Tieres können in einem einzigen Behälter gemeinsam verwahrt werden; diesem Behälter wird ein Gewebeauflösungsmittel zur Auflösung des Stratum corneum zugesetzt. Bestimmte für die Untersuchung bestimmte Gewebe können zur separaten Messung entnommen werden, bevor der Tierkörper auf die resorbierte Körperdosis untersucht wird. Die Körper der einzelnen Tiere sind zu Analysezwecken aufzubewahren. Normalerweise reicht die Analyse des Gesamtgehalts aus. Zur Untersuchung vorgesehene Organe können zur separaten Analyse entnommen werden (wenn dies durch andere Studien angezeigt ist). Der zum Zeitpunkt der Tötung in der Blase enthaltene Urin ist zu dem zuvor gesammelten Urin zu geben. Nachdem die Exkremente zum Zeitpunkt der Tötung aus den Stoffwechselkäfigen gesammelt wurden, sind die Käfige und ihre Auffangvorrichtungen mit einem geeigneten Lösemittel zu waschen. Andere möglicherweise verunreinigte Geräte sind ebenfalls zu analysieren.
1.4.10 Analyse
In allen Studien ist eine ausreichende Rückgewinnung (d. h. ein Mittelwert von 100 ± 10 % der Radioaktivität) anzustreben. Rückgewinnungsraten außerhalb dieses Sollbereichs sind zu begründen. Die Menge der in jeder Probe verabreichten Dosis ist durch in geeigneter Weise validierte Verfahren zu analysieren.
Als Teil der statistischen Untersuchung ist bei den Wiederholungen jeder Anwendung ein Maß für die Streuung zu berücksichtigen.
2. Daten
An sämtlichen Tieren sind bei jeder Probenahme der Testchemikalie und/oder der Stoffwechselprodukte die unten stehenden Messungen durchzuführen. Zusätzlich zu Einzeldaten sind die entsprechend den Probenahmezeiten zu Gruppen zusammengefassten Daten als Mittelwerte in den Bericht aufzunehmen:
- Menge, die den Schutzvorrichtungen zugeordnet wird,
- Menge, die aus der Haut gelöst werden kann,
- Menge in/auf der Haut, die nicht von der Haut abgewaschen werden kann,
- Menge in der Blutprobe,
- Menge in den Exkrementen und der ausgeatmeten Luft (falls maßgeblich),
- im Körper und in zu separaten Analysen entnommenen Organen verbleibende Menge.
Anhand der in den Exkrementen, der ausgeatmeten Luft, im Blut und im Körper enthaltenen Menge der Testsubstanz und/oder der Metaboliten kann die zu den jeweiligen Zeitpunkten resorbierte Gesamtmenge bestimmt werden. Außerdem ist eine Berechnung der Menge der Testchemikalie möglich, die je cm2 der während der Expositionszeit der Testsubstanz ausgesetzten Haut resorbiert wurde.
3. Berichterstattung
3.1 Abschlussbericht
Der Testbericht muss die im Protokoll festgelegten Anforderungen einschließlich einer Begründung für das verwendete Testsystem sowie folgende Angaben enthalten:
Prüfsubstanz
- Kenndaten, z.B. CAS-Nummer, sofern vorhanden, Quelle, Reinheit (radiochemische Reinheit), bekannte Verunreinigungen, Partienummer;
- physikalische Beschaffenheit, physikalischchemische Eigenschaften (z.B. pH-Wert, Flüchtigkeit, Löslichkeit, Stabilität, Molgewicht und log Pow).
Testpräparation:
- Formulierung und Begründung für die Verwendung;
- Details der Testpräparation, aufgetragene Menge, erreichte Konzentration, Vehikel, Stabilität und Homogenität.
Versuchstier:
- Art/Stamm;
- Zahl, Alter und Geschlecht der Tiere;
- Herkunft der Tiere, Unterbringungsbedingungen, Ernährung usw.;
- Gewicht der einzelnen Tiere bei Versuchsbeginn.
Prüfbedingungen:
- Details zur Verabreichung der Testpräparation (Applikationsort, Testmethoden, Okklusion/Nicht-Okklusion, Volumen, Extraktion, Nachweis);
- Angaben zu Futter- und Wasserqualität.
Ergebnisse:
- etwaige Anzeichen von Toxizität;
- in Tabellenform dargestellte Resorptionsdaten (angegeben als Geschwindigkeit, Menge oder Prozentwert);
- Gesamtrückgewinnungsrate aus dem Versuch;
- Auswertung der Ergebnisse, Vergleich mit bereits vorliegenden Daten zur perkutanen Resorption der Testverbindung.
Erörterung der Ergebnisse.
Schlussfolgerungen.
4. Literaturhinweise
1. Testing Method B.45. Skin Absorption: In vitro Method.
2. OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris.
3. ECETOC (1993) Percutaneous Absorption. European Centre for Ecotoxicology and Toxicology of Chemicals, Monograph No. 20.
4. Zendzian R.P. (1989) Skin Penetration Method suggested for Environmental Protection Agency Requirements. J. Am. Coll. Toxicol. 8(5), 829-835.
5. Kemppainen B.W., Reifenrath W.G. (1990) Methods for skin absorption. CRC Press Boca Raton, FL, USA.
6. EPA (1992) Dermal Exposure Assessment: Principles and Applications. Exposure Assessment Group, Office of Health and Environmental Assessment.
7. EPA (1998) Health Effects Test Guidelines, OPPTS 870-7600, Dermal Penetration. Office of Prevention, Pesticides and Toxic Substances.
8. Bronaugh R.L., Wester R.C., Bucks D., Maibach H.I. and Sarason R. (1990) In vivo percutaneous absorption of fragrance ingredients in reshus monkeys and humans. Fd. Chem. Toxic. 28, 369-373.
9. Feldman R.J. and Maibach H.I. (1970) Absorption of some organic compounds through the skin in man.
J. Invest Dermatol. 54, 399-404.
Abbildung 1 Beispiel für die Gestaltung einer typischen Vorrichtung zur Begrenzung und zum Schutz der dermalen Applikationsstelle bei In-vivo-Studien zur perkutanen Resorption
B.45 Hautresorption: In-vitro-Methode
1. Methode
Diese Testmethode entspricht OECD TG 428 (2004).
1.1 Einleitung
Diese Methode soll Aufschluss über die Resorption einer auf ein ausgeschnittenes Hautstück aufgebrachten Testsubstanz geben. Sie kann entweder mit der Hautresorptionsmethode: In-vivo-Methode (1) kombiniert oder separat durchgeführt werden. Für die Gestaltung von Studien auf der Grundlage dieser Methode wird empfohlen, das OECD Guidance Document for the Conduct of Skin Absorption Studies (2) zu Rate zu ziehen. Diese Richtlinie soll Hilfestellung bei der Wahl geeigneter In-vitro-Verfahren für bestimmte Anwendungsfälle leisten, damit die Zuverlässigkeit der durch dieses Verfahren gewonnenen Ergebnisse gewährleistet ist.
Die Methoden zur Messung der Hautresorption und des dermalen Eintrags lassen sich in zwei Kategorien unterteilen: in vivo und in vitro. In-vivo-Methoden zur Untersuchung der Hautresorption sind seit langem etabliert und liefern pharmakokinetische Angaben zu verschiedenen Tierarten. Eine In-vivo-Methode wird in einer weiteren Testmethode (1) beschrieben. In-vitro-Methoden werden seit Jahren ebenfalls zur Messung der Hautresorption eingesetzt. Offizielle Validierungsstudien der In-vitro-Methoden, die unter die vorliegende Testmethode fallen, wurden zwar nicht durchgeführt, doch wurde von den OECD-Experten im Jahr 1999 festgestellt, dass der Umfang der evaluierten Daten als Bestätigung der In-vitro-Methode ausreicht (3). Weitere Details zur Untermauerung dieser Bestätigung - darunter umfangreiche direkte Vergleiche von In-vitro- und In-vivo-Methoden - sind in der unter (2) angegebenen Richtlinie (Guidance Document) enthalten. Dieses Thema wurde bereits in zahlreichen Monografien dargestellt, die auch ausführliche Hintergrundinformationen zum Einsatz von In-vitro-Methoden enthalten (4) (5) (6) (7) (8) (9) (10) (11) (12). Mit In-vitro-Methoden wird die Diffusion von Chemikalien in und durch die Haut in einen Flüssigkeitstank gemessen, wobei an nicht lebensfähiger Haut die reine Diffusion oder an frischer, stoffwechselaktiver Haut gleichzeitig Diffusion und Hautstoffwechsel gemessen werden können. Derartige Methoden finden insbesondere Verwendung als Raster für den Vergleich des Eintrags von Chemikalien aus unterschiedlichen Formulierungen in und durch die Haut und bieten sich darüber hinaus als nützliche Modelle für die Beurteilung der perkutanen Resorption beim Menschen an.
Die In-vitro-Methode eignet sich möglicherweise nicht für sämtliche Situationen und Chemikalienklassen. Möglicherweise kann die In-vitro-Testmethode zur einleitenden qualitativen Evaluierung der Penetration durch die Haut verwendet werden. In bestimmten Fällen muss dies durch In-vivo-Daten untermauert werden. Zur weiteren Vertiefung jener Fälle, in denen die In-vitro-Methode geeignet wäre, ist die unter (2) angegebene Richtlinie (Guidance Document) heranzuziehen. Weitere detaillierte Informationen zur Untermauerung der Entscheidungsfindung sind in (3) nachzulesen.
Die hier beschriebene Methode gibt Aufschluss über die grundlegenden Prinzipien der Messung der dermalen Resorption und des Eintrags einer Testsubstanz auf ausgeschnittenen Hautstücken. Hierfür können Hautproben zahlreicher Säugetierarten - auch menschliche Haut - verwendet werden. Die Permeabilitätseigenschaften der Haut bleiben nach dem Ausschneiden des Hautstücks aus dem Körper erhalten, da die abgestorbene Hornhaut (Stratum corneum) die Hauptdiffusionssperre bildet; ein aktiver Transport von Chemikalien durch die Haut wurde nicht festgestellt. Es wurde nachgewiesen, dass die Haut in der Lage ist, bei der perkutanen Resorption bestimmte Chemikalien in Stoffwechselprodukte umzusetzen (6); allerdings tritt bei diesem Prozess hinsichtlich der tatsächlich resorbierten Dosis keine Begrenzung der Geschwindigkeit ein, jedoch kann die Art des in den Blutkreislauf gelangenden Materials davon beeinflusst werden.
1.2 Definitionen
Unresorbierte Dosis: Dies entspricht der nach der Exposition von der Hautoberfläche abgewaschenen Dosis sowie der ggf. in der nicht okkludierten Abdeckung enthaltenen Dosis, einschließlich etwaiger Dosen, bei denen nachgewiesen wird, dass sie sich während der Exposition auf der Haut verflüchtigen.
Resorbierte Dosis (in vitro): Masse der Testsubstanz, die innerhalb einer bestimmten Zeit in den Rezeptor-Flüssigkeits- oder -Systemkreislauf gelangt.
Resorbierbare Dosis (in vitro): die nach dem Abwaschen auf oder in der Haut vorhandene Dosis.
1.3 Prinzip der Prüfmethode
Die Testsubstanz, die radioaktiv markiert werden kann, wird auf die Oberfläche eines Hautprobestücks aufgetragen, das die beiden Kammern einer Diffusionszelle voneinander trennt. Die Chemikalie verbleibt unter festgelegten Bedingungen eine bestimmte Zeit lang auf der Haut, bevor sie durch ein geeignetes Reinigungsverfahren entfernt wird. Von der Rezeptorflüssigkeit werden zu bestimmten Zeitpunkten während des Versuchs Proben genommen und auf die Testchemikalie und/oder Metaboliten analysiert.
Bei Verwendung stoffwechselaktiver Systeme können die Metaboliten der Testchemikalie durch geeignete Verfahren analysiert werden. Am Ende des Experiments werden die Verteilung der Testchemikalie und ggf. deren Metaboliten quantifiziert.
Unter entsprechenden Bedingungen, die in der vorliegenden Methode und in Richtlinie (2) beschrieben sind, wird durch Analyse der Rezeptorflüssigkeit und der behandelten Haut die während eines bestimmten Zeitraums erfolgte Resorption einer Testsubstanz gemessen. Die auf der Haut verbleibende Testsubstanz ist als resorbiert zu betrachten, es sei denn, es kann nachgewiesen werden, dass die Resorption anhand der Werte der Rezeptorflüssigkeit alleine ermittelt werden kann. Anhand der Analyse der übrigen Bestandteile (von der Haut abgewaschenes und zwischen den Hautschichten verbleibendes Material) kann eine weitere Evaluierung der Daten vorgenommen werden, unter anderem auch die Gesamtverteilung der Testsubstanz und die prozentuale Rückgewinnung.
Als Nachweis für die Leistungsfähigkeit und Zuverlässigkeit des Testsystems müssen die Ergebnisse der relevanten Referenzchemikalien vorliegen und mit dem veröffentlichten Schrifttum zu der verwendeten Methode übereinstimmen. Diese Anforderung könnte erfüllt werden, indem die Tests mit einer geeigneten Referenzsubstanz (deren Lipophilie vorzugsweise den Werten der Testsubstanz näherungsweise entsprechen sollte) zeitgleich mit der Testsubstanz durchgeführt werden oder indem ausreichende historische Daten für verschiedene Referenzsubstanzen unterschiedlicher Lipophilie vorgelegt werden (z.B. Koffein, Benzoesäure und Testosteron).
1.4 Beschreibung der Prüfmethode
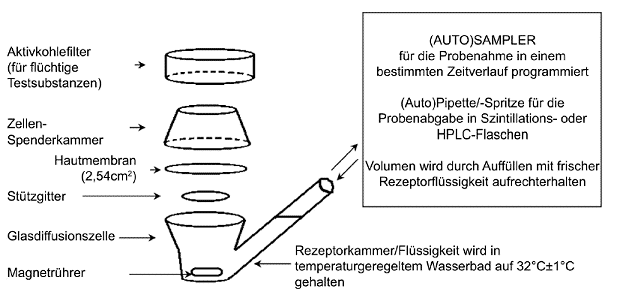
1.4.1 Diffusionszelle
Die Diffusionszelle besteht aus einer Spenderkammer und einer Rezeptorkammer, zwischen denen die Haut angeordnet wird (ein Beispiel für einen typischen Aufbau einer solchen Kammer ist in Abbildung 1 dargestellt). Die Zelle muss so aufgebaut werden, dass die Haut dicht umschlossen wird, die Probenahme auf einfache Weise möglich ist und eine gute Durchmischung der Rezeptorlösung erreicht wird, die mit der Hautunterseite in Berührung kommt, und außerdem eine gute Temperaturregelung der Zelle und ihres Inhalts möglich ist. Es sind sowohl statische Diffusionszellen als auch Durchfluss-Diffusionszellen zulässig. Normalerweise sind die Spenderkammern bei der Exposition gegenüber einer finiten Dosis einer Testpräparation nicht okkludiert. Bei infiniten Applikationen und bestimmten bei finiten Dosen vorkommenden Szenarien können die Spenderkammern jedoch auch okkludiert werden.
1.4.2 Rezeptorflüssigkeit
Vorzugsweise ist eine physiologisch geeignete Rezeptorflüssigkeit zu verwenden, allerdings sind auch andere Flüssigkeitsarten zulässig, sofern deren Verwendung begründet werden kann. Die genaue Zusammensetzung der Rezeptorflüssigkeit ist anzugeben. Eine ausreichende Löslichkeit der Testchemikalie in der Rezeptorflüssigkeit ist nachzuweisen, damit sie nicht als Resorptionssperre wirkt. Außerdem darf die Rezeptorflüssigkeit die Intaktheit des Hautstücks nicht beeinträchtigen. In einem Durchflusssystem darf die Durchflussgeschwindigkeit die Diffusion der Testsubstanz in die Rezeptorflüssigkeit nicht behindern. In einem statischen Zellensystem muss die Flüssigkeit ständig umgerührt werden, und es sind regelmäßig Proben zu entnehmen. Wird der Stoffwechsel untersucht, muss die Rezeptorflüssigkeit so beschaffen sein, dass die Viabilität der Haut während des gesamten Verlaufs des Experiments unterstützt wird.
1.4.3 Herstellung der Hautstücke
Es kann menschliche oder tierische Haut verwendet werden. Natürlich unterliegt die Verwendung menschlicher Haut einzelstaatlichen und internationalen ethischen Kriterien und Auflagen. Vorzugsweise sollte lebensfähige Haut verwendet werden, allerdings ist auch die Verwendung von abgestorbener Haut zulässig, sofern die Intaktheit der Haut nachgewiesen werden kann. Es sind entweder Epidermismembranen (die durch Enzyme, durch Wärme oder auf chemischem Wege separiert wurden) oder geteilte Hautlagen (typischerweise mit einer Dicke von 200-400 µm), die mit einem Dermatom hergestellt wurden, zulässig. Es kann auch die volle Hautdicke verwendet werden, allerdings ist eine übermäßige Hautdicke (ca. > 1 mm) zu vermeiden, sofern dies nicht zur Feststellung der Testchemikalie in den einzelnen Hautlagen erforderlich ist. Die Wahl der Arten, die anatomische Entnahmestelle und das Präparationsverfahren sind zu begründen. Es müssen akzeptable Daten aus mindestens vier Wiederholungs-Gleichtests je Testpräparation vorliegen.
1.4.4 Intakte Beschaffenheit des hergestellten Hautstücks
Eine sachgemäße Herstellung des Hautstücks ist von größter Bedeutung. Durch unsachgemäße Handhabung kann die Hornhaut (Stratum corneum) beschädigt werden; daher ist das hergestellte Hautstück auf intakte Beschaffenheit zu kontrollieren. Bei der Untersuchung des Hautstoffwechsels ist die frisch ausgeschnittene Haut möglichst zeitnah und unter Bedingungen, die nachgewiesenermaßen eine Stoffwechselaktivität unterstützen, zu verwenden. Als Richtwert gilt dabei, dass frisch ausgeschnittene Haut innerhalb von 24 Stunden zu verwenden ist; die zulässige Lagerungsdauer kann jedoch je nach dem an der Metabolisierung beteiligten Enzymsystem und den Lagertemperaturen variieren (13). Wurden die hergestellten Hautstücke vor der Verwendung gelagert, ist nachzuweisen, dass die Barrierefunktion erhalten bleibt.
1.4.5 Testsubstanz
Als Testsubstanz gilt der Stoff, dessen Penetrationseigenschaften untersucht werden sollen. Idealerweise sollte die Testsubstanz radioaktiv markiert werden.
1.4.6 Testpräparation
Die Präparation der Testsubstanz (z.B. reines, verdünntes oder formuliertes Material, das die auf der Haut aufgetragene Testchemikalie enthält) muss der Substanz entsprechen (oder ein realistisches Surrogat hiervon bilden), der Menschen oder andere mögliche Zielarten ausgesetzt sein können. Etwaige Abweichungen von der in der Praxis verwendeten Präparation sind zu begründen.
1.4.7 Konzentrationen und Formulierungen von Testsubstanzen
Normalerweise wird mehr als eine Konzentration der Testsubstanz verwendet; dies deckt die Obergrenze der potenziell beim Menschen auftretenden Expositionswerte ab. Analog ist die Durchführung von Tests mit verschiedenen typischen Formulierungen in Betracht zu ziehen.
1.4.8 Aufbringen auf die Haut
Unter den normalen Bedingungen der Chemikalienexposition beim Menschen kommen üblicherweise finite Dosen vor. Bei Applikationen, bei denen die Exposition beim Menschen nachgestellt wird, sind bei Feststoffen normalerweise 1-5 mg/cm2 Haut und bei Flüssigkeiten bis zu 10 µl/cm2 anzusetzen. Die verwendete Menge ist durch die zu erwartenden Einsatzbedingungen, die Ziele der Studie und die physikalischen Kenngrößen der Testpräparation zu begründen. So kann das Aufbringen auf die Haut beispielsweise in infiniter Form erfolgen, wenn größere Volumina je Flächeneinheit aufgebracht werden.
1.4.9 Temperatur
Die passive Diffusion der Chemikalien (und damit ihre Hautresorption) wird durch die Temperatur beeinflusst. Die Diffusionskammer und die Haut müssen auf konstanter Temperatur in Nähe der normalen Hauttemperatur von 32 ± 1 °C gehalten werden. Bei unterschiedlichem Zellaufbau sind auch unterschiedliche Wasserbad- oder Heizblocktemperaturen erforderlich, damit sich der Rezeptor bzw. die Haut innerhalb der physiologischen Normbedingungen befindet. Die Luftfeuchte sollte möglichst zwischen 30 und 70 % liegen.
1.4.10 Dauer der Exposition und Probenahme
Die Exposition der Haut gegenüber der Testpräparation kann sich über die gesamte Versuchsdauer oder über kürzere Zeiträume erstrecken (z.B. zur Nachahmung einer bestimmten Expositionsart beim Menschen). Überschüssige Rückstände der Testpräparation sind mit einem geeigneten Reinigungsmittel von der Haut abzuwaschen und die Spülrückstände zur Analyse aufzufangen. Das Verfahren zur Entfernung der Testpräparation ist von den erwarteten Einsatzbedingungen abhängig und ist zu begründen. Normalerweise ist eine 24-stündige Probenahmephase erforderlich, um das Resorptionsprofil angemessen charakterisieren zu können. Da bereits nach 24 Stunden eine Verschlechterung der Hautbeschaffenheit eintreten kann, sollte die Probenahmedauer normalerweise einen Zeitraum von 24 Stunden nicht überschreiten. Bei Testsubstanzen, die rasch in die Haut eindringen, ist dies möglicherweise nicht erforderlich, bei Testsubstanzen mit einer sehr langsamen Penetrationsgeschwindigkeit ist evtl. ein längerer Untersuchungszeitraum erforderlich. Die Probenahmefrequenz der Rezeptorflüssigkeit muss so ausgelegt sein, dass das Resorptionsprofil der Testsubstanz grafisch dargestellt werden kann.
1.4.11 Abschließende Schritte
Alle Komponenten des Testsystems sind zu analysieren, und die Rückgewinnungsrate ist zu ermitteln. Hierin einbezogen werden die Spenderkammer, die Spülung der Hautoberfläche, das hergestellte Hautstück und die Rezeptorflüssigkeit/-kammer. In bestimmten Fällen kann die Haut in den der Testsubstanz ausgesetzten Hautbereich und den Hautbereich unter dem Zellenflansch sowie zu separaten Analysen in die Hornhaut, die Epidermis und die Dermis aufgeteilt werden.
1.4.12 Analyse
In sämtlichen Studien sollte eine angemessene Rückgewinnungsrate angestrebt werden (anzustreben ist ein Mittelwert von 100 ± 10 % der radioaktiven Substanz; etwaige Abweichungen sind zu begründen). Die Menge der Testsubstanz in der Rezeptorflüssigkeit, in dem hergestellten Hautstück, den Waschrückständen von der Hautoberfläche und dem Spülwasser aus dem Versuchsaufbau sind durch ein geeignetes Verfahren zu analysieren.
2. Daten
Die Analyse der Rezeptorflüssigkeit, die Verteilung der Testsubstanz im Testsystem und der zeitliche Verlauf des Resorptionsprofils sind darzustellen. Im Fall einer Exposition mit finiten Dosen sind die von der Haut abgewaschene Menge, die der Haut zugeordnete Menge (und die Menge in den verschiedenen Lagen der Haut, sofern diese analysiert werden) und die in der Rezeptorflüssigkeit enthaltene Menge (Geschwindigkeit und Menge bzw. Prozentsatz der applizierten Dosis) zu berechnen. Die Hautresorption kann in bestimmten Fällen auch nur anhand der Rezeptorflüssigkeitsdaten alleine ausgedrückt werden. Verbleibt die Testsubstanz am Ende der Studie jedoch in der Haut, muss sie evtl. in die resorbierte Gesamtmenge einbezogen werden (siehe Abschnitt 66 in Quelle (3)). Im Fall einer Exposition mit infiniten Dosen kann mithilfe der Daten die Permeabilitätskonstante (Kp) ermittelt werden. Unter den letzteren Bedingungen ist der resorbierte Prozentsatz nicht relevant.
3. Berichterstattung
3.1 Abschlussbericht
Der Testbericht muss die im Protokoll angegebenen Anforderungen erfüllen und eine Begründung für das verwendete Testsystem sowie folgende Einzelangaben enthalten:
Testsubstanz:
- physikalische Beschaffenheit, physikalischchemische Eigenschaften (zumindest Molgewicht und log Pow), Reinheit (radiochemische Reinheit);
- Kenndaten (z.B. Chargennummer);
- Löslichkeit in der Rezeptorflüssigkeit.
Testpräparation:
- Formulierung und Begründung für die Verwendung;
- Homogenität.
Prüfbedingungen:
- Herkunft und Lage der Haut, Herstellungsmethode, Lagerungsbedingungen vor der Verwendung, etwaige Vorbehandlung (Reinigung, Antibiotikabehandlungen usw.), Messung der Unversehrtheit der Haut, Stoffwechselstatus, Begründung der Verwendung;
- Zellenaufbau, Zusammensetzung der Rezeptorflüssigkeit, Durchflussrate der Rezeptorflüssigkeit bzw. Probenahmezeiten und -verfahren;
- Details für die Aufbringung der Testpräparation und Quantifizierung der aufgebrachten Dosis;
- Expositionsdauer;
- Details zur Entfernung der Testpräparation von der Haut, z.B. Spülen der Haut;
- Details zur Hautanalyse und zu den zum Nachweis der Verteilung in der Haut verwendeten Fraktionierungstechniken;
- Verfahren zum Waschen der Zelle und der Geräte;
- Testmethoden, Extraktionsverfahren, Detektionsgrenzen und Validierung der Analysenmethoden.
Ergebnisse:
- Gesamtrückgewinnungsrate aus dem Experiment (aufgebrachte Dosis = Waschrückstände von der Haut + Haut + Rezeptorflüssigkeit + Waschrückstände aus der Zelle);
- tabellarische Darstellung der rückgewonnenen Mengen für die einzelnen Zellen in jeder Zellenkammer;
- Resorptionsprofil;
- tabellarische Darstellung der Resorptionsdaten (als Rate, Betrag oder Prozentwert angegeben).
Erörterung der Ergebnisse.
Schlussfolgerungen.
4. Literaturhinweise
1. Testing Method B.44. Skin Absorption: In vivo Method.
2. OECD (2002). Guidance Document for the Conduct of Skin Absorption Studies. OECD, Paris.
3. OECD (2000). Report of the Meeting of the OECD Extended Steering Committee for Percutaneous Absorption Testing, Annex 1 to ENV/JM/TG(2000)5. OECD, Paris.
4. Kemppainen B.W and Reifenrath W.G. (1990). Methods for skin absorption. CRC Press, Boca Raton.
5. Bronaugh R.L. and Collier, S.W. (1991). Protocol for In vitro Percutaneous Absorption Studies, in In vitro Percutaneous Absorption: Principles, Fundamentals and Applications, R.L. Bronaugh and H.I. Maibach, Eds., CRC Press, Boca Raton, 237-241.
6. Bronaugh R.L. and Maibach H.I.. (1991). In vitro Percutaneous Absorption: Principles, Fundamentals and Applications. CRC Press, Boca Raton.
7. European Centre for Ecotoxicology and Toxicology of Chemicals (1993). Monograph No. 20, Percutaneous Absorption, ECETOC, Brussels.
8. Diembeck W., Beck H., Benech-Kieffer F., Courtellemont P., Dupuis J., Lovell W., Paye M., Spengler J., Steiling W. (1999). Test Guidelines for In Vitro Assessment of Dermal Absorption and Percutaneous Penetration of Cosmetic Ingredients, Fd Chem Tox, 37, 191-205.
9. Recommended Protocol for In vitro Percutaneous Absorption Rate Studies (1996). US Federal Register, Vol. 61, No. 65.
10. Howes D., Guy R., Hadgraft J., Heylings J.R. et al. (1996). Methods for assessing Percutaneous absorption. ECVAM Workshop Report ATLA 24, 81 R10.
11. Schaefer H. and Redelmeier T.E. (1996). Skin barrier: principles of percutaneous absorption. Karger, Basel.
12. Roberts M.S. and Walters K.A. (1998). Dermal absorption and toxicity assessment. Marcel Dekker, New York.
13. Jewell, C., Heylings, J.R., Clowes, H.M. and Williams, F.M. (2000). Percutaneous absorption and metabolism of dinitrochlorobenzene in vitro. Arch Toxicol 74: 356-365.
Abbildung 1 Beispiel für den typischen Aufbau einer statischen Diffusionszelle für perkutane In-vitro-Resorptionsstudien
B.46. In vitro-Hautreizung: Prüfmethode Mit rekonstruierter humaner Epidermis 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
1. Diese Prüfmethode (PM) entspricht der OECD-Prüfrichtlinie 439 (2015). Als Hautreizung wird das Auslösen einer reversiblen Hautschädigung nach Applikation einer Prüfchemikalie für die Dauer von bis zu 4 Stunden [ Definition nach dem Global Harmonisierten System (GHS) zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN)](1) und nach Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen (CLP-Verordnung) 1 bezeichnet. Die vorliegende Prüfmethode besteht in einem Invitro-Verfahren, das zur Ermittlung schädlicher Wirkungen von hautreizenden Chemikalien (Stoffen und Gemischen) gemäß UN GHS und CLP-Verordnung Kategorie 2 (2) verwendet werden kann. In Regionen, in denen die optionale UN-GHS-Kategorie 3 (leichte Reizstoffe) nicht übernommen wurde, kann diese Prüfmethode auch zur Identifizierung nicht klassifizierter Chemikalien verwendet werden. Je nach Rechtsrahmen und verwendetem Klassifizierungssystem kann diese Prüfmethode daher im Rahmen einer Prüfstrategie zur Ermittlung des Hautreizungspotenzials von Chemikalien entweder als eigenständige Ersatzprüfung zur Invivo-Untersuchung auf Hautreizungen oder als teilweise Ersatzprüfung verwendet werden (3). 2. Die Bewertung von Hautreizungen erfolgte in der Regel unter Verwendung von Labortieren [PM B.4, entsprechend OECD TG 404, ursprünglich angenommen im Jahr 1981 und geändert in den Jahren 1992, 2002 und 2015] (4). Für die Untersuchung auf Hautverätzungen wurden drei validierte Invitro-Prüfmethoden als EU PM B.40 (entsprechend OECD TG 430), PM B.40bis (entsprechend OECD TG 431) und PM B.65 (entsprechend OECD TG 435) angenommen (5) (6) (7). In einem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA = Integrated Approaches to Testing and Assessment) zur Untersuchung von Hautverätzungen und -reizungen werden mehrere Module beschrieben, in denen Informationsquellen und Analyseinstrumente zu Gruppen zusammengefasst werden. Sie enthalten i) Leitlinien dazu, wie vorhandene Daten aus Prüfungen und aus anderen Quellen integriert und verwendet werden können, um das Hautreizungs- und das Hautverätzungspotenzial von Chemikalien zu bewerten, und ii) einen Vorschlag zur Durchführung ggf. erforderlicher weiterer Prüfungen (3). 3. Mit dieser Prüfmethode werden Hautreizungen mit dem Endpunkt der menschlichen Gesundheit untersucht. Sie beruht auf dem Invitro-Prüfsystem zur Untersuchung rekonstruierter humaner Epidermis (RhE), die die biochemischen und physiologischen Eigenschaften der menschlichen Oberhaut (d. h. der Epidermis) weitgehend widerspiegelt. Im RhE-Prüfsystem werden menschliche nicht transformierte Keratinozyten als zelluläres Ausgangsmaterial zur Rekonstruktion eines Epidermismodells mit repräsentativer Histologie und Zellarchitektur verwendet. Leistungsstandards (PS) erleichtern die Validierung und Bewertung ähnlicher und modifizierter Prüfmethoden, die auf der Verwendung von RhE und der Einhaltung der Grundsätze des OECD-Leitliniendokuments Nr. 34 beruhen (8)(9). Die entsprechende OECD-Prüfrichtlinie wurde ursprünglich 2010 angenommen und 2013 unter Berücksichtigung weiterer Prüfmethoden unter Verwendung von RhE-Modellen zunächst im Jahr 2013 und anschließend nochmals im Jahr 2015 unter Verweis auf das IATA-Leitliniendokument und unter Einführung der Verwendung eines alternativen Verfahrens zur Messung der Viabilität aktualisiert. 4. Zu vier im Handel erhältlichen Invitro-Prüfmodellen wurden auf dem RhE-Prüfsystem (Sensitivität 80 %, Spezifizität 70 % und Genauigkeit 75 %) beruhende Vorvalidierungs-, Optimierungs- und Validierungsstudien durchgeführt (10) (11) (12) (13) (14) (15) (16) (17) (18) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28). Diese vier Prüfmodelle werden in diese Prüfmethode aufgenommen und in Anlage 2 genannt, die auch Informationen zur Art der zur Validierung der jeweiligen Prüfmethoden verwendeten Validierungsstudien enthält. Wie in Anlage 2 erläutert, wurden diese Prüfmethode und die Leistungsstandards mit der validierten Referenzmethode (VRM) entwickelt (8). 5. Die gegenseitige Anerkennung der Daten gemäß dem OECD-Übereinkommen wird nur dann für nach den Leistungsstandards (8) validierte Prüfmodelle garantiert, wenn diese Prüfmodelle von der OECD geprüft und angenommen wurden. Die in diese Prüfmethode aufgenommenen Prüfmodelle und die entsprechende OECD TG können gleichermaßen verwendet werden, um sowohl länderbezogene Anforderungen an die Prüfergebnisse der Invitro-Prüfmethoden zur Untersuchung von Hautreizungen zu erfüllen als auch die gegenseitige Anerkennung von Daten beanspruchen zu können. 6. In diesem Dokument gelten die Begriffsbestimmungen in Anlage 1. Ausgangsüberlegungen und Begrenzungen 7. Eine Begrenzung der Prüfmethode, wie durch die umfassende prospektive Validierungsstudie zur Bewertung und Beschreibung von RhE-Prüfmethoden (16) nachgewiesen, besteht insofern, als keine Einstufung von Stoffen in die optionale Kategorie 3 (leichte Reizstoffe) des UN-GHS-Systems möglich ist (1). Daher wird mit dem Rechtsrahmen in den Mitgliedstaaten festgelegt, wie diese Prüfmethode zu verwenden ist. Die EU hat Kategorie 3 nicht in die CLP-Verordnung aufgenommen. Eine umfassende Evaluierung lokaler Auswirkungen auf die Haut nach einer einmaligen dermalen Exposition ist dem OECD-Leitliniendokument über integrierte Prüfungs- und Bewertungsansätze (IATA) zu entnehmen (3). Die Verwendung menschlicher Haut unterliegt anerkanntermaßen ethischen Überlegungen und Bedingungen auf nationaler und internationaler Ebene. 8. Mit dieser Prüfmethode werden Hautreizungen mit dem Endpunkt der menschlichen Gesundheit untersucht. Obwohl diese Prüfmethode keine angemessenen Informationen über Hautätzungen liefert, wird darauf hingewiesen, dass PM B.40bis (entsprechend OECD-Prüfrichtlinie 431) über Hautätzung auf demselben Prüfsystem auf der Grundlage eines RhE-Modells beruht, aber ein anderes Protokoll vorsieht (Kapitel 6). Die vorliegende Prüfmethode beruht auf Modellen unter Verwendung menschlicher Keratinozyten, die in vitro das Zielorgan der zu schützenden Spezies repräsentieren. Darüber hinaus bildet sie den Anfangsschritt der Entzündungskaskade bzw. des Wirkmechanismus ab (Zell- und Gewebeschädigung mit resultierendem lokalisiertem Trauma), der während der Reizung in vivo auftritt. Für die dieser Prüfmethode zugrunde liegende Validierung wurden Chemikalien unterschiedlicher Klassen untersucht. In der dieser Prüfmethode zugrunde liegenden Validierung wurden zahlreiche Chemikalien geprüft, und die Datenbank der Validierungsstudie umfasste insgesamt 58 Chemikalien (16) (18) (23). Die Prüfmethode ist auf Feststoffe, Flüssigkeiten, halbfeste Stoffe und Wachse anwendbar. Flüssigkeiten können wässrig oder nicht wässrig und Feststoffe in Wasser löslich oder unlöslich sein. Sofern möglich, sollten Feststoffe vor der Applikation zu Feinpulver gemahlen werden; eine weitere Behandlung der Probe ist nicht nötig. Gase und Aerosole wurden bislang in keiner Validierungsstudie bewertet (29). Obwohl deren Prüfung mit der RhE-Technologie prinzipiell vorstellbar ist, erlaubt die vorliegende Prüfmethode das Untersuchen von Gasen und Aerosolen nicht. 9. Bevor die Prüfmethode für die Generierung von Daten für einen bestimmten Regulierungszweck verwendet wird, ist zu prüfen, ob sie für den beabsichtigten Zweck angemessene Ergebnisse liefern kann und, wenn dem so ist, warum. Solche Erwägungen entfallen, wenn die Prüfung des Gemischs rechtlich vorgeschrieben ist. Da Gemische jedoch vielfältigen Kategorien zuzurechnen sein und unterschiedliche Zusammensetzungen haben können, und da gegenwärtig nur begrenzte Informationen über die Prüfung von Gemischen verfügbar sind, sollten Prüfmethoden, bei denen nachgewiesen werden kann, dass sie für eine bestimmte Kategorie von Gemischen nicht geeignet sind (beispielsweise durch ein Verfahren entsprechend dem von Eskes u. a. 2012 vorgeschlagenen Verfahren (30)), für die betreffende Kategorie von Gemischen nicht verwendet werden. Vorsicht ist ebenfalls geboten, wenn festgestellt wird, dass diese Prüfmethode für spezifische Chemikalienklassen oder physikalisch-chemische Eigenschaften nicht geeignet ist. 10. Prüfchemikalien, die Licht im selben Spektrum absorbieren können wie MTT-Formazan, und Prüfchemikalien, die in der Lage sind, den lebenswichtigen Farbstoff MTT zu reduzieren (zu MTT-Formazan), können die Messungen der Zellviabilität stören und erfordern die Verwendung geeigneter Kontrollen zur Korrektur (Nummern 28-34). 11. Vorausgesetzt die Prüfung liefert ein eindeutiges Ergebnis, genügt ein einziger Prüflauf mit drei parallel geprüften Replikat-Geweben. Bei Grenzergebnissen, wie z.B. nicht übereinstimmenden Replikatmessungen und/oder einer mittleren prozentualen Viabilität von 50 ± 5 %, sollte ein zweiter Prüflauf in Betracht gezogen werden bzw. ein dritter bei abweichenden Ergebnissen der ersten beiden Prüfläufe. Prinzip der Prüfmethode 12. Die Prüfchemikalie wird oberflächlich auf ein dreidimensionales RhE-Modell aufgetragen, das aus nicht veränderten humanen epidermalen Keratinozyten besteht, die zu einem mehrschichtigen, ausdifferenzierten Modell humaner Epidermis kultiviert wurden. Das Modell besteht aus geordneten Basal-, Stachel- und Körnerzellschichten und einer mehrlagigen Hornschicht (stratum corneum), die interzelluläre lamellare Fettschichten enthält, deren dominierende Lipidklassen dem in vivo gefundenen Lipidmuster entsprechen. 13. Durch Chemikalien hervorgerufene Reizungen der Haut manifestieren sich hauptsächlich als Erytheme und Ödeme. Diese Symptome sind das Ergebnis einer Kaskade von Ereignissen, an deren Beginn das Eindringen der Chemikalien in das stratum corneum steht, in dem die Chemikalien die darunter liegenden Keratinozytenschichten schädigen können. Die beschädigten Zellen können entweder Entzündungsmediatoren freisetzen oder eine Entzündungskaskade induzieren, die auch auf die Zellen der Dermis wirken, insbesondere auf die Stromal- und Endothelzellen der Blutgefäße. Die Dilation und erhöhte Durchlässigkeit der Endothelzellen erzeugen letztendlich das festgestellte Erythem bzw. Ödem (29). Mit den Prüfmethoden unter Verwendung von RhE (ohne Vaskularisierung im Invitro-Prüfsystem) werden anhand der Zellviabilität die auslösenden Ereignisse in der Kaskade (z.B. Zell- bzw. Gewebeschäden) (16) (17) gemessen. 14. Die Zellviabilität an Modellen rekonstruierter menschlicher Epidermis wird durch Enzymkonversion des Vitalfarbstoffs MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazoliumbromid, Thiazolyl-Blau; CAS-Nummer 298-93-1] zu einem blauen Formazan-Salz gemessen, das nach seiner Extraktion aus Geweben quantifiziert wird (31). Reizstoffe werden anhand ihrer Fähigkeit erkannt, die Zellviabilität unter vorgegebene Schwellenwerte zu senken (d. h. ≤ 50 % bei Reizstoffen der UN-GHS-/CLP-Kategorie 2). Je nach Rechtsrahmen und Anwendbarkeit der Prüfmethode können Chemikalien, die eine Zellviabilität oberhalb des vorgegebenen Grenzwerts erzeugen, als nichtreizend eingestuft werden (d. h. > 50 %, keine Einstufung). Nachweis der Leistungsfähigkeit 15. Vor der routinemäßigen Anwendung eines der vier validierten RhE-Prüfmodelle nach dieser Prüfmethode (Anlage 2) sollten Labors die erforderliche technische Befähigung durch eine ordnungsgemäße Klassifizierung der zehn in Tabelle 1 empfohlenen Leistungsstoffe nachweisen. Wenn beispielsweise ein dort genannter Stoff nicht verfügbar ist, kann ein anderer Stoff verwendet werden, für den geeignete Invivo- und Invitro-Referenzdaten verfügbar sind (z.B. aus der Liste der Referenzchemikalien (8)), sofern die in Tabelle 1 beschriebenen Auswahlkriterien angewendet werden. Die Verwendung alternativer Leistungsstoffe sollte gerechtfertigt sein. 16. Im Rahmen des Nachweises der Leistungsfähigkeit sollten die Benutzer die vom Hersteller des Hautmodells spezifizierten Barriereeigenschaften der Gewebe nach Erhalt überprüfen. Dies ist besonders dann wichtig, wenn die Gewebe über große Entfernungen/Zeiträume transportiert werden. Sobald eine Methode erfolgreich etabliert und ihre Leistungsfähigkeit festgestellt und nachgewiesen wurde, ist diese Überprüfung nicht mehr routinemäßig erforderlich. Allerdings empfiehlt es sich auch bei routinemäßig angewandten Prüfmethoden, die Barriereeigenschaften in regelmäßigen Abständen zu kontrollieren. Tabelle 1 Leistungsstoffe 1
Verfahren 17. Im Folgenden werden die Elemente und Verfahren einer RhE-Prüfmethode zur Bewertung von Hautreizungen beschrieben. (Zu den Parametern der einzelnen Prüfmodelle siehe auch Anlage 3.) Für die vier Modelle, die die Anforderungen dieser Prüfmethode erfüllen, sind Standardarbeitsweisen (SOPs) verfügbar (32) (33) (34) (35). Elemente der RHE-Prüfmethode Allgemeine Bedingungen 18. Die Epithelschicht sollte aus normalen menschlichen Keratinozyten gebildet werden. Unter der funktionsfähigen Hornschicht (stratum corneum) sollten mehrere Lagen lebensfähiger Epithelzellen (Basalzellschicht, stratum spinosum, stratum granulosum) vorhanden sein. Die Hornschicht sollte mehrlagig sein und das zur Erzeugung einer funktionsfähigen Barriere essenzielle Lipidprofil aufweisen. Die Barriere muss robust genug sein, um das schnelle Eindringen zytotoxischer Referenzchemikalien wie Natriumdodecylsulfat (SDS) oder Triton X-100 zu verhindern. Die Barrierefunktion kann entweder durch Bestimmung der Konzentration, bei der eine Referenzchemikalie die Viabilität der Gewebe nach einer vorgegebenen Expositionsdauer um 50 % verringert (IC50), oder durch die Bestimmung der Expositionszeit bewertet werden, die erforderlich ist, um die Zellviabilität bei Anwendung der Referenzchemikalie in einer vorgegebenen festen Konzentration um 50 % zu reduzieren (ET50). Die Rückhalteeigenschaften des RhE-Modells müssen ausschließen, dass Material rund um die Hornschicht in lebensfähiges Gewebe eindringt und die Modellierung der Hautexposition beeinträchtigt. Das RhE-Modell sollte nicht mit Bakterien, Viren, Mykoplasma oder Pilzen kontaminiert sein. Funktionale Bedingungen Viabilität 19. Die Größenordnung der Viabilität wird mit der MTT-Prüfung bestimmt (31). Die lebensfähigen Zellen des RhE-Gewebemodells können den lebenswichtigen Farbstoff MTT zu einem blauen MTT-Formazan-Niederschlag reduzieren, der dann mit Isopropanol (oder einem ähnlichen Lösungsmittel) aus dem Gewebe extrahiert wird. Die optische Dichte (OD) des Extraktionslösungsmittels allein sollte ausreichend gering sein, d. h. OD < 0,1. Das extrahierte MTT-Formazan kann durch eine Standard-(OD)-Absorptionsmessung oder mit einem HPLC/UPLC-Spektrometrieverfahren (36) quantifiziert werden. Die Anwender des RhE-Modells sollten sicherstellen, dass jede Charge des verwendeten Modells die vorgegebenen Kriterien für die Negativkontrolle erfüllt. Der Entwickler/Hersteller des Hautmodells sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für die OD-Werte der Negativkontrolle (unter den Bedingungen der Prüfmethode zur Untersuchung von Hautreizungen) festlegen. Die Akzeptanzspanne der OD-Werte für die vier validierten RhE-Prüfmodelle dieser Prüfmethode sind Tabelle 2 zu entnehmen. Bei Durchführung einer HPLC/UPLC-Spektrophotometrie sollten die in Tabelle 2 genannten OD-Spannen der Negativkontrolle als Akzeptanzkriterium für die Negativkontrolle verwendet werden. Es sollte dokumentiert werden, dass die mit der Negativkontrolle behandelten Gewebe über den gesamten Expositionszeitraum stabil bleiben (bzw. vergleichbare Viabilitätsmessungen ergeben). Tabelle 2 Akzeptanzbereiche für OD-Werte der Negativkontrolle der Prüfmodelle dieser Prüfmethode
Barrierefunktion 20. Das stratum corneum und die Zusammensetzung seiner Fette sollten das schnelle Eindringen zytotoxischer Referenzchemikalien wie z.B. SDS oder Triton X-100 verhindern. Dies ist anhand der Werte für IC50 oder ET50 abzuschätzen (Tabelle 3). Morphologie 21. Bei der histologischen Untersuchung des RhE-Modells sollte nachgewiesen werden, dass das Modell eine der menschlichen Epidermis ähnliche Struktur (einschließlich mehrlagigem stratum corneum) aufweist. Reproduzierbarkeit 22. Die Ergebnisse der Positivkontrolle und der Negativkontrollen der Prüfmethode sollen die Reproduzierbarkeit über längere Zeit belegen. Qualitätskontrolle (QK) 23. Das RhE-Modell sollte nur dann verwendet werden, wenn der Entwickler/Hersteller nachweist, dass jede Charge des verwendeten Hautmodells bestimmten Freigabekriterien genügt, von denen die Kriterien der Viabilität (Nummer 19), der Barrierefunktion (Nummer 20) und der Morphologie (Nummer 21) die wichtigsten sind. Diese Daten sollten den Benutzern der Prüfmethode zur Verfügung gestellt werden, damit diese sie in ihre Prüfberichte aufnehmen können. Der Entwickler/Hersteller des Hautmodells (oder - bei Verwendung eines hauseigenen Modells - der Prüfer) sollte eine Akzeptanzspanne (oberer und unterer Grenzwert) für IC50 bzw. ET50 festlegen. Nur mit geeigneten Geweben erzielte Ergebnisse können für eine zuverlässige Vorhersage für die Einstufung von Reizwirkungen akzeptiert werden. Die Akzeptanzspanne der vier RhE-Prüfmodelle dieser Prüfmethode sind Tabelle 3 zu entnehmen. Tabelle 3 Beispiele für Chargenfreigabekriterien der Prüfmodelle dieser Prüfmethode im Rahmen der Qualitätskontrolle
Applikation der Prüf- und Kontrollchemikalien 24. Für jede Prüfchemikalie und für die Kontrollen bei jedem Prüflauf sollten mindestens drei Replikate verwendet werden. Bei flüssigen und festen Chemikalien sollte eine ausreichende Menge der Prüfchemikalie gleichmäßig auf die gesamte Oberfläche der Epidermis aufgetragen werden; überschüssige Dosen sind zu vermeiden, d. h. es sollten 26 bis 83 µl/cm2 bzw. mg/cm2 verwendet werden (siehe Anlage 3). Bei festen Chemikalien sollte die Epidermis-Oberfläche vor der Applikation mit deionisiertem oder destilliertem Wasser angefeuchtet werden, um guten Hautkontakt zu gewährleisten. Feststoffe sollten nach Möglichkeit als Feinpulver geprüft werden. In manchen Fällen kann ein Nylon-Maschenfilter zum gleichmäßigen Verteilen verwendet werden (siehe Anlage 3). Am Ende der Expositionszeit sollte die Prüfchemikalie mit wässriger Pufferlösung oder 0,9 % NaCl von der Hautoberfläche abgewaschen werden. Je nach verwendeten RhE-Prüfmodellen betragen die Expositionszeiträume zwischen 15 und 60 Minuten bei einer Inkubationstemperatur zwischen 20 und 37 °C. Diese Expositionszeiträume und -temperaturen wurden für die jeweiligen RhE-Prüfmethoden optimiert und tragen den unterschiedlichen inhärenten Merkmalen der Prüfmodelle (z.B. der Barrierefunktion) Rechnung (siehe Anlage 3). 25. Bei jedem Prüflauf sollten gleichzeitig Negativkontrollen und Positivkontrollen (PK) verwendet werden, um nachweisen zu können, dass die Viabilität (mit der NK), die Barrierefunktion und die daraus resultierende Empfindlichkeit der Gewebe (mit der PK) innerhalb einer vorgegebenen historischen Akzeptanzspanne liegen. Als PK-Chemikalie wird 5 %iges wässriges SDS empfohlen. Als NK empfehlen sich Wasser oder Phosphatpuffer (PBS). Zellviabilitätsmessungen 26. Bei dem Prüfverfahren ist von wesentlicher Bedeutung, dass die Viabilitätsmessung nicht unmittelbar nach der Exposition gegenüber der Prüfchemikalie, sondern nach einer ausreichend langen Inkubationszeit des gewaschenen Gewebes (nach der Behandlung) in einem frischen Medium erfolgt. Während dieser Zeit kann sich das Gewebe von milden zytotoxischen Wirkungen erholen bzw. deutliche zytotoxische Effekte können sich herausbilden. Bei der Optimierung von zwei der RhE-Prüfmodelle dieser Prüfmethode hat sich eine 42-stündige Inkubation nach der Behandlung als optimal erwiesen (11) (12) (13) (14) (15). 27. Die MTT-Prüfung ist eine standardisierte quantitative Methode, die zur Messung der Zellviabilität nach dieser Prüfmethode angewandt werden sollte. Sie ist zur Anwendung in einem dreidimensionalen Gewebemodell geeignet. Die Gewebeprobe wird für 3 Stunden in eine MTT-Lösung mit geeigneter Konzentration (z.B. 0,3-1 mg/ml) gelegt. Das MTT wird von den lebensfähigen Zellen in blaues Formazan umgewandelt. Der blaue Formazan-Niederschlag wird sodann mit Hilfe eines Lösungsmittels (z.B. Isopropanol, Säure-Isopropanol) aus dem Gewebe extrahiert, und die Formazankonzentration wird durch Bestimmung des OD-Werts bei 570 nm innerhalb einer Filter-Bandbreite von maximal ± 30 nm oder mit einem HPLC/UPLC-Spektrophotometrieverfahren gemessen (Nummer 34) (36). 28. Optische Eigenschaften der Prüfchemikalie oder ihre chemische Wirkung auf MTT (z.B. die Tatsache, dass Chemikalien Färbungen nicht nur bewirken, sondern auch verhindern oder umkehren können) können die Versuche beeinträchtigen und damit zu falschen Viabilitätsschätzungen führen. Dies kann der Fall sein, wenn eine bestimmte Prüfchemikalie nicht komplett von dem Gewebe abgewaschen wurde oder wenn sie in die Epidermis eindringt. Wirkt sich die Prüfchemikalie unmittelbar auf MTT aus (MTT-Reduktionsmittel), ist sie natürlich gefärbt oder verfärbt sie sich während der Gewebebehandlung, so sollten zum Nachweis und zur Korrektur einer etwaigen Interferenz der Prüfchemikalie mit der Viabilitätsmesstechnik zusätzliche Kontrollen verwendet werden (Nummern 29 und 33). Für eine genaue Beschreibung der richtigen Durchführung der Korrektur der direkten MTT-Reduktion und der Interferenzen durch Färbemittel siehe die Standardarbeitsanweisungen für die vier validierten Prüfmodelle dieser Prüfmethode (32) (33) (34) (35). 29. Um direkte MTT-Reduktionsmittel zu identifizieren, sollten die Prüfchemikalien jeweils zu frisch hergestellter MTT-Lösung hinzugegeben werden. Wenn sich das MTT-Gemisch mit der Prüfchemikalie blau/violett verfärbt, ist davon auszugehen, dass die Prüfchemikalie das MTT direkt reduziert; anschließend sollte unabhängig von der Durchführung der Standardabsorptions-(OD-)Messung oder einer HPLC/UPLC-Spektrophotometrie eine weitere Funktionsprüfung der nicht lebensfähigen RhE-Gewebe vorgenommen werden. Bei dieser zusätzlichen Funktionsprüfung werden abgetötete Gewebe mit nur noch residualer metabolischer Aktivität eingesetzt, die die Prüfchemikalie in ähnlicher Weise wie lebensfähige Gewebe absorbieren. Jede MTT reduzierende Prüfchemikalie wird auf mindestens zwei abgetötete Gewebereplikate aufgetragen, die dem gesamten Prüfverfahren unterzogen werden, um eine nicht spezifische MTT-Reduktion (NSMTT) zu bewirken (32) (33) (34) (35). Pro Prüfchemikalie ist unabhängig von der Anzahl der durchgeführten unabhängigen Prüfungen/Prüfläufe eine einzelne NSMTT-Kontrolle ausreichend. Anschließend wird die tatsächliche Viabilität des Gewebes als Prozentanteil der bei lebenden Geweben nach Exposition gegenüber dem MTT-Reduktionsmittel gemessenen Viabilität abzüglich des Prozentanteils der nicht spezifischen MTT-Reduktion durch dasselbe MTT-Reduktionsmittel bei abgetöteten Geweben bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung durchgeführte Negativkontrolle (%NSMTT) berechnet. 30. Um potenzielle Interferenzen durch Prüffarbstoffe oder durch Prüfchemikalien zu ermitteln, die sich verfärben, wenn sie mit Wasser oder mit Isopropanol in Berührung kommen, und um über die Notwendigkeit weiterer Kontrollen entscheiden zu können, sollte die Prüfchemikalie in Wasser (Umgebung während der Exposition) und/oder Isopropanol (Extraktionslösung) einer Spektralanalyse unterzogen werden. Wenn die Prüfchemikalie in Wasser und/oder Isopropanol Licht im Bereich von 570 ± 30 nm absorbiert, sollten weitere Farbkontrollen durchgeführt oder eine HPLC/UPLC-Spektrophotometrie vorgenommen werden. Im letztgenannten Fall sind diese Kontrollen nicht erforderlich (Nummern 33 und 34). Bei Durchführung der Standardabsorptions-(OD-)Messung wird jeder interferierende Prüffarbstoff auf mindestens zwei lebensfähige Gewebereplikate aufgetragen; diese Replikate werden dem vollständigen Prüfverfahren unterzogen, aber während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert, um eine nicht spezifische Farbkontrolle (NSClebend) herzustellen. Die NSClebend-Kontrolle muss gleichzeitig mit der Prüfung der gefärbten Prüfchemikalie durchgeführt werden, und bei mehreren Prüfungen ist wegen der inhärenten biologischen Variabilität lebender Gewebe für jede durchgeführte Prüfung (bei jedem Prüflauf) eine unabhängige NSClebend-Kontrolle durchzuführen. Die tatsächliche Viabilität des Gewebes wird dann als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit der MTT-Lösung abzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die bei gleichzeitig mit der zu korrigierenden Prüfung untersuchten lebenden Geweben nach Exposition gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist (%NSClebend). 31. Wenn festgestellt wurde, dass Prüfchemikalien sowohl eine direkte MTT-Reduktion (Nummer 29) als auch Farbinterferenzen (Nummer 30) bewirken, wird bei der Durchführung der Standardabsorptions-(OD-)Messung neben den in vorstehenden Nummern beschriebenen Kontrollen (NSMTT und NSClebend) eine dritte Kontrollreihe benötigt. Dies ist gewöhnlich bei dunklen Prüfchemikalien der Fall, die beim MTT-Versuch Interferenzen verursachen (z.B. blau, violett oder schwarz), weil deren Fähigkeit zur direkten MTT-Reduktion durch die inhärente Farbqualität dieser Chemikalien beeinträchtigt wird (Nummer 29). Diese Prüfchemikalien können Bindungen sowohl mit lebenden als auch mit abgetöteten Geweben eingehen. Daher kann bei der NSMTT-Kontrolle eine Korrektur nicht nur der potentiellen direkten MTT-Reduktion durch die Prüfchemikalie, sondern auch der Farbinterferenz infolge der Bindung der Prüfchemikalie an abgetötete Gewebe erfolgen. Dies kann eine doppelte Korrektur der Farbinterferenz zur Folge haben, da mit der NSClebend-Kontrolle bereits die Farbinterferenz aufgrund der Bindung der Prüfchemikalie an lebende Gewebe erfolgt. Um eine mögliche doppelte Korrektur von Farbinterferenzen zu vermeiden, muss eine dritte Kontrolle mit nicht spezifischen Farben mit abgetöteten Geweben (NSCabgetötet) untersucht werden. Bei dieser zusätzlichen Kontrolle wird die Prüfchemikalie auf mindestens zwei abgetötete Gewebereplikate aufgetragen, die dem vollständigen Prüfverfahren zu unterziehen sind, wobei die Gewebe jedoch während der MTT-Inkubation nicht mit der MTT-Lösung, sondern mit einem Medium inkubiert werden. Unabhängig von der Anzahl der durchgeführten unabhängigen Prüfungen/Prüfläufe ist pro Prüfchemikalie eine einzige NSCabgetötet-Kontrolle hinreichend. Diese Kontrolle sollte jedoch gleichzeitig mit der NSMTT-Kontrolle sowie möglichst mit derselben Gewebecharge durchgeführt werden. Die tatsächliche Viabilität des Gewebes wird dann als Prozentanteil der ermittelten Viabilität bei lebendem Gewebe nach der Exposition gegenüber der Prüfchemikalie abzüglich %NSMTT und abzüglich %NSClebend zuzüglich des Prozentanteils der nicht spezifischen Farbe berechnet, die nach Exposition der abgetöteten Gewebe gegenüber der interferierenden Prüfchemikalie und nach Inkubation mit einem Medium ohne MTT entstanden ist; dieser Prozentanteil wird bezogen auf die gleichzeitig mit der zu korrigierenden Prüfung untersuchte Kontrolle (%NSCabgetötet) ermittelt. 32. Wichtig ist die Feststellung, dass nicht spezifische MTT-Reduktionen und nicht spezifische Farbinterferenzen bei den Gewebextrakten Ergebnisse oberhalb der Linearitätsspanne des Photospektrometers begünstigen können. Daher sollte jedes Labor vor der Untersuchung von Prüfchemikalien für rechtliche Zwecke die Linearitätsspanne seines Spektrophotometers mit im Handel bezogenem MTT-Formazan (CAS-Nr. 57360-69-7) ermitteln. Durch die Standardabsorptions-(OD-)Messung mit einem Spektrophotometer können direkte MTT-Reduktionsmittel und Farbinterferenzen verursachende Prüfchemikalien in den Fällen untersucht werden, in denen die mit einer Prüfchemikalie ermittelten und nicht um die direkte MTT-Reduktion und/oder um Farbinterferenzen korrigierten ODs der Gewebeextrakte innerhalb der Linearitätsspanne des Spektrophotometers liegen oder in denen der nicht korrigierte Prozentanteil der Viabilität der Prüfchemikalie bereits ≤ 50 % ist. Ergebnisse bei Prüfchemikalien mit %NSMTT und/oder %NSClebend > 50 % der Negativkontrolle sind jedoch mit Sorgfalt zu bewerten, da dies der Schwellenwert ist, nach dem zwischen klassifizierten und nicht klassifizierten Chemikalien unterschieden wird (Nummer 36). 33. Wenn die Ergebnisse von Prüffarbstoffen wegen übermäßiger Interferenzen beim MTT-Versuch mit der Standardabsorptions-(OD-)Messung nicht vereinbar sind, kann alternativ MTT-Formazan durch HPLC/UPLC-Spektrophotometrie gemessen werden (Nummer 34) (36). Bei der HPLC/UPLC-Spektrophotometrie kann das MTT-Formazan vor der Quantifizierung von der Prüfchemikalie getrennt werden (36). Daher werden unabhängig von der zu prüfenden Chemikalie keine NSClebend- oder NSCabgetötet-Kontrollen benötigt, wenn eine HPLC/UPLC-Spektrophotometrie vorgenommen wird. NSMTT-Kontrollen sollten jedoch verwendet werden, wenn vermutet wird, dass eine Prüfchemikalie MTT direkt reduziert oder wenn die Farbe einer Prüfchemikalie die Beurteilung ihrer Fähigkeit zur direkten MTT-Reduktion beeinträchtigt (Nummer 29). Wenn MTT-Formazan durch HPLC/UPLC-Spektrophotometrie gemessen wird, ist der Prozentanteil der Viabilität des Gewebes als Prozentanteil der MTT-Formazan-Peak-Fläche, die mit lebenden Geweben nach Exposition gegenüber der Prüfchemikalie ermittelt wurde, bezogen auf die MTT-Formazan-Peak-Fläche der gleichzeitigen Negativkontrolle zu berechnen. Bei Prüfchemikalien, die MTT direkt reduzieren können, wird die tatsächliche Gewebeviabilität als Prozentanteil der Gewebeviabilität von lebenden Geweben nach der Exposition gegenüber der Prüfchemikalie abzüglich %NSMTT berechnet. Und schließlich ist festzustellen, dass auch direkte MTT-Reduktionsmittel (die nach der Behandlung in den Geweben gebunden sein und MTT so stark reduzieren können, dass es (bei Standard-OD-Messungen) zu ODs oder (bei der Untersuchung der Gewebeextracte durch UPLC-/HPLC-Spektrophotometrie) zu Peakflächen außerhalb der Linearitätsspanne des Spektrophotometers und zu Farbinterferenzen kommen kann) nicht bewertet werden können. Solche Fälle sind allerdings sehr selten. 34. MTT-Formazan-Messungen können bei allen Arten von Prüfchemikalien (gefärbt, nicht gefärbt, MTT-Reduktionsmittel und Mittel, die keine MTT-Reduktion bewirken) auch durch HPLC/UPLC-Spektrophotometrie vorgenommen werden (36). Wegen der Vielfalt der HPLC/UPLC-Spektrophotometriesysteme sollte die Eignung des jeweiligen HPLC/UPLC-Spektrophotometriesystems vor der Verwendung zur Quantifizierung von MTT-Formazan in Gewebextrakten durch Erfüllung der Akzeptanzkriterien für eine Reihe von Standard-Qualifikationsparametern auf der Grundlage der Branchenleitlinien der US-amerikanischen Food and Drug Administration (FDA) für die Validierung bioanalytischer Methoden nachgewiesen werden (36)(37). Diese Schlüsselparameter sowie die jeweiligen Akzeptanzkriterien sind Anlage 4 zu entnehmen. Wenn die in Anlage 4 beschriebenen Akzeptanzkriterien erfüllt sind, wird das betreffende HPLC/UPLC-Spektrophotometriesystem als geeignet für die Messung von MTT-Formazan unter den in dieser Prüfmethode beschriebenen Versuchsbedingungen betrachtet. Akzeptanzkriterien 35. Bei jeder Prüfung mit validen Chargen des betreffenden RhE-Modells (siehe Absatz 23) sollten mit der Negativkontrolle behandelte Gewebe OD-Werte aufweisen, die die Gewebequalität nach abgeschlossenen Beförderungs- und Annahmevorgängen und sämtlichen Schritten des Protokolls belegen. Die OD-Werte der Kontrollen sollten nicht unter historisch etablierten unteren Grenzwerten liegen. Ähnlich sollte auch nachgewiesen werden, dass mit der PK, d. h. mit 5 %igem wässrigen SDS, behandelte Gewebe unter den Bedingungen der Prüfmethode (siehe Anlage 3 sowie die Standardarbeitsanweisungen für die vier Prüfmodelle dieser Prüfmethode (32) (33) (34) (35)) auf eine hautreizende Chemikalie reagieren können. Entsprechende und geeignete Maße der Variabilität zwischen Gewebereplikaten (d. h. Standardabweichungen (SD)) sollten innerhalb der für das verwendete Prüfmodell festgelegten Akzeptanzgrenzen liegen (siehe Anlage 3). Auswertung der Ergebnisse und Prädiktionsmodell 36. Die für jede Prüfchemikalie erhaltenen OD-Werte können zur Berechnung einer prozentualen Zellviabilität im Vergleich zur Negativkontrolle, die auf 100 % festgesetzt ist, herangezogen werden. Wenn eine HPLC/UPLC-Spektrophotometrie durchgeführt wird, ist der Prozentanteil der Viabilität des Gewebes als Prozentanteil der MTT-Formazan-Peak-Fläche, die mit lebenden Geweben nach Exposition gegenüber der Prüfchemikalie ermittelt wurde, bezogen auf die MTT-Formazan-Peak-Fläche der gleichzeitigen Negativkontrolle zu berechnen. Der Grenzwert der prozentualen Zellviabilität, der zwischen Reizstoff und bisher nicht klassifizierten Prüfchemikalien unterscheidet, und das (die) statistische(n) Verfahren zur Auswertung der Ergebnisse und zur Identifizierung von Reizstoffen sollten genau definiert und dokumentiert werden und nachweislich geeignet sein (weitere Informationen siehe Standardarbeitsanweisungen zu den Prüfmodellen). Die Grenzwerte für die Vorhersage der Reizung sind nachstehend angegeben:
Daten und Berichterstattung Daten 37. Für jeden Prüflauf sollten Daten aus einzelnen Replikatgeweben (z.B. OD-Werte und Daten über die berechnete prozentuale Zellviabilität für jede Prüfchemikalie, einschließlich Einstufung), gegebenenfalls auch Daten aus Wiederholungsversuchen, tabellarisch zusammengefasst werden. Darüber hinaus sollten für jeden Prüflauf die Mittelwerte ± Standardabweichung übermittelt werden. Außerdem sollten für jede geprüfte Chemikalie festgestellte Interaktionen mit dem MTT-Reagens und Prüffarbstoffen berichtet werden. Prüfbericht 38. Der Prüfbericht sollte folgende Angaben enthalten: Prüfchemikalien und Kontrollchemikalien
Verwendetes RhE-Modell und verwendetes Protokoll und Gründe für die Auswahl (wenn relevant) Prüfbedingungen:
Prüfverfahren:
Akzeptanzkriterien für Prüfläufe und Prüfungen:
Ergebnisse:
Erörterung der Ergebnisse Schlussfolgerungen Literatur (1) Vereinte Nationen (UN) (2013). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), fünfte überarbeitete Ausgabe, UN New York und Genf, 2013. Abrufbar unter:http://www.unece.org/trans/danger/publi/ghs/ghs_rev05/05files_e.html. (2) EURL-ECVAM (2009). Erklärung zur "Performance Under UN GHS of Three In Vitro Assays for Skin Irritation Testing and the Adaptation of the Reference Chemicals and Defined Accuracy Values of the ECVAM Skin Irritation Performance Standards", herausgegeben vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM (ESAC31), 9. April 2009. Abrufbar unter:https://eurl-ecvam.jrc.ec.europa.eu/about-ecvam/archive-publications/publication//ESAC31_skin-irritation-statement_20090922.pdf. (3) OECD (2014). Guidance Document on Integrated Approaches to Testing and Assessment for Skin Irritation/Corrosion. Environment, Health and Safety Publications, Series on Testing and Assessment (No 203), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (4) Kapitel B.4 dieses Anhangs, Akute Hautreizung/-verätzung. (5) Kapitel B.40 dieses Anhangs, Invitro-Hautverätzung: TER-Prüfmethode. (6) Kapitel B.40bis dieses Anhangs, Invitro-Hautverätzung: Prüfmethode mit rekonstruierter humaner Epidermis (RHE). (7) Kapitel B.65 dieses Anhangs, Invitro-Methode zur Untersuchung der Membranbarriere. (8) OECD (2015). Performance Standards for the Assessment of Proposed Similar or Modified In Vitro Reconstructed Human Epidermis (RhE) Test Methods for Skin Irritation in Relation to TG 439. Environment, Health, and Safety Publications, Series on Testing and Assessment (No 220). Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (9) OECD (2005). Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment. Environment, Health and Safety Publications, Series on Testing and Assessment (No 34), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (10) Fentem, J.H., Briggs, D., Chesné, C., Elliot, G.R., Harbell, J.W., Heylings, J.R., Portes, P., Roguet, R., van de Sandt, J.J. M. und Botham, P. (2001). A Prevalidation Study on In Vitro Tests for Acute Skin Irritation, Results and Evaluation by the Management Team, Toxicol. in Vitro 15, 57-93. (11) Portes, P., Grandidier, M.-H., Cohen, C. und Roguet, R. (2002). Refinement of the EPISKIN Protocol for the Assessment of Acute Skin Irritation of Chemicals: Follow-Up to the ECVAM Prevalidation Study, Toxicol. in Vitro 16, 765-770. (12) Kandárová, H., Liebsch, M., Genschow, E., Gerner, I., Traue, D., Slawik, B. und Spielmann, H. (2004). Optimisation of the EpiDerm Test Protocol for the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests, ALTEX 21, 107-114. (13) Kandárová, H., Liebsch, M., Gerner, I., Schmidt, E., Genschow, E., Traue, D. und Spielmann, H. (2005), The EpiDerm Test Protocol for the Upcoming ECVAM Validation Study on In Vitro Skin Irritation Tests - An Assessment of the Performance of the Optimised Test, ATLA 33, 351-367. (14) Cotovio, J., Grandidier, M.-H., Portes, P., Roguet, R. und Rubinsteen, G. (2005). The In Vitro Acute Skin Irritation of Chemicals: Optimisation of the EPISKIN Prediction Model Within the Framework of the ECVAM Validation Process, ATLA 33, 329-349. (15) Zuang, V., Balls, M., Botham, P.A., Coquette, A., Corsini, E., Curren, R.D., Elliot, G.R., Fentem, J.H., Heylings, J.R., Liebsch, M., Medina, J., Roguet, R., van De Sandt, J.J.M., Wiemann, C. und Worth, A. (2002). Follow-Up to the ECVAM Prevalidation Study on In Vitro Tests for Acute Skin Irritation, The European Centre for the Validation of Alternative Methods Skin Irritation Task Force report 2, ATLA 30, 109-129. (16) Spielmann, H., Hoffmann, S., Liebsch, M., Botham, P., Fentem, J., Eskes, C., Roguet, R., Cotovio, J., Cole, T., Worth, A., Heylings, J., Jones, P., Robles, C., Kandárová, H., Gamer, A., Remmele, M., Curren, R., Raabe, H., Cockshott, A., Gerner, I. und Zuang, V. (2007). The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Report on the Validity of the EPISKIN and EpiDerm Assays and on the Skin Integrity Function Test, ATLA 35, 559-601. (17) Hoffmann, S. (2006). ECVAM Skin Irritation Validation Study Phase II: Analysis of the Primary Endpoint MTT and the Secondary Endpoint IL1-±. (18) Eskes, C., Cole, T., Hoffmann, S., Worth, A., Cockshott, A., Gerner, I. und Zuang, V. (2007). The ECVAM International Validation Study on In Vitro Tests for Acute Skin Irritation: Selection of Test Chemicals, ATLA 35, 603-619. (19) Cotovio, J., Grandidier, M.-H., Lelièvre, D., Roguet, R., Tinois-Tessonneaud, E. und Leclaire, J. (2007). In Vitro Acute Skin Irritancy of Chemicals Using the Validated EPISKIN Model in a Tiered Strategy - Results and Performances with 184 Cosmetic Ingredients, ALTEX, 14, 351-358. (20) EURL-ECVAM (2007). Statement on the Validity of In Vitro Tests for Skin Irritation, Issued by the ECVAM Scientific Advisory Committee (ESAC26), 27. April 2007. Abrufbar unter:https://eurl-ecvam.jrc.ec.europa.eu/about-ecvam/archive-publications/publication//ESAC26_statement_SkinIrritation_20070525_C.pdf. (21) EURL-ECVAM. (2007). Performance Standards for Applying Human Skin Models to In Vitro Skin Irritation Testing. Hinweis: Dies sind die ursprünglichen Leistungsstandards für die Validierung von zwei Prüfmethoden. Diese Leistungsstandards sollten nicht mehr verwendet werden, da inzwischen eine aktualisierte Fassung (8) verfügbar ist. (22) EURL-ECVAM. (2008). Statement on the Scientific Validity of In Vitro Tests for Skin Irritation Testing, Issued by the ECVAM Scientific Advisory Committee (ESAC29), 5. November 2008.https://eurl-ecvam.jrc.ec.europa.eu/about-ecvam/archive-publications/publication/ESAC_Statement_SkinEthic-EpiDerm-FINAL-0812-01.pdf. (23) OECD (2010). Explanatory Background Document to the OECD Draft Test Guideline on In Vitro Skin Irritation Testing. Environment, Health and Safety Publications. Environment, Health and Safety Publications, Series on Testing and Assessment, (No 137), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (24) Katoh, M., Hamajima, F., Ogasawara, T. und Hata, K. (2009). Assessment of Human Epidermal Model LabCyte EPI-MODEL for In Vitro Skin Irritation Testing According to European Centre for the Validation of Alternative Methods (ECVAM)-Validated Protocol, J Toxicol Sci, 34, 327-334 (25) Katoh, M. und Hata, K. (2011). Refinement of LabCyte EPI-MODEL24 Skin Irritation Test Method for Adaptation to the Requirements of OECD Test Guideline 439, AATEX, 16, 111-122 (26) OECD (2011). Validation Report for the Skin Irritation Test Method Using LabCyte EPI-MODEL24. Environment, Health and Safety Publications, Series on Testing and Assessment (No 159), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (27) OECD (2011). Peer Review Report of Validation of the Skin Irritation Test Using LabCyte EPI-MODEL24. Environment, Health and Safety Publications, Series on Testing and Assessment (No 155), Organisation für wirtschaftliche Zusammenarbeit und Entwicklung, Paris. (28) Kojima, H., Ando, Y., Idehara, K., Katoh, M., Kosaka, T., Miyaoka, E., Shinoda, S., Suzuki, T., Yamaguchi, Y., Yoshimura, I., Yuasa, A., Watanabe, Y. und Omori, T. (2012). Validation Study of the In Vitro Skin Irritation Test with the LabCyte EPI-MODEL24, Altern Lab Anim, 40, 33-50. (29) Welss, T., Basketter, D.A. und Schröder, K.R. (2004). In Vitro Skin Irritation: Fact and Future. State of the Art Review of Mechanisms and Models, Toxicol. In Vitro 18:231-243. (30) Eskes, C., u. a. (2012). Regulatory Assessment of In Vitro Skin Corrosion and Irritation Data within the European Framework: Workshop Recommendations. Regul.Toxicol.Pharmacol. 62, 393-403). (31) Mosmann, T. (1983). Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays, J. Immunol. Methods 65, 55-63. (32) EpiSkinTM (Februar 2009). SOP, Version 1.8ECVAM Skin Irritation Validation Study: Validation of the EpiSkinTM Test Method 15 min - 42 hours for the Prediction of acute Skin Irritation of Chemicals (33) EpiDermTM (geändert im März 2009). SOP, Version 7.0, Protocol for: In Vitro EpiDermTM Skin Irritation Test (EPI-200-SIT), for Use with MatTek Corporation"s Reconstructed Human Epidermal Model EpiDerm (EPI-200). (34) SkinEthicTM RHE (February 2009) SOP, Version 2.0, SkinEthic Skin Irritation Test-42bis Test Method for the Prediction of Acute Skin Irritation of Chemicals: 42 Minutes Application + 42 Hours Post-Incubation. (35) LabCyte (June 2011). EPI-MODEL24 SIT SOP, Version 8.3, Skin Irritation Test Using the Reconstructed Human Model "LabCyte EPI-MODEL24" (36) Alépée, N., Barroso, J., De Smedt, A., De Wever, B., Hibatallah, J., Klaric, M., Mewes, K.R., Millet, M., Pfannenbecker, U., Tailhardat, M., Templier, M. und McNamee, P. Use of HPLC/UPLC-Spectrophotometry for Detection of MTT Formazan in In Vitro Reconstructed Human Tissue (RhT)-Based Test Methods Employing the MTT Assay to Expand their Applicability to Strongly Coloured Test Chemicals. Manuskript in Vorbereitung. (37) US FDA (2001). Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration. Mai 2001. Abrufbar unter: [http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf]. (38) Harvell, J.D., Lamminstausta, K. und Maibach, H.I. (1995). Irritant Contact Dermatitis, in: Practical Contact Dermatitis, S. 7-18 (Hrsg. Guin, J. D.). Mc Graw-Hill, New York. (39) EURL-ECVAM (2009). Performance Standards for in vitro skin irritation test methods based on Reconstructed human Epidermis (RhE). Hinweis: Dies ist die aktuelle Fassung der ECVAM-Leistungsstandards, aktualisiert im Jahr 2009 angesichts der Einführung des UN GHS. Diese Leistungsstandards sollten nicht mehr verwendet werden, da inzwischen eine aktualisierte Fassung (8) verfügbar ist. (40) EURL-ECVAM. (2009). ESAC Statement on the Performance Standards (PS) for In Vitro Skin Irritation Testing Using Reconstructed Human Epidermis, Issued by the ECVAM Scientific Advisory Committee (ESAC31), 8. Juli 2009. (41) EG (2001). Richtlinie 2001/59/EG der Kommission vom 6. August 2001 zur 28. Anpassung der Richtlinie 67/548/EWG des Rates zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten für die Einstufung, Verpackung und Kennzeichnung gefährlicher Stoffe an den technischen Fortschritt, Amtsblatt der Europäischen Union L 255, S. 1.
1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. L 353 vom 31.12.2008 S. 1). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.47 Trübungs- und Durchlässigkeitstest an der Rinderhornhaut zwecks Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern 17 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 437 (2013). Der Trübungs- und Durchlässigkeitstest an der Rinderhornhaut (Bovine Corneal Opacity and Permeability, BCOP) wurde vom Organisationsübergreifenden Koordinationsausschuss zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods, ECVAM) und des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese Centre for the Validation of Alternative Methods, JaCVAM) in den Jahren 2006 und 2010 evaluiert (1) (2). Im Rahmen der ersten Evaluierung wurde der BCOP-Test im Hinblick auf seine Eignung zur Identifizierung von Chemikalien (Stoffen und Gemischen) überprüft, die schwere Augenschäden verursachen 1. Im Rahmen der zweiten Evaluierung wurde der BCOP-Test im Hinblick auf seine Eignung zum Nachweis von Chemikalien (Stoffen und Gemischen) überprüft, die nicht als augenreizende oder schwer augenschädigende Stoffe eingestuft wurden (2). Die BCOP-Validierungsdatenbank enthielt insgesamt 113 Stoffe und 100 Gemische (2)(3). Auf der Grundlage dieser Evaluierungen und zugehöriger Peer-Reviews wurde die Schlussfolgerung gezogen, dass sich die Prüfmethode sowohl zum Nachweis von Chemikalien (Stoffen und Gemischen) eignet, die schwere Augenschäden verursachen (Kategorie 1), als auch von Chemikalien, bei denen keine Einstufung aufgrund von Augenreizung oder schweren Augenschäden gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (4) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 erforderlich ist, weshalb sie für beide Zwecke als wissenschaftlich gültig anerkannt wurde. Unter schweren Augenschäden sind Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges zu verstehen, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Prüfchemikalien, die schwere Augenschäden hervorrufen, werden als Stoffe der UN-GHS-Kategorie 1 eingestuft. Chemikalien, die nicht als augenreizende oder schwer augenschädigende Stoffe eingestuft werden, sind als Stoffe definiert, die nicht den Anforderungen zur Einstufung in die UN-GHS-Kategorie 1 oder 2 (2A oder2B) entsprechen, d. h. sie werden als Stoffe der UN-GHS-Kategorie "Keine Einstufung" bezeichnet. Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des BCOP-Tests auf Grundlage der zugehörigen Evaluierungen. Zu den wichtigsten Unterschieden zwischen der ursprünglichen Fassung der OECD-Prüfrichtlinie aus dem Jahr 2009 und der aktualisierten Fassung von 2013 gehören u. a. die Verwendung der BCOP-Prüfmethode zur Identifizierung von Chemikalien, die keine Einstufung nach dem UN-GHS-Klassifizierungssystem erfordern (Nummern 2 und 7), Klarstellungen zur Anwendbarkeit der BCOP-Prüfmethode für die Prüfung von Alkoholen, Ketonen und Feststoffen (Nummern 6 und 7) sowie von Stoffen und Gemischen (Nummer 8), Klarstellungen zur Art der Prüfung von oberflächenaktiven Stoffen und tensidhaltigen Gemischen (Nummer 28), Aktualisierungen und Klarstellungen zu Positivkontrollen (Nummern 39 und 40), eine Aktualisierung der Entscheidungskriterien der BCOP-Prüfmethode (Nummer 47), eine Aktualisierung der Gültigkeitskriterien der Studie (Nummer 48), eine Aktualisierung der Bestandteile des Prüfberichts (Nummer 49), eine Aktualisierung der Anlage 1 zu Definitionen, die Ergänzung von Anlage 2 zur Vorhersagefähigkeit der BCOP-Prüfmethode unter verschiedenen Klassifizierungssystemen, eine Aktualisierung der Anlage 3 zur Liste der Leistungschemikalien und eine Aktualisierung der Anlage 4 zum BCOP-Hornhauthalter (Nummer 1) und zum Opazimeter (Nummern 2 und 3). Gegenwärtig herrscht allgemeiner Konsens darüber, dass der In-vivo-Draize-Augentest in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Augenreizungen für verschiedene Chemikalienklassen vorherzusagen. Unter Umständen ist es jedoch möglich, den Draize-Augentest durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie zu ersetzen (5). Der Top-Down-Ansatz (5) wird verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie ein hohes Reizpotenzial aufweist. Umgekehrt wird der Bottom-Up-Ansatz (5) verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie keine ausreichende Augenreizung erzeugt und somit keine Einstufung erfordert. Die BCOP-Prüfmethode ist eine In-vitro-Prüfmethode, die unter bestimmten Bedingungen und mit bestimmten Grenzen zur Einstufung und Kennzeichnung von Chemikalien mit augengefährdender Wirkung angewendet werden kann. Auch wenn der Test den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird die BCOP-Prüfmethode als erster Schritt im Rahmen einer Prüfstrategie wie des von Scott et al. vorgeschlagenen Top-Down-Ansatzes (5) empfohlen, um ohne weitere Tests (4) schwer augenschädigende Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, zu identifizieren. Die BCOP-Prüfmethode wird auch für die Identifizierung von Chemikalien empfohlen, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS ("Keine Einstufung") (4) erfordern, und kann daher im Rahmen einer Prüfstrategie z.B. mit Bottom-up-Ansatz verwendet werden (5). Im Falle einer Chemikalie, bei der anhand der BCOP-Prüfmethode nicht vorhergesagt werden kann, dass sie schwere Augenschäden verursacht oder keine Einstufung als augenreizend/schwer augenschädigend erfordert, müssten jedoch weitere Tests (in vitro und/oder in vivo) durchgeführt werden, um eine eindeutige Einstufung vornehmen zu können. Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie, gemessen als ihre Fähigkeit, bei isolierten Rinderhornhäuten Trübungs- und verstärkte Durchlässigkeitseffekte hervorzurufen. Toxische Effekte für die Hornhaut sind messbar durch: i) verminderte Lichtübertragung (Trübung) und ii) den verstärkten Durchtritt von Fluorescein-Natrium-Farbstoff (Durchlässigkeit). Die Trübungs- und Durchlässigkeitsbewertungen der Hornhaut nach Applikation einer Prüfchemikalie ergeben in Kombination einen In-vitro-Reizwert (In Vitro Irritancy Score, IVIS), der für die Einstufung des Reizpotenzials der Prüfchemikalie maßgeblich ist. Definitionen sind Anlage 1 zu entnehmen. Vorbemerkungen und Einsatzgrenzen Diese Prüfmethode basiert auf dem BCOP-Testprotokoll des Organisationsübergreifenden Koordinationsausschusses zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) (6)(7), das ursprünglich auf Informationen des Instituts für In-vitro- Forschung (Institute for In Vitro Sciences, IIVS) und auf dem INVITTOX-Protokoll 124 (8) beruht. Letzteres Protokoll wurde für die von der Europäischen Gemeinschaft finanzierte Prävalidierungsstudie aus den Jahren 1997-1998 herangezogen. Beide Protokolle beruhen auf der BCOP-Prüfmethodik, wie sie von Gautheron et al. (9) erstmals beschrieben wurde. Die BCOP-Prüfmethode kann zur Identifizierung von schwer augenschädigenden Chemikalien gemäß UN-GHS-Definition, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, angewendet werden (4). Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist die BCOP-Prüfmethode für diesen Zweck eine allgemeine Genauigkeit von 79 % (150/191), eine Falsch-Positiv-Rate von 25 % (32/126) und eine Falsch-Negativ-Rate von 14 % (9/65) auf (3) (siehe Anlage 2, Tabelle 1). Werden Prüfchemikalien einer bestimmten Chemikalienklasse (Alkohole, Ketone) oder mit bestimmten physikalischen Eigenschaften (Feststoffe) aus der Datenbank ausgeschlossen, so liegt die allgemeine Genauigkeit der BCOP-Prüfmethode gemessen am UN-GHS-Klassifizierungssystem bei 85 % (111/131), die Falsch-Positiv-Rate bei 20 % (16/81) und die Falsch-Negativ-Rate bei 8 % (4/50) (3). Potenzielle Mängel der BCOP-Prüfmethode bei der Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) gründen auf hohen Falsch-Positiv-Raten für Alkohole und Ketone und hohen Falsch-Negativ-Raten für Feststoffe, die in den Validierungsdaten beobachtet wurden (1)(2)(3). Da jedoch nicht für alle Alkohole und Ketone durch die BCOP-Prüfmethode zu hohe Werte vorhergesagt werden und einige korrekt als Stoffe der UN-GHS-Kategorie 1 eingestuft werden, werden diese beiden organischen funktionellen Gruppen nicht als Stoffe außerhalb des Anwendungsbereichs dieser Prüfmethode betrachtet. Es muss also bei der Anwendung dieser Prüfmethode entschieden werden, ob eine gegebenenfalls zu hohe Vorhersage für einen Alkohol oder ein Keton vertretbar ist oder ob weitere Tests mit einem evidenzbasierten Bewertungsansatz durchgeführt werden sollten. Bezüglich der Falsch-Negativ-Raten für Feststoffe ist zu beachten, dass Feststoffe beim In-vivo-Draize-Augenreizungstest zu variablen und extremen Expositionsbedingungen führen können, aus denen sich möglicherweise irrelevante Vorhersagen über das tatsächliche Reizungspotenzial ableiten (10). Es ist ferner zu beachten, dass keine der Falsch-Negativ-Raten, die in den ICCVAM-Validierungsdaten (2)(3) im Zusammenhang mit der Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) festgestellt wurden, im Ergebnis zu einem IVIS ≤ 3 führten, dem Kriterium zur Identifizierung einer Prüfchemikalie als Stoff der UN-GHS-Klasse "Keine Einstufung" . Des Weiteren gelten Falsch-Negativ-Raten der BCOP-Prüfmethode in diesem Zusammenhang nicht als kritisch, da alle Prüfchemikalien mit einem Wert von 3 < IVIS ≤ 55 nachfolgend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequentiellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz, anhand weiterer, angemessen validierter In-vitro-Tests oder als letzte Option an Kaninchen getestet werden würden. Angesichts der Tatsache, dass einige feste Chemikalien anhand der BCOP-Prüfmethode korrekterweise als Stoffe der UN-GHS-Kategorie 1 vorhergesagt werden, wird auch nicht davon ausgegangen, dass diese physikalischen Eigenschaften außerhalb des Anwendungsbereichs dieser Prüfmethode liegen. Prüfer können diese Prüfmethode für alle Arten von Chemikalien einsetzen und ein IVIS-Wert > 55 sollte als Indikator für eine schwer augenschädigende Wirkung gelten, nach der eine Einstufung in UN-GHS-Kategorie 1 ohne weitere Tests vorzunehmen ist. Wie bereits erwähnt, sollten Positivergebnisse bei Verwendung von Alkoholen oder Ketonen angesichts des Risikos der Vorhersage zu hoher Werte jedoch mit einer gewissen Zurückhaltung interpretiert werden. Die BCOP-Prüfmethode kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN-GHS-Klassifizierungssystem erfordern (4). Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist die BCOP-Prüfmethode für diesen Zweck eine allgemeine Genauigkeit von 69 % (135/196), eine Falsch-Positiv-Rate von 69 % (61/89) und eine Falsch-Negativ-Rate von 0 % (0/107) auf (3) (siehe Anlage 2, Tabelle 2). Die erhaltene Falsch-Positiv-Rate (In-vivo-Chemikalien der UN-GHS-Klasse "Keine Einstufung" mit einem IVIS-Wert > 3, siehe Nummer 47) ist zwar recht hoch, gilt in diesem Zusammenhang jedoch nicht als kritisch, da alle Prüfchemikalien mit einem Wert von 3 < IVIS ≤ 55 nachfolgend, je nach den gesetzlichen Anforderungen und unter Verwendung einer sequentiellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz, anhand weiterer, angemessen validierter In-vitro-Tests oder als letzte Option an Kaninchen getestet werden würden. Die BCOP-Prüfmethode weist keine nennenswerten Mängel für die Untersuchung von Alkoholen, Ketonen und Feststoffen auf, sofern das Ziel die Identifizierung von Chemikalien ist, die keine Einstufung als augenreizend oder schwer augenschädigend (UN-GHS-Klasse "Keine Einstufung") erfordern (3). Prüfer können diese Prüfmethode jedoch für alle Arten von Chemikalien einsetzen, und ein Negativergebnis (IVIS ≤ 3) sollte als Indikator dafür gelten, dass keine Einstufung vorzunehmen ist (UN-GHS-Klasse "Keine Einstufung"). Da mit der BCOP-Prüfmethode nur 31 % der Chemikalien richtig identifiziert werden können, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern, sollte diese Prüfmethode nicht die erste Wahl sein, wenn als erster Schritt ein Bottom-up-Ansatz verwendet werden soll (5), sofern andere validierte und akzeptierte In-vitro-Verfahren mit ähnlich hoher Empfindlichkeit, jedoch einer höheren Spezifität zur Verfügung stehen. Die BCOP-Validierungsdatenbank enthielt insgesamt 113 Stoffe und 100 Gemische (2)(3). Die BCOP-Prüfmethode gilt demnach als für die Prüfung von Stoffen und Gemischen gleichermaßen anwendbar. Aufgrund der beträchtlichen Anzahl von Chemikalien der UN-GHS-Kategorie 1, die zu niedrig in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, und von Chemikalien, die keine Einstufung erfordern, jedoch zu hoch in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, wird die BCOP-Prüfmethode nicht für die Identifizierung von Prüfchemikalien empfohlen, die als augenreizend (d. h. UN-GHS-Kategorie 2 oder Kategorie 2A) oder leicht augenreizend (UN-GHS-Kategorie 2B) eingestuft werden sollten (2)(3). Diesbezüglich können weitere Tests mit einer anderen geeigneten Methode erforderlich sein. Bei allen Verfahren, die Rinderaugen und Rinderhornhäute involvieren, sollten die geltenden Regeln und Verfahrensvorschriften der Prüfanstalt für den Umgang mit Tiermaterial (u. a. Gewebe und Gewebeflüssigkeiten) eingehalten werden. Universelle Laborregeln sollten beachtet werden (11). Bindehaut- und Irisverletzungen werden bei der BCOP-Prüfmethode zwar außer Acht gelassen, doch werden Auswirkungen auf die Hornhaut berücksichtigt, die wesentliche Einflussfaktoren für die In-vivo-Klassifizierung nach dem UN-GHS-Klassifizierungssystem bilden. Die Reversibilität von Hornhautläsionen lässt sich per se mit der BCOP-Prüfmethode nicht beurteilen. Doch wurde ausgehend von Kaninchenaugenstudien vorgeschlagen, eine Bewertung der anfänglichen Tiefe der Hornhautverletzung heranzuziehen, um einige Arten irreversibler Wirkungen zu identifizieren (12). Allerdings sind weitere wissenschaftliche Erkenntnisse erforderlich, um zu verstehen, wie irreversible Wirkungen auftreten, die nicht mit einer anfänglichen starken Verletzung im Zusammenhang stehen. Schließlich sei auch erwähnt, dass das Potenzial für eine mit der Augenexposition verbundene systemische Toxizität nach der BCOP-Methode nicht bewertet werden kann. Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Beispielsweise können histopathologische Befunde potenziell nützlich sein, wenn eine umfassendere Charakterisierung der Hornhautschädigung erforderlich ist. Wie im OECD-Leitfaden Nr. 160 (13) umrissen, sollten Anwender Hornhäute aufbewahren und histopathologische Proben zubereiten, die zum Aufbau einer Datenbank und zur Entwicklung von Entscheidungskriterien herangezogen werden können, mit denen sich die Genauigkeit dieser Prüfmethode weiter verbessern lässt. Laboratorien, die diese Prüfmethode zum ersten Mal anwenden, sollten die in Anlage 3 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung der BCOP-Prüfmethode nachzuweisen, bevor sie BCOP-Testdaten für Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen. Die BCOP-Prüfmethode ist ein organtypisches Modell, das die normalen physiologischen und biochemikalischen Funktionen von Rinderhornhäuten in vitro kurzfristig aufrechterhält. Bei dieser Methode werden durch die Prüfchemikalie hervorgerufene Schäden bewertet, indem Veränderungen von Trübung und Durchlässigkeit der Hornhaut mithilfe eines Opazimeters bzw. eines VIS-Spektrofotometers quantitativ gemessen werden. Die beiden Messwerte dienen der Berechnung eines In-vitro-Reizwertes (IVIS), der seinerseits herangezogen wird, um zwecks Prädiktion des Augenreizpotenzials einer Prüfchemikalie am lebenden Objekt (in vivo) eine Zuordnung zu einer Gefahrenkategorie aufgrund des In-vitro-Reizungspotenzials des Stoffes vorzunehmen (siehe Entscheidungskriterien unter Nummer 48). Für die BCOP-Prüfmethode werden isolierte Hornhäute der Augen frisch geschlachteter Rinder verwendet. Die Hornhauttrübung wird quantitativ bestimmt als Menge der die Hornhaut durchdringenden Lichtstrahlung. Die Durchlässigkeit der Hornhaut wird quantitativ bestimmt als die Menge an Natrium-Fluorescein, die die gesamte Dicke der Hornhaut durchdringt und im Medium in der Hinterkammer nachgewiesen wird. Die Applikation der Prüfchemikalien auf die Epitheloberfläche der Hornhaut erfolgt durch Zugabe der Stoffe in die Vorderkammer des Hornhauthalters. Anlage 4 enthält eine Beschreibung sowie ein Schaubild einer Halterung, wie sie für die BCOP-Prüfmethode verwendet wird. Hornhauthalter sind im Handel erhältlich oder können als Bausatz bezogen werden. Bezugsquelle und Alter der Rinderaugen und Auswahl der Tiere Schlachtrinder werden in der Regel für den menschlichen Verzehr oder für andere gewerbliche Zwecke getötet. Nur gesunde und für die menschliche Nahrungskette geeignet befundene Tiere kommen als Bezugsquelle für Augenhornhäute für den BCOP-Test in Frage. Da Rinder je nach Rasse, Alter und Geschlecht eine große Gewichtsspanne aufweisen, gibt es keine Gewichtsempfehlung für die Tiere zum Zeitpunkt der Schlachtung. Augenhornhäute von Tieren verschiedener Altersklassen sind mitunter unterschiedlich groß. Hornhäute mit einem horizontalen Durchmesser > 30,5 mm und einer zentralen Hornhautdicke (CCT) mit Werten ≥ 1 100 µm stammen in der Regel von über acht Jahre alten Rindern, während Hornhäute mit einem horizontalen Durchmesser < 28,5 mm und CCT-Werten < 900 µm im Allgemeinen von weniger als fünf Jahre alten Tieren stammen (14). Deshalb werden Augen von über 60 Monate alten Tieren in der Regel nicht verwendet. Traditionsgemäß werden keine Augen von weniger als 12 Monate alten Rindern verwendet, da die Augen dieser Tiere noch nicht ausgewachsen sind und Hornhautdicke sowie Hornhautdurchmesser wesentlich kleiner sind als bei ausgewachsenen Rindern. Hornhäute von Jungtieren (d. h. Tiere im Alter von sechs bis 12 Monaten) sind aufgrund ihrer Vorteile jedoch zulässig: Sie sind beispielsweise leichter erhältlich, die Altersspanne ist geringer und das Laborpersonal ist in geringerem Maße BSE-gefährdet (15). Da eine weitere Evaluierung des Einflusses von Hornhautgröße oder Hornhautdicke auf die Wirkung verätzender oder reizender Stoffe nützlich wäre, sollten Anwender das geschätzte Alter und/oder das Gewicht der Tiere, von denen die für eine Studie verwendeten Hornhäute stammen, mitteilen. Gewinnung und Beförderung der Augen zum Labor Die Augen werden im Schlachthof gewonnen. Um jede mechanische und sonstige Schädigung der Augen auf ein Minimum zu beschränken, sollten die Augen nach der Tötung des Tieres so bald wie möglich ausgeschält und unmittelbar danach sowie während der Beförderung gekühlt werden. Damit die Augen nicht mit potenziellen Reizstoffen in Berührung kommen, sollte das Schlachthofpersonal zum Abspülen des Schädels keine Detergenzien verwenden. Die Augen sollten in einem angemessen großen Behältnis vollständig in HBSS eingelegt und so zum Labor befördert werden, dass sich ihr Zustand möglichst nicht verschlechtert und/oder es möglichst nicht zu einer bakteriellen Kontamination kommt. Da die Augen während des Schlachtprozesses gewonnen werden, könnten sie mit Blut und anderen biologischen Stoffen wie Bakterien und sonstigen Mikroorganismen in Berührung kommen. Deshalb muss unbedingt sichergestellt werden, dass das Kontaminationsrisiko auf ein Mindestmaß begrenzt wird (z.B. indem das für die Beförderung der Augen verwendete Behältnis auf Nasseis gelagert und die zur Lagerung der Augen während der Beförderung verwendete HBSS-Lösung mit Antibiotika (z.B. 100 IU/ml Penizillin und 100 µg/ml Streptomycin) angereichert wird). Der Zeitabstand zwischen der Gewinnung der Augen und der Verwendung der Hornhäute im BCOP-Test sollte möglichst gering sein (im Idealfall sollten die Hornhäute am selben Tag gewonnen und verwendet werden) und die Testergebnisse nachweislich nicht kompromittieren. Letztere basieren auf den Auswahlkriterien für die Augen sowie auf den Reaktionen der Positiv- und Negativkontrollen. Alle für die Prüfung verwendeten Augen sollten aus einer an ein und demselben Tag gewonnenen Partie Augen stammen. Auswahlkriterien für Augen, die im BCOP-Test eingesetzt werden Unmittelbar nach ihrer Ankunft im Labor werden die Augen sorgfältig auf Mängel wie unter anderem verstärkte Trübung, Kratzer und Neovaskularisation untersucht. Nur Hornhäute von Augen, die keine derartigen Mängel aufweisen, dürfen verwendet werden. Die Qualität jeder Hornhaut wird auch in späteren Testphasen geprüft. Hornhäute, die nach einer ersten einstündigen Äquilibrierung mehr als sieben Trübungseinheiten oder einen vergleichbaren Wert für das verwendete Opazimeter und die verwendeten Hornhauthalterungen aufweisen (ANMERKUNG: der Opazimeter sollte nach den Trübungsnormen kalibriert werden, die auch zur Festlegung der Trübungseinheiten verwendet werden; siehe Anlage 4), sind zu verwerfen. Jede Behandlungsgruppe (Prüfchemikalie sowie gleichzeitige Negativ- und Positivkontrollen) umfasst mindestens drei Augen. Für die Negativkontrollen des BCOP-Tests sollten drei Hornhäute verwendet werden. Da die Hornhäute operativ vom Augapfel entfernt und in die Hornhautkammern eingespannt werden, kann es infolge dieser Handgriffe bei einzelnen Hornhäuten zu veränderten Trübungs- und Durchlässigkeitswerten kommen (auch bei der Negativkontrolle). Außerdem werden die Trübungs- und Durchlässigkeitswerte von Hornhäuten der Negativkontrolle dazu verwendet, die Trübungs- und Durchlässigkeitswerte der mit der Prüfchemikalie behandelten Hornhäute und der Hornhäute der Positivkontrolle bei den IVIS-Berechnungen zu korrigieren. Vorbereitung der Augen Unbeschädigte Hornhäute werden seziert, wobei ein 2 bis 3 mm breiter Sklera-Rand belassen wird, um das anschließende Manipulieren zu erleichtern; dabei ist darauf zu achten, dass die Epithel- und Endothelzellschicht der Hornhaut nicht beschädigt wird. Die isolierten Hornhäute werden in speziell entwickelte Hornhauthalter eingespannt, die aus Vorder- und Hinterkammern bestehen, welche die Schnittstellen zur Epithel- bzw. Endothelseite der Hornhäute bilden. Beide Kammern (Hinterkammer zuerst) werden bis zum Überlaufen mit vorgewärmtem phenolrotfreiem EMEM-Medium gefüllt, wobei sicherzustellen ist, dass sich keine Luftblasen bilden. Das Gerät wird sodann bei 32 ± 11°C für mindestens eine Stunde äquilibriert, um die Hornhäute mit dem Medium zu äquilibrieren und so weit wie möglich eine normale Stoffwechseltätigkeit zu gewährleisten (die ungefähre Temperatur der Hornhautoberfläche beträgt in vivo 321 °C). Nach der Äquilibrierung wird frisches vorgewärmtes, phenolrotfreies EMEM-Medium in beide Kammern gegeben, und für jede Hornhaut werden Referenztrübungswerte abgelesen. Hornhäute, die makroskopische Gewebeschäden (z.B. Kratzer, Pigmentierung, Neovaskularisation) oder über sieben Trübungseinheiten bzw. einen für das verwendete Opazimeter und die verwendeten Hornhauthalterungen vergleichbaren Wert aufweisen, werden verworfen. Mindestens drei Hornhäute werden für die Negativ-(oder Lösungsmittel-)Kontrolle ausgewählt. Die restlichen Hornhäute werden in Behandlungs- und Positivkontrollgruppen aufgeteilt. Da die Wärmekapazität von Wasser höher ist als die Wärmekapazität von Luft, bietet Wasser stabilere Temperaturbedingungen für die Inkubation. Deshalb wird ein Wasserbad empfohlen, um Hornhauthalter samt Inhalt auf einer Temperatur von 32 ± 11 °C zu halten. Es können jedoch auch Luftinkubatoren verwendet werden, sofern alle erforderlichen Vorkehrungen getroffen wurden, um die Temperatur stabil zu halten (z.B. durch Vorwärmen der Halterungen und Medien). Applikation der Prüfchemikalie Es werden zwei unterschiedliche Behandlungsprotokolle verwendet - eines für Flüssigkeiten und Tenside (Feststoffe oder Flüssigkeiten) und eines für nichttensidische Feststoffe. Flüssigkeiten werden unverdünnt, halbfeste Stoffe, Cremes und Wachse werden in der Regel als Flüssigkeiten getestet. Unverdünnte Tenside werden in einer Konzentration von 10 % w/v in 0,9 % Natriumchloridlösung, destilliertem Wasser oder einem anderen Lösungsmittel, welches das Testsystem nachweislich nicht beeinträchtigt, getestet. Sofern alternative Lösungskonzentrationen verwendet werden, ist dies angemessen zu begründen. Tensidhaltige Gemische können unverdünnt getestet oder zunächst auf eine geeignete Konzentration entsprechend der jeweiligen In-vivo-Exposition verdünnt werden. Die getestete Konzentration ist angemessen zu begründen. Die Hornhäute werden den Flüssigkeiten und Tensiden für die Dauer von zehn Minuten ausgesetzt. Bei anderen Expositionszeiten sollten diese wissenschaftlich begründet werden. Vgl. Anlage 1 bezüglich einer Definition für "Tensid" und "tensidhaltiges Gemisch" . Nichttensidische Feststoffe werden in der Regel als Lösungen oder Suspensionen in einer Konzentration von 20 % w/v in 0,9 % Natriumchloridlösung, destilliertem Wasser oder einem anderem Lösungsmittel, welches das Testsystem nachweislich nicht beeinträchtigt, getestet. Unter bestimmten Bedingungen und sofern wissenschaftlich begründet, können Feststoffe auch unverdünnt durch direkte Applikation auf die Hornhautoberfläche getestet werden, wobei die offene Methode (open chamber method, siehe Nummer 32) angewandt wird. Die Hornhäute werden den Feststoffen für die Dauer von vier Stunden ausgesetzt; soweit wissenschaftlich fundiert können jedoch auch, wie bei Flüssigkeiten und Tensiden, alternative Expositionszeiten verwendet werden. Je nach den physikalischen und chemischen Eigenschaften der Prüfchemikalie (z.B. fest, flüssig, zähflüssig oder nicht zähflüssig) können unterschiedliche Behandlungsmethoden angewandt werden. Der kritische Faktor besteht darin sicherzustellen, dass die Prüfchemikalie die Epitheloberfläche angemessen bedeckt und während der Spülungen sachgemäß entfernt wird. Die geschlossene Methode (closed chamber method) wird in der Regel für nicht zähflüssige bis leicht zähflüssige Prüfchemikalien eingesetzt, während die offene Methode eher für halbzähflüssige und zähflüssige Prüfchemikalien sowie für unverdünnte Feststoffe verwendet wird. Bei der geschlossenen Methode wird über die Dosieröffnungen auf der Oberseite der Kammer so viel Prüfchemikalie (750 µl) in die Vorderkammer gegeben, dass die Epithelseite der Hornhaut bedeckt ist; die ¦ffnungen werden anschließend für die gesamte Expositionsdauer mit den Kammerverschlüssen abgedichtet. Es muss unbedingt sichergestellt werden, dass alle Hornhäute der Prüfchemikalie für eine angemessene Dauer ausgesetzt werden. Bei der offenen Methode werden vor der Behandlung Scheibenverschlussring und Glasscheibe von der Vorderkammer entfernt. Die Kontroll- oder die Prüfchemikalie (750 µl bzw. genÌgend Prüfchemikalie, um die Hornhaut komplett zu bedecken) wird mit einer Mikropipette direkt auf die Epitheloberfläche der Hornhaut appliziert. Erweist sich das Pipettieren einer Prüfchemikalie als schwierig, so kann diese zur leichteren Dosierung mittels Druckausgleich in eine Direktverdrängungspipette geladen werden. Die Spitze der Direktverdrängungspipette wird in die Dosierspitze der Spritze eingeführt, damit das Material unter Druck in die Spitze der Verdrängungspipette geladen werden kann. Lässt man den Bedienknopf langsam zurückgleiten, fährt der Kolben der Pipette nach oben. Treten in der Pipettenspitze Luftbläschen auf, wird die Prüfchemikalie verworfen (ausgestoßen) und der Prozess wird so lange wiederholt, bis die Spitze ohne Luftblasen gefüllt ist. Erforderlichenfalls kann eine normale Spritze (ohne Nadel) verwendet werden, denn sie gestattet das Abmessen einer akkuraten Menge Prüfchemikalie und erleichtert die Applikation auf die Epitheloberfläche der Hornhaut. Nach der Dosierung wird die Glasscheibe wieder auf die Vorderkammer gesetzt, um das System wieder zu schließen. Inkubation nach der Exposition Nach Ablauf der Expositionszeit werden Prüfchemikalie, Negativkontrolle oder Positivkontrolle aus der Vorderkammer entfernt, und das Epithel wird mindestens drei Mal (oder bis keine Prüfchemikalie mehr sichtbar ist) mit (phenolrothaltigem) EMEM-Medium gewaschen. Phenolrot wird zum Abspülen verwendet, da sich aufgrund der Farbveränderung bei Phenolrot die Wirksamkeit des Abspülens saurer oder alkalischer Prüfchemikalien bestimmen lässt. Die Hornhäute werden mehr als drei Mal gewaschen, wenn die Farbveränderung (ins Gelbe oder Lila) anhält oder wenn die Prüfchemikalie nach wie vor sichtbar ist. Sobald das Medium frei von der Prüfchemikalie ist, werden die Hornhäute ein letztes Mal mit EMEM-Medium (ohne Phenolrot) abgespült. Das EMEM-Medium (ohne Zusatz von Phenolrot) wird als letzte Spülung verwendet, um sicherzustellen, dass der Phenolrot-Farbstoff vor der Trübungsmessung aus der Vorderkammer entfernt wurde. Die Vorderkammer wird sodann wieder mit nicht phenolrothaltigem frischem EMEM-Medium aufgefüllt. Bei Flüssigkeiten oder Tensiden werden die Hornhäute nach dem Abspülen für weitere zwei Stunden bei 32 ± 11 °C inkubiert. Unter bestimmten Umständen könnte sich nach der Exposition eine längere Inkubationszeit als zweckdienlich erweisen; dies sollte auf Fallbasis geprüft werden. Mit Feststoffen behandelte Hornhäute werden nach Ablauf der vierstündigen Exposition gründlich abgespült; eine längere Inkubation ist nicht erforderlich. Nach Ablauf der auf die Exposition folgenden Inkubation (Flüssigkeiten und Tenside) bzw. nach Ablauf der vierstündigen Exposition (nichttensidische Feststoffe) werden Trübung und Durchlässigkeit der einzelnen Hornhäute aufgezeichnet. Außerdem wird jede Hornhaut visuell geprüft, und relevante Befunde (z.B. Gewebeabschälung, Reste der Prüfchemikalie, uneinheitliche Trübungsmuster) werden aufgezeichnet. Diese Daten könnten insofern wichtig sein, als sie Abweichungen der Opazimeter-Messwerte bestätigen könnten. Kontrollchemikalien Jeder Versuch beinhaltet gleichzeitige Negativ- oder Lösungsmittel-/Vehikelkontrollen und Positivkontrollen. Für Tests von Flüssigstoffen zu 100 % sieht der BCOP-Test eine gleichzeitige Negativkontrolle (z.B. 0,9 % Natriumchloridlösung oder destilliertes Wasser) vor, damit unspezifische Veränderungen im Testsystem festgestellt werden können und ein Referenzwert für die Test-Endpunkte ermittelt werden kann. Auf diese Weise wird auch sichergestellt, dass die Testbedingungen nicht zu einer unerwünschten Reizwirkung führen. Für Tests von verdünnten Flüssigkeiten, Tensiden oder Feststoffen sieht der BCOP-Test eine Gruppe gleichzeitiger Lösungsmittel-/Vehikelkontrollen vor, damit unspezifische Veränderungen im Testsystem festgestellt werden können und ein Referenzwert für die Test-Endpunkte ermittelt werden kann. Es sind nur Lösungsmittel/Vehikel zulässig, die das Testsystem nachweislich nicht beeinträchtigen. Jeder Versuch beinhaltet als gleichzeitige Positivkontrolle eine bekannte augenreizende Chemikalie, damit die Integrität des Testsystems und seine korrekte Durchführung überprüft werden können. Um sicherzustellen, dass im Zeitverlauf auftretende Wirkungsschwankungen bei der Positivkontrolle bewertet werden können, sollte die Reaktion jedoch nicht zu heftig sein. Beispiele für Positivkontrollen für flüssige Prüfchemikalien sind 100 %iges Ethanol oder 100 %iges Dimethylformamid. Für feste Prüfchemikalien käme 20 %iges (Gewichtsprozent) Imidazol in 0,9 % Natriumchloridlösung als Positivkontrolle in Frage. Referenzchemikalien sind nützlich für die Evaluierung des Augenreizpotenzials unbekannter Chemikalien einer bestimmten Chemikalien- oder Produktklasse oder zur Evaluierung des relativen Reizpotenzials eines Augenreizstoffes innerhalb einer spezifischen Spanne von Reizwirkungen. Gemessene Endpunkte Die Trübung wird durch Messung der durch die Hornhaut durchgehenden Lichtstrahlung bestimmt. Die quantitative Messung der Hornhauttrübung erfolgt mithilfe eines Opazimeters, eines Messgeräts, bei dem die Trübungswerte kontinuierlich gemessen werden. Die Durchlässigkeit wird durch Messung der Menge Fluorescein-Natrium bestimmt, die alle Zellschichten der Hornhaut (d. h. von der Epithelzellschicht auf der äußeren Hornhautoberfläche bis zur Endothelzellschicht auf der inneren Hornhautfläche) passiert. Die Vorderkammer des Hornhauthalters, die die Verbindung zur Epithelseite der Hornhaut bildet, wird mit 1 ml Fluorescein-Natrium-Lösung (4 oder 5 mg/ml bei Flüssigkeiten und Tensiden bzw. nichttensidischen Feststoffen) aufgefüllt, während die Hinterkammer, die die Verbindung zur Endothelseite der Hornhaut bildet, mit frischem EMEM-Medium gefüllt wird. Die Halterung wird sodann in horizontaler Position für 90 ± 5 Minuten bei 32 ± 11 °C inkubiert. Die Menge an Fluorescein-Natrium, die in die Hinterkammer eindringt, wird mithilfe eines UV/VIS-Spektrofotometers gemessen. Bei 490 nm ausgewertete spektrofotometrische Messungen werden als Werte für optische Dichte (OD490) oder Absorbanzwerte aufgezeichnet, die kontinuierlich gemessen werden. Zur Bestimmung der Fluorescein-Durchlässigkeit werden OD490-Werte herangezogen, die mithilfe eines VIS-Spektrofotometers bei einer Standardpfadlänge von 1 cm ermittelt wurden. Alternativ kann ein Plattenleser für 96-Mulden-Mikrotiterplatten verwendet werden, sofern i) der lineare Messbereich des Plattenlesers für die Bestimmung der Fluorescein- OD490-Werte festgelegt werden kann und ii) in der 96-Mulden-Mikrotiterplatte Fluorescein-Proben in der richtigen Menge verwendet werden, um OD490-Werte zu ergeben, die der Standardpfadlänge von 1 cm entsprechen (dies könnte vollständig gefüllte Mulden [in der Regel 360 µl] erfordern). Datenauswertung Sobald die Trübungs- und die mittleren Durchlässigkeits-(OD490-)Werte um die Hintergrundtrübungswerte und die OD490-Durchlässigkeitswerte der Negativkontrolle korrigiert wurden, werden die mittleren Trübungs- und OD490-Durchlässigkeitswerte für jede Behandlungsgruppe in einer empirisch abgeleiteten Formel kombiniert, um für jede Behandlungsgruppe einen In-vitro-Reizwert (IVIS) zu berechnen: IVIS = mittlerer Trübungswert + (15 x mittlerer OD490-Durchlässigkeitswert) Nach Sina et al. (16) wurde diese Formel aus Labor- und Interlaborstudien abgeleitet. Die für eine Serie von 36 Verbindungen in einer Multilaborstudie generierten Daten wurden einer multivariaten Analyse unterzogen, um die Best-fit-Gleichung zwischen In-vivo- und In-vitro-Daten zu ermitteln. Diese Analyse wurde von Wissenschaftlern zweier separater Unternehmen durchgeführt, die zu quasi identischen Gleichungsergebnissen gelangten. Die Trübungs- und Durchlässigkeitswerte sollten auch unabhängig voneinander bestimmt werden, um feststellen zu können, ob eine Prüfchemikalie für nur einen der beiden Endpunkte (siehe Entscheidungskriterien) eine augenverätzende oder stark augenreizende Wirkung verursachte. Entscheidungskriterien Die IVIS-Schwellenwerte zur Identifizierung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) und Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern (UN-GHS-Klasse "Keine Einstufung") sind nachfolgend aufgeführt:
Studienakzeptanzkriterien Ein Test gilt als akzeptabel, wenn die Positivkontrolle einen IVIS-Wert innerhalb von zwei Standardabweichungen des geltenden historischen Mittelwertes ergibt, der mindestens alle drei Monate bzw. immer dann aktualisiert werden muss, wenn Laboratorien, die nur selten Testungen vornehmen (d. h. weniger als einmal im Monat), einen akzeptablen Test durchführen. Die Negativ- oder Lösungsmittel-/Vehikelkontrollen sollten Trübungs- und Durchlässigkeitswerte ergeben, die geringer sind als die Obergrenzen, die für Hintergrundtrübungs- und Durchlässigkeitswerte für Rinderhornhäute vorgegeben sind, welche mit der jeweiligen Negativ- bzw. Lösungsmittel-/Vehikelkontrolle behandelt wurden. Kann eine eindeutige Klassifizierung vorgenommen werden, so ist ein einziger Testlauf, der aus mindestens drei Hornhäuten besteht, ausreichend. Kommt es jedoch im ersten Testlauf zu grenzwertigen Ergebnissen, sollte ein zweiter Testlauf in Erwägung gezogen werden (ist jedoch nicht unbedingt erforderlich) und darüber hinaus ein dritter, sollte es beim ersten und zweiten Testlauf zu abweichenden mittleren IVIS-Ergebnissen kommen. In diesem Zusammenhang gilt ein Ergebnis im ersten Testlauf als grenzwertig, wenn die Vorhersagen aus den 3 Hornhäuten nicht übereinstimmend waren, und zwar wie folgt:
Prüfbericht Der Prüfbericht sollte die folgenden Informationen umfassen, soweit sie für die Studie relevant sind: Prüfchemikalien und Kontrollchemikalien
Informationen zu Auftraggeber und Prüfanstalt
Testbedingungen
Testakzeptanzkriterien
Gewinnung und Vorbereitung der Augen
Testverfahren
Ergebnisse
Erörterung der Ergebnisse Schlussfolgerung (1) ICCVAM (2006). Test Method Evaluation Report - In Vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm. (2) ICCVAM (2010). ICCVAM Test Method Evaluation Report: Current Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products. NIH-Veröffentlichung Nr. 10-7553: Research Triangle Park, NC: National Institute of Environmental Health Sciences. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm. (3) OECD (2013). Streamlined Summary Document supporting the Test Guideline 437 for eye irritation/corrosion. Series on Testing and Assessment, No. 189, OECD, Paris. (4) UN (2011). United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), ST/SG/AC.10/30 Rev 4, New York und Genf: United Nations. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev04/04files_e.html. (5) Scott, L., Eskes, C., Hoffman, S., Adriaens, E., Alepee, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Hamernik, K., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., Mcnamee, P., Osborn, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielmann, H., Stokes, W., Trouba, K., Vassallo, M., Van den Berghe, C., Van Goethem, F., Vinardell, P., Zuang, V (2010), A proposed eye irritation testing strategy to reduce and replace in vivo studies using Bottom-Up and Top-Down approaches. Toxicol. in Vitro 24:1-9. (6) ICCVAM (2006). ICCVAM Recommended BCOP Test Method Protocol. In: ICCVAM Test Method Evaluation Report - in vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm. (7) ICCVAM (2010). ICCVAM Recommended BCOP Test Method Protocol. In: ICCVAM Test Method Evaluation Report - Current Validation Status of In Vitro Test Methods Proposed for Identifying Eye Injury Hazard Potential of Chemicals and Products. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 10-7553A. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm. (8) INVITTOX (1999). Protokoll Nr. 124: Bovine Corneal Opacity and Permeability Assay - SOP of Microbiological Associates Ltd. Ispra, Italien: European Centre for the Validation of Alternative Methods (ECVAM). (9) Gautheron, P., Dukic, M., Alix, D. and Sina, J.F. (1992). Bovine corneal opacity and permeability test: An in vitro assay of ocular irritancy. Fundam. Appl. Toxicol. 18:442-449. (10) Prinsen, M.K. (2006). The Draize Eye Test and in vitro alternatives; a left-handed marriage? Toxicol. in Vitro 20:78-81. (11) Siegel, J.D., Rhinehart, E., Jackson, M., Chiarello, L. und das Healthcare Infection Control Practices Advisory Committee (2007), Guideline for Isolation Precautions: Preventing Transmission of Infectious Agents in Healthcare Settings. Verfügbar unter: [http://www.cdc.gov/ncidod/dhqp/pdf]. (12) Maurer, J.K., Parker, R.D. und Jester, J.V. (2002). Extent of corneal injury as the mechanistic basis for ocular irritation: key findings and recommendations for the development of alternative assays. Reg. Tox. Pharmacol., 36:106-117. (13) OECD (2011). Guidance Document on The Bovine Corneal Opacity and Permeability (BCOP) and Isolated Chicken Eye (ICE) Test Methods: Collection of Tissues for Histological Evaluation and Collection of Data on Non-severe Irritants. Series on Testing and Assessment, No. 160. Angenommen am 25. Oktober2011. Paris: Organisation for Economic Co-operation and Development. (14) Doughty, M.J., Petrou, S. and Macmillan, H. (1995). Anatomy and morphology of the cornea of bovine eyes from a slaughterhouse. Can. J. Zool. 73:2159-2165. (15) Collee, J. and Bradley, R. (1997). BSE: A decade on - Part I. The Lancet 349: 636-641. (16) Sina, J.F., Galer, D.M., Sussman, R.S., Gautheron, P.D., Sargent, E.V., Leong, B., Shah, P.V., Curren, R.D., and Miller, K. (1995). A collaborative evaluation of seven alternatives to the Draize eye irritation test using pharmaceutical intermediates. Fundam. Appl. Toxicol. 26:20-31. (17) Kapitel B.5 dieses Anhangs, Akute Augenreizung/-verätzung. (18) ICCVAM (2006). Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Bovine Corneal Opacity and Permeability Test Method. NIH-Veröffentlichung Nr. 06-4512. Research Triangle Park: National Toxicology Program. Verfügbar unter: [http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_brd_bcop.htm]. (19) OECD (1998). Series on Good Laboratory Practice and Compliance Monitoring. No. 1: OECD Principles on Good Laboratory Practice (revised in 1997). Verfügbar unter: http://www.oecd.org/document/63/0,3343,en_2649_34381_2346175_1_1_1_1,00.html _____ 1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
Definitionen Augenreizung: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Austauschbar mit "Reversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 2" (4). Bottom-Up-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie keine Einstufung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zuerst Chemikalien, die keine Einstufung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden. Chemikalie: Stoff oder Gemisch. Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Prüfchemikalie entscheiden zu können und diese Entscheidung zu untermauern. Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen. Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (4). Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode. Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann. Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (4). Hornhaut: Der Iris und Pupille überdeckende transparente vordere Teil des Augapfels, über den Licht ins Augeninnere übertragen wird. Hornhautdurchlässigkeit: Quantitativer Messwert für die Schädigung der Epithelzellschicht der Hornhaut, ermittelt durch Bestimmung der Menge an Fluorescein-Natrium, die alle Zellschichten der Hornhaut durchdringt. Hornhauttrübung: Messwert für die Undurchsichtigkeit der Hornhaut nach Applikation einer Prüfchemikalie. Eine verstärkte Hornhauttrübung ist ein Indikator für die Schädigung der Hornhaut. Die Trübung kann subjektiv (Draize-Kaninchenaugentest) oder objektiv (mithilfe eines Messgeräts, z.B. eines Opazimeters) bestimmt werden. In-vitro-Reizwert (In Vitro Irritancy Score, IVIS): Eine bei der BCOP-Prüfmethode verwendete empirisch abgeleitete Formel, bei der der mittlere Trübungs- und der mittlere Durchlässigkeitswert für jede Behandlungsgruppe in einem einzigen In-vitro-Wert zusammengefasst werden. IVIS = mittlerer Trübungswert + (15 x mittlerer Durchlässigkeitswert). Irreversible Wirkungen am Auge: Siehe "Schwere Augenschädigung" Keine Einstufung nach UN-GHS: Chemikalien, die die Voraussetzungen für eine Einstufung in die UN-GHS-Kategorien 1 oder 2 (2A oder2B) nicht erfüllen. Austauschbar mit "Nicht eingestuft". Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert. Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert. Nicht eingestuft: Chemikalien, die nicht als augenreizend (UN-GHS-Kategorie 2, 2A oder2B) oder schwer augenschädigend (UN-GHS-Kategorie 1) eingestuft sind. Austauschbar mit der UN-GHS-Klasse "Keine Einstufung". Opazimeter: Ein Instrument zur Messung der "Hornhauttrübung" durch quantitative Evaluierung der Lichtübertragung durch die Hornhaut. Das Gerät besteht in der Regel aus zwei Kammern, beide mit eigener Lichtquelle und Fotozelle. Eine Kammer wird für die behandelte Hornhaut verwendet, die zweite zur Kalibrierung und Nulleinstellung des Instruments. Licht aus einer Halogenleuchte wird durch eine Kontrollkammer (leere Kammer ohne Fenster oder Flüssigkeit) zu einer Fotozelle geleitet und mit dem Lichtstrahl verglichen, der durch die Versuchskammer, welche die Kammer mit der Hornhaut enthält, zu einer Fotozelle geleitet wird. Der Unterschied bei der Lichtübertragung aus den Fotozellen wird errechnet und als numerischer Trübungswert digital angezeigt. Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird. Referenzchemikalie: Eine zum Vergleich mit einer Prüfchemikalie verwendete Bezugsgröße. Eine Referenzchemikalie sollte die folgenden Eigenschaften aufweisen: i) beständige und zuverlässige Quelle(n); ii) strukturelle und funktionelle Ähnlichkeit zur Klasse der geprüften Chemikalien; iii) bekannte physikalische/chemische Eigenschaften; iv) unterstützende Daten zu bekannten Wirkungen und v) bekannte Potenz innerhalb der gewünschten Wirkungsspanne. Reversible Wirkungen am Auge: Siehe "Augenreizung". Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Austauschbar mit "Irreversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 1" (4). Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (4). Tensid: Auch als oberflächenaktiver Stoff bezeichnet. Hierbei handelt es sich um Stoffe, wie waschaktive Substanzen (Detergenzien), die die Oberflächenspannung einer Flüssigkeit herabsetzen und so die Bildung von Schaum oder das Eindringen in feste Stoffe ermöglichen; auch bekannt als Netzmittel. Tensidhaltiges Gemisch: Im Kontext dieser Prüfmethode ein Gemisch mit einem oder mehreren Tensiden in einer Endkonzentration von > 5 %. Top-Down-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zuerst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden. UN-GHS-Kategorie 1: Siehe "Schwere Augenschädigung" UN-GHS-Kategorie 2: Siehe "Augenreizung". Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden. Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
Tabelle 1 Vorhersagefähigkeit der BCOP-Prüfmethode für die Ermittlung von Chemikalien, die schwere Augenschäden verursachen ([UN-GHS/EU-CLP Kategorie 1 vs. nicht Kategorie 1 (Kategorie 2 + Keine Einstufung); US-EPA-Kategorie I vs. nicht Kategorie I (Kategorie II + Kategorie III + Kategorie IV)]
Tabelle 2 Vorhersagefähigkeit der BCOP-Prüfmethode für die Ermittlung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern ("Stoffe ohne Reizwirkung") [UN-GHS/EU-CLP Keine Einstufung vs. nicht Keine Einstufung (Kategorie 1 + Kategorie 2); US-EPA-Kategorie IV vs. nicht Kategorie IV (Kategorie I + Kategorie II + Kategorie III)]
Vor der routinemäßigen Anwendung dieser Prüfmethode sollten Laboratorien ihre technische Kompetenz nachweisen, indem sie die 13 in Tabelle 1 empfohlenen Chemikalien in die richtige Augengefährdungsklasse einstufen. Die Chemikalien wurden so ausgewählt, dass sie die Bandbreite von Augengefährdungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405) (17) und dem UN-GHS-Klassifizierungssystem (d. h. den Kategorien 1, 2A, 2B oder "Nicht eingestuft") basieren (4). Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger In-vitro-Daten aus der BCOP-Prüfmethode. Referenzdaten können aus dem Streamlined Summary Document (3) und den Background Review Documents des ICCVAM für die BCOP-Prüfmethode bezogen werden (2) (18). Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des BCOP-Tests
BCOP-Hornhauthalter sind aus einem trägen Material (z.B. Polypropylen) gefertigt. Sie bestehen aus zwei Hälften (einer Vorder- und einer Hinterkammer) sowie zwei identischen zylindrischen Innenkammern. Jede Kammer hat ein Fassungsvermögen von ca. 5 ml und schließt mit einer Glasscheibe ab, durch die die Trübungsmesswerte abgelesen werden. Jede Innenkammer hat einen Durchmesser von 1,7 cm und ist 2,2 cm tief 1. Ein auf der Hinterkammer positionierter Dichtungsring verhindert Leckagen. Die Hornhäute werden mit der Endothel-Seite nach unten auf den Dichtungsring der Hinterkammern platziert, während die Vorderkammern auf die Epithel-Seite der Hornhäute gesetzt werden. Die Kammern werden mit drei Schrauben aus rostfreiem Stahl an den Außenkanten der Kammer fixiert. Jede Kammer schließt mit einer Glasscheibe ab, die für den leichten Zugang zur Hornhaut abgenommen werden kann. Um Leckagen zu verhindern, befindet sich zwischen Glasscheibe und Kammer ein weiterer Dichtungsring. Zwei Öffnungen auf der Oberseite jeder Kammer gestatten den Ein- und Auslass des Mediums und der Prüfchemikalien. Sie werden während der Behandlung und der Inkubation mit Gummistöpseln verschlossen. Die Lichtübertragung durch die Hornhauthalter kann sich potenziell verändern, da Lichtstreuung oder Reflexion durch Abnutzung oder Ablagerung bestimmter chemischer Rückstände an den Bohrungen der Innenkammer oder an der Glasscheibe beeinflusst werden können. Dies könnte höhere oder niedrigere Referenzwerte für die Lichtübertragung durch die Hornhauthalter (und umgekehrt niedrigere oder höhere Referenztrübungswerte) zur Folge haben und sich als deutliche Veränderungen bei den erwarteten ersten Referenzmessungen der Hornhauttrübung in den einzelnen Kammern bemerkbar machen (d. h. die anfänglichen Hornhauttrübungswerte bei spezifischen einzelnen Hornhauthaltern können regelmäßig um mehr als zwei oder drei Trübungseinheiten von den erwarten Referenzwerten abweichen). Jedes Labor sollte in Erwägung ziehen, ein Programm zur Evaluierung der Veränderungen der Lichtübertragung durch die Hornhauthalter einzurichten, das die Art der geprüften Chemikalien und die Häufigkeit des Gebrauchs der Kammern berücksichtigt. Zur Festlegung von Referenzwerten können die Hornhauthalter vor dem regelmäßigen Einsatz durch Messung der Referenztrübungswerte (oder Lichtübertragung) der vollständig mit dem Medium gefüllten Kammern, ohne Hornhäute, geprüft werden. Anschließend werden die Hornhauthalter während ihres Einsatzes in regelmäßigen Abständen auf Veränderungen der Lichtübertragung kontrolliert. Jedes Labor kann die Häufigkeit der Kontrollen der Hornhauthalter abhängig von der Art der geprüften Chemikalien, der Häufigkeit des Gebrauchs und der beobachteten Veränderungen der Referenzwerte für die Hornhauttrübung festlegen. Werden deutliche Veränderungen in der Lichtübertragung durch die Hornhauthalter festgestellt, sind eine Reinigung und/oder Polierung der Innenfläche der Hornhauthalter mit geeigneten Verfahren oder ein Austausch in Betracht zu ziehen. Hornhauthalter: Explosionsdarstellung _____
Opazimeter Der Opazimeter ist ein Gerät zur Messung der Lichtübertragung. Bei dem zur Validierung des BCOP-Tests verwendeten Gerät OP-KIT von Electro Design (Riom, Frankreich) wird beispielsweise Licht aus einer Halogenleuchte durch eine Kontrollkammer (leere Kammer ohne Fenster oder Flüssigkeit) zu einer Fotozelle geleitet und mit dem Lichtstrahl verglichen, der durch die Versuchskammer, welche die Kammer mit der Hornhaut enthält, zu einer Fotozelle geleitet wird. Der Unterschied bei der Lichtübertragung aus den Fotozellen wird errechnet und als numerischer Trübungswert digital angezeigt. Die Trübungseinheiten sind vorgegeben. Andere Arten von Opazimetern mit einem anderen Aufbau (bei denen z.B. keine parallelen Messungen der Kontrollkammer und Versuchskammer erforderlich sind) können verwendet werden, wenn sie nachweislich ähnliche Ergebnisse wie das validierte Gerät liefern. Der Opazimeter sollte eine lineare Reaktion innerhalb eines Bereichs von Trübungsmesswerten ergeben, der den Schwellenwerten für die vom Vorhersagemodell beschriebenen unterschiedlichen Klassifizierungen (d. h. bis zu dem Schwellenwert, der die verätzende/stark reizende Wirkung bestimmt) Rechnung trägt. Um lineare und akkurate Messwerte bis zu 75-80 Trübungseinheiten zu gewährleisten, muss der Opazimeter mit einer Reihe von Kalibratoren geeicht werden.
Die Kalibratoren werden in die Kalibrierkammer (eine für die Aufnahme der Kalibratoren konzipierte Hornhautkammer) platziert und auf dem Opazimeter abgelesen.
Die Kalibrierkammer ist derart konzipiert, dass die Kalibratoren in ungefähr demselben Abstand zu Lichtquelle und Fotozelle gehalten werden, in dem sich die Hornhäute bei der Trübungsmessung befinden würden.
Die Referenzwerte und der anfängliche Sollwert sind von der Art des verwendeten Geräts abhängig.
Die Linearität der Trübungsmessungen sollte durch geeignete (instrumentspezifische) Verfahren sichergestellt werden.
Beim Gerät OP-KIT von Electro Design (Riom, Frankreich) wird der Opazimeter beispielsweise zunächst auf null Trübungseinheiten geeicht, wobei die Kalibrierkammer ohne Kalibrator verwendet wird.
Anschließend werden nacheinander drei unterschiedliche Kalibratoren in die Kalibrierkammer gesetzt, und die Trübungswerte werden gelesen.
Die Kalibratoren 1, 2 und 3 sollten Trübungswerte ergeben, die ihren eingestellten Werten von 75, 150 bzw. 225 Trübungseinheiten ± 5 % entsprechen. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.48 Test am isolierten Hühnerauge zur Identifizierung von i) Chemikalien, die schwere Augenschäden verursachen, und ii) Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern 17 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 438 (2013). Der Test am isolierten Hühnerauge (Isolated Chicken Eye, ICE) wurde vom Organisationsübergreifenden Koordinationsausschuss zur Validierung alternativer Methoden (Interagency Coordinating Committee on the Validation of Alternative Methods, ICCVAM) unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods, ECVAM) und des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese Centre for the Validation of Alternative Methods, JaCVAM) in den Jahren 2006 und 2010 evaluiert (1) (2) (3). Bei der ersten Evaluierung wurde der ICE-Test als wissenschaftlich fundierte Prüfmethode für den Einsatz als Screening-Test zur Identifizierung von Chemikalien (Stoffen und Gemischen), die schwere Augenschäden (Kategorie 1) gemäß Definition im Globalen Harmonisierten System zur Einstufung und Kennzeichnung von Chemikalien (GHS) der Vereinten Nationen (UN) (1) (2) (4) und der Verordnung (EG) Nr. 1272/2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen 1 verursachen, anerkannt. Bei der zweiten Evaluierung wurde der ICE-Test im Hinblick auf seine Eignung als Screening-Test zur Identifizierung von Chemikalien, die nicht als augenreizend oder schwer augenschädigend gemäß UN-GHS einzustufen sind, beurteilt (3) (4). Die Ergebnisse der Validierungsstudie und die Empfehlungen der Peer-Review-Gruppe bestätigten die ursprüngliche Empfehlung, den ICE-Test für die Einstufung von schwer augenschädigenden Chemikalien (UN-GHS-Kategorie 1) zu verwenden, da sich die verfügbare Datenbank seit der ursprünglichen Validierung des ICCVAM nicht geändert hat. In jener Phase wurden keine weiteren Empfehlungen für eine Erweiterung des Anwendungsbereichs des ICE-Tests auch auf andere Kategorien ausgesprochen. Es wurde eine Neubewertung des in der Validierungsstudie verwendeten In-vitro- und In-vivo-Datensatzes durchgeführt. Der Schwerpunkt lag dabei auf der Evaluierung der Zweckmäßigkeit des ICE-Tests für die Identifizierung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend (5) erfordern. Die Neubewertung führte zu dem Ergebnis, dass der ICE-Test auch verwendet werden kann, um Chemikalien zu identifizieren, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS (4) (5) erfordern. Diese Prüfmethode berücksichtigt die empfohlenen Einsatzbereiche und Einsatzgrenzen des ICE-Tests auf Grundlage dieser Evaluierungen. Die Hauptunterschiede zwischen der ursprünglichen Fassung der OECD-Prüflinie aus dem Jahr2009 und der aktualisierten Fassung von 2013 sind u. a. die Verwendung des ICE-Tests zur Identifizierung von Chemikalien, die keine Einstufung nach dem UN-GHS-Klassifizierungssystem erfordern, eine Aktualisierung der Elemente des Prüfberichts, eine Aktualisierung der Definitionen in Anlage 1 sowie eine Aktualisierung der Leistungschemikalien in Anlage 2. Gegenwärtig herrscht allgemeiner Konsens darüber, dass der In-vivo-Draize-Augentest in absehbarer Zukunft nicht durch einen einzigen In-vitro-Augenreizungstest ersetzt werden kann, der in der Lage ist, das gesamte Spektrum an Augenreizungen für verschiedene Chemikalienklassen vorherzusagen. Unter Umständen ist es jedoch möglich, den Draize-Augentest durch strategische Kombinationen mehrerer alternativer Prüfmethoden im Rahmen einer (gestuften) Prüfstrategie zu ersetzen (6). Der Top-Down-Ansatz (7) wird verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie ein hohes Reizpotenzial aufweist. Umgekehrt wird der Bottom-Up-Ansatz (7) verwendet, wenn auf Basis der vorliegenden Informationen davon auszugehen ist, dass eine Chemikalie keine ausreichende Augenreizung für eine Einstufung hervorruft. Der ICE-Test ist eine In-vitro-Prüfmethode, die unter bestimmten Bedingungen und mit bestimmten Grenzen, wie unter den Nummern 8 bis 10 erläutert, zur Einstufung und Kennzeichnung von Chemikalien mit augengefährdender Wirkung angewendet werden kann. Auch wenn der Test den In-vivo-Kaninchenaugentest nicht absolut ersetzen kann, wird der ICE-Test als erster Schritt im Rahmen einer Prüfstrategie wie des von Scott et al. vorgeschlagenen Top-Down-Ansatzes (7) empfohlen, um ohne weitere Tests (4) schwer augenschädigende Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 einzustufen sind, zu identifizieren. Der ICE-Test wird auch für die Identifizierung von Chemikalien empfohlen, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß UN-GHS (Keine Einstufung (No Category), NC) (4) erfordern, und kann daher als erster Schritt im Rahmen einer Prüfstrategie mit Bottom-up-Ansatz verwendet werden (7). Im Falle einer Chemikalie, bei der mit dem ICE-Test nicht vorhergesagt werden kann, dass sie schwere Augenschäden verursacht oder keine Einstufung als augenreizend/schwer augenschädigend erfordert, müssten jedoch weitere Tests (in vitro und/oder in vivo) durchgeführt werden, um eine eindeutige Einstufung festzulegen. Außerdem sollten die zuständigen Regulierungsbehörden konsultiert werden, bevor der ICE-Test in einem Bottom-Up-Ansatz unter einer anderen Klassifizierungsregelung als dem UN-GHS angewendet wird. Diese Prüfmethode beschreibt die Verfahrensschritte für die Beurteilung des augengefährdenden Potenzials einer Prüfchemikalie, gemessen als ihre Fähigkeit, im isolierten Hühnerauge eine toxische Wirkung hervorzurufen oder nicht. Toxische Wirkungen auf die Hornhaut werden gemessen durch i) qualitative Bewertung der Trübung, ii) qualitative Bewertung der Schädigung der Epithel-Zellschicht durch Applikation von Fluorescein auf das Auge (Fluorescein-Verfärbung), iii) quantitative Messung verstärkter Dicke (Schwellung) und iv) qualitative Beurteilung makroskopischer morphologischer Oberflächenschädigungen. Hornhauttrübungen, Hornhautschwellungen und Hornhautschädigungen nach Applikation einer Prüfchemikalie werden zunächst einzeln bewertet und anschließend zwecks Klassifizierung des Augenreizwertes (Eye Irritancy Classification) kombiniert. Definitionen sind Anlage 1 zu entnehmen. Vorbemerkungen und Einsatzgrenzen Diese Prüfmethode basiert auf dem im OECD Guidance Document 160 (8) empfohlenen Protokoll, das im Anschluss an eine internationale Validierungsstudie des ICCVAM (1) (3) (9) und unter Mitwirkung des Europäischen Zentrums zur Validierung alternativer Methoden (European Centre for the Validation of Alternative Methods), des Japanischen Zentrums zur Validierung alternativer Methoden (Japanese center for the Validation of Alternative Methods) und der Abteilung Toxikologie und angewandte Pharmakologie des niederländischen Forschungsinstituts TNO Quality of Life entwickelt wurde. Das Protokoll beruht auf Informationen aus veröffentlichten Protokollen und auf dem von TNO derzeit verwendeten Protokoll (10) (11) (12) (13) (14). Bei der dieser Prüfmethode zugrunde liegenden Validierung wurde ein breites Spektrum von Chemikalien getestet. Die empirische Datenbank der Validierungsstudie umfasste 152 Chemikalien (72 Stoffe und 80 Gemische) (5). Die Prüfmethode ist auf Feststoffe, Flüssigkeiten, Emulsionen und Gele anwendbar. Die Flüssigkeiten können wässrig oder nichtwässrig, die Feststoffe wasserlöslich oder wasserunlöslich sein. Gase und Aerosole wurden in einer Validierungsstudie bisher noch nicht bewertet. Der ICE-Test kann zur Identifizierung von schwer augenschädigenden Chemikalien, d. h. Chemikalien, die in die UN-GHS-Kategorie 1 (4) einzustufen sind, angewendet werden. In diesem Anwendungsbereich beruhen die anerkannten Einsatzgrenzen des ICE-Tests auf den hohen Falsch-Positiv-Raten für Alkohole und den hohen Falsch-Negativ-Raten für Feststoffe und Tenside (1) (3) (9). Die Falsch-Negativ-Raten sind in diesem Kontext (UN-GHS-Kategorie 1 wird als nicht UN-GHS-Kategorie 1 erkannt) jedoch nicht maßgeblich, da alle Prüfchemikalien mit negativem Ergebnis anschließend im Rahmen anderer angemessen validierter In-vitro-Tests oder als letzte Möglichkeit - je nach Vorschriften - an Kaninchen anhand einer sequenziellen Prüfstrategie mit einem evidenzbasierten Bewertungsansatz (weight-of-evidence approach) geprüft werden. Es ist zu beachten, dass Feststoffe beim In-vivo-Draize-Augenreizungstest zu variablen und extremen Expositionsbedingungen führen können, aus denen sich möglicherweise irrelevante Vorhersagen über das tatsächliche Reizungspotenzial ableiten (15). Prüfer können diese Prüfmethode jedoch für alle Arten von Chemikalien einsetzen und ein Positivergebnis als Indikator für eine schwer augenschädigende Wirkung, d. h. eine Einstufung in UN-GHS-Kategorie 1, ohne weitere Tests akzeptieren. Positivergebnisse bei Verwendung von Alkohol sollten angesichts des Risikos von Falsch-Positiv-Prognosen (over-prediction) jedoch mit einer gewissen Zurückhaltung interpretiert werden. Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist der ICE-Test hinsichtlich der Identifizierung von Chemikalien mit schwer augenschädigender Wirkung (UN-GHS-Kategorie 1) eine allgemeine Genauigkeit von 86 % (120/140), eine Falsch-Positiv-Rate von 6 % (7/113) und eine Falsch-Negativ-Rate von 48 % (13/27) auf (4) (5). Der ICE-Test kann auch zur Identifizierung von Chemikalien angewendet werden, die keine Einstufung als augenreizend oder schwer augenschädigend gemäß dem UN-GHS-Klassifizierungssystem erfordern (4). Bevor der ICE-Test in einem Bottom-Up-Ansatz unter einer anderen Klassifizierungsregelung angewendet wird, sollten die zuständigen Regulierungsbehörden konsultiert werden. Diese Prüfmethode kann für alle Arten von Chemikalien eingesetzt werden, wobei ein Negativergebnis als Indikator für die Nichteinstufung einer Chemikalie als augenreizend oder schwer augenschädigend akzeptiert werden könnte. Auf der Basis eines Ergebnisses aus der Validierungsdatenbank besteht bei Bewuchsschutzfarben mit organischen Lösungsmitteln jedoch das Risiko von Falsch-Negativ-Prognosen (under-prediction) (5). Gemessen an Daten aus In-vivo-Kaninchenaugentests, die nach dem UN-GHS-Klassifizierungssystem eingestuft wurden, weist der ICE-Test hinsichtlich der Identifizierung von Chemikalien, die keine Einstufung als augenreizend oder schwer augenschädigend erfordern, eine allgemeine Genauigkeit von 82 % (125/152), eine Falsch-Positiv-Rate von 33 % (26/79) und eine Falsch-Negativ-Rate von 1 % (1/73) auf (4) (5). Werden Stoffe einer bestimmten Chemikalienklasse (d. h. Bewuchsschutzfarben mit organischen Lösungsmitteln) aus der Datenbank ausgeschlossen, so liegt die Genauigkeit des ICE-Tests gemessen am UN-GHS-Klassifizierungssystem bei 83 % (123/149), die Falsch-Positiv-Rate bei 33 % (26/78) und die Falsch-Negativ-Rate bei 0 % (0/71) (4) (5). Aufgrund der beträchtlichen Anzahl von Chemikalien der UN-GHS-Kategorie 1, die zu niedrig in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, und von Chemikalien, die keine Einstufung erfordern, jedoch zu hoch in die UN-GHS-Kategorien 2, 2A oder2B eingestuft sind, wird der ICE-Test nicht für die Identifizierung von Prüfchemikalien empfohlen, die als augenreizend (d. h. UN-GHS-Kategorie 2 oder Kategorie 2A) oder leicht augenreizend (UN-GHS-Kategorie 2B) eingestuft werden sollten. Diesbezüglich können weitere Tests mit einer anderen geeigneten Methode erforderlich sein. Bei allen Verfahren, die Hühneraugen involvieren, sind die geltenden Regeln und Verfahrensvorschriften der Prüfanstalt für den Umgang mit Human- bzw. Tiermaterial (u. a. Gewebe und Gewebeflüssigkeiten) einzuhalten. Universelle Laborregeln sollten beachtet werden (16). Die im Kaninchenaugentest evaluierten Bindehaut- und Irisverletzungen werden beim ICE-Test zwar außer Acht gelassen, doch werden bei der Prüfmethode Auswirkungen auf die Hornhaut berücksichtigt, die wesentliche Einflussfaktoren für die In-vivo-Klassifizierung nach dem UN-GHS-Klassifizierungssystem bilden. Auch wurde - obwohl sich die Reversibilität von Hornhautläsionen per se mit dem ICE-Test nicht beurteilen lässt - ausgehend von Kaninchenaugenstudien vorgeschlagen, dass eine Bewertung der anfänglichen Tiefe der Hornhautverletzung herangezogen werden kann, um einige Arten irreversibler Wirkungen zu identifizieren (17). Insbesondere sind weitere wissenschaftliche Erkenntnisse erforderlich, um zu verstehen, wie irreversible Wirkungen auftreten, die nicht mit einer anfänglichen starken Verletzung im Zusammenhang stehen. Schließlich sei auch erwähnt, dass das Potenzial für eine mit der Augenexposition verbundene systemische Toxizität mit der ICE-Methode nicht bewertet werden kann. Diese Prüfmethode wird regelmäßig aktualisiert, um neue Informationen und Daten zu berücksichtigen. Beispielsweise können histopathologische Befunde potenziell nützlich sein, wenn eine umfassendere Charakterisierung der Hornhautschädigung erforderlich ist. Zur Evaluierung dieser Möglichkeit sollten Anwender die Augen aufbewahren und histopathologische Proben zubereiten, die zum Aufbau einer Datenbank und zur Entwicklung von Entscheidungskriterien herangezogen werden können, mit denen sich die Genauigkeit dieser Prüfmethode weiter verbessern lässt. Die OECD hat einen Leitfaden (Guidance Document) für die Anwendung von Prüfmethoden zur Untersuchung der okularen Toxizität erarbeitet. Dieser enthält ausführliche Verfahrensanweisungen für die Entnahme histopathologischer Proben und Angaben darüber, wo die Proben und/oder histopathologischen Daten einzureichen sind (8). Laboratorien, die diese Prüfmethode erstmals anwenden, sollten die in Anlage 2 genannten Leistungschemikalien verwenden. Laboratorien können diese Chemikalien verwenden, um ihre technische Kompetenz zur Durchführung des ICE-Tests nachzuweisen, bevor sie ICE-Testdaten zum Zwecke der vorschriftsmäßigen Gefahrenklassifizierung einreichen. Die ICE-Prüfmethode ist ein organtypisches Modell, das die Zellstruktur des Hühnerauges in vitro kurzfristig funktionsfähig hält. Bei dieser Prüfmethode werden durch die Prüfchemikalie hervorgerufene Schäden als Hornhautschwellungen, Hornhauttrübung und korneale Fluorescein-Verfärbung festgestellt. Während die beiden letztgenannten Parameter eine qualitative Bewertung erfordern, muss die Hornhautschwellung quantitativ bestimmt werden. Jedes Messergebnis wird entweder in einen quantitativen Wert umgerechnet, der seinerseits für die Berechnung eines Gesamtreizindexes (Overall Irritancy Index) zugrunde gelegt wird, oder einer Qualitätskategorie zugeordnet, die wiederum für die Einordnung in Klassen für Chemikalien mit in vitro augengefährdender Wirkung (UN-GHS-Kategorie 1 oder keine Einstufung nach UN-GHS) herangezogen wird. Jedes dieser Ergebnisse kann dann verwendet werden, um das Potenzial einer Prüfchemikalie, in vivo schwere Augenschäden hervorzurufen oder keine Einstufung als augengefährdend zu erfordern, vorherzusagen (siehe Entscheidungskriterien). Allerdings ist keine Einstufung bei Chemikalien möglich, bei denen mit dem ICE-Test nicht vorhergesagt wird, dass sie schwere Augenschäden verursachen oder nicht einzustufen sind (siehe Nummer 11). Bezugsquelle und Alter der Hühneraugen Traditionell werden für diesen Test Augen von Hühnern verwendet, die in einer Schlächterei für den menschlichen Verzehr geschlachtet wurden; auf diese Weise wird der Einsatz von Versuchstieren vermieden. Nur Augen von gesunden Tieren, die als für die Nahrungskette geeignet angesehen werden, dürfen verwendet werden. Es wurde keine kontrollierte Studie zur Bestimmung des optimalen Alters der Hühner durchgeführt, doch werden für diesen Test bisher Hühner verwendet, bei denen es sich nach Alter und Gewicht um Junghühner handelt (d. h. Hühner im Alter von ungefähr sieben Wochen mit einem Gewicht von 1,5 bis 2,5 kg), die in der Regel in einer Geflügelschlächterei geschlachtet werden. Gewinnung und Beförderung der Augen zum Labor Die Köpfe sollten unmittelbar nach dem Betäuben der Tiere, normalerweise durch Elektroschock, und nach dem Entbluten durch Nackenstich abgesetzt werden. Sie sollten aus einer in Nähe des Labors gelegenen Quelle bezogen werden, um die Köpfe möglichst schnell vom Schlachthof zum Labor befördern zu können, damit sich ihr Zustand nicht verschlechtert und/oder Bakterienkontaminationen auf ein Mindestmaß begrenzt werden. Der Zeitabstand zwischen der Gewinnung der Hühnerköpfe und ihrem Einsetzen in die Superfusionskammer nach der Ausschälung muss möglichst gering sein (d. h. höchstens zwei Stunden betragen), damit die Akzeptanzkriterien des Tests erfüllt werden. Alle für die Prüfung verwendeten Augen sollten aus einer an ein und demselben Tag gewonnenen Partie Augen stammen. Da die Augen im Labor seziert werden, sind die intakten Köpfe in Kunststoffbehältnissen, die mit Tüchern, welche in isotonischer Kochsalzlösung getränkt wurden, ausgeschlagen sind, und bei Umgebungstemperatur (normalerweise zwischen 18 1 °C und 25 1 °C) vom Schlachthof zum Labor zu befördern. Auswahlkriterien und Anzahl der Augen, die im ICE-Test eingesetzt werden Augen mit starker Ausgangs-Fluorescein-Verfärbung (d. h. > 0,5) oder mit starker Hornhauttrübung (d. h. > 0,5) nach der Ausschälung werden verworfen. Jede Behandlungsgruppe und die gleichzeitige Positivkontrolle umfassen mindestens drei Augen. Die Negativkontrollen bzw. die Lösungsmittelkontrolle (wenn ein anderes Lösungsmittel als Kochsalzlösung verwendet wird) bestehen aus mindestens einem Auge. Im Fall von Feststoffen mit dem GHS-Ergebnis "Keine Einstufung" (No Category, NC) wird ein zweiter Durchlauf mit drei Augen empfohlen, um das Negativergebnis zu bestätigen oder zu verwerfen. Vorbereitung der Augen Die Augenlider werden sorgfältig weggeschnitten, wobei darauf zu achten ist, dass die Hornhaut nicht beschädigt wird. Die Unversehrtheit der Hornhaut wird mithilfe eines Tropfens von 2 %igem (w/v) Natrium-Fluorescein, der für einige Sekunden auf die Hornhautoberfläche appliziert und anschließend mit isotonischer Kochsalzlösung abgespült wird, schnell überprüft. Fluorescein-behandelte Augen werden sodann mit einem Spaltlampenmikroskop auf Hornhautschädigung untersucht (d. h. Fluorescein-Verfärbung und Hornhauttrübungswerte müssen ≤ 0,5 betragen). Liegt keine Schädigung vor, wird das Auge weiter freigelegt, wobei dafür Sorge zu tragen ist, dass die Hornhaut nicht beschädigt wird. Der Augapfel wird aus der Augenhöhle herausgezogen, indem die Nickhaut mithilfe einer chirurgischen Zange festgehalten und die Augenmuskulatur mit einer gebogenen, stumpfendigen Schere durchtrennt wird. Dieser Schritt ist wichtig, um zu vermeiden, dass die Hornhaut durch übermäßigen Druck geschädigt wird (Kompressionsartefakte). Beim Herauslösen des Auges aus der Augenhöhle sollte ein sichtbarer Teil des Sehnervs noch anhaften. Das Auge wird anschließend auf eine saugfähige Unterlage gesetzt, und Nickhaut sowie anderes Bindegewebe werden weggeschnitten. Das auf diese Weise ausgeschälte Auge wird in einer Klemme aus rostfreiem Stahl fixiert, wobei die Hornhaut vertikal positioniert sein muss. Die Klemme wird sodann in eine Kammer des Superfusionsgeräts gesetzt (18). Im Superfusionsgerät sind die Klemmen so zu positionieren, dass die gesamte Hornhaut mit der Kochsalzinfusion (3-4 Tropfen pro Minute bzw. 0,1-0,15 ml/min) versorgt wird. Die Temperatur in den Kammern des Superfusionsgeräts sollte auf 32 ± 1,5°C gehalten werden. Anlage 3 zeigt ein Schaubild eines typischen Superfusionsgeräts mit Augenklemmen; das Gerät ist im Handel fertig oder als Bausatz erhältlich. Es kann den Bedürfnissen des jeweiligen Labors angepasst werden (z.B. um Augen in verschiedener Anzahl aufzunehmen). Nach dem Einsetzen ins Superfusionsgerät werden die Augen erneut mit einem Spaltlampenmikroskop untersucht, um sicherzustellen, dass sie während des Sezierens nicht beschädigt wurden. Bei dieser Gelegenheit sollte mit dem Pachometeraufsatz des Spaltlampenmikroskops auch die Hornhautdicke im Apex gemessen werden. Augen mit i) einer Fluorescein-Verfärbung von > 0,5, ii) einer Hornhauttrübung von > 0,5 oder iii) etwaigen anderen Anzeichen einer Schädigung sind zu ersetzen. Einzelne Augen, auf die keines der genannten Kriterien zutrifft, werden verworfen, wenn sie eine Hornhautdicke aufweisen, die mehr als 10 % vom mittleren Dickenwert aller Augen zusammengerechnet abweicht. Anwender sollten sich darüber im Klaren sein, dass Spaltlampenmikroskope mit unterschiedlicher Spaltbreiteneinstellung unterschiedliche Dickenmesswerte ergeben können. Die Spaltbreite sollte auf 0,095 mm eingestellt sein. Sobald alle Augen untersucht und für einwandfrei befunden wurden, werden sie zwecks Äquilibrierung des Testsystems vor Applikation der Prüfchemikalie für ungefähr 45 bis 60 Minuten inkubiert. Nach der Äquilibrierung werden Referenzmessungen von Hornhautdicke und Hornhauttrübung zum Zeitpunkt Null vorgenommen, welche als Referenzszenario dienen (d. h. Zeitpunkt = 0). Der beim Sezieren ermittelte Fluorescein-Wert wird als Referenzmesswert für diesen Endpunkt verwendet. Applikation der Prüfchemikalie Unmittelbar nach den Referenzmessungen zum Null-Zeitpunkt wird das Auge (in seiner Halterung) aus dem Superfusionsgerät entnommen und zur Applikation der Prüfchemikalie auf die Hornhaut horizontal positioniert. Flüssige Prüfchemikalien werden in der Regel unverdünnt getestet, können jedoch verdünnt werden, sofern dies für notwendig gehalten wird (z.B. als Teil des Studienkonzepts). Bevorzugtes Lösungsmittel für verdünnte Prüfchemikalien ist physiologische Kochsalzlösung. Unter kontrollierten Bedingungen können auch alternative Lösungsmittel verwendet werden, deren Eignung jedoch nachzuweisen ist. Flüssige Prüfchemikalien werden so auf die Hornhaut appliziert, dass die gesamte Hornhautoberfläche gleichmäßig von der Prüfchemikalie bedeckt ist; das Standardvolumen beträgt 0,03 ml. Feste Prüfchemikalien sollten wenn möglich im Mörser oder mit einem vergleichbaren Zerkleinerungsgerät so fein wie möglich gemahlen werden. Das Pulver wird so auf die Hornhaut appliziert, dass die Oberfläche von der Prüfchemikalie gleichmäßig bedeckt ist; die Standardmenge beträgt 0,03 g. Die Prüfchemikalie (flüssig oder fest) wird für 10 Sekunden appliziert und anschließend mit isotonischer Kochsalzlösung (ungefähr 20 ml) bei Umgebungstemperatur vom Auge abgespült. Das Auge (in seiner Halterung) wird anschließend wieder in aufrechter Ausgangsstellung in das Superfusionsgerät eingesetzt. Bei Bedarf können nach der 10-sekündlichen Applikation und zu späteren Zeitpunkten (z.B. bei der Feststellung von Rückständen der Prüfchemikalie auf der Hornhaut) weitere Spülungen vorgenommen werden. Im Allgemeinen ist die Menge an Kochsalzlösung, die zusätzlich für die Spülungen verwendet wird, nicht maßgeblich; wichtig ist jedoch die Beobachtung von Anhaftungen der Chemikalie an der Hornhaut. Kontrollchemikalien Jeder Versuch sollte gleichzeitige Negativ- oder Lösungsmittel-/Vehikelkontrollen und gleichzeitige Positivkontrollen umfassen. Beim Prüfen unverdünnter (100 %iger) Flüssigkeiten oder Feststoffe wird als gleichzeitige Negativkontrolle im ICE-Test physiologische Kochsalzlösung verwendet, um unspezifische Veränderungen im Testsystem festzustellen und um sicherzustellen, dass die Testbedingungen keine ungerechtfertigten Reizwirkungen hervorrufen. Zum Testen verdünnter Flüssigkeiten umfasst der Test auch eine Gruppe gleichzeitiger Lösungsmittel-/Vehikelkontrollen, um unspezifische Veränderungen im Testsystem nachzuweisen und um sicherzustellen, dass die Testbedingungen keine ungerechtfertigten Reizwirkungen hervorrufen. Wie unter Nummer 31 erwähnt, sind nur Lösungsmittel/Vehikel zulässig, die das Testsystem nachweislich nicht beeinträchtigen. Jeder Versuch umfasst als gleichzeitige Positivkontrolle einen bekannten Augenreizstoff, damit überprüft werden kann, ob eine adäquate Wirkung eintritt. Da der ICE-Test bei dieser Prüfmethode eingesetzt wird, um verätzende oder stark reizende Stoffe zu identifizieren, sollte es sich bei der Positivkontrolle um eine Referenzchemikalie handeln, die bei dieser Prüfmethode eine starke Wirkung hervorruft. Um zu gewährleisten, dass die Veränderlichkeit der Reaktion der Positivkontrolle im Zeitverlauf bewertet werden kann, sollte die Reaktionsstärke jedoch nicht allzu heftig sein. Es sollten genügend In-vitro-Daten für die Positivkontrolle generiert werden, damit eine statistisch vorgegebene vertretbare Spanne für die Positivkontrolle berechnet werden kann. Liegen für eine bestimmte Positivkontrolle keine angemessenen historischen Daten zur ICE-Prüfmethode vor, so können Studien erforderlich werden, um die notwendigen Daten zu generieren. Beispiele für Positivkontrollen für flüssige Prüfchemikalien sind 10 %ige Essigsäure oder 5 %iges Benzalkoniumchlorid, während für Positivkontrollen für feste Prüfchemikalien Natriumhydroxid oder Imidazol in Frage kommen. Referenzchemikalien sind nützlich für die Evaluierung des Augenreizpotenzials unbekannter Chemikalien einer bestimmten Chemikalien- oder Produktklasse oder zur Evaluierung des relativen Reizpotenzials eines Augenreizstoffes innerhalb einer spezifischen Spanne von Reizwirkungen. Gemessene Endpunkte Behandelte Hornhäute werden vor der Behandlung evaluiert sowie in Abständen von 30, 75, 120, 180 und 240 Minuten (± 5 Minuten) nach dem Abspülen im Anschluss an die Behandlung. Diese Zeitreihe gestattet eine angemessene Anzahl von Messungen während des vierstündigen Behandlungszeitraums und lässt genügend Zeit zwischen den Messungen, um alle Augen vorschriftsgemäß beobachten zu können. Die evaluierten Endpunkte sind Hornhauttrübung, Hornhautschwellung, Fluorescein-Verfärbung und morphologische Effekte (z.B. durchlöchertes oder abgelöstes Epithel). Alle Endpunkte mit Ausnahme der Fluorescein-Verfärbung (die ausschließlich vor der Behandlung sowie 30 Minuten nach Applikation der Prüfchemikalie ermittelt wird) werden zu jedem der vorgenannten Zeitpunkte bestimmt. Hornhauttrübung, Fluorescein-Verfärbung, morphologische Effekte und, sofern sie vorliegen, histopathologische Befunde sollten fotografiert werden. Nach der abschließenden Untersuchung am Ende des vierstündigen Behandlungszeitraums sind die Augen in einer geeigneten Fixierlösung (z.B. neutrales gepuffertes Formalin) aufzubewahren, damit sie gegebenenfalls histopathologisch untersucht werden können (Einzelheiten siehe Nummer 14 und Literaturhinweis (8)). Hornhautschwellungen werden durch Hornhautdickenmessungen bestimmt, die mit dem optischen Pachometeraufsatz eines Spaltlampenmikroskops vorgenommen werden. Der Schwellungsmesswert wird als Prozentsatz ausgedrückt und auf Basis der Hornhautdickenmesswerte nach folgender Formel berechnet: Für alle getesteten Augen wird die mittlere Hornhautschwellung (in Prozent) für sämtliche Beobachtungszeitpunkte berechnet. Auf Basis des höchsten Mittelwertes für die Hornhautschwellung (beliebiger Beobachtungszeitpunkt) wird anschließend jeder Prüfchemikalie ein Gesamtwert für die Kategorie (overall category score) zugeordnet (siehe Nummer 51). Die Hornhauttrübung wird anhand der Hornhautfläche bewertet, die am stärksten getrübt ist (siehe Tabelle 1). Für alle getesteten Augen wird der mittlere Trübungswert für jeden Beobachtungszeitpunkt berechnet. Auf Basis des höchsten Mittelwertes für die Hornhauttrübung (beliebiger Beobachtungszeitpunkt) wird anschließend jeder Prüfchemikalie ein Gesamtwert für die Kategorie (overall category score) zugeordnet (siehe Nummer 51). Tabelle 1 Hornhauttrübungswerte
Die Fluorescein-Verfärbung wird nur für den 30-minütigen Beobachtungszeitpunkt bewertet (siehe Tabelle 2). Anschließend wird für alle getesteten Augen die mittlere Fluorescein-Verfärbung für den 30-minütigen Beobachtungszeitpunkt berechnet und zur Ermittlung des jeder Prüfchemikalie zugeordneten Gesamtwertes für die Kategorie herangezogen (siehe Nummer 51). Tabelle 2 Fluorescein-Verfärbungswerte
Morphologische Effekte umfassen "durchlöcherte" Hornhaut-Epithelzellen, "abgelöste"Epithelzellen, "aufgeraute" Hornhautoberfläche und "Verklebung" der Hornhaut mit der Prüfchemikalie. Diese Befunde können unterschiedlich stark ausgeprägt sein und gleichzeitig auftreten. Ihre Einstufung erfolgt subjektiv je nach Auswertung des Prüfers. Datenauswertung Die Ergebnisse für die Hornhauttrübung, die Hornhautschwellung und die Fluorescein-Verfärbung sind einzeln zu evaluieren, um für jeden Endpunkt eine ICE-Klasse zu generieren. Kombiniert ergeben die ICE-Klassen für die einzelnen Endpunkte eine Reizklasse (Irritancy Classification) für die einzelnen Prüfchemikalien. Entscheidungskriterien Nach der Evaluierung jedes Endpunktes können die ICE-Klassen entsprechend einer vorgegebenen Spanne zugeordnet werden. Die Auswertung der Hornhautschwellung (Tabelle 3), der Hornhauttrübung (Tabelle 4) und der Fluorescein-Verfärbung (Tabelle 5) und ihre Einstufung in vier ICE-Klassen erfolgen anhand der nachstehenden Bewertungsskalen. Die Werte für die Hornhautschwellung in Tabelle 3 sind nur gültig, wenn die Dicke mit einem Spaltlampenmikroskop (z.B. Haag-Streit BP900) mit Tiefenmessgerät Nr. 1 und einer Spaltbreiteneinstellung von 9,5 (= 0,095 mm) gemessen wird. Anwender sollten sich darüber im Klaren sein, dass Spaltlampenmikroskope mit unterschiedlicher Spaltbreiteneinstellung unterschiedliche Dickenmesswerte ergeben können. Tabelle 3 Kriterien für die Einstufung in ICE-Klassen - Hornhautschwellung
Tabelle 4 Kriterien für die Einstufung in ICE-Klassen - Hornhauttrübung
Tabelle 5 Kriterien für die Einstufung in ICE-Klassen - mittlere Fluorescein-Verfärbung
Die In-vitro-Klassifizierung einer Prüfchemikalie richtet sich nach der GHS-Klasse, die der Kombination von Kategorien entspricht, die für die Hornhautschwellung, die Hornhauttrübung und die Fluorescein-Verfärbung ermittelt wurden, und wird gemäß Tabelle 6 vorgenommen. Tabelle 6 In-vitro-Gesamtklassifizierung
Studienakzeptanzkriterien Ein Test gilt als akzeptabel, wenn die gleichzeitigen Negativ- oder Vehikel-/Lösungsmittelkontrollen sowie die gleichzeitigen Positivkontrollen "Keine Einstufung" nach GHS bzw. GHS-Kategorie 1 ergeben. Prüfbericht Der Prüfbericht sollte die folgenden Informationen umfassen, soweit sie für die Studie relevant sind: Prüfchemikalien und Kontrollchemikalien
Informationen zu Auftraggeber und Prüfanstalt
Testbedingungen
Gewinnung und Vorbereitung der Augen
Testverfahren
Ergebnisse
Erörterung der Ergebnisse Schlussfolgerung (1) ICCVAM (2007). Test Method Evaluation Report - In Vitro Ocular Toxicity Test Methods for Identifying Ocular Severe Irritants and Corrosives. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 07-4517. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_tmer.htm. (2) ESAC (2007). Statement on the conclusion of the ICCVAM retrospective study on organotypic in vitro assays as screening tests to identify potential ocular corrosives and severe eye irritants. Verfügbar unter: http://ecvam.jrc.it/index.htm. (3) ICCVAM (2010). ICCVAM Test Method Evaluation Report - Current Status of in vitro Test Methods for Identifying Mild/Moderate Ocular Irritants: The Isolated Chicken Eye (ICE) Test Method. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program (NTP) Interagency center for the Evaluation of Alternative Toxicological Methods (NICEATM). NIH-Veröffentlichung Nr. 10-7553A. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/MildMod-TMER.htm. (4) Vereinte Nationen (UN) (2011). Globally Harmonized System of Classification and Labelling of Chemicals (GHS), vierte überarbeitete Ausgabe, UN New York und Genf, 2011. Verfügbar unter: http://www.unece.org/trans/danger/publi/ghs/ghs_rev04/04files_e.html. (5) Streamlined Summary Document Supporting OECD Test Guideline 438 on the Isolated Chicken Eye for Eye Irritation/Corrosion. Series on Testing and Assessment Nr. 188 (Part 1 und Part 2), OECD, Paris. (6) Kapitel B.5 dieser Anlage, Akute Augenreizung/-verätzung. (7) Scott, L., Eskes, C., Hoffman, S., Adriaens, E., Alepee, N., Bufo, M., Clothier, R., Facchini, D., Faller, C., Guest, R., Hamernik, K., Harbell, J., Hartung, T., Kamp, H., Le Varlet, B., Meloni, M., Mcnamee, P., Osborn, R., Pape, W., Pfannenbecker, U., Prinsen, M., Seaman, C., Spielmann, H., Stokes, W., Trouba, K., Vassallo, M., Van den Berghe, C., Van Goethem, F., Vinardell, P., Zuang, V (2010). A proposed Eye Irritation Testing Strategy to Reduce and Replace in vivo Studies Using Bottom-up and Top-down Approaches. Toxicology in Vitro, 24, 1-9. (8) OECD (2011). Guidance Document on The Bovine Corneal Opacity and Permeability (BCOP) and Isolated Chicken Eye (ICE) Test Methods: Collection of Tissues for Histological Evaluation and Collection of Data on Non-Severe Irritants. Series on Testing and Assessment, Nr. 160, OECD, Paris. (9) ICCVAM (2006). Background review document: Current Status of In Vitro Test Methods for Identifying Ocular Corrosives and Severe Irritants: Isolated Chicken Eye Test Method. NIH-Veröffentlichung Nr. 06-4513. Research Triangle Park: National Toxicology Program. Verfügbar unter: http://iccvam.niehs.nih.gov/methods/ocutox/ivocutox/ocu_brd_ice.htm. (10) Prinsen, M.K. und Koëter, B.W.M. (1993). Justification of the enucleated eye test with eyes of slaughterhouse animals as an alternative to the Draize eye irritation test with rabbits. Fd. Chem. Toxicol. 31:69-76. (11) DB-ALM (INVITTOX) (2009). Protokoll Nr. 80: Chicken enucleated eye test (CEET) / Isolated Chicken Eye Test, 13pp. Verfügbar unter: http://ecvam-dbalm.jrc.ec.europa.eu/. (12) Balls, M., Botham, P.A., Bruner, L.H. und Spielmann H. (1995). The EC/HO international validation study on alternatives to the Draize eye irritation test. Toxicol. in Vitro, 9:871-929. (13) Prinsen, M.K. (1996). The chicken enucleated eye test (CEET): A practical (pre)screen for the assessment of eye irritation/corrosion potential of test materials. Food Chem. Toxicol. 34:291-296. (14) Chamberlain, M., Gad, S.C., Gautheron, P. und Prinsen, M.K. (1997). IRAG-Arbeitsgruppe I: Organotypic models for the assessment/prediction of ocular irritation. Food Chem. Toxicol. 35:23-37. (15) Prinsen, M.K. (2006). The Draize Eye Test and in vitro alternatives; a left-handed marriage? Toxicology in Vitro, 20:78-81. (16) Siegel, J.D., Rhinehart, E., Jackson, M., Chiarello, L. und das Healthcare Infection Control Practices Advisory Committee (2007), Guideline for Isolation Precautions: Preventing Transmission of Infectious Agents in Healthcare Settings. Verfügbar unter: http://www.cdc.gov/ncidod/dhqp/pdf/isolation2007.pdf. (17) Maurer, J.K., Parker, R.D. und Jester, J.V. (2002). Extent of corneal injury as the mechanistic basis for ocular irritation: key findings and recommendations for the development of alternative assays. Reg. Tox. Pharmacol., 36:106-117. (18) Burton, A.B.G., M. York und R.S. Lawrence (1981). The in vitro assessment of severe irritants. Fd. Cosmet.- Toxicol., 19:471-480. _____ 1) Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
Definitionen Augenreizung: Erzeugen von Veränderungen am Auge nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation vollständig reversibel sind. Austauschbar mit "Reversible Wirkungen am Auge" und mit der "UN-GHS-Kategorie 2" (4). Bottom-Up-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie keine Einstufung als augenreizend oder schwer augenschädigend erfordert. Dabei werden zuerst Chemikalien, die keine Einstufung erfordern (negatives Ergebnis), von anderen Chemikalien (positives Ergebnis) unterschieden. Chemikalie: Stoff oder Gemisch. Evidenzbasierte Bewertung: Prüfung der Stärken und Schwächen verschiedener Informationen, um über das Gefahrenpotenzial einer Chemikalie entscheiden zu können und diese Entscheidung zu untermauern. Falsch-Negativ-Rate: Der Anteil aller positiven Chemikalien, die von einer Prüfmethode fälschlicherweise als negativ identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. Falsch-Positiv-Rate: Der Anteil aller negativen Chemikalien, die von einer Prüfmethode fälschlicherweise als positiv identifiziert werden. Sie ist ein Leistungsindikator der Prüfmethode. Fluorescein-Verfärbung: Subjektiver Messwert im Rahmen des ICE-Tests für die Fluorescein-Natrium-Verfärbung der Epithel-Zellen in der Hornhaut nach Applikation einer Prüfchemikalie. Das Ausmaß der Fluorescein-Verfärbung ist ein Indikator der Schädigung des Hornhautepithels. Gefahr: Inhärente Eigenschaft eines Stoffes oder eines Umfelds mit dem Potenzial, einen Organismus, ein System oder eine (Sub)population bei Exposition gegenüber diesem Stoff zu schädigen. Gemisch: Gemisch oder Lösung aus zwei oder mehr Stoffen, die nicht miteinander reagieren (4). Genauigkeit: Der Grad der Übereinstimmung zwischen Testergebnissen und anerkannten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der "Relevanz". Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode. Gestufte Prüfstrategie: Eine schrittweise Prüfstrategie, bei der alle vorhandenen Informationen über eine Prüfchemikalie in einer vorgegebenen Reihenfolge überprüft werden, wobei auf jeder Stufe nach dem evidenzbasierten Bewertungsansatz (weight-of-evidence) vorgegangen wird, um feststellen zu können, ob genügend Informationen für eine Gefahrenklassifizierung vorliegen, bevor zur nächsten Stufe übergegangen wird. Wenn das Reizpotenzial einer Prüfchemikalie auf Basis der vorliegenden Informationen zugeordnet werden kann, sind keine weiteren Testungen erforderlich. Ist dies nicht der Fall, müssen schrittweise sequenzielle Tierversuche durchgeführt werden, bis eine eindeutige Klassifizierung vorgenommen werden kann. Globales Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien der Vereinten Nationen (UN-GHS): Ein System zur Klassifizierung von Chemikalien (Stoffen und Gemischen) nach standardisierten Typen und Stufen physikalischer, gesundheitlicher und ökologischer Gefahren und zur entsprechenden Kennzeichnung durch Piktogramme, Signalwörter, Gefahrenhinweise, Sicherheitshinweise und Sicherheitsdatenblätter, um zum Schutz des Menschen (einschließlich Arbeitgeber, Arbeiter, Spediteure, Verbraucher und Notfall-Einsatzkräfte) und der Umwelt Informationen über die schädlichen Wirkungen der betreffenden Chemikalien zu verbreiten (4). Hornhaut: Der Iris und Pupille überdeckende transparente vordere Teil des Augapfels, über den Licht ins Augeninnere übertragen wird. Hornhautschwellung: Objektiver Messwert im Rahmen des ICE-Tests für die Umfangszunahme der Hornhaut nach Applikation einer Prüfchemikalie. Der Wert wird als Prozentsatz ausgedrückt und auf Basis von Referenz-Hornhautdickenmessungen (Messung vor der Applikation) und der in regelmäßigen Abständen nach der Applikation der Prüfchemikalie im ICE-Test aufgezeichneten Dicke berechnet. Das Ausmaß der Hornhautschwellung ist ein Indikator der Hornhautschädigung. Hornhauttrübung: Messwert für die Undurchsichtigkeit der Hornhaut nach Applikation einer Prüfchemikalie. Eine verstärkte Hornhauttrübung ist ein Indikator für die Schädigung der Hornhaut. Irreversible Wirkungen am Auge: siehe "Schwere Augenschädigung" und "UN-GHS-Kategorie 1". Keine Einstufung nach UN-GHS: Stoffe, die die Voraussetzungen für eine Einstufung in die UN-GHS-Kategorien 1 oder2 (2A oder2B) nicht erfüllen. Austauschbar mit "Nicht eingestuft". Lösungsmittel-/Vehikelkontrolle: Eine unbehandelte Probe, die alle Komponenten eines Testsystems enthält, einschließlich des Lösungsmittels oder Vehikels, und die mit den prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt wird, um die Referenzreaktion für die mit der Prüfchemikalie behandelten Proben, die im selben Lösungsmittel oder Vehikel aufgelöst wurden, zu bestimmen. Bei der Testung mit einer gleichzeitigen Negativkontrolle zeigt diese Probe außerdem an, ob das Lösungsmittel oder Vehikel mit dem Testsystem interagiert. Negativkontrolle: Ein unbehandeltes Replikat, das alle Komponenten eines Testsystems enthält. Diese Probe wird mit prüfchemikalienbehandelten Proben und anderen Kontrollproben mitgeführt, um festzustellen, ob das Lösungsmittel mit dem Testsystem interagiert. Nicht eingestuft: Stoffe, die nicht als augenreizend (UN-GHS-Kategorie 2) oder schwer augenschädigend (UN-GHS-Kategorie 1) eingestuft sind. Austauschbar mit der UN-GHS-Klasse "Keine Einstufung". Positivkontrolle: Ein Replikat, das alle Komponenten eines Testsystems enthält und mit einer Chemikalie behandelt wird, die bekanntermaßen eine positive Reaktion hervorruft. Um sicherzustellen, dass Abweichungen bei der Positivkontrollreaktion im Zeitverlauf bewertet werden können, sollte die Reaktion nicht zu heftig sein. Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird. Referenzchemikalie: Eine zum Vergleich mit einer Prüfchemikalie verwendete Bezugsgröße. Eine Referenzchemikalie sollte die folgenden Eigenschaften aufweisen: i) beständige und zuverlässige Quelle(n); ii) strukturelle und funktionelle Ähnlichkeit zur Klasse der geprüften Chemikalien; iii) bekannte physikalische/chemische Eigenschaften; iv) unterstützende Daten zu bekannten Wirkungen und v) bekannte Potenz innerhalb der gewünschten Wirkungsspanne. Reversible Wirkungen am Auge: siehe "Augenreizung" und "UN-GHS-Kategorie 2". Schwere Augenschädigung: Erzeugen von Gewebeschäden im Auge oder eine schwerwiegende Verschlechterung des Sehvermögens nach Applikation einer Prüfchemikalie auf die Oberfläche des Auges, die innerhalb von 21 Tagen nach der Applikation nicht vollständig reversibel sind. Austauschbar mit "Irreversible Wirkungen am Auge" und mit "UN-GHS-Kategorie 1" (4). Spaltlampenmikroskop: Ein Instrument zur direkten Untersuchung des Auges, vergrößert mit einem binokularen Aufrechtbild-Stereomikroskop. Beim ICE-Test wird dieses Instrument eingesetzt, um die Vorderkammern des Hühnerauges visuell zu untersuchen und um die Hornhautdicke anhand eines Pachometer-Aufsatzes objektiv zu messen. Stoff: Chemische Elemente und ihre Verbindungen in natürlicher Form oder durch ein Produktionsverfahren hergestellt, einschließlich der zur Wahrung der Produktstabilität notwendigen Zusatzstoffe und der bei der Herstellung entstehenden Verunreinigungen, mit Ausnahme von Lösungsmitteln, die von dem Stoff ohne Beeinträchtigung seiner Stabilität und ohne Änderung seiner Zusammensetzung abgetrennt werden können (4). Tensid: Auch als oberflächenaktiver Stoff bezeichnet. Hierbei handelt es sich um Stoffe, wie waschaktive Substanzen (Detergenzien), die die Oberflächenspannung einer Flüssigkeit herabsetzen und so die Bildung von Schaum oder das Eindringen in feste Stoffe ermöglichen; auch bekannt als Netzmittel. Top-Down-Ansatz: Schrittweiser Ansatz für eine Chemikalie, von der vermutet wird, dass sie schwere Augenschäden verursacht. Dabei werden zuerst Chemikalien, die schwere Augenschäden verursachen (positives Ergebnis), von anderen Chemikalien (negatives Ergebnis) unterschieden. UN-GHS-Kategorie 1: siehe "Schwere Augenschädigung" und/oder "Irreversible Wirkungen am Auge". UN-GHS-Kategorie 2: siehe "Augenreizung" und/oder "Reversible Wirkungen am Auge". Validierte Prüfmethode: Eine Prüfmethode, für die zwecks Bestimmung ihrer Relevanz (einschließlich Genauigkeit) und Zuverlässigkeit für einen bestimmten Zweck Validierungsstudien abgeschlossen wurden. Es wird darauf hingewiesen, dass eine validierte Prüfmethode möglicherweise nicht genau und zuverlässig genug ist, um für den vorgeschlagenen Zweck akzeptiert zu werden. Zuverlässigkeit: Maß der Reproduzierbarkeit einer Prüfmethode innerhalb von und zwischen Laboratorien über einen längeren Zeitraum und bei einheitlichem Protokoll. Die Zuverlässigkeit wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit und Intralabor-Wiederholbarkeit bewertet.
Vor der routinemäßigen Anwendung eines Tests, der den Anforderungen der vorliegenden Prüfmethode genügt, haben Laboratorien ihre technische Kompetenz nachzuweisen, indem sie die 13 in Tabelle 1 empfohlenen Chemikalien in die richtige Augengefährdungsklasse einstufen. Die Chemikalien wurden so ausgewählt, dass sie die Bandbreite von Augengefährdungen repräsentieren, die auf den Ergebnissen des In-vivo-Kaninchenaugentests (TG 405) und dem UN-GHS-Klassifizierungssystem (d. h. den UN-GHS-Kategorien 1, 2A, 2B oder "Keine Einstufung") basieren (4) (6). Weitere Auswahlkriterien betrafen die Erhältlichkeit der Chemikalien im Handel, die Verfügbarkeit hochwertiger In-vivo-Referenzdaten und das Vorhandensein hochwertiger Daten aus dem ICE-In-vitro-Test. Referenzdaten können aus dem SSD (5) und den Background Review Documents des ICCVAM für den ICE-Test bezogen werden (9). Tabelle 1 Empfohlene Chemikalien für den Nachweis der technischen Kompetenz von Laboratorien zur Durchführung des ICE-Tests
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.49 In-vitro-Mikronukleustest an Säugetierzellen 17 24
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
Diese Prüfmethode entspricht der OECD-Prüfrichtlinie 487 (2016) und ist Teil einer Reihe von Prüfmethoden zur genetischen Toxikologie. Es wurde ein OECD-Dokument erstellt, das kurz gefasste und hilfreiche Informationen zu Untersuchungen zur genetischen Toxikologie sowie eine Übersicht über die jüngsten Änderungen dieser Prüfrichtlinien enthält (1). Der In-vitro-Mikronukleustest (MNvit) ist ein Genotoxizitätstest zum Nachweis von Mikronuklei im Zytoplasma von Interphasezellen. Mikronuklei oder Mikrokerne können aus azentrischen Chromosomenfragmenten (d. h. Chromosomen, denen ein Zentromer fehlt) oder aus ganzen Chromosomen entstehen, die während der Anaphase der Zellteilung nicht zu den Polen wandern können. Der MNvit-Test ist daher eine In-vitro-Methode, die eine breite Basis für die In-vitro-Erforschung potenzieller Chromosomenschädigungen bildet, da sowohl Aneugene als auch Klastogene (2) (3) in Zellen nachgewiesen werden können, die während oder nach Kontakt mit der Prüfchemikalie eine Zellteilung durchlaufen haben (weitere Einzelheiten siehe Nummer 13). Mikrokerne stellen auf Tochterzellen übertragene Schäden dar, während in Metaphasezellen festgestellte Chromosomenaberrationen unter Umständen nicht übertragen werden. In beiden Fällen sind die Veränderungen möglicherweise nicht mit dem Überleben der Zellen kompatibel. Die vorliegende Prüfmethode gestattet die Verwendung von Protokollen mit und ohne den Aktin-Polymerisationsinhibitor Cytochalasin B (cytoB). Durch Beigabe von cytoB vor der Mitose entstehen Zellen mit zwei Kernen. Dies ermöglicht die Identifizierung und selektive Analyse von Mikrokernen in Zellen, die eine Mitose durchlaufen haben (4) (5). Diese Prüfmethode gestattet auch die Verwendung von Protokollen ohne Zytokinese-Block, vorausgesetzt, es gibt Beweise dafür, dass die analysierte Zellpopulation eine Mitose vollzogen hat. Zusätzlich zum MNvit-Test zur Identifizierung von Chemikalien, die Mikrokerne erzeugen, können auch die immunchemische Markierung von Kinetochoren oder die Hybridisierung mit Zentromer- bzw. Telomer-Sonden (Fluoreszenz-in-situ-Hybridisierung (FISH)) zusätzliche Informationen über die Mechanismen der Chromosomenschädigung und der Bildung von Mikrokernen liefern (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17). Diese Markierungs- und Hybridisierungsverfahren können angewandt werden, wenn eine verstärkte Mikrokernbildung festgestellt wird und der Prüfer erkennen will, ob diese Zunahme das Ergebnis klastogener und/oder aneugener Ereignisse ist. Da Mikrokerne in Zellen im Stadium der Interphase mit relativer Objektivität bewertet werden können, muss das Laborpersonal nur bestimmen, wie viele Zellen zwei Kerne (bei Hinzufügung von cytoB) bzw. (in allen anderen Fällen) wie viele Zellen einen Mikrokern enthalten. Infolgedessen können die Objektträger relativ schnell bewertet werden, und die Analyse lässt sich automatisieren. Dies macht es praktisch möglich, Tausende statt Hunderte von Zellen pro Behandlung zu bewerten und so die Aussagekraft des Tests zu erhöhen. Da schließlich die Mikrokerne von verzögert transportierten Chromosomen herrühren können, besteht die Möglichkeit, Aneuploidie induzierende Agenzien nachzuweisen, deren Untersuchung in konventionellen Chromosomenaberrationstests nur schwer möglich ist, z.B. Kapitel B.10 dieses Anhangs (18). Allerdings gestattet der in dieser Prüfmethode beschriebene MNvit-Test die Differenzierung zwischen Veränderungen der Chromosomenzahl und/oder Polyploidie induzierenden Chemikalien und Klastogenizität verursachenden Chemikalien nur, wenn besondere Techniken wie die unter Nummer 4 genannte FISH eingesetzt werden. Der MNvit-Test ist zuverlässig und kann bei einer Vielfalt von Zelltypen, mit oder ohne Zusatz von cytoB, durchgeführt werden. Umfassende Daten belegen die Validität des MNvit-Tests bei Verwendung unterschiedlicher Zelltypen (kultivierter Zelllinien oder primärer Zellkulturen) (19) (20) (21) (22) (23) (24) (25) (26) (27) (28) (29) (30) (31) (32) (33) (34) (35) (36). Hierzu zählen insbesondere die internationalen Validierungsstudien, koordiniert durch die Société Française de Toxicologie Génétique (SFTG) (19) (20) (21) (22) (23), und die Berichte des International Workshop on Genotoxicity Testing (5) (17). Die verfügbaren Daten wurden außerdem vom Europäischen Zentrum zur Validierung von Alternativmethoden (ECVAM) der Europäischen Kommission in einer retrospektiven Validierungsstudie nach dem Weight-of-Evidence-Ansatz neu bewertet, und die Prüfmethode wurde vom Wissenschaftlich Beratenden Ausschuss (ESAC) des ECVAM als wissenschaftlich validiert anerkannt (37) (38) (39). Beim MNvit-Test an Säugetierzellen können kultivierte Zelllinien oder primäre Zellkulturen menschlichen Ursprungs oder von Nagetieren zum Einsatz kommen. Da die Hintergrundfrequenz von Mikrokernen die Empfindlichkeit des Tests beeinflusst, empfiehlt es sich, Zelltypen mit einer stabilen und festgelegten Hintergrundfrequenz der Mikrokernbildung zu verwenden. Die verwendeten Zellen werden unter dem Gesichtspunkt der Wachstumsfähigkeit in Kultur, der Karyotypstabilität (einschließlich Chromosomenzahl) und der spontanen Häufigkeit von Mikrokernen ausgewählt (40). Zum gegenwärtigen Zeitpunkt lassen die verfügbaren Daten keine konkreten Empfehlungen zu, deuten jedoch darauf hin, dass es bei der Bewertung chemischer Gefahren wichtig ist, den p53-Status, die genetische (Karyotyp-)Stabilität, die DNA-Reparaturfähigkeit und die Herkunft (Nagetier oder Mensch) der für die Tests ausgewählten Zellen zu berücksichtigen. Anwendern dieser Prüfmethode wird daher empfohlen, den Einfluss dieser und anderer Zelleigenschaften auf die Leistung einer Zelllinie bei der Erkennung der Mikrokerninduktion zu berücksichtigen, da sich die Erkenntnisse auf diesem Gebiet ständig weiterentwickeln. Es gelten die Definitionen gemäß Anlage 1. Vorbemerkungen und Einsatzgrenzen In vitro durchgeführte Tests erfordern in der Regel den Zusatz eines exogenen Stoffwechselaktivierungssystems, sofern die Zellen nicht im Hinblick auf die Prüfchemikalien metabolisch kompetent sind. Mit diesem exogenen Stoffwechselaktivierungssystem lassen sich die In-vivo-Bedingungen jedoch nicht gänzlich nachvollziehen. Es sind auch Bedingungen zu vermeiden, die zu künstlich positiven Ergebnissen führen, die nicht die Genotoxizität der Prüfchemikalie widerspiegeln. Dazu gehören unter anderem Veränderungen des pH-Wertes (41) (42) (43) bzw. der Osmolalität, Wechselwirkung mit dem Zellkulturmedium (44) (45) oder hochgradige Zytotoxizität (siehe Nummer 29). Zur Analyse der Mikrokerninduktion ist es wesentlich, dass sowohl die behandelten als auch die unbehandelten Kulturen eine Mitose durchlaufen haben. Das informativste Stadium für die Auswertung von Mikrokernen liegt in Zellen vor, die eine Mitose während oder nach der Behandlung mit der Prüfchemikalie vollzogen haben. Bei hergestellten Nanomaterialien sind besondere Anpassungen dieser Prüfmethode erforderlich, die hier jedoch nicht beschrieben werden. Bevor die Prüfmethode auf ein Gemisch angewendet wird, um zu Regulierungszwecken Daten zu gewinnen, sollte geprüft werden, ob, und falls ja, warum, sie für diesen Zweck geeignete Ergebnisse liefern kann. Diese Überlegungen erübrigen sich, sofern die Durchführung von Tests für das Gemisch gesetzlich vorgeschrieben ist. Zellkulturen menschlichen Ursprungs oder von Säugetieren werden sowohl mit als auch ohne exogene Stoffwechselaktivierung mit der Prüfchemikalie in Kontakt gebracht, außer wenn Zellen mit eigener adäquater Metabolisierungskapazität verwendet werden (siehe Nummer 19). Während oder nach dem Kontakt mit der Prüfchemikalie werden die Zellen so lange kultiviert, dass Chromosomenschäden oder andere Wirkungen auf den Zellzyklus/die Zellteilung zur Bildung von Mikrokernen in Interphasezellen führen können. Zur Induktion einer Aneuploidie sollte die Prüfchemikalie während der Mitose vorhanden sein. Gewonnene und gefärbte Interphasezellen werden auf das Vorhandensein von Mikrokernen untersucht. Im Idealfall sollten Mikrokerne nur in Zellen ausgewertet werden, die während des Kontakts mit der Prüfchemikalie oder ggf. während der Zeit nach der Behandlung eine Mitose durchlaufen haben. In Kulturen, die mit einem Zytokinese-Blocker behandelt wurden, ist dies ohne Weiteres möglich, indem nur zweikernige Zellen ausgewertet werden. Wenn kein Zytokinese-Blocker verwendet wurde, ist es wichtig nachzuweisen, dass die analysierten Zellen aufgrund einer Zunahme der Zellpopulation wahrscheinlich während oder nach dem Kontakt mit der Prüfchemikalie eine Zellteilung durchlaufen haben. Bei allen Protokollen ist es wichtig nachzuweisen, dass sowohl in den Kontrollkulturen als auch in den behandelten Kulturen eine Zellproliferation stattgefunden hat. Das Ausmaß der von der Prüfchemikalie induzierten Zytotoxizität oder Zytostase sollte in allen Kulturen bewertet werden, die auf das Vorhandensein von Mikrokernen untersucht werden. Zellen Es können kultivierte primäre Lymphozyten aus dem peripheren Blut von Menschen oder anderen Säugetieren (7) (20) (46) (47) sowie verschiedene Zelllinien von Nagetieren wie CHO, V79, CHL/IU und L5178Y oder menschliche Zelllinien wie TK6 verwendet werden (19) (20) (21) (22) (23) (26) (27) (28) (29) (31) (33) (34) (35) (36) (siehe Nummer 6). Andere Zelllinien wie HT29 (48), Caco-2 (49), HepaRG (50) (51), HepG2-Zellen (52) (53), A549 und primäre Embryonalzellen des Syrischen Hamsters (54) wurden in Mikronukleustests verwendet, sind zu diesem Zeitpunkt jedoch nicht umfassend validiert worden. Die Verwendung dieser Zelllinien und -typen sollte daher anhand ihrer nachgewiesenen Leistung im Test begründet werden, wie im Abschnitt "Akzeptanzkriterien" beschrieben. CytoB kann Berichten zufolge das Wachstum von L5178Y-Zellen beeinträchtigen und wird daher bei dieser Zelllinie nicht empfohlen (23). Wenn aus Gründen des Tierschutzes primäre Zellen verwendet werden, sollten, soweit möglich, Zellen menschlichen Ursprungs in Betracht gezogen und nach humanethischen Grundsätzen und Vorschriften beprobt werden. Lymphozyten aus dem peripheren Blut von Menschen sollten jungen (ca. 18- bis 35-jährigen) nicht rauchenden Personen ohne bekannte Erkrankung entnommen werden, die bekanntermaßen in letzter Zeit nicht mit genotoxischen Einwirkungen (z.B. in Form von Chemikalien oder ionisierender Strahlung) in einer Intensität in Kontakt gekommen sind, die zu einem Anstieg der Hintergrundrate von Mikrokernzellen führen würde. Dies stellt eine niedrige und gleichmäßige Hintergrundrate von Mikrokernzellen sicher. Die Hintergrundrate von Mikrokernzellen nimmt mit fortschreitendem Alter zu. Dieser Trend ist bei Frauen ausgeprägter als bei Männern (55). Wenn Zellen von mehreren Spendern zur Verwendung gepoolt werden, ist die Anzahl der Spender anzugeben. Es muss nachgewiesen werden, dass sich die Zellen seit dem Beginn der Behandlung mit der Prüfchemikalie bis zur Zellentnahme geteilt haben. Zellkulturen werden in einer Phase des exponentiellen Wachstums gehalten (Zelllinien) oder zur Teilung angeregt (primäre Lymphozytenkulturen), damit die Zellen in unterschiedlichen Stadien des Zellzyklus exponiert werden, da die Empfindlichkeit der Zellstadien gegenüber den Prüfchemikalien möglicherweise nicht bekannt ist. Die Primärzellen, die mit mitogenen Stoffen zur Teilung stimuliert werden müssen, werden in der Regel während der Behandlung mit der Prüfchemikalie nicht weiter synchronisiert (z.B. humane Lymphozyten nach einer 48-stündigen mitogenen Stimulierung). Die Verwendung synchronisierter Zellen während der Behandlung mit der Prüfchemikalie wird nicht empfohlen, kann jedoch zulässig sein, sofern sie begründet wird. Kulturmedien und Inkubationsbedingungen Zur Kultivierung sind geeignete Kulturmedien und Inkubationsbedingungen (Kulturgefäße, ggf. befeuchtete Atmosphäre mit einer CO2-Konzentration von 5 %, Temperatur von 371 °C) zu verwenden. Zelllinien sind routinemäßig auf Stabilität der modalen Chromosomenzahl und Mykoplasmaverunreinigung zu überprüfen, und Zellen sollten bei Verunreinigung oder bei veränderter modaler Chromosomenzahl nicht herangezogen werden. Die normale Dauer des Zellzyklus bei den gewählten Zelllinien oder der im Prüflabor verwendeten primären Kulturen sollte bekannt sein und mit den veröffentlichten Zelleigenschaften übereinstimmen. Vorbereitung der Kulturen Zelllinien: Die Zellen werden aus Stammkulturen gewonnen und im Kulturmedium in einer solchen Dichte überimpft, dass die Zellen in Suspensions- oder Monolayerkultur bis zum Zeitpunkt der Gewinnung weiterhin exponentiell wachsen (z.B. sollte eine Konfluenz bei in Monolayerkultur gezüchteten Zellen vermieden werden). Lymphozyten: Mit einem Antikoagulans (z.B. Heparin) behandeltes Vollblut oder separierte Lymphozyten werden einem Kulturmedium beigegeben (z.B. bei humanen Lymphozyten für die Dauer von 48 Stunden), das ein Mitogen (z.B. Phytohämagglutinin (PHA) im Fall von humanen Lymphozyten) enthält, um eine Zellteilung vor der Behandlung mit der Prüfchemikalie und cytoB herbeizuführen. Stoffwechselaktivierung Bei Zellen mit ungeeigneter Stoffwechselkapazität sollten exogene metabolisierende Systeme eingesetzt werden. Das am häufigsten verwendete, standardmäßig empfohlene System, sofern nicht anders begründet, ist eine durch Ko-Faktoren ergänzte post-mitochondriale Fraktion (S9) aus der Leber von Nagetieren (in der Regel Ratten), die mit enzyminduzierenden Agenzien wie Aroclor 1254 (56) (57) oder einem Gemisch aus Phenobarbital und b-Naphtoflavon (58) (59) (69) vorbehandelt wurde. Das letztgenannte Gemisch verstößt nicht gegen das Stockholmer Übereinkommen über persistente organische Schadstoffe (61) und hat sich bei der Induktion von Mischfunktionsoxidasen als ebenso wirksam wie Aroclor 1254 erwiesen (58) (59) (60). Die S9-Fraktion wird im Endmedium in der Regel in Konzentrationen von 1 bis 2 % v/v verwendet, kann jedoch auf 10 % v/v erhöht werden. Die Verwendung von Produkten, die den Mitoseindex senken, insbesondere Komplexbildnern für Calcium (62), sollte während der Behandlung vermieden werden. Die Wahl der Art und Konzentration des exogenen Metabolisierungssystems oder metabolischen Agens ist möglicherweise von der geprüften chemischen Klasse abhängig. Vorbereitung der Prüfchemikalie Feste Prüfchemikalien sollten vor der Zellbehandlung in geeigneten Lösungsmitteln gelöst und ggf. verdünnt werden. Flüssige Prüfchemikalien können dem Versuchssystem vor der Behandlung direkt beigegeben und/oder verdünnt werden. Gasförmige oder flüchtige Prüfchemikalien sind mithilfe geeigneter Änderungen an den Standardprotokollen, wie z.B. durch Behandlung in hermetisch verschlossenen Gefäßen (63) (64) (65), zu prüfen. Zubereitungen der Prüfchemikalie sollten kurz vor der Behandlung hergestellt werden, es sei denn, die Stabilität der Chemikalie bei Lagerung wird nachgewiesen. Prüfbedingungen Lösungsmittel Das Lösungsmittel sollte so gewählt werden, dass eine optimale Löslichkeit der Prüfchemikalie gewährleistet ist, ohne dass die Durchführung des Versuchs negativ beeinträchtigt wird, z.B. durch Änderung des Zellwachstums, Beeinträchtigung der Integrität der Prüfchemikalie, Reaktion mit Kulturgefäßen, Behinderung des Stoffwechselaktivierungssystems. Es ist zu empfehlen, als erste Wahl möglichst die Verwendung eines wässrigen Lösungsmittels (oder Kulturmediums) in Erwägung zu ziehen. Gründlich erprobte Lösungsmittel sind Wasser oder Dimethylsulfoxid (DMSO). In der Regel sollen organische Lösungsmittel 1 % v/v nicht übersteigen. Wenn cytoB in DMSO gelöst wird, darf die für die Prüfchemikalie und cytoB verwendete Gesamtmenge an organischen Lösungsmitteln nicht mehr als 1 % v/v betragen; anderenfalls sind unbehandelte Kontrollen zu verwenden, um sicherzustellen, dass der Anteil an organischen Lösungsmitteln keine schädliche Wirkung hat. Wässrige Lösungsmittel (Kochsalzlösung oder Wasser) sollen 10 % v/v im Endmedium nicht übersteigen. Kommen weniger gründlich erprobte Lösungsmittel zur Verwendung (z. B Ethanol oder Aceton), ist deren Verwendung durch Daten zu stützen, die ihre Verträglichkeit mit der Prüfchemikalie, mit dem Versuchssystem sowie ihre mangelnde Genotoxizität in der verwendeten Konzentration belegen. Liegen keine Daten vor, die dies belegen, ist es wichtig, unbehandelte Kontrollen (siehe Anlage 1) sowie Lösungsmittelkontrollen einzubeziehen, um nachzuweisen, dass durch die gewählten Lösungsmittel keine schädlichen oder chromosomalen Wirkungen (z.B. Aneuploidie oder Klastogenizität) ausgelöst werden. Verwendung von cytoB als Zytokinese-Blocker Eine der wichtigsten Überlegungen bei der Durchführung des MNvit-Tests gilt der Vergewisserung, dass die bewerteten Zellen während der Behandlung oder ggf. während der Inkubationszeit nach der Behandlung eine Mitose durchlaufen haben. Die Auswertung von Mikrokernen soll daher auf Zellen beschränkt werden, die während oder nach der Behandlung eine Mitose vollzogen haben. CytoB ist das am häufigsten verwendete Agens zur Zytokinese-Blockierung, da es den Aktinaufbau hemmt und somit die Trennung der Tochterzellen nach der Mitose verhindert, was wiederum zur Bildung zweikerniger Zellen führt (6) (66) (67). Zugleich kann bei Verwendung von cytoB die Auswirkung der Prüfchemikalie auf die Zellproliferationskinetik gemessen werden. CytoB sollte bei der Verwendung menschlicher Lymphozyten als Zytokinese-Blocker eingesetzt werden, da die Zellzyklendauer zwischen einzelnen Spendern variiert und nicht alle Lymphozyten auf Phytohämagglutinin reagieren. Bei anderen Zelltypen ist cytoB nicht zwingend erforderlich, sofern nachgewiesen werden kann, dass diese eine Teilung durchlaufen haben, wie unter Nummer 27 beschrieben. Außerdem wird cytoB im Allgemeinen nicht verwendet, wenn die Proben mithilfe durchflusszytometrischer Methoden auf Mikrokerne untersucht werden. Für jeden Zelltyp sollte das Labor die geeignete cytoB-Konzentration festlegen, um in den Lösungsmittelkontrollkulturen eine optimale Frequenz der zweikernigen Zellen zu erreichen. Ferner sollte die Konzentration nachweislich eine gute Ausbeute an zweikernigen Zellen zur Auswertung ergeben. Die geeignete cytoB-Konzentration liegt in der Regel zwischen 3 und 6 µ/ml (19). Messung von Zellproliferation und Zytotoxizität und Wahl der Behandlungskonzentrationen Bei der Festlegung der höchsten zu testenden Konzentration der Prüfchemikalie sind Konzentrationen zu vermeiden, die zu künstlich positiven Reaktionen führen können, wie z.B. zu übermäßiger Zytotoxizität (siehe Nummer 29), Ausfällungen im Kulturmedium (siehe Nummer 30) oder ausgeprägten Änderungen des pH-Werts oder der Osmolalität (siehe Nummer 9). Sofern die Prüfchemikalie zum Zeitpunkt der Zugabe den pH-Wert des Mediums erheblich verändert, lässt sich der pH-Wert auch durch Anwendung eines Puffers im Endmedium einstellen, damit künstlich positive Reaktionen vermieden und geeignete Kulturbedingungen aufrechterhalten werden. Es werden Messungen der Zellproliferation vorgenommen, um sicherzustellen, dass genügend behandelte Zellen während des Tests eine Mitose durchlaufen haben und dass die Behandlungen bei geeigneten Zytotoxizitätsniveaus durchgeführt werden (siehe Nummer 29). Die Zytotoxizität sollte im Hauptversuch mit und ohne Stoffwechselaktivierung und unter Verwendung eines geeigneten Indikators für Zelltod und -wachstum bestimmt werden (siehe Nummern 26 und 27). Wenngleich die Bewertung der Zytotoxizität im Rahmen eines ersten Vorversuchs nützlich sein kann, um die im Hauptversuch verwendeten Konzentrationen besser bestimmen zu können, ist ein Vorversuch nicht zwingend erforderlich. Wird er durchgeführt, ersetzt er nicht die Messung der Zytotoxizität im Hauptversuch. Die Behandlung von Kulturen mit cytoB und die Messung der relativen Häufigkeit von einkernigen, zweikernigen und mehrkernigen Zellen in der Kultur sind ein genaues Verfahren zur Quantifizierung des Effekts auf die Zellproliferation und die zytotoxische oder zytostatische Wirkung einer Behandlung (6) und stellen sicher, dass nur Zellen mikroskopisch bewertet werden, die während oder nach der Behandlung eine Teilung vollzogen haben. Es wird empfohlen, die zytotoxische und zytostatische Wirkung einer Behandlung mittels des Zytokinese-Block-Proliferationsindex (CBPI) (6) (27) (68) oder des Replikationsindex (RI) bei mindestens 500 Zellen pro Kultur abzuleiten (Formeln siehe Anlage 2). Hierzu werden die Werte in behandelten Kulturen und Kontrollkulturen miteinander verglichen. Die Bewertung anderer Zytotoxizitäts-Indikatoren (z.B. Zellintegrität, Apoptose, Nekrose, Metaphasezählung, Zellzyklus) könnte ebenfalls nützliche Informationen liefern, darf jedoch nicht als Ersatz für den CBPI oder RI verwendet werden. In Studien ohne cytoB muss nachgewiesen werden, dass sich die Zellen in der Kultur geteilt haben, sodass ein erheblicher Teil der bewerteten Zellen während oder nach Behandlung mit der Prüfchemikalie eine Teilung durchlaufen hat. Anderenfalls kann es zu falsch negativen Reaktionen kommen. Die Messung der relativen Populationsverdopplung (RPD) oder der relativen Erhöhung der Zellzahl (RICC) sind empfohlene Verfahren zur Schätzung der zytostatischen Wirkung eines Behandlung (17) (68) (69) (70) (71) (Formeln siehe Anlage 2). Bei längeren Probenahmephasen (z.B. Behandlung über einen Zeitraum von 1,5 bis 2 normalen Zellzykluslängen und Gewinnung nach weiteren 1,5 bis 2 normalen Zellzykluslängen, woraus sich eine Probenahmezeit von insgesamt mehr als 3 bis 4 normalen Zellzykluslängen ergibt, wie unter Nummern 38 und 39 beschrieben) könnte es bei der RPD zu einer Unterschätzung der Toxizität kommen (71). Unter diesen Umständen ist die RICC möglicherweise ein besseres Verfahren. Anderenfalls erhält man anhand einer Bewertung der Zytotoxizität nach 1,5 bis 2 normalen Zellzykluslängen einen hilfreichen Schätzwert. Die Bewertung anderer Marker für Zytotoxizität oder Zytostase (z.B. Zellintegrität, Apoptose, Nekrose, Metaphasezählung, Proliferationsindex (PI), Zellzyklus, nukleoplasmische Brücken oder Kernknospen) könnten nützliche Informationen liefern, dürfen jedoch nicht als Ersatz für die RPD oder die RICC verwendet werden. Es sollten mindestens drei Versuchskonzentrationen (ausgenommen das Lösungsmittel und Positivkontrollen), die die Akzeptanzkriterien erfüllen (geeignete Zytotoxizität, Anzahl der Zellen usw.), ausgewertet werden. Unabhängig von der Art der Zellen (Zelllinien oder Primärkulturen von Lymphozyten) können für jede überprüfte Konzentration Replikat- oder Einfachkulturen herangezogen werden. Wenngleich die Verwendung von Zweifachkulturen ratsam ist, sind Einfachkulturen auch zulässig, vorausgesetzt, es wird für Einfach- oder Zweifachkulturen jeweils die gleiche Gesamt-Zellpopulation ausgewertet. Die Verwendung von Einzelkulturen ist insbesondere dann relevant, wenn mehr als drei Konzentrationen bewertet werden (siehe Nummern 44 und 45). Die Ergebnisse aus den unabhängigen Replikatkulturen bei einer festgelegten Konzentration können zu Datenanalysezwecken gepoolt werden. Bei Prüfchemikalien mit geringer oder keiner Zytotoxizität sind in der Regel Konzentrationsintervalle mit zwei- bis dreifacher Konzentration geeignet. Wenn Zytotoxizität auftritt, sollten die Prüfkonzentrationen einen Bereich ausgehend vom Wert, bei dem Zytotoxizität auftritt (siehe Beschreibung unter Nummer 29), bis zu Konzentrationen mit mäßiger oder geringer oder nicht vorhandener Toxizität umfassen. Viele Prüfchemikalien zeigen steile Konzentrations-Wirkungs-Kurven. Um Daten zu mäßiger oder geringer Toxizität zu erhalten oder um die Dosis-Wirkungs-Beziehungen im Einzelnen auszuwerten, wird es daher erforderlich sein, Konzentrationen mit kleineren Abständen und/oder mehr als drei Konzentrationen zu verwenden (Einfach- oder Replikatkulturen), insbesondere in Fällen, in denen ein Wiederholungsversuch erforderlich ist (siehe Nummer 60). Beruht die höchste Konzentration auf der Zytotoxizität, sollte die Höchstkonzentration darauf abzielen, unter Verwendung der empfohlenen Zytotoxizitätsparameter (d. h. Verringerung der RICC und RPD bei Zelllinien, wenn cytoB nicht verwendet wird, und Verringerung des CBPI oder RI, wenn cytoB verwendet wird, auf 45 ± 5 % der gleichzeitigen Negativkontrollen) eine Zytotoxizität von 55 ± 5 % zu erreichen (72). Ausschließlich am oberen Ende dieses Zytotoxizitätsbereichs von 55 ± 5 % anzutreffende positive Ergebnisse sollten mit Vorsicht interpretiert werden (71). Im Falle schwer löslicher Chemikalien, die bei Konzentrationen unterhalb der niedrigsten unlöslichen Konzentration nicht zytotoxisch sind, sollte die höchste analysierte Konzentration am Ende der Behandlung mit der Prüfchemikalie eine Trübung oder eine mit bloßem Auge oder mithilfe eines Inversmikroskops erkennbare Ausfällung bewirken. Auch wenn Zytotoxizität oberhalb der niedrigsten unlöslichen Konzentration auftritt, ist es ratsam, nur eine Konzentration zu testen, die eine Trübung oder sichtbare Ausfällung hervorruft, da artefaktische Wirkungen eine Folge dieser Ausfällung sein könnten. Bei der Konzentration, bei der es zu einer Ausfällung kommt, ist unbedingt sicherzustellen, dass die Ausfällung nicht die Durchführung des Versuchs beeinträchtigt (z.B. Färbung oder Auswertung). Es ist möglicherweise sinnvoll, die Löslichkeit im Kulturmedium vor dem Versuch zu bestimmen. Wenn keine Ausfällung bzw. keine begrenzende Zytotoxizität festgestellt wird, sollte die Höchstkonzentration bei Versuchen 10 mM, 2 mg/ml oder2 µl/ml betragen, je nachdem, welcher Wert am niedrigsten ist (73) (74) (75). Sofern die Zusammensetzung der Prüfchemikalie nicht genau definiert ist, z.B. ein Stoff mit unbekannter oder schwankender Zusammensetzung, komplexe Reaktionsprodukte oder biologische Materialien (UVCB) (76), ein Umweltextrakt usw., muss die höchste Konzentration möglicherweise höher angesetzt werden (z.B. bei 5 mg/ml), sofern keine ausreichende Zytotoxizität vorhanden ist, um die Konzentration der einzelnen Bestandteile zu erhöhen. Es sei jedoch darauf hingewiesen, dass diese Anforderungen sich von denen für Humanpharmazeutika unterscheiden können (93). Kontrollen Bei jedem Gewinnungszeitpunkt sind parallel laufende Negativkontrollen (siehe Nummer 21) anzulegen, bei denen das Behandlungsmedium lediglich Lösungsmittel enthält und die auf die gleiche Weise wie die Behandlungskulturen bearbeitet werden. Gleichzeitige Positivkontrollen müssen durchgeführt werden, um die Eignung des Labors zum Nachweis von Klastogenen und Aneugenen unter den Bedingungen des verwendeten Prüfprotokolls sowie ggf. die Wirksamkeit des exogenen Stoffwechselaktivierungssystems nachzuweisen. Beispiele für Positivkontrollen sind nachstehender Tabelle 1 zu entnehmen. Es können andere geeignete Positivkontrollchemikalien verwendet werden, sofern dies begründet ist. Zum gegenwärtigen Zeitpunkt sind keine Aneugene bekannt, die zu ihrer genotoxischen Wirkung eine Stoffwechselaktivierung benötigen (17). Da In-vitro-Tests auf Genotoxizität in Säugetierzellen für die gleichzeitigen Kurzzeitbehandlungen mit und ohne Zusatz eines Stoffwechselaktivierungssystems über die gleiche Behandlungsdauer ausreichend standardisiert sind, kann sich die Hinzuziehung von Positivkontrollen auf ein Klastogen beschränken, das eine Stoffwechselaktivierung erfordert. In diesem Fall wird durch die Reaktion einer einzelnen Positivkontrolle sowohl die Aktivität des Stoffwechselaktivierungssystems als auch die Reaktionsfähigkeit des Testsystems nachgewiesen. Im Falle einer Langzeitbehandlung (ohne S9) sollte jedoch eine gesonderte Positivkontrolle erfolgen, da die Behandlungsdauer beim Test mit Stoffwechselaktivierung eine andere ist. Wird als einzige Positivkontrolle für die Kurzzeitbehandlung mit und ohne Stoffwechselaktivierung ein Klastogen ausgewählt, sollte für die Langzeitbehandlung ohne Stoffwechselaktivierung ein Aneugen ausgewählt werden. Positivkontrollen für Klastogenizität und Aneugenizität sollten in metabolisch kompetenten Zellen verwendet werden, die kein S9 benötigen. Jede Positivkontrolle sollte bei einer oder mehreren Konzentrationen durchgeführt werden, die voraussichtlich eine reproduzierbare und erkennbare Zunahme gegenüber dem Hintergrund ergeben, womit sich die Empfindlichkeit des Versuchssystems nachweisen lässt (d. h. die Wirkungen sind eindeutig, lassen aber beim Ablesen nicht sofort die Identität der kodierten Objektträger erkennen), und die Wirkung sollte nicht durch einen Zytotoxizitätswert beeinträchtigt werden, der die in dieser Prüfmethode vorgegebenen Grenzen überschreitet. Tabelle 1 Zur Beurteilung der Eignung des Labors und zur Wahl der Positivkontrollen empfohlene Referenzchemikalien
Behandlungsplan Um die Wahrscheinlichkeit zu maximieren, dass ein Aneugen oder Klastogen in einem bestimmten Stadium des Zellzyklus erkannt wird, muss eine ausreichende Anzahl an Zellen während aller Stadien ihrer Zellzyklen mit der Prüfchemikalie behandelt werden. Alle Behandlungen sollten während des exponentiellen Wachstums der Zellen beginnen und enden, und die Zellen sollten bis zum Zeitpunkt der Probenahme weiter wachsen. Der Behandlungsplan für Zelllinien und primäre Zellkulturen kann daher in gewisser Weise von dem für Lymphozyten abweichen, die für den Beginn ihres Zellzyklus einer mitogenen Stimulation bedürfen (17). Bei Lymphozyten ist es am wirksamsten, die Behandlung mit der Prüfchemikalie 44 bis 48 Stunden nach der PHA-Stimulation zu beginnen, wenn sich die Zellen asynchron teilen (6). Veröffentlichte Daten (19) legen nahe, dass die meisten Aneugene und Klastogene entdeckt werden, wenn eine kurze Behandlung von 3 bis 6 Stunden mit oder ohne Vorhandensein von S9 erfolgt, woraufhin die Prüfchemikalie entfernt wird und die Zellen in einem Zeitraum beprobt werden, der etwa der 1,5-fachen bis 2-fachen Dauer des normalen Zellzyklus nach Behandlungsbeginn entspricht (7). Für eine gründliche Bewertung, die erforderlich wäre, um auf ein negatives Ergebnis zu schließen, sollten bei Durchführung einer Kurzzeitbehandlung mit und ohne Stoffwechselaktivierung und einer Langzeitbehandlung ohne Stoffwechselaktivierung alle drei der folgenden Versuchsbedingungen angewandt werden, und zwar eine (siehe Nummern 56, 57 und 58):
Führt eine der oben genannten Versuchsbedingungen zu einem positiven Befund, ist es möglicherweise nicht erforderlich, Untersuchungen anhand der anderen Behandlungsverfahren durchzuführen. Wenn bekannt ist oder der Verdacht besteht, dass die Prüfchemikalie die Dauer des Zellzyklus beeinflusst (z.B. bei der Prüfung von Nucleosidanalogen), was insbesondere für p53-kompetente Zellen gilt (35) (36) (77), können die Zeiträume für die Probenahme oder Regenerierung um bis zu eine weitere 1,5-fache bis 2-fache Dauer des normalen Zellzyklus (d. h. insgesamt die 3-fache bis 4-fache Dauer des Zellzyklus nach Beginn der Kurzzeit- und Langzeitbehandlungen) ausgedehnt werden. Diese Optionen beziehen sich auf Situationen, in denen mögliche Wechselwirkungen zwischen der Prüfchemikalie und cytoB befürchtet werden. Bei verlängerten Probenahmezeiten (d. h. einer Kulturzeit von insgesamt der 3-fachen bis 4-fachen Zellzyklusdauer) ist unbedingt sicherzustellen, dass sich die Zellen noch aktiv teilen. Bei Lymphozyten beispielsweise kann das exponentielle Wachstum 96 Stunden nach der Stimulation nachlassen, und Monolayerkulturen können konfluent werden. Die empfohlenen Pläne für die Zellbehandlung sind in Tabelle 2 zusammengefasst. Diese allgemeinen Behandlungspläne können je nach Stabilität oder Reaktivität der Prüfchemikalie oder den besonderen Wachstumseigenschaften der verwendeten Zellen (mit entsprechender Begründung) modifiziert werden. Tabelle 2 Zellbehandlung und Entnahmezeitpunkte beim MNvit-Test
Bei Monolayerkulturen können am Ende der 3- bis 6-stündigen Behandlung mitotische Zellen vorhanden sein (erkennbar daran, dass sie rund sind und sich von der Oberfläche ablösen). Da diese mitotischen Zellen sich leicht lösen, können sie bei Entfernung des Mediums mit der Prüfchemikalie verloren gehen. Kann nachgewiesen werden, dass die Zahl der mitotischen Zellen gegenüber den Kontrollen erheblich zugenommen hat, was mit hoher Wahrscheinlichkeit auf einen mitotischen Arrest hinweist, sollten die Zellen durch Zentrifugieren gewonnen und anschließend der Kultur wieder zugeführt werden, um zu vermeiden, dass Zellen verlorengehen, die sich in der Mitose befinden und zum Zeitpunkt der Gewinnung dem Risiko einer Mikrokernbildung/Chromosomenaberration ausgesetzt sind. Zellgewinnung und Präparation des Objektträgers Jede Kultur wird gesondert gewonnen und aufbereitet. Die Zellpräparation kann eine Behandlung mit hypotoner Lösung beinhalten, dieser Schritt ist jedoch nicht notwendig, wenn eine angemessene Ausbreitung der Zellen anderweitig sichergestellt wird. Bei der Präparation der Objektträger können verschiedene Techniken angewandt werden, vorausgesetzt, es werden Zellpräparate hoher Qualität zur Auswertung bereitgestellt. Dabei sollten Zellen mit intakter Zellmembran und intaktem Zytoplasma zurückgehalten werden, um die Erkennung von Mikrokernen sowie (bei der Zytokinese-Block-Methode) die zuverlässige Identifizierung zweikerniger Zellen zu ermöglichen. Die Objektträger können mit verschiedenen Verfahren angefärbt werden, beispielsweise mit Giemsa oder fluoreszierenden DNA-spezifischen Farbstoffen. Durch Verwendung eines DNA-spezifischen Farbstoffs (z.B. Acridin Orange (78) oder Hoechst 33258 Plus Pyronin-Y (79)) lassen sich bestimmte Artefakte vermeiden, die bei der Verwendung eines nicht DNA-spezifischen Farbstoffs auftreten. Zur Identifizierung des Inhalts (ganze Chromosomen werden eingefärbt, azentrische Chromosomenfragmente nicht) von Mikrokernen können Anti-Kinetochor-Antikörper, FISH mit panzentromerischen DNA-Sonden oder eine präparierte In-situ-Markierung mit panzentromer-spezifischen Primern, zusammen mit der geeigneten DNA-Gegenfärbung, verwendet werden, wenn mechanistische Informationen zu ihrer Bildung benötigt werden (16) (17). Andere Methoden zur Unterscheidung zwischen Klastogenen und Aneugenen können ebenfalls angewandt werden, wenn sie sich als wirksam erwiesen haben. Beispielsweise könnten bei bestimmten Zelllinien die Messungen von sub-2N-Kernen als hypodiploide Ereignisse mittels Techniken wie Bildanalyse, Laser-Scan-Zytometrie oder Durchflusszytometrie ebenfalls nützliche Informationen liefern (80) (81) (82). Morphologische Beobachtungen von Kernen könnten ebenfalls auf eine mögliche Aneuploidie hinweisen. Ein Test auf Chromosomenaberrationen in der Metaphase, vorzugsweise am gleichen Zelltyp und unter Verwendung eines Protokolls mit vergleichbarer Empfindlichkeit, könnte ebenfalls eine nützliche Methode sein, um festzustellen, ob die Mikrokerne auf einen Chromosomenbruch zurückzuführen sind (in dem Wissen, dass ein Chromosomenverlust mit dem Test auf Chromosomenaberrationen nicht erkannt würde). Analyse Alle Objektträger, auch die für das Lösungsmittel und die unbehandelten Kontrollen (sofern verwendet) und die Positivkontrollen, sind vor der mikroskopischen Untersuchung der Mikrokernfrequenzen von unabhängiger Seite zu kodieren. Bei der Verwendung eines automatisierten Auswertungssystems, wie Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse, sollten geeignete Techniken zum Ausgleich systematischer Messfehler verwendet werden. Unabhängig davon, welche automatisierte Plattform zur Zählung der Mikrokerne verwendet wird, sind CBPI, RI, RPD oder RICC parallel auszuwerten. In Kulturen, die mit cytoB behandelt wurden, sind die Mikrokernfrequenzen von mindestens 2.000 zweikernigen Zellen pro Konzentration und Kontrolle (83) zu analysieren, die, sofern Replikate verwendet werden, gleichmäßig auf die Replikate verteilt sein sollten. Bei der Verwendung von Einfachkulturen pro Dosis (siehe Nummer 28) sollten in einer solchen Kultur mindestens 2.000 zweikernige Zellen pro Kultur (83) ausgewertet werden. Wenn wesentlich weniger als 1.000 zweikernige Zellen pro Kultur (bei Zweifachkulturen) bzw. 2.000 (bei Einfachkulturen) in jeder Konzentration für die Auswertung zur Verfügung stehen und wenn keine signifikante Zunahme an Mikrokernen festgestellt wird, ist der Test mit einer höheren Anzahl an Zellen oder mit weniger zytotoxischen Konzentrationen zu wiederholen, je nachdem, was angemessen ist. Es ist darauf zu achten, dass keine zweikernigen Zellen ausgewertet werden, die unregelmäßige Formen aufweisen oder deren zwei Kerne in der Größe beträchtlich voneinander abweichen. Außerdem dürfen zweikernige Zellen nicht mit schlecht auf dem Träger verteilten mehrkernigen Zellen verwechselt werden. Zellen mit mehr als zwei Hauptkernen werden nicht auf Mikrokerne untersucht, da in diesen Zellen die zugrunde liegende Mikrokernfrequenz höher sein könnte (84). Die Auswertung einkerniger Zellen ist akzeptabel, wenn die Prüfchemikalie die Wirkung von cytoB beeinträchtigt. In diesen Fällen könnte ein Wiederholungstest ohne cytoB sinnvoll sein. Die Auswertung einkerniger Zellen zusätzlich zu den zweikernigen Zellen könnte nützliche Informationen liefern (85) (86), ist jedoch nicht zwingend erforderlich. In Zelllinien, die ohne cytoB-Behandlung getestet wurden, sind die Mikrokerne in mindestens 2.000 Zellen pro Prüfkonzentration und Kontrolle (83) zu analysieren, die, sofern Replikate verwendet werden, gleichmäßig auf die Replikate verteilt sein sollten. Bei der Verwendung von Einzelkulturen je Konzentration (siehe Nummer 28) sind in dieser Kultur mindestens 2.000 Zellen auszuwerten. Wenn wesentlich weniger als 1.000 Zellen pro Kultur (bei Zweifachkulturen) bzw. 2.000 Zellen (bei Einfachkulturen) in jeder Konzentration für die Auswertung zur Verfügung stehen und wenn keine signifikante Zunahme an Mikrokernen festgestellt wird, ist der Test mit einer höheren Anzahl an Zellen oder mit weniger zytotoxischen Konzentrationen zu wiederholen, je nachdem, was angemessen ist. Wenn cytoB verwendet wird, ist von mindestens 500 Zellen pro Kultur ein CBPI oder RI zur Bewertung der Zellproliferation (siehe Anlage 2) festzulegen. Werden die Behandlungen ohne cytoB durchgeführt, muss unbedingt nachgewiesen werden, dass die Zellen in der Kultur eine Zellteilung vollzogen haben, wie unter den Nummern 24 bis 28 erläutert. Kompetenz des Labors Um ausreichende Erfahrungen mit der Prüfung zu sammeln, bevor routinemäßige Tests erfolgen, sollte das Labor eine Reihe von Versuchen mit positiven Referenzchemikalien durchgeführt haben, die sich unterschiedlicher Mechanismen (mindestens einer mit und einer ohne Stoffwechselaktivierung sowie einer mit einem aneugenen Wirkmechanismus, ausgewählt aus den in Tabelle 1 aufgeführten Chemikalien) und verschiedener Negativkontrollen (einschließlich unbehandelter Kulturen und verschiedener Lösungsmittel/Vehikel) bedienen. Die Reaktionen dieser Positiv- und Negativkontrollen sollten mit der Literatur im Einklang stehen. Dies gilt nicht für erfahrene Labors, d. h. für Labors, die über eine Datenbank mit historischen Daten gemäß der Definition unter den Nummern 49 bis 52 verfügen. Eine Auswahl von Positivkontrollchemikalien (siehe Tabelle 1) sollte anhand von Kurz- und Langzeitbehandlungen ohne Stoffwechselaktivierung und darüber hinaus anhand einer Kurzzeitbehandlung mit Stoffwechselaktivierung untersucht werden, um die Fähigkeit des Labors zum Nachweis klastogener und aneugener Chemikalien, zur Bestimmung der Wirksamkeit des Stoffwechselaktivierungssystems und des Nachweises der Eignung der Auswertungsverfahren (visuelle mikroskopische Untersuchung, Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse) zu belegen. Zum Nachweis der Empfindlichkeit und dynamischen Bandbreite des Versuchssystems sollte eine Spanne der Konzentrationen der ausgewählten Chemikalien festgelegt werden, um reproduzierbare und konzentrationsbezogene Zunahmen gegenüber dem Hintergrund zu erhalten. Historische Kontrolldaten Das Labor sollte Folgendes bestimmen:
Bei der erstmaligen Erfassung von Daten zur Verteilung einer historischen Negativkontrolle sollten gleichzeitige Negativkontrollen veröffentlichten Negativkontrolldaten entsprechen, sofern solche vorhanden sind. Werden weitere Versuchsdaten zur Verteilung der Kontrollen hinzugefügt, sollten gleichzeitige Negativkontrollen idealerweise innerhalb von 95 % der Kontrollgrenzen der gewählten Verteilung liegen (87) (88). Die Datenbank des Labors zu historischen Negativkontrollen sollte zunächst mit mindestens 10 Versuchen angelegt werden. Vorzugsweise sollte sie jedoch aus mindestens 20 Versuchen bestehen, die unter vergleichbaren Versuchsbedingungen durchgeführt wurden. Labors sollten Qualitätskontrollverfahren anwenden, wie z.B. Qualitätsregelkarten (z.B. C-Karten oder X-Bar-Karten (88)), um zu ermitteln, wie variabel ihre Positiv- und Negativkontrolldaten sind, und um nachzuweisen, dass die Methodik in ihrem Labor "unter Kontrolle" ist (83). Weitere Empfehlungen zum Aufbau und zur Verwendung von Sammlungen historischer Daten (d. h. Kriterien für die Aufnahme und den Ausschluss von Daten in bzw. aus historischen Datensätzen und die Gültigkeitskriterien für einen bestimmten Versuch) sind den Literaturangaben zu entnehmen (87). Sämtliche Änderungen am Versuchsprotokoll sind auf Übereinstimmung mit den bereits vorhandenen historischen Kontrolldaten zu prüfen. Bei größeren Inkonsistenzen sollte eine neue historische Kontrolldatenbank erstellt werden. Negativkontrolldaten sollten das Auftreten von Zellen mit Mikrokernen aus einer Einzelkultur oder aus einer Summe von Replikatkulturen erfassen (vgl. Beschreibung unter Nummer 28). Gleichzeitige Negativkontrollen sollten idealerweise innerhalb der Kontrollgrenzen von 95 % der gewählten Verteilung in der Datenbank des Labors zu historischen Negativkontrollen liegen (87) (88). Sofern gleichzeitige Negativkontrolldaten außerhalb der Kontrollgrenzen von 95 % liegen, ist es zulässig, sie in die historische Kontrollverteilung aufzunehmen, solange es sich bei den Daten nicht um "extreme Ausreißer" handelt und nachgewiesen werden kann, dass das Testsystem "unter Kontrolle" ist (siehe Nummer 50) und nachweislich kein technisches oder menschliches Versagen vorliegt. Interpretation der Ergebnisse Bei Verwendung des Zytokinese-Block-Verfahrens werden nur die Frequenzen zweikerniger Zellen mit Mikrokernen (unabhängig von der Anzahl der Mikrokerne pro Zelle) zur Bewertung der Mikrokern-Induktion herangezogen. Die Auswertung der Anzahl an Zellen mit einem, zwei oder mehr Mikrokernen kann separat ausgewiesen werden und könnte nützliche Informationen liefern, ist jedoch nicht zwingend notwendig. Festzulegen sind auch Maßnahmen, die gleichzeitig zur Bestimmung der Zytotoxizität und/oder Zytostase aller behandelten Kulturen und aller Negativ- und Positivkontrollkulturen durchgeführt werden (16). Der CBPI oder der RI ist für alle behandelten Kulturen und Kontrollkulturen zu berechnen, da sich bei Anwendung der Zytokinese-Block-Methode die Messungen des Zellzyklus verzögern. Kommt cytoB nicht zum Einsatz, sollte die relative Populationsverdopplung (RPD) oder die relative Erhöhung der Zellzahl (RICC) herangezogen werden (siehe Anlage 2). Es sind die Daten für die einzelnen Kulturen zu dokumentieren. Zusätzlich sollten alle Daten in tabellarischer Form zusammengefasst werden. Gültigkeitskriterien Die Akzeptanz eines Versuchs beruht auf folgenden Kriterien:
Bewertung und Interpretation der Ergebnisse Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig positiv, wenn unter den geprüften Versuchsbedingungen (siehe Nummern 36-39)
Sind alle diese Kriterien erfüllt, wird davon ausgegangen, dass die Prüfchemikalie in diesem Testsystem Chromosomenbrüche und/oder Gewinn bzw. Verlust von Chromosomenmaterial auslösen kann. Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (90) (91) (92). Unter der Voraussetzung, dass alle Akzeptanzkriterien erfüllt sind, gilt eine Prüfchemikalie als eindeutig negativ, wenn unter allen geprüften Versuchsbedingungen (siehe Nummern 36-39)
In diesem Fall wird davon ausgegangen, dass die Prüfchemikalie keine Chromosomenbrüche und/oder Gewinn bzw. Verlust von Chromosomenmaterial in diesem Testsystem auslösen kann. Empfehlungen zu den am besten geeigneten statistischen Methoden sind der Literatur zu entnehmen (90) (91) (92). Bei einer eindeutig positiven oder negativen Reaktion ist eine Verifizierung nicht erforderlich. In den Fällen, in denen die Reaktion, wie oben beschrieben, weder eindeutig negativ noch eindeutig positiv ist, und/oder um die biologische Relevanz eines Ergebnisses zu untermauern, sollten die Daten durch eine fachkundige Beurteilung und/oder anhand weiterer Untersuchungen bewertet werden. Die Auswertung weiterer Zellen oder die Durchführung eines Wiederholungsversuchs, möglicherweise unter veränderten Versuchsbedingungen (z.B. Abstände der Konzentrationen, andere Stoffwechselaktivierungsbedingungen (d. h. Konzentration oder Herkunft von S9), könnte gegebenenfalls hilfreich sein. In seltenen Fällen erlaubt der Datensatz selbst nach weiteren Untersuchungen keine definitive Aussage zu positiven oder negativen Ergebnissen, und es wird daher die Schlussfolgerung gezogen, dass die Reaktion nicht eindeutig ist. Wenn Prüfchemikalien im MNvit-Test Mikrokerne induzieren, kann dies darauf zurückzuführen sein, dass sie Chromosomenbrüche, Chromosomenverlust oder eine Kombination aus beidem verursachen. Deshalb können weitere Analysen unter Einsatz von Anti-Kinetochor-Antikörpern, zentromer-spezifischen In-situ-Sonden oder mithilfe sonstiger Methoden vorgenommen werden, um festzustellen, ob der Mechanismus der Mikrokern-Induktion von einer klastogenen und/oder aneugenen Wirkung herrührt. Testbericht Der Prüfbericht muss folgende Angaben enthalten: Prüfchemikalie:
Einkomponentige Substanz:
Mehrkomponentige Substanz, UVCB-Stoffe und Gemische:
Lösungsmittel:
Zellen:
Prüfbedingungen:
Ergebnisse:
Diskussion der Ergebnisse: Schlussfolgerungen (1) OECD (2016). Overview of the set of OECD Genetic Toxicology Test Guidelines and updates performed in 2014-2015. ENV Publications. Series on Testing and Assessment, No. 234, OECD, Paris. (2) Kirsch-Volders, M. (1997). Towards a validation of the micronucleus test. Mutation Research, Vol. 392/1-2, 1-4. (3) Parry, J.M., A. Sors (1993). The detection and assessment of the aneugenic potential of environmental chemicals: the European Community aneuploidy project. Mutation Research, Vol. 287/1, 3-15. (4) Fenech, M., A.A. Morley (1985). Solutions to the kinetic problem in the micronucleus assay. Cytobios, Vol. 43/172-173, 233-246. (5) Kirsch-Volders, M. et al. (2000). Report from the In Vitro Micronucleus Assay Working Group. Environmental and Molecular Mutagenesis, Vol. 35/3, 167-172. (6) Fenech, M. (2007). Cytokinesis-block micronucleus cytome assay. Nature Protocols, Vol. 2/5, 1084-1104. (7) Fenech, M., A.A. Morley (1986). Cytokinesis-block micronucleus method in human lymphocytes: effect of in-vivo ageing and low dose X-irradiation. Mutation Research, Vol. 161/2, 193-198. (8) Eastmond, D.A., J.D. Tucker (1989). Identification of aneuploidy-inducing agents using cytokinesis-blocked human lymphocytes and an antikinetochore antibody. Environmental and Molecular Mutagenesis, Vol. 13/1, 34-43. (9) Eastmond, D.A., D. Pinkel (1990). Detection of aneuploidy and aneuploidy-inducing agents in human lymphocytes using fluorescence in-situ hybridisation with chromosome-specific DNA probe. Mutation Research, Vol. 234/5, 9-20. (10) Miller, B.M. et al. (1991). Classification of micronuclei in murine erythrocytes: immunofluorescent staining using CREST antibodies compared to in situ hybridization with biotinylated gamma satellite DNA. Mutation Research, Vol. 6/4, 297-302. (11) Farooqi, Z., F. Darroudi, A. T. Natarajan (1993). The use of fluorescence in-situ hybridisation for the detection of aneugens in cytokinesis-blocked mouse splenocytes. Mutation Research, Vol. 8/4, 329-334. (12) Henderson, L. et al. (1993). Cytogenetic damage induced in human lymphocytes by four vanadium compounds and micronucleus analysis by fluorescence in situ hybridization with a centromeric probe. Mutation Research, Vol. 319/3, 205-213. (13) Norppa, H., L. Renzi, C. Lindholm (1993). Detection of whole chromosomes in micronuclei of cytokinesis-blocked human lymphocytes by antikinetochore staining and in situ hybridization. Mutation Research, Vol. 8/6, 519-525. (14) Eastmond, D.A, D.S. Rupa, L.S. Hasegawa (1994). Detection of hyperdiploidy and chromosome breakage in interphase human lymphocytes following exposure to the benzene metabolite hydroquinone using multicolor fluorescence in situ hybridization with DNA probes. Mutation Research, Vol. 322/1, 9-20. (15) Marshall, R.R. et al. (1996). Fluorescence in situ hybridisation (FISH) with chromosome-specific centromeric probes: a sensitive method to detect aneuploidy. Mutation Research, Vol. 372/2, 233-245. (16) Zijno, P. et al. (1996). Analysis of chromosome segregation by means of fluorescence in situ hybridization: application to cytokinesis-blocked human lymphocytes. Mutation Research, Vol. 372/2, 211-219. (17) Kirsch-Volders et al. (2003). Report from the in vitro micronucleus assay working group. Mutation Research, Vol. 540/2, 153-163. (18) Kapitel B.10: In-vitro-Test auf Chromosomenaberrationen in Säugetierzellen. (19) Lorge, E. et al. (2006). SFTG International collaborative Study on in vitro micronucleus test. I. General conditions and overall conclusions of the study. Mutation Research, Vol. 607/1, 13-36. (20) Clare, G. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test. II. Using human lymphocytes. Mutation Research, Vol. 607/1, 37-60. (21) Aardema, M.J. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, III. Using CHO cells. Mutation Research, Vol. 607/1, 61-87. (22) Wakata, A. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, IV. Using CHO/IU cells. Mutation Research, Vol. 607/1, 88-124. (23) Oliver, J. et al. (2006). SFTG International collaborative study on the in vitro micronucleus test, V. Using L5178Y cells. Mutation Research, Vol. 607/1, 125-152. (24) Albertini, S. et al. (1997). Detailed data on in vitro MNT and in vitro CA: industrial experience. Mutation Research, Vol. 392/1-2, 187-208. (25) Miller, B. et al. (1997). Comparative evaluation of the in vitro micronucleus test and the in vitro chromosome aberration test: industrial experience. Mutation Research, Vol. 392/1-2, 45-59. (26) Miller, B. et al. (1998). Evaluation of the in vitro micronucleus test as an alternative to the in vitro chromosomal aberration assay: position of the GUM Working Group on the in vitro micronucleus test. Gesellschaft für Umwelt-Mutationsforschung, Mutation Research, Vol. 410, 81-116. (27) Kalweit, S. et al. 1999. Chemically induced micronucleus formation in V79 cells - comparison of three different test approaches. Mutation Research, Vol. 439/2, 183-190. (28) Kersten, B. et al. 1999. The application of the micronucleus test in Chinese hamster V79 cells to detect drug-induced photogenotoxicity. Mutation Research, Vol. 445/1, 55-71. (29) von der Hude, W. et al. (2000). In vitro micronucleus assay with Chinese hamster V79 cells - results of a collaborative study with in situ exposure to 26 chemical substances. Mutation Research, Vol. 468/2, 137-163. (30) Garriott, M.L., J.B. Phelps, W.P. Hoffman (2002). A protocol for the in vitro micronucleus test, I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity. Mutation Research, Vol. 517/1-2, 123-134. (31) Matsushima, T. et al. 1999. Validation study of the in vitro micronucleus test in a Chinese hamster lung cell line (CHL/IU). Mutation Research, Vol. 14/6, 569-580. (32) Elhajouji, A., E. Lorge (2006). Sonderausgabe: SFTG International collaborative study on in vitro micronucleus test. Mutation Research, Vol. 607/1, 1-152. (33) Kirkland, D. (2010). Evaluation of different cytotoxic and cytostatic measures for the in vitromicronucleus test (MNVit): Introduction to the collaborative trial. Mutation Research, Vol. 702/2, 139-147. (34) Hashimoto K. et al. (2011). Comparison of four different treatment conditions of extended exposure in the in vitro micronucleus assay using TK6 lymphoblastoid cells. Regulatory Toxicology and Pharmacology, Vol. 59/1, 28-36. (35) Honma, M., M. Hayashi (2011). Comparison of in vitro micronucleus and gene mutation assay results for p53-competent versus p53-deficient human lymphoblastoid cells. Environmental and Molecular Mutagenesis, Vol. 52/5, 373-384. (36) Zhang, L.S. et al. (1995). A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test. Mutation Research Letters, Vol. 347/3-4, 105-115. (37) ECVAM (2006). Statement by the European Centre for the Validation of Alternative Methods (ECVAM) Scientific Advisory Committee (ESAC) on the scientific validity of the in vitro micronucleus test as an alternative to the in vitro chromosome aberration assay for genotoxicity testing. 25. Sitzung des ESAC, 16./17. November2006, verfügbar unter: http://ecvam.jrc.it/index.htm. (38) ESAC (2006). Peer-Review des Wissenschaftlich Beratenden Ausschusses (ESAC) des ECVAM, Retrospective Validation of the In Vitro Micronucleus Test, Summary and Conclusions of the Peer Review Panel. Verfügbar unter: http://ecvam.jrc.it/index.htm. (39) Corvi, R. et al. (2008). ECVAM Retrospective Validation of in vitro Micronucleus Test (MNT). Mutagenesis, Vol. 23/4, 271-283. (40) Publikation des ILSI (Entwurf). Lorge, E., M.M. Moore, J. Clements, M. O"Donovan, M. Honma, A. Kohara, J. van Benthem, S. Galloway, M.J. Armstrong, A. Sutter, V. Thybaud, B. Gollapudi, M. Aardema, J. Young-Tannir. Standardized Cell Sources and Recommendations for Good Cell Culture Practices in Genotoxicity Testing. Mutation Research. (41) Scott, D. et al. (1991). International Commission for Protection Against Environmental Mutagens and Carcinogens, Genotoxicity under extreme culture conditions. A report from ICPEMC Task Group 9. Mutation Research, Vol. 257/2, 147-205. (42) Morita, T. et al. (1992). Clastogenicity of low pH to various cultured mammalian cells. Mutation Research, Vol. 268/2, 297-305. (43) Brusick, D. (1986). Genotoxic effects in cultured mammalian cells produced by low pH treatment conditions and increased ion concentrations. Environmental Mutagenesis, Vol. 8/6, 789-886. (44) Long, L.H. et al. (2007). Different cytotoxic and clastogenic effects of epigallocatechin gallate in various cell-culture media due to variable rates of its oxidation in the culture medium. Mutation Research, Vol. 634/1-2, 177-183. (45) Nesslany, F. et al. (2008). Characterization of the Genotoxicity of Nitrilotriacetic Acid. Environmental and Molecular Mutation, Vol. 49, 439-452. (46) Fenech, M., A.A. Morley (1985). Measurement of micronuclei in lymphocytes. Mutation Research, Vol. 147/1-2, 29-36. (47) Fenech, M. (1997). The advantages and disadvantages of cytokinesis-blood micronucleus method. Mutation Research, Vol. 392, 11-18. (48) Payne, C.M. et al. (2010). Hydrophobic bile acid-induced micronuclei formation, mitotic perturbations, and decreases in spindle checkpoint proteins: relevance to genomic instability in colon carcinogenesis. Nutrition and Cancer, Vol. 62/6, 825-840. (49) Bazin, E. et al. (2010). Genotoxicity of a Freshwater Cyanotoxin, Cylindrospermopsin, in Two Human Cell Lines: Caco-2 and HepaRG. Environmental and Molecular Mutagenesis, Vol. 51/3, 251-259. (50) Le Hegarat, L. et al. (2010). Assessment of the genotoxic potential of indirect chemical mutagens in HepaRG cellsby the comet and the cytokinesis-block micronucleus assays. Mutagenesis, Vol. 25/6, 555-560. (51) Josse, R. et al. (2012). An adaptation of the human HepaRG cells to the in vitro micronucleus assay. Mutagenesis, Vol. 27/3, 295-304. (52) Ehrlich, V. et al. (2002). Fumonisin B1 is genotoxic in human derived hepatoma (HepG2) cells. Mutation Research, Vol. 17/3, 257-260. (53) Knasmüller, S. et al. (2004). Use of human-derived liver cell lines for the detection of environmental and dietary genotoxicants; current state of knowledge. Toxicology, Vol. 198/1-3, 315-328. (54) Gibson, D.P. et al. (1997). Induction of micronuclei in Syrian hamster embryo cells: comparison to results in the SHE cell transformation assay for National Toxicology Program test chemicals. Mutation Research, Vol. 392/1-2, 61-70. (55) Bonassi, S. et al. (2001). HUman Micro Nucleus Project: international database comparison for results with the cytokinesis-block micronucleus assay in human lymphocytes, I. Effect of laboratory protocol, scoring criteria and host factors on the frequency of micronuclei. Environmental and Molecular Mutagenesis, Vol. 37/1, 31-45. (56) Maron, D.M., B.N. Ames (1983). Revised methods for the Salmonella mutagenicity test. Mutation Research, Vol. 113/3-4, 173-215. (57) Ong, T.-m. et al. (1980). Differential effects of cytochrome P450-inducers on promutagen activation capabilities and enzymatic activities of S-9 from rat liver. Journal of Environmental Pathology and Toxicology, Vol. 4/1, 55-65. (58) Elliott, B.M. et al. (1992). Alternatives to Aroclor 1254-induced S9 in in-vitro genotoxicity assays. Mutagenesis, Vol. 7, 175-177. (59) Matsushima, T. et al. (1976)."A Safe Substitute for Polychlorinated Biphenyls as an Inducer of Metabolic Activation Systems", in In Vitro Metabolic Activation in Mutagenesis Testing, de Serres, F.J. et al. (eds), Elsevier, North-Holland, 85-88. (60) Johnson, T.E., D. R. Umbenhauer, S.M. Galloway (1996). Human liver S-9 metabolic activation: proficiency in cytogenetic assays and comparison with phenobarbital/beta-naphthoflavone or Aroclor 1254 induced rat S-9. Environmental and Molecular Mutagenesis, Vol. 28, 51-59. (61) UNEP (2001). Stockholmer Übereinkommen über persistente organische Schadstoffe, Umweltprogramm der Vereinten Nationen (UNEP). Verfügbar unter: http://www.pops.int/ (62) Tucker, J.D., M.L. Christensen (1987). Effects of anticoagulants upon sister-chromatid exchanges, cell-cycle kinetics, and mitotic index in human peripheral lymphocytes. Mutation Research, Vol. 190/3, 225-8. (63) Krahn, D.F., F.C. Barsky, K.T. McCooey (1982)."CHO/HGPRT Mutation Assay: Evaluation of Gases and Volatile Liquids", in Genotoxic Effects of Airborne Agents. Tice, R.R., D.L. Costa, K.M. Schaich (eds.), Plenum, New York, 91-103. (64) Zamora, P.O. et al. (1983). Evaluation of an exposure system using cells grown on collagen gels for detecting highly volatile mutagens in the CHO/HGPRT mutation assay. Environmental Mutagenesis, Vol. 5/6, 795-801. (65) Asakura, M. et al. (2008). An improved system for exposure of cultured mammalian cells to gaseous compounds in the chromosomal aberration assay. Mutation Research, Vol. 652/2, 122-130. (66) Fenech, M. (1993). The cytokinesis-block micronucleus technique: a detailed description of the method and its application to genotoxicity studies in human populations. Mutation Research, Vol. 285/1, 35-44. (67) Phelps, J.B., M.L. Garriott, W.P. Hoffman (2002). A protocol for the in vitro micronucleus test. II. Contributions to the validation of a protocol suitable for regulatory submissions from an examination of 10 chemicals with different mechanisms of action and different levels of activity. Mutation Research, Vol. 521/1-2, 103-112. (68) Kirsch-Volders, M. et al. (2004). Corrigendum to "Report from the in vitro micronucleus assay working group" . Mutation Research, 564, 97-100. (69) Lorge, E. et al. (2008). Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test. I. Theoretical aspects. Mutation Research, Vol. 655/1-2, 1-3. (70) Surralles, J. et al. (1995). Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures. Mutation Research, Vol. 341/3, 169-184. (71) Honma, M. (2011). Cytotoxicity measurement in in vitro chromosome aberration test and micronucleus test. Mutation Research, Vol. 724, 86-87. (72) Pfuhler, S. et al. (2011). In vitro genotoxicity test approaches with better predictivity: Summary of an IWGT workshop. Mutation Research, Vol. 723/2, 101-107. (73) OECD (2014). Document supporting the WNT decision to implement revised criteria for the selection of the top concentration in the in vitro mammalian cell assays on genotoxicity (Test Guidelines 473, 476 and 487). ENV/JM/TG(2014)17. Auf Anfrage erhältlich. (74) Morita T., M. Honma, K. Morikawa (2012). Effect of reducing the top concentration used in the in vitro chromosomal aberration test in CHL cells on the evaluation of industrial chemical genotoxicity. Mutation Research, Vol. 741, 32-56. (75) Brookmire, L., J.J. Chen, D.D. Levy (2013). Evaluation of the Highest Concentrations Used in the in vitro Chromosome Aberrations Assay. Environmental and Molecular Mutagenesis, Vol. 54/1, 36-43. (76) EPA, Office of Chemical Safety and Pollution Prevention (2011). Chemical Substances of Unknown or Variable Composition, Complex Reaction Products and Biological Materials: UVCB Substances. http://www.epa.gov/opptintr/newchems/pubs/uvcb.txt. (77) Sobol, Z. et al. (2012). Development and validation of an in vitro micronucleus assay platform in TK6 cells. Mutation Research, Vol. 746/1, 29-34. (78) Hayashi, M., T. Sofuni, M. Jr. Ishidate (1983). An Application of Acridine Orange Fluorescent Staining to the Micronucleus Test. Mutation Research, Vol. 120/4, 241-247. (79) MacGregor, J. T., C.M. Wehr, R.G. Langlois (1983). A Simple Fluorescent Staining Procedure for Micronuclei and RNA in Erythrocytes Using Hoechst 33258 and Pyronin Y. Mutation Research, Vol. 120/4, 269-275. (80) Bryce, S.M. et al. (2011). Miniaturized flow cytometry-based CHO-K1 micronucleus assay discriminates aneugenic and clastogenic modes of action. Environmental and Molecular Mutagenesis, Vol. 52/4, 280-286. (81) Nicolette, J. et al. (2011). in vitro micronucleus screening of pharmaceutical candidates by flow cytometry in Chinese hamster V79 cells. Environmental and Molecular Mutagenesis, Vol. 52/5, 355-362. (82) Shi, J., R. Bezabhie, A. Szkudlinska (2010). Further evaluation of a flow cytometric in vitro micronucleus assay in CHO-K1 cells: a reliable platform that detects micronuclei and discriminates apoptotic bodies. Mutagenesis, Vol. 25/1, 33-40. (83) OECD (2014). Statistical analysis supporting the revision of the genotoxicity Test Guidelines. OECD Environment, Health and Safety Publications (EHS), Series on Testing and Assessment Nr. 198, OECD Publishing, Paris. (84) Fenech, M. et al. (2003). HUMN project: detailed description of the scoring criteria for the cytokinesis-block micronucleus assay using isolated human lymphocyte cultures. Mutation Research, Vol. 534/1-2, 65-75. (85) Elhajouji, A., M. Cunha, M. Kirsch-Volders (1998). Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis-block assay. Mutagenesis, Vol. 13/2, 193-8. (86) Kirsch-Volders, M. et al. (2011). The in vitro MN assay in 2011: origin and fate, biological significance, protocols, high throughput methodologies and toxicological relevance. Archives of Toxicology, Vol. 85/8, 873-99. (87) Hayashi, M. et al. (2010). Compilation and use of genetic toxicity historical control Data. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 723/2, 87-90. (88) Ryan, T. P. (2000). Statistical Methods for Quality Improvement. 2. Auflage, John Wiley and Sons, New York (89) Hoffman, W.P., M.L. Garriott, C. Lee (2003)."In vitro micronucleus test", in Encyclopedia of Biopharmaceutical Statistics, 2. Auflage. Chow, S. (Hrsg.), Marcel Dekker, Inc. New York, 463-467. (90) Fleiss, J. L., B. Levin, M.C. Paik (2003). Statistical Methods for Rates and Proportions, 3. Auflage, John Wiley & Sons, New York. (91) Galloway, S.M. et al. (1987). Chromosome aberration and sister chromatid exchanges in Chinese hamster ovary cells: Evaluation of 108 chemicals. Environmental and Molecular Mutagenesis, Vol. 10/suppl. 10, 1-175. (92) Richardson, C. et al. (1989). Analysis of Data from in vitro Cytogenetic Assays, in Statistical Evaluation of Mutagenicity Test Data, Kirkland, D.J. (Hrsg.), Cambridge University Press, Cambridge, 141-154. (93) International Conference on Harmonisation (ICH) Guidance S2 (R1) on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended For Human Use.
Definitionen Aneugen: Chemikalie oder Prozess, die/der durch Wechselwirkung mit den Komponenten des mitotischen und meiotischen Zellteilungszyklus-Apparats zu Aneuploidie in Zellen oder Organismen führt. Aneuploidie: Abweichung von der normalen diploiden (oder haploiden) Chromosomenzahl durch ein einziges Chromosom oder mehr, nicht aber durch einen ganzen (oder mehrere) Chromosomensatz/-sätze (Polyploidie). Apoptose: Programmierter Zelltod, der durch eine Reihe von Schritten charakterisiert ist, an deren Ende eine Desintegration von Zellen in membranumschlossene Partikel steht, die schließlich durch Phagozytose oder Shedding abgebaut werden. Chemikalie: Stoff oder Gemisch. Genotoxisch: Allgemeine Bezeichnung für alle Arten von DNA- oder Chromosomenschäden, einschließlich Brüchen, Deletionen, Addukten, Nukleotidmodifikationen und -verknüpfungen, Reunionen, Genmutationen, Chromosomenaberrationen und Aneuploidie. Nicht alle genotoxischen Effekte führen zu Mutationen oder stabilen Chromosomenschäden. Kinetochor: Proteinhaltige Struktur, die sich am Zentromer eines Chromosoms sammelt, an der während der Zellteilung die Spindelfasern anhaften, wodurch die ordnungsgemäße Beförderung der Tochterchromosomen zu den Polen der Tochterzellen ermöglicht wird. Klastogen: Chemikalie oder Ereignis, die/das strukturelle Chromosomenaberrationen in Zellpopulationen oder eukaryontischen Organismen auslöst. Konzentrationen: Beziehen sich auf Endkonzentrationen der Prüfchemikalie im Kulturmedium. Interphasezellen: Zellen, die sich nicht im Stadium der Mitose befinden. Lösungsmittelkontrolle: allgemeiner Begriff zur Bezeichnung der Kontrollkulturen, die nur mit dem Lösungsmittel behandelt werden, in dem die Prüfchemikalie gelöst wird. Mikronuclei/Mikrokerne: Kleine Kerne zusätzlich zu den Hauptkernen der Zellen und von diesen getrennt, die während der Telophase der Mitose oder Meiose durch zurückgebliebene Chromosomenteile oder ganze Chromosomen gebildet werden. Mitose: Teilung des Zellkerns, die in der Regel in Prophase, Prometaphase, Metaphase, Anaphase und Telophase gegliedert ist. Mitoseindex: Anteil der Zellen einer Zellpopulation, die sich zum Beobachtungszeitpunkt in Metaphase befinden; ein Hinweis auf den Grad der Zellproliferation dieser Population. Mutagen: Auslöser einer Erbgutveränderung der DNA-Basenpaarsequenz(en) in Genen oder in der Chromosomenstruktur (Chromosomenaberrationen). Non-Disjunktion: Unvermögen der paarweise angeordneten Chromatiden, sich zu trennen und sich auf die in Entwicklung begriffenen Tochterzellen aufzuteilen. Dabei entstehen Tochterzellen mit abweichenden Chromosomenzahlen. p53-Status: Das p53-Protein ist an der Regulierung des Zellzyklus, der Apoptose und der DNA-Reparatur beteiligt. Zellen, denen ein funktionales p53-Protein fehlt und die nicht in der Lage sind, den Zellzyklus aufzuhalten oder beschädigte Zellen über Apoptose oder andere Mechanismen (z.B. Einleitung einer DNA-Reparatur) im Zusammenhang mit Aufgaben des p53-Proteins als Reaktion auf DNA-Schäden zu beseitigen, sollten theoretisch eher zu Genmutationen oder Chromosomenaberrationen neigen. Polyploidie: Zahlenmäßige Chromosomenaberrationen in Zellen oder Organismen, von denen ein oder mehrere ganze Chromosomensätze betroffen sind, im Gegensatz zur Aneuploidie, bei der nur ein oder mehrere einzelne Chromosomen betroffen sind. Proliferationsindex (PI): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2). Prüfchemikalie: Stoff oder Gemisch, der bzw. das nach dieser Prüfmethode getestet wird. Relative Erhöhung der Zellzahl (RICC): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2). Relative Populationsverdopplung (RPD): Verfahren zur Messung der Zytotoxizität, wenn kein cytoB verwendet wird (Formel siehe Anlage 2). Replikationsindex (RI): Anteil der abgeschlossenen Zellteilungszyklen in einer behandelten Kultur im Verhältnis zur nicht behandelten Kontrollgruppe während der Expositions- und Regenerationsphase (Formel siehe Anlage 2). S9-Gemisch: Gemisch aus der S9-Leberfraktion und den für die metabolische Enzymaktivität notwendigen Ko-Faktoren. S9-Leberfraktion: Überstand des Leberhomogenats nach Zentrifugieren bei 9.000 g, d. h. Rohleberextrakt. Unbehandelte Kontrollen: Kulturen, die nicht behandelt werden (d. h. weder mit einer Prüfchemikalie noch mit Lösungsmittel), jedoch gleichzeitig in gleicher Weise aufbereitet werden wie die Kulturen, die mit der Prüfchemikalie behandelt werden. Zellproliferation: Zunahme der Anzahl von Zellen als Ergebnis der mitotischen Zellteilung. Zentromer: DNA-Bereich eines Chromosoms, an dem die beiden Chromatiden zusammengehalten werden und an dem beide Kinetochoren Seite an Seite angeordnet sind. Zytokinese: Prozess der Zellteilung unmittelbar nach der Mitose, bei dem zwei Tochterzellen, jeweils mit einem einzigen Kern, gebildet werden. Zytokinese-Block-Proliferationsindex (CBPI): Anteil der Zellen aus zweiter Teilung in der behandelten Population im Verhältnis zur nicht behandelten Kontrollgruppe (Formel siehe Anlage 2). Zytostase: Hemmung des Zellwachstums (Formel siehe Anlage 2). Zytotoxizität: Bei den unter diese Prüfmethode fallenden Versuchen, die mit Zusatz von Cytochalasin B durchgeführt werden, bezeichnet Zytotoxizität eine Verringerung des Zytokinese-Block-Proliferationsindex (CBPI) oder Replikationsindex (RI) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 26 und Anlage 2). Bei den unter diese Prüfmethode fallenden Versuche, die ohne Zusatz von Cytochalasin B durchgeführt werden, bezeichnet Zytotoxizität eine Verringerung der relativen Populationsverdopplung (RPD) bzw. der relativen Erhöhung der Zellzahl (RICC) der behandelten Zellen, verglichen mit der Negativkontrolle (siehe Nummer 27 und Anlage 2).
Wenn cytoB zum Einsatz kommt, sollte die Bewertung der Zytotoxizität auf dem Zytokinese-Block-Proliferationsindex (CBPI) oder dem Replikationsindex (RI) beruhen (17) (69). Der CBPI gibt die mittlere Zahl der Kerne pro Zelle an und kann zur Berechnung der Zellproliferation eingesetzt werden. Der RI gibt die relative Zahl der Zellzyklen pro Zelle während der Exposition gegenüber cytoB in behandelten Kulturen im Vergleich zu Kontrollkulturen an und kann zur Berechnung der Zytostase in % verwendet werden: % Zytostase = 100 -100{(CBPIT -1) ÷ (CBPIC -1)} und: T = mit der Prüfchemikalie behandelte Kultur C = Vehikel-Kontrollkultur Dabei gilt:
Damit entspricht ein CBPI von 1 (alle Zellen sind einkernig) einer Zytostase von 100 %. Zytostase = 100 - RI
T = behandelte Kulturen C = Kontrollkulturen Ein RI von 53 % bedeutet somit, dass sich in der behandelten Kultur gegenüber der Anzahl der Zellen in der Kontrollkultur, die sich geteilt und zwei- und mehrkernige Zellen gebildet haben, nur 53 % geteilt haben, was eine Zytostase von 47 % bedeutet. Wenn kein cytoB zum Einsatz kommt, wird eine Bewertung der Zytotoxizität auf der Basis der relativen Erhöhung der Zellzahl (RICC) oder der relativen Populationsverdopplung (RPD) empfohlen (69), da bei beiden der Anteil der Zellpopulation berücksichtigt wird, der eine Teilung durchlaufen hat.
Dabei gilt: Populationsverdopplung = [log ((Zellzahl nach Behandlung ÷ Anfängliche Zellzahl)] ÷ log 2 Somit zeigt eine RICC oder eine RPD von 53 % eine Zytotoxizität/Zytostase von 47 % an. Bei Verwendung eines Proliferationsindex (PI) kann die Zytotoxizität durch das Zählen aller Klone bewertet werden, die aus 1 Zelle (cl1), 2 Zellen (cl2), 3 bis 4 Zellen (cl4) sowie 5 bis 8 Zellen (cl8) bestehen.
Der PI wurde als aussagefähiger und zuverlässiger Parameter für die Zytotoxizität auch bei Zelllinien verwendet, die in vitro ohne cytoB kultiviert wurden (35) (36) (37) (38) und kann als nützlicher zusätzlicher Parameter angesehen werden. In jedem Fall sollen die behandelten Kulturen und die Negativkontrollkulturen vor der Behandlung die gleiche Zellzahl aufweisen. Wenngleich der RCC-Wert (d. h. Zellzahl in behandelten Kulturen/Zellzahl in Kontrollkulturen) in der Vergangenheit als Bestimmungsgröße für die Zytotoxizität herangezogen wurde, wird er nicht mehr empfohlen, da er zu einer Unterschätzung der Toxizität führen kann. Bei der Verwendung automatisierter Auswertungssysteme, wie Durchflusszytometrie, Laser-Scan-Zytometrie oder Bildanalyse, kann die Zellzahl in der Formel durch die Anzahl der Kerne ersetzt werden. In den Negativkontrollkulturen sollte die Populationsverdopplung bzw. der Replikationsindex mit den Anforderungen an die Probenahme von Zellen nach der Behandlung zu einem Zeitpunkt, der etwa der 1,5-bis 2-fachen Dauer des normalen Zellzyklus entspricht, übereinstimmen. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
B.50. Hautsensibilisierung: Lokaler Lymphknotentest: DA 12
1. Die OECD-Leitlinien für die Prüfung von Chemikalien und die EU-Prüfmethoden werden regelmäßig überarbeitet, um dem wissenschaftlichen Fortschritt, sich ändernden Rechtsvorgaben und Belangen des Tierschutzes gerecht zu werden. Die ursprüngliche Prüfmethode (B.42) zur Bestimmung der Hautsensibilisierung bei der Maus, der Lokale Lymphknotentest (Local Lymph Node Assay bzw. LLNA; OECD-Prüfrichtlinie 429) wurde inzwischen überarbeitet (1). Verschiedene Veröffentlichungen (2) (3) (4) (5) (6) (7) (8) (9) gehen genauer auf die Validierung des LLNA und auf die mit ihm verbundenen wissenschaftlichen Arbeiten ein. Bei dem LLNA wird zur Messung der Lymphozytenproliferation radioisotopisches Thymidin oder Jod verwendet. Der Test hat daher Beschränkungen, wenn der Erwerb, die Verwendung oder die Entsorgung radioaktiver Materialien mit Problemen verbunden sind. Bei dem LLNA: DA (entwickelt von Daicel Chemical Industries, Ltd.) handelt es sich um eine nichtradioaktive Modifikation des LLNA, die den Gehalt an Adenosintriphosphat (ATP) mittels Biolumineszenz als Indikator für die Lymphozytenproliferation bestimmt. Die Prüfmethode LLNA: DA wurde durch ein internationales Gremium für Sachverständigengutachten (peer review) validiert, geprüft und zur Identifizierung sensibilisierender und nicht sensibilisierender Chemikalien mit einigen Beschränkungen als geeignet befunden (10) (11) (12) (13). Diese Prüfmethode wurde zur Bewertung des Hautsensibilisierungspotenzials von Chemikalien (Stoffen und Gemischen) bei Tieren entwickelt. Kapitel B.6 dieses Anhangs und OECD-Prüfrichtlinie 406 stützen sich auf Meerschweinchen-Tests, insbesondere den Meerschweinchen-Maximierungstest und den Bühler-Test (14). Der LLNA (Kapitel B.42 dieses Anhangs; OECD-Prüfrichtlinie 429) und die beiden nichtradioaktiven Modifikationen, LLNA: DA (Kapitel B.50 dieses Anhangs; OECD-Prüfrichtlinie 442 A) und LLNA: BrdU-ELISA (Kapitel B.51 dieses Anhangs; OECD-Prüfrichtlinie 442 B) haben insofern Vorteile gegenüber den Meerschweinchen-Tests in B.6 und OECD-Prüfrichtlinie 406 (14), als sie einen verringerten und gezielteren Einsatz von Versuchstieren ermöglichen.
2. Ebenso wie im LLNA wird bei dem LLNA: DA die Induktionsphase der Hautsensibilisierung untersucht. Der Test liefert quantitative Daten für die Bewertung von Dosis-Wirkungs-Beziehungen. Durch den möglichen Nachweis von hautsensibilisierenden Stoffen ohne die Verwendung radioaktiver Markierungen für die DNA können die berufliche Exposition gegenüber Radioaktivität und Entsorgungsprobleme vermieden werden. Dies wiederum ermöglicht eine verstärkte Verwendung von Mäusen zur Bestimmung hautsensibilisierender Stoffe und somit eine geringere Verwendung von Meerschweinchen zur Bewertung des hautsensibilisierenden Potenzials (B.6; OECD-Prüfrichtlinie 406) (14).
3. Die verwendeten Begriffsbestimmungen sind in Anlage 1 aufgeführt.
Ausgangsüberlegungen und Begrenzungen
4. Der LLNA: DA ist eine modifizierte LLNA-Methode zum Nachweis von Chemikalien mit potenziell hautsensibilisierenden Eigenschaften, mit bestimmten Beschränkungen. Dies bedeutet nicht notwendigerweise, dass der LLNA: DA zwingend in jedem Fall anstelle des LLNA oder der Meerschweinchen-Tests (B.6; OECD-Prüfrichtlinie) (14) eingesetzt werden muss. Vielmehr erweist sich der Test als gleichermaßen wertvoll und kann als Alternative gewählt werden, bei der im Allgemeinen keine weitere Bestätigung von positiven oder negativen Ergebnissen erforderlich ist. (10) (11). Das Prüflabor sollte vor der Durchführung der Studie alle verfügbaren Informationen über die Prüfsubstanz auswerten und berücksichtigen. Zu diesen Informationen zählen die Identität und die chemische Struktur der Prüfsubstanz, ihre physikalisch-chemischen Eigenschaften, die Ergebnisse aller sonstigen in vitro- oder in vivo-Toxizitätsprüfungen der Prüfsubstanz sowie toxikologische Daten zu strukturell verwandten Chemikalien. Diese Informationen sind zu berücksichtigen, um die Eignung des LLNA: DA für die jeweilige Prüfsubstanz festzustellen (bestimmte Arten chemischer Stoffe sind mit dem LLNA: DA nicht kompatibel [siehe Absatz 5]) und die Festlegung der Dosis zu erleichtern.
5. Beim LLNA: DA handelt es sich um eine in vivo-Methode. Das bedeutet, dass die kontaktsensibilisierenden Eigenschaften weiterhin an Tieren bewertet werden. Der Test erlaubt es jedoch, im Vergleich zu den Meerschweinchen-Tests (B.6; OECD-Prüfrichtlinie 406) die Anzahl der für diesen Zweck benötigten Tiere zu reduzieren (14). Zudem ist der LLNA: DA eine wesentlich verfeinerte Methode im Hinblick auf die Verwendung der Tiere (weniger Schmerzen und Leiden) bei den Tests zur Kontraktsensibilisierung, da es beim LLNA: DA, anders als bei B.6 und OECD-Prüfrichtlinie 406, nicht erforderlich ist, provozierte Überempfindlichkeitsreaktionen der Haut auszulösen. Trotz der Vorteile, die der LLNA: DA gegenüber B.6 und OECD-Prüfrichtlinie 406 (14) aufweist, ist er mit gewissen Einschränkungen behaftet, die die Anwendung von B.6 oder von OECD-Prüfrichtlinie 406 erforderlich machen können (z.B. die Prüfung bestimmter Metalle, falsch positive Ergebnisse bei bestimmten hautreizenden Stoffen [wie etwa bei tensidähnlichen Chemikalien] (6) (1 und Kapitel B.42 in diesem Anhang), oder die Löslichkeit der Prüfsubstanz). Zudem können chemische Klassen oder Substanzen mit Funktionsgruppen, die erwiesenermaßen als potenzielle Störfaktoren wirken können (16), den Einsatz von Meerschweinchen-Tests erforderlich machen (B.6; OECD-Prüfrichtlinie 406) (14)). Beschränkungen, die für den LLNA festgestellt wurden (1, und Kapitel B.42 dieses Anhangs), empfehlen sich auch für den LLNA: DA (10). Zudem könnte die Verwendung des LLNA: DA ungeeignet sein für die Prüfung von Substanzen mit Auswirkungen auf das ATP-Niveau (z.B. Substanzen, die als ATP-Hemmer wirken) oder Substanzen, die die genaue Messung der intrazellulären ATP- Messung beeinträchtigen (z.B. Vorhandensein ATP abbauender Enzyme, Vorhandensein extrazellulären ATPs im Lymphknoten). Abgesehen von diesen bekannten Beschränkungen dürfte der LLNA: DA für die Prüfung aller Substanzen geeignet sein, sofern mit diesen Substanzen keine Eigenschaften verbunden sind, die die Genauigkeit des LLNA: DA beeinträchtigen könnten. Außerdem sollte die Möglichkeit positiver Grenzergebnisse in Betracht gezogen werden, wenn für den Stimulationsindex (SI) Werte zwischen 1,8 und 2,5 ermittelt werden (siehe Absätze 31-32). Dies beruht auf der Validierungs-Datenbasis von 44 Substanzen mit einem SI ≥ 1,8 (siehe Absatz 6), bei denen der LLNA: DA alle 32 LLNA-Sensibilisatoren richtig identifizierte, jedoch drei von 12 LLNA-Nichtsensibilisatoren mit SI-Werten zwischen 1,8 und 2,5 (d. h. positive Grenzwerte) (10) nicht korrekt identifizierte. Da allerdings derselbe Datensatz für die Einstellung der SI-Werte und die Berechnung der prädiktiven Eigenschaften des Tests verwendet wurde, könnten die festgestellten Ergebnisse auch eine Überbewertung der tatsächlichen prädiktiven Eigenschaften sein.
6. Der LLNA: DA beruht auf dem Prinzip, dass sensibilisierende Stoffe eine Lymphozytenproliferation in den drainierenden Lymphknoten an der Stelle der Applikation des chemischen Stoffes induzieren. Diese Proliferation verläuft proportional zur Dosis und zur Wirksamkeit des applizierten Allergens und bietet sich als einfache Möglichkeit an, eine quantitative Messung der Sensibilisierung zu erhalten. Die Proliferation wird gemessen, indem man die mittlere Proliferation jeder Prüfgruppe mit der mittleren Proliferation der mit Vehikel behandelten Kontrollgruppe (VK) vergleicht. Das Verhältnis der mittleren Proliferation in jeder Behandlungsgruppe zu dem in der gleichzeitigen Vehikelkontrollgruppe, auch als SI bezeichnet, wird festgelegt und sollte ≥ 1,8 betragen, ehe eine weitere Bewertung einer Prüfsubstanz als potenzieller Hautsensibilisator erfolgt. Die hier beschriebenen Methoden beruhen auf der Messung des ATP-Gehalts durch Biolumineszenz (die bekanntermaßen mit der Anzahl lebender Zellen korreliert) (17), um einen erhöhten Anteil proliferierender Zellen in den drainierenden aurikulären Lymphknoten nachzuweisen (18) (19). Die Biolumineszenz-Methode stützt sich auf das Enzym Luciferase, um die Bildung von Licht aus ATP und Luciferin mittels der folgenden Reaktion zu katalysieren:
ATP + Luciferin + O2  Oxyluciferin + AMP + PPi + CO2 + Light Oxyluciferin + AMP + PPi + CO2 + Light |
Die emittierte Lichtintensität steht in linearer Beziehung zur ATP-Konzentration und wird mit einem Luminometer gemessen. Bei dem Luciferin-Luciferase-Test handelt es sich um eine sensible Methode zur ATP-Quantifizierung, die in einer breiten Palette von Anwendungen zum Einsatz kommt (20).
Auswahl von Versuchstierarten
7. Für diesen Test ist die Maus die Spezies der Wahl. Validierungsstudien für den LLNA: DA wurden ausschließlich mit dem CBA/J-Stamm durchgeführt, der daher als bevorzugter Stamm gilt (12) (13). Es werden junge erwachsene weibliche Mäuse verwendet, die weder geworfen haben noch trächtig sind. Bei Versuchsbeginn sollten die Tiere 8- 12 Wochen alt sein, die Gewichtsunterschiede minimal sein und 20 % des mittleren Gewichts nicht übersteigen. Alternativ können Tests an anderen Stämmen und männlichen Tieren erfolgen, wenn anhand von hinlänglich großen Datenangaben nachgewiesen werden kann, dass keine signifikanten stamm- und/oder geschlechtsspezifischen Unterschiede in der Reaktion auf den LLNA: DA bestehen.
Haltungs- und Fütterungsbedingungen
8. Mäuse sollten in Gruppen gehalten werden (21), wenn nicht angemessene wissenschaftliche Begründungen eine Einzelhaltung nahelegen. Die Temperatur im Versuchsraum sollte 22 ± 3 °C betragen. Obwohl die relative Luftfeuchtigkeit mindestens 30 % betragen und zu anderen Zeiten als während der Reinigung vorzugsweise nicht über 70 % liegen soll, ist ein Wert von 50-60 % anzustreben. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten.
Vorbereitung der Tiere
9. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert (aber nicht am Ohr) und vor Beginn der Dosierung für einen Zeitraum von mindestens 5 Tagen in ihren Käfigen an die Laborbedingungen gewöhnt. Vor Behandlungsbeginn werden alle Tiere untersucht, um sicherzustellen, dass keine sichtbaren Hautverletzungen bestehen.
Vorbereitung der Dosierlösungen
10. Feste Chemikalien sollten vor der Applikation in ein Mäuseohr in Lösungsmitteln/Vehikeln gelöst oder suspendiert und ggf. verdünnt werden. Flüssige Chemikalien können direkt appliziert oder zuvor verdünnt werden. Unlösliche Chemikalien, wie sie in der Regel in Medizinprodukten vorliegen, sollten vor der Applikation in ein Mäuseohr in einem geeigneten Lösungsmittel einer übertrieben starken Extraktion unterzogen werden, um alle extrahierbaren Inhaltsstoffe vor der Prüfung sichtbar zu machen. Die Prüfsubstanzen sollten täglich zubereitet werden, es sei denn, die Stabilität der Substanz bei Lagerung wird nachgewiesen.
Überprüfung der Zuverlässigkeit:
11. Anhand von positiven Kontrollchemikalien (PC) wird die ordnungsgemäße Leistung des Tests nachgewiesen. Hierzu ist eine angemessene und reproduzierbare Empfindlichkeit der Reaktion auf eine sensibilisierende Prüfsubstanz erforderlich, wobei die Reaktionsstärke gut charakterisiert ist. Es wird die Einbeziehung einer gleichzeitigen Positivkontrolle empfohlen, da diese die Fähigkeit des Labors belegt, jeden Test erfolgreich durchzuführen und eine Bewertung der Wiederholbarkeit und Vergleichbarkeit zwischen verschiedenen Labors ermöglicht. Einige Regulierungsbehörden schreiben eine Positivkontrolle für jede Studie vor. Daher wird Anwendern empfohlen, vor Durchführung des LLNA: DA die zuständigen Behörden zu Rate zu ziehen. Dementsprechend wird die routinemäßige Verwendung einer gleichzeitigen Positivkontrolle angeregt, um die Notwendigkeit zusätzlicher Tierversuche zur Erfüllung von Anforderungen zu vermeiden, die aus der Verwendung einer nur periodischen Positivkontrolle resultieren könnten (siehe Absatz 12). Die Positivkontrolle sollte eine positive Reaktion auf den LLNA: DA bei einem Expositionsniveau hervorrufen, das einen Anstieg des Stimulationsindex (SI ≥ 1,8 im Vergleich zur Negativkontrollgruppe, NK) bewirkt. Die positive Kontrolldosis soll so gewählt werden, dass sie keine übermäßige Hautreizung oder systemische Toxizität verursacht und die Induktion reproduzierbar, aber nicht exzessiv ist (z.B. ein SI > 10 würde als exzessiv gelten). Bevorzugte positive Kontrollstoffe sind 25 % Hexylcinnaminaldehyd (Chemical Abstracts Service [CAS]-Nr. 101-86-0) und 25 % Eugenol (CAS-Nummer 97-53-0) in Aceton: Olivenöl (4:1, v/v). Unter bestimmten Umständen können in ausreichend begründeten Fällen andere Kontrollsubstanzen eingesetzt werden, die den genannten Kriterien entsprechen.
12. Obwohl die Einbeziehung einer gleichzeitigen Positivkontrollgruppe empfohlen wird, können in gewissen Situationen periodische Prüfungen (d. h. in Abständen ≤ 6 Monaten) der Positivkontrolle für Laboratorien ausreichen, die den LLNA: DA regelmäßig (d. h. mindestens einmal pro Monat) durchführen und über eine etablierte historische Kontrolldatenbasis verfügen, die die Fähigkeit des Labors bestätigt, reproduzierbare und genaue Ergebnisse mit Positivkontrollen zu erzielen. Eine angemessene Beherrschung des LLNA: DA kann erfolgreich nachgewiesen werden, indem durchgehend positive Ergebnisse mit den positiven Kontrollchemikalien in mindestens 10 unabhängigen Prüfungen erzielt werden, die innerhalb eines angemessenen Zeitraums (d. h. in weniger als einem Jahr) durchgeführt wurden.
13. Eine gleichzeitige Positivkontrollgruppe sollte immer einbezogen werden, wenn Verfahrensänderungen im LLNA: DA auftreten (z.B. Wechsel des geschulten Personals, Änderung der Materialien bei den Prüfmethoden und/oder der Reagenzien, Änderung der Ausrüstung, Änderung der Herkunft der Versuchstiere). Solche Änderungen sollten in Laborberichten dokumentiert werden. Ferner ist der Einfluss dieser Änderungen auf die Aussagefähigkeit der zuvor eingerichteten historischen Datenbank zu bedenken. Dabei sollte die Notwendigkeit überdacht werden, eine neue historische Datenbank einzurichten, um die gleichbleibende Qualität der positiven Kontrollergebnisse zu dokumentieren.
14. Die Prüfer sollten sich der Tatsache bewusst sein, dass die Entscheidung, eine PC-Studie periodisch statt gleichzeitig durchzuführen, Auswirkungen auf die Aussagefähigkeit und Akzeptanz negativer Studienergebnisse haben kann, die ohne gleichzeitige Positivkontrolle im Zeitraum zwischen den einzelnen periodischen PC-Studien auftreten können. Wenn zum Beispiel ein falsch negatives Ergebnis in der periodischen PC-Studie auftritt, können negative Ergebnisse für die Prüfsubstanz, die in dem Zeitraum zwischen der letzten akzeptablen periodischen PC- Studie und der inakzeptablen periodischen PC-Studie aufgetreten waren, in Frage gestellt werden. Die Auswirkungen solcher Ergebnisse sollten sorgfältig bedacht werden, wenn entschieden wird, ob gleichzeitige oder nur periodische PC-Studien durchgeführt werden sollen. Auch ist die Verwendung einer geringeren Anzahl an Versuchstieren bei der gleichzeitigen Positivkontroll-Gruppe zu bedenken, wenn dies wissenschaftlich zu begründen ist und wenn das Labor auf der Grundlage laborspezifischer historischer Daten nachweist, dass eine geringere Anzahl an Mäusen ausreicht (22).
15. Obwohl eine positive Kontrollchemikalie in einem Vehikel getestet werden sollte, das bekanntermaßen eine gleichbleibende Reaktion auslöst (z.B. Aceton: Olivenöl; 4:1, v/v), können in bestimmten Rechtssituationen außerdem Prüfungen in einem Nichtstandard-Vehikel (klinisch/chemisch relevante Formulierung) erforderlich sein (23). Wenn die gleichzeitige Positivkontrolle in einem anderen Vehikel als der Prüfsubstanz getestet wird, sollte eine gesonderte Vehikelkontrolle für die gleichzeitige PC einbezogen werden.
16. In Fällen, in denen Prüfsubstanzen einer bestimmten chemischen Klasse oder eine Reihe von Reaktionen bewertet werden, können Referenzsubstanzen nützlich sein, um nachzuweisen, dass die Prüfmethode zur Feststellung des Hautsensibilisierungspotenzials dieser Art von Prüfsubstanzen ordnungsgemäß funktioniert. Geeignete Referenzsubstanzen sollten die folgenden Eigenschaften haben:
- strukturelle und funktionale Ähnlichkeit mit der Klasse der getesteten Prüfsubstanz;
- bekannte physikalische/chemische Eigenschaften; - zugrunde liegende Daten aus dem LLNA: DA;
- zugrunde liegende Daten aus anderen Tiermodellen und/oder von Menschen.
Anzahl der Versuchstiere und Dosierungen
17. Mindestens vier Tiere pro Dosisgruppe mit jeweils mindestens drei Konzentrationen der Prüfsubstanz werden benötigt; zusätzlich braucht man eine Negativkontrollgruppe, die nur mit dem Vehikel für die Prüfsubstanz behandelt wird, sowie eine Positivkontrolle (gleichzeitig oder jüngeren Datums, auf der Grundlage der Laborvorschriften in den relevanten Abschnitten 11-15). Tests mit Mehrfachdosen der PC sollten in Erwägung gezogen werden, insbesondere wenn die Positivkontrolle intermittierend getestet wird. Bis auf die Verabreichung der Prüfsubstanz sind die Tiere der Kontrollgruppen ebenso zu behandeln wie die Tiere der Behandlungsgruppen.
18. Die Auswahl der Dosis und des Vehikels soll anhand der Empfehlungen in (2) und (24) erfolgen. Für aufeinanderfolgende Dosierungen werden normalerweise geeignete abgestufte Konzentrationen gewählt, wie z.B. 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % usw. Die Auswahl der Konzentrationsfolge sollte angemessen wissenschaftlich begründet werden. Alle vorhandenen toxikologischen Angaben (z.B. über die akute Toxizität und Hautreizung) sowie strukturelle und physiochemische Angaben zu der jeweiligen Prüfsubstanz (und/oder strukturverwandten Substanzen) sollten bei der Festlegung von drei aufeinander folgenden Konzentrationen berücksichtigt werden, so dass bei der Höchstkonzentration einerseits die Exposition maximiert und andererseits eine systemische Toxizität und/oder eine übermäßige lokale Hautreizung ausgeschlossen werden (24) (25). Mangels solcher Informationen kann ein anfänglicher Dosisfindungstest erforderlich sein (siehe Absätze 21-24).
19. Das Vehikel sollte das Testergebnis nicht beeinflussen oder beeinträchtigen und sollte so gewählt werden, dass die Löslichkeit zur Erzielung einer möglichst hohen Konzentration maximiert und gleichzeitig eine für das Applizieren der Prüfsubstanz geeignete Lösung/Suspension hergestellt werden kann. Empfohlene Vehikel sind Aceton: Olivenöl (4:1, v/v), N,N-Dimethylformamid, Methylethylketon, Propylenglykol und Dimethylsulphoxid (6), wobei mit hinreichender wissenschaftlicher Begründung auch andere Vehikel verwendet werden können. Unter bestimmten Umständen muss ein klinisch relevantes Lösungsmittel oder die handelsübliche Zubereitung, in der die Prüfsubstanz vermarktet wird, als zusätzliche Kontrolle genutzt werden. Besondere Sorgfalt sollte darauf verwendet werden, zu gewährleisten, dass in das Vehikelsystem durch die Verwendung geeigneter Lösungsvermittler (z.B. 1 % Pluronic® L92) hydrophile Stoffe eingearbeitet werden, die die Haut befeuchten und nicht sofort ablaufen. Folglich sind vollständig wässrige Vehikel zu vermeiden.
20. Die Bearbeitung von Lymphknoten einzelner Mäuse ermöglicht die Bewertung der Variabilität von Tieren sowie einen statistischen Vergleich zwischen der Prüfsubstanz und Messungen an der Vehikelkontrollgruppe (siehe Absatz 33). Die Möglichkeit, die Anzahl der Mäuse in der Positivkontrollgruppe zu verringern, ist nur realistisch, wenn Einzeltierdaten erhoben werden (22). Außerdem verlangen einige Regulierungsbehörden die Erhebung von Daten einzelner Tiere. Die regelmäßige Erhebung von Einzeltierdaten stellt im Hinblick auf den Tierschutz einen Vorteil dar, indem Doppeltests vermieden werden. Diese wären notwendig, wenn die ursprünglich gemeinsam gesammelten Ergebnisse für die Prüfsubstanz (z.B. über gepoolte Daten) später von Regulierungsbehörden mit anderen Vorschriften (z.B. Einzeltierdaten) geprüft würden.
Dosisfindungstest
21. Wenn über die höchst mögliche Dosierung keine Informationen vorliegen (siehe Absatz 18), sollte ein Dosisfindungstest durchgeführt werden, um die geeignete Dosierung für den LLNA: DA festzulegen. Der Dosisfindungstest soll eine Orientierung bei der Auswahl der höchst möglichen Dosisstufe für die Hauptuntersuchung des LLNA: DA in Fällen liefern, in denen keine Informationen über Konzentrationen vorliegen, die zur systemischen Toxizität (siehe Absatz 24) und/oder zu übermäßiger Hautreizung führen (siehe Absatz 23). Die höchsten geprüften Dosisstufen sollten 100 % der Prüfsubstanz bei Flüssigkeiten bzw. die höchst möglichen Konzentrationen bei Feststoffen oder Suspensionen sein.
22. Der Dosisfindungstest wird unter Bedingungen durchgeführt, die denen der Hauptstudie des LLNA: DA genau entsprechen. Allerdings gibt es hier keine Bewertung der Lymphknotenproliferation und es können weniger Tiere pro Dosisgruppe eingesetzt werden. Es werden ein oder zwei Tiere pro Dosisgruppe empfohlen. Alle Mäuse werden täglich auf klinische Zeichen für systemische Toxizität oder lokale Reizungen an der Applikationsstelle untersucht. Das Körpergewicht wird vor dem Test und vor dem Abschluss (Tag 8) protokolliert. Beide Ohren jeder Maus werden auf Anzeichen für Erytheme untersucht und mit Hilfe von Tabelle 1 bewertet (25). Die Ohrdicke wird mit Hilfe eines Ohrdickenmessgeräts (z.B. digitaler Mikrometer oder Peacock Dickenmessuhr) an Tag 1 vor der Dosierung, Tag 3 (ca. 48 Stunden nach der ersten Dosis), Tag 7 (24 Stunden vor Abschluss) und Tag 8 bestimmt. Zusätzlich kann an Tag 8 die Ohrdicke durch die Gewichtsbestimmung von Ohrstanzproben ermittelt werden. Dies sollte nach der tierschutzgerechten Tötung der Tiere erfolgen. Eine übermäßige lokale Hautreizung wird durch eine Erythem-Punktzahl von ≥ 3 und/oder eine Zunahme der Ohrdicke ≥ 25 % an jedem beliebigen Messtag angezeigt (26) (27). Als höchste Dosis für die Hauptuntersuchung des LLNA: DA wird die nächst niedrigere Dosis in der Konzentrationsreihe des Dosisfindungstests gewählt (siehe Absatz 18), die keine systemische Toxizität und/oder übermäßige lokale Hautreizung verursacht.
Tabelle 1: Erythem-Klassifizierung
| Beobachtung | Punktzahl |
| Kein Erythem | 0 |
| Sehr leichtes Erythem (kaum wahrnehmbar) | 1 |
| Klar abgegrenztes Erythem | 2 |
| Mäßiges bis ausgeprägtes Erythem | 3 |
| Schweres Erythem (dunkelrot) bis hin zur Schorfbildung, so dass eine Bewertung nicht möglich ist | 4 |
23. Zusätzlich zu einer Zunahme der Ohrdicke um 25 % (26) (27) wurde ein statistisch signifikanter Anstieg der Ohrdicke bei den behandelten Mäusen im Vergleich zu den Kontrollmäusen zugrunde gelegt, um Reizstoffe in dem LLNA zu identifizieren (28) (29) (30) (31) (32) (33) (34). Obwohl ein statistisch signifikanter Anstieg auch bei einer Ohrdicke von weniger als 25 % auftreten kann, konnte kein spezifischer Zusammenhang mit einer übermäßigen Hautreizung festgestellt werden (30) (31) (32) (33) (34).
24. Die folgenden klinischen Beobachtungen können, sofern sie Bestandteil einer Gesamtbewertung sind, auf systemische Toxizität hinweisen (35) und somit die höchst mögliche Dosisstufe für die Hauptuntersuchung des LLNA: DA anzeigen: Änderungen der Funktionen des Nervensystems (z.B. Piloerektion, Ataxie, Tremor und Krämpfe); Verhaltensänderungen (z.B. Aggressivität, Änderungen bei der Fellpflege, auffallende Änderung des Aktivitätsniveaus); Änderungen des Atemmusters (d. h. Änderung der Häufigkeit und Intensität des Atmens wie Dyspnoe, Keuchen, rasselnder Atem) sowie Änderungen der Futter- und Wasseraufnahme. Zusätzlich sollten Anzeichen von Lethargie und/oder Teilnahmslosigkeit sowie alle klinischen Anzeichen für mehr als leichte oder momentane Schmerzen und Qual, eine Gewichtsreduktion > 5 % zwischen Tag 1 und Tag 8 sowie die Sterblichkeit in der Bewertung berücksichtigt werden. Moribunde Tiere oder Tiere, die Anzeichen starker und andauernder Qualen zeigen, sollten tierschutzgerecht getötet werden (36).
Versuchsplan der Hauptuntersuchung
25. Der Versuchsplan des Tests ist wie folgt:
- Tag 1: Jedes Tier wird einzeln gekennzeichnet und das Gewicht sowie jede klinische Beobachtung protokolliert. Es wird 1 % Natriumlaurylsulfat (NLS) in wässriger Lösung auf die Rückseite jedes Ohrs aufgetragen. Dies erfolgt mit vier bis fünf Strichen einer in die NLS-Lösung getauchten Bürste, so dass die gesamte Rückseite jedes Ohrs bedeckt ist. Eine Stunde nach der NLS-Behandlung werden 25 µL der Prüfsubstanz in der jeweiligen Verdünnung, des Vehikels allein oder der PK (gleichzeitig oder jüngeren Datums) gemäß den Laborvorschriften in den relevanten Abschnitten 11-15) auf die Rückseite jedes Ohrs appliziert.
- Tage 2, 3 und 7: Die an Tag 1 erfolgte Vorbehandlung mit 1 % NLS wässriger Lösung und die Applikation der Prüfsubstanz werden wiederholt.
- Tage 4, 5 und 6: Keine Behandlung.
- Tag 8: Das Gewicht jedes Tiers sowie jede klinische Beobachtung werden protokolliert. Ca. 24 bis 30 Stunden nach Beginn der Applikation an Tag 7 werden die Tiere tierschutzgerecht getötet. Die drainierenden aurikulären Lymphknoten jedes Mäuseohrs werden entfernt und separat (für jedes Tier einzeln) in phosphatgepufferte Kochsalzlösung (PBS) gegeben. Einzelheiten und Diagramme der Lymphknotenidentifikation und -entfernung sind unter Referenz (22) aufgeführt. Zur weiteren Kontrolle der lokalen Hautreaktion in der Hauptuntersuchung können zusätzliche Parameter wie die Auswertung des Ohr-Erythems oder Ohrdickemessungen (mittels eines Dickenmessgeräts oder durch die Gewichtsbestimmung von Ohrstanzproben bei der Nekropsie) in das Untersuchungsprotokoll aufgenommen werden.
Vorbereitung der Zellsuspensionen
26. Für jede Maus wird eine Einzelzellsuspension der beidseitig entnommenen Lymphknotenzellen (LNC) erstellt, indem die Lymphknoten zwischen zwei Glasträger geklemmt werden und leichter Druck ausgeübt wird, um die Knoten zu zerquetschen. Nach der Vergewisserung, dass das Gewebe sich dünnflächig verteilt hat, können die beiden Träger voneinander gelöst werden. Das Gewebe auf beiden Trägern wird in PBS gelöst, indem jeder Träger im Winkel über die Petrischale gehalten wird und mit PBS gespült wird, während gleichzeitig das Gewebe mit einem Zellschaber von dem Träger abgeschabt wird. Die Lymphknoten bei Tieren der Negativkontrollgruppe sind klein; daher ist Vorsicht geboten, um eine ungewollte Beeinflussung der SI-Werte zu vermeiden. Für das Spülen beider Träger sollte ein Gesamtvolumen von 1 mL PBS verwendet werden. Die Lymphknoten-Lösung in der Petrischale sollte mit dem Zellschaber leicht homogenisiert werden. Anschließend wird mit einer Mikropipette ein 20 µL Aliquot der Lymphknotenlösung entnommen (Achtung: nicht die für das Auge sichtbare Membran entnehmen) und mit 1,98 mL PBS gemischt, sodass sich eine Probe von 2 mL ergibt. Darauf wird eine zweite Probe von 2 mL mittels desselben Verfahrens zubereitet, sodass für jedes Tier zwei Proben vorliegen.
Bestimmung der Zellproliferation (Messung des ATP-Gehalts der Lymphozyten)
27. Erhöhungen des ATP-Gehalts in den Lymphknoten werden mittels der Luciferin-/Luciferase-Methode mit einem ATP-Mess-Kit bestimmt, das die Biolumineszenz in Relativen Lumineszenz-Einheiten (Relative Luminescence Units; RLU) misst. Die Testzeit zwischen dem Zeitpunkt der Tiertötung bis zur Messung des ATP-Gehalts für jedes einzelne Tier sollte gleich bleiben und bei bis zu ca. 30 Minuten liegen, da davon ausgegangen wird, dass der ATP- Gehalt nach der Tiertötung allmählich abnimmt (12). Folglich sollten die verschiedenen Verfahrensschritte ab der Entnahme der aurikulären Lymphknoten bis zur ATP-Messung nach einem vorher festgelegten Zeitplan, der für jedes Tier gleich ist, innerhalb von 20 Minuten abgeschlossen werden. Die ATP-Lumineszenz sollte in jeder Probe von 2 mL gemessen werden, so dass für jedes Tier zwei ATP-Messungen vorliegen. Anschließend wird die mittlere ATP-Lumineszenz bestimmt und in den folgenden Berechnungen verwendet (siehe Absatz 30).
Klinische Beobachtungen
28. Jede Maus sollte mindestens einmal täglich sorgfältig auf klinische Zeichen, d. h. lokale Reizung an der Applikationsstelle, oder auf systemische Toxizität untersucht werden. Alle Beobachtungen werden systematisch in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Die Überwachungspläne sollten Kriterien beinhalten, anhand derer Mäuse mit systemischer Toxizität, übermäßiger lokaler Hautreizung oder Hautätzung schnell zu Zwecken der schmerzlosen Tötung identifiziert werden können (36).
Körpergewicht
29. In Abschnitt 25 wurde bereits ausgeführt, dass das Körpergewicht der einzelnen Tiere zu Versuchsbeginn und zum Zeitpunkt der tierschutzgerechten Tötung der Tiere laut Versuchsplan festgestellt werden soll.
30. Die Ergebnisse für jede Behandlungsgruppe werden als mittlerer Stimulationsindex (SI) angegeben. Den SI erhält man durch Teilen der mittleren RLU pro Maus innerhalb jeder Prüfgruppe und der Positivkontrollgruppe durch die mittleren RLU pro Maus für die Lösungsmittel-/Vehikelkontrollgruppe. Der durchschnittliche SI beträgt demnach für die mit Vehikel behandelten Kontrollen 1.
31. In dem Entscheidungsprozess gilt ein Ergebnis als positiv, wenn SI ≥ 1,8 ist (10). Die Stärke der Dosis-Wirkung, die statistische Signifikanz und die gleichbleibende Qualität der Lösungsmittel-/Vehikel-Reaktion sowie der Reaktion der Positivkontrolle können aber auch benutzt werden, um festzulegen, ob ein Grenzergebnis (d. h. SI-Wert zwischen 1,8 und 2,5) als positiv bezeichnet werden soll (2) (3) (37).
32. Bei einer positiven Grenzwertreaktion mit einem SI zwischen 1,8 und 2,5 können Anwender zusätzliche Informationen berücksichtigen, wie z.B. die Dosis-Wirkungs-Beziehung, das Vorliegen einer systemischen Toxizität oder übermäßigen Hautreizung, sowie gegebenenfalls die statistische Signifikanz zusammen mit den SI-Werten, um zu bestätigen, dass die Ergebnisse positiv sind (10). Zu beachten sind ferner die verschiedenen Eigenschaften der Prüfsubstanz, wobei es unter anderem zu klären gilt, ob ein struktureller Zusammenhang mit bekannten Hautsensibilisatoren besteht, ob sie eine übermäßige lokale Hautreizung bei der Maus hervorruft und wie die festgestellte Art der Reaktion bezüglich der Dosis ist. Diese und weitere Aspekte werden an anderer Stelle ausführlich erörtert (4).
33. Werden die Daten für jede einzelne Maus erhoben, kann eine statistische Analyse zu Vorhandensein und Grad des Dosis-Wirkungs-Verhältnisses in den Daten durchgeführt werden. Jede statistische Auswertung könnte eine Bewertung der Dosis-Wirkungs-Beziehung sowie geeignete Testgruppen-Vergleiche beinhalten (z.B. Gruppe mit paarweiser Dosierung vs. gleichzeitige Lösungsmittel-/Vehikelkontrollgruppen). Die statistischen Analysen könnten z.B. die lineare Regression, den Williams-Test zur Bewertung von Dosis-Wirkungs-Trends oder den Dunnett-Test für paarweise Vergleiche beinhalten. Bei der Auswahl einer geeigneten Methode für die statistische Analyse sollte sich der Prüfer möglicher ungleicher Varianzen und anderer damit zusammenhängender Probleme stets bewusst sein, denn diese erfordern unter Umständen eine Datentransformation oder ein nichtparametrisches statistisches Verfahren. Auf jeden Fall muss der Prüfer unter Umständen SI-Berechnungen und statistische Analysen mit und ohne bestimmte Datenpunkte (manchmal als "Ausreißer" bezeichnet) vornehmen.
Daten
34. Die Daten sind in tabellarischer Form zusammenzufassen. Sie sollten die RLU-Einzelwerte, die mittleren RLU-Gruppenwerte pro Tier, die damit verbundene Fehlervariable (z.B. SD, SEM) und den mittleren SI für jede Dosisgruppe, verglichen mit der gleichzeitigen Lösungsmittel-/Vehikelkontrollgruppe angeben.
Prüfbericht
35. Der Testbericht sollte folgende Angaben enthalten:
Prüf- und Kontrollsubstanzen:
- Angaben zur Identität (z.B. CAS-Nummer und EG-Nummer, falls vorhanden; Bezugsquelle; Reinheit; bekannte Verunreinigungen; Chargennummer);
- physikalische Beschaffenheit und physikalisch-chemische Eigenschaften (z.B. Flüchtigkeit, Stabilität, Löslichkeit);
- bei Mischungen Zusammensetzung und relative Anteile der Bestandteile in Prozent;
Lösungsmittel/Vehikel:
- Angaben zur Identität (Reinheit, Konzentration, gegebenenfalls verwendete Mengen);
- Begründung für die Wahl des Vehikels;
Versuchstiere:
- Herkunft der CBA-Mäuse;
- mikrobiologischer Status der Tiere, soweit bekannt;
- Anzahl und Alter der Tiere;
- Herkunft der Tiere, Haltungsbedingungen, Futter usw.;
Prüfbedingungen:
- Herkunft, Chargennummer und Daten des Herstellers zur Qualitätssicherung/Qualitätskontrolle für das ATP- Kit;
- Angaben zur Vorbereitung und Applikation der Prüfsubstanz;
- Begründung der gewählten Dosierung (mit Ergebnissen des eventuell durchgeführten Dosisfindungstests);
- verwendete Konzentrationen des Vehikels und der Prüfsubstanz sowie Gesamtmenge der applizierten Prüfsubstanz;
- Angaben über Futter- und Wasserqualität (einschließlich Art/Herkunft des Futters, Wasserquelle);
- nähere Angaben zum Behandlungs- und Stichprobenentnahmeplan;
- Methoden zur Bestimmung der Toxizität;
- Kriterien zur Einstufung der Studien als positiv oder negativ;
- Einzelheiten zu eventuellen Protokollabweichungen und eine Erklärung dazu, wie sich diese Abweichung auf Prüfdesign und Ergebnisse auswirkt.
Überprüfung der Zuverlässigkeit:
- Zusammenfassende Darstellung der Ergebnisse der letzten Überprüfung der Zuverlässigkeit mit Angaben zu verwendeten Prüfsubstanzen, Konzentration und Vehikel;
- gleichzeitige und/oder historische PC und gleichzeitige Daten der Negativkontrolle (Lösungsmittel/Vehikel) für das Prüflabor;
- wenn keine gleichzeitige PC eingeschlossen war, Datum und Laborbericht über die jüngste periodische PC sowie ein Bericht mit ausführlichen Angaben über die historischen positiven Kontrolldaten des Labors mit Angabe der Gründe, warum keine gleichzeitige PC durchgeführt wurde;
Ergebnisse:
- Gewicht der einzelnen Tiere bei Beginn der Prüfung und zum Zeitpunkt der Tötung laut Versuchsplan, sowie mittlere und zugehörige Fehlervariable (z.B. SD, SEM) für jede Behandlungsgruppe;
- Beginn und zeitlicher Verlauf der toxischen Erscheinungen für jedes Tier, einschließlich gegebenenfalls Hautreizungen an der Stelle der Verabreichung;
- Zeitpunkt der Tötung und Zeitpunkt der ATP-Messungen für jedes Tier;
- Tabelle der RLU-Werte für jede Maus und der SI-Werte für jede Dosisgruppe;
- mittlere verbundene Fehlervariable (z.B. SD, SEM) für RLU/Maus für jede Behandlungsgruppe sowie die Ergebnisse der Ausreißer-Analyse für jede Behandlungsgruppe;
- berechneter SI und geeignete Messung der Variabilität, welche die Variabilität der Tiere sowohl in der Behandlungsgruppe als auch in der Kontrollgruppe berücksichtigt;
- Dosis-Wirkungs-Beziehung;
- gegebenenfalls statistische Analysen.
Diskussion der Ergebnisse:
- Kurze Kommentierung der Ergebnisse, der Dosis-Wirkungs-Beziehung und gegebenenfalls der statistischen Analysen mit Schlussfolgerungen zur Frage, ob die Prüfsubstanz als Hautsensibilisator eingestuft werden soll.
(1) OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
(2) Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem, Toxicol., 34, 999-1002.
(3) Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem, Toxicol., 34, 985-997.
(4) Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-333.
(5) Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59.
(6) ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/ llna/llnarep.pdf]
(7) Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol. 34, 258-273.
(8) Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol. 34, 274-286.
(9) Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process.Reg. Toxicol. Pharmacol. 34, 249-257.
(10) ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: modified by Daicel Chemical Industries, Ltd., based on ATP content test method protocol (LLNA: DA). NIH Publication No. 10-7551A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-DA/TMER.htm]
(11) ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf].
(12) Idehara, K., Yamagishi, G., Yamashita, K. and Ito, M. (2008), Characterization and evaluation of a modified local lymph node assay using ATP content as a nonradio isotopic endpoint.J. Pharmacol. Toxicol. Meth., 58, 1-10.
(13) Omori, T., Idehara, K., Kojima, H., Sozu, T., Arima, K., Goto, H., Hanada, T., Ikarashi, Y., Inoda, T., Kanazawa, Y., Kosaka, T., Maki, E., Morimoto, T., Shinoda, S., Shinoda, N., Takeyoshi, M., Tanaka, M., Uratani, M., Usami, M., Yamanaka, A., Yoneda, T., Yoshimura, I. and Yuasa, A. (2008), Interlaboratory validation of the modified murine local lymph node assay based on adenosine triphosphate measurement. J. Pharmacol. Toxicol. Meth., 58, 11-26.
(14) OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
(15) Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904.
(16) Basketter, D., Ball, N., Cagen, S., Carrillo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96.
(17) Crouch, S.P., Kozlowski, R., Slater, K.J. and Fletcher J. (1993), The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Meth., 160, 81-88.
(18) Ishizaka, A., Tonooka, T. and Matsumoto, S. (1984), Evaluation of the proliferative response of lymphocytes by measurement of intracellular ATP. J. Immunol. Meth., 72, 127-132.
(19) Dexter, S.J., Cämara, M., Davies, M. and Shakesheff, K.M. (2003), Development of a bioluminescent ATP assay to quantify mammalian and bacterial cell number from a mixed population. Biomat., 24, 27-34.
(20) Lundin A. (2000), Use of firefly luciferase in ATP-related assays of biomass, enzymes, and metabolites. Meth. Enzymol., 305, 346-370.
(21) ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press.
(22) ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay, NIH Publication Number 09-7357, Research Triangle Park, NC: National Institute of Environmental Health Science. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llnaps/LLNAPerfStds.pdf]
(23) McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89.
(24) Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31.
(25) OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
(26) Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235.
(27) ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf].
(28) Hayes, B.B., Gerber, P.C., Griffey, S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug. Chem. Toxicol., 21, 195-206.
(29) Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemicalinduced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94.
(30) Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256.
(31) Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug Chem. Toxicol., 22, 491-506.
(32) Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European interlaboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68.
(33) Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728.
(34) Patterson, R.M., Noga, E. and Germolec, D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028.
(35) ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam. niehs.nih.gov/methods/acutetox/Tox_workshop.htm]
(36) OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
(37) Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ. Health, 53 563-79.
(38) OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
| Anlage 1 |
Genauigkeit: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (38).
Vergleichssubstanz: Eine sensibilisierende oder nicht sensibilisierende Substanz, die als Standard zu Vergleichszwecken für eine Prüfsubstanz verwendet wird. Eine Vergleichssubstanz sollte die folgenden Eigenschaften haben: i) gleichbleibende und verlässliche Quelle(n); ii) strukturelle und funktionelle Ähnlichkeit mit der Klasse der zu prüfenden Stoffe; iii) bekannte physikalisch-chemische Eigenschaften; iv) unterstützende Daten zu bekannten Effekten und v) bekannte Wirksamkeit im Bereich der erwünschten Reaktion.
Falsch negativ: Eine Substanz, die durch eine Prüfmethode fälschlich als negativ oder nicht wirksam charakterisiert wird, obwohl sie in Wirklichkeit positiv bzw. wirksam ist.
Falsch positiv: Eine Substanz, die durch eine Prüfmethode fälschlich als positiv oder wirksam charakterisiert wird, obwohl sie in Wirklichkeit negativ bzw. nicht wirksam ist.
Gefahr: Potenzial eines schädlichen Effekts für Gesundheit oder Umwelt. Die schädliche Wirkung manifestiert sich nur, wenn es zu einem ausreichenden Expositionsniveau kommt.
Inter-Labor-Reproduzierbarkeit: Das Ausmaß, in dem unterschiedliche qualifizierte Laboratorien, die dasselbe Protokoll verwenden und dieselben Prüfsubstanzen testen, qualitativ und quantitativ vergleichbare Ergebnisse erzielen können. Die Inter-Labor-Reproduzierbarkeit wird während der Prävalidierungs- und Validierungsverfahren ermittelt und zeigt das Maß an, in dem ein Test erfolgreich zwischen Laboratorien übertragen werden kann. Im englischen Sprachgebrauch spricht man in diesem Zusammenhang auch von "Betweenlaboratory reproducibility" (38).
Intra-Labor-Reproduzierbarkeit: Das Ausmaß, in dem qualifizierte Personen innerhalb desselben Labors, die dasselbe spezifische Protokoll zu unterschiedlichen Zeiten verwenden, erfolgreich dieselben Ergebnisse replizieren können. In diesem Zusammenhang spricht man auch von laborinterner Reproduzierbarkeit (38).
Ausreißer: Ein Ausreißer ist ein Messwert, der sich beträchtlich von anderen Werten in einem zufällig ausgewählten Muster in einer Population unterscheidet.
Qualitätssicherung: Ein Managementprozess, mittels dessen die Einhaltung von Laborprüfnormen, Anforderungen und Aufzeichnungsverfahren, sowie die Genauigkeit des Datentransfers durch Individuen bewertet wird, die von den testenden Personen unabhängig sind.
Zuverlässigkeit: Maß der Verlässlichkeit der Reproduzierbarkeit der Prüfmethode innerhalb von und zwischen Laboratorien in einem bestimmten Zeitintervall bei einheitlichem Protokoll. Sie wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet (38).
Hautsensibilisierung: Ein immunologischer Prozess, der auftritt, wenn ein empfindliches Individuum oberflächlich einem induzierenden chemischen Allergen ausgesetzt ist, das eine kutane Immunreaktion auslöst, die zur Entwicklung einer Kontaktsensibilisierung führen kann.
Stimulationsindex (SI): Ein Wert, der zur Bewertung des Hautsensibilisierungspotenzials einer Prüfsubstanz berechnet wird. Der SI ist das Verhältnis der Proliferation in behandelten Gruppen zu dem der gleichzeitigen Vehikelkontrollgruppe.
Prüfsubstanz (auch Prüfchemikalie): Jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird.
B.51. Hautsensibilisierung: Lokaler Lymphknotentest: BRDU-ELISA 12 23
Die vollständige Beschreibung dieser Prüfmethode wurde gestrichen.
Die gleichwertige internationale Prüfmethode ist in Teil 0 Tabelle 2 aufgeführt.
| Einleitung
1. Die OECD-Leitlinien für die Prüfung von Chemikalien und die EU-Prüfmethoden werden regelmäßig überarbeitet, um dem wissenschaftlichen Fortschritt, sich ändernden Rechtsvorgaben und Belangen des Tierschutzes gerecht zu werden. Die ursprüngliche Prüfmethode (B.42) zur Bestimmung der Hautsensibilisierung bei der Maus, der Lokale Lymphknotentest (Local Lymph Node Assay bzw. LLNA; OECD-Prüfrichtlinie 429) wurde inzwischen überarbeitet (1, und Kapitel B.42 in diesem Anhang). Verschiedene Veröffentlichungen (2) (3) (4) (5) (6) (7) (8) (9) gehen genauer auf die Validierung des LLNA und auf die mit ihm verbundenen wissenschaftlichen Arbeiten ein. Bei dem LLNA wird zur Messung der Lymphozytenproliferation radioisotopisches Thymidin oder Jod verwendet. Der Test hat daher Beschränkungen, wenn der Erwerb, die Verwendung oder die Entsorgung radioaktiver Materialien mit Problemen verbunden sind. Die Prüfmethode LLNA: BrdU-ELISA [Enzymgebundener Immunoassay] ist eine nicht- radioaktive Modifikation der LLNA-Prüfmethode, bei der nicht radioaktiv gekennzeichnetes 5-Bromo-2-deoxyuridin (BrdU) (Chemical Abstracts Service [CAS-] Nr. 59-14-3) in einem ELISA-basierten Testsystem zur Messung der Lymphozytenproliferation zum Einsatz kommt. Die Prüfmethode LLNA: BrdU-ELISA wurde durch ein internationales wissenschaftliches Gremium für Sachverständigengutachten (peer review) validiert, geprüft und zur Identifizierung sensibilisierender und nicht sensibilisierender Chemikalien mit einigen Beschränkungen als geeignet befunden (10) (11) (12). Diese Prüfmethode wurde zur Bewertung des Hautsensibilisierungspotenzials von Chemikalien (Stoffen und Gemischen) bei Tieren entwickelt. Kapitel B.6 dieses Anhangs und OECD-Prüfrichtlinie 406 stützen sich auf Meerschweinchen-Tests, insbesondere den Meerschweinchen-Maximierungstest und den Bühler- Test (13). Der LLNA (Kapitel B.42 dieses Anhangs; OECD-Prüfrichtlinie 429) und die beiden nichtradioaktiven Modifikationen, LLNA: BrdU-ELISA (Kapitel B.51 dieses Anhangs; OECD-Prüfrichtlinie 442 B) und LLNA: DA (Kapitel B.50 dieses Anhangs; OECD-Prüfrichtlinie 442 A) haben insofern Vorteile gegenüber den Meerschweinchen-Tests in B.6 und OECD-Prüfrichtlinie 406 (13), als sie einen verringerten und gezielteren Einsatz von Versuchstieren ermöglichen. 2. Ähnlich wie der LLNA untersucht der LLNA: BrdU-ELISA die Induktionsphase der Hautsensibilisierung, und der Test liefert quantitative Daten für die Bewertung von Dosis-Wirkungs-Beziehungen. Durch den möglichen Nachweis von hautsensibilisierenden Stoffen ohne die Verwendung radioaktiver Markierungen für die DNA können die berufliche Exposition gegenüber Radioaktivität und Entsorgungsprobleme vermieden werden. Dies wiederum ermöglicht eine verstärkte Verwendung von Mäusen zur Bestimmung hautsensibilisierender Stoffe und somit eine geringere Verwendung von Meerschweinchen zur Bewertung des hautsensibilisierenden Potenzials (B.6; OECD-Prüfrichtlinie 406) (13). 3. Die verwendeten Begriffsbestimmungen sind in Anlage 1 aufgeführt. Ausgangsüberlegungen und Begrenzungen 4. Die Prüfmethode LLNA: BrdU-ELISA ist eine modifizierte LLNA-Methode zum Nachweis von Chemikalien mit potenziell hautsensibilisierenden Eigenschaften, mit bestimmten Beschränkungen. Dies bedeutet nicht notwendigerweise, dass der LLNA: BrdU-ELISA zwingend in jedem Fall anstelle des LLNA oder der Meerschweinchen-Tests (B.6; OECD-Prüfrichtlinie 406) (13) eingesetzt werden muss. Vielmehr erweist sich der Test als gleichermaßen wertvoll und kann als Alternative gewählt werden, bei der im Allgemeinen keine weitere Bestätigung von positiven wie auch negativen Ergebnissen erforderlich ist (10) (11). Das Prüflabor sollte vor der Durchführung der Studie alle verfügbaren Informationen über die Prüfsubstanz auswerten und berücksichtigen. Zu diesen Informationen zählen die Identität und die chemische Struktur der Prüfsubstanz, ihre physikalisch-chemischen Eigenschaften, die Ergebnisse aller sonstigen in vitro- oder in vivo-Toxizitätsprüfungen der Prüfsubstanz sowie toxikologische Daten zu strukturell verwandten Chemikalien. Dies gilt es zu berücksichtigen, um die Eignung des LLNA: BrdU-ELISA für die jeweilige Prüfsubstanz festzustellen (bestimmte Arten chemischer Stoffe sind mit dem LLNA: BrdU-ELISA nicht kompatibel [siehe Absatz 5]) und die Festlegung der Dosis zu erleichtern. 5. Die Prüfmethode LLNA: BrdU-ELISA ist eine in vivo-Methode. Das bedeutet, dass die kontaktsensibilisierenden Eigenschaften weiterhin an Tieren bewertet werden. Der Test erlaubt es jedoch, im Vergleich zu den Meerschweinchen-Tests (B.6; OECD-Prüfrichtlinie 406) die Anzahl der für diesen Zweck benötigten Tiere zu reduzieren (13). Ferner stellt der LLNA: BrdU-ELISA eine substanzielle Verfeinerung in der Behandlung von Versuchstieren bei Tests auf allergische Kontaktsensibilisierung dar, da anders als B.6 und OECD-Prüfrichtlinie 406 der LLNA: BrdU- ELISA keine Auslösung von provozierten Überempfindlichkeitsreaktionen der Haut erfordert. Außerdem erfordert der LLNA: BrdU-ELISA, anders als der Meerschweinchen-Maximierungstest, keinen Einsatz von Adjuvanzien (Kapitel B.6 dieses Anhangs, 13). Damit verringert der LLNA: BrdU-ELISA das Leiden der Tiere. Trotz der Vorteile des LLNA: BrdU-ELISA gegenüber B.6 und OECD-Prüfrichtlinie 406 (13) ist er mit gewissen Einschränkungen behaftet, die die Anwendung von B.6 oder von OECD-Prüfrichtlinie 406 erforderlich machen können (z.B. die Prüfung bestimmter Metalle, falsch positive Ergebnisse bei bestimmten hautreizenden Stoffen [wie etwa bei tensidähnlichen Chemikalien] (6) (1 und Kapitel B.42 in diesem Anhang), oder die Löslichkeit der Prüfsubstanz). Zudem können chemische Klassen oder Substanzen mit Funktionsgruppen, die erwiesenermaßen als potenzielle Störfaktoren wirken können (15), den Einsatz von Meerschweinchen-Tests erforderlich machen (B.6; OECD-Prüfrichtlinie 406 (13)). Beschränkungen, die für den LLNA festgestellt wurden (1 und Kapitel B.42 dieses Anhangs), empfehlen sich auch für den LLNA: BrdU-ELISA (10). Abgesehen von diesen bekannten Beschränkungen dürfte der LLNA: BrdU-ELISA für die Prüfung aller Chemikalien geeignet sein, sofern mit diesen Chemikalien keine Eigenschaften verbunden sind, die die Genauigkeit des LLNA: BrdU-ELISA beeinträchtigen könnten. Außerdem sollte die Möglichkeit positiver Grenzergebnisse in Betracht gezogen werden, wenn für den Stimulationsindex (SI) Werte zwischen 1,6 und 1,9 ermittelt werden (siehe Absätze 31-32). Dies beruht auf der Validierungs-Datenbasis von 43 Substanzen mit einem SI ≥ 1,6 (siehe Absatz 6), bei denen der LLNA: BrdU-ELISA alle 32 LLNA- Sensibilisatoren richtig identifizierte, jedoch zwei von 11 LLNA-Nichtsensibilisatoren mit SI-Werten zwischen 1,6 und 1,9 (d. h. positive Grenzwerte) nicht korrekt identifizierte (10). Da allerdings derselbe Datensatz für die Einstellung der SI-Werte und die Berechnung der prädiktiven Eigenschaften des Tests verwendet wurde, könnten die festgestellten Ergebnisse auch eine Überbewertung der tatsächlichen prädiktiven Eigenschaften sein. 6. Der LLNA: BrdU-ELISA beruht auf dem Prinzip, dass sensibilisierende Stoffe eine Lymphozytenproliferation in den drainierenden Lymphknoten an der Stelle der Applikation des chemischen Stoffes induzieren. Diese Proliferation verläuft proportional zur Dosis und zur Wirksamkeit des applizierten Allergens und bietet sich als einfache Möglichkeit an, eine quantitative Messung der Sensibilisierung zu erhalten. Die Proliferation wird gemessen, indem man die mittlere Proliferation jeder Prüfgruppe mit der mittleren Proliferation der mit Vehikel behandelten Kontrollgruppe vergleicht. Das Verhältnis der mittleren Proliferation in jeder Behandlungsgruppe zu dem in der gleichzeitigen Vehikelkontrollgruppe, auch als SI bezeichnet, wird festgelegt und sollte ≥ 1,6 betragen, ehe eine weitere Bewertung einer Prüfsubstanz als potenzieller Hautsensibilisator erfolgt. Die hier beschriebenen Methoden beruhen auf der Messung des BrdU-Gehalts zur Anzeige einer erhöhten Anzahl proliferierender Zellen in den drainierenden aurikulären Lymphknoten. BrdU ist eine Nachbildung von Thymidin und wird ähnlich in die DNA proliferierender Zellen integriert. Die Integration von BrdU wird durch ELISA mittels eines für BrdU spezifischen Antikörpers gemessen, der auch mit Peroxidase markiert wird. Bei Hinzufügung des Substrats reagiert die Peroxidase mit diesem und es entsteht ein gefärbtes Produkt, das mittels eines Mikrotiterplatten-Lesegeräts auf ein bestimmtes Absorptionsmaß gemessen wird. Auswahl von Versuchstierarten 7. Für diesen Test ist die Maus die Spezies der Wahl. Validierungsstudien für den LLNA: BrdU-ELISA wurden ausschließlich mit dem CBA/JN-Stamm durchgeführt, der daher als bevorzugter Stamm gilt (10) (12). Es werden junge erwachsene weibliche Mäuse verwendet, die weder geworfen haben noch trächtig sind. Bei Versuchsbeginn sollten die Tiere 8-12 Wochen alt sein, die Gewichtsunterschiede minimal sein und 20 % des mittleren Gewichts nicht übersteigen. Alternativ können Tests an anderen Stämmen und männlichen Tieren erfolgen, wenn anhand von hinlänglich großen Datenangaben nachgewiesen werden kann, dass keine signifikanten stamm- und/oder geschlechtsspezifischen Unterschiede in der Reaktion auf den LLNA: BrdU-ELISA bestehen. Haltungs- und Fütterungsbedingungen 8. Mäuse sollten in Gruppen gehalten werden (16), wenn nicht angemessene wissenschaftliche Begründungen eine Einzelhaltung nahelegen. Die Temperatur im Versuchsraum sollte 22 ± 3 °C betragen. Obwohl die relative Luftfeuchtigkeit mindestens 30 % betragen und zu anderen Zeiten als während der Reinigung vorzugsweise nicht über 70 % liegen soll, ist ein Wert von 50-60 % anzustreben. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln. An die Versuchstiere kann herkömmliches Laborfutter verfüttert werden, und eine unbegrenzte Trinkwasserversorgung ist zu gewährleisten. Vorbereitung der Tiere 9. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert (aber nicht am Ohr) und vor Beginn der Dosierung für einen Zeitraum von mindestens 5 Tagen in ihren Käfigen an die Laborbedingungen gewöhnt. Vor Behandlungsbeginn werden alle Tiere untersucht, um sicherzustellen, dass keine sichtbaren Hautverletzungen bestehen. Vorbereitung der Dosierlösungen 10. Feste Chemikalien sollten vor der Applikation in ein Mäuseohr in Lösungsmitteln/Vehikeln gelöst oder suspendiert und ggf. verdünnt werden. Flüssige Chemikalien können direkt appliziert oder zuvor verdünnt werden. Unlösliche Chemikalien, wie sie in der Regel in Medizinprodukten vorliegen, sollten vor der Applikation in ein Mäuseohr in einem geeigneten Lösungsmittel einer übertrieben starken Extraktion unterzogen werden, um alle extrahierbaren Inhaltsstoffe vor der Prüfung sichtbar zu machen. Die Prüfsubstanzen sollten täglich zubereitet werden, es sei denn, die Stabilität der Substanz bei Lagerung wird nachgewiesen. Überprüfung der Zuverlässigkeit: 11. Anhand von positiven Kontrollchemikalien (PC) wird die ordnungsgemäße Leistung des Tests nachgewiesen. Hierzu ist eine angemessene und reproduzierbare Empfindlichkeit der Reaktion auf eine sensibilisierende Prüfsubstanz erforderlich, wobei die Reaktionsstärke gut charakterisiert ist. Es wird die Einbeziehung einer gleichzeitigen Positivkontrolle empfohlen, da diese die Fähigkeit des Labors belegt, jeden Test erfolgreich durchzuführen und eine Bewertung der Wiederholbarkeit und Vergleichbarkeit zwischen verschiedenen Labors ermöglicht. Einige Regulierungsbehörden schreiben auch eine Positivkontrolle für jede Studie vor. Daher wird Anwendern empfohlen, vor Durchführung des LLNA: BrdU-ELISA die zuständigen Behörden zu Rate zu ziehen. Dementsprechend wird die routinemäßige Verwendung einer gleichzeitigen Positivkontrolle angeregt, um die Notwendigkeit zusätzlicher Tierversuche zur Erfüllung von Anforderungen zu vermeiden, die aus der Verwendung einer nur periodischen Positivkontrolle resultieren könnten (siehe Absatz 12). Die Positivkontrolle sollte eine positive Reaktion auf den LLNA: BrdU-ELISA bei einem Expositionsniveau hervorrufen, das einen Anstieg des Stimulationsindex (SI ≥ 1,6 im Vergleich zur Negativkontrollgruppe, NK) bewirkt. Die positive Kontrolldosis soll so gewählt werden, dass sie keine übermäßige Hautreizung oder systemische Toxizität verursacht und die Induktion reproduzierbar, aber nicht exzessiv ist (z.B. ein SI > 14 würde als exzessiv gelten). Bevorzugte positive Kontrollstoffe sind 25 % Hexylcinnaminaldehyd (CAS-Nr. 101-86-0) und 25 % Eugenol (CAS-Nr. 97-53-0) in Aceton: Olivenöl (4:1, v/v). Unter bestimmten Umständen können in ausreichend begründeten Fällen andere Kontrollsubstanzen eingesetzt werden, die den genannten Kriterien entsprechen. 12. Obwohl die Einbeziehung einer gleichzeitigen Positivkontrollgruppe empfohlen wird, können in gewissen Situationen periodische Prüfungen (d. h. in Abständen ≤ 6 Monaten) der Positivkontrolle für Laboratorien ausreichen, die denen der LLNA: BrdU-ELISA regelmäßig (d. h. mindestens einmal pro Monat) durchgeführt wird und die über eine etablierte historische Kontrolldatenbasis verfügen, die die Fähigkeit des Labors bestätigt, reproduzierbare und genaue Ergebnisse mit Positivkontrollen zu erzielen. Eine angemessene Beherrschung des LLNA: BrdU-ELISA kann erfolgreich nachgewiesen werden, indem durchgehend positive Ergebnisse mit den positiven Kontrollchemikalien in mindestens 10 unabhängigen Prüfungen erzielt werden, die innerhalb eines angemessenen Zeitraums (d. h. in weniger als einem Jahr) durchgeführt wurden. 13. Eine gleichzeitige Positivkontrollgruppe sollte immer einbezogen werden, wenn Verfahrensänderungen im LLNA: BrdU-ELISA auftreten (z.B. Wechsel des geschulten Personals, Änderung der Materialien bei den Prüfmethoden und/oder der Reagenzien, Änderung der Ausrüstung, Änderung der Herkunft der Versuchstiere). Solche Änderungen sollten in Laborberichten dokumentiert werden. Ferner ist der Einfluss dieser Änderungen auf die Aussagefähigkeit der zuvor eingerichteten historischen Datenbank zu bedenken. Dabei sollte die Notwendigkeit überdacht werden, eine neue historische Datenbank einzurichten, um die gleichbleibende Qualität der positiven Kontrollergebnisse zu dokumentieren. 14. Die Prüfer sollten sich der Tatsache bewusst sein, dass die Entscheidung, eine PC-Studie periodisch statt gleichzeitig durchzuführen, Auswirkungen auf die Aussagefähigkeit und Akzeptanz negativer Studienergebnisse haben kann, die ohne gleichzeitige Positivkontrolle im Zeitraum zwischen den einzelnen periodischen PC-Studien auftreten können. Wenn zum Beispiel ein falsch negatives Ergebnis in der periodischen PC-Studie auftritt, können negative Ergebnisse für die Prüfsubstanz, die in dem Zeitraum zwischen der letzten akzeptablen periodischen PC- Studie und der inakzeptablen periodischen PC-Studie aufgetreten waren, in Frage gestellt werden. Die Auswirkungen solcher Ergebnisse sollten sorgfältig bedacht werden, wenn entschieden wird, ob gleichzeitige oder nur periodische PC-Studien durchgeführt werden sollen. Auch ist die Verwendung einer geringeren Anzahl an Versuchstieren bei der gleichzeitigen Positivkontrollgruppe zu bedenken, wenn dies wissenschaftlich zu begründen ist und wenn das Labor auf der Grundlage laborspezifischer historischer Daten demonstriert, dass eine geringere Anzahl an Mäusen ausreicht (17). 15. Obwohl eine positive Kontrollchemikalie in dem Vehikel geprüft werden sollte, von dem bekannt ist, dass es eine gleichbleibende Reaktion hervorruft (z.B. Aceton: Olivenöl; 4:1, v/v), können in bestimmten Rechtssituationen auch Prüfungen in einem Nichtstandard-Vehikel (klinisch/chemisch relevante Formulierung) erforderlich sein (18). Wenn die gleichzeitige Positivkontrolle in einem anderen Vehikel als der Prüfsubstanz getestet wird, sollte eine gesonderte Vehikelkontrolle für die gleichzeitige PC einbezogen werden. 16. In Fällen, in denen Prüfsubstanzen einer bestimmten chemischen Klasse oder eine Reihe von Reaktionen bewertet werden, können Referenzsubstanzen nützlich sein, um nachzuweisen, dass die Prüfmethode zur Feststellung des Hautsensibilisierungspotenzials dieser Art von Substanzen ordnungsgemäß funktioniert. Geeignete Referenzsubstanzen sollten die folgenden Eigenschaften haben:
Anzahl der Versuchstiere und Dosierungen 17. Mindestens vier Tiere pro Dosisgruppe mit jeweils mindestens drei Konzentrationen der Prüfsubstanz werden benötigt; zusätzlich braucht man eine Negativkontrollgruppe, die nur mit dem Vehikel für die Prüfsubstanz behandelt wird, sowie eine Positivkontrolle (gleichzeitig oder jüngeren Datums, auf der Grundlage der Laborvorschriften in den relevanten Abschnitten 11-15). Tests mit Mehrfachdosen der PC sollten in Erwägung gezogen werden, insbesondere wenn die Positivkontrolle intermittierend getestet wird. Bis auf die Verabreichung der Prüfsubstanz sind die Tiere der Kontrollgruppen ebenso zu behandeln wie die Tiere der Behandlungsgruppen. 18. Die Auswahl von Dosierung und Vehikel sollte auf den Empfehlungen in den Referenzen 2 und 19 beruhen. Für aufeinanderfolgende Dosierungen werden normalerweise geeignete abgestufte Konzentrationen gewählt, wie z.B. 100 %, 50 %, 25 %, 10 %, 5 %, 2,5 %, 1 %, 0,5 % usw. Die Auswahl der Konzentrationsfolge sollte angemessen wissenschaftlich begründet werden. Alle vorhandenen toxikologischen Angaben (z.B. über die akute Toxizität und Hautreizung) sowie strukturelle und physiochemische Angaben zu der jeweiligen Prüfsubstanz (und/oder strukturverwandten Substanzen) sollten bei der Festlegung von drei aufeinander folgenden Konzentrationen berücksichtigt werden, so dass bei der Höchstkonzentration einerseits die Exposition maximiert und andererseits eine systemische Toxizität und/oder eine übermäßige lokale Hautreizung ausgeschlossen werden (19) (20 und Kapitel B.4 dieses Anhangs). Mangels solcher Informationen kann ein anfänglicher Dosisfindungstest erforderlich sein (siehe Absätze 21-24). 19. Das Vehikel sollte das Testergebnis nicht beeinflussen oder beeinträchtigen und sollte so gewählt werden, dass die Löslichkeit zur Erzielung einer möglichst hohen Konzentration maximiert und gleichzeitig eine für das Applizieren der Prüfsubstanz geeignete Lösung/Suspension hergestellt werden kann. Empfohlene Vehikel sind Aceton: Olivenöl (4:1, v/v), N,N-Dimethylformamid, Methylethylketon, Propylenglykol und Dimethylsulphoxid (6), wobei mit hinreichender wissenschaftlicher Begründung auch andere Vehikel verwendet werden können. Unter bestimmten Umständen muss ein klinisch relevantes Lösungsmittel oder die handelsübliche Zubereitung, in der die Prüfsubstanz vermarktet wird, als zusätzliche Kontrolle genutzt werden. Besondere Sorgfalt sollte darauf verwendet werden, zu gewährleisten, dass in das Vehikelsystem durch die Verwendung geeigneter Lösungsvermittler (z.B. 1 % Pluronic® L92) hydrophile Stoffe eingearbeitet werden, die die Haut befeuchten und nicht sofort ablaufen. Folglich sind vollständig wässrige Vehikel zu vermeiden. 20. Die Arbeit an Lymphknoten einzelner Mäuse ermöglicht die Bewertung der Variabilität von Tieren sowie einen statistischen Vergleich zwischen der Prüfsubstanz und Messungen an der Vehikelkontrollgruppe (siehe Absatz 33). Die Möglichkeit, die Anzahl der Mäuse in der Positivkontrollgruppe zu verringern, ist nur realistisch, wenn Einzeltierdaten erhoben werden (17). Außerdem verlangen einige Regulierungsbehörden die Erhebung von Daten einzelner Tiere. Die regelmäßige Erhebung von Einzeltierdaten stellt im Hinblick auf den Tierschutz einen Vorteil dar, indem Doppeltests vermieden werden. Diese wären notwendig, wenn die ursprünglich gemeinsam gesammelten Ergebnisse für die Prüfsubstanz (z.B. über gepoolte Daten) später von Regulierungsbehörden mit anderen Vorschriften (z.B. Einzeltierdaten) geprüft würden. Dosisfindungstest 21. Wenn über die höchst mögliche Dosierung keine Informationen vorliegen (siehe Absatz 18), sollte ein Dosisfindungstest durchgeführt werden, um die geeignete Dosierung für den LLNA: BrdU-ELISA festzulegen. Der Dosisfindungstest soll eine Orientierung bei der Auswahl der höchst möglichen Dosisstufe für die Hauptuntersuchung des LLNA: BrdU-ELISA geben, wenn keine Informationen darüber vorliegen, welche Konzentration systemische Toxizität (siehe Absatz 24) und/oder übermäßige lokale Hautreizung (siehe Absatz 23) verursacht. Die höchste geprüfte Dosisstufe sollte eine Konzentration von 100 % der Prüfsubstanz bei Flüssigkeiten bzw. die höchst mögliche Konzentration bei Feststoffen oder Suspensionen sein. 22. Der Dosisfindungstest wird unter Bedingungen durchgeführt, die denen der Hauptstudie des LLNA: BrdU-ELISA genau entsprechen. Allerdings gibt es hier keine Bewertung der Lymphknotenproliferation und es können weniger Tiere pro Dosisgruppe eingesetzt werden. Es werden ein oder zwei Tiere pro Dosisgruppe empfohlen. Alle Mäuse werden täglich auf klinische Zeichen für systemische Toxizität oder lokale Reizungen an der Applikationsstelle untersucht. Das Körpergewicht wird vor dem Test und vor dem Abschluss (Tag 6) protokolliert. Beide Ohren jeder Maus werden auf Anzeichen für Erytheme untersucht und mit Hilfe von Tabelle 1 (20 und Kapitel B.4 dieses Anhangs) bewertet. Die Ohrdicke wird mit Hilfe eines Ohrdickenmessgeräts (z.B. digitaler Mikrometer oder Peacock Dickenmessuhr) an Tag 1 vor der Dosierung, Tag 3 (ca. 48 Stunden nach der ersten Dosis) und Tag 6 bestimmt. Zusätzlich kann an Tag 6 die Ohrdicke durch die Gewichtsbestimmung von Ohrstanzproben ermittelt werden. Dies sollte nach der tierschutzgerechten Tötung der Tiere erfolgen. Eine übermäßige lokale Hautreizung wird durch eine Erythem-Punktzahl von ≥ 3 und/oder eine Zunahme der Ohrdicke ≥ 25 % an jedem beliebigen Messtag angezeigt (21) (22). Als höchste Dosis für die Hauptuntersuchung des LLNA: BrdU-ELISA wird die nächst niedrigere Dosis in der Konzentrationsreihe des Dosisfindungstests gewählt (siehe Absatz 18), die keine systemische Toxizität und/oder übermäßige lokale Hautreizung verursacht. Tabelle 1: Erythem-Klassifizierung
23. Zusätzlich zu einer Zunahme der Ohrdicke um 25 % (21) (22) wurde ein statistisch signifikanter Anstieg der Ohrdicke bei den behandelten Mäusen im Vergleich zu den Kontrollmäusen zugrundelegt, um Reizstoffe in dem LLNA zu identifizieren (22) (23) (24) (25) (26) (27) (28). Obwohl ein statistisch signifikanter Anstieg auch bei einer Ohrdicke von weniger als 25 % auftreten kann, konnte kein spezifischer Zusammenhang mit einer übermäßigen Hautreizung festgestellt werden (25) (26) (27) (28) (29). 24. Die folgenden klinischen Beobachtungen können, sofern sie Bestandteil einer Gesamtbewertung sind, auf systemische Toxizität hinweisen (30) und somit die höchst mögliche Dosisstufe für die Hauptuntersuchung des LLNA: BrdU-ELISA anzeigen: Änderungen der Funktionen des Nervensystems (z.B. Piloerektion, Ataxie, Tremor und Krämpfe); Verhaltensänderungen (z.B. Aggressivität, Änderungen bei der Fellpflege, auffallende Änderung des Aktivitätsniveaus); Änderungen des Atemmusters (d. h. Änderung der Häufigkeit und Intensität des Atmens wie Dyspnoe, Keuchen, rasselnder Atem) sowie Änderungen der Futter- und Wasseraufnahme. Zusätzlich sollten Anzeichen von Lethargie und/oder Teilnahmslosigkeit sowie alle klinischen Anzeichen für mehr als leichte oder momentane Schmerzen und Qual, eine Gewichtsreduktion von > 5 % zwischen Tag 1 und Tag 6 und die Sterblichkeit in der Bewertung berücksichtigt werden. Moribunde Tiere oder Tiere, die Anzeichen starker und andauernder Qualen zeigen, sollten tierschutzgerecht getötet werden (31). Versuchsplan der Hauptuntersuchung 25. Der Versuchsplan des Tests ist wie folgt:
Vorbereitung der Zellsuspensionen 26. Für jede Maus wird aus den paarweise entnommenen Lymphknotenzellen (LNC) durch vorsichtigen mechanischen Aufschluss in einem Edelstahlfilter mit einer Maschenweite von 200 Mikron oder mittels einer anderen geeigneten Technik (z.B. Verwendung eines wegwerfbaren Kunststoff-Stößels zum Zerkleinern der Lymphknoten, die anschließend durch ein #70 Nylon-Gewebe passiert werden) eine Einzelzellsuspension hergestellt. Das Verfahren zur Zubereitung der Lymphknotensuspension ist für diesen Test von entscheidender Bedeutung. Deshalb sollte jeder Prüfer diese Technik im Voraus gut beherrschen erlernen. Die Lymphknoten bei Tieren der Negativkontrollgruppe sind klein; daher ist Vorsicht geboten, um eine ungewollte Beeinflussung der SI-Werte zu vermeiden. In jedem Fall sollte das Zielvolumen der LNC-Lösung an ein vorgegebenes Optimalvolumen (ca. 15 mL) angepasst werden. Das Optimalvolumen beruht auf der Erzielung eines mittleren Absorptionsmaßes der Negativkontrollgruppe zwischen 0,1 und 0,2. Bestimmung der Zellproliferation (Messung des BrdU-Gehalts in der DNA der Lymphozyten) 27. Die Messung des BrdU erfolgt im ELISA mit einem im Handel erhältlichen Mess-Kit (z.B. Roche Applied Science, Mannheim, Deutschland, Katalog-Nr. 11 647 229 001). Darauf werden kurz 100 µL der Lymphknoten-Suspension in die Vertiefungen einer flachgrundigen Mikroplatte in dreifacher Ausfertigung gegeben. Nach der Fixierung und Denaturierung der Lymphknoten-Suspension werden in jede Vertiefung Anti-BrdU-Antikörper gegeben und zur Reaktion gebracht. Die Anti-BrdU-Antikörper werden anschließend durch Spülen entfernt. Die Substratlösung wird hinzugefügt und kann Chromogen erzeugen. Anschließend wird das Absorptionsmaß bei 370 nm mit einer Referenzwellenlänge von 492 nm gemessen. In allen Fällen sollten die Prüfbedingungen des Tests optimiert werden (siehe Absatz 26). Klinische Beobachtungen 28. Jede Maus sollte mindestens einmal täglich sorgfältig auf klinische Zeichen, d. h. lokale Reizung an der Applikationsstelle, oder auf systemische Toxizität untersucht werden. Alle Beobachtungen werden systematisch in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Die Überwachungspläne sollten Kriterien beinhalten, anhand derer Mäuse mit systemischer Toxizität, übermäßiger lokaler Hautreizung oder Hautätzung schnell zu Zwecken der schmerzlosen Tötung identifiziert werden können (31). Körpergewicht 29. In Abschnitt 25 wurde bereits ausgeführt, dass das Körpergewicht der einzelnen Tiere zu Versuchsbeginn und zum Zeitpunkt der tierschutzgerechten Tötung der Tiere laut Versuchsplan festgestellt werden soll. 30. Die Ergebnisse für jede Behandlungsgruppe werden als mittlerer Stimulationsindex (SI) angegeben. Den SI erhält man durch Teilen des mittleren BrdU-Markierungsindex pro Maus innerhalb jeder Prüfgruppe und der Positivkontrollgruppe durch den mittleren BrdU-Markierungsindex pro Maus für die Lösungsmittel/Vehikelkontrollgruppe. Der durchschnittliche SI beträgt demnach für die mit Vehikel behandelten Kontrollen 1. Der BrdU-Markierungsindex ist definiert als: BrdU-Markierungsindex = (ABSem - ABS blankem) - (ABSref - ABS blankref) Dabei gilt: em = Emissionswellenlänge und ref = Referenzwellenlänge. 31. In dem Entscheidungsprozess gilt ein Ergebnis als positiv, wenn SI ≥ 1,6 ist (10). Die Stärke der Dosis-Wirkung, die statistische Signifikanz und die gleichbleibende Qualität der Lösungsmittel-/Vehikel-Reaktion sowie der Reaktion der Positivkontrolle können aber auch benutzt werden, um festzulegen, ob ein Grenzergebnis (d. h. SI-Wert zwischen 1,6 und 1,9) als positiv bezeichnet werden soll (3) (6) (32). 32. Bei einer positiven Grenzwertreaktion mit einem SI zwischen 1,6 und 1,9 können Anwender zusätzliche Informationen berücksichtigen, wie z.B. die Dosis-Wirkungs-Beziehung, das Vorliegen einer systemischen Toxizität oder übermäßigen Hautreizung, sowie gegebenenfalls die statistische Signifikanz zusammen mit den SI-Werten, um zu bestätigen, dass die Ergebnisse positiv sind (10). Zu beachten sind ferner die verschiedenen Eigenschaften der Prüfsubstanz, wobei es unter anderem zu klären gilt, ob ein struktureller Zusammenhang mit bekannten Hautsensibilisatoren besteht, ob sie eine übermäßige lokale Hautreizung bei der Maus hervorruft und wie die festgestellte Art der Dosis-Wirkungs-Beziehung ist. Diese und weitere Aspekte werden an anderer Stelle ausführlich erörtert (4). 33. Werden die Daten für jede einzelne Maus erhoben, kann eine statistische Analyse zu Vorhandensein und Grad des Dosis-Wirkungs-Verhältnisses in den Daten durchgeführt werden. Jede statistische Auswertung könnte eine Bewertung der Dosis-Wirkungs-Beziehung sowie geeignete Testgruppen-Vergleiche beinhalten (z.B. Gruppe mit paarweiser Dosierung vs. gleichzeitige Lösungsmittel-/Vehikelkontrollgruppen). Die statistischen Analysen könnten z.B. die lineare Regression, den Williams-Test zur Bewertung von Dosis-Wirkungs-Trends oder den Dunnett-Test für paarweise Vergleiche beinhalten. Bei der Auswahl einer geeigneten Methode für die statistische Analyse sollte sich der Prüfer möglicher ungleicher Varianzen und anderer damit zusammenhängender Probleme stets bewusst sein, denn diese erfordern unter Umständen eine Datentransformation oder ein nichtparametrisches statistisches Verfahren. Auf jeden Fall muss der Prüfer unter Umständen SI-Berechnungen und statistische Analysen mit und ohne bestimmte Datenpunkte (manchmal als "Ausreißer" bezeichnet) vornehmen. Daten 34. Die Daten sind in tabellarischer Form zusammenzufassen. Sie sollten die BrdU-Index-Werte pro Tier, den mittleren BrdU-Markierungsindex pro Tier für jede Gruppe, die damit verbundene Fehlervariable (z.B. SD, SEM) und den mittleren SI für jede Dosisgruppe, verglichen mit der gleichzeitigen Lösungsmittel-/Vehikelkontrollgruppe angeben. Prüfbericht 35. Der Testbericht sollte folgende Angaben enthalten: Prüf- und Kontrollsubstanzen: (1) OECD (2010), Skin Sensitisation: Local Lymph Node Assay, Test Guideline No. 429, Guidelines for the Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines] (2) Chamberlain, M. and Basketter, D.A. (1996), The local lymph node assay: status of validation. Food Chem. Toxicol., 34, 999-1002. (3) Basketter, D.A., Gerberick, G.F., Kimber, I. and Loveless, S.E. (1996), The local lymph node assay: A viable alternative to currently accepted skin sensitisation tests. Food Chem. Toxicol., 34, 985-997. (4) Basketter, D.A., Gerberick, G.F. and Kimber, I. (1998), Strategies for identifying false positive responses in predictive sensitisation tests. Food Chem. Toxicol., 36, 327-33. (5) Van Och, F.M.M., Slob, W., De Jong, W.H., Vandebriel, R.J. and Van Loveren, H. (2000), A quantitative method for assessing the sensitising potency of low molecular weight chemicals using a local lymph node assay: employment of a regression method that includes determination of uncertainty margins. Toxicol., 146, 49-59. (6) ICCVAM (1999), The murine local lymph node Assay: A test method for assessing the allergic contact dermatitis potential of chemicals/compounds: The results of an independent peer review evaluation coordinated by the Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM) and the National Toxicology Program center for the Evaluation of Alternative Toxicological Methods (NICETAM). NIH Publication No: 99-4494. Research Triangle Park, N.C. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llna/llnarep.pdf] (7) Dean, J.H., Twerdok, L.E., Tice, R.R., Sailstad, D.M., Hattan, D.G., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: II. Conclusions and recommendations of an independent scientific peer review panel. Reg. Toxicol. Pharmacol., 34(3), 258-273. (8) Haneke, K.E., Tice, R.R., Carson, B.L., Margolin, B.H., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: III. Data analyses completed by the national toxicology program interagency center for the evaluation of alternative toxicological methods. Reg. Toxicol. Pharmacol., 34(3), 274-286. (9) Sailstad, D.M., Hattan, D., Hill, R.N., Stokes, W.S. (2001), ICCVAM evaluation of the murine local lymph node assay: I. The ICCVAM review process. Reg. Toxicol. Pharmacol., 34(3), 249-257. (10) ICCVAM (2010), ICCVAM Test Method Evaluation Report. Nonradioactive local lymph node assay: BrdU-ELISA Test Method Protocol (LLNA: BrdU-ELISA). NIH Publication No. 10-7552A/B. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/methods/immunotox/llna-ELISA/TMER.htm] (11) ICCVAM (2009), Independent Scientific Peer Review Panel Report: Updated validation status of new versions and applications of the murine local lymph node assay: a test method for assessing the allergic contact dermatitis potential of chemicals and products. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/LLNAPRPRept2009.pdf] (12) Takeyoshi, M., Iida, K., Shiraishi, K. and Hoshuyama, S. (2005), Novel approach for classifying chemicals according to skin sensitising potency by nonradioisotopic modification of the local lymph node assay. J. Appl. Toxicol., 25, 129-134. (13) OECD (1992), Skin Sensitisation, Test Guideline No. 406, Guidelines for Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines] (14) Kreiling, R., Hollnagel, H.M., Hareng, L., Eigler, L., Lee, M.S., Griem, P., Dreessen, B., Kleber, M., Albrecht, A., Garcia, C. and Wendel, A. (2008), Comparison of the skin sensitising potential of unsaturated compounds as assessed by the murine local lymph node assay (LLNA) and the guinea pig maximization test (GPMT). Food Chem. Toxicol., 46, 1896-1904. (15) Basketter, D., Ball, N., Cagen, S., Carrilo, J.C., Certa, H., Eigler, D., Garcia, C., Esch, H., Graham, C., Haux, C., Kreiling, R. and Mehling, A. (2009), Application of a weight of evidence approach to assessing discordant sensitisation datasets: implications for REACH. Reg. Toxicol. Pharmacol., 55, 90-96. (16) ILAR (1996), Institute of Laboratory Animal Research (ILAR) Guide for the Care and Use of Laboratory Animals. 7th ed. Washington, DC: National Academies Press. (17) ICCVAM (2009), Recommended Performance Standards: Murine Local Lymph Node Assay. NIH Publication Number 09-7357. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/docs/immunotox_docs/llnaps/LLNAPerfStds.pdf] (18) McGarry, H.F. (2007), The murine local lymph node assay: regulatory and potency considerations under REACH. Toxicol., 238, 71-89. (19) Kimber, I., Dearman, R.J., Scholes E.W. and Basketter, D.A. (1994), The local lymph node assay: developments and applications. Toxicol., 93, 13-31. (20) OECD (2002), Acute Dermal Irritation/Corrosion, Test Guideline No. 404, Guidelines for Testing of Chemicals, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines] (21) Reeder, M.K., Broomhead, Y.L., DiDonato, L. and DeGeorge, G.L. (2007), Use of an enhanced local lymph node assay to correctly classify irritants and false positive substances. Toxicologist, 96, 235. (22) ICCVAM (2009), Nonradioactive Murine Local Lymph Node Assay: Flow Cytometry Test Method Protocol (LLNA: BrdU-FC) Revised Draft Background Review Document. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam.niehs.nih.gov/methods/immunotox/fcLLNA/BRDcomplete.pdf]. (23) Hayes, B.B., Gerber, P.C., Griffey, S. and Meade, B.J. (1998), Contact hypersensitivity to dicyclohexylcarbodiimide and diisopropylcarbodiimide in female B6C3F1 mice. Drug Chem. Toxicol., 21, 195-206. (24) Homey, B., von Schilling, C., Blumel, J., Schuppe, H.C., Ruzicka, T., Ahr, H.J., Lehmann, P. and Vohr, V.W. (1998), An integrated model for the differentiation of chemicalinduced allergic and irritant skin reactions. Toxicol. Appl. Pharmacol., 153, 83-94. (25) Woolhiser, M.R., Hayes, B.B. and Meade, B.J. (1998), A combined murine local lymph node and irritancy assay to predict sensitisation and irritancy potential of chemicals. Toxicol. Meth., 8, 245-256. (26) Hayes, B.B. and Meade, B.J. (1999), Contact sensitivity to selected acrylate compounds in B6C3F1 mice: relative potency, cross reactivity, and comparison of test methods. Drug. Chem. Toxicol., 22, 491-506. (27) Ehling, G., Hecht, M., Heusener, A., Huesler, J., Gamer, A.O., van Loveren, H., Maurer, T., Riecke, K., Ullmann, L., Ulrich, P., Vandebriel, R. and Vohr, H.W. (2005), A European inter- laboratory validation of alternative endpoints of the murine local lymph node assay: first round. Toxicol., 212, 60-68. (28) Vohr, H.W. and Ahr, H.J. (2005), The local lymph node assay being too sensitive? Arch. Toxicol., 79, 721-728. (29) Patterson, R.M., Noga, E. and Germolec D. (2007), Lack of evidence for contact sensitisation by Pfiesteria extract. Environ. Health Perspect., 115, 1023-1028. (30) ICCVAM (2009), Report on the ICCVAM-NICEATM/ECVAM/JaCVAM Scientific Workshop on Acute Chemical Safety Testing: Advancing In Vitro Approaches and Humane Endpoints for Systemic Toxicity Evaluations. Research Triangle Park, NC: National Institute of Environmental Health Sciences. Abrufbar unter: [http://iccvam. niehs.nih.gov/methods/acutetox/Tox_workshop.htm]. (31) OECD (2000), Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation, Environmental Health and Safety Monograph Series on Testing and Assessment No. 19, ENV/JM/MONO(2000)7, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines] (32) Kimber, I., Hilton, J., Dearman, R.J., Gerberick, G.F., Ryan, C.A., Basketter, D.A., Lea, L., House, R.V., Ladies, G.S., Loveless, S.E. and Hastings, K.L. (1998), Assessment of the skin sensitisation potential of topical medicaments using the local lymph node assay: An interlaboratory exercise. J. Toxicol. Environ.1 Health, 53, 563-79. (33) OECD (2005), Guidance Document on the Validation and International Acceptance of New or Updated Test Methods for Hazard Assessment, Environment, Health and Safety Monograph Series on Testing and Assessment No. 34, ENV/JM/MONO(2005)14, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines
Genauigkeit: Der Grad an Übereinstimmung zwischen Testergebnissen und akzeptierten Referenzwerten. Die Genauigkeit ist ein Maß der Leistung der Prüfmethode und ein Aspekt der Relevanz. Der Begriff wird oft im Sinne von "Übereinstimmung" verwendet und bezeichnet den Anteil der korrekten Ergebnisse einer Prüfmethode (33). Vergleichssubstanz: Eine sensibilisierende oder nicht sensibilisierende Substanz, die als Standard zu Vergleichszwecken für eine Prüfsubstanz verwendet wird. Geeignete Vergleichssubstanzen sollten die folgenden Eigenschaften haben: (i) gleichbleibende und verlässliche Quelle(n); (ii) strukturelle und funktionelle Ähnlichkeit mit der Klasse der zu prüfenden Stoffe; (iii) bekannte physikalisch-chemische Eigenschaften; (iv) unterstützende Daten zu bekannten Effekten und (v) bekannte Wirksamkeit im Bereich der erwünschten Reaktion. Falsch negativ: Eine Prüfsubstanz, die durch eine Prüfmethode fälschlich als negativ oder nicht wirksam charakterisiert wird, obwohl sie in Wirklichkeit positiv bzw. wirksam ist (33). Falsch positiv: Eine Prüfsubstanz, die durch eine Prüfmethode fälschlich als positiv oder wirksam charakterisiert wird, obwohl sie in Wirklichkeit negativ bzw. nicht wirksam ist (33). Gefahr: Potenzial eines schädlichen Effekts für Gesundheit oder Umwelt. Die schädliche Wirkung manifestiert sich nur, wenn es zu einem ausreichenden Expositionsniveau kommt. Inter-Labor-Reproduzierbarkeit: Das Ausmaß, in dem unterschiedliche qualifizierte Laboratorien, die dasselbe Protokoll verwenden und dieselben Prüfsubstanz testen, qualitativ und quantitativ vergleichbare Ergebnisse erzielen können. Die Inter-Labor-Reproduzierbarkeit wird während der Prävalidierungs- und Validierungsverfahren ermittelt und zeigt das Maß an, in dem ein Test erfolgreich zwischen Laboratorien übertragen werden kann. Im englischen Sprachgebrauch spricht man in diesem Zusammenhang auch von "Betweenlaboratory reproducibility" (33). Intra-Labor-Reproduzierbarkeit: Das Ausmaß, in dem qualifizierte Personen innerhalb desselben Labors, die dasselbe spezifische Protokoll zu unterschiedlichen Zeiten verwenden, erfolgreich dieselben Ergebnisse replizieren können. In diesem Zusammenhang spricht man auch von laborinterner Reproduzierbarkeit (33). Ausreißer: Ein Ausreißer ist ein Messwert, der sich beträchtlich von anderen Werten in einem zufällig ausgewählten Muster in einer Population unterscheidet. Qualitätssicherung: Ein Managementprozess, mittels dessen die Einhaltung von Laborprüfnormen, Anforderungen und Aufzeichnungsverfahren, sowie die Genauigkeit des Datentransfers durch Individuen bewertet wird, die von den testenden Personen unabhängig sind. Zuverlässigkeit: Maß der Verlässlichkeit der Reproduzierbarkeit der Prüfmethode innerhalb von und zwischen Laboratorien in einem bestimmten Zeitintervall bei einheitlichem Protokoll. Sie wird durch Berechnung der Intra- und Interlabor-Reproduzierbarkeit bewertet (33). Hautsensibilisierung: Ein immunologischer Prozess, der auftritt, wenn ein empfindliches Individuum oberflächlich einem induzierenden chemischen Allergen ausgesetzt ist, das eine kutane Immunreaktion auslöst, die zur Entwicklung einer Kontaktsensibilisierung führen kann. Stimulationsindex (SI): Ein Wert, der zur Bewertung des Hautsensibilisierungspotenzials einer Prüfsubstanz berechnet wird. Der SI ist das Verhältnis der Proliferation in behandelten Gruppen zu dem der gleichzeitigen Vehikelkontrollgruppe. Prüfsubstanz (auch Prüfchemikalie): Jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird. |
B.52 Akute Inhalationstoxizität - Akut-toxische Klassenmethode 14
1. Diese Prüfmethode entspricht der OECD-Prüfrichtlinie (TG) 436 (2009). Die erste Methode zur Prüfung auf akute Inhalationstoxizität TG 403 wurde 1981 angenommen und seitdem überarbeitet (siehe Kapitel B.2 dieses Anhangs (1)). Nach der Annahme der überarbeiteten akut-toxischen Klassenmethode (ATK) mit oraler Verabreichung (Kapitel B.1 tris dieses Anhangs) (5) wurde es für angebracht gehalten, eine akut-toxische Klassenmethode mit Inhalation (2) (3) (4) zu entwickeln. Eine retrospektive Leistungsbewertung der ATK-Methode zur Prüfung auf akute Inhalationstoxizität hat gezeigt, dass die Methode für Zwecke der Einstufung und Kennzeichnung geeignet ist (6). Bei der ATK-Inhalationsprüfmethode kann die Toxizität der Prüfsubstanz mithilfe aufeinander folgender Stufen festgelegter Zielkonzentrationen eingestuft werden. Der wichtigste Endpunkt ist Letalität, aber Tiere, bei denen starke Schmerzen oder schweres Leiden festgestellt werden, oder Tiere, deren Tod bevorsteht, sollten zur Minimierung ihres Leidens tierschutzgerecht getötet werden. Hinweise zu humanen Endpunkten sind im OECD Guidance Document No. 19 (7) zu finden.
2. Das Guidance Document No. 39 on Acute Inhalation Toxicity Testing (GD 39 (8)) enthält Hinweise zur Durchführung und Auswertung dieser Prüfmethode.
3. Die im Zusammenhang mit dieser Prüfmethode verwendeten Begriffe werden in Anlage 1 und im GD 39 (8) definiert.
4. Die Prüfmethode gibt Aufschluss über die gefährlichen Eigenschaften der Prüfsubstanz und ermöglicht ihre Einstufung gemäß der Verordnung (EG) Nr. 1272/2008 zur Einstufung chemischer Stoffe mit akut toxischer Wirkung (9). Falls Punktschätzungen von LC50-Werten oder Analysen der Konzentrations-Wirkungs-Beziehung erforderlich sind, ist Kapitel B.2 dieses Anhangs (1) die geeignete Prüfmethode. Weitere Hinweise zur Wahl der Prüfmethode sind im GD 39 (8) zu finden. Diese Prüfmethode ist nicht speziell für die Prüfung von Spezialmaterialien wie schwer löslichen isometrischen oder Fasermaterialien oder hergestellten Nanomaterialien bestimmt.
5. Bevor Versuche nach dieser Prüfmethode in Betracht gezogen werden, sollte das Prüflabor alle verfügbaren Informationen über die Prüfsubstanz, einschließlich bereits vorliegender Studien, deren Ergebnisse darauf hindeuten, dass auf weitere Versuche verzichtet werden kann, auswerten, damit möglichst wenig Tiere verwendet werden. Für die Auswahl der in Bezug auf Art, Stamm und Geschlecht am besten geeigneten Tiere sowie der geeigneten Expositionsart und Prüfkonzentrationen sollten u. a. Informationen wie die Identität, die chemische Struktur und die physikalisch-chemischen Eigenschaften der Prüfsubstanz, Ergebnisse jeglicher In-vitro- oder In-vivo-Toxizitätsprüfungen, vorgesehene Verwendungen und die Möglichkeit der Exposition des Menschen, (Q)SAR- Daten und toxikologische Daten über strukturverwandte Substanzen herangezogen werden. Konzentrationen, die wegen ätzender 1 oder stark reizender Wirkungen voraussichtlich starke Schmerzen und Qualen verursachen, sollten mit dieser Prüfmethode nicht geprüft werden [siehe GD 39 (8)].
6. Das Prinzip der Prüfung besteht darin, in einem schrittweisen Verfahren genügend Informationen über die akute Toxizität der Prüfsubstanz nach Inhalation über einen Expositionszeitraum von 4 Stunden zu gewinnen, damit sie eingestuft werden kann. Für spezifische Regulierungszwecke sind andere Expositionszeiten möglich. Bei jeder der festgelegten Konzentrationsstufen werden je drei Tiere beider Geschlechter geprüft. Je nach Mortalität und/ oder moribundem Zustand der Tiere können zwei Stufen ausreichen, um eine Beurteilung der akuten Toxizität der Prüfsubstanz zu ermöglichen. Gibt es Anhaltspunkte dafür, dass ein Geschlecht empfindlicher reagiert als das andere, kann die Prüfung mit dem empfindlicheren Geschlecht allein fortgesetzt werden. Das Ergebnis einer Stufe bestimmt die jeweils folgende Stufe:
- keine weitere Prüfung erforderlich,
- Prüfung von drei Tieren je Geschlecht oder
- Prüfung von sechs Tieren des empfindlicheren Geschlechts, d. h. die Schätzungen der Untergrenze der Toxizitätsklasse sollte unabhängig vom Geschlecht auf sechs Tieren je Prüfkonzentrationsgruppe basieren.
7. Moribunde Tiere oder Tiere, die offensichtlich unter Schmerzen leiden oder Anzeichen von schwerem und anhaltendem Leiden zeigen, sollten auf tierschutzgerechte Weise getötet werden und sind bei der Auswertung der Testergebnisse auf die gleiche Weise zu werten wie während des Tests gestorbene Tiere. Kriterien für die Entscheidung, moribunde oder schwer leidende Tiere zu töten, sowie Hinweise zur Erkennung des absehbaren oder bevorstehenden Todes sind Gegenstand des OECD Guidance Document No. 19 on Humane Endpoints (7).
Auswahl von Versuchstierarten
8. Es sollten junge, gesunde, adulte Tiere aus üblicherweise eingesetzten Laborstämmen verwendet werden. Bevorzugtes Versuchstier ist die Ratte; werden andere Tierarten eingesetzt, ist dies zu begründen.
Vorbereitung der Tiere
9. Die weiblichen Tiere dürfen weder bereits geworfen haben noch momentan trächtig sein. Am Tag der Exposition sollten die jungen, adulten Tiere 8 bis 12 Wochen alt sein; ihr Körpergewicht sollte innerhalb von ± 20 % des mittleren Gewichts für jedes Geschlecht aller zuvor exponierten Tiere desselben Alters liegen. Die Tiere werden nach Zufallskriterien ausgewählt, zur individuellen Identifizierung markiert und vor Beginn der Prüfung für einen Zeitraum von mindestens fünf Tagen in ihren Käfigen an die Laborbedingungen gewöhnt. Die Tiere sollten auch für einen kurzen Zeitraum vor der Prüfung an die Versuchsapparatur gewöhnt werden, da dies den Stress durch die Umsetzung in eine neue Umgebung verringert.
Tierhaltung
10. Die Temperatur in dem Raum, in dem die Versuchstiere gehalten werden, sollte 22 ± 3 °C betragen. Die relative Luftfeuchtigkeit sollte im Idealfall im Bereich zwischen 30 und 70 % liegen; bei Verwendung von Wasser als Vehikel könnte dies jedoch unmöglich sein. Die Tiere sollten vor und nach den Expositionen im Allgemeinen nach Geschlecht und Konzentration in Käfigen gruppiert werden, wobei aber die Anzahl der Tiere pro Käfig noch eine genaue Beobachtung der einzelnen Tiere ermöglichen muss und Verluste aufgrund von Kannibalismus oder Kämpfen minimiert werden sollten. Wenn die Tiere der Prüfsubstanz nur mit der Nase ausgesetzt werden sollen, müssen sie möglicherweise an die Restrainer gewöhnt werden. Die Restrainer sollten die Tiere weder körperlich noch in Bezug auf Wärme oder Fixierung übermäßig beeinträchtigen. Die Fixierung kann physiologische Endpunkte wie Körpertemperatur (Hyperthermie) und/oder das Atemminutenvolumen beeinflussen. Wenn generische Daten zeigen, dass keine derartigen Veränderungen in nennenswertem Ausmaß vorkommen, ist eine Eingewöhnung an die Restrainer nicht erforderlich. Bei der Ganzkörperexposition gegen ein Aerosol sollten die Tiere während der Exposition einzeln untergebracht sein, damit sie die Prüfsubstanz nicht durch das Fell ihrer Käfiggenossen filtriert einatmen. Außer während der Exposition kann herkömmliches und zertifiziertes Labortierfutter verwendet werden bei uneingeschränkter Versorgung mit Trinkwasser. Die Beleuchtung sollte künstlich sein und die Hell- und Dunkelphasen sollten sich im Abstand von 12 Stunden abwechseln.
Inhalationskammern
11. Bei der Auswahl einer Inhalationskammer sind die Art der Prüfsubstanz und das Ziel der Prüfung zu berücksichtigen. Das bevorzugte Verfahren ist die "Nose-only"-Exposition (dieser Begriff umfasst "nur Kopf", "nur Nase" oder "nur Schnauze"). Für die Untersuchung von Flüssigkeits- oder Feststoffaerosolen und für Dämpfe, die zu Aerosolen kondensieren können, wird im Allgemeinen die "Nose-only"-Exposition bevorzugt. Besondere Ziele der Untersuchung können möglicherweise mit einer Ganzkörperexposition besser erreicht werden, doch dies sollte im Prüfbericht begründet werden. Um bei Verwendung einer Ganzkörperkammer die Stabilität der Atmosphäre sicherzustellen, sollte das Gesamtvolumen der Versuchstiere 5 % des Volumens der Kammer nicht übersteigen. Die Grundzüge der "Nose-only"- und der Ganzkörperexposition sowie ihre jeweiligen Vor- und Nachteile sind im GD 39 (8) beschrieben.
Verabreichung der Konzentrationen
12. Es wird eine feste Expositionsdauer von 4 Stunden - ohne die zur Erreichung des Gleichgewicht erforderliche Zeit - empfohlen. Um spezifische Erfordernisse zu erfüllen, sind auch andere Expositionszeiten möglich; diese sind jedoch im Prüfbericht zu begründen [siehe GD 39 (8)]. Bei Ganzkörperexpositionen sollten die Tiere einzeln untergebracht sein, um eine Aufnahme der Prüfsubstanz durch das Putzen von Käfiggenossen zu verhindern. Während der Exposition sollte kein Futter verabreicht werden. Wasser kann während einer Ganzkörperexposition jederzeit angeboten werden.
13. Die Tiere werden der Prüfsubstanz in Form von Gas, Dampf, Aerosol oder einer Kombination dieser Formen ausgesetzt. Der zu prüfende Aggregatzustand hängt von den physikalisch-chemischen Eigenschaften der Prüfsubstanz, der gewählten Konzentration und/oder der physikalischen Form ab, in der die Prüfsubstanz bei der Handhabung und Verwendung am wahrscheinlichsten vorliegt. Hygroskopische und chemisch reaktive Prüfsubstanzen sollten bei geringer Luftfeuchtigkeit geprüft werden. Dabei ist darauf zu achten, dass keine explosionsfähigen Konzentrationen erzeugt werden.
Partikelgrößenverteilung
14. Bei allen Aerosolen und bei Dämpfen, die zu Aerosolen kondensieren können, sollte die Partikelgröße bestimmt werden. Damit alle relevanten Regionen der Atemwege der Prüfsubstanz ausgesetzt werden, werden mittlere aerodynamische Massendurchmesser (Mass Median Aerodynamic Diameter - MMAD) von 1 bis 4 μm mit einer geometrischen Standardabweichung (σg) von 1,5 bis 3,0 empfohlen (8) (13) (14). Wenngleich nach Möglichkeit versucht werden sollte, diese Werte zu erreichen, ist Fachwissen erforderlich, falls sie nicht erzielt werden können. Metalldämpfe können z.B. unter diesen Werten liegen, und geladene Partikel, Fasern und hygroskopische Stoffe (die sich in der feuchten Umgebung der Atemwege ausdehnen) können diese Werte überschreiten.
Vorbereitung der Prüfsubstanz in einem Vehikel
15. Um die gewünschte Konzentration und Partikelgröße der Prüfsubstanz in der Atmosphäre herzustellen, kann ein Vehikel verwendet werden. Hierbei ist in der Regel Wasser zu bevorzugen. Partikel können durch mechanische Prozesse auf die erforderliche Größenverteilung gebracht werden. Dabei ist darauf zu achten, dass die Prüfsubstanz nicht zersetzt oder verändert wird. Wenn angenommen wird, dass die Zusammensetzung der Prüfsubstanz durch mechanische Prozesse verändert wurde (z.B. hohe Temperatur aufgrund von Reibung durch übermäßiges Mahlen), sollte die Zusammensetzung der Prüfsubstanz analytisch überprüft werden. Es ist darauf zu achten, dass die Prüfsubstanz nicht kontaminiert wird. Nicht brüchige Granulate, die speziell so formuliert sind, dass sie nicht eingeatmet werden können, brauchen nicht geprüft zu werden. Mit einem Abriebtest sollte nachgewiesen werden, dass beim Umgang mit dem Granulat keine lungengängigen Partikel entstehen. Entstehen bei einem Abriebtest lungengängige Partikel, so sollte eine Inhalationstoxizitätsprüfung durchgeführt werden.
Kontrolltiere
16. Eine negative Kontrollgruppe (Luft) ist nicht erforderlich. Wenn zur Erzeugung der Prüfatmosphäre ein anderes Vehikel als Wasser verwendet wird, sollte nur dann eine Vehikelkontrollgruppe verwendet werden, wenn keine historischen Daten über Inhalationstoxizität vorliegen. Ergibt eine Toxizitätsstudie einer in einem Vehikel formulierten Prüfsubstanz, dass keine Toxizität vorliegt, ist das Vehikel folglich in der geprüften Konzentration nicht toxisch. Daher ist keine Vehikelkontrolle erforderlich.
Überwachung der Expositionsbedingungen
Luftstrom in der Inhalationskammer
17. Der Luftstrom durch die Kammer sollte während jeder Exposition sorgfältig geregelt, kontinuierlich überwacht und mindestens stündlich protokolliert werden. Die Überwachung der Konzentration (oder Stabilität) der Prüfatmosphäre ist eine integrale Messung aller dynamischen Parameter und gibt indirekt die Möglichkeit, alle relevanten dynamischen Parameter der Erzeugung der Prüfatmosphäre zu messen. Es sollte besonders darauf geachtet werden, das erneute Einatmen in "Nose-only"-Expositionskammern zu vermeiden, wenn die Luftströmung durch das Expositionssystem nicht ausreicht, um eine dynamische Strömung der Prüfsubstanzatmosphäre zu erreichen. Es gibt festgelegte Methoden, mit denen nachgewiesen werden kann, dass es unter den gewählten Bedingungen nicht zu erneutem Einatmen kommt (8) (15). Die Sauerstoffkonzentration sollte mindestens 19 % betragen, und die Kohlendioxidkonzentration sollte 1 % nicht überschreiten. Gibt es Grund zu der Annahme, dass diese Werte nicht eingehalten werden können, sind die Sauerstoff- und Kohlendioxidkonzentrationen zu messen.
Temperatur und relative Luftfeuchtigkeit in der Inhalationskammer
18. Die Temperatur in der Inhalationskammer sollte 22 ± 3 °C betragen. Sowohl bei der "Nose-only"- als auch bei der Ganzkörperexposition sollte die relative Luftfeuchtigkeit im Atembereich der Tiere mindestens dreimal für Zeiträume von bis zu 4 Stunden und für kürzere Zeiträume stündlich überwacht und dokumentiert werden. Die relative Luftfeuchtigkeit sollte im Idealfall zwischen 30 und 70 % liegen, was jedoch möglicherweise nicht erreichbar ist (z.B. bei der Prüfung von wasserbasierten Mischungen) oder wegen chemischer Interferenz mit der Prüfmethode nicht gemessen werden kann.
Prüfsubstanz: nominale Konzentration
19. Die nominale Konzentration in der Expositionskammer sollte möglichst berechnet und protokolliert werden. Die nominale Konzentration ist die Masse der erzeugten Prüfsubstanz dividiert durch das Gesamtvolumen der durch das Kammersystem geleiteten Luft. Sie wird nicht zur Beschreibung der Exposition der Tiere verwendet; vielmehr gibt ein Vergleich der nominalen Konzentration und der tatsächlichen Konzentration Aufschluss über die Effizienz des Prüfsystems bei der Erzeugung der Prüfkonzentration, und kann daher für die Aufdeckung von Problemen bei dieser Erzeugung verwendet werden.
Prüfsubstanz: tatsächliche Konzentration
20. Die tatsächliche Konzentration ist die Konzentration der Prüfsubstanz im Atembereich der Tiere in einer Inhalationskammer. Die tatsächlichen Konzentrationen können entweder durch spezifische Methoden (z.B. direkte Probenahme, adsorptive Methoden oder chemische Reaktionsverfahren mit anschließender analytischer Charakterisierung) oder durch unspezifische Methoden wie Gravimetrie bestimmt werden. Die gravimetrische Methode ist lediglich für Aerosole mit nur einem Bestandteil in Pulverform oder Aerosole von Flüssigkeiten mit geringer Flüchtigkeit akzeptabel und sollte sich auf geeignete, vor der Studie zu erstellende und für die Prüfsubstanz spezifische Beschreibungen stützen. Die Konzentration von Aerosolen mit mehreren Bestandteilen in Pulverform kann ebenfalls gravimetrisch bestimmt werden. Hierzu muss jedoch mit Analysedaten belegt werden, dass die Schwebstoffe eine ähnliche Zusammensetzung haben wie das Ausgangsmaterial. Liegen diese Angaben nicht vor, muss die Prüfsubstanz (im Idealfall im Schwebezustand) möglicherweise im Verlauf der Studie in regelmäßigen Abständen neu analysiert werden. Bei aerosolisierten Agenzien, die verdunsten oder sublimieren können, sollte gezeigt werden, dass alle Phasen von der gewählten Methode erfasst wurden. Im Prüfbericht sollten die Zielkonzentration sowie die nominale und die tatsächliche Konzentration angegeben werden, aber nur die tatsächlichen Konzentrationen werden für statistische Analysen zur Berechnung letaler Konzentrationswerte verwendet.
21. Es sollte möglichst eine Partie der Prüfsubstanz verwendet werden; die Probe sollte unter Bedingungen aufbewahrt werden, die ihre Reinheit, Homogenität und Stabilität gewährleisten. Die Prüfsubstanz sollte vor Beginn der Studie mit Angaben zur Reinheit und, falls technisch machbar, zur Identität sowie zu Mengen identifizierter Schadstoffe und Verunreinigungen beschrieben werden. Hierzu können unter anderem die folgenden Daten verwendet werden: Retentionszeit und relative Peakfläche, durch Massenspektrometrie oder Gaschromatographie bestimmtes Molekulargewicht oder andere Werte. Das Prüflabor ist zwar nicht für die Identität der Probe verantwortlich, doch es kann ratsam sein, dass es die Beschreibung des Auftraggebers zumindest in gewissen Grenzen (z.B. Farbe, physikalische Beschaffenheit usw.) überprüft.
22. Die Expositionsatmosphäre ist so konstant wie möglich zu halten und je nach Analysemethode kontinuierlich und/oder intermittierend zu überwachen. Bei der intermittierenden Probenahme sollten in einer vierstündigen Studie mindestens zweimal Proben der Atmosphäre in der Kammer genommen werden. Ist dies wegen begrenzter Luftdurchflussraten oder niedriger Konzentrationen nicht möglich, kann während der gesamten Expositionszeit eine einzige Probe genommen werden. Weichen die einzelnen Proben stark voneinander ab, sollten bei den nächsten geprüften Konzentrationen vier Proben je Exposition gezogen werden. Die einzelnen Proben der Konzentration in der Kammer sollten bei Gasen und Dämpfen nicht mehr als ± 10 % und bei Flüssig- oder Feststoffaerosolen nicht mehr als ± 20 % von der mittleren Kammerkonzentration abweichen. Die Zeit bis zum Erreichen eines Gleichgewichts in der Kammer (t95) ist zu berechnen und zu dokumentieren. Die Expositionsdauer erstreckt sich über den Zeitraum, in dem die Prüfsubstanz erzeugt wird; dazu gehört die zur Erreichung von t95 erforderliche Zeit. GD 39 (8) enthält Hinweise zur Einschätzung von t95.
23. Bei sehr komplexen Mischungen aus Dämpfen/Gasen und Aerosolen (z.B. Verbrennungsatmosphären und Prüfsubstanzen, die aus hierzu bestimmten Endverbraucherprodukten/-geräten gesprüht werden) kann sich jede Phase in einer Inhalationskammer anders verhalten, so dass mindestens eine Indikatorsubstanz (Analyt), normalerweise der wichtigste Wirkstoff in der Mischung, von jeder Phase (Dampf/Gas und Aerosol) ausgewählt werden sollte. Wenn die Prüfsubstanz eine Mischung ist, sollte die Analysekonzentration für die gesamte Mischung und nicht nur für den Wirkstoff oder den Bestandteil (Analyt) dokumentiert werden. Weitere Informationen zu tatsächlichen Konzentrationen sind im GD 39 (8) zu finden.
Prüfsubstanz: Partikelgrößenverteilung
24. Die Partikelgrößenverteilung von Aerosolen sollte während jeder 4-stündigen Exposition mindestens zweimal mit einem Kaskaden-Impaktor oder einem anderen Messgerät wie einem APS bestimmt werden. Kann nachgewiesen werden, dass die mit einem Kaskaden-Impaktor und einem alternativen Messgerät erzielten Ergebnisse gleichwertig sind, so kann das alternative Instrument während der gesamten Studie verwendet werden. Parallel zum Hauptinstrument ist ein zweites Gerät wie ein Gravimetriefilter oder eine Gaswaschflasche zu verwenden, um den Abscheidegrad des Hauptinstruments zu bestätigen. Die durch die Partikelgrößenanalyse bestimmte Massenkonzentration sollte innerhalb vertretbarer Grenzen um die durch die Filteranalyse bestimmte Massenkonzentration liegen [siehe DG 39 (8)]. Wenn die Gleichwertigkeit zu Beginn der Studie nachgewiesen werden kann, kann auf weitere bestätigende Messungen verzichtet werden. Aus Tierschutzgründen sollten Vorkehrungen getroffen werden, um unklare Daten zu minimieren, die dazu führen könnten, dass eine Exposition wiederholt werden muss. Wenn die Möglichkeit besteht, dass Dampfkondensation zur Bildung eines Aerosols führen kann, oder wenn in einer Dampfatmosphäre mit dem Potenzial für gemischte Phasen Partikel nachgewiesen werden, sollte eine Partikelgrößenbestimmung für Dämpfe vorgenommen werden (siehe Nummer 14).
Hauptversuch
25. Für jede Stufe werden drei Tiere je Geschlecht oder sechs Tiere des empfindlicheren Geschlechts verwendet. Wenn für eine "Nose-only"-Exposition andere Nagetierarten als Ratten verwendet werden, kann die maximale Expositionsdauer angepasst werden, um artenspezifisches Leiden zu minimieren. Die als Startdosis zu verwendende Konzentrationsstufe wird aus vier festen Dosiswerten gewählt; dabei sollte die Stufe gewählt werden, bei der die Wahrscheinlichkeit, dass sie bei einigen der behandelten Tiere Toxizität hervorruft, am größten ist. Die Prüfschemata für Gase, Dämpfe und Aerosole (Anlagen 2, 3 und 4) veranschaulichen die Prüfung mit den Berücksichtigungsgrenzwerten der GHS-Kategorien 1-4 (9) für Gase (100, 500, 2.500, 20.000 ppm/4 Std.) (Anlage 2), für Dämpfe (0,5, 2, 10, 20 mg/l/4 Std.) (Anlage 3) und für Aerosole (0,05, 0,5, 1, 5 mg/l/4 Std.) (Anlage 4). Kategorie 5, die in der Verordnung (EG) Nr. 1272/2008 (9) nicht umgesetzt ist, bezieht sich auf Konzentrationen über den jeweiligen Grenzkonzentrationen. Für jede Startkonzentration gilt das entsprechende Prüfschema. Der Verlauf des Prüfverfahrens folgt je nach Anzahl der tierschutzgerecht getöteten oder gestorbenen Versuchstiere den angegebenen Pfeilen, bis eine Einstufung vorgenommen werden kann.
26. Der Zeitabstand zwischen den einzelnen Expositionsgruppen richtet sich nach dem Einsetzen, der Dauer und dem Schweregrad der toxischen Zeichen. Die Tiere sollten erst dann der nächsten Konzentrationsstufe ausgesetzt werden, wenn mit angemessener Gewissheit vom Überleben der zuvor getesteten Tiere ausgegangen werden kann. Es wird empfohlen, zwischen den Versuchen mit den einzelnen Konzentrationsstufen einen Abstand von drei bis vier Tagen einzuhalten, damit eine verzögerte Toxizität festgestellt werden kann. Dieser Zeitabstand kann nach Bedarf angepasst werden, wenn z.B. die beobachteten Reaktionen keine Schlussfolgerungen zulassen.
Limit-Test
27. Der Limit-Test wird verwendet, wenn bekannt oder zu erwarten ist, dass die Prüfsubstanz praktisch nicht toxisch ist, d. h. nur über der regulatorisch festgelegten Grenzkonzentration eine toxische Wirkung hervorruft. Informationen über die Toxizität der Prüfsubstanz können aus Kenntnissen über ähnliche geprüfte Stoffe oder ähnliche Mischungen gewonnen werden, wobei Art und prozentualer Anteil der Komponenten zu berücksichtigen sind, deren toxikologische Relevanz bekannt ist. In den Fällen, in denen nur wenige oder keine Informationen über die Toxizität der Prüfsubstanz vorliegen oder in denen von einer Toxizität der Prüfsubstanz ausgegangen wird, sollte der Hauptversuch durchgeführt werden [weitere Hinweise sind GD 39 (8) zu entnehmen].
28. Bei der normalen Vorgehensweise werden je drei Tiere beider Geschlechter oder sechs Tiere des empfindlicheren Geschlechts Konzentrationen von 20.000 ppm im Fall von Gasen, 20 mg/l im Fall von Dämpfen und 5 mg/l im Fall von Stäuben/Nebel ausgesetzt. Wenn diese Konzentrationen erreicht werden, dienen sie als Limit-Test für diese Prüfmethode. Bei der Prüfung von Aerosolen sollte das Hauptziel darin bestehen, eine lungengängige Partikelgröße (d. h. ein MMAD von 1-4 pm) zu erreichen. Dies ist bei den meisten Prüfsubstanzen bei einer Konzentration von 2 mg/l der Fall. Aerosole sollten nur dann bei mehr als 2 mg/l geprüft werden, wenn eine lungengängige Partikelgröße erreicht werden kann [siehe GD 39 (8)]. In Übereinstimmung mit dem GHS (16) wird aus Tierschutzgründen von Prüfungen über einer Grenzkonzentration abgeraten. Prüfungen in der GHS- Kategorie 5 (16), die in der Verordnung (EG) Nr. 1272/2008 (9) nicht umgesetzt ist, sollten nur dann in Betracht gezogen werden, wenn es sehr wahrscheinlich ist, dass die Ergebnisse einer solchen Prüfung von unmittelbarer Relevanz für den Schutz der menschlichen Gesundheit sind. Im Studienbericht ist eine Begründung anzugeben. Bei potenziell explosiven Prüfsubstanzen ist darauf zu achten, dass keine explosionsfördernden Bedingungen geschaffen werden. Um eine unnötige Verwendung von Versuchstieren zu vermeiden und sicherzustellen, dass die Kammerbedingungen für einen Limit-Test erreicht werden können, sollte vor dem Limit-Test ein Probedurchlauf ohne Tiere vorgenommen werden.
29. Die Tiere sollten während der Exposition häufig auf klinische Zeichen beobachtet werden. Nach der Exposition sollten klinische Beobachtungen mindestens zweimal am Tag der Exposition oder, falls es aufgrund der Reaktion der Tiere auf die Behandlung angezeigt erscheint, häufiger und danach für einen Zeitraum von insgesamt 14 Tagen mindestens einmal täglich vorgenommen werden. Die Länge des Beobachtungszeitraums ist nicht festgelegt; sie sollte nach Art und Zeitpunkt des Einsetzens klinischer Zeichen und der Länge der Erholungsphase bestimmt werden. Der Zeitpunkt, zu dem die Toxizitätszeichen auftreten und wieder abklingen, ist von Bedeutung, insbesondere dann, wenn Anzeichen für ein verzögertes Auftreten von Toxizitätszeichen erkennbar sind. Sämtliche Beobachtungen werden systematisch in Einzelprotokollen dokumentiert, die für jedes Tier geführt werden. Tiere, bei denen ein moribunder Zustand festgestellt wird, sowie Tiere, die starke Schmerzen haben oder anhaltende Anzeichen von schwerem Leiden zeigen, sollten aus Tierschutzgründen getötet werden. Bei den Untersuchungen auf klinische Toxizitätszeichen ist darauf zu achten, dass ein anfänglich schlechtes Aussehen und vorübergehende Atemveränderungen, die auf das Expositionsverfahren zurückzuführen sind, nicht mit behandlungsbedingten Wirkungen verwechselt werden. Die im Humane Endpoints Guidance Document (7) zusammengefassten Prinzipien und Kriterien sind zu berücksichtigen. Wenn Tiere aus Tierschutzgründen getötet werden oder ihr Tod festgestellt wird, sollte der Todeszeitpunkt so genau wie möglich registriert werden.
30. Bei den Beobachtungen ist auf Veränderungen von Haut, Fell, Augen und Schleimhäuten sowie Atmung, Kreislauf, autonomes und zentrales Nervensystem, Somatomotorik und Verhaltensmuster zu achten. Soweit möglich, ist auf Differenzierungen zwischen lokalen und systemischen Wirkungen zu achten. Besonderes Augenmerk ist auf Tremor, Konvulsionen, Salivation, Diarrhö, Lethargie, Schlaf und Koma zu richten. Die Messung der Rektaltemperatur kann zusätzliche Belege für mit der Behandlung oder Unterbringung zusammenhängende Reflex- Bradypnoe oder Hypo-/Hyperthermie liefern.
Körpergewicht
31. Das Körpergewicht der einzelnen Tiere sollte einmal während der Eingewöhnungszeit, am Tag der Exposition vor der Exposition (Tag 0) und mindestens an den Tagen 1, 3 und 7 (und danach wöchentlich) sowie zum Zeitpunkt des Todes oder der Tötung, falls später als Tag 1, dokumentiert werden. Das Körpergewicht gilt als kritischer Indikator für Toxizität; Tiere, die gegenüber ihrem Gewicht vor der Prüfung eine dauerhafte Abnahme um ≥ 20 % aufweisen, sollten sorgfältig überwacht werden. Die überlebenden Tiere werden gewogen und am Ende der Post-Expositionsphase auf tierschutzgerechte Weise getötet.
Pathologie
32. Alle Versuchstiere (einschließlich der Tiere, die während des Tests sterben oder aus Tierschutzgründen getötet und aus der Studie genommen werden) sind auf makroskopische Veränderungen zu untersuchen. Kann die Nekropsie nicht unmittelbar nach Auffinden eines toten Tieres erfolgen, sollte der Körper auf eine Temperatur gekühlt (nicht eingefroren) werden, die tief genug ist, um die Autolyse zu minimieren. Die Nekropsie ist baldmöglichst, in der Regel innerhalb von einem oder zwei Tagen durchzuführen. Alle makroskopischen Veränderungen sollten für jedes Tier protokolliert werden, wobei besonders auf Veränderungen der Atemwege zu achten ist.
33. Es kann in Betracht gezogen werden, von vorneherein zusätzliche Untersuchungen in die Studienauslegung aufzunehmen, die die Aussagekraft der Studie erhöhen, z.B. die Messung des Lungengewichts überlebender Ratten und/oder den Nachweis einer Reizwirkung durch mikroskopische Untersuchung der Atemwege. Untersucht werden können auch diejenigen Organe, die bei 24 Stunden oder länger überlebenden Tieren makroskopische Befunde aufweisen, sowie Organe, die bekanntermaßen oder vermutlich betroffen sind. Eine mikroskopische Untersuchung des gesamten Atemtrakts kann nützliche Informationen über Prüfsubstanzen liefern, die mit Wasser reagieren, z.B. Säuren und hygroskopische Prüfsubstanzen.
Daten
34. Das Körpergewicht der einzelnen Tiere und Sektionsbefunde sollten angegeben werden. Die Daten der klinischen Beobachtung sollten in tabellarischer Form zusammengefasst werden. Daraus müssen für jede Prüfgruppe die Anzahl der verwendeten Tiere, die Anzahl der Tiere mit spezifischen Toxizitätszeichen, die Anzahl der Tiere, die während der Prüfung tot aufgefunden oder vorzeitig getötet wurden, der Todeszeitpunkt der einzelnen Tiere, eine Beschreibung und der zeitliche Verlauf der toxischen Wirkungen und deren Reversibilität sowie die Sektionsbefunde ersichtlich sein.
Prüfbericht
35. Der Prüfbericht sollte, soweit zutreffend, die folgenden Informationen enthalten:
Versuchstiere und Tierhaltung
- Beschreibung der Haltungsbedingungen mit Angaben zu Anzahl (oder Veränderung der Anzahl) der Tiere je Käfig, Einstreu, Umgebungstemperatur und relativer Luftfeuchtigkeit, Photoperiode und Futter,
- Art/Stamm und Begründung für die Verwendung einer anderen Art als der Ratte,
- Anzahl, Alter und Geschlecht der Tiere,
- Randomisierungsmethode,
- Angaben über Futter- und Wasserqualität (einschließlich Art/Herkunft des Futters, Wasserquelle),
- Beschreibung etwaiger Vorbereitung vor der Prüfung, einschließlich Ernährung, Quarantäne und Behandlung von Krankheiten.
Prüfsubstanz
- physikalische Beschaffenheit, Reinheit und, wenn maßgeblich, physikalisch-chemische Eigenschaften (einschließlich Isomerisierung),
- Angaben zur Identifikation und CAS-Nummer (Chemical Abstract Services), falls bekannt.
Vehikel
- Begründung für die Verwendung eines Vehikels sowie für die Wahl des Vehikels (falls nicht Wasser),
- historische oder parallel erzeugte Daten, die belegen, dass das Vehikel keinen Einfluss auf das Ergebnis der Studie hat.
Inhalationskammer
- Beschreibung der Inhalationskammer mit Angabe von Abmessungen und Volumen,
- Herkunft und Beschreibung der für die Exposition der Tiere sowie für die Erzeugung der Atmosphäre verwendeten Ausrüstung,
- Ausrüstung für die Messung von Temperatur, Luftfeuchtigkeit, Partikelgröße und tatsächlicher Konzentration,
- Herkunft der Luft, Behandlung der eingeleiteten/entzogenen Luft sowie Klimatisierungssystem,
- für die Kalibrierung der Ausrüstung verwendete Methoden, um eine homogene Prüfatmosphäre sicherzustellen,
- Druckunterschied (positiv oder negativ),
- Expositions-Öffnungen je Kammer ("Nose-only"-Exposition); Anordnung der Tiere im System (Ganzkörper),
- zeitliche Homogenität/Stabilität der Prüfatmosphäre,
- Lage von Temperatur- und Feuchtigkeitssensoren und Ort der Probenahme der Prüfatmosphäre in der Kammer,
- Luftdurchflussraten, Luftdurchflussrate/Expositions-Öffnung ("Nose-only"-Exposition) oder Anzahl der Tiere je Kammer (Ganzkörperexposition),
- Angaben zur Ausrüstung für die Messung des Sauerstoff- und Kohlendioxidgehalts, falls zutreffend,
- bis zum Erreichen des Gleichgewichts in der Inhalationskammer benötigte Zeit (t95),
- Zahl der Volumenänderungen pro Stunde,
- Messgeräte (falls zutreffend).
Expositionsdaten
- Begründung für die Wahl der Zielkonzentration in der Hauptstudie,
- nominale Konzentrationen (Gesamtmasse der in die Inhalationskammer eingeleiteten Prüfsubstanz dividiert durch das Volumen der durch die Kammer geleiteten Luft),
- im Atembereich der Tiere ermittelte tatsächliche Konzentrationen der Prüfsubstanz; bei Prüfgemischen mit heterogenen physikalischen Formen (Gase, Dämpfe, Aerosole) kann jede Form getrennt analysiert werden,
- alle Luftkonzentrationen sollten in Masseneinheiten angegeben werden (z.B. mg/l, mg/m3 usw.), Volumeneinheiten können in Klammern ebenfalls angegeben werden (z.B. ppm, ppb),
- Partikelgrößenverteilung, mittlerer aerodynamischer Massendurchmesser (MMAD) und geometrische Standardabweichung (σg), einschließlich der Berechnungsmethoden. Einzelne Partikelgrößenanalysen sind zu protokollieren.
Prüfbedingungen
- Angaben zur Vorbereitung der Prüfsubstanz, einschließlich Angaben zu den Verfahren zur Reduzierung der Partikelgröße von Feststoffen oder zur Herstellung von Lösungen der Prüfsubstanz. Wenn die Zusammensetzung der Prüfsubstanz durch mechanische Prozesse verändert worden sein kann, sind die Ergebnisse der Analysen zur Überprüfung der Zusammensetzung der Prüfsubstanz beizufügen.
- Eine Beschreibung (möglichst mit Diagramm) der Ausrüstung, die zur Erzeugung der Prüfatmosphäre und zur Exposition der Tiere gegen die Prüfatmosphäre verwendet wurde.
- Angaben zur verwendeten chemischen Analysemethode und zur Validierung der Methode (einschließlich der Effizienz der Wiederfindung der Prüfsubstanz im Medium).
- Begründung für die Wahl der Prüfkonzentrationen.
Ergebnisse
- tabellarische Darstellung von Temperatur, Luftfeuchtigkeit und Luftstrom in der Kammer,
- tabellarische Darstellung von Daten zur nominalen und tatsächlichen Konzentration in der Kammer,
- tabellarische Darstellung der Partikelgrößendaten einschließlich Daten zur analytischen Probenahme, Partikelgrößenverteilung und Berechnung des MMAD und der σg,
- tabellarische Erfassung der erhobenen Daten und der Konzentration für jedes einzelne Tier (d. h. Tiere, die Anzeichen für Toxizität zeigen, einschließlich Mortalität sowie Art, Schweregrad und Dauer der Wirkungen),
- Körpergewicht der einzelnen Tiere, erfasst an Prüfungstagen, Datum und Zeitpunkt des Todes, falls vor geplanter Tötung, zeitlicher Verlauf des Einsetzens von Toxizitätsanzeichen sowie Angabe, ob diese reversibel waren, für jedes einzelne Tier,
- Sektionsbefunde und histopathologische Ergebnisse für jedes einzelne Tier, falls vorhanden,
- GHS-Kategorieeinstufung und LC50-Berücksichtigungsgrenzwert.
Diskussion und Auswertung der Ergebnisse
- Besondere Aufmerksamkeit sollte der Beschreibung der Methoden gelten, die verwendet wurden, um die Kriterien dieser Prüfmethode zu erfüllen, z.B. die Grenzkonzentration oder die Partikelgröße.
- Die Lungengängigkeit von Partikeln vor dem Hintergrund der Gesamtbefunde sollte behandelt werden, insbesondere, wenn die Partikelgrößenkriterien nicht erfüllt werden konnten.
- Die Kohärenz der Methoden zur Bestimmung der nominalen und der tatsächlichen Konzentration sowie die Beziehung der tatsächlichen Konzentration zur nominalen Konzentration sind in die Gesamtbewertung der Studie aufzunehmen.
- Die wahrscheinliche Todesursache und die vorherrschende Wirkungsweise (systemisch oder lokal) sollten behandelt werden.
- Falls Tiere, die unter Schmerzen litten oder Zeichen für schweres und anhaltendes Leiden aufwiesen, auf tierschutzgerechte Weise getötet werden mussten, ist eine Erklärung auf der Grundlage der Kriterien im OECD Guidance Document on Humane Endpoints (7) zu geben.
1. Kapitel B.2 dieses Anhangs, Akute Inhalationstoxizität.
2. Holzhütter H-G, Genschow E, Diener W, and Schlede E (2003). Dermal and Inhalation Acute Toxicity Class Methods: Test Procedures and Biometric Evaluations for the Globally Harmonized Classification System. Arch. Toxicol. 77: 243-254.
3. Diener W, Kayser D and Schlede E (1997). The Inhalation Acute-Toxic-Class Method; Test Procedures and Biometric Evaluations. Arch. Toxicol. 71: 537-549.
4. Diener W and Schlede E (1999). Acute Toxic Class Methods: Alternatives to LD/LC 50 Tests. ALTEX 1: 129-134.
5. Kapitel B.1.tris dieses Anhangs, Akute orale Toxizität - Akut toxische Klassenmethode.
6. OECD (2009). Report on Biostatistical Performance Assessment of the Draft TG 436 Acute Toxic Class Testing Method for Acute Inhalation Toxicity. Environmental Health and Safety Monograph Series on Testing and Assessment No. 105, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
7. OECD (2000). Guidance Document on the Recognition, Assessment and Use of Clinical Signs as Humane Endpoints for Experimental Animals Used in Safety Evaluation. Environmental Health and Safety Monograph Series on Testing and Assessment No. 19. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
8. OECD (2009). Guidance Document on Acute Inhalation Toxicity Testing. Environmental Health and Safety Monograph Series on Testing and Assessment No. 39, OECD, Paris. Abrufbar unter: [http://www.oecd.org/ env/testguidelines]
9. Verordnung (EG) Nr. 1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006 (ABl. Nr. L 353 vom 31.12.2008 S. 1).
10. Kapitel B.40 dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: TER-Test (transcutaneous electrical resistance test).
11. Kapitel B.40.bis dieses Anhangs, In-vitro-Prüfung auf hautätzende Wirkung: Test mit menschlichem Hautmodell.
12. OECD (2005). In Vitro Membrane Barrier Test Method for Skin Corrosion. OECD Guideline for testing of chemicals No. 435, OECD, Paris. Abrufbar unter: [http://www.oecd.org/env/testguidelines]
13. Phalen RF (2009). Inhalation Studies: Foundations and Techniques. (2nd Edition) Informa Healthcare, New York.
14. SOT (1992). Technical Committee of the Inhalation Specialty Section, Society of Toxicology (SOT). Recommendations for the Conduct of Acute Inhalation Limit Tests. Fund. Appl. Toxicol. 18: 321-327.
15. Pauluhn J and Thiel A (2007). A Simple Approach to Validation of Directed-Flow Nose-Only Inhalation Chambers. J. Appl. Toxicol. 27: 160-167
16. UN (2007), United Nations Globally Harmonized System of Classification and Labelling of Chemicals (GHS), ST/SG/AC.10/30, UN New York and Geneva. Abrufbar unter: [http://www.unece.org/trans/danger/publi/ghs/ghs_welcome_e.html]
____
1) Die Bewertung der Ätzwirkung könnte auf dem Urteil von Experten beispielsweise anhand folgender Belege beruhen: Erfahrungswerte bei Mensch und Tier, vorliegende (In-vitro-)Daten, z.B. Kapitel B.40 (10), B.40.bis (11) dieses Anhangs oder OECD TG 435 (12), pH- Werte sowie Informationen über ähnliche Substanzen und andere sachdienliche Daten.
| Definition | Anlage 1 |
Prüfsubstanz: jeder Stoff oder jedes Gemisch, der/das mit dieser Prüfmethode getestet wird.
| Verfahrensweise für jede Startkonzentration von Gasen (ppm/4 Std.) | Anlage 2 |
Allgemeine Hinweise 1
Die Prüfschemata in dieser Anlage geben für jede Startkonzentration die jeweils zu beachtende Vorgehensweise an.
Anlage 2a: Startkonzentration 100 ppm
Anlage 2b: Startkonzentration 500 ppm
Anlage 2c: Startkonzentration 2.500 ppm
Anlage 2d: Startkonzentration 20.000 ppm
Der Testverlauf folgt je nach Anzahl der tierschutzgerecht getöteten oder gestorbenen Versuchstiere den angegebenen Pfeilen.
______
1) In den folgenden Tabellen wird auf das GHS (Global Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien verwiesen.
Die entsprechende Rechtsvorschrift in der EU ist die Verordnung (EG) Nr. 1272/2008. Im Fall der akuten Inhalationstoxizität wird die Kategorie 5 in der Verordnung (EG) Nr. 1272/2008 (9) nicht umgesetzt.
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 100 ppm/4 Std. für Gase | Anlage 2a |
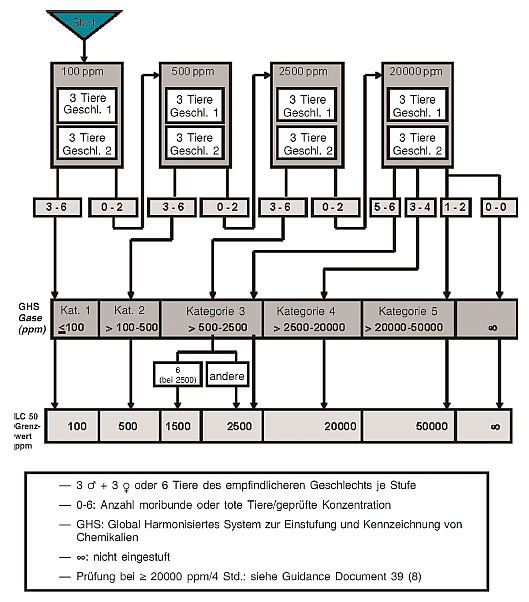
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 500 ppm/4 Std. für Gase | Anlage 2b |
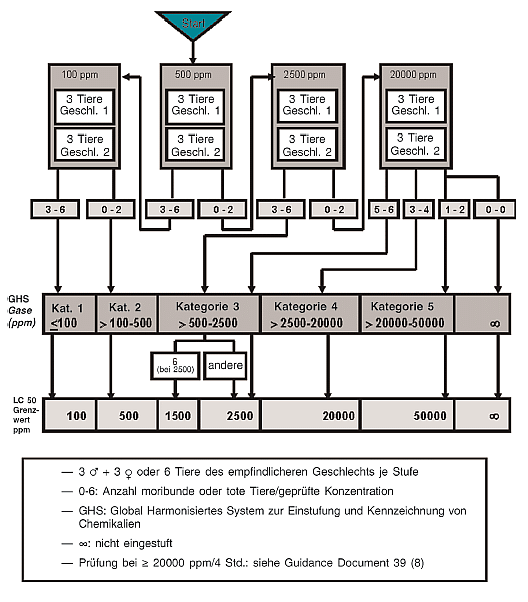
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 2.500 ppm/4 Std. für Gase | Anlage 2c |
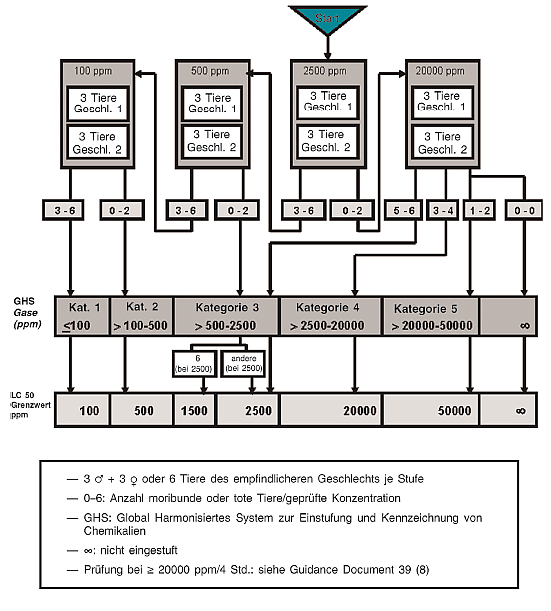
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 20.000 ppm/4 Std. für Gase | Anlage 2d |
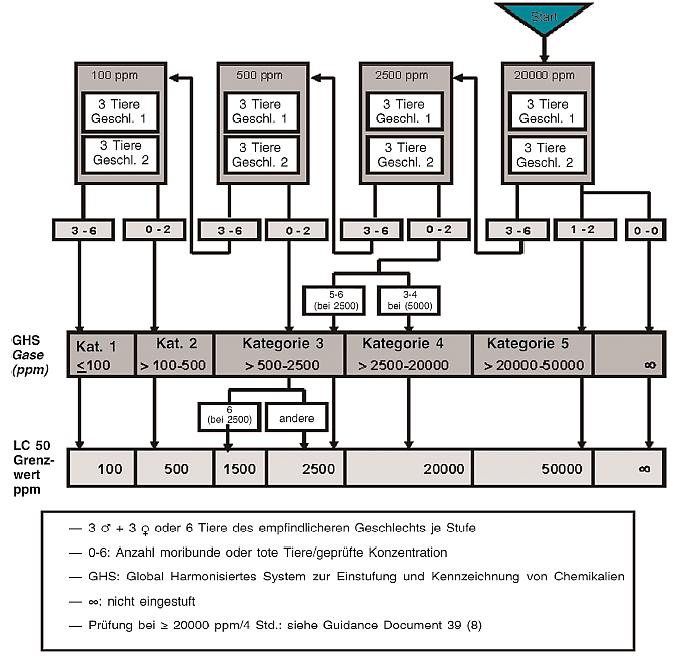
| Verfahrensweise für jede Startkonzentration von Dämpfen (mg/l/4 Std.) | Anlage 3 |
Allgemeine Hinweise 1
Die Prüfschemata in dieser Anlage geben für jede Startkonzentration die jeweils zu beachtende Vorgehensweise an.
Anlage 3a: Startkonzentration 0,5 mg/l
Anlage 3b: Startkonzentration 2,0 mg/l
Anlage 3c: Startkonzentration 10 mg/l
Anlage 3d: Startkonzentration 20 mg/l
Der Testverlauf folgt je nach Anzahl der tierschutzgerecht getöteten oder gestorbenen Versuchstiere den angegebenen Pfeilen.
______
1) In den folgenden Tabellen wird auf das GHS (Global Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien verwiesen.
Die entsprechende Rechtsvorschrift in der EU ist die Verordnung (EG) Nr. 1272/2008. Im Fall der akuten Inhalationstoxizität wird die Kategorie 5 in der Verordnung (EG) Nr. 1272/2008 (9) nicht umgesetzt.
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 0,5 mg/l/4 Std. für Dämpfe | Anlage 3a |
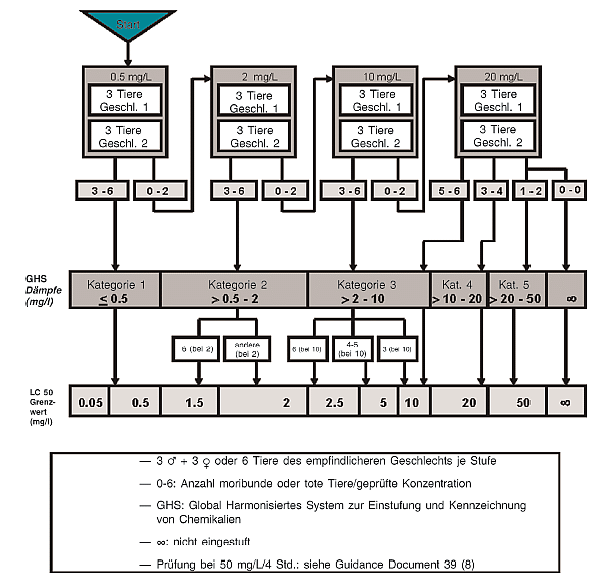
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 2 mg/l/4 Std. für Dämpfe | Anhang 3b |
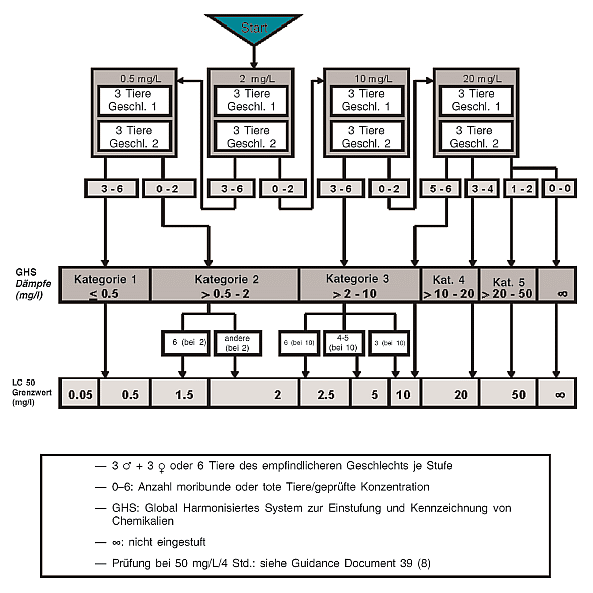
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 10 mg/l/4 Std. für Dämpfe | Anlage 3c |
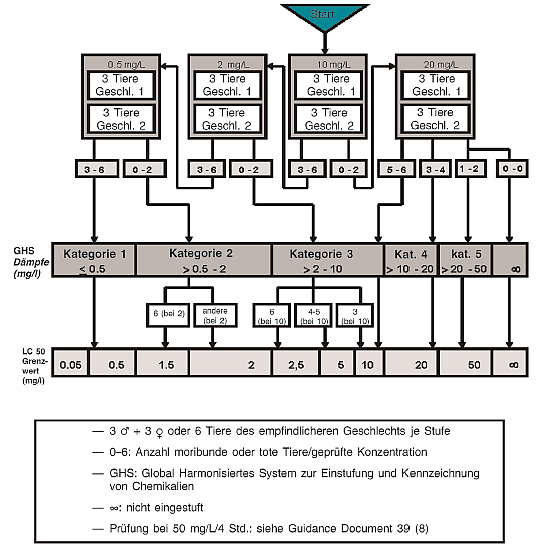
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 20 mg/l/4 Std. für Dämpfe | Anlage 3d |
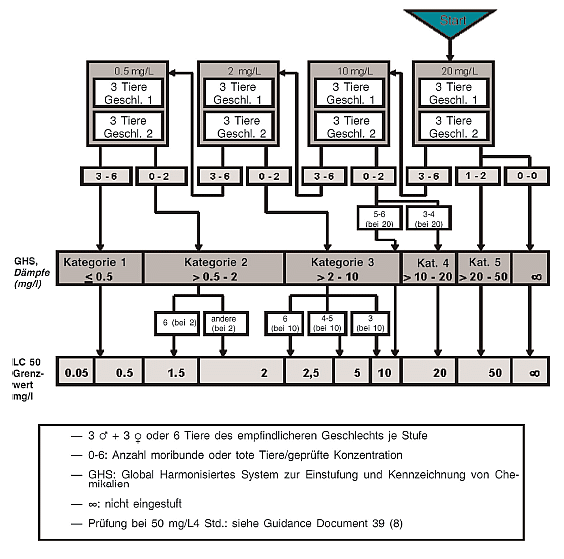
| Verfahrensweise für jede Startkonzentration von Aerosolen (mg/l/4 Std.) | Anlage 4 |
Allgemeine Hinweise 1
Die Prüfschemata in dieser Anlage geben für jede Startkonzentration die jeweils zu beachtende Vorgehensweise an.
Anlage 4a: Startkonzentration 0,05 mg/l
Anlage 4b: Startkonzentration 0,5 mg/l
Anlage 4c: Startkonzentration 1 mg/l
Anlage 4d: Startkonzentration 5 mg/l
Der Testverlauf folgt je nach Anzahl der tierschutzgerecht getöteten oder gestorbenen Versuchstiere den angegebenen Pfeilen.
_______
1) In den folgenden Tabellen wird auf das GHS (Global Harmonisiertes System zur Einstufung und Kennzeichnung von Chemikalien verwiesen.
Die entsprechende Rechtsvorschrift in der EU ist die Verordnung (EG) Nr. 1272/2008. Im Fall der akuten Inhalationstoxizität wird die Kategorie 5 in der Verordnung (EG) Nr. 1272/2008 (9) nicht umgesetzt.
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 0.05 mg/l/4 Std. für Dämpfe | Anlage 4a |
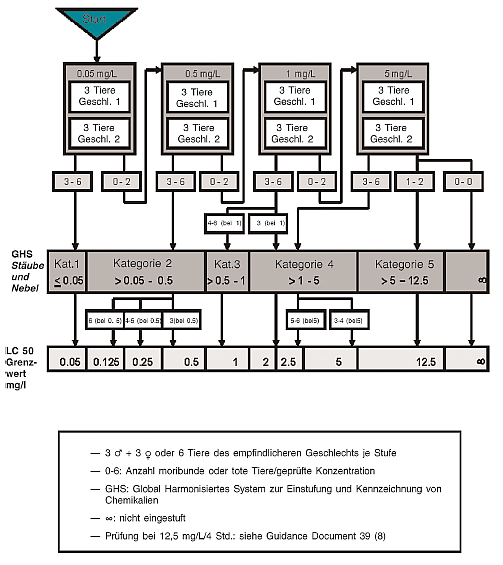
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 0,5 mg/l/4Std. für Aerosole | Anlage 4b |

| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 1 mg/l/4 Std. für Aerosole | Anlage 4c |
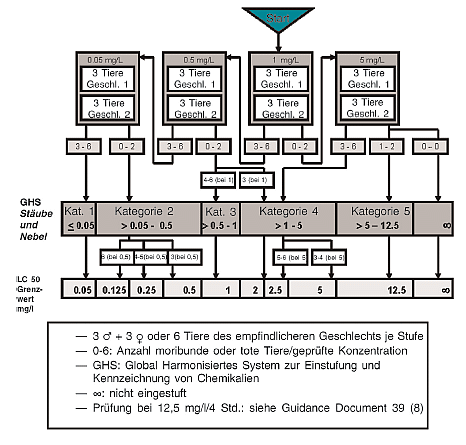
| Akute Inhalationstoxizität: Prüfverfahren mit einer Ausgangskonzentration von 5 mg/l/4 Std. für Aerosole | Anlage 4d |
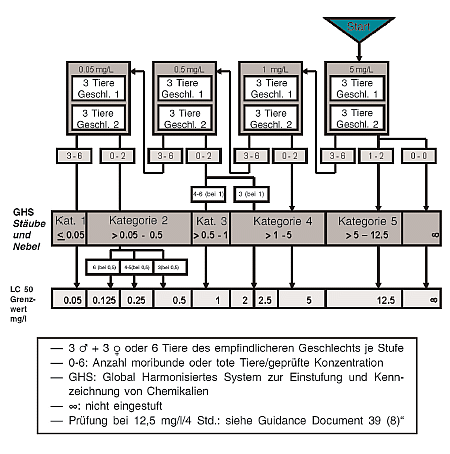
| weiter . |  |
...
X
⍂
↑
↓