

| zurück | |
| Analysemethoden zur Untersuchung von Futtermitteln auf unerwünschte Stoffe | Anhang V 24 |
A. Bestimmung des Gehalts an Dioxinen (PCDD/PCDF) und PCB
Kapitel I
Probenahmeverfahren und Auswertung von Analyseergebnissen
1. Zweck und Anwendungsbereich
Die Proben für die amtliche Untersuchung des Gehalts an polychlorierten Dibenzo-p-dioxinen (PCDD), polychlorierten Dibenzofuranen (PCDF) sowie dioxinähnlichen polychlorierten Biphenylen (PCB) 1 und nicht dioxinähnlichen PCB in Futtermitteln werden nach den in Anhang I beschriebenen Verfahren genommen. Hinsichtlich der Untersuchung auf Stoffe oder Erzeugnisse, die in Futtermitteln gleichmäßig verteilt sind, gelten die quantitativen Anforderungen gemäß Anhang I Nummer 5.1. Die mit diesem Verfahren gewonnenen Sammelproben sind als repräsentativ für die betreffenden Partien oder Teilpartien anzusehen. Anhand der bei den Laborproben bestimmten Gehalte wird festgestellt, ob die in der Richtlinie 2002/32/EG festgesetzten Höchstgehalte eingehalten wurden.
Für die Zwecke dieses Teils gelten die Begriffsbestimmungen in Anhang I der Durchführungsverordnung (EU) 2021/808 der Kommission 2.
Darüber hinaus gelten für die Zwecke dieses Teils folgende Begriffsbestimmungen:
| "Screening-Verfahren" sind Verfahren, die zur Auswahl derjenigen Proben genutzt werden, deren Gehalt an PCDD/F und dioxinähnlichen PCB die Höchstgehalte oder die Aktionsgrenzwerte überschreitet. Sie müssen auf kostengünstige Weise einen hohen Probendurchsatz erlauben, was die Chancen erhöht, neue Vorfälle mit hoher Exposition und Gesundheitsgefahren für die Verbraucher aufzudecken. Screening-Verfahren beruhen auf bioanalytischen oder GC-MS-Verfahren. Ergebnisse von Proben, in denen der Cut-off-Wert für die Überprüfung der Konformität mit dem Höchstgehalt überschritten wird, sind durch eine erneute vollständige Analyse der ursprünglichen Probe mittels eines Bestätigungsverfahrens zu überprüfen. |
| "Bestätigungsverfahren" sind Verfahren, die vollständige oder ergänzende Daten liefern, mit denen die PCDD/F und dioxinähnlichen PCB am Höchstgehalt oder erforderlichenfalls am Aktionsgrenzwert eindeutig identifiziert und quantifiziert werden können. Bei diesen Verfahren kommen Gaschromatografie/hochauflösende Massenspektrometrie (GC-HRMS) oder Gaschromatografie/Tandem-Massenspektrometrie (GC-MS/MS) zum Einsatz. |
2. Übereinstimmung der Partie bzw. Teilpartie mit den Anforderungen in Bezug auf die Höchstgehalte
2.1. Nicht dioxinähnliche PCB
Die Partie oder Teilpartie entspricht den Anforderungen in Bezug auf den Höchstgehalt, wenn das Ergebnis der Untersuchung für die Summe von PCB 28, PCB 52, PCB 101, PCB 138, PCB 153 und PCB 180 (im Folgenden "nicht dioxinähnliche PCB") den in der Richtlinie 2002/32/EG festgelegten Höchstgehalt unter Berücksichtigung der erweiterten Messunsicherheit nicht überschreitet 3. Die Partie oder Teilpartie entspricht nicht den Anforderungen in Bezug auf den Höchstgehalt gemäß der Richtlinie 2002/32/EG, wenn das Mittel der durch Zweitanalyse 4 ermittelten Obergrenzen ("upper-bound") 5 zweier Untersuchungsergebnisse unter Berücksichtigung der erweiterten Messunsicherheit den Höchstgehalt zweifelsfrei überschreitet, d. h. zur Beurteilung, ob die Höchstgehalte eingehalten werden, wird die gemessene Konzentration nach Abzug der erweiterten Messungenauigkeit herangezogen.
Die erweiterte Messunsicherheit wird mithilfe eines Erweiterungsfaktors von 2 berechnet, der zu einem Grad des Vertrauens von ca. 95 % führt. Eine Partie oder Teilpartie entspricht nicht den Vorgaben, wenn das Mittel der gemessenen Werte abzüglich der erweiterten Messunsicherheit des Mittels über dem Höchstgehalt liegt.
Die in den vorstehenden Absätzen unter dieser Nummer genannten Bestimmungen gelten für das Untersuchungsergebnis der für die amtliche Kontrolle entnommenen Probe. Im Falle einer Analyse für die Zwecke eines zweiten Sachverständigengutachtens oder für Referenzwecke gelten die einzelstaatlichen Bestimmungen.
2.2. Für PCDD/F und dioxinähnliche PCB
Die Partie oder Teilpartie entspricht den Anforderungen in Bezug auf die Höchstgehalte, wenn das Ergebnis einer einzelnen Untersuchung,
Für Screening-Assays ist ein Cut-off-Wert festzulegen, anhand dessen entschieden wird, ob eine Probe den jeweiligen Höchstgehalten für PCDD/F bzw. die Summe von PCDD/F und dioxinähnlichen PCB entspricht.
Die Partie oder Teilpartie entspricht nicht den Anforderungen in Bezug auf den Höchstgehalt gemäß der Richtlinie 2002/32/EG, wenn das Mittel der durch Zweitanalyse 6 mit einem Bestätigungsverfahren ermittelten Obergrenzen ("upper-bound") 7 zweier Untersuchungsergebnisse unter Berücksichtigung der erweiterten Messunsicherheit den Höchstgehalt zweifelsfrei überschreitet, d. h., zur Beurteilung, ob die Höchstgehalte eingehalten werden, wird die gemessene Konzentration nach Abzug der erweiterten Messungenauigkeit herangezogen.
Die erweiterte Messunsicherheit wird mithilfe eines Erweiterungsfaktors von 2 berechnet, der zu einem Grad des Vertrauens von ca. 95 % führt. Eine Partie oder Teilpartie entspricht nicht den Vorgaben, wenn das Mittel der gemessenen Werte abzüglich der erweiterten Messunsicherheit des Mittels über dem Höchstgehalt liegt.
Die Summe der geschätzten erweiterten Messunsicherheiten der getrennten Analyseergebnisse der PCDD/F und dioxinähnlichen PCB ist für die Summe der PCDD/F und dioxinähnlichen PCB zu verwenden.
Die in den vorstehenden Absätzen unter dieser Nummer genannten Bestimmungen gelten für das Untersuchungsergebnis der für die amtliche Kontrolle entnommenen Probe. Im Falle einer Analyse für Rechtfertigungs- oder Referenzwecke gelten die einzelstaatlichen Bestimmungen.
3. Ergebnisse, die die Aktionsgrenzwerte gemäß Anhang II der Richtlinie 2002/32/EG überschreiten
Aktionsgrenzwerte dienen als Instrument zur Auswahl der Proben in Fällen, in denen eine Kontaminationsquelle gefunden wird und Maßnahmen zu deren Eindämmung oder Beseitigung getroffen werden müssen. Durch Screening-Verfahren sind die geeigneten Cut-off-Werte für die Auswahl dieser Proben festzulegen. Wenn die Bestimmung einer Kontaminationsquelle und deren Eindämmung oder Beseitigung erhebliche Anstrengungen erfordert, ist es angezeigt, die Überschreitung der Aktionsgrenzwerte durch eine Zweitanalyse im Bestätigungsverfahren und unter Berücksichtigung der erweiterten Messunsicherheit zu bestätigen 8.
Kapitel II
Probenvorbereitung und Anforderungen an Untersuchungsverfahren zur amtlichen Kontrolle des Gehalts an Dioxinen (PCDD/F) und dioxinähnlichen PCB in Futtermitteln
1. Anwendungsbereich
Die in diesem Kapitel beschriebenen Anforderungen gelten, wenn Futtermittel zur amtlichen Kontrolle des Gehalts an 2,3,7,8-substituierten PCDD/F und dioxinähnlichen PCB sowie für die Probenvorbereitung und Untersuchungsanforderungen zu anderen regulatorischen Zwecken, darunter die Kontrollen der Futtermittelunternehmer zur Gewährleistung der Vorschriftsmäßigkeit gemäß der Verordnung (EG) Nr. 183/2005 des Europäischen Parlaments und des Rates 9, untersucht werden.
Die Überwachung von Futtermitteln auf Vorhandensein von PCDD/F und dioxinähnlichen PCB kann mit zwei unterschiedlichen Verfahrensarten erfolgen:
a) Screening-Verfahren
Ziel der Screening-Verfahren ist die Auswahl derjenigen Proben, deren Gehalt an PCCD/F und dioxinähnlichen PCB die Höchstgehalte oder die Aktionsgrenzwerte überschreitet. Screening-Verfahren sollen auf kostengünstige Weise einen hohen Probendurchsatz erlauben, was die Chancen erhöht, neue Vorfälle mit hoher Exposition und Gesundheitsgefahren für die Verbraucher aufzudecken. Ihre Anwendung hat insbesondere die Vermeidung falsch-negativer Ergebnisse zum Ziel. Sie können bioanalytische und GC-MS-Verfahren umfassen.
Bei Screening-Verfahren wird das Analyseergebnis mit einem Cut-off-Wert verglichen und angegeben, ob der Höchstgehalt oder Aktionsgrenzwert möglicherweise überschritten wird oder nicht. Die Konzentration von PCDD/F und der Summe von PCDD/F und dioxinähnlichen PCB in Proben, in denen eine Überschreitung des Höchstgehalts vermutet wird, muss durch ein Bestätigungsverfahren ermittelt oder bestätigt werden.
Darüber hinaus können Screening-Verfahren Hinweise auf den in der Probe vorhandenen Gehalt an PCDD/F und dioxinähnlichen PCB liefern. Bei bioanalytischen Screening-Verfahren wird das Ergebnis in bioanalytischen Äquivalenten (BEQ) und bei physikalisch-chemischen GC-MS-Verfahren in Toxizitätsäquivalenten (TEQ) ausgedrückt. Die in Zahlen ausgedrückten Ergebnisse von Screening-Verfahren sind geeignet, die Konformität bzw. die vermutete Nichtkonformität oder das Überschreiten von Aktionsgrenzwerten anzuzeigen und weisen außerdem für den Fall einer Weiterverfolgung mittels Bestätigungsverfahren auf die Gehaltsbereiche hin. Sie sind nicht für Zwecke wie die Bewertung von Hintergrundkonzentrationen, die Schätzung der Aufnahme, die Verfolgung der zeitlichen Entwicklung von Gehalten oder die Neubewertung von Aktionsgrenzwerten und Höchstgehalten geeignet.
b) Bestätigungsverfahren
Bestätigungsverfahren ermöglichen die eindeutige Identifizierung und Quantifizierung von in einer Probe vorhandenen PCDD/F und dioxinähnlichen PCB und liefern vollständige Informationen über den Gehalt der einzelnen Kongenere. Sie erlauben somit die Kontrolle von Höchstgehalten und Aktionsgrenzwerten, einschließlich der Bestätigung von mittels Screening-Verfahren erzielten Ergebnissen. Außerdem können die Ergebnisse für weitere Zwecke genutzt werden, wie die Bestimmung der Belastung im niedrigen Hintergrundbereich bei der Futtermittelüberwachung, die Verfolgung der zeitlichen Entwicklung, die Expositionsbewertung und für den Aufbau einer Datenbank im Hinblick auf eine mögliche Neubewertung der Aktionsgrenzwerte und Höchstgehalte. Sie sind außerdem bei der Feststellung von Kongeneren-Mustern zur Bestimmung der Quelle einer möglichen Kontamination von Bedeutung. Solche Verfahren verwenden GC-HRMS. Zur Bestätigung der Konformität oder Nichtkonformität mit dem Höchstgehalt kann auch die GC-MS/MS angewendet werden.
2. Hintergrund
Zur Berechnung der TEQ-Konzentrationen werden die Konzentrationen der einzelnen Stoffe in einer bestimmten Probe mit den jeweiligen Toxizitätsäquivalenzfaktoren (TEF) (siehe Fußnote 1 in Kapitel I) multipliziert und dann addiert, woraus sich die Gesamtkonzentration an dioxinähnlichen Verbindungen, ausgedrückt in TEQ, ergibt.
Für die Zwecke dieses Teils A entspricht die akzeptierte spezifische Bestimmungsgrenze eines einzelnen Kongeners dem niedrigsten Analytgehalt, der sich mit angemessener statistischer Zuverlässigkeit quantifizieren lässt und die Identifizierungskriterien erfüllt, wie sie in international anerkannten Normen, z.B. in der Norm EN 16215:2012 (Futtermittel - Bestimmung von Dioxinen und dioxin-ähnlichen PCBs mittels GC/HRMS und von Indikator-PCBs mittels GC/HRMS) und/oder in den überarbeiteten EPA-Methoden 1613 und 1668, beschrieben sind.
Die Bestimmungsgrenze eines einzelnen Kongeners lässt sich bestimmen als
Mit bioanalytischen Methoden erhält man kein Ergebnis auf Ebene der Kongeneren, sondern lediglich einen Hinweis 10 auf den TEQ-Gehalt, der in BEQ ausgedrückt wird, wodurch der Tatsache Rechnung getragen wird, dass möglicherweise nicht alle in einem Probenextrakt vorliegenden Verbindungen, die ein Signal erzeugen, allen Voraussetzungen des TEQ-Prinzips genügen.
Screening- und Bestätigungsverfahren können nur dann zur Kontrolle einer bestimmten Matrix angewendet werden, wenn sie empfindlich genug sind, Gehalte im Bereich der Aktionsgrenzwerte oder Höchstgehalte zuverlässig zu bestimmen.
3. Anforderungen an die Qualitätssicherung
3.1. Auf jeder Stufe des Probenahme- und Analyseverfahrens sind Maßnahmen zur Vermeidung von Kreuzkontamination zu treffen.
3.2. Die Proben sind in hierfür geeigneten Glas-, Aluminium-, Polypropylen- oder Polyethylen-Behältnissen zu lagern und zu transportieren, sodass der Gehalt der Proben an PCDD/F und dioxinähnlichen PCB nicht verfälscht wird. Spuren von Papierstaub sind vom Probenbehälter zu entfernen.
3.3. Lagerung und Transport der Proben haben so zu erfolgen, dass die Futtermittelprobe unversehrt erhalten bleibt.
3.4. Sofern zutreffend, sind die einzelnen Laborproben mithilfe eines Verfahrens, mit dem nachweislich eine vollständige Homogenisierung erreicht wird, fein zu mahlen und gründlich zu mischen (z.B. so fein gemahlen, dass die Probe durch ein Sieb mit einer Maschenweite von 1 mm passt). Falls die Proben einen zu hohen Feuchtigkeitsgehalt aufweisen, sind sie vor dem Zermahlen zu trocknen.
3.5. Die Reagenzien, Glasgeräte und die weitere Ausrüstung sind auf Faktoren, die möglicherweise die TEQ- und BEQ-basierten Ergebnisse verfälschen könnten, zu kontrollieren.
3.6. Es ist eine Methodenleerwert-Untersuchung durchzuführen, bei der das gesamte Analyseverfahren durchgeführt und nur die Probe weggelassen wird.
3.7. Bei Anwendung bioanalytischer Methoden ist zu überprüfen, dass sämtliche Glasgeräte und Lösungsmittel, die bei der Analyse verwendet werden, frei von Verbindungen sind, die die Erkennung der Zielverbindungen im Arbeitsbereich beeinträchtigen könnten. Glasgeräte sind mit Lösungsmitteln zu spülen oder auf Temperaturen zu erhitzen, die geeignet sind, alle Spuren von PCDD/F, dioxinähnlichen Verbindungen und interferierenden Verbindungen von der Oberfläche der Geräte zu entfernen.
3.8. Die Menge der für die Extraktion verwendeten Probe muss ausreichend groß sein, um die Anforderungen hinsichtlich eines ausreichend niedrigen Arbeitsbereichs, der den Konzentrationsbereich der Höchstgehalte oder Aktionsgrenzwerte beinhaltet, zu erfüllen.
3.9. Zur spezifischen Vorbereitung der Proben der zu untersuchenden Erzeugnisse sind Verfahren gemäß international anerkannten Leitlinien, d. h. EN ISO 6498, anzuwenden.
4. Anforderungen an Laboratorien
4.1. Gemäß den Bestimmungen der Verordnung (EU) 2017/625 müssen die Laboratorien von einer anerkannten Stelle akkreditiert sein, die nach ISO/IEC Guide 58 arbeitet, damit sichergestellt ist, dass die Laboratorien bei der Untersuchung Qualitätssicherungsverfahren anwenden. Die Laboratorien müssen gemäß der Norm EN ISO/IEC 17025 akkreditiert sein. Es sind die in den technischen Leitlinien für die Schätzung der Messunsicherheit und der Bestimmungsgrenzen für die Untersuchung auf PCDD/F und PCB beschriebenen Grundsätze zu befolgen 11.
4.2. Die Laborleistung ist durch die kontinuierliche erfolgreiche Teilnahme an Laborvergleichsuntersuchungen zur Ermittlung des Gehalts an PCDD/F und dioxinähnlichen PCB in den entsprechenden Futtermittelmatrices und Konzentrationsbereichen unter Beweis zu stellen.
4.3. Laboratorien, die zur Routinekontrolle von Proben Screening-Verfahren anwenden, müssen eng mit Laboratorien zusammenarbeiten, die Bestätigungsverfahren anwenden, sowohl zur Qualitätssicherung als auch zur Bestätigung der Ergebnisse verdächtiger Proben.
5. Grundlegende Anforderungen an Verfahren zur Untersuchung auf Dioxine (PCDD/F) und dioxinähnliche PCB
5.1. Niedriger Arbeitsbereich und niedrige Bestimmungsgrenzen
Bei PCDD/F müssen die nachweisbaren Mengen wegen der extrem hohen Toxizität einiger dieser Verbindungen im oberen Femtogramm-Bereich (10-15g) liegen. Bei den meisten PCB-Kongeneren ist eine Bestimmungsgrenze im Bereich Nanogramm (10-9g) bereits ausreichend. Zur Messung der toxischeren dioxinähnlichen PCB-Kongenere (insbesondere der non-orthosubstituierten Kongenere) muss das untere Ende des Arbeitsbereichs bis in den unteren Pikogramm-Bereich (10-12g) reichen. Bei allen anderen PCB-Kongeneren ist eine Bestimmungsgrenze im Nanogramm-Bereich (10-9g) ausreichend.
5.2. Hohe Selektivität (Spezifizität)
5.2.1. PCDD/F und dioxinähnliche PCB müssen von einer Vielzahl anderer, gemeinsam extrahierter und möglicherweise interferierender Verbindungen unterschieden werden, die in Konzentrationen von bis zu mehreren Größenordnungen höher als diejenigen der interessierenden Analyten vorhanden sind. Bei GC-MS-Verfahren ist eine Unterscheidung zwischen verschiedenen Kongeneren erforderlich, beispielsweise zwischen toxischen (z.B. die siebzehn 2,3,7,8-substituierten PCDD/F sowie die zwölf dioxinähnlichen PCB) und anderen Kongeneren.
5.2.2. Bioanalytische Methoden müssen einen Nachweis der Zielverbindungen als Summe der PCDD/F und/oder dioxinähnlichen PCB ermöglichen. Ziel der Probenaufreinigung muss es sein, Verbindungen, die falsch-positive Ergebnisse verursachen könnten, oder Verbindungen, die das Signal schwächen und dadurch falsch-negative Ergebnisse verursachen könnten, zu entfernen.
5.3. Hohe Genauigkeit (Richtigkeit und Präzision, beobachtete Bioassay-Wiederfindung)
5.3.1. Bei Anwendung von GC-MS-Verfahren muss die Bestimmung eine valide Schätzung der tatsächlichen Konzentration in einer Probe ermöglichen. Hohe Genauigkeit ist notwendig, damit die Zurückweisung eines Ergebnisses einer Probenuntersuchung aufgrund der geringen Zuverlässigkeit des bestimmten TEQ-Gehalts vermieden wird. Die Genauigkeit des Analyseverfahrens wird angegeben durch die Richtigkeit (Differenz zwischen dem gemessenen Mittelwert eines Analyten in einem zertifizierten Material und seinem zertifizierten Wert, ausgedrückt als Prozentsatz dieses Wertes) und die Präzision (RSDR; relative Standardabweichung, berechnet aus unter Vergleichbarkeitsbedingungen ermittelten Ergebnissen).
5.3.2. Für bioanalytische Methoden ist die beobachtete Bioassay-Wiederfindung zu bestimmen. Die beobachtete Bioassay-Wiederfindung ist der BEQ-Gehalt, berechnet anhand einer TCDD-Kalibrierkurve oder einer PCB-126-Kalibrierkurve nach Korrektur um den Blindwert, geteilt durch den mittels Bestätigungsverfahren bestimmten TEQ-Wert. Dadurch sollen Faktoren wie der Verlust von PCDD/F und dioxinähnlichen Verbindungen während der einzelnen Extraktions- bzw. Reinigungsschritte, die Verstärkung oder Abschwächung des Signals durch mitextrahierte Verbindungen (agonistische bzw. antagonistische Wirkung), die Qualität der Kurvenanpassung oder Unterschiede zwischen TEF- und REP-(Relative-Potency-)Werten korrigiert werden. Die beobachtete Bioassay-Wiederfindung wird anhand geeigneter Referenzproben berechnet, die im Bereich der interessierenden Konzentration liegen und repräsentative Kongeneren-Muster aufweisen.
5.4. Validierung im Bereich des Höchstgehalts und allgemeine Qualitätssicherungsmaßnahmen
5.4.1. Die Laboratorien haben im Rahmen der Validierung und während der Routineanalytik den Nachweis der Leistungsfähigkeit eines Verfahrens im Bereich des Höchstgehalts, z.B. 0,5x, 1x und 2x Höchstgehalt mit einem akzeptablen Variationskoeffizienten für wiederholte Untersuchung, zu führen.
5.4.2. Als interne Qualitätssicherungsmaßnahmen müssen regelmäßige Methodenleerwert-Kontrollen und Experimente mit dotierten Proben oder Analysen von Kontrollproben (sofern erhältlich, vorzugsweise zertifiziertes Referenzmaterial) durchgeführt werden. Für Methodenleerwert-Kontrollen, Experimente mit dotierten Proben und Analysen von Kontrollproben sind Qualitätskontroll-Charts anzufertigen und zu prüfen, damit sichergestellt ist, dass die Analyseleistungsfähigkeit den Anforderungen genügt.
5.5. Bestimmungsgrenze
5.5.1. Bei einem bioanalytischen Screening-Verfahren ist eine Ermittlung der Bestimmungsgrenze (LOQ) nicht unbedingt erforderlich; es muss jedoch nachgewiesen sein, dass mit dem Verfahren eine Unterscheidung zwischen dem Methodenleerwert und dem Cut-off-Wert möglich ist. Wird ein BEQ-Gehalt angegeben, ist eine Konzentration festzulegen, ab der Ergebnisse mitgeteilt werden (Meldewert), um Proben, die ein Ergebnis unterhalb davon aufweisen, entsprechend einstufen zu können. Der Meldewert muss sich mindestens um den Faktor 3 von Methodenleerwert-Proben mit einem Signal unterhalb des Arbeitsbereichs unterscheiden. Er ist daher auf der Grundlage von Proben zu berechnen, die ungefähr den erforderlichen Mindestgehalt an Zielverbindungen enthalten, und nicht aus dem Signal/Rausch-Verhältnis oder einem Assay-Leerwert.
5.5.2. Die LOQ liegt beim Bestätigungsverfahren bei etwa einem Fünftel des Höchstgehalts.
5.6. Analysekriterien
Damit die Ergebnisse der Bestätigungs- oder Screening-Verfahren zuverlässig sind, müssen folgende Kriterien im Bereich des Höchstgehalts für den TEQ-Wert bzw. den BEQ-Wert erfüllt sein, entweder bestimmt als Gesamt-TEQ bzw. Gesamt-BEQ (Summe der PCDD/F und dioxinähnlichen PCB) oder separat für PCDD/F und dioxinähnliche PCB.
| Screening mit bioanalytischen oder physikalisch-chemischen Verfahren | Bestätigungsverfahren | |
| Falsch-negativ-Rate * | < 5 % | |
| Richtigkeit | -20 % bis +20 % | |
| Wiederholbarkeit (RSDr) | < 20 % | |
| Laborpräzision (RSDR) | < 25 % | < 15 % |
| *) Bezogen auf die Höchstgehalte. | ||
5.7. Besondere Anforderungen an Screening-Verfahren
5.7.1. Sowohl GC-MS-Verfahren als auch bioanalytische Methoden dürfen zum Screening angewendet werden. Bei GC-MS-Verfahren sind die unter Nummer 6 festgelegten Anforderungen zu erfüllen. Für zellbasierte bioanalytische Methoden sind unter Nummer 7 spezifische Anforderungen festgelegt.
5.7.2. Laboratorien, die zur Routinekontrolle von Proben Screening-Verfahren verwenden, müssen eng mit Laboratorien zusammenarbeiten, die das Bestätigungsverfahren anwenden.
5.7.3. Der Nachweis der Leistungsfähigkeit des Screening-Verfahrens ist während der Routineanalyse durch Qualitätssicherung und permanente Methodenvalidierung zu erbringen. Es muss ein kontinuierliches Programm zur Kontrolle der als konform beurteilten Analysenergebnisse geben.
5.7.4. Prüfung auf eine mögliche Unterdrückung der Zellantwort und auf Zytotoxizität:
20 % der Probenextrakte sind während des Routine-Screenings sowohl ohne als auch mit Zusatz einer dem Höchstgehalt oder dem Aktionsgrenzwert entsprechenden Menge von 2,3,7,8-TCDD zu analysieren, damit überprüft werden kann, ob das Signal möglicherweise durch interferierende Stoffe im Probenextrakt unterdrückt wird. Die gemessene Konzentration der dotierten Probe ist mit der Summe aus der Konzentration der nicht dotierten Probe und der Konzentration der Dotierung zu vergleichen. Liegt die gemessene Konzentration mehr als 25 % unter der berechneten (Summen-)Konzentration, ist dies ein Hinweis auf eine mögliche Signalunterdrückung und die entsprechende Probe ist einem GC-HRMS-Bestätigungsverfahren zu unterziehen. Die Ergebnisse sind anhand von Qualitätskontroll-Charts zu überwachen.
5.7.5. Qualitätssicherung bei als konform beurteilten Proben:
Etwa 2-10 % der konformen Proben sind, je nach Probenmatrix und Laborerfahrung, mittels GC-HRMS zu bestätigen.
5.7.6. Bestimmung der Falsch-negativ-Raten auf Grundlage der Qualitätssicherungsdaten:
Die Rate falsch-negativer Ergebnisse beim Screening von Proben unter- und oberhalb der Höchstgehalte oder Aktionsgrenzwerte ist zu bestimmen. Der tatsächliche Anteil der falsch-negativen Ergebnisse muss unter 5 % liegen. Wenn im Rahmen der Qualitätssicherung je Matrix/Matrixgruppe mindestens 20 Proben bestätigt wurden, ist aus dieser Datenbasis die Falsch-negativ-Rate zu ermitteln. Die zur Ermittlung der Falsch-negativ-Rate mindestens erforderlichen 20 Ergebnisse können auch die Ergebnisse von in Ringversuchen oder im Rahmen eines Kontaminationsereignisses untersuchten Proben mit einschließen, die einen Konzentrationsbereich von beispielsweise bis zum doppelten Höchstgehalt abdecken. Die Proben müssen die häufigsten Kongeneren-Muster abdecken, die verschiedene Kontaminationsquellen repräsentieren.
Obwohl Screening-Verfahren hauptsächlich auf die Ermittlung derjenigen Proben abzielen, in denen der Aktionsgrenzwert überschritten wird, ist das ausschlaggebende Kriterium zur Bestimmung der Falsch-negativ-Rate der Höchstgehalt, unter Berücksichtigung der erweiterten Messunsicherheit des Bestätigungsverfahrens.
5.7.7. Alle Proben, die im Screening-Verfahren als möglicherweise nicht konform beurteilt werden, müssen durch eine erneute vollständige Untersuchung der ursprünglichen Probe im Rahmen eines Bestätigungsverfahrens überprüft werden. Diese Proben dürfen auch zur Bewertung der Rate der falsch-positiven Ergebnisse herangezogen werden. Im Rahmen von Screening-Verfahren entspricht die Falsch-Positiv-Rate dem Anteil derjenigen Ergebnisse, von denen sich im Bestätigungsverfahren herausstellt, dass sie konform sind, nachdem sie zunächst als möglicherweise nicht konform eingestuft worden waren. Die Vorteile des Screening-Verfahrens sind auf Grundlage eines Vergleichs zwischen der Zahl der falsch-positiven Proben und der Gesamtzahl der untersuchten Proben zu bewerten. Dabei muss der Anteil der falsch-positiven Proben so gering sein, dass ein Screening vorteilhaft ist.
5.7.8. Unter Validierungsbedingungen müssen bioanalytische Methoden einen stichhaltigen Hinweis auf den TEQ-Gehalt ergeben, berechnet und ausgedrückt als BEQ.
Bei unter wiederholten Bedingungen angewandten bioanalytischen Methoden wäre in der Regel die laborinterne Wiederholbarkeit RSDr geringer als die unter Vergleichbarkeitsbedingungen (RSDR).
6. Besondere Anforderungen an GC-MS-Verfahren für Screening- oder Bestätigungszwecke
6.1. Annehmbare Differenzen zwischen Obergrenze ("upper-bound") und Untergrenze ("lower-bound") (WHO-TEQ-Ergebnisse)
Zur Bestätigung der Überschreitung von Höchstgehalten oder erforderlichenfalls von Aktionsgrenzwerten darf die Differenz zwischen Ober- und Untergrenze nicht mehr als 20 % betragen.
6.2. Kontrolle der Wiederfindungsrate
6.2.1. Die Zugabe von 13C-markierten 2,3,7,8-chlorsubstituierten internen PCDD/F-Standards und 13C-markierten internen dioxinähnlichen PCB-Standards ist ganz zu Anfang des Analyseverfahrens, z.B. vor der Extraktion, durchzuführen, damit das Analyseverfahren validiert werden kann. Bei jeder der tetra- bis octachlorierten homologen Gruppen von PCDD/F und bei jeder der homologen Gruppen von dioxinähnlichen PCB muss mindestens ein Kongener zugegeben werden (alternativ dazu mindestens ein Kongener je massenspektrometrisch ausgewählter Ionenaufzeichnungsfunktion zur Überwachung von PCDD/F und dioxinähnlichen PCB). Im Fall der Bestätigungsverfahren sind alle 17 13C-markierten 2,3,7,8-substituierten internen PCDD/F-Standards und alle 12 13C-markierten internen dioxinähnlichen PCB-Standards zu verwenden.
6.2.2. Die relativen Responsefaktoren sind mittels geeigneter Kalibrierlösungen auch für diejenigen Kongenere zu bestimmen, bei denen kein 13C-markiertes Analogon zugegeben ist.
6.2.3. Bei Futtermitteln pflanzlichen Ursprungs und Futtermitteln tierischen Ursprungs, die weniger als 10 % Fett enthalten, ist die Zugabe der internen Standards vor der Extraktion obligatorisch. Bei Futtermitteln tierischen Ursprungs, die mehr als 10 % Fett enthalten, sind die internen Standards entweder vor oder nach der Fettextraktion zuzugeben. Die Extraktionseffizienz ist auf geeignete Weise zu validieren, abhängig davon, auf welcher Stufe die internen Standards zugegeben werden.
6.2.4. Vor der GC-MS-Analyse sind 1 oder 2 Wiederfindungs-(Surrogat-)Standard(s) zuzugeben.
6.2.5. Es ist eine Kontrolle der Wiederfindungsrate erforderlich. Bei Bestätigungsverfahren müssen die Wiederfindungsraten der einzelnen internen Standards zwischen 60 und 120 % liegen. Geringere oder höhere Wiederfindungsraten für einzelne Kongenere, insbesondere für einige hepta- und octachlorierte Dibenzo-p-dioxine und Dibenzofurane, werden unter der Bedingung akzeptiert, dass ihr Beitrag zum TEQ-Wert 10 % des gesamten TEQ-Werts (basierend auf der Summe von PCDD/F und dioxinähnlichen PCB) nicht übersteigt. Bei GC-MS-Screening-Verfahren müssen die Wiederfindungen zwischen 30 und 140 % liegen.
6.3. Entfernung interferierender Stoffe
6.4. Kalibrierung mittels Standardkurve
Die Kalibrierkurve muss alle jeweils relevanten Bereiche der Höchstgehalte oder Aktionsgrenzwerte abdecken.
6.5. Besondere Kriterien für Bestätigungsverfahren
Bei der HRMS muss die Auflösung für den gesamten Massenbereich bei 10 % Tal in der Regel gleich oder größer als 10.000 sein.
Erfüllung weiterer Identifizierungs- und Bestätigungskriterien, wie sie in international anerkannten Normen, z.B. in der Norm EN 16215:2012 (Futtermittel - Bestimmung von Dioxinen und dioxin-ähnlichen PCBs mittels GC/HRMS und von Indikator-PCBs mittels GC/HRMS) und/oder in den überarbeiteten EPA-Methoden 1613 und 1668, beschrieben sind.
Messung von mindestens 2 spezifischen Vorläufer-Ionen, jeweils mit einem spezifischen zugehörigen Übergangsprodukt-Ion für alle markierten und nicht markierten Analyten im Untersuchungsbereich.
Zulässige Höchsttoleranz für relative Ionenintensitäten von ± 15 % für ausgewählte Übergangs-Produkt-Ionen im Vergleich zu berechneten oder gemessenen Werten (Mittelwert aus Kalibrierstandards) unter Anwendung identischer MS/MS-Bedingungen, insbesondere Kollisionsenergie und Kollisionsgasdruck, für jeden Übergang eines Analyts.
Festlegung der Auflösung für jeden Quadrupol gleichwertig oder besser als die Einheitsmassenauflösung (Einheitsmassenauflösung: ausreichende Auflösung zur Auftrennung zweier Peaks, die sich um eine Masseneinheit unterscheiden), um mögliche Auswirkungen von Interferenzen auf die interessierenden Analyten zu minimieren.
Erfüllung der weiteren Kriterien, wie sie in international anerkannten Normen, z.B. in der Norm EN 16215:2012 (Futtermittel - Bestimmung von Dioxinen und dioxin-ähnlichen PCBs mittels GC/HRMS und von Indikator-PCBs mittels GC/HRMS) und/oder in den überarbeiteten EPA-Methoden 1613 und 1668, beschrieben sind, die Pflicht zur Verwendung von GC-HRMS ausgenommen.
7. Besondere Anforderungen an bioanalytische Methoden
Bioanalytische Methoden sind Verfahren, die auf der Anwendung biologischer Grundsätze beruhen, beispielsweise zellbasierte Assays, Rezeptor-Assays oder Immunoassays. Unter dieser Nummer werden allgemeine Anforderungen an bioanalytische Methoden festgelegt.
Mit einem Screening-Verfahren wird eine Probe prinzipiell entweder als konform oder als vermutlich nicht konform eingestuft. Dazu wird der berechnete BEQ-Wert mit dem Cut-off-Wert verglichen (siehe Nummer 7.3). Proben, die unter dem Cut-off-Wert liegen, gelten als konform, Proben, die dem Cut-off-Wert entsprechen oder diesen überschreiten, gelten als vermutlich nicht konform und müssen mit einem Bestätigungsverfahren untersucht werden. In der Praxis kann ein BEQ-Gehalt, der zwei Drittel des Höchstgehalts entspricht, als Cut-off-Wert dienen, sofern eine Falsch-negativ-Rate von unter 5 % sowie eine annehmbare Rate von falsch-positiven Ergebnissen gewährleistet wird. Da für PCDD/F und für die Summe von PCDD/F und dioxinähnliche PCB unterschiedliche Höchstgehalte gelten, ist zur Prüfung der Konformität der Proben ohne Fraktionierung ein geeigneter Bioassay-Cut-off-Wert für PCDD/F erforderlich. Zur Überprüfung von Proben, in denen die Aktionsgrenzwerte überschritten werden, eignet sich ein entsprechender Prozentsatz des jeweiligen Aktionsgrenzwerts als Cut-off-Wert.
Wird ein ungefährer Gehalt in BEQ ausgedrückt, müssen die Probenergebnisse angegeben werden, die im Arbeitsbereich liegen und den Meldewert überschreiten (siehe Nummern 7.1.1 und 7.1.6).
7.1. Signalauswertung
7.1.1. Allgemeine Anforderungen
7.1.2. Kalibrierung
7.1.2.1. Kalibrierung mittels Standardkurve
7.1.2.2. Kalibrierung anhand von Referenzproben
Alternativ kann eine Kalibrierkurve auf Grundlage von mindestens vier Referenzproben verwendet werden (siehe Nummer 7.2.4): eine Matrixleerwert-Probe sowie 3 Referenzproben, die jeweils 0,5x, 1x und 2x den Höchstgehalt oder den Aktionsgrenzwert enthalten, wodurch keine Notwendigkeit zur Korrektur um Blindwerte und Wiederfindung besteht, wenn sich die Matrixeigenschaften bei Referenzproben und unbekannten Proben decken. In diesem Fall kann das Signal, das zwei Drittel des Höchstgehalts entspricht (siehe Nummer 7.3), direkt auf Grundlage dieser Proben berechnet und als Cut-off-Wert verwendet werden. Zur Überprüfung von Proben, in denen die Aktionsgrenzwerte überschritten werden, eignet sich ein entsprechender Prozentsatz dieser Aktionsgrenzwerte als Cut-off-Wert.
7.1.3. Separate Bestimmung von PCDD/F und dioxinähnlichen PCB
Extrakte können in Fraktionen, welche PCDD/F und dioxinähnliche PCB enthalten, aufgetrennt werden, sodass PCDD/F-TEQ und TEQ der dioxinähnlichen PCB-Verbindungen (jeweils als BEQ) getrennt angegeben werden können. Zur Bewertung der Ergebnisse für die Fraktion, die dioxinähnliche PCB enthält, ist vorzugsweise eine PCB-126-Standardkalibrierkurve zu verwenden.
7.1.4. Beobachtete Bioassay-Wiederfindung
Die "beobachtete Bioassay-Wiederfindung" ist auf Grundlage geeigneter Referenzproben mit repräsentativen Kongeneren-Mustern im Bereich des Höchstgehalts oder des Aktionsgrenzwert zu berechnen und wird als Prozentsatz des BEQ-Gehalts im Vergleich zum TEQ-Gehalt ausgedrückt. Je nachdem, welche Art von Assay oder TEF 12 verwendet wird, können die Unterschiede zwischen TEF- und REP-Faktoren in dioxinähnlichen PCB zu niedrigeren Wiederfindungswerten für dioxinähnliche PCB im Vergleich zu PCDD/F führen. Daher muss die beobachtete Bioassay-Wiederfindung bei einer getrennten Bestimmung von PCDD/F und dioxinähnlichen PCB für dioxinähnliche PCB 20 bis 60 % und für PCDD/F 50 bis 130 % betragen (bei Verwendung einer TCDD-Kalibrierkurve). Der Beitrag der dioxinähnlichen PCB zur Summe der PCDD/F und dioxinähnlichen PCB kann je nach Matrices und Proben unterschiedlich sein; dies spiegelt sich in den Bereichen der beobachteten Bioassay-Wiederfindung für die Summe der PCDD/F und dioxinähnlichen PCB wider, die zwischen 30 und 130 % liegen müssen. Jeder Niederschlag wesentlicher Änderungen der TEF-Werte auf die Rechtsvorschriften der Union über PCDD/F und dioxinähnliche PCB erfordert eine Überarbeitung dieser Bereiche.
7.1.5. Kontrolle der Wiederfindung nach Reinigung der Probenextrakte
Der Verlust von Verbindungen während der Reinigung ist im Rahmen der Validierung zu überprüfen. Eine Matrixleerprobe, dotiert mit einem Gemisch verschiedener Kongenere, ist dem Reinigungsverfahren zu unterziehen (mindestens n = 3), und Wiederfindung und Streuung sind mittels eines Bestätigungsverfahrens zu untersuchen. Die Wiederfindung muss zwischen 60 und 120 % betragen, insbesondere für Kongenere, die in verschiedenen Gemischen jeweils mehr als 10 % des TEQ-Gehalts ausmachen.
7.1.6. Meldegrenze
Werden BEQ-Gehalte angegeben, ist auf der Grundlage relevanter Matrix-Proben, die typische Kongeneren-Muster aufweisen, eine Meldegrenze zu ermitteln; dabei ist die Kalibrierkurve der Standards aufgrund ihrer geringen Präzision im unteren Bereich nicht heranzuziehen. Die Einflüsse aus Extraktion und Reinigung müssen berücksichtigt werden. Die Meldegrenze muss mindestens um den Faktor 3 über den Methodenleerwerten liegen.
7.2. Verwendung von Referenzproben
7.2.1. Referenzproben müssen repräsentativ für Probenmatrix, Kongeneren-Muster und Konzentrationen für PCDD/F und dioxinähnliche PCB im Bereich des Höchstgehalts oder des Aktionsgrenzwerts sein.
7.2.2. Bei jeder Test-Reihe ist eine Matrixleerprobe, oder, sofern dies nicht möglich ist, eine Methodenleerwert-Probe, sowie eine Referenzprobe im Bereich des Höchstgehalts oder des Aktionsgrenzwerts einzubeziehen. Diese Proben müssen zur gleichen Zeit und unter identischen Bedingungen extrahiert und analysiert werden. Die Referenzprobe muss im Vergleich zu der Matrixleerprobe ein deutlich höheres Signal aufweisen, wodurch die Eignung des Analyseverfahrens gewährleistet ist. Solche Proben können zur Korrektur um Leerwert und Wiederfindung verwendet werden.
7.2.3. Referenzproben, die zur Korrektur um die Wiederfindung herangezogen werden, müssen repräsentativ für die Analysenproben sein, d. h., die Kongeneren-Muster dürfen nicht zu einer Unterschätzung der Gehalte führen.
7.2.4. Zusätzliche Referenzproben, deren Konzentration z.B. das 0,5- und 2-Fache des Höchstgehalts oder des Aktionsgrenzwerts beträgt, können einbezogen werden, damit die ordnungsgemäße Durchführung der Untersuchungen in dem für die Kontrolle des Höchstgehalts oder des Aktionsgrenzwerts relevanten Bereich nachgewiesen werden kann. In Kombination können diese Proben zur Berechnung der BEQ-Gehalte in den untersuchten Proben verwendet werden (siehe Nummer 7.1.2.2).
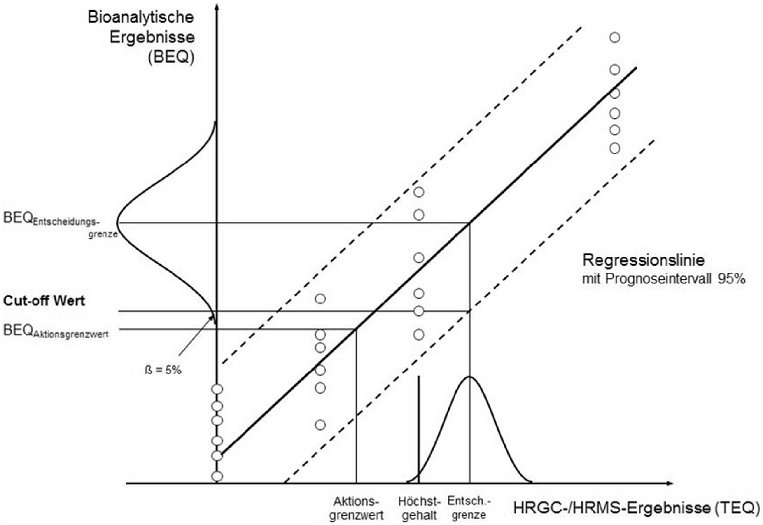
7.3. Bestimmung der Cut-off-Werte
Die Beziehung zwischen den in BEQ ausgedrückten Ergebnissen der bioanalytischen Methode und den in TEQ ausgedrückten Ergebnissen von Bestätigungsverfahren ist zu ermitteln, z.B. durch matrixbezogene Kalibrierexperimente unter Verwendung von Referenzproben, die mit 0, 0,5x, 1x und 2x Höchstgehalt dotiert sind und auf jeder Konzentrationsstufe jeweils 6 Mal untersucht werden (n = 24). Korrekturfaktoren (Leerwert und Wiederfindung) können auf der Grundlage dieses Verhältnisses geschätzt werden, sind jedoch gemäß Nummer 7.2.2 zu überprüfen.
Für die Entscheidung, ob eine Probe den Höchstgehalten entspricht, oder gegebenenfalls zur Überprüfung der Aktionsgrenzwerte sind Cut-off-Werte zu ermitteln, wobei die jeweiligen Höchstgehalte bzw. Aktionsgrenzwerte entweder einzeln für PCDD/F und dioxinähnliche PCB oder aber für die Summe von PCDD/F und dioxinähnlichen PCB festgelegt sein können. Sie stellen den unteren Endpunkt der Verteilung bioanalytischer Ergebnisse (korrigiert um Leerwert und Wiederfindung) von Proben dar, die Gehalte an der Entscheidungsgrenze des Bestätigungsverfahrens aufweisen, berechnet auf Grundlage eines Vertrauensniveaus von 95 %, was eine Falsch-negativ-Rate von < 5 % impliziert, und auf Basis einer RSDR unter 25 %. Die Entscheidungsgrenze des Bestätigungsverfahrens entspricht dem Höchstgehalt zuzüglich der erweiterten Messunsicherheit.
Der Cut-off-Wert (in BEQ) kann gemäß einem der unter den Nummern 7.3.1, 7.3.2 oder 7.3.3 beschriebenen Ansätze berechnet werden (siehe Abbildung 1).
7.3.1. Verwendung des unteren Bands des Prognoseintervalls von 95 % an der Entscheidungsgrenze des Bestätigungsverfahrens
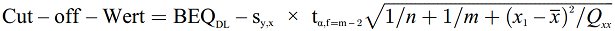
Dabei ist
| BEQDL | der BEQ-Wert, der der Entscheidungsgrenze des Bestätigungsverfahrens entspricht, die wiederum dem Höchstgehalt unter Berücksichtigung der erweiterten Messunsicherheit entspricht. |
| sy,x | die Reststandardabweichung |
| tα,f = m-2 | der Student-Faktor (α = 5 %, f = Freiheitsgrade, einseitig) |
| m | die Gesamtzahl der Kalibrierpunkte (Laufzahl j) |
| n | die Anzahl der Wiederholungen auf jeder Ebene |
| xi | die Probenkonzentration (in TEQ) des Kalibrierpunkts i, durch ein Bestätigungsverfahren ermittelt |
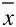 | der Mittelwert der Konzentrationen (in TEQ) aller Kalibrierproben |
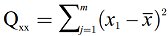 | Quadratsummenparameter, i = Laufzahl des Kalibrierpunkts i |
7.3.2. Berechnung aus bioanalytischen Ergebnissen (korrigiert um Leerwert und Wiederfindung) aus der Mehrfachuntersuchung (n ≥ 6) von Proben, die Gehalte an der Entscheidungsgrenze des Bestätigungsverfahrens aufweisen, als unterer Endpunkt der Datenverteilung am entsprechenden BEQ-Mittelwert:
Cut - off - Wert =BEQDL - 1,64 × SDR
Dabei ist
| SDR | die Standardabweichung der Bioassay-Ergebnisse am BEQDL, gemessen unter laborinternen Vergleichbarkeitsbedingungen. |
7.3.3. Berechnung als Mittelwert der bioanalytischen Ergebnisse (in BEQ, korrigiert um Leerwert und Wiederfindung) auf der Grundlage mehrfacher Untersuchungen (n ≥ 6) von Proben, die mit zwei Dritteln des Höchstgehalts oder des Aktionsgrenzwerts kontaminiert sind, auf Grundlage der Beobachtung, dass dieser Wert in der Nähe des gemäß Nummer 7.3.1 oder 7.3.2 bestimmten Cut-off-Wertes liegt.
Berechnung der Cut-off-Werte auf der Grundlage eines Vertrauensniveaus von 95 %, was eine Falsch-negativ-Rate von < 5 % impliziert, und auf Basis einer RSDR < 25 %:
7.3.4. Beschränkungen der Cut-off-Werte:
Auf BEQ basierende Cut-off-Werte, die anhand der im Rahmen der Validierung und unter Verwendung einer begrenzten Anzahl von Proben mit unterschiedlichen Matrix-/Kongeneren-Mustern erzielten RSDR berechnet wurden, können höher sein als die auf TEQ basierenden Höchstgehalte oder Aktionsgrenzwerte, da hier die Präzision höher ist, als es in einer Routine möglich ist, in der ein unbekanntes Spektrum möglicher Kongeneren-Muster überprüft werden muss. In solchen Fällen ist der Berechnung der Cut-off-Werte eine RSDR = 25 % zugrunde zu legen, oder aber es sind zwei Drittel des Höchstgehalts oder des Aktionsgrenzwerts als Cut-off-Wert zu verwenden.
7.4. Leistungsmerkmale
7.4.1. Da bei bioanalytischen Methoden keine internen Standards verwendet werden können, sind Tests zur Wiederholbarkeit bioanalytischer Methoden durchzuführen, um Informationen über die Standardabweichung innerhalb einer Testreihe bzw. zwischen Testreihen zu erhalten. Die Wiederholbarkeit muss unter 20 % liegen, die laborinterne Vergleichbarkeit unter 25 %. Grundlage dafür müssen die nach Korrektur um Blindwert und Wiederfindung als BEQ berechneten Konzentrationen sein.
7.4.2. Während der Validierung muss nachgewiesen werden, dass mit dem Testverfahren zwischen einer Leerprobe und einem Gehalt in Höhe des Cut-off-Werts unterschieden werden kann, sodass Proben, deren Gehalt über dem entsprechenden Cut-off-Wert liegt, identifiziert werden können (siehe Nummer 7.1.2).
7.4.3. Die zu bestimmenden Verbindungen, mögliche auftretende Störungen und der maximal akzeptable Leerwert müssen festgelegt werden.
7.4.4. Die Standardabweichung in Prozent, mit der die Signalwerte oder die aus den Signalwerten berechneten Konzentrationen (nur möglich im Arbeitsbereich) behaftet sind, darf bei einer dreifachen Bestimmung eines Probenextrakts nicht mehr als 15 % betragen.
7.4.5. Zur Bewertung der Leistungsfähigkeit einer bioanalytischen Methode über einen konstanten Zeitraum hinweg sind die unkorrigierten Ergebnisse der Referenzprobe(n), ausgedrückt in BEQ (Leerwert und Höchstgehalt oder Aktionsgrenzwert), heranzuziehen.
7.4.6. Für Leerwert-Proben und für jede Art von Referenzproben sind Qualitätskontroll-Charts anzufertigen und zu prüfen, damit sichergestellt ist, dass die analytische Leistungsfähigkeit den Anforderungen genügt, insbesondere bei Methodenleerwert-Proben im Hinblick auf den erforderlichen Mindestabstand zum unteren Ende des Arbeitsbereichs und für Referenzproben hinsichtlich der laborinternen Vergleichbarkeit. Methodenleerwert-Proben sind so zu prüfen, dass falsch-negative Ergebnisse bei Abzug der Werte vermieden werden.
7.4.7. Die Ergebnisse der in Bezug auf verdächtige Proben durchgeführten Bestätigungsverfahren und von 2 bis 10 % der konformen Proben (mindestens 20 Proben je Matrix) sind zu sammeln und zur Bewertung der Leistungsfähigkeit des Screening-Verfahrens und der Beziehung zwischen BEQ und TEQ zu verwenden. Diese Datenbank kann zur Neubewertung der Cut-off-Werte für Routineproben der validierten Matrices genutzt werden.
7.4.8. Die Leistungsfähigkeit eines Verfahrens kann auch durch Teilnahme an Ringversuchen nachgewiesen werden. Die Ergebnisse von in Ringversuchen analysierten Proben, die einen Konzentrationsbereich bis zum doppelten Höchstgehalt abdecken, können zur Bewertung der Falsch-negativ-Rate herangezogen werden, wenn ein Laboratorium seine Leistungsfähigkeit unter Beweis gestellt hat. Die Proben müssen die häufigsten Kongeneren-Muster abdecken, die verschiedene Kontaminationsquellen repräsentieren.
7.4.9. Bei Kontaminationsfällen können die Cut-off-Werte neu ermittelt werden, um den Besonderheiten von Matrix und Kongeneren-Muster des jeweiligen Zwischenfalls Rechnung zu tragen.
8. Bericht über die Ergebnisse
8.1. Bestätigungsverfahren
8.1.1. Die Untersuchungsergebnisse müssen die Werte der einzelnen Kongenere von PCDD/F und dioxinähnlichen PCB enthalten, und die TEQ-Werte müssen als Untergrenze ("lower-bound"), Obergrenze ("upper-bound") und Mittelwert ("medium-bound") gemeldet werden, damit möglichst viele Informationen in den Untersuchungsberichten enthalten sind und die Ergebnisse somit entsprechend den speziellen Anforderungen interpretiert werden können.
8.1.2. In dem Bericht muss das zur Extraktion der PCDD/F und dioxinähnlichen PCB angewendete Verfahren genannt werden.
8.1.3. Die Wiederfindungsraten der einzelnen internen Standards sind zur Verfügung zu stellen, falls die Wiederfindungen außerhalb des unter Nummer 6.2.5 genannten Bereichs liegen, falls die Gehalte in den Proben den Höchstgehalt überschreiten (in diesem Fall die Wiederfindungen aus einer der beiden Untersuchungen) sowie in anderen Fällen auf Anfrage.
8.1.4. Da bei der Entscheidung über die Konformität einer Probe die erweiterte Messunsicherheit zu berücksichtigen ist, ist dieser Parameter vorzulegen. Das Analyseergebnis ist als x ± U anzugeben, wobei x das Analyseergebnis darstellt und U die erweiterte Messunsicherheit unter Verwendung eines Erweiterungsfaktors von 2, was einem Vertrauensniveau von ca. 95 % entspricht. Bei einer getrennten Bestimmung des Gehalts an PCDD/F und dioxinähnlichen PCB ist die Summe der geschätzten erweiterten Messunsicherheit der getrennten Analyseergebnisse der PCDD/F und der dioxinähnlichen PCB für die Berechnung der Summe der PCDD/F und dioxinähnlichen PCB zu verwenden.
8.1.5. Die Ergebnisse sind in denselben Einheiten und mit mindestens derselben Anzahl signifikanter Stellen anzugeben wie die in der Richtlinie 2002/32/EG festgelegten Höchstgehalte.
8.2. Bioanalytische Screening-Verfahren
8.2.1. Das Ergebnis des Screenings ist anzugeben als "konform" oder "vermutlich nicht konform" ("verdächtig").
8.2.2. Außerdem können annähernde Ergebniswerte für PCDD/F und/oder dioxinähnliche PCB in BEQ, nicht in TEQ, angegeben werden.
8.2.3. Proben, deren Gehalt unterhalb der Meldegrenze liegt, sind als solche zu bezeichnen. Proben, deren Gehalt "oberhalb des Arbeitsbereichs" liegt, sind mit dieser Angabe zu melden, und der der Obergrenze des Arbeitsbereichs entsprechende Gehalt ist in BEQ anzugeben.
8.2.4. In dem Bericht muss für jede Art von Probenmatrix der Höchstgehalt oder der Aktionsgrenzwert genannt werden, auf dem die Bewertung beruht.
8.2.5. Aus dem Bericht müssen die Art des verwendeten Tests, die grundlegenden Testprinzipien und die Art der Kalibrierung hervorgehen.
8.2.6. In dem Bericht muss das zur Extraktion der PCDD/F und dioxinähnlichen PCB angewendete Verfahren genannt werden.
8.2.7. Für Proben, die vermutlich nicht konform sind, muss der Bericht einen Hinweis auf die zu ergreifenden Maßnahmen enthalten. Die Konzentration von PCDD/F und der Summe von PCDD/F und dioxinähnlichen PCB in diesen Proben mit erhöhten Gehalten muss durch ein Bestätigungsverfahren ermittelt/bestätigt werden.
8.2.8. Nicht konforme Ergebnisse werden nur gemeldet, wenn sie in einem Bestätigungsverfahren ermittelt wurden.
8.3. Physikalisch-chemische Screening-Verfahren
8.3.1. Das Ergebnis des Screenings ist anzugeben als "konform" oder "vermutlich nicht konform" ("verdächtig").
8.3.2. In dem Bericht muss für jede Art von Probenmatrix der Höchstgehalt oder der Aktionsgrenzwert genannt werden, auf dem die Bewertung beruht.
8.3.3. Zudem können Werte der einzelnen Kongenere von PCDD/F und dioxinähnlichen PCB sowie als Untergrenze, Obergrenze und Mittelwert gemeldete TEQ-Werte angegeben werden. Die Ergebnisse sind in denselben Einheiten und mit mindestens derselben Anzahl signifikanter Stellen anzugeben wie die in der Richtlinie 2002/32/EG festgelegten Höchstgehalte.
8.3.4. Die Wiederfindungsraten der einzelnen internen Standards sind zur Verfügung zu stellen, falls die Wiederfindungen außerhalb des unter Nummer 6.2.5 genannten Bereichs liegen, falls die Gehalte in den Proben den Höchstgehalt überschreiten (in diesem Fall die Wiederfindungen aus einer der beiden Untersuchungen) sowie in anderen Fällen auf Anfrage.
8.3.5. Aus dem Bericht muss hervorgehen, welches GC-MS-Verfahren angewendet wurde.
8.3.6. In dem Bericht muss das zur Extraktion der PCDD/F und dioxinähnlichen PCB angewendete Verfahren genannt werden.
8.3.7. Für Proben, die vermutlich nicht konform sind, muss der Bericht einen Hinweis auf die zu ergreifenden Maßnahmen enthalten. Die Konzentration von PCDD/F und der Summe von PCDD/F und dioxinähnlichen PCB in diesen Proben mit erhöhten Gehalten muss durch ein Bestätigungsverfahren ermittelt/bestätigt werden.
8.3.8. Ob ein Wert nicht konform ist, kann nur in einem Bestätigungsverfahren entschieden werden.
Kapitel III
Probenvorbereitung und Anforderungen an Untersuchungsverfahren zur amtlichen Kontrolle des Gehalts an nicht dioxinähnlichen PCB in Futtermitteln
1. Anwendungsbereich
Die in diesem Kapitel beschriebenen Anforderungen gelten, wenn Futtermittel zur amtlichen Kontrolle des Gehalts an nicht dioxinähnlichen PCB sowie für die Probenvorbereitung und Untersuchungsanforderungen zu anderen regulatorischen Zwecken, darunter die Kontrollen der Futtermittelunternehmer zur Gewährleistung der Vorschriftsmäßigkeit gemäß der Verordnung (EG) Nr. 183/2005, untersucht werden.
2. Anzuwendende Nachweisverfahren
Gaschromatografie/Elektroneneinfangdetektor (GC-ECD), GC-LRMS, GC-MS/MS, GC-HRMS oder gleichwertige Verfahren.
3. Bestimmung und Bestätigung der interessierenden Analyten
3.1. Relative Retentionszeit im Verhältnis zu internen Standards oder Referenzstandards (akzeptable Abweichung ± 0,25 %).
3.2. Gaschromatografische Trennung der nicht dioxinähnlichen PCB von interferierenden Stoffen, insbesondere von koeluierenden PCB und insbesondere dann, wenn die Gehalte der Proben an der gesetzlichen Grenze liegen und bestätigt werden muss, dass sie nicht konform sind 13.
3.3. Anforderungen an GC-MS-Techniken
Messung von mindestens der folgenden Anzahl an Molekül-Ionen oder charakteristischen Ionen des Molekül-Clusters:
Zulässige Höchsttoleranzen für das Isotopenhäufigkeitsverhältnis für ausgewählte Massenfragmente:
Relative Abweichung des Isotopenhäufigkeitsverhältnisses ausgewählter Massenfragmente von der theoretischen Häufigkeit oder dem Kalibrierstandard für das Ziel-Ion (das am häufigsten vorkommende Ion) und das/die Qualifizier-Ion(en): ± 15 %.
3.4. Anforderungen an GC-ECD-Techniken
Ergebnisse, die den Höchstgehalt überschreiten, sind anhand von zwei GC-Säulen mit stationären Phasen unterschiedlicher Polarität zu bestätigen.
4. Nachweis der Leistungsfähigkeit des Verfahrens
Die Leistungsfähigkeit der Methode im Bereich des Höchstgehalts (0,5- bis 2facher Höchstgehalt) mit einem akzeptablen Variationskoeffizienten für wiederholte Untersuchung (siehe Anforderungen an die Laborpräzision unter Nummer 9) ist zu validieren.
5. Bestimmungsgrenze
Die Summe der Bestimmungsgrenzen (LOQ) 14 nicht dioxinähnlicher PCB darf ein Drittel des Höchstgehalts nicht übersteigen 15.
6. Qualitätssicherung
Regelmäßige Blindkontrollen, Analysen dotierter Proben, Qualitätssicherungsproben, Teilnahme an Laborvergleichsuntersuchungen zu relevanten Matrices.
7. Kontrolle der Wiederfindungsrate
7.1. Es sind geeignete interne Standards mit physikalisch-chemikalischen Eigenschaften, die denen der interessierenden Analyten vergleichbar sind, zu verwenden.
7.2. Zugabe interner Standards:
Zugabe zu Erzeugnissen (vor Extraktion und Clean-up).
7.3. Anforderungen an Verfahren, bei denen alle sechs isotopenmarkierten nicht dioxinähnlichen PCB-Kongenere verwendet werden:
7.4. Anforderungen an Verfahren, bei denen nicht alle sechs isotopenmarkierten internen Standards oder andere interne Standards verwendet werden:
7.5. Die Wiederfindungen nicht markierter Kongenere sind mittels dotierter Proben oder Qualitätskontrollproben mit Konzentrationen im Bereich des Höchstgehalts zu prüfen. Für diese Kongenere sind Wiederfindungsraten zwischen 60 und 120 % akzeptabel.
8. Anforderungen an Laboratorien
Gemäß den Bestimmungen der Verordnung (EU) 2017/625 müssen die Laboratorien von einer anerkannten Stelle akkreditiert sein, die nach ISO/IEC Guide 58 arbeitet, damit sichergestellt ist, dass die Laboratorien bei der Untersuchung Qualitätssicherungsverfahren anwenden. Die Laboratorien müssen gemäß der Norm EN ISO/IEC 17025 akkreditiert sein. Zudem sind die in den technischen Leitlinien für die Schätzung der Messunsicherheit und der Bestimmungsgrenzen für die Untersuchung auf PCB beschriebenen Grundsätze zu befolgen 16.
9. Leistungsmerkmale: Kriterien für die Summe der nicht dioxinähnlichen PCB im Bereich des Höchstgehalts
| Isotopenverdünnungs-Massenspektrometrie * | Andere Techniken | |
| Richtigkeit | -20 bis + 20 % | -30 bis + 30 % |
| Laborpräzision (RSD %) | ≤ 15 % | ≤ 20 % |
| Differenz zwischen berechneter Obergrenze ("upper-bound") und Untergrenze ("lower-bound") | ≤ 20 % | ≤ 20 % |
| *) Alle sechs 13C-markierten Analoga müssen als interne Standards verwendet werden. | ||
10. Bericht über die Ergebnisse
10.1. Die Untersuchungsergebnisse müssen die Werte der einzelnen nicht dioxinähnlichen PCB und der Summe solcher PCB-Kongenere enthalten, angegeben als Untergrenze ("lower-bound"), Obergrenze ("upper-bound") und Mittelwert ("medium-bound"), damit möglichst viele Informationen in den Untersuchungsberichten enthalten sind und die Ergebnisse somit entsprechend den speziellen Anforderungen interpretiert werden können.
10.2. In dem Bericht muss das zur Extraktion der PCB angewendete Verfahren genannt werden.
10.3. Die Wiederfindungsraten der einzelnen internen Standards sind zur Verfügung zu stellen, falls die Wiederfindungen außerhalb des unter Nummer 7 genannten Bereichs liegen, falls die Gehalte in den Proben den Höchstgehalt überschreiten sowie in anderen Fällen auf Anfrage.
10.4. Da bei der Entscheidung über die Konformität einer Probe die erweiterte Messunsicherheit zu berücksichtigen ist, ist dieser Parameter ebenfalls vorzulegen. Das Analyseergebnis ist als x ± U anzugeben, wobei x das Analyseergebnis darstellt und U die erweiterte Messunsicherheit unter Verwendung eines Erweiterungsfaktors von 2, was einem Vertrauensniveau von ca. 95 % entspricht.
10.5. Die Ergebnisse sind in denselben Einheiten und mit mindestens derselben Anzahl signifikanter Stellen anzugeben wie die in der Richtlinie 2002/32/EG festgelegten Höchstgehalte.
B. Europäische Normen (EN)
Für die Anwendung von Artikel 34 Absatz 2 Buchstabe a der Verordnung (EU) 2017/625 sind die folgenden europäischen Normen maßgeblich:
| EN 17194: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Deoxynivalenol, Aflatoxin B1, Fumonisin B1 und B2, T-2- und HT-2-Toxine, Zearalenon und Ochratoxin A in Einzelfuttermitteln und Mischfuttermitteln mittels LC-MS/MS |
| EN 17270: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Theobromin in Einzel- und Mischfuttermitteln, einschließlich aus Kakao gewonnenen Bestandteilen, mittels Flüssigchromatographie |
| EN 17504: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Gossypol in Baumwollsamen und Futtermitteln mittels LC-MS/MS |
| EN 17362: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Pentachlorphenol (PCP) in Futtermittel und Mischfuttermittel mittels LC-MS/MS |
| EN 16279: Futtermittel - Bestimmung des Fluoridgehaltes nach Salzsäure-Behandlung mit ionensensitiver Elektrode (ISE) |
| EN 17053: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Spurenelementen, Schwermetallen und anderen Elementen in Futtermitteln mittels ICP-MS (Multimethode) |
| EN 15550: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von Cadmium und Blei mittels Graphitrohrofen-Atomabsorptionsspektrometrie (GF-AAS) nach Druckaufschluss |
| EN 16206: Futtermittel - Bestimmung von Arsen mit Atomabsorptionsspektrometrie-Hydridtechnik (HD-AAS) nach Mikrowellen-Druckaufschluss (Aufschluss mit 65 % Salpetersäure und 30 % Wasserstoffperoxid) |
| EN 16277: Futtermittel - Bestimmung von Quecksilber mit Kaltdampf-Atomabsorptionsspektrometrie (KD-AAS) nach Mikrowellen-Druckaufschluss (Extraktion mit 65 % Salpetersäure und 30 % Wasserstoffperoxid) |
| EN 16278: Futtermittel - Bestimmung von anorganischem Arsen mit Atomabsorptionsspektrometrie-Hydridtechnik (HD-AAS) nach Mikrowellen-Extraktion und Trennung durch Festphasenextraktion (SPE) |
| EN 17374: Futtermittel - Probenahme- und Untersuchungsverfahren - Bestimmung von anorganischem Arsen in Futtermittel mittels Anionenaustausch HPLC-ICP-MS |
| Kongener | TEF-Wert | Kongener | TEF-Wert |
| Dibenzo-p-dioxine (PCDD) und Dibenzo-p-furane (PCDF) | "Dioxinähnliche PCB" Non-ortho-PCB + Mono-ortho-PCB | ||
| 2,3,7,8-TCDD | 1 | ||
| 1,2,3,7,8-PeCDD | 1 | Non-ortho PCB | |
| 1,2,3,4,7,8-HxCDD | 0,1 | PCB 77 | 0,0001 |
| 1,2,3,6,7,8-HxCDD | 0,1 | PCB 81 | 0,0003 |
| 1,2,3,7,8,9-HxCDD | 0,1 | PCB 126 | 0,1 |
| 1,2,3,4,6,7,8-HpCDD | 0,01 | PCB 169 | 0,03 |
| OCDD | 0,0003 | Mono-ortho PCB | |
| 2,3,7,8-TCDF | 0,1 | PCB 105 | 0,00003 |
| 1,2,3,7,8-PeCDF | 0,03 | PCB 114 | 0,00003 |
| 2,3,4,7,8-PeCDF | 0,3 | PCB 118 | 0,00003 |
| 1,2,3,4,7,8-HxCDF | 0,1 | PCB 123 | 0,00003 |
| 1,2,3,6,7,8-HxCDF | 0,1 | PCB 156 | 0,00003 |
| 1,2,3,7,8,9-HxCDF | 0,1 | PCB 157 | 0,00003 |
| 2,3,4,6,7,8-HxCDF | 0,1 | PCB 167 | 0,00003 |
| 1,2,3,4,6,7,8-HpCDF | 0,01 | PCB 189 | 0,00003 |
| 1,2,3,4,7,8,9-HpCDF | 0,01 | ||
| OCDF | 0,0003 | ||
Abkürzungen: "T" = tetra; "Pe" = penta; "Hx" = hexa; "Hp" = hepta; "O" = octa; "CDD" = Chlordibenzodioxin; "CDF" = Chlordibenzofuran; "CB" = Chlorbiphenyl.
2) Durchführungsverordnung (EU) 2021/808 der Kommission vom 22. März 2021 über Leistungskriterien für Analysemethoden für Rückstände pharmakologisch wirksamer Stoffe in zur Lebensmittelerzeugung genutzten Tieren und über die Auswertung von Ergebnissen sowie über die für Probenahmen anzuwendenden Methoden und zur Aufhebung der Entscheidungen 2002/657/EG und 98/179/EG (ABl. L 180 vom 21.05.2021 S. 84).
3) Falls zutreffend, sind die im "Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry" (https://food.ec.europa.eu/system/files/2017-05/animal-feed-guidance_document_pcdd-f_pcb_en.pdf) beschriebenen Grundsätze zu befolgen.
4) "Zweitanalyse": getrennte Untersuchung der interessierenden Analyten anhand eines zweiten Aliquots derselben homogenisierten Probe. Grundsätzlich gelten für Zweitanalysen die Anforderungen gemäß Anhang II Kapitel C Nummer 3. Bei Verfahren, bei denen 13C-markierte interne Standards für die relevanten Analyten verwendet werden, ist die Zweitanalyse jedoch nur erforderlich, wenn das Ergebnis der ersten Bestimmung nicht konform ist. Die Zweitanalyse ist erforderlich, um eine interne Kreuzkontamination oder eine versehentliche Vertauschung der Proben auszuschließen. Bei einer Untersuchung im Verlauf eines Kontaminationsfalls kann auf die Bestätigung durch Zweitanalyse verzichtet werden, wenn sich die untersuchten Proben auf den Kontaminationsfall zurückverfolgen lassen und der gemessene Wert deutlich über dem Höchstgehalt liegt.
5) Nach dem Konzept der "Obergrenze" ("upper-bound") wird der Beitrag jedes nicht quantifizierten Kongeners der Bestimmungsgrenze gleichgesetzt. Beim Konzept der "Untergrenze" ("lower-bound") wird der Beitrag jedes nicht quantifizierten Kongeners gleich null gesetzt. Beim Konzept des "Mittelwerts" ("medium-bound") wird der Beitrag jedes nicht quantifizierten Kongeners der Hälfte der Bestimmungsgrenze gleichgesetzt.
6) Grundsätzlich gelten für Zweitanalysen die Anforderungen gemäß Anhang II Kapitel C Nummer 3. Bei Bestätigungsverfahren, bei denen 13C-markierte interne Standards für die relevanten Analyten verwendet werden, ist die Zweitanalyse jedoch nur erforderlich, wenn das Ergebnis der ersten Bestimmung nicht konform ist. Die Zweitanalyse ist erforderlich, um eine interne Kreuzkontamination oder eine versehentliche Vertauschung der Proben auszuschließen. Bei einer Untersuchung im Verlauf eines Kontaminationsfalls kann auf die Bestätigung durch Zweitanalyse verzichtet werden, wenn sich die untersuchten Proben auf den Kontaminationsfall zurückverfolgen lassen und der gemessene Wert deutlich über dem Höchstgehalt liegt.
7) Nach dem Konzept der "Obergrenze" ("upper-bound") wird der Beitrag jedes nicht quantifizierten Kongeners zum TEQ der Bestimmungsgrenze gleichgesetzt. Beim Konzept der "Untergrenze" ("lower-bound") wird der Beitrag jedes nicht quantifizierten Kongeners zum TEQ gleich null gesetzt. Beim Konzept des "Mittelwerts" ("medium-bound") wird der Beitrag jedes nicht quantifizierten Kongeners zum TEQ der Hälfte der Bestimmungsgrenze gleichgesetzt.
8) Für Zweitanalysen zur Kontrolle der Aktionsgrenzwerte gelten die gleichen Erklärungen und Anforderungen wie die für Höchstgehalte in Fußnote 33 genannten.
9) Verordnung (EG) Nr. 183/2005 des Europäischen Parlaments und des Rates vom 12. Januar 2005 mit Vorschriften für die Futtermittelhygiene (ABl. L 35 vom 08.02.2005 S. 1).
10) Bioanalytische Methoden sind nicht spezifisch für diejenigen Kongenere, die in das TEF-Schema fallen. Im Probenextrakt können auch andere, strukturverwandte AhR-aktive Verbindungen vorliegen, die zur Gesamt-Zellantwort beitragen. Daher erlauben bioanalytische Ergebnisse keine Schätzung des TEQ-Gehalts, sondern stellen eher einen Hinweis auf den TEQ-Gehalt in einer Probe dar.
11) "Guidance Document on Measurement Uncertainty for Laboratories performing PCDD/F and PCB Analysis using Isotope Dilution Mass Spectrometry" [https://food.ec.europa.eu/system/files/2017-05/animal-feed-guidance_document_pcdd-f_pcb_en.pdf], "Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food" [https://food.ec.europa.eu/system/files/2016-10/cs_contaminants_sampling_analysis-report_2004_en.pdf].
12) Die derzeitigen Anforderungen basieren auf den in M. Van den Berg et al, Toxicol Sci 93 (2), 223-241 (2006) veröffentlichten TEF.
13) Kongenere, die oft koeluieren, sind beispielsweise PCB 28/31, PCB 52/69 und PCB 138/163/164. Bei GC-MS-Verfahren muss auch die Möglichkeit von Störungen durch Fragmente höher chlorierter Kongenere berücksichtigt werden.
14) Falls zutreffend, sind die im "Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food" (https://data.europa.eu/doi/10.2787/8931) beschriebenen Grundsätze zu befolgen.
15) Ein geringerer Beitrag der Reagenzienleerwerte zum Kontaminationsgehalt der Probe ist äußerst empfehlenswert. Das Labor ist dafür zuständig, die Variation der Leerwerte zu überwachen, insbesondere, wenn die Leerwerte abgezogen werden.
16) Siehe Fußnote 37.
| Analysemethoden zur Bestimmung der Bestandteile tierischen Ursprungs bei der amtlichen Untersuchung von Futtermitteln | Anhang VI 13 20 22 |
1. Zweck und Anwendungsbereich 22
Die Bestimmung von Bestandteilen tierischen Ursprungs in Futtermitteln wird nach den Bestimmungen dieses Anhangs mithilfe der Lichtmikroskopie oder der Polymerase-Kettenreaktion (PCR) vorgenommen.
Mit diesen beiden Methoden können Bestandteile tierischen Ursprungs in Vormischungen, Einzelfuttermitteln und Mischfuttermitteln nachgewiesen werden. Die Berechnung der Menge solcher Bestandteile in Vormischungen, Einzelfuttermitteln und Mischfuttermitteln ist mit ihnen jedoch nicht möglich. Bei beiden Methoden liegt die Nachweisgrenze unter 0,1 % (m/m).
Mit der PCR-Methode lässt sich die taxonomische Gruppe der in Vormischungen, Einzelfuttermitteln und Mischfuttermitteln vorhandenen Bestandteile tierischen Ursprungs ermitteln.
Die Methoden werden eingesetzt, um die Anwendung der Verbote gemäß Artikel 7 Absatz 1 der Verordnung (EG) Nr. 999/2001 des Europäischen Parlaments und des Rates 1, Anhang IV der genannten Verordnung sowie Artikel 11 Absatz 1 der Verordnung (EG) Nr. 1069/2009 des Europäischen Parlaments und des Rates 1a zu überwachen.
Abhängig von der Art des zu untersuchenden Futtermittels können diese Methoden für eine Untersuchung entweder einzeln oder kombiniert nach der Standardarbeitsanweisung (SOP) angewandt werden, die das EU-Referenzlabor für tierische Proteine in Futtermitteln (EURL-AP) erstellt und auf seiner Website 1b veröffentlicht hat.
2. Methoden
2.1.1. Grundsatz
Die Bestandteile tierischen Ursprungs, die in zur Analyse versandten Vormischungen, Einzelfuttermitteln und Mischfuttermitteln vorhanden sein können, werden anhand charakteristischer und mikroskopisch erkennbarer Merkmale, zum Beispiel Muskelfasern und andere Fleischpartikel, Knorpel, Knochen, Horn, Haare, Borsten, kutikuläre Fragmente von Wirbellosen, tracheale Strukturen von Insekten, Blutprodukte, Milchkügelchen, Laktosekristalle, Federn, Eierschalen, Gräten und Schuppen identifiziert.
Mikroskopische Untersuchungen werden nach Vorbereitung der Proben mittels Sedimentation durchgeführt.
Die Proben durchlaufen eine Sedimentationsphase wie folgt:
2.1.2. Reagenzien und Geräte 22
2.1.2.1. Reagenzien
2.1.2.1.1. Konzentrationsmittel
2.1.2.1.2. Nachweisreagenz
2.1.2.1.3. Einbettungsmedien
2.1.2.1.4. Färbende Einbettungsmedien
2.1.2.1.5. Spülmittel
2.1.2.1.6. Bleichmittel
2.1.2.2. Geräte
Gläserner Scheidetrichter von 250 ml mit konischem Boden, unten verschlossen mit einem Absperrhahn aus Teflon oder Schliffglas. Öffnung im Absperrhahn ≥ 4 mm Durchmesser. Alternativ kann - nur für eine einmalige Sedimentation mit TCE - auch ein Absetzbecher mit konischem Boden verwendet werden, wenn das Labor bewiesen hat, dass die Nachweisgrenzen denjenigen bei Verwendung des Scheidetrichters gleichwertig sind.
Scheidetrichter
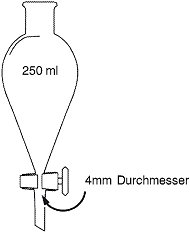
Scheidetrichter
2.1.3. Probenahme und Probenvorbereitung 22
2.1.3.1. Probenahme
Untersucht wird eine repräsentative Probe, die gemäß Anhang I entnommen wurde.
2.1.3.1.1. Trocknung der Proben
Proben mit einem Feuchtigkeitsgehalt > 14 % werden vor Gebrauch gemäß Anhang III getrocknet.
2.1.3.1.2. Vorsieben der Proben
Um Informationen über eine mögliche Verunreinigung der Futtermittel durch die Umwelt (Umweltkontamination) zu erhalten, wird empfohlen, pelletierte Futtermittel und Kerne bis zu einer Größe von 1 mm vorzusieben und die beiden Fraktionen, die als unterschiedliche Proben zu betrachten sind, dann getrennt zu präparieren, zu untersuchen und zu melden.
2.1.3.2. Vorsichtsmaßnahmen
Um eine Kreuzkontamination im Labor zu vermeiden, sind sämtliche wiederverwendbaren Geräte vor Gebrauch sorgfältig zu reinigen. Scheidetrichter werden vor dem Reinigen in ihre Einzelteile zerlegt. Glas- und sonstige Teile der Scheidetrichter werden von Hand vorgewaschen und dann in der Spülmaschine gewaschen. Siebe sind mit einer Bürste mit steifen Synthetikborsten zu reinigen. Nach dem Sieben von fetthaltigem Material wie Fischmehl ist eine abschließende Reinigung der Siebe mit Aceton und Druckluft zu empfehlen.
2.1.3.3. Vorbereitung von Proben aus Fett oder Öl
Für die Vorbereitung von Proben aus Fett gilt folgender Ablauf:
Derselbe Untersuchungsablauf - mit Ausnahme des ersten und des vierten Gedankenstrichs - ist bei der Vorbereitung von Proben aus Öl zu befolgen.
2.1.3.4. Vorbereitung anderer Proben als Fett oder Öl
2.1.3.4.1. Herstellung von Teilproben und Zerkleinern: Von mindestens 50 g der Probe werden Teilproben für die Untersuchung hergestellt und anschließend zerkleinert.
2.1.3.4.2. Vorbereitung von Ausgangsmaterial: Eine Menge von mindestens 5 g der zerkleinerten Teilprobe ist zu präparieren. Das Material wird auf eine Partikelgröße von 0,25 mm heruntergesiebt; beide Fraktionen werden untersucht.
2.1.3.4.3. Einmalige Sedimentation mit TCE zum Nachweis anderer Bestandteile tierischen Ursprungs als von wirbellosen Landtieren.
10 g (bis auf 0,01 g genau) der zerkleinerten Teilprobe werden in den Scheidetrichter bzw. Absetzbecher mit konischem Boden gegeben und mit 50 ml TCE ergänzt. Bei Fischmehl oder anderen reinen tierischen Erzeugnissen, mineralischen Zutaten oder Vormischungen mit mehr als 10 % Sediment wird nur eine Menge von 3 g in den Trichter gegeben. Das Gemisch mindestens 30 s lang kräftig schütteln; dann vorsichtig mindestens 50 ml TCE hinzugeben, wobei darauf zu achten ist, dass von der Innenwand des Trichters sämtliche anhaftenden Partikel abgespült werden. Die entstandene Lösung mindestens 5 min stehen lassen und dann das Sediment durch Öffnen des Absperrhahns abscheiden.
Bei Gebrauch eines Absetzbechers mit konischem Boden das Gemisch mindestens 15 s kräftig schütteln; an der Innenseite des Bechers haftende Partikel mit mindestens 10 ml reinem TCE sorgfältig in das Gefäß hineinspülen. Die Lösung 3 min stehen lassen und wieder 15 s schütteln; noch an der Innenseite des Bechers haftende Partikel mit mindestens 10 ml reinem TCE sorgfältig hineinspülen. Die entstandene Lösung mindestens 5 min stehen lassen; dann die flüssige Fraktion durch vorsichtiges Abgießen trennen und beseitigen, wobei das Sediment vollständig erhalten bleiben muss.
Das Sediment wird auf einem in einen Trichter eingelegten Filterpapier aufgefangen, damit das verbleibende TCE getrennt werden kann, ohne dass sich Fett in das Sediment ablagert. Das Sediment wird getrocknet. Es wird empfohlen, das Sediment anschließend (auf 0,001 g genau) auszuwiegen, um die Sedimentationsphase zu kontrollieren. Schließlich wird das Sediment auf eine Partikelgröße von 0,25 mm heruntergesiebt und beide Fraktionen werden untersucht, es sei denn, das Sieben wird nicht für notwendig erachtet.
Nach Erhalt des Sediments mit der oben beschriebenen Methode müssen zwei Phasen im Scheidetrichter verbleiben: eine aus TCE bestehende flüssige Phase und eine aus aufschwimmendem Material bestehende feste Phase. Diese feste Phase ist das Flotat, das gewonnen wird, indem man das TCE durch Öffnen des Absperrhahns vollständig ablaufen lässt. Das Flotat wird aus dem Scheidetrichter in eine große Petrischale gekippt und im Abzug luftgetrocknet. Es wird auf eine Partikelgröße von 0,25 mm heruntergesiebt, und beide Fraktionen werden untersucht.
Zur korrekten Bestimmung der Bestandteile tierischen Ursprungs kann der Untersuchende bei der Probenvorbereitung Färbereagenzien verwenden, wie vom EURL-AP in den auf seiner Website veröffentlichten Leitlinien beschrieben.
Bei Verwendung von Alizarinrot-Lösung zum Färben des Sediments gilt folgender Ablauf:
2.1.3.4.4. Zweimalige Sedimentation mit PE/TCE zum Nachweis von Bestandteilen wirbelloser Landtiere
Alle Phasen müssen in einem gläsernen Scheidetrichter von 250 ml mit konischem Boden stattfinden, wie unter Nummer 2.1.2.2 vierter Gedankenstrich beschrieben.
2.1.4. Mikroskopische Untersuchung 22
2.1.4.1. Vorbereitung der Objektträger
Von dem Sediment und, je nach Präferenz des Untersuchenden, von dem Flotat oder dem Ausgangsmaterial werden Objektträger präpariert. Gegebenenfalls sind - ausschließlich zum Nachweis von Bestandteilen wirbelloser Landtiere - auch Objektträger von dem gemäß der Beschreibung unter Nummer 2.1.3.4.4 gewonnenen endgültigen Flotat zu präparieren. Die beiden entstandenen Fraktionen (fein und grob) werden präpariert. Die zur Untersuchung auf die Träger gestrichenen Teile der Fraktionen sind repräsentativ für die gesamte Fraktion.
Die Zahl der präparierten Träger muss ausreichen, damit ein kompletter Untersuchungsablauf nach Nummer 2.1.4.2 ausgeführt werden kann.
Die Objektträger werden nach der vom EURL-AP ausgearbeiteten und auf seiner Website veröffentlichten SOP mit dem passenden Einbettungsmedium eingedeckt. Auf den Trägern werden Deckgläser platziert.
2.1.4.2. Untersuchungsablaufplan für den Nachweis tierischer Partikel in Mischfuttermitteln, Einzelfuttermitteln und Vormischungen
Die präparierten Objektträger werden gemäß den Untersuchungsablaufplänen in den Abbildungen 1 und 2 untersucht.
Die mikroskopische Untersuchung des Sediments und, je nach Präferenz des Untersuchenden, des Flotats oder des Ausgangsmaterials wird mit dem zusammengesetzten Mikroskop durchgeführt. Im Fall des Nachweises von Bestandteilen wirbelloser Landtiere sind zusätzlich mikroskopische Untersuchungen des gemäß der Beschreibung unter Nummer 2.1.3.4.4 entsprechend Abbildung 3 gewonnenen endgültigen Flotats durchzuführen. Für die groben Fraktionen kann zusätzlich auch ein Stereomikroskop verwendet werden. Jedes Präparat wird mit unterschiedlicher Vergrößerung vollständig abgesucht. Genaue Erläuterungen zur Verwendung der Ablaufpläne sind in einer SOP enthalten, die das EURL-AP erstellt und auf seiner Website veröffentlicht hat.
In jedem Schritt des Untersuchungsablaufplans ist die festgelegte Mindestzahl von Präparaten zu untersuchen, es sei denn, das gesamte Material der Fraktion reicht dafür nicht aus, beispielsweise wenn kein Sediment erzielt wird. Bei jeder Bestimmung werden höchstens 6 Präparate zur Aufzeichnung der Partikelzahl untersucht.
Werden von dem Flotat oder dem Ausgangsmaterial zusätzliche Objektträger mit einem spezifischeren färbenden Einbettungsmedium gemäß Nummer 2.1.2.1.4 präpariert, um Strukturen (z.B. Federn, Haare, Muskel- oder Blutpartikel) näher zu charakterisieren, die auf mit anderen Einbettungsmedien gemäß Nummer 2.1.2.1.3 präparierten Objektträgern nachgewiesen wurden, so wird die Anzahl der Partikel auf der Grundlage einer Anzahl von höchstens 6 Objektträgern pro Bestimmung gezählt, einschließlich der zusätzlichen Objektträger mit einem spezifischeren Einbettungsmedium. Die zusätzlichen von dem gemäß der Beschreibung unter Nummer 2.1.3.4.4 gewonnenen endgültigen Flotat präparierten Objektträger zum Nachweis von Bestandteilen wirbelloser Landtiere werden für die Bestimmung anderer Arten (Landwirbeltiere und Fische) nicht berücksichtigt.
Für die Bestimmung von Art und Ursprung der Partikel kann der Untersuchende Hilfsinstrumente wie Systeme zur Unterstützung der Entscheidungsfindung, Bildarchive und Referenzproben hinzuziehen.
Abbildung 1
Untersuchungsablaufplan nach einmaliger Sedimentation mit TCE für den Nachweis anderer tierischer Partikel als von wirbellosen Landtieren in Mischfuttermitteln, Einzelfuttermitteln und Vormischungen (erste Bestimmung)
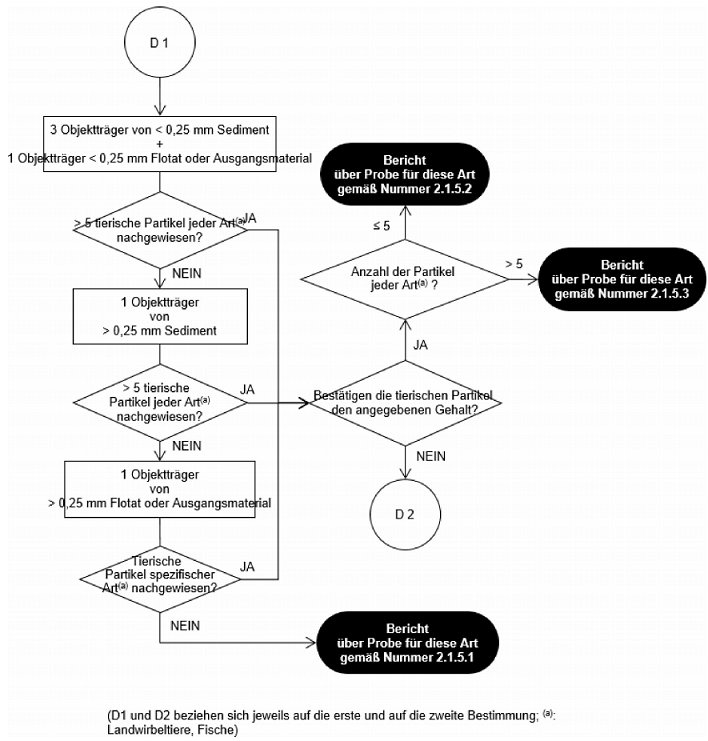
Abbildung 2
Untersuchungsablaufplan nach einmaliger Sedimentation mit TCE für den Nachweis anderer tierischer Partikel als von wirbellosen Landtieren in Mischfuttermitteln, Einzelfuttermitteln und Vormischungen (zweite Bestimmung)
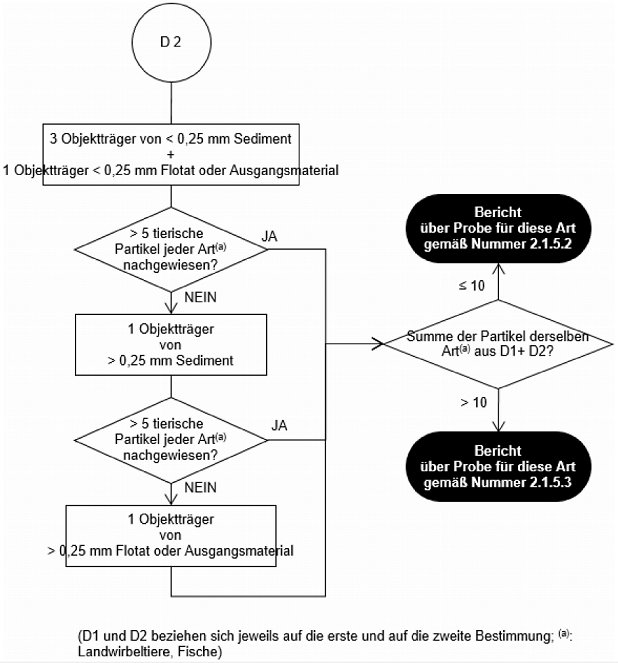
Abbildung 3
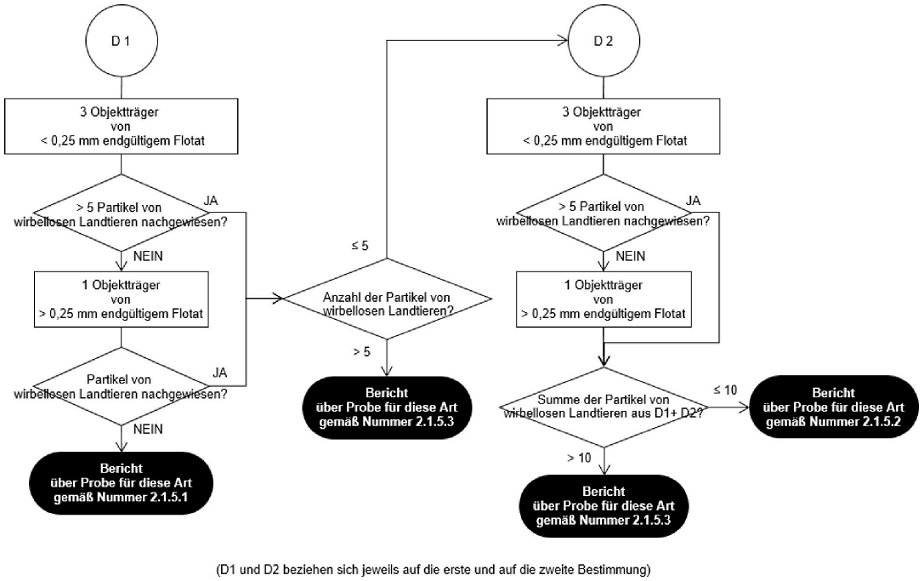
Untersuchungsablaufplan nach zweimaliger Sedimentation mit PE/TCE für den Nachweis von Bestandteilen wirbelloser Landtiere in Mischfuttermitteln, Einzelfuttermitteln und Vormischungen
2.1.4.3. Anzahl der Bestimmungen
Die Bestimmungen sind mit verschiedenen Teilproben von jeweils 50 g durchzuführen.
Werden bei der ersten Bestimmung gemäß dem Untersuchungsablaufplan in Abbildung 1 bzw. Abbildung 3 keine tierischen Partikel nachgewiesen, so ist keine weitere Bestimmung erforderlich, und über das Ergebnis der Analyse wird unter Verwendung der Formulierung in Nummer 2.1.5.1 berichtet.
Werden bei der ersten Bestimmung gemäß dem Untersuchungsablaufplan in Abbildung 1 ein oder mehrere tierische Partikel spezifischer Art (d. h. von Landwirbeltieren oder Fischen) nachgewiesen und bestätigt die Art der gefundenen Partikel den für die Probe angegebenen Gehalt, so ist keine zweite Bestimmung erforderlich. Liegt bei der ersten Bestimmung die Anzahl der nachgewiesenen tierischen Partikel spezifischer Art über 5, so wird über das Ergebnis der Analyse pro Art der Tiere unter Verwendung der Formulierung in Nummer 2.1.5.3 berichtet. Andernfalls wird über das Ergebnis der Analyse pro Art der Tiere unter Verwendung der Formulierung in Nummer 2.1.5.2 berichtet.
Werden bei der ersten Bestimmung gemäß dem Untersuchungsablaufplan in Abbildung 3 mehr als 5 Partikel wirbelloser Landtiere nachgewiesen, so ist keine zweite Bestimmung erforderlich, und über das Ergebnis der Analyse für diese Art wird unter Verwendung der Formulierung in Nummer 2.1.5.3 berichtet.
In allen anderen Fällen, einschließlich wenn dem Labor keine Angabe zum Gehalt vorgelegt wurde, wird eine zweite Bestimmung mit einer neuen Teilprobe durchgeführt. Liegt nach der zweiten Bestimmung gemäß dem Untersuchungsablaufplan in Abbildung 2 bzw. Abbildung 3 die Summe der in den zwei Durchgängen nachgewiesenen tierischen Partikel spezifischer Art über 10, so wird über das Ergebnis der Analyse pro Art der Tiere unter Verwendung der Formulierung in Nummer 2.1.5.3 berichtet. Andernfalls wird über das Ergebnis der Analyse pro Art der Tiere unter Verwendung der Formulierung in Nummer 2.1.5.2 berichtet.
2.1.5. Formulierung der Ergebnisse 20 22
In seinem Bericht über die Ergebnisse gibt das Labor an, welche Art von Material (Sediment, Flotat, endgültiges Flotat oder Ausgangsmaterial) analysiert wurde. Aus dem Bericht muss eindeutig hervorgehen, wie viele Bestimmungen durchgeführt wurden und ob die Fraktionen vor der Vorbereitung der Objektträger nicht gemäß Nummer 2.1.3.4.3 erster Gedankenstrich dritter Absatz oder Nummer 2.1.3.4.4 dritter Gedankenstrich gesiebt wurden.
Der Laborbericht enthält zumindest Informationen über das Vorhandensein von Bestandteilen, die von Landwirbeltieren und von Fischen stammen.
Die verschiedenen Fälle werden wie folgt dargestellt:
2.1.5.1. Kein tierisches Partikel spezifischer Art nachgewiesen:
2.1.5.2. Zwischen 1 und 5 tierische Partikel spezifischer Art nachgewiesen, wenn nur eine Bestimmung durchgeführt wurde, oder zwischen 1 und 10 Partikel spezifischer Art nachgewiesen, wenn zwei Bestimmungen durchgeführt wurden (die Anzahl der festgestellten Partikel liegt unter der Entscheidungsgrenze, die in den SOP des EURL-AP festgelegt und auf seiner Website veröffentlicht wurde):
Wenn nur eine Bestimmung durchgeführt wurde:
Wenn zwei Bestimmungen durchgeführt wurden:
Außerdem gilt Folgendes:
2.1.5.3. Mehr als 5 tierische Partikel spezifischer Art nachgewiesen, wenn nur eine Bestimmung durchgeführt wurde, oder mehr als 10 Partikel spezifischer Art nachgewiesen, wenn zwei Bestimmungen durchgeführt wurden:
Wenn nur eine Bestimmung durchgeführt wurde:
Wenn zwei Bestimmungen durchgeführt wurden:
Außerdem gilt Folgendes:
2.2. PCR
2.2.1. Grundsatz
Aus Desoxyribonucleinsäure (DNA) bestehende Fragmente tierischen Ursprungs, die in Einzelfuttermittel und Mischfuttermitteln vorhanden sein können, werden durch PCR mit einer Genamplifikationstechnik nachgewiesen, die nach tierartspezifischen DNA-Sequenzen sucht.
Für die PCR-Methode muss zunächst DNA extrahiert werden. Der Amplifikationsschritt wird anschließend an dem so erhaltenen DNA-Extrakt durchgeführt, um die Tierart nachzuweisen, auf die untersucht wird.
2.2.2. Reagenzien und Geräte
2.2.2.1. Reagenzien
2.2.2.1.1. Reagenzien für die DNA-Extraktion
Nur vom EURL-AP genehmigte und auf dessen Website veröffentlichte Reagenzien dürfen verwendet werden.
2.2.2.1.2. Reagenzien für die genetische Amplifikation
2.2.2.1.2.1. Primer und Sonden
Nur Primer und Sonden mit vom EURL-AP validierten Oligonucleotid-Sequenzen dürfen verwendet werden 2.
2.2.2.1.2.2. Master Mix
Nur Master-Mix-Lösungen ohne Reagenzien, die zu falschen Ergebnissen führen können, dürfen verwendet werden 3.
2.2.2.1.2.3. Dekontaminations-Reagenzien
2.2.2.1.2.3.1. Salzsäure-Maßlösung (0,1 N)
2.2.2.1.2.3.2. Bleichmittel (Natriumhypochlorit-Lösung, 0,15 % aktives Chlor)
2.2.2.1.2.3.3. Nicht ätzende Reagenzien für die Dekontamination kostspieliger Geräte wie Analysewaagen (z.B. DNA EraseTM von MP Biomedicals)
2.2.2.2. Geräte
2.2.2.2.1. Analysenwaage mit einer Genauigkeit von 0,001 g
2.2.2.2.2. Zerkleinerungsgeräte
2.2.2.2.3. Thermocycler für Echtzeit-PCR
2.2.2.2.4. Mikrozentrifuge für entsprechende Reaktionsgefäße
2.2.2.2.5. Satz von Mikropipetten für das Pipettieren von 1 bis 1 000 µl
2.2.2.2.6. In der Molekularbiologie übliche Verbrauchsmaterialien: Mikrozentrifugen-Röhrchen, Kunststofffilterspitzen für Mikropipetten, geeignete Reaktionsgefäße für den Thermocycler
2.2.2.2.7. Kühlschränke für die Lagerung von Proben und Reagenzien
2.2.3. Probenahme und Probenvorbereitung
2.2.3.1. Probenahme
Untersucht wird eine repräsentative Probe, die nach der Beschreibung in Anhang I entnommen wurde.
2.2.3.2. Probenvorbereitung
Die Vorbereitung von Laborproben bis hin zur Extraktion der DNA entspricht den Anforderungen in Anhang II. Von mindestens 50 g der Probe werden Teilproben für die Untersuchung hergestellt und anschließend zerkleinert.
Die Proben werden gemäß ISO 24276 in einem anderen als den Räumen vorbereitet, in denen DNA extrahiert und vermehrt wird.
Es werden zwei Testmengen zu je mindestens 100 mg vorbereitet.
2.2.4. Extraktion der DNA
Die DNA wird nach der vom EURL-AP ausgearbeiteten und auf seiner Website veröffentlichten SOP aus beiden Testmengen extrahiert.
Für jede Extraktionsserie werden nach ISO 24276 zwei Extraktionskontrollen vorbereitet:
2.2.5. Genetische Amplifikation
Die genetische Amplifikation geschieht anhand von Methoden, die für die zu bestimmenden Tierarten validiert wurden. Diese Methoden sind in der vom EURL-AP ausgearbeiteten und auf seiner Website veröffentlichten SOP beschrieben. Jedes DNA-Extrakt ist in mindestens zwei unterschiedlichen Verdünnungen zu untersuchen, um die Inhibition zu bewerten.
Für jede Zieltierart werden nach ISO 24276 zwei Amplifikationskontrollen vorbereitet:
2.2.6. Interpretation und Formulierung der Ergebnisse
Bei der Berichterstattung über die Ergebnisse gibt das Labor zumindest das Gewicht der Testmengen, die Extraktionstechnik, die Zahl der durchgeführten Bestimmungen und die Nachweisgrenze der Methode an.
Ergebnisse werden nicht interpretiert und berichtet, wenn die positive DNA-Extraktionskontrolle und die positiven DNA-Zielkontrollen keine positiven Ergebnisse für das gesuchte Ziel ergeben und die Amplifikationsreagenz-Kontrolle gleichzeitig negativ ist.
Stimmen die Ergebnisse der beiden Testmengen nicht überein, ist zumindest die genetische Amplifikation zu wiederholen. Geht das Labor davon aus, dass dies an den DNA-Extrakten liegen kann, wird vor der Interpretation der Ergebnisse erneut DNA extrahiert und vermehrt.
Die abschließende Formulierung der Ergebnisse stützt sich auf die Integration und Interpretation der Ergebnisse der beiden Testmengen nach der vom EURL-AP ausgearbeiteten und auf seiner Website veröffentlichten SOP.
2.2.6.1. Negatives Ergebnis
Über ein negatives Ergebnis wird wie folgt berichtet:
In der vorliegenden Probe wurde keine DNA von X nachgewiesen (wobei X die Tierart oder Gruppe von Tierarten ist, auf die untersucht wird).
2.2.6.2. Positives Ergebnis
Über ein positives Ergebnis wird wie folgt berichtet:
In der vorliegenden Probe wurde DNA von X nachgewiesen (wobei X die Tierart oder Gruppe von Tierarten ist, auf die untersucht wird).
_______
1) Verordnung (EG) Nr. 999/2001 des Europäischen Parlaments und des Rates vom 22. Mai 2001 mit Vorschriften zur Verhütung, Kontrolle und Tilgung bestimmter transmissibler spongiformer Enzephalopathien (ABl. L 147 vom 31.05.2001 S. 1).
1a) Verordnung (EG) Nr. 1069/2009 des Europäischen Parlaments und des Rates vom 21. Oktober 2009 mit Hygienevorschriften für nicht für den menschlichen Verzehr bestimmte tierische Nebenprodukte und zur Aufhebung der Verordnung (EG) Nr. 1774/2002 (Verordnung über tierische Nebenprodukte) (ABl. L 300 vom 14.11.2009 S. 1).
1b) https://www.eurl.craw.eu/legal-sources-and-sops/method-of-reference-and-sops/
2) Die Liste dieser Primer und Sonden für die einzelnen gesuchten Tierarten findet sich auf der Website des EURL-AP.
3) Beispiele brauchbarer Master Mixes finden sich auf der Website des EURL-AP.
| Methode zur Berechnung des Energiegehalts von Futtermitteln für Geflügel | Anhang VII 24 |
1. Berechnungsmethode und Formel des Energiegehalts
Der Energiegehalt von Mischfuttermitteln für Geflügel muss anhand der prozentualen Anteile bestimmter analytischer Bestandteile der Futtermittel nach nachstehender Formel berechnet werden. Dieser Wert wird in Megajoules (MJ) umsetzbarer, N-korrigierter Energie (ME) je kg Mischfuttermittel ausgedrückt und nach folgender Formel berechnet:
MJ ME/kg = 0,1551 × % Rohprotein + 0,3431 × % Rohfett + 0,1669 × % Stärke + 0,1301 × % Gesamtzucker (ausgedrückt als Saccharose).
2. Toleranzen auf die angegebenen Gehalte
Ergibt sich bei den amtlichen Untersuchungen eine energieerhöhende oder energievermindernde Abweichung zwischen dem Kontrollergebnis und dem angegebenen Energiegehalt, so gilt eine Toleranz von 0,4 MJ ME/kg.
3. Angabe der Ergebnisse
Das nach vorstehender Formel errechnete Ergebnis ist mit nur einer Dezimalstelle anzugeben.
4. Probenahmeverfahren und Analysemethoden
Die Probenahme beim Mischfuttermittel und die Bestimmung des Gehalts an der Berechnungsmethode zugrunde liegenden analytischen Bestandteilen erfolgt in Übereinstimmung mit den Probenahmeverfahren bzw. Analysemethoden der Union für die amtliche Untersuchung von Futtermitteln.
Anzuwenden sind:
Methode zur Berechnung des Energiegehalts von Futtermittel-Ausgangserzeugnissen und Mischfuttermitteln für Katzen und Hunde
Der Energiegehalt von Futtermittel-Ausgangserzeugnissen und Mischfuttermitteln für Katzen und Hunde ist gemäß folgender Norm zu berechnen: EN 16967: Futtermittel - Probenahme- und Untersuchungsverfahren - Schätzgleichungen für umsetzbare Energie in Futtermittel-Ausgangserzeugnissen und Mischfuttermitteln (Heimtierfutter) für Katzen und Hunde, einschließlich Diätfuttermittel.
| - gestrichen - | Anhang VIII 24 |
| Entsprechungstabellen gemäss Artikel 6 | Anhang IX |
| Richtlinie 71/250/EWG | Vorliegende Verordnung |
| Artikel 1 Unterabsatz 1 | Artikel 3 |
| Artikel 1 Unterabsatz 2 | Artikel 2 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | Anhang II |
| Anhang Teil 2 | - |
| Anhang Teil 3 | - |
| Anhang Teil 4 | Anhang III Teil O |
| Anhang Teil 5 | Anhang III Teil M |
| Anhang Teil 6 | Anhang III Teil N |
| Anhang Teil 7 | Anhang III Teil Q |
| Anhang Teil 9 | Anhang III Teil K |
| Anhang Teil 10 | - |
| Anhang Teil 11 | - |
| Anhang Teil 12 | Anhang III Teil J |
| Anhang Teil 14 | Anhang III Teil D |
| Anhang Teil 16 | - |
| Richtlinie 71/393/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil I | Anhang III Teil A |
| Anhang Teil II | Anhang III Teil E |
| Anhang Teil III | Anhang III Teil P |
| Anhang Teil IV | Anhang III Teil H |
| Richtlinie 72/199/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang I Teil 1 | Anhang III Teil L |
| Anhang I Teil 2 | Anhang III Teil C |
| Anhang I Teil 2 | - |
| Anhang I Teil 3 | - |
| Anhang I Teil 3 | Anhang V Teil A |
| Anhang II | - |
| Richtlinie 73/46/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang I Teil 1 | Anhang III Teil B |
| Anhang I Teil 2 | - |
| Anhang I Teil 3 | Anhang III Teil I |
5. Richtlinie 76/371/EWG
| Richtlinie 76/371/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 1 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang I |
| Richtlinie 76/372/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
| Richtlinie 78/633/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | - |
| Anhang Teil 2 | - |
| Anhang Teil 3 | Anhang IV Teil C |
8. Richtlinie 81/715/EWG
| Richtlinie 81/715/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
9. Richtlinie 84/425/EWG
| Richtlinie 84/425/EWG | Vorliegende Verordnung |
| Artikel 1 | - |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | - |
10. Richtlinie 86/174/EWG
| Richtlinie 86/174/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 4 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang VII |
11. Richtlinie 93/70/EWG
| Richtlinie 93/70/EWG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang | Anhang IV Teil D |
12. Richtlinie 93/117/EG
| Richtlinie 93/117/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Anhang Teil 1 | Anhang IV Teil E |
| Anhang Teil 2 | Anhang VIII Teil A |
13. Richtlinie 98/64/EG
| Richtlinie 98/64/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang Teil A | Anhang III Teil F |
| Anhang Teil C | Anhang VIII Teil B |
14. Richtlinie 1999/27/EG
| Richtlinie 1999/27/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 und 5 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Artikel 6 | - |
| Artikel 7 | - |
| Anhang Teil A | Anhang VIII Teil C |
| Anhang Teil B | Anhang IV Teil F |
| Anhang Teil C | Anhang VIII Teil D |
15. Richtlinie 1999/76/EG
| Richtlinie 1999/76/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang | Anhang IV Teil G |
16. Richtlinie 2000/45/EG
| Richtlinie 2000/45/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Anhang Teil A | Anhang IV Teil A |
| Anhang Teil B | Anhang IV Teil B |
| Anhang Teil C | Anhang III Teil G |
17. Richtlinie 2002/70/EG
| Richtlinie 2002/70/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 1 |
| Artikel 2 | Artikel 2 und 3 |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Anhang I | Anhang I und Anhang V Teil B(I) |
| Anhang II | Anhang II und Anhang V Teil B(II) |
| Richtlinie 2003/126/EG | Vorliegende Verordnung |
| Artikel 1 | Artikel 3 |
| Artikel 2 | - |
| Artikel 3 | - |
| Artikel 4 | - |
| Artikel 5 | - |
| Artikel 6 | - |
| Anhang | Anhang VI |
| ENDE | |