

umwelt-online: VerfO - Verfahrensordnung des Gemeinsamen Bundesausschusses (2)
| zurück | |
5. Kapitel 11a 18c 19a
Bewertung des Nutzens und der Kosten von Arzneimitteln nach § § 35a und 35b SGB V
1. Abschnitt
Bewertung des Nutzens von Arzneimitteln
1. Titel
Geltungsbereich und Begriffsdefinitionen
(1) Dieses Kapitel regelt auf der Grundlage der Arzneimittel-Nutzenbewertungsverordnung (AM-NutzenV) das Verfahren der Nutzenbewertung von erstattungsfähigen Arzneimitteln mit neuen Wirkstoffen nach § 35a Absatz 1 SGB V, insbesondere die Beratung, die Anforderungen an die für den Beleg des Nutzens erforderlichen Nachweise (Dossier), das Verfahren der Anhörungen und die Umsetzung der Nutzenbewertung in die Arzneimittel-Richtlinie.
(2) Die Nutzenbewertung nach § 35a Absatz 1 SGB V wird durchgeführt für erstattungsfähige Arzneimittel mit neuen Wirkstoffen und neuen Wirkstoffkombinationen,
(2a) Die Nutzenbewertung nach § 35a Absatz 6 SGB V wird auf Veranlassung des Gemeinsamen Bundesausschusses nach § 16 durchgeführt für erstattungsfähige Arzneimittel mit Wirkstoffen oder Wirkstoffkombinationen, die keine neuen Wirkstoffe oder Wirkstoffkombinationen im Sinne des § 2 Absatz 1 sind, sowie für Arzneimittel mit einem neuen Wirkstoff oder Wirkstoffkombinationen im Sinne des § 2 Absatz 1, wenn für das Arzneimittel eine neue Zulassung mit neuem Unterlagenschutz erteilt wird.
(3) Die Regelungen des 4. Kapitels der Verfahrensordnung (VerfO) bleiben hiervon unberührt.
§ 2 Arzneimittel mit neuen Wirkstoffen
(1) Arzneimittel mit neuen Wirkstoffen sind Arzneimittel, die Wirkstoffe enthalten, deren Wirkungen bei der erstmaligen Zulassung in der medizinischen Wissenschaft nicht allgemein bekannt sind. Ein Arzneimittel mit neuen Wirkstoffen gilt so lange als ein Arzneimittel mit einem neuen Wirkstoff, wie für das erstmalig zugelassene Arzneimittel mit dem Wirkstoff Unterlagenschutz besteht. Als Arzneimittel im Sinne von Satz 1 gelten auch
(2) Ein neues Anwendungsgebiet ist ein Anwendungsgebiet, für das nach § 29 Absatz 3 Nummer 3 des Arzneimittelgesetzes (AMG) eine neue Zulassung erteilt wird oder das als größere Änderung des Typs 2 nach Anhang 2 Nummer 2 Buchstabe a der Verordnung (EG) Nr. 1234/ 2008 der Kommission vom 24. November 2008 über die Prüfung von Änderungen der Zulassungen von Human- und Tierarzneimitteln (ABl. L 334 vom 12.12.2008 S. 7) eingestuft wird. Ein Anwendungsgebiet ist im Vergleich zu dem bereits zugelassenen Anwendungsgebiet eines Arzneimittels insbesondere neu, wenn sich der Indikationsanspruch des Anwendungsgebietes auf einen Patientenkreis bezieht, der von bereits zugelassenen Anwendungsgebieten abweicht, eine Indikation hinzugefügt wird, die einem anderen therapeutischen Bereich (Behandlung, Diagnose oder Prophylaxe) zuzurechnen ist, oder die Indikation in einen anderen therapeutischen Bereich (Behandlung, Diagnose oder Prophylaxe) verlagert wird.
§ 3 Nutzen und Zusatznutzen
(1) Der Nutzen eines Arzneimittels ist der patientenrelevante therapeutische Effekt insbesondere hinsichtlich der Verbesserung des Gesundheitszustands, der Verkürzung der Krankheitsdauer, der Verlängerung des Überlebens, der Verringerung von Nebenwirkungen oder einer Verbesserung der Lebensqualität.
(2) Der Zusatznutzen eines Arzneimittels ist ein Nutzen nach Absatz 1, der qualitativ oder quantitativ höher ist als der Nutzen, den die zweckmäßige Vergleichstherapie aufweist.
§ 4 Zuständigkeit für die Durchführung der Nutzenbewertung
(1) Für die Durchführung des Bewertungsverfahrens ist der Unterausschuss Arzneimittel zuständig. Er richtet hierzu Arbeitsgruppen ein, die insbesondere mit der Durchführung folgender Aufgaben beauftragt werden können:
(2) Über die Durchführung der Nutzenbewertung wird eine zusammenfassende Dokumentation erstellt. Die zusammenfassende Dokumentation enthält:
(3) Der Unterausschuss berät auf der Basis eines Berichts der Arbeitsgruppe und legt dem Plenum das Ergebnis seiner Bewertung sowie einen Beschlussentwurf vor.
2. Titel
Nachweis des Zusatznutzens und Bestimmung der Vergleichstherapie
§ 5 Anforderungen an den Nachweis des Zusatznutzens durch den pharmazeutischen Unternehmer 18
(1) Der Zusatznutzen ist vom pharmazeutischen Unternehmer im Dossier nach § 9 nachzuweisen. Der Gemeinsame Bundesausschuss hat keine Amtsermittlungspflicht.
(2) Für erstattungsfähige Arzneimittel mit neuen Wirkstoffen gemäß 4. Kap § § 19 bis 22 VerfO, die pharmakologisch-therapeutisch vergleichbar mit Festbetragsarzneimitteln sind, ist der medizinische Zusatznutzen als therapeutische Verbesserung entsprechend § 35 Absatz 1 b Satz 1 bis 5 SGB V nachzuweisen. Der Nachweis einer therapeutischen Verbesserung erfolgt aufgrund der Fachinformationen und durch Bewertung von klinischen Studien nach den internationalen Standards der evidenzbasierten Medizin. Vorrangig sind klinische Studien, insbesondere direkte Vergleichsstudien mit anderen Arzneimitteln dieser Festbetragsgruppe mit patientenrelevanten Endpunkten, insbesondere Mortalität, Morbidität und Lebensqualität, zu berücksichtigen.
(3) Für Arzneimittel mit neuen Wirkstoffen, die die Voraussetzungen nach Absatz 2 nicht erfüllen, erfolgt der Nachweis eines Zusatznutzens indikationsspezifisch im Vergleich zu der nach § 6 bestimmten zweckmäßigen Vergleichstherapie auf der Grundlage von Unterlagen zum Nutzen des Arzneimittels in den zugelassenen Anwendungsgebieten. Basis sind die arzneimittelrechtliche Zulassung, die behördlich genehmigten Produktinformationen sowie Bekanntmachungen von Zulassungsbehörden und die Bewertung von klinischen Studien nach den internationalen Standards der evidenzbasierten Medizin. Sofern es unmöglich oder unangemessen ist, Studien höchster Evidenzstufe durchzuführen oder zu fordern, sind mit besonderer Begründung des pharmazeutischen Unternehmers Nachweise der best verfügbaren Evidenzstufe einzureichen. Darüber hinaus hat er darzulegen, inwieweit die von ihm als best verfügbar eingereichte Evidenz zum Nachweis eines Zusatznutzens geeignet ist. Die Anerkennung des Zusatznutzens auf Grundlage von Unterlagen einer niedrigeren Evidenzstufe bedarf jedoch umso mehr einer Begründung, je weiter von der Evidenzstufe I abgewichen wird.
(4) Im Dossier ist unter Angabe der Aussagekraft der Nachweise darzulegen, mit welcher Wahrscheinlichkeit und in welchem Ausmaß ein Zusatznutzen vorliegt; hinsichtlich der Wahrscheinlichkeit ist darzulegen, mit welcher Sicherheit eine Aussage über das Vorhandensein eines Zusatznutzens getroffen werden kann (Ergebnissicherheit). Diese Angaben zur Aussagekraft der Ergebnisse sollen sowohl bezogen auf die Anzahl der Patientinnen und Patienten als auch bezogen auf die Größe des Zusatznutzens erfolgen.
(5) Für Arzneimittel nach Absatz 3 wird der Zusatznutzen gegenüber der zweckmäßigen Vergleichstherapie festgestellt als Verbesserung der Beeinflussung patientenrelevanter Endpunkte zum Nutzen gemäß § 3 Absatz 1; bei der Bewertung des Zusatznutzens von Antibiotika soll die Resistenzsituation berücksichtigt werden. Vorrangig sind für den Nachweis des Zusatznutzens randomisierte, verblindete und kontrollierte direkte Vergleichsstudien zu berücksichtigen, deren Methodik internationalen Standards und der evidenzbasierten Medizin entspricht und die an Populationen oder unter Bedingungen durchgeführt sind, die für die übliche Behandlungssituation repräsentativ und relevant sind sowie gegenüber einer zweckmäßigen Vergleichstherapie gemäß § 6 durchgeführt wurden. Liegen keine direkten Vergleichsstudien für das neue Arzneimittel gegenüber der zweckmäßigen Vergleichstherapie vor oder lassen diese keine Aussagen über den Zusatznutzen zu, können verfügbare klinische Studien, vorrangig randomisierte, verblindete und kontrollierte Studien, für die zweckmäßige Vergleichstherapie herangezogen werden, die sich für einen indirekten Vergleich gegenüber dem Arzneimittel mit neuen Wirkstoffen und somit für den Nachweis eines Zusatznutzens durch indirekten Vergleich eignen. Können zum Zeitpunkt der Bewertung valide Daten zu patientenrelevanten Endpunkten noch nicht vorliegen, erfolgt die Bewertung auf Grundlage der best verfügbaren Evidenz unter Berücksichtigung der Studienqualität mit Angabe der Wahrscheinlichkeit für den Beleg eines Zusatznutzens.
(6) Die Aussagekraft der Nachweise ist unter Berücksichtigung der Studienqualität, der Validität der herangezogenen Endpunkte sowie der Evidenzstufe darzulegen und es ist zu bewerten, mit welcher Wahrscheinlichkeit und in welchem Ausmaß ein Zusatznutzen vorliegt; Absatz 4 Satz 1 Halbs. gilt entsprechend. Die vorgelegten Studien werden hinsichtlich ihrer Planungs-, Durchführungs- und Auswertungsqualität und ihrer Aussagekraft zur Relevanz des Zusatznutzens bewertet. Im Dossier ist für alle eingereichten Unterlagen darzulegen, auf welcher Evidenzstufe diese erbracht werden. Es gelten folgende Evidenzstufen:
(7) Für Arzneimittel nach Absatz 3 sind das Ausmaß des Zusatznutzens und die therapeutische Bedeutung des Zusatznutzens unter Berücksichtigung des Schweregrades der Erkrankung gegenüber der zweckmäßigen Vergleichstherapie wie folgt zu quantifizieren:
§ 6 Zweckmäßige Vergleichstherapie
(1) Zweckmäßige Vergleichstherapie ist diejenige Therapie, deren Nutzen mit dem Nutzen eines Arzneimittels mit neuen Wirkstoffen für die Nutzenbewertung nach § 35a SGB V verglichen wird.
(2) Die zweckmäßige Vergleichstherapie ist regelhaft zu bestimmen nach Maßstäben, die sich aus den internationalen Standards der evidenzbasierten Medizin ergeben.
(3) Die zweckmäßige Vergleichstherapie muss eine nach dem allgemein anerkannten Stand der medizinischen Erkenntnisse zweckmäßige Therapie im Anwendungsgebiet sein (§ 12 SGB V), vorzugsweise eine Therapie, für die Endpunktstudien vorliegen und die sich in der praktischen Anwendung bewährt hat, soweit nicht Richtlinien nach § 92 Absatz 1 SGB V oder das Wirtschaftlichkeitsgebot dagegen sprechen. Bei der Bestimmung der zweckmäßigen Vergleichstherapie sind insbesondere folgende Kriterien zu berücksichtigen:
(4) Sind nach den Absätzen 1 bis 3 mehrere Alternativen für die Vergleichstherapie gleichermaßen zweckmäßig, kann der Zusatznutzen gegenüber einer dieser Therapien nachgewiesen werden.
(5) Für Arzneimittel einer Wirkstoffklasse ist unter Berücksichtigung von Absatz 3 die gleiche zweckmäßige Vergleichstherapie heranzuziehen, um eine einheitliche Bewertung zu gewährleisten. Die zweckmäßige Vergleichstherapie muss auch geeignet sein für Bewertungen von Arzneimitteln auf Veranlassung des Gemeinsamen Bundesausschusses nach § 35a Absatz 6 SGB V, die vor dem 1. Januar 2011 in den Verkehr gebracht worden sind.
(1) Der Gemeinsame Bundesausschuss berät den pharmazeutischen Unternehmer aufgrund einer schriftlichen Anforderung auf Grundlage der eingereichten Unterlagen nach Satz 6 insbesondere zu konkreten Inhalten der vorzulegenden Unterlagen und Studien sowie zur zweckmäßigen Vergleichstherapie. Beratungen zum Inhalt von abgeschlossenen Verfahren sowie anhängigen Rechtsverfahren sind grundsätzlich ausgeschlossen. Es findet keine Vorprüfung von Daten im Hinblick auf eine zukünftige Dossiereinreichung statt. Für die Anforderung ist das Formular gemäß Anlage I (Anforderungsformular) zu verwenden. In dem Anforderungsformular (Anlage I) sind die Fragen in deutscher Sprache zu übermitteln, die im Beratungsgespräch erörtert werden sollen. Der pharmazeutische Unternehmer übermittelt dem Gemeinsamen Bundesausschuss die für die Erstellung eines Dossiers zur Nutzenbewertung bedeutsamen Unterlagen und Informationen, über die er zu diesem Zeitpunkt verfügt, in deutscher oder englischer Sprache. Die Beratungen werden innerhalb von acht Wochen nach Einreichen der Unterlagen durchgeführt. Übermittelt der pharmazeutische Unternehmer die für die Durchführung der Beratung erforderlichen Unterlagen nicht, kann der Gemeinsame Bundesausschuss eine Beratung ablehnen. Die Beratung wird durch die Geschäftsstelle des Gemeinsamen Bundesausschusses durchgeführt, sofern er nichts anderes beschließt. Eine Beratung vor Beginn von Zulassungsstudien der Phase drei oder zur Planung klinischer Prüfungen soll unter Beteiligung des Bundesinstituts für Arzneimittel und Medizinprodukte oder des Paul-Ehrlich-Instituts stattfinden.
(1a) Die Anforderung ist in elektronischer Form einzureichen; die Einreichung erfolgt mit einem Anschreiben in Schriftform oder elektronischer Form unter Verwendung einer qualifizierten elektronischen Signatur. Als Datenträger ist eine Digital Versatile Disc (DVD) zu verwenden, sofern der Gemeinsame Bundesausschuss nicht andere Verfahren zur elektronischen Einreichung von Unterlagen zur Nutzenbewertung nach § 35a SGB V zur Verfügung stellt. Die Datenträger dürfen nicht kopiergeschützt sein. Für alle einzureichenden Dateien gilt, dass diese nicht geschützt sein dürfen, das heißt sie müssen ohne Kennworteingabe lesbar, speicherbar und druckbar sein. Die Anforderung ist zudem in deutscher Sprache einzureichen.
(2) Die im Rahmen der Beratung übermittelten Informationen sind vertraulich zu behandeln. Der pharmazeutische Unternehmer erhält eine Niederschrift über das Beratungsgespräch. Der Gemeinsame Bundesausschuss kann über die im Beratungsgespräch erörterten Themen Vereinbarungen mit dem pharmazeutischen Unternehmer treffen. Die vom Gemeinsamen Bundesausschuss im Rahmen einer Beratung erteilten Auskünfte zu Beratungsthemen nach Absatz 1 Satz 1 sind nicht verbindlich.
(3) Die Beratung wird in deutscher Sprache durchgeführt.
(4) Für die Beratung werden Gebühren erhoben. Das Nähere zur Höhe der Gebühren ist in der Gebührenordnung geregelt.
(5) Bei Nutzenbewertungen nach § 16 ist eine Beratung anzubieten, bevor der Gemeinsame Bundesausschuss den pharmazeutischen Unternehmer zur Einreichung eines Dossiers auffordert
3. Titel
Bewertungsverfahren
§ 8 Beginn des Bewertungsverfahrens 18 18c
(1) Das Bewertungsverfahren beim Gemeinsamen Bundesausschuss beginnt zu folgenden Zeitpunkten:
(2) Der Gemeinsame Bundesausschuss kann zum Zwecke der Zusammenlegung von Bewertungsverfahren den Zeitpunkt für die Vorlage der erforderlichen Nachweise auf Antrag des pharmazeutischen Unternehmers abweichend von Absatz 1 Nummer 1 oder Nummer 2 bestimmen, wenn innerhalb eines Zeitraums von sechs Monaten ab dem nach Absatz 1 Nummer 1 oder Nummer 2 maßgeblichen Zeitpunkt die Zulassung von mindestens einem neuen Anwendungsgebiet oder weiteren neuen Anwendungsgebieten zu erwarten ist. Der vom Gemeinsamen Bundesausschuss bestimmte Zeitpunkt darf nicht mehr als sechs Monate nach dem maß- geblichen Zeitpunkt nach Absatz 1 Nummer 1 oder Nummer 2 liegen. Der pharmazeutische Unternehmer hat den Antrag nach Satz 1 spätestens drei Monate vor dem maßgeblichen Zeitpunkt nach Absatz 1 Nummer 1 oder Nummer 2 zu stellen. Dem Antrag sind begründende Unterlagen zu den zu erwartenden Zulassungszeitpunkten beizufügen. Der Gemeinsame Bundesausschuss entscheidet über den Antrag innerhalb von acht Wochen. Der nach Satz 1 bestimmte Zeitpunkt gilt als neuer maßgeblicher Zeitpunkt für den Verfahrensbeginn der zusammenzulegenden Verfahren.
§ 9 Anforderungen an das Dossier 18a 18c 19a
(1) Das Dossier dient der Bewertung des Nutzens des Arzneimittels. Das Dossier ist in deutscher Sprache einzureichen, soweit sich aus den Vorgaben für das Dossier nichts anderes ergibt. Das Dossier ist in elektronischer Form einzureichen; die Einreichung erfolgt mit einem Anschreiben in Schriftform oder elektronischer Form unter Verwendung einer qualifizierten elektronischen Signatur . Als Datenträger ist eine Digital Versatile Disc (DVD) zu verwenden, sofern der Gemeinsame Bundesausschuss nicht andere Verfahren zur elektronischen Einreichung von Unterlagen zur Nutzenbewertung nach § 35a SGB V zur Verfügung stellt. Die Datenträger dürfen nicht kopiergeschützt sein. Für alle einzureichenden Dateien gilt, dass diese nicht geschützt sein dürfen, das heißt sie müssen ohne Kennworteingabe lesbar, speicherbar und druckbar sein. In dem Dossier hat der pharmazeutische Unternehmer nach Maßgabe des § 5 und der Vorgaben in Absatz 2 den Zusatznutzen des Arzneimittels gegenüber der zweckmäßigen Vergleichstherapie nachzuweisen. Hierzu muss es die folgenden Angaben enthalten:
(2) Für die Zusammenstellung der Unterlagen ist die Dossier-Vorlage in Anlage II zu verwenden. Die Daten nach den Absätzen 1, 4 bis 8 sind entsprechend der in den Modulen 1 bis 5 festgelegten Anforderungen aufzubereiten und einzureichen. Die Module 1 bis 4 enthalten die Grundlagen, auf die sich die Bewertung stützt, und werden vollständig auf der Internetseite des Gemeinsamen Bundesausschusses veröffentlicht. Unterlagen, die Betriebs- und Geschäftsgeheimnisse enthalten, müssen in Modul 5 vom pharmazeutischen Unternehmer gekennzeichnet werden.
(3) Auch wenn der pharmazeutische Unternehmer unter Berufung auf § 10 einer Veröffentlichung von Dokumenten in Modul 5 widerspricht, hat er dennoch zu gewährleisten, dass alle Angaben zu Studienmethodik und -ergebnissen vollständig zur Veröffentlichung im Dossier in Modul 1 bis 4 nach Maßgabe von Absatz 2 Satz 2 zur Verfügung gestellt werden. Entspricht das Dossier nicht diesen Anforderungen, kann der Nachweis des Zusatznutzens als nicht erbracht angesehen werden.
(4) Für das zu bewertende Arzneimittel legt der pharmazeutische Unternehmer im Dossier den Ergebnisbericht der Zulassungsstudien einschließlich der Studienprotokolle und des Bewertungsberichtes der Zulassungsbehörde vor, sowie alle Studien, die der Zulassungsbehörde übermittelt worden sind. Darüber hinaus werden alle Ergebnisse, Studienberichte und Studienprotokolle von Studien mit dem Arzneimittel übermittelt, für die der Unternehmer Sponsor war, sowie alle verfügbaren Angaben über laufende oder abgebrochene Studien mit dem Arzneimittel, für die der Unternehmer Sponsor ist oder auf andere Weise finanziell beteiligt ist, und entsprechende Angaben über Studien von Dritten, soweit diese verfügbar sind. Das Datum der erforderlichen Literatur- und Studienregisterrecherche sowie des Studienstatus soll nicht mehr als drei Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
(5) Das Dossier soll auch die Zulassungsnummer, das Datum der Zulassung, den Zulassungsinhaber, die Pharmazentralnummer, die Zuordnung zur anatomisch-therapeutisch-chemischen (ATC) Klassifikation und die Bezeichnung des Arzneimittels enthalten.
(6) Für die zweckmäßige Vergleichstherapie übermittelt der pharmazeutische Unternehmer im Dossier alle verfügbaren Ergebnisse von klinischen Studien einschließlich von Studienprotokollen, die geeignet sind, Feststellungen über den Zusatznutzen des zu bewertenden Arzneimittels zu treffen. Liegen keine klinischen Studien zum direkten Vergleich mit dem zu bewertenden Arzneimittel vor oder lassen diese keine ausreichenden Aussagen über den Zusatznutzen zu, können im Dossier indirekte Vergleiche vorgelegt werden.
(7) Der pharmazeutische Unternehmer hat die Kosten für die gesetzliche Krankenversicherung gemessen am Apothekenabgabepreis und die den Krankenkassen tatsächlich entstehenden Kosten zu übermitteln; sofern eine Darlegung der Kosten gemessen am Apothekenabgabepreis nicht möglich ist, sind die Kosten auf Basis anderer geeigneter Angaben darzulegen. Die Angabe der Kosten erfolgt sowohl für das zu bewertende Arzneimittel als auch für alle vom Gemeinsamen Bundesausschuss als zweckmäßige Vergleichstherapie bestimmten Therapien. Maßgeblich sind die direkten Kosten der gesetzlichen Krankenversicherung über einen bestimmten Zeitraum. Bestehen bei Anwendung der Arzneimittel entsprechend der Fach- oder Gebrauchsinformation regelhaft Unterschiede bei der notwendigen Inanspruchnahme ärztlicher Behandlung oder bei der Verordnung sonstiger Leistungen zwischen dem zu bewertenden Arzneimittel und der zweckmäßigen Vergleichstherapie, sind die damit verbundenen Kostenunterschiede für die Feststellung der den Krankenkassen tatsächlich entstehenden Kosten zu berücksichtigen.
(8) Für Arzneimittel im Sinne des § 5 Absatz 2 Satz 1 ist für den Nachweis des medizinischen Zusatznutzens als therapeutische Verbesserung abweichend von den Absätzen 2 bis 7 die Dossier-Vorlage in Anlage VI zu verwenden.
(9) Im Dossier macht der pharmazeutische Unternehmer Angaben zu der Frage, ob gemäß § 87 Absatz 5b Satz 5 SGB V die Fachinformation des Arzneimittels zu seiner Anwendung eine zwingend erforderliche Leistung vorsieht, die eine Anpassung des einheitlichen Bewertungsmaßstabs für ärztliche Leistungen erforderlich macht. Die Angaben sind nach Maßgabe der in Modul 3 festgelegten Anforderungen aufzubereiten und einzureichen.3Sie sind nicht Gegenstand der Bewertung des Nutzens von Arzneimitteln nach den Vorschriften dieses Kapitels einschließlich der Beschlussfassung nach § 20.
§ 10 Offenlegung
(1) Das Dossier wird gleichzeitig mit der Nutzenbewertung nach § 18 Absatz 5 auf der Internetseite des Gemeinsamen Bundesausschusses veröffentlicht, sofern nicht Betriebs- und Geschäftsgeheimnisse, der Schutz des geistigen Eigentums oder der Schutz personenbezogener Daten dagegen sprechen. Die Veröffentlichung enthält die Grundlagen, auf die sich die Bewertung stützt.
(2) Der pharmazeutische Unternehmer kennzeichnet Betriebs- und Geschäftsgeheimnisse im Dossier; § 9 Absatz 2 und 3 bleibt unberührt. Diese Kennzeichnung darf der Pflicht zur Offenlegung der Studienergebnisse nicht entgegenstehen.
(3) Der Gemeinsame Bundesausschuss kann mit den maßgeblichen Verbänden der pharmazeutischen Industrie und mit pharmazeutischen Unternehmern das Nähere durch Vereinbarung regeln.
§ 11 Vorlage des Dossiers
(1) Das Dossier ist spätestens zu den in § 8 bestimmten Zeitpunkten zu übermitteln. Der Gemeinsame Bundesausschuss hat nur fristgerecht eingereichte Unterlagen zu berücksichtigen. Dokumente der Zulassungsbehörden, die dem pharmazeutischen Unternehmer zu dem für die Einreichung maßgeblichen Zeitpunkt noch nicht vorgelegen haben, sind zu berücksichtigen, sofern sich dadurch die Nutzenbewertung nicht verzögert.
(2) Der pharmazeutische Unternehmer kann das Dossier dem Gemeinsamen Bundesausschuss auch vor den in § 8 genannten Zeitpunkten übermitteln. Legt der pharmazeutische Unternehmer das Dossier drei Wochen vor dem jeweiligen Zeitpunkt beim Gemeinsamen Bundesausschuss vor, führt die Geschäftsstelle des Gemeinsamen Bundesausschusses eine formale Vorprüfung auf Vollständigkeit des Dossiers durch. Ist das Dossier unvollständig, teilt die Geschäftsstelle des Gemeinsamen Bundesausschusses dem pharmazeutischen Unternehmer in der Regel innerhalb von zwei Wochen mit, welche zusätzlichen Angaben erforderlich sind. Die inhaltliche Prüfung des Dossiers bleibt davon unberührt.
(3) Der Gemeinsame Bundesausschuss fordert den betroffenen pharmazeutischen Unternehmer zur rechtzeitigen und vollständigen Einreichung des Dossiers auf. Der Gemeinsame Bundesausschuss kann den pharmazeutischen Unternehmer auch nach einer Beratung gemäß § 7 zur fristgerechten Vorlage eines vollständigen Dossiers auffordern.
§ 12 Anforderungen an das Dossier für Arzneimittel für seltene Leiden (sog. Orphan Drugs)
Für Arzneimittel, die zur Behandlung eines seltenen Leidens nach der Verordnung (EG) Nr. 141/2000 des Europäischen Parlaments und des Rates vom 16. Dezember 1999 über Arzneimittel für seltene Leiden zugelassen sind (Arzneimittel für seltene Leiden), gelten die Vorschriften dieses Kapitels mit folgenden Maßgaben:
§ 13 Anforderung eines Dossiers durch den Gemeinsamen Bundesausschuss wegen neuer wissenschaftlicher Erkenntnisse 18
(1) Der Gemeinsame Bundesausschuss kann auf Antrag seiner Mitglieder oder der in § 139b Absatz 1 Satz 2 SGB V genannten Organisationen und Institutionen wegen neuer wissenschaftlicher Erkenntnisse frühestens ein Jahr nach dem Beschluss über eine Nutzenbewertung nach § 20 eine erneute Nutzenbewertung eines nach den Vorschriften dieses Abschnitts bewerteten Arzneimittels beschließen. Dies gilt auch, wenn das Anwendungsgebiet des Arzneimittels durch die zuständigen Zulassungsbehörden eingeschränkt worden ist.
(2) Mit der Zustellung des Beschlusses werden dem pharmazeutischen Unternehmer die Gründe für die Nutzenbewertung mitgeteilt. Das Dossier ist innerhalb von drei Monaten nach Aufforderung durch den Gemeinsamen Bundesausschuss vorzulegen. Vor der Aufforderung ist dem pharmazeutischen Unternehmer eine Beratung nach § 7 anzubieten.
§ 14 Aufforderung zur Vorlage eines Dossiers aufgrund eines Antrags des pharmazeutischen Unternehmers auf erneute Nutzenbewertung wegen Vorliegen neuer wissenschaftlicher Erkenntnisse 18 18a
(1) Für ein Arzneimittel, für das ein Beschluss nach § 20 vorliegt, kann der pharmazeutische Unternehmer beim Gemeinsamen Bundesausschuss eine erneute Nutzenbewertung beantragen, wenn er die Erforderlichkeit wegen neuer wissenschaftlicher Erkenntnisse nachweist.
(1a) Der Antrag ist schriftlich oder in elektronischer Form unter Verwendung einer qualifizierten elektronischen Signatur und die zu seiner Begründung erforderlichen Unterlagen sind in elektronischer Form einzureichen. Als Datenträger ist eine Digital Versatile Disc (DVD) zu verwenden, sofern der Gemeinsame Bundesausschuss nicht andere Verfahren zur elektronischen Einreichung von Freistellungsanträgen zur Verfügung stellt. Die Datenträger dürfen nicht kopiergeschützt sein. Für alle einzureichenden Dateien gilt, dass diese nicht geschützt sein dürfen, das heißt sie müssen ohne Kennworteingabe lesbar, speicherbar und druckbar sein.
(2) Der Gemeinsame Bundesausschuss beschließt über den Antrag innerhalb von acht Wochen. Hält der Gemeinsame Bundesausschuss den Antrag für begründet, fordert er den pharmazeutischen Unternehmer auf, die für die Nutzenbewertung nach den Vorschriften dieses Abschnitts erforderlichen Nachweise zu übermitteln. Das Dossier ist innerhalb von drei Monaten nach Aufforderung durch den Gemeinsamen Bundesausschuss vorzulegen. Vor der Aufforderung ist dem pharmazeutischen Unternehmer eine Beratung nach § 7 anzubieten.
§ 15 Freistellung von der Nutzenbewertung nach § 35a Absatz 1a SGB V 16b 18c
(1) Der pharmazeutische Unternehmer kann spätestens drei Monate vor dem nach § 8 Nummer 1, 3 und 7 maßgeblichen Zeitpunkt beim Gemeinsamen Bundesausschuss beantragen, ihn von der Verpflichtung zur Vorlage von Nachweisen nach § 9 und das Arzneimittel von der Nutzenbewertung nach den Vorschriften dieses Abschnitts freizustellen, wenn zu erwarten ist, dass den gesetzlichen Krankenkassen nur geringfügige Ausgaben für das Arzneimittel entstehen werden. Die Beurteilung der Geringfügigkeit erfolgt auf der Grundlage von Angaben des pharmazeutischen Unternehmers zu den zu erwartenden Ausgaben der gesetzlichen Krankenkassen für das Arzneimittel, grundsätzlich auf Basis der Apothekenverkaufspreise. Soweit die dauerhaft zu erwartenden Ausgaben auf Basis der Apothekenverkaufspreise einschließlich Umsatzsteuer einen Betrag in Höhe von 1.000 000 Euro innerhalb von 12 Kalendermonaten nicht überschreiten, gelten die Ausgaben als geringfügig. Wenn eine Ermittlung der zu erwartenden Ausgaben auf Basis der Apothekenverkaufspreise nicht möglich ist, ist stattdessen auf Basis anderer geeigneter Angaben darzulegen, dass den gesetzlichen Krankenkassen nur geringfügige Ausgaben für das Arzneimittel entstehen werden.
(2) Der pharmazeutische Unternehmer hat die Gründe für den Freistellungsantrag nach Absatz 1 nachzuweisen. Zur Beurteilung der Geringfügigkeit der Ausgaben macht er dem Gemeinsamen Bundesauschuss zureichende Angaben.
(2a) § 14 Absatz 1a gilt entsprechend.
(3) Der Gemeinsame Bundesausschuss beschließt innerhalb von acht Wochen über den Antrag.
(4) Übersteigen die Ausgaben der gesetzlichen Krankenkassen nach Absatz 1 Satz 2 oder Satz 4 innerhalb von zwölf Kalendermonaten einen Betrag von 1.000 000 Euro, hat der pharmazeutische Unternehmer innerhalb von drei Monaten nach Aufforderung durch den Gemeinsamen Bundesausschuss Nachweise nach § 5 Absatz 1 bis 6 zu übermitteln und darin den Zusatznutzen gegenüber der zweckmäßigen Vergleichstherapie nachzuweisen. Die Ausgaben sind aufgrund der Angaben nach § 84 Absatz 5 Satz 4 SGB V zu ermitteln, soweit die Beurteilung der Geringfügigkeit nach Absatz 1 Satz 2 erfolgt; anderenfalls sind die Ausgaben der gesetzlichen Krankenkassen für das Arzneimittel aufgrund anderer geeigneter Angaben zu ermitteln.
(5) Bescheide über die Freistellung eines Arzneimittels von der Nutzenbewertung nach § 35a SGB V ergehen mit der Maßgabe, dass ihre Geltungsdauer mit der Erhöhung des Abgabepreises des pharmazeutischen Unternehmers (gemäß § 8 Nummer 1 Satz 2) endet. Sofern nicht spätestens drei Monate vor der Erhöhung des Abgabepreises des pharmazeutischen Unternehmers ein erneuter Antrag auf Freistellung gestellt wird, beginnt das Bewertungsverfahren nach § 35a SGB V zum Zeitpunkt der Veröffentlichung der Erhöhung des Abgabepreises (gemäß § 8 Nummer 1 Satz 2). Hat der pharmazeutische Unternehmer zu diesem Zeitpunkt ein Dossier nicht oder nicht vollständig vorgelegt, gilt § 17 Absatz 1 Satz 2 entsprechend.
(6) Absatz 5 gilt entsprechend, wenn der pharmazeutische Unternehmer für das freigestellte Arzneimittel eine neue Darreichungsform, Wirkstärke, Dosierung oder Packungsgröße in den Verkehr bringt.
(7) Für einen Antrag nach Absatz 1 Satz 1 ist Anlage V zu verwenden.
§ 16 Nutzenbewertung von Arzneimitteln mit bekannten Wirkstoffen mit neuer Zulassung und neuem Unterlagenschutz 18
(1) Für ein Arzneimittel mit Wirkstoffen, die keine neuen Wirkstoffe im Sinne des § 2 Absatz 1 sind (bekannte Wirkstoffe), kann der Gemeinsame Bundesausschuss eine Nutzenbewertung nach den Vorschriften dieses Kapitels veranlassen, wenn für das Arzneimittel eine neue Zulassung mit neuem Unterlagenschutz erteilt wird. Eine Nutzenbewertung kann insbesondere für Arzneimittel veranlasst werden, deren Anwendungsgebiet von dem Anwendungsgebiet der Arzneimittel mit denselben bekannten Wirkstoffen abweicht. Eine Abweichung kann sich insbesondere aus Veränderungen in einem Anwendungsgebiet ergeben, die im Vergleich zu dem Anwendungsgebiet des Arzneimittels mit demselben bekannten Wirkstoff einem anderen Therapiegebiet zuzuordnen sind, indem:
(2) Absatz 1 gilt entsprechend für Arzneimittel mit einem neuen Wirkstoff im Sinne des § 2 Absatz 1, welche vor dem 1. Januar 2011 in Verkehr gebracht worden sind, wenn für das Arzneimittel eine neue Zulassung mit neuem Unterlagenschutz erteilt wird.
§ 17 Entscheidung über die Durchführung der Nutzenbewertung 19a
(1) Der Gemeinsame Bundesausschuss prüft, ob der pharmazeutische Unternehmer die ihm nach dem Gesetz obliegende Verpflichtung zur fristgerechten Vorlage eines vollständigen Dossiers erfüllt hat. Die Prüfung erfolgt nach Maßgabe einer formalen Prüfung auf Vollständigkeit der vom pharmazeutischen Unternehmer nach § 9 vorzulegenden Unterlagen; § 11 Abs. 2 Satz 4 gilt entsprechend. Hat der pharmazeutische Unternehmer das Dossier trotz Aufforderung, spätestens zu den nach § 8 maßgeblichen Zeitpunkten nicht vorgelegt,trifft der Gemeinsame Bundesausschuss die Feststellung, dass der Zusatznutzen des Arzneimittels als nicht belegt gilt; die Bewertung des Nutzens des Arzneimittels bleibt hiervon unberührt. Entsprechendes gilt, wenn einem fristgerecht eingereichten Dossier ein oder mehrere Module nach § 9 Abs. 2 fehlen. Hat der pharmazeutische Unternehmer das Dossier trotz Aufforderung nicht vollständig vorgelegt, teilt der Gemeinsame Bundesausschuss dem pharmazeutischen Unternehmer mit, welche erforderlichen Angaben nachzureichen sind; § 11 Abs. 1 bleibt unberührt. Legt der pharmazeutische Unternehmer sämtliche in der Mitteilung nach Satz 5 als erforderlich bezeichneten Angaben nicht innerhalb einer Frist von fünf Werktagen ab Zugang der Mitteilung vor, gilt Satz 3 entsprechend.
(2) Der Gemeinsame Bundesausschuss entscheidet, ob er die Nutzenbewertung selbst durchführt oder hiermit das IQWiG oder Dritte beauftragt.
(3) Soweit die Bewertung des Nutzens eines Arzneimittels auf der Grundlage einer Bewertung des IQWiG oder Dritter erfolgen soll, ist der Auftrag mit der Maßgabe zu versehen, dass diese
(4) Zum Zwecke der Aufgabenerfüllung nach § 87 Absatz 5b Satz 5 und 6 SGB V informiert der G-BA den Bewertungsausschuss regelhaft zum maßgeblichen Zeitpunkt nach § 8 VerfO über den Beginn des Nutzenbewertungsverfahrens und leitet ihm die nach § 9 Absatz 9 verfügbaren Angaben zur Prüfung eines Anpassungsbedarfs des einheitlichen Bewertungsmaßstabs für ärztliche Leistungen nach § 87 Absatz 5b Satz 5 SGB V zu. Vor Beschlussfassung nach § 35a Absatz 3 SGB V sind dem G-BA die Ergebnisse der Prüfung hinsichtlich der Erforderlichkeit einer Anpassung des einheitlichen Bewertungsmaßstabs für ärztliche Leistungen zuzuleiten.
(1) Bei der Nutzenbewertung wird geprüft, ob für das Arzneimittel ein Zusatznutzen gegenüber der zweckmäßigen Vergleichstherapie belegt ist, welcher Zusatznutzen für welche Patientengruppen in welchem Ausmaß belegt ist, wie die vorliegende Evidenz zu bewerten ist und mit welcher Wahrscheinlichkeit der Beleg jeweils erbracht wird. Bei der Bewertung des Zusatznutzens von Antibiotika soll die Resistenzsituation berücksichtigt werden. Die Nutzenbewertung erfolgt auf der Grundlage des Dossiers. Ergibt die Nutzenbewertung, dass die Aufbereitung der Unterlagen im Dossier in einem Ausmaß von den in § 9 festgelegten Anforderungen abweicht, welches einer sachgerechten Bewertung des Zusatznutzens entgegensteht, kann der Gemeinsame Bundesausschuss die Feststellung treffen, dass der Zusatznutzen nicht belegt ist
(2) Mit der Nutzenbewertung wird die Validität und Vollständigkeit der Angaben im Dossier geprüft. Dabei werden die Unterlagen hinsichtlich ihrer Planungs-, Durchführungs- und Auswertungsqualität im Hinblick auf ihre Aussagekraft für Wahrscheinlichkeit und Ausmaß des Zusatznutzens und hinsichtlich der Angaben zu den Therapiekosten bewertet. Die Nutzenbewertung enthält auch eine Zusammenfassung der wesentlichen Aussagen als Bewertung der Angaben im Dossier nach § 9 Absatz 1. Maßstab für die Beurteilung ist der allgemein anerkannte Stand der medizinischen Erkenntnisse. Grundlage sind die internationalen Standards der evidenzbasierten Medizin und der Gesundheitsökonomie.
(3) Für die erstmalige Bewertung nach § 35a SGB V zum Zeitpunkt der Markteinführung sind für die Bewertung des Arzneimittels mit neuen Wirkstoffen grundsätzlich die Zulassungsstudien zugrunde zu legen. Reichen die Zulassungsstudien nicht aus, kann der Gemeinsame Bundesausschuss weitere Nachweise verlangen.
(4) Können zum Zeitpunkt der Bewertung valide Daten zu patientenrelevanten Endpunkten noch nicht vorliegen, erfolgt die Bewertung auf Grundlage der best verfügbaren Evidenz unter Berücksichtigung der Studienqualität mit Angabe der Wahrscheinlichkeit für den Beleg eines Zusatznutzens. Sind für den Beleg eines Zusatznutzens valide Daten zu patientenrelevanten Endpunkten erforderlich, kann der Gemeinsame Bundesausschuss bei der Beschlussfassung nach § 20 eine Frist bestimmen, bis wann diese Daten vorgelegt werden sollen.
(5) Die Nutzenbewertung wird spätestens innerhalb von drei Monaten ab den maßgeblichen Zeitpunkten gemäß § 8 abgeschlossen und im Internet veröffentlicht.
§ 19 Gesetzliches Stellungnahmeverfahren
(1) Mit Veröffentlichung der Nutzenbewertung auf der Internetseite des Gemeinsamen Bundesausschusses wird den Sachverständigen der medizinischen und pharmazeutischen Wissenschaft und Praxis sowie den für die Wahrnehmung der wirtschaftlichen Interessen gebildeten maßgeblichen Spitzenorganisationen der pharmazeutischen Unternehmer, den betroffenen pharmazeutischen Unternehmern, den Berufsvertretungen der Apotheker und den maßgeblichen Dachverbänden der Ärztegesellschaften der besonderen Therapierichtungen auf Bundesebene Gelegenheit gegeben, zu der Nutzenbewertung des Arzneimittels schriftlich Stellung zu nehmen unter Verwendung der Vorgaben in Anlage III. Die Stellungnahmefrist beträgt drei Wochen.
(2) Im Anschluss an das schriftliche Stellungnahmeverfahren und vor einer Beschlussfassung über die Nutzenbewertung nach § 92 Absatz 1 Satz 2 Nummer 6 SGB V gibt der Gemeinsame Bundesausschuss den Stellungnahmeberechtigten nach Absatz 1 Gelegenheit, zu der Nutzenbewertung auch mündlich Stellung zu nehmen. Soweit sie eine schriftliche Stellungnahme nach Absatz 1 abgegeben haben, können an der mündlichen Anhörung die Sachverständigen sowie jeweils maximal zwei Vertreter der nach Absatz 1 stellungnahmeberechtigten Organisationen und betroffenen Unternehmer teilnehmen. Die mündliche Stellungnahme ersetzt nicht die nach Absatz 1 abgegebene Stellungnahme. Sie dient dazu, insbesondere zu
(3) Die schriftlich und mündlich abgegebenen Stellungnahmen nach Absatz 1 und 2 werden in die Entscheidung über die Beschlussfassung der Nutzenbewertung nach § 92 Absatz 1 Satz 2 Nummer 6 SGB V ein bezogen. Für die Auswertung der Stellungnahmen gilt 1. Kapitel § 10 Absatz 3 VerfO.
4. Titel
Beschlussfassung und Umsetzung der Nutzenbewertung in die Arzneimittel-Richtlinie
§ 20 Beschlussfassung über die Nutzenbewertung 18
(1) Der Gemeinsame Bundesausschuss beschließt über die Nutzenbewertung innerhalb von drei Monaten nach ihrer Veröffentlichung. Der Beschluss wird im Internet veröffentlicht. Er ist Teil der Arzneimittel-Richtlinie nach § 92 Absatz 1 Satz 2 Nummer 6 SGB V und wird im Bundesanzeiger bekannt gemacht. § 94 Absatz 1 SGB V gilt nicht.
(2) Der Beschluss ist Grundlage für Vereinbarungen für alle Arzneimittel mit dem Wirkstoff nach § 130b SGB V über Erstattungsbeträge und für die Bestimmung von Anforderungen an die Zweckmäßigkeit, Qualität und Wirtschaftlichkeit der Verordnung sowie für die Anerkennung als Praxisbesonderheit oder für die Zuordnung von Arzneimitteln ohne Zusatznutzen zu einer Festbetragsgruppe nach § 35 SGB V.
(3) Auf der Grundlage der Nutzenbewertung trifft der Gemeinsame Bundesausschuss mit dem Beschluss nach § 35a Absatz 3 SGB V Feststellungen in der Arzneimittel-Richtlinie zur wirtschaftlichen Verordnungsweise des Arzneimittels, insbesondere
(4) Besteht im Nachgang zu einem gefassten Beschluss nach § 35a Absatz 3 SGB V über die Nutzenbewertung von Arzneimitteln (Anlage XII der Arzneimittel-Richtlinie) Änderungsbedarf im Sinne einer sachlichrechnerischen Richtigstellung hinsichtlich der Angaben nach § 20 Absatz 3 Nummer 2 zu der Anzahl der Patienten bzw. der Abgrenzung der für die Behandlung infrage kommenden Patientengruppen oder der Angaben nach § 20 Absatz 3 Nummer 4 zu den Therapiekosten, kann der Unterausschuss durch einvernehmlichen Beschluss die entsprechenden Änderungen dieses Beschlusses in der Anlage XII zur Arzneimittel-Richtlinie vornehmen soweit dadurch der Kerngehalt der Richtlinie nicht berührt wird.
(5) Der Gemeinsame Bundesausschuss beschließt nach § 35a Absatz 1 Satz 5 in Verbindung mit Absatz 3 SGB V, dass ein Zusatznutzen als nicht belegt gilt, wenn die Voraussetzungen nach § 17 Absatz 1 Satz 3, 4 oder Satz 6 vorliegen.
§ 21 Arzneimittel ohne Zusatznutzen
Ergibt die Nutzenbewertung, dass für das Arzneimittel mit dem neuen Wirkstoff ein therapierelevanter Zusatznutzen nach dem allgemein anerkannten Stand der medizinischen Erkenntnisse nicht belegt ist, ist nach Maßgabe der Nummern 1 und 2 zu prüfen, ob das Arzneimittel einer Festbetragsgruppe nach § 35 Absatz 1 SGB V zugeordnet werden kann:
§ 22 Arzneimittel mit Zusatznutzen
Ergibt die Nutzenbewertung, dass für das Arzneimittel ein Zusatznutzen nach dem allgemein anerkannten Stand der medizinischen Erkenntnisse belegt ist, stellt der Gemeinsame Bundesausschuss dies durch Beschluss nach § 35a Absatz 3 SGB V in der Arzneimittel-Richtlinie mit Angaben zum Ausmaß des Zusatznutzens fest.
2. Abschnitt
Kosten-Nutzen-Bewertung von Arzneimitteln nach § 35b SGB V
1. Titel
Einleitung des Verfahrens und Vorbereitung eines Auftrags zur Kosten-Nutzen-Bewertung von Arzneimitteln nach § 35b SGB V
(1) Die Bewertung des Kosten-Nutzen-Verhältnisses von Arzneimitteln nach § 35b SGB V erfolgt auf Antrag eines Antragsberechtigten nach Absatz 2.
(2) Antragsberechtigt sind:
(3) Der Antrag und die erforderlichen Unterlagen sind schriftlich oder elektronisch unter Verwendung einer qualifizierten elektronischen Signatur zu stellen und bei der Geschäftsstelle des Gemeinsamen Bundesausschusses einzureichen. Zu den erforderlichen Unterlagen gehören:
Es ist das Antragsformular in Anlage VII zu verwenden. Nach Vorliegen eines vollständigen Antrags benachrichtigt die Geschäftsstelle des Gemeinsamen Bundesausschusses den jeweils anderen Antragsberechtigten nach Absatz 2 über die Antragstellung und übermittelt ihm die eingereichten Unterlagen.
(4) Der Antrag ist innerhalb von einem Jahr nach Zustellung des Schiedsspruchs oder im Falle einer Antragstellung nach § 35a Absatz 5a SGB V innerhalb von einem Jahr nach Veröffentlichung des Beschlusses des Gemeinsamen Bundesausschusses gemäß § 35a Absatz 3 SGB V zu stellen.
(5) Das Plenum hat den Antrag anzunehmen, soweit
Sind die zur Begründung des Antrags erforderlichen Angaben unvollständig, teilt die Geschäftsstelle des Gemeinsamen Bundesausschusses dem Antragsteller mit, welche erforderlichen Angaben nachzureichen sind. Legt der Antragsteller sämtliche in der Mitteilung als erforderlich bezeichneten Angaben nicht innerhalb einer Frist von vier Wochen ab Zugang der Mitteilung vor, kann der Antrag abgelehnt werden. Das Plenum entscheidet über einen Antrag spätestens innerhalb von acht Wochen nach Ablauf der Frist nach Satz 3.
(6) Nach Annahme eines Antrags durch das Plenum ist eine weitere Antragstellung nicht möglich.
(7) Nach Annahme des Antrags beauftragt das Plenum den Unterausschuss Arzneimittel mit der Vorbereitung einer Beauftragung des IQWiG mit der Kosten-Nutzen-Bewertung des zu bewertenden Arzneimittels. Sofern eine Kosten-Nutzen-Bewertung von beiden Antragsberechtigten nach § 23 Absatz 2 beantragt wird, sind die Anträge in einer Beauftragung zusammenzufassen.
§ 23a Einstellung der Kosten-Nutzen-Bewertung
(1) Ein Antrag kann vom Antragsteller ohne Begründung zurückgenommen werden. Durch Rücknahme des Antrags kann das Verfahren nur bis zur Annahme des Antrags durch das Plenum beendet werden. Sofern beide Antragsberechtigte eine Kosten-Nutzen-Bewertung beantragt haben, endet das Verfahren nur dann, wenn beide Anträge zurückgenommen werden.
(2) Ein Bewertungsverfahren kann auch ohne Rücknahme des Antrags auf Beschluss des Plenums eingestellt werden. Der Einstellungsbeschluss ist zu begründen.
(3) Ein Einstellungsbeschluss ist mit seiner Begründung im Internet zu veröffentlichen.
§ 24 Bestimmung des Auftragsinhalts zur Kosten-Nutzen-Bewertung
(1) In dem Auftrag zur Kosten-Nutzen-Bewertung des zu bewertenden Arzneimittels ist insbesondere festzulegen,
(2) Bei der Bewertung ist grundsätzlich die Perspektive der Versichertengemeinschaft der gesetzlichen Krankenversicherung (§ 1 SGB V) zugrunde zu legen.
(3) Der Vorschlag über die Auftragsinhalte nach § 23 Absatz 3 Nummer 3 wird in die Erstellung des Auftrags einbezogen.
§ 25 Stellungnahmeverfahren
(1) Vor einer Beauftragung des IQWiG mit einer Kosten-Nutzen-Bewertung wird den Stellungnahmeberechtigten nach § 92 Absatz 3a SGB V Gelegenheit zur Stellungnahme zum Auftrag gegeben. Die Stellungnahmefrist beträgt vier Wochen. Es ist die Vorlage in Anlage III zu verwenden.
(2) Der Unterausschuss soll innerhalb von acht Wochen über die Einleitung des Stellungnahmeverfahrens zur Beauftragung des IQWiG beschließen.
(3) Im Anschluss an das schriftliche Stellungnahmeverfahren gibt der Gemeinsame Bundesausschuss den Stellungnahmeberechtigten nach Absatz 1 Satz 1 Gelegenheit, zur Beauftragung des IQWiG mit einer Kosten-Nutzen-Bewertung auch mündlich Stellung zu nehmen. Soweit sie eine schriftliche Stellungnahme nach Absatz 1 abgegeben haben, können an der mündlichen Anhörung die Sachverständigen sowie jeweils maximal zwei Vertreter der nach Absatz 1 Satz 1 stellungnahmeberechtigten Organisationen und betroffenen Unternehmen teilnehmen. Die mündliche Stellungnahme ersetzt nicht die nach Absatz 1 abgegebene Stellungnahme. Sie dient dazu, insbesondere zu neueren wissenschaftlichen Erkenntnissen, die sich zeitlich nach Erstellung des Auftrags ergeben haben, Stellung zu nehmen.
(4) Die Stellungnahmen nach Absatz 1 und 3 werden in die Entscheidung über die Beauftragung des IQWiG mit einer Kosten-Nutzen-Bewertung nach § 35b Absatz 1 SGB V einbezogen. Für die Auswertung der Stellungnahmen gilt 1. Kapitel § 10 Absatz 3 VerfO.
2. Titel
Beauftragung und Durchführung der Kosten-Nutzen-Bewertung
§ 26 Entscheidung über die Beauftragung des IQWiG
(1) Das Plenum beschließt über die Beauftragung des IQWiG sowie über die Berücksichtigung von Versorgungsstudien gemäß § § 35 bis 38 innerhalb von vier Monaten nach Einleitung des Stellungnahmeverfahrens gemäß § 25 Absatz 1. Der Auftrag enthält neben den Inhalten nach § 24
(2) Die Frist für die Erstellung der Kosten-Nutzen-Bewertung ab Einreichung des Dossiers bis zur Einleitung des Stellungnahmeverfahrens nach § 35b Absatz 1 Satz 6 SGB V ist auftragsbezogen in Abstimmung mit dem IQWiG festzulegen. Sie soll in der Regel zwölf Monate nicht überschreiten.
(3) Die Tragenden Gründe des Beschlusses werden im Internet veröffentlicht.
§ 27 Maßgeblicher Zeitpunkt für die Einreichung des Dossiers
(1) Zeitgleich zur Beauftragung des IQWiG fordert der Gemeinsame Bundesausschuss den pharmazeutischen Unternehmer auf, ein vollständiges Dossier für die Kosten-Nutzen-Bewertung für das zu bewertende Arzneimittel vorzulegen. Das Dossier ist innerhalb von drei Monaten nach Zustellung des Beschlusses vom pharmazeutischen Unternehmer bei der Geschäftsstelle des Gemeinsamen Bundesausschusses einzureichen.
(2) Soweit der Gemeinsame Bundesausschuss mit dem pharmazeutischen Unternehmer eine Vereinbarung über die Durchführung von Versorgungsstudien getroffen hat, ist das Dossier innerhalb von drei Monaten ab dem Zeitpunkt einzureichen, an dem die Frist für die Vorlage der Versorgungsstudien endet.
§ 28 Formale Vorprüfung auf Vollständigkeit des Dossiers
Der pharmazeutische Unternehmer kann das Dossier dem Gemeinsamen Bundesausschuss auch vor den in den in § 27 Absatz 1 und 2 genannten Zeitpunkten übermitteln. Legt der pharmazeutische Unternehmer das Dossier drei Wochen vor dem jeweiligen Zeitpunkt beim Gemeinsamen Bundesausschuss vor, führt die Geschäftsstelle des Gemeinsamen Bundesausschusses eine formale Vorprüfung auf Vollständigkeit des Dossiers durch. Ist das Dossier unvollständig, teilt die Geschäftsstelle des Gemeinsamen Bundesausschusses dem pharmazeutischen Unternehmer in der Regel innerhalb von zwei Wochen mit, welche zusätzlichen Angaben erforderlich sind. Die inhaltliche Prüfung des Dossiers bleibt davon unberührt.
§ 29 Entscheidung über die Durchführung der Kosten-Nutzen-Bewertung
(1) Der Gemeinsame Bundesausschuss prüft, ob der pharmazeutische Unternehmer die ihm nach dieser Verfahrensordnung obliegende Verpflichtung zur Vorlage eines vollständigen Dossiers zum maßgeblichen Zeitpunkt nach § 27 erfüllt hat und übermittelt die eingereichten Unterlagen an das IQWiG. Die Prüfung erfolgt nach Maßgabe einer formalen Prüfung auf Vollständigkeit der vom pharmazeutischen Unternehmer nach § 39 vorzulegenden Unterlagen; § 11 Absatz 2 Satz 4 gilt entsprechend.
(2) Legt der pharmazeutische Unternehmer das Dossier nicht zum maßgeblichen Zeitpunkt nach § 27 vor, besteht keine Verpflichtung des Gemeinsamen Bundesausschusses, vom pharmazeutischen Unternehmer später eingereichte Unterlagen zur Bewertung des Kosten-Nutzen-Verhältnisses für das zu bewertende Arzneimittel zu berücksichtigen. Entsprechendes gilt, wenn einem fristgerecht eingereichten Dossier ein oder mehrere Module nach § 39 Absatz 2 fehlen. Hat der pharmazeutische Unternehmer das Dossier trotz Aufforderung nicht vollständig vorgelegt, teilt der Gemeinsame Bundesausschuss dem pharmazeutischen Unternehmer mit, welche erforderlichen Angaben nachzureichen sind; § 27 Absatz 1 und 2 bleibt unberührt. Legt der pharmazeutische Unternehmer sämtliche in der Mitteilung nach Satz 3 als erforderlich bezeichneten Angaben nicht innerhalb einer Frist von fünf Werktagen ab Zugang der Mitteilung vor, gilt Satz 1 entsprechend.
§ 30 Aussetzung der Kosten-Nutzen-Bewertung
(1) Sofern der Gemeinsame Bundesausschuss zu einem Arzneimittel, zu dem eine Kosten-Nutzen-Bewertung anhängig ist, gemäß § § 13 oder 14 ein Verfahren auf erneute Nutzenbewertung nach § 35a SGB V einleitet, kann er beschließen, die Kosten-Nutzen-Bewertung auszusetzen, bis die Nutzenbewertung nach § 35a SGB V abgeschlossen ist.
(2) Nach Abschluss des Verfahrens auf erneute Nutzenbewertung entscheidet der Gemeinsame Bundesausschuss über die Fortsetzung der Kosten-Nutzen-Bewertung.
§ 31 Übermittlung der Kosten-Nutzen-Bewertung durch das IQWiG
Das IQWiG übermittelt dem Gemeinsamen Bundesausschuss die Kosten-Nutzen-Bewertung innerhalb von drei Monaten nach Einleitung des Stellungnahmeverfahrens nach § 35b Absatz 1 Satz 6 SGB V. Die Kosten-Nutzen-Bewertung und das Dossier werden auf der Internetseite des Gemeinsamen Bundesausschusses veröffentlicht.
§ 32 Bewertung der Ergebnisse der Kosten-Nutzen-Bewertung
(1) Nach Abschluss der Kosten-Nutzen-Bewertung durch das IQWiG überprüft der Gemeinsame Bundesausschuss die Empfehlung des IQWiG im Rahmen einer Plausibilitätskontrolle nach Maßgabe des 4. Kapitels § 8 Absatz 2 VerfO.
(2) Auf der Grundlage der Empfehlung des IQWiG nimmt der Gemeinsame Bundesausschuss eine Gewichtung der Ergebnisse unter Berücksichtigung ihrer Bedeutung für die Versorgung der Versicherten vor.
(3) Im Anschluss an die Gewichtung nach Absatz 2 bewertet der Gemeinsame Bundesausschuss auf der Grundlage der Empfehlung des IQWiG die Wirtschaftlichkeit des Arzneimittels auch unter Berücksichtigung der Angemessenheit und Zumutbarkeit einer Kostenübernahme durch die Versichertengemeinschaft. Die Bewertung der Angemessenheit und Zumutbarkeit einer Kostenübernahme erfolgt in Hinblick darauf, ob unter Beachtung des Grundsatzes der Verhältnismäßigkeit eine begründbare Relation zwischen den Kosten und dem Nutzen des Arzneimittels besteht. Bei dieser Abwägung sind insbesondere Feststellungen zu berücksichtigen
§ 33 Stellungnahmeverfahren
Vor einer Beschlussfassung über die Umsetzung der Kosten-Nutzen-Bewertung in die Arzneimittel-Richtlinie gibt der Gemeinsame Bundesausschuss innerhalb von drei Monaten nach Veröffentlichung der Kosten-Nutzen-Bewertung den Stellungnahmeberechtigten nach § 92 Absatz 3a SGB V Gelegenheit zur Stellungnahme. Die Stellungnahmefrist beträgt vier Wochen. Es ist die Vorlage in Anlage III zu verwenden. Im Anschluss an das schriftliche Stellungnahmeverfahren gibt der Gemeinsame Bundesausschuss den Stellungnahmeberechtigten nach Satz 1 nach Maßgabe des § 25 Absatz 3 Satz 2 bis 4 auch Gelegenheit zur mündlichen Stellungnahme. Die abgegebenen Stellungnahmen werden in die Entscheidung über die Beschlussfassung der Kosten-Nutzen-Bewertung einbezogen. Für die Auswertung der Stellungnahmen gilt 1. Kapitel § 10 Absatz 3 VerfO.
§ 34 Beschluss über die Kosten-Nutzen-Bewertung
(1) Der Gemeinsame Bundesausschuss beschließt über die Kosten-NutzenBewertung innerhalb von neun Monaten nach ihrer Veröffentlichung. Mit dem Beschluss über die Kosten-Nutzen-Bewertung werden insbesondere Feststellungen zum Zusatznutzen auch im Verhältnis zu den Therapiekosten bei Anwendung des jeweiligen Arzneimittels getroffen. Der Beschluss wird im Internet veröffentlicht. Der Beschluss ist Teil der Arzneimittel-Richtlinie nach § 92 Absatz 1 Satz 2 Nummer 6 SGB V und wird im Bundesanzeiger bekannt gemacht. § 94 Absatz 1 SGB V gilt nicht.
(2) Der Beschluss ist Grundlage für Vereinbarungen nach § 130b Absatz 8 Satz 3 SGB V für das jeweilige Arzneimittel.
3.Titel
Inhaltliche Anforderungen an die Bewertung des Kosten-Nutzen-Verhältnisses von Arzneimitteln
§ 35 Anforderungen an die Bewertung des Kosten-Nutzen- Verhältnisses von Arzneimitteln
(1) Die Bewertung des Kosten-Nutzen-Verhältnisses von Arzneimitteln erfolgt nach Maßgabe des Auftragsinhalts durch Vergleich mit anderen Arzneimitteln und Behandlungsformen, insbesondere der zweckmäßigen Vergleichstherapie unter Berücksichtigung des therapeutischen Zusatznutzens für die Patienten im Verhältnis zu den Kosten. Grundlage der Bewertung sind
(2) Versorgungsstudien werden in die Bewertung einbezogen, sofern der Gemeinsame Bundesausschuss
§ 36 Anforderungen an die Anerkennung von Versorgungsstudien
Der Gemeinsame Bundesausschuss entscheidet über die Anerkennung einer Untersuchung als Versorgungsstudie nach Maßgabe insbesondere folgender Anforderungen:
§ 37 Anforderungen an die Vereinbarung einer Versorgungsstudie
(1) Wird vom pharmazeutischen Unternehmer eine Vereinbarung über die Durchführung einer Versorgungsstudie angestrebt, sind folgende Unterlagen vorzulegen:
(2) Die Frist zur Vorlage dieser Studien bemisst sich nach der Indikation und dem nötigen Zeitraum für die Bereitstellung valider Daten; sie soll drei Jahre nicht überschreiten. Die Studien sind auf Kosten des pharmazeutischen Unternehmers bevorzugt in Deutschland durchzuführen. § 36 Nummer 1 bis 3 gelten entsprechend.
§ 38 Entscheidung über die Berücksichtigung von Versorgungsstudien
(1) Anträge über die Anerkennung von Versorgungsstudien oder Unterlagen zur Vereinbarung von Versorgungsstudien sind spätestens mit Ablauf des schriftlichen Stellungnahmeverfahrens über die Beauftragung des IQWiG mit einer Kosten-Nutzen-Bewertung beim Gemeinsamen Bundesausschuss einzureichen.
(2) Im Zusammenhang mit der Beauftragung des IQWIG entscheidet das Plenum auch über die Anerkennung oder den Abschluss einer Vereinbarung von Versorgungsstudien.
Voraussetzung für den Abschluss einer Vereinbarung nach § 35 Absatz 2 Nummer 2 ist, dass eine Einigung über die wesentlichen Inhalte des Prüfplans nach § 37 Absatz 1 Nummer 5 erzielt wird. Die Wirksamkeit der Vereinbarung steht unter dem Vorbehalt, dass der pharmazeutische Unternehmer die in § 37 Absatz 1 Nummer 1 bis 5 genannten Unterlagen dem Gemeinsamen Bundesausschuss innerhalb von sechs Monaten ab Abschluss der Vereinbarung vorlegt.
§ 39 Anforderungen an das Dossier
(1) Das Dossier dient der Bewertung des Kosten-Nutzen-Verhältnisses des Arzneimittels. Das Dossier ist in deutscher Sprache einzureichen, soweit sich aus den Vorgaben für das Dossier nichts anderes ergibt. In dem Dossier hat der pharmazeutische Unternehmer auf der Grundlage der in dem Auftrag enthaltenen Angaben und der Vorgaben in Absatz 2 das Kosten-Nutzen-Verhältnis des Arzneimittels zu bewerten. Hierzu muss es die folgenden Angaben enthalten:
(2) Für die Zusammenstellung der Unterlagen ist die Dossier-Vorlage in Anlage VIII zu verwenden. Die Daten nach Absatz 1 sind entsprechend der in den Modulen K1 bis K5 festgelegten Anforderungen aufzubereiten und einzureichen. Die Module K1 bis K4 sowie das entscheidungsanalytische Modell in Modul K5 enthalten die Grundlagen, auf die sich die Bewertung stützt, und werden vollständig auf der Internetseite des Gemeinsamen Bundesausschusses veröffentlicht. Unterlagen, die Betriebs- und Geschäftsgeheimnisse enthalten, müssen in Modul K5 vom pharmazeutischen Unternehmer gekennzeichnet werden. § 10 Absatz 1 und 2 gilt entsprechend.
(3) Auch wenn der pharmazeutische Unternehmer unter Berufung auf § 10 Absatz 1 und 2 einer Veröffentlichung von Dokumenten in Modul K5 widerspricht, hat er dennoch zu gewährleisten, dass alle Angaben zu Studienmethodik und -ergebnissen vollständig zur Veröffentlichung nach Maßgabe von Absatz 2 Satz 2 zur Verfügung gestellt werden. § 9 Absätze 4, 5, 6 und 8 gelten entsprechend.
§ 40 Übergangsregelung zu § 23 Absatz 4 - Antragsfrist
In den Fällen einer Antragsberechtigung nach § 23 vor Inkrafttreten dieses Abschnitts beginnt die Frist nach § 23 Absatz 4 mit Inkrafttreten dieses Abschnitts.
| Anforderungsformular für eine Beratung | Anlage I (zum 5. Kapitel) |
Anforderungsformular
| 1) | Pharmazeutischer Unternehmer | |
| a) Name des pharmazeutischen Unternehmers | << >> | |
| b) Anschrift | << >> | |
| 2) | Ansprechpartner beim pharmazeutischen Unternehmer | |
| a) Name | << >> | |
| b) Abteilung und Funktion | << >> | |
| c) Adresse | << >> | |
| d) Email | << >> | |
| e) Telefon- und Faxnummer | << >> | |
Informationen zur Art der Beratung
| 3) | Angaben zum Arzneimittel/Wirkstoff | |
| a) Neuer Wirkstoff | Ja/Nein | |
| b) Neues Anwendungsgebiet | Ja/Nein | |
| c) Bezeichnung des arzneilich wirksamen Bestandteils (INN) | << >> | |
| d) Bezeichnung des Fertigarzneimittels | << >> | |
| e) Darreichungsform | << >> | |
| f) Anwendungsart | << >> | |
| g) Indikation (zugelassene oder geplante Indikation bzw. geplantes neues Anwendungsgebiet) | << >> | |
| 4) | Zulassungsstatus des Arzneimittels | |
a) Nicht zugelassen
1. Zulassung beantragt b) Zugelassen 1. Inverkehrbringen vorgesehen für Datum c) Ist der Wirkstoff in einem anderen Land bereits zugelassen? Wenn ja, wo und für welche Indikation(en) | << >> JA/NEIN | |
| 5) | Anlagen | |
| 1. Unterlagen und Informationen gemäß § 7 VerfO | ||
| 2. Wenn Beratungsgespräche bei Zulassungsbehörden stattgefunden haben, sind die Protokolle beizufügen. | ||
| 3. Für Studien, auf die in den Fragen Bezug genommen wird, sind Registereinträge in der Anlage zu übermitteln. | ||
| 6) | Fragen, die im Beratungsgespräch erörtert werden sollen | |
| (Hinweis: Zu jeder Frage sollte der pharmazeutische Unternehmer seine Position und ggf. Begründung formulieren. Des Weiteren soll die Dokumentation entsprechend dem Fragenkatalog gegliedert werden.) | ||
| 1 | << >> | |
| 2 | << >> | |
| 3 | << >> | |
| 4 | << >> | |
(Bitte weitere Felder einfügen sofern benötigt.)
| Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards | Anlage II (zum 5. Kapitel) |
| Erstellung und Einreichung eines Dossiers zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.1 |
Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards
Stand: 18. April 2013
Abbildungsverzeichnis
Abbildung 1: Modularer Aufbau des Dossiers zur Nutzungsbewertung nach § 35a SGB V
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| ASCII | American Standard Code for Information Interchange |
| CTD | Common Technical Document |
| DVD | Digital Versatile Disc |
| EPAR | European Public Assessment Report |
| GKV | Gesetzliche Krankenversicherung |
| ICH | International Conference on Harmonisation |
| Portable Document Format | |
| RIS | Research Information System |
| SGB | Sozialgesetzbuch |
1 Aufbau des Dossiers zur Nutzenbewertung nach § 35a SGB V
Das Dossier zur Nutzenbewertung nach § 35a SGB V ist modular aufgebaut. Die nachfolgende Abbildung 1 zeigt den modularen Aufbau in der Übersicht.
Abbildung 1: Modularer Aufbau des Dossiers zur Nutzenbewertung nach § 35a SGB V
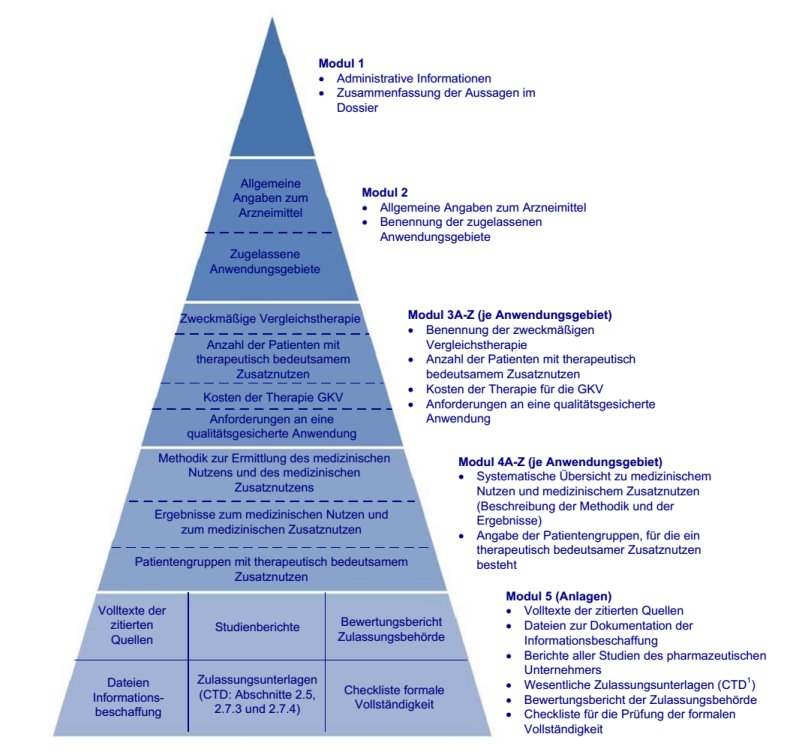
1.1 Inhalte der Module 1 bis 4 - Übersicht
Die Module 1 bis 4 bestehen aus Dokumenten, die auf Basis bereitgestellter Dokumentvorlagen für diese Module zu erstellen sind (siehe Kapitel 2).
Modul 1 des Dossiers enthält administrative Angaben sowie die Zusammenfassung der Aussagen aus den Modulen 2, 3 und 4.
Modul 2 enthält die Beschreibung des zu bewertenden Arzneimittels und Angaben über die zugelassenen Anwendungsgebiete.
Modul 3 enthält für jedes zu bewertende Anwendungsgebiet Angaben zur zweckmäßigen Vergleichstherapie, Angaben zur Anzahl der Patienten, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, Angaben zu den Kosten der Therapie für die gesetzliche Krankenversicherung (GKV) sowie Angaben zu den Anforderungen an eine qualitätsgesicherte Anwendung.
Modul 4 enthält für jedes zu bewertende Anwendungsgebiet die Angaben zum medizinischen Nutzen und zum medizinischen Zusatznutzen im Vergleich zur zweckmäßigen Vergleichstherapie sowie Angaben zu den Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen.
Für die Module 1 und 2 ist jeweils ein Dokument auf Basis der jeweils bereitgestellten Dokumentvorlage zu erstellen. Für die Module 3 und 4 ist für jedes zu bewertende Anwendungsgebiet jeweils ein Dokument auf Basis der jeweils bereitgestellten Dokumentvorlage zu erstellen. Die Anwendungsgebiete erhalten jeweils eine Kodierung mit den Buchstaben A bis Z. Diese Kodierung wird im Dokument für Modul 2 vorgenommen und ist durchgängig für das gesamte Dossier zu verwenden. Wird ein Dossier für ein einzelnes Anwendungsgebiet erstellt, ist auch dieses Anwendungsgebiet zu kodieren, und zwar mit dem Buchstaben A.
1.2 Inhalt von Modul 5 - Übersicht
Modul 5 enthält Dokumente, die für die Aussagen in den Modulen 2 bis 4 herangezogen werden, sowie eine Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers als Anlage zu Modul 1. Eine detaillierte Darstellung des Inhalts von Modul 5 findet sich in Kapitel 3 des vorliegenden Dokuments.
1.3 Orphan Drugs
Im Rahmen der frühen Nutzenbewertung von Arzneimitteln mit neuen Wirkstoffen gelten für Orphan Drugs folgende Regelungen: Gemäß der gesetzlichen Vorgaben (§ 35a Absatz 1 Satz 10 SGB V) gilt für diese Medikamente der medizinische Zusatznutzen bereits durch die Zulassung als belegt; Nachweise zum medizinischen Nutzen und zum medizinischen Zusatznutzen im Verhältnis zur zweckmäßigen Vergleichstherapie müssen nicht vorgelegt werden. Lediglich das Ausmaß des Zusatznutzens ist für die Anzahl der Patienten und Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, nachzuweisen. Ausgehend von dieser gesetzlichen Vorgabe bestimmt der Gemeinsame Bundesausschuss bei Orphan Drugs, die den Umsatz von 50 Millionen Euro in den letzten zwölf Kalendermonaten nicht übersteigen, das Ausmaß des Zusatznutzens auf der Grundlage der Zulassung und der die Zulassung begründenden Studien. In diesem Fall sind daher abweichend folgende Dossierunterlagen vom pharmazeutischen Unternehmer einzureichen:
Modul 1
Nicht auszufüllen sind die Abschnitte 1.4 (zweckmäßige Vergleichstherapie) und 1.5 (medizinischer Nutzen und Zusatznutzen).
Modul 2
Nicht auszufüllen ist der Abschnitt 2.2.3 (Zulassungsstatus international).
Modul 3
Nicht auszufüllen ist der Abschnitt 3.1 (Bestimmung der zweckmäßigen Vergleichstherapie).
Modul 4
Es sind nur die Abschnitte 4.4.2 und 4.4.3 auszufüllen.
Dabei ist zu berücksichtigen, dass die Angaben zum Ausmaß des Zusatznutzens auf der Grundlage der Zulassung und der die Zulassung begründenden Studien erfolgen müssen.
Entgegen den Erläuterungen in den Abschnitten 4.4.2 und 4.4.3 müssen für die Darstellung dieser Ergebnisse die Abschnitte 4.3.1 und 4.3.2 nicht ausgefüllt werden, sie können jedoch (wie die übrigen Abschnitte von Modul 4 auch) zur strukturierten Darstellung der Ergebnisse ausgefüllt werden.
Modul 5
Vorzulegen sind Studienberichte einschließlich Studienprotokollen zu Zulassungsstudien sowie alle im Anwendungsgebiet durchgeführten Studien, die der Zulassungsbehörde übermittelt worden sind (vgl. 5. Kapitel § 9 Absatz 4 Satz 1 VerfO), die Dokumente der Zulassungsbehörden (u. a. der EPAR) sowie die weiteren Unterlagen/Volltexte zu den Modulen 1 bis 4.
2 Erstellung der Dokumente für die Module 1 bis 4 des Dossiers
Für die Module 1 bis 4 werden Dokumentvorlagen für das Standardtextverarbeitungsprogramm "Microsoft Word" 2) auf der Website des Gemeinsamen Bundesausschusses (http://www.g-ba.de) bereitgestellt. Die Dokumentvorlagen sind bei der Erstellung des Dossiers zu verwenden. Die Struktur der Dokumente einschließlich der Benennung der Abschnitte, Abbildungen und Tabellen soll nur angepasst werden, wenn an der entsprechenden Stelle in der Dokumentvorlage darauf hingewiesen wird. Die Dokumente sind in deutscher Sprache zu erstellen.
Folgende Elemente sind in den Dokumentvorlagen enthalten:
Erläuterungen/Beispiele für den jeweiligen Abschnitt (hellgrau unterlegt mit Rahmen)
Die Elemente sollen bei der Erstellung des Dossiers nicht aus den Dokumenten entfernt werden. Ausnahmen sind Elemente, bei denen in den Dokumentvorlagen selbst an der jeweiligen Stelle darauf verwiesen wird, dass sie fragestellungsbezogen anzupassen sind (z.B. Tabellenüberschriften), und Beispielzeilen in Tabellen (diese sollen überschrieben werden).
Stellen, an denen Platzhalter hinterlegt sind, sind grundsätzlich mit Angaben des pharmazeutischen Unternehmers zu füllen, es sei denn, dass explizit darauf verwiesen wird, dass dies nur in bestimmten Fällen erforderlich ist (z.B. Darstellung weiterer Unterlagen zum medizinischen Nutzen und Zusatznutzen in Modul 4). Die Felder auf dem Deckblatt sollen wie folgt gefüllt werden:
In den Dokumenten verwendete Abkürzungen sind in das jeweilige Abkürzungsverzeichnis aufzunehmen. Sofern Abbildungen oder Tabellen verwendet werden, sind diese im jeweiligen Abbildungs- bzw. Tabellenverzeichnis aufzuführen.
Nach Fertigstellung der Dokumente für die Module 1 bis 4 sind von diesen PDF-Dateien zu erstellen. Die PDF-Dokumente müssen navigierbar sein, d. h., Verweise auf Abschnitte, Abbildungen und Tabellen innerhalb des jeweiligen Dokuments müssen als klickbare Verlinkungen enthalten sein. Auch die Verweise in den Verzeichnissen (Abbildungs-, Inhalts- und Tabellenverzeichnis) müssen klickbare Verlinkungen zu den entsprechenden Abbildungen, Abschnitten bzw. Tabellen darstellen. Verweise zwischen verschiedenen Dokumenten (d. h. auf andere Module) müssen nicht als klickbare Verlinkungen enthalten sein.
Bei der Erstellung der PDF-Dateien ist darauf zu achten, dass Seitenzahlen, Beschriftungen von Abbildungen und Tabellen, Querverweise auf Abbildungen und Tabellen sowie Verzeichnisse (Inhaltsverzeichnis, Tabellenverzeichnis, Abbildungsverzeichnis) im PDF-Dokument richtig dargestellt sind. Die PDF-Dateien dürfen keine Wasserzeichen enthalten und nicht geschützt werden; die Dokumente müssen elektronisch kommentierbar und die Inhalte elektronisch entnehmbar sein.
3 Inhalt von Modul 5 des Dossiers im Detail
Modul 5 enthält Dokumente, die für die Aussagen in den Modulen 2 bis 4 herangezogen werden, sowie eine Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers als Anlage zu Modul 1. Die Dokumente, die für die Aussagen in den Modulen 2 bis 4 herangezogen werden, können in deutscher oder englischer Sprache (ggf. als Übersetzung) beigelegt werden. Die Checkliste zur Prüfung der formalen Vollständigkeit ist in deutscher Sprache zu erstellen.
3.1 Dokumente, die für Aussagen in den Modulen 2 bis 4 herangezogen werden
Die nachfolgende Liste zeigt in der Übersicht, welche Dokumente für die Aussagen in den Modulen 2 bis 4 herangezogen werden und deshalb in Modul 5 abzulegen sind.
3.1.1 Studienberichte
Gemäß Arzneimittel-Nutzenbewertungsverordnung vom 28. Dezember 2010 legt der pharmazeutische Unternehmer im Dossier den Ergebnisbericht der Zulassungsstudien einschließlich der Studienprotokolle und des Bewertungsberichtes der Zulassungsbehörde vor, sowie alle Studien, die der Zulassungsbehörde übermittelt worden sind. Darüber hinaus werden alle Ergebnisse, Studienberichte und Studienprotokolle von Studien zum Arzneimittel übermittelt, für die der Unternehmer Sponsor war, sowie alle verfügbaren Angaben über laufende oder abgebrochene Studien mit dem Arzneimittel, für die der Unternehmer Sponsor ist oder auf andere Weise finanziell beteiligt ist, und entsprechende Angaben über Studien von Dritten, soweit diese verfügbar sind.
In der Dossiervorlage zu Modul 4 des Dossiers ist konkretisiert, welche Studien des pharmazeutischen Unternehmers aufzulisten sind:
Dem entsprechend müssen für alle abgeschlossenen und abgebrochenen Studien, die in Modul 4 als Studien des pharmazeutischen Unternehmers mit dem zu bewertenden Arzneimittel im jeweiligen Anwendungsgebiet aufgeführt sind, die Studienberichte und Studienprotokolle beigelegt werden, unabhängig davon, ob sie in die Bewertung eingeschlossen oder aus der Bewertung begründet ausgeschlossen wurden. Gleiches gilt für Studien von Dritten, soweit diese Unterlagen verfügbar sind.
Für die genannten Studien sind die vollständigen Studienberichte nach ICH E3 einschließlich der zugehörigen Appendizes beizulegen. Appendizes, die individuelle Patienteninformationen (patient data listings) bzw. andere individuelle personenbezogene Angaben (z.B. Angaben zu Prüfärzten) enthalten, müssen nicht beigelegt werden und können gegebenenfalls entfernt oder unkenntlich gemacht werden. Das Inhaltsverzeichnis zu den Appendizes, mit den Angaben zu allen zugehörigen Appendizes, darf weder entfernt noch anderweitig abgeändert werden. Ist das Studienprotokoll zu einer Studie nicht im zugehörigen Appendix enthalten, ist es gesondert beizulegen. Sind Berichte nach ICH E3 nicht verfügbar, sind Studienberichte beizulegen, die einen ähnlichen Detaillierungsgrad wie Studienberichte nach ICH E3 besitzen.
Einreichung von Studienberichten, die nicht in deutscher oder englischer Sprache vorliegen
Gemäß Verfahrensordnung des Gemeinsamen Bundesausschusses müssen alle Dokumente, die in Modul 5 enthalten sind, in deutscher oder englischer Sprache vorgelegt werden. Studienberichte nach ICH E3 können einschließlich Appendizes mehrere tausend Seiten umfassen, so dass für diese speziellen Dokumente ein erheblicher Übersetzungsaufwand entstehen kann, wenn sie nicht in deutscher oder englischer Sprache erstellt wurden. Für Studienberichte, die nicht in deutscher oder englischer Sprache erstellt wurden, gelten daher folgende vereinfachte Regeln:
Format und Gliederung, einzureichende Unterlagen, technische Standards
3.1.2 Bewertungsbericht der Zulassungsbehörden
Unter "Bewertungsbericht der Zulassungsbehörden" sind veröffentlichte und unveröffentlichte Dokumente des abschließenden Bewertungsberichts aus dem für Deutschland gültigen Zulassungsverfahren zu verstehen, die dem pharmazeutischen Unternehmer vorliegen. Diese sind für das zu bewertende Arzneimittel für alle zu bewertenden Anwendungsgebiete vollständig beizulegen. Für zentrale Zulassungsverfahren sind folgende Bewertungsberichte einzureichen 23:
3.2 Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers als Anlage zu Modul 1
Für die Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers wird auf der Website des Gemeinsamen Bundesausschusses (http://www.g-ba.de) eine Dokumentvorlage bereitgestellt, die zu verwenden ist. Nach Fertigstellung der Checkliste ist von dieser eine PDF-Datei zu erstellen. Die PDF-Datei darf nicht geschützt werden; das Dokument darf kein Wasserzeichen enthalten, es muss elektronisch kommentierbar und die Inhalte müssen elektronisch entnehmbar sein.
3.3 Kennzeichnung von Betriebs- und Geschäftsgeheimnissen
In Modul 5 hinterlegte Dokumente, die aus Sicht des pharmazeutischen Unternehmers Betriebs- und Geschäftsgeheimnisse enthalten und deshalb nicht veröffentlicht werden sollen, sind vom pharmazeutischen Unternehmer entsprechend zu kennzeichnen. Die Kennzeichnung erfolgt, indem der Name und der Ablageort dieser Dokumente in einem speziell hierfür vorgesehenen PDF-Dokument anzugeben sind. Näheres hierzu ist im Abschnitt 4.1 beschrieben.
4 Hinweise zur elektronischen Einreichung des Dossiers
Die Einreichung des Dossiers hat elektronisch zu erfolgen. Als Datenträger ist eine Digital Versatile Disc (DVD) zu verwenden. Die Datenträger dürfen nicht kopiergeschützt sein. Das Dossier ist in zweifacher Ausfertigung einzureichen.
Die Struktur der einzureichenden DVD ist im Abschnitt 4.1 beschrieben. Der Verzeichnisbaum liegt auch auf der Website des Gemeinsamen Bundesausschusses (http://www.g-ba.de) als ladbare Datei vor. Bei der Erstellung der DVD ist darauf zu achten, dass der längste Dateipfad (einschließlich Dateiname) höchstens 160 Zeichen lang ist.
Für alle einzureichenden Dateien gilt, dass diese ohne Kennworteingabe lesbar, speicherbar und druckbar sein müssen. PDF-Dokumente des pharmazeutischen Unternehmers (insbesondere Module 1 bis 4, Checkliste zur Prüfung der formalen Vollständigkeit und Studienberichte inkl. Anlagen) müssen darüber hinaus navigierbar (siehe auch Abschnitt 2), elektronisch kommentierbar und die Inhalte elektronisch entnehmbar sein. Zudem dürfen diese Dokumente kein Wasserzeichen enthalten.
Sonderfälle bei der Einreichung (z.B. nicht ausreichende Speicherkapazität bei Verwendung einer einzelnen DVD) sind in Abschnitt 4.2 beschrieben.
4.1 Struktur der einzureichenden DVD
Die erste Ebene der DVD enthält jeweils ein Verzeichnis für die Module 1 bis 5:

Im Verzeichnis "Modul1" ist Modul 1, im Verzeichnis "Modul2" ist Modul 2 des Dossiers zu hinterlegen, und zwar jeweils im PDF-Format. Die Dateien sind wie folgt zu benennen:
#JJJJ-MM-TT#_Modul1_#Wirkstoff#.pdf
bzw.
#JJJJ-MM-TT#_Modul2_#Wirkstoff#.pdf
Dabei ist "#JJJJ-MM-TT#" jeweils durch den Stand des Dossiers zu ersetzen, der auch auf dem Deckblatt der einzelnen Module hinterlegt ist. "#Wirkstoff#" ist durch den Namen des zu bewertenden Wirkstoffs zu ersetzen, wie er auch auf dem Deckblatt der einzelnen Module hinterlegt ist.
Modul 3
Im Verzeichnis "Modul3" ist für jedes zu bewertende Anwendungsgebiet das zugehörige Modul 3 des Dossiers im PDF-Format zu hinterlegen. Die Dateien sind wie folgt zu benennen:
#JJJJ-MM-TT#_Modul3#A-Z#_#Wirkstoff#.pdf
Dabei ist "#JJJJ-MM-TT#" jeweils durch den Stand des Dossiers zu ersetzen, der auch auf dem Deckblatt der einzelnen Module hinterlegt ist. "#A-Z#" ist durch die Kodierung für das jeweilige Anwendungsgebiet zu ersetzen, wie auf dem Deckblatt des jeweiligen Moduls hinterlegt (Buchstabe A bis Z); die Kodierung der einzelnen Anwendungsgebiete wird in Modul 2 vorgenommen. "#Wirkstoff#" ist durch den Namen des zu bewertenden Wirkstoffs zu ersetzen, wie er auch auf dem Deckblatt der einzelnen Module hinterlegt ist.
Modul 4
Im Verzeichnis "Modul4" ist für jedes zu bewertende Anwendungsgebiet das zugehörige Modul 4 des Dossiers im PDF-Format zu hinterlegen. Die Dateien sind wie folgt zu benennen:
#JJJJ-MM-TT#_Modul4#A-Z#_#Wirkstoff#.pdf
Dabei ist "#JJJJ-MM-TT#" jeweils durch den Stand des Dossiers zu ersetzen, der auch auf dem Deckblatt der einzelnen Module hinterlegt ist. "#A-Z#" ist durch die Kodierung für das jeweilige Anwendungsgebiet zu ersetzen, wie auf dem Deckblatt des jeweiligen Moduls hinterlegt (Buchstabe A bis Z); die Kodierung der einzelnen Anwendungsgebiete wird in Modul 2 vorgenommen. "#Wirkstoff#" ist durch den Namen des zu bewertenden Wirkstoffs zu ersetzen, wie er auch auf dem Deckblatt der einzelnen Module hinterlegt ist.
Modul 5 (Anlagen)
Modul 5 enthält Dokumente, die für die Aussagen in den Modulen 2 bis 4 herangezogen werden, eine Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers als Anlage zu Modul 1 sowie ein Dokument, in dem Dokumente aus Modul 5, die aus Sicht des pharmazeutischen Unternehmers Betriebs- und Geschäftsgeheimnisse enthalten, gekennzeichnet werden können. Das Verzeichnis "Modul5" enthält fünf Unterverzeichnisse, vier für die Dateien zu den Modulen 1 bis 4 und eins für die Kennzeichnung der Dokumente mit Betriebs- und Geschäftsgeheimnissen:
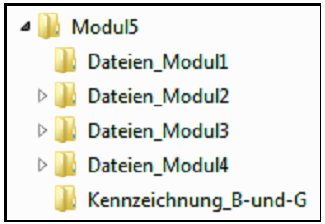
Dateien für Modul 1
Im Verzeichnis "Dateien-fuer-Modul1" ist die ausgefüllte Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers im PDF-Format zu hinterlegen. Die Datei ist wie folgt zu benennen:
M l_Checkliste-Vollstaendigkeit.pdf
Dateien für Modul 2
Das Verzeichnis "Dateien-fuer-Modul2" enthält ein Unterverzeichnis "Volltexte", in dem alle in Modul 2 zitierten und in der zugehörigen Referenzliste aufgeführten Quellen als Volltexte zu hinterlegen sind.

Die einzelnen Volltexte sind im PDF-Format zu hinterlegen. Die Benennung der Dateien ist wie folgt:
#Zitat-Nr#_#Erstautor#_#JJJJ#.pdf
Dabei ist "#Zitat-Nr#" durch die Nummer zu ersetzen, unter der der betreffende Volltext in der Referenzliste aufgeführt wird. Für "#Erstautor#" ist der Nachname des Erstautors der Publikation anzugeben. Ist für die Quelle kein Erstautor genannt, kann "Anonym" oder, falls zutreffend, die für die Quelle verantwortliche Institution genannt werden. "#JJJJ#" ist durch das Jahr der Publikation oder, falls zutreffend, der Erstellung der Quelle zu ersetzen. Falls weder Publikations- noch Erstellungsdatum bekannt sind, kann "0000" angegeben werden.
Zusätzlich ist in dem Unterverzeichnis "Volltexte" eine RIS-Datei der Referenzliste zu Modul 2 zu hinterlegen. Die Benennung dieser Datei ist wie folgt:
M2_Referenzliste.ris
Dateien für Modul 3
Das Verzeichnis "Dateien_Modul3" enthält für jedes zu bewertende Anwendungsgebiet ein Unterverzeichnis "AWG_#A-Z#", wobei "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen ist. Die Kodierung der einzelnen Anwendungsgebiete wird in Modul 2 vorgenommen.
Jedes dieser Unterverzeichnisse "AWG_#A-Z#" enthält wiederum 4 Unterverzeichnisse, in denen jeweils die in den Abschnitten 3.1 bis 3.4 von Modul 3 zitierten und in der jeweiligen Referenzliste aufgeführten Quellen als Volltexte zu hinterlegen sind 24.
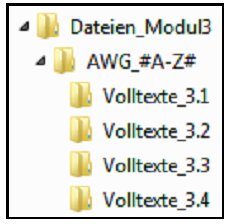
Die einzelnen Volltexte sind im PDF-Format zu hinterlegen. Die Benennung der Dateien ist wie folgt:
für Abschnitt 3.1: #Zitat-Nr#_#Erstautor#_#JJJJ#.pdf
für Abschnitt 3.2: #Zitat-Nr#_#Erstautör#_#JJJJ#.pdf
für Abschnitt 3.3: #Zitat-Nr# JErstautor#_#JJJJ#.pdf
für Abschnitt 3.4: #Zitat-Nr#_#Erstautor# _#JJJJ#.pdf
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. "#Zitat-Nr#" ist durch die Nummer zu ersetzen, unter der der betreffende Volltext in der Referenzliste aufgeführt wird. Für "#Erstautor#" ist der Nachname des Erstautors der Publikation anzugeben. Ist für die Quelle kein Erstautor genannt, kann "Anonym" oder, falls zutreffend, die für die Quelle verantwortliche Institution genannt werden. "#JJJJ#" ist durch das Jahr der Publikation oder, falls zutreffend, der Erstellung der Quelle zu ersetzen. Falls weder Publikations- noch Erstellungsdatum bekannt sind, kann "0000" angegeben werden.
Zusätzlich ist in jedem Unterverzeichnis eine RIS-Datei der jeweiligen Referenzliste zu hinterlegen. Die Benennung ist wie folgt:
für Abschnitt 3.1: M3#A-Z#_3-1_Referenzliste.ris
für Abschnitt 3.2: M3#A-Z#_3-2_Referenzliste.ris
für Abschnitt 3.3: M3#A-Z#_3-3_Referenzliste.ris
für Abschnitt 3.4: M3#A-Z#_3-4_Referenzliste.ris
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen.
Dateien für Modul 4
Das Verzeichnis "Dateien_Modul4" enthält für jedes zu bewertende Anwendungsgebiet ein Unterverzeichnis "AWG_#A-Z#", wobei "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen ist. Die Kodierung der einzelnen Anwendungsgebiete wird in Modul 2 vorgenommen.
Jedes dieser Unterverzeichnisse "AWG_#A-Z#" enthält wiederum 6 Unterverzeichnisse, in denen die verschiedenen Anlagen von Modul 4 zu hinterlegen sind:
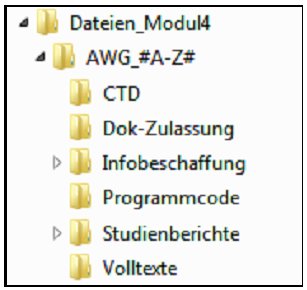
CTD
Im Verzeichnis "CTD" sind die Abschnitte 2.5, 2.7.3 25 und 2.7.4 des Zulassungsdossiers nach CTD als PDF-Dateien zu hinterlegen. Die Benennung der Dateien ist wie folgt:
M4#A-Z#_CTD-Abschnitt2-5.pdf
M4#A-Z#_CTD-Abschnitt2-7-3.pdf
M4#A-Z#_CTD-Abschnitt2-7-4.pdf
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen.
Dokumente der Zulassungsbehörden
Im Verzeichnis "Dok-Zulassung" sind veröffentlichte und unveröffentlichte Dokumente des abschließenden Bewertungsberichts aus dem für Deutschland gültigen Zulassungsverfahren, die dem pharmazeutischen Unternehmer vorliegen, vollständig im PDF-Format zu hinterlegen (siehe auch Abschnitt 3.1.2). Die Benennung ist dabei wie folgt:
M4#A-Z#_DokZB_#Benennung#.pdf
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. "#Benennung#" ist durch eine eindeutige, möglichst sprechende Bezeichnung des jeweiligen Dokuments zu ersetzen (z.B. "EPAR" für den European Public Assessment Report oder "Tag-150-Bericht" für den Rapporteurs' Day 150 Joint Response Assessment Report).
Informationsbeschaffung
Im Verzeichnis "Infobeschaffung" sind Dateien zur Dokumentation der Informationsbeschaffung für Modul 4 zu hinterlegen. Dies betrifft sowohl die bibliografische Literaturrecherche als auch die Recherche in Studienregistern. Die Ablage erfolgt für einzelne Recherchen und Datenbanken getrennt, analog der Strukturierung in den Anhängen 4-A, 4-B, 4-C und 4-D von Modul 4.
Anhang 4-A
Im Verzeichnis "Anhang-4-A" sind RIS-Dateien zur bibliografischen Literaturrecherche abzulegen. Die Ablage erfolgt getrennt nach Thema der Recherche (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.; siehe auch Anhang 4-A von Modul 4). Hierzu enthält das Verzeichnis "Anhang-4-A" 4 Unterverzeichnisse ("Anhang-4-A1", "Anhang-4-A2", "Anhang-4-A3" und "Anhang-4-A4"). Für jede durchsuchte Datenbank ist eine separate RIS-Datei abzulegen, die alle Treffer der Suche in dieser Datenbank zum jeweiligen Thema enthält. Diese ist wie folgt zu benennen:
M4#A-Z#_#Datenbankname#_Anhang_4-A#Thema#.ris
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. "#Datenbankname#" ist durch den Namen der durchsuchten Datenbank zu ersetzen (z.B. EMBASE). "#Thema#" ist die Nummer des Themas analog der Struktur in Anhang 4-A von Modul 4 (z.B. "1" für die Suche nach RCT mit dem zu bewertenden Arzneimittel, "2" für die Suche nach RCT für indirekte Vergleiche etc.).
Anhang 4-B
Im Verzeichnis "Anhang-4-B" sind RIS-Dateien zur Suche in Studienregistern abzulegen. Die Ablage erfolgt getrennt nach Thema der Recherche (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.; siehe auch Anhang 4-B von Modul 4). Hierzu enthält das Verzeichnis "Anhang-4-B" 4 Unterverzeichnisse ("Anhang-4-B1", "Anhang-4-B2", "Anhang-4-B3" und "Anhang-4-B4"). Für jedes durchsuchte Studienregister ist eine separate RIS-Datei abzulegen, die alle Treffer der Suche in diesem Register zum jeweiligen Thema enthält. Diese ist wie folgt zu benennen:
M4#A-Z#_#Registername#_Anhang_4-B#Thema#.ris
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. "#Registername#" ist durch den Namen des durchsuchten Studienregisters zu ersetzen (z.B. clinicaltrials.gov). "#Thema#" ist die Nummer des Themas analog der Struktur in Anhang 4-B von Modul 4 (z.B. "1" für die Suche nach RCT mit dem zu bewertenden Arzneimittel, "2" für die Suche nach RCT für indirekte Vergleiche etc.).
Anhang 4-C
Im Verzeichnis "Anhang-4-C" sind alle im Anhang 4-C von Modul 4 zitierten Quellen als Volltexte im PDF-Format zu hinterlegen. Die Ablage erfolgt getrennt nach Thema der Recherche (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.; siehe auch Anhang 4-C von Modul 4). Hierzu enthält das Verzeichnis "Anhang-4-C" 4 Unterverzeichnisse ("Anhang-4-C1", "Anhang-4-C2", "Anhang-4-C3" und "Anhang-4-C4"). Die Dateien sind wie folgt zu benennen:
#Zitat-Nr#_#Erstautor#_#JJJJ#.pdf
"#Zitat-Nr#" ist durch die Nummer zu ersetzen, unter der der betreffende Volltext in der jeweiligen Referenzliste aufgeführt wird. Für "#Erstautor#" ist der Nachname des Erstautors der Publikation anzugeben. Ist für die Quelle kein Erstautor genannt, kann "Anonym" oder, falls zutreffend, die für die Quelle verantwortliche Institution genannt werden. "#JJJJ#" ist durch das Jahr der Publikation oder, falls zutreffend, der Erstellung der Quelle zu ersetzen. Falls weder Publikations- noch Erstellungsdatum bekannt sind, kann "0000" angegeben werden.
Anhang 4-D
Im Verzeichnis "Anhang-4-D" sind alle im Anhang 4-D von Modul 4 zitierten Quellen als Volltexte im PDF-Format zu hinterlegen (bei Registereinträgen z.B. Screenshots, bei Ergebnisberichten die Ergebnisberichte selbst). Die Ablage erfolgt getrennt nach Thema der Recherche (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.; siehe auch Anhang 4-D von Modul 4). Hierzu enthält das Verzeichnis "Anhang-4-D" 4 Unterverzeichnisse ("Anhang-4-D1", "Anhang-4-D2", "Anhang-4-D3" und "Anhang-4-D4"). Die Dateien sind wie folgt zu benennen:
#Zitat-Nr#_#Studie#_#JJJJ-MM#.pdf
"#Zitat-Nr#" ist durch die Nummer zu ersetzen, unter der der betreffende Volltext in der jeweiligen Referenzliste aufgeführt wird. Für "#Studie#" ist eine Studienbezeichnung einzugeben, die sich aus dem jeweiligen Eintrag in das Studienregister ergibt. Dies kann ein Studienkürzel sein (z.B. ALLHAT) oder die Nummer des Registereintrags (z.B. NCT00000542). "#JJJJ-MM#" ist durch den Stand des Registereintrags (Monat und Jahr) zu ersetzen. Falls der Stand unbekannt ist, kann "00-0000" angegeben werden.
Programmcode
Im Verzeichnis "Programmcode" sind Programmcodes (im ASCII-Format) zu hinterlegen, die für die Durchführung indirekter Vergleiche in Modul 4 verwendet wurden. Die Benennung der Dateien ist wie folgt:
M4#A-Z#_Programmcode_#Benennung#.#xxx#
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. #Benennung# ist durch eine eindeutige, möglichst sprechende Bezeichnung des jeweiligen Programmcodes zu ersetzen. #xxx# ist durch die jeweilige Dateiendung zu ersetzen.
Zusätzlich ist ein PDF-Dokument zu hinterlegen, in dem formlos unter Verweis auf die jeweiligen Abschnitte in Modul 4 dokumentiert wird, welcher indirekte Vergleich in den Dateien jeweils abgebildet ist (bzw. ggf. die Angabe, dass keine indirekten Vergleiche durchgeführt wurden). In diesem Dokument ist auch für jede Datei anzugeben, welche Software in welcher Version zur Ausführung des Programms verwendet wurde (ggf. inkl. Spezifizierung von Modulen, Prozeduren, Packages etc.). Die Benennung des PDF-Dokuments ist wie folgt:
M4#A-Z#_Dokumentation-indirekte-Vergleiche.pdf
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen.
Studienberichte
Im Verzeichnis "Studienberichte" sind die Studienberichte einschließlich der zugehörigen Studienprotokolle der Studien des pharmazeutischen Unternehmers zu hinterlegen (siehe auch Abschnitt 3.1.1). Hierzu ist für jede Studie ein separates Unterverzeichnis anzulegen:

Dabei ist "#Studienname#" durch eine eindeutige Bezeichnung der jeweiligen Studie zu ersetzen. Sofern es sich um eine eingeschlossene und daher in Abschnitt 4.6 von Modul 4 aufgeführte Studie handelt, soll die Bezeichnung der im Dossier verwendeten Bezeichnung für diese Studie entsprechen.
Im Unterverzeichnis "Studie-#Studienname#" ist der Studienbericht im PDF-Format einschließlich der Appendizes zu hinterlegen. Dabei kann der Bericht auf mehrere PDF-Dateien aufgeteilt sein. Appendizes, die individuelle Patienteninformationen enthalten (patient data listings), müssen nicht beigelegt werden. Sofern das Studienprotokoll nicht Bestandteil des Studienberichts einschließlich seiner Appendizes ist, ist dieses separat beizulegen.
Die Benennung der Dateien ist wie folgt:
M4#A-Z#_#Studienname#_#Dokumenttyp#.pdf
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen. "#Studienname#" ist durch die Bezeichnung der Studie zu ersetzen, die auch für die Benennung des Verzeichnisses gewählt wurde. "#Dokumenttyp#" ist durch eine eindeutige, möglichst sprechende Bezeichnung des Typs des jeweiligen Dokuments zu ersetzen (z.B. "Studienbericht", "Studienprotokoll", "Appendix16.1.1", "AppendixA").
Volltexte
Im Verzeichnis "Volltexte" sind alle in Modul 4 zitierten und in der Referenzliste (Abschnitt 4.7 von Modul 4) aufgeführten Quellen als Volltexte im PDF-Format zu hinterlegen (Ausnahme Studienberichte, s. u.). Die Benennung der Dateien ist wie folgt:
#Zitat-Nr#_#Erstautor#_#JJJJ#.pdf
Dabei ist "#Zitat-Nr#" durch die Nummer zu ersetzen, unter der der betreffende Volltext in der Referenzliste aufgeführt wird. Für "#Erstautor#" ist der Nachname des Erstautors der Publikation anzugeben. Ist für die Quelle kein Erstautor genannt, kann "Anonym" oder, falls zutreffend, die für die Quelle verantwortliche Institution genannt werden. "#JJJJ#" ist durch das Jahr der Publikation oder, falls zutreffend, der Erstellung der Quelle zu ersetzen. Falls weder Publikations- noch Erstellungsdatum bekannt sind, kann "0000" angegeben werden.
Für in der Referenzliste zitierte Dokumente, die bereits im Verzeichnis "Studienberichte" abgelegt wurden, ist keine erneute Ablage im Verzeichnis "Volltexte" erforderlich. Hierfür ist die Ablage einer nach obigem Schema (#Zitat-Nr#_#Erstautor#_#JJJJ#.pdf) benannten PDF-Datei ausreichend, in der formlos der Ablageort des zitierten Dokuments auf der DVD genannt wird (siehe auch Abschnitt 4.2.2).
Zusätzlich sind im Verzeichnis "Volltexte" eine RIS-Datei der Referenzliste in Abschnitt 4.7 von Modul 4 sowie eine RIS-Datei für die in Abschnitt 4.6 von Modul 4 zitierten Dokumente zu hinterlegen. Die Benennung dieser Dateien ist wie folgt:
M4#A-Z#_Referenzliste_Abschnitt4-7.ris M4#A-Z#_Zitate_Abschnitt4-6.ris
Dabei ist "#A-Z#" durch die Kodierung des Anwendungsgebiets zu ersetzen.
Kennzeichnung von Betriebs- und Geschäftsgeheimnissen
Im Verzeichnis "Kennzeichnung_B-und-G" ist ein PDF-Dokument zu hinterlegen, in dem alle Dokumente aus Modul 5 aufgeführt werden, die aus Sicht des pharmazeutischen Unternehmers Betriebs- und Geschäftsgeheimnisse enthalten und deshalb nicht veröffentlicht werden sollen. Die Benennung dieses PDF-Dokuments ist wie folgt:
M5_Kennzeichnung_B- und-G.pdf
Die Kennzeichnung der Dokumente aus Modul 5 mit Betriebs- und Geschäftsgeheimnissen erfolgt in diesem PDF-Dokument formlos durch Angabe ihres Namens und ihres Ablageortes (Pfades) auf der DVD. Das PDF-Dokument ist auch dann zu erstellen, wenn keins der Dokumente aus Modul 5 aus Sicht des pharmazeutischen Unternehmers Betriebs- und Geschäftsgeheimnisse enthält. In dem PDF-Dokument soll dann die Feststellung "Modul 5 enthält keine Dokumente mit Betriebs- und Geschäftsgeheimnissen" getroffen werden.
4.2 Sonderfälle bei der Einreichung
4.2.1 Nicht ausreichende Speicherkapazität bei Verwendung einer einzelnen DVD
Reicht die Speicherkapazität einer einzelnen DVD nicht für das Dossier aus, können die Dokumente auf zwei, bei Bedarf auch auf mehr als zwei DVDs verteilt werden. Jede DVD soll dabei nur diejenigen Verzeichnisbäume enthalten, in denen Dokumente hinterlegt sind. Die DVDs sind fortlaufend zu nummerieren. Auf der ersten DVD ist im Ursprungsverzeichnis ("root") ein PDF-Dokument zu hinterlegen, in dem formlos die Aufteilung der Dokumente auf die verschiedenen DVDs beschrieben wird.
4.2.2 Mehrfache Ablage von Dokumenten
Nach dem in Abschnitt 4.1 dargestellten Schema kann es notwendig sein, dass einzelne Dokumente an verschiedenen Stellen auf der DVD abgelegt werden müssen. Dies kann z.B. dann vorkommen, wenn einzelne Studien und damit deren Studienberichte für mehrere Anwendungsgebiete relevant sind. Nach der in Abschnitt 4.1 beschriebenen Struktur sind diese Dokumente je Anwendungsgebiet abzulegen.
Optional kann auf die Mehrfachablage von Dokumenten verzichtet werden. Das jeweilige Dokument ist dann einmalig an einer korrekten Stelle gemäß Abschnitt 4.1 abzulegen. An den übrigen Stellen sind PDF-Dokumente mit korrekter Benennung gemäß Abschnitt 4.1 abzulegen, in denen formlos auf den Ablageort des eigentlichen Dokuments innerhalb der DVD-Struktur verwiesen wird. Zusätzlich ist auf der DVD (bzw. bei Einreichung mehrerer DVDs auf der ersten DVD) im Ursprungsverzeichnis ("root") ein PDF-Dokument zu hinterlegen, in dem formlos beschrieben wird, welche der mehrfach abzulegenden Dokumente nur einmalig abgelegt wurden und von welchen Stellen aus auf diesen Ablageort verwiesen wird.
| Dossier zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.2 |
<<Wirkstoff>> (<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
Zusammenfassung der Aussagen
im Dossier
Stand: <TT.MM.JJJJ>
Tabellenverzeichnis
Tabelle 1-1: Für das Dossier verantwortliches pharmazeutisches Unternehmen
Tabelle 1-2: Zuständige Kontaktperson des für das Dossier verantwortlichen pharmazeutischen Unternehmens
Tabelle 1-3: Zulassungsinhaber des zu bewertenden Arzneimittels
Tabelle 1-4: Allgemeine Angaben zum zu bewertenden Arzneimittel
Tabelle 1-5: Zugelassene Anwendungsgebiete, auf die sich das Dossier bezieht
Tabelle 1-6: Weitere in Deutschland zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels
Tabelle 1-7: Zweckmäßige Vergleichstherapie (Angabe je Anwendungsgebiet)
Tabelle 1-8: Angaben zur Beanspruchung eines Zusatznutzens (Angabe je Anwendungsgebiet)
Tabelle 1-9: Anzahl der GKV-Patienten in der Zielpopulation (Angabe je Anwendungsgebiet)
Tabelle 1-10: Patientengruppen und Anzahl der Patienten, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, einschließlich Ausmaß des Zusatznutzens (Angabe je Anwendungsgebiet)
Tabelle 1-11: Jahrestherapiekosten für das zu bewertende Arzneimittel in der Zielpopulation (Angabe je Anwendungsgebiet)
Tabelle 1-12: Jahrestherapiekosten für das zu bewertende Arzneimittel in der Zielpopulation (Summe über alle Anwendungsgebiete)
Tabelle 1-13: Jahrestherapiekosten für das zu bewertende Arzneimittel - Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (Angabe je Anwendungsgebiet)
Tabelle 1-14: Jahrestherapiekosten für das zu bewertende Arzneimittel - Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (Summe über alle Anwendungsgebiete)
Tabelle 1-15: Jahrestherapiekosten für die zweckmäßige Vergleichstherapie - alle Populationen/Patientengruppen (Angabe je Anwendungsgebiet)
Abbildungsverzeichnis
Es konnten keine Einträge für ein Abbildungsverzeichnis gefunden werden.
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| ATC-Code | Anatomisch-Therapeutisch-Chemischer Code |
| GKV | Gesetzliche Krankenversicherung |
1 Modul 1- allgemeine Informationen
Modul 1 enthält administrative Informationen zum für das Dossier verantwortlichen pharmazeutischen Unternehmer und zum Zulassungsinhaber sowie die Zusammenfassung der Aussagen aus den Modulen 2, 3 und 4. Von den Modulen 3 und 4 liegen dabei ggf. mehrere Ausführungen vor, und zwar jeweils eine je zu bewertendes Anwendungsgebiet. Die Kodierung der Anwendungsgebiete (A-Z) ist in Modul 2 zu hinterlegen. Sie ist je Anwendungsgebiet einheitlich für die übrigen Module des Dossiers zu verwenden.
Im Dokument verwendete Abkürzungen sind in das Abkürzungsverzeichnis aufzunehmen. Sofern Sie für Ihre Ausführungen Abbildungen oder Tabellen verwenden, sind diese im Abbildungs- bzw. Tabellenverzeichnis aufzuführen.
1.1 Administrative Informationen
Benennen Sie in den nachfolgenden Tabellen (Tabelle 1-1 bis Tabelle 1-3) das für das Dossier verantwortliche pharmazeutische Unternehmen, die zuständige Kontaktperson sowie den Zulassungsinhaber des zu bewertenden Arzneimittels.
Tabelle 1-1: Für das Dossier verantwortliches pharmazeutisches Unternehmen
| Name des pharmazeutischen Unternehmens: | |
| Anschrift: |
Tabelle 1-2: Zuständige Kontaktperson des für das Dossier verantwortlichen pharmazeutischen Unternehmens
| Name: | |
| Position: | |
| Adresse: | |
| Telefon: | |
| Fax: | |
| E-Mail: |
Tabelle 1-3: Zulassungsinhaber des zu bewertenden Arzneimittels
| Name des pharmazeutischen Unternehmens: | |
| Anschrift: |
1.2 Allgemeine Angaben zum Arzneimittel
In diesem Abschnitt werden die Angaben aus Modul 2, Abschnitt 2.1 (Allgemeine Angaben zum Arzneimittel) zusammengefasst.
Geben Sie in Tabelle 1-4 den Namen des Wirkstoffs, den Handelsnamen und den ATC-Code für das zu bewertende Arzneimittel an. (Referenz: Modul 2, Abschnitt 2.1.1)
Tabelle 1-4: Allgemeine Angaben zum zu bewertenden Arzneimittel
| Wirkstoff: | |
| Handelsname: | |
| ATC-Code: |
Beschreiben Sie zusammenfassend (maximal 1.500 Zeichen) den Wirkmechanismus des zu bewertenden Arzneimittels. Beschreiben Sie dabei auch, ob und inwieweit sich der Wirkmechanismus des zu bewertenden Arzneimittels vom Wirkmechanismus anderer bereits in Deutschland zugelassener Arzneimittel unterscheidet. (Referenz: Modul 2, Abschnitt 2.1.2)
<< Angaben des pharmazeutischen Unternehmers >>
1.3 Zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels
In diesem Abschnitt werden die Angaben aus Modul 2, Abschnitt 2.2 (Zugelassene Anwendungsgebiete) zusammengefasst.
Benennen Sie in der nachfolgenden Tabelle 1-5 die Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht, einschließlich der Kodierung, die im Dossier für jedes Anwendungsgebiet verwendet wird. Geben Sie hierzu den Wortlaut der Fachinformation an; sofern im Abschnitt "Anwendungsgebiete" der Fachinformation Verweise enthalten sind, führen Sie auch den Wortlaut an, auf den verwiesen wird. Fügen Sie für jedes Anwendungsgebiet eine neue Zeile ein. (Referenz: Modul 2, Abschnitt 2.2.1)
Tabelle 1-5: Zugelassene Anwendungsgebiete, auf die sich das Dossier bezieht
| Anwendungsgebiet (Wortlaut der Fachinformation inkl. Wortlaut bei Verweisen) | Datum der Zulassungserteilung | Kodierung im Dossier a |
| a: Angabe "A" bis "Z". | ||
Falls es sich um ein Dossier zu einem neuen Anwendungsgebiet eines bereits zugelassenen Arzneimittels handelt, benennen Sie in der nachfolgenden Tabelle 1-6 die weiteren in Deutschland zugelassenen Anwendungsgebiete des zu bewertenden Arzneimittels. Geben Sie hierzu den Wortlaut der Fachinformation an; sofern im Abschnitt "Anwendungsgebiete" der Fachinformation Verweise enthalten sind, führen Sie auch den Wortlaut an, auf den verwiesen wird. Fügen Sie dabei für jedes Anwendungsgebiet eine neue Zeile ein. Falls es kein weiteres zugelassenes Anwendungsgebiet gibt oder es sich nicht um ein Dossier zu einem neuen Anwendungsgebiet eines bereits zugelassenen Arzneimittels handelt, fügen Sie in der ersten Zeile unter "Anwendungsgebiet" "kein weiteres Anwendungsgebiet" ein. (Referenz: Modul 2, Abschnitt 2.2.2)
Tabelle 1-6: Weitere in Deutschland zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels
| Anwendungsgebiet (Wortlaut der Fachinformation inkl. Wortlaut bei Verweisen) | Datum der Zulassungserteilung |
1.4 Zweckmäßige Vergleichstherapie
In diesem Abschnitt werden die Angaben aus Modul 3, Abschnitt 3.1 (Bestimmung der zweckmäßigen Vergleichstherapie) zusammengefasst, und zwar für alle Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht.
Benennen Sie in der nachfolgenden Tabelle 1-7 die zweckmäßige Vergleichstherapie. Unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten, auf die sich das vorliegende Dossier bezieht. Fügen Sie für jedes Anwendungsgebiet eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.1.1)
Tabelle 1-7: Zweckmäßige Vergleichstherapie (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Bezeichnung der zweckmäßigen Vergleichstherapie | |
| Kodierung a | Kurzbezeichnung | |
| a: Angabe der im Dossier verwendeten Kodierung. | ||
Begründen Sie zusammenfassend die Wahl der zweckmäßigen Vergleichstherapie (maximal 1.500 Zeichen je Anwendungsgebiet). (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.1.2)
<< Angaben des pharmazeutischen Unternehmers >>
1.5 Medizinischer Nutzen, medizinischer Zusatznutzen
In diesem Abschnitt werden die Angaben aus Modul 4, Abschnitt 4.3 (Ergebnisse zum medizinischen Nutzen und zum medizinischen Zusatznutzen) und Abschnitt 4.4.2 (Beschreibung des Zusatznutzens einschließlich dessen Wahrscheinlichkeit und Ausmaß) zusammengefasst, und zwar für alle Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht.
Fassen Sie die Aussagen zum medizinischen Nutzen und zum medizinischen Zusatznutzen zusammen; unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten, auf die sich das Dossier bezieht (maximal 3.000 Zeichen je Anwendungsgebiet). Geben Sie auch die Effektmaße einschließlich der zugehörigen Konfidenzintervalle an. (Referenz: Modul 4 [alle Anwendungsgebiete], Abschnitt 4.3)
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in Tabelle 1-8 für alle Anwendungsgebiete, auf die sich das Dossier bezieht, jeweils an, ob Sie die Anerkennung eines Zusatznutzens im Vergleich zur zweckmäßigen Vergleichstherapie beanspruchen. Fügen Sie dabei für jedes Anwendungsgebiet eine neue Zeile ein. (Referenz: Modul 4 [alle Anwendungsgebiete], Abschnitt 4.4.2)
Tabelle 1-8: Angaben zur Beanspruchung eines Zusatznutzens (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Anerkennung eines Zusatznutzens wird beansprucht b | |
| Kodierung a | Kurzbezeichnung | |
| a: Angabe der im Dossier verwendeten Kodierung.
b: Angabe "ja" oder "nein". | ||
Begründen Sie für alle Anwendungsgebiete, für die die Anerkennung eines Zusatznutzens beansprucht wird, warum sich aus der Zusammenschau der Ergebnisse zu den einzelnen Endpunkten insgesamt ein Zusatznutzen ergibt und worin der Zusatznutzen besteht (maximal 5.000 Zeichen je Anwendungsgebiet). Stellen Sie dabei die Wahrscheinlichkeit für das Vorliegen eines Zusatznutzens unter Berücksichtigung der Ergebnissicherheit dar und kategorisieren Sie das Ausmaß des Zusatznutzens (erheblich, beträchtlich, gering, nicht quantifizierbar). Berücksichtigen Sie bei den Aussagen ggf. nachgewiesene Unterschiede zwischen verschiedenen Patientengruppen. (Referenz: Modul 4 [alle Anwendungsgebiete], Abschnitt 4.4.2)
<< Angaben des pharmazeutischen Unternehmers >>
1.6 Anzahl der Patienten und Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht
In diesem Abschnitt werden die Angaben aus Modul 3, Abschnitt 3.2 (Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen) sowie aus Modul 4, Abschnitt 4.4.3 (Angabe der Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht) zusammengefasst, und zwar für alle Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht.
Charakterisieren Sie zusammenfassend die Patientengruppen, für die die Behandlung mit dem Arzneimittel im Rahmen der im Dossier bewerteten Anwendungsgebiete gemäß Zulassung infrage kommt (Zielpopulation); unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten (maximal 1.500 Zeichen je Anwendungsgebiet). (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.2.1)
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie zusammenfassend, welcher therapeutische Bedarf über die bereits vorhandenen Behandlungsmöglichkeiten hinaus in den Anwendungsgebieten, auf die sich das Dossier bezieht, jeweils besteht (maximal 1.500 Zeichen je Anwendungsgebiet). Beschreiben Sie dabei, ob und wie dieser Bedarf durch das zu bewertende Arzneimittel gedeckt werden soll. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.2.2)
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle 1-9 die Anzahl der Patienten in der gesetzlichen Krankenversicherung (GKV) an, für die eine Behandlung mit dem zu bewertenden Arzneimittel gemäß Zulassung infrage kommt (Zielpopulation), und zwar getrennt für jedes Anwendungsgebiet. Fügen Sie je Anwendungsgebiet eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.2.4)
Tabelle 1-9: Anzahl der GKV-Patienten in der Zielpopulation (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Anzahl der GKV-Patienten in der Zielpopulation | |
| Kodierung a | Kurzbezeichnung | |
| a: Angabe der im Dossier verwendeten Kodierung. | ||
Beschreiben Sie in Tabelle 1-10 für jedes Anwendungsgebiet, bei welchen Patientengruppen ein therapeutisch bedeutsamer Zusatznutzen besteht und welche Ausprägung dieser Zusatznutzen jeweils hat, und geben Sie die zugehörige Anzahl der Patienten in der GKV an. Fügen Sie für jedes Anwendungsgebiet und jede Patientengruppe eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.2.5 und Modul 4 [alle Anwendungsgebiete], Abschnitt 4.4.3)
Tabelle 1-10: Patientengruppen und Anzahl der Patienten, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, einschließlich Ausmaß des Zusatznutzens (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Bezeichnung der Patientengruppe mit therapeutisch bedeutsamem Zusatznutzen | Ausmaß des Zusatznutzens | Anzahl der Patienten in der GKV | |
| Kodierung a | Kurzbezeichnung | |||
| a: Angabe der im Dossier verwendeten Kodierung. | ||||
1.7 Kosten der Therapie für die gesetzliche Krankenversicherung
In diesem Abschnitt werden die Angaben aus Modul 3, Abschnitt 3.3 (Kosten der Therapie für die gesetzliche Krankenversicherung) zusammengefasst, und zwar für alle Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht.
Geben Sie in Tabelle 1-11 an, welche Jahrestherapiekosten der GKV durch die Behandlung mit dem zu bewertenden Arzneimittel innerhalb der Zielpopulation (alle Patienten, für die die Behandlung mit dem neuen Arzneimittel infrage kommt) entstehen. Unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten. Fügen Sie für jedes Anwendungsgebiet eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.3.5)
Tabelle 1-11: Jahrestherapiekosten für das zu bewertende Arzneimittel in der Zielpopulation (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Jahrestherapie- kosten pro Patient in Euro | Jahrestherapiekosten GKV insgesamt in Euro | |
| Kodierung a | Kurzbezeichnung | ||
| a Angabe der im Dossier verwendeten Kodierung. | |||
Geben Sie in Tabelle 1-12 für das zu bewertende Arzneimittel die Summe der Jahrestherapiekosten (GKV insgesamt) über alle Anwendungsgebiete in der Zielpopulation an. Summieren Sie dazu die entsprechenden Angaben in Tabelle 1-11.
Tabelle 1-12: Jahrestherapiekosten für das zu bewertende Arzneimittel in der Zielpopulation (Summe über alle Anwendungsgebiete)
| Jahrestherapiekosten GKV insgesamt in Euro |
Geben Sie in Tabelle 1-13 an, welche Jahrestherapiekosten der GKV durch die Behandlung mit dem zu bewertenden Arzneimittel innerhalb der Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen entstehen. Unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten. Fügen Sie für jedes Anwendungsgebiet und jede Patientengruppe eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.3.5)
Tabelle 1-13: Jahrestherapiekosten für das zu bewertende Arzneimittel - Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Bezeichnung der Patientengruppe | Jahrestherapiekosten pro Patient in Euro | Jahrestherapiekosten GKV insgesamt in Euro | |
| Kodierung a | Kurzbezeichnung | |||
| a: Angabe der im Dossier verwendeten Kodierung. | ||||
Geben Sie in Tabelle 1-14 für das zu bewertende Arzneimittel die Summe der Jahrestherapiekosten (GKV insgesamt) über alle Anwendungsgebiete für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen an. Summieren Sie dazu die entsprechenden Angaben in Tabelle 1-13.
Tabelle 1-14: Jahrestherapiekosten für das zu bewertende Arzneimittel - Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (Summe über alle Anwendungsgebiete)
| Jahrestherapiekosten GKV insgesamt in Euro |
Geben Sie in Tabelle 1-15 an, welche Jahrestherapiekosten der GKV durch die Behandlung mit der zweckmäßigen Vergleichstherapie entstehen. Unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten und den verschiedenen Populationen bzw. Patientengruppen. Fügen Sie für jedes Anwendungsgebiet, jede Therapie und jede Population bzw. Patientengruppe eine neue Zeile ein. (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.3.5)
Tabelle 1-15: Jahrestherapiekosten für die zweckmäßige Vergleichstherapie - alle Populationen/Patientengruppen (Angabe je Anwendungsgebiet)
| Anwendungsgebiet | Bezeichnung der Therapie (zweckmäßige Vergleichstherapie) | Bezeichnung der Population/ Patientengruppe | Jahres- therapie- kosten pro Patient in Euro | Jahrestherapiekosten GKV insgesamt in Euro | |
| Kodierung a | Kurzbezeichnung | ||||
| a: Angabe der im Dossier verwendeten Kodierung. | |||||
1.8 Anforderungen an eine qualitätsgesicherte Anwendung
In diesem Abschnitt werden die Angaben aus Modul 3, Abschnitt 3.4 (Anforderungen an eine qualitätsgesicherte Anwendung) zusammengefasst, und zwar für alle Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht.
Beschreiben Sie zusammenfassend, ob und, wenn ja, welche Anforderungen an eine qualitätsgesicherte Anwendung des zu bewertenden Arzneimittels bestehen. Unterscheiden Sie dabei zwischen den verschiedenen Anwendungsgebieten, auf die sich das Dossier bezieht (maximal 3.000 Zeichen je Anwendungsgebiet). (Referenz: Modul 3 [alle Anwendungsgebiete], Abschnitt 3.4)
<< Angaben des pharmazeutischen Unternehmers >>
| Dossier zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.3 |
<<Wirkstoff>>(<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
Modul 1 Anhang
Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers
Stand: <<TT.MM.JJJJ>>
| Anhang zu Modul 1: Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers - allgemeine Informationen, Ausfüllhinweise | Anhang 1-A: |
Modul 1 Anhang: Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers enthält folgende Checklisten:
Jede in den jeweiligen Checklisten enthaltene Zeile zu Modul 3, 4 sowie ggf. zu Modul 5 (sofern der Hinweis "A-Z Kodierung eintragen" in der zweiten Spalte enthalten ist) ist für jedes kodierte Anwendungsgebiet (A-Z) separat auszufüllen. Hierzu ist die jeweilige Zeile zu kopieren und "A-Z Kodierung eintragen" mit der entsprechenden Kodierung zu überschreiben.
| Checkliste zur formalen Vollständigkeitsprüfung des Dossiers zur Nutzenbewertung, Vorlage der Module 1 bis 4 des Dossiers | Anhang 1-A.1: |
| Thema Modul | Anforderung | Anforderung erfüllt (ankreuzen, falls ja) | Prüfvermerk G-BA |
| Vollständige Vorlage der Module 1 bis 4 des Dossiers | |||
| Modul 1 | Modul 1 liegt vor | [ ] | |
| Modul 2 | Modul 2 liegt vor | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Modul 3 <A-Z Kodierung eintragen> liegt vor | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Modul 4 <A-Z Kodierung eintragen> liegt vor | [ ] | |
| Checkliste zur formalen Vollständigkeitsprüfung des Dossiers zur Nutzenbewertung, Vollständigkeit der Inhalte und Anhänge | Anhang 1-A.2: |
| Thema Modul | Anforderung | Anforderung erfüllt (ankreuzen, falls ja) | Prüfvermerk G-BA |
| Administrative Angaben | |||
| Modul 1 | Abschnitt 1.1 "Administrative Informationen" ist vollständig ausgefüllt | [ ] | |
| Modul 5 (Anlagen) | Die Anlage zu Modul 1 enthält folgendes Dokument:
- Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers | [ ] | |
| Angaben zum Arzneimittel und zugelassene Anwendungsgebiete | |||
| Modul 1 | Abschnitt 1.2 "Allgemeine Angaben zum Arzneimittel" ist vollständig ausgefüllt | [ ] | |
| Modul 1 | Abschnitt 1.3 "Zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels" ist vollständig ausgefüllt | [ ] | |
| Modul 2 | Abschnitt 2.1 "Allgemeine Angaben zum Arzneimittel" ist vollständig ausgefüllt | [ ] | |
| Modul 2 | Abschnitt 2.2 "Zugelassene Anwendungsgebiete" ist vollständig ausgefüllt | [ ] | |
| Modul 2 | Abschnitt 2.3 "Beschreibung der Informationsbeschaffung für Modul 2" ist ausgefüllt; | [ ] | |
| im Abschnitt 2.4 "Referenzliste für Modul 2" ist eine Referenzliste der in Modul 2 zitierten Literatur enthalten | [ ] | ||
| Modul 5 (Anlagen) | Die Anlagen zu Modul 2 enthalten folgende Dokumente: | ||
| - Volltexte für alle, in der Referenzliste für Modul 2 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Modul 2 | [ ] | ||
| Zweckmäßige Vergleichstherapie | |||
| Modul 1 | Abschnitt 1.4 "Zweckmäßige Vergleichstherapie" ist vollständig ausgefüllt | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.1 "Bestimmung der zweckmäßigen Vergleichstherapie": Unterabschnitte 3.1.1 und 3.1.2 sind vollständig ausgefüllt | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.1.3 "Beschreibung der Informationsbeschaffung für Abschnitt 3.1" ist ausgefüllt; | [ ] | |
| im Abschnitt 3.1.4 "Referenzliste für Abschnitt 3.1" ist eine Referenzliste der in Abschnitt 3.1 zitierten Literatur enthalten | [ ] | ||
| Modul 5 (Anlagen) | Die Anlagen zu Modul 3 <A-Z Kodierung eintragen> enthalten folgende Dokumente: | ||
| - Volltexte für alle in der Referenzliste für Abschnitt 3.1 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 3.1 | [ ] | ||
| Medizinischer Nutzen und Zusatznutzen | |||
| Modul 1 | Abschnitt 1.5 "Medizinischer Nutzen, medizinischer Zusatznutzen" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.1 "Zusammenfassung der Inhalte von Modul 4" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.2 "Methodik" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.3.1 "Ergebnisse randomisierter kontrollierter Studien mit dem zu bewertenden Arzneimittel" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.4.1 "Beurteilung der Aussagekraft der Nachweise" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.4.2 "Beschreibung des Zusatznutzens einschließlich dessen Wahrscheinlichkeit und Ausmaß" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.4.3 "Angabe der Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht" ist ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Abschnitt 4.5 "Begründung für die Vorlage weiterer Unterlagen und Surrogatendpunkte" ist vollständig ausgefüllt | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | In Abschnitt 4.6 "Liste der eingeschlossenen Studien" sind Studien gelistet | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Im Abschnitt 4.7 "Referenzliste" ist eine Referenzliste der in Modul 4 zitierten Literatur enthalten | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Angaben zur bibliografischen Literaturrecherche und zur Suche in Studienregistern (Suche nach RCT mit dem zu bewertenden Arzneimittel): | ||
| - Im Anhang 4-A1 sind Angaben zur Suchstrategie für die bibliografische Literaturrecherche enthalten | |||
| - Im Anhang 4-B1 sind Angaben zur Suchstrategie für die Suche in Studienregistern enthalten | |||
| - Anhang 4-C1 enthält eine Liste der im Volltext gesichteten und ausgeschlossenen Dokumente mit Ausschlussgrund (bibliografische Literaturrecherche) | |||
| - Anhang 4-D1 enthält eine Liste der ausgeschlossenen Studien mit Ausschlussgrund (Suche in Studienregistern) | |||
| Modul 4
<A-Z Kodierung eintragen> | Falls eine Bewertung mittels indirekter Vergleiche auf Basis von RCT (Abschnitt 4.3.2.1 von Modul 4) durchgeführt wurde, Angaben zur bibliografischen Literaturrecherche und zur Suche in Studienregistern: | ||
| - Im Anhang 4-A2 sind Angaben zur Suchstrategie für die bibliografische Literaturrecherche enthalten | |||
| - Im Anhang 4-B2 sind Angaben zur Suchstrategie für die Suche in Studienregistern enthalten | |||
| - Anhang 4-C2 enthält eine Liste der im Volltext gesichteten und ausgeschlossenen Dokumente mit Ausschlussgrund (bibliografische Literaturrecherche) | |||
| - Anhang 4-D2 enthält eine Liste der ausgeschlossenen Studien mit | |||
| Modul 4
<A-Z Kodierung eintragen> | Falls eine Bewertung auf Basis nicht randomisierter vergleichender Studien (Abschnitt 4.3.2.2 von Modul 4) durchgeführt wurde, Angaben zur bibliografischen Literaturrecherche und zur Suche in Studienregistern: | ||
| - Im Anhang 4-A3 sind Angaben zur Suchstrategie für die bibliografische Literaturrecherche enthalten | |||
| - Im Anhang 4-B3 sind Angaben zur Suchstrategie für die Suche in Studienregistern enthalten | |||
| - Anhang 4-C3 enthält eine Liste der im Volltext gesichteten und ausgeschlossenen Dokumente mit Ausschlussgrund (bibliografische Literaturrecherche) | |||
| - Anhang 4-D3 enthält eine Liste der ausgeschlossenen Studien mit Ausschlussgrund (Suche in Studienregistern) | |||
| Modul 4
<A-Z Kodierung eintragen> | Falls eine Bewertung auf Basis weiterer Untersuchungen (Abschnitt 4.3.2.3 von Modul 4) durchgeführt wurde, Angaben zur bibliografischen Literaturrecherche und zur Suche in Studienregistern: | ||
| - Im Anhang 4-A4 sind Angaben zur Suchstrategie für die bibliografische Literaturrecherche enthalten | |||
| - Im Anhang 4-B4 sind Angaben zur Suchstrategie für die Suche in Studienregistern enthalten | |||
| - Anhang 4-C4 enthält eine Liste der im Volltext gesichteten und ausgeschlossenen Dokumente mit Ausschlussgrund (bibliografische Literaturrecherche) | |||
| - Anhang 4-D4 enthält eine Liste der ausgeschlossenen Studien mit Ausschlussgrund (Suche in Studienregistern) | |||
| Modul 4
<A-Z Kodierung eintragen> | Falls im Abschnitt 4.2.3.2 "Bibliografische Literaturrecherche" eine durchgeführte Recherche beschrieben wurde: | ||
| - Im Anhang 4-A sind Angaben zur Suchstrategie der bibliografischen Literaturrecherche enthalten | [ ] | ||
| - Anhang 4-C enthält eine Liste der im Volltext gesichteten und ausgeschlossenen Studien mit Ausschlussgrund | [ ] | ||
| Modul
<A-Z Kodierung eintragen> | Anhang 4-E enthält eine Tabelle zur Methodik jeder eingeschlossenen, randomisierten kontrollierten Studie | [ ] | |
| Modul 4
<A-Z Kodierung eintragen> | Anhang 4-F enthält einen Bewertungsbogen zur Bewertung von Verzerrungsaspekten für jede eingeschlossene, randomisierte kontrollierte und nicht randomisierte vergleichende Studie | [ ] | |
| Modul 5 (Anlagen) | Anlagen zu Modul 4 <A-Z Kodierung eintragen> enthalten jeweils folgende Dokumente: | ||
| Unterverzeichnis Dokumente der Zulassungsbehörden: | |||
| - veröffentlichte und unveröffentlichte Dokumente des abschließenden Bewertungsberichts aus dem für Deutschland gültigen Zulassungsverfahren (näheres siehe Abschnitt 3.1.2 des Dokuments zur Erstellung und Einreichung eines Dossiers) | [ ] | ||
| Unterverzeichnis Informationsbeschaffung: | |||
| - Im Unterverzeichnis Anhang-4-A sind folgende Dokumente abzulegen:
o Für jedes Thema je durchsuchter Datenbank eine RIS-Datei aller durch die bibliografische Literaturrecherche identifizierten Literaturstellen (alle Treffer) | |||
| - Im Unterverzeichnis Anhang-4-B sind folgende Dokumente abzulegen:
o Für jedes Thema je durchsuchtem Studienregister eine RIS-Datei aller durch die Registerrecherche identifizierten Studien (alle Treffer) | [ ] | ||
| - Im Unterverzeichnis Anhang-4-C sind folgende Dokumente anzulegen:
o Volltexte für alle in den Referenzlisten in Anhang 4-C genannten Literaturzitate | [ ] | ||
| - Im Unterverzeichnis Anhang-4-D sind folgende Dokumente anzulegen:
o Volltexte für alle in den Referenzlisten in Anhang 4-D genannten Studien | [ ] | ||
| Unterverzeichnis Programmcode: | |||
| - Falls im Abschnitt 4.3.2.1 "Indirekte Vergleiche auf Basis randomisierter kontrollierter Studien" indirekte Vergleiche dargestellt wurden: | |||
| - Programmcodes, die für die Durchführung indirekter Vergleiche verwendet wurden | [ ] | ||
| - PDF-Dokument zur Dokumentation der durchgeführten indirekten Vergleiche | [ ] | ||
| Unterverzeichnis Studienberichte: | |||
| - Ergebnisberichte einschließlich der Studienprotokolle aller abgeschlossenen und abgebrochenen Studien des pharmazeutischen Unternehmers | [ ] | ||
| Unterverzeichnis Volltexte: | |||
| - Volltexte für alle in der Referenzliste (Abschnitt 4.7) ür Modul 4 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Modul 4 (Abschnitt 4.7) | [ ] | ||
| - RIS-Datei der in Abschnitt 4.6 von Modul 4 zitierten Dokumente | [ ] | ||
| Unterverzeichnis Weitere Zulassungsunterlagen: | [ ] | ||
| - CTD-Modul 2, Abschnitt 2.5 (Clinical Overview) | [ ] | ||
| - CTD-Modul 2, Abschnitt 2.7.3 (Clinical Summary Efficacy); falls Tabelle 2.7.3.1 nicht Bestandteil des Abschnitts 2.7.3 des CTD ist, ist diese Tabelle separat abzulegen | [ ] | ||
| - CTD-Modul 2, Abschnitt 2.7.4 (Clinical Summary Safety) | |||
| Anzahl der Patienten und Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht | |||
| Modul 1 | Abschnitt 1.6 "Anzahl der Patienten und Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht" ist vollständig ausgefüllt | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.2 "Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen": Unterabschnitte 3.2.1 bis 3.2.5 sind beantwortet | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.2.6 "Beschreibung der Informationsbeschaffung für Abschnitt 3.2" ist ausgefüllt; | [ ] | |
| im Abschnitt 3.2.7 "Referenzliste für Abschnitt 3.2" ist eine Referenzliste der in Abschnitt 3.2 zitierten Literatur enthalten | [ ] | ||
| Modul 5 (Anlagen) | Die Anlagen zu Modul 3 <A-Z Kodierung eintragen> enthalten folgende Dokumente: | ||
| - Volltexte für alle in der Referenzliste für Abschnitt 3.3 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 3.3 | [ ] | ||
| Anforderungen an eine qualitätsgesicherte Anwendung | |||
| Modul 1 | Abschnitt 1.8 "Anforderungen an eine qualitätsgesicherte Anwendung" ist vollständig ausgefüllt | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.4 "Anforderungen an eine qualitätsgesicherte Anwendung": Unterabschnitte 3.4.1 bis 3.4.5 sind vollständig ausgefüllt | [ ] | |
| Modul 3
<A-Z Kodierung eintragen> | Abschnitt 3.4.6 "Beschreibung der Informationsbeschaffung für Abschnitt 3.4" ist ausgefüllt; | [ ] | |
| im Abschnitt 3.4.7 "Referenzliste für Abschnitt 3.4" ist eine Referenzliste der in Abschnitt 3.4 zitierten Literatur enthalten | [ ] | ||
| Modul 5 (Anlagen) | Die Anlagen zu Modul 3 <A-Z Kodierung eintragen> enthalten folgende Dokumente: | ||
| - Volltexte für alle in der Referenzliste für Abschnitt 3.4 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 3.4 | [ ] | ||
| Kennzeichnung von Betriebs- und Geschäftsgeheimnissen | |||
| Modul 5 (Anlagen) | Modul 5 enthält ein PDF-Dokument, in dem die Dokumente aus Modul 5, die aus Sicht des pharmazeutischen Unternehmers Betriebs- und Geschäftsgeheimnisse enthalten, benannt sind | ||
| Dossier zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.4 |
.<<Wirkstoff>> (<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
Modul 2
Allgemeine Angaben zum Arzneimittel, zugelassene Anwendungsgebiete
Stand: <<TT.MM.JJJJ>>
Tabellenverzeichnis
Tabelle 2-1: Allgemeine Angaben zum zu bewertenden Arzneimittel
Tabelle 2-2: Pharmazentralnummem und Zulassungsnummern für das zu bewertende Arzneimittel
Tabelle 2-3: Zugelassene Anwendungsgebiete, auf die sich das Dossier bezieht
Tabelle 2-4: Weitere in Deutschland zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels
Abbildungsverzeichnis
Es konnten keine Einträge für ein Abbildungsverzeichnis gefunden werden.
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| ATC-Code | Anatomisch-Therapeutisch-Chemischer Code |
| PZN | Pharmazentralnummer |
2. Modul 2 - allgemeine Informationen
Modul 2 enthält folgende Informationen:
Alle in den Abschnitten 2.1 und 2.2 getroffenen Aussagen sind zu begründen. Die Quellen (z.B. Publikationen), die für die Aussagen herangezogen werden, sind in Abschnitt 2.4 (Referenzliste) eindeutig zu benennen. Das Vorgehen zur Identifikation der Quellen ist im Abschnitt 2.3 (Beschreibung der Informationsbeschaffung) darzustellen.
Im Dokument verwendete Abkürzungen sind in das Abkürzungsverzeichnis aufzunehmen. Sofern Sie für Ihre Ausführungen Tabellen oder Abbildungen verwenden, sind diese im Tabellen- bzw. Abbildungsverzeichnis aufzuführen.
2.1 Allgemeine Angaben zum Arzneimittel
2.1.1 Administrative Angaben zum Arzneimittel
Geben Sie in Tabelle 2-1 den Namen des Wirkstoffs, den Handelsnamen und den ATC-Code für das zu bewertende Arzneimittel an.
Tabelle 2-1: Allgemeine Angaben zum zu bewertenden Arzneimittel
| Wirkstoff: | |
| Handelsname: | |
| ATC-Code: |
Geben Sie in der nachfolgenden Tabelle 2-2 an, welche Pharmazentralnummern (PZN) und welche Zulassungsnummern dem zu bewertenden Arzneimittel zuzuordnen sind, und benennen Sie dabei die zugehörige Wirkstärke und Packungsgröße. Fügen Sie für jede Pharmazentralnummer eine neue Zeile ein.
Tabelle 2-2: Pharmazentralnummern und Zulassungsnummern für das zu bewertende Arzneimittel
| Pharmazentralnummer (PZN) | Zulassungsnummer | Wirkstärke | Packungsgröße |
2.1.2 Angaben zum Wirkmechanismus des Arzneimittels
Beschreiben Sie den Wirkmechanismus des zu bewertenden Arzneimittels. Begründen Sie Ihre Angaben unter Nennung der verwendeten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob und inwieweit sich der Wirkmechanismus des zu bewertenden Arzneimittels vom Wirkmechanismus anderer bereits in Deutschland zugelassener Arzneimittel unterscheidet. Differenzieren Sie dabei zwischen verschiedenen Anwendungsgebieten, für die das zu bewertende Arzneimittel zugelassen ist. Begründen Sie Ihre Angaben unter Nennung der verwendeten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
2.2 Zugelassene Anwendungsgebiete
2.2.1 Anwendungsgebiete, auf die sich das Dossier bezieht
Benennen Sie in der nachfolgenden Tabelle 2-3 die Anwendungsgebiete, auf die sich das vorliegende Dossier bezieht. Geben Sie hierzu den Wortlaut der Fachinformation an. Sofern im Abschnitt "Anwendungsgebiete" der Fachinformation Verweise enthalten sind, führen Sie auch den Wortlaut an, auf den verwiesen wird. Fügen Sie für jedes Anwendungsgebiet eine neue Zeile ein, und vergeben Sie eine Kodierung (fortlaufende Bezeichnung von "A" bis "Z") [Anmerkung: Diese Kodierung ist für die übrigen Module des Dossiers entsprechend zu verwenden].
Tabelle 2-3: Zugelassene Anwendungsgebiete, auf die sich das Dossier bezieht
| Anwendungsgebiet (Wortlaut der Fachinformation inkl. Wortlaut bei Verweisen) | orphan (ja/nein) | Datum der Zulassungserteilung | Kodierung im Dossier a |
| a: Fortlaufende Angabe "A" bis "Z" | |||
Benennen Sie die den Angaben in Tabelle 2-3 zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
2.2.2 Weitere in Deutschland zugelassene Anwendungsgebiete
Falls es sich um ein Dossier zu einem neuen Anwendungsgebiet eines bereits zugelassenen Arzneimittels handelt, benennen Sie in der nachfolgenden Tabelle 2-4 die weiteren in Deutschland zugelassenen Anwendungsgebiete des zu bewertenden Arzneimittels. Geben Sie hierzu den Wortlaut der Fachinformation an; sofern im Abschnitt "Anwendungsgebiete" der Fachinformation Verweise enthalten sind, führen Sie auch den Wortlaut an, auf den verwiesen wird. Fügen Sie dabei für jedes Anwendungsgebiet eine neue Zeile ein. Falls es kein weiteres zugelassenes Anwendungsgebiet gibt oder es sich nicht um ein Dossier zu einem neuen Anwendungsgebiet eines bereits zugelassenen Arzneimittels handelt, fügen Sie in der ersten Zeile unter "Anwendungsgebiet" "kein weiteres Anwendungsgebiet" ein.
Tabelle 2-4: Weitere in Deutschland zugelassene Anwendungsgebiete des zu bewertenden Arzneimittels
| Anwendungsgebiet (Wortlaut der Fachinformation inkl. Wortlaut bei Verweisen) | Datum der Zulassungserteilung |
Benennen Sie die den Angaben in Tabelle 2-4 zugrunde gelegten Quellen. Falls es kein weiteres zugelassenes Anwendungsgebiet gibt oder es sich nicht um ein Dossier zu einem neuen Anwendungsgebiet eines bereits zugelassenen Arzneimittels handelt, geben Sie "nicht zutreffend" an.
<< Angaben des pharmazeutischen Unternehmers >>
2.3 Beschreibung der Informationsbeschaffung für Modul 2
Erläutern Sie an dieser Stelle das Vorgehen zur Identifikation der im Abschnitt 2.1 und im Abschnitt 2.2 genannten Quellen (Informationsbeschaffung). Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
<< Angaben des pharmazeutischen Unternehmers>>
Benennen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie in den vorhergehenden Abschnitten angegeben haben. Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard).
<< Angaben des pharmazeutischen Unternehmers >>
| Dossier zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.5 |
<<Wirkstoff>> (<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
<<Anwendungsgebiet>>
Zweckmäßige Vergleichstherapie, Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen, Kosten der Therapie für die GKV, Anforderungen an eine qualitätsgesicherte Anwendung
Stand: <<TT.MM.JJJJ>>
Tabellenverzeichnis
Tabelle 3-1: Anzahl der GKV-Patienten in der Zielpopulation
Tabelle 3-2: Anzahl der Patienten, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, mit Angabe des Ausmaßes des Zusatznutzens (zu bewertendes Arzneimittel)
Tabelle 3-3: Angaben zum Behandlungsmodus (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
Tabelle 3-4: Behandlungstage pro Patient pro Jahr (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
Tabelle 3-5: Jahresdurchschnittsverbrauch pro Patient (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
Tabelle 3-6: Kosten des zu bewertenden Arzneimittels und der zweckmäßigen Vergleichstherapie
Tabelle 3-7: Zusätzlich notwendige GKV-Leistungen bei Anwendung der Arzneimittel gemäß Fach- und Gebrauchsinformation (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
Tabelle 3-8: Zusätzlich notwendige GKV-Leistungen - Kosten pro Einheit
Tabelle 3-9: Zusätzlich notwendige GKV-Leistungen - Zusatzkosten für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie pro Jahr (pro Patient und für die jeweilige Population/Patientengruppe insgesamt)
Tabelle 3-10: Jahrestherapiekosten für die GKV für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie (pro Patient und insgesamt)
Tabelle 3-11: Alle ärztlichen Leistungen, die gemäß aktuell gültiger Fachinformation des zu bewertenden Arzneimittels zu seiner Anwendung angeführt sind
Abbildungsverzeichnis
Es konnten keine Einträge für ein Abbildungsverzeichnis gefunden werden.
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| DDD | Defined Daily Dose |
| EPAR | European Public Assessment Report |
| EU | Europäische Union |
| G-BA | Gemeinsamer Bundesausschuss |
| GKV | Gesetzliche Krankenversicherung |
| IU | International Unit |
| SGB | Sozialgesetzbuch |
3 Modul 3 - allgemeine Informationen
Modul 3 enthält folgende Angaben:
Alle in diesen Abschnitten getroffenen Aussagen und Kalkulationsschritte sind zu begründen. In die Kalkulation eingehende Annahmen sind darzustellen. Die Berechnungen müssen auf Basis der Angaben nachvollziehbar sein und sollen auch Angaben zur Unsicherheit enthalten.
Die Abschnitte enthalten jeweils einen separaten Abschnitt zur Beschreibung der Informationsbeschaffung sowie eine separate Referenzliste.
Für jedes zu bewertende Anwendungsgebiet ist eine separate Version des vorliegenden Dokuments zu erstellen. Die Kodierung der Anwendungsgebiete ist in Modul 2 hinterlegt. Sie ist je Anwendungsgebiet einheitlich für die übrigen Module des Dossiers zu verwenden.
Im Dokument verwendete Abkürzungen sind in das Abkürzungsverzeichnis aufzunehmen. Sofern Sie für Ihre Ausführungen Abbildungen oder Tabellen verwenden, sind diese im Abbildungs- bzw. Tabellenverzeichnis aufzuführen.
3.1 Bestimmung der zweckmäßigen Vergleichstherapie
Zweckmäßige Vergleichstherapie ist diejenige Therapie, deren Nutzen mit dem Nutzen des zu bewertenden Arzneimittels verglichen wird. Näheres hierzu findet sich in der Verfahrensordnung des Gemeinsamen Bundesausschusses.
Die zweckmäßige Vergleichstherapie ist regelhaft zu bestimmen nach Maßstäben, die sich aus den internationalen Standards der evidenzbasierten Medizin ergeben. Bei mehreren Alternativen ist die wirtschaftlichere Therapie zu wählen, vorzugsweise eine Therapie, für die ein Festbetrag gilt. Die zweckmäßige Vergleichstherapie muss eine nach dem allgemein anerkannten Stand der medizinischen Erkenntnisse zweckmäßige Therapie im Anwendungsgebiet sein, vorzugsweise eine Therapie, für die Endpunktstudien vorliegen und die sich in der praktischen Anwendung bewährt hat, soweit nicht Richtlinien oder das Wirtschaftlichkeitsgebot dagegen sprechen.
Bei der Bestimmung der Vergleichstherapie sind insbesondere folgende Kriterien zu berücksichtigen:
Für Arzneimittel einer Wirkstoffklasse ist unter Berücksichtigung der oben genannten Kriterien die gleiche zweckmäßige Vergleichstherapie heranzuziehen, um eine einheitliche Bewertung zu gewährleisten. Die zweckmäßige Vergleichstherapie muss auch geeignet sein für Bewertungen von Arzneimitteln auf Veranlassung des Gemeinsamen Bundesausschusses nach § 35a Absatz 6 SGB V, die vor dem 1. Januar 2011 in den Verkehr gebracht worden sind.
Zur zweckmäßigen Vergleichstherapie kann ein Beratungsgespräch mit dem Gemeinsamen Bundesausschuss stattfinden. Näheres dazu findet sich in der Verfahrensordnung des Gemeinsamen Bundesausschusses.
3.1.1 Benennung der zweckmäßigen Vergleichstherapie
Benennen Sie die zweckmäßige Vergleichstherapie für das Anwendungsgebiet, auf das sich das vorliegende Dokument bezieht.
<< Angaben des pharmazeutischen Unternehmers >>
3.1.2 Begründung für die Wahl der zweckmäßigen Vergleichstherapie
Geben Sie an, ob ein Beratungsgespräch mit dem Gemeinsamen Bundesausschuss zum Thema "zweckmäßige Vergleichstherapie" stattgefunden hat. Falls ja, geben Sie das Datum des Beratungsgesprächs und die vom Gemeinsamen Bundesausschuss übermittelte Vorgangsnummer an, und beschreiben Sie das Ergebnis dieser Beratung hinsichtlich der Festlegung der zweckmäßigen Vergleichstherapie. Sofern ein Beratungsprotokoll erstellt wurde, benennen Sie dieses als Quelle (auch in Abschnitt 3.1.4).
<< Angaben des pharmazeutischen Unternehmers >>
Falls ein Beratungsgespräch mit dem Gemeinsamen Bundesausschuss zum Thema "zweckmäßige Vergleichstherapie" nicht stattgefunden hat oder in diesem Gespräch keine Festlegung der zweckmäßigen Vergleichstherapie erfolgte oder Sie trotz Festlegung der zweckmäßigen Vergleichstherapie in dem Beratungsgespräch eine andere zweckmäßige Vergleichstherapie für die vorliegende Bewertung ausgewählt haben, begründen Sie die Wahl der Ihrer Ansicht nach zweckmäßigen Vergleichstherapie. Benennen Sie die vorhandenen Therapieoptionen im Anwendungsgebiet, auf das sich das vorliegende Dossier bezieht. Äußern Sie sich bei der Auswahl der zweckmäßigen Vergleichstherapie aus diesen Therapieoptionen explizit zu den oben genannten Kriterien 1 bis 5. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.1.3 Beschreibung der Informationsbeschaffung für Abschnitt 3.1
Erläutern Sie das Vorgehen zur Identifikation der in Abschnitt 3.1.2 genannten Quellen (Informationsbeschaffung). Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
<< Angaben des pharmazeutischen Unternehmers >>
3.1.4 Referenzliste für Abschnitt 3.1
Listen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie in den Abschnitten 3.1.2 und 3.1.3 angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
<< Angaben des pharmazeutischen Unternehmers >>
3.2 Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen
3.2.1 Beschreibung der Erkrankung und Charakterisierung der Zielpopulation
Geben Sie einen kurzen Überblick über die Erkrankung (Ursachen, natürlicher Verlauf), zu deren Behandlung das zu bewertende Arzneimittel eingesetzt werden soll und auf die sich das vorliegende Dokument bezieht. Insbesondere sollen die wissenschaftlich anerkannten Klassifikationsschemata und Einteilungen nach Stadien herangezogen werden. Berücksichtigen Sie dabei, sofern relevant, geschlechts- und altersspezifische Besonderheiten. Charakterisieren Sie die Patientengruppen, für die die Behandlung mit dem Arzneimittel gemäß Zulassung infrage kommt (im Weiteren "Zielpopulation" genannt). Die Darstellung der Erkrankung in diesem Abschnitt soll sich auf die Zielpopulation konzentrieren. Begründen Sie Ihre Aussagen durch Angabe von Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.2.2 Therapeutischer Bedarf innerhalb der Erkrankung
Beschreiben Sie zusammenfassend, welcher therapeutische Bedarf über alle bereits vorhandenen medikamentösen und nicht medikamentösen Behandlungsmöglichkeiten hinaus innerhalb der Erkrankung besteht. Beschreiben Sie dabei im Überblick, ob und wie dieser Bedarf durch das zu bewertende Arzneimittel gedeckt werden soll. An dieser Stelle ist keine datengestützte Darstellung des Nutzens oder des Zusatznutzens des Arzneimittels vorgesehen, sondern eine allgemeine Beschreibung des therapeutischen Ansatzes. Begründen Sie Ihre Aussagen durch Angabe von Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.2.3 Prävalenz und Inzidenz der Erkrankung in Deutschland
Geben Sie eine Schätzung für die Prävalenz und Inzidenz der Erkrankung bzw. der Stadien der Erkrankung in Deutschland an, für die das Arzneimittel laut Fach- und Gebrauchsinformation zugelassen ist. Geben Sie dabei jeweils einen üblichen Populationsbezug und zeitlichen Bezug (z.B. Inzidenz pro Jahr, Perioden- oder Punktprävalenz jeweils mit Bezugsjahr) an. Bei Vorliegen alters- oder geschlechtsspezifischer Unterschiede oder von Unterschieden in anderen Gruppen sollen die Angaben auch für Altersgruppen, Geschlecht bzw. andere Gruppen getrennt gemacht werden. Weiterhin sind Angaben zur Unsicherheit der Schätzung erforderlich. Verwenden Sie hierzu eine tabellarische Darstellung. Begründen Sie Ihre Aussagen durch Angabe von Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie nachfolgend an, ob und, wenn ja, welche wesentlichen Änderungen hinsichtlich Prävalenz und Inzidenz der Erkrankung in Deutschland innerhalb der nächsten 5 Jahre zu erwarten sind. Verwenden Sie hierzu eine tabellarische Darstellung. Benennen Si die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.2.4 Anzahl der Patienten in der Zielpopulation
Geben Sie in der nachfolgenden Tabelle 3-1 die Anzahl der Patienten in der GKV an, für die eine Behandlung mit dem zu bewertenden Arzneimittel in dem Anwendungsgebiet, auf das sich das vorliegende Dokument bezieht, gemäß Zulassung infrage kommt (Zielpopulation). Die Angaben sollen sich auf einen Jahreszeitraum beziehen. Berücksichtigen Sie auch, dass das zu bewertende Arzneimittel ggf. an bisher nicht therapierten Personen zur Anwendung kommen kann; eine lediglich auf die bisherige Behandlung begrenzte Beschreibung der Zielpopulation kann zu einer Unterschätzung der Zielpopulation führen.
Generell soll für die Bestimmung des Anteils der Versicherten in der GKV folgende Quelle verwendet werden: Gesetzliche Krankenversicherung - Kennzahlen und Faustformeln - (http://www.bmg.bund.de/fileadmin/dateien/Downloads/Statistiken/GKV/Kennzahlen_Daten/ Kennzahlen_und_Faustformeln_GKV_2001-2012_120903.pdf). Gibt es Hinweise, dass sich dies in einem Krankheitsbild anders verhält, kann unter Angabe der Gründe und entsprechender Nachweise davon abgewichen werden.
Tabelle 3-1: Anzahl der GKV-Patienten in der Zielpopulation
| Bezeichnung der Therapie (zu bewertendes Arzneimittel) | Anzahl der Patienten in der Zielpopulation (inklusive Angabe der Unsicherheit) | Anzahl der GKV-Patienten in der Zielpopulation (inklusive Angabe der Unsicherheit) |
Begründen Sie die Angaben in Tabelle 3-1 unter Nennung der verwendeten Quellen. Ziehen Sie dabei auch die Angaben zu Prävalenz und Inzidenz (wie oben angegeben) heran. Alle Annahmen und Kalkulationsschritte sind darzustellen und zu begründen. Die Berechnungen müssen auf Basis dieser Angaben nachvollzogen werden können. Machen Sie auch Angaben zur Unsicherheit, z.B. Angabe einer Spanne.
<< Angaben des pharmazeutischen Unternehmers >>
3.2.5 Angabe der Anzahl der Patienten mit therapeutisch bedeutsamem Zusatznutzen
Geben Sie in der nachfolgenden Tabelle 3-2 die Anzahl der Patienten an, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, und zwar innerhalb des Anwendungsgebiets, auf das sich das vorliegende Dokument bezieht. Die hier dargestellten Patientengruppen sollen sich unmittelbar aus der Nutzenbewertung in Modul 4 ergeben. Ziehen Sie hierzu die Angaben aus Modul 4, Abschnitt 4.4.3 heran und differenzieren Sie ggf. zwischen Patientengruppen mit unterschiedlichem Ausmaß des Zusatznutzens. Fügen Sie für jede Patientengruppe eine neue Zeile ein.
Tabelle 3-2: Anzahl der Patienten, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, mit Angabe des Ausmaßes des Zusatznutzens (zu bewertendes Arzneimittel)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel) | Bezeichnung der Patientengruppe mit therapeutisch bedeutsamem Zusatznutzen | Ausmaß des Zusatznutzens | Anzahl der Patienten in der GKV |
Begründen Sie die Angaben in Tabelle 3-2 unter Nennung der verwendeten Quellen. Ziehen Sie dabei auch die Angaben zu Prävalenz und Inzidenz (wie im Abschnitt 3.2.3 angegeben) heran.
<< Angaben des pharmazeutischen Unternehmers >>
3.2.6 Beschreibung der Informationsbeschaffung für Abschnitt 3.2
Erläutern Sie das Vorgehen zur Identifikation der in den Abschnitten 3.2.1 bis 3.2.5 genannten Quellen (Informationsbeschaffung). Im Allgemeinen sollen deutsche Quellen bzw. Quellen, die über die epidemiologische Situation in Deutschland Aussagen erlauben, herangezogen werden. Weiterhin sind bevorzugt offizielle Quellen zu nutzen. Aktualität und Repräsentativität sind bei der Auswahl zu berücksichtigen und ggf. zu diskutieren. Sofern erforderlich können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen nennen.
Wenn eine Recherche in offiziellen Quellen oder in bibliografischen Datenbanken durchgeführt wurde, sollen Angaben zu den Suchbegriffen, den Datenbanken/ Suchoberflächen, dem Datum der Recherche nach den üblichen Vorgaben gemacht werden. Die Ergebnisse der Recherche sollen dargestellt werden, damit nachvollziehbar ist, welche Daten bzw. Publikationen berücksichtigt bzw. aus- und eingeschlossen wurden. Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
Wenn eine (hier optionale) systematische bibliografische Literaturrecherche durchgeführt wurde, soll eine vollständige Dokumentation erfolgen. Die entsprechenden Anforderungen an die Informationsbeschaffung sollen nachfolgend analog den Vorgaben in Modul 4 (siehe Abschnitte 4.2.3.2 Bibliografische Literaturrecherche, 4.3.1.1.2 Studien aus der bibliografischen Literaturrecherche, Anhang 4-A, 4-C) umgesetzt werden.
<< Angaben des pharmazeutischen Unternehmers>>
3.2.7 Referenzliste für Abschnitt 3.2
Listen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie in den Abschnitten 3.2.1 bis 3.2.6 angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
<< Angaben des pharmazeutischen Unternehmers >>
3.3 Kosten der Therapie für die gesetzliche Krankenversicherung
Im Abschnitt 3.3 wird an mehreren Stellen gefordert, Spannen anzugeben, wenn dies an den entsprechenden Stellen zutrifft. Mit diesen Spannen ist in den nachfolgenden Tabellen konsequent weiterzurechnen, sodass daraus in Tabelle 3-10 Angaben für Jahrestherapiekosten pro Patient und für die GKV insgesamt mit einer Unter- und Obergrenze resultieren.
Therapieabbrüche sind in den Tabellen 3-1 bis 3-10 nicht zu veranschlagen; sie sind im Abschnitt 3.3.6 darzustellen.
3.3.1 Angaben zur Behandlungsdauer
Geben Sie in der nachfolgenden Tabelle 3-3 an, nach welchem Behandlungsmodus (z.B. kontinuierlich, in Zyklen, je Episode, bei Bedarf) das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie eingesetzt werden. Machen Sie diese Angaben getrennt für die Zielpopulation sowie für die Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (siehe Abschnitt 3.2). Geben Sie die Anzahl der Behandlungen pro Patient pro Jahr, die Behandlungsdauer je Behandlung in Tagen sowie die daraus resultierenden Behandlungstage pro Jahr an. Falls eine Therapie länger als ein Jahr dauert, jedoch zeitlich begrenzt ist, soll zusätzlich die Gesamttherapiedauer angegeben werden. Fügen Sie für jede Therapie, Behandlungssituation und jede Population bzw. Patientengruppe eine neue Zeile ein.
Zur Ermittlung der Kosten der Therapie müssen Angaben zur Behandlungsdauer auf Grundlage der Fachinformation gemacht werden. Zunächst ist auf Grundlage der Fachinformation zu prüfen, ob es unterschiedliche Behandlungssituationen oder Behandlungsdauern gibt. Mit einer Behandlungssituation ist gemeint, dass für Patienten aufgrund unterschiedlicher Eigenschaften unterschiedliche Behandlungsdauern veranschlagt werden, z.B. 12 Wochen vs. 24 Wochen. Mit Behandlungsdauer ist hier gemeint, dass unabhängig von diesen in der Fachinformation vorgegebenen Patienteneigenschaften eine Spanne der Behandlungsdauer gewählt werden kann, z.B. 12 bis 15 Wochen. Die Angaben sind für jede Behandlungssituation einzeln zu machen. Ist für eine Behandlungssituation keine eindeutige Behandlungsdauer angegeben, sondern eine Zeitspanne, dann ist die jeweilige Unter- und Obergrenze anzugeben und bei den weiteren Berechnungen zu verwenden. Wenn aus der Fachinformation keine maximale Behandlungsdauer hervorgeht, ist die Behandlung grundsätzlich für ein Jahr anzusetzen, ansonsten die zulässige Anzahl an Gaben, z.B. maximal mögliche Anzahl der Zyklen pro Jahr.
Tabelle 3-3: Angaben zum Behandlungsmodus (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Behandlungsmodus | Anzahl Behandlungen pro Patient pro Jahr (ggf. Spanne) | Behandlungsdauer je Behandlung in Tagen (ggf. Spanne) |
| Wenn eine Behandlung nicht dauerhaft, aber länger als ein Jahr, z.B. bei einer Infektionskrankheit, durchgeführt werden muss, ist dies anzumerken. In den folgenden Tabellen müssen die Kosten dann sowohl für ein Jahr als auch für die gesamte Behandlungsdauer pro Patient und die entsprechende Patientengruppe angegeben werden. | ||||
Begründen Sie die Angaben in Tabelle 3-3 unter Nennung der verwendeten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle 3-4 die Behandlungstage pro Patient pro Jahr für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie an. Machen Sie diese Angaben getrennt für die Zielpopulation und die Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen. Die Behandlungstage pro Patient pro Jahr ergeben sich aus der Anzahl der Behandlungen pro Patient pro Jahr und der Behandlungsdauer je Behandlung (siehe Tabelle 3-3). Fügen Sie für jede Therapie, Behandlungssituation und jede Population bzw. Patientengruppe eine neue Zeile ein.
Tabelle 3-4: Behandlungstage pro Patient pro Jahr (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Behandlungsmodus | Behandlungstage pro Patient pro Jahr (ggf. Spanne) |
| Wenn eine Behandlung nicht dauerhaft, aber länger als ein Jahr, z.B. bei einer Infektionskrankheit, durchgeführt werden muss, ist dies anzumerken. In den folgenden Tabellen müssen die Kosten dann sowohl für ein Jahr als auch für die gesamte Behandlungsdauer pro Patient und die entsprechende Patientengruppe angegeben werden. | |||
3.3.2 Angaben zum Verbrauch für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie
Geben Sie in der nachfolgenden Tabelle 3-5 den Jahresdurchschnittsverbrauch pro Patient für das zu bewertende Arzneimittel sowie für die zweckmäßige Vergleichstherapie in DDD (Defined Daily Dose) an, d. h. Anzahl DDDs pro Jahr. Zusätzlich ist die festgelegte bzw. den Berechnungen zugrunde liegende Maßeinheit der jeweiligen DDD (z.B. 10 mg) anzugeben. Falls die zweckmäßige Vergleichstherapie eine nichtmedikamentöse Behandlung ist, geben Sie ein anderes im jeweiligen Anwendungsgebiet international gebräuchliches Maß für den Jahresdurchschnittsverbrauch der zweckmäßigen Vergleichstherapie an. Fügen Sie für jede Therapie eine neue Zeile ein.
Tabelle 3-5: Jahresdurchschnittsverbrauch pro Patient (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Behandlungs- tage pro Patient pro Jahr (ggf. Spanne) | Verbrauch pro Gabe (ggf. Spanne) | Jahresdurchschnittsverbrauch pro Patient (DDD; im Falle einer nichtmedikamentösen Behandlung Angabe eines anderen im jeweiligen Anwendungsgebiet international gebräuchlichen Maßes) |
Begründen Sie die Angaben in Tabelle 3-5 unter Nennung der verwendeten Quellen. Nehmen Sie ggf. Bezug auf andere Verbrauchsmaße, die im Anwendungsgebiet gebräuchlich sind (z.B. IU [International Unit], Dosierung je Quadratmeter Körperoberfläche, Dosierung je Kilogramm Körpergewicht).
<< Angaben des pharmazeutischen Unternehmers >>
3.3.3 Angaben zu Kosten des zu bewertenden Arzneimittels und der zweckmäßigen Vergleichstherapie
Geben Sie in Tabelle 3-6 an, wie hoch die Apothekenabgabepreise für das zu bewertende Arzneimittel sowie für die zweckmäßige Vergleichstherapie sind. Generell soll(en) die für die Behandlungsdauer zweckmäßigste(n) und wirtschaftlichste(n) verordnungsfähige(n) Packungsgröße(n) gewählt werden. Sofern Festbeträge vorhanden sind, müssen diese angegeben werden. Sofern keine Festbeträge bestehen, soll das günstigste Arzneimittel gewählt werden. Importarzneimittel sollen nicht berücksichtigt werden. Geben Sie zusätzlich die den Krankenkassen tatsächlich entstehenden Kosten an. Dazu ist der Apothekenabgabepreis nach Abzug der gesetzlich vorgeschriebenen Rabatte (siehe § 130 und § 130a SGB V mit Ausnahme der in § 130a Absatz 8 SGB V genannten Rabatte) anzugeben. Im Falle einer nichtmedikamentösen zweckmäßigen Vergleichstherapie sind entsprechende Angaben zu deren Vergütung aus GKV-Perspektive zu machen. Fügen Sie für jede Therapie eine neue Zeile ein.
Tabelle 3-6: Kosten des zu bewertenden Arzneimittels und der zweckmäßigen Vergleichstherapie
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Kosten pro Packung (Apothekenabgabepreis in Euro nach Wirkstärke, Darreichungsform und Packungsgröße, für nichtmedikamentöse Behandlungen Angaben zu deren Vergütung aus GKV-Perspektive) | Kosten nach Abzug gesetzlich vorgeschriebener Rabatte in Euro |
Begründen Sie die Angaben in Tabelle 3-6 unter Nennung der verwendeten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.3.4 Angaben zu Kosten für zusätzlich notwendige GKV-Leistungen
Bestehen bei Anwendung des zu bewertenden Arzneimittels und der zweckmäßigen Vergleichstherapie entsprechend der Fach- oder Gebrauchsinformation regelhaft Unterschiede bei der notwendigen Inanspruchnahme ärztlicher Behandlung oder bei der Verordnung sonstiger Leistungen zwischen dem zu bewertenden Arzneimittel und der zweckmäßigen Vergleichstherapie, sind diese bei den den Krankenkassen tatsächlich entstehenden Kosten zu berücksichtigen. Im nachfolgenden Abschnitt werden die Kosten dieser zusätzlich notwendigen GKV-Leistungen dargestellt.
Geben Sie in der nachfolgenden Tabelle 3-7 an, welche zusätzlich notwendigen GKV-Leistungen (notwendige Inanspruchnahme ärztlicher Behandlung oder Verordnung sonstiger Leistungen zulasten der GKV) bei Anwendung der Arzneimittel entsprechend der Fach- oder Gebrauchsinformation entstehen. Geben Sie dabei auch an, wie häufig die Verordnung zusätzlich notwendiger GKV-Leistungen pro Patient erforderlich ist, und zwar sowohl bezogen auf eine Episode, einen Zyklus etc. als auch bezogen auf ein Jahr. Machen Sie diese Angaben sowohl für das zu bewertende Arzneimittel als auch für die zweckmäßige Vergleichstherapie sowie getrennt für die Zielpopulation und die Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen (siehe Abschnitt 3.2). Fügen Sie für jede Therapie, jede Population bzw. Patientengruppe und jede zusätzlich notwendige GKV-Leistung eine neue Zeile ein.
Tabelle 3-7: Zusätzlich notwendige GKV-Leistungen bei Anwendung der Arzneimittel gemäß Fach- und Gebrauchsinformation (zu bewertendes Arzneimittel und zweckmäßige Vergleichstherapie)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Bezeichnung der zusätzlichen GKV-Leistung | Anzahl der zusätzlich notwendigen GKV-Leistungen je Episode, Zyklus etc. | Anzahl der zusätzlich notwendigen GKV-Leistungen pro Patient pro Jahr |
Begründen Sie die Angaben in Tabelle 3-7 unter Nennung der verwendeten Quellen. Ziehen Sie dabei auch die Angaben zur Behandlungsdauer (wie im Abschnitt 3.3.1 angegeben) heran.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle 3-8 an, wie hoch die Kosten der in Tabelle 3-7 benannten zusätzlich notwendigen GKV-Leistungen pro Einheit jeweils sind. Geben Sie, so zutreffend, EBM-Ziffern oder OPS Codes an. Fügen Sie für jede zusätzlich notwendige GKV-Leistung eine neue Zeile ein.
Tabelle 3-8: Zusätzlich notwendige GKV-Leistungen - Kosten pro Einheit
| Bezeichnung der zusätzlich notwendigen GKV-Leistung | Kosten pro Leistung in Euro |
Begründen Sie die Angaben in Tabelle 3-8 unter Nennung der verwendeten Quellen.
<< Angaben des pharmazeutischen Unternehmers>>
Geben Sie in Tabelle 3-9 an, wie hoch die zusätzlichen Kosten bei Anwendung der Arzneimittel gemäß Fach- oder Gebrauchsinformation pro Jahr sind, und zwar pro Patient sowie für die jeweilige Population/ Patientengruppe insgesamt. Führen Sie hierzu die Angaben aus Tabelle 3-7 (Anzahlzusätzlich notwendiger GKV-Leistungen), Tabelle 3-8 (Kosten für zusätzlich notwendige GKV-Leistungen je Einheit), Tabelle 3-1 (Anzahl der Patienten in der Zielpopulation) und Tabelle 3-2 (Anzahl Patienten mit therapeutisch bedeutsamem Zusatznutzen) zusammen. Fügen Sie für jede Therapie und Population bzw. Patientengruppe sowie jede zusätzlich notwendige GKV-Leistung eine neue Zeile ein.
Tabelle 3-9: Zusätzlich notwendige GKV-Leistungen - Zusatzkosten für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie pro Jahr (pro Patient und für die jeweilige Population/Patientengruppe insgesamt)
| Bezeichnung der Therapie (zu bewerten- des Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Bezeichnung der zusätzlich notwendigen GKV-Leistung | Zusatzkosten pro Patient pro Jahr in Euro | Zusatzkosten für die Population bzw. Patientengruppe insgesamt in Euro |
3.3.5 Angaben zu Jahrestherapiekosten
Geben Sie in Tabelle 3-10 die Jahrestherapiekosten für die GKV durch Zusammenführung der in den Abschnitten 3.3.1 bis 3.3.4 entwickelten Daten an, und zwar getrennt für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie sowie getrennt für die Zielpopulation und die Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen. Weisen Sie die Jahrestherapiekosten sowohl bezogen auf einen einzelnen Patienten als auch für die GKV insgesamt (d. h. für die gesamte jeweilige Population bzw. Patientengruppen nach Abschnitt 3.2.3, Tabelle 3-1, sowie Abschnitt 3.2.5, Tabelle 3-2) aus. Fügen Sie für jede Therapie, Behandlungssituation und jede Population bzw. Patientengruppe eine neue Zeile ein. Unsicherheit sowie variierende Behandlungsdauern sollen in Form von Spannen ausgewiesen werden.
Tabelle 3-10: Jahrestherapiekosten für die GKV für das zu bewertende Arzneimittel und die zweckmäßige Vergleichstherapie (pro Patient und insgesamt)
| Bezeichnung der Therapie (zu bewertendes Arzneimittel, zweckmäßige Vergleichstherapie) | Bezeichnung der Population bzw. Patientengruppe | Jahrestherapie- kosten pro Patient in Euro | Jahrestherapiekosten GKV insgesamt in Euro a |
| a: Als Jahrestherapiekosten GKV insgesamt sollen die Kosten ausgewiesen werden, die der GKV entstehen, wenn die in Abschnitt 3.2.3, Tabelle 3-1, sowie Abschnitt 3.2.5, Tabelle 3-2 dargestellte Zielpopulation bzw. Patientengruppen vollständig mit dem zu bewertenden Arzneimittel behandelt werden. | |||
3.3.6 Angaben zu Versorgungsanteilen
Beschreiben Sie unter Bezugnahme auf die in Abschnitt 3.2.3 dargestellten Daten zur aktuellen Prävalenz und Inzidenz, welche Versorgungsanteile für das zu bewertende Arzneimittel innerhalb des Anwendungsgebiets, auf das sich das vorliegende Dokument bezieht, zu erwarten sind. Nehmen Sie bei Ihrer Begründung auch Bezug auf die derzeit gegebene Versorgungssituation mit der zweckmäßigen Vergleichstherapie. Beschreiben Sie insbesondere auch, welche Patientengruppen wegen Kontraindikationen nicht mit dem zu bewertenden Arzneimittel behandelt werden sollten. Weiterhin ist zu erläutern, welche Raten an Therapieabbrüchen in den Patientengruppen zu erwarten sind. Im Weiteren sollten bei dieser Abschätzung auch der Versorgungskontext und Patientenpräferenzen berücksichtigt werden. Differenzieren Sie nach ambulantem und stationärem Versorgungsbereich. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie auf Basis der von Ihnen erwarteten Versorgungsanteile, ob und, wenn ja, welche Änderungen sich für die in Abschnitt 3.3.5 beschriebenen Jahrestherapiekosten ergeben. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
3.3.7 Beschreibung der Informationsbeschaffung für Abschnitt 3.3
Erläutern Sie das Vorgehen zur Identifikation der in den Abschnitten 3.2.1 bis 3.2.5 genannten Quellen (Informationsbeschaffung). Im Allgemeinen sollen deutsche Quellen bzw. Quellen, die über die epidemiologische Situation in Deutschland Aussagen erlauben, herangezogen werden. Weiterhin sind bevorzugt offizielle Quellen zu nutzen. Aktualität und Repräsentativität sind bei der Auswahl zu berücksichtigen und ggf. zu diskutieren. Sofern erforderlich können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen nennen.
Wenn eine Recherche in offiziellen Quellen oder in bibliografischen Datenbanken durchgeführt wurde, sollen Angaben zu den Suchbegriffen, den Datenbanken/ Suchoberflächen, dem Datum der Recherche nach den üblichen Vorgaben gemacht werden. Die Ergebnisse der Recherche sollen dargestellt werden, damit nachvollziehbar ist, welche Daten bzw. Publikationen berücksichtigt bzw. aus- und eingeschlossen wurden. Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
Wenn eine (hier optionale) systematische bibliografische Literaturrecherche durchgeführt wurde, soll eine vollständige Dokumentation erfolgen. Die entsprechenden Anforderungen an die Informationsbeschaffung sollen nachfolgend analog den Vorgaben in Modul 4 (siehe Abschnitte 4.2.3.2 Bibliografische Literaturrecherche, 4.3.1.1.2 Studien aus der bibliografischen Literaturrecherche, Anhang 4-A, 4-C) umgesetzt werden.
<< Angaben des pharmazeutischen Unternehmers >>
3.3.8 Referenzliste für Abschnitt 3.3
Listen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie in den Abschnitten 3.3.1 bis 3.3.7 angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
<< Angaben des pharmazeutischen Unternehmers >>
3.4 Anforderungen an eine qualitätsgesicherte Anwendung
3.4.1 Anforderungen aus der Fach- und Gebrauchsinformation
Benennen Sie Anforderungen, die sich aus der Fach- und Gebrauchsinformation des zu bewertenden Arzneimittels für eine qualitätsgesicherte Anwendung ergeben. Beschreiben Sie insbesondere Anforderungen an die Diagnostik, die Qualifikation der Ärzte und Ärztinnen und des weiteren medizinischen Personals, die Infrastruktur und die Behandlungsdauer. Geben Sie auch an, ob kurz- oder langfristige Überwachungsmaßnahmen durchgeführt werden müssen, ob die behandelnden Personen oder Einrichtungen für die Durchführung spezieller Notfallmaßnahmen ausgerüstet sein müssen und ob Interaktionen mit anderen Arzneimitteln oder Lebensmitteln zu beachten sind. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen abweichende Anforderungen als die zuvor genannten bestehen und, wenn ja, welche dies sind.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.2 Bedingungen oder Einschränkungen für den sicheren und wirksamen Einsatz des Arzneimittels
Benennen Sie Anforderungen, die sich aus Annex IIb (Bedingungen der Genehmigung für das Inverkehrbringen) des European Assessment Reports (EPAR) des zu bewertenden Arzneimittels für eine qualitätsgesicherte Anwendung ergeben. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen abweichende Anforderungen als die zuvor genannten bestehen und, wenn ja, welche dies sind.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.3 Informationen zum Risk-Management-Plan
Sofern im zentralen Zulassungsverfahren für das zu bewertende Arzneimittel ein Annex IV (Bedingungen oder Einschränkungen für den sicheren und wirksamen Einsatz des Arzneimittels, die von den Mitgliedsstaaten umzusetzen sind) des EPAR erstellt wurde, benennen Sie die dort genannten Anforderungen. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen abweichende Anforderungen als die zuvor genannten bestehen und, wenn ja, welche dies sind.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.4 Weitere Anforderungen an eine qualitätsgesicherte Anwendung
Benennen Sie die vorgeschlagenen Maßnahmen zur Risikominimierung ("proposed risk minimization activities"), die in der Zusammenfassung des EU-Risk-Management-Plans beschrieben und im European Public Assessment Report (EPAR) veröffentlicht sind. Machen Sie auch Angaben zur Umsetzung dieser Maßnahmen. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen abweichende Anforderungen als die zuvor genannten bestehen und, wenn ja, welche dies sind.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.5 Beschreibung der Informationsbeschaffung für Abschnitt 3.4
Benennen Sie weitere Anforderungen, die sich aus Ihrer Sicht hinsichtlich einer qualitätsgesicherten Anwendung des zu bewertenden Arzneimittels ergeben, insbesondere bezüglich der Dauer eines Therapieversuchs, des Absetzens der Therapie und ggf. notwendiger Verlaufskontrollen. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie, ob für Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen abweichende Anforderungen als die zuvor genannten bestehen und, wenn ja, welche dies sind.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.6 Referenzliste für Abschnitt 3.4
Erläutern Sie das Vorgehen zur Identifikation der in den Abschnitten 3.4.1 bis 3.4.5 genannten Quellen (Informationsbeschaffung). Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
<< Angaben des pharmazeutischen Unternehmers >>
3.4.7 Referenzliste für Abschnitt 3.4
Listen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie in den Abschnitten 3.4.1 bis 3.4.6 angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
<< Angaben des pharmazeutischen Unternehmers >>
3.5 Angaben zur Prüfung der Erforderlichkeit einer Anpassung des einheitlichen Bewertungsmaßstabs für ärztliche Leistungen (EBM) gemäß § 87 Absatz 5b Satz 5 SGB V 19a
Die Angaben in diesem Abschnitt betreffen die Regelung in § 87 Absatz 5b Satz 5 SGB V, nach der der EBM zeitgleich mit dem Beschluss nach § 35a Absatz 3 Satz 1 SGB V anzupassen ist, sofern die Fachinformation des Arzneimittels zu seiner Anwendung eine zwingend erforderliche Leistung vorsieht, die eine Anpassung des EBM erforderlich macht.
Geben Sie in der nachfolgenden Tabelle 3-11 zunächst alle ärztlichen Leistungen an, die laut aktuell gültiger Fachinformation des zu bewertenden Arzneimittels zu seiner Anwendung angeführt sind. Berücksichtigen Sie auch solche ärztlichen Leistungen, die gegebenenfalls nur bestimmte Patientenpopulationen betreffen oder nur unter bestimmten Voraussetzungen durchzuführen sind. Geben Sie für jede identifizierte ärztliche Leistung durch das entsprechende Zitat aus der Fachinformation den Empfehlungsgrad zur Durchführung der jeweiligen Leistung an. Sofern dieselbe Leistung mehrmals angeführt ist, geben Sie das Zitat mit dem jeweils stärksten Empfehlungsgrad an, auch wenn dies gegebenenfalls nur bestimmte Patientenpopulationen betrifft. Geben Sie in Tabelle 3-11 zudem für jede ärztliche Leistung an, ob diese aus Ihrer Sicht für die Anwendung des Arzneimittels als zwingend erforderliche und somit verpflichtende Leistung einzustufen ist.
Tabelle 3-11: Alle ärztlichen Leistungen, die gemäß aktuell gültiger Fachinformation des zu bewertenden Arzneimittels zu seiner Anwendung angeführt sind
| Nr. | Bezeichnung der ärztlichen Leistung | Zitat(e) aus der Fachinformation mit dem jeweils stärksten Empfehlungsgrad (kann/sollte/soll/muss/ist etc.) und Angabe der genauen Textstelle (Seite, Abschnitt) | Einstufung aus Sicht des pharmazeutischen Unternehmers, ob es sich um eine zwingend erforderliche Leistung handelt (ja/nein) |
Geben Sie den Stand der Information der Fachinformation an.
<< Angaben des pharmazeutischen Unternehmers >>
Benennen Sie nachfolgend solche zwingend erforderlichen ärztlichen Leistungen aus Tabelle 3-11, die Ihrer Einschätzung nach bisher nicht oder nicht vollständig im aktuell gültigen EBM abgebildet sind. Begründen Sie jeweils Ihre Einschätzung. Falls es Gebührenordnungspositionen gibt, mittels derer die ärztliche Leistung bei anderen Indikationen und/oder anderer methodischer Durchführung erbracht werden kann, so geben Sie diese bitte an. Behalten Sie bei Ihren Angaben die Nummer und Bezeichnung der ärztlichen Leistung aus Tabelle 3-11 bei.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie die verwendete EBM-Version (Jahr/Quartal) an.
<< Angaben des pharmazeutischen Unternehmers >>
Legen Sie nachfolgend für jede der zwingend erforderlichen ärztlichen Leistungen, die Ihrer Einschätzung nach bisher nicht (vollständig) im aktuell gültigen EBM abgebildet sind, detaillierte Informationen zu Art und Umfang der Leistung dar. Benennen Sie Indikationen für die Durchführung der ärztlichen Leistung sowie die Häufigkeit der Durchführung für die Zeitpunkte vor, während und nach Therapie. Falls die ärztliche Leistung nicht für alle Patienten gleichermaßen erbracht werden muss, benennen und definieren sie abgrenzbare Patientenpopulationen.
Stellen Sie detailliert Arbeits- und Prozessschritte bei der Durchführung der ärztlichen Leistung sowie die gegebenenfalls notwendigen apparativen Anforderungen dar. Falls es verschiedene Verfahren gibt, so geben Sie bitte alle an. Die Angaben sind durch Quellen (z.B. Publikationen, Methodenvorschriften, Gebrauchsanweisungen) zu belegen, so dass die detaillierten Arbeits- und Prozessschritte zweifelsfrei verständlich werden.
<< Angaben des pharmazeutischen Unternehmers >>
3.5.1 Referenzliste für Abschnitt 3.5
Listen Sie nachfolgend alle Quellen (z.B. Publikationen, Methodenvorschriften, Gebrauchsanweisungen), die Sie im Abschnitt 3.5 angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Sämtliche Quellen sind im Volltext beizufügen.
<< Angaben des pharmazeutischen Unternehmers >>
| Dossier zur Nutzenbewertung gemäß § 35a SGB V | Anlage II.6 |
<<Wirkstoff>> (<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
Modul 4 <<Kodierung A-Z>>
<<Anwendungsgebiet>>
Medizinischer Nutzen und
medizinischer Zusatznutzen,
Patientengruppen mit therapeutisch
bedeutsamem Zusatznutzen
Stand: <TT.MM.JJJJ>
Tabellenverzeichnis
Tabelle 4-1: Liste der Studien des pharmazeutischen Unternehmers - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-2: Studien des pharmazeutischen Unternehmers, die nicht für die Nutzenbewertung herangezogen wurden - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-3: Relevante Studien aus der Suche in Studienregistern - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-4: Studienpool - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-5: Charakterisierung der eingeschlossenen Studien - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-6: Charakterisierung der Interventionen - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-7: Charakterisierung der Studienpopulationen - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-8: Verzerrungspotenzial auf Studienebene - RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-9: Matrix der Endpunkte in den eingeschlossenen RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-10: Operationalisierung von <Endpunkt xxx>
Tabelle 4-11: Bewertung des Verzerrungspotenzials für <Endpunkt xxx> in RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-12: Ergebnisse für <Endpunkt xxx> aus RCT mit dem zu bewertenden Arzneimittel
Tabelle 4-13: Matrix der Endpunkte in den eingeschlossenen RCT für indirekte Vergleiche
Tabelle 4-14: Zusammenfassung der verfügbaren Vergleiche in den Studien, die für den indirekten Vergleich herangezogen wurden
Tabelle 4-15: Operationalisierung von <Endpunkt xxx>
Tabelle 4-16: Bewertung des Verzerrungspotenzials für <Endpunkt xxx> in RCT für indirekte Vergleiche
Tabelle 4-17: Ergebnisse für <Endpunkt xxx> aus RCT für indirekte Vergleiche
Tabelle 4-18: Verzerrungsaspekte auf Studienebene - nicht randomisierte vergleichende Interventionsstudien
Tabelle 4-19: Operationalisierung von <Endpunkt xxx>
Tabelle 4-20: Verzerrungsaspekte für <Endpunkt xxx> - nicht randomisierte vergleichende Studien
Tabelle 4-21: Operationalisierung von <Endpunkt xxx> - weitere Untersuchungen
Tabelle 4-22: Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, einschließlich Ausmaß des Zusatznutzens
Tabelle 4-23 (Anhang): Studiendesign und -methodik für Studie <Studienbezeichnung>
Tabelle 4-24 (Anhang): Bewertungsbogen zur Beschreibung von Verzerrungsaspekten für Studie <Studienbezeichnung>
Abbildungsverzeichnis
Abbildung 1: Flussdiagramm der bibliografischen Literaturrecherche - Suche nach randomisierten kontrollierten Studien mit dem zu bewertenden Arzneimittel
Abbildung 2: Meta-Analyse für <Endpunkt xxx> aus RCT; <zu bewertendes Arzneimittel> versus <Vergleichstherapie>
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| CONSORT | Consolidated Standards of Reporting Trials |
| DIMDI | Deutsches Institut für Medizinische Dokumentation |
| EG | Europäische Gemeinschaft |
| ITT | Intention to treat |
| MTC | Mixed Treatment Comparison |
| RCT | Randomized Controlled Trial |
| SGB | Sozialgesetzbuch |
| STE | Surrogate Threshold Effects |
| STROBE | Strengthening the Reporting of Observational Studies in Epidemiology |
| TREND | Transparent Reporting of Evaluation with Non-Randomized Design |
| WHO | World Health Organization |
4 Modul 4 - allgemeine Informationen
Modul 4 enthält folgende Angaben:
Für jedes zu bewertende Anwendungsgebiet ist eine separate Version des vorliegenden Dokuments zu erstellen. Die Kodierung der Anwendungsgebiete ist in Modul 2 hinterlegt. Sie ist je Anwendungsgebiet einheitlich für die Module 3, 4 und 5 zu verwenden.
Im Dokument verwendete Abkürzungen sind in das Abkürzungsverzeichnis aufzunehmen. Sofern Sie für Ihre Ausführungen Tabellen und Abbildungen verwenden, sind diese im Tabellen- bzw. Abbildungsverzeichnis aufzuführen.
4.1 Zusammenfassung der Inhalte von Modul 4
Stellen Sie eine strukturierte Zusammenfassung der Inhalte von Modul 4 zur Verfügung.
Fragestellung
<< Angaben des pharmazeutischen Unternehmers>>
Datenquellen
<< Angaben des pharmazeutischen Unternehmers >>
Ein-/Ausschlusskriterien für Studien
<< Angaben des pharmazeutischen Unternehmers >>
Methoden zur Bewertung der Aussagekraft der Nachweise und zur Synthese von Ergebnissen
<< Angaben des pharmazeutischen Unternehmers >>
Ergebnisse zum medizinischen Nutzen und medizinischen Zusatznutzen
<< Angaben des pharmazeutischen Unternehmers >>
Schlussfolgerungen zum Zusatznutzen und zum therapeutisch bedeutsamen Zusatznutzen
<< Angaben des pharmazeutischen Unternehmers >>
4.2 Methodik
Abschnitt 4.2 soll die Methodik der im Dossier präsentierten Bewertung des medizinischen Nutzens und des medizinischen Zusatznutzens beschreiben. Der Abschnitt enthält Hilfestellungen für die Darstellung der Methodik sowie einige Vorgaben, die aus den internationalen Standards der evidenzbasierten Medizin abgeleitet sind. Eine Abweichung von diesen methodischen Vorgaben ist möglich, bedarf aber einer Begründung.
4.2.1 Fragestellung
Nach den internationalen Standards der evidenzbasierten Medizin soll eine Bewertung unter einer definierten Fragestellung vorgenommen werden, die mindestens folgende Komponenten enthält:
Unter Endpunkte sind dabei alle für die frühe Nutzenbewertung relevanten Endpunkte anzugeben (d. h. nicht nur solche, die ggf. in den relevanten Studien untersucht wurden).
Geben Sie die Fragestellung der vorliegenden Aufarbeitung von Unterlagen zur Untersuchung des medizinischen Nutzens und des medizinischen Zusatznutzens des zu bewertenden Arzneimittels an. Begründen Sie Abweichungen von den oben beschriebenen Vorgaben.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.2 Kriterien für den Einschluss von Studien in die Nutzenbewertung
Die Untersuchung der in Abschnitt 4.2.1 benannten Fragestellung soll auf Basis von klinischen Studien vorgenommen werden. Für die systematische Auswahl von Studien für diese Untersuchung sollen Ein- und Ausschlusskriterien für die Studien definiert werden. Dabei ist zu beachten, dass eine Studie nicht allein deshalb ausgeschlossen werden soll, weil keine in einer Fachzeitschrift veröffentlichte Vollpublikation vorliegt. Eine Bewertung der Studie kann beispielsweise auch auf Basis eines ausführlichen Ergebnisberichts aus einem Studienregister erfolgen, während ein Kongressabstract allein in der Regel nicht für eine Studienbewertung ausreicht.
Benennen Sie die Ein- und Ausschlusskriterien für Studien zum medizinischen Nutzen und Zusatznutzen. Machen Sie dabei mindestens Aussagen zur Patientenpopulation, zur Intervention, zur Vergleichstherapie, zu den Endpunkten, zum Studientyp und zur Studiendauer und begründen Sie diese. Stellen Sie die Ein- und Ausschlusskriterien zusammenfassend in einer tabellarischen Übersicht dar.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.3 Informationsbeschaffung
In den nachfolgenden Abschnitten ist zu beschreiben, nach welcher Methodik Studien identifiziert wurden, die für die Bewertung des medizinischen Nutzens und des medizinischen Zusatznutzens in dem in diesem Dokument bewerteten Anwendungsgebiet herangezogen werden. Dies bezieht sich sowohl auf publizierte als auch auf unpublizierte Studien. Die Methodik muss dazu geeignet sein, die relevanten Studien (gemäß den in Abschnitt 4.2.2 genannten Kriterien) systematisch zu identifizieren (systematische Literaturrecherche).
4.2.3.1 Studien des pharmazeutischen Unternehmers
Für die Identifikation der Studien des pharmazeutischen Unternehmers ist keine gesonderte Beschreibung der Methodik der Informationsbeschaffung erforderlich. Die vollständige Auflistung aller Studien, die an die Zulassungsbehörde übermittelt wurden (Zulassungsstudien), sowie aller Studien, für die der pharmazeutische Unternehmer Sponsor ist oder war oder auf andere Weise finanziell beteiligt ist oder war, erfolgt in den Abschnitten 4.3.1 und 4.3.2, jeweils im Unterabschnitt "Studien des pharmazeutischen Unternehmers". Die Darstellung soll auf Studien mit Patienten in dem Anwendungsgebiet, für das das vorliegende Dokument erstellt wird, beschränkt werden.
4.2.3.2 Bibliografische Literaturrecherche
Die Durchführung einer bibliografischen Literaturrecherche ist erforderlich, um sicherzustellen, dass ein vollständiger Studienpool in die Bewertung einfließt.
Eine bibliografische Literaturrecherche muss für RCT mit dem zu bewertenden Arzneimittel (Abschnitt 4.3.1) immer durchgeführt werden. Für indirekte Vergleiche auf Basis von RCT (Abschnitt 4.3.2.1), nicht randomisierte vergleichende Studien (Abschnitt 4.3.2.2) sowie weitere Untersuchungen (Abschnitt 4.3.2.3) muss eine bibliografische Literaturrecherche immer dann durchgeführt werden, wenn auf Basis solcher Studien der medizinische Zusatznutzen bewertet wird.
Das Datum der Recherche soll nicht mehr als 3 Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
Die bibliografische Literaturrecherche soll mindestens in den Datenbanken MEDLINE und EMBASE sowie in der Cochrane-Datenbank "Cochrane Central Register of Controlled Trials (Clinical Trials)" durchgeführt werden. Optional kann zusätzlich eine Suche in weiteren themenspezifischen Datenbanken (z.B. CINAHL, PsycINFO etc.) durchgeführt werden.
Die Suche soll in jeder Datenbank einzeln und mit einer für die jeweilige Datenbank adaptierten Suchstrategie durchgeführt werden. Die Suchstrategien sollen jeweils in Blöcken, insbesondere getrennt nach Indikation, Intervention und ggf. Studientypen, aufgebaut werden. Wird eine Einschränkung der Strategien auf bestimmte Studientypen vorgenommen (z.B. randomisierte kontrollierte Studien), sollen aktuelle validierte Filter hierfür verwendet werden. Alle Suchstrategien sind in Anhang 4-A zu dokumentieren.
Beschreiben Sie nachfolgend für alle durchgeführten Recherchen, in welchen Datenbanken eine bibliografische Literaturrecherche durchgeführt wurde. Begründen Sie Abweichungen von den oben beschriebenen Vorgaben. Geben Sie auch an, wenn bei der Recherche generelle Einschränkungen vorgenommen wurden (z.B. Sprach- oder Jahreseinschränkungen), und begründen Sie diese.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.3.3 Suche in Studienregistern
Eine Suche in öffentlich zugänglichen Studienregistern ist grundsätzlich durchzuführen, um sicherzustellen, dass laufende Studien sowie abgeschlossene Studien auch von Dritten vollständig identifiziert werden und in Studienregistern vorliegende Informationen zu Studienmethodik und -ergebnissen in die Bewertung einfließen.
Eine Suche in Studienregistern muss für RCT mit dem zu bewertenden Arzneimittel (Abschnitt 4.3.1) immer durchgeführt werden. Für indirekte Vergleiche auf Basis von RCT (Abschnitt 4.3.2.1), nicht randomisierte vergleichende Studien (Abschnitt 4.3.2.2) sowie weitere Untersuchungen (Abschnitt 4.3.2.3) muss eine Suche in Studienregistern immer dann durchgeführt werden, wenn auf Basis solcher Studien der medizinische Zusatznutzen bewertet wird.
Das Datum der Recherche soll nicht mehr als 3 Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
Die Suche soll mindestens in den Studienregistern clinicaltrials.gov (www.clinicaltrials.gov), EU Clinical Trials Register (EU-CTR, www.clinicaltrialsregister.eu), Klinische Prüfungen PharmNet.Bund (http://www.pharmnet-bund.de/dynamic/de/klinische-pruefungen/index.htm) sowie über das International Clinical Trials Registry Platform Search Portal (ICTRP Search Portal, Suchportal der WHO: http://apps.who.int/trialsearch/) durchgeführt werden. Optional kann zusätzlich eine Suche in weiteren themenspezifischen Studienregistern (z.B. krankheitsspezifische Studienregister oder Studienregister einzelner pharmazeutischer Unternehmen) durchgeführt werden. Die Suche in Studienregistern anderer pharmazeutischer Unternehmer ist insbesondere bei indirekten Vergleichen sinnvoll, wenn Studien zu anderen Arzneimitteln identifiziert werden müssen.
Die Suche soll in jedem Studienregister einzeln und mit einer für das jeweilige Studienregister adaptierten Suchstrategie durchgeführt werden. Die Suche soll abgeschlossene, abgebrochene und laufende Studien erfassen. Alle Suchstrategien sind in Anhang 4-B zu dokumentieren.
Beschreiben Sie nachfolgend für alle durchgeführten Recherchen, in welchen Studienregistern die Suche durchgeführt wurde. Begründen Sie dabei Abweichungen von den oben beschriebenen Vorgaben. Geben Sie auch an, wenn bei der Recherche generelle Einschränkungen vorgenommen wurden (z.B. Jahreseinschränkungen), und begründen Sie diese.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.3.4 Selektion relevanter Studien
Beschreiben Sie das Vorgehen bei der Selektion relevanter Studien aus dem Ergebnis der in den Abschnitten 4.2.3.2 und 4.2.3.3 beschriebenen Rechercheschritte. Begründen Sie das Vorgehen, falls die Selektion nicht von zwei Personen unabhängig voneinander durchgeführt wurde.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.4 Bewertung der Aussagekraft der Nachweise
Zur Bewertung der Aussagekraft der im Dossier vorgelegten Nachweise sollen Verzerrungsaspekte der Ergebnisse für jede eingeschlossene Studie beschrieben werden, und zwar separat für jeden patientenrelevanten Endpunkt. Dazu sollen insbesondere folgende endpunktübergreifende (A) und endpunktspezifische (B) Aspekte systematisch extrahiert werden (zur weiteren Erläuterung der einzelnen Aspekte siehe Bewertungsbogen in Anhang 4-F):
A: Verzerrungsaspekte der Ergebnisse auf Studienebene
B: Verzerrungsaspekte der Ergebnisse auf Endpunktebene
Für randomisierte Studien soll darüber hinaus das Verzerrungspotenzial bewertet und als "niedrig" oder "hoch" eingestuft werden. Ein niedriges Verzerrungspotenzial liegt dann vor, wenn mit großer Wahrscheinlichkeit ausgeschlossen werden kann, dass die Ergebnisse relevant verzerrt sind. Unter einer relevanten Verzerrung ist zu verstehen, dass sich die Ergebnisse bei Behebung der verzerrenden Aspekte in ihrer Grundaussage verändern würden.
Eine zusammenfassende Bewertung der Verzerrungsaspekte soll nicht für nicht randomisierte Studien erfolgen.
Für die Bewertung eines Endpunkts soll für randomisierte Studien zunächst das Verzerrungspotenzial endpunktübergreifend anhand der unter A aufgeführten Aspekte als "niedrig" oder "hoch" eingestuft werden. Falls diese Einstufung als "hoch" erfolgt, soll das Verzerrungspotenzial für den Endpunkt in der Regel auch als "hoch" bewertet werden, Abweichungen hiervon sind zu begründen. Ansonsten sollen die unter B genannten endpunktspezifischen Aspekte Berücksichtigung finden.
Eine Einstufung des Verzerrungspotenzials des Ergebnisses für einen Endpunkt als "hoch" soll nicht zum Ausschluss der Daten führen. Die Klassifizierung soll vielmehr der Diskussion heterogener Studienergebnisse und der Einschätzung der Aussagekraft der Nachweise dienen. Für nicht randomisierte Studien können für solche Diskussionen einzelne Verzerrungsaspekte herangezogen werden.
Beschreiben Sie die für die Bewertung der Verzerrungsaspekte und des Verzerrungspotenzials eingesetzte Methodik. Begründen Sie, wenn Sie von der oben beschriebenen Methodik abweichen.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.5 Informationssynthese und -analyse
4.2.5.1 Beschreibung des Designs und der Methodik der eingeschlossenen Studien
Das Design und die Methodik der eingeschlossenen Studien soll in den Abschnitten 4.3.1 und 4.3.2, jeweils in den Unterabschnitten "Charakteristika der in die Bewertung eingeschlossenen Studien" und den dazugehörigen Anhängen, dargestellt werden. Die Darstellung der Studien soll für randomisierte kontrollierte Studien mindestens die Anforderungen des CONSORT-Statements erfüllen (Items 2b bis 14, Informationen aus dem CONSORT-Flow-Chart) 5. Die Darstellung nicht randomisierter Interventionsstudien und epidemiologischer Beobachtungsstudien soll mindestens den Anforderungen des TREND 6 bzw. STROBE-Statements 7 folgen. Design und Methodik weiterer Untersuchungen sollen gemäß den verfügbaren Standards dargestellt werden.
Beschreiben Sie, nach welchen Standards und mit welchen Informationen (Items) Sie das Design und die Methodik der eingeschlossenen Studien in Modul 4 dargestellt haben. Begründen Sie Abweichungen von den oben beschriebenen Vorgaben.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.5.2 Gegenüberstellung der Ergebnisse der Einzelstudien
Die Ergebnisse der einzelnen Studien sollen in den Abschnitten 4.3.1 und 4.3.2 in den entsprechenden Unterabschnitten zunächst für jede eingeschlossene Studie separat dargestellt werden. Die Darstellung soll die Charakteristika der Studienpopulationen sowie die Ergebnisse zu allen in den eingeschlossenen Studien berichteten patientenrelevanten Endpunkten (Verbesserung des Gesundheitszustands, Verkürzung der Krankheitsdauer, Verlängerung des Überlebens, Verringerung von Nebenwirkungen, Verbesserung der Lebensqualität) umfassen. Anforderungen an die Darstellung werden in den Unterabschnitten beschrieben.
Benennen Sie die Patientencharakteristika und patientenrelevanten Endpunkte, die in den relevanten Studien erhoben wurden. Begründen Sie, wenn Sie von den oben benannten Vorgaben abgewichen sind. Beschreiben Sie für jeden Endpunkt, warum Sie ihn als patientenrelevant einstufen, und machen Sie Angaben zur Validität des Endpunkts (z.B. zur Validierung der eingesetzten Fragebögen). Geben Sie für den jeweiligen Endpunkt an, ob unterschiedliche Operationalisierungen innerhalb der Studien und zwischen den Studien verwendet wurden. Benennen Sie die für die Bewertung herangezogene(n) Operationalisierung(en) und begründen Sie die Auswahl. Beachten Sie bei der Berücksichtigung von Surrogatendpunkten Abschnitt 4.5.4.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.5.3 Meta-Analysen
Sofern mehrere Studien vorliegen, sollen diese in einer Meta-Analyse quantitativ zusammengefasst werden, wenn die Studien aus medizinischen (z.B. Patientengruppen) und methodischen (z.B. Studiendesign) Gründen ausreichend vergleichbar sind. Es ist jeweils zu begründen, warum eine Meta-Analyse durchgeführt wurde oder warum eine Meta-Analyse nicht durchgeführt wurde bzw. warum einzelne Studien ggf. nicht in die Meta-Analyse einbezogen wurden. Für Meta-Analysen soll die im Folgenden beschriebene Methodik eingesetzt werden.
Für die statistische Auswertung sollen primär die Ergebnisse aus Intention-to-treat-Analysen, so wie sie in den vorliegenden Dokumenten beschrieben sind, verwendet werden. Die Meta-Analysen sollen in der Regel auf Basis von Modellen mit zufälligen Effekten 8) erfolgen. In begründeten Ausnahmefällen sollen zusätzlich Modelle mit festen Effekten eingesetzt werden. Falls die für eine Meta-Analyse notwendigen Schätzer für Lage und Streuung in den Studienunterlagen nicht vorliegen, sollen diese nach Möglichkeit aus den vorhandenen Informationen eigenständig berechnet beziehungsweise näherungsweise bestimmt werden.
Für kontinuierliche Variablen soll die Mittelwertdifferenz, gegebenenfalls standardisiert mittels Hedges' g, als Effektmaß eingesetzt werden. Bei binären Variablen sollen Meta-Analysen primär sowohl anhand des Odds Ratios als auch des Relativen Risikos durchgeführt werden. In begründeten Ausnahmefällen können auch andere Effektmaße zum Einsatz kommen. Bei kategorialen Variablen soll ein geeignetes Effektmaß in Abhängigkeit vom konkreten Endpunkt und den verfügbaren Daten verwendet 9 werden.
Die Effektschätzer und Konfidenzintervalle aus den Studien sollen mittels Forest Plots zusammenfassend dargestellt werden. Anschließend soll die Einschätzung einer möglichen Heterogenität der Studienergebnisse anhand des Maßes I und des statistischen Tests auf Vorliegen von Heterogenität 10) erfolgen. Ist die Heterogenität der Studienergebnisse nicht bedeutsam, soll der gemeinsame (gepoolte) Effekt inklusive Konfidenzintervall dargestellt werden. Bei bedeutsamer Heterogenität sollen die Ergebnisse nur in begründeten Ausnahmefällen gepoolt werden. Außerdem soll untersucht werden, welche Faktoren diese Heterogenität möglicherweise erklären könnten. Dazu zählen methodische Faktoren (siehe Abschnitt 4.2.5.4) und klinische Faktoren, sogenannte Effektmodifikatoren (siehe Abschnitt 4.2.5.5).
Beschreiben Sie die für Meta-Analysen eingesetzte Methodik. Begründen Sie, wenn Sie von der oben beschriebenen Methodik abweichen.
<< Angaben des pharmazeutischen Unternehmers>>
4.2.5.4 Sensitivitätsanalysen
Zur Einschätzung der Robustheit der Ergebnisse sollen Sensitivitätsanalysen hinsichtlich methodischer Faktoren durchgeführt werden. Die methodischen Faktoren bilden sich aus den im Rahmen der Informationsbeschaffung und -bewertung getroffenen Entscheidungen, zum Beispiel die Festlegung von Cut-off-Werten für Erhebungszeitpunkte oder die Wahl des Effektmaßes. Insbesondere die Einstufung des Verzerrungspotenzials der Ergebnisse in die Kategorien "hoch" und "niedrig" soll für Sensitivitätsanalysen verwendet werden.
Das Ergebnis der Sensitivitätsanalysen kann die Einschätzung der Aussagekraft der Nachweise beeinflussen.
Begründen Sie die durchgeführten Sensitivitätsanalysen oder den Verzicht auf Sensitivitätsanalysen. Beschreiben Sie die für Sensitivitätsanalysen eingesetzte Methodik. Begründen Sie, wenn Sie von der oben beschriebenen Methodik abweichen.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.5.5 Subgruppenmerkmale und andere Effektmodifikatoren
Die Ergebnisse sollen hinsichtlich potenzieller Effektmodifikatoren, das heißt klinischer Faktoren, die die Effekte beeinflussen, untersucht werden. Dies können beispielsweise direkte Patientencharakteristika (Subgruppenmerkmale) sowie Spezifika der Behandlungen (z.B. die Dosis) sein. Im Gegensatz zu den in Abschnitt 4.2.5.4 beschriebenen methodischen Faktoren für Sensitivitätsanalysen besteht hier das Ziel, mögliche Effektunterschiede zwischen Patientengruppen und Behandlungsspezifika aufzudecken. Eine potenzielle Effektmodifikation soll anhand von Homogenitäts- bzw. Interaktionstests oder von Interaktionstermen aus Regressionsanalysen (mit Angabe von entsprechenden Standardfehlern) untersucht werden. Subgruppenanalysen auf der Basis individueller Patientendaten haben in der Regel eine größere Ergebnissicherheit als solche auf Basis von Meta-Regressionen oder Meta-Analysen unter Kategorisierung der Studien bezüglich der möglichen Effektmodifikatoren, sie sind deshalb zu bevorzugen. Es sollten, soweit sinnvoll, folgende Faktoren bezüglich einer möglichen Effektmodifikation berücksichtigt werden:
Sollten sich aus den verfügbaren Informationen Anhaltspunkte für weitere mögliche Effektmodifikatoren ergeben, können diese ebenfalls begründet einbezogen werden. Die Ergebnisse von in Studien a priori geplanten und im Studienprotokoll festgelegten Subgruppenanalysen für patientenrelevante Endpunkte sind immer darzustellen.
Bei Identifizierung möglicher Effektmodifikatoren kann gegebenenfalls eine Präzisierung der aus den für die Gesamtgruppe beobachteten Effekten abgeleiteten Aussagen erfolgen. Ergebnisse von Subgruppenanalysen können die Identifizierung von Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen unterstützen.
Benennen Sie die durchgeführten Subgruppenanalysen. Begründen Sie die durchgeführten Subgruppenanalysen bzw. die Untersuchung von Effektmodifikatoren oder den Verzicht auf solche Analysen. Beschreiben Sie die für diese Analysen eingesetzte Methodik. Begründen Sie, wenn Sie von der oben beschriebenen Methodik abweichen.
<< Angaben des pharmazeutischen Unternehmers >>
4.2.5.6 Indirekte Vergleiche
Zurzeit sind international Methoden in der Entwicklung, um indirekte Vergleiche zu ermöglichen. Nicht adjustierte indirekte Vergleiche (d.h. Vergleiche einzelner Behandlungsgruppen aus verschiedenen Studien ohne Bezug zu einem gemeinsamen Komparator) stellen dabei keine valide Analysemethode dar, der Einsatz einfacher adjustierter indirekter Vergleiche ist möglich 11. Komplexe Verfahren für den simultanen Vergleich von mehr als zwei Therapien unter Berücksichtigung sowohl direkter als auch indirekter Vergleiche werden in der Literatur unterschiedlich bezeichnet, z.B. als "Mixed-Treatment-Comparison (MTC)-Meta-Analysen" 12, "Multiple-Treatment-Meta-Analysen" 13 oder auch "Netzwerk-Meta-Analysen" 14, sie gehen aber im Prinzip von denselben wesentlichen Annahmen aus.
Grundannahme für solche komplexen Analysen ist die Annahme der Konsistenz innerhalb des zu analysierenden Netzwerkes. Als Inkonsistenz wird dabei die Diskrepanz zwischen dem Ergebnis eines direkten und eines oder mehreren indirekten Vergleichen verstanden, die nicht mehr nur durch Zufallsfehler oder Heterogenität erklärbar ist 15.
Insgesamt ist es notwendig, die zugrunde liegende Methodik genau und reproduzierbar zu beschreiben und die Annahme der Konsistenz zu untersuchen 16.
Beschreiben Sie detailliert und vollständig die zugrunde liegende Methodik des indirekten Vergleichs. Dabei sind mindestens folgende Angaben notwendig:
<< Angaben des pharmazeutischen Unternehmers >>
4.3 Ergebnisse zum medizinischen Nutzen und zum medizinischen Zusatznutzen
In den nachfolgenden Abschnitten sind die Ergebnisse zum medizinischen Nutzen und zum medizinischen Zusatznutzen zu beschreiben. Abschnitt 4.3.1 enthält dabei die Ergebnisse aus randomisierten kontrollierten Studien, die mit dem zu bewertenden Arzneimittel durchgeführt wurden (Evidenzstufen Ia/lb).
Abschnitt 4.3.2 enthält weitere Unterlagen anderer Evidenzstufen, sofern diese aus Sicht des pharmazeutischen Unternehmers zum Nachweis des Zusatznutzens erforderlich sind. Diese Unterlagen teilen sich wie folgt auf:
4.3.1 Ergebnisse randomisierter kontrollierter Studien mit dem zu bewertenden Arzneimittel
4.3.1.1 Ergebnis der Informationsbeschaffung - RCT mit dem zu bewertenden Arzneimittel
4.3.1.1.1 Studien des pharmazeutischen Unternehmers
Nachfolgend sollen alle Studien (RCT), die an die Zulassungsbehörde übermittelt wurden (Zulassungsstudien), sowie alle Studien (RCT), für die der pharmazeutische Unternehmer Sponsor ist oder war oder auf andere Weise finanziell beteiligt ist oder war, benannt werden. Beachten Sie dabei folgende Konkretisierungen:
Es sollen alle RCT, die der Zulassungsbehörde im Zulassungsdossier übermittelt wurden und deren Studienberichte im Abschnitt 5.3.5 des Zulassungsdossiers enthalten sind, aufgeführt werden. Darüber hinaus sollen alle RCT, für die der pharmazeutische Unternehmer Sponsor ist oder war oder auf andere Weise finanziell beteiligt ist oder war, aufgeführt werden.
Benennen Sie in der nachfolgenden Tabelle nur solche RCT, die ganz oder teilweise innerhalb des in diesem Dokument beschriebenen Anwendungsgebiets durchgeführt wurden. Fügen Sie dabei für jede Studie eine neue Zeile ein.
Folgende Informationen sind in der Tabelle darzulegen: Studienbezeichnung, Angabe "Zulassungsstudie ja/nein", Angabe über die Beteiligung (Sponsor ja/nein), Studienstatus (abgeschlossen, abgebrochen, laufend), Studiendauer und Therapiearme. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Tabellenzeile.
Tabelle 4-1: Liste der Studien des pharmazeutischen Unternehmers -RCT mit dem zu bewertenden Arzneimittel
| Studienbezeichnung | Zulassungsstudie (ja/nein) | Sponsor (ja/nein) | Status (abgeschlossen/ abgebrochen/ laufend) | Studiendauer | Therapiearme |
| <Studie 1> | ja | ja | abgeschlossen | 12 Monate | Medikament A, Medikament B, Placebo |
Geben Sie an, welchen Stand die Information in Tabelle 4-1 hat, d. h. zu welchem Datum der Studienstatus abgebildet wird. Das Datum des Studienstatus sollte nicht mehr als 3 Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle an, welche der in Tabelle 4-1 genannten Studien nicht für die Nutzenbewertung herangezogen wurden. Begründen Sie dabei jeweils die Nichtberücksichtigung. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-2: Studien des pharmazeutischen Unternehmers, die nicht für die Nutzenbewertung herangezogen wurden - RCT mit dem zu bewertenden Arzneimittel
| Studienbezeichnung | Begründung für die Nichtberücksichtigung der Studie |
4.3.1.1.2 Studien aus der bibliografischen Literaturrecherche
Beschreiben Sie nachfolgend das Ergebnis der bibliografischen Literaturrecherche. Illustrieren Sie den Selektionsprozess und das Ergebnis der Selektion mit einem Flussdiagramm. Geben Sie dabei an, wie viele Treffer sich insgesamt (d. h. über alle durchsuchten Datenbanken) aus der bibliografischen Literaturrecherche ergeben haben, wie viele Treffer sich nach Entfernung von Dubletten ergeben haben, wie viele Treffer nach Sichtung von Titel und, sofern vorhanden, Abstract als nicht relevant angesehen wurden, wie viele Treffer im Volltext gesichtet wurden, wie viele der im Volltext gesichteten Treffer nicht relevant waren (mit Angabe der Ausschlussgründe) und wie viele relevante Treffer verblieben. Geben Sie zu den relevanten Treffern an, wie vielen Einzelstudien diese zuzuordnen sind. Listen Sie die im Volltext gesichteten und ausgeschlossenen Dokumente unter Nennung des Ausschlussgrunds in Anhang 4-C.
[Anmerkung: "Relevanz" bezieht sich in diesem Zusammenhang auf die im Abschnitt 4.2.2 genannten Kriterien für den Einschluss von Studien in die Nutzenbewertung.]
Geben Sie im Flussdiagramm auch das Datum der Recherche an. Die Recherche soll nicht mehr als 3 Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
Orientieren Sie sich bei der Erstellung des Flussdiagramms an dem nachfolgenden Beispiel.
Abbildung 1: Flussdiagramm der bibliografischen Literaturrecherche - Suche nach randomisierten kontrollierten Studien mit dem zu bewertenden Arzneimittel
4.3.1.1.3 Studien aus der Suche in Studienregistern
Beschreiben Sie in der nachfolgenden Tabelle alle relevanten Studien, die durch die Suche in Studienregistern identifiziert wurden. Geben Sie dabei an, in welchem Studienregister die Studie identifiziert wurde und welche Dokumente dort zur Studie jeweils hinterlegt sind (z.B. Studienregistereintrag, Bericht über Studienergebnisse etc.). Geben Sie auch an, ob die Studie in der Liste der Studien des pharmazeutischen Unternehmers enthalten ist (siehe Tabelle 4-1) und ob die Studie auch durch die bibliografische Literaturrecherche identifiziert wurde. Fügen Sie für jede Studie eine neue Zeile ein. Listen Sie die ausgeschlossenen Studien unter Nennung des Ausschlussgrunds in Anhang 4-D.
[Anmerkung: "Relevanz" bezieht sich in diesem Zusammenhang auf die in Abschnitt 4.2.2 genannten Kriterien für den Einschluss von Studien in die Nutzenbewertung.]
Orientieren Sie sich bei Ihren Angaben an der beispielhaften ersten Tabellenzeile.
Tabelle 4-3: Relevante Studien aus der Suche in Studienregistern -RCT mit dem zu bewertenden Arzneimittel
| Studienbezeichnung | Identifikationsorte (Name der Studienregister und Angabe der Zitate a) | Studie in Liste der Studien des pharmazeutischen Unternehmers enthalten (ja/nein) | Studie durch bibliografische Literaturrecherche identifiziert (ja/nein) | Status (abgeschlossen/ abgebrochen/ laufend) |
| <Studie 1> | clinicaltrials.gov [6, 7] | ja | nein | abgeschlossen |
| a: Zitat des Studienregistereintrags sowie, falls vorhanden, der im Studienregister aufgelisteten Berichte über Studiendesign und/oder -ergebnisse. | ||||
Geben Sie an, welchen Stand die Information in Tabelle 4-3 hat, d. h. zu welchem Datum die Recherche durchgeführt wurde. Das Datum der Recherche soll nicht mehr als 3 Monate vor dem für die Einreichung des Dossiers maßgeblichen Zeitpunkt liegen.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.1.1.4 Resultierender Studienpool: RCT mit dem zu bewertenden Arzneimittel
Benennen Sie in der nachfolgenden Tabelle den aus den verschiedenen Suchschritten (Abschnitte 4.3.1.1.1, 4.3.1.1.2 und 4.3.1.1.3) resultierenden Pool relevanter Studien (exklusive laufender Studien) für das zu bewertende Arzneimittel, auch im direkten Vergleich zur zweckmäßigen Vergleichstherapie. Führen Sie außerdem alle relevanten Studien einschließlich der verfügbaren Quellen in Abschnitt 4.6 auf. Alle durch die vorhergehenden Schritte identifizierten und in der Tabelle genannten Quellen der relevanten Studien sollen für die Bewertung dieser Studien herangezogen werden.
Folgende Informationen sind in der Tabelle darzulegen: Studienbezeichnung, Studienkategorie und verfügbare Quellen. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Tabellenzeile. Hierbei sollen die Studien durch Zwischenzeilenüberschriften ggf. sinnvoll angeordnet werden, beispielsweise nach Therapieschema (Akut-/Langzeitstudien) und jeweils separat nach Art der Kontrolle (Placebo, zweckmäßige Vergleichstherapie, beides). Sollten Sie eine Strukturierung des Studienpools vornehmen, berücksichtigen Sie diese auch in den weiteren Tabellen in Modul 4.
Tabelle 4-4: Studienpool - RCT mit dem zu bewertenden Arzneimittel
| Studie | Studienkategorie | verfügbare Datenquellen a | ||||
| Studie zur Zulassung des zu bewertenden Arzneimittels | gesponserte Studie b | Studie Dritter | Studien- bericht | Registereintrag c | Publikation | |
| (ja/nein) | (ja/nein) | (ja/nein) | (ja/nein [Zitat]) | (ja/nein [Zitat]) | (ja/nein [Zitat]) | |
| ggf. Zwischenüberschrift zur Strukturierung des Studienpools | ||||||
| placebokontrolliert | ||||||
| <Studie 1> | ja | ja | nein | ja [5] | ja [6, 7] | ja [8] |
| aktivkontrolliert, zweckmäßige Vergleichstherapie(n) | ||||||
| a: Bei Angabe "ja" sind jeweils die Zitate der Quelle(n) (z.B. Publikationen, Studienberichte, Studienregistereinträge) mit anzugeben, und zwar als Verweis auf die in Abschnitt 4.7 genannte Referenzliste.
Darüber hinaus ist darauf zu achten, dass alle Quellen, auf die in dieser Tabelle verwiesen wird, auch in Abschnitt 4.6 (Liste der eingeschlossenen Studien) aufgeführt werden.
b: Studie, für die der Unternehmer Sponsor war c: Zitat der Studienregistereinträge sowie, falls vorhanden, der in den Studienregistern aufgelisteten Berichte über Studiendesign und/oder -ergebnisse. | ||||||
4.3.1.2 Charakteristika der in die Bewertung eingeschlossenen Studien - RCT mit dem zu bewertenden Arzneimittel
4.3.1.2.1 Studiendesign und Studienpopulationen
Beschreiben Sie das Studiendesign und die Studienpopulation der in die Bewertung eingeschlossenen Studien mindestens mit den Informationen in den folgenden Tabellen. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Tabellenzeile. Fügen Sie für jede Studie eine neue Zeile ein.
Weitere Informationen zu Studiendesign, Studienmethodik und Studienverlauf sind in Anhang 4-E zu hinterlegen.
Tabelle 4-5: Charakterisierung der eingeschlossenen Studien - RCT mit dem zu bewertenden Arzneimittel
| Studie | Studiendesign <RCT, doppelblind/einfach verblindet/offen, parallel/cross-over etc.> | Population <relevante Charakteristika, z.B. Schwere- grad> | Interventionen (Zahl der randomisierten Patienten) | Studiendauer <ggf. Run-in, Behandlung, Nachbeobachtung> | Ort und Zeitraum der Durchführung | Primärer Endpunkt; patientenrelevante sekundäre Endpunkte |
| <Studie 1> | RCT, doppelblind, parallel | Jugendliche und Erwachsene, leichtes bis mittelschweres Asthma | <Gruppe 1> (n = 354) <Gruppe 2> (n = 347) | Run-in: 2 Wochen Behandlung: 6 Monate | Europa (Deutsch- land, Frankreich, Polen) 9/2003-12/2004 | FEV1; Asthma-Symptome, Exazerbationen, gesundheitsbezogene Lebensqualität, unerwünschte Ereignisse |
Tabelle 4-6: Charakterisierung der Interventionen - RCT mit dem zu bewertenden Arzneimittel
| Studie | <Gruppe 1> | <Gruppe 2> | ggf. weitere Spalten mit Behandlungscharakteristika z.B. Vorbehandlung, Behandlung in der Run-in-Phase etc. |
| <Studie 1> | xxx 250 µg, 1 Inhalation bid + Placebo 2 Inhalationen bid | yyy 200 µg, 2 Inhalationen bid + Placebo 1 Inhalation bid | Vorbehandlung: zzz 1.000 µg pro Tag, 4 Wochen vor Studienbeginn Bedarfsmedikation: aaa |
Tabelle 4-7: Charakterisierung der Studienpopulationen - RCT mit dem zu bewertenden Arzneimittel
| Studie Gruppe | N | Alter (Jahre) | Geschlecht w/m (%) | ggf. weitere Spalten mit Populationscharakteristika z.B. Dauer der Erkrankung, Schweregrad, weitere Basisdaten projektabhängig |
| <Studie 1>
<Gruppe 1> | ||||
Beschreiben Sie die Studien zusammenfassend. Machen Sie auch Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext.
Sollte es Unterschiede zwischen den Studien geben, weisen Sie in einem erläuternden Text darauf hin.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.1.2.2 Verzerrungspotenzial auf Studienebene
Bewerten Sie das Verzerrungspotenzial der RCT auf Studienebene mithilfe des Bewertungsbogens in Anhang 4-F. Fassen Sie die Bewertung mit den Angaben in der folgenden Tabelle zusammen. Fügen Sie für jede Studie eine neue Zeile ein.
Dokumentieren Sie die Einschätzung für jede Studie mit einem Bewertungsbogen in Anhang 4-F.
Tabelle 4-8: Verzerrungspotenzial auf Studienebene - RCT mit dem zu bewertenden Arzneimittel
| Verblindung | |||||||
| Studie | Adäquate Erzeugung der Randomisierungssequenz | Verdeckung der Gruppenzuteilung | Patient | Behandelnde Personen | Ergebnisunabhängige Berichterstattung | Keine sonstigen Aspekte | Verzerrungs- potenzial auf Studienebene |
| <Studie 1> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein> | <hoch/niedrig> |
Begründen Sie für jede Studie die abschließende Einschätzung.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.1.3 Ergebnisse aus randomisierten kontrollierten Studien
Geben Sie in der folgenden Tabelle einen Überblick über die patientenrelevanten Endpunkte, auf denen Ihre Bewertung des medizinischen Nutzens und Zusatznutzens beruht. Geben Sie dabei an, welche dieser Endpunkte in den relevanten Studien jeweils untersucht wurden. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Tabellenzeile. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-9: Matrix der Endpunkte in den eingeschlossenen RCT mit dem zu bewertenden Arzneimittel
| Studie | <Mortalität> | <Gesundheitsbezogene Lebensqualität> | <Endpunkt> | <Endpunkt> | <Endpunkt> |
| <Studie 1> | nein | ja | ja | ja | nein |
4.3.1.3.1 <Endpunkt xxx> - RCT
Die Ergebnisdarstellung für jeden Endpunkt umfasst 3 Abschnitte. Zunächst soll für jede Studie das Verzerrungspotenzial auf Endpunkt-ebene in einer Tabelle zusammengefasst werden. Dann sollen die Ergebnisse der einzelnen Studien zu dem Endpunkt tabellarisch dargestellt und in einem Text zusammenfassend beschrieben werden. Anschließend sollen die Ergebnisse, wenn möglich und sinnvoll, in einer Meta-Analyse zusammengefasst und beschrieben werden.
Die tabellarische Darstellung der Ergebnisse für den jeweiligen Endpunkt soll mindestens die folgenden Angaben enthalten:
Bei Überlebenszeitanalysen soll die Kaplan-Meier-Kurve einschließlich Angaben zu den Patienten unter Risiko im Zeitverlauf (zu mehreren Zeitpunkten) abgebildet werden.
Falls für die Auswertung eine andere Population als die ITT-Population herangezogen wird, soll diese benannt (z.B. Safety-Population) und definiert werden.
Sofern mehrere Studien vorliegen, sollen diese in einer Meta-Analyse zusammengefasst werden, wenn die Studien aus medizinischen (z.B. Patientengruppen) und methodischen (z.B. Studiendesign) Gründen ausreichend vergleichbar sind. Es ist jeweils zu begründen, warum eine Meta-Analyse durchgeführt wurde oder warum eine Meta-Analyse nicht durchgeführt wurde bzw. warum einzelne Studien ggf. nicht in die Meta-Analyse einbezogen wurden. Sofern die vorliegenden Studien für eine Meta-Analyse geeignet sind, sollen die Meta-Analysen als Forest-Plot dargestellt werden. Die Darstellung soll ausreichende Informationen zur Einschätzung der Heterogenität der Ergebnisse zwischen den Studien in Form von geeigneten statistischen Maßzahlen enthalten (siehe Abschnitt 4.2.5.3). Eine Gesamtanalyse aller Patienten aus mehreren Studien ohne Berücksichtigung der Studienzugehörigkeit (z.B. Gesamt-Vierfeldertafel per Addition der Einzel-Vierfeldertafeln) soll vermieden werden, da so die Heterogenität nicht eingeschätzt werden kann.
Beschreiben Sie die Operationalisierung des Endpunkts für jede Studie in der folgenden Tabelle. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-10: Operationalisierung von <Endpunkt xxx>
| Studie | Operationalisierung |
| <Studie 1> | |
Bewerten Sie das Verzerrungspotenzial für den in diesem Abschnitt beschriebenen Endpunkt mithilfe des Bewertungsbogens in Anhang 4-F. Fassen Sie die Bewertung mit den Angaben in der folgenden Tabelle zusammen. Fügen Sie für jede Studie eine neue Zeile ein.
Dokumentieren Sie die Einschätzung für jede Studie mit einem Bewertungsbogen in Anhang 4-F.
Tabelle 4-11: Bewertung des Verzerrungspotenzials für <Endpunkt xxx> in RCT mit dem zu bewertenden Arzneimittel
| Studie | Verzerrungspotenzial auf Studienebene | Verblindung Endpunkterheber | Adäquate Umsetzung des ITT-Prinzips | Ergebnisunabhängige Berichterstattung | Keine sonstigen Aspekte | Verzerrungspotenzial Endpunkt |
| <Studie 1> | <hoch/niedrig> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein> | <hoch/niedrig> |
Begründen Sie für jede Studie die abschließende Einschätzung.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die Ergebnisse für den Endpunkt xxx für jede einzelne Studie in tabellarischer Form dar. Fügen Sie für jede Studie eine neue Zeile ein. Beschreiben Sie die Ergebnisse zusammenfassend.
Tabelle 4-12: Ergebnisse für <Endpunkt xxx> aus RCT mit dem zu bewertenden Arzneimittel
| Studie | Tabellarische Präsentation in geeigneter Form (Anforderungen siehe Erläuterung oben) |
| <Studie 1> | |
<< Angaben des pharmazeutischen Unternehmers >>
Sofern die vorliegenden Studien bzw. Daten für eine Meta-Analyse medizinisch und methodisch geeignet sind, fassen Sie die Einzelergebnisse mithilfe von Meta-Analysen quantitativ zusammen und stellen Sie die Ergebnisse der Meta-Analysen (in der Regel als Forest-Plot) dar. Beschreiben Sie die Ergebnisse zusammenfassend. Begründen Sie, warum eine Meta-Analyse durchgeführt wurde bzw. warum eine Meta-Analyse nicht durchgeführt wurde bzw. warum einzelne Studien ggf. nicht in die Meta-Analyse einbezogen wurden. Machen Sie auch Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext.
<Abbildung Meta-Analyse>
Abbildung 2: Meta-Analyse für <Endpunkt xxx> aus RCT; <zu bewertendes Arzneimittel> versus <Vergleichstherapie>
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die in diesem Abschnitt beschriebenen Informationen für jeden weiteren Endpunkt aus RCT mit dem zu bewertenden Arzneimittel fortlaufend in einem eigenen Abschnitt dar.
4.3.1.3.2 Subgruppenanalysen - RCT
Für die tabellarische Darstellung der Ergebnisse aus Subgruppenanalysen gelten die gleichen Anforderungen wie für die tabellarische Darstellung von Ergebnissen aus Gesamtpopulationen in Abschnitt 4.3.1.3.1.
Beschreiben Sie die Ergebnisse von Subgruppenanalysen (einschließlich der Interaktionsterme). Stellen Sie dabei die Ergebnisse in den Subgruppen zunächst für jede einzelne Studie in tabellarischer Form dar. Diese Anforderung gilt sowohl für Subgruppenanalysen auf Basis individueller Patientendaten als auch für solche auf Basis aggregierter Daten. Begründen Sie die Wahl von Trennpunkten, wenn quantitative Merkmale kategorisiert werden. Verwenden Sie dabei nach Möglichkeit die in dem jeweiligen Gebiet gebräuchlichen Einteilungen und begründen Sie etwaige Abweichungen. Kennzeichnen Sie in einzelnen Studien a priori geplante Subgruppenanalysen.
Sofern die vorliegenden Studien bzw. Daten für eine Meta-Analyse medizinisch und methodisch geeignet sind, fassen Sie die Ergebnisse mithilfe einer Meta-Analyse quantitativ zusammen und stellen Sie die Ergebnisse der Meta-Analyse (als Forest-Plot) dar.
Beschreiben Sie die Ergebnisse zusammenfassend. Begründen Sie Ihr Vorgehen, wenn Sie keine Meta-Analyse durchführen bzw. wenn Sie nicht alle Studien in die Meta-Analyse einschließen.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.1.3.3 Zusammenfassung der Ergebnisse aus randomisierten kontrollierten Studien
Der vorliegende Abschnitt soll einen Überblick über die Ergebnisse zum medizinischen Nutzen und medizinischen Zusatznutzen aus randomisierten kontrollierten Studien geben. Die Zusammenfassung soll Aussagen zu allen in Abschnitt 4.3.1.3 präsentierten Endpunkten und Subgruppenanalysen enthalten. Dabei sollen, soweit verfügbar, numerische Ergebnisse aus Meta-Analysen einschließlich Konfidenzintervallen dargestellt werden.
Fassen Sie die Ergebnisse zum medizinischen Nutzen und zum medizinischen Zusatznutzen aus randomisierten kontrollierten Studien zusammen.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2 Weitere Unterlagen
4.3.2.1 Indirekte Vergleiche auf Basis randomisierter kontrollierter Studien
Hinweis: Die nachfolgenden Unterabschnitte sind nur dann auszufüllen, wenn indirekte Vergleiche als Nachweis für einen Zusatznutzen herangezogen werden sollen. Das ist dann möglich, wenn keine direkten Vergleichsstudien für das zu bewertende Arzneimittel gegenüber der zweckmäßigen Vergleichstherapie vorliegen oder diese keine ausreichenden Aussagen über den Zusatznutzen zulassen.
4.3.2.1.1 Ergebnis der Informationsbeschaffung - Studien für indirekte Vergleiche
Beschreiben Sie nachfolgend das Ergebnis der Informationsbeschaffung zu Studien für indirekte Vergleiche. Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.1 (Ergebnis der Informationsbeschaffung - RCT mit dem zu bewertenden Arzneimittel) und stellen Sie Informationen analog Abschnitt 4.3.1.1 zur Verfügung (einschließlich tabellarischer Darstellungen, Angabe eines Flussdiagramms etc.). Benennen Sie
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.1.2 Charakteristika der Studien für indirekte Vergleiche
Charakterisieren Sie nachfolgend die Studien, die für indirekte Vergleiche herangezogen wurden, und bewerten Sie deren Verzerrungspotenzial. Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.2 und stellen Sie Informationen analog Abschnitt 4.3.1.2 zur Verfügung.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.1.3 Ergebnisse aus indirekten Vergleichen
Geben Sie in der folgenden Tabelle einen Überblick über die patientenrelevanten Endpunkte, auf denen Ihre Bewertung des medizinischen Nutzens und Zusatznutzens aus indirekten Vergleichen beruht. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Zeile. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-13: Matrix der Endpunkte in den eingeschlossenen RCT für indirekte Vergleiche
| Studie | <Mortalität> | <Gesundheitsbezogene Lebensqualität> | <Endpunkt> | <Endpunkt> | <Endpunkt> |
| <Studie 1> | nein | ja | ja | ja | nein |
4.3.2.1.3.1 <Endpunkt xxx> - indirekte Vergleiche aus RCT
Für die indirekten Vergleiche soll zunächst für jeden Endpunkt eine Übersicht über die verfügbaren Vergleiche gegeben werden. Anschließend soll die Darstellung der Ergebnisse in 3 Schritten erfolgen: 1) Bewertung des Verzerrungspotenzials auf Endpunktebene pro Studie, 2) tabellarische Darstellung der Ergebnisse der einzelnen Studien, 3) Darstellung des indirekten Vergleichs. Für die Punkte 1 und 2 gelten die gleichen Anforderungen wie für die Darstellung der Ergebnisse der direkten Vergleiche in Abschnitt 4.3.1.3.1.
Geben Sie für den im vorliegenden Abschnitt präsentierten Endpunkt einen Überblick über die in den Studien verfügbaren Vergleiche. Beispielhaft wäre folgende Darstellung denkbar:
Tabelle 4-14: Zusammenfassung der verfügbaren Vergleiche in den Studien, die für den indirekten Vergleich herangezogen wurden
| Anzahl Studien | Studien | Intervention | <Vergleichstherapie 1> | <Vergleichstherapie 2> | <Vergleichstherapie 3> |
| 1 | <Studie 1> | • | • | • | |
| 2 | <Studie 2> | • | • | ||
| <Studie 3> | • | • | |||
| 1 | <Studie 4> | • | • | • | |
| etc. | etc. | etc. | etc. |
Stellen Sie zusätzlich die Netzwerkstruktur des indirekten Vergleichs grafisch dar.
<< Angaben des pharmazeutischen Unternehmers >>
Beschreiben Sie die Operationalisierung des Endpunkts für jede Studie in der folgenden Tabelle. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-15: Operationalisierung von <Endpunkt xxx>
| Studie | Operationalisierung |
| <Studie 1> | |
Bewerten Sie das Verzerrungspotenzial für den in diesem Abschnitt beschriebenen Endpunkt mithilfe des Bewertungsbogens in Anhang 4-F. Fassen Sie die Bewertung mit den Angaben in der folgenden Tabelle zusammen. Fügen Sie für jede Studie eine neue Zeile ein.
Dokumentieren Sie die Einschätzung für jede Studie mit einem Bewertungsbogen in Anhang 4-F.
Tabelle 4-16: Bewertung des Verzerrungspotenzials für <Endpunkt xxx> in RCT für indirekte Vergleiche
| Studie | Verzerrungspotenzial auf Studienebene | Verblindung Endpunkterheber | Adäquate Umsetzung des ITT-Prinzips | Ergebnisunabhängige Berichterstattung | Keine sonstigen Aspekte | Verzerrungspotenzial Endpunkt |
| <Studie 1> | <hoch/niedrig> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein> | <hoch/niedrig> |
Begründen Sie für jede Studie die abschließende Einschätzung.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die Ergebnisse für den Endpunkt xxx für jede einzelne Studie in tabellarischer Form dar. Fügen Sie für jede Studie eine neue Zeile ein. Beschreiben Sie die Ergebnisse zusammenfassend.
Tabelle 4-17: Ergebnisse für <Endpunkt xxx> aus RCT für indirekte Vergleiche
| Studie | Tabellarische Präsentation in geeigneter Form (Anforderungen siehe Erläuterung in Abschnitt 4.3.1.3.1) |
| <Studie 1> | |
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die Ergebnisse der indirekten Vergleiche in tabellarischer Form dar. Optional können die Ergebnisse zusätzlich auch grafisch illustriert werden. Orientieren Sie sich dabei an der üblichen Darstellung metaanalytischer Ergebnisse. Gliedern Sie die Ergebnisse nach folgenden Punkten:
Machen Sie darüber hinaus Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die in diesem Abschnitt beschriebenen Informationen für jeden weiteren Endpunkt, für den ein indirekter Vergleich vorgenommen wird, fortlaufend in einem eigenen Abschnitt dar.
4.3.2.1.3.2 Subgruppenanalysen - indirekte Vergleiche aus RCT
Beschreiben Sie nachfolgend die Ergebnisse von Subgruppenanalysen auf Basis indirekter Vergleiche aus RCT. Berücksichtigen Sie dabei die Anforderungen gemäß Abschnitt 4.3.1.3.2.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.2 Nicht randomisierte vergleichende Studien
Hinweis: Die nachfolgenden Unterabschnitte sind nur dann auszufüllen, wenn nicht randomisierte vergleichende Studien als Nachweis für einen Zusatznutzen herangezogen werden sollen.
4.3.2.2.1 Ergebnis der Informationsbeschaffung - nicht randomisierte vergleichende Studien
Beschreiben Sie nachfolgend das Ergebnis der Informationsbeschaffung zu nicht randomisierten vergleichenden Studien. Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.1 (Ergebnis der Informationsbeschaffung - RCT mit dem zu bewertenden Arzneimittel) und stellen Sie Informationen analog Abschnitt 4.3.1.1 zur Verfügung (einschließlich tabellarischer Darstellungen, Angabe eines Flussdiagramms etc.). Benennen Sie
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.2.2 Charakteristika der nicht randomisierten vergleichenden Studien
Charakterisieren Sie nachfolgend die nicht randomisierten vergleichenden Studien. Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.2 und stellen Sie Informationen analog Abschnitt 4.3.1.2 zur Verfügung.
Beschreiben Sie die Verzerrungsaspekte der nicht randomisierten vergleichenden Studie auf Studienebene mithilfe des Bewertungsbogens in Anhang 4-F. Fassen Sie die Beschreibung mit den Angaben in der folgenden Tabelle zusammen. Fügen Sie für jede Studie eine neue Zeile ein.
Dokumentieren Sie die Einschätzung für jede Studie mit einem Bewertungsbogen in Anhang 4-F.
Tabelle 4-18: Verzerrungsaspekte auf Studienebene - nicht randomisierte vergleichende Interventionsstudien
| Verblindung | ||||||
| Studie | Zeitliche Parallelität der Gruppen | Vergleichbarkeit der Gruppen bzw. adäquate Berücksichtigung von prognostisch relevanten Faktoren | Patient | Behandelnde Personen | Ergebnisunabhängige Berichterstattung | Keine sonstigen Aspekte |
| <Studie 1> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein/unklar> | <ja/nein> |
Beschreiben Sie zusammenfassend die Bewertungsergebnisse zu Verzerrungsaspekten auf Studienebene.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.2.3 Ergebnisse aus nicht randomisierten vergleichenden Studien
4.3.2.2.3.1 <Endpunkt xxx> - nicht randomisierte vergleichende Studien
Beschreiben Sie die Operationalisierung des Endpunkts für jede Studie in der folgenden Tabelle. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-19: Operationalisierung von <Endpunkt xxx>
| Studie | Operationalisierung |
| <Studie 1> | |
Beschreiben Sie die Verzerrungsaspekte für den in diesem Abschnitt beschriebenen Endpunkt mithilfe des Bewertungsbogens in Anhang 4-F. Fassen Sie die Bewertung mit den Angaben in der folgenden Tabelle zusammen. Fügen Sie für jede Studie eine neue Zeile ein.
Dokumentieren Sie die Einschätzung für jede Studie mit einem Bewertungsbogen in Anhang 4-F.
Tabelle 4-20: Verzerrungsaspekte für <Endpunkt xxx> - nicht randomisierte vergleichende Studien
| Studie | Endpunkterheber Verblindung | Adäquate Umsetzung des ITT-Prinzips | Ergebnisunabhängige Berichterstattung | Keine sonstigen Aspekte |
| <Studie 1> | ja/nein/ unklar | ja/nein/ unklar | ja/nein/ unklar | ja/nein |
Beschreiben Sie zusammenfassend die Bewertungsergebnisse zu Verzerrungsaspekten auf Endpunktebene.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die Ergebnisse der nicht randomisierten vergleichenden Studien gemäß den Anforderungen des TREND- bzw. des STROBE-Statements dar. Machen Sie dabei auch Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die in diesem Abschnitt beschriebenen Informationen für jeden weiteren Endpunkt aus nicht randomisierten vergleichenden Studien fortlaufend in einem eigenen Abschnitt dar.
4.3.2.2.3.2 Subgruppenanalysen - nicht randomisierte vergleichende Studien
Beschreiben Sie nachfolgend die Ergebnisse von Subgruppenanalysen aus nicht randomisierten vergleichenden Studien. Berücksichtigen Sie dabei die Anforderungen gemäß Abschnitt 4.3.1.3.2.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.3 Weitere Untersuchungen
Hinweis: Die nachfolgenden Unterabschnitte sind nur dann auszufüllen, wenn über die in den Abschnitten 4.3.1, 4.3.2.1 und 4.3.2.2 genannten Studien hinausgehende Untersuchungen als Nachweis für einen Zusatznutzen herangezogen werden sollen.
4.3.2.3.1 Ergebnis der Informationsbeschaffung - weitere Untersuchungen
Beschreiben Sie nachfolgend das Ergebnis der Informationsbeschaffung nach Untersuchungen, die nicht in den Abschnitten 4.3.1, 4.3.2.1 und 4.3.2.2 aufgeführt sind. Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.1 (Ergebnis der Informationsbeschaffung - RCT mit dem zu bewertenden Arzneimittel) und stellen Sie Informationen analog Abschnitt 4.3.1.1 zur Verfügung (einschließlich tabellarischer Darstellungen, Angabe eines Flussdiagramms etc.). Benennen Sie
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.3.2 Charakteristika der weiteren Untersuchungen
Charakterisieren Sie nachfolgend die weiteren Untersuchungen und bewerten Sie deren Verzerrungspotenzial.
Ergebnisse nicht randomisierter Studien, die keine kontrollierten Interventionsstudien sind, gelten aufgrund ihres Studiendesigns generell als potenziell hoch verzerrt. Trifft das auf die von Ihnen vorgelegten Studien nicht zu, begründen Sie Ihre Einschätzung.
Strukturieren Sie diesen Abschnitt analog Abschnitt 4.3.1.2 und stellen Sie Informationen analog Abschnitt 4.3.1.2 zur Verfügung.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.3.3 Ergebnisse aus weiteren Untersuchungen
4.3.2.3.3.1 <Endpunkt xxx> - weitere Untersuchungen
Beschreiben Sie die Operationalisierung des Endpunkts für jede Studie in der folgenden Tabelle. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 4-21: Operationalisierung von <Endpunkt xxx> - weitere Untersuchungen
| Studie | Operationalisierung |
| <Studie 1> | |
Bewerten Sie das Verzerrungspotenzial für den in diesem Abschnitt beschriebenen Endpunkt Ergebnisse nicht randomisierter Studien, die keine kontrollierten Interventionsstudien sind, gelten aufgrund ihres Studiendesigns generell als potenziell hoch verzerrt. Trifft das auf die von Ihnen vorgelegten Studien nicht zu, begründen Sie Ihre Einschätzung.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die Ergebnisse der weiteren Untersuchungen gemäß den jeweils gültigen Standards für die Berichterstattung dar. Begründen Sie dabei die Auswahl des Standards für die Berichterstattung. Machen Sie darüber hinaus Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext.
<< Angaben des pharmazeutischen Unternehmers >>
Stellen Sie die in diesem Abschnitt beschriebenen Informationen für jeden weiteren Endpunkt aus weiteren Untersuchungen fortlaufend in einem eigenen Abschnitt dar.
4.3.2.3.3.2 Subgruppenanalysen - weitere Untersuchungen
Beschreiben Sie nachfolgend die Ergebnisse von Subgruppenanalysen aus weiteren Untersuchungen. Berücksichtigen Sie dabei die Anforderungen gemäß Abschnitt 4.3.1.3.2.
<< Angaben des pharmazeutischen Unternehmers >>
4.3.2.4 Zusammenfassung der Ergebnisse aus weiteren Unterlagen
Der vorliegende Abschnitt soll einen Überblick über die Ergebnisse aus weiteren Unterlagen (Abschnitte 4.3.2.1, 4.3.2.2 und 4.3.2.3) geben. Die Zusammenfassung soll Aussagen zu allen in diesen Abschnitten präsentierten Endpunkten und Subgruppenanalysen enthalten. Dabei sollen, soweit verfügbar, numerische Ergebnisse aus Meta-Analysen einschließlich Konfidenzintervallen dargestellt werden.
Fassen Sie die Ergebnisse aus weiteren Unterlagen zusammen.
<< Angaben des pharmazeutischen Unternehmers >>
4.4 Abschließende Bewertung der Unterlagen zum Nachweis des Zusatznutzens
4.4.1 Beurteilung der Aussagekraft der Nachweise
Legen Sie für alle im Dossier eingereichten Unterlagen die Evidenzstufe dar. Beschreiben Sie zusammenfassend auf Basis der in den Abschnitten 4.3.1 und 4.3.2 präsentierten Ergebnisse die Aussagekraft der Nachweise für einen Zusatznutzen unter Berücksichtigung der Studienqualität, der Validität der herangezogenen Endpunkte sowie der Evidenzstufe.
<< Angaben des pharmazeutischen Unternehmers >>
4.4.2 Beschreibung des Zusatznutzens einschließlich dessen Wahrscheinlichkeit und Ausmaß
Führen Sie die in den Abschnitten 4.3.1 und 4.3.2 beschriebenen Ergebnisse zum Zusatznutzen auf Ebene einzelner Endpunkte zusammen und leiten Sie ab, ob sich aus der Zusammenschau der Ergebnisse zu den einzelnen Endpunkten insgesamt ein Zusatznutzen des zu bewertenden Arzneimittels im Vergleich zur zweckmäßigen Vergleichstherapie ergibt. Berücksichtigen Sie dabei auch die Angaben zur Übertragbarkeit der Studienergebnisse auf den deutschen Versorgungskontext. Liegt ein Zusatznutzen vor, beschreiben Sie, worin der Zusatznutzen besteht.
Stellen Sie die Wahrscheinlichkeit des Zusatznutzens dar, d. h., beschreiben und begründen Sie unter Berücksichtigung der in Abschnitt 4.4.1 dargelegten Aussagekraft der Nachweise die Ergebnissicherheit der Aussage zum Zusatznutzen.
Beschreiben Sie außerdem das Ausmaß des Zusatznutzens unter Verwendung folgender Kategorisierung (in der Definition gemäß AM-NutzenV):
Berücksichtigen Sie bei den Aussagen zum Zusatznutzen ggf. nachgewiesene Unterschiede zwischen verschiedenen Patientengruppen.
<< Angaben des pharmazeutischen Unternehmers >>
4.4.3 Angabe der Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht
Geben Sie auf Basis der in den Abschnitten 4.3.1 und 4.3.2 beschriebenen Ergebnisse und unter Berücksichtigung des in Abschnitt 4.4.2 dargelegten Zusatznutzens sowie dessen Wahrscheinlichkeit und Ausmaß in der nachfolgenden Tabelle an, für welche Patientengruppen ein therapeutisch bedeutsamer Zusatznutzen besteht. Benennen Sie das Ausmaß des Zusatznutzens in Patientengruppen mit therapeutisch bedeutsamem Zusatznutzen. Fügen Sie für jede Patientengruppe mit therapeutisch bedeutsamem Zusatznutzen eine neue Zeile ein.
Tabelle 4-22: Patientengruppen, für die ein therapeutisch bedeutsamer Zusatznutzen besteht, einschließlich Ausmaß des Zusatznutzens
| Bezeichnung der Patientengruppen | Ausmaß des Zusatznutzens |
4.4.4 (aufgehoben)
4.5 Begründung für die Vorlage weiterer Unterlagen und Surrogatendpunkte
4.5.1 Begründung für die Vorlage indirekter Vergleiche
Sofern mit dem Dossier indirekte Vergleiche (Abschnitt 4.3.2.1) eingereicht wurden, begründen Sie dies. Begründen Sie dabei auch, warum sich die ausgewählten Studien jeweils für einen indirekten Vergleich gegenüber dem zu bewertenden Arzneimittel und damit für den Nachweis eines Zusatznutzens durch indirekten Vergleich eignen.
<< Angaben des pharmazeutischen Unternehmers >>
4.5.2 Begründung für die Vorlage nicht randomisierter vergleichender Studien und weiterer Untersuchungen
Sofern mit dem Dossier nicht randomisierte vergleichende Studien (Abschnitt 4.3.2.2) oder weitere Untersuchungen (Abschnitt 4.3.2.3) eingereicht wurden, nennen Sie die Gründe, nach denen es unmöglich oder unangemessen ist, zu den in diesen Studien bzw. Untersuchungen behandelten Fragestellungen Studien höchster Evidenzstufe (randomisierte klinische Studien) durchzuführen oder zu fordern.
<< Angaben des pharmazeutischen Unternehmers >>
4.5.3 Begründung für die Bewertung auf Grundlage der verfügbaren Evidenz, da valide Daten zu patientenrelevanten Endpunkten noch nicht vorliegen
Falls aus Ihrer Sicht valide Daten zu patientenrelevanten Endpunkten zum Zeitpunkt der Bewertung noch nicht vorliegen können, begründen Sie dies.
<< Angaben des pharmazeutischen Unternehmers >>
4.5.4 Verwendung von Surrogatendpunkten
Die Verwendung von Surrogatendpunkten bedarf einer Begründung (siehe Abschnitt 4.5.3). Zusätzlich soll dargelegt werden, ob und warum die verwendeten Surrogatendpunkte im betrachteten Kontext valide Surrogatendpunkte darstellen bzw. Aussagen zu patientenrelevanten Endpunkten zulassen.
Eine Validierung von Surrogatendpunkten bedarf in der Regel einer Meta-Analyse von Studien, in denen sowohl Effekte auf den Surrogatendpunkt als auch Effekte auf den interessierenden patientenrelevanten Endpunkt untersucht wurden (Burzykowski 2005 17), Molenberghs 2010 18)). Diese Studien müssen bei Patientenkollektiven und Interventionen durchgeführt worden sein, die Aussagen für das dem vorliegenden Antrag zugrunde liegende Anwendungsgebiet und das zu bewertende Arzneimittel sowie die Vergleichstherapie erlauben.
Eine Möglichkeit der Verwendung von Surrogatendpunkten ohne abschließende Validierung stellt die Anwendung des Konzepts eines sogenannten Surrogate-Threshold-Effekts (STE) (Burzykowski 2006 19)) dar. Daneben besteht die Möglichkeit einer Surrogatvalidierung in der quantitativen Betrachtung geeigneter Korrelationsmaße von Surrogatendpunkt und interessierendem patientenrelevanten Endpunkt ("individuelle Ebene") sowie von Effekten auf den Surrogatendpunkt und Effekten auf den interessierenden patientenrelevanten Endpunkt ("Studienebene"). Dabei ist dann zu zeigen, dass die unteren Grenzen der entsprechenden 95%-Konfidenzintervalle fiir solche Korrelationsmaße ausreichend hoch sind. Die Anwendung alternativer Methoden zur Surrogatvalidierung (siehe Weir 2006 20)) soll ausreichend begründet werden, insbesondere dann, wenn als Datengrundlage nur eine einzige Studie verwendet werden soll.
Berichten Sie zu den Studien zur Validierung oder zur Begründung für die Verwendung von Surrogatendpunkten mindestens folgende Informationen:
Sofern Sie im Dossier Ergebnisse zu Surrogatendpunkten eingereicht haben, benennen Sie die Gründe für die Verwendung von Surrogatendpunkten. Beschreiben Sie, ob und warum die verwendeten Surrogatendpunkte im betrachteten Kontext valide Surrogatendpunkte darstellen bzw. Aussagen zu patientenrelevanten Endpunkten zulassen.
<< Angaben des pharmazeutischen Unternehmers >>
4.6 Liste der eingeschlossenen Studien
Listen Sie alle für die Nutzenbewertung berücksichtigten Studien und Untersuchungen unter Angabe der im Dossier verwendeten Studienbezeichnung und der zugehörigen Quellen (z.B. Publikationen, Studienberichte, Studienregistereinträge).
<< Angaben des pharmazeutischen Unternehmers >>
4.7 Referenzliste
Listen Sie nachfolgend alle Quellen (z.B. Publikationen, Studienberichte, Studienregistereinträge), die Sie im vorliegenden Dokument angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
<< Angaben des pharmazeutischen Unternehmers >>
| Suchstrategien - bibliografische Literaturrecherche | Anhang 4-A |
Geben Sie nachfolgend die Suchstrategien für die bibliografische(n) Literaturrecherche(n) an, und zwar getrennt für die einzelnen Recherchen (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.). Für jede durchsuchte Datenbank ist die verwendete Strategie separat darzustellen.
Geben Sie dabei zunächst jeweils den Namen der durchsuchten Datenbank (z.B. EMBASE), die verwendete Suchoberfläche (z.B. DIMDI, Ovid etc.), das Datum der Suche, das Zeitsegment (z.B.: "1980 to 2010 week 50") und die gegebenenfalls verwendeten Suchfilter (mit Angabe einer Quelle) an. Listen Sie danach die Suchstrategie einschließlich der resultierenden Trefferzahlen auf. Orientieren Sie sich bei Ihren Angaben an dem nachfolgenden Beispiel (eine umfassende Suche soll Freitextbegriffe und Schlagwörter enthalten):
| Datenbankname
Suchoberfläche Datum der Suche Zeitsegment Suchfilter | EMBASE
Ovid 08.12.2010 1980 to 2010 week 50 Filter für randomisierte kontrollierte Studien nach Wong 2006 [Quelle 26] - Strategy minimizing difference between sensitivity and specificity |
| # | Suchbegriffe | Ergebnis |
| 1 | Meglitinide/ | 848 |
| 2 | Nateglinide/ | 1686 |
| 3 | Repaglinide/ | 2118 |
| 4 | (glinid* or meglitinid* or nateglinid* or repaglinid*).ab,ti. | 1069 |
| 5 | (starlix or novonorm or novo norm or prandin).ab,ti. | 32 |
| 6 | (105816-04-4 or 135062-02-1).rn. | 2854 |
| 7 | or/1-6 | 3467 |
| 8 | Diabetes mellitus/ | 224164 |
| 9 | Non Insulin dependent Diabetes mellitus/ | 91081 |
| 10 | (diabet* or niddm or t2dm).ab,ti. | 379777 |
| 11 | or/8-10 | 454517 |
| 12 | (random* or double-blind*).tw. | 650136 |
| 13 | placebo*.mp. | 243550 |
| 14 | or/12-13 | 773621 |
| 15 | and/7,11,14 | 719 |
| Suche nach RCT mit dem zu bewertenden Arzneimittel | Anhang 4-A1 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach RCT für indirekte Vergleiche | Anhang 4-A2 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach nicht randomisierten vergleichenden Studien | Anhang 4-A3 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach weiteren Untersuchungen | Anhang 4-A4 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suchstrategien - Suche in Studienregistern | Anhang 4-B: |
Geben Sie nachfolgend die Suchstrategien für die Suche(n) in Studienregistern an. Machen Sie die Angaben getrennt für die einzelnen Recherchen (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.) wie unten angegeben.
Für jedes durchsuchte Studienregister ist eine separate Strategie darzustellen.
Geben Sie dabei jeweils den Namen des durchsuchten Studienregisters (z.B. clinicaltrials.gov), die Internetadresse, unter der das Studienregister erreichbar ist (z.B. http://www.clinicaltrials.gov), das Datum der Suche, die verwendete Suchstrategie und die resultierenden Treffer an. Orientieren Sie sich bei Ihren Angaben an dem nachfolgenden Beispiel:
| Studienregister | clinicaltrials.gov |
| Internetadresse | http://www.clinicaltrials.gov |
| Datum der Suche | 8. Dezember 2010 |
| Suchstrategie | (Starlix OR Novonorm OR Prandin OR Nateglinid OR Repaglinid) [ALL-FIELDS] AND ("Phase II" OR "Phase III" OR "Phase IV") [PHASE] |
| Treffer | 23 |
| Suche nach RCT mit dem zu bewertenden Arzneimittel | Anhang 4-B1 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach RCT für indirekte Vergleiche | Anhang 4-B2 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach nicht randomisierten vergleichenden Studien | Anhang 4-B3 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach weiteren Untersuchungen | Anhang 4-B4 |
<< Angaben des pharmazeutischen Unternehmers >>
| Liste der im Volltext gesichteten und ausgeschlossenen Studien mit Ausschlussgrund | Anhang 4-C: |
Listen Sie nachfolgend die im Volltext gesichteten und ausgeschlossenen Dokumente aus der / den bibliografischen Literaturrecherche(n) auf. Machen Sie die Angaben getrennt für die einzelnen Recherchen (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.) wie unten angegeben. Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard) und nummerieren Sie die Zitate fortlaufend. Geben Sie jeweils einen Ausschlussgrund an und beziehen Sie sich dabei auf die im Abschnitt 4.2.2 genannten Ein- und Ausschlusskriterien.
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach RCT mit dem zu bewertenden Arzneimittel | Anhang 4-C1 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach RCT für indirekte Vergleiche | Anhang 4-C2 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach nicht randomisierten vergleichenden Studien | Anhang 4-C3 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach weiteren Untersuchungen | Anhang 4-C4 |
<< Angaben des pharmazeutischen Unternehmers >>
| Liste der abgebrochenen Studien mit Ausschlussgrund (Suche in Studienregistern) | Anhang 4-D: |
Listen Sie nachfolgend die durch die Studienregistersuche(n) identifizierten, aber ausgeschlossenen Studien auf. Machen Sie die Angaben getrennt für die einzelnen Recherchen (Suche nach RCT mit dem zu bewertenden Arzneimittel, Suche nach RCT für indirekte Vergleiche etc.) wie unten angegeben. Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard) und nummerieren Sie die Zitate fortlaufend. Geben Sie jeweils einen Ausschlussgrund an und beziehen Sie sich dabei auf die im Abschnitt 4.2.2 genannten Ein- und Ausschlusskriterien.
| Suche nach RCT mit dem zu bewertenden Arzneimittel | Anhang 4-D1 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach RCT für indirekte Vergleiche | Anhang 4-D2 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach nicht randomisierten vergleichenden Studien | Anhang 4-D3 |
<< Angaben des pharmazeutischen Unternehmers >>
| Suche nach weiteren Untersuchungen | Anhang 4-D4 |
<< Angaben des pharmazeutischen Unternehmers >>
| Methodik der eingeschlossenen Studien - RCT | Anhang 4-E: |
Beschreiben Sie nachfolgend die Methodik jeder eingeschlossenen, in Abschnitt 4.3.1.1.4 genannten Studie. Erstellen Sie hierfür je Studie eine separate Version der nachfolgend dargestellten Tabelle 4-23 inklusive eines Flow-Charts für den Patientenfluss.
Sollten Sie im Dossier indirekte Vergleiche präsentieren, beschreiben Sie ebenfalls die Methodik jeder zusätzlich in den indirekten Vergleich eingeschlossenen Studie (Abschnitt 4.3.2.1). Erstellen Sie hierfür je Studie eine separate Version der nachfolgend dargestellten Tabelle 4-23 inklusive eines Flow-Charts für den Patientenfluss.
Benennen Sie in der nachfolgenden Tabelle die laufenden Studien aus dem durch die verschiedenen Suchschritte in Abschnitt 4.3.1.1 identifizierten Studienpool.
Tabelle 4-23: (Anhang): Studiendesign und -methodik für Studie <Studienbezeichnung>
| Item a | Charakteristikum | Studieninformation |
| Studienziel | ||
| 2b | Genaue Ziele, Fragestellung und Hypothesen | |
| Methoden | ||
| 3 | Studiendesign | |
| 3a | Beschreibung des Studiendesigns (z.B. parallel, faktoriell) inklusive Zuteilungsverhältnis | |
| 3b | Relevante Änderungen der Methodik nach Studienbeginn (z.B. Ein-/Ausschlusskriterien), mit Begründung | |
| 4 | Probanden / Patienten | |
| 4a | Ein-/Ausschlusskriterien der Probanden / Patienten | |
| 4b | Studienorganisation und Ort der Studiendurchführung | |
| 5 | Interventionen
Präzise Angaben zu den geplanten Interventionen jeder Gruppe und zur Administration etc. | |
| 6 | Zielkriterien | |
| 6a | Klar definierte primäre und sekundäre Zielkriterien, Erhebungszeitpunkte, ggf. alle zur Optimierung der Ergebnisqualität verwendeten Erhebungsmethoden (z.B. Mehrfachbeobachtungen, Training der Prüfer) und ggf. Angaben zur Validierung von Erhebungsinstrumenten | |
| 6b | Änderungen der Zielkriterien nach Studienbeginn, mit Begründung | |
| 7 | Fallzahl | |
| 7a | Wie wurden die Fallzahlen bestimmt? | |
| 7b | Falls notwendig, Beschreibung von Zwischenanalysen und Kriterien für einen vorzeitigen Studienabbruch | |
| 8 | Randomisierung, Erzeugung der Behandlungsfolge | |
| 8a | Methode zur Generierung der zufälligen Zuteilung | |
| 8b | Einzelheiten (z.B. Blockrandomisierung, Stratifizierung) | |
| 9 | Randomisierung, Geheimhaltung der Behandlungsfolge (allocation concealment)
Durchführung der Zuteilung (z.B. nummerierte Behälter; zentrale Randomisierung per Fax / Telefon), Angabe, ob Geheimhaltung bis zur Zuteilung gewährleistet war | |
| 10 | Randomisierung, Durchführung
Wer hat die Randomisierungsliste erstellt, wer nahm die Probanden/Patienten in die Studie auf und wer teilte die Probanden/Patienten den Gruppen zu? | |
| 11 | Verblindung | |
| 11a | Waren a) die Probanden / Patienten und / oder b) diejenigen, die die Intervention / Behandlung durchführten, und / oder c) diejenigen, die die Zielgrößen beurteilten, verblindet oder nicht verblindet, wie wurde die Verblindung vorgenommen? | |
| 11b | Falls relevant, Beschreibung der Ähnlichkeit von Interventionen | |
| 12 | Statistische Methoden | |
| 12a | Statistische Methoden zur Bewertung der primären und sekundären Zielkriterien | |
| 12b | Weitere Analysen, wie z.B. Subgruppenanalysen und adjustierte Analysen | |
| Resultate | ||
| 13 | Patientenfluss (inklusive Flow-Chart zur Veranschaulichung im Anschluss an die Tabelle) | |
| 13a | Anzahl der Studienteilnehmer für jede durch Randomisierung gebildete Behandlungsgruppe, die
| |
| 13b | Für jede Gruppe: Beschreibung von verlorenen und ausgeschlossenen Patienten nach Randomisierung mit Angabe von Gründen | |
| 14 | Aufnahme / Rekrutierung | |
| 14a | Nähere Angaben über den Zeitraum der Studienaufnahme der Probanden / Patienten und der Nachbeobachtung | |
| 14b | Informationen, warum die Studie endete oder beendet wurde | |
| a: nach CONSORT 2010. | ||
Stellen Sie für jede Studie den Patientenfluss in einem Flow-Chart gemäß CONSORT dar.
<< Angaben des pharmazeutischen Unternehmers >>
Tabelle 4-25 (aufgehoben)
| Bewertungsbögen zur Einschätzung von Verzerrungsaspekten | Anhang 4-F: |
Der nachfolgend dargestellte Bewertungsbogen dient der Dokumentation der Einstufung des Potenzials der Ergebnisse für Verzerrungen (Bias). Für jede Studie soll aus diesem Bogen nachvollziehbar hervorgehen, inwieweit die Ergebnisse für die einzelnen Endpunkte als möglicherweise verzerrt bewertet wurden, was die Gründe für die Bewertung waren und welche Informationen aus den Quellen dafür Berücksichtigung fanden.
Der Bogen gliedert sich in zwei Teile:
Für jedes Kriterium sind unter "Angaben zum Kriterium" alle relevanten Angaben aus den Quellen zur Bewertung einzutragen (Stichworte reichen ggf., auf sehr umfangreiche Informationen in den Quellen kann verwiesen werden).
Grundsätzlich sollen die Bögen studienbezogen ausgefüllt werden. Wenn mehrere Quellen zu einer Studie vorhanden sind, müssen die herangezogenen Quellen in der folgenden Tabelle genannt und jeweils mit Kürzeln (z.B. A, B, C ...) versehen werden. Quellenspezifische Angaben im weiteren Verlauf sind mit dem jeweiligen Kürzel zu kennzeichnen.
Hinweis: Der nachfolgend dargestellte Bewertungsbogen ist die Blankoversion des Bogens. Dieser Blankobogen ist für jede Studie heranzuziehen. Im Anschluss daran ist ein Bewertungsbogen inklusive Ausfüllhinweisen abgebildet, der als Ausfüllhilfe dient, aber nicht als Vorlage verwendet werden soll.
Beschreiben Sie nachfolgend die Verzerrungsaspekte jeder eingeschlossenen Studie (einschließlich der Beschreibung für jeden berücksichtigten Endpunkt). Erstellen Sie hierfür je Studie eine separate Version des nachfolgend dargestellten Bewertungsbogens.
Tabelle 4-24 (Anhang): Bewertungsbogen zur Beschreibung von Verzerrungsaspekten für Studie <Studienbezeichnung>
| Bewertungsbögen zur Einschätzung von Verzerrungsaspekten | Anhang 4-G: |
Studie: ___________________________________________
Tabelle:
Liste der für die Bewertung herangezogenen Quellen
| Genaue Benennung der Quelle | Kürzel |
A: Verzerrungsaspekte auf Studienebene:
Einstufung als randomisierte Studie
[ ] ja ℜ→ Bewertung der Nummer 1 und 2 für randomisierte Studien
[ ] nein: Bewertung der Punkte 1 und 2 für nicht randomisierte Studien
ℜ→ Bewertung der Nummer 1 und 2 für nicht randomisierte Studien
Angaben zum Kriterium:
...............................................................................................................................................................
...............................................................................................................................................................
1. für randomisierte Studien:
Adäquate Erzeugung der Randomisierungssequenz
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
für nicht randomisierte Studien:
Zeitliche Parallelität der Gruppen
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
2. für randomisierte Studien:
Verdeckung der Gruppenzuteilung ("allocation concealment")
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
für nicht randomisierte Studien:
Vergleichbarkeit der Gruppen bzw. adäquate Berücksichtigung von prognostisch relevanten Faktoren
[ ] ja:
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
3. Verblindung von Patienten und Behandlern
Patient
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
behandelnde bzw. weiterbehandelnde Personen:
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
4. Ergebnisunabhängige Berichterstattung aller relevanten Endpunkte
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
5. Keine sonstigen (endpunktübergreifenden) Aspekte, die zu Verzerrungen führen können
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse auf Studienebene (ausschließlich für randomisierte Studien durchzuführen):
[ ] niedrig
[ ] hoch
Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
B: Verzerrungsaspekte auf Endpunktebene pro Endpunkt:
Endpunkt: ..............................................................................
1. Verblindung der Endpunkterheber
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung
...............................................................................................................................................................
...............................................................................................................................................................
2. Adäquate Umsetzung des ITT-Prinzips
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
3. Ergebnisunabhängige Berichterstattung dieses Endpunkts alleine
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
4. Keine sonstigen (endpunktspezifischen) Aspekte, die zu Verzerrungen führen können
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse des Endpunkts (ausschließlich für randomisierte Studien durchzuführen):
[ ] niedrig
[ ] hoch
Begründung für die Einstufung:
...............................................................................................................................................................
...............................................................................................................................................................
Hinweis: Der nachfolgend dargestellte Bewertungsbogen mit Ausfüllhinweisen dient nur als Ausfüllhilfe für den Blankobogen. Er soll nicht als Vorlage verwendet werden.
Bewertungsbogen zur Beschreibung von Verzerrungsaspekten (Ausfüllhilfe)
Anhand der Bewertung der folgenden Kriterien soll das Ausmaß möglicher Ergebnisverzerrungen eingeschätzt werden (A: endpunktübergreifend; B: endpunktspezifisch).
A Verzerrungsaspekte auf Studienebene:
Einstufung als randomisierte Studie
[ ] ja ℜ→ Bewertung der Punkte 1 und 2 für randomisierte Studien
[ ] nein: Aus den Angaben geht klar hervor, dass es keine randomisierte Zuteilung gab, oder die Studie ist zwar als randomisiert beschrieben, es liegen jedoch Anzeichen vor, die dem widersprechen (z.B. wenn eine alternierende Zuteilung erfolgte). Eine zusammenfassende Bewertung der Verzerrungsaspekte soll für nicht randomisierte Studien nicht vorgenommen werden.
ℜ→ Bewertung der Punkte 1 und 2 für nicht randomisierte Studien
Angaben zum Kriterium:___________________________
1. für randomisierte Studien:
Adäquate Erzeugung der Randomisierungssequenz
[ ] ja: Die Gruppenzuteilung erfolgte rein zufällig, und die Erzeugung der Zuteilungssequenz ist beschrieben und geeignet (z.B. computergenerierte Liste).
[ ] unklar: Die Studie ist zwar als randomisiert beschrieben, die Angaben zur Erzeugung der Zuteilungssequenz fehlen jedoch oder sind ungenügend genau.
[ ] nein: Die Erzeugung der Zuteilungssequenz war nicht adäquat.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:______________________________
für nicht randomisierte Studien:
Zeitliche Parallelität der Gruppen
[ ] ja: Die Gruppen wurden zeitlich parallel verfolgt.
[ ] unklar: Es finden sich keine oder ungenügend genaue diesbezügliche Angaben.
[ ] nein: Die Gruppen wurden nicht zeitlich parallel verfolgt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:_______________
2. für randomisierte Studien:
Verdeckung der Gruppenzuteilung ("allocation concealment")
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Die Angaben der Methoden zur Verdeckung der Gruppenzuteilung fehlen oder sind ungenügend genau.
[ ] nein: Die Gruppenzuteilung erfolgte nicht verdeckt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:_____________
für nicht randomisierte Studien:
Vergleichbarkeit der Gruppen bzw. adäquate Berücksichtigung von prognostisch relevanten Faktoren
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Die Angaben zur Vergleichbarkeit der Gruppen bzw. zur Berücksichtigung von Einflussgrößen fehlen oder sind ungenügend genau.
[ ] nein: Die Vergleichbarkeit ist nicht gegeben und diese Unterschiede werden in den Auswertungen nicht adäquat berücksichtigt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:____________
3. Verblindung von Patienten und behandelnden Personen
Patient:
[ ] ja: Die Patienten waren verblindet.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass die Patienten nicht verblindet waren.
Medizinischer Nutzen, medizinischer Zusatznutzen, Patientengruppen mit therap. bedeutsamem Zusatznutzen Angaben zum Kriterium; obligate Begründung für die Einstufung:
behandelnde bzw. weiterbehandelnde Personen:
[ ] ja: Das behandelnde Personal war bzgl. der Behandlung verblindet. Wenn es, beispielsweise bei chirurgischen Eingriffen, offensichtlich nicht möglich ist, die primär behandelnde Person (z.B. Chirurg) zu verblinden, wird hier beurteilt, ob eine angemessene Verblindung der weiteren an der Behandlung beteiligten Personen (z.B. Pflegekräfte) stattgefunden hat.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass die behandelnden Personen nicht verblindet waren.
Angaben zum Kriterium; obligate Begründung für die Einstufung:___________
4. Ergebnisunabhängige Berichterstattung aller relevanten Endpunkte
Falls die Darstellung des Ergebnisses eines Endpunkts von seiner Ausprägung (d. h. vom Resultat) abhängt, können erhebliche Verzerrungen auftreten. Je nach Ergebnis kann die Darstellung unterlassen worden sein (a), mehr oder weniger detailliert (b) oder auch in einer von der Planung abweichenden Weise erfolgt sein (c).
Beispiele zu a und b:
Beispiele zu c: Ergebnisgesteuerte Auswahl in der Auswertung verwendeter
Zur Einschätzung einer potenziell vorhandenen ergebnisgesteuerten Berichterstattung sollten folgende Punkte - sofern möglich - berücksichtigt werden:
Anzumerken ist, dass Anzeichen für eine ergebnisgesteuerte Darstellung eines Endpunkts zu Verzerrungen der Ergebnisse der übrigen Endpunkte führen kann, da dort ggf. auch mit einer selektiven Darstellung gerechnet werden muss. Insbesondere bei Anzeichen dafür, dass die Ergebnisse einzelner Endpunkte selektiv nicht berichtet werden, sind Verzerrungen für die anderen Endpunkte möglich. Eine von der Planung abweichende selektive Darstellung des Ergebnisses eines Endpunkts führt jedoch nicht zwangsläufig zu einer Verzerrung der anderen Endpunkte; in diesem Fall ist die ergebnisgesteuerte Berichterstattung endpunktspezifisch unter Punkt B.3 (siehe unten) einzutragen.
Des Weiteren ist anzumerken, dass die Berichterstattung von unerwünschten Ereignissen üblicherweise ergebnisabhängig erfolgt (es werden nur Häufungen / Auffälligkeiten berichtet) und dies nicht zur Verzerrung anderer Endpunkte führt.
[] ja: Eine ergebnisgesteuerte Berichterstattung ist unwahrscheinlich, unklar: Die verfügbaren Angaben lassen eine Einschätzung nicht zu.
[] nein: Es liegen Anzeichen für eine ergebnisgesteuerte Berichterstattung vor, die das Verzerrungspotenzial aller relevanten Endpunkte beeinflusst.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:___________
5. Keine sonstigen (endpunktübergreifenden) Aspekte, die zu Verzerrung führen können
z.B.:
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:____________
Einstufung des Verzerrungspotenzials der Ergebnisse auf Studienebene (ausschließlich für randomisierte Studien durchzuführen):
Die Einstufung des Verzerrungspotenzials der Ergebnisse erfolgt unter Berücksichtigung der einzelnen Bewertungen der vorangegangenen Punkte A.1 bis A.5. Eine relevante Verzerrung bedeutet hier, dass sich die Ergebnisse bei Behebung der verzerrenden Aspekte in ihrer Grundaussage verändern würden.
[ ] niedrig: Es kann mit großer Wahrscheinlichkeit ausgeschlossen werden, dass die Ergebnisse durch diese endpunktübergreifenden Aspekte relevant verzerrt sind.
[ ] hoch: Die Ergebnisse sind möglicherweise relevant verzerrt.
Begründung für die Einstufung:___________________________
B Verzerrungsaspekte auf Endpunktebene pro Endpunkt:
Die folgenden Punkte B1 bis B4 dienen der Einschätzung der endpunktspezifischen Aspekte für das Ausmaß möglicher Ergebnisverzerrungen. Diese Punkte sollten i. d. R. für jeden relevanten Endpunkt separat eingeschätzt werden (ggf. lassen sich mehrere Endpunkte gemeinsam bewerten, z.B. Endpunkte zu unerwünschten Ereignissen).
Endpunkt:__________________________
1. Verblindung der Endpunkterheber
Für den Endpunkt ist zu bestimmen, ob das Personal, welches die Zielkriterien erhoben hat, bzgl. der Behandlung verblindet war.
In manchen Fällen kann eine Verblindung auch gegenüber den Ergebnissen zu anderen Endpunkten (z.B. typischen unerwünschten Ereignissen) gefordert werden, wenn die Kenntnis dieser Ergebnisse Hinweise auf die verabreichte Therapie gibt und damit zu einer Entblindung führen kann.
[ ] ja: Der Endpunkt wurde verblindet erhoben.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass keine verblindete Erhebung erfolgte.
Angaben zum Kriterium; obligate Begründung für die Einstufung:
2. Adäquate Umsetzung des ITT-Prinzips
Kommen in einer Studie Patienten vor, die die Studie entweder vorzeitig abgebrochen haben oder wegen Protokollverletzung ganz oder teilweise aus der Analyse ausgeschlossen wurden, so sind diese ausreichend genau zu beschreiben (Abbruchgründe, Häufigkeit und Patientencharakteristika pro Gruppe) oder in der statistischen Auswertung angemessen zu berücksichtigen (i. d. R. ITT-Analyse, siehe Äquivalenzstudien). Bei einer ITT("intention to treat`)-Analyse werden alle randomisierten Patienten entsprechend ihrer Gruppenzuteilung ausgewertet (ggf. müssen fehlende Werte für die Zielkriterien in geeigneter Weise ersetzt werden). Zu beachten ist, dass in Publikationen der Begriff ITT nicht immer in diesem strengen Sinne Verwendung findet. Es werden häufig nur die randomisierten Patienten ausgewertet, die die Therapie zumindest begonnen haben und für die mindestens ein PostBaseline-Wert erhoben worden ist ("full analysis set`). Dieses Vorgehen ist in begründeten Fällen Guideline-konform, eine mögliche Verzerrung sollte jedoch, insbesondere in nicht verblindeten Studien, überprüft werden. Bei Äquivalenz- und Nichtunterlegenheitsstudien ist es besonders wichtig, dass solche Patienten sehr genau beschrieben werden und die Methode zur Berücksichtigung dieser Patienten transparent dargestellt wird.
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Aufgrund unzureichender Darstellung ist der adäquate Umgang mit Protokollverletzern un Lost-to-follow-up-Patienten nicht einschätzbar.
[ ] nein: Keines der unter "ja" genannten drei Merkmale trifft zu.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
3. Ergebnisunabhängige Berichterstattung dieses Endpunkts alleine
Beachte die Hinweise zu Punkt A.4!
[ ] ja: Eine ergebnisgesteuerte Berichterstattung ist unwahrscheinlich.
[ ] unklar: Die verfügbaren Angaben lassen eine Einschätzung nicht zu.
[ ] nein: Es liegen Anzeichen für eine ergebnisgesteuerte Berichterstattung vor.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:____________
4. Keine sonstigen (endpunktspezifischen) Aspekte, die zu Verzerrungen führen können
z.B.
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
Einstufung des Verzerrungspotenzials der Ergebnisse des Endpunkts (ausschließlich für randomisiert Studien durchzuführen):
Die Einstufung des Verzerrungspotenzials erfolgt unter Berücksichtigung der einzelnen Bewertungen der vorangegangenen endpunktspezifischen Punkte B.1 bis B.4 sowie der Einstufung des Verzerrungspotenzials auf Studienebene. Falls die endpunktübergreifende Einstufung mit "hoch" erfolgte, ist das Verzerrungspotenzial für den Endpunkt i. d. R. auch mit "hoch" einzuschätzen. Eine relevante Verzerrung bedeutet hier, dass sich die Ergebnisse bei Behebung der verzerrenden Aspekte in ihrer Grundaussage verändern würden.
[ ] niedrig: Es kann mit großer Wahrscheinlichkeit ausgeschlossen werden, dass die Ergebnisse für diesen Endpunkt durch die endpunktspezifischen sowie endpunktübergreifenden Aspekte relevant verzerrt sind.
[ ] hoch: Die Ergebnisse sind möglicherweise relevant verzerrt.
Begründung für die Einstufung:______________________________
Tabelle 4-26 (aufgehoben)
Tabelle 4-27 (Anhang): (aufgehoben)
| Vorlage zur Abgabe einer schriftlichen Stellungnahme zur Nutzenbewertung nach § 35a SGB V | Anlage III (zum 5. Kapitel) |
| Datum | < TT.MM.JJJJ > |
| Stellungnahme zu | < Wirkstoff/Markenname > |
| Stellungnahme von | < Firma/Institution > |
Die Stellungnahme inkl. der Literatur im Volltext und weiterer Anhänge ist dem G-BA elektronisch zu übermitteln. Das ausgefüllte Dokument ist dem G-BA im Word-Format einzureichen.
Bitte verwenden Sie zur Auflistung der zitierten Literatur eine nummerierte Referenzliste und behalten Sie diese Nummerierung bei der Benennung der Dateien bei.
1. Stellungnahme zu allgemeinen Aspekten
Stellungnehmer:
| Allgemeine Anmerkung | Ergebnis nach Prüfung (wird vom G-BA ausgefüllt) |
(Bitte fügen Sie weitere Zeilen an, falls dies notwendig sein sollte.)
2. Stellungnahme zu spezifischen Aspekten
Stellungnehmer
| Seite, Zeile | Stellungnahme mit Begründung sowie vorgeschlagene Änderung
Falls Literaturstellen zitiert werden, müssen diese eindeutig benannt und im Anhang im Volltext beigefügt werden. | Ergebnis nach Prüfung (wird vom G-BA ausgefüllt) |
| Anmerkung: Vorgeschlagene Änderung: | ||
| Anmerkung: Vorgeschlagene Änderung: | ||
| Anmerkung: Vorgeschlagene Änderung: |
(Bitte fügen Sie weitere Zeilen an, falls dies notwendig sein sollte.)
Literaturverzeichnis
| Gebührenordnung nach § 35a Absatz 7 Satz 5 in Verbindung mit 5. Kapitel § 7 VerfO | Anlage IV 11b 16f (zum 5. Kapitel) |
§ 1 Regelungsbereich
(1) Der Gemeinsame Bundesausschuss erhebt für Beratungen nach 5. Kapitel § 7 VerfO Gebühren nach dieser Gebührenordnung.
(2) Eine gebührenpflichtige Beratung erfolgt aufgrund einer schriftlichen Anforderung nach 5. Kapitel § 7 VerfO. Als Beratungsleistungen gelten auch schriftliche Auskünfte, die sich auf einen Beratungsgegenstand nach § 35a Absatz 1 SGB V beziehen.
§ 2 Gebühren bei Rücknahme der Beratungsanforderung
Wird eine Beratungsanforderung zurückgenommen, nachdem mit der sachlichen Bearbeitung begonnen worden ist, so kann sich die Gebühr bis zu einem Viertel der vorgesehenen Gebühr ermäßigen oder es kann von ihrer Erhebung abgesehen werden, wenn dies der Billigkeit entspricht.
§ 3 Höhe der Gebühren
(1) Die Beratungsleistungen werden nach Maßgabe der folgenden Gebühren abgerechnet:
Allgemeine Anfragen zur Verfahrensordnung oder im Aufwand vergleichbare sonstige Anfragen
Anfragen zu den vorzulegenden Unterlagen und Studien zur Bewertung des Nutzens von Arzneimitteln und Zusammenstellung der Dossierunterlagen nach 5. Kapitel § § 5 und 9 VerfO oder im Aufwand vergleichbare sonstige Anfragen
Anfragen zu einer zweckmäßigen Vergleichstherapie oder im Aufwand vergleichbare sonstige Anfragen
(2) Die Beratung wird von der Zahlung eines Vorschusses in Höhe von 5.000 Euro abhängig gemacht.
§ 4 Erhöhungen und Ermäßigungen
(1) Hat die Beratung im Einzelfall einen außergewöhnlich hohen Aufwand erfordert, so kann die Gebühr bis auf das Doppelte der vorgesehenen Gebühr erhöht werden. Der Gebührenschuldner ist zu hören, wenn mit einer Erhöhung der Gebühren zu rechnen ist.
(2) Die Gebühr kann bis auf die Hälfte der vorgesehenen Gebühr ermäßigt werden, wenn der mit der Beratung verbundene Personal- und Sachaufwand einerseits und die Bedeutung des Arzneimittels für den pharmazeutischen Unternehmer andererseits dies rechtfertigen.
§ 5 Festsetzung der Gebühren, Fälligkeit
(1) Die Gebühren werden durch schriftlichen Bescheid festgesetzt.
(2) Die Gebühren werden mit der Bekanntgabe der Gebührenentscheidung an den Gebührenschuldner fällig, wenn nicht der Gemeinsame Bundesausschuss einen späteren Zeitpunkt bestimmt.
§ 6 Säumniszuschlag
(1) Werden bis zum Ablauf eines Monats nach dem Fälligkeitstag Gebühren nicht entrichtet, so kann für jeden angefangenen Monat der Säumnis ein Säumniszuschlag von eins vom Hundert des rückständigen Betrages erhoben werden, wenn dieser 50 Euro übersteigt.
(2) Absatz 1 gilt nicht, wenn Säumniszuschläge nicht rechtzeitig entrichtet werden.
(3) Für die Berechnung des Säumniszuschlages wird der rückständige Betrag auf volle 50 Euro nach unten abgerundet.
(4) Als Tag, an dem eine Zahlung entrichtet worden ist, gilt
§ 7 Rechtsbehelf
Die Entscheidung über die Gebühren kann mit dem Rechtsbehelf des Widerspruchs angefochten werden.
| Antrag auf Freistellung von der Nutzenbewertung nach § 35a Absatz 1a SGB V in Verbindung mit § 15 VerfO wegen Geringfügigkeit für Fertigarzneimittel | Anlage V 18c (zum 5. Kapitel) |
1 Pharmazeutischer Unternehmer
| a) Name des pharmazeutischen Unternehmers | << >> |
| b) Anschrift | << >> |
Ansprechpartner beim pharmazeutischen Unternehmer
| a) Name | << >> |
| b) Abteilung und Funktion | << >> |
| c) Adresse | << >> |
| d) E-Mail | << >> |
| e) Telefon- und Telefaxnummer | << >> |
2 Allgemeine Angaben zum Arzneimittel
| a) Wirkstoff | << >> * |
| b) Markenname | << >> * |
| c) Darreichungsform | << >> * |
| d) ATC-Code | << >> * |
| e) Falls "positive opinion" vorhanden, hier Datum angeben | << Datum >> |
| f) (voraussichtliches) Datum der Zulassungserteilung | << Datum >> |
| g) Orphan designation erhalten/beantragt | << >> * |
| h) Anwendungsgebiete) | << >> * |
| *) Falls das Arzneimittel bereits zugelassen, aber noch nicht im Verkehr ist: Wortlaut lt. Fachinformation. Falls für das Arzneimittel eine "positive opinion" vorhanden ist: Wortlaut lt. positive opinion. Falls weder die Zulassung noch eine "positive opinion" vorliegen: Wortlaut lt. Zulassungsantrag. | |
3 Prävalenz und Inzidenz der Erkrankung in Deutschland
Geben Sie eine Schätzung für die Prävalenz und Inzidenz der Erkrankung in Deutschland an. Geben Sie dabei jeweils einen üblichen Populationsbezug und zeitlichen Bezug an. Bei Vorliegen alters- oder geschlechtsspezifischer Unterschiede oder von Unterschieden in anderen Gruppen sollen die Angaben auch für Altersgruppen, Geschlecht bzw. andere Gruppen getrennt erfolgen. Verwenden Sie hierzu eine tabellarische Darstellung. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie nachfolgend an, ob und, wenn ja, welche wesentlichen Änderungen hinsichtlich Prävalenz und Inzidenz der Erkrankung in Deutschland innerhalb der nächsten fünf Jahre zu erwarten sind. Verwenden Sie hierzu eine tabellarische Darstellung. Benennen Sie die zugrunde gelegten Quellen.
<< Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle 3-1 die Anzahl der Patienten in der GKV an, für die eine Behandlung mit dem Arzneimittel in dem Anwendungsgebiet (gemäß Nummer 2 Buchstabe h) infrage kommt (Zielpopulation). Die Angaben sollen sich, sofern geeignet, auf einen Jahreszeitraum beziehen.
Tabelle 3-1 Anzahl der GKV-Patienten in der Zielpopulation
| Bezeichnung des Arzneimittels | Anzahl der GKV-Patienten in der Zielpopulation |
Begründen Sie die Angaben in Tabelle 3-1 unter Nennung der verwendeten Quellen. Ziehen Sie dabei auch die Angaben zu Prävalenz und Inzidenz (wie oben angegeben) heran.
<< Angaben des pharmazeutischen Unternehmers >>
4 Anzahl der Patienten, die ambulant und stationär behandelt werden
Geben Sie in der nachfolgenden Tabelle 4-1 an, wie viele Patienten voraussichtlich behandelt werden und begründen Sie Ihre Angaben. Die Angaben können separat für stationär und ambulant behandelte Patienten dargestellt werden, sofern dies relevant ist.
Tabelle 4-1 Anzahl der GKV-Patienten, die behandelt werden.
| Bezeichnung des Arzneimittels | Anzahl der GKV-Patienten |
Begründen Sie die Angaben in Tabelle 4-1 unter Nennung der verwendeten Quellen. Alle Annahmen und Kalkulationsschritte für die Herleitung der Patientenzahlen sind darzustellen und zu begründen. Die Berechnungen müssen auf Basis dieser Angaben nachvollzogen werden können. Machen Sie auch Angaben zur Unsicherheit, z.B. Angabe einer Spanne.
<< Angaben des pharmazeutischen Unternehmers >>
5 Erwartete Kosten für die gesetzliche Krankenversicherung
Bitte beziehen Sie sich im Folgenden auf die Anzahl der GKV-Patienten, für die eine Behandlung mit dem Arzneimittel in der Zielpopulation infrage kommt.
5.1 Angaben zur Behandlungsdauer
Geben Sie in der nachfolgenden Tabelle 5-1.a an, nach welchem Behandlungsmodus (z.B. kontinuierlich, in Zyklen, je Episode, bei Bedarf) das Arzneimittel eingesetzt wird. Geben Sie die Anzahl der Behandlungen (z.B. Zyklen, Episoden) pro Patient pro Jahr an (bei kontinuierlicher Behandlung ist in der Spalte "Anzahl Behandlungen pro Patient pro Jahr" "kontinuierlich" anzugeben). Geben Sie jeweils auch die Behandlungsdauer in Tagen an (bei kontinuierlicher Behandlung: 365 Tage bei täglicher Behandlung, 182 bei zweitäglicher Behandlung etc.; sonst Angabe als Mittelwert und Spannweite). Sofern sich diese Angaben für verschiedene Populationen und Patientengruppen unterscheiden (z.B. bei verschiedenen Anwendungsgebieten oder Schweregraden), fügen Sie für jede Population bzw. Patientengruppe eine neue Zeile ein und geben Sie deren Größen) und Anteil (%) an der Zielpopulation an.
Tabelle 5-1.a: Angaben zum Behandlungsmodus
| Arzneimittel | Bezeichnung der Population bzw. Patientengruppe | Größe(n) der Population bzw. Patientengruppe und deren Anteil (%) an der Zielpopulation | Behandlungsmodus | Anzahl Behandlungen pro Patient pro Jahr | Behandlungsdauer je Behandlung (Tage) |
Begründen Sie die Angaben in Tabelle 5-1.a unter Nennung der verwendeten Quellen. << Angaben des pharmazeutischen Unternehmers >>
Geben Sie in der nachfolgenden Tabelle 5-1.b die Behandlungstage pro Patient pro Jahr für das Arzneimittel an. Die Behandlungstage pro Patient pro Jahr ergeben sich aus der Anzahl der Behandlungen pro Patient pro Jahr und der Behandlungsdauer je Behandlung (siehe Tabelle 5-1.a).
Tabelle 5-1.b: Behandlungstage pro Patient pro Jahr
| Arzneimittel | Bezeichnung der Population bzw. Patientengruppe | Behandlungstage pro Patient pro Jahr |
5.2 Angaben zum Verbrauch für das Arzneimittel
Geben Sie in der nachfolgenden Tabelle 5-2 den Jahresdurchschnittsverbrauch pro Patient für das Arzneimittel pro Anwendungsgebiet und Ausprägung* an.
Tabelle 5-2: Jahresdurchschnittsverbrauch pro Patient
| Arzneimittel | Jahresdurchschnittsverbrauch pro Patient pro Anwendungsgebiet und Ausprägung * |
| *) Ausprägung ist die Kombination aus Wirkstärke, Darreichungsform und Packungsgröße. | |
Begründen Sie die Angaben in Tabelle 5-2 unter Nennung der verwendeten Quellen. Nehmen Sie ggf. Bezug auf andere Verbrauchsmaße, die im Anwendungsgebiet gebräuchlich sind (z.B. IU [ International Unit], Dosierung je Quadratmeter Körperoberfläche, Dosierung je Kilogramm Körpergewicht).
<< Angaben des pharmazeutischen Unternehmers >>
5.3 Angaben zu Kosten des Arzneimittels
Geben Sie in Tabelle 5-3 an, wie hoch die Apothekenabgabepreise für das Arzneimittel sind. Falls keine Angaben zu den Apothekenabgabepreisen gemacht werden können, sind andere geeignete Angaben zu den Preisen und zu den erwarteten Ausgaben für GKV-Patienten zu machen.
Tabelle 5-3: Kosten des Arzneimittels
| Arzneimittel | Apothekenabgabepreis oder andere geeignete Angaben in Euro nach Wirkstärke, Darreichungsform und Packungsgröße |
Begründen Sie die Angaben in Tabelle 5-3 unter Nennung der verwendeten Quellen. << Angaben des pharmazeutischen Unternehmers >>
5.4 Angaben zu Jahrestherapiekosten
Geben Sie in Tabelle 5-4 die Jahrestherapiekosten für die GKV durch Zusammenführung der in Tabelle 5.1 bis 5.3 entwickelten Daten an. Weisen Sie die Jahrestherapiekosten sowohl bezogen auf einen einzelnen Patienten als auch für die GKV insgesamt aus. Die Berechnung ist darzustellen.
Tabelle 5-4: Jahrestherapiekosten für die GKV für das Arzneimittel pro Patient und insgesamt für die Zielpopulation
| Arzneimittel | Jahrestherapiekosten pro Anwendungsgebiet und Ausprägung* des Arzneimittels pro Patient in Euro | Jahrestherapiekosten GKV insgesamt in Euro |
| *) Ausprägung ist die Kombination aus Wirkstärke, Darreichungsform und Packungsgröße. | ||
6 Informationsbeschaffung
6.1 Beschreibung der Informationsbeschaffung für die Nummern 3 bis 5
Erläutern Sie das Vorgehen zur Identifikation der in den Nummern 3 bis 5 genannten Quellen (Informationsbeschaffung). Sofern erforderlich, können Sie zur Beschreibung der Informationsbeschaffung weitere Quellen benennen.
<< Angaben des pharmazeutischen Unternehmers >>
6.2 Referenzliste für die Nummern 3 bis 5
Benennen Sie nachfolgend alle Quellen (z.B. Publikationen, gegebenenfalls vorläufige Fachinformation), die Sie in den Nummern 3 bis 5 angegeben haben. Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Fügen Sie als Anhang zu diesem Dokument alle zitierten Quellen als Volltext (in PDF) an. Alle Dateien sind wie folgt zu benennen: #Zitat-Nr#_#Erstautor#_#JJJJ#.pdf
<< Angaben des pharmazeutischen Unternehmers >>
| Dossier zur Nutzenbewertung gemäß § 35a SGB V (für mit Festbetragsarzneimitteln pharmakologisch-therapeutisch vergleichbare Arzneimittel) | Anlage VI (zum 5. Kapitel) |
Dokumentvorlage, Version vom 18.04.2013
Dossier zur Nutzenbewertung gemäß § 35a SGB V
(für mit Festbetragsarzneimitteln pharmakologisch-therapeutisch vergleichbare Arzneimittel)
<<Wirkstoff>> (<<Handelsname>>)
<<Pharmazeutischer Unternehmer>>
Stand: <<TT.MM.JJJJ >>
Abbildungsverzeichnis
Tabellenverzeichnis
Tabelle 1-1: Für das Dossier verantwortliches pharmazeutisches Unternehmen
Tabelle 1-2: Zuständige Kontaktperson des für das Dossier verantwortlichen pharmazeutischen Unternehmens
Tabelle 1-3: Zulassungsinhaber des zu bewertenden Arzneimittels
Tabelle 1-4: Allgemeine Angaben zum zu bewertenden Arzneimittel
Tabelle 1-5: Pharmazentralnummern und Zulassungsnummern des zu bewertenden Arzneimittels
Tabelle 1-6: Angaben zur Festbetragsgruppe
Tabelle 3-1: Charakterisierung der eingeschlossenen Studien - RCT mit dem zu bewertenden Arzneimittel
Tabelle 3-2: Tabellarische Darstellung der Bewertung der Treffer in der/den Recherche(n)
Abkürzungsverzeichnis
| Abkürzung | Bedeutung |
| ATC-Code | Anatomisch-Therapeutisch-Chemischer Code |
| CONSORT | Consolidated Standards of Reporting Trials |
| DDD | Defined Daily Dose |
| DIMDI | Deutsches Institut für Medizinische Dokumentation |
| EG | Europäische Gemeinschaft |
| EPAR | European Public Assessment Report |
| EU | Europäische Union |
| G-BA | Gemeinsamer Bundesausschuss |
| GKV | Gesetzliche Krankenversicherung |
| ITT | Intention to treat |
| IU | International Unit |
| PZN | Pharmazentralnummer |
| RCT | Randomized Controlled Trial |
| SGB | Sozialgesetzbuch |
| WHO | World Health Organization |
Hinweise zur Erstellung und Einreichung des Dossiers
Gliederung des Dossiers
Das Dossier gliedert sich in drei Abschnitte und einen Anhang. Der erste Abschnitt enthält administrative Informationen zum für das Dossier verantwortlichen pharmazeutischen Unternehmer, zum Zulassungsinhaber, zu dem zu bewertenden Arzneimittel und zu der Festbetragsgruppe mit pharmakologisch-therapeutisch vergleichbaren Arzneimitteln. Die Abschnitte 2 und 3 beinhalten die Begründung und die entsprechenden Nachweise für die therapeutische Verbesserung des zu bewertenden Arzneimittels im Vergleich zu anderen Arzneimitteln derselben Festbetragsgruppe.
In den Anlagen sind Dokumente enthalten, die für die Aussagen in den Abschnitten 2 und 3 herangezogen werden, sowie eine Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers.
Erstellung der Dokumente
Für das Dossier werden die Dokumentvorlagen für das Standardtextverarbeitungsprogramm "Microsoft Word" auf der Website des Gemeinsamen Bundesausschusses (http://www.gba.de) bereitgestellt. Die Dokumentvorlagen sind bei der Erstellung des Dossiers zu verwenden. Die Struktur der Dokumente einschließlich der Benennung der Abschnitte und Tabellen soll nur angepasst werden, wenn an der entsprechenden Stelle in der Dokumentvorlage darauf hingewiesen wird.
Die Dokumente sind in deutscher Sprache zu erstellen.
Folgende Elemente sind in den Dokumentvorlagen enthalten:
Die Elemente sollen bei der Erstellung des Dossiers nicht aus den Dokumenten entfernt werden. Ausnahmen sind Elemente, bei denen in den Dokumentvorlagen selbst an der jeweiligen Stelle darauf verwiesen wird, dass sie fragestellungsbezogen anzupassen sind (z.B. Tabellenüberschriften), und Beispielzeilen in Tabellen (diese sollen überschrieben werden).
Stellen, an denen Platzhalter hinterlegt sind, sind grundsätzlich mit Angaben des pharmazeutischen Unternehmers zu füllen, es sei denn, dass explizit darauf verwiesen wird, dass dies nur in bestimmten Fällen erforderlich ist.
Die Felder auf dem Deckblatt sollen wie folgt gefüllt werden:
Im Dokument verwendete Abkürzungen sind in das Abkürzungsverzeichnis aufzunehmen. Sofern Sie für Ihre Ausführungen Abbildungen oder Tabellen verwenden, sind diese im Abbildungs- bzw. Tabellenverzeichnis aufzuführen.
Nach Fertigstellung der Dokumente sind von diesen PDF-Dateien zu erstellen. Die PDF-Dokumente müssen navigierbar sein, d. h., Verweise auf Abschnitte, Abbildungen und Tabellen innerhalb des jeweiligen Dokuments müssen als klickbare Verlinkungen enthalten sein. Auch die Verweise in den Verzeichnissen (Abbildungs-, Inhalts- und Tabellenverzeichnis) müssen klickbare Verlinkungen zu den entsprechenden Abbildungen, Abschnitten bzw. Tabellen darstellen. Bei der Erstellung der PDF-Dateien ist darauf zu achten, dass Seitenzahlen, Beschriftungen von Abbildungen und Tabellen, Querverweise auf Abbildungen und Tabellen sowie Verzeichnisse (Inhaltsverzeichnis, Tabellenverzeichnis, Abbildungsverzeichnis) im PDF-Dokument richtig dargestellt sind. Die PDF-Dateien dürfen keine Wasserzeichen enthalten und nicht geschützt werden; die Dokumente müssen elektronisch kommentierbar und die Inhalte elektronisch entnehmbar sein.
Ablage und Benennung der Dateien
Die Einreichung des Dossiers hat elektronisch zu erfolgen. Als Datenträger ist eine Digital Versatile Disc (DVD) zu verwenden. Die Datenträger dürfen nicht kopiergeschützt sein. Das Dossier ist in zweifacher Ausfertigung einzureichen. Für alle einzureichenden Dateien gilt, dass diese nicht geschützt sein dürfen, d.h., sie müssen ohne Kennworteingabe lesbar, speicherbar und druckbar sein. Die Dokumente können in deutscher oder englischer Sprache beigelegt werden. Die Checkliste zur Prüfung der formalen Vollständigkeit ist in deutscher Sprache zu erstellen.
Alle Dokumente (Darlegung der Rechercheergebnisse, Bewertung der relevanten Publikationen, Volltexte) sind in elektronisch lesbarer Form im Word- oder PDF-Format zu übermitteln.
Die DVD enthält zwei Unterverzeichnisse, mit weiteren Unterordnern

Die nachfolgende Liste zeigt in der Übersicht, welche Dokumente in den Unterordnern abzulegen sind.
Festbetragsarzneimitteln pharmakologisch-therapeutisch vergleichbare
Arzneimittel
Für die Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers wird auf der Website des Gemeinsamen Bundesausschusses (http://www.gba.de) eine Dokumentvorlage bereitgestellt, die zu verwenden ist. Nach Fertigstellung der Checkliste ist von dieser eine PDF-Datei zu erstellen.
Es kann notwendig sein, dass einzelne Dokumente an verschiedenen Stellen auf der DVD abgelegt werden müssen. Optional kann auf die Mehrfachablage von Dokumenten verzichtet werden. Das jeweilige Dokument ist dann einmalig an einer korrekten Stelle abzulegen. An den übrigen Stellen sind PDF-Dokumente abzulegen, in denen formlos auf den Ablageort des eigentlichen Dokuments innerhalb der DVD-Struktur verwiesen wird.
Hinweis: Zusätzlich zum Dossier ist das Original einer beglaubigten Kopie des erteilten Patentes in Papierform einzureichen.
Benennung der Dateien
| Volltexte | #Zitat-Nr#_#Erstautor#_#JJJJ#.pdf |
| Betriebs- und Geschäftsgeheimnisse | #Kennzeichnung_B-und-G# |
| Dossier | #JJJJ-MM-TT#_Dossier_#Wirkstoff#.pdf
Dabei ist "#JJJJ-MM-TT#" jeweils durch den Stand des Dossiers und "#Wirkstoff#" durch den Namen des zu bewertenden Wirkstoffs zu ersetzen. |
| Checkliste | Checkliste-Vollstaendigkeit.pdf |
| Referenzlisten für Abschnitt 2 und 3 | 2_Referenzliste.ris 3_Referenzliste.ris |
| Dokumente der Zulassungsbehörden | #Benennung#_#Wirkstoff#.pdf
Dabei ist "#Benennung#" jeweils durch die entsprechende Benennung und "#Wirkstoff#" durch den Namen des zu bewertenden Wirkstoffs zu ersetzen. Benennung z.B. European Public Assessment Report: EPAR, Fachinformation: FI |
| Informationsbeschaffung | Informationsbeschaffung_alle-Referenzen.ris |
| Studien | #Studienname#_#Dokumenttyp#.pdf
"#Dokumenttyp#" ist durch eine eindeutige, möglichst sprechende Bezeichnung des Typs des jeweiligen Dokuments zu ersetzen (z.B. "Studienbericht", "Studienprotokoll", "Appendix1") |
Abschnitt 1
allgemeine Informationen
Abschnitt 1 enthält administrative Informationen zum für das Dossier verantwortlichen pharmazeutischen Unternehmer, zum Zulassungsinhaber, zu dem zu bewertenden Arzneimittel und zu der Festbetragsgruppe mit pharmakologisch-therapeutisch vergleichbaren Arzneimitteln.
Benennen Sie das für das Dossier verantwortliche pharmazeutische Unternehmen, die zuständige Kontaktperson und den Zulassungsinhaber des zu bewertenden Arzneimittels.
Tabelle 1-1: Für das Dossier verantwortliches pharmazeutisches Unternehmen
| Name des pharmazeutischen Unternehmens: | |
| Anschrift: |
Tabelle 1-2: Zuständige Kontaktperson des für das Dossier verantwortlichen pharmazeutischen Unternehmens
| Name: | |
| Position: | |
| Adresse: | |
| Telefon: | |
| Telefax: | |
| E-Mail: |
Tabelle 1-3: Zulassungsinhaber des zu bewertenden Arzneimittels
| Name des pharmazeutischen Unternehmens: | |
| Anschrift: |
Geben Sie den Namen des Wirkstoffs, den Handelsnamen des zu bewertenden Arzneimittels, den zugehörigen ATC-Code, die Darreichungsform und die in Deutschland zugelassenen Anwendungsgebiete des zu bewertenden Arzneimittels an. Sofern im Abschnitt "Anwendungsgebiete" der Fachinformation Verweise enthalten sind, führen Sie auch den Wortlaut an, auf den verwiesen wird. Zusätzlich ist das Datum der Zulassungserteilung anzugeben.
Tabelle 1-4: Allgemeine Angaben zum zu bewertenden Arzneimittel
| Wirkstoff: | |
| Handelsname: | |
| ATC-Code: | |
| Darreichungsform: | |
| in Deutschland zugelassene Anwendungsgebiete (Wortlaut der Fachinformation inkl. Wortlaut bei Verweisen): | |
| Datum der Zulassungserteilung: |
Geben Sie an, welche Pharmazentralnummern (PZN) und welche Zulassungsnummern dem zu bewertenden Arzneimittel zuzuordnen sind, und benennen Sie dabei die zugehörige Wirkstärke und Packungsgröße. Fügen Sie für jede Pharmazentralnummer eine neue Zeile ein.
Tabelle 1-5: Pharmazentralnummern und Zulassungsnummern des zu bewertenden Arzneimittels
| Pharmazentralnummer (PZN) | Zulassungsnummer | Wirkstärke | Packungsgröße |
Tragen Sie in die nachfolgende Tabelle die in dem Aufforderungsschreiben zur Dossiereinreichung enthaltenen Angaben zur Festbetragsgruppe mit pharmakologisch-therapeutisch vergleichbaren Wirkstoffen und zu den gemeinsamen Anwendungsgebieten der Festbetragsgruppe sowie die gemeinsamen Anwendungsgebiete, auf die Sie sich im Hinblick auf den Nachweis der therapeutischen Verbesserung beziehen, ein.
Tabelle 1-6: Angaben zur Festbetragsgruppe
| Festbetragsgruppe: | |
| Gemeinsame(s) Anwendungsgebiet(e) der Festbetragsgruppe laut Aufforderungsschreiben | |
| Gemeinsame(s) Anwendungsgebiet(e) der Festbetragsgruppe für die eine therapeutische Verbesserung nachgewiesen werden soll |
1.1 Patentschutz des Wirkstoffes
Ausgenommen von der Gruppenbildung nach § 35 Absatz 1 Satz 2 Nummer 2 und 3 SGB V sind Arzneimittel mit patentgeschützten Wirkstoffen, deren Wirkungsweise neuartig ist oder die eine therapeutische Verbesserung, auch wegen geringerer Nebenwirkungen, bedeuten. Ausgenommen von der Gruppenbildung nach § 35 Absatz 1a SGB V sind patentgeschützte
Wirkstoffe, die eine therapeutische Verbesserung, auch wegen geringerer Nebenwirkungen, bedeuten.
Als neuartig gilt ein Wirkstoff, solange derjenige Wirkstoff, der als Erster dieser Gruppe in Verkehr gebracht worden ist, unter Patentschutz steht. Die Neuartigkeit der Wirkungsweise ist nur relevant für Festbetragsgruppen nach § 35 Absatz 1 Satz 2 Nummer 2 und 3 SGB V.
Das zu bewertende Arzneimittel ist nur dann aufgrund einer therapeutischen Verbesserung von einer Gruppenbildung ausgenommen, wenn für den Wirkstoff des Arzneimittels ein gültiges Wirkstoffpatent besteht. Ein Wirkstoffpatent (Basispatent) schließt z.B. Modifikationen und Erzeugnisformen ein. Für die Prüfung dieser Voraussetzung ist deshalb zusätzlich zum Dossier das Original einer beglaubigten Kopie des erteilten Patents einzureichen. Anderenfalls muss davon ausgegangen werden, dass für den Wirkstoff des betreffenden Arzneimittels kein gültiges Wirkstoffpatent vorliegt, mit der Folge, dass die Möglichkeit, das Arzneimittel von einer Festbetragsgruppenbildung wegen einer therapeutischen Verbesserung auszunehmen, nicht besteht.
1.2 Unterlagen der Zulassungsbehörden
Die Feststellung, ob eine therapeutische Verbesserung dem allgemeinen anerkannten Stand der medizinischen Erkenntnisse entspricht, erfolgt grundsätzlich auf der Basis
Neben den in "Abschnitt 3 - Nachweise für eine therapeutische Verbesserung" vorzulegenden klinischen Studien sind die aktuellen Fachinformationen und die öffentlich zugänglichen Bewertungsberichte der Zulassungsbehörde für das zu bewertende Arzneimittel beizufügen.
Abschnitt 2
Begründung der therapeutischen Verbesserung
Ein Arzneimittel mit einem patentgeschützten Wirkstoff zeigt im Vergleich zu anderen Arzneimitteln derselben Festbetragsgruppe eine therapeutische Verbesserung i. S. des § 35 Absatz 1 Satz 3 und Absatz 1a Satz 2 SGB V, wenn es einen therapierelevanten höheren Nutzen als andere Arzneimittel dieser Wirkstoffgruppe hat und deshalb als zweckmäßige Therapie regelmäßig oder auch für relevante Patientengruppen oder Indikationsbereiche den anderen Arzneimitteln dieser Gruppe vorzuziehen ist. Entsprechende Bewertungen erfolgen für gemeinsame Anwendungsgebiete der Arzneimittel der Festbetragsgruppe. Eine therapeutische Verbesserung kann sich insbesondere daraus ergeben, dass das Arzneimittel einen therapierelevanten höheren Nutzen hat
Als Nebenwirkung bezeichnet man eine Reaktion, die schädlich und unerwünscht ist und bei Dosierungen auftritt, wie sie normalerweise beim Menschen zur Prophylaxe, Diagnose oder Therapie von Krankheiten oder für die Änderung einer physiologischen Funktion verwendet werden. Die Nebenwirkungen werden in der Regel der Organklassensystematik der WHO zugeordnet und nach Häufigkeitsklassen (entsprechend der SPC-Guideline) quantifiziert. Art und Ausmaß beschreiben den Schweregrad der Nebenwirkungen. Eine Nebenwirkung ist schwerwiegend, wenn sie tödlich oder lebensbedrohend ist, zu Arbeitsunfähigkeit führt oder einer Behinderung oder eine stationäre Behandlung oder Verlängerung einer stationären Behandlung zur Folge hat.
Für die Anerkennung von "geringeren Nebenwirkungen" als therapeutische Verbesserung ist erforderlich, dass die Verringerung von Nebenwirkungen quantitativ (Verringerung der Häufigkeit) oder qualitativ (Verringerung des Schweregrades therapierelevanter Nebenwirkungen) ein therapeutisch relevantes Ausmaß aufweist.
2.1 Therapeutische Verbesserung aufgrund einer überlegenen Wirksamkeit oder einer Verringerung von Nebenwirkungen
Beschreiben Sie, ob eine therapeutische Verbesserung
Unveröffentlichte Studien können nur berücksichtigt werden, wenn die unveröffentlichten Studien in einem Format zur Verfügung gestellt werden, die dem PRISMA-Statement genügen. Studienberichte nach ICH E3 genügen i. d. R. diesen Anforderungen. Zudem haben Sie schriftlich zuzustimmen, dass der G-BA diese Studien der Öffentlichkeit durch Einstellung auf der Internetseite des G-BA zur Verfügung stellen kann.
<< Argumentation des pharmazeutischen Unternehmers >>
2.2 Referenzliste für Abschnitt 2
Listen Sie nachfolgend alle Quellen (z.B. Publikationen), die Sie im vorliegenden Dokument angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
Abschnitt 3
Nachweise für eine therapeutische Verbesserung
Die Feststellung, ob eine therapeutische Verbesserung dem allgemeinen anerkannten Stand der medizinischen Erkenntnisse entspricht, erfolgt grundsätzlich auf der Basis
Die therapeutische Verbesserung soll in randomisierten, verblindeten und kontrollierten direkten Vergleichsstudien, deren Methodik internationalen Standards entspricht, nachgewiesen sein und ein therapeutisch bedeutsames Ausmaß aufweisen. Sie sollen an Populationen oder unter Bedingungen durchgeführt sein, die für die übliche Behandlungssituation repräsentativ und relevant sind. Sie sollen gegenüber Standardmitteln der Vergleichsgruppe durchgeführt werden, um die mögliche Überlegenheit der therapeutischen Verbesserung mit ausreichender Sicherheit prüfen zu können.
Liegen direkte Vergleichsstudien nicht vor, ist zu prüfen, ob placebokontrollierte Studien verfügbar sind, die sich für einen indirekten Nachweis einer therapeutischen Verbesserung eignen und den oben beschriebenen Qualitätsanforderungen entsprechen.
Die vorgelegten Studien sind hinsichtlich ihrer Planungs-, Durchführungs- und Auswertungsqualität zu prüfen. Unter Berücksichtigung der aktuellen Fachinformation des zu bewertenden Arzneimittels ist ihre Aussagekraft zur Relevanz der therapeutischen Verbesserung zu bewerten.
In diesem Abschnitt ist anzugeben, auf Basis welcher Nachweise sich eine therapeutische Verbesserung des zu bewertenden Arzneimittels
Beschreiben Sie das Studiendesign und die Studienpopulation der in die Bewertung eingeschlossenen Studien mindestens mit den Informationen in Tabelle 3-1. Orientieren Sie sich dabei an der beispielhaften Angabe in der ersten Tabellenzeile. Fügen Sie für jede Studie eine neue Zeile ein.
Tabelle 3-1: Charakterisierung der eingeschlossenen Studien - RCT mit dem zu bewertenden Arzneimittel
| Studie | Studiendesign <RCT, doppelblind/einfach verblindet/offen, parallel/cross-over etc.> | Population <relevante Charakteristika, z.B. Schweregrad> | Interventionen (Zahl der randomisierten Patienten) | Studiendauer <ggf. Run-in, Behandlung, Nachbeobachtung> | Ort und Zeitraum der Durchführung | Primärer Endpunkt; patientenrelevante sekundäre Endpunkte |
| <Studie 1> | RCT, doppelblind, parallel | Jugendliche und Erwachsene, leichtes bis mittelschweres Asthma | <Gruppe 1> (n= 354) <Gruppe 2> (n= 347) | Run-in: 2 Wochen
Behandlung: 6 Monate | Europa (Deutschland, Frankreich, Polen)
9/2003 - 12/2004 | FEV1; Asthma-Symptome, Exazerbationen, gesundheitsbezogene Lebensqualität, unerwünschte Ereignisse |
Datenbankrecherche(n):
Für Einzelstudien und systematische Übersichten:
Mindestens eine Suche in der Datenbank Medline mit Darlegung der Suchstrategie, des Datenbankanbieters und des Datums der Recherche, der Treffer, Angabe von Ein- und Ausschlusskriterien, Bewertung der Treffer anhand der benannten Ein- und Ausschlusskriterien in tabellarischer Form für die im Volltext gesichteten Publikationen, siehe Tabelle 3-2, Darstellung einer Übersicht in Form eines Flussdiagramms (analog dem PRISMA-Statement: http://www.prisma-statement.org/statement.htm
Für Leitlinien:
Mindestens eine Suche nach deutschen Versorgungsleitlinien (http://www.versorgungsleitlinien.de), bei der Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (AWMF) und Evidence based Guidelines (http://ebmg.wiley.com) mit Darlegung der Suchstrategie, bei weiteren Suchen mit Angaben des Datenbankanbieters, Angabe des Datums der Recherche, Angabe von Ein- und Ausschlusskriterien, Bewertung anhand der benannten Ein- und Ausschlusskriterien in tabellarischer Form. (siehe Tabelle 3-2)
Für HTA-Berichte:
Mindestens eine Suche bei der Deutschen Agentur für Health Technology Assessment (DAHTA) bei DIMDI (http://www.dimdi.de) und Centre for Reviews and Dissemination (CRD) (http://www.york.ac.uk/inst/crd) und der Cochrane Datenbank für systematische Übersichten (http://www.thecochranelibrary.com) mit Darlegung der Suchstrategie, bei weiteren Datenbänken mit Angabe des Datenbankanbieters, Angabe des Datums der Recherche, Angabe von Ein- und Ausschlusskriterien, Bewertung anhand der benannten Ein- und Ausschlusskriterien in tabellarischer Form. (siehe Tabelle 3-2)
Einzelne klinische Studien, die nach dem obigen Vorgehen gefunden und als relevant erachtet wurden, sind nach dem beigefügten Bogen (Anlage 3-a) zu extrahieren und systematische Übersichten sind nach dem Bogen Anlage 3-c) zu extrahieren. Alle Treffer sind gemäß Verfahrensordnung des Gemeinsamen Bundesausschusses zu klassifizieren.
Die Verzerrungsaspekte jeder eingeschlossenen Studie (einschließlich der Beschreibung für jeden berücksichtigten Endpunkt) sind anhand des Bewertungsbogens (Anlage 3-b) darzustellen.
Studienregister:
Eine Suche in öffentlich zugänglichen Studienregistern ist grundsätzlich durchzuführen, um sicherzustellen, dass laufende Studien sowie abgeschlossene Studien von Dritten vollständig identifiziert werden.
Die Suche soll mindestens in den Studienregistern clinicaltrials.gov (www.clinicaltrials.gov), EU Clinical Trials Register (EU-CTR, www.clinicaltrialsregister.eu), Klinische Prüfungen PharmNet.Bund (http://www.pharmnet-bund.de/dynamic/de/klinische-pruefungen/index.htm) sowie über das International Clinical Trials Registry Platform Search Portal (ICTRP Search Portal, Suchportal der WHO: http://apps.who.int/trialsearch/) durchgeführt werden. Optional kann zusätzlich eine Suche in weiteren themenspezifischen Studienregistern (z.B. krankheitsspezifische Studienregister oder Studienregister einzelner pharmazeutischer Unternehmen) durchgeführt werden. Bewertung anhand der benannten Ein- und Ausschlusskriterien in tabellarischer Form (siehe Tabelle 3-2).
Die Suche soll in jedem Studienregister einzeln und mit einer für das jeweilige Studienregister adaptierten Suchstrategie durchgeführt werden. Geben Sie dabei auch an, ob bei der Recherche generelle Einschränkungen vorgenommen wurden (z.B. Jahreseinschränkungen), und begründen Sie diese. Die Suche soll abgeschlossene, abgebrochene und laufende Studien erfassen. Eine gemeinsame Suche nach Studien zu mehreren Fragestellungen (z.B. direkt vergleichende Studien sowie Studien für einen indirekten Vergleich) ist möglich.
Alle Suchstrategien sind in Anlage 3-d zu dokumentieren.
<< Angaben des pharmazeutischen Unternehmers >>
Tabelle 3-2: Tabellarische Darstellung der Bewertung der Treffer in der/den Recherche(n)
| Treffer | Bewertung |
| Treffernummer, Autor, Titel, Quelle | Stichwortartige Angabe zum Ausschlussgrund, ansonsten Angabe "eingeschlossen" |
3.1 Referenzliste für Abschnitt 3
Listen Sie nachfolgend alle Quellen (z.B. Publikationen, Studienberichte etc.), die Sie im vorliegenden Dokument angegeben haben (als fortlaufend nummerierte Liste). Verwenden Sie hierzu einen allgemein gebräuchlichen Zitierstil (z.B. Vancouver oder Harvard). Geben Sie bei Fachinformationen immer den Stand des Dokuments an.
Anlage 3-a: Studienextraktionsbogen für Einzelstudien
| Nr. | Feld | |
| 1. | Autor | |
| 2. | Titel | |
| 3. | Quelle | |
| 4. | Indikation | |
| 5. | Bezugsrahmen | |
| 6. | Fragestellung / Zielsetzung | |
| 7. | relevante Ein-, Ausschlusskriterien | |
| 8. | Prüf-Intervention | |
| 9. | Vergleichsintervention | |
| 10. | evtl. weitere Behandlungsgruppen | |
| 11. | Design | |
| 12. | Zahl der Zentren | |
| 13. | Details, falls > 1 | |
| 14. | Randomisierung | analog Bewertungsbogen |
| 15. | Concealment | analog Bewertungsbogen |
| 16. | Verblindung | analog Bewertungsbogen |
| 17. | Beobachtungsdauer | |
| 18. | primäre Zielkriterien* | |
| 19. | sekundäre Zielkriterien* | |
| 20. | Anzahl zu behandelnder Patienten | |
| 21. | Anzahl eingeschlossener und ausgewerteter Patienten | |
| 22. | Vergleichbarkeit der Gruppen | |
| 23. | Darstellung nach CONSORT-Schema der eingeschlossenen Patienten | |
| 24. | Ergebnisse* zu primären und sekundären Zielkriterien | |
| 25. | Ergebnisse zu unerwünschten Ereignissen | |
| 26. | Fazit der Autoren | |
| 27. | Bewertung der methodischen Qualität | |
| * Bei Erhebung der Lebensqualität sind Angaben zur Validität der Messinstrumente erforderlich | ||
Für quantitative Zielkriterien
| Zielkriterium | Verum | Kontrolle | Maß für Gruppenunterschied | Schätzer und 95% Konfidenzintervall für Gruppenunterschied | p-Wert (optional) | ||||
| n | χ' | SD | n | χ' | SD | ||||
χ' = Mittelwert
Für dichotome Zielkriterien
| Zielkriterium | Anzahl Patienten mit Ereignis / Anzahl aller Patienten | Maß für Gruppenunterschied | Schätzer und 95% Konfidenzintervall für Gruppenunterschied | p-Wert (optional) | |
| Verum | Kontrolle | ||||
Anlage 3-b: Bewertungsbögen zur Einschätzung von Verzerrungsaspekten
Beschreiben Sie nachfolgend die Verzerrungsaspekte jeder eingeschlossenen Studie (einschließlich der Beschreibung für jeden berücksichtigten Endpunkt). Erstellen Sie hierfür je Studie eine separate Version des nachfolgend dargestellten Bewertungsbogens.
Grundsätzliche Hinweise zum Ausfüllen des Bewertungsbogens
Der nachfolgend dargestellte Bewertungsbogen dient der Dokumentation der Einstufung des Potenzials der Ergebnisse für Verzerrungen (Bias). Für jede Studie soll aus diesem Bogen nachvollziehbar hervorgehen, inwieweit die Ergebnisse für die einzelnen Endpunkte als möglicherweise verzerrt bewertet wurden, was die Gründe für die Bewertung waren und welche Informationen aus den Quellen dafür Berücksichtigung fanden.
Der Bogen gliedert sich in zwei Teile:
Für jedes Kriterium sind unter "Angaben zum Kriterium" alle relevanten Angaben aus den Quellen zur Bewertung einzutragen (Stichworte reichen ggf., auf sehr umfangreiche Informationen in den Quellen kann verwiesen werden).
Grundsätzlich sollen die Bögen studienbezogen ausgefüllt werden. Wenn mehrere Quellen zu einer Studie vorhanden sind, müssen die herangezogenen Quellen in der folgenden Tabelle genannt und jeweils mit Kürzeln (z.B. A, B, C ...) versehen werden. Quellenspezifische Angaben im weiteren Verlauf sind mit dem jeweiligen Kürzel zu kennzeichnen.
Hinweis: Der nachfolgend dargestellte Bewertungsbogen ist die Blankoversion des Bogens. Dieser Blankobogen ist für jede Studie heranzuziehen. Im Anschluss daran ist ein Bewertungsbogen inklusive Ausfüllhinweisen abgebildet, der als Ausfüllhilfe dient, aber nicht als Vorlage verwendet werden soll.
Studie:______________________
Tabelle: Liste der für die Bewertung herangezogenen Quellen
| Genaue Benennung der Quelle | Kürzel |
A Verzerrungsaspekte auf Studienebene:
Einstufung als randomisierte Studie
[ ] ja ℜ→Bewertung der Punkte 1 und 2 für randomisierte Studien
[ ] nein ℜ→Bewertung der Punkte 1 und 2 für nicht randomisierte Studien
Angaben zum Kriterium:
............................................................................................................................................
1. für randomisierte Studien: Adäquate Erzeugung der Randomisierungssequenz
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
für nicht randomisierte Studien: Zeitliche Parallelität der Gruppen
ja [ ]
unklar [ ]
nein [ ]
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
2. für randomisierte Studien: Verdeckung der Gruppenzuteilung ("allocation concealment")
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
für nicht randomisierte Studien: Vergleichbarkeit der Gruppen bzw. adäquate Berücksichtigung von prognostisch relevanten Faktoren
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
3. Verblindung von Patienten und behandelnden Personen Patient:
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
behandelnde bzw. weiterbehandelnde Personen:
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
4. Ergebnisunabhängige Berichterstattung aller relevanten Endpunkte:
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
5. Keine sonstigen (endpunktübergreifenden) Aspekte, die zu Verzerrungen führen können
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
............................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse auf Studienebene (ausschließlich für randomisierte Studien durchzuführen):
[ ] niedrig
[ ] hoch
Begründung für die Einstufung:
............................................................................................................................................
B Verzerrungsaspekte auf Endpunktebene pro Endpunkt: Endpunkt:
1. Verblindung der Endpunkterheber
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
2. Adäquate Umsetzung des ITT-Prinzips
[ ] ja
[ ] unklar
[ ]nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
3. Ergebnisunabhängige Berichterstattung dieses Endpunkts alleine
[ ] ja
[ ] unklar
[ ] nein
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
4. Keine sonstigen (endpunktspezifischen) Aspekte, die zu Verzerrungen führen können
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
............................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse des Endpunkts (ausschließlich für randomisierte Studien durchzuführen):
[ ] niedrig
[ ] hoch
Begründung für die Einstufung:
............................................................................................................................................
Hinweis: Der nachfolgend dargestellte Bewertungsbogen mit Ausfüllhinweisen dient nur als Ausfüllhilfe für den Blankobogen. Er soll nicht als Vorlage verwendet werden.
Bewertungsbogen zur Beschreibung von Verzerrungsaspekten (Ausfüllhilfe)
A Verzerrungsaspekte auf Studienebene: Einstufung als randomisierte Studie
[ ] ja ℜ→ Bewertung der Punkte 1 und 2 für randomisierte Studien
[ ] nein:
Aus den Angaben geht klar hervor, dass es keine randomisierte Zuteilung gab, oder die Studie ist zwar als randomisiert beschrieben, es liegen jedoch Anzeichen vor, die dem widersprechen (z.B. wenn eine alternierende Zuteilung erfolgte). Eine zusammenfassende Bewertung der Verzerrungsaspekte soll für nicht randomisierte Studien nicht vorgenommen werden.
ℜ→ Bewertung der Punkte 1 und 2 für nicht randomisierte Studien
Angaben zum Kriterium:
............................................................................................................................................
1. für randomisierte Studien:
Adäquate Erzeugung der Randomisierungssequenz
[ ] ja: Die Gruppenzuteilung erfolgte rein zufällig, und die Erzeugung der Zuteilungssequenz ist beschrieben und geeignet (z.B. computergenerierte Liste).
[ ] unklar: Die Studie ist zwar als randomisiert beschrieben, die Angaben zur Erzeugung der Zuteilungssequenz fehlen jedoch oder sind ungenügend genau.
[ ] nein: Die Erzeugung der Zuteilungssequenz war nicht adäquat.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
für nicht randomisierte Studien:
Zeitliche Parallelität der Gruppen
[ ] ja: Die Gruppen wurden zeitlich parallel verfolgt.
[ ] unklar: Es finden sich keine oder ungenügend genaue diesbezügliche Angaben.
[ ] nein: Die Gruppen wurden nicht zeitlich parallel verfolgt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
2. für randomisierte Studien:
Verdeckung der Gruppenzuteilung ("allocation concealment")
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Die Angaben der Methoden zur Verdeckung der Gruppenzuteilung fehlen oder sind ungenügend genau.
[ ] nein: Die Gruppenzuteilung erfolgte nicht verdeckt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
für nicht randomisierte Studien:
Vergleichbarkeit der Gruppen bzw. adäquate Berücksichtigung von prognostisch relevanten Faktoren
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Die Angaben zur Vergleichbarkeit der Gruppen bzw. zur Berücksichtigung von Einflussgrößen fehlen oder sind ungenügend genau.
[ ] nein: Die Vergleichbarkeit ist nicht gegeben und diese Unterschiede werden in den Auswertungen nicht adäquat berücksichtigt.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
3. Verblindung von Patienten und behandelnden Personen
Patient
[ ] ja: Die Patienten waren verblindet.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass die Patienten nicht verblindet waren.
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
behandelnde bzw. weiterbehandelnde Personen
[ ] ja: Das behandelnde Personal war bzgl. der Behandlung verblindet. Wenn es, beispielsweise bei chirurgischen Eingriffen, offensichtlich nicht möglich ist, die primär behandelnde Person (z.B. Chirurg) zu verblinden, wird hier beurteilt, ob eine angemessene Verblindung der weiteren an der Behandlung beteiligten Personen (z.B. Pflegekräfte) stattgefunden hat.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass die behandelnden Personen nicht verblindet waren.
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
4. Ergebnisunabhängige Berichterstattung aller relevanten Endpunkte
Falls die Darstellung des Ergebnisses eines Endpunkts von seiner Ausprägung (d. h. vom Resultat) abhängt, können erhebliche Verzerrungen auftreten. Je nach Ergebnis kann die Darstellung unterlassen worden sein (a), mehr oder weniger detailliert (b) oder auch in einer von der Planung abweichenden Weise erfolgt sein (c).
Beispiele zu a und b:
Beispiele zu c: Ergebnisgesteuerte Auswahl in der Auswertung verwendeter
Zur Einschätzung einer potenziell vorhandenen ergebnisgesteuerten Berichterstattung sollten folgende Punkte - sofern möglich - berücksichtigt werden:
Anzumerken ist, dass Anzeichen für eine ergebnisgesteuerte Darstellung eines Endpunkts zu Verzerrungen der Ergebnisse der übrigen Endpunkte führen kann, da dort ggf. auch mit einer selektiven Darstellung gerechnet werden muss. Insbesondere bei Anzeichen dafür, dass die Ergebnisse einzelner Endpunkte selektiv nicht berichtet werden, sind Verzerrungen für die anderen Endpunkte möglich. Eine von der Planung abweichende selektive Darstellung des Ergebnisses eines Endpunkts führt jedoch nicht zwangsläufig zu einer Verzerrung der anderen Endpunkte; in diesem Fall ist die ergebnisgesteuerte Berichterstattung endpunktspezifisch unter Punkt B.3 (siehe unten) einzutragen.
Des Weiteren ist anzumerken, dass die Berichterstattung von unerwünschten Ereignissen üblicherweise ergebnisabhängig erfolgt (es werden nur Häufungen / Auffälligkeiten berichtet) und dies nicht zur Verzerrung anderer Endpunkte führt.
[ ] ja: Eine ergebnisgesteuerte Berichterstattung ist unwahrscheinlich, unklar: Die verfügbaren Angaben lassen eine Einschätzung nicht zu.
[ ] nein: Es liegen Anzeichen für eine ergebnisgesteuerte Berichterstattung vor, die das Verzerrungspotenzial aller relevanten Endpunkte beeinflusst.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
5. Keine sonstigen (endpunktübergreifenden) Aspekte, die zu Verzerrung führen können
z.B.
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
............................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse auf Studienebene (ausschließlich für randomisierte Studien durchzuführen):
Die Einstufung des Verzerrungspotenzials der Ergebnisse erfolgt unter Berücksichtigung der einzelnen Bewertungen der vorangegangenen Punkte A.1 bis A.5. Eine relevante Verzerrung bedeutet hier, dass sich die Ergebnisse bei Behebung der verzerrenden Aspekte in ihrer Grundaussage verändern würden.
[ ] niedrig: Es kann mit großer Wahrscheinlichkeit ausgeschlossen werden, dass die Ergebnisse durch diese endpunktübergreifenden Aspekte relevant verzerrt sind.
[ ] hoch: Die Ergebnisse sind möglicherweise relevant verzerrt.
Begründung für die Einstufung:
............................................................................................................................................
B Verzerrungsaspekte auf Endpunktebene pro Endpunkt:
Die folgenden Punkte B.1 bis B.4 dienen der Einschätzung der endpunktspezifischen Aspekte für das Ausmaß möglicher Ergebnisverzerrungen. Diese Punkte sollten i. d. R. für jeden relevanten Endpunkt separat eingeschätzt werden (ggf. lassen sich mehrere Endpunkte gemeinsam bewerten, z.B. Endpunkte zu unerwünschten Ereignissen).
Endpunkt:
1. Verblindung der Endpunkterheber
Für den Endpunkt ist zu bestimmen, ob das Personal, welches die Zielkriterien erhoben hat, bzgl. der Behandlung verblindet war.
In manchen Fällen kann eine Verblindung auch gegenüber den Ergebnissen zu anderen Endpunkten (z.B. typischen unerwünschten Ereignissen) gefordert werden, wenn die Kenntnis dieser Ergebnisse Hinweise auf die verabreichte Therapie gibt und damit zu einer Entblindung führen kann.
[ ] ja: Der Endpunkt wurde verblindet erhoben.
[ ] unklar: Es finden sich keine diesbezüglichen Angaben.
[ ] nein: Aus den Angaben geht hervor, dass keine verblindete Erhebung erfolgte.
Angaben zum Kriterium; obligate Begründung für die Einstufung:
............................................................................................................................................
2. Adäquate Umsetzung des ITT-Prinzips
Kommen in einer Studie Patienten vor, die die Studie entweder vorzeitig abgebrochen haben oder wegen Protokollverletzung ganz oder teilweise aus der Analyse ausgeschlossen wurden, so sind diese ausreichend genau zu beschreiben (Abbruchgründe, Häufigkeit und Patientencharakteristika pro Gruppe) oder in der statistischen Auswertung angemessen zu berücksichtigen (i d. R. ITT-Analyse, siehe Äquivalenzstudien). Bei einer ITT ("intention to treat")-Analyse werden alle randomisierten Patienten entsprechend ihrer Gruppenzuteilung ausgewertet (ggf. müssen fehlende Werte für die Zielkriterien in geeigneter Weise ersetzt werden). Zu beachten ist, dass in Publikationen der Begriff ITT nicht immer in diesem strengen Sinne Verwendung findet. Es werden häufig nur die randomisierten Patienten ausgewertet, die die Therapie zumindest begonnen haben und für die mindestens ein PostBaseline-Wert erhoben worden ist ("full analysis set"). Dieses Vorgehen ist in begründeten Fällen Guideline-konform, eine mögliche Verzerrung sollte jedoch, insbesondere in nicht verblindeten Studien, überprüft werden. Bei Äquivalenz- und Nichtunterlegenheitsstudien ist es besonders wichtig, dass solche Patienten sehr genau beschrieben werden und die Methode zur Berücksichtigung dieser Patienten transparent dargestellt wird.
[ ] ja: Eines der folgenden Merkmale trifft zu:
[ ] unklar: Aufgrund unzureichender Darstellung ist der adäquate Umgang mit Protokollverletzern und Lost-to-follow-up-Patienten nicht einschätzbar.
[ ] nein: Keines der unter "ja" genannten drei Merkmale trifft zu.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
3. Ergebnisunabhängige Berichterstattung dieses Endpunkts alleine
Beachte die Hinweise zu Punkt A.4!
[ ] ja: Eine ergebnisgesteuerte Berichterstattung ist unwahrscheinlich.
[ ] unklar: Die verfügbaren Angaben lassen eine Einschätzung nicht zu.
[ ] nein: Es liegen Anzeichen für eine ergebnisgesteuerte Berichterstattung vor.
Angaben zum Kriterium; falls unklar oder nein, obligate Begründung für die Einstufung:
............................................................................................................................................
4. Keine sonstigen (endpunktspezifischen) Aspekte, die zu Verzerrungen führen können
z.B.
[ ] ja
[ ] nein
Angaben zum Kriterium; falls nein, obligate Begründung für die Einstufung:
............................................................................................................................................
Einstufung des Verzerrungspotenzials der Ergebnisse des Endpunkts (ausschließlich für randomisierte Studien durchzuführen):
Die Einstufung des Verzerrungspotenzials erfolgt unter Berücksichtigung der einzelnen Bewertungen der vorangegangenen endpunktspezifischen Punkte B.1 bis B.4 sowie der Einstufung des Verzerrungspotenzials auf Studienebene. Falls die endpunktübergreifende Einstufung mit "hoch" erfolgte, ist das Verzerrungspotenzial für den Endpunkt i. d. R. auch mit "hoch" einzuschätzen. Eine relevante Verzerrung bedeutet hier, dass sich die Ergebnisse bei Behebung der verzerrenden Aspekte in ihrer Grundaussage verändern würden.
[ ] niedrig: Es kann mit großer Wahrscheinlichkeit ausgeschlossen werden, dass die Ergebnisse für diesen Endpunkt durch die endpunktspezifischen sowie endpunktübergreifenden Aspekte relevant verzerrt sind.
[ ] hoch: Die Ergebnisse sind möglicherweise relevant verzerrt.
Begründung für die Einstufung:
............................................................................................................................................
Anlage 3-c: Studienextraktionsbogen für systematische Übersichten
| Nr. | Feld | Hinweise |
| 1. | Autor | |
| 2. | Titel | |
| 3. | Quelle | |
| 4. | Bezugsrahmen | |
| 5. | Fragestellung / Zielsetzung | |
| 6. | Krankheit | |
| 7. | Intervention | |
| 8. | Einschlusskriterien | The selection criteria (i.e. population, intervention, outcome, and study design): methods for validity assessment, data abstraction, and study characteristics, and quantitative data synthesis in sufficient detail to permit replication |
| 9. | Ausschlusskriterien | |
| 10. | Ergebnis der Recherche | Characteristics of the RCTs included and excluded: qualitative and quantitative findings (i.e. point estimates and confidence intervals); and sub-group analyses |
| 11. | Einführung | The explicit clinical problem, biological rationale for the intervention and rationale for the review |
| 12. | Beschreibung der Suche | The information sources in detail (e.g. databases, registers, personal files, expert informants, agencies, hand-searching), and any restrictions (years considered, publication status, language of publication) |
| 13. | Methodische Beschreibung des Vorgehens | Data abstraction: The process or processes used (e.g. completed independently, in duplicate) |
| 14. | Validität | The criteria and process used (e.g. masked conditions, quality assessment, and their findings) |
| 15. | Charakterisierung der Studien | The type of study design, participants' characteristics, details of intervention, outcome definitions, and how clinical heterogeneity was assessed |
| 16. | Quantitative Ergebnisse der Synthese | The principal measures of effect (e.g. relative risk), method of combining results (statistical testing and confidence intervals), handling of missing data; how statistical heterogeneity was assessed; a rationale for any apriori sensitivity and sub-group analyses; and any assessment of publication bias |
| 17. | Darstellung in einem Flussdiagramm | Trial flow: Provide a meta-analysis profile summarising trial flow |
| 18. | Charakterisierung der gefunden Studien | Present descriptive data for each trial (e.g. age, sample size, intervention, dose, duration, follow-up period) |
| 19. | Nebenwirkungen | |
| 20. | Schlussfolgerung | The main results |
| 21. | Bewertung der methodischen Qualität | Summarise key findings; discuss clinical inferences based on internal and external validity; interpret the results in the light of the totality of available evidence; describe potential biases in the review process e.g. publication bias); and suggest a future re-search agenda |
Anlage 3-d: Suchstrategien - Suche in Studienregistern
Geben Sie nachfolgend die Suchstrategien für die Suche in Studienregistern an, und zwar einzeln für jedes Studienregister. Geben Sie dabei jeweils den Namen des durchsuchten Studienregisters (z.B. clinicaltrials.gov), die Internetadresse, unter der das Studienregister erreichbar ist (z.B. http://www.clinicaltrials.gov), das Datum der Suche, die verwendete Suchstrategie und die resultierenden Treffer an.
Orientieren Sie sich bei Ihren Angaben an dem nachfolgenden Beispiel:
Studienregister: clinicaltrials.gov
Internetadresse: http://www.clinicaltrials.gov
Datum der Suche: 08.12.2010
Suchstrategie: (Starlix OR Novonorm OR Prandin OR Nateglinid OR Repaglinid)
[ALL-FIELDS] AND ("Phase II" OR "Phase III" OR "Phase IV") [PHASE]
Treffer: 23
<< Angaben des pharmazeutischen Unternehmers >>
Als Anhang sind die Dokumente, die für die Aussagen in den Abschnitten 2 und 3 herangezogen werden, sowie eine Checkliste für die Prüfung der formalen Vollständigkeit des Dossiers beizufügen. Siehe dazu auch die Hinweise zur Erstellung und Einreichung des Dossiers Seite 6 bis 10)
Dokumentvorlage, Version vom 18.04.2013
Dossier zur Nutzenbewertung gemäß § 35a SGB V
(für mit Festbetragsarzneimitteln pharmakologisch-therapeutisch vergleichbare Arzneimittel)
<<Pharmazeutischer Unternehmer>>
(<<Handelsname>>)
<<Wirkstoff>>
Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers (für mit Festbetragsarzneimitteln pharmakologisch-therapeutisch vergleichbare Arzneimittel)
Stand: <<TT.MM.JJJJ>>
Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers
| Abschnitt | Anforderung | Anforderung erfüllt (ankreuzen, falls ja) | Prüfvermerk G-BA |
| Abschnitt 1 | Abschnitt "allgemeine Informationen" (Tabelle 1-1 bis 1-6) ist vollständig ausgefüllt. | [ ] | |
| Abschnitt 1.1 | Beglaubigte Kopie der Patentschrift ist in Papierform eingereicht. | [ ] | |
| Abschnitt 1.2 | Unterlagen der Zulassungsbehörden sind abgelegt. | [ ] | |
| Abschnitt 2.1 | "Begründung der therapeutischen Verbesserung" ist vollständig ausgefüllt. | [ ] | |
| Abschnitt 2.2 | "Referenzliste für Abschnitt 2" ist eine Referenzliste der in Abschnitt 2 zitierten Literatur enthalten. | [ ] | |
| Abschnitt 3 | - Tabelle Charakterisierung der eingeschlossenen Studien ist ausgefüllt. | [ ] | |
| - Darstellung der Datenbankrecherche in Form eines Flussdiagrammes. | [ ] | ||
| - Für jede Literaturrecherche (Datenbankrecherche, Leitlinienrecherche, HTA-Berichte, Studienregister) ist eine Bewertung der Treffer in Tabelle 3-2 eingetragen. | [ ] | ||
| - Anlage 3-a (Extraktionsbogen für Einzelstudien) ist ausgefüllt. | [ ] | ||
| - Anlage 3-b (Bewertungsbogen zur Einschätzung von Verzerrungsaspekten) ist ausgefüllt. | [ ] | ||
| - Anlage 3-c (Extraktionsbogen für systematische Übersichtsarbeiten) ist ausgefüllt. | [ ] | ||
| - Anlage 3-d (Suchstrategie für jede Literaturrecherche) ist ausgefüllt. | [ ] | ||
| Abschnitt 3.1 | "Referenzliste für Abschnitt 3" ist eine Referenzliste der in Abschnitt 3 zitierten Literatur enthalten. | [ ] | |
| Anlagen | Die Anlagen zu Abschnitt 3 enthalten folgende Dokumente: |
| Abschnitt | Anforderung | Anforderung erfüllt (ankreuzen, falls ja) | Prüfvermerk G-BA |
| - Volltexte für alle in der Referenzliste für Abschnitt 2 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 2 | [ ] | ||
| Anhang | Der Ordner Volltexte zu Abschnitt 2 enthält folgende Dokumente: | ||
| - Volltexte für alle in der Referenzliste für Abschnitt 2 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 2 | [ ] | ||
| Der Ordner Volltexte zu Abschnitt 3 enthält folgende Dokumente: | |||
| - Volltexte für alle in der Referenzliste für Abschnitt 3 genannten Literaturzitate | [ ] | ||
| - RIS-Datei der Referenzliste zu Abschnitt 3 | [ ] | ||
| Der Ordner Informationsbeschaffung enthält folgende Dokumente: | |||
| - RIS-Dateien die Dokumentation der Rechercheergebnisse enthaltend | [ ] | ||
| Der Ordner Informationsbeschaffung enthält die entsprechenden Zulassungs-Dokumente. | [ ] | ||
| Der Ordner Studienberichte enthält für jede Studie, für die der Unternehmer Sponsor, einen separaten Unterordner, enthaltend den entsprechenden Studienbericht inklusive Appendizes und das Studienprotokoll der Studie. | [ ] | ||
| Der Ordner "weitere Unterlagen" enthält folgende Dokumente: | |||
| - Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers (ausgefüllt) | [ ] | ||
| - Kennzeichnung Betriebs und Geschäftsgeheimnisse | [ ] | ||
| - Niederschrift zum Beratungsgespräch | [ ] |
| Antragsformular | Anlage VII |
[ ] Antrag des Spitzenverbands Bund der Krankenkassen gemäß § 130b Absatz 8 SGB V
| Ansprechpartner beim GKV-Spitzenverband | |
| a) Name | << >> |
| b) Adresse | << >> |
| c) E-Mail | << >> |
| d) Telefon- und Faxnummer | << >> |
[ ] Antrag eines pharmazeutischen Unternehmers
| Pharmazeutischer Unternehmer | ||
| a) | Name des pharmazeutischen Unternehmers | << >> |
| b) | Anschrift | << >> |
| Ansprechpartner beim pharmazeutischen Unternehmer | ||
| c) | Name | << >> |
| d) | Abteilung und Funktion | << >> |
| e) | Adresse | << >> |
| f) | << >> | |
| g) | Telefon- und Faxnummer | << >> |
Informationen zum Arzneimittel, für das eine Kosten-Nutzen-Bewertung beantragt wird
| 1) Angaben zum Arzneimittel | |
| a) Wirkstoff | << >> |
| b) Anwendungsgebiet(e) | << >> |
| c) Das Arzneimittel ist im Geltungsbereich des Arzneimittelgesetzes in Verkehr | Ja / Nein |
| 2) | Beschluss des Gemeinsamen Bundesausschusses nach § 35a Absatz 3 SGB V | |
| a) | Datum | << >> |
| b) | Vorgangsnummer | << >> |
| 3) | Anlagen gemäß 5. Kapitel § 23 Absatz 3 VerfO |
| 1. | Entscheidung der Schiedsstelle einschließlich ihrer Begründung (bei Antragstellung nach § 130b Absatz 8 SGB V) |
| 2. | Verpflichtungserklärung des pharmazeutischen Unternehmers über die Kostentragung (bei Antragstellung nach § 35a Absatz 5a SGB V) |
| 3. | Vorschlag über die Auftragsinhalte gemäß 5. Kapitel § 24 VerfO |
Datum Unterschrift
| Anlage VIII |
Anlage VIII.a: Format und Gliederung des Dossiers, einzureichende Unterlagen, Vorgaben für technische Standards
Anlage VIII.b: Modul K1 - Zusammenfassung der Aussagen im Dossier
Anlage VIII.c: Modul K1 Anhang - Checkliste zur Prüfung der formalen Vollständigkeit des Dossiers
Anlage VIII.d: Modul K2 - Allgemeine Angaben zum Arzneimittel, zugelassene Anwendungsgebiete
Anlage VIII.e: Modul K3 - Ergebnisse zum Nutzen und Zusatznutzen
Anlage VIII.f: Modul K4 - Kosten-Nutzen-Bewertung
6. Kapitel
Verfahren für Richtlinienbeschlüsse nach § 137f SGB V
§ 1 Regelungsbereich
Dieses Kapitel regelt das Verfahren für die Erarbeitung und Aktualisierung der Richtlinienbeschlüsse nach § 137f SGB V. Der Gemeinsame Bundesausschuss legt in Richtlinien nach § 137f Abs. 1 SGB V geeignete chronische Krankheiten fest, für die strukturierte Behandlungsprogramme entwickelt werden sollen. Hierzu erlässt er gemäß § 137f Abs. 2 SGB V Richtlinien zu den Anforderungen an die Ausgestaltung der strukturierten Behandlungsprogramme. Seine Richtlinien hat der Gemeinsame Bundesausschuss gemäß § 137f Abs. 2 SGB V regelmäßig zu überprüfen.
§ 2 Arbeitsweise und Zusammensetzung der Arbeitsgruppen 16a
(1) Der für die Erarbeitung der Richtlinien nach § 1 zuständige Unterausschuss setzt zur Vorbereitung und Bearbeitung von Beschlüssen sowie zur Klärung wissenschaftlicher Fragestellungen Arbeitsgruppen ein.
(2) Die Arbeitsgruppen bearbeiten konkrete Fragestellungen, die vom Unterausschuss im Rahmen eines Arbeitsauftrages präzise formuliert werden. In Abhängigkeit von der Fragestellung sind die Zusammensetzung der Arbeitsgruppen und die zu veranschlagende Dauer der Beratung im Unterausschuss abzustimmen. Sofern sich aus dem Arbeitsauftrag oder der wissenschaftlichen Fragestellung keine Besonderheiten hinsichtlich der Besetzung ergeben, setzen sich die Arbeitsgruppen in der Regel zusammen aus:
(3) Für spezifische Fragestellungen können im Einvernehmen der AG auch Vertreter des Institutes für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG), der Institution nach § 137a SGB V und des Bundesversicherungsamtes sowie gemäß § 20 Abs. 6 GO bestellte andere Sachverständige an der Beratung teilnehmen.
§ 3 Identifikation geeigneter chronischer Erkrankungen für strukturierte Behandlungsprogramme
Grundlage für die Vorgehensweise bei der Auswahl neuer chronischer Erkrankungen, die für eine Richtlinie nach § 137f Abs. 1 SGB V in Betracht kommen, sind die Kriterien gemäß § 137f Abs. 1 Satz 2 SGB V.
§ 4 Erarbeitung von Richtlinien für die Anforderungen an die Ausgestaltung von strukturierten Behandlungsprogrammen nach § 137f SGB V
(1) Die Richtlinien für die Anforderungen an die Ausgestaltung von strukturierten Behandlungsprogrammen sollen die evidenzbasierte Behandlung unter Berücksichtigung des jeweiligen Versorgungssektors, Qualitätssicherungsmaßnahmen, die Voraussetzungen für Einschreibung und Teilnahme, Schulungsmaßnahmen, die Dokumentation einschließlich der für die Durchführung der Programme erforderlichen personenbezogenen Daten und deren Aufbewahrungsfristen, die Evaluation und Vorgaben für die Qualitätsberichte der Krankenkassen berücksichtigen. Die Anforderungen sollen sich dabei insbesondere auf diejenigen Aspekte der Versorgung beziehen, für die begründete Hinweise auf relevante Versorgungsdefizite bestehen und für die entsprechende Versorgungsziele definiert werden können. Für die Beseitigung von Versorgungsdefiziten bzw. die Erreichung von Versorgungszielen sollen gleichermaßen evidenzbasierte Grundlagen im Hinblick auf Wirksamkeit und Sicherheit verfügbar sein. Gleichzeitig ist auf die Umsetzbarkeit von Maßnahmen unter den Bedingungen der RSAV zu achten.
(2) Das Verfahren zur Erarbeitung der Richtlinien besteht aus folgenden Schritten:
Die Aufbewahrungsfristen der für die Durchführung der strukturierten Behandlungsprogramme erforderlichen personenbezogenen Daten sind festzulegen.
(3) Bei der erstmaligen Erarbeitung von Richtlinien für die Anforderungen an die Ausgestaltung von strukturierten Behandlungsprogrammen sollen Leitlinienrecherche, Leitlinienauswahl und -bewertung sowie die Extraktion der relevanten Inhalte durch das IQWiG erfolgen. Hierzu ist jeweils eine Beauftragung durch den Gemeinsamen Bundesausschuss erforderlich. Die Einzelheiten des Auftrags (Umfang der Beauftragung, Auswahl relevanter Leitlinien und Inhalte etc.) werden für jedes strukturierte Behandlungsprogramm vom Unterausschuss im Rahmen der Auftragskonkretisierung als Beschlussvorlage für das Plenum erarbeitet.
(4) Anforderungen zu neuen Indikationen, die in Form eines Moduls in bereits bestehende Programme integriert werden sollen, sind auf ihre Umsetzbarkeit in der Versorgung zu prüfen und entsprechend der in Absatz 2 vorgegebenen Verfahrensweise zu erarbeiten. Die bei der Implementierung gesammelten Erfahrungen sollen in die Weiterentwicklung des Verfahrens einfließen.
§ 5 Überprüfung von Richtlinien für bestehende strukturierte Behandlungsprogramme
(1) Der Unterausschuss überprüft die bestehenden Richtlinien regelmäßig und passt sie bei festgestelltem Änderungsbedarf an. Die regelmäßige Überprüfung gemäß § 137f Abs. 2 Satz 6 SGB V umfasst die Überprüfung auf einen möglichen Änderungsbedarf gemäß Abs. 2, die Feststellung eines ggf. bestehenden Aktualisierungsbedarfs gemäß Abs. 3 und endet - sofern erforderlich - mit dem Verfahren der Überarbeitung gemäß Abs. 4.
(2) Die Informationsgrundlagen für die Überprüfung auf einen möglichen Änderungsbedarf bilden:
Diese Informationen sollen spezifisch, mit Quellen unterlegt und mit klarem Bezug zu den bereits bestehenden Anforderungen erfolgen. Unaufgefordert eingegangene Informationen werden an den Unterausschuss weitergeleitet.
(3) Stellt der Unterausschuss bei der Überprüfung einvernehmlich einen Aktualisierungsbedarf fest, leitet er unmittelbar ein Überarbeitungsverfahren ein. Ergeben sich außerhalb der regelmäßigen Überprüfung Hinweise auf das Vorliegen neuer Erkenntnisse, die einen maßgeblichen Einfluss auf die Ausgestaltung bestehender Empfehlungen haben, leitet der Unterausschuss eine außerplanmäßige Aktualisierung ein.
(4) Das Verfahren der Überarbeitung besteht aus folgenden Schritten:
§ 6 Bewertung von Evidenz
(1) Die Ermittlung des Stellenwertes von diagnostischen und therapeutischen Methoden, die in die Richtlinien zur Ausgestaltung strukturierter Behandlungsprogramme eingehen sollen, orientiert sich im Vordergrund an evidenzbasierten Leitlinien. Sofern eine Bewertung der Evidenz, die einer Empfehlung zugrunde liegt, vorgenommen wird, erfolgt diese nach den Methoden der evidenzbasierten Medizin. Soweit der Gemeinsame Bundesausschuss in Bewertungsverfahren gemäß Kapitel 2, 3, 4 und 5 dieser Verfahrensordnung (VerfO) eine Evidenzprüfung vorgenommen hat, sind die Ergebnisse dieser Evidenzprüfung bei der Formulierung von Anforderungen für strukturierte Behandlungsprogramme in die Prüfung mit einzubeziehen.
(2) Bei jedem Bearbeitungsschritt ist zu prüfen, welche Tiefe der Evidenzprüfung angemessen ist. Die Klassifizierung von Unterlagen zu diagnostischen und therapeutischen Methoden erfolgt entsprechend 2. Kapitel § 11 Abs. 2 und 3 VerfO.
§ 7 (aufgehoben)
7. Kapitel 16f
Verfahren für Richtlinienbeschlüsse nach § 92 Absatz 1 Satz 2
Nummer 15 i. V. m. § 20i Absatz 1 SGB V
1. Abschnitt
Regelungsbereich und allgemeine Vorschriften
Dieser Abschnitt regelt das Verfahren für Richtlinienbeschlüsse zur Schutzimpfungs-Richtlinie nach § 20i Absatz 1 SGB V. Für die Durchführung des Verfahrens nach Satz 1 ist der Unterausschuss Arzneimittel (im Weiteren Unterausschuss) des Gemeinsamen Bundesausschusses zuständig.
§ 2 Anwendbarkeit anderer Vorschriften der Verfahrensordnung
Soweit in diesem Kapitel keine speziellen Regelungen getroffen sind, finden die Vorschriften des 1. Kapitels (Allgemeiner Teil) der Verfahrensordnung Anwendung.
2. Abschnitt 16f
Bewertung von Schutzimpfungen nach § 20i Absatz 1 SGB V
1. Titel
Verordnungsvoraussetzungen
§ 3 Verordnungsvoraussetzung
(1) Versicherte haben Anspruch auf Leistungen für Schutzimpfungen, die vom Gemeinsamen Bundesausschuss auf der Grundlage der Empfehlungen der STIKO in die Schutzimpfungs-Richtlinie (SI-RL) aufgenommen wurden.
(2) Der Anspruch umfasst für in der SI-RL geregelte Impfungen auch die Nachholung von Impfungen sowie die Vervollständigung des Impfschutzes, bei Jugendlichen spätestens bis zum vollendeten 18. Lebensjahr.
(3) Von der Leistungspflicht ausgeschlossen sind Schutzimpfungen, die wegen eines durch einen nicht beruflichen Auslandsaufenthalt erhöhten Gesundheitsrisikos indiziert sind (sog. Reiseschutzimpfungen), es sei denn, dass nach Anlage 1 der SI-RL zum Schutz der öffentlichen Gesundheit ein besonderes Interesse daran besteht, der Einschleppung einer übertragbaren Krankheit in die Bundesrepublik Deutschland vorzubeugen.
2. Titel
Allgemeine Grundzüge des Verfahrens
§ 4 Entscheidungen des Gemeinsamen Bundesausschusses 16f
(1) Der Gemeinsame Bundesausschuss kann von den Empfehlungen der STIKO mit besonderer Begründung abweichen. Zum Nutzen von seitens der STIKO nicht empfohlenen Schutzimpfungen kann der Gemeinsame Bundesausschuss keine eigenen Bewertungen vornehmen.
(2) Zu Änderungen der Empfehlungen der STIKO hat der Gemeinsame Bundesausschuss gemäß § 20i Absatz 1 Satz 5 SGB V innerhalb von 3 Monaten nach ihrer Veröffentlichung eine Entscheidung zu treffen. Die Entscheidungsfrist beginnt mit Veröffentlichung der Empfehlungen einschließlich der ausführlichen wissenschaftlichen Begründungen. Unbenommen davon kann der Unterausschuss seine Beratungen über Einzelheiten zu Voraussetzungen, Art und Umfang der Leistungen nach Maßgabe des 3. Titels mit dem Zurverfügungstellen der ausführlichen wissenschaftlichen Begründungen beginnen.
(3) Bei Beschlüssen über die Schutzimpfungs-Richtlinie, deren Gegenstand die Berufsausübung der Ärzte berührt, wird der Bundesärztekammer Gelegenheit zur Stellungnahme gegeben. Bei Beschlüssen, die die Erhebung, Verarbeitung oder Nutzung personenbezogener oder personenbeziehbarer Daten regeln oder voraussetzen, wird dem Bundesbeauftragten für den Datenschutz und die Informationsfreiheit Gelegenheit zur Stellungnahme gegeben.
§ 5 Aktualisierung der Richtlinie
Die Schutzimpfungs-Richtlinie nach § 2 Abs. 1 Satz 3 SGB V hat dem allgemein anerkannten Stand der medizinischen Erkenntnisse zu entsprechen und ist deshalb in geeigneten Zeitabständen zu überprüfen.
3. Titel
Verfahren zur Umsetzung von STIKO-Empfehlungen
§ 6 Verfahren zur Umsetzung von STIKO-Empfehlungen 16f
(1) Der Unterausschuss berät nach § 20i Absatz 1 SGB V über die Voraussetzungen sowie Art und Umfang der Leistungen bei der Umsetzung der Empfehlungen der STIKO.
(2) Der Unterausschuss prüft bei der Umsetzung in die Schutzimpfungs-Richtlinie die Empfehlungen der STIKO dahingehend, ob sie in sich schlüssig und nachvollziehbar sind. Neben den von der STIKO veröffentlichten ausführlichen wissenschaftlichen Begründungen bezieht der Unterausschuss ggf. eigene Recherchen in seine Bewertung ein.
(3) Bei der Umsetzung ist die Zuständigkeit anderer Kostenträger (z.B. nach der Verordnung zur arbeitsmedizinischen Vorsorge - ArbMedVV) zu berücksichtigen.
§ 7 Nutzen und Notwendigkeit
Der Unterausschuss bewertet den Nutzen und die Notwendigkeit der seitens der STIKO empfohlenen Schutzimpfungen für die Versicherten unter besonderer Berücksichtigung der Bedeutung der Schutzimpfungen für die öffentliche Gesundheit.
§ 8 Wirtschaftlichkeit
Der Unterausschuss bewertet die Wirtschaftlichkeit von der STIKO empfohlener Impfungen nach den Maßgaben § § 2, 12, 70 und 72 SGB V.
8. Kapitel
Verfahren für Richtlinienbeschlüsse sowie sonstige Beschlüsse und Aufgaben zur Qualitätssicherung
1. Abschnitt
Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten
§ 1 Regelungsbereich und Begriffsbestimmung
Der Gemeinsame Bundesausschuss (G-BA) ermöglicht Dritten nach Maßgabe dieser Regelung für Zwecke der wissenschaftlichen Forschung oder der Weiterentwicklung der Qualitätssicherung Auswertungen der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten durch hierzu vom Gemeinsamen Bundesausschuss beauftragte Stellen (sekundäre Datennutzung).
§ 2 Verfahrensablauf
Das Verfahren untergliedert sich in
§ 3 Beauftragte Stelle
(1) G-BA beauftragt das Institut für Qualitätssicherung und Transparenz im Gesundheitswesen (IQTIG) oder eine andere an der einrichtungsübergreifenden Qualitätssicherung beteiligte Stelle, Anträge Dritter auf Auswertung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten für Zwecke der wissenschaftlichen Forschung oder der Weiterentwicklung der Qualitätssicherung anzunehmen, die Vorprüfung und Einschätzung vorzunehmen und nach positiver Entscheidung durch den G-BA zu bearbeiten. Der G-BA informiert auf seinen Internetseiten über die von ihm beauftragten Stellen.
(2) Der Unterausschuss Qualitätssicherung des G-BA beschließt eine Beschreibung der bei der jeweiligen beauftragten Stelle für die sekundäre Datennutzung zur Verfügung stehenden Daten sowie zu deren Art und Struktur (Datensatzbeschreibungen) und veröffentlicht diese für jedes Erfassungsjahr auf den Internetseiten des G-BA.
§ 4 Anforderungen an den Datenschutz
(1) Gegenstand der sekundären Datennutzung sind die bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten. Das Zusammenführen mit anderen Daten als denen nach Satz 1 zum Zwecke einer Auswertung ist möglich, soweit diese Daten ausschließlich anonym sind und sichergestellt werden kann, dass eine Identifizierung einzelner Personen oder Leistungserbringer ausgeschlossen ist.
(2) Die Antragstellerin oder der Antragsteller dürfen keinen Zugriff auf die erhobenen Daten erhalten. Die jeweilige beauftragte Stelle stellt sicher, dass die Auswertung nur in geschützter Umgebung in ihren eigenen Räumen stattfindet, die Daten ausschließlich durch Mitarbeiterinnen oder Mitarbeiter der beauftragten Stellen bearbeitet werden und keine Möglichkeit zur Übermittlung der Daten besteht.
(3) Die Auswertungsergebnisse werden der Antragstellerin oder dem Antragsteller nur anonymisiert und in aggregierter Form zur Verfügung gestellt. Die Anonymisierung hat so zu erfolgen, dass eine Identifizierung einzelner Personen oder Leistungserbringer auch unter Nutzung von Zusatzwissen der Antragstellerin oder des Antragstellers sicher ausgeschlossen ist.
(4) Die jeweilige beauftragte Stelle erstellt ein Datenschutzkonzept, welches auch die in den Absätzen 1 bis 3 genannten Anforderungen umfasst. Das Datenschutzkonzept ist auf Betreiben der jeweiligen beauftragten Stelle oder nach Aufforderung durch den G-BA zu aktualisieren. Der G-BA nimmt das Datenschutzkonzept ab, nachdem er es einem unabhängigen Gutachter zur Prüfung und Bewertung vorgelegt hat und dieser eine Abnahme empfiehlt. Der G-BA veröffentlicht das Ergebnis der Prüfung und Bewertung auf seinen Internetseiten. Eine sekundäre Datennutzung ohne vorliegendes und abgenommenes Datenschutzkonzept ist ausgeschlossen.
§ 5 Antragsberechtigung und Antragstellung
(1) Antragsberechtigt ist jede natürliche oder juristische Person.
(2) Der Antrag ist bei der jeweiligen beauftragten Stelle gemäß § 3 zu stellen. Für die Antragstellung einschließlich der Selbsterklärung zu potentiellen Interessenkonflikten sind die Formulare nach Anlage I und II zu verwenden. Der Antrag ist von der Person zu stellen, die die Datenauswertungen nutzen will.
(3) Dem vollständig ausgefüllten Antrag ist ein Dokument (Exposé) beizufügen, in dem der Forschungskontext oder der Weiterentwicklungsbedarf zur Qualitätssicherung dargelegt wird sowie ausführlich die wissenschaftliche oder für die Qualitätssicherung relevante Fragestellung dargestellt, gegebenenfalls Hypothesen formuliert, die methodische Herangehensweise detailliert beschrieben sowie ein Zeitplan hinzugefügt werden. Anhand der Datensatzbeschreibung nach § 3 Absatz 2 ist durch die Antragstellerin oder den Antragsteller darzulegen, wie eine Analyse der Daten im Kontext einer spezifischen Fragestellung mit hinreichender Wahrscheinlichkeit erfolgversprechend sein kann. Die Antragstellerin oder der Antragsteller muss zudem auf Basis der Datensatzbeschreibung die konkreten Auswertungsziele benennen und den konkreten Auswertungsplan einreichen und in dem Antrag (Anlage I) entsprechende Angaben zu den hierfür benötigten Daten sowie ihrer Auswertung machen. Auf Grundlage des Exposés muss der jeweiligen beauftragten Stelle die antragsgemäße Durchführung der Auswertung möglich sein.
§ 5a Antragstellung durch das Bundesministerium für Gesundheit
Antragsberechtigt im Sinne von § 5 Absatz 1 ist auch das Bundesministerium für Gesundheit (BMG). Die Vorgaben zur Antragstellung in § 5 Absatz 2 und 3 sowie die Regelungen in §§ 4, 6 bis 11 gelten auch für die Anträge des BMG.
§ 5b Antragstellung durch die beauftragte Stelle
(1) Antragsberechtigt im Sinne von § 5 Absatz 1 ist auch die jeweilige beauftragte Stelle. Die Vorgaben zur Antragstellung in § 5 Absatz 2 und 3 sowie die Regelungen in §§ 6, 8 und 10 gelten auch für Anträge der jeweiligen beauftragten Stelle. 3 Die Vorprüfung und Einschätzung nach § 7 entfällt.
(2) Die Regelungen zum Datenschutz nach § 4 sowie zur Auswertung der Daten nach § 9 finden auch dann Anwendung, wenn der Antrag durch die jeweilige beauftragte Stelle oder deren Mitarbeiter oder Mitarbeiterinnen gestellt wird. Dazu sind im Datenschutzkonzept nach § 4 Absatz 4 die erforderlichen Vorgaben zu regeln. Soweit sich aus der Identität von Antragstellerin und Antragsteller auf der einen und beauftragter Stelle auf der anderen Seite besondere datenschutzrechtliche Vorgaben ergeben, sind diese ebenfalls im Datenschutzkonzept nach § 4 Absatz 4 aufzunehmen. Im Datenschutzkonzept ist insbesondere zu regeln, dass Mitarbeiterinnen und Mitarbeiter der jeweiligen beauftragten Stelle, die einen Antrag gestellt haben, für diesen nicht an der Vorprüfung gemäß § 7 und an der Bearbeitung gemäß § 9 beteiligt werden und insoweit keinen Zugriff auf die auszuwertenden Daten erhalten. Satz 4 gilt entsprechend für die Mitarbeiterinnen und Mitarbeiter, die für die jeweilige beauftragte Stelle bei deren Antragstellung tätig werden.
(3) Die Kosten für den durch die Auswertung der Daten bei Anträgen der beauftragten Stelle im Sinne von Absatz 1 entstandenen Personal- und Sachaufwand sind von dieser selbst zu tragen. Die Regelung zur Vorauszahlung sowie zur Kostenübernahme nach § 11 findet keine Anwendung. 3 Bei Anträgen, die das IQTIG als beauftragte Stelle zur Erfüllung von Aufträgen nach § 137a Absatz 4 Satz 2 SGB V stellt, trägt das BMG die Kosten für den entstandenen Personal- und Sachaufwand; es gelten die Regelungen nach § 11.
§ 6 Voraussetzungen für die Gewährung sekundärer Datennutzung
Der G-BA gibt einem Antrag auf sekundäre Datennutzung statt, wenn
§ 7 Vorprüfung und Einschätzung durch die beauftragte Stelle
(1) Die jeweilige beauftragte Stelle nimmt Anträge auf Gewährung sekundärer Datennutzung entgegen, bestätigt unverzüglich der Antragstellerin oder dem Antragssteller den Eingang und führt eine Vorprüfung durch,
(2) Fehlende oder unzureichende Angaben oder Unterlagen fordert die jeweilige beauftragte Stelle bei der Antragstellerin oder dem Antragsteller unverzüglich nach. Werden bis sechs Monate nach Aufforderung keine weiteren Unterlagen eingereicht, gilt der Antrag als zurückgenommen. Der Antragstellerin oder dem Antragsteller wird der Eingang eines formal vollständigen Antrags durch die jeweilige beauftragte Stelle unverzüglich bestätigt.
(3) Sofern die Vorauszahlung für die Prüfung des Antrags gemäß § 11 i.V.m. Anlage III getätigt wurde, gibt die jeweilige beauftragte Stelle eine Stellungnahme ab, in der sie das Ergebnis der Vorprüfung nach Absatz 1 abbildet sowie eine zusammenfassende Einschätzung abgibt, ob das Vorhaben der sekundären Datennutzung durchführbar ist. Die jeweilige beauftragte Stelle übermittelt die Stellungnahme zusammen mit den Antragsunterlagen und einer Prognose des bei der Durchführung der sekundären Datennutzung zu erwartenden Personal- und Sachaufwandes sowie der angefallenen Kosten für die Antragsprüfung inklusive der anfallenden gesetzlichen Mehrwertsteuer gemäß § 11 in Verbindung mit Anlage III spätestens acht Wochen nach Vorliegen des formal vollständigen Antrags elektronisch an die Geschäftsstelle des G-BA.
§ 8 Verfahren der Entscheidung durch den Gemeinsamen Bundesausschuss
(1) Der sektorenübergreifend besetzte Unterausschuss Qualitätssicherung (UA QS) prüft den Antrag gemäß § 6 unter Berücksichtigung der Stellungnahme nach § 7 Absatz 3 und soll in der nächsten fristgerecht erreichbaren Sitzung entscheiden.
(2) Ober die Entscheidung der beantragten Gewährung der sekundären Datennutzung ist ein Bescheid zu erteilen der die Durchführung durch die jeweilige beauftragte Stelle gegenüber der Antragstellerin oder dem Antragsteller genehmigt. Diesem Bescheid werden die Prognose des zu erwartenden Personal- und Sachaufwandes sowie eine Aufstellung über die angefallenen Kosten für die Antragsprüfung gemäß § 7 Absatz 3 Satz 2 zur Information der Antragstellerin oder des Antragstellers beigefügt.
(3) Die Geschäftsstelle des G-BA informiert die jeweilige beauftragte Stelle über die Entscheidung und veranlasst diese bei stattgebender Entscheidung (genehmigter Antrag), die sekundäre Datennutzung durchzuführen.
§ 9 Verfahren der Auswertung der Daten durch die beauftragte Stelle
(1) Für einen genehmigten Antrag wertet die jeweilige beauftragte Stelle die Daten antragsgemäß aus und übermittelt die Auswertungsergebnisse an die Antragstellerin oder den Antragsteller unter Einhaltung der Anforderungen an den Datenschutz nach § 4.
(2) Die jeweilige beauftragte Stelle informiert die Geschäftsstelle des G-BA über die Durchführung der sekundären Datennutzung und den dabei entstandenen Personal- und Sachaufwand gemäß § 11 in Verbindung mit Anlage III.
§ 10 Veröffentlichung
(1) Das IQTIG veröffentlicht alle genehmigten Anträge und Veröffentlichungen aus der sekundären Datennutzung auf dessen Internetseiten gemäß Absatz 2 und 3. Die notwendigen Informationen werden vom G-BA an das IQTIG weitergeleitet, sofern das IQTIG nicht selbst die beauftragte Stelle ist.
(2) Der G-BA und das IQTIG veröffentlichen nach Genehmigung des Antrags durch den G-BA die Kontaktdaten der Antragstellerin oder des Antragstellers, den Titel sowie eine Kurzdarstellung des Forschungsprojekts entsprechend der Anlage I zum 8. Kapitel der VerfO zusammen mit der Selbsterklärung der Antragstellerin oder des Antragstellers zu potentiellen Interessenkonflikten auf ihren Internetseiten.
(3) Die Antragstellerin oder der Antragsteller ist verpflichtet, dem G-BA die veröffentlichten Ergebnisse in Form wissenschaftlicher Publikationen, die aus der Nutzung der Daten resultieren oder diese zum Gegenstand haben, unverzüglich nach Veröffentlichung zur Verfügung zu stellen. Die Antragstellerin oder der Antragsteller stimmt einer Veröffentlichung der Publikation - soweit die Rechte Dritter nicht berührt werden - oder zumindest des Quellennachweises der oben genannten Ergebnisse auf den Internetseiten des IQTIG zu. In Fällen, bei denen nach Ablauf von 2 Jahren nach Übermittlung der Auswertungsergebnisse an die Antragstellerin oder den Antragsteller keine Informationen über Publikationen der Ergebnisse durch die Antragstellerin oder den Antragsteller an den G-BA eingegangen sind, wird das IQTIG diese zur Einreichung vorhandener Publikationen auffordern. Das IQTIG veröffentlicht diese Aufforderung sowie die Rückmeldung der Antragstellerin oder des Antragstellers auf seinen Internetseiten. In jeder Publikation und Präsentation (z.B. Vortrag) ist wie folgt auf die Datenquelle hinzuweisen: "Es wurden Daten aus Qualitätssicherungsverfahren gemäß § 136 SGB V des Gemeinsamen Bundesausschusses verwendet."
§ 11 Kosten
Die Kosten für den durch die Vorprüfung und Einschätzung des Antrags gemäß § 7 sowie die Durchführung der Auswertung gemäß § 9 entstandenen Personal- und Sachaufwand sind von der Antragstellerin oder vom Antragsteller zu tragen. Diese tatsächlich anfallenden Kosten können die der Antragstellerin oder dem Antragsteller gemäß § 8 Absatz 2 mitgeteilten prognostizierten Kosten übersteigen. Die jeweilige beauftragte Stelle macht die nach Satz 1 anfallenden Kosten gegenüber der Antragstellerin oder dem Antragsteller geltend. Nach Abschluss der Auswertung gemäß § 9 wird dazu von der jeweiligen beauftragten Stelle eine Schlussrechnung erstellt. Die bereits mit Antragstellung fällige Vorauszahlung wird in der Schlussrechnung mit den Kosten nach Satz 1 verrechnet. Die jeweilige beauftragte Stelle ist berechtigt, Zwischenrechnungen zu erstellen. Das Nähere ist in der Kostenordnung (Anlage III) geregelt.
§ 12 Auswertung
Zum Zwecke der Optimierung des oben beschriebenen Verfahrens zur sekundären Datennutzung kann der G-BA die jeweils beauftragte Stelle mit einer Auswertung beauftragen.
2. Abschnitt 18b
Verfahren zur Festlegung von Mindestmengen gemäß § 136b Absatz 1 Nummer 2 SGB V
1. Titel
Geltungsbereich und Zuständigkeiten
§ 13 Geltungsbereich
Dieser Abschnitt regelt gemäß § 136b Absatz 3 Satz 2 SGB V das Verfahren des G-BA für Beschlüsse nach § 136b Absatz 1 Satz 1 Nummer 2 SGB V, mit dem Ziel, das Verfahren und die Entscheidung transparent zu machen. Geregelt werden insbesondere das Nähere zur Auswahl einer planbaren Leistung sowie zur Festlegung der Höhe einer Mindestmenge.
§ 14 Grundzüge des Verfahrens und Zuständigkeit
(1) Dieses Verfahren untergliedert sich insbesondere in folgende Schritte:
(2) Für die Durchführung des Verfahrens ist der Unterausschuss Qualitätssicherung zuständig. Er nimmt unter regelhafter Einbeziehung vorbereitender Arbeitsgruppen die Aufgaben nach Absatz 1 zur Beratung wahr und legt dem Plenum seine Beschlussempfehlungen zur Entscheidung vor.
2. Titel
Verfahren zur Auswahl planbarer Leistungen und Festlegung von Mindestmengen
§ 15 Antrag
(1) Einen Antrag zur Festlegung einer Mindestmenge können folgende Personen oder Organisationen stellen:
(2) Die Ländervertreter haben das Recht, Mindestmengen zur Beratung auf die Tagesordnung setzen zu lassen.
(3) Der Antrag ist in Textform beim G-BA einzureichen. Die Geschäftsstelle prüft die Antragsberechtigung und die formale Vollständigkeit der Angaben nach Absatz 4 .Sie wirkt auf die formale Vollständigkeit des Antrags hin. 4Sie legt dem Unterausschuss den Antrag zusammen mit dem Ergebnis der Prüfung in der nächsten fristgerecht erreichbaren Sitzung zur Beratung vor.
(4) Der Antrag muss
(5) Der Unterausschuss prüft die Zulässigkeit und Eignung der Begründung des Antrags und gibt eine Empfehlung an das Plenum zur Einleitung des Beratungsverfahrens oder die Ablehnung des Antrags ab. Vor einer Ablehnungsempfehlung kann der Unterausschuss die Antragstellerin oder den Antragssteller zur Ergänzung oder Präzisierung des Antrags innerhalb einer angemessenen Frist auffordern. Der Unterausschuss prüft die Notwendigkeit der Einbindung fachlich unabhängiger wissenschaftlicher Institute oder Experten und gibt hierzu ebenfalls eine Empfehlung an das Plenum ab. Wenn der Unterausschuss in der Sitzung, in der über den Antrag erstmalig beraten wird, noch keine Empfehlung an das Plenum gemäß Satz 1 und Satz 3 abgeben kann, soll er in dieser Sitzung das Vorgehen zur weiteren Bearbeitung des Antrags und Vorbereitung der Beschlussempfehlung an das Plenum festlegen.
(6) Das Plenum entscheidet über den Antrag durch Beschluss. Das Plenum kann ein bereits eingeleitetes Beratungsverfahren an jedem Punkt des Verfahrens beenden.
§ 16 Feststellung der Mindestmengenfähigkeit einer Leistung
(1) Mindestmengen können gemäß § 136b Absatz 1 Satz 1 Nummer 2 SGB V für planbare Leistungen festgelegt werden, bei denen die Qualität des Behandlungsergebnisses von der Menge der erbrachten Leistungen abhängig ist.
(2) Eine Leistung im Sinne von Absatz 1 ist planbar, wenn sie in der Regel in dafür vorgesehenen Krankenhäusern medizinisch sinnvoll und für die Patientinnen und Patienten zumutbar erbracht werden kann. Dies setzt voraus, dass die Aufnahme und Durchführung der gebotenen stationären Behandlung in einem dafür vorgesehenen Krankenhaus unter Berücksichtigung zu überwindender räumlicher und zeitlicher Distanzen ohne unzumutbares Risiko für die Patientinnen und Patienten erfolgen kann.
(3) Die erforderliche Abhängigkeit im Sinne von Absatz 1 setzt voraus, dass eine Studienlage besteht, die auf einen wahrscheinlichen Zusammenhang zwischen Behandlungsmenge und Ergebnisqualität der Leistung hinweist. Erforderlich ist, dass der aktuelle Erkenntnisstand gemäß Absatz 5 insbesondere eine Reduzierung von Behandlungsrisiken und Steigerung der Patientensicherheit erwarten lässt.
(4) Die Leistung, für die eine Mindestmenge festgelegt werden soll, ist konkret und eindeutig zu benennen. Sie ist in der Regel durch Verwendung der Medizinischen Klassifikationssysteme ICD-10-GM (Diagnosen) oder OPS (Prozeduren) und sofern erforderlich auf Basis ausgewählter Merkmale aus der Datensatzbeschreibung gemäß § 301 SGB V zu operationalisieren.
(5) Über die Erfüllung der Anforderungen an eine Leistung nach den Absätzen 1 bis 3 wird auf Grundlage folgender Informationen entschieden:
Bei der Entscheidung über die Mindestmengenfähigkeit einer Leistung sind die Güte und der Evidenzgrad der Informationen nach Satz 1 zu berücksichtigen.
§ 17 Festlegung der Höhe und des Bezugs von Mindestmengen
(1) Bezugspunkt einer Mindestmengenfestlegung ist entweder die Ärztin oder der Arzt oder der Standort eines Krankenhauses oder eine Kombination von Ärztin oder Arzt und Krankenhausstandort. Für die Festlegung des Bezugspunktes sind die Informationen nach § 16 Absatz 5 zu nutzen. Eine nur auf die Ärztin oder den Arzt bezogene Mindestmenge kann festgelegt werden, wenn vorrangig deren oder dessen besondere Qualifikation und Erfahrung für die Qualität des Behandlungsergebnisses maßgeblich sind. Eine auf den Standort des Krankenhauses bezogene Mindestmenge kann festgelegt werden, wenn die interdisziplinäre Versorgung der Patientin oder des Patienten im Team für die Qualität des Behandlungsergebnisses maßgeblich ist. Eine Kombination einer Ärztin- oder Arztbezogenen mit einer Krankenhausstandortbezogenen Mindestmenge kann gewählt werden, wenn sowohl die besondere Qualifikation und Erfahrung der einzelnen Ärztin oder des einzelnen Arztes als auch die interdisziplinäre Versorgung im Team für die Qualität des Behandlungsergebnisses der Leistung maßgeblich sind. Die Festsetzung des Bezugspunktes erfolgt bei Festlegung einer Mindestmenge. Die für diesen Bezugspunkt maßgebliche Zählweise der jeweiligen Leistung zur Darrlegung der Prognose legt der G-BA in den Mindestmengenregelungen und deren Anlage fest. Für Änderungen des Bezugspunktes in einer bestehenden Mindestmengenfestlegung gilt § 21.
(2) Für die Festlegung der Höhe der Mindestmenge einer gemäß § 16 Absätze 1 bis 3 mindestmengenfähigen Leistung sind die jeweils durch die Regelung konkret betroffenen Belange gegeneinander und untereinander abzuwägen. Hierzu werden die Belange, die für die Abwägung von Bedeutung sind, auf der Grundlage der Informationen nach § 16 Absatz 5 ermittelt und in einer Gesamtschau bewertet. Für die Bewertung können insbesondere folgende Belange von Bedeutung sein:
Der G-BA prüft, ob zur Durchführung einer Folgenabschätzung eine Beauftragung des Instituts nach § 137a SGB V erforderlich ist.
(3) Bei Hinweisen auf eine Reduzierung von Behandlungsrisiken und Steigerung der Patientensicherheit soll der G-BA bei der Festlegung der Höhe der Mindestmenge zumindest eine Gelegenheitsversorgung ausschließen.
(4) Festlegungen zu neuen Mindestmengen oder Änderungen bestehender Mindestmengen treten zum 1. Januar eines Kalenderjahres in Kraft.
§ 18 Ausnahmetatbestände und Übergangsregelungen
Der G-BA soll gemäß § 136b Absatz 3 Satz 1 SGB V bei Mindestmengenfestlegungen Ausnahmetatbestände und Übergangsregelungen vorsehen. Das Nähere bestimmen die Mindestmengenregelungen.
§ 19 Begleitevaluation
(1) Der G-BA soll gemäß § 136b Absatz 3 Satz 3 SGB V möglichst zeitnah mit Festlegung einer neuen Mindestmenge eine wissenschaftliche Begleitevaluation beauftragen. Er kann hiervon nur dann absehen, wenn die Studienlage eindeutig ist und die Mindestmenge auf eine klare wissenschaftliche, durch hochwertige Studien gesicherte Evidenz zu den Versorgungsvor- und -nachteilen gestützt werden kann. Die Evaluation durch ein fachlich unabhängiges Institut dient
(2) Die Auswirkungen der Festlegung neuer Mindestmengen sollen vom G-BA in regelmäßigen Abständen, erstmalig drei Jahre nach Beschluss einer neuen Mindestmenge, anhand der Ergebnisse der Begleitevaluation bewertet werden.
(3) Die grundsätzliche Verpflichtung des G-BA zur Evaluation bereits bestehender Mindestmengen gemäß § 136d SGB V bleibt hiervon unberührt.
(4) Der Abschlussbericht über die wissenschaftliche Begleitevaluation ist nach Freigabe durch den G-BA durch das beauftragte fachlich unabhängige Institut zu veröffentlichen.
§ 20 Zusammenfassende Dokumentation
Über das Verfahren ist eine zusammenfassende Dokumentation zu erstellen. Die zusammenfassende Dokumentation enthält:
Unterschiedliche Positionen der Träger nach § 91 Absatz 1 Satz 1 SGB V und der Patientenvertretung werden bei der Dokumentation der Abschnitte zu Satz 1 Nummer 3 dargestellt. Abweichende Beschlussentwürfe werden zusammen mit ihrer Begründung in die zusammenfassende Dokumentation aufgenommen.
3. Titel
Wiederaufnahme der Beratungen
§ 21 Überprüfung der Regelungen für bestehende Mindestmengen und Überarbeitungsverfahren
(1) Der Unterausschuss überprüft die bestehenden Mindestmengenregelungen in regelmäßigen Abständen auf einen möglichen Änderungsbedarf. Ein Änderungsbedarf kann sich insbesondere aus
ergeben.
(2) Erkennt der Unterausschuss einen Änderungsbedarf, empfiehlt er dem Plenum die Wiederaufnahme des Beratungsverfahrens.
(3) Eine Wiederaufnahme der Beratungen kann auch durch Personen oder Organisationen gemäß § 15 Absatz 1 beantragt werden. Der Antrag ist in Textform beim G-BA einzureichen und soll eine Begründung für den Änderungsbedarf im Sinne von Absatz 1 Satz 2 enthalten. Absatz 2 gilt entsprechend.
(4) Über die erforderlichen OPS- sowie ICD-Anpassungen durch die jährliche Aktualisierung des Deutschen Instituts für Medizinische Dokumentation und Information entscheidet der Unterausschuss, soweit gemäß 1. Kapitel § 4 Absatz 2 Satz 2 der Verfahrensordnung der Kerngehalt der Mindestmengenregelungen nicht berührt wird.
(5) Für das wiederaufgenommene Beratungsverfahren (Überarbeitungsverfahren) gelten die §§ 16 bis 20 entsprechend.
| Antrag auf Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten | Anlage I |
Allgemeine Hinweise:
Beauftragte Stelle (vom Antragsteller einzutragen):
Ich beantrage die Gewährung der sekundären Datennutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten und stimme den Regelungen des 8. Kapitels Abschnitt "Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten" zu. Ich verpflichte mich damit insbesondere zur Kostentragung für die Vorprüfung und Einschätzung des Antrags sowie die Auswertung der Daten durch die jeweils beauftragte Stelle gemäß 8. Kapitel § 11 VerfO in Verbindung mit Anlage 3 (Kostenordnung - Anlage 3 zum 8. Kapitel VerfO).
| _______________________________ Datum, Ort | _______________________________ Unterschrift |
Ich stimme Folgendem zu:
| _______________________________ Datum, Ort | _______________________________ Unterschrift |
| Antragssteller | Name, Vorname, Titel des Antragstellers oder der Antragstellerin * | |
| Name der Institution oder Organisation (sofern möglich) * | ||
| Funktion des Antragstellers oder der Antragstellerin in der Institution oder Organisation | ||
| Abteilung oder Bereich (sofern möglich) | ||
| Straße und Hausnummer | ||
| Postleitzahl und Ort * | ||
| Telefon | ||
| Telefax (optional) | ||
| E-Mail * | ||
| Homepage (optional) | ||
| Titel und Kurzdarstellung des Projektes und der Fragestellung (max. 2000 Zeichen) für die Veröffentlichung gemäß 8. Kapitel § 10 VerfO* | ||
| Projektbeschreibung und Verwendungszweck | Angabe des Verwendungszwecks der Daten
Hier muss der Bezug a) zur wissenschaftlichen Forschung oder b) zur Weiterentwicklung der Qualitätssicherung dargelegt werden. | |
| Ausführliches Exposé des Projekts zur wissenschaftlichen Forschung oder zur Weiterentwicklung der Qualitätssicherung | ||
Von der Antragstellerin oder dem Antragsteller wird erwartet, dass sie oder er
Auf Grundlage des Exposés muss der beauftragten Stelle die konkrete Durchführung des Antrags möglich sein. (Bitte Exposé als Anlage beifügen). | ||
*) Diese Angaben werden gemäß 8. Kapitel § 10 Absatz 2 Satz 1 VerfO veröffentlicht.
Bitte beschreiben Sie Ihr Projekt einschließlich seiner Zielsetzung und Methodik ausführlich in einem Exposé.
Folgende Unterlagen sind meinem Antrag als Anlage beigefügt:
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
__________________________________________________________________________________________
Hiermit bestätige ich, dass meine Angaben nach bestem Wissen wahrheitsgemäß und vollständig sind.
| _______________________________ Datum, Ort | _______________________________ Unterschrift |
| Selbsterklärung zu Potentiellen Interessenkonflikten zu Anträgen auf Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten | Anlage II |
Allgemeine Hinweise:
Selbsterklärung zu potentiellen Interessenkonflikten:
Ich willige ein, dass diese Selbsterklärung zu potentiellen Interessenkonflikten gemäß 8. Kapitel § 10 Absatz 2 und 3 VerfO auf den Internetseiten des IQTIG veröffentlicht wird.
| _______________________________ Datum, Ort | _______________________________ Unterschrift |
Hiermit bestätige ich, dass meine Angaben nach bestem Wissen wahrheitsgemäß und vollständig sind.
| _______________________________ Datum, Ort | _______________________________ Unterschrift |
___
1) Hierbei sind finanzielle oder geldwerte Vorteile von über 250 Euro zu berücksichtigen.
| Kostenordnung für die Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten | Anhang III |
§ 1 Regelungsbereich
(1) Der Gemeinsame Bundesausschuss (G-BA) hat nach § 137a Absatz 10 Satz 4 SGB V ein Verfahren zur Kostenübernahme für Leistungen nach 8. Kapitel Abschnitt (Gewährung der sekundären Nutzung der bei den verpflichtenden Maßnahmen der Qualitätssicherung nach § 136 Absatz 1 Satz 1 Nummer 1 SGB V erhobenen Daten) zu regeln. Mit der hier vorliegenden Kostenordnung wird dieser Regelungsauftrag durch den G-BA umgesetzt.
(2) Nach § 137a Absatz 10 Satz 3 SGB V setzt die Gewährung der sekundären Datennutzung die Übernahme der entstehenden Kosten durch die Antragstellerin oder den Antragsteller gemäß 8. Kapitel § 5 Absatz 1 i.V.m. § 11 VerfO voraus. Die zu übernehmenden Kosten umfassen den entstandenen Personal- und Sachaufwand für
§ 2 Kosten der Antragsprüfung
(1) Mit der Antragstellung hat die Antragstellerin oder der Antragsteller eine Vorauszahlung für die Prüfung des Antrags in Höhe von 500 Euro zzgl. der gesetzlichen Mehrwertsteuer (MwSt.) zu entrichten. Ein ermäßigter Betrag von 250 Euro zzgl. MwSt. wird für Studierende erhoben. Die jeweilige beauftragte Stelle erteilt der Antragstellerin oder dem Antragsteller mit dem Eingang des Antrags gemäß 8. Kapitel § 7 Absatz 1 VerfO zusätzlich zur Eingangsbestätigung eine den umsatzsteuerrechtlichen Vorgaben genügende Rechnung über die entrichtete Vorauszahlung. Der Eingang dieser Vorauszahlung bei der jeweiligen beauftragten Stelle ist gemäß 8. Kapitel § 7 Absatz 3 Satz 1 i.V.m. § 8 Absatz 1 und 3 VerfO Bedingung für die Prüfung des Antrags und den Beginn der Auswertung der Daten. Die tatsächlichen Kosten der Antragsprüfung und die Kostenprognose nach § 3 werden der Antragstellerin oder dem Antragssteller mit der Entscheidung des G-BA über den Antrag mitgeteilt.
(2) Die jeweilige beauftragte Stelle hat auf ihrer Internetseite die Bankverbindung und die weiteren Zahlungsmodalitäten für die Vorauszahlung zu veröffentlichen und den Eingang der Vorauszahlung gegenüber der Antragstellerin oder dem Antragsteller zu bestätigen.
§ 3 Prognose des entstehenden Personal- und Sachaufwandes
(1) Die jeweils beauftragte Stelle hat gemäß 8. Kapitel § 7 Absatz 3 eine Prognose des bei der Durchführung der sekundären Datennutzung zu erwartenden Personal- und Sachaufwandes und die angefallenen Kosten für die Antragsprüfung abzugeben. Diese Prognose hat auf der Grundlage des von der jeweiligen beauftragten Stelle zu erarbeitenden Preisblattes zu erfolgen. Im Preisblatt hat die jeweilige beauftragte Stelle die einzelnen Nettokostenpositionen (wie Stundensätze, Materialkosten usw.) konkret aufzulisten.
(2) Sollte der tatsächliche Aufwand wesentlich die Prognose des entstehenden Personal- und Sachaufwandes überschreiten, so ist die Antragstellerin oder der Antragsteller durch die jeweils beauftragte Stelle hierüber zeitnah zu unterrichten.
(3) Die Erstfassung des Preisblattes sowie jede nachfolgende inhaltliche Änderung sind dem Unterausschuss Qualitätssicherung des G-BA zur Kenntnis zu geben. Die jeweilige beauftragte Stelle hat das Preisblatt auf ihrer Internetseite zu veröffentlichen.
§ 4 Rechnungsstellung
(1) Nach erfolgter Auswertung der Daten gemäß 8. Kapitel § 9 VerfO hat die jeweilige beauftragte Stelle eine Schlussrechnung über die insgesamt angefallenen Kosten entsprechend dem tatsächlich entstandenen Personal- und Sachaufwand auf der Grundlage des Preisblattes zu erstellen, die den Anforderungen der umsatzsteuerrechtlichen Regelungen an eine Rechnung genügt. Übersteigt die Vorauszahlung gemäß § 2 die Kosten des bis dahin entstandenen Personal- und Sachaufwandes, wird der überschießende Betrag erstattet.
(2) Die jeweilige beauftragte Stelle ist berechtigt, Zwischenrechnungen zu erstellen.
(3) Die Kosten werden mit Übersendung der Zwischen- und Schlussrechnung an die Antragstellerin oder den Antragsteller zzgl. MwSt. geltend gemacht. Dabei erfolgt die Anrechnung der bereits getätigten Vorauszahlung gemäß § 2. Die Zahlungsfrist wird in der jeweiligen Rechnung mitgeteilt.
§ 5 Kosten bei Rücknahme und Ablehnung des Antrags
(1) Wird ein Antrag auf Gewährung der sekundären Datennutzung nach § 137a Absatz 10 SGB V nach Beginn und vor Abschluss der Antragsbearbeitung zurückgenommen, so wird der bis dahin angefallene Personal- und Sachaufwand unter Anrechnung der bereits getätigten Vorauszahlung gemäß § 2 in Rechnung gestellt. Übersteigt die Vorauszahlung gemäß § 2 die Kosten des bis dahin entstandenen Personal- und Sachaufwandes, wird der überschießende Betrag erstattet.
(2) Wird ein Antrag auf Gewährung der sekundären Datennutzung nach § 137a Absatz 10 SGB V vom zuständigen Unterausschuss Qualitätssicherung abgelehnt, so wird der bis dahin angefallene Personal- und Sachaufwand unter Anrechnung der bereits getätigten Vorauszahlung gemäß § 2 in Rechnung gestellt. Absatz 1 Satz 2 gilt entsprechend.
Protokollnotizen
Protokollnotiz zu 1. Kapitel § 5 Abs. 4:
Der Gemeinsame Bundesausschuss strebt an, die von ihm verfassten und für die breite Öffentlichkeit bestimmten Dokumente - insbesondere die tragenden Gründe - möglichst allgemeinverständlich zu verfassen, um vor allem bei Versicherten das Verständnis seiner Entscheidungen zu erhöhen.
Protokollnotiz zu 1. Kapitel § 20 Abs. 1:
Im beidseitigen Interesse von Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen und Gemeinsamem Bundesausschuss sollen der Auftrag, die Ergebnisse der Auftragsbearbeitung und Informationen, die im Rahmen der Auftragsbearbeitung erhalten wurden, nicht vor Abschluss des Auftrages (Empfehlung) veröffentlicht werden. Soweit die wissenschaftliche Bearbeitung eines Auftrages es aber nach den Methoden des Instituts für Qualität und Wirtschaftlichkeit im Gesundheitswesen erforderlich macht, kann das Institut Vorberichte veröffentlichen. Diese Veröffentlichungen und die Veröffentlichungen von Empfehlungen sollen dem Gemeinsamen Bundesausschuss mit angemessenem Vorlauf angekündigt werden.
Protokollnotiz zu 2. Kapitel § 11 Abs. 6:
Als allgemein anerkannte Empfehlungen betrachtet der Gemeinsame Bundesausschuss bei Verabschiedung der Verfahrensordnung die internationalen Standards nach CONSORT, STARD und GRADE.
Protokollnotiz zu 3. Kapitel § 7:
Notwendigkeit wird i. S. d."medizinischen Notwendigkeit" nach § 116b Abs. 4 Satz 2 SGB V verstanden.
______
1) Das Common Technical Document (CTD) ist das international harmonisierte Format für em Zulassungsdossier.
Die genannten Abschnitte enthalten die Informationen über die klinischen Studien, die für die Zulassung eingereicht wurden.
2) Microsoft Word Version 2010
4) ASCII: American Standard Code for Information Interchange.
5) Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010; 340: c332.
6) Des Jarlais DC, Lyles C, Crepaz N. Improving the reporting quality of nonrandomized evaluations of behavioral and public health interventions: the TREND statement. Am J Publ Health 2004; 94(3): 361-366.
7) Von Elm E, Altman DG, Egger M, Pocock SJ, Geitsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Am Intern Med 2007; 147(8): 573-577.
8) DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7(3): 177-188.
9) Deeks JJ, Higgins JPT, Altman DG. Analysing data and undertaking metaanalyses. In: Higgins JPT, Green S (Ed). Cochrane handbook for systematic reviews of interventions. Chichester: Wiley; 2008. S. 43-296.
10) Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-560.
11) Glenny AM, Altman DG, Song F, Sakarovitch C, Deeks JJ, DAmico R et al. Indirect comparisons of competing interventions. Health Technol Assess 2005; 9(26): 1-148.
12) Lu G, Ades AE. Combination of direct and indirect evidence in mixed treatment comparisons. Stat Med 2004; 23(20): 3105-3124.
13) Caldwell DM, Ades AE, Higgins JP. Simultaneous comparison of multiple treatments: combining direct and indirect evidence. BMJ 2005; 331(7521): 897-900.
14) Salanti G, Higgins JPT, Ades AE, Ionnnidis JPA. Evaluation of networks ofrandomized trials. Stat Methods Med Res 2008;17(3): 279-301.
15) B. Schöttker, D. Lühmann, D. Boulkhemair, and H. Raspe. Indirekte Vergleiche von Therapieverfahren. Schriftenreihe Health Technology Assessment Band 88, DIMDI, Köln, 2009.
16) Song F, Loke YK, Walsh T, Glenny AM, Eastwood AJ, Altman DJ. Methodological problems in the use of indirect comparisons for evaluating healthcare interventions: survey of published systematic reviews. BMJ 2009; 338: b1147.
17) Burzykowski T (Ed.: The evaluation of surrogate endpoints. New York: Springer; 2005.
18) Molenberghs G, Burzykowski T, Alonso A, Assam P, Tilahun A, Buyse M: A unified framework for the evaluation of surrogate endpoints in mentalhealth clinical trials. Stat Methods Med Res 2010; 19(3): 205-236.
19) Burzykowski T, Buyse M: Surrogate threshold effect: an alternative measure for meta-analytic surrogate endpoint validation. Pharm Stat 2006; 5(3): 173-186.
20) Weir CJ, Walley RJ. Statistical evaluation of biornarkers as surrogate endpoints: a literature review. Stat Med 2006; 25(2): 183-203.
21) Das Zitat zu dem hier beispielhaft angegebenen Suchfilter lautet wie folgt: Wong SSL, Wilczynski NL, Haynes RB. Comparison of top-performing search strategies for detecting clinically sound treatment studies and systematic reviews in MEDLINE and EMBASE. J Med Libr Assoc 2006; 94(4): 451-455.
22) Dazu gehört auch die Tabelle 2.7.3.1 "Description of Clinical Efficacy and Safety Studies", auch wenn sie nicht in Abschnitt 2.7.3, sondern in einem Appendix abgelegt ist.
23) Dabei sind nur die für das betreffende Verfahren tatsächlich erstellten Bewertungsberichte zu berücksichtigen, auf Basis der im Abschlussbericht beschriebenen Schritte bei der Bewertung. Diese umfassen nicht immer einen Bewertungsbericht zu Tag 180.
24) Sofern für die Abschnitte 3.2 oder 3.3 von Modul 3 eine bibliografische Literaturrecherche durchgeführt wurde, können weitere Unterordner zur Dokumentation der Informationsbeschaffung ergänzt werden. Die Dokumentation sollte dann analog der Dokumentation der bibliografischen Literaturrecherche für Modul 4 erfolgen (s. u.).
25) Falls Tabelle 2.7.3.1 nicht Bestandteil des Abschnitts 2.7.3 ist, ist diese Tabelle separat abzulegen (Benennung: M4#A-Z#_CTD-Tabelle2-7-3-1.pdf; siehe auch Abschnitt 3.1.
26) Das Zitat zu dem hier beispielhaft angegebenen Suchfilter lautet wie folgt: Wong SSL, Wilczynski NL, Haynes RB. Comparison of top-performing search strategies for detecting clinically sound treatment studies and systematic reviews in MEDLINE and EMBASE. J Med Libr Assoc 2006; 94(4): 451-455. Hinweis: Für die Suche in der Cochrane-Datenbank "Cochrane Central Register of Controlled Trials (Clinical Trials)" sollte kein Studienfilter verwendet werden.
| ENDE | |