 Für einen individuellen Ausdruck passen Sie bitte die
Für einen individuellen Ausdruck passen Sie bitte dieEinstellungen in der Druckvorschau Ihres Browsers an. Regelwerk

| Für einen individuellen Ausdruck passen Sie bitte die Einstellungen in der Druckvorschau Ihres Browsers an. Regelwerk | |
Handbuch für GLP-Inspektionen
Die Durchführung von GLP-Inspektionen in Deutschland
- BLAC-AS GLP -
11. Auflage Januar 2018
(Publikationen BLAC)
Bund/Länder-Arbeisgemeinschaft Chemikaliensicherheit
BLAC-AS GLP - Ausschuss "GLP und andere Qualitätssicherungs-Systeme"
Mit der Novellierung des Chemikaliengesetzes im Jahr 1990 wurde die Gute Laborpraxis in deutsches Recht überführt. Das erste Handbuch erschien 1997 und sollte eine Anleitung zur Durchführung von GLP-Inspektionen sein. Dazu wurden verschiedene GLP-Dokumente erfasst, die zur Präzisierung von Fragestellungen erstellt worden waren. Es stellte einen Überblick über die gesetzlichen Regelwerke verbunden mit dem Erfahrungsschatz von Inspektorinnen und Inspektoren aus der Praxis dar. Seit dieser Zeit erfolgten etliche Änderungen und Neuregelungen im gesetzlichen Bereich. Gleichzeitig kam von Seiten der Inspektorinnen und Inspektoren eine Vielzahl von Anregungen und Hinweise, so dass es nach 20 Jahren an der Zeit war, eine umfassende Überarbeitung vorzunehmen. Diese bestand im Wesentlichen aus der Aktualisierung der rechtlichen Grundlagen, der Überarbeitung der Anhänge und einer Neugliederung. Nach wie vor soll das Handbuch die Inspektorinnen und Inspektoren bei der Vorbereitung und Durchführung von GLP-Inspektionen unterstützen.
Hinweis
Das vorliegende Handbuch einschließlich seiner Anlagen erhebt keinen Anspruch auf Vollständigkeit und besitzt keinen rechtsverbindlichen Charakter. Rechtlich verbindlich ist nur der Wortlaut der jeweiligen Rechtsvorschriften.
BLAC-AS GLP, AG "GLP-Handbuch"
Die AG bedankt sich herzlich bei allen Kolleginnen und Kollegen, die die Grundlagen für das Handbuch gelegt haben, und bei denen, die für die Überarbeitung wertvolle Beiträge geleistet haben.
Anmerkung zur Nummerierung:
Um das Zitieren von einzelnen Passagen des Handbuchs zu erleichtern, sind die Kapitel nummeriert. Auch der Text ist in nummerierte Abschnitte unterteilt.
Die ersten drei Nummerierungsebenen sind den Überschriften vorbehalten, die vierte Nummerierungsebene dem Text. Dies ist konsequent durchgehalten, auch wenn dadurch in manchen Kapiteln nicht alle Nummerierungsebenen genutzt werden.
| AB | Abschlussbericht |
| AV | Archiwerantwortliche/r |
| DV | Datenverarbeitung |
| GLP | Gute Laborpraxis |
| IC | In Compliance (den GLP-Grundsätzen entsprechend) |
| LPE | Leitung der Prüfeinrichtung (Management) |
| LPSt | Leitung des Prüfstandorts |
| MS | Master Schedule |
| MSP | Multi-Site-Prüfung |
| NIC | Not in Compliance (nicht den GLP-Grundsätzen entsprechend) |
| PE | Prüfeinrichtung (Test Facility) |
| PP | Prüfplan |
| PSt | Prüfstandort (Test Site) |
| PL | Prüfleiter/in (Study Director) |
| PI | Principal Investigator (örtliche/r Versuchsleiter/in) |
| PRG | Prüf- und Referenzgegenstände |
| QS | Qualitätssicherung (Quality Assurance) |
| SOP | Standardarbeitsanweisung (Standard Operating Procedure) |
I. GLP-Regelwerk (National und International)
Das internationale Regelwerk der Guten Laborpraxis (GLP) basiert auf den OECD-Ratsentscheidungen von 1981 und 1989, die mit ihren Anhängen (Grundsätze der Guten Laborpraxis, Leitfaden für die Verfahren zur Überwachung der Einhaltung der GLP, Leitlinien für die Durchführung von Inspektionen einer Prüfeinrichtung und die Überprüfung von Prüfungen) mit den Richtlinien 2004/10/EG (87/18/EWG) und 2004/9/EG (88/320/EWG) in europäisches Recht übernommen wurden. Die Umsetzung in deutsches Recht erfolgte durch das Chemikaliengesetz 2013 (1990) und die Allgemeine Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis 2011 (1990).
Ausführliche international abgestimmte Erläuterungen und Interpretationen zur GLP sind in der OECD Series on GLP zusammengefasst, die u. a. auf der Internetseite der OECD veröffentlicht sind. Hierzu gehören neben den für OECD-Mitgliedstaaten völkerrechtlich verbindlichen OECD-Ratsentscheidungen (OECD-GLP-Dokumente 1, 2, 3) eine Anzahl weiterer Dokumente, die wenn auch nicht per se rechtlich verpflichtend, so doch durch unterschiedliche Einbindung verschiedener Adressaten und einstimmiger Beschlussfassung auf OECD-Ebene von großer Aussagekraft sind.
OECD Series on GLP
GLP-Grundsätze (1)
Compliance Monitoring (2,3)
Konsensdokumente (4, 5, 6, 7, 8, 10, 13)
Advisory Dokumente (11, 12, 14, 15, 16, 17)
Position Papers (18, sowie Accreditation (1994) und Outsourcing inspection functions (2006))
Frequently Asked Questions (FAQs) (seit 2014 kontinuierlich ergänzt)
II Durchführung von GLP-Inspektionen
Gemäß Verwaltungsvorschrift ist die GLP-Inspektion eine vor Ort durchgeführte Besichtigung der Prüfeinrichtung (PE). Die Befragung von Beschäftigten und die Einsichtnahme in die Dokumentation sind die wichtigsten Tätigkeiten. Die nachfolgenden Ausführungen sollen die Durchführung des Verfahrens unterstützen und einzelne Punkte näher erörtern.
1. Vorbereitung einer Inspektion
1.1 Fragen an die Prüfeinrichtung/den Prüfstandort vor einer Inspektion
1.1.1.1. Typ, Größe der PE (z.B. Anzahl der Beschäftigten, Prüfungsumfang, evtl. Auftraggeber)
1.1.1.2. Art der Prüfungen/Prüfkategorien (evtl. Spezifizierung bzw. Einschränkungen von Prüfkategorien), siehe auch Erläuterungen in Anhang 7 (Prüfkategorien)
1.1.1.3. Erklärung, ob Prüfungen nach § 19a bzw. § 19b ChemG stattfinden (GLP-Pflicht bzw. berechtigtes Interesse)
1.1.1.4. Organisationsstruktur (Veränderungen seit der letzten Inspektion):
1.2. Bildung des Inspektionsteams
1.2.1.1. Mindestens zwei Inspektorinnen/Inspektoren (Leitung festlegen); i. d. R. fachlich ausgerichtet auf die Art der Prüfungen, die in der PE durchgeführt werden
1.2.1.2. Bei Tierversuchen: i. d. R. Teilnahme einer Tierärtin/eines Tierarztes bzw. einer Inspektorin/eines Inspektors mit Ausbildung als Veterinär/in oder vorherige Prüfung der Einhaltung der Tierschutzbestimmungen durch die zuständige Behörde
1.2.1.3. Ggf. Teilnahme von Sachverständigen (Vertraulichkeit muss gewahrt und schriftlich abgesichert sein)
1.2.1.4. Ggf. um Amtshilfe ersuchen, wenn abhängige PSt, eigenständige PSt oder PE in anderen Bundesländern an Prüfungen beteiligt sind. Falls PE bzw. PSt im Ausland einbezogen werden sollen, ist die GLP-Bundesstelle zu beteiligen.
1.3. Anforderung von Unterlagen und organisatorische Absprachen
Der Versand von Unterlagen elektronisch oder auf dem Postweg muss in Übereinstimmung mit Maßnahmen zur Wahrung der Vertraulichkeit erfolgen.
Zur Vorbereitung der Inspektion sollen insbesondere angefordert werden:
1.3.1.1. Master Schedule (MS): vollständige Liste aller laufenden, abgeschlossenen und abgebrochenen Prüfungen mit Angaben zu: Prüfungscodierung, Prüfgegenstand, Prüfsystem, Art der Prüfung, PL, Prüfungsbeginn, Prüfungsende bzw. Status der Prüfung, Auftraggeber, Anforderungen bzgl. Multi-Site-Prüfungen (MSP) (siehe Anhang 4) mindestens seit der letzten Inspektion (falls notwendig: GLP/nicht GLP-pflichtige Prüfungen)
1.3.1.2. Organigramme (Firmen-/GLP-Struktur; IT-Organisationsstruktur mit Darstellung der An- und Einbindung des IT-Managements in die GLP-Struktur), ggf. abhängige bzw. eigenständige PSt berücksichtigen, Liste des GLP-Personals
1.3.1.3. Standardarbeitsanweisungen (SOP) aus dem Bereich QS
1.3.1.4. Verzeichnis aller SOP, evtl. Kopien der wichtigsten SOP (siehe Anhang 1).
1.3.1.5. Liste der wichtigen Geräte (Geräte, einschließlich validierter computergestützter Systeme, die zur Gewinnung, Erfassung und Wiedergabe von Daten und zur Kontrolle der für die Prüfung bedeutsamen Umweltbedingungen verwendet werden) - siehe hierzu auch 1.3.1.7
1.3.1.6. Gebäude-/Lagepläne (Markierung von GLP-Bereichen); ggf. auch für abhängige PSt
1.3.1.7. Liste aller im Zusammenhang mit GLP-Prüfungen verwendeten computergestützten Systeme sowie Software (siehe Anhang 3)
1.3.1.8. Übersicht über IT-Struktur
Die Dokumente werden zusammen mit bereits vorliegenden Informationen, wie z.B. früheren Inspektionsberichten, Dokumentation zu Mängelbeseitigungen, Änderungsmitteilungen etc. überprüft, um sich mit der PE vertraut zu machen.
Die Informationen zu den Punkten 1.3.1.7 und 1.3.1.8 müssen nicht unbedingt vorab angefordert werden. Sie sollten jedoch zum Zeitpunkt der Vorinspektion vorliegen, um den Aufwand für die Inspektion von IT-Systemen im Rahmen der Hauptinspektion besser einschätzen zu können.
1.4. Vorinspektion
Eine Vorinspektion in der PE ist vor erstmaligen Inspektionen als technischorganisatorische Vorbereitung notwendig. Sie kann auch nach wesentlichen Änderungen in der PE hilfreich sein. Falls nicht alle unter 1.3 aufgeführten Dokumente vorab vorliegen, können diese bei der Vorinspektion in der PE eingesehen werden. Dies kann z.B. Informationen zu den Punkten 1.3.1.7 und 1.3.1.8 zutreffen, um den Aufwand für die Inspektion von IT-Systemen im Rahmen der Hauptinspektion besser einschätzen zu können.
1.4.1. Einführungsbesprechung
1.4.1.1. Teilnehmer: Inspektionsteam, LPE, PL, Leitung der QS, AV, ggf. PI/LPSt
1.4.1.2. Besprechung der angeforderten Unterlagen, Zuständigkeiten, Vertretungsregelungen, Aufgabenspektrum
1.4.1.3. Festlegung der genauen Bezeichnung der PE/PSt
1.4.1.4. Beteiligung von Externen an der Durchführung von Prüfungen
1.4.1.5. Festlegung der Bereiche der PE, die inspiziert werden sollen
1.4.2. Orientierende Begehung der Räume
1.4.3. Abschlussbesprechung
Wenn die Voraussetzungen für eine Hauptinspektion gegeben sind, wird abgeklärt:
1.4.3.1. Zeitpunkt und Umfang der Inspektion (Absprache über laufende Prüfungen während der Inspektion, ggf. Entscheidung über Inspektionen abhängiger PSt bzw. eigenständiger PSt)
1.4.3.2. Prüfkategorien (Art der Prüfungen) für die GLP-Bescheinigung
1.4.3.3. Gesprächspartner für die Inspektion
1.4.3.4. Bereitstellung eines separaten Arbeitsraumes und technischer Möglichkeiten zur Fertigung bzw. Vervielfältigung von Dokumenten (PC, Kopierer, Drucker) für das Inspektionsteam während der Inspektion
Bereits bei der Vorinspektion festgestellte Mängel sollten der PE mitgeteilt werden.
1.4.4. Anforderung von Unterlagen vor der Hauptinspektion
1.4.4.1. MS (ggf. aktualisiert, s. 1.3.1.1)
1.4.4.2. Muster eines Prüfplanes (PP), bzw. PP und Abschlussbericht (AB) einer oder mehrerer durchgeführter Prüfungen (s. 2.8.2)
1.4.4.3. Ausgewählte SOP (s. Anhang 1)
2. Inspektion einer Prüfeinrichtung bzw. eines Prüfstandortes
Bei Inspektionen von PSt sind relevante Punkte entsprechend zu inspizieren.
Der Ablauf der Inspektion hängt stark von den Gegebenheiten in der PE ab und kann, falls bei der Inspektion bestimmte Schwerpunkte gesetzt werden sollen, variieren. Vor der Vereinbarung des Inspektionstermins ist mit der PE abzuklären, dass
In der Regel ist folgender allgemeiner Ablauf einer Inspektion gängige Praxis.
Falls keine Vorinspektion vor Ort durchgeführt wurde, sind die in Abschnitt 1.4 genannten Punkte während der Inspektion abzuhandeln.
In der Regel führt das Inspektionsteam die Begehung gemeinsam durch (mindestens Zweiergruppen).
Befragung des GLP-Personals z.B. zu:
2.1. Organisation und Personal
2.1.1. Organisation
2.1.1.1. Darstellung der Organisationsstruktur unter GLP-Gesichtspunkten (insbesondere
Unabhängigkeit der einzelnen GLP-Funktionen, schriftlich ausgewiesene LPE, ggf. LPSt), z.B. Organigramm, Verfahren und Dokumentation zu Vertretungsregelungen
2.1.1.2. Anzahl der GLP-/ nicht GLP-Prüfungen
2.1.1.3. Anzahl der Beschäftigten (PL, sonstige akademische, technische, sonstige), davon Teilzeitbeschäftigte
2.1.1.4. MS zum Abschätzen der Arbeitsbelastung des GLP-Personals und des Anwendungsbereiches der PE; Führung und Archivierung des MS
2.1.2. Personal
2.1.2.1. Qualifikation und Tätigkeitsbereiche des Personals:
2.1.2.2. Sicherheits- und Gesundheitsvorkehrungen; Ausschluss von Beschäftigten, deren Gesundheitszustand sich nachteilig auf die Prüfung auswirken kann (z.B. auf biol. Prüfsysteme)
2.1.2.3. Namens-/Unterschriften-/Kürzelliste
2.2. Qualitätssicherung
Da sich spezielle Fragen an die QS häufig erst im Verlauf der Inspektion ergeben, ist es i.d.R. sinnvoll, die Inspektion der QS erst vor der Abschlussbesprechung durchzuführen. Anforderungen an die QS, die sich aus der Durchführung von MSP ergeben, sind in Anhang 4 aufgeführt. Die Tätigkeit der QS gliedert sich in prüfungs-, einrichtungs- und verfahrensbezogene Tätigkeiten.
2.2.1. Qualitätssicherungs-Programm
2.2.1.1. Anzahl und Qualifikation der Beschäftigten in der QS, Berücksichtigung von abhängigen PSt bzw. eigenständigen PSt
2.2.1.2. Unabhängigkeit der QS von der Prüfungsdurchführung
2.2.1.3. Vertretungsregelung
2.2.1.4. Arbeitsumfang:
2.2.1.5. Motivation/Akzeptanz der QS (Unterstützung der QS durch die LPE und die PL)
2.2.1.6. Vorliegen aller aktuellen SOP (sowie SOP-Liste mit Versionsnummern) bei der QS
2.2.1.7. Art der Beteiligung der QS bei der Abfassung, Überarbeitung und Aktualisierung von SOP
2.2.2. Einrichtungsbezogene Tätigkeit der Qualitätssicherung
2.2.2.1. Überprüfung der Organisation und des Personals:
2.2.2.2. Planung und Durchführung von prüfungs-, verfahrens- und PE-bezogenen Inspektionen:
2.2.2.3. Überprüfung von SOP:
2.2.2.4. Dokumentation jeder Tätigkeit der QS
2.2.3. Allgemeine prüfungsbezogene Tätigkeit der Qualitätssicherung
2.2.3.1. Überprüfung von PP:
2.2.3.2. Überprüfung von AB:
2.2.4. Qualitätssicherung und Tierhaltung
2.2.4.1. Überprüfung der Tierhaltung (Quarantäne, Akklimatisierung und tierärztliche Eingangsuntersuchung), Futterlager sowie der Einhaltung der Sicherheits- und Gesundheitsvorkehrungen durch die QS
2.2.4.2. Kontakt zu Tierschutzbeauftragten
2.2.4.3. Kritische Phasen sind z.B.:
2.2.5. Qualitätssicherung und Freiland
Aufgrund der Vielzahl der PSt/Außenstellen können sich hier besondere Probleme ergeben; evtl. zusätzliches QS-Personal vor Ort.
2.2.5.1. Inspektionen der QS im Freiland bei kritischen Phasen wie:
2.2.5.2. Präzisierung der Termine
2.2.5.3. Definierte Kommunikationswege zwischen LPE, PL, QS und ggf. PI; Einhaltung und Dokumentation (siehe auch Anhang 4)
2.2.6. Qualitätssicherung und Datenverarbeitung
2.2.6.1. Überprüfung der Organisation von DV-Systemen durch die QS:
2.2.6.2. direkter Lesezugriff (read only) der QS auf alle Rohdaten, Nachvollziehbarkeit aller Eingaben und korrigierten Eingaben für die QS, z.B. anhand eines Audit Trails
2.2.6.3. Kontrolle aller Vorgänge der manuellen oder nicht validierten Online-Dateneingabe, -verarbeitung und -ausgabe der DV-Anlage in ausreichenden Stichproben durch die QS
2.2.6.4. Überprüfung, soweit erforderlich, der Validierung und Revalidierung des DV-Systems durch die QS
2.2.6.5. Überprüfung der Evaluation von alten DV-Geräten auf GLP-Konformität durch die QS
2.2.6.6. genügende DV-Grundkenntnisse der QS zur Beurteilung der Validierung des DV-Systems, ggf. Hinzuziehen von externen Sachverständigen
2.2.7. Qualitätssicherung und instrumentelle Analytik / physikalisch-chemische Prüfungen
2.2.7.1. Unterrichtung der QS bei kurzen kritischen Phasen über den genauen Zeitpunkt (Uhrzeit) der Durchführung bzw. Änderung von Terminen
2.2.7.2. Kritische Phasen sind z.B.:
Zur Verfahrensweise bei Kurzzeitprüfungen siehe Anhang 2
2.3. Räumlichkeiten / Einrichtungen
2.3.1.1. Zweckentsprechende Größe, Konstruktion, Funktionalität und Lage anhand aktueller Gebäude-/Lagepläne (inkl. PSt)
2.3.1.2. Überprüfung der Funktion der Räume auf Übereinstimmung mit den Bezeichnungen in den Gebäudeplänen
2.3.1.3. Überprüfung der Eignung der Räume hinsichtlich der Größe, Ausstattung, Funktionalität, Ordnung, Sauberkeit etc.
2.3.1.4. Separate Räume oder Raumbereiche für:
2.3.1.5. Ausreichende Trennung der GLP-Prüfungen von anderen Prüfungen Wenn keine räumliche Trennung möglich ist, muss eine geeignete organisatorische Trennung festgelegt sein
2.3.1.6. Überprüfung der Raum-/Umweltbedingungen:
und deren Überwachung gemäß SOP, Regelungen für unvorhersehbare Ereignisse, Alarmvorrichtungen (Regelungen für Feiertage und Wochenenden)
2.3.1.7. Verhalten bei unvorhersehbaren Ereignissen, z.B. Stromausfall (ist ein Notstromaggregat vorhanden, werden Funktionstests durchgeführt und dokumentiert?)
2.4. Geräte, Reagenzien und Materialien
2.4.1.1. Geräte, die zur Gewinnung, Erfassung und Wiedergabe von Daten und zur Kontrolle der für die Prüfung bedeutsamen Umweltbedingungen verwendet werden, sind zweckmäßig unterzubringen und müssen eine geeignete Konstruktion und ausreichende Leistungsfähigkeit aufweisen
2.4.1.2. Überprüfung der Aufzeichnungen zu Wartung, Reparatur und Freigabe, Kalibrierung und Justierung von Geräten (Geräte-/Logbücher), Verfahren bei Überschreitung der Toleranzgrenzen
2.4.1.3. Überprüfung der ordnungsgemäßen Kennzeichnung der Behältnisse von Prüf- und Referenzgegenständen (mindestens Verfalldatum, besondere Lagerungshinweise, evtl. Öffnungsdatum und Haltbarkeitsfrist) und von Reagenzien (Herkunft, Identität, Konzentration, Angaben über die Stabilität, Herstellungs- und Verfalldatum, besondere Lagerungshinweise, ggf. Gefahrenpiktogramme)
2.4.1.4. Entsorgung von Materialien und Reagenzien
2.4.1.5. Geräte-SOP:
2.5. Prüfsysteme
2.5.1. Physikalische und chemische Prüfsysteme
2.5.1.1. Überprüfung von z.B.:
2.5.1.2. Eindeutige und dauerhafte Kennzeichnung von Geräten und Gerätemodulen.
2.5.1.3. Eindeutige Zuordnung von:
2.5.1.4. Lückenlose Dokumentation der Berechnungen von den Rohdaten bis zu den Ergebnissen im AB
2.5.1.5. Zuordnung prüfungsübergreifender Rohdaten (beispielsweise ein Standard für mehrere Prüfungen) zu den einzelnen Prüfungen (z.B. durch authentifizierte Kopien)
2.5.1.6. Verfahren zur organisatorischen bzw. räumlichen Trennung von GLP- und nicht GLP-Prüfungen
2.5.2. Mikrobielle, zelluläre und subzelluläre Prüfsysteme
2.5.2.1. Falls erforderlich, liegen vor:
Behördliche Erlaubnis zum Umgang mit pathogenen Mikroorganismen gemäß Infektionsschutzgesetz
Behördliche Genehmigung zum Umgang mit pathogenen Mikroorganismen gemäß Tierseuchenerregerverordnung
Behördliche Genehmigung oder Anzeige bzw. Anmeldung von gentechnischen Arbeiten gemäß Gentechnikgesetz
2.5.2.2. Bechäftigte werden ggf. über den Umgang mit gefährlichen biologischen Prüfsystemen hinreichend geschult
2.5.2.3. Für diese Prüfsysteme existieren Vorschriften über:
2.5.2.4. Charakterisierung des Prüfsystems (Herkunft, Spezies, Stamm)
2.5.2.5. Entsorgung der biologischen Prüfsysteme (Sammlung, Lagerung, Dekontaminierungs- und Transportverfahren)
2.5.2.6. Dokumentierte Festlegung von Prüfgegenstand und Prüfsystem
2.5.3. Prüfungen an Tieren
2.5.3.1. Tierschutz:
2.5.3.2. Qualifikation des Tierpflegepersonals
2.5.3.3. Dokumentation der Herkunft der Tiere
2.5.3.4. Quarantäne für neu eingetroffene Tiere; Eingangsuntersuchungen sowie laufende Dokumentation des Gesundheitsstatus (bei Erkrankungen evtl. Zurückweisung mit Begründung)
2.5.3.5. Randomisierung/Zuordnung von Tieren
2.5.3.6. Umgang mit überzähligen Tieren
2.5.3.7. Unverwechselbare Kennzeichnung von Tieren und Käfigen/Behältern
2.5.3.8. Gesundheitsüberwachung der Tiere, ggf. Verabreichung von Arzneimitteln
2.5.3.9. Qualität und Reinheit der Futtermittel (Lieferschein, Zertifikat über Zusammensetzung und Verunreinigungen)
2.5.3.10. Überprüfung der Futter- und Wasserqualität, Verfahren bei Mängelfeststellung
2.5.3.11. Lagerbedingungen von Futtermitteln (z.B. Schädlingsbekämpfung)
2.5.3.12. Qualität und Reinheit von Einstreu, Substrat usw.
2.5.3.13. Dokumentation sämtlicher Schritte der Prüfung für jedes Tier (z.B. Applikation, Probenahme, Sektion)
2.5.3.14. Der Pathologie steht ein geeignetes Verfahren zur Aufzeichnung von Daten zur Verfügung
2.5.3.15. Entsorgung von Tierkörpern
2.5.3.16. Allgemeine Sauberkeit
2.5.3.17. Reinigung von Käfigen/Behältern, Futtergefäßen und sonstigem Zubehör
2.5.3.18. Trennung von reinen und unreinen Bereichen (Käfigwaschanlagen, Tierräume)
2.5.3.19. Verhalten bei unvorhersehbaren Ereignissen
2.5.3.20. Dokumentation bei Versand von Materialien (Schnitte, Blöcke usw.) an abhängige PSt bzw. eigenständige PSt oder andere PE
2.5.4. Prüfungen an Pflanzen
Prüfungen im Gewächshaus und im Halbfreiland:
2.5.4.1. Prüfsystem:
2.5.4.2. Substrat:
2.5.4.3. Pflege des Prüfsystems:
2.5.4.4. Nur im Gewächshaus:
Sauberkeit, Klimatisierung, Registrierung von Temperatur und Feuchtigkeit, Beleuchtung (Stärke, Dauer), Schattierung (Dauer), Belüftung (mechanischer Schutz gegen Eindringen von Fremd- und Schadorganismen), ausreichende Größe zur getrennten Haltung der Prüfsysteme (bei Pflanzen: Art der Standflächen und der Bewässerung, evtl. Maßnahmen gegen Wasserhärte; bei Tieren: Art und Größe der Behälter/Gehäuse)
2.5.4.5. Prüfgegenstand: Art und Zeitpunkt der Applikation
2.5.4.6. Entsorgung des Prüfsystems und des Substrats
2.5.4.7. Sicherheitsmaßnahmen für das Personal existieren, deren Kenntnis ist gewährleistet und sie werden befolgt (z.B. Gesundheitsschutz), ggf. bestehen Überwachungssysteme
Prüfungen im Freiland:
2.5.4.8. Beschreibung des Prüfsystems (Standort, Historie der Parzelle, Parzellengröße, Lage zur Windrichtung, Nachbarflächen, Kennzeichnung, Wiederauffindbarkeit, Kartierung; bei Pflanzen: Art, Sorte, Herkunft des Saatguts oder der Pflanzen, Gesundheitszustand, Pflegemaßnahmen wie Schnitt, Bodenbearbeitung, Düngung, Bewässerung, Pflanzenschutz; bei Böden: Bodenart, org. Substanzen, pH-Wert, Bearbeitung)
2.5.4.9. Applikation des Prüfgegenstandes (Berechnung der Dosierung; Informationen zu Haltbarkeit und Verhalten bei bestimmten pH-Werten des Wassers; Art der Ausbringung, z.B. Gießen, Sprühen, Streuen, Stäuben; Geräteart, Düsentyp, Gestänge; Erproben der Applikation; Häufigkeit und Zeitpunkte, ggf. Präzisierung von Terminen aufgrund phänomenologischer Angaben; Erfassen von Temperatur und Windgeschwindigkeit während der Applikation; Beseitigung von Flüssigkeitsresten; Maßnahmen zur Verhinderung von Kontaminationen, u. a. durch Abdrift)
2.5.4.10. Kennzeichnung, Wartung und Reinigung der Geräte (z.B. Waagen, Pipetten, Applikationsgeräte einschließlich Düsen, Kühlgeräte, Geräte zur Erfassung von Wetterdaten)
2.5.4.11. Proben für Rückstandsbestimmungen (Art und Termin der Probenentnahme, Kennzeichnung und Aufbewahrung der Proben, Kontrolle der Lagerungsbedingungen, geeigneter Transport zum analytischen Labor, Kühlkette einhalten)
2.5.4.12. Verhinderung von Kontaminationen (ausreichende Trennung des Prüfgegenstandes von Proben bei Lagerung und beim Umgang; Maßnahmen bei Geräten, Arbeitstischen, Behältnissen, Räumlichkeiten)
2.5.4.13. Vorliegen von SOP, PP und Formblättern in der Außenstelle, Aufzeichnungen der Kommunikation (Telefonate, Fax), Zwischenlager von Rohdaten in der Außenstelle
2.5.4.14. Sauberkeit (Geräte, Behälter, Räumlichkeiten)
2.5.4.15. Entsorgung von Resten des Prüfgegenstandes und des Prüfsystems, Dokumentation
2.5.4.16. Kommunikation zwischen LPE, PL, PI, ggf. LPSt und QS; Verfahren zur Information der QS über kritische Phasen wie z.B. Applikation des Prüfgegenstandes, Probenentnahme, -aufbereitung, -lagerung und -transport (Präzisierung der Termine im PP) (siehe auch Anhang 4)
2.6. Prüf- und Referenzgegenstände
2.6.1.1. Verfahren zur Übergabe des Prüfgegenstandes vom Auftraggeber an die PE (z.B. sichere Handhabung, Umweltbedingungen wie Temperatur und rel. Luftfeuchtigkeit, Lagerbedingungen)
2.6.1.2. Authentizität des Prüfgegenstandes bei Eingang in die PE:
Verfahren zur Verifizierung der Identität (OECD-Grundsätze der Guten Laborpraxis Kap. 6.2 Nr. 2 und 3: Für jede Prüfung müssen Identität, einschließlich Chargennummer, Reinheit, Zusammensetzung, Konzentration oder sonstige Eigenschaften zur Charakterisierung jeder Charge der Prüf- oder Referenzgegenstände bekannt sein. Bei der Lieferung des Prüfgegenstandes durch einen Auftraggeber ist in Zusammenarbeit zwischen Auftraggeber und PE ein Verfahren festzulegen, auf welche Weise die Identität des Prüfgegenstandes, der in der Prüfung eingesetzt wird, eindeutig bestätigt wird).
In Ausnahmefällen Rücksendung eines Musters an den Hersteller zur Bestätigung bzw. andere nachvollziehbare Rückverfolgbarkeit über die Art der Identitätsfeststellung; andernfalls ist ein deutlicher Hinweis im PP und AB erforderlich
2.6.1.3. Mikrobiologische Prüfungen: Festlegung PG/Prüfsystem
2.6.1.4. Kennzeichnung als Prüf- oder Referenzgegenstand, eindeutige Codierung
2.6.1.5. Verteilung des Prüfgegenstandes in der PE; Dokumentation
2.6.1.6. Buchführung über die erhaltene Menge, Verbrauch und Entsorgung des Prüfgegenstandes (evtl. Rückgabe an den Hersteller)
2.6.1.7. Archivierung von Rückstellmustern von Prüf- und Referenzgegenständen (PRG)
2.6.1.8. Zubereitung der Applikationsfornn der PRG:
2.6.1.9. Kennzeichnung, Homogenität, Stabilität und Lagerung der Applikationsform
2.7. Standardarbeitsanweisungen
2.7.1.1. Abdeckung der Prüfbereiche in den zu bescheinigenden Prüfkategorien durch SOP
2.7.1.2. SOP über Form, Erstellung, Änderung, Aktualisierung, Indexierung, Autorisierung, Verteilung, Archivierung von SOP
2.7.1.3. SOP zum QS-Programm
2.7.1.4. Alle prüfungsübergreifenden SOP (z.B. Eingang des Prüfgegenstandes, Codierung von Prüfungen, Archivierung usw.), siehe Anhang 1
2.7.1.5. Prüfungen der SOP vor Ort:
2.8. Prüfungsablauf
Vergleiche hierzu auch Anhang 4 - Anforderungen an Multi-Site-Prüfungen.
2.8.1. Prüfplan
Der Prüfplan (PP) muss (soweit aufgrund der Art der Prüfung zutreffend) die im Folgenden aufgeführten Angaben enthalten. Die Anforderungen an standardisierte PP bei Kurzzeitprüfungen sind in Anhang 2 beschrieben.
Festlegungen für Freiland-Prüfsysteme:
2.8.2. Überprüfung laufender Prüfungen (Study Audit)
Ein wichtiger Teil der Vor-Ort-Besichtigung ist die Inspektion einer laufenden Prüfung. Dabei kann beobachtet werden, wie das Personal mit der Durchführung der Prüfung vertraut ist, ob schriftliche PP vorliegen und die Durchführung der Prüfung mit den GLP-Grundsätzen übereinstimmen bzw. die Vorgaben in den SOP umgesetzt werden.
2.8.2.1. Das Inspektionsteam sollte sich u. a. vergewissern, dass
2.8.3. Überprüfung von abgeschlossenen Prüfungen (Study Audit)
2.8.3.1. Möglichst kurzfristige Auswahl vor oder zu Beginn der Inspektion (s. 1.4.4.2)
2.8.3.2. Prüfungen sollen relevant für die beantragten Prüfkategorien und nach Möglichkeit für die Vorlage bei einer Bewertungsbehördevorgesehen sein.
2.8.3.3. Nach Möglichkeit sollten auch abgebrochene Prüfungen überprüft werden.
2.8.3.4. Prüfungen sollen nach GLP durchgeführt, abgeschlossen, archiviert und i. d. R. nicht älter als die vorhergehende Inspektion sein.
2.8.4. Vorbereitung der Überprüfung von Prüfungen
2.8.4.1. Anfertigung von Kopien von PP und ggf. AB für Notizen, Vergleiche etc., ggf. auch Übersetzungen.
2.8.4.2. Ggf. Rücksprache mit der/dem PL der jeweiligen Prüfung und/oder der QS.
2.8.4.3. Überprüfung auf Vollständigkeit der Unterlagen (evtl. Inhaltsverzeichnis, Paginierung), Ordnungskriterien und einheitliche Codierung
2.8.4.4. Überprüfung der datierten Unterschriften im Original
2.8.4.5. Überprüfung des PP
2.8.4.6. Überprüfung der Rohdaten (ggf. stichprobenartig) auf:
2.8.4.7. Im Falle abgeschlossener Prüfungen: Überprüfung des AB (siehe 2.9)
Wurden Teile der Prüfung nicht nach GLP durchgeführt, so ist dies im statement of compliance zu vermerken
2.8.4.8. Überprüfung auf Kongruenz von PP, Rohdaten und ggf. AB (systematische Vorgehensweise vom PP über die Rohdaten ggf. zum AB oder umgekehrt), z.B.:
2.8.4.9. Ggf. ergänzende Überprüfung von:
2.8.4.10. Falls die Überprüfung von Prüfungen nicht im Rahmen einer Erst- oder Wiederholungsinspektion gem. ChemVwV-GLP erfolgt, sondern auf Grund von Anfragen der Bewertungsbehörden, sind folgende Punkte zu beachten:
2.8.4.11. Evtl. bestimmte Verfahrensweisen in einer vergleichbaren laufenden Prüfung ansehen
2.8.5. Besprechung der Ergebnisse der Überprüfung von Prüfungen
2.8.5.1. Absprache innerhalb des Inspektorenteams
2.8.5.2. Besprechung mit PL, QS, LPE und ggf. weiteren Personen, z.B. PI
2.8.5.3. Forderung eines Nachtrags zum AB bei relevanten Mängeln
2.8.5.4. Bei schwerwiegenden Mängeln Einstufung der Prüfung als nicht GLP-gerecht mit umgehender Meldung an die GLP-Bundesstelle (s. 2.14)
2.9. Bericht über die Prüfergebnisse
Der Bericht über die Prüfergebnisse (Abschlussbericht, AB) muss bestimmte Angaben enthalten.
Für Kurzzeitprüfungen sind dabei bestimmte Vereinfachungen möglich
(Standardabschlussbericht, s. Anhang 2).
2.10. Archivierung von Aufzeichnungen und Materialien
Vergleiche hierzu auch Konsensdokument der Bund-/Länderarbeitsgruppe Gute Laborpraxis zur Archivierung und Aufbewahrung von Aufzeichnungen und Materialien.
2.10.1. Allgemeine Aspekte zur Archivierung
2.10.1.1. Bauliche Gegebenheiten, z.B. Schutz vor Feuer (feuerhemmende Ausführung), Wasser, Diebstahl und sonstigen nachteiligen Einwirkungen
2.10.1.2. Zugangsregelung, Archiv-Verantwortlichkeit, Vertretung der AV
2.10.1.3. Bestandsverzeichnis sowie Ordnungs- und Indexierungssystem für archivierte Unterlagen und Materialien (auch von abgebrochenen Prüfungen), schnelle Wiederauffindbarkeit
2.10.1.4. QS-Inspektion des Daten- und Materialarchivs
2.10.1.5. Verfahren bei der Anfertigung mehrerer Originale von AB
2.10.2. Datenarchiv
2.10.2.1. Überprüfung der Unterlagen auf Vollständigkeit, z.B. PP, Rohdaten, AB, sonstige Unterlagen; Hinweis auf Fundstellen prüfungsübergreifender Daten wie Waagen-, Klimakontrolldaten, Gerätekalibrierung, evtl. autorisierte Kopien
2.10.2.2. Paginierung bzw. adäquate Verfahrensweise zur schnellen Wiederauffindbarkeit sowie zum Schutz gegen Verlust und Austausch der zu einer Prüfung gehörenden Rohdaten, ggf. unter Einbeziehung von PP, AB und sonstiger Daten
2.10.2.3. Inhaltsverzeichnis der archivierten Unterlagen jeder Prüfung
2.10.2.4. Regelung für Entnahme und Rückgabe von archivierten Unterlagen (i.d.R. Kopien anfertigen)
2.10.2.5. Sicherung der Lesbarkeit bei Aufbewahrung von Daten auf magnetischen Datenträgern
2.10.2.6. Verfahren zur Mikroverfilmung von Daten, Überprüfung
2.10.2.7. Sichere Zwischenlagerung von Daten bei abhängigen PSt bzw. eigenständigen PSt sowie deren Übermittlung an PL bzw. Archiv (auch Auftragsarchiv)
2.10.2.8. Archivierung von QS-Unterlagen
2.10.2.9. Chronologische Sammlung aller SOP
2.10.2.10. Archivierung prüfungsübergreifender Daten (z.B. Logbücher, Aufzeichnungen über Klimakontrollen u. a).
2.10.2.11. Archivierung von Gebäude- und Stockwerksgrundrissen
2.10.2.12. Archivierung von Organigrammen, Personalunterlagen, MS
2.10.3. Materialarchiv
2.10.3.1. Archiv für Rückstellmuster von PRG, Kennzeichnung, Aufbewahrungsdauer
2.10.3.2. Archiv für Proben, Temperaturkontrolle bei gekühlter Aufbewahrung
2.10.3.3. Archiv für Nassasservate, spezielle Anforderungen z.B. für Formaldehyd-Präparate
2.10.3.4. Archiv für Paraffinblöcke, Schnitte, Ausstriche
2.10.3.5. Zwischenlagerung von Proben bei abhängigen bzw. eigenständigen PSt; Kühlung, Erhaltung der Kühlkette bei Transport
2.10.3.6. Anzeige bei der QS vor der Auslagerung zum Ende der Aufbewahrungsfrist
2.11. Elektronische Datenverarbeitungssysteme
Siehe Anhang 3 - Inspektion von Datenverarbeitungssystemen
2.12. Multi-Site-Prüfungen
Siehe Anhang 4 - Multi-Site-Prüfungen
2.13. Abschlussbesprechung
2.13.1.1. Teilnehmer: Inspektionsteann, LPE, PL, Leitung der QS, evtl. AV, ggf. PI und LPSt
2.13.1.2. Zusammenfassung des Inspektionsergebnisses inkl. Mitteilung der festgestellten Mängel, i.d.R. in schriftlicher Form (Kurzbericht)
2.13.1.3. Festlegung der Frist zur Abstellung der Mängel
2.13.1.4. Ankündigung einer Nachinspektion bei schwerwiegenden Mängeln
2.13.1.5. Verbindliche Bezeichnung der PE
2.13.1.6. Festlegung der Prüfkategorien und deren spezielle Ausgestaltung (siehe auch 1.1.1.2, Anhang 5 und Anhang 6)
2.13.1.7. evtl. vorläufiges Votum des Inspektionsteams
Eine schriftliche Zusammenfassung des Inspektionsergebnisses und der festgestellten Mängel erfolgt entweder im Rahmen der Abschlussbesprechung oder zeitnah im Nachgang der Inspektion. Ggf. ist eine Bestätigung der Kenntnisnahme, z.B. durch Gegenzeichnung durch den LPE, sinnvoll.
2.14. Mitteilungspflichten aus der Inspektion
Wenn bei der Inspektion festgestellt wurde, dass einzelne Prüfungen nicht in Übereinstimmung mit den GLP-Grundsätzen (NIC) durchgeführt wurden, die Integrität der ermittelten Daten nicht gegeben ist bzw. einzuschätzen ist, dass die PE / der PSt insgesamt nicht gemäß den GLP-Regeln arbeitet, muss dieses so zeitnah wie möglich nach Abschluss der Inspektion an die GLP-Bundesstelle berichtet werden. Für diese Berichterstattung ist das Excel-Formular "OECD Template for non_compliance.xls" zu verwenden.
3. Inspektionsbericht
Inn Folgenden werden die Mindestanforderungen bei der Erstellung von Inspektionsberichten aufgeführt. Grundlage sind die inhaltlichen Anforderungen des Anhangs 1 ChemG (Grundsätze der Guten Laborpraxis) in Verbindung mit den Vorgaben des OECD Advisory Documents No. 9 (Guidance for the Preparation of GLP Inspection Reports).
Eine Arbeitsgruppe aus mehreren Bundesländern hat auf diesen Grundlagen einen Musterentwurf eines GLP-Inspektionsberichts erarbeitet und vorgestellt. Dieser harmonisierte Inspektionsbericht wurde bei der 38. Sitzung der BLAC angenommen. Seitens der BLAC wird den Ländern die Verwendung des GLP-Inspektionsberichtes als Muster empfohlen. Der Berichtsentwurf ist als Formular ausgeführt und an länderspezifische Gegebenheiten anpassbar. Das Muster ist auf den internen Seiten der BLAC eingestellt, unter:
BLAC/BLAC-Intern/Ausschuss GLP/Vollzugshilfen GLP
Inhalt des Inspektionsberichts:
1. Zusammenfassung
2. Einleitung
2.1. Identifizierung der Prüfeinrichtung (PE)2.2. Art der GLP-Überwachungsmaßnahme
2.3. Charakterisierung der Prüfeinrichtung
2.4. Inspektionsteam und Inspektionsdatum
3. Inspektion und Feststellungen
3.1. Organisation und Personal der PE3.2. Qualitätssicherungsprogramm
3.3. Räumlichkeiten und Einrichtungen
3.4. Geräte, Reagenzien und Materialien
3.5. Prüfsysteme
3.5.1. Physikalische und chemische Prüfsysteme3.5.2. Mikrobielle, zelluläre und subzelluläre Prüfsysteme
3.5.3. Prüfungen an Tieren
3.5.4. Prüfungen an Pflanzen
3.6. Prüf- und Referenzgegenstände (PRG)3.7. Standardarbeitsanweisungen (SOP)
3.8. Prüfungsablauf
3.8.1. Überprüfung von Prüfungen (Study Audit)
3.9. Bericht über Prüfergebnisse
3.10. Archivierung und Aufbewahrung von Aufzeichnungen und Materialien
3.11. Elektronische Datenverarbeitungssysteme (EDV-Systeme)
3.12. Multisite-Verfahren
4. Abschlussbesprechung
5. Ergebnis
6. Anlagen
4. Publikationen und grundlegende Dokumente zur GLP
4.1. OECD
Die OECD-Dokumente sind verfügbar auf der OECD-GLP-Internetseite und der Internetseite der GLP-Bundesstelle.
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 1: OECD-Grundsätze der Guten Laborpraxis.
(Neufassung 1997)
OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring, Number 2 (revised): Guidance for GLP-Monitoring Authorities, Revised Guides for Compliance Monitoring Procedures for Good Laboratory Practice. (1995)
(deutsche Übersetzung als Anhang 1 der EU Richtlinie 2004/9/EG Abschnitt A)
OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring, Number 3 (revised): Guidance for GLP-Monitoring Authorities, Revised Guidance for the Conduct of Laboratory Inspections and Study Audits. (1995)
(deutsche Übersetzung als Anhang 1 der EU Richtlinie 2004/9/EG Abschnitt A)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 4: QS und Gute Laborpraxis.
(1999 überarbeitet)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 5: Einhaltung der GLP-Grundsätze durch Lieferanten.
(1999 überarbeitet)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 6: Die Anwendung der GLP-Grundsätze auf Freilandprüfungen.
(1999 überarbeitet)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 7: Die Anwendung der GLP-Grundsätze auf Kurzzeit-Prüfungen.
(1999 überarbeitet)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 8: Die Rolle und Verantwortlichkeiten des Prüfleiters bei GLP-Prüfungen.
(1999 überarbeitet)
OECD series on Principles of Good Laboratory Practice and Compliance Monitoring,
Number 9: Guidance for GLP-Monitoring Authorities, Guidance for the Preparation of GLP Inspection Reports.
(keine deutsche Übersetzung vorgesehen)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung Ihrer Einhaltung, GLP-Konsensdokument
Nummer 10: Die Anwendung der GLP-G rundsätze auf Computergestützte Systeme.
(2016 ersetzt durch Nummer 17)
OECD series on Principles of Good Laboratory Practice and Compliance Monitoring,
Number 11: Advisory Document of the Panel on GLP,
The Roles and Responsibilities of the Sponsor in the Application of the Principles of GLP. (1998)
(keine deutsche Übersetzung vorgesehen)
OECD series on Principles of Good Laboratory Practice and Compliance Monitoring,
Number 12: Advisory Document of the Working Group on Good Laboratory Practice, Requesting and Carrying Out Inspections and Study Audits in Another Country. (2000)
(keine deutsche Übersetzung vorgesehen)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung.
Konsensdokument der Arbeitsgruppe Gute Laborpraxis,
Nummer 13: Die Anwendung der OECD GLP-Grundsätze auf Organisation und Management von Multi-Site-Prüfungen. (2002)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung.
Beratungsdokument der Arbeitsgruppe Gute Laborpraxis
Nummer 14: Die Anwendung der OECD GLP-Grundsätze auf in vitro Prüfungen (2004)
OECD-Schriftenreihe über Grundsätze der Guten Laborpraxis und Überwachung ihrer Einhaltung,
Nummer 15: Einrichtung und Betrieb von Archiven in Übereinstimmung mit den Grundsätzen der Guten Laborpraxis. (2007)
OECD series on Principles of Good Laboratory Practice and Compliance Monitoring,
Number 16: Advisory Document of the Working Group on Good Laboratory Practice, Guidance on the GLP - Requirements for Peer Review of Histopathology. (2014)
OECD series on Principles of Good Laboratory Practice and Compliance Monitoring,
Number 17: Advisory Document of the Working Group on Good Laboratory Practice, Application of GLP Principles t- Computerised Systems. (2016)
(ersetzt das Konsensdokument Nr. 10)
Position Papers:
Number 18: OECD Position Paper Regarding the Relationship between the OECD Principles of GLP and ISO/IEC 17025 (2016)
The Use of Laboratory Accreditation with reference t- GLP Compliance Monitoring (1994)
"Outsourcing" of Inspection Functions by GLP Compliance Monitoring Authorities (2006)
Frequently Asked Questions on Technical issues (FAQs): OECD GLP Website (kontinuierlich seit 2014)
4.2. Europäische Union
Die Dokumente sind verfügbar auf der EU-GLP-Internetseite und der Internetseite der GLP-Bundesstelle.
Richtlinie 2004/10/EG des Europäischen Parlaments und des Rates vom 11. Februar 2004 zur Angleichung der Rechts- und Verwaltungsvorschriften für die Anwendung der Grundsätze der Guten Laborpraxis und zur Kontrolle ihrer Anwendung bei Versuchen mit chemischen Stoffen (kodifizierte Fassung)
Richtlinie 2004/9/EG des Europäischen Parlaments und des Rates vom 11. Februar 2004 über die Inspektion und Überprüfung der Guten Laborpraxis (GLP) (kodifizierte Fassung)
Good Laboratory Practicies - EU Productspecific legal acts (EU-GLP-Internetseite, kontinuierlich)
Guidance on crosscontamination of control samples with test item in animal studies (EU-GLP-Internetseite, 2004)
http://ec.europa.eu/DocsRoom/documents/13222
Guidance for GLP facilities on the implementation and maintenance of a riskbased quality assurance programme (EU-GLP-Internetseite, 2017)
http://ec.europa.eu/DocsRoom/documents/22262
Guidance on GLP compliance monitoring of international multisite studies (EU-GLP-Internetseite, 2017)
http://ec.europa.eu/DocsRoom/documents/20821
Questions & Answers (EU-GLP-Internetseite, 2017)
http://ec.europa.eu/DocsRoom/documents/22261
4.3. Bundesrepublik Deutschland
Neufassung des Chemikaliengesetzes (ChemG) vom 28. August 2013; Bundesgesetzblatt Teil I, S. 3498 - 3991, in der jeweils gültigen Fassung
GLP-Konsensdokument: Die Anwendung der GLP-Grundsätze auf computergestützte Systeme; Bundesanzeiger Nr. 231, Jahrgang 1996, S. 12749-12753
Neufassung Allgemeine Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis (ChemVwV-GLP) in der Fassung vom 15. Mai 1997; Gemeinsames Ministerialblatt vom 09. Juni 1997, 48. Jahrgang, Nr. 17, S. 257-264
Allgemeine Verwaltungsvorschrift zur Änderung der Allgemeinen Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis vom 02. September 2011, Gemeinsames Ministerialblatt vom 14. Dezember 2011, 62. Jahrgang, Nr. 48, S. 967 - 968
Bekanntmachung eines Konsens-Dokuments der Bund-Länder-Arbeitsgruppe Gute Laborpraxis zur Archivierung und Aufbewahrung von Aufzeichnungen und Materialien vom 05.05.1998; Bundesanzeiger Nr. 98, S. 7439-7440
Leitfaden zur Harmonisierung des GLP-Überwachungsverfahrens in der Bundesrepublik Deutschland des BLAC-AK "GLP und andere Qualitätssicherungssysteme", UMK-Umlaufbeschlüsse 4/2007 u. 5/2007
4.4. Andere Dokumente
Schlottmann/Kayser (Hrsg.), GLP Gute Laborpraxis, Textsammlung und Einführung, Behr"s Verlag Hamburg, 3. Auflage 1997
G.A. Christ, S.J. Harston, H.W. Hembeck, K.A. Opfer, GLP - Handbuch für Praktiker, 2. Auflage 1998, GIT-Verlag Darmstadt
PIC/S, Draft PIC/S Guidance, Good practices for computerised systems in regulated "GXP" Environments, PI 011-, 20. August 2003
FDA, Guidance for Industry, Part 11, Electronic Records; Electronic Signatures- Scope and Application, August 2003
5. Begriffsbestimmungen
Quellen:
[1] OECD Nr. 1 - Grundsätze, 1998.
[2] OECD Nr. 6 - Freilandprüfungen, 1999.
[3] OECD Nr. 14 - In-Vitro-Prüfungen, 2004.
[4] OECD Nr. 13 - Multi-Site-Prüfungen, 2002.
[5] Konsensdokument zur Archivierung, 1998.
[6] OECD Nr. 4 - Qualitätssicherung und QS, 1999.
[7] OECD Nr. 15 - Archive, 2007.
[8] OECD Nr. 17 - Computerised Systems, 2016.
Grundbegriffe - Organisation
Auftraggeber (sponsor) ist eine natürliche oder juristische Person, die eine nichtklinische gesundheits- und umweltrelevante Sicherheitsprüfung in Auftrag gibt, unterstützt oder einreicht. [1]
Gute Laborpraxis (Good Laboratory Practice) ist ein Qualitätssicherungssystem, das sich mit dem organisatorischen Ablauf und den Rahmenbedingungen befasst, unter denen nichtklinische gesundheits- und umweltrelevante Sicherheitsprüfungen geplant, durchgeführt und überwacht werden sowie mit der Aufzeichnung, Archivierung und Berichterstattung der Prüfungen. [1]
Leitung der Prüfeinrichtung (test facility management) bezeichnet diejenige Person (Personengruppe), die die Zuständigkeit und formale Verantwortung für die Organisation und das Funktionieren der Prüfeinrichtung gemäß diesen Grundsätzen der Guten Laborpraxis besitzt. [1]
Leitung eines Prüfstandortes (test site management) - sofern benannt - bezeichnet diejenige Person (Personengruppe), die sicherzustellen hat, dass diejenigen Phasen der Prüfung, für die sie die Verantwortung übernommen hat, nach diesen Grundsätzen der Guten Laborpraxis durchgeführt werden. [1]
Master Schedule (master schedule) ist eine Zusammenstellung von Informationen, die der Abschätzung der Arbeitsbelastung und der Verfolgung des Ablaufs von Prüfungen in einer Prüfeinrichtung dient. [1]
Principal Investigator (Principal Investigator) bezeichnet diejenige Person, die, im Falle einer Multi-Site-Prüfung, im Auftrag des Prüfleiters bestimmte Verantwortlichkeiten für die ihr übertragenen Phasen von Prüfungen übernimmt. [1]
Prüfeinrichtung (test facility) umfasst die Personen, Räumlichkeiten und Arbeitseinheit(en), die zur Durchführung von nichtklinischen gesundheits- und umweltrelevanten Sicherheitsprüfungen notwendig sind. Bei Prüfungen, die in Phasen an mehr als einem Standort durchgeführt werden, sogenannte Multi-Site-Prüfungen, umfasst der Begriff Prüfeinrichtung sowohl den Standort, an dem der Prüfleiter angesiedelt ist, als auch alle anderen individuellen Prüfstandorte. Die Prüfstandorte können sowohl in ihrer Gesamtheit als auch jeweils einzeln als Prüfeinrichtung definiert werden. [1]
Prüfleiter/Prüfleiterin (Study Director) ist diejenige Person, die für die Gesamtleitung der nichtklinischen gesundheits- und umweltrelevanten Sicherheitsprüfung verantwortlich ist. [1]
Prüfstandort (test site) ist der Ort, an dem eine oder mehrere Phase(n) einer Prüfung durchgeführt werden. [1]
Standardarbeitsanweisungen (Standard Operating Procedures) sind dokumentierte Verfahrensanweisungen über die Durchführung derjenigen Untersuchungen oder Tätigkeiten, die in der Regel in Prüfplänen oder Prüfrichtlinien nicht in entsprechender Ausführlichkeit beschrieben sind. [1]
Grundbegriffe - Prüfung
Abschluss einer Prüfung ist der Tag, an dem der Prüfleiter den Abschlussbericht unterschreibt. [1]
Beginn der experimentellen Phase ist der Tag, an dem die ersten prüfungsspezifischen Rohdaten erhoben werden. [1]
Beginn einer Prüfung ist der Tag, an dem der Prüfleiter den Prüfplan unterschreibt. [1]
Ende der experimentellen Phase ist der letzte Tag, an dem noch prüfungsspezifische Rohdaten erhoben werden. [1]
Freilandprüfung umfasst experimentelle Tätigkeiten, die außerhalb des gewöhnlichen Laborbetriebes durchgeführt werden, wie z.B. auf Freilandflächen, in Teichen oder Gewächshäusern, oft in Kombination oder mit sich anschließenden Laboruntersuchungen. [2]
In vitro Prüfungen sind Prüfungen, die nicht an vollständigen mehrzelligen Organismen durchgeführt werden, sondern die als Prüfsysteme Mikroorganismen, lsolate aus Organismen oder entsprechende Simulationen davon benutzen. Sie sind oftmals Kurzzeitprüfungen. [3]
Kritische Phasen: Einzelne definierte Prozeduren oder Tätigkeiten innerhalb einer Prüfung, von deren korrekter Ausführung die Qualität, Aussagekraft und Verlässlichkeit der Prüfung entscheidend abhängig ist. [3]
Kurzzeitprüfung ist eine Prüfung von kurzer Dauer, die nach weithin gebräuchlichen Routinemethoden durchgeführt wird. [1]
Multi-Site-Prüfung ist eine Prüfung, bei der Phasen an mehr als einem Standort durchgeführt werden. Eine Durchführung als Multi-Site-Prüfung wird notwendig, wenn bei einer Prüfung der Bedarf besteht, Standorte zu beteiligen, die geografisch voneinander entfernt liegen, organisatorisch voneinander unabhängig oder anderweitig getrennt sind. [4]
Nichtklinische gesundheits- und umweltrelevante Sicherheitsprüfung (Prüfung, study) ist eine Untersuchung oder eine Reihe von Untersuchungen, die mit einem Prüfgegenstand unter Labor- oder Umweltbedingungen durchgeführt wird, um Daten über seine Eigenschaften und/oder über seine Unbedenklichkeit zu gewinnen, mit der Absicht diese den zuständigen Bewertungsbehörden einzureichen. [1]
Phase ist eine bestimmte Aufgabe oder eine Reihe von Aufgaben im Rahmen der Durchführung einer Prüfung. [4]
Proben (specimen) sind Materialien, die zur Untersuchung, Auswertung oder Aufbewahrung aus dem Prüfsystem entnommen werden. [1]
Prüfplan (study plan) ist ein Dokument, das die Ziele und experimentelle Gesamtplanung zur Durchführung der Prüfung beschreibt; es schließt sämtliche Prüfplanänderungen ein. [1]
Prüfplanabweichung (study plan deviation) ist ein unbeabsichtigtes Abweichen vom Prüfplan nach Beginn der Prüfung. [1]
Prüfplanänderung (study plan amendment) ist eine geplante Veränderung des Prüfplans nach Beginn der Prüfung in Form einer Ergänzung. [1]
Prüfsystem (test system) ist jedes biologische, chemische oder physikalische System - oder eine Kombination daraus -, das bei einer Prüfung verwendet wird. [1] Im Zusammenhang mit Freilandprüfungen sind auch komplexe ökologische Systeme eingeschlossen. [2]
Rohdaten (raw data) sind alle ursprünglichen Aufzeichnungen und Unterlagen der Prüfeinrichtung oder deren überprüfte Kopien, die als Ergebnis der ursprünglichen Beobachtungen oder Tätigkeiten bei einer Prüfung anfallen. Zu den Rohdaten zählen beispielsweise Fotografien, Mikrofilm- oder Mikrofichekopien, computerlesbare Medien, diktierte Beobachtungen, aufgezeichnete Daten von automatisierten Geräten oder irgendwelche anderen Daten auf Speichermedien, die anerkanntermaßen geeignet sind, Informationen über einen festgelegten Zeitraum sicher zu speichern. [1]
Grundbegriffe - Prüfgegenstand
Charge (batch) ist eine bestimmte Menge oder Partie eines Prüf- oder Referenzgegenstandes, die in einem bestimmten Herstellungsgang derart gefertigt wurde, dass einheitliche Eigenschaften zu erwarten sind; sie wird als solche gekennzeichnet. [1]
Prüfgegenstand (test item) ist ein Objekt, das der Prüfung unterliegt. [1]
Referenzgegenstand (Vergleichsgegenstand, reference (control) item) ist ein Objekt, das zum Vergleich mit dem Prüfgegenstand verwendet wird. [1] Im Zusammenhang mit Freilandprüfungen können das auch analytische Standards sein. [2] Bei in vitro Prüfungen kann der Referenzgegenstand als deckungsgleich mit den Begriffen "positive, negative und/oder Trägerstoff-Kontrollen" angesehen werden. [3]
Rückstellmuster (sample) Das Muster (Rückstellmuster) dient der nachträglichen Identitätsprüfung einer Prüfsubstanz. Menge und Aufbewahrungsbedingungen der Muster sind nach dieser Zweckbestimmung festzulegen. [5]
Trägerstoff (vehicle) ist ein Stoff, mit dem der Prüf- oder Referenzgegenstand gemischt, dispergiert oder aufgelöst wird, um die Anwendung am Prüfsystem zu erleichtern. [1]
Grundbegriffe - Qualitätssicherung
Einrichtungsbezogene Inspektionen (facilitybased inspections) stützen sich nicht auf bestimmte Prüfungen, sondern beinhalten die allgemeinen Einrichtungen und Tätigkeiten in einem Labor (Technische Anlagen, Hilfsdienste, Computersysteme, Schulung, Umweltüberwachung, Wartung, Kalibrierung, usw.). [6]
Prüfungsbezogene Inspektionen (studybased inspections) werden nach dem zeitlichen Ablauf einer vorgegebenen Prüfung durchgeführt, gewöhnlich nach vorheriger Identifikation der kritischen Phasen einer Prüfung. [6]
Qualitätssicherungsprogramm ist ein definiertes System, dessen Personal von der Prüfungsdurchführung unabhängig ist, und das der Leitung der Prüfeinrichtung Gewissheit gibt, dass die Grundsätze der Guten Laborpraxis eingehalten werden. [1]
Verfahrensbezogene Inspektionen (processbased inspections) werden unabhängig von bestimmten Prüfungen durchgeführt. Sie dienen zur Überwachung von sich wiederholenden Verfahren und Prozessen und werden gewöhnlich stichprobenartig durchgeführt. [6]
Grundbegriffe - Archiv und computergestützte Systeme
Archiv ist eine bestimmte Räumlichkeit oder ein Bereich (z.B. Schrank, Raum, Gebäude oder computergestütztes System) zur sicheren Archivierung und Aufbewahrung von Aufzeichnungen und Materialien. [7]
Archivpersonal sind Personen die unter der Aufsicht des/der Archivverantwortlichen für Routinetätigkeiten und Verfahren bei der Archivierung verantwortlich sind. [7]
Archivverantwortliche/Archivverantwortlicher ist eine von der Leitung der Prüfeinrichtung oder des Prüfstandortes bestimmte Person, die für die Führung des Archivs verantwortlich ist, z.B. für Tätigkeiten und Verfahren bei der Archivierung. [7]
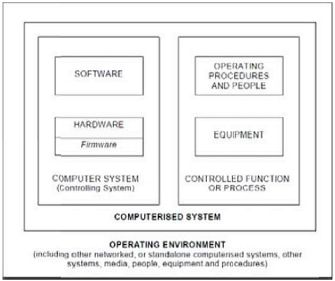
Computerised System is a function (process or operation) integrated with a computer system and performed by trained personnel. The function is controlled by the computer system. The controlling computer system is comprised of hardware and software. The controlled function is comprised of equipment to be controlled and operating procedures performed by personnel." PIC/S PI 11-3 "Good Practices for Computerised Systems in Regulated GxP Environments" [8]
Elektronische Archive: Einrichtungen und Systeme, die für den Erhalt elektronischer Aufzeichnungen zur Verfügung stehen. [7]
Elektronische Aufzeichnungen: Alle ursprünglichen Aufzeichnungen und Unterlagen einschließlich direkt in einen Computer eingegebene Daten, die als Ergebnis der ursprünglichen Beobachtungen und Tätigkeiten bei einer Prüfung anfallen und zur Rekonstruktion und zur Bewertung der Berichterstattung dieser Prüfung notwendig sind. [7]
Life cycle is an approach t- computerised system development that begins with identification of the user"s requirements, continues through design, integration, qualification, user validation, control and maintenance, and ends when use of the system is retired. [8]
Metadaten: Informationen, die Merkmale anderer Daten beschreiben. Meistens sind dies Informationen, die die Struktur, die Datenbestandteile, das Wirkungsgefüge und andere Charakteristika von elektronischen Aufzeichnungen beschreiben. [7]
Migration: Übertragung elektronischer Aufzeichnungen von einem Format, Datenträger oder computergestützten System auf ein anderes. [7]
Qualification is the action of proving that any equipment including software operates correctly and is fit for its purpose. [8]
Systemeigner: Die Leitung oder ein von ihr Beauftragter der am meisten betroffenen Abteilung oder hauptsächlichen Nutzer des Systems. [7]
Validation is the action of proving that a process leads t- the expected results. Validation of a computerised system requires ensuring and demonstrating the fitness for its purpose. [8]
III. Anhänge
| Standardarbeitsanweisungen (SOP) Stand: Januar 2018 | Anhang 1 |
1. Anforderungen
Die PE muss über schriftliche SOP verfügen, die von der LPE genehmigt sind. Die SOP müssen grundsätzlich in deutscher Sprache vorliegen. In Ausnahmefällen können fremdsprachige SOP akzeptiert werden, wenn die PE nachweisen kann, dass alle Beschäftigten, die mit den SOP arbeiten müssen, die Sprache hinreichend gut beherrschen. Die Überwachungsbehörde und das Inspektionsteam haben gemäß Verwaltungsverfahrensgesetz § 23 jederzeit das Recht zu verlangen, dass ihnen die für das Überwachungsverfahren notwendigen Dokumente auf Deutsch vorgelegt werden.
SOP müssen vor Ort vorhanden sein. Es muss sichergestellt sein, dass es sich jeweils nur um die aktuelle Fassung handelt. Außer Kraft gesetzte Fassungen müssen entweder entfernt (eingesammelt) oder als ungültig gekennzeichnet werden.
Fachbücher etc. und Bedienungsanleitungen für Geräte können ergänzend verwendet werden. Wenn sie einen Bestandteil der Standardarbeitsanweisung darstellen, sind sie mit dieser zu archivieren. Dies ist nicht erforderlich, wenn in der Standardarbeitsanweisung nur darauf verwiesen wird und im Übrigen die wesentlichen Informationen des Fachbuches/der Bedienungsanleitung bereits enthalten sind.
1.1. Formale Anforderungen
1.2. Weitere Anforderungen
Angaben von Fundstellen bzw. Bedienungsanleitungen der Gerätehersteller.
Ergänzungen und Änderungen müssen genehmigt und datiert sein. (Gewährleistung der Änderungen auf allen autorisierten Exemplaren)
Verfahren zur Überprüfung und Aktualisierung müssen vorliegen. Eine historische Ablage aller SOP muss erfolgen. Eine Auflistung aller GLP-relevanten SOP ist hilfreich.
Standardarbeitsanweisungen für die Überprüfung von Geräten, Einrichtungen und dgl. müssen Anweisungen für den Fall enthalten, dass die vorgegebenen Toleranzen oder Bedingungen nicht erfüllt sind.
Auf dem Sektor "Organisation und Personal" bedürfen die im Folgenden aufgeführten Punkte einer adäquaten schriftlichen Regelung, jedoch nicht unbedingt in Form von Standardarbeitsanweisungen:
2. Abzudeckende Bereiche
Mindestens folgende Bereiche müssen durch SOP abgedeckt sein, wobei jeweils mehrere der aufgeführten Punkte in einer Anweisung zusammengefasst sein können:
| Kurzzeitprüfungen Stand: November 2017 | Anhang 2 |
Für Kurzzeitprüfungen, die gemäß den OECD-GLP-Richtlinien durchgeführt werden, wird ein vereinfachtes Verfahren ermöglicht. Dies wird beschrieben im GLP-Konsensdokument Nummer 7, Anwendung der GLP-Grundsätze bei Kurzzeitprüfungen.
1. Definition Kurzzeitprüfung
OECD-Grundsätze der Guten Laborpraxis / ChemG Anh. 1:
Eine Kurzzeitprüfung ist eine Prüfung von kurzer Dauer, die nach weithin gebräuchlichen Routinemethoden durchgeführt wird.
Beschreibung im OECD-Konsensdokument Nr. 7:
Es existiert weder eine präzise Definition noch eine umfassende Liste von Kurzzeitprüfungen. Der Begriff "kurz" kann für biologische Prüfungen anders interpretiert werden als für physikalisch-chemische. Daher erfolgt die Einstufung vor allem aufgrund von Parametern wie:
Daraus folgt, dass ein und dieselbe Prüfung nicht unbedingt in jeder PE als Kurzzeitprüfung gelten kann. Es müssen ggf. Einzelfallentscheidungen getroffen werden, die von der PE zu dokumentieren sind. Auch eine sehr einfache Prüfung wie z.B. eine Schmelzpunktbestimmung ist ggf. keine Kurzzeitprüfung, wenn sie in einer PE nur sehr selten durchgeführt wird.
2. Anforderungen
2.1. Qualitätssicherung
Die QS muss nicht für jede Kurzzeitprüfung eine Inspektion einer kritischen Phase durchführen. Stattdessen können verfahrensbezogene Inspektionen durchgeführt werden.
Das verfahrensbezogene Inspektionsprogramm muss jede einzelne Prüfungsart einschließen, die von der PE als Kurzzeitprüfung definiert wurde und die nicht prüfungsbezogen inspiziert wird (OECD 7, 11.2.2.1.). Die Frequenz ist in einer SOP festzulegen und richtet sich nach Anzahl, Häufigkeit und Komplexität der Prüfungen in jeder Prüfungsart.
In der QS-Erklärung jeder Prüfung muss angegeben werden, welche Art von Inspektion (prüfungs- oder verfahrensbezogene) wann durchgeführt wurde.
Jeder einzelne Abschlussbericht muss von der QS geprüft werden. Aus der QS-Erklärung muss dies eindeutig hervorgehen.
2.2. Standard-Prüfplan und -Abschlussbericht
Es können ein Standard-Prüfplan (PP) und-Abschlussbericht (AB) erstellt werden, die jeweils die sich wiederholenden Informationen enthalten.
Wesentlich dabei ist, dass das Standard-Dokument zusammen mit der jeweiligen prüfungsspezifischen Ergänzung alle geforderten Inhalte für PP bzw. AB enthält. Wenn eine Prüfungsart z.B. immer für den gleichen Auftraggeber durchgeführt wird, könnte dessen Adresse in den Standard-PP aufgenommen werden.
Unterschriftenregelung:
Standard-PP: LPE, alle in Frage kommenden PL, QS
Prüfungsspezifische Ergänzung zum PP: aktuelle/aktueller PL, Kenntnisnahme QS
Standard-AB: LPE, alle in Frage kommenden PL
Prüfungsspezifische Ergänzung zum AB: aktueller PL, QS-Erklärung
Abweichungen vom Standard-PP sind wie Prüfplanänderungen zu handhaben. Abweichungen vom Standard-AB sind wie Korrekturen des AB zu handhaben (Nachtrag mit deutlicher Begründung und datierter Unterschrift des PL).
2.3. Geräte, Prüfsysteme, PRG
Es müssen Regelungen dafür getroffen werden, dass Tätigkeiten, die nicht für jede einzelne Kurzzeitprüfung durchgeführt werden (Kalibrierungen, Charakterisierung von PRG, Charakterisierung von mikrobiologischen Prüfsystemen...), regelmäßig und GLP-konform durchgeführt und dokumentiert werden.
3. Zusammenfassung der wichtigsten Punkte
| Inspektion von Datenverarbeitungs(DV)-Systemen | Anhang 3 |
(Über BLAC nicht publiziert)
| Multi-Site-Prüfungen Stand: Januar 2018 | Anhang 4 |
Nichtklinische gesundheits- und umweltrelevante Sicherheitsprüfungen können aus einer Vielzahl von Gründen als Multi-Site-Prüfungen (MSP) durchgeführt werden. Bei einer MSP werden Phasen dieser Prüfung aufgrund geographischer oder organisatorischer Bedingungen oder aufgrund der Anwendung spezieller Verfahren an mehr als einem Prüfstandort durchgeführt. Im OECD Konsensdokument Nr. 13 über MSP wird die Phase einer Prüfung als "... eine bestimmte Aufgabe oder eine Reihe von Aufgaben im Rahmen der Durchführung einer Prüfung" definiert. Unter dem Begriff "Aufgaben" sind sämtliche Einzelschritte einer
GLP-Prüfung zu verstehen. Dazu zählen u. a.:
Aufgrund der Tatsache, dass verschiedene Prüftätigkeiten an unterschiedlichen Prüfstandorten durchgeführt werden, sind die Planung, die eindeutige Zuordnung von Verantwortlichkeiten, eine wirkungsvolle Kommunikation und eine nachvollziehbare Kontrolle der Prüfung von entscheidender Bedeutung.
Auch wenn eine MSP sich aus einer Reihe von Untersuchungen und Tätigkeiten zusammensetzt, die an mehr als einem Prüfstandort durchgeführt werden, handelt es sich dennoch um eine einzige Prüfung. Das bedeutet, dass ein einziger Prüfplan (PP) vorhanden ist, ein/e einzige/r PL die Verantwortung für die Gesamtprüfung übernimmt und ein einziger Abschlussbericht (AB) erstellt wird. Mit der Definition zu MSP im Konsensdokument wird jedoch nicht ausgeschlossen, dass auch weiterhin komplexe Prüfungen (Untersuchungen) in einzelne eigenständige GLP-Prüfungen gesplittet werden können.
In der Regel ist der/die PL am Standort der LPE angesiedelt. Ist dies nicht der Fall, sollte das Inspektionsteam bei der Auditierung einer Prüfung im Rahmen einer Inspektion der PE dennoch darauf bestehen, dass der/die PL befragt werden kann. Es sollte überprüft werden, ob der/die PL die Fähigkeiten hat, alle Phasen einer Prüfung verantwortlich zu beaufsichtigen und ob die Voraussetzungen dafür in der PE gegeben sind.
Jede Phase einer Multi-Site-Prüfung muss in der Regel unter Einhaltung der GLP-Grundsätze durchgeführt werden und durch eine Inspektionskommission überprüfbar sein. Ein beteiligter PSt verfügt daher entweder über eine eigene GLP-Bescheinigung oder wird als abhängiger PSt der PE tätig. Falls Phasen einer Prüfung an einem PSt nicht unter GLP-Bedingungen durchgeführt wurden, ist dieses im statement of compliancezu berichten.
Im Folgenden werden Fragen formuliert, die bei der Inspektion von Multi-Site-Prüfungen von Bedeutung sein können:
1. Organisation und Personal
1.1.1.1. Wurden im Rahmen der Prüfungsplanung zwischen der auftraggebenden PE und
dem auftragnehmenden PSt schriftliche Vereinbarungen u. a. hinsichtlich der Festlegung von Verantwortlichkeiten, der Art und des Umfangs erforderlicher Informationen, einzuhaltender Kommunikationswege, der Sicherstellung angemessener Maßnahmen der QS, der Gestaltung des Abschlussberichtes, der Archivierungsmodalitäten und der einzuleitenden Maßnahmen bei unvorhergesehenen Ereignissen getroffen?
1.1.1.2. Wann wurde der/die PL von der LPE benannt? Wo ist der/die PL angesiedelt?
1.1.1.3. Wurden bei der Auswahl des PSt und der Vergabe von Prüfungsphasen PL und QS beteiligt? Hat sich der/die PL davon überzeugt, dass der PSt die GLP-Grundsätze einhalten kann?
1.1.1.4. Wurden geeignete Kommunikationswege im Voraus festgelegt, eingerichtet und getestet?
1.1.1.5. Fand die Kommunikation zwischen den Beteiligten direkt statt und wurde diese dokumentiert?
1.1.1.6. Wurde vor Beginn der Prüfung bzw. falls notwendig vor einer entsprechenden Prüfungsphase ein oder mehrere PI benannt? Wie hat der/die PL die Aufsicht über Prüfungsphasen sichergestellt, sofern kein/keine PI benannt wurde?
1.1.1.7. Wurde ein Verfahren für das Ersetzen einer/eines PI festgelegt?
1.1.1.8. Wurden allen beteiligten Personen die Erfordernisse der Prüfung bekannt gemacht?
1.1.1.9. Liegen für alle Personen (auch für befristet Beschäftigte, sofern diese prüfungsrelevante Tätigkeiten durchführen) Qualifikationsnachweise und Aufgabenbeschreibungen vor?
1.1.1.10. Enthält das Master Schedule der PE Angaben über beteiligte PSt, PI, Prüfungsphasen ggf. mit entsprechender Codierung, den Beginn und das Ende der Gesamtprüfung?
1.1.1.11. Enthält das Master Schedule eines PStes Angaben über PE, PL, PI, Prüfungsphase ggf. mit Codierung der Prüfung, Beginn und Ende der entsprechenden Prüfungsphase?
2. Qualitätssicherung
2.1.1.1. Wurde eine federführende QS benannt?
2.1.1.2. Wo ist die federführende QS angesiedelt?
2.1.1.3. Wurde zwischen der federführenden QS und ggf. den an den PSten tätigen QS ein gemeinsamer Inspektionsplan vor Beginn der Prüfung erstellt?
2.1.1.4. Wurde das beteiligte QS-Personal benannt?
2.1.1.5. Wurden die Verantwortlichkeiten und der Umfang der Überwachungsaufgaben der beteiligten QS festgelegt?
2.1.1.6. Welche SOP werden für das Überwachungsprogramm zugrunde gelegt?
2.1.1.7. Liegen den an den PSt beteiligten QS Kopien der Prüfpläne und ggf. der Prüfplanänderungen vor?
2.1.1.8. Wie erfolgt die Berichterstattung durch die an den PSt verantwortlichen QS?
2.1.1.9. Wurden die Inspektionsergebnisse am PSt unverzüglich anPI, LPSt, PL, LPE und federführende QS berichtet?
3. Prüfplan
3.1.1.1. Wurde die Kenntnisnahme des Prüfplans durch den PI dokumentiert?
3.1.1.2. Wurde eine dokumentierte Vereinbarung dahingehend getroffen, dass der/die PI die ihm/ihr übertragene Prüfungsphase in Übereinstimmung mit dem Prüfplan und den GLP-Grundsätzen durchführt?
3.1.1.3. Enthält der Prüfplan Angaben über alle beteiligten PSt (Name und Anschrift), die
dort durchgeführten Prüfungsphasen inklusive der Terminvorgaben sowie Name und Anschrift einschließlich Telefonnummern etc. der entsprechenden PI?
3.1.1.4. Werden alle beteiligten QS-Einheiten benannt?
3.1.1.5. Wurde im Prüfplan, sofern an einem PSt kein/keine PI ernannt wurde, das Personal aufgeführt mit dem sich der/die PL an diesem Prüfstandort unmittelbar verständigt?
3.1.1.6. Werden die Prüfungsphasen, die an einem PSt durchgeführt werden sollen, ausführlich im Prüfplan oder in einer Änderung zum Prüfplan dargelegt?
3.1.1.7. Werden im Prüfplan die für die Durchführung der entsprechenden Prüfungsphasen relevanten SOP bzw. die anzuwendenden Verfahren benannt?
3.1.1.8. Wurden Änderungen zum Prüfplan ausschließlich durch den/die PL begründet und genehmigt?
3.1.1.9. Enthält der Prüfplan Angaben darüber, wie die an den PSt erzeugten Daten dem/der PL zur Aufnahme in den Abschlussbericht übermittelt werden?
3.1.1.10. Enthält der Prüfplan Angaben über alle Orte der Archivierung?
3.1.1.11. Hat die federführende QS den Prüfplan überprüft?
3.1.1.12. Wurden die Teile der Prüfpläne die sich auf die Tätigkeiten an den PSten beziehen durch die jeweils verantwortliche QS überprüft?
3.1.1.13. Bei Zusammenarbeit von PE/PSt mit unterschiedlichen Sprachen: Enthält der originale Prüfplan Angaben über erforderliche Übersetzungen? Liegen die Übersetzungen dem Prüfplan bei?
3.1.1.14. Wie wurde die Richtigkeit und Vollständigkeit eines übersetzten Prüfplans sichergestellt?
4. Standardarbeitsanweisungen
4.1.1.1. Hat das Personal an den PSt Zugriff auf alle anzuwendenden SOP?
4.1.1.2. Falls am PSt nach SOP der PE gearbeitet werden soll: Hat die LPSt der Verwendung der SOP der PE schriftlich zugestimmt?
4.1.1.3. Wurde sichergestellt, dass an den PSt nur die aktuellen Fassungen der SOP der PE vorliegen? Erfolgt ein Austausch neuer Versionen?
4.1.1.4. Bei Zusammenarbeit von PE/PSt mit unterschiedlichen Sprachen: Wie wurde die Richtigkeit und Vollständigkeit bei notwendigen Übersetzungen sichergestellt?
5. Prüfungsablauf
5.1.1.1. Sind ggf. interne Codierungen der Prüfungsphasen an den PSt auf die ursprüngliche Codierung der Prüfung rückführbar?
5.1.1.2. Informieren die PI den/die PL schriftlich über den Fortgang der betreffenden Phasen der Prüfung?
5.1.1.3. Wurden dokumentierte Verfahren für den Transfer von Daten und Materialien etabliert, die deren Integrität gewährleisten? Erfolgt eine lückenlose Dokumentation? Wurden entsprechende Zuständigkeiten festgeschrieben?
5.1.1.4. Wurden Regelungen über Lagerung, Rückgabe und Entsorgung überschüssiger Prüf- und Referenzgegenstände getroffen?
5.1.1.5. Wurden Abweichungen vom Prüfplan oder von SOP zeitnah an den/die PL gemeldet?
5.1.1.6. Wurden diese am PSt dokumentiert und durch den/die PI bestätigt? Hat der/die PL die Kenntnisnahme bestätigt und erforderliche Maßnahmen durchgeführt?
5.1.1.7. Hat der/die PI nach Abschluss der Prüfungsphase u. a. alle Rohdaten, Proben etc. an den/die PL weitergeleitet oder diese gemäß Prüfplan archiviert? Wurde der/die PL über die Archivierung in Kenntnis gesetzt?
5.1.1.8. Wurden am PSt Proben entsorgt? Liegt eine schriftliche Zustimmung durch den/die PLvor?
5.1.1.9. Kennt das Personal an den PSt alle aktuell anzuwendenden Verfahren? Wurden entsprechende Schulungsmaßnahmen durchgeführt? Wurden diese dokumentiert?
6. Abschlussbericht
6.1.1.1. Enthält der Abschlussbericht Angaben über alle beteiligten PSte, die PI und die an sie delegierten Prüfungsphasen und deren Ergebnisse sowie alle Aufgaben im Rahmen der Gesamtprüfung?
6.1.1.2. Wurden Teilberichte durch PI erstellt? Wurden diese in den Gesamtbericht integriert, durch die entsprechenden PI unterschrieben und wurde erklärt, inwieweit die Phase der Prüfung unter Einhaltung der GLP-Grundsätze durchgeführt wurde? Wurde der Teilbericht durch die am PSt verantwortliche QS überprüft?
6.1.1.3. Wurden im Abschlussbericht alle Prüfungsphasen und alle Beiträge der PI berücksichtigt? Enthalten die Beiträge der PI schriftliche Zusicherungen, dass die GLP-Grundsätze eingehalten wurden?
6.1.1.4. Hat der/die PL den Abschlussbericht datiert unterschrieben und die Verantwortung für die Zuverlässigkeit aller Daten übernommen, indem er erklärt hat, inwieweit die Gesamtprüfung mit den GLP-Grundsätzen übereinstimmt?
6.1.1.5. Enthält der Abschlussbericht Angaben über alle Aufbewahrungsorte prüfungsbezogener Rohdaten, Unterlagen, Muster von Prüf- und Referenzgegenständen und Proben?
6.1.1.6. Wurde der Abschlussbericht durch die federführende QS inspiziert?
6.1.1.7. Liegt dem Abschlussbericht eine unterzeichnete Erklärung der federführenden QS bei?
Enthält diese Angaben über die durchgeführten Inspektionen an allen PSt oder wird auf Erklärungen der an den PSt verantwortlichen QS verwiesen?
6.1.1.8. Wurden Korrekturen und Ergänzungen eines Abschlussberichtes in Form von Nachträgen durch den/die PL vorgenommen? Erfolgte eine Abstimmung zwischen PL und PI, sofern es sich um einen Nachtrag zu einer delegierten Phase einer Prüfung handelt?
| Anwendungsbereiche der GLP Stand: Januar 2018 | Anhang 5 |
1. GLP-Anforderungen in EU-Rechtsnormen
Dieser Abschnitt ist angelehnt an das Dokument "EU legislation with Good Laboratory Practice (GLP) provisions" der Europäischen Kommission von März 2016 (herunterzuladen auf der Internetseite der Europäischen Kommission).
Ergänzungen und Aktualisierungen, die sich seit der Veröffentlichung dieses Dokuments bis Juli 2017 ergeben haben, sind kursiv gesetzt.
1.1. Chemikalien
1.1.1. Richtlinie (EG) Nr. 2004/10 (GLP)
Richtlinie (EG) Nr. 2004/10 vom 11. Februar 2004 zur Angleichung der Rechts- und Verwaltungsvorschriften für die Anwendung der Grundsätze der Guten Laborpraxis und zur Kontrolle ihrer Anwendung bei Versuchen mit chemischen Stoffen (kodifizierte Fassung)
1.1.2. Verordnung (EG) Nr.1907/2006 (REACH)
Verordnung (EG) Nr.1907/2006 des Europäischen Parlaments und des Rates vom 18. Dezember 2006 zur Registrierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) zur Schaffung einer Europäischen Chemikalienagentur, zur Änderung der Richtlinie 1999/45/EG und zur Aufhebung der Verordnung (EWG) Nr. 793/93 des Rates, der Verordnung (EG) Nr. 1488/94 der Kommission, der Richtlinie 76/769/EWG des Rates sowie der Richtlinien 91/155/EWG, 93/67/EWG, 93/105/EG und 2000/21/EG der Kommission
Anmerkung: Die REACH-Verordnung lässt zwar neben GLP auch andere internationale Standards, die von der Kommission oder von der Europäischen Chemikalienagentur (ECHA) als gleichwertig anerkannt sind, zu. Derartige Standards sind derzeit aber nicht verfügbar (siehe auch Q&A der ECHA, ID number 0117).
1.1.3. Verordnung (EG) Nr. 1272/2008 (CLP)
Verordnung (EG) Nr.1272/2008 des Europäischen Parlaments und des Rates vom 16. Dezember 2008 über die Einstufung, Kennzeichnung und Verpackung von Stoffen und Gemischen, zur Änderung und Aufhebung der Richtlinien 67/548/EWG und 1999/45/EG und zur Änderung der Verordnung (EG) Nr. 1907/2006
1.2. Biozide und Pflanzenschutzmittel
1.2.1. Verordnung (EU) Nr. 528/2012 (Biozidprodukte)
Verordnung (EU) Nr. 528/2012 des Europäischen Parlaments und des Rates vom 22. Mai 2012 über die Bereitstellung auf dem Markt und die Verwendung von Biozidprodukten
Anmerkung: Die Biozid-Verordnung lässt zwar neben GLP auch andere internationale Standards, die von der Kommission oder von der Europäischen Chemikalienagentur (ECHA) als gleichwertig anerkannt sind, zu. Derartige Standards sind derzeit aber nicht verfügbar (siehe auch Q&A der ECHA, ID number 0989).
1.2.2. Verordnung (EG) Nr. 1107/2009 (Pflanzenschutzmittel)
Verordnung (EG) Nr. 1107/2009 des Europäischen Parlaments und des Rates vom 21. Oktober 2009 über das Inverkehrbringen von Pflanzenschutzmitteln und zur Aufhebung der Richtlinien 79/117/EWG und 91/414/EWG des Rates in Verbindung mit der Verordnung (EU) Nr. 283/2013 und Verordnung (EU) Nr. 284/2013
Verordnung (EU) Nr. 283/2013 der Kommission vom 1. März 2013 zur Festlegung der Datenanforderungen für Wirkstoffe gemäß der Verordnung (EG) Nr. 1107/2009 des Europäischen Parlaments und des Rates über das Inverkehrbringen von Pflanzenschutzmitteln
Verordnung (EU) Nr. 284/2013 der Kommission vom 1. März 2013 zur Festlegung der Datenanforderungen für Pflanzenschutzmittel gemäß der Verordnung (EG) Nr. 1107/2009 des Europäischen Parlaments und des Rates über das Inverkehrbringen von Pflanzenschutzmitteln
1.3. Lebensmittel/Futtermittel
1.3.1. Verordnung (EG) Nr. 429/2008 (Futtermittelzusatzstoffe)
Verordnung (EG) Nr. 429/2008 der Kommission vom 25. April 2008 mit Durchführungsbestimmungen zur Verordnung (EG) Nr. 1831/2003 des Europäischen Parlaments und des Rates hinsichtlich der Erstellung und Vorlage von Anträgen sowie der Bewertung und Zulassung von Futtermittelzusatzstoffen
Weitere Ausführungen zu GLP-Erfordernis sind in den Abschnitt 2 und 3 des Anhangs II Verordnung (EG) Nr. 429/2008 zu finden, unter anderem zu toxikologischen Untersuchungen
1.3.2. Verordnung (EU) Nr. 234/2011 (Lebensmittelzusatzstoffe)
Verordnung (EU) Nr. 234/2011 der Kommission vom 10. März 2011 zur Durchführung der Verordnung (EG) Nr. 1331/2008 des Europäischen Parlaments und des Rates über ein einheitliches Zulassungsverfahren für Lebensmittelzusatzstoffe, -enzyme und -aromen
1.3.3. Durchführungsverordnung (EU) Nr. 503/2013 (Genetisch veränderte Lebens- und Futtermittel)
Durchführungsverordnung (EU) Nr. 503/2013 der Kommission vom 3. April 2013 über Anträge auf Zulassung genetisch veränderter Lebens- und Futtermittel gemäß der Verordnung (EG) Nr. 1829/2003 des Europäischen Parlaments und des Rates und zur Änderung der Verordnungen (EG) Nr. 641/2004 und (EG) Nr. 1981/2006 der Kommission
1.3.4. Empfehlung der Kommission 97/618/EG vom 29. Juli 1997 (Neuartige Lebensmittel)
Empfehlung der Kommission vom 29. Juli 1997 zu den wissenschaftlichen Aspekten und zur Darbietung der für Anträge auf Genehmigung des Inverkehrbringens neuartiger Lebensmittel und Lebensmittelzutaten erforderlichen Informationen sowie zur Erstellung der Berichte über die Erstprüfung gemäß der Verordnung (EG ) Nr. 258/97 des Europäischen Parlaments und des Rates (97/618/EG)
1.4. Arzneimittel und Medizinprodukte
1.4.1. Richtlinie 2003/63/EG der Kommission (Humanarzneimittel)
Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates vom 6. November 2001 zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel in Verbindung mit der Richtlinie 2003/63/EG der Kommission vom 25. Juni 2003 zur Änderung der Richtlinie 2001/83/EG des Europäischen Parlaments und des Rates zur Schaffung eines Gemeinschaftskodexes für Humanarzneimittel
1.4.2. Verordnung (EU) Nr. 536/2014 (Klinische Prüfungen)
Verordnung (EU) Nr. 536/2014 des Europäischen Parlaments und des Rates vom 16. April 2014 über klinische Prüfungen mit Humanarzneimitteln und zur Aufhebung der Richtlinie 2001/20/EG
1.4.3. Richtlinie 2009/9/EG (Tierarzneimittel)
Richtlinie 2009/9/EG der Kommission vom 10. Februar 2009 zur Änderung der Richtlinie 2001/82/EG des Europäischen Parlaments und des Rates zur Schaffung eines Gemeinschaftskodexes für Tierarzneimittel
1.4.4. Verordnung (EU) 2017/745 (Medizinprodukte)
Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates vom 5. April 2017 über Medizinprodukte, zur Änderung der Richtlinie 2001/83/EG, der Verordnung (EG) Nr. 178/2002 und der Verordnung (EG) Nr. 1223/2009 und zur Aufhebung der Richtlinien 90/385/EWG und 93/42/EWG des Rates
Anmerkung: Die Verordnung ist am 25. Mai 2017 in Kraft getreten. Sie gilt ab dem 26. Mai 2020 (Abweichungen s. Artikel 123 Absatz 3 der VO)
Nach Anhang II "Technische Dokumentation" Nr. 6.1 (Vorklinische und klinische Daten) ist zu bestimmten Prüfungen gegebenenfalls die Übereinstimmung mit den GLP-Grundsätzen nachzuweisen.
1.5. Kosmetische Mittel
1.5.1. Verordnung (EG) Nr. 1223/2009
Verordnung (EG) Nr. 1223/2009 des Europäischen Parlaments und des Rates vom 30. November 2009 über kosmetische Mittel
Anmerkung: Die Verordnung lässt zwar neben GLP auch andere internationale Standards, die von der Kommission oder von der Europäischen Chemikalienagentur (ECHA) als gleichwertig anerkannt sind, zu. Derartige Standards sind derzeit aber nicht verfügbar.
1.6. Detergenzien
1.6.1. Verordnung (EG) Nr. 648/2004
Verordnung (EG) Nr. 648/2004 des Europäischen Parlaments und des Rates vom 31. März 2004 über Detergenzien
2. GLP-Pflicht im deutschen Recht
Im Chemikaliengesetz wird die Einhaltung der GLP-Grundsätze für nichtklinische gesundheits- und umweltrelevante Sicherheitsprüfungen von Stoffen oder Gemischen, deren Ergebnisse die behördliche Bewertung möglicher Gefahren für Mensch und Umwelt ermöglichen sollen, verbindlich vorgeschrieben.
In der Allgemeinen Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis (ChemVwV-GLP) 1 wird in Nummer 2 der Anwendungsbereich für Biozide (1), Chemikalien (2), Pflanzenschutzmittel (3), Arzneimittel (4), Sprengstoffe (5) und Lebensmittel- und Futtermittelzusatzstoffe (6) konkretisiert. Dem Anwendungsbereich des § 19a Absatz 1 ChemG unterliegen insbesondere folgende nichtklinische experimentelle Prüfungen:
Auf die nicht dem Anwendungsbereich des § 19a Absatz 1 ChemG unterliegenden Prüfungen ist diese Verwaltungsvorschrift entsprechend anzuwenden, wenn die Prüfungen aufgrund von Rechtsakten eines Organs der Europäischen Union nach den Grundsätzen der Guten Laborpraxis zu erfolgen haben.
____
1) Neufassung Allgemeine Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis vom 15. Mai 1997 und Allgemeine Verwaltungsvorschrift zur Änderung der Allgemeinen Verwaltungsvorschrift zum Verfahren der behördlichen Überwachung der Einhaltung der Grundsätze der Guten Laborpraxis vom 16. November 2011
| Prüfkategorien Stand: November 2017 | Anhang 6 |
| Prüfka- tegorie | Anwendungsbereich | |
| 1 | Prüfungen zur Bestimmung der physikalisch-chemischen Eigenschaften und Gehaltsbestimmungen | Physicalchemical testing |
| 2 | Prüfungen zur Bestimmung der toxikologischen Eigenschaften | Toxicity studies |
| 3 | Prüfungen zur Bestimmung der erbgutverändernden Eigenschaften (in vitro und in vivo) | Mutagenicity studies |
| 4 | Ökotoxikologische Prüfungen zur Bestimmung der Auswirkungen auf aquatische und terrestrische Organismen | Environmental toxicity studies on aquatic and terrestrial organisms |
| 5 | Prüfungen zum Verhalten im Boden, im Wasser und in der Luft; Prüfungen zur Bioakkumulation und zur Metabolisierung | Studies on behaviour in water, soil and air; bioaccumulation |
| 6 | Prüfungen zur Bestimmung von Rückständen | Residue studies |
| 7 | Prüfungen zur Bestimmung der Auswirkungen auf Mesokosmen und natürliche Ökosysteme | Studies on effects on mesocosms and natural ecosystems |
| 8 | Analytische Prüfungen an biologischen Materialien | Analytical and clinical chemistry testing |
| 9 | Sonstige Prüfungen (mit Erläuterung) | other studies (specify) |
Mit Inkrafttreten der Neufassung der "Allgemeinen Verwaltungsvorschrift GLP (ChemVwV-GLP)" vom Mai 1997 ist in Deutschland die Aufteilung der durchgeführten GLP-Prüfungen in neun Prüfkategorien eingeführt worden.
Die Ausgestaltung des Begriffs Prüfung ist in den GLP-Grundsätzen sehr offen gehalten, so dass ein Spielraum für die nationalen Behörden und die Industrie bei der praktischen Umsetzung besteht. So kann eine Freilandprüfung, die an mehreren Orten stattfindet, als eine Gesamtprüfung durchgeführt werden, oder es kann eine Aufsplittung in mehrere abgeschlossene Einzelprüfungen erfolgen. Für jede komplette Prüfung muss ein Prüfleiter/eine Prüfleiterin benannt sein sowie ein Prüfplan, und ein Abschlussbericht vorliegen. Vorgaben für die Definition einer Prüfung werden z. T. von den zuständigen Bewertungsbehörden gemacht, letztendlich definiert aber die Prüfeinrichtung selbst, was sie als Prüfung im Sinne der GLP-Grundsätze betrachtet.
Zweck der Prüfkategorien ist es, den Informationsaustausch zwischen den OECD-Mitgliedstaaten zu verbessern, und Bewertungsbehörden sowie potentielle Auftraggeber über die bei den staatlichen GLP-Inspektionen berücksichtigten Bereiche zu informieren. Prüfungen, die nicht eindeutig in die angegebenen Prüfkategorien einzuordnen sind, werden von den Bewertungsbehörden nicht aus formalen Gründen zurückgewiesen. Sollten bei den Bewertungsbehörden ernste Bedenken bestehen, ob die vorgelegten Prüfungen durch die in der GLP-Bescheinigung / Liste aufgeführten Prüfkategorien abgedeckt sind, kann dies im Einzelfall über die Inspektionsberichte bzw. Nachfrage bei den zuständigen Überwachungsbehörden geklärt werden.
Alle Teilbereiche einer Untersuchung, wie z.B. bei toxikologischen Prüfungen die Tieraufzucht, Tierhaltung, Analytik etc. sind in der entsprechenden Prüfkategorie enthalten. Falls eine Prüfeinrichtung neben der vollständigen Durchführung von Prüfungen auch Teilbereiche von Prüfungen derselben Prüfkategorie, z.B. im Auftrag anderer Prüfeinrichtungen vornimmt, sind diese ebenfalls abgedeckt, auch wenn in diesem Fall nicht die gesamte Prüfung durchgeführt wird. So ist z.B. die Analytik in den Prüfkategorien 2 - 7 enthalten und in diesen Fällen muss die Kategorie 8 nicht noch zusätzlich vergeben werden.
Eine Besonderheit besteht bei spezialisierten Analytiklaboratorien, die häufig Teile/Phasen von GLP-Prüfungen für verschiedene Auftraggeber durchführen. Erfolgt dies in Form eines Unterauftrages, müssen die Analytiklaboratorien bei einer GLP-Inspektion der Auftraggeber jedes Mal miteinbezogen werden. Hier kann eine eigene GLP-Bescheinigung der einzelnen Analytiklaboratorien praktikabler und sinnvoller sein. Aus diesem Grund wurde von der OECD die Prüfkategorie 8 eingeführt, obwohl es sich bei der Analytik selten um komplette, sondern eher um Teilbereiche GLP-pflichtiger Prüfungen handelt. Eine eigene GLP-Bescheinigung setzt dabei die vollständige Implementierung der GLP-Grundsätze voraus.
Auch wenn nicht das komplette Spektrum einer Prüfkategorie durchgeführt wird, sollte in der Regel die dazugehörige Kategorie insgesamt erteilt werden. Die mögliche zwischenzeitliche Erweiterung des Prüfumfangs innerhalb einer Prüfkategorie sollte dann bei der Folgeinspektion besonders berücksichtigt werden. Lediglich wenn begründete Bedenken bestehen, z.B. wegen eines sehr geringen Prüfumfanges einer Prüfeinrichtung eine komplette Prüfkategorie zu bescheinigen, können unter Prüfkategorie 9 auch engbegrenzte Prüfbereiche der Kategorien 1 - 7 aufgeführt werden.
Im Folgenden werden einige Erläuterungen und Beispiele zu den einzelnen OECD-Prüfkategorien gegeben:
Prüfkategorie 1: Prüfungen zur Bestimmung der physikalisch-chemischen Eigenschaften und Gehaltsbestimmungen
In diese Kategorie sind Prüfungen subsumiert, deren Ergebnisse ausschließlich der Bestimmung physikalischer, chemischer und physikalisch-chemischer Parameter dienen. Diese Prüfungen sind in der VO 440/2008/EG - Teil A: Methoden zur Bestimmung der physikalisch-chemischen Eigenschaften aufgeführt.
Prüfkategorie 2: Prüfungen zur Bestimmung der toxikologischen Eigenschaften
Voraussetzung für die Einordnung in diese Prüfkategorie ist, dass ein Prüfgegenstand am Tier appliziert wird, und die Ergebnisse der Untersuchungen für die Bewertung toxischer Auswirkungen auf den Menschen herangezogen werden sollen. Alle Bereiche, die zur Prüfungsdurchführung notwendig sind, wie z.B. Aufzucht, Haltung und Pflege der Versuchstiere und die begleitende Analytik sind durch diese Kategorie mit abgedeckt. Diese Prüfungen sind in der VO 440/2008/EG - Teil B: Methoden zur Bestimmung der Toxizität und sonstiger Auswirkungen aufgeführt.
Beispiele sind Prüfungen zu:
akuter Toxizität (oral, dermal, inhalativ); subakuter Toxizität (28 Tage); subchronischer Toxizität (90 Tage); chronischer Toxizität; Haut- und Augenreizung; Sensibilisierung; verhaltensstörenden Eigenschaften; Karzinogenität; Reproduktionstoxizität; Teratogenität; Embryotoxizität; Toxikokinetik.
Prüfungen zur Bestimmung der erbgutverändernden Eigenschaften (in vitro und in vivo)
Unter diese Prüfkategorie fallen die Untersuchungen zur Gentoxizität, insbesondere die in vitro-Prüfungen zur Mutagenität. Bei den in vivo-Untersuchungen bestehen Überschneidungen mit der Prüfkategorie 2. Auch diese Prüfungen sind in der VO 440/2008/EG - Teil B: Methoden zur Bestimmung der Toxizität und sonstiger Auswirkungen aufgeführt.
Beispiele sind Prüfungen zu/an:
Rückmutation (E. coli, S. typhimurium); Genmutation, Mitotischer Rekombination (Saccharomyces cerevisiae); Letalmutation (Drosophila melanogaster); Säugetierzellen in vitro (DNS-Schädigung und -Reparatur, Schwesterchromatidaustausch, Zell-Transformation); Säugern in vivo (Mikrokerntest, Dominant-Letal-Test, Keimzellzytogenetik, Fellfleckentest, Translokationstest).
Prüfkategorie 4: Ökotoxikologische Prüfungen zur Bestimmung der Auswirkungen auf aquatische und terrestrische Organismen
In diese Kategorie werden alle ökotoxikologischen Prüfungen an Einzelspezies zusammengefasst, die zur Bewertung von Risiken für die Umwelt herangezogen werden. Im Vordergrund der Untersuchungen stehen die Auswirkungen der Substanz auf Organismen.
Aufgrund ihrer stark voneinander abweichenden Struktur wird zwischen aquatischen und terrestrischen Lebensräumen bzw. Ökosystemen unterschieden. Für aquatische Lebensräume ist das Medium Wasser biotopbestimmend. Der terrestrische Lebensraum wird zumindest von Boden und Luft bestimmt, die zudem auch als Teillebensräume des terrestrischen Bereichs verstanden werden. Der Begriff terrestrische Organismen ist somit weit auszulegen und entspricht nicht dem englischen Begriff "soil organism" im Sinne von Bodenorganismen. Insofern umfasst die Kategorie 4 z.B. auch Nützlinge, Bienen, Insekten und Vögel. Diese Prüfungen sind in der VO 440/2008/EG - Teil C: Methoden zur Bestimmung der Ökotoxizität aufgeführt.
Beispiele sind Prüfungen zur Toxizität/Auswirkungen an:
Fischen (akut, verlängert), Daphnien (akut, verlängert); Vögeln, Bakterien, Bodenorganismen (Bodenfauna); Bodenmikroflora; höheren Pflanzen; Grünalgen; Honigbienen; sonstigen Nutzorganismen. Fütterungsstudien mit Untersuchungen zum Stoffwechsel und zur Kinetik (Metabolismus) von Rückständen im Futter zur Beurteilung der Gesundheitsbeeinträchtigung von landwirtschaftlichen Nutztieren oder Wildtieren sind ebenfalls durch diese Kategorie abgedeckt.
Prüfkategorie 5: Prüfungen zum Verhalten im Boden, im Wasser und in der Luft; Prüfungen zur Bioakkumulation und zur Metabolisierung
Im Vordergrund der Prüfungen steht die Beurteilung des Verhaltens der Substanz, wie Verflüchtigung, Umsetzung, Verbleib, Bindung und Verteilung. Damit wird die Verfügbarkeit einer Substanz in der Umwelt untersucht, die Einfluss auf Art und Dauer möglicher Auswirkungen haben kann und die Grundlage für eine Expositionsanalyse darstellt. Die direkten schädigenden Auswirkungen auf Organismen sind nicht Gegenstand der Prüfungen in dieser Kategorie.
Beispiele sind Prüfungen zum/zur: Verbleib im Boden, im Wasser und in der Luft, Photolyse, Flüchtigkeit aus Pflanzen und aus dem Boden, photochemischoxidativen Abbau, Adsorption/Desorption, Bioakkumulation bei Fischen sowie zu Lysimeterversuchen.
Prüfkategorie 6: Prüfungen zur Bestimmung von Rückständen
In die Prüfkategorie 6 fallen vornehmlich Rückstandsversuche im Sinne der Richtlinien der Biologischen Bundesanstalt für die Prüfung von Pflanzenschutzmitteln im Zulassungsverfahren. Sofern als fester Bestandteil dieser Versuche auch Untersuchungen auf Rückstände im Wasser und Boden durchgeführt werden müssen, sind sie hier zu subsumieren. Alle Teilbereiche dieser Untersuchungen, wie z.B. Applikation, Probenahme, Probenaufbereitung und Analytik sind abgedeckt.
Prüfkategorie 7: Prüfungen zur Bestimmung der Auswirkungen auf Mesokosmen und natürliche Ökosysteme
Bei diesen Prüfungen werden Eintrag, Verbleib und ökologische Auswirkungen von Prüfgegenständen in künstlich angelegten komplexen Ökosystemen oder auch direkt im Freiland untersucht. Im Rahmen des Zulassungsverfahrens von Pflanzenschutzmitteln können diese Untersuchungen wesentlich sein, wenn der Vergleich von Expositionsanalyse und ökotoxikologischen Ergebnissen an Einzelspezies keine abschließende Risikoanalyse erlauben. Das Prüfungsdesign variiert entsprechend der speziellen Fragestellung. Für den aquatischen Bereich sind die sogenannten "Pond studies", in denen mehrere künstlich angelegte Teiche parallel mit unterschiedlichen Konzentrationen der Testsubstanz appliziert werden, die verbreitetste Art von Prüfungen an Mesokosmen. In den USA werden Prüfungen an aquatischen und terrestrischen Ökosystemen, entsprechend dem "Federal Insecticide, Fungicide and Rodenticide Act (FIFRA)" seit 1989 nach GLP durchgeführt.
Prüfkategorie 8: Analytische Prüfungen an biologischen Materialien
Die Kategorie 8 betrifft jene Prüfeinrichtungen, die ausschließlich die Analytik in den Prüfkategorien 2 - 7 vornehmen.
Prüfkategorie 9: Sonstige Prüfungen (mit Erläuterung)
Unter diese Kategorie fallen alle Prüfungen, die nicht durch die Kategorien 1 - 8 abgedeckt werden. Dies können auch Untersuchungen sein, die in Deutschland nicht unter die GLP-Pflicht (§ 19a ChemG) fallen, aber aufgrund eines "berechtigten Interesses" entsprechend § 19b Abs. 1 ChemG auf Einhaltung der GLP-Grundsätze inspiziert wurden. Weiterhin können unter diese Kategorie auch engbegrenzte Prüfbereiche der Kategorien 1 - 8 subsumiert werden, wenn die Einteilung der entsprechenden Gesamt-Prüfkategorie zu weitreichend erscheint.
Eine aktuelle Übersicht der GLP-Prüfeinrichtungen und der bescheinigten Prüfungsarten in der Prüfkategorie 9 kann dem "Verzeichnis der Prüfeinrichtungen/Prüfstandorte mit GLP-Bescheinigung in Deutschland" entnommen werden. Dieses wird einmal im Jahr im Bundesanzeiger veröffentlich. Die jeweils aktuelle Fassung kann auf der Internetseite des Bundesinstituts für Risikobewertung heruntergeladen werden.
| Prüfmethoden Stand: November 2017 | Anhang 7 |
Gemäß Verordnung 440/2008/EG der Kommission vom 30. Mai 2008 zur Festlegung von Prüfmethoden gemäß der Verordnung (EG) Nr. 1907/2006 des Europäischen Parlaments und des Rates zur Regierung, Bewertung, Zulassung und Beschränkung chemischer Stoffe (REACH) - Stand 14. Februar 2017 /EU 2017/735):
Teil A: Methoden zur Bestimmung der physikalisch-chemischen Eigenschaften
| A.1 | SCHMELZ-/GEFRIERTEMPERATUR |
| A.2 | SIEDETEMPERATUR |
| A.3 | RELATIVE DICHTE |
| A.4 | DAMPFDRUCK |
| A.5 | OBERFLÄCHENSPANNUNG |
| A.6 | WASSERLÖSLICHEKEIT |
| A.8 | VERTEILUNGSKOEFFIZIENT |
| A.9 | FLAMMPUNKT |
| A.10 | ENTZÜNDLICHKEIT (FESTE STOFFE) |
| A.11 | ENTZÜNDLICHKEIT (GASE) |
| A.12 | ENTZÜNDLICHKEIT (BERÜHRUNG MIT WASSER) |
| A.13 | PYROPHORE EIGENSCHAFTEN VON FESTEN UND FLÜSSIGEN STOFFEN |
| A.14 | EXPLOSIONSGEFAHR |
| A.15 | ZÜNDTEMPERATUR (FLÜSSIGKEIT UND GASE) |
| A.16 | RELATIVE SELBENTZÜNDUNGSTEMPERATUR FÜR FESTSTOFFE |
| A.17 | BRANDFÖRDERNDE EIGENSCHAFTEN (FESTSTOFFE) |
| A.18 | ZAHLENGEMITTELTE MOLMASSE UND MOLMASSENVERTEILUNG VON POLYMEREN |
| A.19 | NIEDERMOLEKULARER ANTEIL VON POLYMEREN |
| A.20 | LÖSUNGS-/EXTRAKTIONSVERHALTEN VON POLYMEREN IN WASSER |
| A.21 | BRANDFÖRDERNDE EIGENSCHAFTEN (FLÜSSIGE STOFFE) |
| A.22 | ETRISCHER DURCHMESSER VON FASERN |
| A.23 | 1-OCTANOL/WASSER-VERTEILUNGSKOEFFIZIENT: ME |
| A.24 | VERTEILUNGSKOEFFIZIENT (N-OCTANOL/WASSER), HOCHLEISTUNGS-FL (HPLC-METHODE) |
| A.25 | DISSOZIATIONSKONSTANTEN IN WASSER (TITRATIONSVERFAHREN - SPEKTROMETRISCHES VERFAHREN - KONDUKTOMETRISCHES VERFAHREN) |
Teil B: Methoden zur Bestimmung der Toxizität und sonstiger Auswirkungen auf die Gesundheit
| B.1 bis | AKUTE ORALE TOXIZITÄR - FEST-DOSIS-METHODE |
| B.1 tris | AKUTE ORALE TOXIZITÄT - AKUT-TOXISCHE KLASSENMETHODE |
| B.2 | AKUTE INHALATIONSTOXIZITÄT |
| B.3 | AKUTE TOXIZITÄT (DERMAL) |
| B.4 | AKUTE TOXIZITÄT; HAUTREIZUNG/-VERÄTZUNG |
| B.5 | AKUTE AUGENREZUNG/-VERÄTZUNG |
| B.6 | SENSIBILISIERUNG DER HAUT |
| B.7 | 28-TAGE-TOXIZITÄTSSTUDIE MIT WIEDERHOLTER ORALER VERABREICHUG AN NAGETIEREN |
| B.8 | PRÜFUNG AUF SUBAKUTE TOXIZITÄT NACH IN HALATION - 28-TAGE-TEST |
| B.9 | TOXIZITÄT NACH 28-TÄGIGER GABE (DERMAL) |
| B.10 | IN-VITRO-TEST AUF CHROMOSOMENABERRA |
| B.11 | TEST AUF CHROMOSOMENABERRATIONEN |
| B.12 | ERYTHROZYTEN-MIKROKERNTEST BEI SÄUGERN |
| B.13/14 | MUTAGENITÄT - RÜCKMUTATIONSTEST UNTER VERWENDUNG VON BAKTERIEN |
| B.17 | MUTAGENITÄT - IN VITRO-GENMUTATIONSTEST AN SÄUGETIERZELLEN |
| B.21 | IN-VITRO-ZELLTRANSFORMATIONSTEST |
| B.22 | SÄUGER-IN-VIVO-DOMINANT-LETAHL-TEST |
| B.23 | SPERMATOGONIEN-CHROMOSOMENABER |
| B.25 | IN-VIVO-SÄUGER-TRANSLOKATIONSTEST |
| B.26 | PRÜFUNG AUF SUB-CHRONISCHE ORALE TOXIZITÄT - 90-TAGE-TOXIZITÄTSSTUDIE BEI WIEDERHOLTER ORALER VERABREICHUNG AN NAGETIEREN |
| B.27 | PRÜFUNG AUF SUB-CHRONISCHE ORALE TOXIZITÄT - 90-TAGE-TOXIZITÄTSSTUDIE BEI WIEDERHOLTER ORALER VERABREICHUNG AN NICHT-NAGETIEREN |
| B.28 | PRÜFUNG AUF SUB-CHRONISCHE TOXIZITÄT NACH DERMALER APPLIKATION - 90-TAGE-TEST MIT NAGERN |
| B.29 | PRÜFUNG AUF SUB-CHRONISCHE TOXIZITÄT NACH INHALATION - 90-TAGE-TEST MIT NAGERN |
| B.30 | PRÜFUNG AUF CHRONISCHE TOXIZITÄT |
| B.31 | STUDIE ZUR PRÜFUNG AUF PRÄNATALE ENTWICKLUNGSTOXIZITÄT |
| B.32 | PRÜFUNG AUF KANZEROGENITÄT |
| B.33 | KOMBINIERTE STUDIEN ZUR PRÜFUNG AUF CHRONISCHE TOXIZITÄT UND KANZEROGENITÄT |
| B.34 | PRÜFUNG AUF REPRODUKTIONSTOXIZITÄT WÄHREND EINER GENERATION |
| B.35 | ZWEIGENERATIONENSTUDIE ZUR PRÜFUNG`AUF REPRODUKTIONSTOXIZITÄT |
| B.36 | TOXIKOKINETIK |
| B.37 | VERZÖGERTE NAUROTOXIZITÄT PHOSPHORORGANISCHER SUBSTANZEN NACH AKUTER EXPOSITION |
| B.38 | VERZÖGERTE NEUROTOXIZITÄT PHOSPHORORGANISCHER SUBSTANZEN BEI WIEDERHOLTER GABE ÜER 28 TAGE |
| B.39 | IN-VIVO-TEST ZUR UNPLANMÄSSIGEN DANN-SYNTHESE (UDS) IN SÄUGETIERLEBERZELLEN |
| B.40 | IN-VITRO-PRÜFUNG AUF HAUTÄTZENDE WIRKUNG: TER-TEST (TRANSCUTANEOUS ELECTRICAL RESISTANCE TEST) |
| B.40 bis | IN-VITRO-PRÜFUNG AUF HAUTÄTZENDE WIRKUNG: TEST MIT MENSCHLICHEM HAUTMODELL |
| B.41 | IN-VITRO-3T3-NRU-FOTOTOXIZITÄTSTEST |
| B.42 | HAUTSENSIBILISIERUNG: LOKALER LYMPHKNOTENTEST |
| B.43 | PRÜFUNG AUF NEUROTOXIZITÄT BEI NAGETIEREN |
| B.44 | HAUTRESORPTION: IN-VIVO-METHODE |
| B.45 | HAUTRESORPTION: IN-VITRO-METHODE |
| B.46 | IN-VITRO-HAUTREIZUNG: TEST AN REKONSTRUIERTEN MODELLEN HUMANER EPIDERMIS |
| B.47 | TRÜBUNGS- UND DURCHLÄSSIGKEITSTEST AN DER RINDERHORNHAUT ZWECKS IDENTIFIZIERUNG VON I) CHEMIKALIEN, DIE SCHWERE AUGENSCHÄDEN VERURSACHEN, UND II) CHEMIKALIEN, DIE KEINE EINSTUFUNG ALS AUGENREIZEND ODER SCHWER AUGENSCHÄDIGEND ERFORDERN |
| B.48 | TEST AM ISOLIERTEN HÜHNERAUGE ZUR IDENTIFIZIERUNG VON I) CHEMIKALIEN, DIE SCHWERE AUGENSCHÄDEN VERURSACHEN, UND II) CHEMIKALIEN, DIE KEINE EINSTUFUNG ALS AUGENREIZEND ODER SCHWER AUGENSCHÄDIGEND ERFORDERN |
| B.49 | IN-VITRO-MIKRONUKLEUSTEST AN SÄUGETIERZELLEN |
| B.50 | HAUTSENSIBILISIERUNG: LOKALER LYMPHKNOTENTEST: DA |
| B.51 | HAUTSENSIBILISIERUNG: LOKALER LYMPHKNOTENTEST: BRDU-ELISA |
| B.52 | AKUTE INHALATIONSTOXIZITÄT - AKUT-TOXISCHE KLASSENMETHODE |
| B.53 | PRÜFUNG AUF ENTWICKLUNGSNEUROTOXIZITÄT |
| B.54 | UTEROTROPHER BIOASSAY MIT NAGERN: EIN KURZZEIT-SCREENING-EIGENSCHAFTEN |
| B.55 | HERSHBERGER-BIOASSAY MIT RATTEN: EIN KURZZEIT- SCREENING-TEST AUF (ANTI-)ANDROGENE EIGENSCHAFTEN |
| B.56 | ERWEITERTE EIN-GENERATIONEN-PRÜFUNG AUF REPRODUKTIONSTOXIZITÄT |
| B.57 | H295R-STEROIDGENESE-ASSAY |
| B.58 | GENMUTATIONS-ASSAYS AN SOMATISCHEN ZELLEN UND KEIMZELLEN TRANSGENER NAGETIERE |
| B.59 | IN-CHEMICO-HAUTSENSIBILISIERUNG: DIREKT STEST (DPRA) |
Teil C: Methoden zur Bestimmung der Ökotoxizität
| C.1 | AKUTE TOXIZITÄT FÜR FISCHE |
| C.2 | DAPHNIA-SP-TEST AUF AKUTE SCHWIMMUNFÄHIGKEIT |
| C.3 | SÜSSWASSERALGEN UND CYANOBAKTERIEN: WACHSTUMSINHIBITIONSTEST |
| C.4 | BIOLOGISCHE ABBAUBARKEIT - BESTIMMUNG DER "LEICHTEN" BIOLOGISCHEN ABBAUBARKEIT |
| TEIL I | ALLGEMEINES |
| TEIL II | DOC-DIE-AWAY-TEST - ABNAHME VON GELÖSTEM ORGANISCHEM KOHLENSTOFF (DOC) (Methode C.4-A) |
| TEIL III | MODIFIZIERTER OECD-SCREENING-TEST (Methode C.4-B) |
| TEIL IV | CO2-ENTWICKLUNGSTEST (Methode C.4-C) |
| TEIL V | MANOMETRISCHER RESPIRATIONSTEST (Methode C.4-D) |
| TEIL VI | GESCHLOSSENER FLASCHENTEST (Methode C.4-E) |
| TEIL VII | MITI-TEST (Methode C.4-F) |
| C.5 | ABBAUBARKEIT - BIOCHEMISCHER SAUERSTOFFBEDARF |
| C.6 | ABBAUBARKEIT - CHEMISCHER SAUERSTOFFBEDARF |
| C.7 | ABBAUBARKEIT - ABIOTISCHER ABBAU: HYDROLYSE IN ABHÄNGIGKEIT VOM pH-WERT |
| C.8 | TOXIZITÄT FÜR REGENWÜRMER |
| C.9 | BIOLOGISCHE ABBAUBARKEIT - ZAHN-WELLENS-TEST |
| C.10 | SIMULATION DER AEROBEN ABWASSERBEHANDLUNG: C.10-A: BELEBTSCHLAMM - C.10-B: BIOFILME |
| C.11 | BELEBTSCHLAMM, PRÜFUNG DER ATMUNGSHEMMUNG (KOHLENSTOFF- UND AMMONIUMOXIDATION) |
| C.12 | BIOLOGISCHE ABBAUBARKEIT - MODIFIZIERTER SCAS-TEST |
| C.13 | BIOAKKUMULATIONSPRÜFUNG AM FISCH MIT AQUATISCHER EXPOSITION UND EXPOSITION ÜBER DAS FUTTER |
| C.14 | WACHSTUMSTEST AN JUNGFISCHEN |
| C.15 | FISCHE, KURZFRISTIGE TOXIZITÄTSPRÜFUNG AN EMBRYONEN UND JUNGFISCHEN MIT DOTTERSACK |
| C.16 | HONIGBIENEN - AKUTE ORALE TOXIZITÄTSPRÜFUNG |
| C.17 | HONIGBIENEN - AKUTE KONTAKTTOXIZITÄTSPRÜFUNG |
| C.18 | ADSORPTION/DESORPTION NACH EINER SCHÜTTELMETHODE |
| C.19 | SCHÄTZUNG DES ADSORPTIONSKOEFFIZIENTEN (Koc) IM BODEN UND IN KLÄRSCHLAMM MITTELS DER HOCHDRUCK- FLÜSSIGCHROMATOGRAFIE (HPLC) |
| C.20 | DAPHNIA-MAGNA-REPRODUKTIONSTEST |
| C.21 | BODENMIKROORGANISMEN: STICKSTOFFTRANSFORMATIONSTEST |
| C.22 | BODENMIKROORGANISMEN: KOHLENSTOFFTRANSFORMATIONSTEST |
| C.23 | AEROBE UND ANAEROBE TRANSFORMATION IM BODEN |
| C.24 | AEROBE UND ANAEROBE TRANSFORMATION IN WASSER-SEDIMENT-SYSTEMEN |
| C.25 | AEROBE MINERALISATION IN OBERFLÄCHENWASSER - SIMULATIONSTEST ZUR BIOLOGISCHEN ABBAUBARKEIT |
| C.26 | LEMNA SP. - WACHSTUMSINHIBITIONSTEST |
| C.27 | CHIRONOMIDEN-TOXIZITÄTSTEST IN SEDIMENT-WASSER-SYSTEMEN MIT GESPIKTEM SEDIMENT |
| C.28 | CHIRONOMIDEN-TOXIZITÄTSTEST IN SEDIMENT-WASSER-SYSTEMEN MIT GESPIKTEM WASSER |
| C.29 | LEICHTE BIOLOGISCHE ABBAUBARKEIT - BESTIMMUNG VON CO2 IN GESCHLOSSENEN FLASCHEN (HEADSPACE-TEST) |
| C.30 | BIOAKKUMULATION IN TERRESTRISCHEN OLIGOCHAETEN |
| C.31 | WACHSTUMSTEST BEI LANDPFLANZEN: UNTERSUCHUNG VON AUFLAUF UND WACHSTUM VON KEIMLINGEN |
| C.32 | ENCHYTRAEEN-REPRODUKTIONSTEST |
| C.33 | REPRODUKTIONSTEST MIT REGENWÜRMERN (EISENIA FETIDA/EISENIA ANDREI) |
| C.34 | BESTIMMUNG DER HEMMUNG ANAEROBER BAKTERIEN - REDUKTION DER GASPRODUKTION VON ANAEROBEM FAULSCHLAMM |
| C.35 | SEDIMENT-WASSER-TOXIZITÄTSSTUDIE MIT DOTIERTEM SEDIMENT AN LUMBRICULUS |
| C.36 | REPRODUKTIONSTEST MIT RAUBMILBEN (HYPOASPIS (GEOLAELAPS) ACULEIFER) IN BODENPROBEN |
| C.37 | 21-TAGE FISCH-SCREENING-ASSAY: EIN KURZZEITTEST ZUR BESTIMMUNG DER ÖSTROGENEN UND ANDROGENEN AKTIVITÄT UND DER AROMATASEHEMMUNG |
| C.38 | DER AMPHIBIEN-METAMORPHOSE-ASSAY (AMA) |
| C.39 | COLLEMBOLEN-REPRODUKTIONSTESTS IN BÖDEN |
| C.40 | LEBENSZYKLUS-TOXIZITÄTSTESTS BEI CHIRONOMIDEN IN SEDIMENT-WASSER-SYSTEMEN MIT DOTIERTEM SEDIMENT |
| C.41 | FISH SEXUAL DEVELOPMENT TEST (TEST ZUR GESCHLECHTSENTWICKLUNG BEI FISCHEN) |
| C.42 | BIOLOGISCHE ABBAUBARKEIT IN MEERWASSER |
| C.43 | ANAEROBE BIOLOGISCHE ABBAUBARKEIT ORGANISCHER STOFFE IN FAULSCHLAMM: BESTIMMUNG DURCH MESSUNG DER GASPRODUKTION |
| C.44 | VERSICKERUNG IN BODENSÄULEN |
| C.45 | ABSCHÄTZUNG DER EMISSIONEN VON MIT HOLZSCHUTZMITTELN BEHANDELTEM HOLZ IN DIE UMWELT: LABORMETHODE FÜR UNBESCHICHTETE HOLZPRODUKTE, DIE MIT SÜSSWASSER ODER MIT MEERWASSER IN BERÜHRUNG KOMMEN |
| C.46 | BIOAKKUMULATION IN SEDIMENTBEWOHNENDEN BENTHISCHEN OLIGOCHAETEN |
| C.47 | TOXIZITÄTSPRÜFUNG AN FISCHEN IM FRÜHEN ENTWICKLUNGSSTADIUM |
 | ENDE | |